 Minchuan Zhang
Minchuan Zhang Kong-Peng Lam1,2,3*
Kong-Peng Lam1,2,3* Shengli Xu
Shengli Xu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 09 August 2023
Sec. NK and Innate Lymphoid Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1207276
This article is part of the Research TopicChallenges in NK Cell Therapeutic DevelopmentView all 8 articles
Natural Killer (NK) cells are a type of innate lymphoid cells that play a crucial role in immunity by killing virally infected or tumor cells and secreting cytokines and chemokines. NK cell-mediated immunotherapy has emerged as a promising approach for cancer treatment due to its safety and effectiveness. NK cell engagers (NKCEs), such as BiKE (bispecific killer cell engager) or TriKE (trispecific killer cell engager), are a novel class of antibody-based therapeutics that exhibit several advantages over other cancer immunotherapies harnessing NK cells. By bridging NK and tumor cells, NKCEs activate NK cells and lead to tumor cell lysis. A growing number of NKCEs are currently undergoing development, with some already in clinical trials. However, there is a need for more comprehensive studies to determine how the molecular design of NKCEs affects their functionality and manufacturability, which are crucial for their development as off-the-shelf drugs for cancer treatment. In this review, we summarize current knowledge on NKCE development and discuss critical factors required for the production of effective NKCEs.
Immunotherapy has profoundly impacted cancer treatment, spurring a multi-billion-dollar industry devoted to discovering and developing novel immunotherapeutic drugs. Unlike conventional chemotherapy or radiotherapy, which directly targets tumor cells, immunotherapy harnesses immune cells to combat cancer. Therapeutic antibodies against immune checkpoint receptors, such as PD-1 and CTLA-4, along with adoptive transfer of genetically engineered chimeric antigen-receptor (CAR)-T cells, have demonstrated tremendous success in treating some cancers. Specifically, checkpoint inhibitors have been notably effective against skin and lung cancers, while CAR-T therapy has been remarkably successful in the treatment of some haematological malignancies, with both unleashing the full antitumor activities of T cells (1).
While T-cell immunotherapies exhibit remarkable advantages, they also present some constraints and limitations. For example, although anti-PD-1 and anti-CTLA-4 monoclonal antibodies show spectacular clinical outcomes (>70% efficacy) against cancers such as melanoma, they can only achieve less than 20-30% efficacy in most other cancer types, exhibiting considerable variability among patient responses and often leading to relapse in initial responders (2, 3). Likewise, the USA FDA-approved CAR-T cell therapy, such as Yescarta and Kymriah, was initially perceived as a medical miracle as a single dose could cure certain blood cancers. Still, ongoing research uncovers several challenges, including disease relapse, limited success in solid tumors, and severe side effects such as cytokine release syndrome (CRS) and neurotoxicity, which hinder their broad adoption (4–7). Additionally, CAR-T therapy is limited by the quantity and quality of autologous T cells obtained from cancer patients, who are often immune compromised as a result of the disease or treatments. The prolonged and complex process of scaling up the production of autologous CAR-T cells for patients with limited life span and its daunting price tag also pose significant hurdles (8, 9).
These challenges underscore the urgent need to develop new immunotherapeutic modalities and the exploration of other types of immune cells, such as Natural Killer (NK) cells. NK cells could potentially circumvent some of the inherent limitations of T-cell immunotherapy. As one of the initial defences in antitumor immunity, NK cells naturally possess the ability to kill tumor cells without prior sensitization and modulate antitumor functions via cytokine secretion (10). Therapeutic antibodies inducing NK cell antibody-dependent cellular cytotoxicity (ADCC) and genetically engineered CAR-NK cells have been developed for cancer treatment. However, these two modalities bear some inherent limitations. For instance, polymorphism at positions 48 and 158 of CD16a has been reported to impact human IgG1 binding and subsequently influence NK cell ADCC (11–14). Moreover, it is well-known that CAR-NK cells present significant challenges in genetic manipulation, and their antitumor efficacy is limited by the short life of NK cells (15, 16). Another class of immunotherapeutic modalities harnessing NK cells, known as NK cell engagers (NKCEs), has recently emerged and exhibited several advantages over NK cell ADCC and CAR-NK cell strategies. In this review, we will discuss various aspects with regard to NKCE development, including the selection of target molecules for NK activation by NKCEs, the design of NKCE formats, and factors impacting the functionality and manufacturability of NKCEs. Moreover, we aim to illuminate the challenges and opportunities in developing NKCEs as potent immunotherapeutics for cancer treatment.
NK cells are a type of innate immune cells belonging to the group 1 innate lymphoid cell (ILC) family. They play a crucial role in the body’s first line of defence by identifying and eliminating stressed cells, including those infected by viruses and tumor cells (17, 18). Unlike T cells, NK cells do not express CD3; instead, they express CD56 on their cell surface. They constitute 5% to 15% of circulating lymphocytes in the blood (19). NK cells can be categorized into two major subsets based on their CD56 and CD16a expression: (1) immature CD56brightCD16adim NK cells, known for their high production of cytokines, such as IFNγ, and (2) mature CD56dimCD16bright NK cells, making up 90% of peripheral blood NK cells, primarily exerting cytotoxic function (19).
NK cells recognize membrane-bound ligands on target cells through an array of germline-encoded activating and inhibitory receptors on their cell surface (17). The activation status of NK cells is determined by the balance of activating and inhibitory signals they receive upon contact with other cells. Major histocompatibility complex class I molecules (MHC I)/Human leukocyte antigens (HLA), which are abundantly expressed on healthy cells, inhibit NK cell activation through the killer immunoglobulin-like (KIR) family of inhibitory receptors present on the surface of NK cells. Conversely, NK cells become activated when there is a downregulation of HLA molecules on the target cells or an upregulation of the ligands for NK activating receptors, such as NKp46 and NKG2D. NK cells also get activated when their CD16a receptors are engaged by antigen-bound IgGs. When activating signals prevail over inhibitory signals, NK cells become activated and release cytotoxic granules loaded with perforin and granzymes, causing target cells to lyse. In addition, NK cells can induce apoptosis of target cells through Fas-L or TRAIL (20), especially during the later phases of NK cell serial killing (21).
Compared to T cells, NK cells have several unique advantages in the context of immune cell therapy. As they are not HLA-restricted, NK cells can be sourced allogenically, thereby making them a feasible “off-the-shelf” therapeutic option. KIR-ligand-mismatch between donors and recipients can even potentially enhance the effectiveness of adoptive NK cell therapy by circumventing inhibitory signals on host cells (22, 23). Additionally, unlike T cells, NK cells do not secrete high levels of cytokines that could trigger CRS. They also do not cause graft-versus-host (GVH) reactions, making them a safer treatment option. As of 2021, more than 400 reported clinical trials for cancer treatment based on NK cells have been reported (24) including monotherapies involving NK cells from various sources, such as peripheral blood,umbilical cord blood, hematopoietic stem cell-derived NK, and CAR-NK cells (24). Additionally, combination therapies have been explored, which pair NK cells with other therapeutic agents, such as immune checkpoint inhibitors, antibodies or NK cell engagers (24).
There are three primary approaches directing NK cells for cancer treatment: 1) employing NK cell antibody-dependent cellular cytotoxicity (NK cell ADCC), which is initiated through the Fc receptor CD16a on NK cells and directed by a monoclonal antibody (mAb); 2) genetically engineering CAR-NK cells that specifically target tumor antigens using the CAR and become activated; 3) developing recombinant proteins, known as NK cell engagers (NKCE), that bring tumor and NK cells into proximity and activate the NK cells (Figure 1).
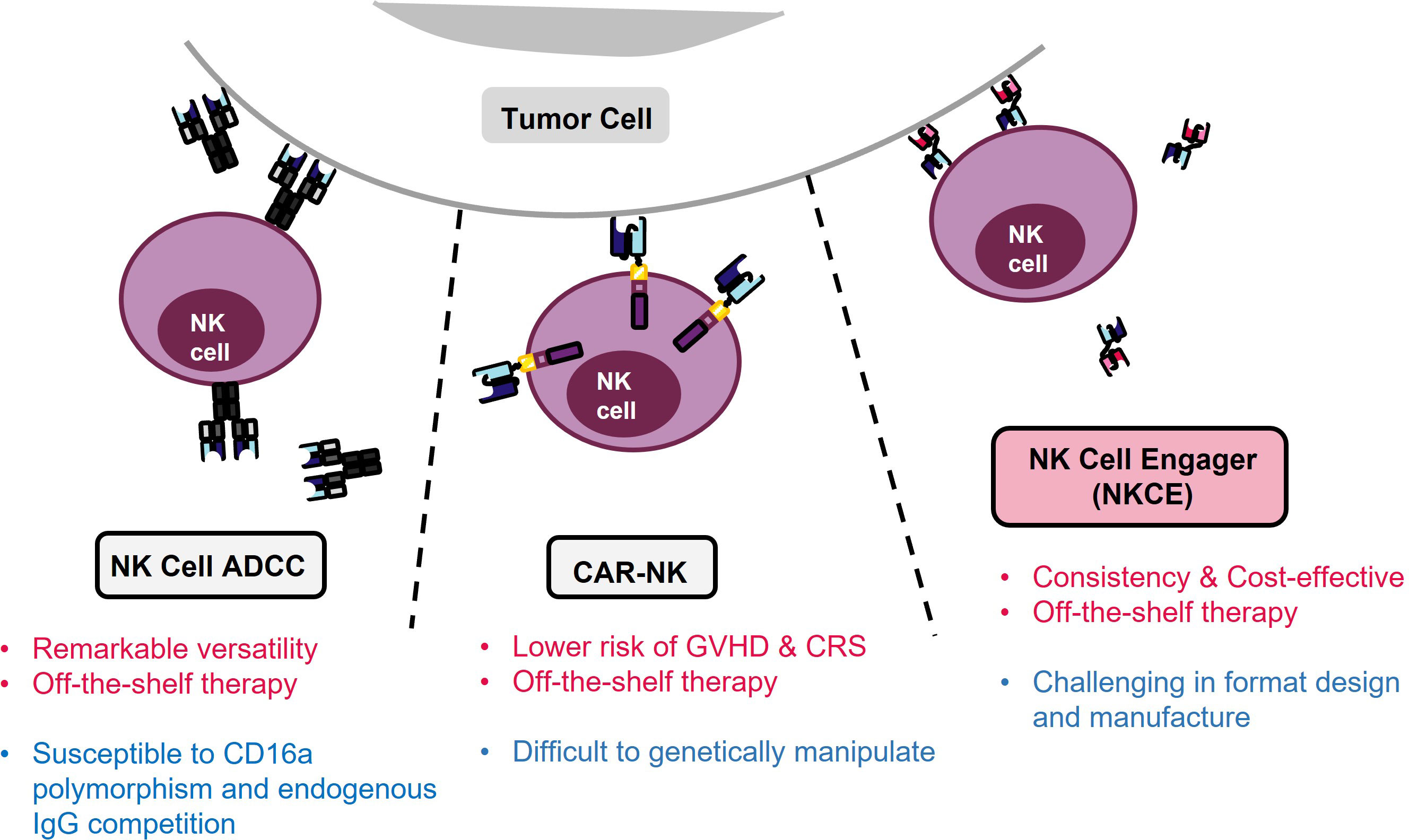
Figure 1 Three strategies for NK immunotherapy. Pros of each strategy are denoted in red, and limitation is marked in blue. NK ADCC targets tumor cells using monoclonal antibodies (mAbs) and activates NK cells through the CD16a Fc receptor. However, the efficacy of NK ADCC can be reduced by polymorphism of CD16a, resulting in lower binding of mAbs. In contrast, CAR-NK therapy avoids the problems associated with CD16a polymorphism but it suffers from difficulties in genetic manipulation of NK cells. On the other hand, NKCE offers a balanced profile and a cost-effective solution for NK cell-based cancer immunotherapy.
NK cell ADCC leverages the antigen-specificity of mAbs to redirect NK cells towards tumor cells. This approach relies on the binding of the Fc region of the mAbs to the Fcγ receptor IIIa (or CD16a) of NK cells, thereby triggering CD16a activation signalling and inducing the lysis of the mAb-coated target cells by NK cells (25). Currently, there are many mAbs available on the market that target different cancers, such as Trastuzumab, Pertuzumab, and Margetuximab for HER2+ cancers (25), Rituximab for non-Hodgkin’s lymphoma (26), and Cetuximab for EGFR+ colorectal cancer (27). The versatility of mAbs positions the NK cell ADCC method as an effective approach for various cancer treatments. However, not all patients respond well to this approach. The presence of the 158F variant of CD16a can decrease its binding affinity for IgG mAbs, which is the primary isotype of therapeutic mAbs (28, 29). Remarkably, a majority of the population carries at least one CD16a allele of this variant (15), which could potentially undermine the effectiveness of NK cell ADCC treatment (28, 30). Nevertheless, the issue of CD16a polymorphism could potentially be mitigated through afucoslylation or engineering of the Fc region of mAbs to enhance their binding affinity to CD16a (31).
CAR-NK cell therapy can circumvent the issue of CD16a polymorphism present in NK cell ADCC as the genetically engineered CAR-NK cells recognize and target cancer cells using their antigen-specific CARs, which signal and activate NK cells through their signalling module-bearing cytoplasmic tails (32). Compared to CAR-T cell therapies, CAR-NK cells can be produced more efficiently from an allogenic source (32). Additionally, CAR-NK therapy is safer due to the lower risk of GVHD and CRS (33). A variety of CAR-NK cells are currently undergoing assessment for their efficacy against haematological and solid tumors in both preclinical and clinical studies (24, 32, 34).
Despite its promising potential, CAR-NK cell therapy is still in the early stage of development, with many challenges yet to be addressed. A key issue is the short lifespan of CAR-NK cells, which do not persist in the body for long (35). This necessitates frequent infusion, thereby increasing the treatment costs (32). Therefore, it is crucial to find a solution to extend the in vivo survival of CAR-NK cells. Furthermore, CAR-NK cells present more significant challenges in genetic manipulation compared to CAR-T cells. Their lower transduction efficiency of NK cells necessitates a larger initial NK cell number, followed by sorting and expansion (34), increasing the overall costs and extending the production time.
NKCEs, such as BiKE (bispecific killer cell engager) or TriKE (trispecific killer cell engager), provide several advantages over the earlier two strategies. Compared to bispecific BiKEs, TriKEs possess an additional antigen specificity, enabling more precise targeting and enhanced functionalities. In contrast to tumor-targeting mAbs, CD16a-targeted NKCEs are not necessarily affected by the CD16a polymorphism, as they may recognize regions not influenced by the polymorphism (36). Additionally, NKCEs engaging activating receptors on NK cells other than CD16a are not affected by CD16a polymorphism or its expression level. Unlike in the case of NK cell ADCC, these NKCEs do not compete with endogenous IgG1 for CD16a binding. Besides activating NK cells, NKCEs could also activate other immune cells and redirect them towards tumor cells if they target receptors commonly expressed in NK and other immune cells. For example, NK cell activating receptor NKG2D is also expressed in NKT, some CD8+ T and a subset of γδ T cells (37), thus NKG2D-targeting NKCEs can potentially activate these immune cells in addition to NK cells. Moreover, NKCEs are easier to manufacture and much less expensive than CAR-NK cell therapy. Appropriately designed NKCEs also have an extended retention time in the body (38), making NKCEs a more cost-effective strategy compared to CAR-NK cell therapy (34).
NKCEs can also be combined with adoptive NK cell transfer by forming complexes with allogenic NK cells (15, 39). A study combining AFM13 and cord-blood NK cells has shown that this combination stimulates the NK cells against CD30+ tumor cells (40). This approach circumvents the need for genetic engineering of CAR-NK cells, as retaining NKCEs on the NK surface eliminates the requirement for recurrent genetic modifications of NK cells (41, 42). Several clinical studies are currently in progress to assess the effects of co-administering adoptive NK cell CYNK-101, an NK cell line optimized for IgG binding, with anti-CD38 mAb Daratumumab (43) or anti-HER2 Trastuzumab and Pembrolizumab (NCT05207722) for cancer treatment.
Although NKCEs offer several advantages as an immunotherapeutic modality, substantial efforts towards optimization are required to develop potent and effective NKCEs. Therefore, several critical factors must be carefully considered during NKCE development.
CD16a, also known as Fcγ receptor IIIa (FcγRIIIa), is an activating receptor abundantly expressed in CD56dim NK cells. It is anchored to the plasma membrane of NK cells and is associated with CD3ζ and/or FcϵRIγ chain (44) (Table 1). Upon engagement by IgG antibodies, CD16a molecules cluster together, leading to the phosphorylation of the tyrosine residues of the ITAM motifs of CD3ζ and/or FcϵRIγ chains by Src-family kinases. Subsequently, signalling cascades involving phosphatidyl-inositol-3-OH kinase (PI3K), phospholipase C γ (PLCγ) (45), and Vav (46) are triggered in NK cells. Unlike other NK cell activating receptors that require co-engagement of additional receptors to achieve optimal activation, engagement of CD16a alone can fully activate NK cells (47). The full activation of NK cells by CD16a engagement also differs from the optimal activation of T cells, which necessitates the ligation of both T cell receptors and co-stimulatory receptors. Certain bispecific T cell engagers (BiTEs), which require engagement of the costimulatory receptor CD28 to fully activate T cells and eliminate tumor cells (48). Therefore, NKCEs that engage CD16 are capable of inducing complete activation of NK cells without the need for triggering other NK cell activating receptors.

Table 1 The structures and properties of NK cell activating and inhibitory receptors targeted by NKCEs.
Currently, CD16a is the most popular target for NKCE development, with many products currently in preclinical development or clinical trials (Table 2). For example, AFM13, a tetravalent bispecific anti-CD30xCD16a NKCE, is formed by homodimerization of a tandem diabody (TandAb), which is constructed through a special arrangement of Fv heavy and light chain domains of anti-CD30 and -CD16a antibodies with a 9-amino acid-long peptide linker (Table 2 and Figure 2). This 104 kDa TandAb has demonstrated CD30-dependent activation of NK cells, with a half-life of 19 hours (68). Clinical trials have shown AMF13 to be effective in treating patients with relapsed or brentuximab vedotin-refractory Hodgkin’s lymphoma, and it was well-tolerated during continuous treatment (68, 91, 92). With the proven effectiveness of AFM13, NKCEs with similar structures (AFM22/24/26) (Table 2) have also been developed to target EGFR vIII, EGFR, and BCMA, respectively (93, 94). AFM22 and AFM24 are designed for the treatment of solid tumors. AFM24 is currently undergoing a Phase 1/2a clinical trial for advanced solid cancers (NCT04259450) and is also being tested in combination with Atezolizumab for EGFR-expressing advanced solid malignancies (NCT05109442). On the other hand, AFM22, which specifically targets EGFR vIII is still in the preclinical development stage. In addition, preclinical evaluation of BCMA-targeted AFM26 has shown that it binds to NK cells with high avidity and is largely unaffected by low BCMA expression levels on multiple myeloma (MM) cells (94).
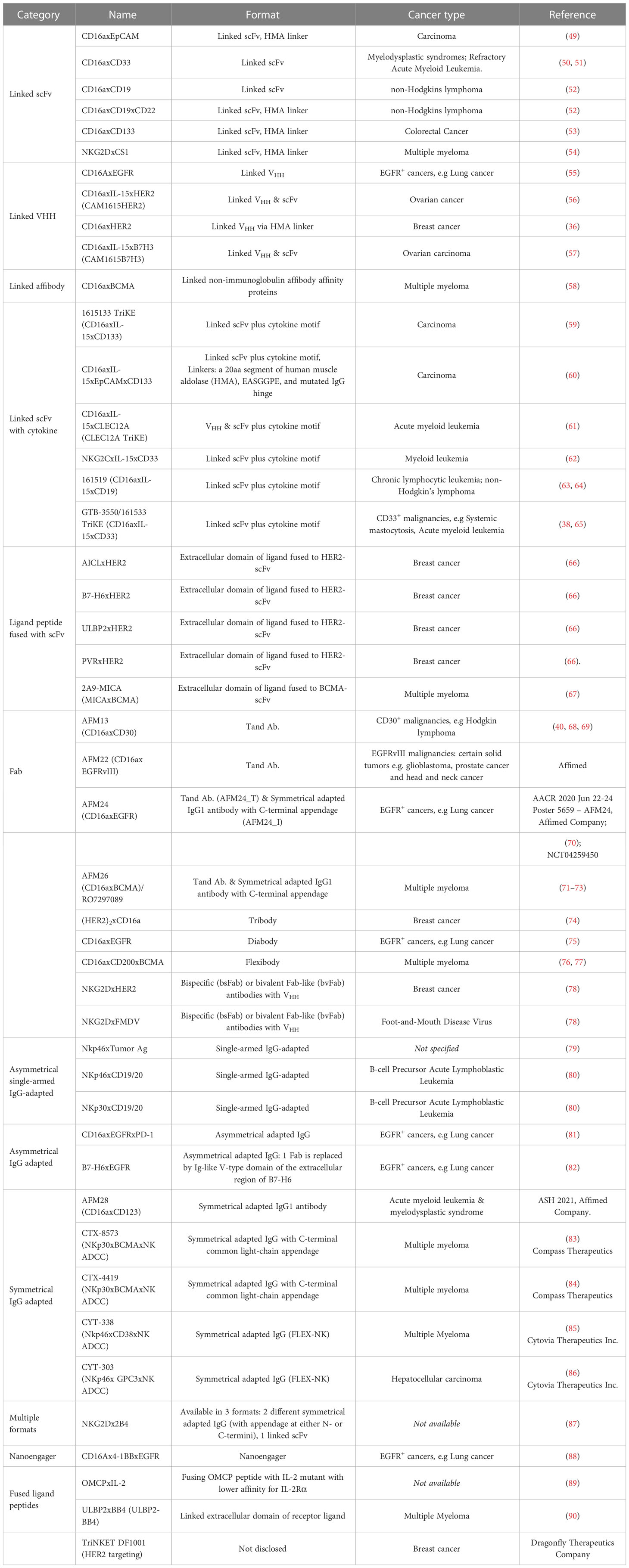
Table 2 NKCEs that are currently undergoing development.
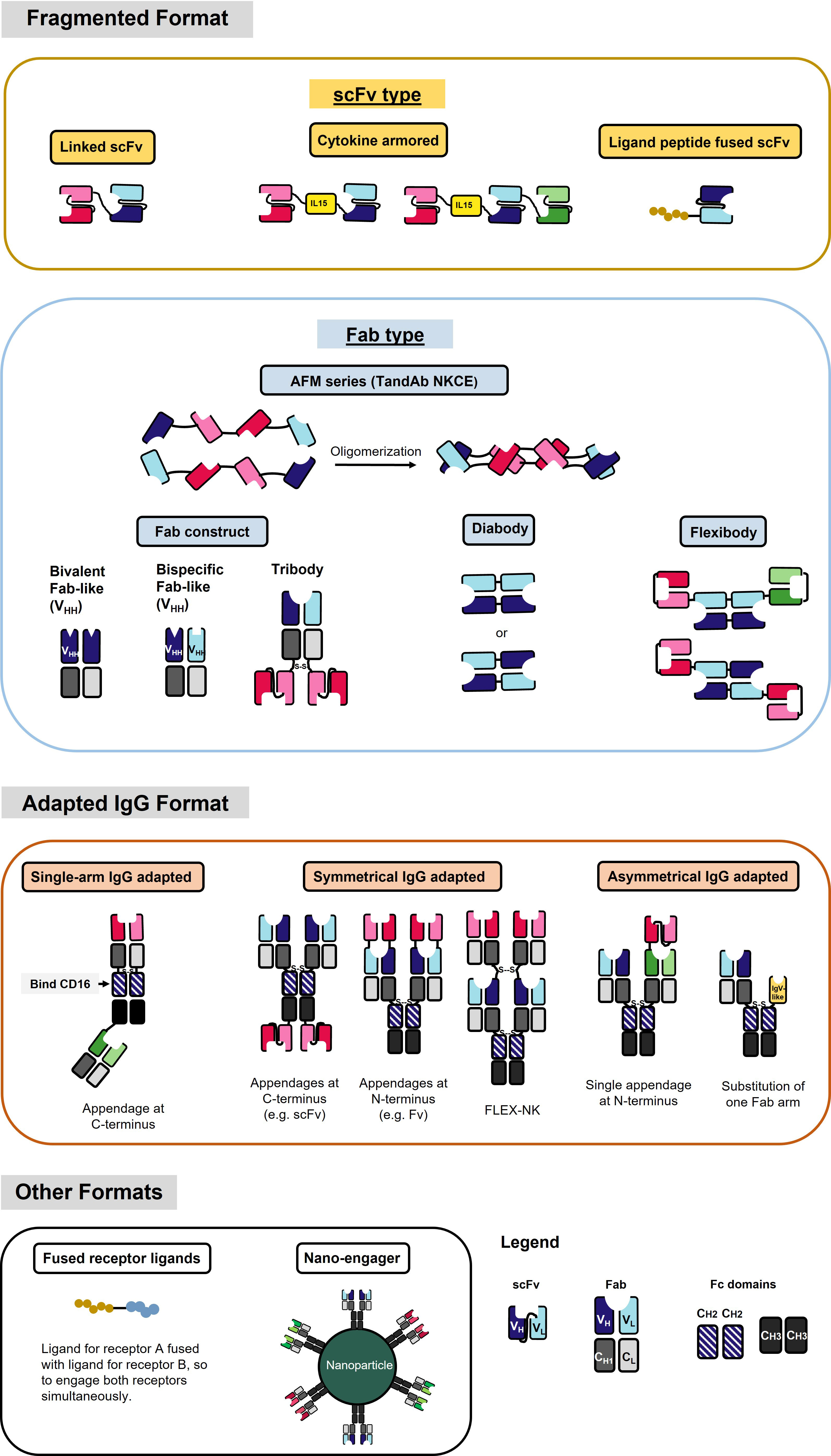
Figure 2 NKCE formats that are currently under development. Different color (red, blue, and green) indicates different antigen specificity. The dark and light colors label the heavy chain (VH) and light chain (VL), respectively. The fragmented format contains a variety of structures by different linkages of the VH and VL, including scFv and Fab. The adapted IgG format contains structures that generally have the constant region of the IgG antibody. Single-arm IgG adapted format uses Fc region CH2 domain to bind to CD16a. Please refer to Table 2 for specific NKCEs.
It is noteworthy that CD16a is rapidly cleaved from NK cells after activation by metalloproteases, such as ADAM17 (95, 96), or matrix metalloproteases (MMPs), like MMP25 (97). The downregulation of CD16a expression may facilitate NK cells to detach from their current target after cytolysis, enabling them to engage the next target cell (98). On the other hand, this process may also pose a disadvantage for CD16a-engaging NKCE development, as CD16a downregulation may make CD16a-targeted NKCEs less effective. The use of MMP inhibitors in combination with CD16a-targeting NKCEs could be a potential strategy to prevent CD16a shedding from NK cells and enhance their activation (99). Noteworthy, NKCEs that target CD16a can also bind to the membrane-bound CD16b receptors on neutrophils, whose extracellular domain is remarkably similar to that of CD16a on NK cells (100). In addition, soluble CD16 present in the serum, primarily consisting CD16b shed from the surface of neutrophils (101) and a smaller amount of CD16a shed from NK cells (96), creates a considerable “sink” effect. This effect could potentially compete with CD16a on NK cells for the binding of CD16a-engaging NKCEs, considerably diminishing their effectiveness.
NKG2D is a disulfide-linked homodimeric C-type lectin-like receptor expressed in NK cells, NKT cells, a subset of CD8+ T cells, and a subset of γδ T cells (37). Although its intracellular domain lacks ITAMs and does not bind with an ITAM-bearing adaptor, NKG2D associates with an adaptor protein known as DAP10. DAP10 contains a tyrosine-based YxxM motif in its cytoplasmic tail. Upon tyrosine phosphorylation by Src-family kinases (102, 103), DAP10 induces the recruitment and activation of PI3K (104) and the Grb2/Vav1/PLCγ signalling complex (102) (Table 1).
The engagement of NKG2D activates the cytotoxic function and cytokine release of NK cells (105, 106). Notably, NKG2D-mediated tumor cell killing occurs more swiftly than that mediated by CD16a (98), although its activation strength is comparatively weaker. Interestingly, NKG2D-activated NK cells demonstrate a higher likelihood of sequential killing tumor cells compared to those activated by CD16a (98). Furthermore, NK cells are less motile in tumors lacking NKG2D ligands than those with NKG2D ligands (107), suggesting that targeted blockage of NKG2D ligands in tumors could help retain tumor-infiltrated NK cells within the tumor.
Several studies have evaluated the effectiveness of NKCEs that target NKG2D for treating multiple myeloma (MM) (Table 2). A linked single-chain variable fragment (scFv) NKCE, known as CS1xNKG2D, which targets CS1 (also called SLAMF7, a tumor antigen expressed on MM), has been demonstrated to induce dose-dependent cytotoxicity against CS1+ MM cells in vitro, stimulate IFN-γ production, and enhance the survival of NSG mice implanted with human MM cells (54). Moreover, various NKG2D+ cytotoxic immune cells, including NK, NKT, and CD8+ T cells, have been demonstrated to lyse MM cells additively when induced by the NKG2D-targeted NKCE (54).
Another NKCE that targets NKG2D, BCMAxMICA (also known as 2A9-MICA), utilizes the MHC I-related chain A (MICA), which is a ligand of NKG2D, to activate NKG2D on NK cells (67). 2A9-MICA is comprised of the extracellular region of human MICA and an scFv against BCMA, connected by a G4S linker (67). It has been reported that adding 2A9-MICA to a co-culture of NK-92 and MM cells increases NK-92 cell degranulation and enhances the cytolysis of BCMA+ MM cells by NK-92 cells. Additionally, 2A9-MICA could significantly suppress tumor growth in a nude mouse model without evident toxicity (67). However, it is noteworthy that several cancers exhibit high levels of soluble MICA (108), which could potentially compete with MICA-bearing NKCEs for binding of NKG2D, thereby reducing their effectiveness.
Bispecific antibodies targeting NKG2D and 2B4 receptors have been evaluated for cytotoxicity and IFNγ production of NK cells (87). These antibodies, designed in various formats, such as linked scFv, and symmetrical IgG with appendage at C- or N-terminus, have demonstrated a superior capability for activating NK cells compared to a combination of monospecific antibodies against NKG2D and 2B4, suggesting that co-engagement of these two receptors could synergize NK cell activation (87). It is thus intriguing to hypothesize that NKCEs, simultaneously engaging NKG2D, 2B4, and a tumor-associated antigen, could induce a more potent NK cell activation and more robust anti-tumor activity.
Nkp30 is an Ig-like transmembrane protein with a single V-type extracellular domain (109). It is constitutively expressed in resting and activated NK cells (110). Similar to CD16a, Nkp30 is also associated with disulfide-linked homodimers or heterodimers of CD3ζ (109) and/or FcϵRIγ (111) chain for signal transduction (Table 1). As a natural cytotoxicity receptor (NCR) family member, Nkp30 triggers NK cell degranulation and cytokine release upon engagement (66, 111). Unlike CD16a, Nkp30 is not rapidly downregulated following NK activation, rendering it a more promising target over CD16a for better efficacy and persistence.
An NKCE incorporating the Nkp30-specific ligand, B7-H6, can adequately activate NK cells and direct them towards tumor cells (66), leading to the secretion of IFN-γ and TNF-α (112). Studies have shown that the cytolysis of HER2+ cells by NK cells, mediated by the Nkp30xHER2 NKCE, is comparable to or slightly superior to that mediated by therapeutic mAbs Trastuzumab and Cetuximab (66). Currently, two Nkp30xBCMA NKCEs, CTX-4419, and CTX-8573, are undergoing preclinical assessment for their effects in treating MM (83, 84). Both NKCEs were constructed using anti- Nkp30 and anti-BCMA antibodies that share a common light chain. The two anti-Nkp30 Fab fragments are appended at the C-terminus of the heavy chain of the anti-BCMA IgG1 antibody, which has an afucosylated Fc for enhanced binding with CD16a (Table 2). These NKCEs have been demonstrated to induce greater tumor cell killing and IFN-γ release than therapeutic mAbs, such as Elotuzumab or Daratumumab (83, 84). Notably, even without CD16a engagement by the Fc region, both NKCEs can still elicit the lysis of tumor cells via Nkp30 (83, 84). Both CTX-4419 and CTX-8573 activate NK cells in the presence of BCMA+ tumor cells, indicating good safety due to minimal tonic activation (83, 84).
However, the lower cell surface expression of Nkp30 might be a concern when considering it as a target for NCKE development. A study has shown that each NK cell has approximately 1000 Nkp30 molecules expressed on the cell surface, which is considerably lower than the density of CD16a (~70,000 molecules per NK cell) (66). Therefore, designing Nkp30-targeted NKCEs to accommodate the larger spacing between antigens might necessitate a more flexible structure.
Nkp46 is an NCR expressed on all mature NK cells and is associated with CD3ζ and/or FcϵRIγ homodimers or heterodimers for signal transduction (113). Despite sharing many characteristics with Nkp30, Nkp46 has two extracellular C-type Ig domains (114), whereas Nkp30 has only one Ig domain (Table 1). Engagement of Nkp46 alone only weakly activates resting NK cells (115), but co-engagement with other activating receptors, such as 2B4, DNAM1, or CD2, can significantly enhance NK cell activation (115). Notably, unlike CD16a, Nkp46 is not downregulated in the tumor-infiltrated NK cells in several cancers, including lung carcinoma (116), acute myeloid leukaemia (117), and breast cancer (118). Therefore, NKP46 holds promise as a target for NKCE development.
A Nkp46xTAA NKCE is constructed using a single-armed adapted IgG format, targeting Nkp46 by the Fab at the C-terminus and the tumor antigen at the N-terminus (79) (Figure 2). Besides activating NK cells by engaging Nkp46, this NKCE also promotes NK cell ADCC through its Fc region via CD16a binding. It has demonstrated better suppression of CD19+ or CD20+ tumor cell growth compared to the therapeutic mAb Rituximab or Obinutuzumab in both in vitro (80) and in vivo studies (79).
Another tetravalent NKCE, Nkp46xCD38 (CYT-338), is also undergoing development. CYT-338 has an IgG1 backbone that can mediate NK cell ADCC through its Fc region. It recognizes Nkp46 through its Fab regions; additional Fab fragments targeting the tumor antigen are symmetrically appended at the N-terminus of heavy chains of the IgG backbone (FLEX-NK, Figure 2). Mutations have been introduced at the IgG backbone’s constant regions (CH1 and CL) to minimize the mispairing of heavy and light chains and facilitate the proper chain pairing. CYT-338 has shown a 3-fold higher binding to MM cell lines than Daratumumab and has demonstrated greater dose-dependent NK cell cytolysis and cytokine production (119). Notably, these NKCEs may link two NK cells together by co-engaging Nkp46 and CD16a or Nkp46 and CD38 on NK cells. Thus, it is yet to be determined if there is a risk of fratricide of two neighbouring NK cells that could potentially be co-engaged by the same NKCE. This is especially pertinent given that the monospecific, bivalent therapeutic mAb Dratumumab, which targets CD38 on NK cells, has been reported to induce NK cell fratricide (120).
Nkp80 (KLRF1) is a homodimeric C-type lectin-like receptor similar to NKG2D (121). It is primarily expressed in NK cells (122) (Table 1). The cytoplasmic tail of Nkp80 bears a hemi-ITAM-like sequence (YxxL), which, upon binding, associates with Lck and Syk kinases (123). The only known natural ligand for Nkp80 is the activation-induced C-type lectin (AICL) (121).
An Nkp80-targeting NKCE, known as AICLxHER2-scFv, has been developed using AICL to engage Nkp80 (66). This NKCE contains the extracellular domain of AICL and the scFv of an anti-HER2 antibody. AICLxHER2-scFv has demonstrated its capability to activate NK cells and direct NK cytolysis towards HER2+ tumor cells, exhibiting synergistic effects with therapeutic mAb Trastuzumab or Cetuximab (66). Interestingly, it has been suggested that simultaneous Nkp80 and DNAM-1 activation could significantly enhance NK activation, whereas Nkp80 does not show synergy with Nkp30 (66). These findings highlight Nkp80 as a promising target for NKCE development. However, the relatively low surface expression Nkp80 (about 5,000 copies per NK cell) (66) may require some special NKCE design considerations such as enhanced flexibility and multivalency to improve the efficacy of Nkp80-targeted NKCE.
NKG2C, also known as CD159a, is an activating receptor that specifically recognizes HLA-E (124). It forms a heterodimer with CD94 and is associated with DAP-12 through a positively charged residue in its transmembrane domain (125). (Table 1) NKG2C is predominantly expressed in mature NK cells that lack NKG2A, and its increased expression signifies a “memory” NK cell phenotype (126).
Similar to CD16a, NKG2C can trigger strong activation of NK cells even without co-activation (127). However, in contrast to CD16a, NKG2C does not rapidly shed off post-NK cell activation, making it a promising candidate for NKCE development. An NKCE, known as NKG2CxIL-15xCD33, comprising an anti-NKG2C scFv and an anti-CD33 scFv, linked by a wild-type IL-15 in the middle (62), has demonstrated satisfactory NK cell activation, expansion, and cytotoxicity against CD33+ myeloid leukaemia cells both in vitro and in vivo in an NSG mouse model (62). Its efficacy in suppressing tumor growth suppression is comparable to 161533 NKCE (62).
However, one potential limitation of NKG2C compared to CD16a is its lower and variable expression level on the surface of NK cells (62, 128). The population of NKG2C+ NK cells correlates with an individual’s history of human cytomegalovirus (CMV) exposure. Individuals with previous CMV exposure will likely have a larger NKG2C+ NK cell population than those who are naïve to the virus (62). Even among CMV-seropositive individuals, the frequency of NKG2C+ NK cells could vary considerably (128, 129). Therefore, NKG2C-NKCE may not be effective for some patients due to a low population of NKG2C+ NK cells.
Unlike most of the killer cell Ig-like receptor (KIR) family members, which are inhibitory, KIR2DS and KIR3DS are two activating receptors of the KIR family (130, 131). Despite sharing two or three Ig-like extracellular domains with other inhibitory KIRs, KIR2DS and KIR3DS possess a short cytoplasmic tail without ITIMs. Instead, they associate with ITAM-bearing adaptor DAP12 (132) (Table 1). Studies have shown that KIR2DS expression in NK cells is positively correlated with improved NK activation and cytokine production (133). Therefore, KIR2DS and KIR3DS are potential candidates for NKCE development. However, the high homology in the extracellular domains of the activating and inhibitory KIRs might make it challenging to generate specific antibodies or CDR sequences that can distinguish these two types of KIRs (131).
CD160 is an Ig-like glycosyl phosphatidyl inositol (GPI)-anchored transmembrane protein, similar to KIR receptors. It is expressed in a fraction of NK cells with high cytotoxicity and a subset of T cells (134) (Table 1). Upon engagement by its ligand HLA-C, CD160 can trigger robust NK cell activation leading to efficient tumor cell lysis (135) and secretion of TNF-α, IFN-γ, and IL-6 (136, 137), making it a potential target for NKCE development. However, CD160 has been shown to undergo downregulation following NK cell activation by phorbol ester (138).
DNAM-1, also known as CD226, is another activating receptor constitutively expressed in NK cells, T cells, and certain myeloid cells. It is a transmembrane glycoprotein with two Ig-like domains (139, 140) (Table 1). Unlike other Ig superfamily members, the cytoplasmic tail of DNAM-1 contains four tyrosine residues (Y293, Y300, Y322, and Y325), one asparagine residue (N324) and one serine residue (S329), which can recruit Src family kinase Fyn, adaptor Grb2 and PKC upon phosphorylation (140). Although engagement of DNAM-1 alone does not increase NK cell cytotoxicity, it promotes NK cell adhesion to the target cell and granule polarization (140) and can synergize with 2B4 to enhance NK cell cytotoxicity (47). DNAM-1 competes with inhibitory receptor TIGIT and CD96 for binding with their shared ligands PVR (CD155) and Nectin-2 (CD112) (141). A study showed that a DNAM-1-engaged NKCE, PVRxHER2-scFv, failed to display potent cytotoxicity, probably due to PVR’s competitive binding with inhibitory receptors (66). Moreover, DNAM-1 has a low surface expression on NK cells. Therefore, future DNAM-1-engaged NKCE development requires more specific design strategies.
2B4 (SLAMF4, CD244) belongs to SLAM (Signaling Lymphocytic Activation Molecule) family and is expressed in NK and CD8+ T cells (142). It has two Ig-like extracellular domains and a cytoplasmic tail with four immunoreceptor tyrosine-based switch motifs (ITSMs), which can recruit SAP and protein tyrosine kinase Fyn, and SHP1/2 (143, 144). Although engagement of 2B4 alone does not activate NK cells, it can synergize with Nkp46, NKG2D, and DNAM-1 to achieve significantly enhanced NK cell activation (47). Therefore, 2B4 is frequently employed as an auxiliary receptor in the design of some multispecific NKCEs.
Stimulatory cytokines can dramatically enhance NK cell proliferation and cytotoxicity. For example, the anti-tumor capabilities of functionally impaired tumor-infiltrated NK cells can be fully restored with interleukin (IL)-2 and IL-15 (145, 146). A clinical trial also revealed that IL-15 infusion in cancer patients could significantly expand NK and memory CD8+ T cell populations (147). These findings suggest that incorporating stimulatory cytokines, such as IL-2 or IL-15, into NKCEs may synergistically enhance their antitumor potency.
The incorporation of an IL-15 moiety into an NKCE that has two scFv-segments, with one binding CD16a on NK cells and the other one binding tumor-associated antigens on tumor cells, can significantly enhance the cytotoxicity of NK cells and their pro-inflammatory cytokine production compared to NKCE without IL-15 (38, 59, 60) (Table 2 and Figure 2). An IL-15-incorporated NKCE against acute myeloid leukaemia (AML), referred to as CLEC12A TriKE or CD16axIL-15xCLEC12A (Table 2), has been shown to specifically expand NK cells, but not T cells, leading to the potent killing of AML cells by NK cells (61). Another study showed that IL-15-incorporated NKCE (161533 TriKE/GTB-3550) rectified the functional defects of NK cells and extended the survival of cancer patients post-hematopoietic stem cell transplantation (38). GTB-3550 has shown promise in treating CD33+ malignancies (NCT03214666). These findings demonstrate the significant clinical values of incorporating IL-15 into NK cell therapy for cancer treatment.
Similar to IL-15, IL-2 is also a potential stimulator for NKCE. However, the unspecific activation of Treg cells by IL-2 (148) can be problematic. The activity of IL-2 can be limited to NK cells by incorporating it into an NKCE but negating its effect on Treg cells, which can suppress many immune cells, including NK cells (149). A fusion protein consisting of NKG2D ligand OMCP and a modified IL-2 has shown the feasibility of this restricted delivery of IL-2, resulting in specific activation of NKG2D+ cytotoxic cells (89).
Additionally, combining IL-15, IL-12, and IL-18 can induce memory-like NK phenotype, leading to extended persistence and superior cytotoxicity of NK cells (150). Hence, it would be interesting to investigate if NKCEs incorporating IL-15, IL-12, and IL-18 can induce more potent NK cell cytotoxicity than NKCEs without cytokine incorporation.
Although NKCEs that engage activating receptors can facilitate the formation of immune synapses between healthy NK and tumor cells, leading to NK cell activation and tumor cell lysis, the immunosuppressive tumor microenvironment (TME) can dampen the expression of these activating receptors on the NK cell surface (151, 152). This may weaken the engagement of tumor-infiltrated NK cells by NKCEs, resulting in reduced NK cell activation and expansion. On the other hand, tumors and other cells in the TME upregulate the expression of ligands for NK cell inhibitory receptors, which is a mechanism that inhibits the antitumor function of NK cells. Preventing the engagement of these inhibitory receptors on NK cells by their ligands can potentiate the antitumor function of NKCEs. For instance, EGFRxCD16axPD-L1 is a trispecific NKCE that activates NK cell-induced killing of EGFR+ tumor cells through CD16a while blocking the binding of PD-L1 to the immune checkpoint receptor PD-1 on activated NK cells, thus reducing NK cell exhaustion (81). Moreover, the anti-PD-L1 arm on NKCE provides additional recognition of tumor cells, as tumor cells often express PD-L1 (153). Studies have demonstrated that the trispecific EGFRxCD16axPD-L1 NKCE exhibits superior antitumor potency compared to the bispecific EGFRxCD16a NKCE or anti-EGFR mAb (81).
However, one potential issue that may arise when integrating checkpoint blockage into NKCEs concerns dosing. Optimal effects from immune checkpoint blockade typically require high dosage levels (154). However, such high dosages could potentially cause severe toxicity, especially considering that the NK-activating component of the NKCE is typically designed to function effectively at low concentrations.
Several other inhibitory receptors are also known to suppress NK cell antitumor function, particularly within the TME of various cancers. These receptors include NKG2A, TIGIT, TIM3, CD96, and KIR2DL/KIR3DL, in addition to PD-1/PD-L1. NKG2A can suppress NK cell function by recruiting SHP-1/2 through its cytoplasmic tail’s ITIMs upon the engagement by non-classical class I MHC molecules HLA-E (155). Monalizumab, an NKG2A-blocking IgG4 mAb, has been shown to boost NK cell activation and synergistically control tumor size with limited toxicity when combined with a PD-L1 blocking mAb in Phase 2 clinical trial (156). A blocking antibody against TIGIT, an inhibitory receptor on NK cells bearing ITIM- and Ig tail-tyrosine (ITT)-like motif, can reverse the exhausted phenotype of TIGIT-upregulated tumor-infiltrated NK cells, constrain tumor growth, and improve the survival tumor-bearing mice in vivo (157, 158). TIM-3, another NK exhaustion receptor, inhibits NK cell function (159, 160). TIM-3 blockade enhances NK cell cytotoxicity against MM cells (161) and cells chronically infected by the hepatitis B virus (162). Moreover, another mAb, IPH4102, which blocks inhibitory KIR3DL2, has demonstrated promising clinical activity in patients with cutaneous T-cell lymphoma (163). CD96 is another inhibitory receptor, also described as TACTILE (164). Human CD96+ NK cells demonstrate an exhausted phenotype with reduced IFN-γ, TNF-α, perforin, and granzyme B expression, and blocking CD96 interaction with its ligands enhances the cytotoxicity of NK cells (165, 166). NK cells in tumor tissue show a higher level of CD96 than those in the peritumoral region (165). Moreover, the combinatory blockade of PD-1 and CD96 has significantly suppressed tumor growth compared to blocking PD-1 alone (167). Therefore, blocking CD96 can be a novel strategy for reviving NK cells from exhaustion.
It is important to note that the blockade of immune checkpoints may not be effective for all types of cancers. For example, a study investigating the blocking of PD-1 and/or TIM-3 in early chronic lymphocytic leukemia patients failed to demonstrate any significant effect (168). Therefore, the effectiveness of immune checkpoint blockade therapy is ultimately contingent on the unique biology of each specific cancer disease.
Engaging the appropriate target receptors is crucial, as it determines the strength and persistence of NK cell activation and the subset of NK cells that get activated. Therefore, substantial efforts are being devoted to selecting more suitable target receptors for NKCE development. However, the ultimate goal of NKCE development for cancer treatment is to generate potent NKCEs with desirable functionality and manufacturability, both of which are also significantly impacted by their molecular structures. In the following sections, we will discuss several critical aspects that influence the functionality and manufacturability of NKCEs.
The formation of a cytolytic immune synapse (IS) between NK and cancer cells is a prerequisite for an NKCE to eradicate cancer cells effectively. The IS formation involves three phases: the recognition and activation phase, the effector/lysis phase, and finally, the detachment or termination phase (107, 169). The crucial role of NKCEs is facilitating IS formation during the early recognition and activation phase by bridging the interaction between NK and target cells. This interaction subsequently triggers NK cell activation through receptor engagement, resulting in cancer cell lysis (169).
Efficient clustering of receptors on both NK and target cells during their early stages of interaction is crucial for IS formation and NK cell activation (170). Enhancing the valency of a monospecific antibody has been shown to potentially improve the target binding by increasing its avidity (171, 172). This principle is equally applicable to bispecific antibodies. For example, the CD20-targeted T-cell bispecific antibody (RG6026, Roche), which has two CD20-binding Fabs and one CD3-binding Fab, has demonstrated superior potency than a similar format with only one CD20-binding Fab (173). Similarly, the loss in binding and killing ability in the T-cell bispecific antibody with low-affinity variants of HER2-binding Fab can be rectified by the bivalent engagement of two low-affinity Fab variants (174). It is noteworthy that receptor density also impacts the avidity-mediated binding of antibodies. Downregulation of some tumor cell surface antigens is a prevalent mechanism for immune escape (175), such as the downregulation of BCMA in MM cells (176). The cell surface BCMA antigens shed from MM cells and become soluble antigens, which might even compete with those membrane-bound BCMA antigens to bind BCMA-targeted NKCEs and reduce their efficacies. Therefore, the efficient interaction of NKCEs with NK and target cells is critical for developing NKCEs with high efficacy.
Spatial constraint in the synaptic cleft is another critical factor for optimal NKCE-meditated IS formation between NK and target cells. The synaptic cleft of most physiological IS is approximately 10-30 nm in size (177). Molecules exceeding this size could be excluded from the synaptic cleft and unable to induce IS formation (178). Thus, there is a size limit for NKCE molecules. Monoclonal antibodies, typically about 10 nm in diameter (179, 180), fit nicely into the synaptic cleft, thereby promoting stable IS formation. However, the addition of antibody moieties in NKCEs, necessary for achieving multispecificity, or increased valency, inevitably increases their size and compromises their ability to mediate stable IS formation due to the synapse structure distortion. Replacing larger antibody fragments with smaller ones, such as scFv or nanobody, may overcome the size constraint. For example, the ability of a NKG2Dx2B4 NKCE to activate NK cells has been assessed in different formats, including modified IgG format and linked scFv (87). The NKG2Dx2B4 NKCE, formatted as an scFv and smaller size than the IgG format, exhibited comparable efficacy of inducing IFN-γ and granzyme B secretion, with only a marginal lesser capacity to induce NK cell degranulation (87). These findings indicate that it is feasible to maintain full functionality even when larger antibody moieties are replaced with smaller ones in the design of NKCEs.
Furthermore, the physical protrusion and conformation of the tumor antigens and NK cell receptors in the extracellular region could affect the design of NKCEs due to the spatial constraints of the IS. The NKCE’s appropriate size is crucial for stable IS formation, bridging the ectodomains of both NK receptors and tumor antigens. For example, CD20 is less protruding from the cell surface compared to CD19 (181, 182), suggesting that different designs may be required when constructing NKCEs that engage with CD19 or CD20, even though both antigens are expressed on the same B cells and the NKCEs target the same NK cells.
To date, various NKCE formats have been evaluated for their functionality and manufacturability, with many adapted from the formats used for T cell engagers. Based on their molecular structure design, NKCE formats can be broadly classified into two major categories: fragmented formats and adapted IgG formats. The fragmented formats generally have smaller sizes than the adapted IgG formats due to the absence of the Fc region (Figure 2).
Most of the fragmented formats are derived from the variable (V) region of the heavy (H) and light (L) chains of IgGs and lack the constant regions (Figure 2). The linker between different V regions is the most common component of these fragmented formats. This linker connects two V regions, typically a pair of VH and VL, to form an scFv and construct the NKCE. The linker’s length between the two V regions determines their pairing (183). Linkers shorter than 10 amino acids do not allow the V regions to rotate and fold, leading to inter-chain pairing, as seen in the diabody (Figure 2). Studies suggest that the scFv’s linker should span at least 3.5 nm to permit the two V regions to fold and pair to form scFv (184). A typical linker is flexible and hydrophilic, minimizing structural interferences during protein folding within the cell. Several linker options are available to bridge the V regions, such as (G4S)n/(G3S)n linker (185), 218s linker (GSTSGSGKPGSGEGSTKG) (186), or HMA linker (PSGQAGAAASESLFVSNHAY) (49) (Table 3). The (G4S)n linker is most commonly used (187, 188), due to its high flexibility, low immunogenicity, and serine residues’ contribution to solubility. Moreover, the 218s linker is reported to be more proteolytic stable and has reduced aggregation (186), while the HMA linker exhibits lower immunogenicity (189). In addition to these conventional linkers, other linker designs are also possible. Phage display can tailor the linker sequence for a specific antibody, especially when conventional linkers do not work well (190).
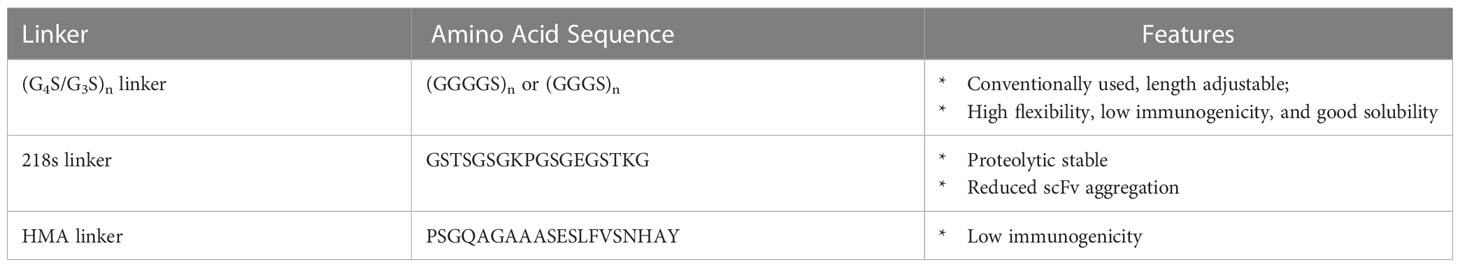
Table 3 Commonly used linkers in NKCE design.
Secondary bonds, like disulfide bonds, are critical to successfully assembling fragmented-type NKCEs. For example, in the tribody format (Fab construct, see Figure 2), the disulfide bond between the heavy and light chains facilitates the pairing. Additionally, introducing a disulfide bond between intra-domain regions can help stabilize the structure of engineered IgG antibodies, particularly when the domain structure lacks intrinsic stability (191).
Adapted IgG formats, which retain the antibody’s Fc region, are larger than fragmented formats (Figure 2). Typical engineering methods of this type of NKCE involve changing the specificity of one arm of the IgG backbone and/or appending an antigen-recognizing moiety, either scFv or Fab, to the N- or C-terminus of the original IgG (Figure 2). Depending on the position of the modifications, adapted IgG formats could be further subdivided into asymmetrical and symmetrical types (Figure 2).
Compared to the relatively simple folding requirement of the fragmented format of NKCEs, asymmetrical adapted IgG NKCEs present more challenges during protein folding. The main issues are the unsought homodimerization of heavy chains and the mispairing of the heavy and light chains of asymmetric IgG NKCEs when different NKCE parts are produced in the same cell (Figure 2). Previously, the production of those asymmetric IgGs through the hybrid hybridomas has encountered low yield due to the undesired homodimers of the heavy chains and mispaired light chains with non-cognate heavy chains (192).
To date, various techniques have been developed to overcome these problems. For example, chimeric rat/mouse-derived quadroma (193), knobs-into-holes (194), strand-exchange engineered domain (SEED) (195), and heterodimerization/electrostatic steering interaction (196) methods are used to enhance the heterodimerization of the heavy chains. CrossMab (197), CH1/CL interface engineering, or IgG/TCR chimaera (198) approaches are employed to minimize the mispairing of heavy and light chains. These technologies create and utilize a higher affinity for one chain to its desired pairing chain over other chains, thereby increasing the yield of NKCEs with the correct chain pairing. Another potential solution to the issue of the mispairing of heavy and light chains could involve using a common light chain. However, this strategy might not be applicable to all the antibodies, particularly those where the CDR sequences of both heavy and light chains are equally critical for determining the antigen-binding specificity.
Alternatively, instead of producing different parts of an NKCE in one single cell, these parts could be synthesized separately and subsequently assembled to form the functional format. This approach, known as co-culture (199), involves producing two distinct half-antibodies in two separate cell lines and combining them to generate the complete structure in a 1:1 molar ratio. While the co-culture method can yield substantial protein quantities, it may escalate costs and contamination risks due to the need for additional purification steps (199).
Due to their symmetrical structures, the symmetrical IgG formats can easily circumvent the chain pairing issues inherent to the asymmetrical formats, potentially reducing production costs. However, a concern for the symmetrical IgG formats is their considerably larger size than the asymmetrical formats when achieving the same multispecificity. The increased size could theoretically compromise the yield and increase the immunogenicity of the symmetrical NKCEs (200, 201).
It is noteworthy that the choice of the antigen recognition moiety can significantly impact the function of NKCEs. Although the Fab format for antigen recognition poses a challenge in correctly pairing heavy and light chains, it is functionally more potent than the scFv format. A study has shown that certain scFvs, which failed to function in vivo, could operate once switched to the Fab format (202).
Some unconventional formats, which are not purely antibody-based, can also be employed in NKCE development (Figure 2). One such format involves fusing the ligands of the corresponding receptors on both the tumor and NK cells to engage the NK cell with the tumor cell (90). This strategy offers a broader range of receptor targets for NKCE engineering; however, it does not prevent competition with the endogenous ligand binding to the receptors. Another approach, known as nano-engager, utilizes the ability of nanoparticles to be coated with multiple antibodies (88) (Figure 2). This method allows a single nanoparticle to display multiple specificities, directing NK cells towards tumor cells.
Nanobodies, also known as VHH (Variable Heavy domain of Heavy chain) antibodies, present a promising alternative for antigen recognition in NKCEs. Unlike scFv or Fab, which is constructed using VH and VL, nanobodies are camelid single-domain antibodies that can achieve specific antigen-binding using heavy chains only (203). Therefore, nanobodies can circumvent the pairing issue of the Fab format and the folding issue of scFv, making them a promising candidate in future NKCE designs. The feasibility of incorporating nanobody into NKCE has been demonstrated by the CD16axIL-15xCLEC12A NKCE (61) (Table 2). In this NKCE, a humanized anti-CD16a VHH fragment, instead of the conventional anti-CD16a scFv, was incorporated into the NKCE and could mediate efficient NK cell activation and tumor cell lysis (61).
Recently, a new form of antigen recognition called affibody has also emerged due to advancements in protein engineering technologies (204). Affibodies are constructed using three-helix subdomains, approximately 7 kDa in size (205). Randomization of the amino acids on two of the helices has been found to generate a large library, from which some potent antigen binders to the antigen can be isolated (205–208). For instance, a CD16axBCMA NKCE developed using affibodies has illustrated the potential of this new format to replace the conventional scFv moieties in the design of NKCEs (Table 2). This compact NKCE (15-23 kDa), consisting of CD16a- and BCMA-specific affibodies connected by a linker, has demonstrated potent efficacy in activating primary NK cells, initiating synapse formation, and specifically lysing MM cells (58).
In addition to the direct effect of format design on the functionality of an NKCE, other factors, such as tissue infiltration and persistence, should also be considered during the development of NKCEs. These factors can significantly influence the therapeutical efficacy of NKCEs.
Currently, most NKCEs undergoing preclinical testing are of fragmented formats (Table 2). Their smaller size (50-100 kDa) affords them excellent tissue infiltration properties and substantially lower immunogenicities. However, these small-size NKCEs often lack in vivo persistence due to renal clearance and degradation (209, 210), requiring frequent infusions to the body to maintain effective drug levels. This will increase treatment costs due to the recurrent patient hospital visit for repeat infusions. Thus, strategies to extend the in vivo lifespan of these small-size NKCEs have been proposed. One such approach is to bind or fuse them to long-lived serum proteins, like albumin (half-life 2-4 weeks) (211, 212), or conjugate them to the Fc region of IgG (212, 213). Additionally, attaching the small-size protein to a chemical polyethene glycol (PEG), which could also prolong its half-life (209), might be feasible to extend the half-life of the smaller NKCEs. Several approved protein drugs, such as interferon-α 2b (IFN-α 2b), granulocyte-colony-stimulating factor (G-CSF), or human growth hormone (hGH), have used this method for longer persistence (214). Another strategy involves multimerizing NKCEs in fragmented format to increase their size. For instance, AFM13, a tetravalent bispecific NKCE that binds to CD16a and CD30 on NK and tumor cells, is achieved by dimerizing two V domains. In a Phase 2 trial, AFM13 demonstrated promising results for treating multi-refractory Hodgkin Lymphoma patients (91, 215).
As mentioned earlier, adapted IgG formats typically have a larger size, usually exceeding 150 kDa (Figure 2). While their larger size may present challenges in tissue penetration, the inclusion of the Fc region significantly enhances their in vivo persistence (216). Additionally, the Fc region could recruit mechanisms such as ADCC, complement-dependent cytotoxicity (CDC), and antibody-dependent cellular phagocytosis (ADCP) in immune cells other than NK cells, adding a bonus to tumor cell killing (217). The most common approach to achieve multispecificity of the adapted IgG format is to add appendages with different antigen specificities to the C- or N-terminus of the IgG protein (81, 193, 218–222). (Figure 2) Nevertheless, incorporating appendages into NKCEs would inevitably increase their size, making tissue penetration more challenging and potentially enhancing immunogenicity (223, 224). Hence, balancing multispecificity, desired tissue penetration, and low immunogenicity is critical for developing NKCEs in the adapted IgG format to achieve satisfactory therapeutical efficacy.
The choice of the production system of NKCE is also an essential factor in their development, as it determines the yield of NKCEs and the necessity for further downstream processing. Generally, two host systems are most commonly employed: bacterial and mammalian systems (refer to Table 4 for specific systems used for individual NKCE). The bacterial system is frequently used for producing fragmented formats, such as linked-scFv type NKCEs, although some are also produced using the mammalian system, such as HEK 293 cells. On the other hand, adapted IgG formats are primarily produced in mammalian cells, such as CHO cells, due to the need for post-translational processing for successful chain pairing.
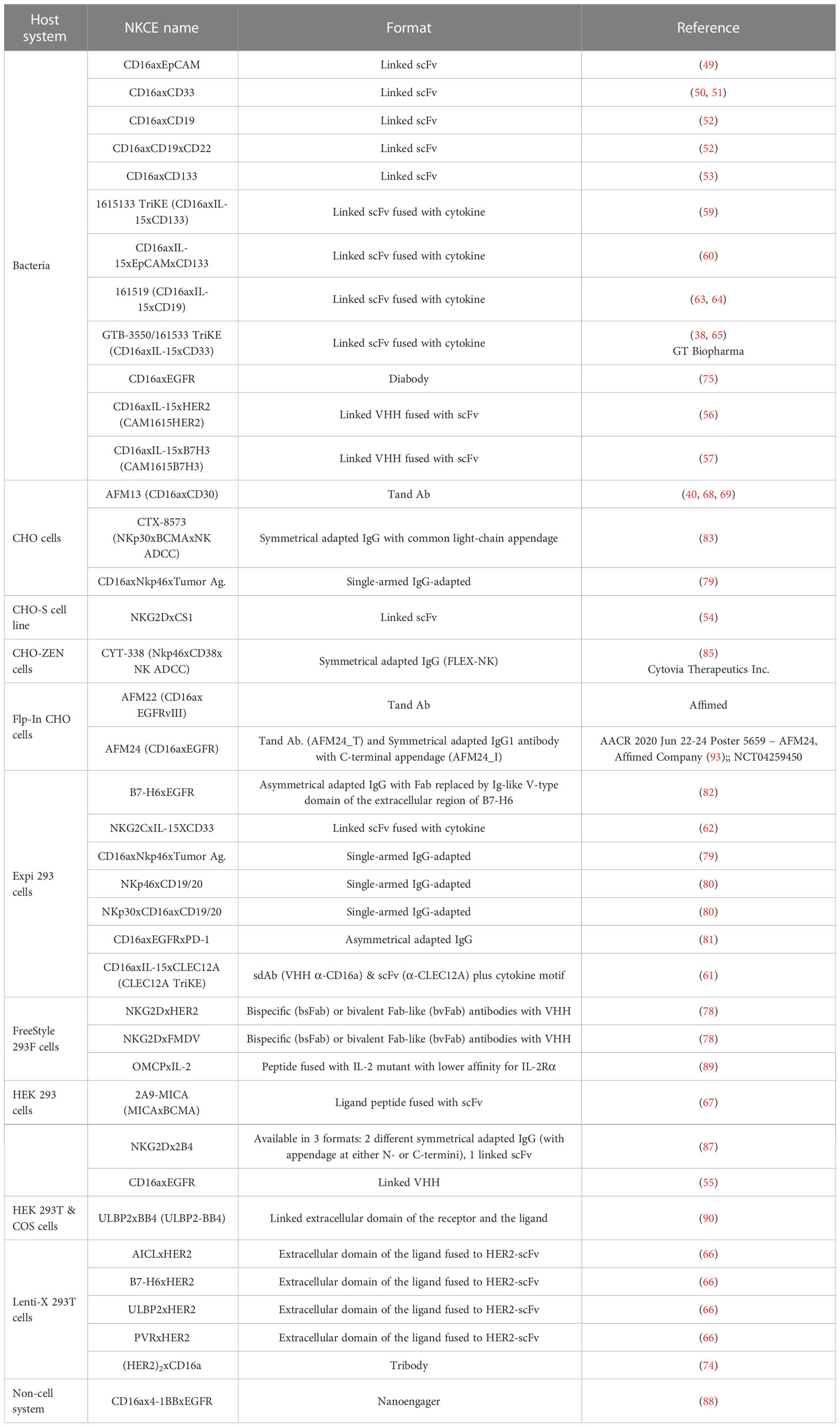
Table 4 Host systems for NKCE production.
The bacterial system allows inexpensive, rapid, and high-volume production of NKCEs, but proteins produced often require refolding (225). Typically, NKCE genes are inserted into an expression plasmid vector with an inducible promoter. When the bacteria culture reaches high density, the NKCE expression is induced. The NKCE proteins produced are isolated from the bacteria’s inclusion body. Harvesting raw NKCE proteins from the bacteria system usually take approximately 1-2 days (49), followed by another 2-3 days for refolding and purification (49).
On the other hand, expressing NKCEs in mammalian cell systems, such as HEK 293T or CHO cells, can bypass the refolding process as these cells can secrete fully functional NKCEs, which can be isolated from the supernatant. However, the yield from the mammalian cell system is typically lower than that of the bacterial system (54, 55). Usually, mammalian cells are co-transfected or transduced with expression vectors encoding the light or heavy chains. After around 6 days of culture, the supernatants are collected for the purification of NKCEs, followed by analyses of chain size and pairing (79). Although HEK 293T cells can offer post-translational modifications closest to those found in the human body (226), they are not as effective as CHO cells for large-scale production due to limitations in growth capacity, yield, and doubling time (227).
NKCEs have exhibited considerable potential as a new immunotherapeutic modality for cancer treatment. While the majority of current NKCEs targets CD16a and NKG2D, other activating receptors also display promising therapeutic potential and deserve further exploration. Moreover, a strategy that combines triggering activating receptors with blocking inhibitory receptors presents an effective approach to achieving enhanced NK cell activation. The inclusion of stimulatory cytokines in NKCEs has been shown to improve their therapeutical efficacies in preclinical studies. However, there is a need for more extensive studies to systematically evaluate how different molecular structures of NKCE impact immune synapse formation, pharmacokinetics, effector functions, and in vivo efficacy. Compared to T cell engagers, the current pool of NKCE formats is limited; hence, further exploration of additional NKCE formats is necessary to achieve the desired antitumor activities. Moreover, challenges such as potential on-target-off-tumor effects, NK cell exhaustion, and poor NK cell survival in the immunosuppressive TME have to be addressed during NKCE development (Figure 3). Therefore, further research is urgently needed to guide the development of NKCEs into off-the-shelf cancer treatment drugs.
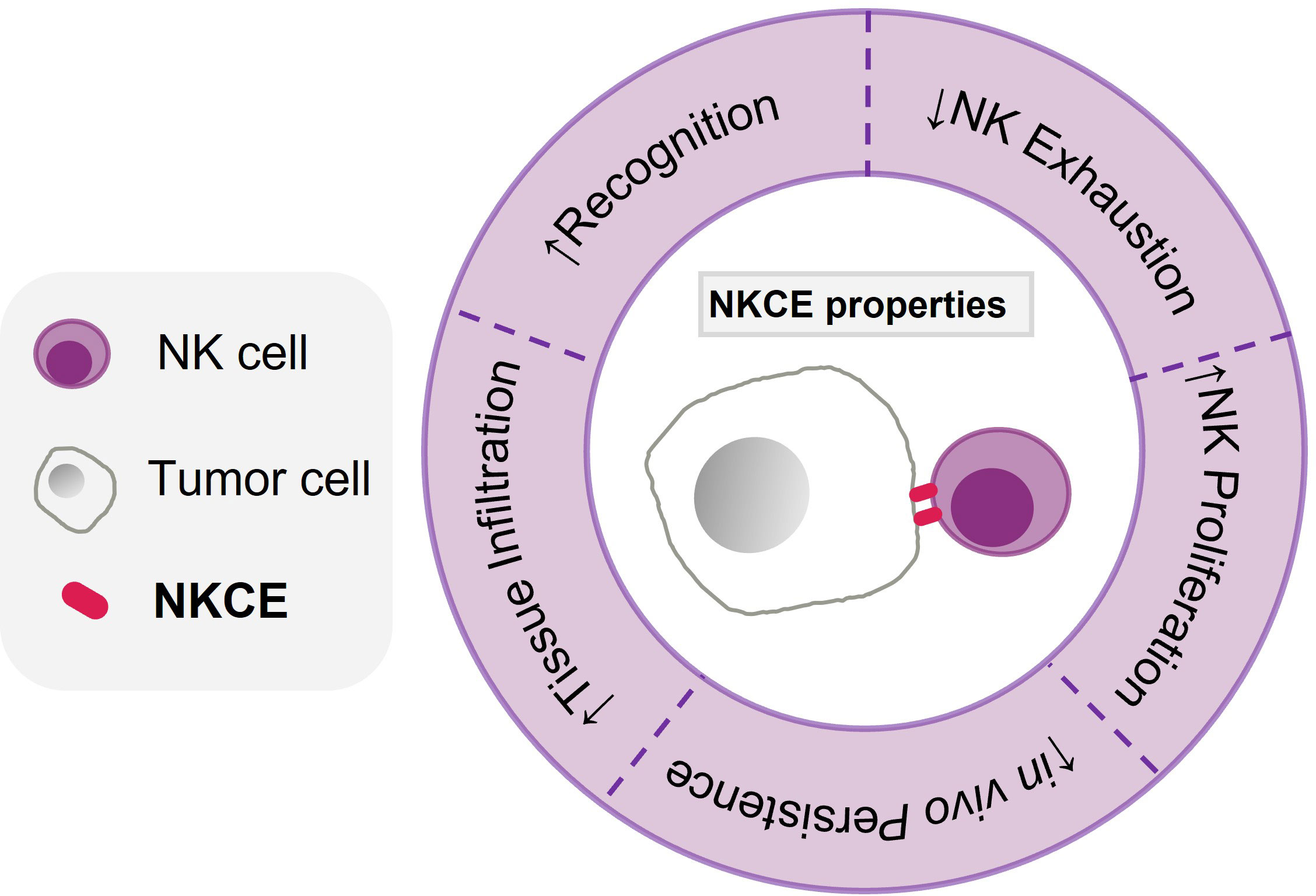
Figure 3 Characteristics of an effective NKCE. The characteristics of an effective NKCE include high specificity and selectivitity to tumor antigen (Recognition), being able to infiltrate into the tumor tissue effectively, enhance NK proliferation (within tumor), reduce NK exhaustion in TME, and persist in vivo for long period.
MZ, K-PL, and SX designed the work. MZ and SX wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Research Foundation (NRF) Competitive Research Programme (CRP) grant (Grant No. CRP26-2021RS-0002) and the core fund of Singapore Immunology Network (SIgN), Agency for Science Technology and Research (A*STAR), Singapore.
We thank members from XSL and LKP groups for the helpful discussions and suggestions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lu J, Jiang G. The journey of CAR-T therapy in hematological malignancies. Mol Cancer (2022) 21(1):194. doi: 10.1186/s12943-022-01663-0
2. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol (2018) 8. doi: 10.3389/fonc.2018.00086
3. Ren D, Hua Y, Yu B, Ye X, He Z, Li C, et al. Predictive biomarkers and mechanisms underlying resistance to PD1/PD-L1 blockade cancer immunotherapy. Mol Cancer (2020) 19(1):19. doi: 10.1186/s12943-020-1144-6
4. Zheng P-P, Kros JM, Li J. Approved CAR T cell therapies: ice bucket challenges on glaring safety risks and long-term impacts. Drug Discov Today (2018) 23(6):1175–82. doi: 10.1016/j.drudis.2018.02.012
5. Wang X, Rivière I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics (2016) 3:16015. doi: 10.1038/mto.2016.15
6. Ma S, Li X, Wang X, Cheng L, Li Z, Zhang C, et al. Current progress in CAR-T cell therapy for solid tumors. Int J Biol Sci (2019) 15(12):2548–60. doi: 10.7150/ijbs.34213
7. Hay AE, Cheung MC. CAR T-cells: costs, comparisons, and commentary. J Med Econ (2019) 22(7):613–5. doi: 10.1080/13696998.2019.1582059
8. Ceppi F, Rivers J, Annesley C, Pinto N, Park JR, Lindgren C, et al. Lymphocyte apheresis for chimeric antigen receptor T-cell manufacturing in children and young adults with leukemia and neuroblastoma. Transfusion (2018) 58(6):1414–20. doi: 10.1111/trf.14569
9. Zhao J, Lin Q, Song Y, Liu D. Universal CARs, universal T cells, and universal CAR T cells. J Hematol Oncol (2018) 11(1):132. doi: 10.1186/s13045-018-0677-2
10. Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer (2016) 16(1):7–19. doi: 10.1038/nrc.2015.5
11. Vance BA, Huizinga TW, Wardwell K, Guyre PM. Binding of monomeric human IgG defines an expression polymorphism of fc gamma RIII on large granular lymphocyte/natural killer cells. J Immunol (1993) 151(11):6429–39. doi: 10.4049/jimmunol.151.11.6429
12. Wu J, Edberg JC, Redecha PB, Bansal V, Guyre PM, Coleman K, et al. A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J Clin Invest (1997) 100(5):1059–70. doi: 10.1172/JCI119616
13. Koene HR, Kleijer M, Algra J, Roos D, von dem Borne AE, de Haas M. Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell fc gammaRIIIa, independently of the fc gammaRIIIa-48L/R/H phenotype. Blood (1997) 90(3):1109–14. doi: 10.1182/blood.V90.3.1109
14. de Haas M, Kleijer \Koene HR, Vries M, Simsek E, Tol van SMJ, et al. A triallelic fc gamma receptor type IIIA polymorphism influences the binding of human IgG by NK cell fc gamma RIIIa. J Immunol (1996) 156(8):2948–55. doi: 10.4049/jimmunol.156.8.2948
15. Pinto S, Pahl J, Schottelius A, Carter PJ, Koch J. Reimagining antibody-dependent cellular cytotoxicity in cancer: the potential of natural killer cell engagers. Trends Immunol (2022) 43(11):932–46. doi: 10.1016/j.it.2022.09.007
16. Pang Z, Wang Z, Li F, Feng C, Mu X. Current progress of CAR-NK therapy in cancer treatment. Cancers (Basel) (2022) 14(17):4318–41. doi: 10.3390/cancers14174318
17. Wu S-Y, Fu T, Jiang Y-Z, Shao Z-M. Natural killer cells in cancer biology and therapy. Mol Cancer (2020) 19(1):120. doi: 10.1158/1557-3125.HIPPO19-IA12
18. Chiossone L, Dumas P-Y, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol (2018) 18(11):671–88. doi: 10.1038/s41577-018-0061-z
19. Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity (2017) 47(5):820–33. doi: 10.1016/j.immuni.2017.10.008
20. Prager I, Watzl C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J Leukocyte Biol (2019) 105(6):1319–29. doi: 10.1002/JLB.MR0718-269R
21. Prager I, Liesche C, Ooijen van H, Urlaub D, Verron Q, Sandström N, et al. NK cells switch from granzyme b to death receptor-mediated cytotoxicity during serial killing. J Exp Med (2019) 216(9):2113–27. doi: 10.1084/jem.20181454
22. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science (2002) 295(5562):2097–100. doi: 10.1126/science.1068440
23. Mehta RS, Rezvani K. Can we make a better match or mismatch with KIR genotyping? Hematol Am Soc Hematol Educ Program (2016) 2016(1):106–18. doi: 10.1182/asheducation-2016.1.106
24. Lamers-Kok N, Panella D, Georgoudaki A-M, Liu H, Özkazanc D, Kučerová L, et al. Natural killer cells in clinical development as non-engineered, engineered, and combination therapies. J Hematol Oncol (2022) 15(1):164. doi: 10.1186/s13045-022-01382-5
25. Li F, Liu S. Focusing on NK cells and ADCC: a promising immunotherapy approach in targeted therapy for HER2-positive breast cancer. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.1083462
26. Lo Nigro C, Macagno M, Sangiolo D, Bertolaccini L, Aglietta M, Merlano MC. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: biological evidence and clinical perspectives. Ann Transl Med (2019) 7(5):105. doi: 10.21037/atm.2019.01.42
27. Messersmith WA, Ahnen DJ. Targeting EGFR in colorectal cancer. N Engl J Med (2008) 359(17):1834–6. doi: 10.1056/NEJMe0806778
28. Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG fc receptor FcγRIIIa gene. Blood (2002) 99(3):754–8. doi: 10.1182/blood.V99.3.754
29. Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, et al. Immunoglobulin G fragment c receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu–positive metastatic breast cancer. J Clin Oncol (2008) 26(11):1789–96. doi: 10.1200/JCO.2007.14.8957
30. Weng W-K, Levy R. Two immunoglobulin G fragment c receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol (2003) 21(21):3940–7. doi: 10.1200/JCO.2003.05.013
31. Pereira NA, Chan KF, Lin PC, Song Z. The "less-is-more" in therapeutic antibodies: afucosylated anti-cancer antibodies with enhanced antibody-dependent cellular cytotoxicity. MAbs (2018) 10(5):693–711. doi: 10.1080/19420862.2018.1466767
32. Zhang L, Meng Y, Feng X, Han Z. CAR-NK cells for cancer immunotherapy: from bench to bedside. biomark Res (2022) 10(1):12. doi: 10.1186/s40364-022-00364-6
33. Lu H, Zhao X, Li Z, Hu Y, Wang H. From CAR-T cells to CAR-NK cells: a developing immunotherapy method for hematological malignancies. Front Oncol (2021) 11. doi: 10.3389/fonc.2021.720501
34. Khawar MB, Sun H. CAR-NK cells: from natural basis to design for kill. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.707542
35. Vogel B, Tennert K, Full F, Ensser A. Efficient generation of human natural killer cell lines by viral transformation. Leukemia (2014) 28(1):192–5. doi: 10.1038/leu.2013.188
36. Nikkhoi SK, Li G, Eleya S, Yang G, Vandavasi VG, Hatefi A. Bispecific killer cell engager with high affinity and specificity toward CD16a on NK cells for cancer immunotherapy. Front Immunol (2022) 13:1039969. doi: 10.3389/fimmu.2022.1039969
37. Wensveen FM, Jelenčić V, Polić B. NKG2D: a master regulator of immune cell responsiveness. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.00441
38. Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU, et al. IL15 trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin Cancer Res (2016) 22(14):3440–50. doi: 10.1158/1078-0432.CCR-15-2710
39. Laskowski TJ, Biederstädt A, Rezvani K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat Rev Cancer (2022) 22(10):557–75. doi: 10.1038/s41568-022-00491-0
40. Kerbauy LN, Marin ND, Kaplan M, Banerjee PP, Berrien-Elliott MM, Becker-Hapak M, et al. Combining AFM13, a bispecific CD30/CD16 antibody, with cytokine-activated blood and cord blood-derived NK cells facilitates CAR-like responses against CD30(+) malignancies. Clin Cancer Res (2021) 27(13):3744–56. doi: 10.1158/1078-0432.CCR-21-0164
41. Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR-NK cells: a promising cellular immunotherapy for cancer. eBioMedicine (2020) 59:102975–84. doi: 10.1016/j.ebiom.2020.102975
42. Reusch U, Ellwanger K, Fucek I, Müller T, Schniegler-Mattox U, Koch J, et al. Cryopreservation of natural killer cells pre-complexed with innate cell engagers. Antibodies (Basel) (2022) 11(1):12–33. doi: 10.3390/antib11010012
43. Raitman I, Gleason J, Rotondo S, He S, Rousseva V, Guo X, et al. Human placental CD34 +-derived natural killer cells with high affinity and cleavage resistant CD16 (CYNK-101) in combination with daratumumab for immunotherapy against CD38 expressing hematological malignancies. Blood (2021) 138:2773. doi: 10.1182/blood-2021-148869
44. Lanier LL, Yu G, Phillips JH. Analysis of fc gamma RIII (CD16) membrane expression and association with CD3 zeta and fc epsilon RI-gamma by site-directed mutation. J Immunol (1991) 146(5):1571–6. doi: 10.4049/jimmunol.146.5.1571
45. Galandrini R, Micucci F, Tassi I, Cifone MG, Cinque B, Piccoli M, et al. Arf6: a new player in FcγRIIIA lymphocyte-mediated cytotoxicity. Blood (2005) 106(2):577–83. doi: 10.1182/blood-2004-10-4100
46. Billadeau DD, Brumbaugh KM, Dick CJ, Schoon RA, Bustelo XR, Leibson PJ. The vav-Rac1 pathway in cytotoxic lymphocytes regulates the generation of cell-mediated killing. J Exp Med (1998) 188(3):549–59. doi: 10.1084/jem.188.3.549
47. Bryceson YT, March ME, Ljunggren H-G, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood (2006) 107(1):159–66. doi: 10.1182/blood-2005-04-1351
48. Arnone CM, Polito VA, Mastronuzzi A, Carai A, Diomedi FC, Antonucci L, et al. Oncolytic adenovirus and gene therapy with EphA2-BiTE for the treatment of pediatric high-grade gliomas. J Immunother Cancer (2021) 9(5):e001930-59. doi: 10.1136/jitc-2020-001930
49. Vallera DA, Zhang B, Gleason MK, Oh S, Weiner LM, Kaufman DS, et al. Heterodimeric bispecific single-chain variable-fragment antibodies against EpCAM and CD16 induce effective antibody-dependent cellular cytotoxicity against human carcinoma cells. Cancer Biother Radiopharm (2013) 28(4):274–82. doi: 10.1089/cbr.2012.1329
50. Gleason MK, Ross JA, Warlick ED, Lund TC, Verneris MR, Wiernik A, et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood (2014) 123(19):3016–26. doi: 10.1182/blood-2013-10-533398
51. Wiernik A, Foley B, Zhang B, Verneris MR, Warlick E, Ross JA, et al. Bi-specific killer cell engagers (BiKEs) signaling through CD16 and targeting CD33 (CD16 × CD33), triggers natural killer (NK) cell cytotoxic and cytokine production against refractory acute myeloid leukemia (AML). Blood (2012) 120(21):256–6. doi: 10.1182/blood.V120.21.256.256
52. Gleason MK, Verneris MR, Todhunter DA, Zhang B, McCullar V, Zhou SX, et al. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol Cancer Ther (2012) 11(12):2674–84. doi: 10.1158/1535-7163.MCT-12-0692
53. Schmohl JU, Gleason MK, Dougherty PR, Miller JS, Vallera DA. Heterodimeric bispecific single chain variable fragments (scFv) killer engagers (BiKEs) enhance NK-cell activity against CD133+ colorectal cancer cells. Target Oncol (2016) 11(3):353–61. doi: 10.1007/s11523-015-0391-8
54. Chan WK, Kang S, Youssef Y, Glankler EN, Barrett ER, Carter AM, et al. A CS1-NKG2D bispecific antibody collectively activates cytolytic immune cells against multiple myeloma. Cancer Immunol Res (2018) 6(7):776–87. doi: 10.1158/2326-6066.CIR-17-0649
55. Toffoli EC, Sheikhi A, Lameris R, King LA, Vliet van A, Walcheck B, et al. Enhancement of NK cell antitumor effector functions using a bispecific single domain antibody targeting CD16 and the epidermal growth factor receptor. Cancers (2021) 13(21):5446. doi: 10.3390/cancers13215446
56. Vallera DA, Oh F, Kodal B, Hinderlie P, Geller MA, Miller JS, et al. A HER2 tri-specific NK cell engager mediates efficient targeting of human ovarian cancer. Cancers (2021) 13(16):3994. doi: 10.3390/cancers13163994
57. Vallera DA, Ferrone S, Kodal B, Hinderlie P, Bendzick L, Ettestad B, et al. NK-Cell-Mediated targeting of various solid tumors using a B7-H3 tri-specific killer engager in vitro and in vivo. Cancers (2020) 12(9):2659. doi: 10.3390/cancers12092659
58. Giang KA, Kanaya M, Boxaspen T, Kanaya M, Krokeide SZ, Diao Y, et al. Affibody-based BCMA x CD16 dual engagers for activation of NK cells towards multiple myeloma. Blood (2022) 140(Supplement 1):10699–700. doi: 10.1182/blood-2022-164753
59. Schmohl JU, Felices M, Oh F, Lenvik AJ, Lebeau AM, Panyam J, et al. Engineering of anti-CD133 trispecific molecule capable of inducing NK expansion and driving antibody-dependent cell-mediated cytotoxicity. Cancer Res Treat (2017) 49(4):1140–52. doi: 10.4143/crt.2016.491
60. Schmohl JU, Felices M, Todhunter D, Taras E, Miller JS, Vallera DA. Tetraspecific scFv construct provides NK cell mediated ADCC and self-sustaining stimuli via insertion of IL-15 as a cross-linker. Oncotarget (2016) 7(45):73830–44. doi: 10.18632/oncotarget.12073
61. Arvindam US, van Hauten PMM, Schirm D, Schaap N, Hobo W, Blazar BR, et al. A trispecific killer engager molecule against CLEC12A effectively induces NK-cell mediated killing of AML cells. Leukemia (2021) 35(6):1586–96. doi: 10.1038/s41375-020-01065-5
62. Chiu E, Felices M, Cichocki F, Davis Z, Wang H, Tuninga K, et al. Anti-NKG2C/IL-15/anti-CD33 killer engager directs primary and iPSC-derived NKG2C(+) NK cells to target myeloid leukemia. Mol Ther (2021) 29(12):3410–21. doi: 10.1016/j.ymthe.2021.06.018
63. Felices M, Kodal B, Hinderlie P, Kaminski MF, Cooley S, Weisdorf DJ, et al. Novel CD19-targeted TriKE restores NK cell function and proliferative capacity in CLL. Blood Adv (2019) 3(6):897–907. doi: 10.1182/bloodadvances.2018029371
64. Cheng Y, Zheng X, Wang X, Chen Y, Wei H, Sun R, et al. Trispecific killer engager 161519 enhances natural killer cell function and provides anti-tumor activity against CD19-positive cancers. Cancer Biol Med (2020) 17(4):1026–38. doi: 10.20892/j.issn.2095-3941.2020.0399
65. Don Yun H, Felices M, Vallera DA, Hinderlie P, Cooley S, Arock M, et al. Trispecific killer engager CD16xIL15xCD33 potently induces NK cell activation and cytotoxicity against neoplastic mast cells. Blood Adv (2018) 2(13):1580–4. doi: 10.1182/bloodadvances.2018018176
66. Peipp M, Derer S, Lohse S, Staudinger M, Klausz K, Valerius T, et al. HER2-specific immunoligands engaging NKp30 or NKp80 trigger NK-cell-mediated lysis of tumor cells and enhance antibody-dependent cell-mediated cytotoxicity. Oncotarget (2015) 6(31):32075–88. doi: 10.18632/oncotarget.5135
67. Wang Y, Li H, Xu W, Pan M, Qiao C, Cai J, et al. BCMA-targeting bispecific antibody that simultaneously stimulates NKG2D-enhanced efficacy against multiple myeloma. J Immunother (2020) 43(6):175–88. doi: 10.1097/CJI.0000000000000320
68. Wu J, Fu J, Zhang M, Liu D. AFM13: a first-in-class tetravalent bispecific anti-CD30/CD16A antibody for NK cell-mediated immunotherapy. J Hematol Oncol (2015) 8(1):96. doi: 10.1186/s13045-015-0188-3
69. Reusch U, Burkhardt C, Fucek I, Gall Le F, Gall Le M, Hoffmann K, et al. A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30+ tumor cells. MAbs (2014) 6(3):728–39. doi: 10.4161/mabs.28591
70. Wingert S, Reusch U, Knackmuss S, Kluge M, Damrat M, Pahl J, et al. Preclinical evaluation of AFM24, a novel CD16A-specific innate immune cell engager targeting EGFR-positive tumors. MAbs (2021) 13(1):1950264. doi: 10.1080/19420862.2021.1950264
71. Gantke T, Reusch U, Ellwanger K, Fucek I, Weichel M, Treder M. Abstract 5671: AFM26 - a novel CD16A-directed bispecific TandAb targeting BCMA for multiple myeloma. Cancer Res (2017) 77(13_Supplement):5671–1. doi: 10.1158/1538-7445.AM2017-5671
72. Cai H, Kakiuchi-Kiyota S, Hendricks R, Zhong S, Liu L, Adedeji AO, et al. Nonclinical pharmacokinetics, pharmacodynamics, and translational model of RO7297089, a novel anti-BCMA/CD16A bispecific tetravalent antibody for the treatment of multiple myeloma. AAPS J (2022) 24(6):100. doi: 10.1208/s12248-022-00744-8
73. Kakiuchi-Kiyota S, Ross T, Wallweber HA, Kiefer JR, Schutten MM, Adedeji AO, et al. A BCMA/CD16A bispecific innate cell engager for the treatment of multiple myeloma. Leukemia (2022) 36(4):1006–14. doi: 10.1038/s41375-021-01478-w
74. Oberg HH, Kellner C, Gonnermann D, Sebens S, Bauerschlag D, Gramatzki M, et al. Tribody [(HER2)2xCD16] is more effective than trastuzumab in enhancing γδ T cell and natural killer cell cytotoxicity against HER2-expressing cancer cells. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.00814
75. Kuwahara A, Nagai K, Nakanishi T, Kumagai I, Asano R. Functional domain order of an anti-EGFR × anti-CD16 bispecific diabody involving NK cell activation. Int J Mol Sci (2020) 21(23):8914. doi: 10.3390/ijms21238914
76. Gantke T, Weichel M, Reusch U, Ellwanger K, Fucek I, Griep R, et al. Trispecific antibodies for selective CD16A-directed NK-cell engagement in multiple myeloma. Blood (2016) 128(22):4513–3. doi: 10.1182/blood.V128.22.4513.4513
77. Gantke T, Weichel M, Herbrecht C, Reusch U, Ellwanger K, Fucek I, et al. Trispecific antibodies for CD16A-directed NK cell engagement and dual-targeting of tumor cells. Protein Engineering Design Selection (2017) 30(9):673–84. doi: 10.1093/protein/gzx043
78. Raynaud A, Desrumeaux K, Vidard L, Termine E, Baty D, Chames P, et al. Anti-NKG2D single domain-based antibodies for the modulation of anti-tumor immune response. Oncoimmunology (2020) 10(1):1854529. doi: 10.1080/2162402X.2020.1854529
79. Gauthier L, Morel A, Anceriz N, Rossi B, Blanchard-Alvarez A, Grondin G, et al. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell (2019) 177(7):1701–1713.e16. doi: 10.1016/j.cell.2019.04.041
80. Colomar-Carando N, Gauthier L, Merli P, Loiacono F, Canevali P, Falco M, et al. Exploiting natural killer cell engagers to control pediatric b-cell precursor acute lymphoblastic leukemia. Cancer Immunol Res (2022) 10(3):291–302. doi: 10.1158/2326-6066.CIR-21-0843
81. Bogen JP, Carrara SC, Fiebig D, Grzeschik J, Hock B, Kolmar H. Design of a trispecific checkpoint inhibitor and natural killer cell engager based on a 2 + 1 common light chain antibody architecture. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.669496
82. Pekar L, Klausz K, Busch M, Valldorf B, Kolmar H, Wesch D, et al. Affinity maturation of B7-H6 translates into enhanced NK cell-mediated tumor cell lysis and improved proinflammatory cytokine release of bispecific immunoligands via NKp30 engagement. J Immunol (2021) 206(1):225–36. doi: 10.4049/jimmunol.2001004
83. Watkins-Yoon J, Guzman W, Oliphant A, Haserlat S, Leung A, Chottin C, et al. CTX-8573, an innate-cell engager targeting BCMA, is a highly potent multispecific antibody for the treatment of multiple myeloma. Blood (2019) 134(Supplement_1):3182–2. doi: 10.1182/blood-2019-128749
84. Draghi M, Schafer JL, Nelson A, Frye Z, Oliphant A, Haserlat S, et al. Abstract 4972: preclinical development of a first-in-class NKp30xBCMA NK cell engager for the treatment of multiple myeloma. Cancer Res (2019) 79(13_Supplement):4972–2. doi: 10.1158/1538-7445.AM2019-4972
85. Lin L, Chang HM, Nakid C, Frankel S, Wu D, Kadouche J, et al. P842: novel multifunctional tetravalent Cd38 Nkp46 flex-nk engagers actively target and kill multiple myeloma cells. Hemasphere (2022) 6(Suppl):736–7. doi: 10.1097/01.HS9.0000846252.68296.03
86. Arulanandam A, Lin L, Chang H-M, Cerutti M, Choblet S, Gao P, et al. Derivation and preclinical characterization of CYT-303, a novel NKp46-NK cell engager targeting GPC3. Cells (2023) 12(7):996. doi: 10.3390/cells12070996
87. Song A-Y, Kim H, Kim JM, Hwang S-H, Ko D-H, Kim HS. Bispecific antibody designed for targeted NK cell activation and functional assessment for biomedical applications. ACS Appl Materials Interfaces (2021) 13(36):42370–81. doi: 10.1021/acsami.1c08986
88. Au KM, Park SI, Wang AZ. Trispecific natural killer cell nanoengagers for targeted chemoimmunotherapy. Sci Adv (2020) 6(27):eaba8564. doi: 10.1126/sciadv.aba8564
89. Ghasemi R, Lazear E, Wang X, Arefanian S, Zheleznyak A, Carreno BM, et al. Selective targeting of IL-2 to NKG2D bearing cells for improved immunotherapy. Nat Commun (2016) 7(1):12878. doi: 10.1038/ncomms12878
90. von Strandmann EP, Hansen HP, Reiners KS, Schnell R, Borchmann P, Merkert S, et al. A novel bispecific protein (ULBP2-BB4) targeting the NKG2D receptor on natural killer (NK) cells and CD138 activates NK cells and has potent antitumor activity against human multiple myeloma in vitro and in vivo. Blood (2006) 107(5):1955–62. doi: 10.1182/blood-2005-05-2177
91. Rothe A, Sasse S, Topp MS, Eichenauer DA, Hummel H, Reiners KS, et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood (2015) 125(26):4024–31. doi: 10.1182/blood-2014-12-614636
92. Sasse S, Bröckelmann PJ, Momotow J, Plütschow A, Hüttmann A, Basara N, et al. AFM13 in patients with relapsed or refractory classical Hodgkin lymphoma: final results of an open-label, randomized, multicenter phase II trial. Leukemia Lymphoma (2022) 63(8):1871–8. doi: 10.1080/10428194.2022.2095623
93. Ellwanger K, Reusch U, Fucek I, Weichel M, Gantke T, Knackmuss S, et al. Abstract 580: anti-EGFRvIII TandAbs recruiting either T or NK cells are highly specific and potent therapeutic antibody candidates for the treatment of EGFRvIII+ tumors. Cancer Res (2016) 76(14_Supplement):580–0. doi: 10.1158/1538-7445.AM2016-580
94. Ross T, Reusch U, Wingert S, Haneke T, Klausz K, Otte A-K, et al. Preclinical characterization of AFM26, a novel b cell maturation antigen (BCMA)-directed tetravalent bispecific antibody for high affinity retargeting of NK cells against myeloma. Blood (2018) 132(Supplement 1):1927–7. doi: 10.1182/blood-2018-99-118970
95. Grzywacz B, Kataria N, Verneris MR. CD56dimCD16+ NK cells downregulate CD16 following target cell induced activation of matrix metalloproteinases. Leukemia (2007) 21(2):356–9. doi: 10.1038/sj.leu.2404499
96. Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood (2013) 121(18):3599–608. doi: 10.1182/blood-2012-04-425397
97. Peruzzi G, Femnou L, Gil-Krzewska A, Borrego F, Weck J, Krzewski K, et al. Membrane-type 6 matrix metalloproteinase regulates the activation-induced downmodulation of CD16 in human primary NK cells. J Immunol (2013) 191(4):1883–94. doi: 10.4049/jimmunol.1300313
98. Srpan K, Ambrose A, Karampatzakis A, Saeed M, Cartwright ANR, Guldevall K, et al. Shedding of CD16 disassembles the NK cell immune synapse and boosts serial engagement of target cells. J Cell Biol (2018) 217(9):3267–83. doi: 10.1083/jcb.201712085
99. Wiernik A, Foley B, Zhang B, Verneris MR, Warlick E, Gleason MK, et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin Cancer Res (2013) 19(14):3844–55. doi: 10.1158/1078-0432.CCR-13-0505
100. Zhang Y, Boesen CC, Radaev S, Brooks AG, Fridman W-H, Sautes-Fridman C, et al. Crystal structure of the extracellular domain of a human FcyRIII. Immunity (2000) 13(3):387–95. doi: 10.1016/S1074-7613(00)00038-8
101. Huizinga TW, de Haas M, Kleijer M, Nuijens JH, Roos D, von dem Borne AE. Soluble fc gamma receptor III in human plasma originates from release by neutrophils. J Clin Invest (1990) 86(2):416–23. doi: 10.1172/JCI114727
102. Watzl C, Long EO. Signal transduction during activation and inhibition of natural killer cells. Curr Protoc Immunol (2010) 90(1):11.9B.1–11.9B.17. doi: 10.1002/0471142735.im1109bs90
103. Billadeau DD, Upshaw JL, Schoon RA, Dick CJ, Leibson PJ. NKG2D-DAP10 triggers human NK cell–mediated killing via a syk-independent regulatory pathway. Nat Immunol (2003) 4(6):557–64. doi: 10.1038/ni929
104. Garrity D, et al. The activating NKG2D receptor assembles in the membrane with two signaling dimers into a hexameric structure. Proc Natl Acad Sci (2005) 102(21):7641–6. doi: 10.1073/pnas.0502439102
105. Sutherland CL, et al. UL16-binding proteins, novel MHC class I-related proteins, bind to NKG2D and activate multiple signaling pathways in primary NK cells. J Immunol (2002) 168(2):671–9. doi: 10.4049/jimmunol.168.2.671
106. Jamieson AM, Diefenbach A, McMahon CW, Xiong N, Carlyle JR, Raulet DH. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity (2002) 17(1):19–29. doi: 10.1016/S1074-7613(02)00333-3
107. Deguine J, Bousso P. Dynamics of NK cell interactions in vivo. Immunol Rev (2013) 251(1):154–9. doi: 10.1111/imr.12015
108. Holdenrieder S, Stieber P, Peterfi A, Nagel D, Steinle A, Salih HR. Soluble MICA in malignant diseases. Int J Cancer (2006) 118(3):684–7. doi: 10.1002/ijc.21382
109. Pende D, Parolini S, Pessino A, Sivori S, Augugliaro R, Morelli L, et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J Exp Med (1999) 190:1505–16. doi: 10.1084/jem.190.10.1505
110. Barrow AD, Martin CJ, Colonna M. The natural cytotoxicity receptors in health and disease. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.00909
111. Pinheiro PF, Justino GC, Marques MM. NKp30 - a prospective target for new cancer immunotherapy strategies. Br J Pharmacol (2020) 177(20):4563–80. doi: 10.1111/bph.15222
112. Phillips M, Romeo F, Bitsaktsis C, Sabatino D. B7H6-derived peptides trigger TNF-α-dependent immunostimulatory activity of lymphocytic NK92-MI cells. Pept Sci (2016) 106(5):658–72. doi: 10.1002/bip.22879
113. Westgaard IH, Berg SF, Vaage JT, Wang LL, Yokoyama WM, Dissen E, et al. Rat NKp46 activates natural killer cell cytotoxicity and is associated with FcεRIγ and CD3ζ. J Leukocyte Biol (2004) 76(6):1200–6. doi: 10.1189/jlb.0903428
114. Foster CE, Colonna M, Sun PD. Crystal structure of the human natural killer (NK) cell activating receptor NKp46 reveals structural relationship to other leukocyte receptor complex immunoreceptors. J Biol Chem (2003) 278(46):46081–6. doi: 10.1074/jbc.M308491200
115. Zamai L, Zotto Del G, Buccella F, Gabrielli S, Canonico B, Artico M, et al. Understanding the synergy of NKp46 and Co-activating signals in various NK cell subpopulations: paving the way for more successful NK-Cell-Based immunotherapy. Cells (2020) 9(3):753–78. doi: 10.3390/cells9030753
116. Platonova S, Cherfils-Vicini J, Damotte D, Crozet L, Vieillard V, Validire P, et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res (2011) 71(16):5412–22. doi: 10.1158/0008-5472.CAN-10-4179
117. Fauriat C, Just-Landi S, Mallet FO, Arnoulet C, Sainty D, Olive D, et al. Deficient expression of NCR in NK cells from acute myeloid leukemia: evolution during leukemia treatment and impact of leukemia cells in NCRdull phenotype induction. Blood (2006) 109(1):323–30. doi: 10.1182/blood-2005-08-027979
118. Mamessier E, Sylvain A, Thibult M-L, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest (2011) 121(9):3609–22. doi: 10.1172/JCI45816
119. Lin L, Chang H-M, Wu D, Titz B, Chen A, Chao Ma M, et al. Biological characterization and differential gene expression analysis of CYT-338 NK cell engager (NKE) against CD38 expressing tumors including multiple myeloma (MM). Blood (2022) 140(Supplement 1):7068–9. doi: 10.1182/blood-2022-170193
120. Wang Y, Zhang Y, Hughes T, Zhang J, Caligiuri MA, Benson DM, et al. Fratricide of NK cells in daratumumab therapy for multiple myeloma overcome by ex vivo–expanded autologous NK cells. Clin Cancer Res (2018) 24(16):4006–17. doi: 10.1158/1078-0432.CCR-17-3117
121. Welte S, Kuttruff S, Waldhauer I, Steinle A. Mutual activation of natural killer cells and monocytes mediated by NKp80-AICL interaction. Nat Immunol (2006) 7(12):1334–42. doi: 10.1038/ni1402
122. Deng G, Zheng X, Zhou J, Wei H, Tian Z, Sun R. Generation and preclinical characterization of an NKp80-fc fusion protein for redirected cytolysis of natural killer (NK) cells against leukemia. J Biol Chem (2015) 290(37):22474–84. doi: 10.1074/jbc.M115.678912
123. Dennehy KM, Klimosch SN, Steinle A. Cutting edge: NKp80 uses an atypical hemi-ITAM to trigger NK cytotoxicity. J Immunol (2011) 186(2):657–61. doi: 10.4049/jimmunol.0904117
124. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol (2019) 16(5):430–41. doi: 10.1038/s41423-019-0206-4
125. Lanier LL, et al. Association of DAP12 with activating CD94/NKG2C NK cell receptors. Immunity (1998) 8(6):693–701. doi: 10.1016/S1074-7613(00)80574-9
126. Della Chiesa M, et al. Features of memory-like and PD-1(+) human NK cell subsets. Front Immunol (2016) 7:351. doi: 10.3389/fimmu.2016.00351
127. Béziat V, et al. Polyclonal expansion of NKG2C+ NK cells in TAP-deficient patients. Front Immunol (2015) 6. doi: 10.3389/fimmu.2015.00507
128. Muntasell A, et al. Relationship of NKG2C copy number with the distribution of distinct cytomegalovirus-induced adaptive NK cell subsets. J Immunol (2016) 196(9):3818–27. doi: 10.4049/jimmunol.1502438
129. Heath J, et al. NKG2C+CD57+ natural killer cell expansion parallels cytomegalovirus-specific CD8+ T cell evolution towards senescence. J Immunol Res (2016) 2016:7470124. doi: 10.1155/2016/7470124
130. Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol (2013) 31:227–58. doi: 10.1146/annurev-immunol-020711-075005
131. Blunt MD, Khakoo SI. Activating killer cell immunoglobulin-like receptors: detection, function and therapeutic use. Int J Immunogenetics (2020) 47(1):1–12. doi: 10.1111/iji.12461
132. Sivori S, Pende D, Quatrini L, Pietra G, Chiesa Della M, Vacca P, et al. NK cells and ILCs in tumor immunotherapy. Mol Aspects Med (2021) 80:100870. doi: 10.1016/j.mam.2020.100870
133. Blunt MD, Pulido Vallejo A, Fisher JG, Graham LV, Doyle ADP, Fulton R, et al. KIR2DS2 expression identifies NK cells with enhanced anticancer activity. J Immunol (2022) 209(2):379–90. doi: 10.4049/jimmunol.2101139
134. Tu TC, Brown NK, Kim TJ, Wroblewska J, Yang X, Guo X, et al. CD160 is essential for NK-mediated IFN-γ production. J Exp Med (2015) 212(3):415–29. doi: 10.1084/jem.20131601
135. Le Bouteiller P, Barakonyi A, Giustiniani J, Lenfant F, Marie-Cardine A, Aguerre-Girr M, et al. Engagement of CD160 receptor by HLA-c is a triggering mechanism used by circulating natural killer (NK) cells to mediate cytotoxicity. Proc Natl Acad Sci (2002) 99(26):16963–8. doi: 10.1073/pnas.012681099
136. Barakonyi A, Rabot M, Marie-Cardine A, Aguerre-Girr M, Polgar B, Schiavon VR, et al. Cutting edge: engagement of CD160 by its HLA-c physiological ligand triggers a unique cytokine profile secretion in the cytotoxic peripheral blood NK cell Subset1. J Immunol (2004) 173(9):5349–54. doi: 10.4049/jimmunol.173.9.5349
137. Le Bouteiller P, Tabiasco J, Polgar B, Kozma N, Giustiniani J, Siewiera J, et al. CD160: a unique activating NK cell receptor. Immunol Lett (2011) 138(2):93–6. doi: 10.1016/j.imlet.2011.02.003
138. Maïza H, Leca G, Mansur IG, Schiavon V, Boumsell L, Bensussan A. A novel 80-kD cell surface structure identifies human circulating lymphocytes with natural killer activity. J Exp Med (1993) 178(3):1121–6. doi: 10.1084/jem.178.3.1121
139. Shibuya A, Campbell D, Hannum C, Yssel H, Franz-Bacon K, McClanahan T, et al. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity (1996) 4(6):573–81. doi: 10.1016/S1074-7613(00)70060-4
140. Zhang Z, Wu N, Lu Y, Davidson D, Colonna M, Veillette A. DNAM-1 controls NK cell activation via an ITT-like motif. J Exp Med (2015) 212(12):2165–82. doi: 10.1084/jem.20150792
141. Pende D, Spaggiari GM, Marcenaro S, Martini S, Rivera P, Capobianco A, et al. Analysis of the receptor-ligand interactions in the natural killer–mediated lysis of freshly isolated myeloid or lymphoblastic leukemias: evidence for the involvement of the poliovirus receptor (CD155) and nectin-2 (CD112). Blood (2005) 105(5):2066–73. doi: 10.1182/blood-2004-09-3548
142. Waggoner S, Kumar V. Evolving role of 2B4/CD244 in T and NK cell responses during virus infection. Front Immunol (2012) 3. doi: 10.3389/fimmu.2012.00377
143. Parolini S, Bottino C, Falco M, Augugliaro R, Giliani S, Franceschini R, et al. X-Linked lymphoproliferative disease. 2B4 molecules displaying inhibitory rather than activating function are responsible for the inability of natural killer cells to kill Epstein-Barr virus-infected cells. J Exp Med (2000) 192(3):337–46. doi: 10.1084/jem.192.3.337
144. Chen R, Relouzat F, Roncagalli R, Aoukaty A, Tan R, Latour S, et al. Molecular dissection of 2B4 signaling: implications for signal transduction by SLAM-related receptors. Mol Cell Biol (2004) 24(12):5144–56. doi: 10.1128/MCB.24.12.5144-5156.2004
145. Rocca YS, Roberti MP, Juliá EP, Pampena MB, Bruno L, Rivero S, et al. Phenotypic and functional dysregulated blood NK cells in colorectal cancer patients can be activated by cetuximab plus IL-2 or IL-15. Front Immunol (2016) 7:413. doi: 10.3389/fimmu.2016.00413
146. Easom NJW, Stegmann KA, Swadling L, Pallett LJ, Burton AR, Odera D, et al. IL-15 overcomes hepatocellular carcinoma-induced NK cell dysfunction. Front Immunol (2018) 9:1009. doi: 10.3389/fimmu.2018.01009
147. Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol (2015) 33(1):74–82. doi: 10.1200/JCO.2014.57.3329
148. Ye C, Brand D, Zheng SG. Targeting IL-2: an unexpected effect in treating immunological diseases. Signal Transduct Target Ther (2018) 3(1):2. doi: 10.1038/s41392-017-0002-5
149. Schmidt A, Oberle N, Krammer P. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol (2012) 3. doi: 10.3389/fimmu.2012.00051
150. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Trans Med (2016) 8(357):357ra123–357ra123. doi: 10.1126/scitranslmed.aaf2341
151. Doubrovina ES, Doubrovin MM, Vider E, Sisson RB, O’Reilly RJ, Dupont B, et al. Evasion from NK cell immunity by MHC class I chain-related molecules expressing colon adenocarcinoma 1. J Immunol (2003) 171(12):6891–9. doi: 10.4049/jimmunol.171.12.6891
152. Pesce S, Tabellini G, Cantoni C, Patrizi O, Coltrini D, Rampinelli F, et al. B7-H6-mediated downregulation of NKp30 in NK cells contributes to ovarian carcinoma immune escape. Oncoimmunology (2015) 4(4):e1001224. doi: 10.1080/2162402X.2014.1001224
153. Zheng Y, Fang YC, Li J. PD-L1 expression levels on tumor cells affect their immunosuppressive activity. Oncol Lett (2019) 18(5):5399–407. doi: 10.3892/ol.2019.10903
154. Jiang M, Hu Y, Lin G, Chen C. Dosing regimens of immune checkpoint inhibitors: attempts at lower dose, less frequency, shorter course. Front Oncol (2022) 12:906251. doi: 10.3389/fonc.2022.906251
155. Creelan BC, Antonia SJ. The NKG2A immune checkpoint [[/amp]]mdash; a new direction in cancer immunotherapy. Nat Rev Clin Oncol (2019) 16(5):277–8. doi: 10.1038/s41571-019-0182-8
156. André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell (2018) 175(7):1731–1743.e13. doi: 10.1016/j.cell.2018.10.014
157. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol (2018) 19(7):723–32. doi: 10.1038/s41590-018-0132-0
158. Sarhan D, Cichocki F, Zhang B, Yingst A, Spellman SR, Cooley S, et al. Adaptive NK cells with low TIGIT expression are inherently resistant to myeloid-derived suppressor cells. Cancer Res (2016) 76(19):5696–706. doi: 10.1158/0008-5472.CAN-16-0839
159. Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev (2017) 276(1):97–111. doi: 10.1111/imr.12520
160. Zheng Y, Li Y, Lian J, Yang H, Li F, Zhao S, et al. TNF-α-induced Tim-3 expression marks the dysfunction of infiltrating natural killer cells in human esophageal cancer. J Trans Med (2019) 17(1):165. doi: 10.1186/s12967-019-1917-0
161. Jiang W, Li F, Jiang Y, Li S, Liu X, Xu Y, et al. Tim-3 blockade elicits potent anti-multiple myeloma immunity of natural killer cells. Front Oncol (2022) 12. doi: 10.3389/fonc.2022.739976
162. Ju Y, Hou N, Meng J, Wang X, Zhang X, Zhao D, et al. T Cell immunoglobulin- and mucin-domain-containing molecule-3 (Tim-3) mediates natural killer cell suppression in chronic hepatitis b. J Hepatol (2010) 52(3):322–9. doi: 10.1016/j.jhep.2009.12.005
163. Bagot M, Porcu P, Marie-Cardine A, Battistella M, William BM, Vermeer M, et al. IPH4102, a first-in-class anti-KIR3DL2 monoclonal antibody, in patients with relapsed or refractory cutaneous T-cell lymphoma: an international, first-in-human, open-label, phase 1 trial. Lancet Oncol (2019) 20(8):1160–70. doi: 10.1016/S1470-2045(19)30320-1
164. Roman Aguilera A, Lutzky VP, Mittal D, Li XY, Stannard K, Takeda K, et al. CD96 targeted antibodies need not block CD96-CD155 interactions to promote NK cell anti-metastatic activity. Oncoimmunology (2018) 7(5):e1424677. doi: 10.1080/2162402X.2018.1424677
165. Sun H, Huang Q, Huang M, Wen H, Lin R, Zheng M, et al. Human CD96 correlates to natural killer cell exhaustion and predicts the prognosis of human hepatocellular carcinoma. Hepatology (2019) 70(1):168–83. doi: 10.1002/hep.30347
166. Liu F, Huang J, He F, Ma X, Fan F, Meng M, et al. CD96, a new immune checkpoint, correlates with immune profile and clinical outcome of glioma. Sci Rep (2020) 10(1):10768. doi: 10.1038/s41598-020-66806-z
167. Blake SJ, Stannard K, Liu J, Allen S, Yong MCR, Mittal D, et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov (2016) 6(4):446–59. doi: 10.1158/2159-8290.CD-15-0944
168. Astaneh M, Rezazadeh H, Hossein-Nataj H, Shekarriz R, Zaboli E, Shabani M, et al. Tim-3 and PD-1 blocking cannot restore the functional properties of natural killer cells in early clinical stages of chronic lymphocytic leukemia: an: in vitro: study. J Cancer Res Ther (2022) 18(3):704–11. doi: 10.4103/jcrt.jcrt_52_21
169. Orange JS. Formation and function of the lytic NK-cell immunological synapse. Nat Rev Immunol (2008) 8(9):713–25. doi: 10.1038/nri2381
170. Zheng P, Bertolet G, Chen Y, Huang S, Liu D. Super-resolution imaging of the natural killer cell immunological synapse on a glass-supported planar lipid bilayer. J Vis Exp (2015) 96:52502–8. doi: 10.3791/52502-v
171. Miller A, Carr S, Rabbitts T, Ali H. Multimeric antibodies with increased valency surpassing functional affinity and potency thresholds using novel formats. MAbs (2020) 12(1):1752529. doi: 10.1080/19420862.2020.1752529
172. Kato Y, Abbott RK, Freeman BL, Haupt S, Groschel B, Silva M, et al. Multifaceted effects of antigen valency on b cell response composition and differentiation in vivo. Immunity (2020) 53(3):548–563.e8. doi: 10.1016/j.immuni.2020.08.001
173. Bacac M, Umaña P, Herter S, Colombetti S, Sam J, Le Clech M, et al. CD20 tcb (RG6026), a novel "2:1" T cell bispecific antibody for the treatment of b cell malignancies. Blood (2016) 128(22):1836. doi: 10.1182/blood.V128.22.1836.1836
174. Slaga D, Ellerman D, Lombana TN, Vij R, Li L, Hristopoulos M, et al. Avidity-based binding to HER2 results in selective killing of HER2-overexpressing cells by anti-HER2/CD3. Sci Trans Med (2018) 10(463):eaat5775. doi: 10.1126/scitranslmed.aat5775
175. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res (2015) 21(4):687–92. doi: 10.1158/1078-0432.CCR-14-1860
176. Shah N, Chari A, Scott E, Mezzi K, Usmani SZ. B-cell maturation antigen (BCMA) in multiple myeloma: rationale for targeting and current therapeutic approaches. Leukemia (2020) 34(4):985–1005. doi: 10.1038/s41375-020-0734-z
177. McCann FE, Vanherberghen B, Eleme K, Carlin LM, Newsam RJ, Goulding D, et al. The size of the synaptic cleft and distinct distributions of filamentous actin, ezrin, CD43, and CD45 at activating and inhibitory human NK cell immune synapses 12. J Immunol (2003) 170(6):2862–70. doi: 10.4049/jimmunol.170.6.2862
178. Cartwright AN, Griggs J, Davis DM. The immune synapse clears and excludes molecules above a size threshold. Nat Commun (2014) 5:5479. doi: 10.1038/ncomms6479
179. Reth M. Matching cellular dimensions with molecular sizes. Nat Immunol (2013) 14(8):765–7. doi: 10.1038/ni.2621
180. Jay JW, Bray B, Qi Y, Igbinigie E, Wu H, Li J, et al. IgG antibody 3D structures and dynamics. Antibodies (Basel) (2018) 7(2):18–37. doi: 10.3390/antib7020018
181. Rougé L, Chiang N, Steffek M, Kugel C, Croll TI, Tam C, et al. Structure of CD20 in complex with the therapeutic monoclonal antibody rituximab. Science (2020) 367(6483):1224–30. doi: 10.1126/science.aaz9356
182. Susa KJ, Seegar TCM, Blacklow SC, Kruse AC. A dynamic interaction between CD19 and the tetraspanin CD81 controls b cell co-receptor trafficking. eLife (2020) 9:e52337. doi: 10.7554/eLife.52337.sa2
183. Shen Z, Yan H, Zhang Y, Mernaugh RL, Zeng X. Engineering peptide linkers for scFv immunosensors. Anal Chem (2008) 80(6):1910–7. doi: 10.1021/ac7018624
184. Huston JS, Mudgett-Hunter M, Tai MS, McCartney J, Warren F, Haber E, et al. Protein engineering of single-chain fv analogs and fusion proteins. Methods Enzymol (1991) 203:46–88. doi: 10.1016/0076-6879(91)03005-2
185. Huston JS, Levinson D, Mudgett-Hunter M, Tai MS, Novotný J, Margolies MN, et al. Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain fv analogue produced in escherichia coli. Proc Natl Acad Sci USA (1988) 85(16):5879–83. doi: 10.1073/pnas.85.16.5879
186. Whitlow M, Bell BA, Feng S-L, Filpula D, Hardman KD, Hubert SL, et al. An improved linker for single-chain fv with reduced aggregation and enhanced proteolytic stability. Protein Engineering Design Selection (1993) 6(8):989–95. doi: 10.1093/protein/6.8.989
187. Holliger P, Prospero T, Winter G. "Diabodies": small bivalent and bispecific antibody fragments. Proc Natl Acad Sci USA (1993) 90(14):6444–8. doi: 10.1073/pnas.90.14.6444
188. Arslan M, Karadag M, Onal E, Gelinci E, Cakan-Akdogan G, Kalyoncu S. Effect of non-repetitive linker on in vitro and in vivo properties of an anti-VEGF scFv. Sci Rep (2022) 12(1):5449. doi: 10.1038/s41598-022-09324-4
189. Felices M, Lenvik TR, Davis ZB, Miller JS, Vallera DA. Generation of BiKEs and TriKEs to improve NK cell-mediated targeting of tumor cells. Methods Mol Biol (2016) 1441:333–46. doi: 10.1007/978-1-4939-3684-7_28
190. Tang Y, Jiang N, Parakh C, Hilvert D. Selection of linkers for a catalytic single-chain antibody using phage display technology. J Biol Chem (1996) 271(26):15682–6. doi: 10.1074/jbc.271.26.15682
191. Hagihara Y, Saerens D. Engineering disulfide bonds within an antibody. Biochim Biophys Acta (BBA) - Proteins Proteomics (2014) 1844(11):2016–23. doi: 10.1016/j.bbapap.2014.07.005
192. Suresh MR, Cuello AC, Milstein C. Bispecific monoclonal antibodies from hybrid hybridomas. In: Methods in enzymology. Academic Press (1986). p. 210–28.
193. Li H, Er Saw P, Song E. Challenges and strategies for next-generation bispecific antibody-based antitumor therapeutics. Cell Mol Immunol (2020) 17(5):451–61. doi: 10.1038/s41423-020-0417-8
194. Ridgway JBB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Engineering Design Selection (1996) 9(7):617–21. doi: 10.1093/protein/9.7.617
195. Davis JH, Aperlo C, Li Y, Kurosawa E, Lan Y, Lo K-M, et al. SEEDbodies: fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Engineering Design Selection (2010) 23(4):195–202. doi: 10.1093/protein/gzp094
196. Gunasekaran K, Pentony M, Shen M, Garrett L, Forte C, Woodward A, et al. Enhancing antibody fc heterodimer formation through electrostatic steering effects: application to bispecific molecules and monovalent IgG. J Biol Chem (2010) 285(25):19637–46. doi: 10.1074/jbc.M110.117382
197. Surowka M, Schaefer W, Klein C. Ten years in the making: application of CrossMab technology for the development of therapeutic bispecific antibodies and antibody fusion proteins. MAbs (2021) 13(1):1967714. doi: 10.1080/19420862.2021.1967714
198. Krah S, Kolmar H, Becker S, Zielonka S. Engineering IgG-like bispecific antibodies–an overview. Antibodies (2018) 7(3):28. doi: 10.3390/antib7030028
199. Spiess C, Merchant M, Huang A, Zheng Z, Yang N-Y, Peng J, et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat Biotechnol (2013) 31(8):753–8. doi: 10.1038/nbt.2621
200. Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol (2010) 10(11):787–96. doi: 10.1038/nri2868
201. Jia R, Guo JH, Fan MW. The effect of antigen size on the immunogenicity of antigen presenting cell targeted DNA vaccine. Int Immunopharmacol (2012) 12(1):21–5. doi: 10.1016/j.intimp.2011.08.016
202. Quintero-Hernández V, Juárez-González VR, Ortíz-León M, Sánchez R, Possani LD, Becerril B. The change of the scFv into the fab format improves the stability and in vivo toxin neutralization capacity of recombinant antibodies. Mol Immunol (2007) 44(6):1307–15. doi: 10.1016/j.molimm.2006.05.009
203. Asaadi Y, Jouneghani FF, Janani S, Rahbarizadeh F. A comprehensive comparison between camelid nanobodies and single chain variable fragments. biomark Res (2021) 9(1):87. doi: 10.1186/s40364-021-00332-6
204. Frejd FY, Kim K-T. Affibody molecules as engineered protein drugs. Exp Mol Med (2017) 49(3):e306–6. doi: 10.1038/emm.2017.35
205. Binz HK, Amstutz P, Plückthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat Biotechnol (2005) 23(10):1257–68. doi: 10.1038/nbt1127
206. Grönwall C, Jonsson A, Lindström S, Gunneriusson E, Ståhl S, Herne N. Selection and characterization of affibody ligands binding to Alzheimer amyloid β peptides. J Biotechnol (2007) 128(1):162–83. doi: 10.1016/j.jbiotec.2006.09.013
207. Kronqvist N, Malm M, Göstring L, Gunneriusson E, Nilsson M, Höidén Guthenberg I, et al. Combining phage and staphylococcal surface display for generation of ErbB3-specific affibody molecules. Protein Engineering Design Selection (2010) 24(4):385–96. doi: 10.1093/protein/gzq118
208. Orlova A, Magnusson M, Eriksson TLJ, Nilsson M, Larsson B, Höidén-Guthenberg I, et al. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res (2006) 66(8):4339–48. doi: 10.1158/0008-5472.CAN-05-3521
209. Kontermann RE. Strategies for extended serum half-life of protein therapeutics. Curr Opin Biotechnol (2011) 22(6):868–76. doi: 10.1016/j.copbio.2011.06.012
210. Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS, et al. Immunopharmacologic response of patients with b-lineage acute lymphoblastic leukemia to continuous infusion of T cell–engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood (2012) 119(26):6226–33. doi: 10.1182/blood-2012-01-400515
211. Müller D, Karle A, Meißburger B, Höfig I, Stork R, Kontermann RE. Improved pharmacokinetics of recombinant bispecific antibody molecules by fusion to human serum albumin. J Biol Chem (2007) 282(17):12650–60. doi: 10.1074/jbc.M700820200
212. Tijink BM, Laeremans T, Budde M, Walsum MS-V, Dreier T, de Haard HJ, et al. Improved tumor targeting of anti–epidermal growth factor receptor nanobodies through albumin binding: taking advantage of modular nanobody technology. Mol Cancer Ther (2008) 7(8):2288–97. doi: 10.1158/1535-7163.MCT-07-2384
213. Kontermann RE. Strategies to extend plasma half-lives of recombinant antibodies. BioDrugs (2009) 23(2):93–109. doi: 10.2165/00063030-200923020-00003
214. Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs (2008) 22(5):315–29. doi: 10.2165/00063030-200822050-00004
215. Nieto Y, Banerjee P, Kaur I, Bassett R, Kerbauy L, Basar R, et al. Abstract CT003: innate cell engager (ICE®) AFM13 combined with preactivated and expanded cord blood (CB)-derived NK cells for patients with refractory/relapsed CD30+ lymphoma. Cancer Res (2022) 82(12_Supplement):CT003–3. doi: 10.1158/1538-7445.AM2022-CT003
216. Roopenian DC, Akilesh S. FcRn: the neonatal fc receptor comes of age. Nat Rev Immunol (2007) 7(9):715–25. doi: 10.1038/nri2155
217. van der Horst HJ, Nijhof IS, Mutis T, Chamuleau MED. Fc-engineered antibodies with enhanced fc-effector function for the treatment of b-cell malignancies. Cancers (Basel) (2020) 12(10):3041–64. doi: 10.3390/cancers12103041
218. Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol (2015) 67(2, Part A):95–106. doi: 10.1016/j.molimm.2015.01.003
219. Labrijn AF, Janmaat ML, Reichert JM, Parren PWHI. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov (2019) 18(8):585–608. doi: 10.1038/s41573-019-0028-1
220. Wang Q, Chen Y, Park J, Liu X, Hu Y, Wang T, et al. Design and production of bispecific antibodies. Antibodies (Basel) (2019) 8(3):43–72. doi: 10.3390/antib8030043
221. Sedykh SE, Prinz VV, Buneva VN, Nevinsky GA. Bispecific antibodies: design, therapy, perspectives. Drug Des Devel Ther (2018) 12:195–208. doi: 10.2147/DDDT.S151282
222. Moon D, Tae N, Park Y, Lee SW, Kim DH. Development of bispecific antibody for cancer immunotherapy: focus on T cell engaging antibody. Immune Netw (2022) 22(1):e4. doi: 10.4110/in.2022.22.e4
223. Hwang WYK, Foote J. Immunogenicity of engineered antibodies. Methods (2005) 36(1):3–10. doi: 10.1016/j.ymeth.2005.01.001
224. Dingman R, Balu-Iyer SV. Immunogenicity of protein pharmaceuticals. J Pharm Sci (2019) 108(5):1637–54. doi: 10.1016/j.xphs.2018.12.014
225. Gopal GJ, Kumar A. Strategies for the production of recombinant protein in escherichia coli. Protein J (2013) 32(6):419–25. doi: 10.1007/s10930-013-9502-5
226. Tan E, Chin CSH, Lim ZFS, Ng SK. HEK293 cell line as a platform to produce recombinant proteins and viral vectors. Front Bioengineering Biotechnol (2021) 9. doi: 10.3389/fbioe.2021.796991
Keywords: cancer, immunotherapy, natural killer (NK) cell, NK cell engager (NKCE), functionality, manufacturability
Citation: Zhang M, Lam K-P and Xu S (2023) Natural Killer Cell Engagers (NKCEs): a new frontier in cancer immunotherapy. Front. Immunol. 14:1207276. doi: 10.3389/fimmu.2023.1207276
Received: 17 April 2023; Accepted: 03 July 2023;
Published: 09 August 2023.
Edited by:
David Wald, Case Western Reserve University, United StatesReviewed by:
Bruce Walcheck, University of Minnesota Twin Cities, United StatesCopyright © 2023 Zhang, Lam and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kong-Peng Lam, bGFtX2tvbmdfcGVuZ0BpbW11bm9sLmEtc3Rhci5lZHUuc2c=; Shengli Xu, eHVfc2hlbmdsaUBpbW11bm9sLmEtc3Rhci5lZHUuc2c=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.