 Anna Karin Hedström
Anna Karin Hedström- Department of Clinical Neuroscience, Karolinska Institute, Stockholm, Sweden
Compelling evidence indicates that Epstein Barr virus (EBV) infection is a prerequisite for multiple sclerosis (MS). The disease may arise from a complex interplay between latent EBV infection, genetic predisposition, and various environmental and lifestyle factors that negatively affect immune control of the infection. Evidence of gene-environment interactions and epigenetic modifications triggered by environmental factors in genetically susceptible individuals supports this view. This review gives a short introduction to EBV and host immunity and discusses evidence indicating EBV as a prerequisite for MS. The role of genetic and environmental risk factors, and their interactions, in MS pathogenesis is reviewed and put in the context of EBV infection. Finally, possible preventive measures are discussed based on the findings presented.
Introduction
Multiple sclerosis (MS) is an inflammatory and neurodegenerative disease of the central nervous system (CNS), with both genetic and environmental factors contributing to its development. While significant progress has been made in identifying genetic risk factors for MS, these genetic variants explain only a proportion of the heritability, which is estimated to be around 50% (1). This missing heritability phenomenon may partly be explained by the influence of multiple genetic variants on the risk of the disease, each with a small or modest effect, making it challenging to identify them. Epigenetic modifications, which can influence gene expression without altering the DNA sequence, may also play a role (2, 3). Furthermore, environmental factors implicated in the disease may interact with genetic and other environmental exposures or lifestyle habits to increase the risk of the disease (4). Thus, the complex interplay between multiple genetic variants, epigenetic modifications, and environmental factors may contribute to explain the missing heritability in MS. Compelling evidence suggests that infection with Epstein-Barr virus (EBV), a gamma-herpes virus that infects more than 90% of the population, is a prerequisite for developing MS. Several aspects of EBV infection, such as high EBV-specific antibodies and infectious mononucleosis (IM), have consistently been associated with increased MS risk (5, 6). Nearly all patients with MS are EBV-seropositive, whereas the risk of the disease in seronegative individuals is extremely low (7). In a seronegative group of young adults in the US military, the risk of MS increased by 32-fold following infection with EBV, strongly indicating a causal link (8). However, since most EBV seropositive individuals will never develop MS, inadequate control of the virus may be another prerequisite for the disease to occur. Several environmental and lifestyle factors have been implicated in MS such as vitamin D, smoking, and obesity (4). According to the sufficient cause model by Rothman, a disease occurs when multiple risk factors act together and reach a threshold level of causality, which then trigger the disease process (9). The model is a useful framework for studying complex diseases as it emphasizes the importance of multiple risk factors interacting to cause disease and can help to identify high-risk populations and guide the development of targeted interventions and preventive strategies. This review gives a short introduction to EBV and host immunity and discusses evidence indicating that EBV is a prerequisite for MS. The role of genetic and environmental risk factors, and their interactions, in the development of MS are reviewed and put in the context of EBV infection. Finally, possible preventive measures are discussed based on the findings presented.
Epstein-Barr virus – background
EBV lytically infects epithelial cells and B lymphocytes. The virus has a double-stranded DNA genome enclosed by an icosahedral capsid, a tegument layer, and an outer lipid envelope containing the viral attachment and entry proteins. Infection of B lymphocytes involves the attachment of the viral envelope glycoprotein gp350/220 to the complement receptor CD21 on B lymphocytes, followed by the interaction of viral gp42 with MCH class II molecules (10). This interaction leads to fusion of the viral envelope with the cellular membrane, allowing the viral capsid to enter the cell. Once inside the cell, the nucleocapsid is transported to the nucleus where viral DNA is released and begins to replicate. Following primary infection, EBV persists latently throughout life, mainly in memory B lymphocytes (11). During latency, the virus enters a state of limited gene expression which enables it to evade host immune surveillance (12). A balance is typically established between viral persistence and immune control, although reactivation of the virus occurs periodically, usually without symptoms (13).
Immune response to EBV
Primary EBV infection is usually asymptomatic in childhood. Subsets of natural killer (NK) cells become activated within one or two days as a first-line defense, playing a crucial role in limiting viral spread. EBV-specific CD8+ T cells are responsible for eliminating proliferating and lytically infected B cells while EBV-specific antibodies neutralize viral infectivity by binding to free virus (14, 15). During latency, the number of infected memory B cells varies between individuals but remains rather stable throughout life, reflecting a dynamic equilibrium between viral persistence and EBV-specific immune responses. Healthy EBV carriers typically show IgG-antibody titers against the viral capsid (VCA) and Epstein-Barr virus nuclear antigens (EBNAs) (16).
When primary EBV infection is delayed beyond childhood, it often manifests as acute IM, indicative of a reduced capacity of the CD8+ T cell response to rapidly control the infection (17). The symptoms of IM are caused by an exaggerated cellular immune response to EBV, which may be influenced by host genetics (18, 19) and an age-related impaired CD8+ T cell function (20–22). Furthermore, early NK cell-mediated control over primary EBV infection decreases during the first decade of life which may contribute to the development of an exaggerated CD8+ T cell response when the infection is acquired at an older age (23, 24).
In IM, the number of latently infected memory B cells can rise to half of the peripheral memory B cell compartment. Latent infection is only achieved after a massive expansion of CD8+ T cells that target EBV proteins from the early stages of the lytic cycle. The CD8+ T cell expansion coincides with the onset of symptoms. In addition, cytotoxic CD4+ T cell populations are also amplified and contribute to the symptoms of acute IM by massive cytokine production (17). Although the total number of CD8+ T cells is not significantly raised in children with asymptomatic primary EBV infection, EBV-specific CD8+ T cells are highly activated and can control the infection without causing excessive expansion (14, 15), confirming that cytotoxic lymphocytes are the cornerstone of EBV-specific immune control, and that IM is an immunopathologic condition.
Epstein-Barr virus as a necessary factor in MS
EBV has a profound influence on the immune system, and recent studies have suggested a causal role of EBV in MS. There is consistent evidence that EBV infection precedes MS onset. EBV-specific antibodies are present in nearly all patients with MS whereas the risk of MS in seronegative individuals is extremely small (7). In a seronegative group of young adults in the US military, the risk of MS increased 32-fold after infection with EBV. The study also demonstrated that serum levels of neurofilament light chain, a biomarker that reflects neuroaxonal damage, increased shortly after seroconversion (8). These findings indicate that EBV is a necessary, but not sufficient, factor in MS development.
Presence of EBV in latency has been found to be considerably more prevalent in MS brain tissue than in non-MS brain, and lytic EBV infection has been detected in chronic MS lesions (25, 26) and in meningeal B cell follicles (27, 28). Apart from infected B lymphocytes, EBV has also been detected in astrocytes and microglia in some MS cases with widespread presence of EBV in brain tissue (26). However, other studies have been unable to detect EBV in MS brain tissue and therefore, the issue of the presence of EBV in MS brain tissue remains unresolved (29–31).
Generally, patients with MS have higher levels of EBV-specific antibodies than control populations (32), which suggests that the control of latent EBV infection may be compromised in these individuals. Several studies have observed a significant increase in EBNA-1 antibody titers many years prior to the clinical onset of MS (33–35). Moreover, the risk of MS increases with increasing EBNA-1 antibody levels (36). Under normal circumstances, the immune system effectively controls EBV infection, leading to a state of viral latency where the virus remains dormant in B cells. However, an increase in the levels of EBNA-1 antibodies implies that the overall immune response may be insufficient to effectively control the virus, resulting in persistent or reactivated infection. The presence of disease-associated oligoclonal Ig-bands in the cerebrospinal fluid (CSF) is a well-established characteristic of MS, indicative of local antibody production within the CNS. In a subset of patients, EBV-specific antibody synthesis occurs within the CNS. However, it is generally observed that the frequency of EBV-specific antibodies in the CSF is lower compared to other viral infections in MS (37), which may be attributed to differences in localization of antibody production, viral tropism, or temporal dynamics of antibody response. EBV-specific CD8+ T cells present in the CNS of MS patients provide further evidence that EBV-infected cells are present in the brain (38, 39). Furthermore, a higher frequency of CD4+ T cells primarily targeting EBNA has been observed in MS patients (40, 41), where a subset of the T cells cross-reacted with CNS-antigens (41). In this context, it is conceivable that the elevated levels of EBNA-1 antibodies reflect a breakdown in immune tolerance, where the immune system fails to distinguish between viral and CNS-antigens. The presence of high levels of EBNA-1 antibodies in MS patients could thus be attributed to various factors, including the complex interplay between EBV infection, immune dysregulation, and potential cross-reactivity with CNS antigens. Further research is needed to elucidate the precise mechanisms underlying these observations and their implications in the pathogenesis of MS.
Several hypotheses have been put forward to explain the role of EBV in MS (42, 43), but the pathogenic mechanisms remain uncertain. During primary EBV infection, there is a high frequency of EBV-infected B lymphocytes. An impaired immune response to the virus may increase the viral load whereby circulating EBV-infected B cells may enter the CNS and initiate proliferation. Failure to eradicate the infection could lead to viral antigens stimulating an anti-EBV immune response with maintenance of local inflammation, intrathecal immunoglobulin synthesis and oligoclonal bands. If EBV-specific CD8+ and CD4+ T cells migrate into the CNS and become reactivated by EBV-infected B lymphocytes, the resulting immune response would lead to bystander damage and the release of CNS-antigens, which could subsequently result in a CNS-directed immune response. Alternatively, EBV-transformed B lymphocytes could activate EBV-specific T cells cross-reactive with CNS antigens. However, the exact mechanisms by which EBV increases the risk of MS are yet to be determined.
Infectious mononucleosis
The risk of MS is increased more than two-fold following IM (5, 6), suggestive of an inadequate cytotoxic CD8+ T cell control of the infection. Several studies have observed impaired EBV-specific CD8 T cell responses in patients with MS (44, 45). Furthermore, CD8+ T cells specific for lytic EBV antigens increase during the active phase of MS whereas CD8+ T cells against latent antigens dominate during inactive disease (46). During primary EBV infection, there is a high frequency of EBV-infected B lymphocytes. An impaired immune response to the virus may, apart from increasing the risk of IM, increase the viral load whereby circulating EBV-infected B cells may enter the CNS and initiate proliferation.
Past IM does not correlate with EBNA-1 antibody levels (36, 47) and both aspects of EBV infection appear to be separate risk factors for MS. A synergistic effect between past IM and high EBNA-1 antibody levels has been observed, which indicates that they are involved in at least one common biological pathway to disease (36). Both past IM and high EBNA-1 antibody levels may thus reflect a deficient control of the EBV infection, for which cell-mediated immune responses play a pivotal role. Since most seropositive individuals will never develop MS, inadequate control of the virus may thus be another prerequisite for developing the disease.
Risk factors for MS onset in the context of EBV
The high prevalence of EBV seropositivity in the general population and the low incidence of MS indicates that a potential pathogenic effect of the virus is influenced by other risk factors for the disease. According to the sufficient cause model by Rothman (9), the development of a disease is the result of a sufficient cause which is a set of conditions or events that act together to produce the disease. A necessary cause is a factor that must be present for the disease to occur, whereas component causes are other factors that contribute to the development of disease but are not sufficient on their own. All component causes in a sufficient cause are required to be either present or have taken place before disease onset. In the case of MS, EBV appears to be a necessary component cause. In every sufficient cause that leads to MS, EBV would thus be required. All component causes in the same sufficient cause act in concert to produce the disease. If EBV is a necessary component cause for MS to occur, it would thus be expected to interact with every other risk factor for the disease (sufficient cause interaction). The sufficient cause model recognizes that multiple pathways can lead to the same disease, and that different combinations of factors can lead to the same outcome (48). The model can be used to guide research and intervention efforts, as it emphasizes the importance of identifying and addressing all factors that contribute to the development of disease.
Interaction between two exposures is present when the effect of one exposure on the outcome depends on the other exposure. Statistical interaction evaluates whether the combined effect of the exposures deviates from either additivity or multiplicativity, depending on the scale of the model (48). Although it is not possible to draw conclusions regarding biologic mechanisms through statistical analysis of observational data, interaction on the additive scale can be used to test for sufficient cause interactions in epidemiologic research when the outcome is rare. Additive interaction (departure from additivity) means that the combined effect of two exposures differs from the sum of the risks conferred by each exposure and can be assessed by using different measures such as the synergy index, the attributable proportion due to interaction, and the relative excess risk due to interaction. The additive scale is preferable from a public health perspective since it implies that some subgroups would obtain a greater absolute risk reduction from preventive measures. Studies investigating additive interactions between risk factors in MS etiology are thus important for the development of effective preventive measures but may also shed light on disease pathogenesis. In the following, risk factors for MS and their interactions will be discussed and put in the context of EBV infection.
Viral infections apart from EBV
In addition to EBV, several other viruses have been implicated in MS, such as human endogenous retroviruses (HERVs), human herpesvirus 6 (HHV-6) and cytomegalovirus (CMV), which all have the capacity of establishing lifelong latent infections (49). The underlying mechanisms are poorly understood. Exposure to prior antigens could result in heterologous immunity in which the immune response to a prior infection provides some degree of protection or enhances response against a different but related infection (50). The specific mechanisms by which prior infections could affect the immune response to EBV are likely complex and multifactorial and may vary depending on factors such as the timing and severity of infections as well as on the host immune status.
HHV-6, associated with increased risk of MS (49), is capable of infecting glia and neurons and is one of the most common viruses detected in the adult brain (51, 52). Regarding risk of MS, an additive interaction has been observed between high antibody levels against EBV and HHV-6A (53). Some studies have reported that HHV-6 can infect and activate EBV-infected B cells, leading to increased viral replication and persistence (54–56). Compared with controls, increased expression of both HHV-6 and HERVs have been found in MS plaques (57–59), and cumulative roles for EBV, HHV6, and HERVs in MS pathogenesis has been suggested (60).
In seroepidemiological studies, CMV has been inversely associated with MS (61, 62). CMV infection may result in changes in the CD8+ T cell repertoire with an accumulation of oligoclonal differentiated T cells with limited proliferative capacity (63), which could potentially limit the immune response to EBV. One study found a reduced proportion of memory B cells, with reduced inflammatory profile, in MS patients with previous CMV infection, compared to those without CMV infection (64). However, potential mechanisms by which viral infections contribute to MS are hypothetical and more research is necessary.
Genetics
The genetic contribution to MS is complex, with hundreds of common genetic variants, identified by genome-wide association studies, contributing to MS heritability (1). The importance of human leukocyte antigen (HLA) class I and II alleles in MS is well established. The strongest genetic factor is the DRB1*15:01 allele in the class II region which increases the risk of MS approximately 3-fold, whereas the HLA class I allele A*02:01 confers a protective effect (65). The class II molecules present peptide ligands to CD4+ lymphocytes, which are thought to play a crucial role in the pathogenesis of MS. In addition to HLA genes, more than 200 non-HLA risk alleles have been identified as implicated in disease susceptibility (1). Most of these are associated with innate or adaptive immune functions or brain-resident immune cells. While individual genetic variants have only small or modest effects on MS risk, recent studies have shown that combining multiple genetic variants into a polygenic risk score can improve the accuracy of predicting the risk of disease (66).
Considering the role of HLA class II for the infection of B lymphocytes, variations in HLA may account for the differences in EBV susceptibility (67). EBV serostatus has been linked to the ability of EBV to bind to B lymphocytes. Since CD21 on B cells is highly conserved, the binding capacity of EBV to B lymphocytes is likely a result of variation in HLA.
Once established, both class I and II-restricted antigen presentation are involved in the control of the EBV infection. Genetic differences in the HLA class I locus have been associated with both the development and outcome of primary EBV infection as well as with viral persistence (19, 68). The frequency of A*02:01 has been shown to be lower among MS patients with past IM compared to both cases without IM history and healthy controls (36). The MS protective allele A*02:01 can present several EBV-derived peptides and mediates an effective immune response against EBV antigens (69, 70), although both B and T cells carrying this allele display a reduced response to type I IFN stimulation (71).
Carriers of DRB1*15:01 have higher EBNA-1 antibody levels compared to DRB1*15:01 negative individuals, both among MS patients and healthy controls (36, 68), indicating a defective immune response to EBV. Non-HLA loci influencing both EBNA-1 antibody levels and risk of MS have also been identified (72, 73). The class II molecules, involved in antigen-presentation to CD4+T cells by antigen presenting cells, are essential in shaping the TCR repertoire. The DRB1*15:01 allele has been associated with a narrower TCR repertoire with T cells of high affinity for specific peptides (74). Epigenetic mechanisms may also be involved, as it has been suggested that DNA methylation mediates the effect of DRB1 expression and could lead to a changed TCR repertoire more prone to CNS-reactive responses (75). In a humanized mouse model, HLA-DRB1*15-restricted CD4+ T clones were less efficient in recognizing EBV-transformed B cell lines, resulting in increased expansion of CD8+ T cells and higher viral load. The CD4+ T cell clones also demonstrated cross-reactivity with CNS-antigens (76). Furthermore, it has been suggested that the DRB1*15:01 haplotype may promote the activation of brain homing autoreactive CD4+ T cells by memory B cells (77). In MS, EBV-specific CD4+ T cells among DRB1*15:01 carriers also show higher reactivity against several CNS-antigens (78).
The findings presented may contribute to explain the interaction between HLA alleles, encoding molecules regulating T cell adaptive immunity, and measures of EBV infection, in the development of MS (79–85). Both past IM and high EBNA1 antibody levels act synergistically with the main MS risk HLA genes, presence of DRB1*15:01 and absence of A*02:01 (85). Carriers of DRB1*15:01 without the protective effect of A*02:01 and with high EBNA-1 antibody levels following IM run an almost 30-fold increased risk of MS, compared to those who have none of these risk factors. Table 1 provides a compilation of studies examining the additive interaction between EBV and HLA-DRB1*15:01 (and absence of HLA-A*02:01) in relation to the development of MS. Studies that investigated the individual effects of each risk factor as well as their combined effects were included. The presented results are directly extracted from the original works referenced.
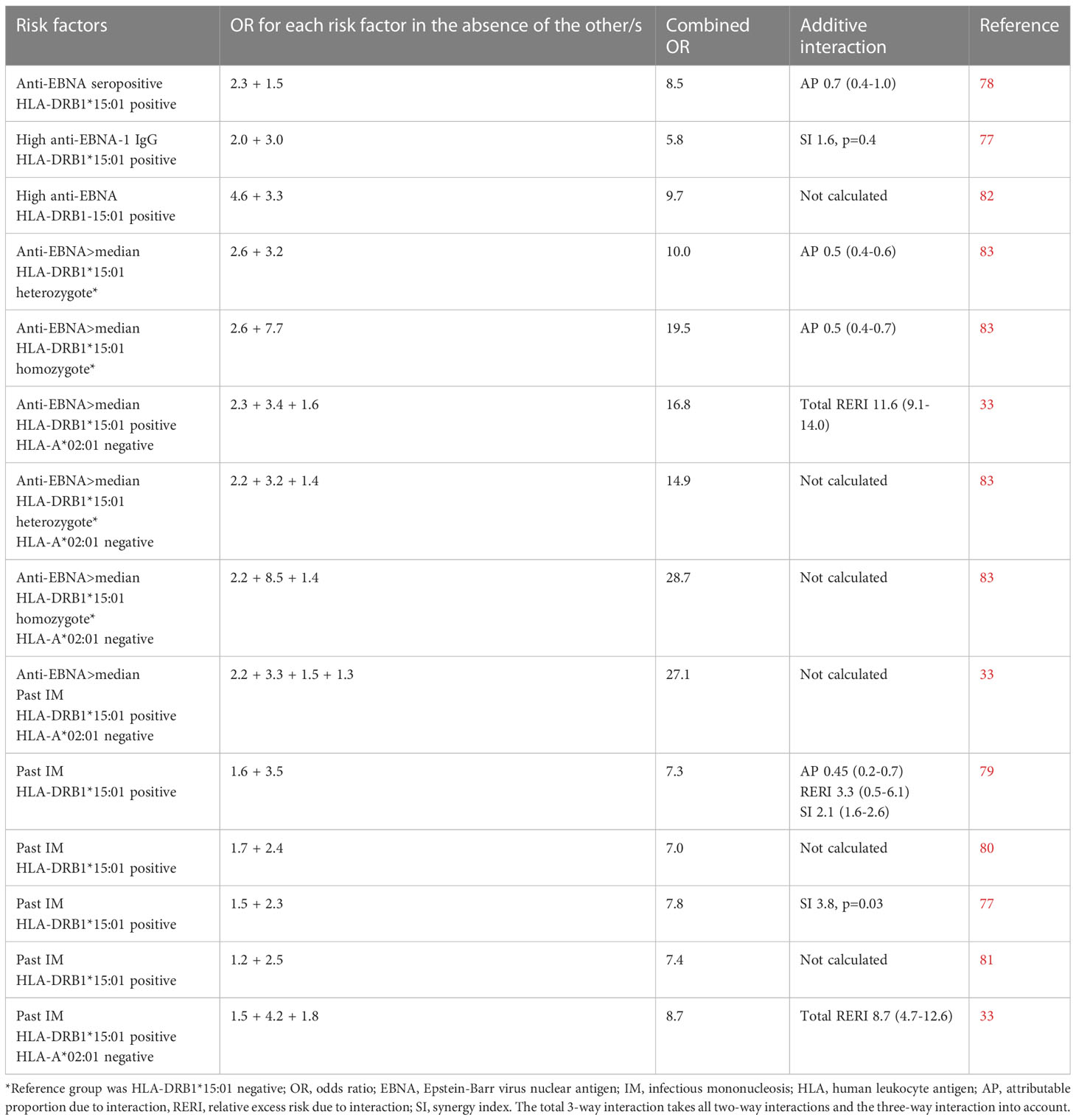
Table 1 Compilation of studies examining the additive interaction between Epstein-Barr virus and HLA-DRB1*15:01 (and absence of HLA-A*02:01) in relation to the development of MS.
Sun exposure and vitamin D
The increasing MS occurrence with distance from the equator (86, 87) has been attributed to lower sun exposure and consequently, lower endogenous production of vitamin D (82, 83). Observational studies have found higher risk of MS with lower sun exposure and with lower vitamin D levels. Although distinguishing between the effects of sun exposure and vitamin D may be challenging in observational studies, independent benefits have been observed when both factors have been considered simultaneously (88, 89). Sun exposure may influence MS risk through both vitamin D and non-vitamin D pathways, both of which impact innate and adaptive immune functions.
Vitamin D is converted enzymatically in the liver to its major circulating form (25-hydroxyvitamin D) and then in the kidney to its active form (1,25-dihydroxyvitamin D). Active vitamin D acts as a transcription factor by binding to the vitamin D receptor (VDR) and modulating the expression of over 200 genes. Vitamin D influences the immune system in multiple ways, including enhancing innate immunity, modulating adaptive immunity, and regulating cytokine production. It increases the differentiation of hematopoietic stem cells into NK cells and may enhance their function (90, 91). It also increases the production of antimicrobial peptides involved in directly killing virus (92). Vitamin D decreases the proliferation and differentiation of T cells and their cytokine production, while promoting the development and function of regulatory T cells. Both in vitro and in vivo studies show that vitamin D results in increased production of the anti-inflammatory cytokine IL-10 (93). The effects of vitamin D on B lymphocyte function are less certain. Although in vitro studies have provided some evidence that vitamin D inhibits the T cell-stimulating capacity of B cells and enhances the activity of regulatory B cells, it is not supported by in vivo data (94).
Exposure to UVR also has effects on the immune system that are independent of vitamin D, including induction of interferons that inhibit viral replication and enhancement of innate immune responses (95). Sun exposure also results in suppression of cell-mediated immunity, independent of vitamin D. Exposure to sun induces regulatory T cells through antigen presentation of sun-damaged Langerhans cells in the lymph nodes, leading to antigen-specific immune tolerance. Exposure to sun also inhibits effector and memory T cell activation, and induces systemic changes in immune mediators, leading to suppression of the adaptive immune response (96, 97). A suppressive effect of UVR through pathways independent of vitamin D have also been confirmed in several experimental studies (98, 99).
In patients with MS, an inverse relationship has been observed between EBNA-1 antibody levels and serum vitamin D levels (100, 101), suggesting that vitamin D deficiency may impair the T cell-mediated response to EBV, leading to viral reactivation and augmented antibody production. Low sun exposure/vitamin D deficiency has also been found to act synergistically with high levels of EBNA-1 antibodies regarding risk of MS (102). There is some evidence that vitamin D levels could affect the interaction between EBV and MS predisposing genes. EBNA2, a transcriptional activator with importance for B cell transformation, binds to MS susceptibility loci in the host genome of infected B cells and may influence their expression (103). An overlap between EBNA2 and VDR binding sites has been observed and it has been suggested that EBNA2 and VDR compete for binding to these susceptibility loci of which the majority is related to immune responses (104). Considering that EBV and vitamin D may have antagonistic effects on B cell function, adequate vitamin D levels could thus have a protective effect against MS by blocking EBNA2 genomic occupancy.
The influence of vitamin D on MS risk has been suggested to be important during a time window. While low vitamin D levels during the first trimester resulted in increased risk of MS in the offspring (105), studies investigating the association between neonatal vitamin D status and risk of subsequently developing MS have been conflicting (106, 107). However, several studies have consistently found decreased risk of MS with increasing vitamin D levels in childhood and young adulthood. In a prospective, nested case-control study among US military personnel, the inverse association between vitamin D levels and risk of MS was particularly strong for vitamin D measured before age 20 years (108).
The latitude gradient in MS occurrence and the association between increased risk of MS and both IM history and vitamin D deficiency have led to the hypothesis that the season of IM could influence MS risk (109). If vitamin D dependent responses against primary EBV infection affects the risk of MS, it would be expected that IM during winter, when vitamin D levels are at their lowest, confers a stronger risk of subsequently developing MS. However, the increased risk of MS following IM does not seem to be dependent on the season of infection (110, 111). One explanation could be that vitamin D deficiency has more long-term effects on the immune system, possibly by reducing the expression of key genes involved in T cell development and function.
Lung-irritating agents
There is strong epidemiological evidence for an association between smoking and other lung-irritating agents and risk of MS. Smoking increases the risk of MS in a dose-response manner (112, 113). A similar dose-response correlation has been observed between increased risk of MS and exposure to passive smoking, organic solvents, and air pollution, respectively (114–117). These lung-irritating agents also display a considerable interaction with MS-associated risk genes (118–121). Since it seems unlikely that MS risk genes would regulate smoking behavior or other exposures to lung-irritating agents, it is an argument for a causal role of pulmonary irritation in MS development.
Tobacco smoking and its oxidative agents results in airway inflammation and increased levels of inflammatory markers (122–124). Alveolar macrophages in the lungs increase in numbers with concurrent impairment of their function, contributing to the inflammatory process. The development and effector function of both innate and adaptive immune cells are affected by smoking, including NK cells, CD8+ and CD4+ T cells as well as memory B and T lymphocytes. The majority of studies in humans have found that smoking increases the number of CD8+ T cells and enhances their functional response while the frequency of CD4+ T cells are reduced (122–124). Smoking also induces changes in the distribution and function of memory B cells, with a higher prevalence of memory B cells in peripheral blood and in the lung, whereas regulatory B cell numbers decrease (125).
Smoking may act synergistically with high levels of EBNA-1 antibodies to increase the risk of MS ( (126–128), Table 2). Apart from inducing proinflammatory responses, smoking results in a relative immune deficiency (123). Smokers have higher EBNA-1 antibody levels than non-smokers both among patients with MS and healthy controls (128–130), and smoking has been associated with frequent reactivation of EBV whereby EBV antibodies against viral antigens become elevated. There is a dose-response relationship between smoking and both oral EBV load, anti-EBV IgG levels, and the frequency of EBV reactivation (131, 132). This is also supported by experimental evidence showing replication of EBV with expression of its lytic-phase genes following exposure to cigarette smoke (133). Smoking-associated DNA methylation and changes in gene expression in immune cell types have been identified and may contribute to EBV reactivation (134).
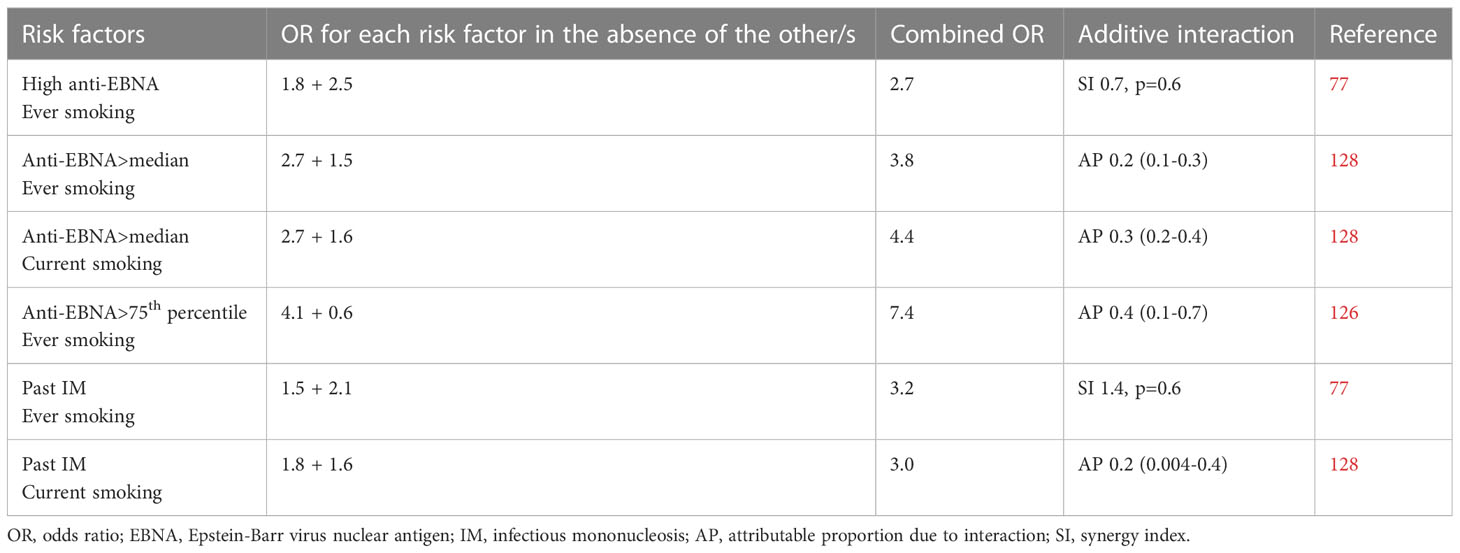
Table 2 Compilation of studies examining the additive interactions between Epstein-Barr virus and smoking in relation to the development of MS.
Smoking-associated pulmonary inflammation could contribute to the activation of tissue-resident immune cells within the lung including potentially autoreactive effector and memory cells. In experimental studies, CNS-directed cells in the lung become activated by local stimulation, whereby they assume a migratory pattern and traffic to the CNS (135). Smoking also affects the immune system barrier function and may promote migration of autoreactive immune cells into the CNS.
While some aspects of childhood or adolescence seem to be critical regarding the impact of several environmental factors on MS risk, smoking increases the risk of MS regardless of age at exposure (113). Following smoking cessation, the detrimental effect abates within a decade, even in heavy smokers with a long duration of smoking (113).
Obesity
Several observational studies have observed that obesity in adolescence and young adulthood confers an approximately doubled risk of MS, compared to normal weight individuals (136, 137). The association is supported by Mendelian randomization studies showing associations between genetic predisposition to obesity and risk of MS (138–140).
Obesity induces a chronic low-grade systemic inflammation due to increased numbers of adipose tissue macrophages that undergo a phenotypic switch with increased production and secretion of inflammatory mediators (141). Several aspects of the adaptive system are affected, including increased proliferation and activation of T cells and reduced numbers of regulatory T cells, which exacerbate adipose tissue inflammation and could promote the onset of autoimmune responses (142, 143). The inflammatory pathways activated by obesity also upregulate pro-inflammatory cytokines and cause a decrease in the blood-brain barrier integrity, allowing for easier immune penetration (144). Apart from increased proinflammatory responses, obesity results in a relative state of immunodeficiency which increases susceptibility to infections (145). Obesity has been associated with a reduced function of NK cells (146), which are critical during primary EBV infection. Obesity could also have impact on the immune response to EBV infection by contributing to dysbiosis (147).
Obesity at young age is associated with subsequent risk of MS (148, 149) while obesity at the time of MS diagnosis does not differ between cases and healthy controls (148). It has also been suggested that obesity during adolescence rather than during childhood, is of importance for adulthood-onset MS, suggesting that obesity at the time of primary EBV infection may be of importance (150). In two studies, adolescent BMI exceeding 25 kg/m2 interacted with both past IM and high EBNA-1 antibody levels regarding risk of MS (Table 3). Past IM in obese individuals carrying the HLA-DRB1*15:01 haplotype rendered a 22-fold increased risk of MS, compared with normal weight DRB1*15:01 negative individuals without history of IM. The interaction between BMI and EBNA-1 status became stronger with increasing EBNA-1 antibody levels (151, 152). The obesity-induced immunodeficiency may thus alter the immune response to EBV and may, particularly in DRB1*15:01 carriers, increase the risk of an autoreactive response directed at CNS-antigens.
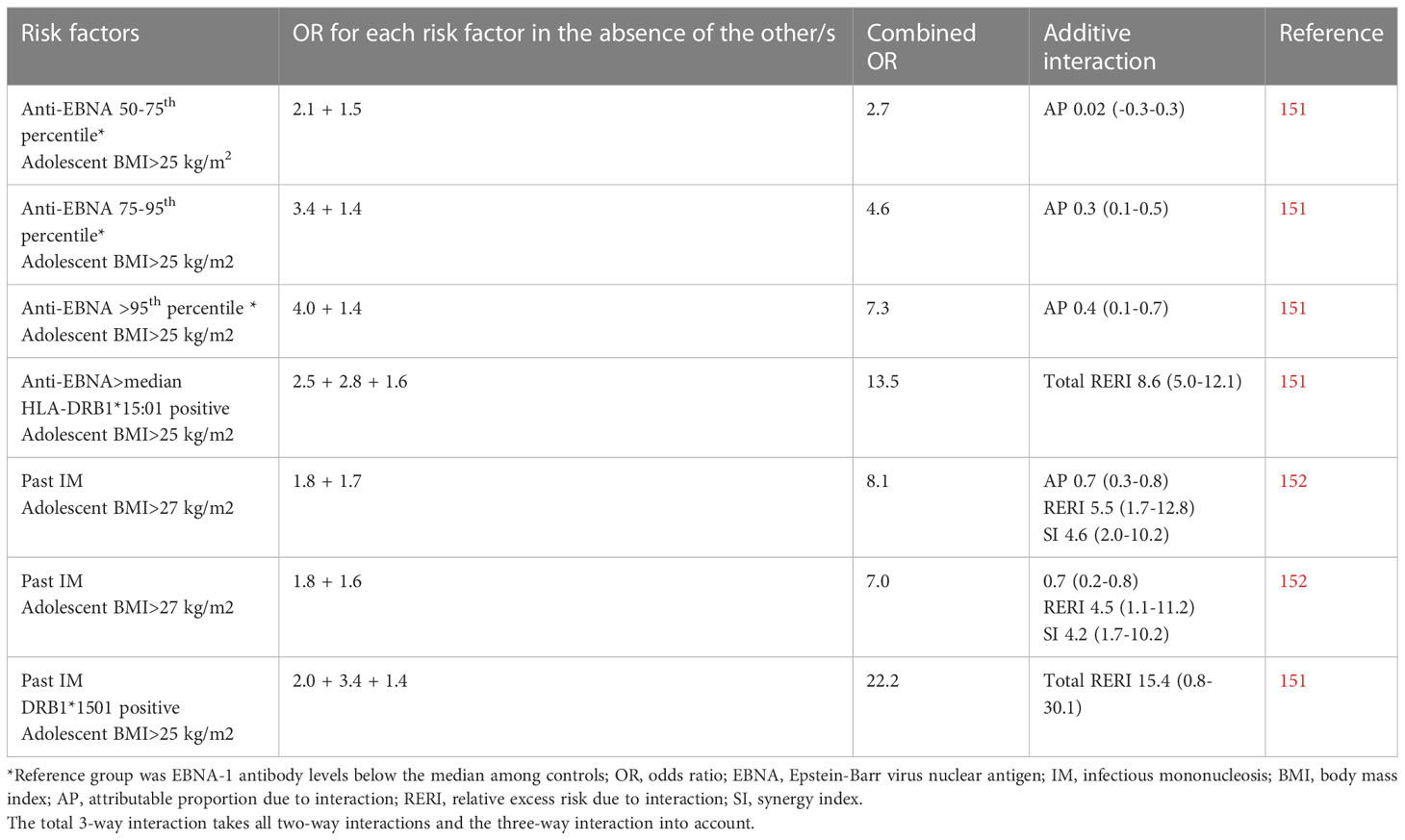
Table 3 Compilation of studies examining the additive interactions between Epstein-Barr virus and overweight/obesity in relation to the development of MS.
Other potential risk factors
The gut microbiota plays a key role in regulating immune functions and has attracted major attention in MS (153). Intestinal microbiota has been shown to promote the triggering of CNS-reactive CD4+ T cells (154) and modulates astrocyte activity and neuroinflammation (155, 156). The microbiota composition in patients with MS has been reported to be distinct compared to healthy controls (157–159) and the transplantation of MS intestinal microbiota has been shown to induce or exacerbate experimental autoimmune encephalomyelitis (158, 159). Dysbiosis, which refers to an imbalance in the gut microbiota, has been associated with changes in the production and metabolism of fatty acids, which have immunomodulatory effects and modulate the differentiation of both innate and adaptive immune cells and their function (160). The level and composition of circulatory and tissue-resident fatty acids have been implicated in MS (160). Although the exact mechanisms by which the microbiome may affect the risk of MS remain uncertain, these findings highlight the importance of further research.
Polyunsaturated fatty acids (PUFAs) possess potent immunomodulatory effects (161). Several observational studies investigating the influence of PUFA dietary sources on the risk of MS have reported that a higher intake of fatty fish or supplementation with cod liver oil is associated with a reduced risk of developing the disease (162–165). However, the findings should be interpreted with caution due to potential sources of bias.
Although studies have not been consistent (165–167), sibship structure has been suggested to decrease the risk of MS by preventing delayed primary EBV infection (167).
Shift work, poor sleep, and psychological stress, all of which can negatively affect the immune response to infection, have also been associated with increased risk of MS (168–170).
Preventive measures
Evidence strongly suggests that EBV is a prerequisite for MS and the most effective way to prevent the disease would be to prevent the primary EBV infection through vaccination. Various immunogens, including both lytic and latent proteins, have been explored for inclusion in prophylactic EBV vaccines (171), and several vaccines are currently undergoing clinical trials. Even if the primary infection is not prevented, a vaccine that prevents the excessive immune responses against primary EBV infection may be effective in reducing the incidence of MS. However, given that the onset of MS may occur decades after primary EBV infection, long-term follow-up studies will be necessary to evaluate the effectiveness of a vaccine in preventing MS, and it is important to identify alternative strategies to reduce the overall risk of the disease.
Several modifiable factors have consistently been associated with increased risk of MS that could be targeted through preventive measures. Based on the presented findings, one approach could be balanced information regarding the risks and benefits of sun exposure. The ultraviolet index can be useful for guiding sun exposure behavior in accordance with WHO guidelines to minimize the risk of skin cancer (172), but sensible sun exposure is essential for vitamin D synthesis and for capturing other vitamin D-independent benefits (95). Reducing the prevalence of smoking requires a multi-faceted approach that involves a combination of strategies, such as governmental regulations, prevention programs in schools, cessation programs for smokers, and increased access to smoking cessation resources. Parents should be informed regarding the importance of protecting their children from secondhand smoke. Childhood obesity is a serious and growing problem in developed countries. Preventive measures include encouraging healthy eating habits, promoting regular physical activity, limiting screen time, and encouraging healthy sleep habits. Healthcare professionals could be consulted when a child is at risk of becoming obese for guidance on appropriate nutrition and physical activity strategies. The health benefits of Mediterranean-style diet for obesity-related comorbidities are well-established (173). The diet is rich in fiber, vitamins, minerals, and antioxidants, and is based on consumption of high amounts of vegetables, fruits, nuts, fish and other seafood, and the use of olive oil (174). These guidelines align with those for general health benefits and may have the potential to lower the risk of MS.
Conclusions
MS may arise from a complex interplay between latent EBV infection, genetic predisposition, and various environmental and lifestyle factors that negatively affect immune control of the infection. Increasing evidence from various research disciplines emphasizes the crucial involvement of EBV in the development of MS. EBV infection as a prerequisite for MS is further supported by evidence indicating that other established risk factors for MS act synergistically with EBV in the development of the disease. However, while numerous genetic variants and environmental factors have been identified that predispose to MS, their biological significance remains uncertain, and their effects are often small or modest. Certain environmental factors could have multiple biological effects that are significant in the pathogenesis, whereas other exposures may impact a small set of shared pathways, leading to comparable biological effects. Despite recent progress, gaining a comprehensive understanding of the various risk factors for MS onset remains a significant challenge. A multidisciplinary approach could provide further insight into the mechanisms of disease pathogenesis and provide important information for disease prevention that could be implemented.
Author contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Patsopoulos NA, Baranzini SE, Santaniello A, Shoostari P, Cotsapas C, Wong G, et al. Multiple sclerosis genomic map implicates perhipheral immune cells and microglia in susceptibility. Science (2019) 365:eaav7188. doi: 10.1126/science.aav7188
2. Zhou Y, Simpson S Jr, Holloway AF, Charlesworth J, van der Mei I, Taylor BV. The potential role of epigenetic modifications in the heritability of multiple sclerosis. Mult Scler (2014) 20:135–40. doi: 10.1177/1352458514520911
3. Andlauer TFM, Buck D, Antony G, Bayas A, Bechmann L, Berthele A, et al. Novel multiple sclerosis susceptiblity loci implicated in epigenetic regulation. Sci Adv (2016) 2:e1501678. doi: 10.1126/sciadv.1501678
4. Zarghami A, Li Y, Claflin SB, van der Mei I, Taylor BV. Role of environmental factors in multiple sclerosis. Expert Rev Neurother (2021) 21:1389–408. doi: 10.1080/14737175.2021.1978843
5. Belbasis L, Bellou V, Evangelou E, Ioannidis JPA, Tzoulaki I. Environmental risk factors and multiple sclerosis: an umbrella review of systematic reviews and meta-analyses. Lancet Neurol (2015) 14:263–73. doi: 10.1016/S1474-4422(14)70267-4
6. Handel AE, Williamson AJ, Disanto G, Handunnetthi L, Giovannoni G, Ramagopalan SV. An updated meta-analysis of risk of multiple sclerosis following infectious mononucleosis. PloS One (2010) 5:e12496. doi: 10.1371/journal.pone.0012496
7. Pakpoor J, Disanto G, Gerber JE, Dobson R, Meier UC, Giovannoni G, et al. The risk of developing multiple sclerosis in individuals seronegative for Epstein-Barr virus: a meta-analysis. Mult Scler (2013) 19:162–6. doi: 10.1177/1352458512449682
8. Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science (2022) 375:296–301. doi: 10.1126/science.abj8222
9. Rothman KJ, Greenland S, Lash TL. Modern epidemiology. 3rd edition. Philadelphia: Lippincott Wolliams & Wilkins (2008).
10. Shannon-Lowe C, Rowe M. Epstein Barr Virus entry; kissing and conjugation. Curr Opion Virol (2014) 4:78–84. doi: 10.1016/j.coviro.2013.12.001
11. Thorley-Lawson DA. EBV persistence – introducing the virus. Curr Top Microbiol Immunol (2015) 390:151–209. doi: 10.1007/978-3-319-22822-8_8
12. Kanda T. EBV-encoded latent genes. Adv Exp Med Biol (2018) 1045:377–94. doi: 10.1007/978-981-10-7230-7_17
13. McKenzie J, El-Guindy A. Epstein-Barr Virus lytic cycle reactivation. Curr Top Microbiol Immunol (2015) 391:237–61. doi: 10.1007/978-3-319-22834-1_8
14. Jayasooriya S, de Silva TI, Njie-jobe J, Sanyang C, Leese AM, Bell AI, et al. Early virological and immunological events in asymptomatic Epstein-Barr virus infection in African children. PloS Pathol (2015) 11:e100474. doi: 10.1371/journal.ppat.1004746
15. Abbott RJ, Pachnio A, Pedroza-Pacheco I, Leese AM, Begum J, Long HM, et al. Asymptomatic primary infection with Epstein–Barr virus: observations on young adult cases. J Virol (2017) 91:e00382. doi: 10.1128/JVI.00382-17
16. Niller HH, Bauer G. Epstein-Barr Virus: clinical diagnostics. Methods Mol Biol (2017) 1532:33–55. doi: 10.1007/978-1-4939-6655-4_2
17. Balfour HH, Dunmire SK, Hogqvuist KA. Infectious mononucleosis. Clin Transl Immunol (2015) 4:e33. doi: 10.1038/cti.2015.1
18. Rubicz R, Yolken R, Drigalenko E, Carless MA, Dyer TD, Bauman L, et al. A genome-wide integrative genomic study localizes genetic factors influencing antibodies against Epstein–Barr virus nuclear antigen 1 (EBNA-1). PloS Genet (2013) 9:e1003147. doi: 10.1371/journal.pgen.1003147
19. McAulay KA, Higgins CD, Macsween KF, Lake A, Jarrett RF, Robertson FL, et al. HLA class I polymorphisms are associated with development of infectious mononucleosis upon primary EBV infection. J Clin Invest (2007) 117:3042–8. doi: 10.1172/JCI32377
20. Fulop T, Larbi A, Pawelec G. Human T cell aging and the impact of persistent viral infections. Front Immunol (2013) 4:271. doi: 10.3389/fimmu.2013.00271
21. Goronzy JJ, Li G, Yang Z, Weyand CM. The janus head of T cell aging–autoimmunity and immunodeficiency. Front Immunol (2013) 4:131. doi: 10.3389/fimmu.2013.00131
22. Tserel L, Kolde R, Limbach M, Tretyakov K, Kasela S, Kisand K, et al. Age-related profiling of DNA methylaltion in CD8+ T cells reveals changes in immune response and transcriptional regular genes. Sci Rep (2015) 5:13107. doi: 10.1038/srep13107
23. Münz C. Role of human natural killer cells during Epstein-Barr virus infection. Crit Rev Immunol (2014) 34:501–7. doi: 10.1615/critrevimmunol.2014012312
24. Azzi T, Lünemann A, Murer A, Ueda S, Béziat V, Malmberg KJ, et al. Role for early-differentiated natural killer cells in infectious mononucleosis. Blood (2014) 124:2533–43. doi: 10.1182/blood-2014-01-553024
25. Moreno MA, Or-Geva N, Aftab BT, Khanna R, Croze E, Steinman L, et al. Molecular signature of Epstein-Barr virus infection in MS brain lesions. Neurol Neuroimmunol Neuroinflamm (2018) 5:e466. doi: 10.1212/NXI.0000000000000466
26. Hassani A, Corboy JR, Al-Salam S, Khan G. Epstein-Barr Virus is present in the brain of most cases of multiple sclerosis and may engage more than just b cells. PloS One (2018) 13:e0192109. doi: 10.1371/journal.pone.0192109
27. Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med (2007) 204:2899–912. doi: 10.1084/jem.20071030
28. Serafini B, Severa M, Columba-Cabezas S, Rosicarelli B, Veroni C, Chiappetta G, et al. Epstein-Barr Virus latent infection and BAFF expression in b cells in the multiple sclerosis brain: implications for viral persistence and intrathecal b-cell activation. J Neuropathol Exp Neurol (2010) 69:677–93. doi: 10.1097/NEN.0b013e3181e332ec
29. Willis SN, Stadelmann C, Rodig SJ, Caron T, Gattenloehner S, Mallozzi SS, et al. Epstein-Barr Virus infection is not a characteristic feature of multiple sclerosis brain. Brain (2009) 132:3318–28. doi: 10.1093/brain/awp200
30. Peferoen LAN, Lamers F, Lodder LNR, Gerritsen WH, Huitinga I, Melief J, et al. Epstein-Barr Virus is not a characteristic feature in the central nervous system in established multiple sclerosis. Brain (2010) 133:e137. doi: 10.1093/brain/awp296
31. Sargsyan SA, Shearer AJ, Ritchie AM, Burgoon MP, Anderson S, Hemmer B, et al. Absence of Epstein-Barr virus in the brain and CSF of patients with multiple sclerosis. Neurology (2010) 74:1127–35. doi: 10.1212/WNL.0b013e3181d865a1
32. Ascherio A, Munger KL. Epstein-Barr virus infection and multiple sclerosis: a review. J Neuroimmune Pharmacol (2010) 5:271–7. doi: 10.1007/s11481-010-9201-3
33. Levin LI, Munger K, Rubertone MV, Peck CA, Lennette ET, Spiegelman D, et al. Temporal relationship between elevation of Epstein-Barr virus antibody titers and initial onset of neurological symptoms in multiple sclerosis. JAMA (2005) 293:2493–500. doi: 10.1001/jama.293.20.2496
34. Sundström P, Juto P, Wadell G, Hallmans G, Svenningsson A, Nyström L, et al. An altered immune response to Epstein–Barr virus in multiple sclerosis: a prospective study. Neurology (2004) 62:2277–82. doi: 10.1212/01.wnl.0000130496.51156.d7
35. DeLorenze GN, Munger KL, Lennette ET, Orentreich N, Vogelman JH, Ascherio A. Epstein-Barr Virus and multiple sclerosis: evidence of association from a prospective study with long-term follow-up. Arch Neurol (2006) 63:839–44. doi: 10.1001/archneur.63.6.noc50328
36. Hedström AK, Huang J, Michel A, Butt J, Brenner N, Hillert J, et al. High levels of Epstein-Barr virus nuclear antigen antigen-1-specific antibodies and infectious mononucleosis act both independently and synergistically to increase multiple sclerosis risk. Front Neurol (2020) 10:1368. doi: 10.3389/fneur.2019.01368
37. Ruprecht K, Wildemann B, Jarius S. Low intrathecal antibody production despite high seroprevalence of Epstein-Barr virus in multiple sclerosis: a review of the literature. J Neurol (2018) 265:239–52. doi: 10.1007/s00415-017-8656-z
38. Serafini B, Rosicarelli B, Veroni C, Mazzola GA, Aloisi F. Epstein-Barr Virus-specific CD8 T cells selectively infiltrate the brain in multiple sclerosis and interact locally with virus-infected cells: clue for a virus-driven immunopathological mechanism. J Virol (2019) 93:e00980. doi: 10.1128/JVI.00980-19
39. Jaquiéry E, Jilek S, Schluep M, Meylan P, Lysandropoulos A, Pantaleo G, et al. Intrathecal immune responses to EBV in early MS. Eur J Immunol (2010) 40:878–87. doi: 10.1002/eji.200939761
40. Lünemann JD, Edwards N, Muraro PA, Hayashi S, Cohen JI, Münz C, et al. Increased frequency and broadened specificity of latent EBV nuclear antigen-1-specific T cells in multiple sclerosis. Brain (2006) 129:1493–506. doi: 10.1093/brain/awl067
41. Lünemann JD, Jelcic I, Roberts S, Lutterotti A, Tackenberg B, Martin R, et al. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-γ and IL-2. J Exp Med (2008) 205:1763–73. doi: 10.1084/jem.20072397
42. Ruprecht K. The role of Epstein-Barr virus in the etiology of multiple sclerosis: a current review. Expert Rev Clin Immunol (2020) 16:1143–57. doi: 10.1080/1744666X.2021
43. Soldan SS, Lieberman PM. Epstein-Barr Virus and multiple sclerosis. Nat Rev Microbiol (2023) 21:51–64. doi: 10.1038/s41579-022-00770-5
44. Pender MP, Csurhes PA, Lenarczyk A, Pfluger CM, Burrows SR. Decreased T cell reactivity to Epstein-Barr virus infected lymphoblastoid cell lines in multiple sclerosis. J Neurol Neurosurg Psychiatry (2009) 80:498–505. doi: 10.1136/jnnp.2008.161018
45. Jilek S, Schluep M, Harari A, Canales M, Lysandropoulos A, Zekeridou A, et al. HLA-B7-restricted EBV-specific CD8+ T cells are dysregulated in multiple sclerosis. J Immunol (2012) 188:4671–80. doi: 10.4049/jimmunol.1103100
46. Angelini DF, Serafini B, Piras E, Severa M, Coccia EM, Rosicarelli B, et al. Increased CD8+ T cell response to Epstein-Barr virus lytic antigens in the active phase of multiple sclerosis. PloS Pathog (2013) 9:e1003220. doi: 10.1371/journal.ppat.1003220
47. Mueller NE, Lennette ET, Dupnik K, Birmann BM. Antibody titers against EBNA1 and EBNA2 in relation to Hodgkin lymphoma and history of infectious mononucleosis. Int J Cancer (2012) 130:2886–91. doi: 10.1002/ijc.26334
48. VanderWeele TJ. Sufficient cause interactions and statistical interactions. Epidemiology (2009) 20:6–13. doi: 10.1097/EDE.0b013e31818f69e7
49. Sedighi S, Gholizadeh O, Yasamineh S, Akbarzadeh S, Amini P, Favakehi P, et al. Comprehensive investigations relationships between viral infections and multiple sclerosis pathogenesis. Curr Microbiol (2022) 80:15. doi: 10.1007/s00284-022-03112-z
50. Welsh RM, Che JW, Brehm MA, Selin LK. Heterologous immunity between viruses. Immunol Rev (2010) 235:244–66. doi: 10.1111/j.0105-2896.2010.00897.x
51. Cuomo L, Trivedi P, Cardillo MR, Gagliardi FM, Vecchione A, Caruso R, et al. Human herpesvirus 6 infection in neoplastic and normal brain tissue. J Med Virol (2001) 63:45–51. doi: 10.1002/1096-9071(200101)63:1<45::AID-JMV1006>3.0.CO;2-K
52. Tuke PW, Hawke S, Grifffiths PD, Clark DA. Distribution and quantification of human herpesvirus 6 in multiple sclerosis and control brains. Mult Scler (2004) 10:355–9. doi: 10.1191/1352458504ms1057oa
53. Engdahl E, Gustafsson R, Huang J, Biström M, Bomfim I, Stridh P, et al. Increased serological response against human herpesvirus 6A is associated with risk for multiple sclerosis. Front Immunol (2019) 10:2715. doi: 10.3389/fimmu.2019.02715
54. Flamand L, Stefanescu I, Ablashi DV, Menezes J. Activation of the Epstein-Barr virus replicative cycle by human herpesvirus 6. J Virol (1993) 67:6768–77. doi: 10.1128/jvi.67.11.6768-6777.1993
55. Flamand L, Menezes J. Cyclic AMP-responsive element-dependent activation of Epstein-Barr virus zebra promoter by human herpesvirus 6. J Virol (1996) 70:1784–91. doi: 10.1128/JVI.70.3.1784-1791.1996
56. Cuomo L, Angeloni A, Zompetta C, Cirone M, Calogero A, Frati L, et al. Human herpesvirus 6 variant a, but not variant b, infects EBV-positive b lymphoid cells, activating the latent EBV genome through a BZLF-1-dependent mechanism. AIDS Res Hum Retroviruses (1995) 11:1241–5. doi: 10.1089/aid.1995.11.1241
57. Challoner PB, Smith KT, Parker JD, MacLeod DL, Coulter SN, Rose TM, et al. Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc Natl Acad Sci U.S.A. (1995) 92:7440–4. doi: 10.1073/pnas.92.16.7440
58. Goodman AD, Mock DJ, Powers JM, Baker JV, Blumberg BM. Human herpesvirus 6 genome and antigen in acute multiple sclerosis lesions. J Infect Dis (2003) 187:1365–76. doi: 10.1086/368172
59. Johnston JB, Silva C, Holden J, Warren KG, Clark AW, Power C. Monocyte activation and differentiation augment human endogenous retrovirus expression: implications for inflammatory brain diseases. Ann Neurol (2001) 50:434–42. doi: 10.1002/ana.1131
60. Meier UC, Cipian RC, Karimi A, Ramasamy R, Middeldorp JM. Cumulative roles for Epstein-Barr virus, human endogenous retroviruses, and human herpes virus-6 in driving an inflammatory cascade underlying MS pathogenesis. Front Immunol (2021) 12:757302. doi: 10.3389/fimmu.2021.757302
61. Sundqvist E, Bergström T, Daialhosein H, Nyström M, Sundström P, Hillert J, et al. Cytomegalovirus seropositivity is negatively associated with multiple sclerosis. Mult Scler (2014) 20:165–73. doi: 10.1177/1352458513494489
62. Waubant E, Mowry EM, Krupp L, Chitnis T, Yeh EA, Kuntz N, et al. Common viruses associated with lower pediatric multiple sclerosis risk. Neurology (2011) 76:1989–95. doi: 10.1212/WNL.0b013e31821e552a
63. Chidrawar S, Khan N, Wei W, McLarnon A, Smith N, Nayak L, et al. Cytomegalovirus-seropositivity has a profound influence on the magnitude of major lymphoid subsets within healthy individuals. Clin Exp Immunol (2009) 155:423–32. doi: 10.1111/j.1365-2249.2008.03785.x
64. Zabalza A, Vera A, Alari-Pahissa E, Munteis E, Moreira A, Yélamos J, et al. Impacto f citomegalovirus infection on b cell differentiation and cytokine production in multiple sclerosis. J Neuroinflamm (2020) 17:161. doi: 10.1186/s12974-020-01840-2
65. Moutsianas L, Jostins L, Beecham AH, Dilthey AT, Xifara DK, Dan M, et al. Class II HLA interactions modulate genetic risk for multiple sclerosis. Nat Genet (2015) 47:1107–13. doi: 10.1038/ng.3395
66. De Jager PL, Chibnik LB, Cui J, Reischl J, Lehr S, Simon KC, et al. Integration of genetic risk factors into a clinical algorithm for multiple sclerosis susceptibility: a weighted genetic risk score. Lancet Neurol (2009) 8:1111–9. doi: 10.1016/S1474-4422(09)70275-3
67. Li Q, Cohen JI. Epstein-Barr Virus and the human leukocyte antigen complex. Curr Clin Microbiol Rep (2019) 6:175–81. doi: 10.1007/s40588-019-00120-9
68. Agostini S, Mancuso R, Guerini FR, D’Alfonso S, Agliardi C, Hernis A, et al. HLA alleles modulate EBV viral load in multiple sclerosis. J Transl Med (2018) 16:80. doi: 10.1186/s12967-018-1450-6
69. Lee SP, Thomas WA, Murray RJ, Khanim F, Kaur S, Young LS, et al. HLA A2.1-restricted cytotoxic T cells recognizing a range of Epstein-Barr virus isolates through a defined epitope in latent membrane protein LMP2. J Virol (1993) 67:7428–35. doi: 10.1128/JVI.67.12.7428-7435.1993
70. Houssaint E, Saulquin X, Scotet E, Bonneville M. Immunodominant CD8 T cell response to Epstein-Barr virus. BioMed Pharmacother (2001) 55:373–80. doi: 10.1016/s0753-3322(01)00082-8
71. Lundtoft C, Pucholt P, Imgenberg-Kreuz J, Carlsson-Almlöf J, Eloranta ML, Syvänen AC, et al. Function of multiple sclerosis-protective HLA class I alleles revealed by genome-wide protein-quantitative trait loci mapping of interferon signalling. PloS Genet (2020) 16:e1009199. doi: 10.1371/journal.pgen.1009199
72. Kreft KL, Van Nierop GP, Scherbeijn SMJ, Janssen M, Verjans GMGM, Hintzen RQ. Elevated EBNA-1 IgG in MS is associated with genetic MS risk variants. Neurol Neuroimmunol Neuroinflamm (2017) 4(6):e406. doi: 10.1212/NXI.0000000000000406
73. Zhou Y, Zhu G, Charlesworth JC, Simpson S Jr, Rubicz R, Göring HH, et al. Genetic loci for Epstein-Barr virus nuclear antigen-1 are associated with risk of multiple sclerosis. Mult Scler (2016) 22:1655–64. doi: 10.1177/1352458515626598
74. Sorosina M, Santoro S, Ferrè L, Mascia E, Clarelli F, Giordano A, et al. Risk HLA variants affect the T-cell repertoire in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm (2023) 10:e200093. doi: 10.1212/NXI.0000000000200093
75. Kular L, Liu Y, Ruhrmann S, Zheleznyakova G, Marabita F, Gomez-Cabrero D, et al. DNA Methylation as a mediator of HLA-DRB1*1501 and a protective variant in multiple sclerosis. Nat Commun (2018) 9:2397. doi: 10.1038/s41467-018-04732-5
76. Zdimerova H, Murer A, Engelmann C, Raykova A, Deng Y, Gujer C, et al. Attenuated immune control of Epstein-Barr virus in humanized mice is associated with the multiple sclerosis risk factor HLA-DR15. Eur J Immunol (2021) 51:64–75. doi: 10.1002/eji.202048655
77. Jelcic I, Nimer F, Wang J, Lentsch V, Planas R, Jelcic I, et al. Memory b cells activate brain-homing, autoreactive CD4+ T cells in multiple sclerosis. Cell (2018) 175:85–100. doi: 10.1016/j.cell.2018.08.011
78. Wang J, Jelcic I, Muhlenbruch L, Haunerdinger V, Toussaint NC, Zhao Y, et al. HLA-DR15 molecules jointly shape an autoreactive T cell repertoire in multiple sclerosis. Cell (2020) 183:1264–1281.e20. doi: 10.1016/j.cell.2020.09.054
79. Van der Mei IAF, Lucas RM, Taylor BV, Valery PC, Dwyer T, Kilpatrick TJ, et al. Population attributable fractions and joint effects of key risk factors for multiple sclerosis. Mult Scler (2016) 22:461–9. doi: 10.1177/1352458515594040
80. Langer-Gould A, Wu J, Lucas R, Smith J, Gonzales E, Amezcua L, et al. Epstein-Barr Virus, cytomegalovirus, and multiple sclerosis susceptibility: a multiethnic study. Neurology (2017) 89:1330–7. doi: 10.1212/WNL.0000000000004412
81. Disanto G, Hall C, Lucas R, Ponsonby AL, Berlanga-Taylor AJ, Giovannoni G, et al. Assessing interactions between HLA-DRB1*15 and infectious mononucleosis on the risk of multiple sclerosis. Mult Scler (2013) 19:1355–8. doi: 10.1177/1352458513477231
82. Nielsen TR, Rostgaard K, Askling J, Steffensen R, Oturai A, Jersild C, et al. Effects of infectious mononucleosis and HLA-DRB1*15 in multiple sclerosis. Mult Scler (2009) 15:431–6. doi: 10.1177/1352458508100037
83. Lucas RM, Ponsonby AL, Dear K, Valery P, Pender MP, Burrows JM, et al. Current and past Epstein-Barr virus infection in risk of initial CNS demyelination. Neurology (2011) 77:371–9. doi: 10.1212/WNL.0b013e318227062a.+113
84. De Jager PL, Simon KC, Munger KL, Rioux JD, Hafler DA, Ascherio A. Integrating risk factors: HLA-DRB1*1501 and Epstein-Barr virus in multiple sclerosis. Neurology (2008) 70:1113–8. doi: 10.1212/01.wnl.0000294325.63006.f8
85. Hedström AK, Hillert J, Brenner N, Butt J, Waterboer T, Strid P, et al. DRB1-environment interactions in multiple sclerosis etiology: results from two Swedish case-control studies. J Neurol Neurosurg Psychiatry (2021) 92:717–22. doi: 10.1136/jnnp-2020-325676
86. Alonso A, Hernan MA. Temporal trends in the incidence of multiple sclerosis: a systematic review. Neurology (2008) 71:129–35. doi: 10.1212/01.wnl.0000316802.35974.34
87. Simpson S Jr, Wang W, Otahal P, Blizzard L, van der Mei IAF, Taylor BV. Latitude continues to be significantly associated with the prevalence of multiple sclerosis: an updated meta-analysis. J Neurol Neurosurg Psychiatry (2019) 90:1193–200. doi: 10.1136/jnnp-2018-320189
88. Lucas RM, Ponsonby AL, Dear K, Valery PC, Pender MP, Taylor BV, et al. Sun exposure and vitamin d are independent risk factors for CNS demyelination. Neurology (2011) 76:540–8. doi: 10.1212/WNL.0b013e31820af93d
89. Hedström AK, Olsson T, Kockum I, Hillert J, Alfredsson L. Low sun exposure increases multiple sclerosis risk both directly and indirectly. J Neurol (2020) 267:1045–52. doi: 10.1007/s00415-019-09677-3
90. Lee KN, Kang HS, Jeon JH, Kim EM, Yoon SR, Song H, et al. VDUP1 is required for the development of natural killer cells. Immunity (2005) 22:195–208. doi: 10.1016/j.immuni.2004.12.012
91. Waddell A, Zhao J, Cantorna MT. NKT cells can help mediate the protective effects of 1,25-dihydroxyvitamin D3 in experimental autoimmune encephalomyelitis in mice. Int Immunol (2015) 27:237–44. doi: 10.1093/intimm/dxu147
92. Gombart AF, Borregaard N, Koeffler HP. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin d receptor and i strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D2. FASEB J (2005) 19:1067–77. doi: 10.1096/fj.04-3284com
93. Galoppin M, Kari S, Soldati S, Pal A, Rival M, Engelhardt B, et al. Full spectrum of vitamin d immunomodulation in multiple sclerosis: mechanisms and therapeutic implications. Brain Commun (2020) 4:fcac171. doi: 10.1093/braincomms/fcac171
94. Rolf L, Muris AH, Hupperts R, Damoiseaux J. Illuminating vitamin d effects on b cells- the multiple sclerosis perspective. Immunol (2016) 147:275–84. doi: 10.1111/imm.12572
95. Erem AS, Razzaque MS. Vitamin d-independent benefits of safe sunlight exposure. J Steroid Biochem Mol Biol (2021) 213:105957. doi: 10.1016/j.jsbmb.2021.105957
96. Lucas RM, Byrne SN, Correale J, Ilschner S, Hart PH. Ultraviolet radiation, vitamin d and multiple sclerosis. Neurodegener Dis Manag (2015) 5:413–24. doi: 10.1111/ene.12986
97. Breuer J, Schwab N, Schneider-Hohendorf T, Marziniak M, Mohan H, Bhatia U, et al. Ultraviolet b light attenuates the systemic immune response in central nervous system autoimmunity. Ann Neurol (2014) 75:739–58. doi: 10.1002/ana.24165
98. Becklund BR, Severson KS, Vang SV, DeLuca HF. UV Radiation suppresses experimental autoimmune encephalomyelitis independent of vitamin d production. Proc Natl Acad Sci U.S.A. (2010) 107:6418–23. doi: 10.1073/pnas.1001119107
99. Irving AA, Marling SJ, Seeman J, Plum LA, DeLuca HF. UV Light suppression of EAE (a mouse model of multiple sclerosis) is independent of vitamin d and its receptor. Proc Natl Acad Sci U.S.A. (2019) 116:22552–5. doi: 10.1073/pnas.1913294116
100. Salzer J, Nyström M, Hallmans G, Stenlund H, Wadell G, Sundström P. Epstein-Barr Virus antibodies and vitamin d in prospective multiple sclerosis biobank samples. Mult Scler (2013) 19:1587–91. doi: 10.1177/1352458513483888
101. Wergeland S, Myhr KM, Løken-Armsrud KI, Beiske AG, Bjerve S, Hovdal H, et al. HLA-DRB1 and Epstein–Barr virus antibody levels in a prospective cohort of multiple sclerosis patients. Eur J Neurol (2016) 23:1064–70. doi: 10.1111/ene.12986
102. Hedström AK, Huang J, Brenner N, Butt J, Kockum I, Waterboer T, et al. Low sun exposure acts synergistically with high Epstein-Barr nuclear antigen 1 (EBNA-1) antibody levels in multiple sclerosis etiology. Eur J Neurol (2021) 28:4146–52. doi: 10.1111/ene.15082
103. Harley JB, Chen XX, Pujato M, Miller D, Maddox A, Forney C, et al. Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat Genet (2018) 50:699–707. doi: 10.1038/s41588-018-0102-3
104. Ricigliano VAG, Handel AE, Sandve GK, Annibali V, Ristori G, Mechelli R, et al. EBNA2 binds to genomic intervals associated with multiple sclerosis and overlaps with vitamin d receptor occupancy. PloS One (2015) 10:e0119605. doi: 10.1371/journal.pone.0119605
105. Munger KL, Aivo J, Hongell K, Soilu-Hanninen M, Surcel HM, Ascherio A. Vitamin d status during pregnancy and risk of multiple sclerosis in offspring of women in the Finnish maternity cohort. JAMA Neurol (2016) 73:515–9. doi: 10.1001/jamaneurol.2015.4800
106. Ueda P, Rafatnia F, Baarnhielm M, Frobom R, Korzunowicz G, Lonnerbro R, et al. Neonatal vitamin d status and risk of multiple sclerosis. Ann Neurol (2014) 76:338–46. doi: 10.1002/ana.24210
107. Nielsen NM, Munger KL, Koch-Henriksen N, Hougaard DM, Magyari M, Jorgensen K, et al. Neonatal vitamin d status and risk of multiple sclerosis: a population-based case-control study. Neurology (2017) 88:44–51. doi: 10.1212/WNL.0000000000003454
108. Munger KL, Levin LI, Hollis BW, Howard NS, Ascherio A. Serum 25-hydroxyvitamin d levels and risk of multiple sclerosis. JAMA (2006) 296:2832–8. doi: 10.1001/jama.296.23.2832
109. Holmoy T. Vitamin d status modulates the immune response to Epstein Barr virus: synergistic effect of risk factors in multiple sclerosis. Med Hypotheses (2008) 70:66–9. doi: 10.1016/j.mehy.2007.04.030
110. Lossius A, Riise T, Pugliatti M, Bjornevik K, Casetta I, Drulovic J, et al. Season of infectious mononucleosis and risk of multiple sclerosis at different latitudes: the EnvIMS study. Mult Scler (2014) 20:669–74. doi: 10.1177/1352458513505693
111. Downham C, Visser E, Vickers M, Counsell C. Season of infectious mononucleosis as a risk factor for multiple sclerosis: a UK primary care case-control study. Mult Scler Relat Disord (2017) 17:103–6. doi: 10.1016/j.msard.2017.07.009
112. Rosso M, Chitnis T. Association between cigarette smoking and multiple sclerosis: a review. JAMA Neurol (2020) 77:245–53. doi: 10.1001/jamaneurol.2019.4271
113. Hedström AK, Hillert J, Olsson T, Alfredsson L. Smoking and multiple sclerosis susceptibility. Eur J Epidemiol (2013) 28:867–74. doi: 10.1007/s10654-013-9853-4
114. Mikaeloff Y, Caridade G, Tardieu M, Suissa S. Parental smoking at home and the risk of childhood-onset multiple sclerosis in children. Brain (2007) 130:2589–95. doi: 10.1093/brain/awm198
115. Hedström AK, Bäärnhielm M, Olsson T, Alfredsson L. Exposure to environmental tobacco smoke is associated with increased risk for multiple sclerosis. Mult Scler (2011) 17:788–93. doi: 10.1177/1352458511399610
116. Hedström AK, Hössjer O, Katsoulis M, Kockum I, Olsson T, Alfredsson L. Organic solvents and MS susceptibility: interaction with MS risk HLA genes. Neurology (2018) 91:e455–62. doi: 10.1212/WNL.0000000000005906
117. Hedström AK, Segersson D, Hillert J, Stridh P, Kockum I, Olsson T, et al. Association between exposure to combustion-related air pollution and multiple sclerosis risk. Int J Epidemiol (2023) 52(3):703–14. doi: 10.1093/ije/dyac234
118. Hedström AK, Sundqvist E, Bäärnhielm M, Nordin N, Hillert J, Kockum I, et al. Smoking and two human leukocyte antigen genes interact to increase the risk for multiple sclerosis. Brain (2011) 134:653–64. doi: 10.1093/brain/awq371
119. Briggs FB, Acuna B, Shen L, Ramsay P, Quach H, Bernstein A, et al. Smoking and risk of multiple sclerosis: evidence of modification by NAT1 variants. Epidemiology (2014) 25:605–14. doi: 10.1097/EDE.0000000000000089
120. Hedström AK, Bomfim IL, Barcellos LF, Briggs F, Schaefer C, Kockum I, et al. Interaction between passive smoking and two HLA genes with regard to multiple sclerosis risk. Int J Epidemiol (2014) 43:1791–8. doi: 10.1093/ije/dyu195
121. Ziaei Y, Graves J, Benson L, Gorman M, Rensel M, Mar S, et al. Gene-environment interactions increase the risk of pediatric-onset multiple sclerosis associated with ozone pollution. Mult Scler (2022) 28:1330–9. doi: 10.1177/13524585211069926
122. Sopori M. Effects of cigarette smoke on the immune system. Nat Rev Immunol (2002) 2:372–7. doi: 10.1038/nri803
123. Arnson Y, Shoenfeld Y, Amital H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J Autoimmun (2010) 34:J258–265. doi: 10.1016/j.jaut.2009.12.003
124. Qui F, Liang CL, Liu H, Zeng YQ, Hou S, Huang S, et al. Impacts of cigarette smoking on immune responsiveness: up and down or upside down? Oncotarget (2017) 8:268–84. doi: 10.18632/oncotarget.13613
125. Brandsma CA, Hylkema MN, Geerlings M, van Geffen WH, Postma DS, Timens W, et al. Increased levels of (class switched) memory b cells in peripheral blood of current smokers. Respir Res (2009) 10:108. doi: 10.1186/1465-9921-10-108
126. Simon KC, van der Mei IA, Munger KL, Ponsonby A, Dickinson J, Dwyer T, et al. Combined effects of smoking, anti-EBNA antibodies, and HLA-DRB1*1501 on multiple sclerosis risk. Neurology (2010) 74:1365–71. doi: 10.1212/WNL.0b013e3181dad57e
127. Salzer J, Stenlund H, Sundström P. The interaction between smoking and Epstein-Barr virus as multiple sclerosis risk factors may depend on age. Mult Scler (2014) 20:747–50. doi: 10.1177/1352458513507820
128. Hedström AK, Huang J, Brenner N, Butt J, Hillert J, Waterboer T, et al. Smoking and Epstein-Barr virus infection in multiple sclerosis development. Sci Rep (2020) 10:10960. doi: 10.1038/s41598-020-67883-w
129. He YQ, Xue WQ, Xu FH, Xu YF, Zhang JB, Lu HL, et al. The relationship between environmental factors and the profile of Epstein–Barr virus antibodies in the lytic and latent infection periods in healthy populations from endemic and non-endemic nasopharyngeal carcinoma areas in China. EBioMedicine (2018) 30:184–91. doi: 10.1016/j.ebiom.2018.02.019
130. Nielsen TR, Pedersen M, Rostgaard K, Frisch M, Hjalgrim H. Correlations between Epstein–Barr virus antibody levels and risk factors for multiple sclerosis in healthy individuals. Mult Scler (2007) 13:420–3. doi: 10.1177/1352458506071470
131. He Y, Liao X, Xue W, Xu YF, Xu FH, Li FF, et al. Association between environmental and oral Epstein-Barr virus DNA loads: a multicenter cross-sectional study in China. J Infect Dis (2019) 219:400–9. doi: 10.1093/infdis/jiy542
132. Hu T, Lin C, Xie S, Chen GH, Lu YQ, Ling W, et al. Smoking can increase nasopharyngeal carcinoma risk by repeatedly reactivation Epstein-Barr virus: an analysis of a prospective study in southern China. Cancer Med (2019) 8:2561–71. doi: 10.1002/cam4.2083
133. Xu FH, Xiong D, Xu YF, Cao SM, Xue WQ, Qin HD, et al. An epidemiological and molecular study of the relationship between smoking, risk of nasopharyngeal carcinoma, and Epstein-Barr virus activation. J Natl Cancer Inst (2012) 104:1396–410. doi: 10.1093/jnci/djs320
134. Su D, Wang X, Campbell MR, Porter DK, Pittman GS, Bennet BD, et al. Distinct epigenetic effects of tobacco smoking in whole blood and among leukocyte subtypes. PloS One (2016) 11:e0166486. doi: 10.1371/journal.pone.0166486
135. Odoardi F, Sie C, Streyl K, Ulaganathan VK, Schläger C, Lodygin D, et al. T Cells become licensed in the lung to enter the central nervous system. Nature (2012) 488:675–9. doi: 10.1038/nature11337
136. Langer-Gould A, Brara SM, Beaber BE, Koebnick C. Childhood obesity and risk of pediatric multiple sclerosis and clinically isolated syndrome. Neurology (2013) 80:548–52. doi: 10.1212/WNL.0b013e31828154f3
137. Munger KL, Bentzen J, Laursen B, Stenager E, Koch-Henriksen N, Sorensen TA, et al. Childhood body mass index and multiple sclerosis risk: a long-term cohort study. Mult Scler (2013) 19:1323–9. doi: 10.1177/1352458513483889
138. Harroud A, Manousaki D, Butler-Laporte G. The relative contributions of obesity, vitamin d, leptin, and adiponectin to multiple sclerosis risk: a mendelian randomization mediation analysis. Mult Scler (2021) 27:1994–2000. doi: 10.1177/1352458521995484
139. Mokry LE, Ross S, Timpson NJ, Sawcer S, Smith GD, Richards JB. Obesity and multiple sclerosis: a mendelian randomization study. PloS Med (2016) 13:e1002053. doi: 10.1371/journal.pmed.1002053
140. Gianfrancesco MA, Stridh P, Rhead B, Shao X, Xu E, Graves JS, et al. Evidence for a causal relationship between low vitamin d, high BMI, and pediatric-onset MS. Neurology (2017) 88:1623–9. doi: 10.1212/WNL.0000000000003849
141. Lumeng CN, Bodzin J, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest (2007) 117:175–84. doi: 10.1172/JCI29881
142. Kwait VR, Reis G, Valera IC, Parvatiyar K, Parvatiyar M. Autoimmunity as a sequela to obesity and systemic inflammation. Front Physiol (2022) 13:e887702. doi: 10.3389/fphys.2022.887702
143. Wang Q, Wang Y, Xu D. The roles of T cells in obese tissue inflammation. Adipocyte (2021) 1:435–45. doi: 10.1080/21623945.2021.1965314
144. Van Dyken P, Lacoste B. Impact of metabolic syndrom on neuroinflammation and the blood-brain barrier. Front Neurosci (2018) 12:930. doi: 10.3389/fnins.2018.00930
145. Karlsson EA, Beck MA. The burden of obesity on infectious disease. Exp Biol Med (2010) 235:1412–24. doi: 10.1258/ebm.2010.010227
146. Viel S, Besson L, Charrier E, Marcias A, Disse E, Bienvenu J, et al. Alteration of natural killer cell phenotype and function in obese individuals. Clin Immunol (2017) 177:12–7. doi: 10.1016/j.clim.2016.01.007
147. Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the immune system. Nat Rev Immunol (2017) 17:219–32. doi: 10.1038/nri.2017.7
148. Hedström AK, Olsson T, Alfredsson L. High body mass index before age 20 is associated with increased risk for multiple sclerosis in both men and women. Mult Scler (2012) 18:1334–6. doi: 10.1177/1352458512436596
149. Munger KL, Chitnis T, Ascherio A. Body size and risk of MS in two cohorts of US women. Neurology (2009) 73:1543–50. doi: 10.1212/WNL.0b013e3181c0d6e0
150. Hedström AK, Olsson T, Alfredsson L. Body mass index during adolescence, rather than childhood, is critical in determining MS risk. Mult Scler (2016) 22:878–3. doi: 10.1177/1352458515603798
151. Hedström AK, Brenner N, Butt J, Hillert J, Waterboer T, Olsson T, et al. Overweight/obesity in young adulthood interacts with aspects of EBV infection in MS etiology. Neurol Neuroimmunol Neuroinflamm (2020) 8:e912. doi: 10.1212/NXI.0000000000000912
152. Hedström AK, Bomfim IL, Hillert J, Olsson T, Alfredsson L. Obesity interacts with infectious mononucleosis in risk of multiple sclerosis. Eur J Neurol (2015) 22:578–e38. doi: 10.1111/ene.12620
153. Kadowaki A, Quintana FJ. The gut-CNS axis in multiple sclerosis. Trends Neurosci (2020) 43:622–34. doi: 10.1016/j.tins.2020.06.002
154. Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature (2011) 479:538–41. doi: 10.1038/nature10554
155. Rothhammer V, Borucki DM, Tjon EC, Takenaka MC, Chao CC, Ardura-Fabregat A, et al. Microglial control of astrocytes in response to microbial metabolites. Nature (2018) 557:724–8. doi: 10.1038/s41586-018-0119-x
156. Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med (2016) 22:5. doi: 10.1038/nm.4106
157. Freedman SN, Shahi SK, Mangalam AK. The “Gut feeling”: breaking down the role of gut microbiome in multiple sclerosis. Neurother (2018) 15:109–25. doi: 10.1007/s13311-017-0588-x
158. Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA, et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc Natl Acad Sci USA (2017) 114:10713–8. doi: 10.1073/pnas.1711235114
159. Berer K, Gerdes LA, Cekanaviciute E, Jia X, Xiao L, Xia Z, et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc Natl Acad Sci USA (2017) 114:10719–24. doi: 10.1073/pnas.1711233114
160. Yu H, Bai S, Hao Y, Guan Y. Fatty acids role in multiple sclerosis as “metabokines”. J Neuroinflamm (2022) 19:157. doi: 10.1186/s12974-022-02502-1
161. Calder PC. Long chain fatty acids and gene expression in inflammation and immunity. Curr Opin Clin Nutr Metab Care (2013) 16:425–33. doi: 10.1097/MCO.0b013e3283620616
162. Langer-Gould A, Black LJ, Waubant E, Smith JB, Wu J, Gonzales EG, et al. Seafood, fatty acid biosynthesis genes, and multiple sclerosis susceptibility. Mult Scler (2020) 26:1476–85. doi: 10.1177/1352458519872652
163. Hoare S, Lithander F, van der Mei I, Ponsonby AL, Lucas RM, Chapman C, et al. Higher intake of omega-3 polyunsaturated fatty acids is associated with a decreased risk of a first clinical diagnosis of central nervous system demyelination: results from the ausimmune study. Mult Scler (2016) 22:884–92. doi: 10.1177/1352458515604380
164. Bjornevik K, Chitnis T, Ascherio A, Munger KL. Polyunsaturated fatty acids and the risk of multiple sclerosis. Mult Scler (2017) 23:1830–8. doi: 10.1177/1352458517691150
165. Cortese M, Riise T, Bjornevik K, Holmoy T, Kampman MT, Magalhaes S, et al. Timing of use of cod liver oil, a vitamin d source, and multiple sclerosis risk: the EnvIMS study. Mult Scler (2015) 21:1856–64. doi: 10.1177/1352458515578770
166. Bager P, Nielsen NM, Bihrmann K, Frisch M, Wohlfart J, Koch-Henriksen N, et al. Sibship characteristics and risk of multiple sclerosis: a nationwide cohort study in Denmark. Am J Epidemiol (2006) 163:1112–7. doi: 10.1093/aje/kwj148
167. Rostgaard K, Nielsen NM, Melbye M, Frisch M, Hjalgrim H. Siblings reduce multiple sclerosis risk by preventing delayed primary Epstein-Barr virus infection. Brain (2022) 146(5):1993-2002. doi: 10.1093/brain/awac401
168. Gustavsen S, Sondergaard HB, Oturai DB, Laursen B, Laursen JH, Magyari M, et al. Shift work at young age is associated with increased risk of multiple sclerosis in a Danish population. Mult Scler Relat Disord (2016) 9:104–9. doi: 10.1016/j.msard.2016.06.010
169. Åkerstedt T, Olsson T, Alfredsson L, Hedström AK. Insufficient sleep during adolescence and risk of multiple sclerosis: results from a Swedish case-control study. J Neurol Neurosurg Psychiatry (2023) 94:331–6. doi: 10.1136/jnnp-2022-330123
170. Saul A, Ponsonby AL, Lucas RM, Taylor BV, Simpson S Jr, Valery P, et al. Stressful life events and the risk of initial nervous system demyelination. Mult Scler (2017) 23:1000–7. doi: 10.1177/1352458516667566
171. Zhong L, Krummenacher C, Zhang W, Hong J, Feng Q, Chen Y, et al. Urgency and necessity of Epstein-Barr virus prophylactic vaccines. NPJ Vaccines (2022) 7:159. doi: 10.1038/s41541-022-00587-6
172. Gies P, van Deventer E, Green AC, Sinclair C, Tinker R. Review of the global solar UV index 2015 workshop report. Health Phys (2018) 114:84–90. doi: 10.1097/HP.0000000000000742
173. Estruch R, Ros E, Salas-Salvadó J, Covas MI, Corella D, Arós F, et al. Primary prevention of cardiovascular disease with a Mediterranean diet supplemented with extra-virgin olive oil or nuts. N Engl J Med (2018) 378:e34. doi: 10.1056/NEJMoa1800389
174. Román GC, Jackson RE, Gadhia R, Román AN, Reis J. Mediterranean Diet: the role of long-chain ω-3 fatty acids in fish; polyphenols in fruits, vegetables, cereals, coffee, tea, cacao and wine; probiotics and vitamins in prevention of stroke, age-related cognitive decline, and Alzheimer disease. Rev Neurol (2019) 175:724–41. doi: 10.1016/j.neurol.2019.08.005
Keywords: multiple sclerosis, Epstein-Barr virus, infectious mononucleosis, human leukocyte antigen, vitamin D, sun exposure, smoking, obesity
Citation: Hedström AK (2023) Risk factors for multiple sclerosis in the context of Epstein-Barr virus infection. Front. Immunol. 14:1212676. doi: 10.3389/fimmu.2023.1212676
Received: 26 April 2023; Accepted: 26 June 2023;
Published: 24 July 2023.
Edited by:
Klemens Ruprecht, Charité University Medicine Berlin, GermanyReviewed by:
Linda Ann Spatz, City University of New York, United StatesShunbin Ning, East Tennessee State University, United States
Copyright © 2023 Hedström. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Karin Hedström, YW5uYS5oZWRzdHJvbUBraS5zZQ==