 Wenhua Wang1
Wenhua Wang1 Zhihui He1,2*
Zhihui He1,2*- 1Department of Intensive Care Unit, the Third Xiangya Hospital, Central South University, Changsha, Hunan, China
- 2Sepsis Translational Medicine Key Laboratory of Hunan Province, Central South University, Changsha, Hunan, China
Sepsis is a hyper-heterogeneous syndrome in which the systemic inflammatory response persists throughout the course of the disease and the inflammatory and immune responses are dynamically altered at different pathogenic stages. Gasdermins (GSDMs) proteins are pore-forming executors in the membrane, subsequently mediating the release of pro-inflammatory mediators and inflammatory cell death. With the increasing research on GSDMs proteins and sepsis, it is believed that GSDMs protein are one of the most promising therapeutic targets in sepsis in the future. A more comprehensive and in-depth understanding of the functions of GSDMs proteins in sepsis is important to alleviate the multi-organ dysfunction and reduce sepsis-induced mortality. In this review, we focus on the function of GSDMs proteins, the molecular mechanism of GSDMs involved in sepsis, and the regulatory mechanism of GSDMs-mediated signaling pathways, aiming to provide novel ideas and therapeutic strategies for the diagnosis and treatment of sepsis.
1 Introduction
Sepsis is a life-threatening organ dysfunction due to host response disorder caused by infection (1). Septic shock is a subtype of sepsis in which severe circulatory, cellular, and metabolic abnormalities occur, resulting in a significantly higher mortality than mono sepsis (2). With high morbidity and mortality (3), sepsis is the leading cause of death in critically ill patients (4). Globally, 48.9 million incident cases of sepsis were recorded in 2017, and 11 million sepsis-related deaths, accounting for 19.7% (18.2 ~ 21.4) of all deaths (5). Moreover, the morbidity of sepsis is increasing year by year with an annual growth rate of approximately 5.7% per year from 2007 to 2013 (6). This is mainly associated with improved diagnostic sensitivity, aging populations, the emergence of antibiotic-resistant strains, and increasing immunodeficient patients. In recent years, with the development of diagnosis and treatment and the standard of intensive care, the mortality of sepsis has decreased slightly (7). However, progress in clinical treatment strategies for sepsis remains slow, mainly due to the complexity of the disease (8). Sepsis can be caused by infections anywhere in the body. Pathogens contributing to sepsis include gram-positive bacteria, gram-negative bacteria, anaerobic, fungi, etc. The pathophysiological mechanisms of sepsis are complex, including imbalance of inflammatory response, endothelial dysfunction, coagulation disorders, imbalance of immune response, etc. (9). Clarifying the pathophysiological process of sepsis is essential for exploring therapeutic approaches.
With the better understanding of sepsis, the role of the dynamic changes of inflammatory reaction and immunosuppression in sepsis progression has become increasingly important, resulting in a highly heterogeneous state (10). In sepsis, pro-inflammatory and anti-inflammatory responses are activated simultaneously. In the early stage, under the stimulation of infection, the immune system is activated and releases pro-inflammatory cytokines and chemokines, presenting hyperinflammation (11). Among them, neutrophils are the earliest innate immune cells which migrate from blood to the infected sites (12). Through phagocytosis, degranulation, and neutrophil extracellular traps (NETs), neutrophils release reactive oxygen species (ROS), proteases, chemokines, and cytokines to kill pathogens and recruit other immune cells, maximizing the host’s immune response (12, 13). GSDMs-mediated cell death and inflammatory factor release aggravated the progress of pro-inflammation (14). If the performances of inhibiting the release of pro-inflammatory factors and regulating the body’s immune system are executed in time at this stage for restoring the balance between pro-inflammation and anti-inflammation, the host may recover to normal (15). Conversely, the interaction between various inflammatory factors and immune cells leads to “cytokine storm”, in which multiple systems such as reticuloendothelial system, complement system, and coagulation system are activated, causing tissue damage, and inducing cell death and even severe immune suppression (10, 11, 15). Furthermore, immature neutrophils suppressed proliferation and cell killing of lymphocytes aggravating the immunosuppression (16). Neutrophil dysfunction is positively correlated with the severity of organ dysfunction in sepsis (17). Specifically, neutrophils over-activated by complement components accumulate extensively and produce excessive pro-inflammatory cytokines, leading to tissue damage (18). NETs produced by neutrophils can damage endothelial cells, resulting in impaired microcirculatory blood flow and procoagulant alterations (12). Additionally, neutrophils promote intravascular coagulation through the NETs-platelet-thrombin axis, leading to microcirculatory dysfunction and tissue damage (19). The formation of NETs is dependent on proteases-activated GSDMs (20). Therefore, early intervention has become the focus of research to prevent sepsis from developing into uncontrollable stages. GSDMs as an important element in the progression of pro-inflammation in sepsis, it is crucial to elucidate its influence in sepsis. GSDMs proteins were first identified as being expressed in gastrointestinal and skin tissues and were named gasdermins (21, 22). Subsequently, studies have shown that GSDMs proteins are widely expressed in various tissues (23). The GSDMs family is a class of effectors that form pores in cell or organelle membranes during cell death and may contribute to multiple physiological and pathological processes such as epithelial cell restitution in inflammatory bowel disease, intestinal epithelium development (24, 25) and inflammation, carcinogenesis, dysregulation of immune response (26, 27). GSDMs proteins are activated by the stimulation of sepsis initiating factors and then cause inflammatory mediators release or inflammatory cell death, further aggravating sepsis progression.
In this review, we summarize the role of GSDMs in sepsis. Through the pathways in which GSDMs function to understand the contribution of GSDMs in the pathogenesis and progression of sepsis, it will be helpful to suggest potentially effective therapeutic strategies for shortening the course of sepsis and improving the prognosis and survival of septic patients.
2 GSDMs
Evidence has shown that the GSDMs family is represented in fish, birds and mammals, with the most studied in humans and mice. Among the proteins encoded by the human genome, GSDMs protein have been classified into six groups: gsdermin A (GSDMA), gsdermin B (GSDMB), gsdermin C (GSDMC), gsdermin D (GSDMD), gsdermin E (GSDME), and Pejvakin (DFNB59). The GSDMs family is relatively conserved (28). Within the mouse genome, it has been divided into Gsdma1-3, Gsdmc1-4, Gsdmd, Gsdme, and Dfnb59 (29) (Figure 1). Except DFNB59, GSDMs are composed of two domains, an N-terminal with pore-forming function and a self-inhibiting C-terminal domain. The middle of the two domains is a linkage region with different lengths and sequences, which is the acting location of different enzymes to activate GSDMs proteins (28). DFNB59 contains only a truncated C-terminal domain, suggesting an entirely distinct activation mechanism from other members (30). Except DFNB59, GSDM proteins are activated for pore formation at the plasma membrane (14). Pyroptosis is the main way in which GSDMs play a role in sepsis (31). Recently, JOHNSON et al. revealed that bacterial genes produce proteins structurally similar to GSDMs proteins in mammals via X-ray crystallography. Both bacterial and human GSDMs are activated by a similar mechanism (32). This important finding revealed pyroptosis as an ancient programmed cell death common in bacteria and animals.
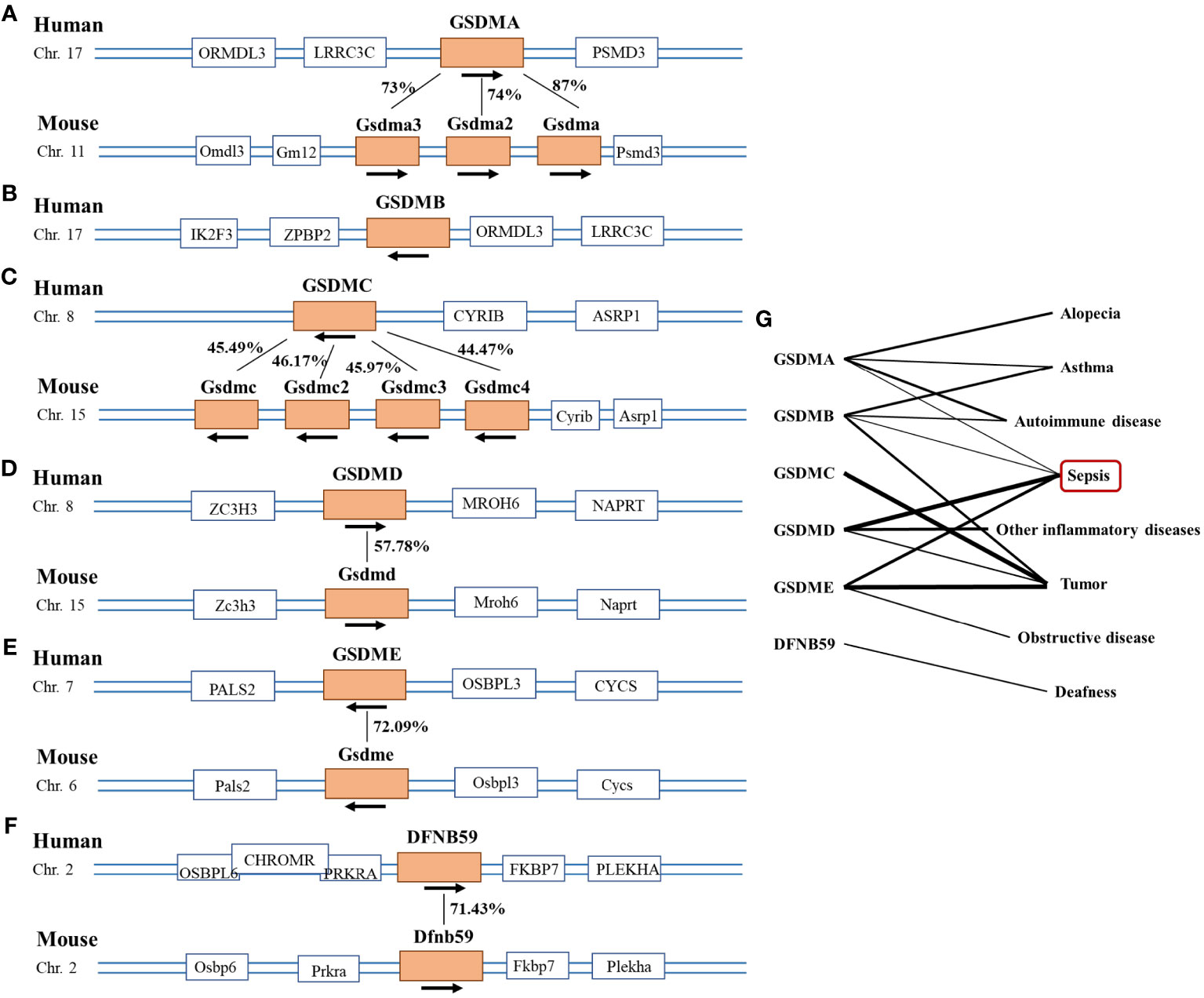
Figure 1 Picture of the Gasdermin family. (A–F) The location of the Gasdermin family on human and mouse chromosomes and the similarity of amino acid sequence between species. Human GSDMA has 87%, 74%, and 73% amino acid sequence similarity with mouse Gsdma, Gsdma2, and Gsdma3 respectively. And human GSDMC has 45.49%, 46.17%, 45.97% and 44.47% amino acid sequence similarity with mouse Gsdmc, Gsdmc2, Gsdmc3 and Gsdmc4 separately, which was 57.78% in GSDMD, 72.09% in GSDME and 71.43% in DFNB59. (G) Diseases caused by abnormal condition of the gasdemin family, the thickness of the lines represents the intensity of the correlation between the gene and the disease. The thicker the line, the stronger the correlation.
2.1 GSDMA
GSDMA is localized on chromosome 17 in humans and chromosome 8 in mice, which is highly expressed in gastrointestinal epithelium, epidermis and hair follicles. Correspondingly, there are three GSDMA alleles Gsdma 1-3 in mice. Gsdma 3 is highest in the skin, Gsdma 2 is highest in the stomach, and Gsdma is abundant in both the stomach and skin (33). GSDMA has been studied in gastric cancer, alopecia and susceptibility to inflammatory diseases (21, 34, 35). In addition, Streptococcus pyogenic exotoxin B (SpeB) could cleave and activate GSDMA (22). Streptococcus pyogenes, a potent toxigenic pathogen, can also cause toxic shock and sepsis through respiratory infection (22, 36). GSDMA has been poorly studied in sepsis, and GSDMA in sepsis needs further exploration.
2.2 GSDMB
GSDMB, also known as GSDML, is present only in the mammals, except in mice (37). The gene location and function of GSDMB and GSDMA are closely correlated, suggesting that both are produced by gene duplication (29). GSDMB has been detected as highly expressed in immune cells such as T cells (38, 39). GSDMB improved cell proliferation and migration, activated immune response, and regulated the processes of cell differentiation and cell death (40). GSDMB is associated with autoimmune disease and tumor progression (24, 41). GSDMB activation is a direct response to natural killer (NK) cells recognizing pathogen-infected cells (42). IpaH7.8 secreted by Shigella flexneri ubiquitinated and targeted GSDMB for 26S proteasome destruction to protect Shigella from the bacteriocidic activity of NK cells (43–45). GSDMB can also contribute to the development of sepsis by activating GSDMD (39). In addition, GSDMB was highly expressed in leukocytes of septic shock patients (39). These indicated the role of GSDMB in sepsis.
2.3 GSDMC
GSDMC is located on chromosome 8 in humans and chromosome 15 in mice, and is mainly expressed in esophagus, trachea, intestine and spleen tissues (46). GSDMC was first found with increased expression in metastatic melanoma and as a marker of melanoma progression (46). GSDMC is mainly related to tumor progression. GSDMC promoted proliferation and migration of tumor cell (35, 47). In macrophages, nuclear programmed death ligand 1 (PD-L1) converted TNF-α-induced apoptosis into pyroptosis by activating GSDMC (48). Gsdmc is the main effector of intestinal type 2 inflammation. After worm infection, GSDMC promoted the secretion of the “alarmin” cytokine interleukin-33 (IL-33) by intestinal epithelial cells to initiate type 2 responses for worm clearance and tolerance (49). Physiologically, Gsdmc N6-adenomethylation (m6A) conserved mitochondrial homeostasis and inhibited apoptotic pathways, critical for intestinal stem cell survival and maintenance of normal colonic epithelial regeneration (50). However, the role of GSDMC in sepsis has not been studied.
2.4 GSDMD
GSDMD is consistently localized on chromosomes with GSDMC in humans and mice, highly expressed in epithelial cells of the upper gastrointestinal and small intestinal mucosa, and immune cells such as macrophages, monocytes, neutrophils, and CD8+T cells (51). GSDMD cleavage was increased in activated CD8+ T cells, and GSDMD deficiency impaired the effector capacity of CD8+T cells (52). Among the gasdermins protein family, GSDMD was the first discovered protein to be cleaved by caspases and then generated an N-terminal domain with the ability to target the cell membrane, ultimately causing pyroptosis (53). Due to its wide distribution and the earliest discovery of mechanisms in pyroptosis, GSDMD is the most extensively studied protein in the pore-forming gasdermin protein family. GSDMD promoted the secretion of intestinal cupped cell mucin and the formation of mucus layer, contributing to homeostasis of the intestinal barrier (54).
GSDMD exerts essential effects on tumors and inflammatory diseases (55). The role of GSDMD in inflammatory diseases is clearly identified, mainly exerting pro-inflammatory effects through inflammatory cell death and the release of inflammatory mediators. Hypoxia/reoxygenation caused GSDMD-mediated cardiomyocyte pyroptosis and release of IL-18 (56). GSDMD contributed to the type II inflammatory response by promoting IL-33 release in pulmonary epithelial cells (57). Many studies have shown that GSDMD may promote the progression of sepsis by inducing pyroptosis and releasing inflammatory mediators (58–60). In neutrophils, GSDMD formed pores in organelle membranes, but not in cytomembrane. For example, GSDMD forming pores in azurophilic granules, elastase was released into the cytoplasm and mediated serine protease-dependent GSDMD cleavage. And forming pores in autophagosomes, GSDMD promoted to release IL-1β through autophagy-dependent pathways (61). In addition, Gsdmd-knockout (KO) mice protected against septic myocardial dysfunction, with a high survival rate (62). Downregulated GSDMD alleviated Candida albicans-associated sepsis (63). GSDMD is regarded as a novel ideal target for sepsis treatment.
2.5 GSDME
GSDME is located on chromosome 8 in humans and chromosome 15 in mice, and is clearly expressed in multiple tissues, including the brain, endometrium, placenta, and intestine (29). GSDME was first described in familial presbycusis, also known as deafness, autosomal dominant 5 (64). GSDME is closely associated with hear impairment, autoimmune diseases, and tumorigenesis (23, 30, 65).
GSDME plays an important role in inflammatory diseases. GSDME exerted protective effects in ultraviolet B-induced skin inflammation by inhibiting neutrophil over-recruitment and activation, thereby inhibiting cutaneous barrier damage (66). Cytokine storm is responsible for high mortality in sepsis patients. The synergism of TNF-α and IFN-β triggered human airway epithelial cells death by activating GSDME-mediated pathway (67). The H7N9 virus activated GSDME-mediated alveolar epithelial cell pyroptosis and inflammatory mediators release in mouse lungs, leading to cytokine storm and mortality in mice (68). GSDME activation is a critical and unique mechanism by which infection contributes to cytokine storm and lethality in sepsis. Thus, GSDME is a potential target for sepsis therapy.
2.6 DFNB59
DFNB59 is located on chromosome 2 in humans and chromosome 2 in mice, and is expressed in inner ear hair cells and other auditory system cells (69). DFNB59 has only been shown to be associated with hearing losses (70). Due to the distinctive structure, it is uncertain whether DFNB59 has the pore-forming capability. Currently, no studies have shown a correlation between DFNB59 and sepsis.
3 GSDMs in sepsis
3.1 GSDMs-mediated pathway
GSDMs proteins are activated by either caspases or granzymes, which is the basis for studies on GSDMs-mediated pathways. However, recent research has shown that GSDMs can also be identified and cleaved by other molecules except caspases and granzymes. Based on this classification, we clarified the following.
3.1.1 GSDMs-mediated pathway including caspases or granzymes
Activated pattern recognition receptors (PRRs) on target cells combined with apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) to form inflammasome complexes containing nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing 1 (NLRP1), NLRP3, NLRC4 and absent in melanoma 2 inflammasome (71). The latter recruited and activated pro-caspase-1 (in the GSDMD-mediated canonical pathway) (72). Pro-caspase-4/-5/-11 (in the GSDMD-mediated noncanonical pathway) (73) and pro-caspase-3 (in the GSDME-mediated pathway) (74) could be activated independently of inflammasome. Caspases can also be activated in other ways. Yersinia triggered the interaction between receptor-interacting serine-threonine protein kinase 1 (RIPK1) and caspase-8, then activated caspase-8 (75). Caspase-11 enhanced the activation of caspase-8 to amplify inflammatory signals associated with tissue damage in sepsis (76). Activated caspases not only cleaved and activated the corresponding GSDMs proteins (35), but also cleaved prototypes of pro-inflammatory factors such as pro-IL-1/-18 (53). GSDME can be activated compensatively when the GSDMD signaling pathway is defective. In Gsdmd-/- macrophages, NLRP3 inflammasome continuously induced caspase-8/-3 and GSDME cleavage and IL-1β maturation. Thus, when classical NLRP3-GSDMD signaling is blocked, the compensatory inflammatory pathway caspase-8/-3-GSDME is activated upon NLRP3 activation (77).
In addition to caspases, granzymes can also activate GSDMs proteins. In septic acute respiratory distress syndrome (ARDS), the expression of both GZMA and GZMB was upregulated (78). Specifically, GZMA secreted by NK cells and cytotoxic T lymphocytes (CTLs) directly activated GSDMB (38). GZMB secreted by CTLs or CAR-T cells could cleave GSDME directly at the site consistent with caspase-3 (79, 80). Also, GZMB cleaved caspase-3 to activate GSDME (81). In addition, GSDMs proteins can interact with each other. For example, GSDMB promotes the activity of caspase-4 by binding to the CRAD domain of caspase-4, thus cleaving GSDMD (39).
3.1.2 GSDMs-mediated pathway except caspases and granzymes
GSDMs can also be cleaved and activated by some other molecules in sepsis, except well-known caspases or granzymes. A recent study showed that SpeB directly and specifically cleaved GSDMA in the junctional region after Gln246 (22). GSDMA functions both as a receptor to recognize exogenous pathogens and as an effector to form pores in the cell membrane and release inflammatory factors to trigger pyroptosis and inflammation. In addition, it has been shown that in lipopolysaccharide (LPS)-induced septic mice, Cathepsin G (Cat G), expressed on myeloid cells, can cleave human or murine GSDMD at Leu274, an upstream site of the caspase effector site Asp276, to form GSDMD-NT distinct from the cleavage of caspase-11 (82).
GSDMs-mediated pathways are regulated by various molecules, such as miRNAs, oxidative stress, chemokines (83–86). Activated GSDMs proteins bound to the plasma membrane and formed pores (87), mediating cell death or inflammatory factors release, functional proteins, etc. (88–90) (Figure 2).
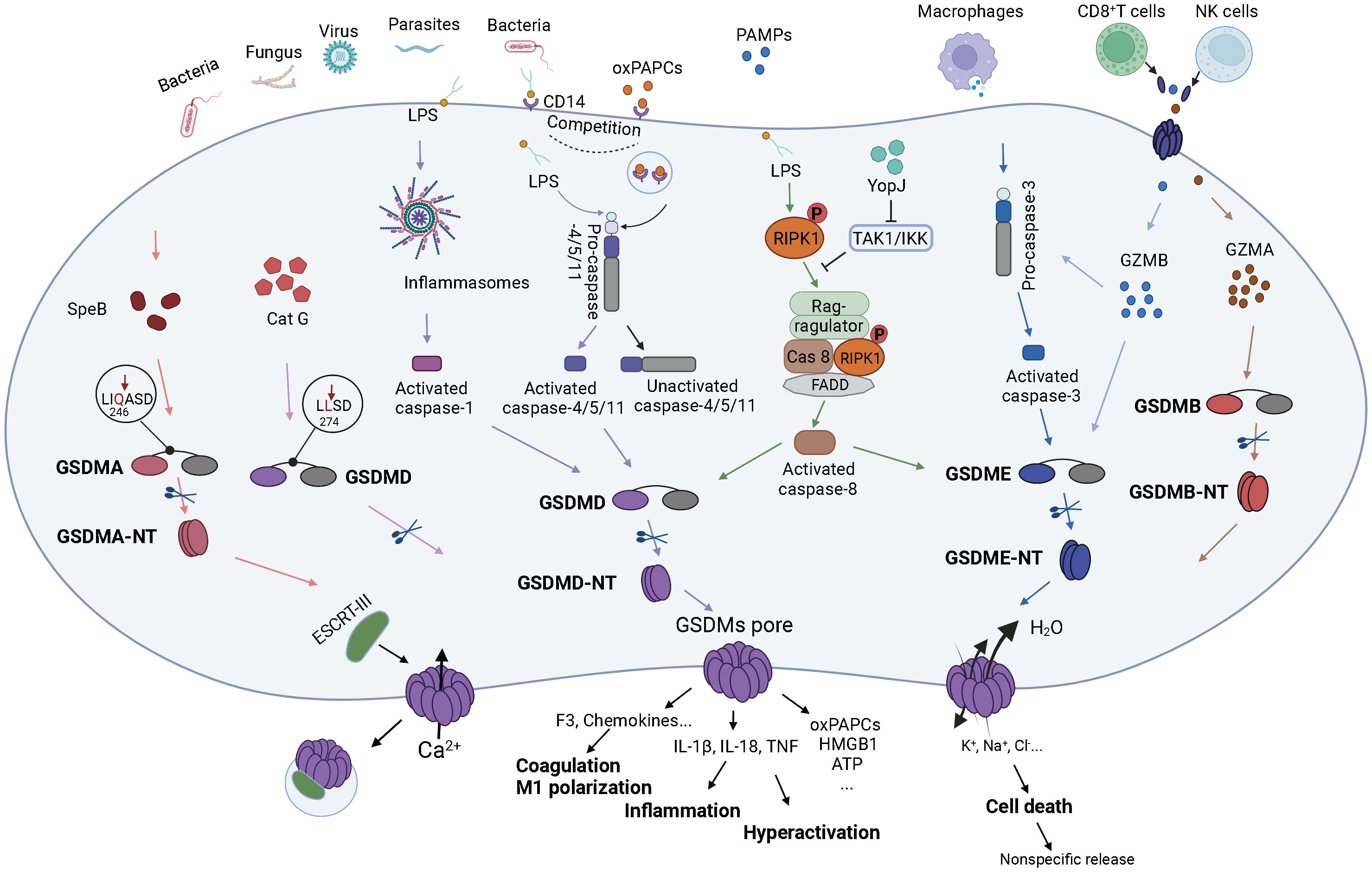
Figure 2 The molecular mechanism of GSDMs in sepsis. Bacterial, fungal, viral or parasitic severe infections or major traumas to body can cause sepsis, which in turn leads to systemic inflammatory response and imbalanced immune regulation. The above factors, as PAMPs or DAMPs, bind to PRRs on the surface of target cells and transmit signals intracellularly, ultimately activating GSDMs-related signaling pathways. SpeB, an exotoxin of Streptococcus pyogenes, can directly act the site after Gln264 in the GSDMA linkage region, which in turn cleaves GSDMA and forms pores in the cell membrane. Perforin and granzyme released by CD8+ T cells and NK cells can also impact the target cells. Specifically, GZMA can interact directly with GSDMB, causing GSDMB to be cleaved and pore formation on the cell membrane. Meanwhile, GZMB can not only activate pro-caspase-3 which in turn activates GSDME, but also can function directly on the cleavage site of GSDME to activate GSDME and thus pore forming. Notably, GSDMD plays the most important effect in sepsis. After the target cells receive the stimulatory signal, the inflammasome complex is assembled and thus activates caspase-1 which cleaves and activates GSDMD and pro-IL-1β. Upon entry into the cytoplasm, LPS exerts on CARD domain of pro-caspase-4/5/11, resulting in activation of caspase-4/5/11, and thus cleaves GSDMD. The oxPAPCs can then compete with LPS to bind the CD14 receptor on the cytosolic membrane and bind the catalytic domain of pro-caspase-4/5/11 to form inactive caspase-4/5/11. In addition, intracellular Cat G can also act directly the site after Leu274 on the GSDMD linkage domain to cleave GSDMD. The virulence of Yersinia, acetyltransferase YopJ-induced inhibition of TAK1 or IKK could recruit the RIPK1- FADD- caspase-8 complex to the Rag-ragulator platform to activate caspase-8 which leaded to macrophages pyroptosis by activating caspase-8/GSDMD or caspase-8/GSDME pathways. Activated GSDMD can also form pores in the cytosolic membrane. GSDMs pores can cause massive H2O molecules into the cell and the ion imbalance of the cell, which in turn leads to cell death and release of cell contents. GSDMs pores can also specifically release molecules, including inflammatory cytokines IL-1β, IL-18, TNF to promote inflammatory responses, coagulation factor F3 and chemokines contributing to the coagulation responses and M1 macrophage polarization, as well as active components such as oxPAPCs, HMGB1, ATP to activate other target cells and initiate the inflammatory cascade. In partial target cells, calcium enters the cells and triggers ESCRT-III to interact with GSDMs pores, causing some GSDMs pores to detach from the cell membrane and preventing cell death while continuously releasing inflammatory substances, which eventually result in over-activation.
3.2 The activation of GSDMs-mediated pathway in sepsis
Sepsis is caused by a dysregulated inflammatory response to the presence of pathogenic microorganisms. PAMPs are highly conserved components of microorganisms’ surface and can be rapidly recognized by PRRs on the host cell surface, which in turn activate signaling pathways associated with GSDMs. PAMPs in sepsis mainly comprise surface membrane components of microorganisms (e.g., lipopolysaccharide, LPS) and microbial nucleic acids (e.g., DNA and RNA) (91). LPS, the main component of the cell wall of gram-negative bacteria, is the most important PAMP and is inextricably related to sepsis.
Clinically, the severity of sepsis has been shown to be correlated with danger-associated molecular patterns (DAMPs) (92–94). DAMPs could be released extensively via GSDMs-mediated pore-forming membrane or cell lysis, and subsequently could be recognized by PRRs and activate the GSDMs-mediated pathway (95). DAMPs are divided into many types, and sepsis-related ones include high mobility group box 1 (HMGB1), histones, ATP, uric acid, DNA, mitochondrial DAMPs, and IL-33 (96, 97). HMGB1 is the first to be identified and also the most intensively studied in sepsis by binding to receptors for advanced glycation end-products (RAGE) and toll-like receptor 2 (TLR2)/TLR4 (91). LPS induced HMGB1 release from hepatocytes into exosomes through the coordinated activities of TLR4 and caspase-11/GSDMD signaling (98). In turn, HMGB1 released from hepatocytes interacted with LPS and is internalized into the lysosomes of macrophages and endothelial cells by binding the RAGE, mediating the caspase-11/GSDMD signaling pathway (99).
3.3 The regulation of GSDMs-mediated pathway in sepsis
The GSDMs-mediated pathway plays a crucial role in sepsis. The main elements of the GSDMs signaling pathway have been well studied (88, 100). However, molecular mechanisms regulating this pathway have not been systematically summarized. In this review, regulation of the GSDMs-mediated pathway was synthesized from multiple perspectives and listed in Table 1.

Table 1 The regulation of GSDMs-mediated pathway in sepsis.
3.3.1 Epigenetic regulation in GSDMs-mediated pathway
Among the many regulatory mechanisms, epigenetic regulation plays an important role. Epigenetic mechanisms are a major way of regulating gene expression, and their core is multiple covalent modifications of nucleic acids and histones reversibly and dynamically, mainly including DNA methylation, non-coding RNA (ncRNA) regulation and histone post-translational modifications (101, 102). Among them, ncRNA regulation and histone post-translational modifications play important roles in GSDMs-mediated signaling pathways in sepsis.
In the human genome, only about 2% of RNAs can be translated into proteins (103). The remaining RNAs are known as ncRNA, particularly long non-coding RNA (lncRNA), microRNA (miRNA) and circular RNA (circRNA) have been identified as critical for regulating the GSDMs pathway in sepsis (104). ncRNA may affect multiple components of the GSDMs pathway in sepsis. First, ncRNA could impact the inflammasome complex. In septic acute lung injury (ALI), the exosomal miR-30d-5p of neutrophils partially activated NF-κB pathway in macrophages and upregulated the expression of NLRP3 to mediate M1 macrophage polarization and pyroptosis (105). miR-21 promoted NLRP3 inflammasome activation to mediate pyroptosis and septic shock by activating NF-κB pathway (106). Circ-HIPK3 upregulated the expression of Krüppel-like factor 6 (KLF6) by competitively binding miR-124-3p. KLF6 not only directly upregulated NLPR3 (107) but also promoted NLRP3 by inhibiting miR-223-3p promoter activity (108) to activate NLRP3/Caspase-1/GSDMD pathway and accentuate septic acute kidney injury (AKI). Exosomal miR-93-5p activated NLRP3 by regulating thioredoxin-interacting protein (TXNIP), contributing to renal epithelial cell pyroptosis in septic mice (109). Downregulated miR-30c-5p triggered NLRP3/caspase-1/GSDMD pathway by promoting expression of TXNIP in septic AKI (110). Meanwhile, the lncRNA XIST/miR-150-5p/c-Fos axis exacerbated septic myocardial injury by regulating the promoter of TXNIP (111). These findings suggest that TXNIP may be a potential therapeutic target in sepsis. miR-34a upregulated expression of ASC protein to activate caspase-1/GSDMD and exacerbate septic ALI (112). In addition, ncRNA may affect the expression of GSDMs. Circ-Katnal1 promoted the expression of GSDMD by influencing miR-31-5p to enhance pyroptosis in septic liver injury (84). LncRNA MEG3 promoted renal tubular epithelial pyroptosis by regulating miR-18a-3p/GSDMD pathway in LPS-induced AKI (113). The studies showed multiple regulation of GSDMs-mediated pathway by ncRNA, suggesting that ncRNA is promising as biomarkers in sepsis.
Post-translational modifications of histones in GSDMs are essential in sepsis. YTH N6-Methyladenosine RNA Binding Protein 1 (YTHDF1), m6A reader protein, induced NLRP3 ubiquitination and inhibited caspase-1-dependent pyroptosis to alleviate sepsis (114). Histone deacetylase 11 (HDAC11) plays an important role in sepsis. In TNF-α-treated human umbilical vein endothelial cells, upregulated HDAC11 decreased the acetylation of ETS-related gene (ERG) to promote NLRP3/caspase-1/GSDMD and caspase-3/GSDME pathway (115). In myeloid cells, acetyltransferase Yersinia outer protein J (YopJ) activated GSDME but not GSDMD by promoting RIPK1/caspase-8 pathway to induce myeloid cell pyroptosis and protect host from Yersinia infection (116). Yet, in another study, YopJ-induced inhibition of TGF-α-activated kinase 1 (TAK1) or IκB kinase (IKK) could activate GSDMD and GSDME by activating caspase-8 to induce macrophages pyroptosis and exacerbate the inflammatory response (75, 117, 118). In infected macrophages, lysine acetyltransferase KAT2B and KAT3B increased the binding of the acetylation of histone H3 Lysine 27 (H3K27ac) to the promoters of caspase-11 and GSDMD to upregulate the expression of caspase-11 and GSDMD and increase caspase-11/GSDMD-mediated pyroptosis (119). Protein s-palmitoylation is a type of lipidation modification, which lipidates cysteine (Cys) residues via thioester bonds to target proteins towards organelles and plasma membranes (120). GSDMD palmitoylation at Cys191/Cys192 (human/mouse) caused GSDMD-NT membrane transposition, and only palmitoylated GSDMD-NT was enabled for membrane transposition and pore formation. GSDMD palmitoylation was modified by palmitoyl acyltransferases zinc finger DHHC domain 5 (zDHHC5) and zDHHC9, promoted by LPS-induced ROS (121, 122). At present, post-translational modifications on histones of GSDMs are still poorly studied in sepsis, and many regulatory mechanisms have yet to be discovered to enrich the role of GSDMs in sepsis.
3.3.2 The regulation of oxidative stress in GSDMs-mediated pathway
Oxidative stress can impair multiple systems in the body by affecting the GSDMs pathway, which is a significant contributor to multiple organ dysfunction in sepsis (123). In macrophages, mitochondrial ROS (mtROS) oxidized the four amino acid residues of GSDMD to promote cleavage of GSDMD (58). Phosphodiesterase 4B (PDE4B) promoted activation of ROS/nuclear factor erythroid2-related factor 2 (Nrf2)/NLRP3 to induce inflammasome activation and pyroptosis in LPS-induced ALI (59). In sepsis, DNA damage may contribute to the activation of Transmembrane protein 173 (TMEM173, also known as STING), an endoplasmic reticulum (ER) stress-associated immune adaptor protein. TMEM173 promoted calcium release from macrophages and monocytes ER, leading to activation of caspase-1/-11/-8 in a bacterial type-dependent manner (e.g., activated caspase-1/-11 in E. coli infection and activated caspase-8 in S. pneumoniae infection). Platelets play an important role in the pathogenesis of sepsis, and the expression of GSDMD in platelets is significantly upregulated in septic patients. Mechanistically, pyroptotic platelet-derived oxidized mitochondrial DNA (ox-mtDNA) promoted the release of S100A8/A9 to activate the caspase-1/GSDMD pathway, inducing pyroptosis of platelets and ultimately forming a positive feedback loop that leads to excessive release of pro-inflammatory cytokines (124). Another in vivo murine experiment demonstrated that PD-L1 knockdown inhibited the expression of caspase-3 to reduce GSMDE-induced IL-1β release and suppressed the activation of integrin αIIbβ3, contributing to alleviate platelet activation in sepsis (125).
3.3.3 Other regulation in GSDMs-mediated pathway
The interferon regulatory factor (IRF) family also had significant effects on the signaling pathway of GSDMs in sepsis. IRF2 can directly drive GSDMD transcription by engaging the promoter of GSDMD to induce pyroptosis in sepsis (126). IRF2-KO septic mice exhibited enhanced survival rates and reduced pathological manifestations (127). IRF1 enhanced caspase-3 expression by binding to its promoter, thereby inducing pyroptosis by activating the GSDME pathway (128). As regulators of the mitochondrial apoptotic pathway, members of the B-cell lymphoma-2 (Bcl-2) family influence the GSDMs pathway in sepsis. In LPS-treated macrophages, Bcl-2 recognized the BH3 domain on GSDMD-NT, promoting caspase directed cleavage of GSDMD at D87, which lacked pore-forming capability, instead of at D275. Furthermore, Bcl-2 impeded NLRP1 oligomerization by reducing the binding of ATP to NLRP, thereby inhibiting the activation of NLRP1 inflammasome (129). Immunometabolism represents a promising therapeutic target in sepsis. Activation of caspase-11 inflammasome and pyroptosis in sepsis was regulated by cAMP metabolism (130). In addition, itaconate, a unique regulatory metabolite, prevented full activation of caspase-1 and GSDMD in LPS-induced macrophages (131).
In sepsis, activation of the complement system results in the generation of membrane attack complex (MAC). MAC promoted ASC oligomerization and NLRP3 inflammasome assembly to activate caspase-1/GSDMD and induce macrophage pyroptosis (132). In addition to acting as PAMPs to initiate the GSDMs signaling pathway, LPS may also affect the expression of GSDMs proteins (133). LPS increased GSDMD expression while decreasing GSDME expression through glycolysis in RAW264.7 cells. And this transcriptional regulation suggested that LPS may contribute to pyroptosis in a GSDMD-dependent manner in high-glucose environments (134). However, the exact mechanism remains unclear and needs to be further explored. Upregulated macrophage migration inhibitory factor (MIF) increased NLRP3 inflammasome mediated cell pyroptosis and aggravated kidney damage by promoting phosphorylation of p65 in septic mice (135). Upregulated IL-6 caused by bacterial infection inhibited caspase-1/-3 activation to suppress GSDMD-/GSDME-mediated pyroptosis and played a partially protective role (136). Notably, the NF-κB signaling pathway exhibits a dual role in sepsis. On the one hand, activated NF-κB pathway reduced mortality in cecal ligation puncture (CLP)-induced septic mice by promoting M1 macrophage polarization and enhancing bacterial phagocytosis of macrophages (137). On the other hand, NF-κB signaling was required for the GSDMD-mediated pyroptosis in LPS-induced adipocytes (138). The role of NF-κB signaling pathway in sepsis remains to be investigated more deeply.
3.4 The effector of GSDMs-mediated pathway in sepsis
After GSDMs are cleaved, GSDMs-NT transfer to the membrane and perform pore-forming functions. This is mainly through selective binding of GSDMs-NT to phosphatidylinositol phosphate, phosphatidylserine and phosphatidic acid of the inner cell membrane, or to cardiolipins of damaged outer mitochondrial and bacterial membranes (28, 87), forming a 10-14 nm pore containing 24-34 symmetrical subunits (139). Cytoplasmic calcium signaling is a prerequisite for of GSDMD-NT translocation to the plasma membrane (133, 140). Mg2+ blocked Ca2+ influx by inhibiting ATP-gated Ca2+ channel P2X7 (140). And phospholipase C increased cytoplasmic calcium to affect GSDMD pore formation (133). Pore-forming cells have two outcomes and exert different effects in vivo.
3.4.1 GSDMs-induced cell death
In sepsis, the most common effect of GSDMs proteins is to induce pyroptosis, which leads to cellular rupture and non-specific release of cellular components, such as HMGB1 and pro-inflammatory cytokines such as IL-1, IL-18 and TNF-α, ultimately triggering inflammatory responses (141). Specifically, after pore formation, the cell membrane loses integrity, the cell ion gradient is depolarized, and the osmotic pressure on both sides of the membrane is imbalanced, which ultimately causes rupture of the cell membrane and release of cell components (100, 142). This result is GSDMs-mediated final effect in most conditions and has been extensively studied.
3.4.2 GSDMs-mediated cellular hyperactivation
In addition to pyroptosis, GSDMs can also cause cells to become hyperactivated (143). It has been shown that in macrophages, dendritic cells and neutrophils, GSDMD pores may induce cell hyperactivation and secrete cytokines (143, 144). This is mainly associated with endosomal sorting complexes required for transport (ESCRT)-dependent repair of plasma membrane pores (145, 146) and stimulation of oxidized phospholipids (oxPAPCs) (147). After GSDMD-NT pore formation at the plasma membrane, ESCRT, specifically ESCRT-III protein, was specifically recruited to the inner layer of the plasma membrane (148). With calcium influx as a signal, ESCRT repaired the damaged membrane region in a punctate pattern, which was verified by calcium chelation increasing macrophage pyroptosis (146). ESCRTs removed GSDMs pores from the plasma membrane by forming ectosomes. After a portion of the GSDMs pores were removed, ESCRT-III began to separate from the pore-forming regions on the plasma membrane (148). Consequently, ESCRT-mediated membrane repair not only maintained cellular activity, but also preserved part of the GSDMs pores to secrete pro-inflammatory cytokines (143, 144).
In addition, GSDMs-mediated cellular hyperactivation is also associated with oxPAPCs. oxPAPCs are components of oxidized low-density lipoprotein that are present in apoptotic cells and are LPS mimic (149). Similar to LPS, oxPAPCs could bind to CD14 on membranes. CD14 transmitted LPS and oxPAPCs to TLR4 receptors on the cell membrane, and then transported LPS and TLR4 into cytosol (150), resulting in CD14 endocytosis and depletion (147, 151). Thus, oxPAPCs competed for CD14 binding with LPS. oxPAPCs acted on the catalytic domain of caspase-11 and generated the cleavage fragment with very low activation activity different from that of LPS. In vitro experiments showed that oxPAPCs blocked LPS-induced caspase-11 cleavage in a dose-dependent manner (152). In addition to the plasma membrane, the GSDMD-NT dynamically binds to abundant intracellular organelle membranes. In neutrophils, GSDMD-NT is transported to azurophilic granules and autophagosomes, releasing IL-1β through autophagy-dependent pathways (61), which provide the rationale for neutrophil hyperactivation in sepsis. Therefore, innate immune cells can achieve different activation states to better defend against infection in sepsis.
3.4.3 Active mediator release via GSDMs pores
In hyperactivated cells, the GSDMs pore preferentially releases mature IL-1β rather than pro-IL-1β. Mature and pro-IL-1β are both significantly smaller than the pore, of size 4.5 nm, suggesting that other factors affect transportation, not size. Pro-IL-1β and GSDMD pore are negatively charged and mutually repulsive. Since caspase-1 has cleaved the acidic domain of pro-IL-1β, IL-1β is positively charged, together with the electrostatic interaction of the pore, mature IL-1β can pass through more easily (139, 153).
In addition to the well-studied pro-inflammatory cytokines IL-1β and IL-18, other active components can be released from GSDMs pores, most commonly DAMPs. DAMPs could be recognized by multiple cells. Activated by DAMPs, innate immune cells can release pro-inflammatory mediators, leading to recruitment of inflammatory cells. DAMP induced the death of non-immune cells, disrupting tissue structure and homeostasis (154, 155) (Table 2).
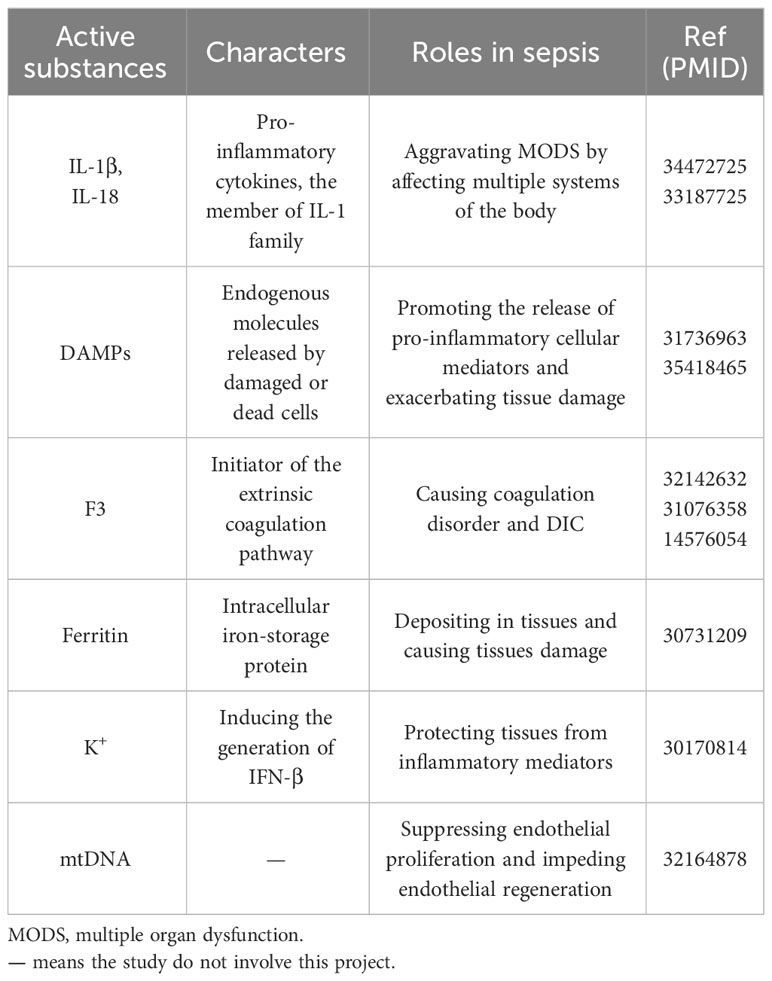
Table 2 Active mediator released by GSDMs pores in sepsis.
In addition, the GSDMs pores can release other substances (Table 2). In sepsis, macrophages or monocytes released coagulation factor III (F3) via the GSDMD pore (60). F3 is an initiator of the extrinsic coagulation pathway and plays an important role in the pathophysiology of disseminated intravascular coagulation (DIC). Blocking F3 prevented endotoxemia and DIC in mice (156, 157). In LPS-induced sepsis, caspase-11/GSDMD pathway activation promoted the release of ferritin from macrophages and increased serum ferritin concentrations (89). Septic patients have a significantly poor prognosis associated with serum ferritin, which may act as a biomarker in sepsis (158). Because serum ferritin is difficult to excrete from the body, increased serum ferritin is deposited in tissues, causing tissue injury and increasing the severity of sepsis. Due to the body’s self-protective mechanisms, GSDMs pores also release some protective signals, such as potassium ion efflux before cell death (159). Although caspase-11/GSDMD activation-mediated potassium efflux is mildly responsible for triggering NLRP3 inflammasome formation and IL-1β activation (160). More importantly, cytoplasmic K+ enhanced the binding of dsDNA to the DNA sensor cyclic GMP-AMP synthase (cGAS) and induced the production of type I interferon (IFN-β) by STING pathway, ultimately causing tissue injury in sepsis. K+ efflux inhibited the generation of IFN-β and alleviated inflammatory response (161). GSDMD pores on mitochondria caused mtDNA release into the cytoplasm, and inhibited cell proliferation via cGAS/STING pathway (142).
3.4.4 The shift of the way of cell death mediated by GSDMs
GSDMs proteins could also mediate the switch between different ways of cell death. Apoptosis and pyroptosis coexist in sepsis (162, 163). Since caspase-3/-8 are both the core of apoptosis and pyroptosis, GSDMs-mediated conversion of cell death between pyroptosis and apoptosis is the most common. Caspase/granzyme-induced apoptosis can be converted to pyroptosis due to the high expression of GSDMs. When the expression of GSDMs is too poor to induce pyroptosis, activated GSDMs may induce apoptosis. AKI-induced autophagy can activate GSDME by activating the caspase-8/-9/-3 apoptotic pathway to trigger pyroptosis. By knocking down autophagy-specific genes atg5 and fip200 or using apoptosis inhibitors, GSDME cleavage was inhibited (164). This research also linked autophagy, apoptosis and pyroptosis, demonstrating the importance of GSDMs proteins in cell death. GSDMs proteins could also promote other ways of cell death. NETosis is a novel type of cell death that differs from apoptosis and necrosis, and the death of neutrophils that occur during NETs formation, including “vital NETosis” and “suicidal NETosis “ (165). The former involves plasma membrane rupture and neutrophil lysis, whereas the latter refers to NETs release from neutrophils while maintaining intact plasma membranes (166). GSDMD is an essential regulator of NETosis (20). Inhibiting GSDMD reduced the production of NETs (167). Studies showed that depletion of GSDMD only on neutrophils worsened the severity of sepsis in mice, while systemic depletion of GSDMD alleviated the severity of sepsis (167, 168), highlighting the crucial role of NETosis in sepsis. In addition, when inflammasomes are activated in macrophages, increased mtROS promoted migration of GSDMDs to the mitochondrial membrane. Mitochondria released mtROS via GSDMD pores, which promoted RIPK1/RIPK3/mixed lineage kinase domain-like-dependent necrosis. The study demonstrated that mitochondrial dysfunction can affect immune outcomes through cell death modality switching (169). Overall, as executors of multiple cell death pathways, GSDMs perform the critical function in sepsis.
3.4.5 Other effects of GSDMs-mediated pathway
Candida albicans promoted itself to escape from macrophages through GSDMD-mediated pyroptosis, which in turn released candidalysin and caused fungal sepsis (63). The GSDMs pathway may also contribute to the polarization of M1 macrophages (105). The deeper mechanisms by which GSDMs affect macrophage polarization still need further study.
4 The therapy targeting GSDMs pathway in sepsis
To date, treatment strategies for septic patients remain limited, relying mainly on antibiotic management and intensive care (170). The pathogenesis of sepsis is complex and multifaceted, affecting almost all bodily systems and organs, and exhibiting diverse pathophysiological presentations during each stage, posing challenges for clinical management (171). A series of specific therapies have been developed based on the pathogenesis of sepsis. Although they have shown significant efficacy in animal models of sepsis or in vitro experiments, none has been conclusively proven effective in clinical trials to date. In addition to the complexity of sepsis, clinical trials have certain inherent limitations, such as inconsistent inclusion and exclusion criteria among clinical trials and some indicators that do not adequately reflect clinical efficacy (172, 173). Current targeted drugs for sepsis focus on TLRs and neutralization of pro-inflammatory factors, but the clinical effects are not significant (174, 175). GSDMs proteins are co-ultimate executors of pyroptosis and inflammatory factors release, so GSDMs proteins are seen as novel and ideal targets for therapeutic agents of sepsis. A number of drugs have been shown to inhibit GSDMs-mediated pathways or GSDMs pore formation (Table 3), and these studies have also contributed to the development of novel effective drug targeted GSDMs proteins for the clinical treatment of sepsis.
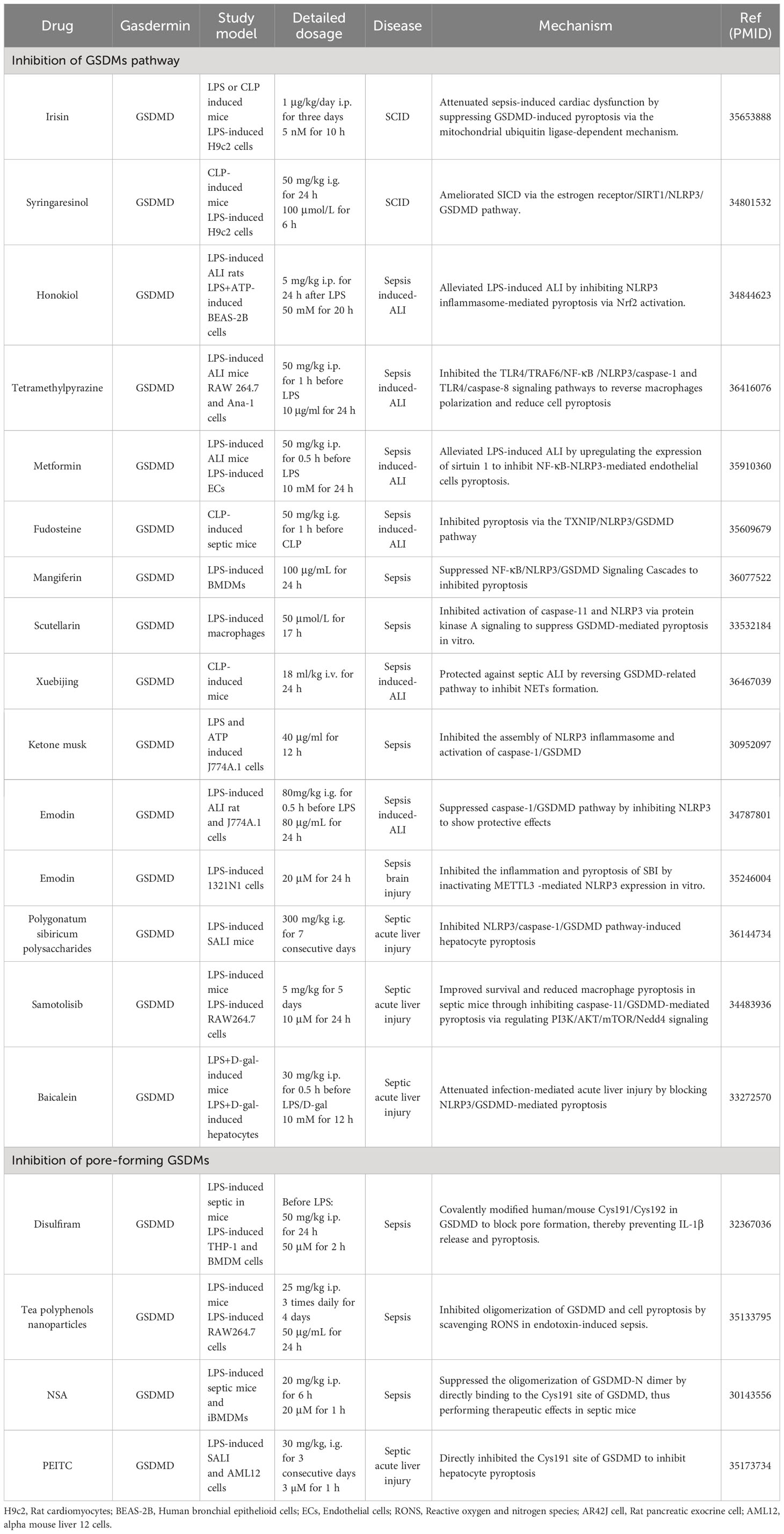
Table 3 The therapy targeting GSDMs in sepsis.
4.1 Inhibition of GSDMs-mediated pathway activation
Melatonin reduced mortality in LPS-induced ALI mice and pyroptosis of human alveolar epithelial cells and macrophages by inhibiting the activation of NLRP3/caspase-1/GSDMD via Nrf2/heme oxygenase 1 pathway (176). In sepsis, damage of the endothelial barrier contributes to tissue ischemia and hypoxia, and one of the target organs is the heart. Irisin attenuated sepsis-induced cardiac dysfunction (SICD) by inhibiting GSDMD-induced pyroptosis through the mitochondrial ubiquitin ligase-dependent mechanism (177). Syringaresinol, a natural abstract, ameliorated SICD via the estrogen receptor/sirtuin 1/NLRP3/GSDMD pathway (178). The lungs are the most vulnerable organ in the progression of sepsis, and septic patients often present with ALI or ARDS at the early stage (179). Honokiol alleviated LPS-induced ALI by inhibiting NLRP3 inflammasome-mediated pyroptosis through Nrf2 activation (180). Tetramethylpyrazine, an effective compound extracted from the umbelliferous plant Chuanxiong, decreased infiltration of inflammatory cells and pro-inflammatory factors in the alveoli and reduced mortality in mice with LPS-induced ALI by inhibiting TLR4/TRAF6/NF-κB pathway to downregulate NLRP3 expression and caspase-1-mediated pyroptosis (181). Metformin alleviated LPS-induced ALI by upregulating the expression of sirtuin 1 to inhibit NF-κB-NLRP3-mediated endothelial cells pyroptosis (182). Fudosteine, a cysteine derivative, attenuated lung inflammation response and oxidative stress in septic mice via the TXNIP/NLRP3/GSDMD pathway (183). Mangiferin, a flavonoid widely distributed in several herbs, inhibited NLRP3/caspase-1/-11-mediated GSDMD activation in sepsis (184). Scutellarin, a flavonoid from Erigeron breviscapus, inhibited activation of caspase-11 and NLRP3 inflammasome via protein kinase A signaling to suppress GSDMD-mediated pyroptosis and the release of inflammatory mediators in LPS-induced macrophages (185). Xuebijing protected against septic ALI by inhibiting GSDMD-associated neutrophil extracellular traps formation (186). Ketone musk and Emodin, respective components of native musk and Chinese medicine Dahuang, enhanced cell viability and inhibited the release of pro-inflammatory cytokines by inhibiting the assembly of NLRP3 inflammasome and activation of caspase-1/GSDMD in LPS-induced macrophages. Emodin showed a significant protective effect against septic ALI (187, 188). In addition, emodin inhibited inflammation and pyroptosis of septic brain injury by inactivating m6A-mediated NLRP3 expression in vitro (189). Liver is the major site of bacterial endotoxin-induced inflammation in sepsis. Polygonatum sibiricum polysaccharides significantly downregulated neutrophil infiltration and pro-inflammatory factor release in liver tissue to reduce 48-hour mortality in septic acute liver injury mice, primarily by inhibiting NLRP3/caspase-1/GSDMD pathway-induced hepatocyte pyroptosis (190). Samotolisib improved survival and reduced macrophage pyroptosis in septic mice by inhibiting caspase-11/GSDMD-mediated pyroptosis (191). Baicalein attenuated infection-mediated acute liver injury by blocking NLRP3/GSDMD-mediated pyroptosis (192).
4.2 Inhibition of pore formation
Disulfiram, a drug for the treatment of alcohol dependence, is an inhibitor of GSDMD pore formation. Disulfiram covalently modified GSDMD at Cys191/Cys192 (human/mouse) to block pore formation without affecting the cleavage of IL-1β and GSDMD, preventing IL-1β release and pyroptosis (193). Tea polyphenols nanoparticles scavenged reactive oxygen and nitrogen species via polyphenols-derived structure to inhibit oligomerization of GSDMD and cell pyroptosis in endotoxin-induced sepsis (194). Necrosulfonamide (NSA) inhibited pyroptosis and pro-inflammatory cytokines release in mice or human LPS and nigericin-induced monocytes/macrophages and reduced lethality in LPS-induced septic mice. NSA suppressed oligomerization of GSDMD-N dimer by directly binding to GSDMD at Cys191, thereby affecting GSDMD pore formation at the plasma membrane (195). Phenethyl isothiocyanate (PEITC), a natural compound found in cruciferous vegetables, may inhibit LPS-induced septic liver injury in a dose-dependent manner. PEITC could directly inhibit GSDMD at Cys191 and thus inhibit hepatocyte pyroptosis, which may provide a potential therapeutic strategy for septic liver injury (196).
5 Conclusion
Since the mortality of sepsis has been persistently high and is a major problem in critical care medicine, many researchers have conducted studies on molecular mechanisms and therapeutic targets in the pathophysiological process of sepsis. GSDMs proteins play important roles in the pathogenesis of sepsis and also aggravate the multi-organ dysfunction induced by sepsis. Among them, four classes are implicated in sepsis, including GSDMA, GSDMB, GSDMD and GSDME, with GSDMD being the most widely studied. More than one pathway may be operational at one time. Many molecules are also involved in the regulation of GSDMs-mediated pathways, such as epigenetic regulation and regulation of oxidative stress. All mechanisms may be involved in the different stages seen in sepsis. The sparking interests of GSDMs in sepsis is acting as pore-forming protein mediating cell pyroptosis or inflammatory factors release. The GSDMs pathway may be a promising therapeutic target for sepsis.
Author contributions
WW conceived the study, data analysis, and drafted the manuscript. ZH conceived the study, its design and critically revised the manuscript. All authors read and approved the final manuscript.
Funding
This study was funded by the National Natural Science Foundation of China (NO: 82272216).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AKI, acute kidney injury; ALI, acute lung injury; ARDS, acute respiratory distress syndrome; ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain; Bcl-2, B-cell lymphoma-2; Cat G, Cathepsin G; cGAS, cyclic GMP-AMP synthase; circRNA, circular RNA; CLP, cecal ligation puncture; CTLs, cytotoxic T lymphocytes; Cys, cysteine; DAMPs, danger-associated molecular patterns; DFNB59, Pejvakin; DIC, disseminated intravascular coagulation; ER, endoplasmic reticulum; ERG, ETS-related gene; ESCRT, endosomal sorting complexes required for transport; F3, coagulation factor III; GSDMA, gsdermin A; GSDMB, gsdermin B; GSDMC, gsdermin C; GSDMD, gsdermin D; GSDME gsdermin E; GSDMs, Gasdermins; H3K27ac, acetylation of histone H3 Lysine 27; HDAC11, Histone deacetylase 11; HMGB1, high mobility group box 1; IFN-β, type I interferon; IKK, IκB kinase; IL-33, interleukin-33; IRF, interferon regulatory factor; KLF6, Krüppel-like factor 6; KO, knockout; lncRNA, long non-coding RNA; LPS, lipopolysaccharide; m6A, N6-adenomethylation; MAC, membrane attack complex; MIF, migration inhibitory factor; miRNA, microRNA; mtROS, mitochondrial ROS; ncRNA, non-coding RNA; NETs, neutrophil extracellular traps; NLRP1, nucleotide-binding oligomerization domain 1; Nrf2, nuclear factor erythroid2-related factor 2; NSA, Necrosulfonamide; ox-mtDNA, oxidized mitochondrial DNA; oxPAPCs, oxidized phospholipids; PDE4B, Phosphodiesterase 4B; PD-L1, programmed death ligand 1; PEITC, Phenethyl isothiocyanate; PRRs, pattern recognition receptors; RAGE, receptors for advanced glycation end-products; RIPK1, receptor-interacting serine-threonine protein kinase 1; ROS, reactive oxygen species; SICD, sepsis-induced cardiac dysfunction; SpeB, Streptococcus pyogenic exotoxin B; TAK1, TGF-α-activated kinase 1; TLR2, toll-like receptor 2; TMEM173/STING, Transmembrane protein 173; TXNIP, thioredoxin-interacting protein; YopJ, Yersinia outer protein J; YTHDF1, YTH N6-Methyladenosine RNA Binding Protein 1; zDHHC5, zinc finger DHHC domain 5
References
1. Evans L, Rhodes A, Alhazzani W, Antonelli M, Coopersmith CM, French C, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock 2021. Intensive Care Med (2021) 47(11):1181–247. doi: 10.1007/s00134-021-06506-y
2. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA (2016) 315(8):801–10. doi: 10.1001/jama.2016.0287
3. Bauer M, Gerlach H, Vogelmann T, Preissing F, Stiefel J, Adam D. Mortality in sepsis and septic shock in Europe, North America and Australia between 2009 and 2019- results from a systematic review and meta-analysis. Crit Care (2020) 24(1):239. doi: 10.1186/s13054-020-02950-2
4. Liu V, Escobar GJ, Greene JD, Soule J, Whippy A, Angus DC, et al. Hospital deaths in patients with sepsis from 2 independent cohorts. JAMA (2014) 312(1):90–2. doi: 10.1001/jama.2014.5804
5. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the Global Burden of Disease Study. Lancet (2020) 395(10219):200–11. doi: 10.1016/S0140-6736(19)32989-7
6. Fleischmann C, Thomas-Rueddel DO, Hartmann M, Hartog CS, Welte T, Heublein S, et al. Hospital incidence and mortality rates of sepsis. Dtsch Arztebl Int (2016) 113(10):159–66. doi: 10.3238/arztebl.2016.0159
7. Kaukonen KM, Bailey M, Suzuki S, Pilcher D, Bellomo R. Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000-2012. JAMA (2014) 311(13):1308–16. doi: 10.1001/jama.2014.2637
8. Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol (2013) 13(12):862–74. doi: 10.1038/nri3552
9. Font MD, Thyagarajan B, Khanna AK. Sepsis and Septic Shock - Basics of diagnosis, pathophysiology and clinical decision making. Med Clin North Am (2020) 104(4):573–85. doi: 10.1016/j.mcna.2020.02.011
10. van der Poll T, Shankar-Hari M, Wiersinga WJ. The immunology of sepsis. Immunity (2021) 54(11):2450–64. doi: 10.1016/j.immuni.2021.10.012
11. Delano MJ, Ward PA. The immune system's role in sepsis progression, resolution, and long-term outcome. Immunol Rev (2016) 274(1):330–53. doi: 10.1111/imr.12499
12. Zhang H, Wang Y, Qu M, Li W, Wu D, Cata JP, et al. Neutrophil, neutrophil extracellular traps and endothelial cell dysfunction in sepsis. Clin Transl Med (2023) 13(1):e1170. doi: 10.1002/ctm2.1170
13. Shen XF, Cao K, Jiang JP, Guan WX, Du JF. Neutrophil dysregulation during sepsis: an overview and update. J Cell Mol Med (2017) 21(9):1687–97. doi: 10.1111/jcmm.13112
14. Xia S, Hollingsworth L, Wu H. Mechanism and regulation of gasdermin-mediated cell death. Cold Spring Harb Perspect Biol (2020) 12(3):a036400. doi: 10.1101/cshperspect.a036400
15. Wiersinga WJ, Leopold SJ, Cranendonk DR, van der Poll T. Host innate immune responses to sepsis. Virulence (2014) 5(1):36–44. doi: 10.4161/viru.25436
16. Venet F, Monneret G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat Rev Nephrol (2018) 14(2):121–37. doi: 10.1038/nrneph.2017.165
17. Patel JM, Sapey E, Parekh D, Scott A, Dosanjh D, Gao F, et al. Sepsis induces a dysregulated neutrophil phenotype that is associated with increased mortality. Mediators Inflamm (2018) 2018:4065362. doi: 10.1155/2018/4065362
18. Polycarpou A, Howard M, Farrar CA, Greenlaw R, Fanelli G, Wallis R, et al. Rationale for targeting complement in COVID-19. EMBO Mol Med (2020) 12(8):e12642. doi: 10.15252/emmm.202012642
19. McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood (2017) 129(10):1357–67. doi: 10.1182/blood-2016-09-741298
20. Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol (2018) 3(26):eaar6689. doi: 10.1126/sciimmunol.aar6689
21. Saeki N, Kuwahara Y, Sasaki H, Satoh H, Shiroishi T. Gasdermin (Gsdm) localizing to mouse Chromosome 11 is predominantly expressed in upper gastrointestinal tract but significantly suppressed in human gastric cancer cells. Mamm Genome (2000) 11(9):718–24. doi: 10.1007/s003350010138
22. Deng W, Bai Y, Deng F, Pan Y, Mei S, Zheng Z, et al. Streptococcal pyrogenic exotoxin B cleaves GSDMA and triggers pyroptosis. Nature (2022) 602(7897):496–502. doi: 10.1038/s41586-021-04384-4
23. Zou J, Zheng Y, Huang Y, Tang D, Kang R, Chen R. The versatile gasdermin family: their function and roles in diseases. Front Immunol (2021) 12:751533. doi: 10.3389/fimmu.2021.751533
24. Rana N, Privitera G, Kondolf HC, Bulek K, Lechuga S, De Salvo C, et al. GSDMB is increased in IBD and regulates epithelial restitution/repair independent of pyroptosis. Cell (2022) 185(2):283–98 e17. doi: 10.1016/j.cell.2021.12.024
25. Fujii T, Tamura M, Tanaka S, Kato Y, Yamamoto H, Mizushina Y, et al. (Gsdmd) is dispensable for mouse intestinal epithelium development. Genesis (2008) 46(8):418–23. doi: 10.1002/dvg.20412
26. Gong W, Yang K, Zhao W, Zheng J, Yu J, Guo K, et al. Intestinal Gasdermins for regulation of inflammation and tumorigenesis. Front Immunol (2022) 13:1052111. doi: 10.3389/fimmu.2022.1052111
27. LaRock DL, Russell R, Johnson AF, Wilde S, LaRock CN. Group A streptococcus infection of the nasopharynx requires proinflammatory signaling through the interleukin-1 receptor. Infect Immun (2020) 88(10):e00356-20. doi: 10.1128/IAI.00356-20
28. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535(7610):111–6. doi: 10.1038/nature18590
29. Broz P, Pelegrin P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol (2020) 20(3):143–57. doi: 10.1038/s41577-019-0228-2
30. Ruan J. Structural insight of gasdermin family driving pyroptotic cell death. Adv Exp Med Biol (2019) 1172:189–205. doi: 10.1007/978-981-13-9367-9_9
31. Gabarin RS, Li M, Zimmel PA, Marshall JC, Li Y, Zhang H. Intracellular and extracellular lipopolysaccharide signaling in sepsis: avenues for novel therapeutic strategies. J Innate Immun (2021) 13(6):323–32. doi: 10.1159/000515740
32. Johnson AG, Wein T, Mayer ML, Duncan-Lowey B, Yirmiya E, Oppenheimer-Shaanan Y, et al. Bacterial gasdermins reveal an ancient mechanism of cell death. Science (2022) 375(6577):221–5. doi: 10.1126/science.abj8432
33. Tanaka S, Mizushina Y, Kato Y, Tamura M, Shiroishi T. Functional conservation of Gsdma cluster genes specifically duplicated in the mouse genome. G3 (Bethesda). (2013) 3(10):1843–50. doi: 10.1534/g3.113.007393
34. Soderman J, Berglind L, Almer S. Gene expression-genotype analysis implicates GSDMA, GSDMB, and LRRC3C as contributors to inflammatory bowel disease susceptibility. BioMed Res Int (2015) 2015:834805. doi: 10.1155/2015/834805
35. Feng S, Fox D, Man SM. Mechanisms of gasdermin family members in inflammasome signaling and cell death. J Mol Biol (2018) 430(18 Pt B):3068–80. doi: 10.1016/j.jmb.2018.07.002
36. Reglinski M, Sriskandan S. The contribution of group A streptococcal virulence determinants to the pathogenesis of sepsis. Virulence (2014) 5(1):127–36. doi: 10.4161/viru.26400
37. Ivanov AI, Rana N, Privitera G, Pizarro TT. The enigmatic roles of epithelial gasdermin B: Recent discoveries and controversies. Trends Cell Biol (2023) 33(1):48–59. doi: 10.1016/j.tcb.2022.06.006
38. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science (2020) 368(6494):965-+. doi: 10.1126/science.aaz7548
39. Chen Q, Shi P, Wang Y, Zou D, Wu X, Wang D, et al. GSDMB promotes non-canonical pyroptosis by enhancing caspase-4 activity. J Mol Cell Biol (2019) 11(6):496–508. doi: 10.1093/jmcb/mjy056
40. Wang L, Wang Y, Wang J, Li L, Bi J. Identification of a prognosis-related risk signature for bladder cancer to predict survival and immune landscapes. J Immunol Res (2021) 2021:3236384. doi: 10.1155/2021/3236384
41. He H, Yi L, Zhang B, Yan B, Xiao M, Ren J, et al. USP24-GSDMB complex promotes bladder cancer proliferation via activation of the STAT3 pathway. Int J Biol Sci (2021) 17(10):2417–29. doi: 10.7150/ijbs.54442
42. Hansen JM, de Jong MF, Wu Q, Zhang LS, Heisler DB, Alto LT, et al. Pathogenic ubiquitination of GSDMB inhibits NK cell bactericidal functions. Cell (2021) 184(12):3178–91 e18. doi: 10.1016/j.cell.2021.04.036
43. Zhong X, Zeng H, Zhou Z, Su Y, Cheng H, Hou Y, et al. Structural mechanisms for regulation of GSDMB pore-forming activity. Nature (2023) 616(7957):598–605. doi: 10.1038/s41586-023-05872-5
44. Wang C, Shivcharan S, Tian T, Wright S, Ma D, Chang J, et al. Structural basis for GSDMB pore formation and its targeting by IpaH7. 8. Nat (2023) 616(7957):590–7. doi: 10.1038/s41586-023-05832-z
45. Yin H, Zheng J, He Q, Zhang X, Li X, Ma Y, et al. Insights into the GSDMB-mediated cellular lysis and its targeting by IpaH7. 8. Nat Commun (2023) 14(1):61. doi: 10.1038/s41467-022-35725-0
46. Watabe K, Ito A, Asada H, Endo Y, Kobayashi T, Nakamoto K, et al. Structure, expression and chromosome mapping of MLZE, a novel gene which is preferentially expressed in metastatic melanoma cells. Jpn J Cancer Res (2001) 92(2):140–51. doi: 10.1111/j.1349-7006.2001.tb01076.x
47. Sun K, Chen RX, Li JZ, Luo ZX. LINC00511/hsa-miR-573 axis-mediated high expression of Gasdermin C associates with dismal prognosis and tumor immune infiltration of breast cancer. Sci Rep (2022) 12(1):14788. doi: 10.1038/s41598-022-19247-9
48. Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol (2020) 22(10):1264–75. doi: 10.1038/s41556-020-0575-z
49. Zhao M, Ren K, Xiong X, Xin Y, Zou Y, Maynard JC, et al. Epithelial STAT6 O-GlcNAcylation drives a concerted anti-helminth alarmin response dependent on tuft cell hyperplasia and Gasdermin C. Immunity (2022) 55(4):623–38 e5. doi: 10.1016/j.immuni.2022.03.009
50. Du J, Sarkar R, Li Y, He L, Kang W, Liao W, et al. N(6)-adenomethylation of GsdmC is essential for Lgr5(+) stem cell survival to maintain normal colonic epithelial morphogenesis. Dev Cell (2022) 57(16):1976–94 e8. doi: 10.1016/j.devcel.2022.07.006
51. Qiu S, Liu J, Xing F. 'Hints' in the killer protein gasdermin D: unveiling the secrets of gasdermins driving cell death. Cell Death Differ (2017) 24(4):588–96. doi: 10.1038/cdd.2017.24
52. Xi G, Gao J, Wan B, Zhan P, Xu W, Lv T, et al. GSDMD is required for effector CD8(+) T cell responses to lung cancer cells. Int Immunopharmacol (2019) 74:105713. doi: 10.1016/j.intimp.2019.105713
53. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
54. Zhang J, Yu Q, Jiang D, Yu K, Yu W, Chi Z, et al. Epithelial Gasdermin D shapes the host-microbial interface by driving mucus layer formation. Sci Immunol (2022) 7(68):eabk2092. doi: 10.1126/sciimmunol.abk2092
55. Li Z, Ji S, Jiang ML, Xu Y, Zhang CJ. The regulation and modification of GSDMD signaling in diseases. Front Immunol (2022) 13:893912. doi: 10.3389/fimmu.2022.893912
56. Shi H, Gao Y, Dong Z, Yang J, Gao R, Li X, et al. GSDMD-mediated cardiomyocyte pyroptosis promotes myocardial I/R injury. Circ Res (2021) 129(3):383–96. doi: 10.1161/CIRCRESAHA.120.318629
57. Chen W, Chen S, Yan C, Zhang Y, Zhang R, Chen M, et al. Allergen protease-activated stress granule assembly and gasdermin D fragmentation control interleukin-33 secretion. Nat Immunol (2022) 23(7):1021–30. doi: 10.1038/s41590-022-01255-6
58. Wang Y, Shi P, Chen Q, Huang Z, Zou D, Zhang J, et al. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J Mol Cell Biol (2019) 11(12):1069–82. doi: 10.1093/jmcb/mjz020
59. Dhar R, Rana MN, Zhang L, Li Y, Li N, Hu Z, et al. Phosphodiesterase 4B is required for NLRP3 inflammasome activation by positive feedback with Nrf2 in the early phase of LPS- induced acute lung injury. Free Radic Biol Med (2021) 176:378–91. doi: 10.1016/j.freeradbiomed.2021.10.007
60. Zhang H, Zeng L, Xie M, Liu J, Zhou B, Wu R, et al. TMEM173 drives lethal coagulation in sepsis. Cell Host Microbe (2020) 27(4):556–70.e6. doi: 10.1016/j.chom.2020.02.004
61. Karmakar M, Minns M, Greenberg EN, Diaz-Aponte J, Pestonjamasp K, Johnson JL, et al. N-GSDMD trafficking to neutrophil organelles facilitates IL-1beta release independently of plasma membrane pores and pyroptosis. Nat Commun (2020) 11(1):2212. doi: 10.1038/s41467-020-16043-9
62. Dai S, Ye B, Zhong L, Chen Y, Hong G, Zhao G, et al. GSDMD mediates LPS-induced septic myocardial dysfunction by regulating ROS-dependent NLRP3 inflammasome activation. Front Cell Dev Biol (2021) 9:779432. doi: 10.3389/fcell.2021.779432
63. Ding X, Kambara H, Guo R, Kanneganti A, Acosta-Zaldivar M, Li J, et al. Inflammasome-mediated GSDMD activation facilitates escape of Candida albicans from macrophages. Nat Commun (2021) 12(1):6699. doi: 10.1038/s41467-021-27034-9
64. Van Laer L, Huizing EH, Verstreken M, van Zuijlen D, Wauters JG, Bossuyt PJ, et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat Genet (1998) 20(2):194–7. doi: 10.1038/2503
65. Tan G, Huang C, Chen J, Chen B, Zhi F. Gasdermin-E-mediated pyroptosis participates in the pathogenesis of Crohn's disease by promoting intestinal inflammation. Cell Rep (2021) 35(11):109265. doi: 10.1016/j.celrep.2021.109265
66. Chen Y, Lian N, Chen S, Xiao T, Ke Y, Zhang Y, et al. GSDME deficiency leads to the aggravation of UVB-induced skin inflammation through enhancing recruitment and activation of neutrophils. Cell Death Dis (2022) 13(10):841. doi: 10.1038/s41419-022-05276-9
67. Sun R, Jiang K, Zeng C, Zhu R, Chu H, Liu H, et al. Synergism of TNF-alpha and IFN-beta triggers human airway epithelial cells death by apoptosis and pyroptosis. Mol Immunol (2023) 153:160–9. doi: 10.1016/j.molimm.2022.12.002
68. Wan X, Li J, Wang Y, Yu X, He X, Shi J, et al. H7N9 virus infection triggers lethal cytokine storm by activating gasdermin E-mediated pyroptosis of lung alveolar epithelial cells. Natl Sci Rev (2022) 9(1):nwab137. doi: 10.1093/nsr/nwab137
69. Liu W, Kinnefors A, Bostrom M, Edin F, Rask-Andersen H. Distribution of pejvakin in human spiral ganglion: An immunohistochemical study. Cochlear Implants Int (2013) 14(4):225–31. doi: 10.1179/1754762812Y.0000000027
70. Collin RW, Kalay E, Oostrik J, Caylan R, Wollnik B, Arslan S, et al. Involvement of DFNB59 mutations in autosomal recessive nonsyndromic hearing impairment. Hum Mutat (2007) 28(7):718–23. doi: 10.1002/humu.20510
71. von Moltke J, Ayres JS, Kofoed EM, Chavarria-Smith J, Vance RE. Recognition of bacteria by inflammasomes. Annu Rev Immunol (2013) 31:73–106. doi: 10.1146/annurev-immunol-032712-095944
72. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol (2010) 11(12):1136–42. doi: 10.1038/ni.1960
73. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479(7371):117–21. doi: 10.1038/nature10558
74. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature (2017) 547(7661):99–103. doi: 10.1038/nature22393
75. Zheng Z, Deng W, Bai Y, Miao R, Mei S, Zhang Z, et al. The lysosomal rag-ragulator complex licenses RIPK1 and caspase-8-mediated pyroptosis by yersinia. Science (2021) 372(6549):1412-+. doi: 10.1126/science.abg0269
76. Mandal P, Feng Y, Lyons JD, Berger SB, Otani S, DeLaney A, et al. Caspase-8 collaborates with caspase-11 to drive tissue damage and execution of endotoxic shock. Immunity (2018) 49(1):42–55.e6. doi: 10.1016/j.immuni.2018.06.011
77. Wang C, Yang T, Xiao J, Xu C, Alippe Y, Sun K, et al. NLRP3 inflammasome activation triggers gasdermin D-independent inflammation. Sci Immunol (2021) 6(64):eabj3859. doi: 10.1126/sciimmunol.abj3859
78. Hashimoto S, Kobayashi A, Kooguchi K, Kitamura Y, Onodera H, Nakajima H. Upregulation of two death pathways of perforin/granzyme and FasL/Fas in septic acute respiratory distress syndrome. Am J Respir Crit Care Med (2000) 161(1):237–43. doi: 10.1164/ajrccm.161.1.9810007
79. Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature (2020) 579(7799):415–20. doi: 10.1038/s41586-020-2071-9
80. Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol (2020) 5(43):eaax7969. doi: 10.1126/sciimmunol.aax7969
81. Catalan E, Jaime-Sanchez P, Aguilo N, Simon MM, Froelich CJ, Pardo J. Mouse cytotoxic T cell-derived granzyme B activates the mitochondrial cell death pathway in a Bim-dependent fashion. J Biol Chem (2015) 290(11):6868–77. doi: 10.1074/jbc.M114.631564
82. Burgener SS, Leborgne NGF, Snipas SJ, Salvesen GS, Bird PI, Benarafa C. Cathepsin G inhibition by serpinb1 and serpinb6 prevents programmed necrosis in neutrophils and monocytes and reduces GSDMD-driven inflammation. Cell Rep (2019) 27(12):3646–56.e5. doi: 10.1016/j.celrep.2019.05.065
83. Lv H, Liu X, Zhou H. Usp25 upregulation boosts gsdmd -mediated pyroptosis of acinar cells in acute pancreatitis. Shock (2022) 58(5):408–16. doi: 10.1097/SHK.0000000000001992
84. Kang K, Li N, Gao Y, Wang C, Chen P, Meng X, et al. circ-Katnal1 Enhances Inflammatory Pyroptosis in Sepsis-Induced Liver Injury through the miR-31-5p/GSDMD Axis. Mediators Inflamm (2022) 2022:8950130. doi: 10.1155/2022/8950130
85. Zhang Y, Yin K, Wang D, Wang Y, Lu H, Zhao H, et al. Polystyrene microplastics-induced cardiotoxicity in chickens via the ROS-driven NF-kappaB-NLRP3-GSDMD and AMPK-PGC-1alpha axes. Sci Total Environ (2022) 840:156727. doi: 10.1016/j.scitotenv.2022.156727
86. Yang B, Zhang T, Wei L, Zhao B, Wang Q, Yao Z, et al. Glucocorticoid induces GSDMD-dependent pyrolysis in PC12 cells via endoplasmic reticulum stress. PloS One (2022) 17(9):e0274057. doi: 10.1371/journal.pone.0274057
87. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature (2016) 535(7610):153–8. doi: 10.1038/nature18629
88. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther (2021) 6(1):128. doi: 10.1038/s41392-021-00507-5
89. Wang D, Yu S, Zhang Y, Huang L, Luo R, Tang Y, et al. Caspse-11-GSDMD pathway is required for serum ferritin secretion in sepsis. Clin Immunol (2019) 205:148–52. doi: 10.1016/j.clim.2018.11.005
90. Bosma EK, van Noorden CJF, Schlingemann RO, Klaassen I. The role of plasmalemma vesicle-associated protein in pathological breakdown of blood-brain and blood-retinal barriers: potential novel therapeutic target for cerebral edema and diabetic macular edema. Fluids Barriers CNS. (2018) 15(1):24. doi: 10.1186/s12987-018-0109-2
91. Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev (2012) 249(1):158–75. doi: 10.1111/j.1600-065X.2012.01146.x
92. Sunden-Cullberg J, Norrby-Teglund A, Rouhiainen A, Rauvala H, Herman G, Tracey KJ, et al. Persistent elevation of high mobility group box-1 protein (HMGB1) in patients with severe sepsis and septic shock. Crit Care Med (2005) 33(3):564–73. doi: 10.1097/01.CCM.0000155991.88802.4D
93. Zhou Y, Dong H, Zhong Y, Huang J, Lv J, Li J. The cold-inducible RNA-binding protein (CIRP) level in peripheral blood predicts sepsis outcome. PloS One (2015) 10(9):e0137721. doi: 10.1371/journal.pone.0137721
94. Ekaney ML, Otto GP, Sossdorf M, Sponholz C, Boehringer M, Loesche W, et al. Impact of plasma histones in human sepsis and their contribution to cellular injury and inflammation. Crit Care (2014) 18(5):543. doi: 10.1186/s13054-014-0543-8
95. Magna M, Pisetsky DS. The alarmin properties of DNA and DNA-associated nuclear proteins. Clin Ther (2016) 38(5):1029–41. doi: 10.1016/j.clinthera.2016.02.029
96. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science (1999) 285(5425):248–51. doi: 10.1126/science.285.5425.248
97. Roh JS, Sohn DH. Damage-associated molecular patterns in inflammatory diseases. Immune Netw (2018) 18(4):e27. doi: 10.4110/in.2018.18.e27
98. Li W, Deng M, Loughran PA, Yang M, Lin M, Yang C, et al. LPS induces active HMGB1 release from hepatocytes into exosomes through the coordinated activities of TLR4 and caspase-11/GSDMD signaling. Front Immunol (2020) 11:229. doi: 10.3389/fimmu.2020.00229
99. Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X, et al. The endotoxin delivery protein HMGB1 mediates caspase-11-dependent lethality in sepsis. Immunity (2018) 49(4):740–53.e7. doi: 10.1016/j.immuni.2018.08.016
100. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci (2017) 42(4):245–54. doi: 10.1016/j.tibs.2016.10.004
101. Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell (2007) 128(4):635–8. doi: 10.1016/j.cell.2007.02.006
102. Hogg SJ, Beavis PA, Dawson MA, Johnstone RW. Targeting the epigenetic regulation of antitumour immunity. Nat Rev Drug Discovery (2020) 19(11):776–800. doi: 10.1038/s41573-020-0077-5
103. Poller W, Dimmeler S, Heymans S, Zeller T, Haas J, Karakas M, et al. Non-coding RNAs in cardiovascular diseases: diagnostic and therapeutic perspectives. Eur Heart J (2018) 39(29):2704–16. doi: 10.1093/eurheartj/ehx165
104. Li Y, Li G, Guo X, Yao H, Wang G, Li C. Non-coding RNA in bladder cancer. Cancer Lett (2020) 485:38–44. doi: 10.1016/j.canlet.2020.04.023
105. Jiao Y, Zhang T, Zhang C, Ji H, Tong X, Xia R, et al. Exosomal miR-30d-5p of neutrophils induces M1 macrophage polarization and primes macrophage pyroptosis in sepsis-related acute lung injury. Crit Care (2021) 25(1):356. doi: 10.1186/s13054-021-03775-3
106. Xue Z, Xi Q, Liu H, Guo X, Zhang J, Zhang Z, et al. miR-21 promotes NLRP3 inflammasome activation to mediate pyroptosis and endotoxic shock. Cell Death Dis (2019) 10(6):461. doi: 10.1038/s41419-019-1713-z
107. Han J, Li W, Zhang J, Guan Y, Huang Y, Li X. Mechanism of circHIPK3-miRNA-124-3p/miRNA-148b-3p-Mediated Inflammatory Responses and Cell Senescence in Candida albicans-Induced Septic Acute Kidney Injury. Gerontology (2022) 68(10):1145–65. doi: 10.1159/000523910
108. Gao M, Li H, Liu Q, Ma N, Zi P, Shi H, et al. KLF6 promotes pyroptosis of renal tubular epithelial cells in septic acute kidney injury. Shock (2022) 57(3):417–26. doi: 10.1097/SHK.0000000000001881
109. Juan CX, Mao Y, Cao Q, Chen Y, Zhou LB, Li S, et al. Exosome-mediated pyroptosis of miR-93-TXNIP-NLRP3 leads to functional difference between M1 and M2 macrophages in sepsis-induced acute kidney injury. J Cell Mol Med (2021) 25(10):4786–99. doi: 10.1111/jcmm.16449
110. Li X, Yao L, Zeng X, Hu B, Zhang X, Wang J, et al. miR-30c-5p alleviated pyroptosis during sepsis-induced acute kidney injury via targeting TXNIP. Inflammation (2021) 44(1):217–28. doi: 10.1007/s10753-020-01323-9
111. Wang X, Li XL, Qin LJ. The lncRNA XIST/miR-150-5p/c-Fos axis regulates sepsis-induced myocardial injury via TXNIP-modulated pyroptosis. Lab Invest. (2021) 101(9):1118–29. doi: 10.1038/s41374-021-00607-4
112. Chen S, Ding R, Hu Z, Yin X, Xiao F, Zhang W, et al. MicroRNA-34a inhibition alleviates lung injury in cecal ligation and puncture induced septic mice. Front Immunol (2020) 11:1829. doi: 10.3389/fimmu.2020.01829
113. Deng J, Tan W, Luo Q, Lin L, Zheng L, Yang J. Long non-coding RNA MEG3 promotes renal tubular epithelial cell pyroptosis by regulating the miR-18a-3p/GSDMD pathway in lipopolysaccharide-induced acute kidney injury. Front Physiol (2021) 12:663216. doi: 10.3389/fphys.2021.663216
114. Zhang S, Guan X, Liu W, Zhu Z, Jin H, Zhu Y, et al. YTHDF1 alleviates sepsis by upregulating WWP1 to induce NLRP3 ubiquitination and inhibit caspase-1-dependent pyroptosis. Cell Death Discovery (2022) 8(1):244. doi: 10.1038/s41420-022-00872-2
115. Yao F, Jin Z, Zheng Z, Lv X, Ren L, Yang J, et al. HDAC11 promotes both NLRP3/caspase-1/GSDMD and caspase-3/GSDME pathways causing pyroptosis via ERG in vascular endothelial cells. Cell Death Discovery (2022) 8(1):112. doi: 10.1038/s41420-022-00906-9
116. Chen KW, Demarco B, Ramos S, Heilig R, Goris M, Grayczyk JP, et al. RIPK1 activates distinct gasdermins in macrophages and neutrophils upon pathogen blockade of innate immune signaling. Proc Natl Acad Sci U.S.A. (2021) 118(28):e2101189118. doi: 10.1073/pnas.2101189118
117. Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science (2018) 362(6418):1064–9. doi: 10.1126/science.aau2818
118. Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci U S A. (2018) 115(46):E10888–E97. doi: 10.1073/pnas.1809548115
119. Li Y, Guo X, Hu C, Du Y, Guo C, Di W, et al. Type I IFN operates pyroptosis and necroptosis during multidrug-resistant A. baumannii infection. Cell Death Differ (2018) 25(7):1304–18. doi: 10.1038/s41418-017-0041-z
120. Jiang H, Zhang X, Chen X, Aramsangtienchai P, Tong Z, Lin H. Protein lipidation: occurrence, mechanisms, biological functions, and enabling technologies. Chem Rev (2018) 118(3):919–88. doi: 10.1021/acs.chemrev.6b00750
121. Du G, Healy LB, David L, Walker C, Fontana P, Dong Y, et al. ROS-dependent palmitoylation is an obligate licensing modification for GSDMD pore formation. bioRxiv (2023). doi: 10.1101/2023.03.07.531538
122. Balasubramanian A, Ghimire L, Hsu AY, Kambara H, Liu X, Hasegawa T, et al. Palmitoylation of gasdermin D directs its membrane translocation and pore formation in pyroptosis. bioRxiv (2023). doi: 10.1101/2023.02.21.529402
123. Joffre J, Hellman J. Oxidative stress and endothelial dysfunction in sepsis and acute inflammation. Antioxid Redox Signal (2021) 35(15):1291–307. doi: 10.1089/ars.2021.0027
124. Su M, Chen C, Li S, Li M, Zeng Z, Zhang Y, et al. Gasdermin D-dependent platelet pyroptosis exacerbates NET formation and inflammation in severe sepsis. Nat Cardiovasc Res (2022) 1(8):732–47. doi: 10.1038/s44161-022-00108-7
125. Li Y, Xin G, Li S, Dong Y, Zhu Y, Yu X, et al. PD-L1 regulates platelet activation and thrombosis via caspase-3/GSDME pathway. Front Pharmacol (2022) 13:921414. doi: 10.3389/fphar.2022.921414
126. Kayagaki N, Lee BL, Stowe IB, Kornfeld OS, O'Rourke K, Mirrashidi KM, et al. IRF2 transcriptionally induces GSDMD expression for pyroptosis. Sci Signal (2019) 12(582):eaax4917. doi: 10.1126/scisignal.aax4917
127. Zhang Y, Zhang Y, Yang A, Xia F. Downregulation of IRF2 Alleviates Sepsis-Related Acute Kidney Injury in vitro and in vivo. Drug Des Devel Ther (2021) 15:5123–32. doi: 10.2147/DDDT.S334518
128. Tan G, Huang C, Chen J, Chen B, Shi Y, Zhi F. An IRF1-dependent pathway of TNFalpha-induced shedding in intestinal epithelial cells. J Crohns Colitis. (2022) 16(1):133–42. doi: 10.1093/ecco-jcc/jjab134
129. Shi CS, Kehrl JH. Bcl-2 regulates pyroptosis and necroptosis by targeting BH3-like domains in GSDMD and MLKL. Cell Death Discovery (2019) 5:151. doi: 10.1038/s41420-019-0230-2
130. Chen R, Zeng L, Zhu S, Liu J, Zeh HJ, Kroemer G, et al. cAMP metabolism controls caspase-11 inflammasome activation and pyroptosis in sepsis. Sci Adv (2019) 5(5):eaav5562. doi: 10.1126/sciadv.aav5562
131. Bambouskova M, Potuckova L, Paulenda T, Kerndl M, Mogilenko DA, Lizotte K, et al. Itaconate confers tolerance to late NLRP3 inflammasome activation. Cell Rep (2021) 34(10):108756. doi: 10.1016/j.celrep.2021.108756
132. Diaz-Del-Olmo I, Worboys J, Martin-Sanchez F, Gritsenko A, Ambrose AR, Tannahill GM, et al. Internalization of the membrane attack complex triggers NLRP3 inflammasome activation and IL-1beta secretion in human macrophages. Front Immunol (2021) 12:720655. doi: 10.3389/fimmu.2021.720655
133. Liu H, Tang D, Zhou X, Yang X, Chen AF. PhospholipaseCgamma1/calcium-dependent membranous localization of Gsdmd-N drives endothelial pyroptosis, contributing to lipopolysaccharide-induced fatal outcome. Am J Physiol Heart Circ Physiol (2020) 319(6):H1482–H95. doi: 10.1152/ajpheart.00731.2019
134. Aki T, Funakoshi T, Unuma K, Uemura K. Inverse regulation of GSDMD and GSDME gene expression during LPS-induced pyroptosis in RAW264.7 macrophage cells. Apoptosis (2022) 27(1-2):14–21. doi: 10.1007/s10495-022-01708-1
135. Li T, Sun H, Li Y, Su L, Jiang J, Liu Y, et al. Downregulation of macrophage migration inhibitory factor attenuates NLRP3 inflammasome mediated pyroptosis in sepsis-induced AKI. Cell Death Discovery (2022) 8(1):61. doi: 10.1038/s41420-021-00803-7
136. Gou X, Xu W, Liu Y, Peng Y, Xu W, Yin Y, et al. IL-6 prevents lung macrophage death and lung inflammation injury by inhibiting GSDME- and GSDMD-mediated pyroptosis during pneumococcal pneumosepsis. Microbiol Spectr (2022) 10(2):e0204921. doi: 10.1128/spectrum.02049-21
137. Lai X, Ding H, Yu R, Bai H, Liu Y, Cao J. CXCL14 protects against polymicrobial sepsis by enhancing antibacterial functions of macrophages. Am J Respir Cell Mol Biol (2022) 67(5):589–601. doi: 10.1165/rcmb.2022-0249OC
138. Liu Z, Gan L, Xu Y, Luo D, Ren Q, Wu S, et al. Melatonin alleviates inflammasome-induced pyroptosis through inhibiting NF-kappaB/GSDMD signal in mice adipose tissue. J Pineal Res (2017) 63(1):e1241. doi: 10.1111/jpi.12414
139. Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature (2021) 593(7860):607–11. doi: 10.1038/s41586-021-03478-3
140. Wang D, Zheng J, Hu Q, Zhao C, Chen Q, Shi P, et al. Magnesium protects against sepsis by blocking gasdermin D N-terminal-induced pyroptosis. Cell Death Differ (2020) 27(2):466–81. doi: 10.1038/s41418-019-0366-x
141. Volchuk A, Ye A, Chi L, Steinberg BE, Goldenberg NM. Indirect regulation of HMGB1 release by gasdermin D. Nat Commun (2020) 11(1):4561. doi: 10.1038/s41467-020-18443-3
142. Barnett KC, Ting JP. Mitochondrial GSDMD pores DAMPen pyroptosis. Immunity (2020) 52(3):424–6. doi: 10.1016/j.immuni.2020.02.012
143. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity (2018) 48(1):35–44.e6. doi: 10.1016/j.immuni.2017.11.013
144. Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, Broz P. The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur J Immunol (2018) 48(4):584–92. doi: 10.1002/eji.201747404
145. Evavold CL, Kagan JC. Defying death: the (W)hole truth about the fate of GSDMD pores. Immunity (2019) 50(1):15–7. doi: 10.1016/j.immuni.2018.12.032
146. Ruhl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science (2018) 362(6417):956–60. doi: 10.1126/science.aar7607
147. Zhivaki D, Kagan JC. Innate immune detection of lipid oxidation as a threat assessment strategy. Nat Rev Immunol (2022) 22(5):322–30. doi: 10.1038/s41577-021-00618-8
148. Jimenez AJ, Maiuri P, Lafaurie-Janvore J, Divoux S, Piel M, Perez F. ESCRT machinery is required for plasma membrane repair. Science (2014) 343(6174):1247136. doi: 10.1126/science.1247136
149. Chang MK, Binder CJ, Miller YI, Subbanagounder G, Silverman GJ, Berliner JA, et al. Apoptotic cells with oxidation-specific epitopes are immunogenic and proinflammatory. J Exp Med (2004) 200(11):1359–70. doi: 10.1084/jem.20031763
150. Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol (2015) 16(4):343–53. doi: 10.1038/ni.3123
151. Zanoni I, Tan Y, Di Gioia M, Springstead JR, Kagan JC. By capturing inflammatory lipids released from dying cells, the receptor CD14 induces inflammasome-dependent phagocyte hyperactivation. Immunity (2017) 47(4):697–709.e3. doi: 10.1016/j.immuni.2017.09.010
152. Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science (2016) 352(6290):1232–6. doi: 10.1126/science.aaf3036
153. Xie WJ, Xia S, Warshel A, Wu H. Electrostatic influence on IL-1 transport through the GSDMD pore. Proc Natl Acad Sci U S A. (2022) 119(6):e2120287119. doi: 10.1073/pnas.2120287119
154. Denning NL, Aziz M, Gurien SD, Wang P. DAMPs and NETs in sepsis. Front Immunol (2019) 10:2536. doi: 10.3389/fimmu.2019.02536
155. Tan C, Reilly B, Jha A, Murao A, Lee Y, Brenner M, et al. Active Release of eCIRP via Gasdermin D Channels to Induce Inflammation in Sepsis. J Immunol (2022) 208(9):2184–95. doi: 10.4049/jimmunol.2101004
156. Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, et al. Inflammasome activation triggers blood clotting and host death through pyroptosis. Immunity (2019) 50(6):1401–11.e4. doi: 10.1016/j.immuni.2019.04.003
157. Pawlinski R, Pedersen B, Schabbauer G, Tencati M, Holscher T, Boisvert W, et al. Role of tissue factor and protease-activated receptors in a mouse model of endotoxemia. Blood (2004) 103(4):1342–7. doi: 10.1182/blood-2003-09-3051
158. Tonial CT, Garcia PCR, Schweitzer LC, Costa CAD, Bruno F, Fiori HH, et al. Cardiac dysfunction and ferritin as early markers of severity in pediatric sepsis. J Pediatr (Rio J). (2017) 93(3):301–7. doi: 10.1016/j.jped.2016.08.006
159. Feltham R, Vince JE. Ion man: GSDMD punches pores to knock out cGAS. Immunity (2018) 49(3):379–81. doi: 10.1016/j.immuni.2018.08.026
160. Swanson KV, Junkins RD, Kurkjian CJ, Holley-Guthrie E, Pendse AA, El Morabiti R, et al. A noncanonical function of cGAMP in inflammasome priming and activation. J Exp Med (2017) 214(12):3611–26. doi: 10.1084/jem.20171749
161. Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, Menoret A, et al. Gasdermin D restrains type I interferon response to cytosolic DNA by disrupting ionic homeostasis. Immunity (2018) 49(3):413–26.e5. doi: 10.1016/j.immuni.2018.07.006
162. Mahidhara R, Billiar TR. Apoptosis in sepsis. Crit Care Med (2000) 28(4 Suppl):N105–13. doi: 10.1097/00003246-200004001-00013
163. Zheng X, Chen W, Gong F, Chen Y, Chen E. The role and mechanism of pyroptosis and potential therapeutic targets in sepsis: A review. Front Immunol (2021) 12:711939. doi: 10.3389/fimmu.2021.711939
164. Liu W, Gan Y, Ding Y, Zhang L, Jiao X, Liu L, et al. Autophagy promotes GSDME-mediated pyroptosis via intrinsic and extrinsic apoptotic pathways in cobalt chloride-induced hypoxia reoxygenation-acute kidney injury. Ecotoxicol Environ Saf. (2022) 242:113881. doi: 10.1016/j.ecoenv.2022.113881
165. Yipp BG, Kubes P. NETosis: how vital is it? Blood (2013) 122(16):2784–94. doi: 10.1182/blood-2013-04-457671
166. Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol (2017) 17(3):151–64. doi: 10.1038/nri.2016.147
167. Silva CMS, Wanderley CWS, Veras FP, Sonego F, Nascimento DC, Goncalves AV, et al. Gasdermin D inhibition prevents multiple organ dysfunction during sepsis by blocking NET formation. Blood (2021) 138(25):2702–13. doi: 10.1182/blood.2021011525
168. Liu F, Ghimire L, Balasubramanian A, Hsu AY, Zhang Z, Yu H, et al. Neutrophil-specific depletion of gasdermin D does not protect against murine sepsis. Blood (2023) 141(5):550–4. doi: 10.1182/blood.2022016931
169. Weindel CG, Martinez EL, Zhao X, Mabry CJ, Bell SL, Vail KJ, et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell (2022) 185(17):3214–31.e23. doi: 10.1016/j.cell.2022.06.038
170. Zhang YY, Ning BT. Signaling pathways and intervention therapies in sepsis. Signal Transduct Target Ther (2021) 6(1):407. doi: 10.1038/s41392-021-00471-0
171. Perner A, Gordon AC, De Backer D, Dimopoulos G, Russell JA, Lipman J, et al. Sepsis: frontiers in diagnosis, resuscitation and antibiotic therapy. Intensive Care Med (2016) 42(12):1958–69. doi: 10.1007/s00134-016-4577-z
172. Grimaldi D, Vincent JL. Clinical trial research in focus: rethinking trials in sepsis. Lancet Respir Med (2017) 5(8):610–1. doi: 10.1016/S2213-2600(17)30268-0
173. Vincent JL, Sakr Y. Clinical trial design for unmet clinical needs: a spotlight on sepsis. Expert Rev Clin Pharmacol (2019) 12(9):893–900. doi: 10.1080/17512433.2019.1643235
174. Tidswell M, Tillis W, Larosa SP, Lynn M, Wittek AE, Kao R, et al. Phase 2 trial of eritoran tetrasodium (E5564), a toll-like receptor 4 antagonist, in patients with severe sepsis. Crit Care Med (2010) 38(1):72–83. doi: 10.1097/CCM.0b013e3181b07b78
175. Shakoory B, Carcillo JA, Chatham WW, Amdur RL, Zhao H, Dinarello CA, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III trial. Crit Care Med (2016) 44(2):275–81. doi: 10.1097/CCM.0000000000001402
176. Kang JY, Xu MM, Sun Y, Ding ZX, Wei YY, Zhang DW, et al. Melatonin attenuates LPS-induced pyroptosis in acute lung injury by inhibiting NLRP3-GSDMD pathway via activating Nrf2/HO-1 signaling axis. Int Immunopharmacol (2022) 109:108782. doi: 10.1016/j.intimp.2022.108782
177. Xiong X, Lu L, Wang Z, Ma J, Shao Y, Liu Y, et al. Irisin attenuates sepsis-induced cardiac dysfunction by attenuating inflammation-induced pyroptosis through a mitochondrial ubiquitin ligase-dependent mechanism. BioMed Pharmacother (2022) 152:113199. doi: 10.1016/j.biopha.2022.113199
178. Wei A, Liu J, Li D, Lu Y, Yang L, Zhuo Y, et al. Syringaresinol attenuates sepsis-induced cardiac dysfunction by inhibiting inflammation and pyroptosis in mice. Eur J Pharmacol (2021) 913:174644. doi: 10.1016/j.ejphar.2021.174644
179. Kumar V. Pulmonary innate immune response determines the outcome of inflammation during pneumonia and sepsis-associated acute lung injury. Front Immunol (2020) 11:1722. doi: 10.3389/fimmu.2020.01722
180. Liu Y, Zhou J, Luo Y, Li J, Shang L, Zhou F, et al. Honokiol alleviates LPS-induced acute lung injury by inhibiting NLRP3 inflammasome-mediated pyroptosis via Nrf2 activation in vitro and in vivo. Chin Med (2021) 16(1):127. doi: 10.1186/s13020-021-00541-z
181. Jiang R, Xu J, Zhang Y, Zhu X, Liu J, Tan Y. Ligustrazine alleviate acute lung injury through suppressing pyroptosis and apoptosis of alveolar macrophages. Front Pharmacol (2021) 12:680512. doi: 10.3389/fphar.2021.680512
182. Zhang Y, Zhang H, Li S, Huang K, Jiang L, Wang Y. Metformin alleviates LPS-induced acute lung injury by regulating the SIRT1/NF-kappaB/NLRP3 pathway and inhibiting endothelial cell pyroptosis. Front Pharmacol (2022) 13:801337. doi: 10.3389/fphar.2022.801337
183. He G, Chen K, Wang H, Li X, Li W, Liu L, et al. Fudosteine attenuates acute lung injury in septic mice by inhibiting pyroptosis via the TXNIP/NLRP3/GSDMD pathway. Eur J Pharmacol (2022) 926:175047. doi: 10.1016/j.ejphar.2022.175047
184. Feng M, Wei S, Zhang S, Yang Y. Anti-inflammation and anti-pyroptosis activities of mangiferin via suppressing NF-kappaB/NLRP3/GSDMD signaling cascades. Int J Mol Sci (2022) 23(17):10124. doi: 10.3390/ijms231710124
185. Ye J, Zeng B, Zhong M, Li H, Xu L, Shu J, et al. Scutellarin inhibits caspase-11 activation and pyroptosis in macrophages via regulating PKA signaling. Acta Pharm Sin B (2021) 11(1):112–26. doi: 10.1016/j.apsb.2020.07.014
186. Shang T, Zhang ZS, Wang XT, Chang J, Zhou ME, Lyu M, et al. Xuebijing injection inhibited neutrophil extracellular traps to reverse lung injury in sepsis mice via reducing Gasdermin D. Front Pharmacol (2022) 13:1054176. doi: 10.3389/fphar.2022.1054176
187. He C, Zhao Y, Jiang X, Liang X, Yin L, Yin Z, et al. Protective effect of Ketone musk on LPS/ATP-induced pyroptosis in J774A.1 cells through suppressing NLRP3/GSDMD pathway. Int Immunopharmacol (2019) 71:328–35. doi: 10.1016/j.intimp.2019.03.054
188. Liu Y, Shang L, Zhou J, Pan G, Zhou F, Yang S. Emodin Attenuates LPS-Induced Acute Lung Injury by Inhibiting NLRP3 Inflammasome-Dependent Pyroptosis Signaling Pathway In vitro and In vivo. Inflammation (2022) 45(2):753–67. doi: 10.1007/s10753-021-01581-1
189. Wang B, Liu Y, Jiang R, Liu Z, Gao H, Chen F, et al. Emodin relieves the inflammation and pyroptosis of lipopolysaccharide-treated 1321N1 cells by regulating methyltransferase-like 3 -mediated NLR family pyrin domain containing 3 expression. Bioengineered (2022) 13(3):6740–9. doi: 10.1080/21655979.2022.2045836
190. Xiao L, Qi L, Zhang G, Liu H, Gu Y, Zhang L, et al. Polygonatum sibiricum Polysaccharides Attenuate Lipopoly-Saccharide-Induced Septic Liver Injury by Suppression of Pyroptosis via NLRP3/GSDMD Signals. Molecules (2022) 27(18):5999. doi: 10.3390/molecules27185999
191. Zhao YY, Wu DM, He M, Zhang F, Zhang T, Liu T, et al. Samotolisib attenuates acute liver injury through inhibiting caspase-11-mediated pyroptosis via regulating E3 ubiquitin ligase nedd4. Front Pharmacol (2021) 12:726198. doi: 10.3389/fphar.2021.726198
192. Xiao T, Cui Y, Ji H, Yan L, Pei D, Qu S. Baicalein attenuates acute liver injury by blocking NLRP3 inflammasome. Biochem Biophys Res Commun (2021) 534:212–8. doi: 10.1016/j.bbrc.2020.11.109
193. Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol (2020) 21(7):736–45. doi: 10.1038/s41590-020-0669-6
194. Chen Y, Luo R, Li J, Wang S, Ding J, Zhao K, et al. Intrinsic radical species scavenging activities of tea polyphenols nanoparticles block pyroptosis in endotoxin-induced sepsis. ACS Nano (2022) 16(2):2429–41. doi: 10.1021/acsnano.1c08913
195. Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol (2018) 3(26):eaat2738. doi: 10.1126/sciimmunol.aat2738
Keywords: sepsis, gasdermin, regulation, cell death, therapy
Citation: Wang W and He Z (2023) Gasdermins in sepsis. Front. Immunol. 14:1203687. doi: 10.3389/fimmu.2023.1203687
Received: 11 April 2023; Accepted: 12 October 2023;
Published: 03 November 2023.
Edited by:
Michal Holub, Charles University, CzechiaReviewed by:
Jianbin Ruan, UCONN Health, United StatesPier Maria Fornasari, Regen Health Solutions, Italy
Copyright © 2023 Wang and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhihui He, aHpoNzAzQGNzdS5lZHUuY24=