 Howard M. Johnson
Howard M. Johnson Chulbul M. Ahmed
Chulbul M. Ahmed
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 01 December 2023
Sec. Vaccines and Molecular Therapeutics
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1208828
This article is part of the Research Topic SOCS: Regulation of the Immune System at a Whole New Level View all 10 articles
The field of antiviral therapeutics is fixated on COVID19 and rightly so as the fatalities at the height of the pandemic in the United States were almost 1,000,000 in a twelve month period spanning parts of 2020/2021. A coronavirus called SARS–CoV2 is the causative virus. Development of a vaccine through molecular biology approaches with mRNA as the inducer of virus spike protein has played a major role in driving down mortality and morbidity. Antivirals have been of marginal value in established infections at the level of hospitalization. Thus, the current focus is on early symptomatic infection of about the first five days. The Pfizer drug paxlovid which is composed of nirmatrelvir, a peptidomimetic protease inhibitor of SARS–CoV2 Mpro enzyme, and ritonavir to retard degradation of nirmatrelvir, is the current FDA recommended treatment of early COVID19. There is no evidence of broad antiviral activity of paxlovid against other diverse viruses such as the influenza virus, poxviruses, as well as a host of respiratory viruses. Although type I interferons (IFNs) are effective against SARS–CoV2 in cell cultures and in early COVID19 infections, they have not been broadly recommended as therapeutics for COVID19. We have developed stable peptidomimetics of both types I and II IFNs based on our noncanonical model of IFN signaling involving the C-terminus of the IFNs. We have also identified two members of intracellular checkpoint inhibitors called suppressors of cytokine signaling (SOCS), SOCS1 and SOCS3 (SOCS1/3), and shown that they are virus induced intrinsic virulence proteins with activity against IFN signaling enzymes JAK2 and TYK2. We developed a peptidomimetic antagonist, based on JAK2 activation loop, against SOCS1/3 and showed that it synergizes with the IFN mimetics for potent broad spectrum antiviral activity without the toxicity of intact IFN molecules. IFN mimetics and the SOCS1/3 antagonist should have an advantage over currently used antivirals in terms of safety and potency against a broad spectrum of viruses.
There is a somewhat Pavlovian response to viral infections that revolves around a very specific approach to development of or identification of antivirals. This approach resides in the chemistry/pharmacology subdiscipline known as medicinal chemistry. Further, there is a penchant for looking for therapeutics via an off-the-shelf approach. Case in point; remdesivir, the broad spectrum pro drug of a nucleotide analog, was approved early by the FDA for treatment of COVID19 in hospitalized patients (1). Remdesivir was originally developed to treat negative stranded RNA Ebola and Marburg virus infections (2). SARS-CoV2 virus is a plus stranded RNA virus, and remdesivir functions as an RNA-dependent RNA polymerase inhibitor, thus it interferes with replication of the virus gene (2, 3). Although FDA approved for treatment of COVID19, it is not clear that remdesivir has much benefit in treating serious illness, particularly regarding moderate to severe disease involving hospitalization beyond early stages of infection (3). On top of this, it can cause liver damage, thus limiting dose and time of treatment (4).
Another drug from the medicinal chemistry tool chest is molnupiravir, a nucleoside analog which like many off-the-shelf antivirals also interferes with SARS-CoV2 RNA-dependent RNA polymerase, but this does not mean that it is specific for SARS (5, 6). It was developed to treat the negative stranded virus of influenza (5). Molnupiravir’s initial metabolite is β-D-N4-hydroxycytidine which has been shown to induce mutations in the DNA of mammalian cells (6). This suggests potential cancer-causing properties of molnupiravir. Still, molnupiravir has been shown to be effective in reducing mild, early diagnosed COVID19 from progressing to severe disease and has been approved by the FDA for treatment of non-hospitalized COVID19 patients. The FDA has recommended, however, that molnuparivir should not be the first choice for treatment of COVID19 because of its potential mutational/carcinogenic side effects. In addition, there are the less concerning gut associated potential side effects of nausea, diarrhea, and dizziness (7, 8).
The Pfizer drug paxlovid is the preferred short term treatment of COVID19 as per the FDA. Paxlovid is really composed of two drugs. These are nirmatrelvir, the active ingredient, and ritonavir which slows down the degradation of nirmatrelvir (9). Nirmatrelvir is a peptidomimetic protease inhibitor that binds covalently to the catalytic cysteine residue of SARS-CoV2 protease Mpro (10). Mpro is involved in cleaving the virus polyproteins that results in the production of key virus nonstructural proteins. Paxlovid has shown remarkable effectiveness in a clinical trial involving oral administration within 5 days of onset of COVID19 symptoms with 89 percent reduction in hospitalizations that included COVID19 high risk older patients (9). Like the SARS-CoV2 virus spike protein, mutations have been observed in the Mpro protease (9, 10). While this has not been a clinical problem with respect to virus resistance to paxlovid so far, such resistance has occurred in the use of protease inhibitors in treatment of AIDS which is caused by the human immunodeficiency virus (HIV) (11). One potential way of dealing with this is to use multiple antivirals against the COVID19 virus as was done with AIDS (11). Finally, it should be noted that remdesivir has shown similar protection under similar conditions (3).
The primary innate host defense against viral infections of all types is the interferon (IFN) family of cytokines (12–15). The question arises as to whether the IFN system plays some significant role in host defense against SARS-CoV2 induced COVID19? Focus on this question has resided in types I and III IFNs. SARS-CoV2 is remarkably contagious and the level of mortality/morbidity is unacceptable, at least in most advanced economies. Most SARS-CoV2 infections are asymptomatic or mildly symptomatic, but nonetheless are of concern as these individuals could be contagious (16). If upper respiratory infection does take hold it most likely will result in mild to moderate COVID19 that resolves on its own in healthy individuals. The real concern is severe disease resulting in hospitalization, intensive care, and the potential of debilitating illness and death. It is thought that types I and III IFNs play key roles in the natural host defense that we see in asymptomatic and mild cases of COVID19. While it has been reported that there is no definitive evidence that type I IFNs have any significant benefit in severe COVID19 (17), a recent report describes a unique type I IFN signature in lung neutrophils from severe COVID patients (18) Further, there is no evidence that any of the therapeutics mentioned earlier, including paxlovid with its nirmatrelvir protease inhibitor, have had any significant effect on severe COVID19 (19). With the above reference to nirmatrelvir, it is thus appropriate to look at type I IFNs in terms of their effects on treatment of COVID19 at the early stages of infection. In this regard, IFN-α2b, IFN-β-1a, and IFN-β-1b have shown promise when administered intranasally or injected early in COVID19 infections (20–22).
Recently, monkeypox, a cousin to the smallpox virus and endemic to West Africa, has been reported virtually worldwide (23). Mortality from this virus is low, but its close relationship to smallpox virus warrants attention. The poxviruses are large double-stranded DNA viruses (24), not prone to significant mutations as are coronaviruses like SARS-CoV2. There are monkeypox vaccines and therapeutics in place. This is possibly because of the historical pandemic damage of the smallpox virus which is estimated to have killed 500 million people worldwide since 1900 alone (25). Massive global vaccination in the latter half of the twentieth century eradicated smallpox. There are a variety of tricks employed by the poxviruses to evade host defense in the unvaccinated, probably the most effective being induction of type I and type II IFN decoy receptors (25). Vaccinia virus is a poxvirus that is closely related to variola, the smallpox virus (24). It is highly lethal in mice due to its production of poxvirus B18R type I IFN decoy receptor (25). B18R is illustrative of the simplicity of the mechanism that is the basis of how a virus virulence factor can have a devastating effect on human life. In the IFN mimetic section, we will show how the IFN molecule can be adapted to deal with poxviruses as well as other lethal viruses, including SARS-CoV2 and influenza virus. We will also show that a key immune checkpoint inhibitor system which can function as intrinsic virus virulence factors and composed of a family of intracellular proteins called suppressors of cytokine signaling (SOCS), can be controlled through a simplified concept that results in strong antiviral activity against SARS-CoV2, influenza virus, herpes viruses, picornaviruses as well as a host of other viruses. As indicated above in the title, there is a simple, effective, and universal approach to dealing with a host of virus pathogens; and it is hiding in plain sight. It consists of IFN mimetics and peptidomimetic antagonist of SOCS1/3 (14, 26–28).
Intrinsically disordered proteins (IDPs) and intrinsically disordered regions (IDRs) in proteins involved in signal transduction have been revealed primarily through the technology of nuclear magnetic resonance (NMR) (29–31). This provided strong support to identification of critical binding sites in proteins in signal transduction that possess the remarkable property of recognition of multiple binding partners at the same time through short continuous and/or contiguous sequences.
Our peptide approach to structure/function predated the discovery that proteins involved in signaling are IDPs and/or proteins with critical IDRs. Our initial approach to studying IFNs via short peptides involved the synthesis of a 20-amino acid peptide corresponding to the N-terminus of human IFNγ (huIFNγ), coupling it to keyhole limpet hemocyanin macroprotein, and using it as an immunogen in rabbits (32). The resultant antibodies interacted specifically with natural huIFNγ, including neutralization of the IFN antiviral activity. The antibodies did not bind to or affect the activity of mouse IFNγ (muIFNγ), huIFNα, or huIFNβ. These results were foundational for the peptide approach to IFN function that has played a central role in our non-canonical model for IFN signaling specifically and cytokine, hormonal signaling in general.
The IFNγ and type I IFN mimetics that have resulted from the model and which are contained in the C-terminus sequences of the IFNs are presented in Table 1. The species sources of the IFNs are indicated; however, all IFN mimetics, contrary to their parent IFNs, are species nonspecific. That is, they are all equally active on human and murine cells as well as in their activity in mice (26, 27). MuIFNγ as well as huIFNγ possesses a functional polycationic nuclear localization sequence (NLS) that is required for function and which can be replaced by SV40 virus prototype polycationic NLS, resulting in full recovery of IFNγ antiviral activity (33). MuIFNγ aa (95-133) and huIFNγ aa (95–134) contain the NLSs, KTGKRK for mouse and RKRKRSR for human, in their C-terminus (See Table 1) (34, 35). MuIFNγ aa (95–133) was the first IFN mimetic discovered (36). Neither mouse IFNGR1 nor IFNGR2 receptor subunits possess a polycationic NLS (34). IFNGR2 remains on the cell surface while IFNGR1, with attached JAKs, undergoes energy-dependent nuclear translocation in IFNγ or IFNγ mimetic treated cells (35, 36). IFNγ binds primarily to IFNGR1 (35). Endocytosis results in polycationation of IFNγ in the acidic endosomes, resulting in movement to cytoplasm and binding to IFNGR1 cytoplasmic domain (36–38). This increases binding affinity of JAK2 for IFNGR1 and thus movement from IFNGR2 (35). IFNGR2 does not play a further role in signaling. IFNγ NLSs are required for nuclear transport and function of IFNγ as a complex of IFNγ, IFNGR1, activated Janus kinase 2 (JAK2), and activated Signal transducer and activator of transcription α (STAT1α). Details of the nuclear transport are summarized elsewhere (26, 37, 39–41). Importantly, the type I and type II IFN mimetics of Table 1 are sufficient for nuclear transport of the complexes and resultant IFN function. These mimetics function like their parent IFNs, including the antiviral activity that is not virus specific, as well as antitumor activity in a B16 mouse model of melanoma, and in treatment of experimental allergic encephalomyelitis (EAE), a mouse model of multiple sclerosis (See Table 1) (26, 27).
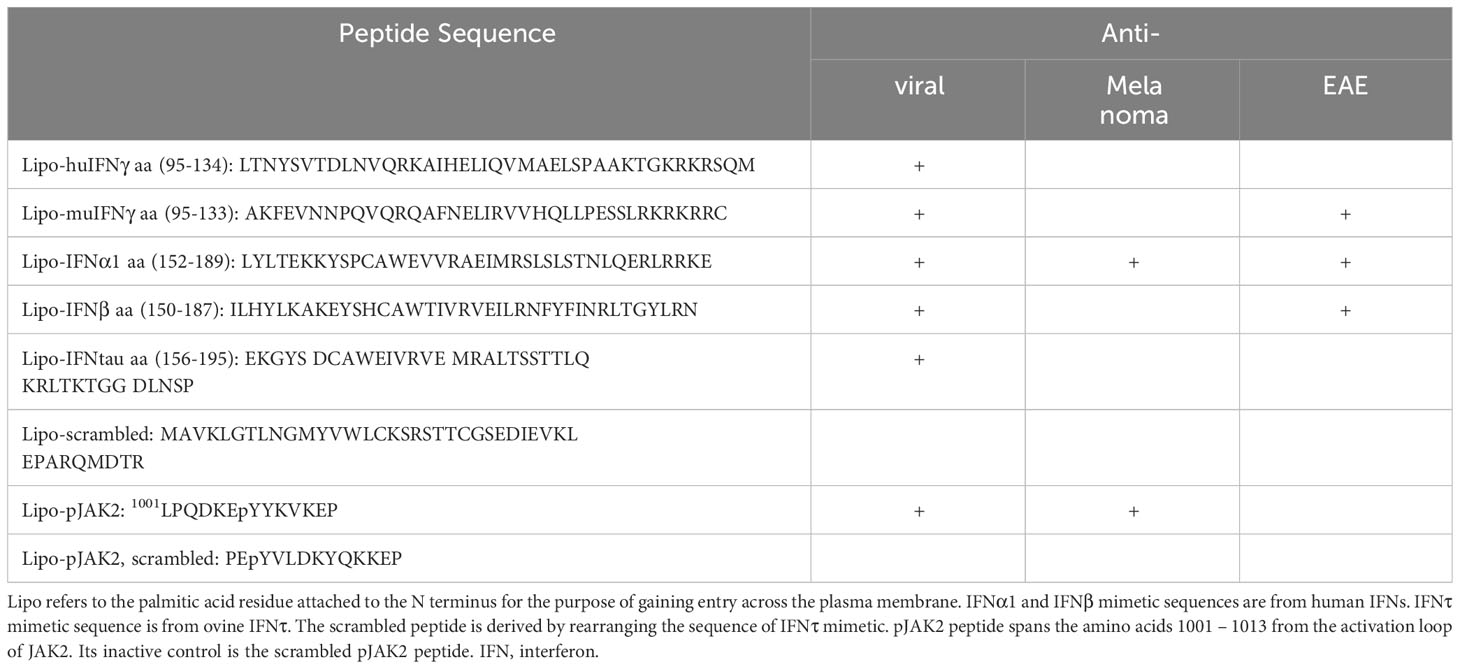
Table 1 IFN Mimetics and SOCS1/3 Antagonist.
The type I IFN mimetics of Table 1 were synthesized in terms of the C-terminal region of the parent IFNs using the IFNγ molecule as the template (42, 43). There is a marked difference, however, between these mimetics and the IFNγ mimetics. There is only one IFNγ for the type II receptor complex of IFNGR1/IFNGR2 (35). There may be up to 20 type I IFNs, all operating through the same IFNAR1/IFNAR2 receptor complex with varying degrees of antiviral and apoptotic effects (44, 45). IFNτ was originally identified as an ovine pregnancy hormone. It was later recognized as an IFN with low apoptotic activity (46). Some of the type I IFN mimetics do not possess an obvious putative polycationic NLS (See Table 1). Nuclear internalization of these mimetics and their parent IFNβ resides in a functional polycationic sequence in receptor subunit IFNAR1; specifically extracellular sequence 382RKIIEKKT of the precursor form of human IFNAR1 (44). Further, both type I receptor subunits IFNAR1 and IFNAR2 undergo active energy dependent nuclear transport complexed to IFN ligand and tyrosine kinase 2 (TYK2) Janus kinase (45). Thus, although IFN mimetic activity resides in the C-terminus of IFNs, there are differences in type I and type II IFN and IFN mimetic signaling in terms of both the ligands and their corresponding receptors. Perhaps related to the diversity of ligands for type I IFN activation relative to a single ligand for IFNGR, it may be evolutionarily advantageous to have the NLS in IFNAR1 rather than in the ligands, given that there are multiple ligands operating through a single receptor. Again, all of these mimetics lack the toxicity associated with the parent IFNs (27, 45). Thus, the relatively simpler IFNγ system has led to our successful discovery of the various type I IFN mimetics.
Viruses have developed a variety of mechanisms to inhibit or blunt the antiviral apparatus of the IFN system. Poxviruses are particularly clever at thwarting the IFN system. These are large double-stranded DNA viruses that replicate in the cytoplasm of the cell. The variola strain of the poxviruses is responsible for smallpox. Worldwide, it is estimated that smallpox has killed up to 500 million people in the 20th century and 400,000 annually in Europe in the 18th century (24). Modern medicine has responded to smallpox by global immunization, but this has been discontinued for many years which has basically resulted in lack of protection of the entire global community to natural or deliberate reintroduction of the virus into society. In addition, the emergence of new poxviruses by recombination within this family of viruses, or by their crossing the species barrier, such as was seen recently in the outbreak with the monkeypox virus from Africa to worldwide are strong reasons for developing effective therapies against this family of viruses.
The assembly of poxviruses in the cytoplasm of infected cells is complex, involving the generation of four types of infectious virus particles. Attachment, internalization, and disassembling of poxviruses is followed by initiation of three waves of mRNA synthesis. The first or early wave codes for virus growth factors and decoy cytokine receptors. Decoy receptors for both type I and type II IFNs are thus produced during early protein synthesis in poxvirus-infected cells. As indicated, the poxvirus decoy receptor for type I IFN is called B18R, while that for type II IFN IFNγ is called B8R (24, 25). Further, smallpox (variola) disease has wracked such havoc on humanity that worldwide immunization has resulted in its eradication by mid twentieth century. If the smallpox virus were to reemerge, we would have a global crisis the likes of COVID19, but with greater mortality and morbidity. This is why the worldwide outbreak of monkeypox, a cousin of smallpox, caused such alarm.
We have shown the mechanism of action of IFN and IFN mimetics (26, 37, 39–41), but we show here that IFN mimetics are quite effective against vaccinia virus in cell culture and in mouse protection. This has implications for treatment of monkeypox. Vaccinia virus, another relative of variola is not harmful to people, but is deadly in a mouse model of lethal poxvirus disease. Intranasal administration of 106 PFU of vaccinia virus (or 107 I.P. administration) results in 100 percent death by day 10. Administration of 20 mg/kg of huIFNα1 aa (152 -189) peptide mimetic I.P. up to 2 days post infection completely protected mice against death (47). It is noteworthy that orally administered muIFNγ aa (95 -133) was also completely protective against vaccinia virus (48). Unlike the parent IFN, the mimetic was nontoxic in mice and in cell culture. The mimetic had a hydrophobic residue (palmitate) attached to facilitate cell penetration. Intact muIFNγ at 5,000 units, in contrast, had no protective effect; all of the mice died by day 10 (47). There is nothing magical about this result. Poxvirus B8R decoy receptor competes with the cell IFNγ receptor, IFNGR1, extracellular domain for IFN. The mimetic binds not to the extracellular IFNGR1 domain but, rather, to the cytoplasmic IFNGR1 binding site, residues 253–287 (26, 37–41). Intact IFNγ after extracellular IFNGR1 binding, binds to the same intracellular IFNGR1 site following endocytosis (26, 37, 39–41). Cell culture experiments with vaccinia virus agree completely with the mouse results (27).
A human type I IFN mimetic, huIFNα1 aa (152–189), was similarly tested in the vaccinia virus mouse model as well as cell culture (27). Conditions were the same as above. As indicated, vaccinia virus like other members of the poxvirus family produces a type I IFN decoy receptor called B18R. HuIFNα1 aa (152–189), administered for the first six days of infection, completely protected mice against a lethal dose of vaccinia, while the intact huIFNα1 was not protective (27). Similar to the IFNγ mimetic, huIFNα1 aa (152–189) was not toxic, causing no loss of weight in contrast to parent huIFNα1 induction of significant weight loss (27). Other type I mimetics (See Table 1), along with huIFNα1 aa (152–189), showed similar anti-vaccinia virus inhibition in cell culture, while intact huIFNα1 did not block vaccinia virus replication (27).
IFNγ mimetic muIFNγ aa (95-133) was also active against a highly lethal strain of the picornavirus encephalomyocarditis (EMC) virus in a mouse model with a lethality similar to that of vaccinia virus (49). The type I IFN mimetic huIFNα1 aa (152–189) was also significantly effective in a very aggressive mouse melanoma (B16F10 cells) where untreated mice all died by 20 to 25 days when injected IP with melanoma cells, while mice treated IP with huIFNα1 aa (152–189) for 14 days showed 40 percent survival (27). HuIFNβ has been used extensively as an FDA approved drug in the treatment of relapsing remitting multiple sclerosis. Experimental allergic encephalomyelitis (EAE) is a widely studied mouse model of multiple sclerosis (27). Treatment of EAE with human IFNβ mimetic, huIFNβ aa (150–187), was as effective as the parent IFNβ in ameliorating paralysis (27). As mentioned in Table 1, IFNα aa (152-189) and IFNτ aa (156-195) mimetics were also effective in preventing the symptoms of EAE (27). Importantly, the mimetic was not toxic as compared with parent IFN, since treated mice did not suffer weight loss or decrease in lymphocytes (27, 45). In cell culture, the IFN mimetic caused less apoptosis than the natural IFN (45). We have demonstrated previously that a greater affinity of the IFN with its receptor subunit was responsible for greater cell cytotoxicity (46). Since the cell penetrating IFN mimetic does not interact with the extracellular surface of the receptor, that may account for lack of apoptosis in cells in culture (45).
The IFN system, including its mimetics, operate in a complex host defense environment. In addition to host protective players like the IFNs, there are the checkpoint inhibitors of the extent of host defense responders such as IFNs. Like their parent IFNs, the IFN mimetics must also contend with these inducible checkpoint inhibitors of host defense. Key checkpoint inhibitors in viral infections are two members of the suppressors of cytokine signaling (SOCS) family. The intracellular SOCS of interest are called SOCS1 and SOCS3 (28, 50, 51). SOCS1 and SOCS3 inhibit IFN signaling, in part, by binding to and inhibiting JAK kinases JAK2 (IFNγ) and TYK2 (types I and III IFNs). SOCS1 and SOCS3 are proteins replete with IDRs, including the N-terminal kinase inhibitory regions (KIRs) (52). We synthesized a short peptide that corresponded to the activation loop region of JAK2 (See Table 1) as well as a peptide that corresponded to KIR of SOCS1 and showed that peptide SOCS1-KIR bound to the Janus kinase 2 (JAK2) activation loop (Reviewed in reference (28). Further, we showed that SOCS1-KIR blocked activated JAK2 (pJAK2) function (28). The activated JAK2 has a phosphorylated tyrosine on residue 1007 of JAK2 (pJAK2). We synthesized this more specific peptide with the lipophilic palmitate attached for cell penetration. The aim was to provide a decoy receptor for KIR of SOCS1 and thus block SOCS1 function (28, 53). Indeed, pJAK2 peptide blocked both SOCS1 and SOCS3 function by binding to their KIRs. The events described here are illustrated in Figure 1.
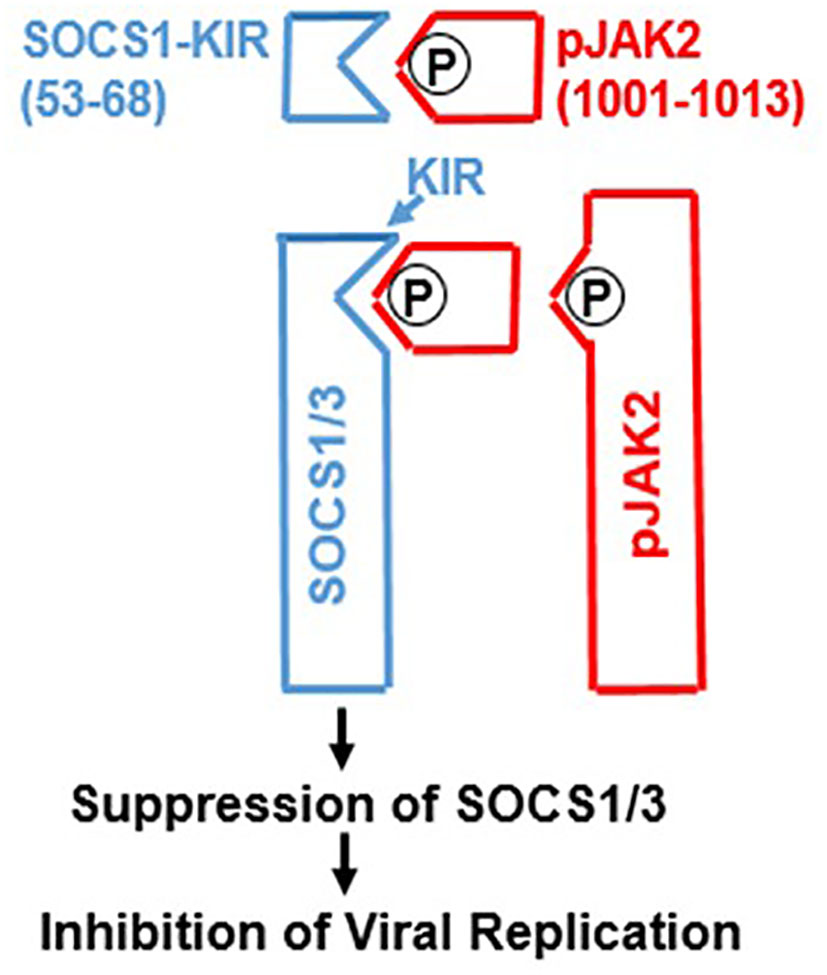
Figure 1 Development of SOCS1/3 antagonist. A peptide spanning amino acids 1001-1013 from the activation loop of tyrosine kinase JAK2, in which tyrosine at 1007 is phosphorylated was synthesized. Attachment of polyarginine (R9) or palmitoyllysine (lipo) to this peptide makes it cell permeable. This peptide binds to the kinase inhibitory region (KIR) of SOCS1 or SOCS3 and thereby allows the activation of JAK2 (or TYK2) for induction of antiviral activity. P, phosphorylation.
The development of the SOCS1/3 antagonist had profound inhibitory effects on cellular growth of poxvirus vaccinia virus, vesicular stomatitis virus, small RNA encephalomyocarditis (EMC) virus, herpes simplex virus, influenza virus, as well as coronaviruses including SARS-CoV2 (COVID19) and OC43 that causes common cold (14, 28, 48, 54, 55). Protection against lethal vaccinia virus, EMC virus, and influenza virus in infected mouse models was also observed (28, 48, 54). This protection was due to blockage of the effects of induced SOCS1 and SOCS3 on endogenous IFNs. Exogenous IFNs and IFN mimetics synergized with SOCS1/3 antagonist for enhanced antiviral effects in culture and in mice (14, 28, 48, 55). SOCS1/3 are thus virus induced endogenous virus virulence factors. An example of IFN mimetic and SOCS1/3 antagonist synergism in antiviral activity is illustrated in Figure 2 (14, 28, 48, 55). In the case of coronavirus OC43, one of the viruses responsible for the common cold, huIFNα1(152 –189) alone significantly inhibited virus induced cell toxicity, consistent with corresponding inhibition of OC43 RNA by inhibition of virus RNA polymerase/helicase activity. SOCS1/3 antagonist had a similar effect; together huIFNα1 aa (152–189) and SOCS1/3 antagonist peptide had a synergistic inhibitory effect on OC43 replication (55). The SOCS1/3 antagonist similarly inhibited replication of SARS-CoV2 RNA gene (55). The IFN mimetic was not tested against SARS-CoV2; however both OC43 and SARS-CoV2 are betacoronaviruses and thus the IFN mimetics and SOCS1/3 antagonist are likely to show synergism against SARS-CoV2. Like other viruses that have been tested, OC43 induces both SOCS1 and SOCS3 (SOCS1/3) in cell cultures (55). These results suggest two things; 1. SOCS1/3 antagonist blocks SOCS1 and SOCS3 inhibition of endogenous IFN in virus infected cells; 2. Addition of IFN mimetics provide excess IFN activity in virus infected cells, adding to that of endogenous IFNs. In addition, SOCS1/3 antagonist was shown to have the following beneficial effects. 1. It enhanced endogenous IFNβ levels; 2. It had adjuvant effects on both cellular and humoral immunity; 3. It was capable of generating immune responses against such poor antigens as bovine serum albumin (56). Likewise, IFNα1 aa (152-189) peptide was also shown to be a strong adjuvant in enhancing both CD4 and CD8 T cell responses (47). A combination of these properties is particularly relevant in the fight against SARS-CoV-2, because it was shown recently that COVID19 infected individuals who were vaccinated and/or exposed to SARS-CoV-2 had a poor response in generating CD8 T cell responses (57). In addition to the viruses mentioned above, there is a number of other viruses of clinical significance that are involved in epidemics/pandemics that induce SOCS1/3 intrinsic virulence factors. Use of the SOCS1/3 antagonist and IFN mimetics should thus prevent and/or blunt the adverse public health effects of these viruses. Viruses in this category include Ebola, Respiratory Syncytial virus, Zika, Dengue and West Nile viruses.
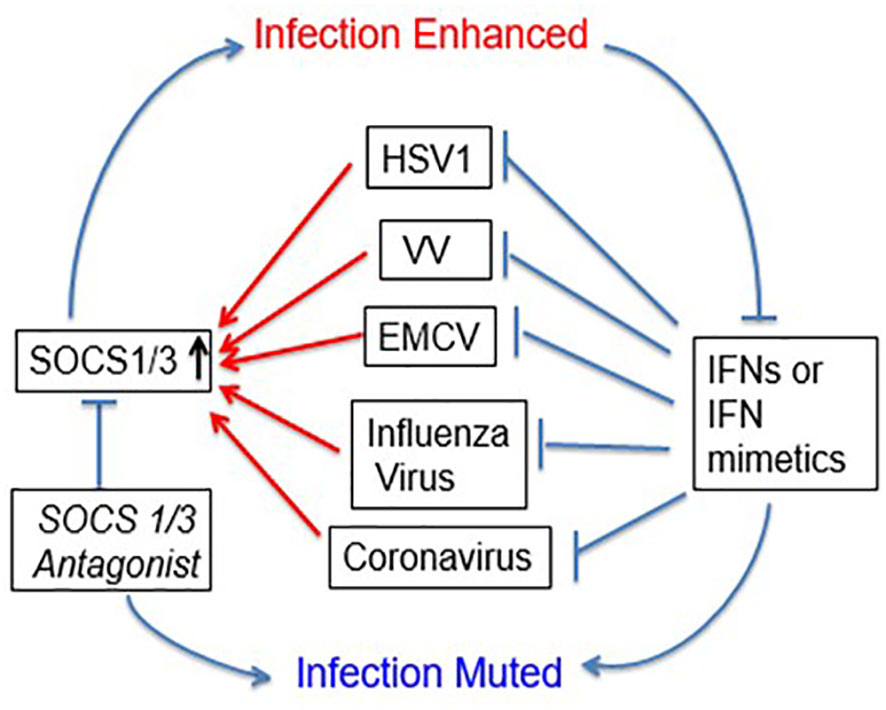
Figure 2 Viral exploitation of host SOCS1/3 to dampen host immune response. Several viruses, including beta coronaviruses have been shown to induce host SOCS1/3 which results in inhibition of signaling through JAK2 and TYK2. A SOCS1/3 antagonist peptide (aka pJAK2) suppresses this activity, resulting in the inhibition of viral replication. IFN mimetic peptide alone, or in synergy with pJAK2 peptide blocks the viral replication.
It has recently been reported that individuals immunocompromised with active AIDS are prone to severe monkeypox infection (58). Related to this, a correlation has been shown between upregulation of SOCS1 and SOCS3 as well as PD-1 in dendritic cells and increased HIV plasma load in AIDS patients (59). A similar correlation has been shown in a methicillin-resistant Staphylococcus aureus (MRSA) mouse model where skin infection was associated with increased SOCS1 (60). Treatment with a SOCS1 antagonist similar to that described here inhibited SOCS1 and improved host defense against MRSA. We consider these findings preliminary, but suggestive of a role for induced SOCS1 and/or SOCS3 in some forms of immune deficiency. Any evidence of induced SOCS1 and/or SOCS3 playing a role in bacterial infections of immunocompromised terminally ill cancer and non-cancer patients would be of profound importance in controlling these infections and extending lives.
While the paxlovid protease inhibitor nirmatrelvir is currently the FDA preferred therapeutic for early mild COVID19, a phenomenon called rebound COVID19 has been reported in paxlovid treated patients (61). This coupled with the recent demonstration of multiple mutations, with varying degrees of resistance in SARS-CoV2 3CL protease in serial cell cultures of nirmatrelvir and SARS-CoV2 variants, portends virus resistance to this therapeutic over time (62). It would seem, therefore, that our system of IFN mimetics and SOCS antagonists combined with nirmatrelvir e is a logical next step in future studies of SARS-CoV2 variants in COVID19 outbreaks.
We have discovered and developed a noncanonical model of the mechanism of action of types I and II IFNs. We have further shown a remarkable similarity of this mechanism to that of steroid/steroid receptor signaling. In this regard, we have unified ligand/membrane receptor signaling with that of cytoplasmic/nuclear receptor signaling. Membrane receptor signaling is conceptually dominated by receptor tyrosine kinases and receptor associated tyrosine kinases. The latter in turn is dominated by the canonical model of JAK/STAT signaling pathway. It is our contention that JAK/STAT signaling in the context of this model is incomplete and replete with inexplicable add-on adjustments that have led to such phrases as “The STAT specificity paradox” (63). The central problem is ascribing the specificity of cytokine signaling to STAT transcription factors, and to be inflexible concerning this even in the face of activated JAKs in the nucleus of cells exposed to cytokines and growth factors (37, 64). That activated JAK2 is involved in the key epigenetics of tyrosine phosphorylation on histone H3 for exposure of DNA stirs little attention (65). The types I and II IFN mimetics described here, as per the non-canonical model of IFN signaling have the potential to be effective therapeutics for viral infections, autoimmune disorders, and cancers. Controlled interactions of IFN mimetics and antagonist of the SOCS1/3 checkpoint inhibitors contribute to IFN mimetic activity. Where tested, our mimetics and antagonist, like the FDA-approved peptidomimetic nirmatrelvir, can be effective antiviral and anticancer immunotherapeutics when administered parenterally or orally.
It is our view that their therapeutic value is hiding in plain sight, waiting to be exploited.
HJ designed the research and wrote the manuscript. CA carried out the experiments. All authors contributed to the article and approved the submitted version.
This research was supported by NIH grant AI 056152 to HJ. HJ and CA are listed as inventors of interferon mimetics and SOCS1/3 antagonist in a patent held by the University of Florida.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the treatment of covid-19 - final report. N Engl J Med (2020) 383(19):1813–26. doi: 10.1056/NEJMoa2007764
2. Tchesnokov EP, Feng JY, Porter DP, Götte M. Mechanism of inhibition of ebola virus RNA-dependent RNA polymerase by remdesivir. Viruses (2019) 11(4):p326. doi: 10.3390/v11040326
3. Gottlieb RL, Vaca CE, Paredes R, Mera J, Webb BJ, Perez G, et al. Early remdesivir to prevent progression to severe covid-19 in outpatients. N Engl J Med (2022) 386(4):305–15. doi: 10.1056/NEJMoa2116846
4. Yang CJ, Wei YJ, Chang HL, Chang PY, Tsai CC, Chen YH, et al. Remdesivir use in the coronavirus disease 2019 pandemic: A mini-review. J Microbiol Immunol Infect (2021) 54(1):27–36. doi: 10.1016/j.jmii.2020.09.002
5. Yip AJW, Low ZY, Chow VTK, Lal SK. Repurposing molnupiravir for COVID-19: the mechanisms of antiviral activity. Viruses (2022) 14(6):p1345. doi: 10.3390/v14061345
6. Hashemian SMR, Pourhanifeh MH, Hamblin MR, Shahrzad MK, Mirzaei H. RdRp inhibitors and COVID-19: Is molnupiravir a good option? BioMed Pharmacother (2022) 146:112517. doi: 10.1016/j.biopha.2021.112517
7. Tian L, Pang Z, Li M, Lou F, An X, Zhu S, et al. Molnupiravir and its antiviral activity against COVID-19. Front Immunol (2022) 13:855496. doi: 10.3389/fimmu.2022.855496
8. Pourkarim F, Pourtaghi-Anvarian S, Rezaee H. Molnupiravir: A new candidate for COVID-19 treatment. Pharmacol Res Perspect (2022) 10(1):e00909. doi: 10.1002/prp2.909
9. Hammond J, Leister-Tebbe H, Gardner A, Abreu P, Bao W, Wisemandle W, et al. Oral nirmatrelvir for high-risk, nonhospitalized adults with covid-19. N Engl J Med (2022) 386(15):1397–408. doi: 10.1056/NEJMoa2118542
10. Greasley SE, Noell S, Plotnikova O, Ferre R, Liu W, Bolanos B, et al. Structural basis for the in vitro efficacy of nirmatrelvir against SARS-CoV-2 variants. J Biol Chem (2022) 298(6):101972. doi: 10.1016/j.jbc.2022.101972
11. Flexner C. HIV-protease inhibitors. N Engl J Med (1998) 338(18):1281–92. doi: 10.1056/NEJM199804303381808
12. Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev (2001) 14(4):778–809. doi: 10.1128/CMR.14.4.778-809.2001
13. Liu G, Gack MU. Insights into pandemic respiratory viruses: manipulation of the antiviral interferon response by SARS-CoV-2 and influenza A virus. Curr Opin Immunol (2022) 78:102252. doi: 10.1016/j.coi.2022.102252
14. Johnson HM, Lewin AS, Ahmed CM. SOCS, intrinsic virulence factors, and treatment of COVID-19. Front Immunol (2020) 11:582102. doi: 10.3389/fimmu.2020.582102
15. Aricò E, Bracci L, Castiello L, Urbani F, Casanova JL, Belardelli F. Exploiting natural antiviral immunity for the control of pandemics: Lessons from Covid-19. Cytokine Growth Factor Rev (2022) 63:23–33. doi: 10.1016/j.cytogfr.2021.12.001
16. Chirico F, Nucera G, Ilesanmi O, Afolabi A, Pruc M, Szarpak L. Identifying asymptomatic cases during the mass COVID-19 vaccination campaign: insights and implications for policy makers. Future Virol (2021). doi: 10.2217/fvl-2021-0243
17. Hung IF, Lung KC, Tso EY, Liu R, Chung TW, Chu MY, et al. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet (2020) 395:1695–704. doi: 10.1016/S0140-6736(20)31042-4
18. Margaroli C, Fram T, Sharma NS, Patel SB, Tipper J, Robison SW, et al. Interferon-dependent signaling is critical for viral clearance in airway neutrophils. JCI Insight (2023) 8(10):e167042. doi: 10.1172/jci.insight.167042
19. Coronavvirus disease 2019 (COVID), treatment guidelines. Available at: https://www.covid19treatmentguidelines.nih.gov/.
20. Wang N, Zhan Y, Zhu L, Hou Z, Liu F, Song P, et al. Retrospective multicenter cohort study shows early interferon therapy is associated with favorable clinical responses in COVID-19 patients. Cell Host Microbe (2020) 28(3):455–64.e2. doi: 10.1016/j.chom.2020.07.005
21. Cojocaru E, Cojocaru C, Antoniu SA, Stafie CS, Rajnoveanu A, Rajnoveanu RM. Inhaled interferons beta and SARS-COV2 infection: a preliminary therapeutic perspective. Expert Rev Respir Med (2022) 16(3):257–61. doi: 10.1080/17476348.2022.2008910
22. Zhou Q, Chen V, Shannon CP, Wei XS, Xiang X, Wang X, et al. Interferon-α2b treatment for COVID-19. Front Immunol (2020) 11:1061. doi: 10.3389/fimmu.2020.01061
23. Lum FM, Torres-Ruesta A, Tay MZ, Lin RTP, Lye DC, Rénia L, et al. Monkeypox: disease epidemiology, host immunity and clinical interventions. Nat Rev Immunol (2022) 22(10):597–613. doi: 10.1038/s41577-022-00775-4
24. Moss B. Poxvirus DNA replication. Cold Spring Harb Perspect Biol (2013) 5(9):a010199. doi: 10.1101/cshperspect.a010199
25. Smith GL, Benfield CTO, Maluquer de Motes C, Mazzon M, Ember SWJ, Ferguson BJ, et al. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J Gen Virol (2013) 94(Pt 11):2367–92. doi: 10.1099/vir.0.055921-0
26. Johnson HM, Noon-Song EN, Dabelic R, Ahmed CM. IFN signaling: how a non-canonical model led to the development of IFN mimetics. Front Immunol (2013) 4:202. doi: 10.3389/fimmu.2013.00202
27. Ahmed CM, Johnson HM. Short Peptide type I interferon mimetics: therapeutics for experimental allergic encephalomyelitis, melanoma, and viral infections. J Interferon Cytokine Res (2014) 34(10):802–9. doi: 10.1089/jir.2014.0041
28. Ahmed CM, Larkin J 3rd, Johnson HM. SOCS1 mimetics and antagonists: A complementary approach to positive and negative regulation of immune function. Front Immunol (2015) 6:183. doi: 10.3389/fimmu.2015.00183
29. Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol (2015) 16(1):18–29. doi: 10.1038/nrm3920
30. Dyson HJ, Wright PE. Perspective: the essential role of NMR in the discovery and characterization of intrinsically disordered proteins. J Biomol NMR (2019) 73(12):651–9. doi: 10.1007/s10858-019-00280-2
31. Dyson HJ, Wright PE. NMR illuminates intrinsic disorder. Curr Opin Struct Biol (2021) 70:44–52. doi: 10.1016/j.sbi.2021.03.015
32. Johnson HM, Langford MP, Lakhchaura B, Chan TS, Stanton GJ. Neutralization of native human gamma interferon (HuIFN gamma) by antibodies to a synthetic peptide encoded by the 5' end of HuIFN gamma cDNA. J Immunol (1982) 129(6):2357–9. doi: 10.4049/jimmunol.129.6.2357
33. Subramaniam PS, Green MM, Larkin J, Torres BA, Johnson HM. Nuclear translocation of IFN-gamma is an intrinsic requirement for its biologic activity and can be driven by a heterologous nuclear localization sequence. J Interferon Cytokine Res (2001) 21(11):951–9. doi: 10.1089/107999001753289569
34. Subramaniam PS, Mujtaba MG, Paddy MR, Johnson HM. The carboxyl terminus of interferon-gamma contains a functional polybasic nuclear localization sequence. J Biol Chem (1999) 274(1):403–7. doi: 10.1074/jbc.274.1.403
35. Larkin J, Johnson HM, Subramaniam PS. Differential nuclear localization of the IFNGR-1 and IFNGR-2 subunits of the IFN-gamma receptor complex following activation by IFN-gamma. J Interferon Cytokine Res (2000) 20(6):565–76. doi: 10.1089/10799900050044769
36. Subramaniam PS, Johnson HM. Lipid microdomains are required sites for the selective endocytosis and nuclear translocation of IFN-gamma, its receptor chain IFN-gamma receptor-1, and the phosphorylation and nuclear translocation of STAT1alpha. J Immunol (2002) 169(4):1959–69. doi: 10.4049/jimmunol.169.4.1959
37. Johnson HM, Noon-Song E, Ahmed CM. Noncanonical IFN signaling, steroids, and STATs: A probable role of V-ATPase. Mediators Inflamm (2019) 2019:4143604. doi: 10.1155/2019/4143604
38. Szente BE, Subramaniam PS, Johnson HM. Identification of IFN-gamma receptor binding sites for JAK2 and enhancement of binding by IFN-gamma and its C-terminal peptide IFN-gamma(95-133). J Immunol (1995) 155(12):5617–22. doi: 10.4049/jimmunol.155.12.5617
39. Johnson HM, Subramaniam PS, Olsnes S, Jans DA. Trafficking and signaling pathways of nuclear localizing protein ligands and their receptors. Bioessays (2004) 26(9):993–1004. doi: 10.1002/bies.20086
40. Johnson HM, Noon-Song EN, Kemppainen K, Ahmed CM. Steroid-like signalling by interferons: making sense of specific gene activation by cytokines. Biochem J (2012) 443(2):329–38. doi: 10.1042/BJ20112187
41. Johnson HM, Ahmed CM. Noncanonical IFN signaling: mechanistic linkage of genetic and epigenetic events. Mediators Inflamm (2016) 2016:9564814. doi: 10.1155/2016/9564814
42. Szente BE, Johnson HM. Binding of IFN gamma and its C-terminal peptide to a cytoplasmic domain of its receptor that is essential for function. Biochem Biophys Res Commun (1994) 201(1):215–21. doi: 10.1006/bbrc.1994.1691
43. Szente BE, Soos JM, Johnson HW. The C-terminus of IFN gamma is sufficient for intracellular function. Biochem Biophys Res Commun (1994) 203(3):1645–54. doi: 10.1006/bbrc.1994.2375
44. Subramaniam PS, Johnson HM. The IFNAR1 subunit of the type I IFN receptor complex contains a functional nuclear localization sequence. FEBS Lett (2004) 578(3):207–10. doi: 10.1016/j.febslet.2004.10.085
45. Ahmed CM, Noon-Song EN, Kemppainen K, Pascalli MP, Johnson HM. Type I IFN receptor controls activated TYK2 in the nucleus: implications for EAE therapy. J Neuroimmunol (2013) 254(1-2):101–9. doi: 10.1016/j.jneuroim.2012.10.006
46. Subramaniam PS, Khan SA, Pontzer CH, Johnson HM. Differential recognition of the type I interferon receptor by interferons tau and alpha is responsible for their disparate cytotoxicities. Proc Natl Acad Sci U S A (1995) 92(26):12270–4. doi: 10.1073/pnas.92.26.12270
47. Ahmed CM, Johnson HM. Type I interferon mimetics bypass vaccinia virus decoy receptor virulence factor for protection of mice against lethal infection. Clin Vaccine Immunol (2014) 21(8):1178–84. doi: 10.1128/CVI.00204-14
48. Ahmed CM, Martin JP, Johnson HM. IFN mimetic as a therapeutic for lethal vaccinia virus infection: possible effects on innate and adaptive immune responses. J Immunol (2007) 178(7):4576–83. doi: 10.4049/jimmunol.178.7.4576
49. Mujtaba MG, Patel CB, Patel RA, Flowers LO, Burkhart MA, Waiboci LW, et al. The gamma interferon (IFN-gamma) mimetic peptide IFN-gamma (95-133) prevents encephalomyocarditis virus infection both in tissue culture and in mice. Clin Vaccine Immunol (2006) 13(8):944–52. doi: 10.1128/CVI.00021-06
50. Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol (2007) 7(6):454–65. doi: 10.1038/nri2093
51. Akhtar LN, Benveniste EN. Viral exploitation of host SOCS protein functions. J Virol (2011) 85(5):1912–21. doi: 10.1128/JVI.01857-10
52. Du Z, Uversky VN. A comprehensive survey of the roles of highly disordered proteins in type 2 diabetes. Int J Mol Sci (2017) 18(10):p2010. doi: 10.3390/ijms18102010
53. Waiboci LW, Ahmed CM, Mujtaba MG, Flowers LO, Martin JP, Haider MI, et al. Both the suppressor of cytokine signaling 1 (SOCS-1) kinase inhibitory region and SOCS-1 mimetic bind to JAK2 autophosphorylation site: implications for the development of a SOCS-1 antagonist. J Immunol (2007) 178(8):5058–68. doi: 10.4049/jimmunol.178.8.5058
54. Ahmed CM, Dabelic R, Bedoya SK, Larkin J 3rd, Johnson HM. A SOCS1/3 antagonist peptide protects mice against lethal infection with influenza A virus. Front Immunol (2015) 6:574. doi: 10.3389/fimmu.2015.00574
55. Ahmed CM, Grams TR, Bloom DC, Johnson HM, Lewin AS. Individual and synergistic anti-coronavirus activities of SOCS1/3 antagonist and interferon α1 peptides. Front Immunol (2022) 13:902956. doi: 10.3389/fimmu.2022.902956
56. Ahmed CM, Dabelic R, Martin JP, Jager LD, Haider SM, Johnson HM. Enhancement of antiviral immunity by small molecule antagonist of suppressor of cytokine signaling. J Immunol (2010) 185(2):1103–13. doi: 10.4049/jimmunol.0902895
57. Gao F, Mallajoysula V, Arunachalam PS, van der Ploeg K, Manohar M, Röltgen K, et al. Spheromers reveal robust T cell responses to the Pfizer/BioNTech vaccine and attenuated peripheral CD8. Immunity (2023) 56(4):864–78.e4. doi: 10.1016/j.immuni.2023.03.005
58. Mitjà O, Alemany A, Marks M, Lezama Mora JI, Rodríguez-Aldama JC, Torres Silva MS, et al. Mpox in people with advanced HIV infection: a global case series. Lancet (2023) 401(10380):939–49. doi: 10.1016/S0140-6736(23)00273-8
59. Sachdeva M, Sharma A, Arora SK. Increased expression of negative regulators of cytokine signaling during chronic HIV disease cause functionally exhausted state of dendritic cells. Cytokine (2017) 91:118–23. doi: 10.1016/j.cyto.2016.08.010
60. Klopfenstein N, Brandt SL, Castellanos S, Gunzer M, Blackman A, Serezani CH. SOCS-1 inhibition of type I interferon restrains Staphylococcus aureus skin host defense. PloS Pathog (2021) 17(3):e1009387. doi: 10.1371/journal.ppat.1009387
61. Anderson AS, Caubel P, Rusnak JM, Investigators E-HT. Nirmatrelvir-ritonavir and viral load rebound in covid-19. N Engl J Med (2022) 387(11):1047–9. doi: 10.1056/NEJMc2205944
62. Iketani S, Mohri H, Culbertson B, Hong SJ, Duan Y, Luck MI, et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature (2022) 613:558–64. doi: 10.1101/2022.08.07.499047
63. Villarino AV, Kanno Y, O'Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol (2017) 18(4):374–84. doi: 10.1038/ni.3691
64. Dawson MA, Bannister AJ, Göttgens B, Foster SD, Bartke T, Green AR, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature (2009) 461(7265):819–22. doi: 10.1038/nature08448
Keywords: monkeypox virus, coronavirus, antivirals, interferon, SOCS antagonist
Citation: Johnson HM and Ahmed CM (2023) Disparate viral pandemics from COVID19 to monkeypox and beyond: a simple, effective and universal therapeutic approach hiding in plain sight. Front. Immunol. 14:1208828. doi: 10.3389/fimmu.2023.1208828
Received: 19 April 2023; Accepted: 16 November 2023;
Published: 01 December 2023.
Edited by:
Ma Luo, Public Health Agency of Canada (PHAC), CanadaReviewed by:
J. Edwin Blalock, University of Alabama at Birmingham, United StatesCopyright © 2023 Johnson and Ahmed. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Howard M. Johnson, am9obnNvbmhAdWZsLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.