
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol. , 10 August 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1210909
This article is part of the Research Topic Multi-targeted Tyrosine Kinase Inhibitors in the Treatment of Cancer and Neurodegenerative Disorders View all 13 articles
 Meng-Ke Liu†
Meng-Ke Liu† Feng Liu†
Feng Liu† Yu-Ting Dai†Xiang-Qin Weng†
Yu-Ting Dai†Xiang-Qin Weng† Li-Li ChengLi-Quan Fan
Li-Li ChengLi-Quan Fan Han Liu
Han Liu Lu Jiang
Lu Jiang Xiao-Jian Sun
Xiao-Jian Sun Hai Fang
Hai Fang Li Wang*
Li Wang* Wei-Li Zhao*
Wei-Li Zhao*Mast cell leukemia is a rare and aggressive disease, predominantly with KIT D816V mutation. With poor response to conventional poly-chemotherapy, mast cell leukemia responded to the midostaurin treatment with a 50% overall response rate (ORR), but complete remission rate is approximately 0%. Therefore, the potential mechanisms of midostaurin resistance and the exact impacts of midostaurin on both gene expression profile and mast cell leukemia microenvironment in vivo are essential for design tailored combination therapy targeting both the tumor cells and the tumor microenvironment. Here we report a 59-year-old male mast cell leukemia patient with KIT F522C mutation treated with midostaurin. Single-cell sequencing of peripheral blood and whole exome sequencing (WES) of bone marrow were performed before and 10 months after midostaurin treatment. In accordance with the clinical response, compared to the pretreatment aberration, the decline of mast cells and increase of T-, NK, B-cells in peripheral blood, and the decrease of the KIT F522C mutation burden in bone marrow were observed. Meanwhile, the emergence of RUNX1 mutation, upregulations of genes expression (RPS27A, RPS6, UBA52, RACK1) on tumor cells, and increased frequencies of T and NK cells with TIGIT, CTLA4, and LAG3 expression were observed after midostaurin treatment, predicting the disease progression of this patient. As far as we know, this is the first case reporting the clinical, immunological, and molecular changes in mast cell leukemia patients before and after midostaurin treatment, illustrating the in vivo mechanisms of midostaurin resistance in mast cell leukemia, providing important clues to develop a sequential option to circumvent tumor progression after targeting oncogene addiction and prolong patients’ survival.
Mast cell leukemia is a rare but aggressive form of systemic mastocytosis (SM) characterized by the expansion of neoplastic mast cells in bone marrow (BM) and peripheral blood (1). Pathogenic mutation of KIT D816V is found in 80% of SM; TET2, SRSF2, ASXL1, RUNX1, JAK2, and RAS are also frequently mutated and related to poor prognosis of mast cell leukemia patients (1, 2). No curative therapy is yet available for mast cell leukemia other than allogeneic hematopoietic stem cell transplantation; a median survival time in mast cell leukemia patients is less than 12 months when treated with chemotherapy or KIT inhibitors (2). Midostaurin is a small molecule inhibitor of multiple tyrosine kinase receptors and has been approved by the FDA and EMA as a KIT inhibitor in treating mast cell leukemia, with an overall remission rate of 50%, a complete remission (CR) rate of 0%, and a median progression-free survival (PFS) of 11.3 months (3). Reasons why mast cell leukemia responded to midostaurin but displayed a low CR rate and the underlying mechanisms behind relapse in vivo after midostaurin treatment were not clearly specified. In our study, we performed whole exome sequencing (WES) of bone marrow and single-cell RNA sequencing of peripheral blood on a mast cell leukemia patient before and after midostaurin treatment to illustrate the in vivo mechanism of midostaurin on mast cell leukemia.
In our study, we reported a 59-year-old male who experienced recurrent diarrhea, severe weight loss, urticaria, anemia and weakness. He came to our hospital in June 2018 and blood tests showed white blood cells (WBCs) 18.4×109/L (3.97-9.15×109/L), red blood cells 1.64×1012/L (4.09-5.74×1012/L), hemoglobin 8g/dl (13.1-17.2 g/dl), platelets 142×109/L (85-303×109/L), and alkaline phosphatase (AKP) 196 U/L (38-126 U/L). BM examinations indicated 84% abnormal mast cells with irregular sizes and positive toluidine blue staining. Peripheral blood aspirate demonstrated neoplastic mast cells with immature nuclear chromatin, abundant tightly packed cytoplasmic granules and high nuclear to cytoplasmic (N:C) ratio (Figure 1A). Flow cytometric analysis was performed on BM sample before treatment to observe mast cells in circulation and used as evidence for diagnosis according to the diagnose criteria of mast cell leukemia (2). As shown in Supplementary Figure 1, a significant population was observed in bone marrow sample showing extremely bright expression of CD117 and positive CD45 with moderate FSC and SSC (red color). Gating on CD117 bright expression cells, the population expressed CD13, CD33, CD203c and CD2 (partially), but not lineage specific markers such as cytoplasmic MPO, CD3, CD79a or the remaining markers. The very bright expression of CD117 combining the positive of CD203c indicated the mast cell origin. The partial expression of CD2 was considered as a common feature of abnormal mast cells. We further stained CD25 and obtained negative result. WES of BM cells revealed KIT F522C mutation and SETD2 mutation, but negative for TET2, ASXL1, BCR-ABL, JAK2, FLT3, and NPM1 mutations (Figure 1B). According to the dose modification instructions of midostaurin (4), the patient was treated with 100 mg/d midostaurin due to anemia and severe fatigue, and achieved a major response 10 months after treatment, with mast cells decreasing to 24% in BM. Meanwhile, blood smears showed mast cells with condensed nuclear chromatin, less granules in cytoplasm and lower N:C ratio (Figure 1A). Upon midostaurin treatment, compared to the pretreatment gene mutation profiles, variant allele frequency (VAF) was also significantly decreased in genes involved with the RAS-MAPK pathway, such as KIT and ERBB3. However, RUNX1 mutation, relating to poor prognosis of mast cell leukemia patients, was only detected after midostaurin treatment, which might indicate subsequent midostaurin resistance. (Figure 1B). Gene set enrichment analysis (GSEA) showed the top 5 downregulated pathways, including PI3K/AKT signaling in cancer, regulation of cell division, MAPK family signaling cascade, cell response to growth factor stimulus, and regulation of cell cycle process (Figure 1C), in which PI3K/AKT and MAPK signaling pathway are the downstream of KIT signaling and are associated with mast cell leukemia progression (5).
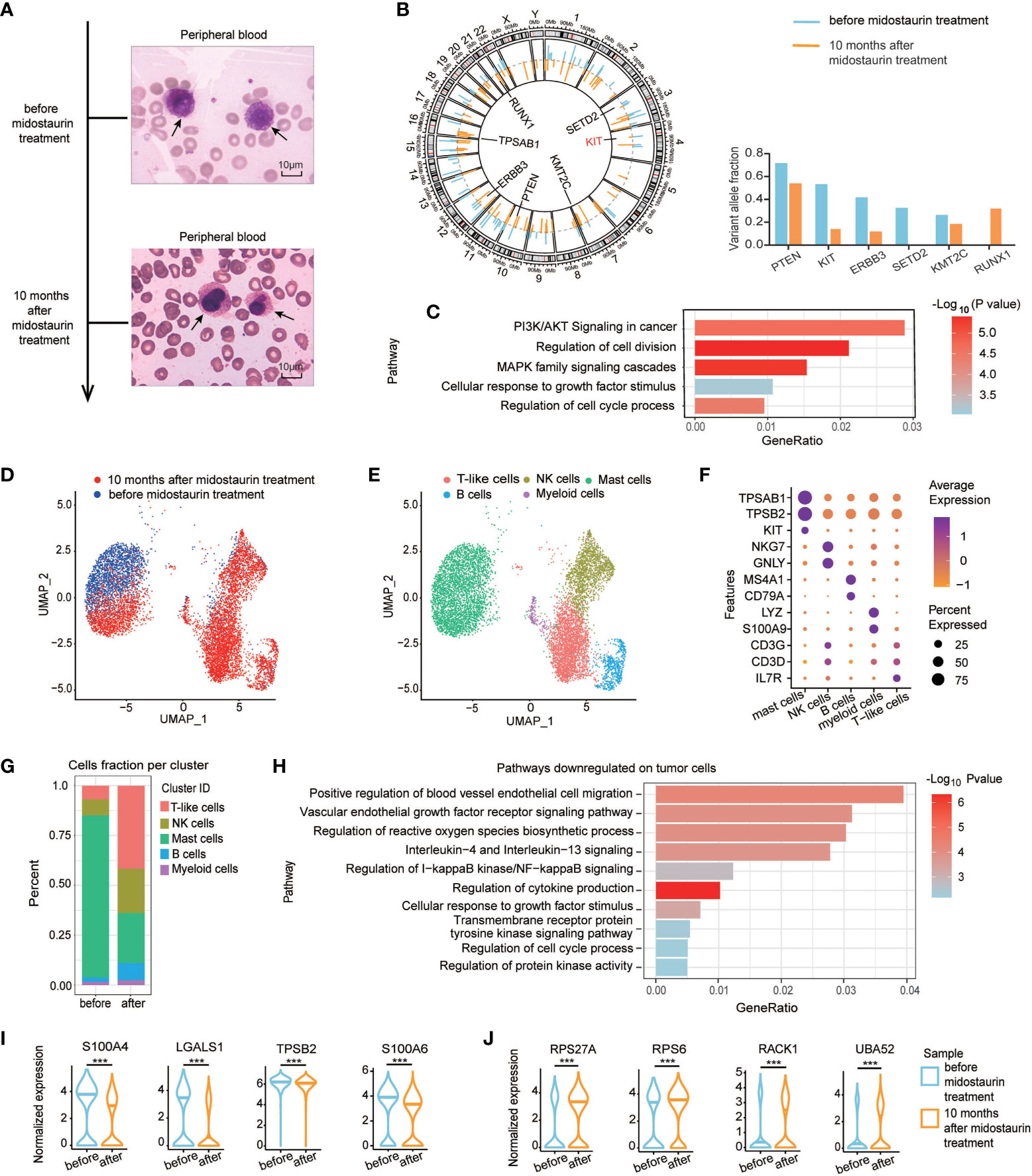
Figure 1 The morphological characteristics, mutation profiles, and single-cell analysis of tumor cells in the patient before and after midostaurin treatment. (A) Peripheral blood aspirate under Wright’s stain, demonstrating neoplastic mast cells with immature nuclear chromatin, abundant tightly packed cytoplasmic granules, high nuclear to cytoplasmic (N:C) ratio before treatment (top) and mast cells with condensed nuclear chromatin, less granules in cytoplasm and lower N:C ratio after 10 months of midostaurin treatment (bottom), bar=10μM. (B) Circos plot of single-nucleotide variations based on WES before and after midostaurin treatment. The outer lines colored in blue represent mutations detected before treatment. The inner lines colored in orange represent mutations detected after treatment. (C) Gene set enrichment analysis of genes decreased with mutation burden after treatment. (D) UMAP of single-cell RNA sequencing experiments of patient PBMCs before and after midostaurin treatment. Each cell represents a cell. Blue and red represent cells collected before or after 10 months of midostaurin treatment. (E) UMAP of single-cell RNA sequencing experiments as in (D) Annotated cell types are distinguished by colors. (F) Dot plot of cell-type-specific marker genes used to annotate the clusters in single-cell RNA sequencing experiments. (G) Fraction of different cell types before and after midostaurin treatment. (H) Pathways downregulated on tumor mast cells after midostaurin treatment. Normalized gene expression of downregulated genes (I), and upregulated genes (J) on tumor cells after midostaurin treatment. ***, represented significant difference of P<0.001.
Before treatment, single-cell RNA sequencing revealed that mast cells were the major fraction (81.40%) in peripheral blood mononuclear cells (PBMC) (Figures 1D–F). After 10 months of midostaurin treatment, the fraction of mast cells in peripheral blood decreased significantly (from 81.40% to 25.10%), while immune cells increased, including T cells (from 6.79% to 41.67%), Natural killer (NK) cells (from 8.07% to 22.16%) and B cells (from 2.18% to 8.72%) (Figure 1G). The top 5 downregulated pathways on mast cells included positive regulation of blood vessel endothelial migration, vascular endothelial growth factor receptor signaling, IL-4 and IL-13 signaling, NF-KB signaling regulation, and cytokine production pathways regulation (Figure 1H). The expression level of genes such as S100A4, LGALS1, TPSB2, and S100A6 were decreased. Instead, the expression level of RPS27A, RPS6, UBA52, and RACK1, involved in tyrosine kinase inhibitors (TKI) resistance, were increased after midostaurin treatment (Figures 1I, J).
Regarding the impact of midostaurin on mast cell leukemia microenvironment, pathways upregulated on T cells mainly included antigen presenting, T cell cytotoxicity, interferon γ signaling, T cell receptor signaling, regulation of immune response, and T cell-mediated immunity (Figure 2A). Among NK cells, antigen presentation, regulation of cell killing, natural killer cells mediated cytotoxicity, leukocyte mediated cytotoxicity, regulation of cell surface receptor, immune response, and cytokine production pathways were upregulated (Figure 2B). However, upon further classifying T and NK cells into 4 clusters, we found that most immune checkpoints such as TIGIT, CTLA4, LAG3, and HAVCR2 (also named TIM3) were highly expressed on cluster 1 and the cell ratios of cluster 1 in both T and NK cells were significantly increased after midostaurin treatment (P<0.05, Figures 2C, D). Further GSEA revealed that as compared to other clusters, the downregulated pathways on cluster 1 cells, including adaptive immune system, T cell activation, antigen processing and presentation pathways on cluster 1 of T cell (Supplementary Figure 2A), as well as vesicle-mediated transport, TCR, endosome to lysosome transport pathways on cluster 1 of NK cell (Supplementary Figure 2B), indicating the impaired functions of antigen presenting and cell cytotoxicity in these T/NK cells.
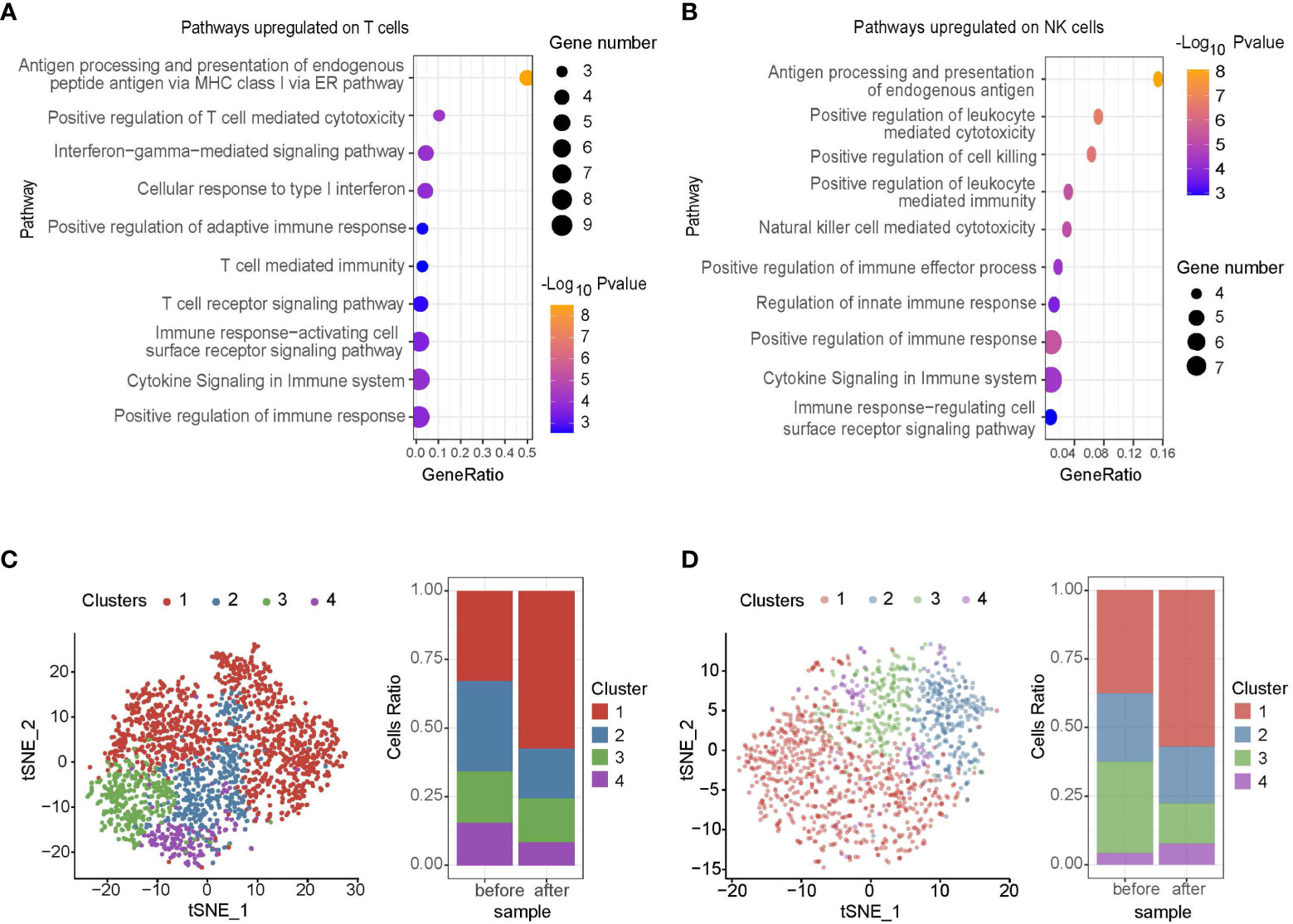
Figure 2 Single-cell RNA sequencing of immune cells in patient before and 10 months after midostaurin treatment. Pathways upregulated on T cells (A) and NK cells (B) after midostaurin treatment. T cell clusters and fraction of clusters (C), NK cell clusters and fraction of clusters (D) before and after midostaurin treatment.
Finally, the patient had disease progression 15 months after midostaurin treatment, with a percentage of mast cells in BM reaching 77.9% in September 2019; the patient was then treated with dasatinib. Unfortunately, he had no response (without any clinical improvement) to dasatinib, but displayed severe pleural effusion. Due to the poor response to dasatinib, the patient received low dose chemotherapy with azacitidine plus homoharringtonine and arsenic trioxide, but showed no response to these treatments. Eventually, the patient died of disease progression in 2020, with severe pleural effusion and ascites, with an OS of 24 months.
Midostaurin has significantly improved the life quality of mast cell leukemia patients with KIT D816V mutation. Few reports focused on its efficacy in KIT F522C mutated patients (3). Our patient with KIT F522C responded well to midostaurin as the first-line therapy and achieved 15 months of progression-free survival. KIT gain-of-function mutation triggers the activation of several downstream signaling, including MAPK, JAK-STAT, and PI3K pathways which regulates the proliferation, survival, and antiapoptotic function in mast cells (5). Consistent with the clinical response, we found the downregulation of PI3K/AKT and MAPK signaling pathways following midostaurin treatment.
However, during the response to midostaurin, emergent TKI resistances were observed not only in tumor cells but also in immune cells. In terms of tumor mast cells, cytokine pathways such as IL-4 and IL-13 signaling pathways, were downregulated after midostaurin treatment, thus indicating mast cells’ inactivation (6). Vascular endothelial-derived growth factor (VEGF) signaling pathway involved in activating blood vessel endothelial migration could also be attenuated through midostaurin treatment (7). These alterations in signaling pathways on tumor cells indicated that midostaurin inhibits tumor activation and metastasis. On the other hand, the upregulation of genes, such as RPS27A, RPS6, UBA52, and RACK1 on tumor cells, involved in TKI resistance (8–11), as well as RUNX1 mutation after midostaurin treatment, all these observations might partially explain the mast cell leukemia progression during midostaurin maintenance and afterward, the resistance to dasatinib, another TKI which has synergetic effect with midostaurin on mast cells (12). Consistent with our observation, in acute myeloid leukemia, midostaurin resistance was mainly attributed to the emergence of new mutations, including either mutation on different locus of KIT, or mutation on a new gene contributing to the activation of downstream signaling pathways (13). In our case, residual tumor cells with KIT F522C mutation after midostaurin treatment or the emergent RUNX1 mutation-containing cells might contribute to the resistance to midostaurin. Indeed, RUNX1 mutation was described as a risk factor in mast cell leukemia patients, and as a transcriptional regulator, RUNX1 could activate the enhancer of c-KIT, trigger the transcriptional regulation of c-KIT to promote the proliferation of malignant cells (14). In addition to RUNX1, increased expression of RPS27A, RPS6, UBA52, and RACK1 on mast cells might also contribute to midostaurin resistance. These emergent/secondary aberrations before and after midostaurin treatment thus suggested multiple mechanisms underlying the relapse of the mast cell leukemia patient. These aberrations could also be served as potential targets in further studies. For example, knockdown of RPS27A could arrest cell growth in TKI-resistant leukemia cell lines (15). PI3K pathway inhibitors could effectively dephosphorylate RPS6 in imatinib-resistant cell lines (9); RACK1 overexpression upregulated protein kinase C (PKC) activity. Therefore, PKC inhibitors also induced cell apoptosis in chemoresistant leukemia cell lines (16). Additionally, novel KIT inhibitor avapritinib, which was licensed by the FDA since 2021 and by the EMA since 2022, has distinct resistance profiles with midostaurin (17), might be an option for midostaurin-resistant patients.
Besides, tumor microenvironment had been improved, with the decrease of mast leukemia cells and increase of T and NK cells, indicating an activated antitumor condition in the patients after midostaurin treatment. In addition, single-cell RNA sequencing revealed an activation in the immune response of T cells, including upregulation of antigen presenting, T cell cytotoxicity and T cell-mediated immunity pathways (18). NK cells mediated cytotoxicity, positive regulation of cell killing, and regulation of cytokine production pathway were also activated on NK cells after treatment (18). Meanwhile, the frequencies of T and NK cells with TIGIT, CTLA4, and LAG3 expression were significantly increased after midostaurin treatment, displaying the resistance mechanisms of microenvironment during the exposure to midostaurin. Consistent with our observations, TIGIT+ T cells or TIGIT+ NK cells had impaired cell function and decreased cytokine secretion, which provided a tumor-supportive microenvironment (19, 20). Because upregulation of these immune checkpoints in peripheral blood were correlated with disease relapse in leukemia (21), an increase of dysfunctional T (e.g., TIGIT+ T) and NK (e.g., TIGIT+ NK) cell clusters might contribute to the relapse of this patient. In the future, therapeutically blockading of TIGIT (e.g., anti-TIGIT antibody) might be introduced to combined therapy in mast cell leukemia to enhance the cytotoxicity of T and NK cells (22).
In this case, we analyzed the genetic and tumor microenvironment changes in the mast cell leukemia patient after midostaurin treatment, in order to illustrate the in vivo mechanisms of aquired resistance to midostaurin. Although the pathogenic gene mutation KIT was repressed by midostaurin when the patient achieved major response, we inferred that the arising of RUNX1 mutation, upregulating genes expression (RPS27A, RPS6, UBA52, RACK1) on tumor cells and the immune suppressive tumor microenvironment with increased dysfunctional T and NK cells could contribute to the subsequent midostaurin resistance and disease progression in the patient. Our observations explain the reasons why mast cell leukemia responded to midostaurin but displayed a low CR rate (0%) and short median PFS (11.3 months) (4), indicating the underlying mechanisms behind relapse in vivo after midostaurin treatment. Furthermore, understanding of these mechanisms can be used to better monitor treatment response and the selection of resistant subclones, providing important clues to develop a sequential option to circumvent mast cell leukemia progression upon midostaurin treatment.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by Shanghai Ruijin Hospital Ethics Board. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
W-LZ and LW supervised the study. M-KL, X-QW, L-LC, L-QF collected clinical data and made the figures. M-KL, FL, Y-TD, HF, HL, LJ, X-JS carried out bioinformatic analysis and W-LZ, LW, M-KL drafted the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by National Natural Science Foundation of China (82170178, 82130004, 81830007 and 82070204), Shanghai Municipal Education Commission Gaofeng Clinical Medicine (20152206 and 20152208), Multicenter Clinical Research Project by Shanghai Jiao Tong University School of Medicine (DLY201601), Clinical Research Plan of Shanghai Hospital Development Center (SHDC2020CR1032B), Chang Jiang Scholars Program, Samuel Waxman Cancer Research Foundation, and the Foundation of National Facility for Translational Medicine (Shanghai, TMSK-2020-115).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1210909/full#supplementary-material
1. Ustun C, Arock M, Kluin-Nelemans HC, Reiter A, Sperr WR, George T, et al. Advanced systemic mastocytosis: from molecular and genetic progress to clinical practice. Haematologica (2016) 101(10):1133–43. doi: 10.3324/haematol.2016.146563
2. Valent P, Akin C and Metcalfe DD. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood (2017) 129(11):1420–7. doi: 10.1182/blood-2016-09-731893
3. Gotlib J, Kluin-Nelemans HC, George TI, Akin C, Sotlar K, Hermine O, et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N Engl J Med (2016) 374(26):2530–41. doi: 10.1056/NEJMoa1513098
4. US FDA. Reference ID: 4090671. RYDAPT (midostaurin) capsules, for oral use . Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/207997s000lbl.pdf.
5. Gilfillan AM, Tkaczyk C. Integrated signalling pathways for mast-cell activation. Nat Rev Immunol (2006) 6(3):218–30. doi: 10.1038/nri1782
6. Nedoszytko B, Arock M, Lyons JJ, Bachelot G, Schwartz LB, Reiter A, et al. Clinical impact of inherited and acquired genetic variants in mastocytosis. Int J Mol Sci (2021) 22(1):411. doi: 10.3390/ijms22010411
7. Fabbro D, Ruetz S, Bodis S, Pruschy M, Csermak K, Man A, et al. PKC412–a protein kinase inhibitor with a broad therapeutic potential. Anticancer Drug Des (2000) 15(1):17–28.
8. Wang H, Xie B, Kong Y, Tao Y, Yang G, Gao M, et al. Overexpression of RPS27a contributes to enhanced chemoresistance of CML cells to imatinib by the transactivated STAT3. Oncotarget (2016) 7(14):18638–50. doi: 10.18632/oncotarget.7888
9. Quentmeier H, Eberth S, ROmani J, Zaborski M and Drexler HG. BCR-ABL1-independent PI3Kinase activation causing imatinib-resistance. J Hematol Oncol (2011) 4:6. doi: 10.1186/1756-8722-4-6
10. Zhang L, Huang Y, Zhuo W, Zhu Y, Zhu B and Chen Z. Identification and characterization of biomarkers and their functions for Lapatinib-resistant breast cancer. Med Oncol (2017) 34(5):89. doi: 10.1007/s12032-017-0953-y
11. Gao X, Xue A, Fang Y, Shu P, Ling J, Hou Y, et al. RACK1 overexpression is linked to acquired imatinib resistance in gastrointestinal stromal tumor. Oncotarget (2016) 7(12):14300–9. doi: 10.18632/oncotarget.7426
12. Gleixner KV, Mayerhofer M, Sonneck K, Gruze A, Samorapoompichit P, Baumgartner C, et al. Synergistic growth-inhibitory effects of two tyrosine kinase inhibitors, dasatinib and PKC412, on neoplastic mast cells expressing the D816V-mutated oncogenic variant of KIT. Haematologica (2007) 92(11):1451–9. doi: 10.3324/haematol.11339
13. Assi R, Ravandi F. FLT3 inhibitors in acute myeloid leukemia: Choosing the best when the optimal does not exist. Am J Hematol (2018) 93(4):553–63. doi: 10.1002/ajh.25027
14. Debaize L, Jakobczyk H, Avner S, Gaudichon J, Rio AG, Serandour AA, et al. Interplay between transcription regulators RUNX1 and FUBP1 activates an enhancer of the oncogene c-KIT and amplifies cell proliferation. Nucleic Acids Res (2018) 46(21):11214–28. doi: 10.1093/nar/gky756
15. Wang H, Yu J, Zhang L, Xiong Y, Chen S, Xing H, et al. RPS27a promotes proliferation, regulates cell cycle progression and inhibits apoptosis of leukemia cells. Biochem Biophys Res Commun (2014) 446(4):1204–10. doi: 10.1016/j.bbrc.2014.03.086
16. Lei J, Li Q, Gao Y, Zhao L and Liu Y. Increased PKCalpha activity by Rack1 overexpression is responsible for chemotherapy resistance in T-cell acute lymphoblastic leukemia-derived cell line. Sci Rep (2016) 6:33717. doi: 10.1038/srep33717
17. Apsel Winger B, Cortopassi WA, Garrido Ruiz D, Ding L, Jang K, Leyte-Vidal A, et al. ATP-competitive inhibitors midostaurin and avapritinib have distinct resistance profiles in exon 17-mutant KIT. Cancer Res (2019) 79(16):4283–92. doi: 10.1158/0008-5472.CAN-18-3139
18. Gajewski TF, Schreiber H and Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol (2013) 14(10):1014–22. doi: 10.1038/ni.2703
19. Liu G, Zhang Q, Yang J, Li X, Xian L, Li W, et al. Increased TIGIT expressing NK cells with dysfunctional phenotype in AML patients correlated with poor prognosis. Cancer Immunol Immunother (2022) 71(2):277–87. doi: 10.1007/s00262-021-02978-5
20. Catakovic K, Gassner FJ, Ratswohl C, Zaborsky N, Rebhandl S, Schubert M, et al. TIGIT expressing CD4+T cells represent a tumor-supportive T cell subset in chronic lymphocytic leukemia. Oncoimmunology (2017) 7(1):e1371399. doi: 10.1080/2162402X.2017.1371399
21. Kong Y, Zhu L, Schell TD, Zhang J, Claxton DF, Ehmann WC, et al. T-cell immunoglobulin and ITIM domain (TIGIT) associates with CD8+ T-cell exhaustion and poor clinical outcome in AML patients. Clin Cancer Res (2016) 22(12):3057–66. doi: 10.1158/1078-0432.CCR-15-2626
Keywords: mast cell leukemia, midostaurin, molecular changes, single-cell RNA sequencing, tumor micoenvironment
Citation: Liu M-K, Liu F, Dai Y-T, Weng X-Q, Cheng L-L, Fan L-Q, Liu H, Jiang L, Sun X-J, Fang H, Wang L and Zhao W-L (2023) Case Report: Molecular and microenvironment change upon midostaurin treatment in mast cell leukemia at single-cell level. Front. Immunol. 14:1210909. doi: 10.3389/fimmu.2023.1210909
Received: 23 April 2023; Accepted: 24 July 2023;
Published: 10 August 2023.
Edited by:
Belgin Sever, Anadolu University, TürkiyeReviewed by:
Azaj Ahmed, The University of Iowa, United StatesCopyright © 2023 Liu, Liu, Dai, Weng, Cheng, Fan, Liu, Jiang, Sun, Fang, Wang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei-Li Zhao, emhhby53ZWlsaUB5YWhvby5jb20=; Li Wang, d2xfd2FuZ2RvbmdAMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.