 Luis Del Pozo-Yauner1*
Luis Del Pozo-Yauner1* Guillermo A. Herrera1
Guillermo A. Herrera1 Julio I. Perez Carreon2
Julio I. Perez Carreon2 Elba A. Turbat-Herrera1,3Francisco J. Rodriguez-Alvarez2Robin A. Ruiz Zamora2
Elba A. Turbat-Herrera1,3Francisco J. Rodriguez-Alvarez2Robin A. Ruiz Zamora2- 1Department of Pathology, University of South Alabama-College of Medicine, Mobile, AL, United States
- 2Instituto Nacional de Medicina Genomica (INMEGEN), Ciudad de México, Mexico
- 3Mitchell Cancer Institute, University of South Alabama-College of Medicine, Mobile, AL, United States
The adaptive immune system of jawed vertebrates generates a highly diverse repertoire of antibodies to meet the antigenic challenges of a constantly evolving biological ecosystem. Most of the diversity is generated by two mechanisms: V(D)J gene recombination and somatic hypermutation (SHM). SHM introduces changes in the variable domain of antibodies, mostly in the regions that form the paratope, yielding antibodies with higher antigen binding affinity. However, antigen recognition is only possible if the antibody folds into a stable functional conformation. Therefore, a key force determining the survival of B cell clones undergoing somatic hypermutation is the ability of the mutated heavy and light chains to efficiently fold and assemble into a functional antibody. The antibody is the structural context where the selection of the somatic mutations occurs, and where both the heavy and light chains benefit from protective mechanisms that counteract the potentially deleterious impact of the changes. However, in patients with monoclonal gammopathies, the proliferating plasma cell clone may overproduce the light chain, which is then secreted into the bloodstream. This places the light chain out of the protective context provided by the quaternary structure of the antibody, increasing the risk of misfolding and aggregation due to destabilizing somatic mutations. Light chain-derived (AL) amyloidosis, light chain deposition disease (LCDD), Fanconi syndrome, and myeloma (cast) nephropathy are a diverse group of diseases derived from the pathologic aggregation of light chains, in which somatic mutations are recognized to play a role. In this review, we address the mechanisms by which somatic mutations promote the misfolding and pathological aggregation of the light chains, with an emphasis on AL amyloidosis. We also analyze the contribution of the variable domain (VL) gene segments and somatic mutations on light chain cytotoxicity, organ tropism, and structure of the AL fibrils. Finally, we analyze the most recent advances in the development of computational algorithms to predict the role of somatic mutations in the cardiotoxicity of amyloidogenic light chains and discuss the challenges and perspectives that this approach faces.
1 Introduction
One factor that influenced the evolution of complex multicellular organisms such as humans is the dual nature of the interaction with bacteria, fungi, viruses, and other microorganisms that coevolved with them, some of which colonized the multicellular organisms to form what is called microbiota (1, 2). While both the multicellular host and its microbiota benefited from the reciprocal interaction (3, 4), the former must avoid succumbing due to the uncontrolled invasion of the later (1, 2). Hence, different strategies and molecular mechanisms evolved in the multicellular organisms to detect and kill pathogenic microbes and parasites (host defend), while maintaining microbiota homeostasis, that is, a balanced interaction with those non-pathogenic microbes that are beneficial (mutualism) (5, 6).
Regardless of their complexity, all mechanisms evolved to protect Metazoans against invading microbes operate on the same biological principle: before any response can be elicited, recognition of an external signal must be achieved (7). The recognition of the external signal, essentially a non-self-molecule, is accomplished by a cell receptor, triggering a cascade of multiple converging events whose ultimate goal is the removal of the “signal” that triggered the response. Plants, which lack a circulatory system and mobile immune cells, evolved an innate immune system that is broadly divided into pathogen-associated molecular pattern- or PAMP-triggered immunity (PTI), the first layer of the immune response, and effector-triggered immunity (ETI). PTI is activated upon perception of molecules with conserved motifs derived from pathogens by surface membrane-anchored pattern recognition receptors (PRRs) (8, 9). Invertebrates also relay on innate defense mechanisms, which use a large diversity of molecules with broad-spectrum bactericidal and fungicidal properties to defend themselves from microbes (7). The evolutionary emergence of vertebrates, about 500 million years ago, was accompanied by a new form of immune protection, the adaptive immune system (10, 11). In vertebrates, the sophisticated but relatively nonspecific innate immunity cooperates with the highly refined adaptive immune system to elicit a highly effective protective response characterized by antigen specificity and immune memory (10). The adaptive immune system uses highly diversified repertoires of antigen receptors located in the membrane of immunocompetent cells, which, remarkably, are structurally different in jawed and jawless vertebrates. In jawless vertebrates, the adaptive immune receptors are termed variable lymphocyte receptors (VLRs) and are constructed from leucine-rich repeat modules. In contrast, the adaptive immune system in jawed vertebrates uses structurally similar antigen receptors, named immunoglobulins and T cell receptors (TCRs), clonally expressed by B and T lymphocytes, respectively (10, 12). Such difference between the adaptive immune receptors in jawless and jaw vertebrates is a consequence of having evolved independently in both groups (7, 10, 12). Immunoglobulins, also known as antibodies, and TCR are generated through combinatorial recombination of gene segments, a process known as V(D)J gene recombination, which theoretically can generate an almost infinite number of antigen-binding specificities. B and T lymphocytes displaying these membrane antigen receptors travel through the circulatory system to detect pathogens or mutated cells. The recognition of a foreign molecule by B and T lymphocytes leads the clonal expansion, which, in the case of B-cell lymphocytes, is accompanied by somatic hypermutation (SHM). This molecular event, unique to B cells, introduces mutations in the variable regions of antibodies that increase the binding affinity for the antigen, greatly expanding the antibody repertoire (13). The end of the road for B lymphocytes activated by antigens is differentiation into plasma cells, which do not display membrane-bound antibodies, but actively secrete them in a soluble form. Antibodies secreted by plasma cells act as mediators of the humoral immune response, promoting different mechanisms of inactivation and/or elimination of foreign molecules and invading pathogenic microbes (10). SHM plays a key role in the antibody response, as it transforms germline-encoded antibodies characterized by low affinity and propensity to cross-react with other antigens, into a more specific antibody with a higher binding affinity (6, 13). While this is key for mounting an effective antibody response, SHM may also be a contributor to the pathogenesis of several human diseases, such as when it generates antibodies that recognize self-antigens (14), or when it promotes misfolding and aggregation of the immunoglobulin light chain (LC) (15, 16). This review focuses on the role of the mechanism for diversification of the human repertoire of antibodies in the molecular pathogenesis of the group of diseases related to misfolding and deposition of monoclonal light chains with an emphasis on light chain-derived (AL) amyloidosis.
2 Structural and genetic basis of antibody function
2.1 Structural bases of antibody function
Antibodies are glycoproteins with a basic structural unit formed by the association of two heavy (HC) and two LCs (Figure 1A). The human genome encodes for five major HC isotypes; γ, δ, α, ε, and µ, which determine the antibody classes IgG, IgD, IgA, IgE, and IgM, respectively. HCs γ, α and δ are ∼450 amino acids long and fold into four domains, the N-terminal variable domain (VH) and three constant domains, designated CH1 to CH3, arranged in tandem. HCs µ and ε are ∼100 residues longer and fold into one additional constant domain (CH4) (Figures 1A–D). Humans produce two different types of LCs, kappa (κ) and lambda (Λ), which are ∼215-225 amino acids long and fold into two domains, the N-terminal variable (VL) and the C-terminal constant (CL) domains (Figure 1E) (17).
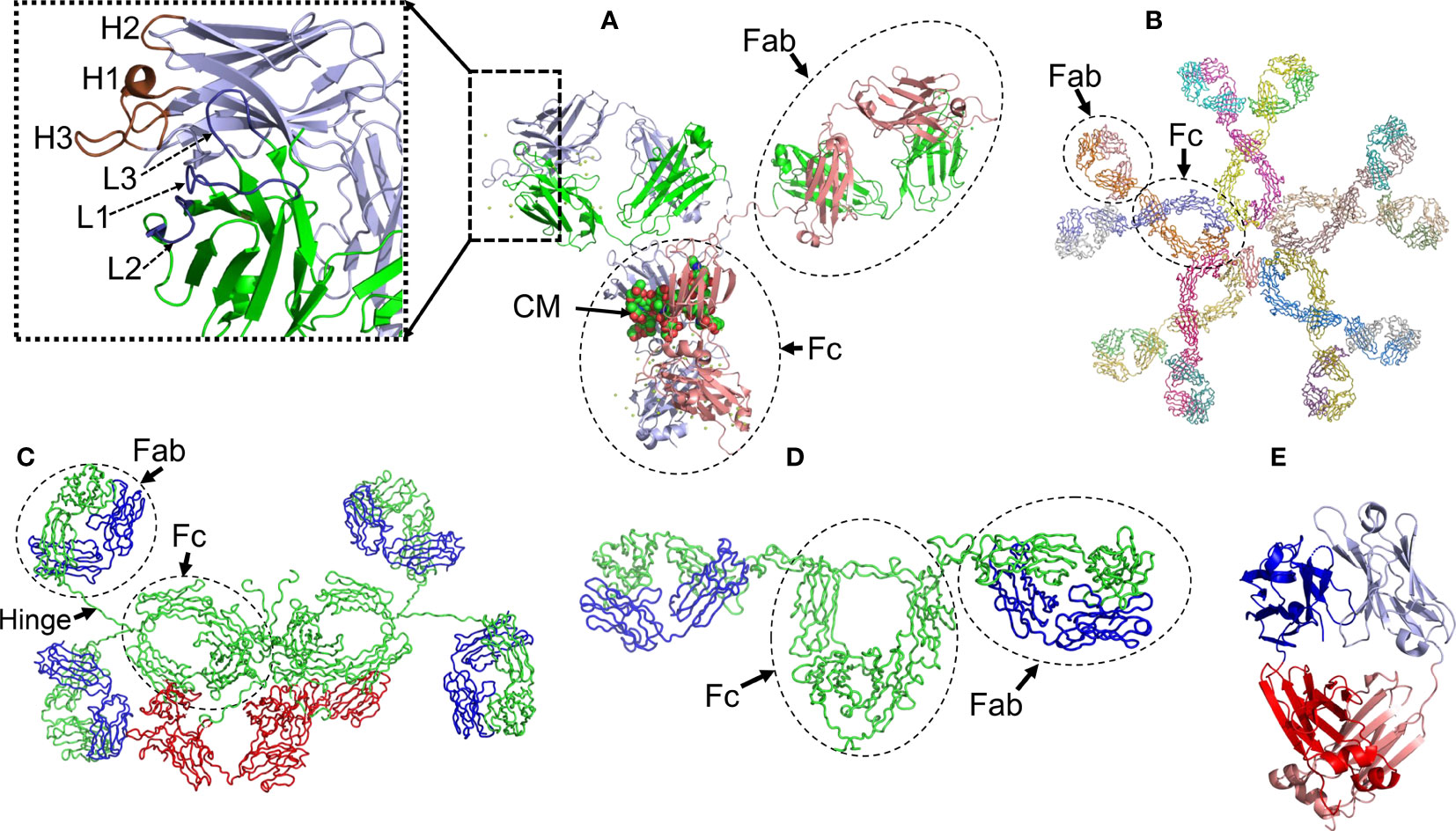
Figure 1 Structural characteristics of human immunoglobulins. (A) Crystal structure of the intact human IgG B12 with broad and potent activity against primary HIV-1 isolates (Method: X-ray diffraction - PDB 1HZH). (B) Solution structure of human Immunoglobulin M (Method: Solution X-ray scattering – PDB 2RCJ) (C) Solution structure of human secretory IgA1 (Method: Solution X-ray scattering – PDB 3CHN). (D) Semi-extended solution structure of human myeloma immunoglobulin D (Method: solution X-ray scattering – PDB 1ZVO). (E) Crystal structure of amyloidogenic and cardiotoxic Bence-Jones Λ2 LC dimer H9 (Method: X-ray crystallography – PDB 5M6A). The insert in A shows the spatial conformation of the light chain (L1 to L3, colored in deep blue) and heavy chain (H1 to H3, colored in brown) CDRs. CM stands for carbohydrate moiety attached to the CH2 domain. The Fab (fragment antigen-binding) and Fc (fragment crystallizing) regions of each antibody are indicated. In C, the secretory component of the dimeric IgA is shown in red. The long hinge connecting the Fab and Fc regions in IgA1 antibodies is also indicated. Due to limitations of the structural method applied, the spatial location of the J chain was not determined. In C and D, the light and heavy chains are shown in blue and green, respectively. In E, the variable domains of the light chain dimer are shown in blue and light blue, while the constant domains are shown in red and salmon. Note that the Bence Jones dimer is stabilized by a LC-LC interface that mimics the LC-HC interface of a functional antibody. All structures were prepared with PyMOL (PyMOL Molecular Graphics System, Version 2.5.2, Schrödinger, LLC).
IgG and IgA antibodies are subdivided into four (IgG1, IgG2, IgG3, and IgG4) and two (IgA1 and IgA2) subclasses, respectively, which reflects the number of γ (γ1 to γ4) and α (α1 and α2) HC genes (isotypes) in the human genome (18, 19). Although the IgG subclasses share ∼90% identity in amino acid sequence, they differ in several biological properties, such as immune complex formation, complement activation, ability to trigger effector cells response, half-life, and placental transport (18). IgA subclasses also share high sequence identity, differing from each other mainly in the structure of their hinge region, located between Val222 and Cys241, and in the number of glycosylation sites. The hinge region of human IgA1 antibodies spans 18 amino acids and contains up to five O-linked glycans on serine and threonine residues. In contrast, IgA2 presents a 5 amino acid hinge and lacks O-linked glycans (20). Structural differences between IgA subclasses translate into distinct effector functions in immune cells, as well as susceptibility to inactivation by cleavage at hinge regions by proteases produced by some pathogenic bacteria (21). Moreover, IgA subclasses are not expressed equal in body fluids, since IgA1 makes up around 80-90% of total serum IgA, but in mucosal surfaces, both isotypes are more evenly distributed (19, 22).
Antibodies of the IgG, IgD, and IgE classes are produced only as monomers of the basic unit, with molecular weight (MW) of ∼150kDa for both IgG and IgD, and 180kDa for IgE. In contrast, IgA antibodies are produced in two formats, as monomers with MW of ∼150kDa, and dimers of ∼385kDa. Monomeric IgA is secreted into the bloodstream, while the dimeric form, known as secretory IgA (SIgA), is secreted to the mucous membranes of the respiratory, genitourinary, and digestive tracks, where it contributes to mucosal immunity and preserving the microbiota homeostasis (6, 22). The dimeric complex of SIgA contains two additional peptides, termed the J-chain and secretory component (23) (Figure 1C). IgM antibodies, on the other hand, are secreted mostly in a pentameric configuration with a MW of 900 kDa (24) (Figure 1). The quaternary structure of antibodies is stabilized by interchain disulfide bonds that link each HC-LC pair and one HC to another, as well as by a myriad of weak non-covalent interactions between HC and LC residues located at the VH-VL and CH1-CL interfaces (25) (Figure 1). Contacts at the LC-HC interface contribute critically to the proper folding of the antibodies in the B lymphocyte. This is particularly relevant for the CH1 domain, which folds via a template-assisted mechanism that requires interactions with key residues in the dimerization interface with a fully folded CL domain (26). The chaperone role of the LC in HC folding is considered part of the control mechanisms in B cells to ensure correct folding and transport of antibodies (26). As will be discussed in more detail later in this review, some hematological disorders are characterized by the presence of a monoclonal LC circulating in the blood in a free state. In this circumstance, the monoclonal LC tends to form dimers that can be detected in serum and urine (Figure 1E).
Both the variable and constant domains of antibodies exhibit the β-sandwich fold, a distinctive trait of the immunoglobulins. The immunoglobulin fold consists of two antiparallel β sheets with a Greek key topology, composed of 4-5 strands, which associate face‐to‐face to form a two-layer sandwich (Figure 1). The core of the sandwich mostly comprises the side chains of non-polar residues, structured around a highly conserved tryptophan residue that tightly packs against the intradomain disulfide bond. The strands of the β sandwich are connected by loops of variable length. In both the VH and VL, three of the connecting loops display high sequence variability and are called Complementarity Determining Region 1 to 3 or CDR1 to CDR3. The regions between the CDRs are called Framework Regions (FRs). The CDRs are spatially oriented in such a way that they form the antibody surface, called the paratope, through which antigen recognition takes place (Figure 1A). The conformational properties of the CDRs, and therefore the nature of their interaction with the antigen, are mainly determined by their sequence but are also influenced by short- and long-distant interactions between the CDRs and other segments of the β-sandwich. This means that mutations in the CDRs but also in the FRs may modulate the affinity and specificity of the antigen-antibody interaction. The antigen-antibody interaction relies on spatial complementarity between paratope and epitope, being stabilized by several types of non-covalent forces: hydrophobic interactions, H-bonds, van der Waals forces, and saline bridge (27, 28). Interfacial water molecules may also be involved in H-bonds, bringing residues at the antigen-antibody interface, and therefore contributing to the overall stability of the antigen-antibody complexes (29, 30).
2.2 Genetics of human antibodies
Functional HC and LC genes are assembled by the combinatorial recombination of gene segments that are located separately in the human genome. The HC gene results from the recombination of four different gene segments, the variable (IGHV), joining (IGHJ), diversity (IGHD), and constant (IGHC) gene segments, of which the first three encode the protein VH domain. On the other hand, the LC gene is assembled by recombination of three gene segments, the variable (IGVL), joining (IGJL), and constant (IGCL) gene segments, with the first two encoding the protein VL domain (Figure 2). The immunoglobulin HC locus (IGH) is located in the long (q) arm of chromosome 14, regions 32.33 (14q32.33), while the immunoglobulin κ (IGK) and Λ (IGL) loci are located in the short (p) arm of chromosome 2 (2p11.2) and long (q) arm of chromosome 22 (22q11.2), respectively. The number of functional gene segments comprised at the IGH, IGK, and IGL loci varies across haplotypes.
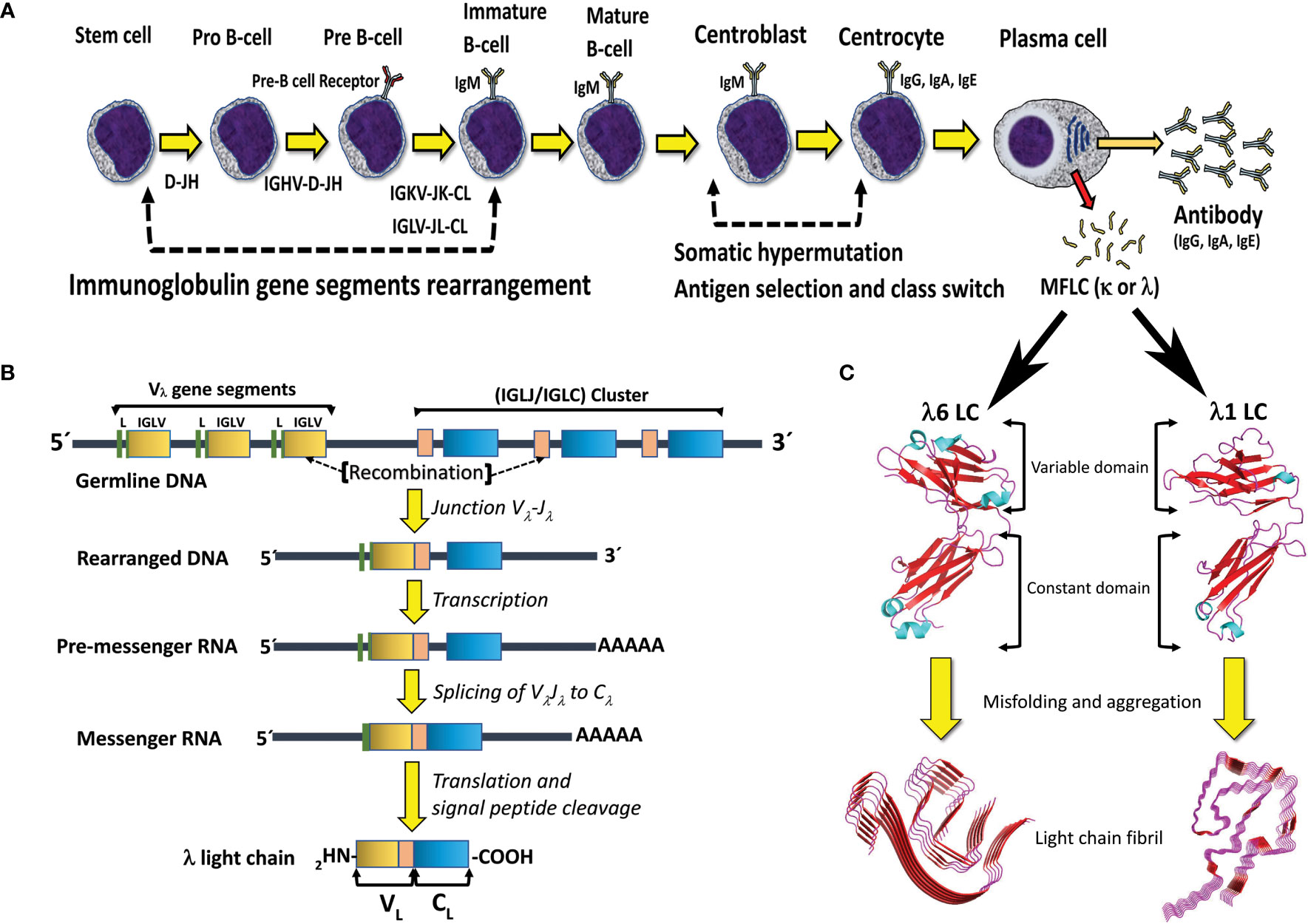
Figure 2 B cell differentiation pathway and mechanisms for generation of antibody diversity. (A) The human polyclonal antibody repertoire is generated by two molecular mechanisms: 1) V(D)J gene segments recombination, and 2) somatic hypermutation, which occurs at specific stages of B-cell differentiation. Plasma cells, the final stage of B cell differentiation, secrete large amounts of the specific antibody (IgG, IgA, or IgE). Under certain circumstances, a clone of plasma cells can overproduce the antibody LC and secret it in a free state, which entails the risk of aggregation and disease. MFLC stands for monoclonal free light chain. (B) Schematic representation of the IGLV-IGLJ-IGLC gene segment recombination in the Λ LC locus. Note that the VL domain of the LC is encoded by the IGLV and IGLJ gene segments, while the CL domain is encoded by the IGLC gene. (C) Structural characteristics of the immunoglobulin LCs and AL amyloid fibrils. The LC structures correspond to the full-length (FL) Λ6 LC of the anti-Hepcidin Fab (PDB 3H0T) and the Λ1 monoclonal free LC (Bence Jones protein) LOCW, both determined by x-ray diffraction. The variable (VL) and constant (CL) domains are indicated. The regions in β-strand conformation are colored red. The AL fibril structures correspond to the ex-vivo Λ6 (PDB 6HUD) and Λ1 (PDB 6IC3) AL fibrils obtained from the cardiac deposits in patients with AL amyloidosis determined by cryo-EM. The β core of the AL fibrils is composed only of segments of the VL domain. The FL LCs and AL fibrils shown in the figure are not related to each other. They were included in the same figure only for comparative purposes. Structures shown in C were prepared with PyMOL (PyMOL Molecular Graphics System, Version 2.5.2, Schrödinger, LLC).
The IGH locus spans ∼1.25 Mb and comprises 38-46 IGHV, 23 IGHJ, 6 IGHD, and 9 IGHC gene segments. The IGK locus spans ∼1.7 Mb and comprises 34-38 IGKV, 5 IGKJ, and only one IGKC gene segment. The IGL locus spans ∼1.1 Mb and comprises 29-33 IGLV, 4-5 IGLJ, and 4-5 IGLC gene segments (31) (http://www.imgt.org/IMGTrepertoire/; data accessed on March 8th, 2023). In the IGK locus, the IGKV gene segments are grouped into two clusters, termed distal (centromeric) and proximal (telomeric) clusters, separated by 800kb. In the telomeric direction, the proximal cluster is followed by the IGKJ cluster and them the only IGKC gene segment (31). The structure of the IGL locus is a little different since the IGLV gene segments are grouped into three clusters, termed C, B, and A (from centromeric to telomeric direction), which are followed by the IGLJ-IGLC cluster that comprises a polymorphic number of highly similar IGLJ-IGLC pairs (31) (Figure 2). Based on the degree of sequence similarity, the IGKV and IGLV gene segments have been subclassified into seven κ (κ1 to κ7) and ten Λ (Λ1 to Λ10) VL subgroups, respectively. These subgroups consist of phylogenetically closely related VL gene segments that were generated in relatively recent events of gene duplication and divergence (32). The number of VL gene segments per subgroup varies from only one, as for subgroups Λ6 and κ4, to twenty in the case of subgroup κ1 (31, 33).
3 Mechanism for generation of antibody diversity
The adaptive immune system of jawed vertebrates evolved to generate a highly diverse repertoire of antibody specificities, which is achieved by two main mechanisms: 1) V(D)J genes combinatorial recombination to generate functional HC and LC genes, and the subsequent combination of the repertoire of HCs and LCs in the repertoire of B lymphocytes, and 2) SHM. V(D)J gene recombination is an antigen-independent mechanism of antibody diversification. In contrast, SHM is an antigen-dependent mechanism since it occurs after the activation of the mature B-cell by the antigen and its subsequent migration to the germinal centers of secondary lymphoid organs. SHM primarily targets the CDRs of antibodies, leading to affinity maturation. The antibody repertoire is further expanded by the addition of non-templated (N) nucleotides into the junctions between the V-D and D-J in HC gene, and although less extensive, also in the V-J junction in LC gene (34), a reaction catalyzed by the terminal deoxynucleotidyl transferase (TdT) (35). Other secondary mechanisms of antibody diversification are the V(DD)J recombination, SHM-associated insertions and deletions, and affinity maturation and antigen contact by non-CDR regions of the antibody (36).
3.1 V(D)J gene recombination
The V(D)J genes recombination begins in pro-B lymphocytes and progresses in discrete steps linked to specific stages of the B cell ontogeny pathway (Figure 2). A key player in this process is a lymphoid-specific enzyme called Recombination-Activating Gene (RAG) recombinase, a Y-shaped heterotetrameric complex made by two distinct subunits RAG1 and RAG2 (37–39). RAG1 is a 1040 amino acid multidomain protein highly homologous to many eukaryotic transposases, or retroviral integrases. It determines most of the catalytic and DNA binding capacity of the RAG complex. On the other hand, RAG2 is 527 amino acids in length and has no homology to any known viral transposase or integrase. Its role in V(D)J gene recombination is not well understood, but the evidence indicates that it is required for RAG complex DNA binding and catalysis (40, 41). Other components of the V(D)J genes recombination machinery are ubiquitous in cells since many of them form part of the non-homologous end-joining (NHEJ) DNA damage repair pathway (42, 43).
V(D)J gene recombination is directed by recombination signal sequences (RSSs), located immediately on the 5’ and/or 3’ flanks of the V, D, and J genes segments. An RSS comprises a highly conserved heptamer (consensus sequence 5’-CACAGTG-3’) and a conserved nonamer (consensus sequence 5’-ACAAAAACC-3’) separated by a poorly conserved spacer sequence of 12 or 23 nucleotides. This architecture determines two types of RSS, 12- and 23-RSS. The RSS located on the 3’ and 5’ flank of the IGHV and IGHJ gene segments, respectively, are of the 23-RSS type, while the IGHD gene segments bear a 12-RSS at both the 5’ and 3’ ends. The V(D)J gene recombination machinery strongly favors the recombination of gene segments with RSS of a different type, which is termed the 12-23 rule. This rule governs the V(D)J gene recombination fidelity, determining that an IGHV cannot directly recombine with an IGHJ gene segment, while an IGHD gene segment can recombine with either an IGHV or an IGHD. However, violation of the 12/23 rule occurs in a fraction of B lymphocytes under physiological conditions, which is believed to contribute to the diversification of the antibody repertoire (36).
RAG recombinase initiates V(D)J gene segments recombination by binding to either a 12-RSS or a 23-RSS and introducing a double-strand break (DSB) in the boundary between the RSS and the coding region of the target gene segment. The generation of a DSB is the signal that activates a group of proteins collectively called the DNA damage response. There is evidence that the High Mobility Group Box 1 (HMGB1) protein plays a critical role in this step, by binding to the RAG-DNA complex (44) and stabilizing a highly bent RSS conformation during catalysis (45, 46). Then, RAG-HMGB1 complex scans the locus and captures an RSS of a different type to form a paired complex, followed by nicking of the DNA chain (39). According to the 12/23 rule, if the RSS bound by the RAG-HMGB1 complex is a 12-RSS, then the RSS that will be captured in this step, and with which the recombination reaction will proceed, is a 23-RSS or vice versa. The DNA nicking generates a free 3’-hydroyl that attacks the opposite strand by transesterification, a reaction that requires Mg2+, and results in a covalently sealed hairpin at gene segment ends and the cleaved RSS ends. Factors in the NHEJ DNA repair pathway, such as the KU70/KU80 heterodimer, DNA PKcs, endonuclease Artemis, DNA polymerases µ and Λ, DNA ligases IV, X-ray cross-complementing Group 4 (XRCC4), TdT, and XRCC4-like factor (XLF) are then recruited to the coding ends. In the first step, the sealed hairpins are opened by the endonuclease activity of the Artemis : DNA-PKcs complex (47, 48), and then, the recombining gene segments are joined, forming imprecise coding joint that contains added nucleotide. The signal ends are also ligated to form a signal joint (42, 43). Most of the previously mentioned factors belong to the canonical NHEJ DNA repair pathway; however, DNA double-break repair can proceed by an alternative NHEJ pathway that involves a different set of factors (42, 43). V(D)J gene recombination results in gene segment joining and the deletion or inversion without deletion of the intervening DNA. Whether the V(D)J gene recombination occurs by a deletional, or inversional mechanism depends on the relative orientation of the two RSS driving the process (43).
Once V(D)J gene recombination has been successfully accomplished, the immature naïve B lymphocytes transcribe the HC and LC genes and express the encoded immunoglobulin as a membrane receptor (Figure 2). Initially, the HC variable region exon is transcribed in association with the immediately downstream Cμ exons, and in some cells, Cδ exons. This is accomplished by alternative splicing of the HC gene transcript, which comprises both µ and δ IGHC exons fused to IGHJ exon (43, 49).
V(D)J gene recombination is linked to specific differentiation stages of B and T cell ontogeny. Mutations that completely suppress the functions of RAG1/2 proteins cause severe combined immune deficiency, due to the developmental arrest at the progenitor stages of both T and B lymphocytes (50–54). In addition, it is important to highlight that V(D)J gene recombination proceeds via DSBs, one of the most toxic DNA lesions, and the rearrangement of relatively large DNA segments. These molecular events imply a latent risk of genome instability and the development of malignant diseases, such as leukemias and lymphomas, if they occur uncontrolled. (55–61). Thus, V(D)J gene recombination is tightly regulated by several mechanisms operating at various levels, to ensure that it occurs at the appropriate cell lineage and stage of development. Regulation is achieved by modulating the expression and activity of the key proteins RAG1/2, as well as through chromatin remodeling and control of the accessibility of the immunoglobulin gene segment locus to the recombination machinery (62).
Chromatin remodeling catalyzed by specific cellular factors turns physically accessible clusters of enhancers and promoter regions of rag1 and rag2 to the cooperative binding of transcription regulators and mediators, such as Pax5, Ets1, Ikaros, and Irf4 for B cells, and Bcl11b, Tcf1, Runx1, Ikaros, and Gata3, for T cells. This results in complex structures known as super-enhancers (SEs), that efficiently promote the transcription of rag1 and rag2, in a lineage- and stage-specific manner (62).
3.2 Somatic hypermutation and class switch recombination
The actual size of the human antibody repertoire remains a matter of debate. Theoretical combinatorial calculations estimate it in the range of 1012 to 1018 different clones (63–65). However, there are several factors that limit the actual size of the antibody repertoire, such as the total number of B cells in a single human body, which has been calculated in 1011 (66). Furthermore, not all Ig gene segments rearrange with the same frequency and, at the protein level, not all HC and LC chains can efficiently pair to form a stable and functional antibody molecule (65, 67). More conservatively, it is estimated that the V(D)J gene segment recombination generates a naive repertoire of the order of 105-106 different antibodies, consisting mainly of low-affinity IgM, a fraction of which are polyreactive (68, 69). This repertoire is further diversified to in excess of 109 different antibodies by two main mechanisms, SHM and antibody class switch recombination (CSR) (70). SHM generates multiple single nucleotide substitutions in and around the productively rearranged V(D)J gene segment, resulting in antibodies with higher antigen binding affinity but the same specificity (13, 43, 70). In contrast to V(D)J gene recombination that occurs in the omentum and fetal liver during B cell development, and in bone marrow in adult individuals, SHM occurs in the germinal centers of the secondary lymphoid organs. There, an iterative alternation of SHM and antigen-mediated selection leads to antibody affinity maturation (13, 43, 70).
The key enzyme in SHM, as well as in CSR, is the activation-induced cytidine deaminase (AID), a 198-amino-acid protein member of the AID/apolipoprotein B mRNA-editing enzyme-catalytic (APOBEC) family (71). AID is a single-strand DNA-specific deaminase that catalyze the conversion of DNA cytidines (dC) to uridines (dUs), with no observable activity on double-strand DNA, RNA, or RNA : DNA hybrids. This enzyme is expressed at high levels in germinal center activated B cells with a very strict temporal and spatial regulation (71). This determines that, in normal conditions, SHM of HC and LC genes occurs during only a narrow window of B cell development (70, 72, 73). SHM occurs through a two-step process. In the first step, AID catalyzes the hydrolytic deamination of dC in single-stranded DNA into dU and ammonia, introducing single nucleotide substitutions in a stepwise manner at a frequency of around 10−3 per base pair per generation (43, 70, 73). This is a million times higher than the normal mutation rates in non-Ig genes (43, 70, 74). AID preferentially targets dC located at hot spot motifs such as WRCY (when W = A or T, R = A or G, Y = C or T), with AGCT being a preferred motif (75). In opposition, the cold spot SYC (S = G/C; Y = C/T) is avoided, being mutated at frequencies 2 to 10-fold lower than the hot spots (70, 75). Codons prone to amino acid change tend to be concentrated in CDRs, as compared to FRs, which are thought to be an evolutionary solution to meet competing demands for diversification and preservation of antibody integrity. However, differences between κ and Λ LCs have been observed. It was found that in germline Λ LCs, codons at the CDRs are prone to replacement mutations, whereas, in the FR, the opposite is true, like in HC. In contrast, in germline κ LCs, codons in both CDR and FR are more prone to replacement mutations (76). It was hypothesized that the observed differences between κ and Λ light chains represent different evolutive strategies to balance diversity and stability in an immune response (76). On the other hand, as we will see later, these differences may also determine different pathogenic behavior of κ and Λ LCs in circumstances in which the LC is secreted in a free state (77, 78).
Generation of dU by AID-dependent deamination of dC results in U:G mismatch. The type of change that results from AID-dependent dC deamination depends on which DNA repair pathway processes the U:G lesion; the mismatch repair (MMR) or the base excision repair (BER) pathway (72). Notably, in SHM, error-prone pathways are favored instead of the canonical pathways that catalyze high-fidelity U:G repair (72). In the process dependent on the MMR pathway, U:G mismatch is detected by MutSα (MSH2/MSH6 heterodimer) (79), which recruits apurinic/apyrimidinic endonuclease 2 (APE2) and exonuclease 1 (Exo1). APE2 then cleaves the DNA 5’ of the mismatch, creating a DNA nick that serves as a point of entry for the Exo1, which initiates resection from the nick going past the mismatch site and creates an extended patch of ssDNA (43, 70, 80). Events in the multi-molecular complex formed at the site of DNA lesion result in the substitution of the high-fidelity DNA pol δ and pol ϵ by error-prone translesion DNA synthesis (TLS) polymerases, such as pol θ, polη, Rev1, and pol ζ (81). Polη is thought to play a major role in this step (73). This factor is recruited through proliferating cell nuclear antigen (PCNA) ubiquitination to resynthesize the DNA gap and introduce mutations, which are mainly at A:T pairs. Polymerase η is a mutator of A/T and C/G pairs in vitro, with one-quarter of its errors being introduced at C/G pairs (73, 82). The average base substitution error rate of human polymerase η is ∼3.5 × 10−2 (82), orders of magnitude higher than those of replicative DNA polymerases, which are in the range from 10−6 to 10−4. After the gap is closed by DNA polymerases, ligase 1 finalizes repair by sealing the break.
When an U:G mismatch is repaired by an error-prone BER pathway, dU is recognized by uracil-DNA glycosylase (hUNG), which removes dU from DNA (83), leaving an abasic site (AP site) that is recognized by APE1/2 (84). APE1 and/or APE2 are believed to generate the strand break by incising the AP site generated by hUNG. Interestingly, a recent study demonstrated that hUNG can catalyze DNA backbone cleavage after uracil excision, which suggests that the first two steps in uracil BER can be performed by this factor (83). In a canonical BER, PARP1 is activated, and scaffold protein XRCC1, Polβ, and DNA ligase IIIα are recruited (85). Polβ then removes the 5’ deoxyribose and inserts a single nucleotide, followed by ligation to complete the repair (84). In non-canonical, error prone BER during SHM, some abasic sites serve as a non-informative template (84). REV, a Y-family DNA polymerase recruited by PCNA ubiquitination as well, inserts dCMP into the new DNA strand opposite the abasic site. After a further round of DNA replication, this can result in a stable transversion mutation at the site of the original C:G base pair (43, 70, 84).
On the other hand, CSR allows the generation of antibody isotypes with different effector functions, which contributes to the effectiveness of the humoral response (86). CSR is a DNA deletional-recombination that removes the µ IGHC exons and places the functional VDJ segment into proximity with the exons of downstream HC constant regions (43, 80). This causes the replacement of membrane-bound IgM and IgD by membrane-bound IgG, IgA, or IgE; better protective antibodies. Such change in antibody class is accomplished without altering the VH(D)JH or VLJL exon assemblies for HC and LC, respectively, which is the base of the one-cell-one-antibody paradigm (87). CSR is driven by switch (S) regions, G-rich repetitive DNA elements located upstream of each CH gene segment, except the CHδ (88). Like SHM, CSR proceeds through AID-introduced DSBs, but in contrast to SHM that mostly targets the VL region, CSR is restricted to the CH locus. As mentioned previously, AID is a ssDNA-specific deaminase, therefore, transcription is essential for the AID action. Each individual CH gene segment, except CHδ, are organized as transcription units, consisting of a cytokine/activation-inducible promoter, followed in downstream direction by an intermediate (I) exon, an S region, and CH exons. Activation of transcription through an individual CH gene segment by cytokine treatment induces CSR to that isotype (89, 90). On the other hand, expression of germline CH µ gene segment transcripts is constitutive and unaffected by mitogen or cytokine treatment (91). At the onset of CSR, the cytokine/activation-inducible promoter of the target CH gene segment is selectively activated, which results in a non-coding germline transcript that initiates at the I exon, proceeds through the S region and terminates downstream of the corresponding CH gene segments. This allows generation of RNA : DNA hybrid structures, such as R-loops, exposing stretches of ssDNA that serve as substrates for AID. There is evidence that AID features an intrinsic preference for G-quadruplex (G4)-containing DNA (92). G4-DNA structures formed by G-rich sequences present in S regions appear to induce CSR by promoting AID binding (93, 94). The binding of AID to the active S region requires the participation of 14-3-3 adaptor proteins (95) and also involves the recruitment of protein kinase A (PKA) (96). PKA phosphorylate AID at Ser38 (96), generating a binding site for RPA, which enhance the deamination activity of AID (97). AID converts dC exposed in the displaced G-rich nontemplate strand within the transcribed targeted S regions to dU, with a specific affinity for WRCY motifs (98). The AID-generated dU:dG mismatch can be processed via either BER or MMR pathways. However, the evidence support the notion that BER pathway is preferred in CSR, in particular when two dU:dG mismatches are closely spaced on opposite DNA strands (98–100). Via BER, DSBs are generated by subsequent dU deglycosylation by UNG that creates an abasic site. Then, the abasic site is recognized by the apurinic/apyrimidic endonuclease APE1, generating a nick. The evidence indicates that MMR pathway plays a less relevant role in CSR than BER (98). It is believed that MMR is involved in the conversion of single-strand breaks (SSBs) on opposite DNA strands that are located distal each other into DSBs (98). It has been suggested that Msh2–Msh6 may bind to dU:dG mismatches that have not been processed by UNG and recruit EXO1 to excise DNA from the nearest 5′-SSB to the mismatch, creating a DSB with a 5′-overhang (98, 101).
End-joining of DSBs between donor and acceptor S regions is carried out mostly by the C-NHEJ DNA DSB repair pathway, which employs the Ku70/80 complex (Ku) for DSB recognition and the XRCC4/DNA ligase 4 (Lig4) complex for ligation. However, A-NHEJ DNA repair pathway is believed to also participate in this step under certain circumstances. The result is the deletion of the expressed IgM/IgD CH gene segments, which are replaced by a new CH gene directly downstream of the VDJ(H) exon.
As for V(D)J gene recombination, B cells use mechanisms operating at different levels to keep both SHM and CSR under strict regulation. At the cell level, SHM is induced by specific stimuli such as crosslinking of the BCR, the engagement of CD40 and co-engagement of CD80 and CD86 on the B-cell surface by CD154 and CD28 expressed on the surface of activated T cells, and cytokines, such as interleukin 4. These stimuli specifically upregulate AID by means of, among others, epigenetic mechanisms mediated by histone-modifying enzymes acting at the promoter regions of AID (43). AID gene is the target of the gene regulatory network composed of several transcription regulators and orchestrated by BACH2 and IRF4 that promotes SHM and CSR of antibodies and simultaneously represses plasma-cell differentiation (102–104). Epigenetic mechanisms induced in response to B cell activating stimulus also activate miRNAs like mir-16, mir-155, and mir-181b that decrease the expression of AID by binding to and degrading complementary sequences of the mRNA (105–107). As mentioned before, AID is also regulated by phosphorylation in Ser38 by cAMP-dependent protein kinase (PKA) that increases AID activity at the Ig HC switch regions (43, 108). The activity of AID can be also regulated by phosphorylation at threonine 140 (Thr140) by protein kinase C (PKC) family members, a mechanism preferentially affecting SHM (109). Serine 3 (Ser3) is another phosphorylation site that causes a reduced activity of AID, an effect that can be reversed by dephosphorylation catalyzed by protein phosphatase 2A (PP2A) (110). AID is also regulated by a complex mechanism that controls the enzyme subcellular localization (111) and stability (112). AID is also cell cycle regulated by processes that involve checkpoint kinase 1 (Chk1) and 2 (Chk2), two factors that regulate both SHM and CSR but with opposite effects (113).
Regardless of the numerous overlapping regulatory mechanisms that control AID, the mutagenic activity of this enzyme can be inappropriately activated by several factors, such as chronic inflammation (114–116), hepatitis C virus infection (117, 118), and estrogen (119, 120). Aberrant activation of AID has been linked to the pathogenesis of autoimmune disorders (121) and several types of cancer (122), including colon cancer (114, 123, 124), hepatocellular carcinoma (125, 126), skin cancer (127, 128), oral squamous cell carcinoma (129), and multiple myeloma and other B cell malignancies (130–133). There is evidence that AID plays a role in the occurrence of chromosomal aberrations in monoclonal gammopathies, such as the translocation of chromosomes 11 and 14 [t(11;14)(q13;32)] (134). This chromosomal alteration is detected in 16%-24% of patients with multiple myeloma (135), a frequency that reaches ∼50%-60% in those with AL amyloidosis, constituting a distinctive characteristic of this disease (136–138). In AL amyloidosis, [t(11;14)(q13;32)] has a significant impact on therapeutic response and overall survival (139–141). AL amyloidosis with [t(11;14)(q13;32)] is associated with low plasma cell count but high serum-free LC level, which may influence the tissue distribution and burden of AL deposits (138).
4 Diseases caused by immunoglobulin light chains misfolding and aggregation
In healthy individuals, most LCs are secreted as part of the antibody (Figure 1), the context in which the LC evolved to carry out its recognition function. The quaternary structure of antibodies represents a protective context for the LC against intrinsic and extrinsic deleterious factors that can promote misfolding and aggregation. The contacts at the HC/LC interface contribute to the LC thermodynamic stability, counterbalancing the destabilizing effect of somatic mutations (142–144). Also, the multidomain structure of the antibody prevents the LC from participating in edge-to-edge intermolecular H-bonding, reducing the risk of aggregation (145, 146). However, in patients with monoclonal gammopathies, the abnormally proliferating plasma cell clone overproduces the LC and secretes it in a free state into the bloodstream (147) (Figure 2A). In such a condition, the monoclonal LC, deprived of its natural protective context, is more susceptible to misfolding and aggregation (77, 78). LCs can form various pathologic aggregates that differ from each other in ultrastructural morphology, internal structure, and biological properties. Some LCs form crystals in the cytoplasm of the cells of the proximal tubules and disturb their absorptive capabilities, causing a disorder known as Fanconi’s syndrome (FS) (148). Other LCs reach distal nephrons, where they co-aggregate with the Tamm-Horsfall protein to form intratubular casts, a disorder termed myeloma (cast) nephropathy that is characterized by renal failure primarily by an obstructive mechanism (149). There is another group of pathogenic LCs that form non-ordered, amorphous aggregates that can deposit in any organ, although affect predominantly the kidneys, causing a disorder known as LC deposition disease (LCDD) (Figure 3). Finally, there is a group of LCs that displays a propensity to deposit in the extracellular spaces of organs and systems in the form of amyloid fibrils, whose distinctive characteristic is a structural core riches in β structure (Figures 2A and 4). This causes LC-derived (AL) amyloidosis, the systemic form of amyloidosis most frequently diagnosed in Western countries (150, 151). As a systemic disorder, AL amyloidosis usually affects various organs, although the heart and kidneys are the most frequently involved.
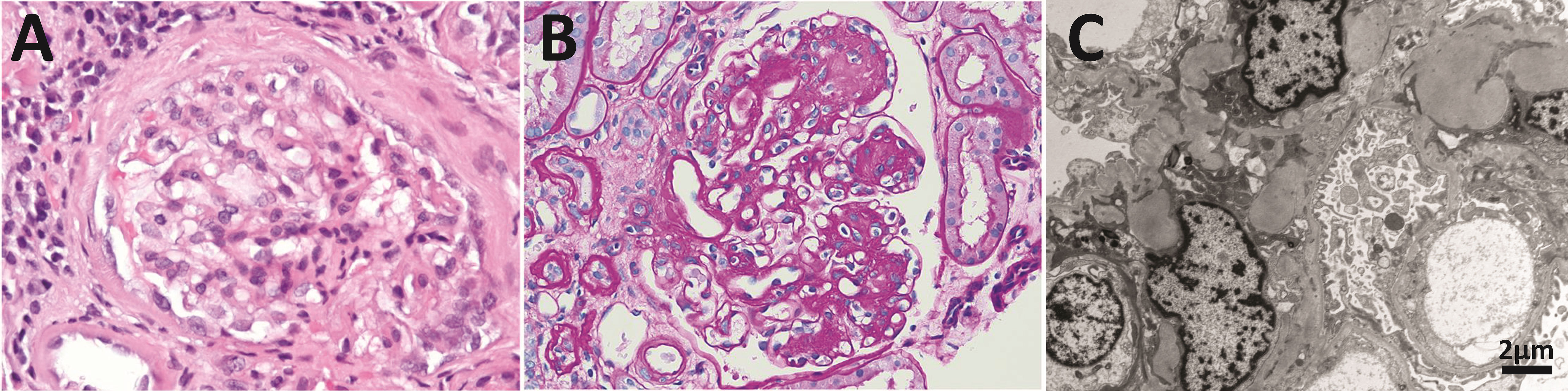
Figure 3 Anatomopathological features of light chain deposition disease (LCDD). (A) Hematoxylin & eosin (H&E) and (B) PAS staining of kidney biopsies from two patients in the early and late stages of the disease, respectively. Note in B the nodular mesangium with a markedly increased tenascin-rich extracellular matrix. (C) Electron microscopy (EM) analysis that shows expanded nodular mesangium with an increased extracellular matrix and punctate, powdery, ground-pepper-like extracellular deposits.
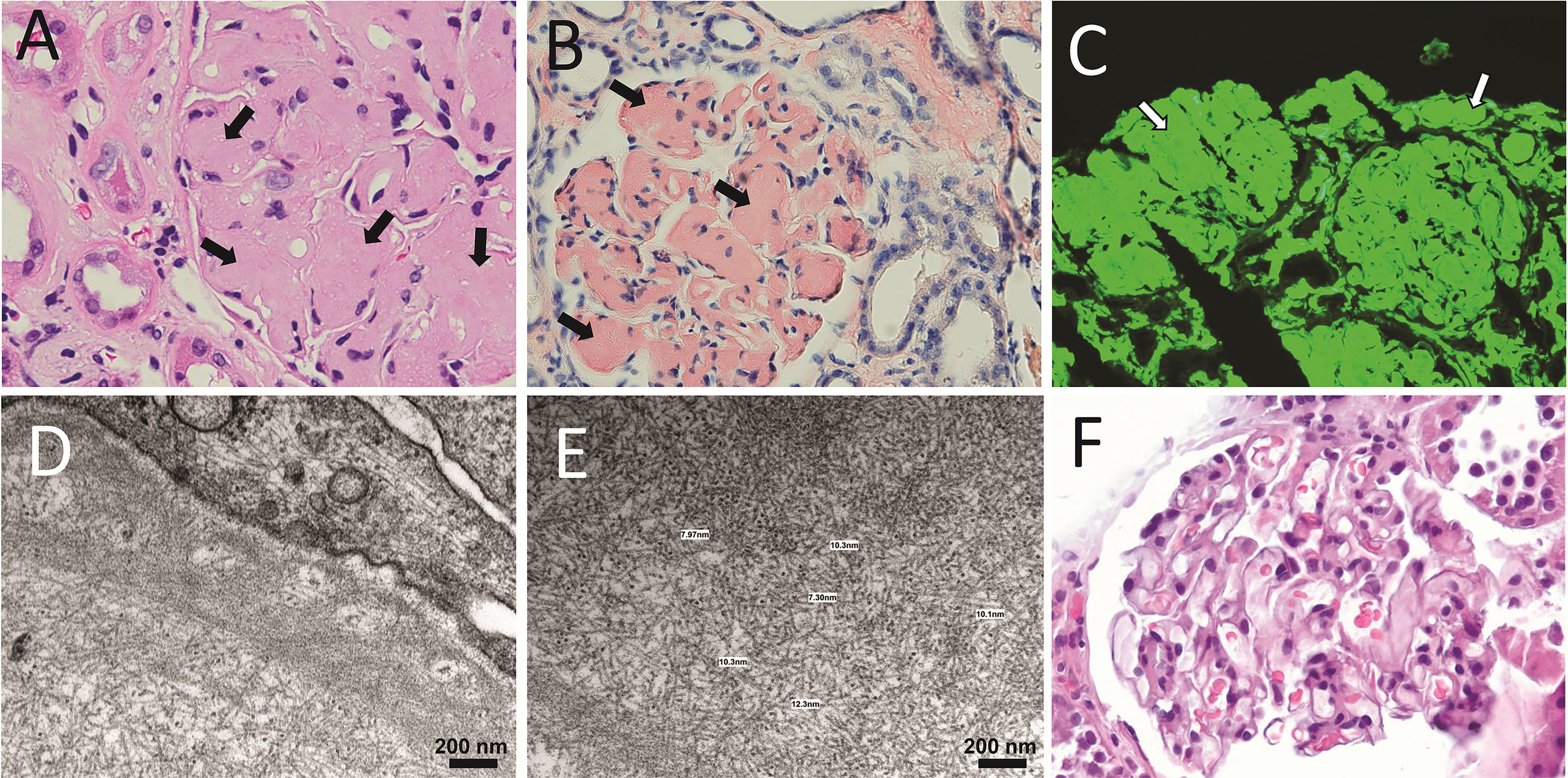
Figure 4 Anatomopathological characteristics of renal AL amyloidosis (A–C) H&E, Congo red, and immunofluorescence analysis for human κ LC of a renal biopsy of a κ AL amyloidosis patient in advance stage of the disease. (D, E) show images taken in EM analysis of the same sample shown in (A–C). (F) The H&E staining of the renal biopsy of a Λ AL amyloidosis in early stage of the disease is presented for comparative purpose. In A, arrows indicate AL amyloid deposits stained with H&E that appear as a waxy, homogenous, eosinophilic material that has almost completely replaced the mesangium. In B and C, arrows indicate the same material stained with Congo red, and detected by the anti-human κ LC antibody, respectively. In C, the primary, and secondary antibodies were goat anti-human κ LC antibody and fluorescein-conjugated rabbit anti-goat IgG, respectively. Note in D and E the abundant randomly disposed fibrils with diameter in the range of 7.3 nm to 12.3 nm, characteristic of the AL amyloid fibrils.
The diversity of pathological aggregates that LCs form is a direct consequence of their sequence diversity and reflects the wide spectrum of conformation that these immunoglobulins can adopt in a context-dependent manner (143, 152–154). In this regard, AL amyloidosis, by far the most clinically heterogeneous of all systemic amyloidoses, is a good example of how LC sequence diversity translates into a wide spectrum of clinical courses (155, 156).
It is important to mention that not all monoclonal LCs aggregate in vivo. Several long-term follow-up studies of patients with Bence Jones idiopathic proteinuria have shown that not all develop AL amyloidosis, or any other disease associated with LC deposition (157–162). This indicates that the secretion of the LC in a free state, although a required condition, is not sufficient for LC aggregation to occur. It is the interaction between the structural and biophysical properties of the LC, both a function of its sequence, and factors of the tissue microenvironment that determine if and how LC aggregation occurs (77, 78, 163–166).
4.1 Genetic and structural factors that drive LC aggregation in human diseases
The intrinsic propensity of a monoclonal LC for pathological aggregation is determined by both the encoding VL gene segment (167–171), and the somatic mutations (54, 152, 164, 172–176). The encoding IGVL gene segment may confer to the LC the propensity to aggregate in a specific form, as well as in a specific organ or tissue (organ tropism). As far as we know, the first report of the association of a VL subgroup with a monoclonal immunoglobulin deposition disease was done by Solomon et al. (177), who observed an overrepresentation of the Λ6 LCs in AL amyloidosis (177). Subsequent studies corroborated Solomon and colleagues’ findings and, furthermore, revealed the bias in the IGVL gene segments usage that characterizes the AL amyloidosis (167, 168, 170, 178–180). Such bias is clearly observed in the collection of AL LCs sequences compiled in the AL-Base (http://albase.bumc.bu.edu/aldb). Currently, this curated database contains the sequence of 570 amyloidogenic LCs, ∼61% of which derive from only five IGLV gene segments (IGLV1-44, IGLV2-14, IGLV3-1, IGLV6-57, and IGKV1-33), These gene segments, in conjunction, account for only ∼7% of the human repertoire of κ and Λ IGVL genes segments, composed by ∼70 elements (31). IGLV6-57, the only member of the Λ6 subgroup, is the most frequently used gene segment in the set of AL-associated LCs compiled in the AL-Base, since it accounts for 17.8% of the whole set (129 κ and 441 Λ) and 23% of those of Λ type (https://wwwapp.bumc.bu.edu/BEDAC_ALBase). Kourelis et al. established the use of immunoglobulin gene segments in a series of 701 and 120 patients with systemic and localized AL amyloidosis, respectively, the largest study to date on this topic (170). They found that, overall, the Λ gene segments IGLV6-57, IGLV2-14, IGLV3-1, and the members of the VL gene segment subgroup κ1, which includes IGKV1-33, constituted 49% of the clones identified in systemic AL amyloidosis. Again, IGLV6-57 was the most frequently used IGVL gene segment, accounting for ∼23% of the systemic AL amyloidosis caused by a Λ LC (170). The strong association of IGLV6-57 gene segment with amyloidosis is supported by two findings. One is the difference between its low frequency of expression (∼2%) in the normal repertoire of polyclonal bone marrow plasma cells expressing a Λ LC (168), and its ten-time higher frequency in AL amyloidosis (170). The second finding is the contrasting difference in the number of amyloidogenic and biopsy-proven non-amyloidogenic monoclonal Λ6 LCs reported in the literature (181). Currently, the AL-Base contains the sequence of 102 Λ6 amyloidogenic LC (Data retrieved on May 18th, 2023). For many years, JTO was the only monoclonal Λ6 LC for which clinical data excluding amyloid deposition was available. This protein was identified in the 90s in a patient with myeloma (cast) nephropathy without clinical evidence of amyloid deposition (182, 183). Nig-48 (Accession number P01722) is another monoclonal Λ6 LC classified in the AL-Base as derived from plasma cell dyscrasias (PCD) other than AL amyloidosis. However, in the original publication that reports this protein, no clinical data of the patient is informed. Therefore, it is unknown whether this protein caused AL amyloidosis (184). The near absence of biopsy-proven non-amyloidogenic monoclonal Λ6 LCs led to suggest a near absolute link between the Λ6 isotype and AL amyloidosis (169, 181). However, very recently, Nau et al., identified 7 monoclonal Λ6 LCs in patients with multiple myeloma without the diagnosis of AL amyloidosis (185). This study was aimed at validating a computational approach using the MiXCR suite of tools to extract complete rearranged IGVL-IGJL sequences from untargeted RNA sequencing data. This approach was successfully applied to whole-transcriptome RNA sequencing data from 766 newly diagnosed patients in the Multiple Myeloma (MM) Research Foundation CoMMpass study (185). While the study of Nau et al. suggests that PCD patients with a circulating monoclonal Λ6 LCs without amyloidosis could be more frequent that previously assumed, we think that it is important consider some facts in analyzing this report (185). Remarkably, the authors reported that only 14 (2%) of 705 patients were reported with the diagnosis of AL amyloidosis, a figure that contrasts with the incidence of secondary amyloidosis in MM patients reported in other studies, which varies from 25% to ∼40% (186–188). In addition, some of the 7 MM patients with a monoclonal LC Λ6 without a diagnosis of AL amyloidosis had signs and symptoms of renal failure and peripheral neuropathy, conditions frequently associated with AL amyloidosis (189). As the authors of this article state, it will be required Congo red staining and immunohistochemistry analysis with LC-specific antibodies in subcutaneous biopsy to rule out the diagnosis of AL amyloidosis in these patients (185).
On the other hand, several studies have consistently found that renal involvement is more frequent in AL patients with an IGLV6-57-derived LC, when compared with patients without (167, 170, 180). Trends suggestive of organ tropism have also been observed in other IGVL gene segments (167, 170, 180).
Although several hypotheses have been proposed, no study has provided straightforward evidence of mechanisms or factors explaining the association of a few IGVL gene segments with AL amyloidosis. In the case of IGLV6-57, structural factors encoded by the germline of this gene seem to play a role, since the rVL protein with the germline sequence of IGLV6-57 was shown to be intrinsically prone to aggregate as amyloid-like fibrils in vitro (169, 181). Indirect evidence obtained in a recent study suggests that the germline IGLV3-1 gene also encodes a VL protein highly prone to aggregate (190). On the other hand, evidence indicates that the association of IGLV1-44 and IGKV1-33 with amyloidosis cannot be explained solely by the properties of the germline VL protein. Other not yet identified factors may be more relevant for these gene segments (181).
The association of certain IGVL gene segments with AL amyloidosis may also reflect the preferential rearrangement of these genes in certain B lymphocyte populations driven by local factors (16, 167). Chronic antigenic challenge, either by foreign or self-antigens, along with local factors, may play a role in promoting the selection and expansion of B lymphocytes that preferentially rearrange these IGVL gene segments (191). However, to our knowledge, there are no reports of antigenic specificity for a plasma cell clone implicated in AL amyloidosis.
Understanding what determines the association of some IGVL genes with amyloidosis deserves further investigation, as mounting evidence indicates that the VL gene segment encoding the amyloidogenic LC influences both the clinical presentation and outcome of patients with AL amyloidosis (167, 170, 179, 180).
Little is known regarding the IGVL gene segment usage in other diseases caused by LC deposition, mainly because of the few numbers of LC sequences reported. The data suggest that the repertoire of IGVL genes in LCDD and Fanconi syndrome is also biased to a few elements. Most of the LCs involved in LCDD are encoded by a restricted set of κ IGVL genes that belong to κ1 (IGKV1-5), κ3 (IGKV3-11 and IGKV3-15) or κ4 (IGKV4-1) subgroups (15, 192). Only two Λ LCDD LCs have been reported so far (193). Most of the LC involved in Fanconi syndrome identified so far were encoded by IGKV1D-33/IGKV1-33 (O8/O18) or IGKV1D-39/IGKV1-39 (O2/O12), two pairs of duplicated gene segments that belong to the κ1 subgroup (194, 195). LCs encoded by the κ3 gene segments IGKV3-11 (L6), IGKV3-15 (L2), and IGKV3-20 (A27) have been reported in Fanconi syndrome patients too. We can conclude that at least part of the structural factors that drive LCs to form pathological aggregates are encoded in the germline IGVL gene segment.
Interestingly, a highly restricted repertoire of IGVL gene segments has been also reported in the monoclonal component present in patients with polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes (POEMS) syndrome. In about 80% of the case, the circulating monoclonal LC is encoded by the Λ1 gene segments IGLV1-40 and IGLV1-44. Furthermore, in almost all cases, the IGJL gene segment has been IGLJ3*02 (196).
4.2 Role of somatic mutations in light chain amyloid aggregation, cytotoxicity, and organ tropism
Early studies found that the amyloidogenic LCs tend to be thermodynamically less stable than their non-pathogenic counterparts (143, 152, 153, 172, 173, 183, 197–200). It has been consistently found that destabilizing somatic mutations, as well as stringent conditions of temperature, pH, or concentration of chaotropic salts, such as urea and guanidinium hydrochloride (GdnHCl), accelerate the kinetic of LC fibrillogenesis (199, 201–204). Thus, thermodynamic stability was early identify as a key driver of LC amyloid aggregation (152, 172, 173, 183, 205–207). It was predicted that a less thermodynamically stable LC is more likely to adopt non-native conformations prone to self-assemble into amyloid. This model confers non-native intermediaries a key role in the mechanism of LC amyloid aggregation, a key concept for understanding AL amyloidosis as a protein misfolding disease (172, 204, 208–210).
The recognition of somatic mutations as main actuators in AL amyloidosis pathogenesis resulted from studies that demonstrated their ability to destabilize the LC and promote its fibrillar aggregation (152, 172, 173, 198).
These studies also showed that the impact of somatic mutations is context-depend. This means that the mechanism by which mutations promote LC aggregation varies depending on the residues that are involved and the structural features of the LC itself (152). Some mutations have been shown to promote LC amyloidogenesis by disrupting the hydrophobic core of the VL domain, destabilizing the domain fold (211). The Λ6 LC AR, the first AL protein whose amino acid sequence was determined, illustrates well this mechanism (212). This protein showed to be highly unstable in both temperature and GdnHCl equilibrium unfolding experiments (205). The VL protein AR also readily aggregated into amyloid-like fibrils at physiological-like conditions of temperature, pH, and ionic strength; with faster kinetics than that of its germline counterpart protein 6aJL2(G25) and the highly amyloidogenic Λ6 VL Wil (205). Protein AR also elongated homologous seeds highly efficiently. According to the thermal unfolding curve, the unfolded fraction of the rVL AR at 37°C was estimated at ∼80%, which can explain its high aggregation propensity (205). AR differs from its germline counterpart in fourteen positions. However, it was shown that the restoratives single- Phe21Ile and double-mutant Phe21Ile/Val104Leu, both located in the hydrophobic core, restored ∼50% and ∼100%, respectively, of the thermodynamic stability lost in the VL AR relative to its germline counterpart (Figure 5). As predicted, both mutants were significantly less fibrillogenic than AR, displaying aggregation kinetics like that of the germline counterpart (211). This study showed how detrimental somatic mutations that disturb the structural core of the VL domain can be. At the same time, it raises the question of the role of this type of mutation in the LC recognition function. It cannot be ruled out that mutations such as those identified in protein AR improve the recognition properties and/or biological function of the antibodies by increasing the conformational flexibility of the VL (214–218).
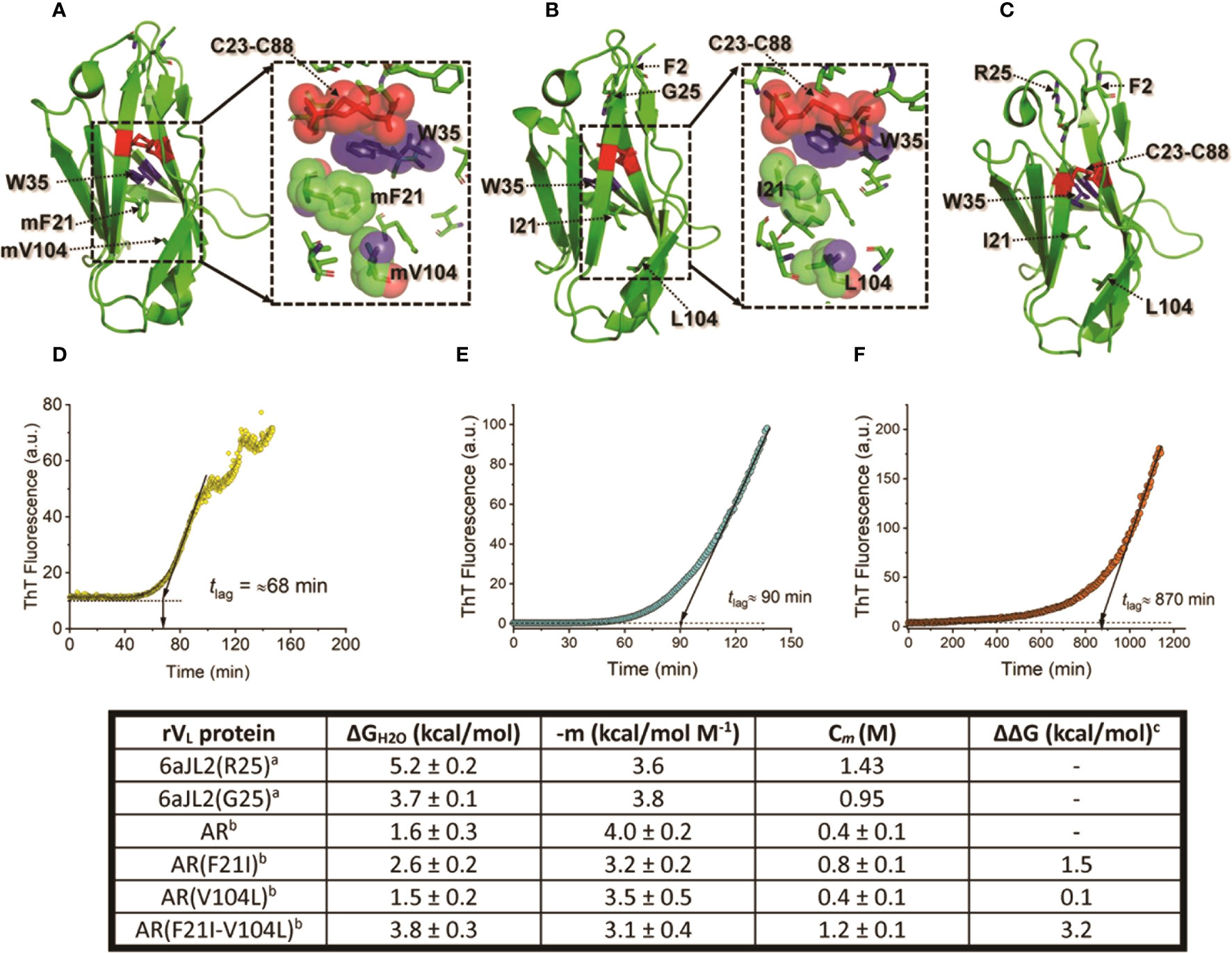
Figure 5 Impact of somatic mutations Ile21Phe and Leu104Val on the structure and biophysical properties of the amyloidogenic Λ6 LC AR. (A) to (C) show the crystallographic structure of the rVL proteins AR, 6aJL2(G25), and 6aJ2(R25). Mutant residues (m) Phe21 and mVal104, and wild type Ile21 and Leu104 are represented in stick format and indicated. For reference purposes, the highly conserved Trp35, the disulfide bond Cys23-Cys88, Phe2, and the residue at position 25 are also represented in stick format and indicated. The inset in A and B show a more detailed representation of the spatial relationship of mPhe21, mVa104, Ile21, and Leu104 with non-polar residues that compose the core of the VL domain fold. The mutant and wild-type residues, Trp35, and the disulfide bond Cys23-Cys88 are represented in a combination of semi-transparent spheres/sticks representation, to highlight the side chain-to-side chain interactions. Proteins 6aJL2(R25) and 6aJL2(G25) have the germline sequence of the IGLV6-57 (Λ6) segments. They have the same sequence, except in position 25, which is an allotypic variation of the IGLV6-57. Variant G25 suppresses a cation-π interaction between Phe2 and Arg25, determining a less thermodynamically stable and more fibrillogenic protein (169). (D-F) show the in vitro fibrillogenesis of the rVL Λ6 proteins AR, 6aJL2(G25), and 6aJL2(R2) performed at 200 µg/ml in presence of 20 µM thioflavin T. The protein solutions were incubated at 37°C with constant stirring of 350 r.p.m. with a 5x2 mm Teflon-coated magnetic micro-stir bar. Amyloid-like fibril formation was detected by serially measuring ThT fluorescence, exciting the sample at 450 nm, and recording the ThT fluorescence at 482 nm. The lag time (tlag) for nucleation was calculated by extrapolating the linear region of the hyperbolic phase back to the abscissa (213). The table in the bottom shows the thermodynamic parameters of the rVL protein AR, its single and double mutants AR(F21I), AR(V104L), and AR(F21I-V104L), and the germline proteins 6aJL2(G25) and 6aJL2(R25) determined by reversible unfolding with increasing concentration of guanidinium hydrochloride (GdnHCl). (A) Data were taken from reference (205). (B, C) Data were taken from reference (211). ΔΔG values were calculated considering AR as the wild type; positive values indicate a higher stability than that of AR. The structures shown in (A) to (C) were prepared with PyMOL (PyMOL Molecular Graphics System, Version 2.5.2, Schrödinger, LLC) using PDB 5IR3 for rVL AR, PDB 5C9K for rVL 6aJL2 (G25), and PDB 2W0K for rVL 6aJL2 (R25).
Other mutations have been shown to promote aggregation by altering the homodimer interface that LCs tend to form in solution (Figure 1E), shifting the equilibrium towards free monomers, which are typically thermodynamically less stable (219–221). This finding is in line with studies showing that the association of the LC in a stable dimer protects it from misfolding and aggregation (222, 223). Hence, stabilizing the LC dimer by small molecules that bind at the dimer interface is one of the strategies being explored to inhibit LC amyloidogenesis (224, 225)
Somatic mutations can also promote amyloidogenesis by shifting the kinetics of unfolding/refolding of the LC, as well as altering the internal dynamic of the protein (175, 226). It is believed that this effect may favor the accumulation of non-native species that nucleate the fibrillogenesis. In a recent study, Oberti et al. compared the structural and physicochemical properties of thirteen sequence-diverse full-length LCs and founds that the ability to form amyloid in vivo correlated with both low thermodynamic stability and high protein dynamics (227). Remarkably, no significant correlation was observed between hydrophobicity, structural rearrangements, and the nature of the LC dimeric association interface with the propensity to form amyloid (227).
On the other hand, Martin-Argany et al. found that highly destabilizing mutations that shift the equilibrium toward the unfolded state can inhibit, rather than promote, the amyloidogenesis of κ1 LCs (207). This finding supports the notion that a partially folded intermediate, and not the totally unfolded state, is the most amyloidogenic species in the LC unfolding pathway. However, evidence suggests that this may not apply to all LCs. It was observed inverse correlation between the unfolded fraction at 37°C, as calculated from the thermal unfolded curve, and the latency time (tlag) of in vitro fibrillogenesis of a group of four Λ6 LCs, which included the highly unstable protein AR and its germline counterpart 6aJL2(G25) (205). As mentioned before, the unfolded fraction of AR at 37°C is ∼80%. This suggests that, at least for Λ6 LCs, highly disordered conformer(s) may be efficient in triggering aggregation in vitro, contrary to what seems to occur in κ1 LC. These differences probably reflect the different contributions of factors such as somatic mutations and structural properties encoded in the germline VL domain, which differ greatly between κ and Λ LC subgroups. (77, 78, 181).
Recently, Peterle et al. published a study aimed at identifying the structural factors driving the high propensity of Λ6 LCs to deposit as amyloid (228). They found that the conservative mutation Asn32Thr destabilized the LC, an effect that propagated across the VL domain increasing the dynamics of regions ∼30Å away from the substitution site. This effect caused the CDR1 to be less protected in hydrogen-deuterium exchange (HDX) mass spectrometry (MS) experiments, indicative of a more flexible loop. In fact, the CDR1 was the region that displayed the lowest protection in HDX-MS experiments in all Λ6 LCs studied by them (228). This is a relevant finding, considering that the CDR1 of the Λ6 LCs contains a highly pro-amyloidogenic hot spot that is predicted to play a key role in the aggregation mechanism (229). Pro-amyloidogenic hot spots are short segments in proteins that can drive amyloid aggregation in a sequence-dependent manner. It is believed that they play a key role in triggering the aggregation reaction, which is why they are considered potential therapeutic targets (230). Structural studies have shown that the CDR1 of Λ6 LCs undergoes extensive conformational adjustment in the amyloid state. This involves a helix-to-β-strand transition that allows the CDR1 to be placed in the inner core of AL fibrils (231, 232) (Figure 6). Therefore, it is plausible that mutations in the CDR1, or in residues that contribute to stabilizing the native fold of this loop, may trigger amyloid aggregation by promoting unprotected exposure of the CDR1 pro-aggregation hot spot to intermolecular contacts, as well as by facilitating the refolding of this loop into its amyloid conformation (78, 231, 232). Since a fibrillogenic hot spot spanning the CDR1 was shown to be also encoded by several κ and Λ germline IGVL gene segments, we anticipate that the above-mentioned mechanism are also relevant to LCs other than Λ6 proteins (229).
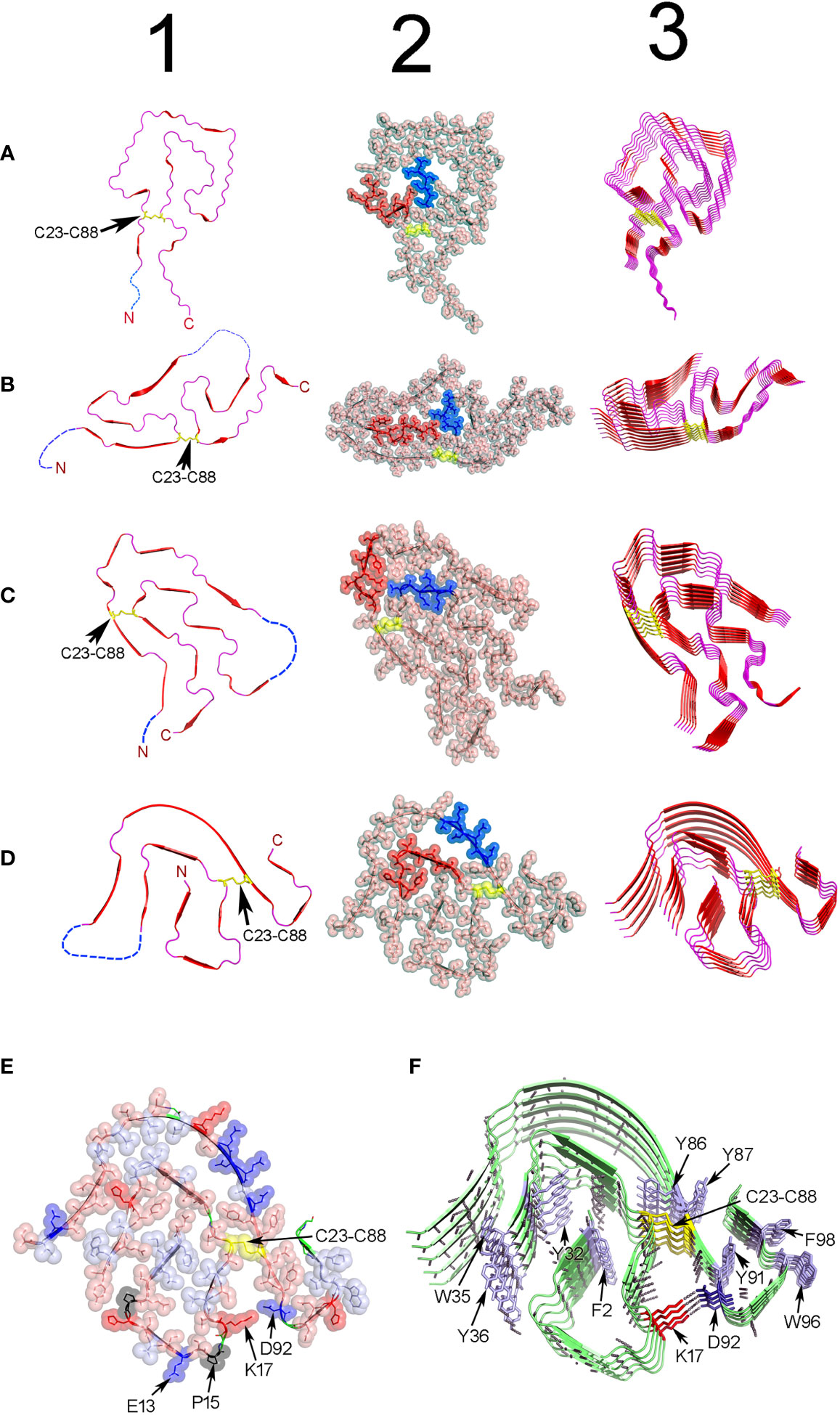
Figure 6 Structure of ex-vivo AL fibrils determined by cryo-EM. (A) AL fibril formed by a Λ1 LC encoded by the VL gene segment IGLV1-44. PDB ID 6IC3 (233). (B) FOR001 AL fibril formed by a Λ1 LC encoded by the VL gene segment IGLV1-51*02. PDB ID 7NSL (234). (C) FOR005 AL fibril formed by a Λ3 LC encoded by the VL gene segment IGLV3-19*01. PDB OD 6Z1O (235). (D) AL fibril formed by the Λ6 LC AL55 encoded by the VL gene segment IGLV6-57*02. PDB ID 6HUD (232). The four AL fibrils were extracted from cardiac deposits of patients with cardiac AL amyloidosis. Column 1 shows cartoon representations of the VL domain polypeptide chain conformation in the amyloid fibrils. Regions in β-strand are shown as arrow and colored red. Disordered regions are represented as dashed lines in an arbitrary arrangement. Column 2 shows the same structures of column 1, but with the projection of the amino acid side chains in a combination of semi-transparent spheres/sticks representation to highlight the side chain-to-side chain interactions that contribute to fibril stability. The amino acid residues spanning the CDR1, and the loop connecting the β-strands E and F (loop E-F) are colored red and blue, respectively. Column 3 shows the assembly of the AL fibrils by stacking the monomers one on top of the other, with parallel in-register arrangement of β strands. In all structures, the conserved intradomain disulfide bond Cys23-Cys88 is represented in sticks or spheres format and colored yellow. N and C stand for N- and C-terminal. (E) and (F) show a cartoon representation of the same AL Λ6 fibril shown in D, with a detailed representation of the different types of interactions that stabilize the fibril structure. In (E), the amino acid side chains are shown in a combined semi-transparent spheres/stick format, using the following color code: blue for negatively charged (acid) amino acids, red for positively charged (basic) amino acids, salmon for polar non-charged amino acids, light blue for non-polar amino acids, black for proline, and yellow for cysteines. Some residues are indicated for structural reference. Note that non-polar side chains tend to cluster buried in the fibril core, while charged and polar non-charged amino acids tend to locate in the solvent exposed surface. The image in (F) shows the stacking of the AL55 LC monomers, one on top of the other, with parallel in-register arrangement of β strands. The H bonds between the peptide backbones of adjacent monomers are shown as black dotted lines. Aromatic amino acids interacting by intermolecular stacking are shown. In both images, the conserved intradomain disulfide bond Cys23-Cys88 (colored yellow) and the fibril-specific salt bridge formed by Lys17 and Asp92 are shown. Note that this salt bridge is intermolecular, that is, it is established between residues located in monomers that are contiguous in the fibrillar structure. Regions in β-strands are represented as arrows. Amino acids are identified using a one-letter code. All structures were prepared with PyMOL (PyMOL Molecular Graphics System, Version 2.5.2, Schrödinger, LLC).
Peterle et al. hypothesized that the observed long-range effects of the somatic mutation Asn32Thr may be relevant to the recognition function of the LC in the antibody (228). Somatic mutations that modulate the antibody conformational flexibility has been suggested to contribute to biologically relevant properties, such as cross-reactivity and broad neutralizing capacity against surface proteins of diverse strains of rapidly evolving viral pathogens, such as SARS-CoV 2 (218, 236, 237). Thus, the study conducted by Peterle et al. highlights the potentially deleterious consequences of somatic mutations which, playing a beneficial role in enhancing antibody recognition properties, can cause highly disabling diseases by promoting LC aggregation (217).
The evidence supports the idea that the VL domain determines the potential of the LC to form amyloid. This put the CL domain out of the focus of most research groups interested in the molecular pathogenesis of AL amyloidosis. However, it has been shown that the CL modulates the unfolding/refolding kinetics of the LC, influencing the mechanism of aggregation (238, 239). Moreover, a recent study showed that a mutation targeting the CL domain may also contribute to the aggregation behavior of the LC (221). Rottenacher et al. reported that a nonconservative valine to glycine replacement in the CL of the amyloidogenic LC FOR005 influenced the amyloid pathway by destabilizing the fold of the CL domain, which impacted the stability of the whole molecule. Consequently, the LC dimer interface was altered, weakening LC dimerization (221). They also showed that this change made the LC monomers more susceptible to proteolytic cleavage, resulting in the release of an isolated amyloidogenic VL domain (221). This study is in line with the evidence indicating that the limiting step for LC fibrillogenesis is the dissociation of the native dimer into misfolded-prone monomers (222, 223, 240–242).
In summary, somatic mutations drive amyloid aggregation of the LCs through different mechanisms that directly or indirectly promote misfolding. It is believed that at least a fraction of the mutations involved in LC amyloid aggregation were positively selected due to their beneficial impact on antibody recognition (243). This contrasts with mutations associated with amyloid aggregation in other amyloid precursors, such as transthyretin, in which a biological advantage linked to the change has not been proven.
4.3 Impact of LC sequence heterogeneity on AL fibrillar structure
Cryo-EM structural model of four ex-vivo AL fibrils, all obtained from patients with cardiac AL amyloidosis, are currently available in the Protein Data Bank (PDB) (https://www.rcsb.org/). Two structures are of fibrils of Λ1 LCs, one identified with the PDB ID 6IC3 (233) and the other with the PDB ID 7NSL (234). The other two cryo-EM structures are of LCs of subgroups Λ3 (PDB ID 6Z1O and 6Z1I) (235) and Λ6 (PDB ID 3HUD) (232), respectively (Figure 6). This is still an insufficient number of structures to issue definitive conclusions regarding the relationship between LC sequence and AL fibril structure. However, they represent a huge step forward in our understanding regarding how the LCs assemble in the amyloid fibril, which forces stabilize the aggregate, and how somatic mutations influence it. In agreement with a previous ssNMR study (231), cryo-EM analyzes showed that AL fibrils are formed by stacking the VL domains, refolded into a quasi-bidimensional, flattened structure, one on top of the other, with the parallel in-register arrangement of the β strands (232, 233). The fibrillar fold is totally different from the β-sandwich topology that characterizes the native VL, indicating that extensive conformational conversion must occur in some step before the LC assembles into AL fibrils in vivo (231). It is important to mention that only the VL domain contributes to the β core of the fibril. Most of the VL segments adopt a defined and stable conformation in the fibril´s β-core, while others, mostly the N- and C-terminals, but also some internal segments, are disordered and exposed to the solvent (Figure 6).
The intradomain disulfide bond Cys23-Cys88 is conserved in all reported AL fibril structures, indicative that LC amyloidogenesis occurs in an oxidative environment (235) (Figure 6). A structural motif common to all AL fibrils is the 180° relative rotation, centered on the Cys23-Cys88 disulfide bond, of the segments corresponding to the native β strands B and F. This rearrangement changes the relative orientation N-term-to-C-term from the parallel, characteristic of the native state, to antiparallel (233).
The primary force stabilizing the AL fibrils seems to be the network of H-bonds between the main chain peptide groups of adjacent monomers (Figure 6). Additionally, the amino acid side chains in the AL fibril´s core interdigitate each other in a tight complementary fashion, forming steric zipper-like unions (244, 245). Nonpolar amino acids tend to cluster and locate buried, while those with polar or charged side chains tend to be solvent exposed. Aromatic amino acids interact through intermolecular pi-stacking, a type of interaction that has been shown to contribute to the stability of both pathogenic and functional amyloids (78, 246, 247).
Apart from some common elements, Cryo-EM analysis confirmed that structural heterogeneity is the hallmark of the AL fibrils. The content of the β structure and the general topology of the fibrillar VL were found to differ widely from one AL fibril to another (Figure 6). Fibril-specific salt bridges, which in some cases resulted from somatic mutations, seen to contribute significantly to the stability of the VL fold in some AL fibrils (234, 248). Cryo-EM studies also suggest that N-glycosylation, one of the post-translational modifications (PTM) found in amyloidogenic LCs, may also contribute to the structural heterogeneity of AL fibrils (234). Radamaker et al. found that the AL fibril FOR001 was N-glycosylated at Asn18, a residue introduced by somatic mutation, (234). Cryo-EM analysis suggested that the carbohydrate moiety, which was exposed at the surface of the FOR001 fibrils, determined the unique fold adopted by FOR001 LC in the fibril (234) (Figure 6).
Studies conducted in the 1980s and 1990s suggested a link between glycosylation and the propensity of LCs to cause amyloidosis and kidney impairment (249–253). Subsequent studies based on the comparative analysis of amyloidogenic and non-amyloidogenic LC sequences showed a preponderance of the consensus glycosylation sequon (AsnXxxSer/Thr) in the FRs of amyloid LCs (254). Based on these analyses, the acquisition of an N-linked glycosylation site through somatic mutations was proposed by Fred J. Stevens as one of four structural risk factors identifying most fibril-forming κ LCs (255). The development of high-throughput procedures based on high-performance liquid chromatography-mass spectrometry (HPLC-MS) for the rapid analysis of LC glycosylation has greatly facilitated the investigation of the impact of glycosylation on LC amyloidogenesis (256, 257). In a recent study, Dispenzieri et al., provided strong evidence that monoclonal LC glycosylation is a potent risk factor for progression of monoclonal gammopathy of undetermined significance (MGUS) to AL amyloidosis, myeloma, and other PCDs (258). More recently, Nevone et al., found evidence for an N-glycosylation hot spot in amyloidogenic κ LCs that differentiates them from their non-amyloidogenic counterparts (259). They found that the majority of N-glycosylation sites in the pool of amyloidogenic κ LC analyzed consisted of the NFT sequon and were located in the FR3, particularly within the β-strand E. Of note, genetic analysis showed that somatic mutations in the context of progenitor glycosylation sites, rather than genomic variants, were responsible for the N-glycosylation hot spot identified in amyloidogenic κ LCs (259). The mechanisms by which glycosylation promotes LC amyloidogenesis are still poorly understood. This posttranslational modification was found to be more frequent in AL κ than in AL Λ LCs (256). It was initially suggested that it may increase the intrinsic aggregation potential of the LC by destabilizing its native fold, and/or decreasing its solubility (255). However, in vitro studies have shown that glycosylation can retard, rather than accelerate, LC aggregation kinetics (234, 260). Based on these findings, Radamaker et al. have proposed that glycosylation may promote amyloid deposition by increasing the resistance of AL fibrils to natural mechanisms that promote the removal of tissue protein aggregates, such as metalloprotease-mediated digestion (234). However, it cannot be ruled out that glycosylation may also promote AL deposition through mechanisms dependent on its impact on the properties of the LC native state.
The monoclonal LC involved in the amyloid deposits is unique for each AL amyloidosis patient, as are the internal structure and pathological properties of the fibrillar and non-fibrillar aggregates that this protein can generate. As stated throughout this review, this is a direct consequence of the genetic mechanisms that generate a diverse repertoire of polyclonal antibodies (143, 261). The LC’s unique structural and physicochemical properties, in interplay with the genetic background and biological momentum of the affected individual, result in a disease that, although named by practice purpose “AL amyloidosis”, is actually a patient-specific disorder (143). At the same time, it is logical to ask whether the AL fibrils formed by the same monoclonal LC in different organs of the same patient are also structurally different. In this regard, Ricagno et al. determined the cryo-EM structure of AL fibrils extracted from the kidney of an AL patient whose cardiac amyloid structure had previously been determined. It was found that the structure of the renal fibril was virtually identical to that reported for the cardiac fibril (262). This finding is highly relevant to understanding the role of the tissue microenvironment in AL fibril formation and may foster the development of fibril-targeted therapeutic approaches, such as those based on antibodies, for AL amyloidosis (263).
4.4 Light chain cardiotoxicity. Is it possible to predict it?
LCs are the amyloid precursor with the greatest sequence diversity, a property that translates into heterogeneity in the clinical course of patients with AL amyloidosis (143, 264). This characteristic, and the fact that in many patients the onset of the disease is insidious, frequently leads to delay in the diagnosis (265). Hence, a significant fraction of patients is diagnosed when they already present signs and symptoms of failure of vital organs, which reduces the therapeutic options and increases the risk of dying (266). The heart and kidneys are the most frequently affected organs, but it is cardiac amyloid infiltration that exerts the most direct impact on the patient’s survival (265, 267–269). This has stimulated research aimed at unraveling the mechanisms by which circulating monoclonal LC and its AL fibrils damage cardiomyocytes. Studies carried out in cell culture demonstrated that soluble LCs obtained from AL patients, but not their non-pathological counterpart, impair the cardiomyocyte contractile function by oxidative stress, inducing apoptosis and inflammatory changes (270–273). The mechanism by which synthetic AL fibrils exert cytotoxicity in cultured cardiomyocytes has also been investigated. Marin-Argany et al. investigated the mechanism of internalization of the amyloidogenic κ1 LC AL-09 into human AC16 cardiomyocytes and found that both the soluble protein and its fibrillar aggregates were internalized via micropinocytosis (274). They found that the external fibrillar aggregates interacted with cell membranes and recruited soluble LCs, promoting their aggregation through seeding. While both soluble LC and fibrils were cytotoxic, fibrils were significantly more so and exerted their toxic effect through different mechanisms than soluble LC. It was shown that soluble protein activated the caspases, suggestive of apoptosis induction (274).
Studies carried out in cell cultures, such as those cited here, are well suited to investigate, under controlled conditions, the complex chain of events that result from the interaction of an amyloidogenic LC, either in the soluble or fibrillar state, with cells. However, this approach fails to provide information regarding the mechanisms of cell damage in the physically restricting 3D structure of the tissue. Radamaker et al. used scanning electron microscopy to determine how AL fibrils interact with cardiomyocytes in an ex-vivo tissue sample of AL cardiac amyloidosis (234). They found that the deposits of AL fibrils infiltrated and disrupted the ordered structure of the cardiomyocytes; the fibrils interacted with the surface of the cardiomyocytes, mainly through the tips of the fibrils, causing in some cases deformations in the cytoplasmic membrane (234). Such physical perturbation likely impaired the contractile function of cardiomyocytes, causing heart failure. Based on this finding, the authors deduced that promoting the remotion of the AL deposits may help restore normal cardiac function (234).
Cardiotoxic LCs can also impair heart function by modulating the expression of proteins that mediate cardiomyocyte damage by indirect mechanisms. Guan et al. reported that mammalian stanniocalcin1 (STC1) expression is elevated in the cardiac tissue of patients with AL cardiomyopathy and that this protein is induced in isolated cardiomyocytes in response to amyloidogenic LC, but not to non-amyloidogenic LC (275). They showed that STC1 overexpression in vitro recapitulated the pathophysiology of AL LC mediated cardiotoxicity, with increased ROS production, contractile dysfunction, and cell death. They corroborated these findings in a zebrafish model (276), where it was found that overexpression of STC1 resulted in significant cardiac dysfunction and cell death, while genetic silencing prevented AL LC-induced cardiotoxicity and protected the animal from an early death. Hence, the authors concluded that STC1 as a critical determinant of AL LC cardiotoxicity (275).
A proteomic-based study showed that amyloidogenic cardiotoxic LCs interact in vitro with specific intracellular proteins involved in viability and metabolism in human cardiac fibroblasts (hCFs) (277). Cardiotoxic LCs, but not non-amyloidogenic non-cardiotoxic controls colocalized with mitochondria and spatially associated with specific cell components such as mitochondrial optic atrophy 1-like protein and peroxisomal acyl-coenzyme A oxidase 1. Additionally, cardiotoxic LC-treated hCFs displayed mitochondrial ultrastructural changes, supporting mitochondrial involvement (277).
Having an animal model that recapitulates the clinical evolution and anatomopathological changes of cardiac amyloidosis would give a great boost to research on this grave complication. In 1992, Solomon et al. reported the successful experimental induction of human AL amyloid deposits in mice by the repeated injection into the animal of Bence Jones proteins obtained from two patients with AL amyloidosis (278). Although this approach was able to produce the characteristic histopathologic lesions of the disease, it suffers from several limitations in terms of reproducibility and ability to replicate the natural course of AL amyloidosis (279). Therefore, several groups have focused their work on the generation of AL amyloidosis models in transgenic mice (280, 281). Although the transgenic mice produced the amyloidogenic LC at serum concentrations equivalent to or higher than the average concentration seen in patients with AL amyloidosis, spontaneous amyloid deposition has not been observed or occurred in small amounts (280, 281). Christophe Sirac’s group has implemented an alternative approach that comprises the intraperitoneal injection of preformed homologous amyloid fibrils in the transgenic mice, as a way of promoting tissue amyloid deposition by seeding. This strategy resulted in amyloid deposition starting at 1-month post-injection, especially in the heart, spleen, liver and, to a lesser extent, in the kidney (279). Although it represents an important step forward, this model still does not fully reproduce the natural progression of the disease, since it suffers from little accumulation of tissue amyloid deposits and slow progression (279, 282). It is not clear what makes mice so resistant to the spontaneous amyloid deposition of LCs demonstrably amyloidogenic in humans. It has been speculated that mice may have developed more effective mechanisms of protein aggregates clearance or a better chaperoning of misfolded proteins than in humans (279).
Other approaches have been explored by other laboratories, such as those based on zebrafish (276, 283) and C. elegans (284, 285). Using a zebrafish model, Guan et al. determined that dysregulation of autophagic flux is critical for mediating amyloidogenic LC cardiac proteotoxicity, an effect that they were able to revert by restoration of autophagic flux by pharmacological intervention using rapamycin (286). They identified impaired lysosomal function as the major cause of defective autophagy and amyloidogenic LC-induced cardiotoxicity (286).
The use of C. elegans as a model of AL cardiac amyloidosis relies in the knowledge that the worm’s pharynx is an “ancestral heart” with the additional ability to recognize stressor compounds (284, 285). Using pharyngeal pumping as a surrogate for cardiac function, Diomede et al. demonstrated that cardiotoxic amyloidogenic LCs directly affect pharyngeal activity. The toxic effect was associated with the release of reactive oxygen species since the use of antioxidant agents restored pharyngeal activity (284).
The same C. elegans model was recently used by Maritan et al. to investigate the role of sequence on LC cardiotoxicity (287). They hypothesized that LC conformational flexibility contributes to cardiotoxic properties. To test this hypothesis, the amino acid sequence of the highly cardiotoxic LC H6 was rationally engineered by introducing three residue mutations, designed to reduce the dynamics of its native state (287). The resulting mutant (mH6) was less toxic than its parent H6 to human cardiac fibroblasts and C. elegans. The comparative structural and biophysical study of H6 and mH6 indicated that H6 featured a poorly cooperative fold, higher flexibility, and kinetic instability, and a higher dynamic state in its native fold (287). This study suggests a close relationship between the general conformational properties of the LC native fold and its ability to cause cardiotoxicity.
Understanding what makes a monoclonal LC cardiotoxic would allow calculating the risk of cardiac damage in newly diagnosed AL amyloidosis patients, a huge step forward in the effort to improve the prognosis and treatment of AL patients. Due to the complexity of this task derived from the high sequence variability of LCs, computational algorithms to predict cardiotoxicity based on the computable properties of LCs are considered a promising strategy. Several algorithms have been developed to predict amyloidogenesis in proteins (288, 289), but to the best of our knowledge, only one algorithm, LICTOR (Λ-LIght-Chain TOxicity predictoR), a machine learning-based tool, was designed with the specific goal of predicting Λ-LC proteotoxicity in mind (290). LICTOR predicts AL Λ-LC toxicity based on the distribution of somatic mutations acquired during clonal selection. This tool achieved a specificity and sensitivity of 0.82 and 0.76, respectively. The calculated area under the receiver operating characteristic curve (AUC) was 0.87. Moreover, LICTOR achieved a prediction accuracy of 83% on an independent set of 12 Λ-LCs sequences with known clinical phenotypes. Using this algorithm, the authors were able to identify two somatic mutations predicted to be responsible for the toxic phenotype, and abolished this property, as tested in the C. elegans model, by reverting the changes to the germline-specific residues (290).
Undoubtedly, LICTOR represents an important advance in the effort to develop the ability to predict the cardiotoxicity of LC, which, if achieved, would open the door to new forms of diagnosis, prognosis, and treatment of AL amyloidosis patients. According to the report of Kourelis et al., ∼65% of patients with systemic AL amyloidosis will eventually develop cardiac involvement, which highlights the need for effective tools for predicting this grave complication (170). At the same time, it is important to point out that acquiring the ability to predict the cardiotoxic potential of a monoclonal LC solves only part of the problem. There are challenges that would have to be undertaken before a tool like LICTOR finds its place in the diagnosis and therapeutic management of AL amyloidosis. Predictors of LC cardiotoxicity may help to identify patients with circulating monoclonal LC at risk of cardiac amyloidosis, which could be a candidate for more aggressive treatments to eliminate the pro-amyloidogenic plasma cell clone. However, the prediction of amyloidogenicity requires the sequence of the LC, which is not easily obtained, especially in the absence of a diagnosis of amyloidosis. Moreover, currently, many patients are still diagnosed when the signs and symptoms of organ damage, including heart failure, are already present (150, 291, 292). Improving our ability to diagnose AL patients at the earliest stages of the disease would increase the relevance of this type of predictive tool.
4.5 Somatic mutations in renal amyloidosis and the role of mesangial cells
Studies conducted in vitro demonstrated that LC amyloid aggregation can be accomplished without the participation of cells (183, 213). These studies critically contributed to our current understanding of the mechanism of LC misfolding and amyloid aggregation and, moreover, showed that the potential of an LC to form amyloid fibrils is determined by its sequence (152, 160, 172, 173, 198). As a rule, in vitro aggregation experiments are performed with the sole presence of the monoclonal LC dissolved in a buffer solution (213). This is a situation far distant from reality in humans where the circulating monoclonal LC must interact with both cells and components of the extracellular matrix in the formation of amyloid fibrils. The evidence supports the notion that it is through the interplay between the physicochemical and structural properties of the LC and factors of the tissue microenvironment that the amyloidogenic potential of a monoclonal LC translates into amyloid deposition (78). Therefore, unraveling the role of the tissue components in LC amyloidogenesis is an inescapable task in the effort to understand the pathogenesis of AL amyloidosis. Studies conducted in the last two decades provided abundant evidence of the involvement of mesangial cells in AL deposition in the renal glomerulus and pericytes/smooth muscle cells in the vasculature (293–298). It is now known that the circulating monoclonal LC interacts with different cell types and the consequences of such interaction depend on two main factors: 1) the structural and physicochemical characteristics of the LC, which are dictated by the LC sequence, and 2) the ability of certain cell types to internalize the LC and promote its aggregation into amyloid fibrils in the mature lysosomal compartment (297, 298).
In healthy individuals, the kidneys catabolize ∼50 mg per day of polyclonal free LCs, which originate from the excess production of LC over the HC that occurs in the normal B cells (299, 300). Once filtered in the glomerulus and delivered to the proximal tubules, the polyclonal LCs are avidly endocytosed by the cubilin-megalin receptor, a unique receptor for low molecular weight proteins located at the microvillous surface of the proximal tubular cells (301–303). Then, the endosomes in the proximal tubules catabolize the internalized LCs, returning the amino acids to circulation. Due to the low concentration and structural heterogeneity of polyclonal free LCs circulating in the blood of normal individuals, tissue deposition rarely occurs (304).
As mentioned before, in a fraction of patients with plasma cell dyscrasias, the abnormally proliferating plasma cell clone produces the monoclonal LC in large excess, which is secreted in a free state to the bloodstream and reaches the kidneys. From 50% to 70% of patients with a circulating monoclonal LC have clinical evidence of renal damage, indicating that it is nephrotoxic (305). Approximately 70% of the nephrotoxic mLC cause tubular injury and are referred to as tubulopathic LCs (TLCs). The remaining 30% are associated with glomerular alterations and are termed glomerulopathic LCs (GLCs). Only a few monoclonal LCs are both TLC and GLC (297, 298).
GLCs produce two diametrically opposite diseases in terms of their renal pathological expressions: AL amyloidosis and LC deposition disease (LCDD) (Figures 3 and 4). As was mentioned previously, in AL amyloidosis, the pathogenic monoclonal LC, most commonly of Λ-type, deposits in the extracellular space in the form of randomly arranged nonbranching fibrils with a diameter of 10-12 nm (306). As renal amyloidosis progresses, the mesangial matrix is destroyed and replaced by AL fibrils (Figure 4). The destruction of the native mesangial matrix occurs because of the direct activation of matrix metalloproteinases (MMPs). In contrast, in LCDD, the monoclonal LC, most commonly of κ type, deposits in the mesangium and along glomerular and tubular basement membranes. Electron microscopy analysis shows mesangial and inner glomerular and outer tubular basement membrane deposits of extracellular punctate, powdery, ground-pepper-like deposits (307) (Figure 3). Activation of TGF-β leads to the formation of mesangial nodules and deposition of tenascin replacing the predominant collagen IV matrix that is present in the normal mesangium (297).
The two diseases have been reproduced in experimental platforms permitting crucial evaluation of the step-by-step mechanisms involved. Three experimental platforms, with distinct levels of complexity, have been used to study interactions between GLCs and MCs. The first uses MCs in culture with or without a matrix, which are incubated with GLC. The second is an ex-vivo platform in which an explanted rat or mice kidneys is perfused with the GLC through the renal artery. The third is an in-vivo animal model that involves penile injections of LCs in mice (297). These experimental platforms have provided evidence that the interaction of the GLCs with MCs involves a receptor: SORL1 (298). The interaction GLC-MC triggers a chain of molecular events that determine the pathophysiological response and fate of the cell. It is important to emphasize that, although both the LCs from patients with AL or LCDD interact with the same membrane receptor on MCs, the cellular response, and ultimate pathological changes that they promote differ significantly (298).
When an AL LC interacts with mesangial cells at the cell surfaces, signals are activated, and the LC is endocytosed into the cells and routed to the mature lysosomal system for processing (293, 294, 296). Rab proteins participate in the migration of the monoclonal LC from the mesangial cell surface to the lysosomes. Once in the lysosomes, the stringent low pH and potentially the presence of lysosomal proteases promote LC misfolding and aggregation into amyloid fibrils (298, 308) (Figure 7). The last step involves the exocytosis of the fibrils into the extracellular compartment. Two factors, C-fos and NF-kB, control the mesangial cell activities leading to amyloid fibril formation (309). They are instrumental in fostering cellular activities that control amyloidogenesis. C-fos is necessary for the phenotypic transformation of mesangial cells into a macrophage phenotype, a crucial event that needs to occur before mesangial cells can engage in active amyloid formation (294, 296). The same is true about vasculature where the AL LC interacts with pericytes/smooth muscle cells to produce amyloid, through a process that is amazingly like that taking place in the renal mesangium (295). In contrast, the incubation of MC with LCDD LCs appears to activate different signaling pathways, since LC internalization does not occur. Moreover, treated MCs undergo a completely different phenotypic transformation, in this case toward a myofibroblasts phenotype, acquiring enhanced protein synthesis function (294).
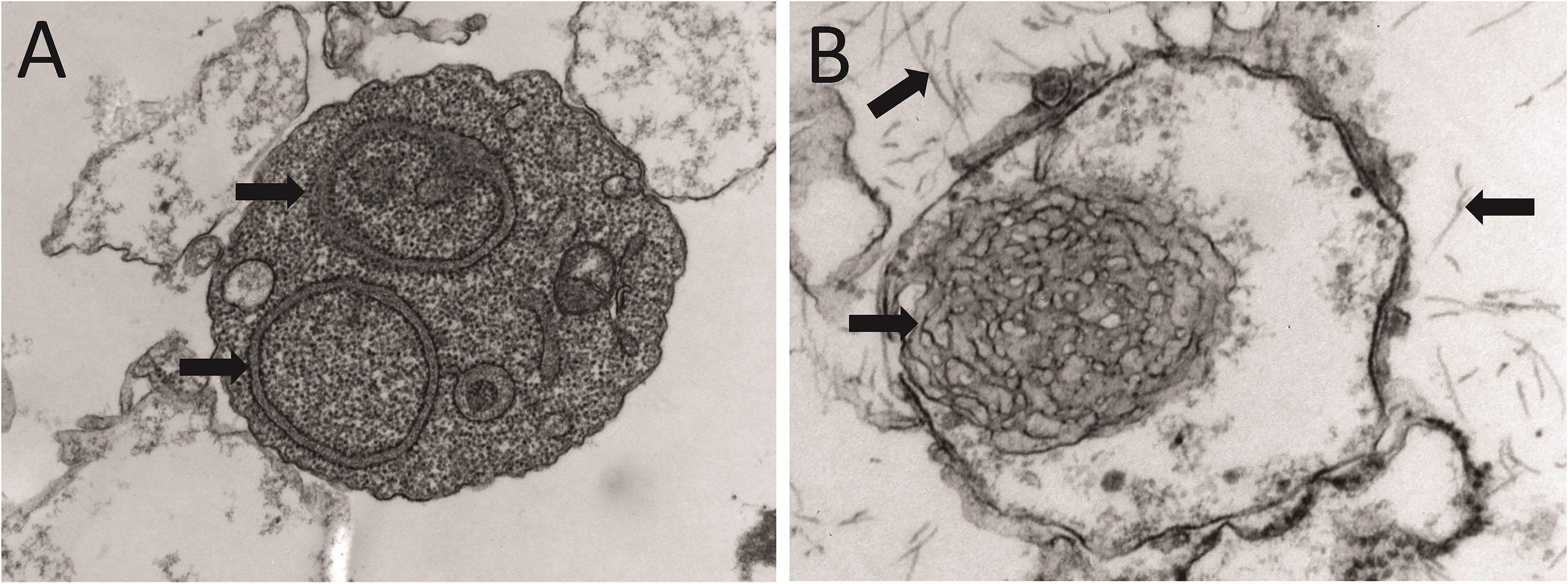
Figure 7 Intralisosomal processing and fibrillar aggregation of an amyloidogenic LC in human mesangial cells. EM micrographs of lysosomes obtained from cultured human mesangial cells that were incubated with an amyloidogenic Λ LC purified from the urine of a patient with AL amyloidosis. After (A) 30 min and (B) 2 h, the cells were harvested, and the lysosomal fraction was obtained by density gradient ultracentrifugation. Note in (A) the intralysosomal amyloid fibrils arranged in perfect circles (black arrows). In (B), the amyloid fibrils show a less orderly arrangement (black arrow inside the lysosome). Note some amyloid fibrils that appear to have escaped from the lysosome (black arrows outside the lysosome).
Overall, these studies strongly suggest that the physicochemical and structural properties of the monoclonal LC influence how MCs respond to the LC interaction. Therefore, both the IGVL gene segment encoding the monoclonal LC and somatic mutations can be anticipated to modulate pathogenic events triggered by GLC-MC interaction, such as cell-mediated LC amyloidogenesis. Determining how the LC sequence modulates the pathogenic response of MCs will significantly expand our understanding of the structural bases of AL amyloidosis and LCDD and may pave the way for developing more effective therapeutic approaches for these devastating diseases.
5 Conclusion
The development of an adaptive immune system in jawed vertebrates gave them the ability to produce antibodies and T-cell receptors specific to virtually any foreign molecule. Such highly diverse repertoire of antibody/T-cell receptor is generated by two main mechanisms, V(D)J gene segment recombination and SHM. Given the very molecular nature of these mechanisms, there is a latent risk of genomic instability that may result in the malignant transformation of the cell. Moreover, the insertion of multiple mutations in the variable region of the antibody LCs by somatic hypermutation can promote misfolding and tissue deposition of this protein if it is secreted in a free state. Important advances have been made in understanding the molecular mechanisms that generate diversity in the adaptive immune system. In the same way, we now know more regarding the causes and mechanisms of the pathological aggregation of LCs, which opens new opportunities for better diagnosis and treatment of patients with AL amyloidosis, LCDD, myeloma (cast) nephropathy, and other disorders caused by LC deposition. However, given the extremely high antibody diversity generated by the aforementioned mechanisms, understanding what governs the pathogenic properties of a LC remains a major challenge.
Author contributions
LP-Y and GH determined the general theme, objectives, and structure of the review, and designed and prepared the figures. All authors contributed to the writing, revision, and correction of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
LP-Y received a seed fund from the University of South Alabama School of Medicine to investigate the mechanism of light chain amyloid aggregation. This fund will be used to pay the publication fees for this review if it is accepted.
Acknowledgments
We thank Adrian Hoff and Jerry W. Resmondo for their valuable contribution to the preparation of figures.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature (2012) 489:231–41. doi: 10.1038/nature11551
2. Amato KR, Jeyakumar T, Poinar H, Gros P. Shifting climates, foods, and diseases: the human microbiome through evolution. Bioessays (2019) 41:e1900034. doi: 10.1002/bies.201900034
3. Murdoch CC, Rawls JF. Commensal microbiota regulate vertebrate innate immunity-insights from the zebrafish. Front Immunol (2019) 10:2100. doi: 10.3389/fimmu.2019.02100
4. Popkes M, Valenzano DR. Microbiota-host interactions shape ageing dynamics. Philos Trans R Soc Lond B Biol Sci (2020) 375:20190596. doi: 10.1098/rstb.2019.0596
5. Zhang H, Luo XM. Control of commensal microbiota by the adaptive immune system. Gut Microbes (2015) 6:156–60. doi: 10.1080/19490976.2015.1031946
6. Xu Z, Takizawa F, Casadei E, Shibasaki Y, Ding Y, Sauters TJC, et al. Specialization of mucosal immunoglobulins in pathogen control and microbiota homeostasis occurred early in vertebrate evolution. Sci Immunol (2020) 5:eaay3254. doi: 10.1126/sciimmunol.aay3254
7. Du Pasquier L. The immune system of invertebrates and vertebrates. Comp Biochem Physiol B Biochem Mol Biol (2001) 129:1–15. doi: 10.1016/S1096-4959(01)00306-2
8. Spoel SH, Dong X. How do plants achieve immunity? defence without specialized immune cells. Nat Rev Immunol (2012) 12:89–100. doi: 10.1038/nri3141
9. Hunter P. No place to run: plants have evolved a dazzling array of chemical defences and regulatory networks that matches the mammalian immune system for complexity. EMBO Rep (2018) 19:e46020. doi: 10.15252/embr.201846020
10. Deng L, Luo M, Velikovsky A, Mariuzza RA. Structural insights into the evolution of the adaptive immune system. Annu Rev Biophys (2013) 42:191–215. doi: 10.1146/annurev-biophys-083012-130422
11. Van Niekerk G, Hattingh SM, Engelbrecht AM. Invertebrates: why no adaptive immune system? Scand J Immunol (2016) 83:160–1. doi: 10.1111/sji.12400
12. Boehm T, Iwanami N, Hess I. Evolution of the immune system in the lower vertebrates. Annu Rev Genomics Hum Genet (2012) 13:127–49. doi: 10.1146/annurev-genom-090711-163747
13. French DL, Laskov R, Scharff MD. The role of somatic hypermutation in the generation of antibody diversity. Science (1989) 244:1152–7. doi: 10.1126/science.2658060
14. Schroeder K, Herrmann M, Winkler TH. The role of somatic hypermutation in the generation of pathogenic antibodies in SLE. Autoimmunity (2013) 46:121–7. doi: 10.3109/08916934.2012.748751
15. Vidal R, Goni F, Stevens F, Aucouturier P, Kumar A, Frangione B, et al. Somatic mutations of the L12a gene in V-kappa(1) light chain deposition disease: potential effects on aberrant protein conformation and deposition. Am J Pathol (1999) 155:2009–17. doi: 10.1016/S0002-9440(10)65520-4
16. Abraham RS, Geyer SM, Ramirez-Alvarado M, Price-Troska TL, Gertz MA, Fonseca R. Analysis of somatic hypermutation and antigenic selection in the clonal b cell in immunoglobulin light chain amyloidosis (AL). J Clin Immunol (2004) 24:340–53. doi: 10.1023/B:JOCI.0000029113.68758.9f
17. Solomon A. Light chains of immunoglobulins: structural-genetic correlates. Blood (1986) 68:603–10. doi: 10.1182/blood.V68.3.603.603
18. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol (2014) 5:520. doi: 10.3389/fimmu.2014.00520
19. Steffen U, Koeleman CA, Sokolova MV, Bang H, Kleyer A, Rech J, et al. IgA subclasses have different effector functions associated with distinct glycosylation profiles. Nat Commun (2020) 11:120. doi: 10.1038/s41467-019-13992-8
20. Sumiyama K, Saitou N, Ueda S. Adaptive evolution of the IgA hinge region in primates. Mol Biol Evol (2002) 19:1093–9. doi: 10.1093/oxfordjournals.molbev.a004167
21. Chintalacharuvu KR, Chuang PD, Dragoman A, Fernandez CZ, Qiu J, Plaut AG, et al. Cleavage of the human immunoglobulin A1 (IgA1) hinge region by IgA1 proteases requires structures in the fc region of IgA. Infect Immun (2003) 71:2563–70. doi: 10.1128/IAI.71.5.2563-2570.2003
22. Patel A, Jialal I. Biochemistry, immunoglobulin a. StatPearls (2022). Treasure Island (FL). Available at: https://pubmed.ncbi.nlm.nih.gov/31855360/
23. Brandtzaeg P, Savilahti E. Further evidence for a role of secretory component (SC) and J chain in the glandular transport of IgA. Adv Exp Med Biol (1978) 107:219–26. doi: 10.1007/978-1-4684-3369-2_26
24. Sathe A, Cusick JK. Biochemistry, immunoglobulin m. StatPearls (2022). Treasure Island (FL). Available at: https://pubmed.ncbi.nlm.nih.gov/32310455/
25. Liu H, May K. Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function. MAbs (2012) 4:17–23. doi: 10.4161/mabs.4.1.18347
26. Feige MJ, Hendershot LM, Buchner J. How antibodies fold. Trends Biochem Sci (2010) 35:189–98. doi: 10.1016/j.tibs.2009.11.005
27. Nguyen MN, Pradhan MR, Verma C, Zhong P. The interfacial character of antibody paratopes: analysis of antibody-antigen structures. Bioinformatics (2017) 33:2971–6. doi: 10.1093/bioinformatics/btx389
28. Yoshida K, Kuroda D, Kiyoshi M, Nakakido M, Nagatoishi S, Soga S, et al. Exploring designability of electrostatic complementarity at an antigen-antibody interface directed by mutagenesis, biophysical analysis, and molecular dynamics simulations. Sci Rep (2019) 9:4482. doi: 10.1038/s41598-019-40461-5
29. Bhat TN, Bentley GA, Boulot G, Greene MI, Tello D, Dall'acqua W, et al. Bound water molecules and conformational stabilization help mediate an antigen-antibody association. Proc Natl Acad Sci USA (1994) 91:1089–93. doi: 10.1073/pnas.91.3.1089
30. Yokota A, Tsumoto K, Shiroishi M, Kondo H, Kumagai I. The role of hydrogen bonding via interfacial water molecules in antigen-antibody complexation. the HyHEL-10-HEL interaction. J Biol Chem (2003) 278:5410–8. doi: 10.1074/jbc.M210182200
31. Giudicelli V, Chaume D, Lefranc MP. IMGT/GENE-DB: a comprehensive database for human and mouse immunoglobulin and T cell receptor genes. Nucleic Acids Res (2005) 33:D256–261. doi: 10.1093/nar/gki010
32. Bentley DL, Rabbitts TH. Evolution of immunoglobulin V genes: evidence indicating that recently duplicated human V kappa sequences have diverged by gene conversion. Cell (1983) 32:181–9. doi: 10.1016/0092-8674(83)90508-1
33. Retter I, Althaus HH, Munch R, Muller W. VBASE2, an integrative V gene database. Nucleic Acids Res (2005) 33:D671–674. doi: 10.1093/nar/gki088
34. Bridges SL Jr. Frequent n addition and clonal relatedness among immunoglobulin lambda light chains expressed in rheumatoid arthritis synovia and PBL, and the influence of V lambda gene segment utilization on CDR3 length. Mol Med (1998) 4:525–53. doi: 10.1007/BF03401757
35. Gilfillan S, Benoist C, Mathis D. Mice lacking terminal deoxynucleotidyl transferase: adult mice with a fetal antigen receptor repertoire. Immunol Rev (1995) 148:201–19. doi: 10.1111/j.1600-065X.1995.tb00099.x
36. Briney BS, Crowe JE Jr. Secondary mechanisms of diversification in the human antibody repertoire. Front Immunol (2013) 4:42. doi: 10.3389/fimmu.2013.00042
37. Grundy GJ, Ramon-Maiques S, Dimitriadis EK, Kotova S, Biertumpfel C, Heymann JB, et al. Initial stages of V(D)J recombination: the organization of RAG1/2 and RSS DNA in the postcleavage complex. Mol Cell (2009) 35:217–27. doi: 10.1016/j.molcel.2009.06.022
38. Kim MS, Lapkouski M, Yang W, Gellert M. Crystal structure of the V(D)J recombinase RAG1-RAG2. Nature (2015) 518:507–11. doi: 10.1038/nature14174
39. Ru H, Chambers MG, Fu TM, Tong AB, Liao M, Wu H. Molecular mechanism of V(D)J recombination from synaptic RAG1-RAG2 complex structures. Cell (2015) 163:1138–52. doi: 10.1016/j.cell.2015.10.055
40. Oettinger MA, Schatz DG, Gorka C, Baltimore D. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science (1990) 248:1517–23. doi: 10.1126/science.2360047
41. Glynn RA, Bassing CH. From RAG2 to T cell riches and future fortunes. J Immunol (2019) 202:1315–6. doi: 10.4049/jimmunol.1900010
42. Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol (2017) 18:495–506. doi: 10.1038/nrm.2017.48
43. Chi X, Li Y, Qiu X. V(D)J recombination, somatic hypermutation and class switch recombination of immunoglobulins: mechanism and regulation. Immunology (2020) 160:233–47. doi: 10.1111/imm.13176
44. Little AJ, Corbett E, Ortega F, Schatz DG. Cooperative recruitment of HMGB1 during V(D)J recombination through interactions with RAG1 and DNA. Nucleic Acids Res (2013) 41:3289–301. doi: 10.1093/nar/gks1461
45. Ciubotaru M, Trexler AJ, Spiridon LN, Surleac MD, Rhoades E, Petrescu AJ, et al. RAG and HMGB1 create a large bend in the 23RSS in the V(D)J recombination synaptic complexes. Nucleic Acids Res (2013) 41:2437–54. doi: 10.1093/nar/gks1294
46. Thwaites DT, Carter C, Lawless D, Savic S, Boyes JM. A novel RAG1 mutation reveals a critical in vivo role for HMGB1/2 during V(D)J recombination. Blood (2019) 133:820–9. doi: 10.1182/blood-2018-07-866939
47. Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell (2002) 108:781–94. doi: 10.1016/S0092-8674(02)00671-2
48. Niewolik D, Pannicke U, Lu H, Ma Y, Wang LC, Kulesza P, et al. DNA-PKcs dependence of Artemis endonucleolytic activity, differences between hairpins and 5' or 3' overhangs. J Biol Chem (2006) 281:33900–9. doi: 10.1074/jbc.M606023200
49. Hwang JK, Alt FW, Yeap LS. Related mechanisms of antibody somatic hypermutation and class switch recombination. Microbiol Spectr (2015) 3:MDNA3–0037-2014. doi: 10.1128/9781555819217.ch15
50. Villa A, Santagata S, Bozzi F, Imberti L, Notarangelo LD. Omenn syndrome: a disorder of Rag1 and Rag2 genes. J Clin Immunol (1999) 19:87–97. doi: 10.1023/A:1020550432126
51. Tabori U, Mark Z, Amariglio N, Etzioni A, Golan H, Biloray B, et al. Detection of RAG mutations and prenatal diagnosis in families presenting with either T-b- severe combined immunodeficiency or omenn's syndrome. Clin Genet (2004) 65:322–6. doi: 10.1111/j.1399-0004.2004.00227.x
52. Marrella V, Maina V, Villa A. Omenn syndrome does not live by V(D)J recombination alone. Curr Opin Allergy Clin Immunol (2011) 11:525–31. doi: 10.1097/ACI.0b013e32834c311a
53. Valeri L, Lugli L, Iughetti L, Soresina A, Giliani S, Porta F, et al. Omenn syndrome due to RAG1 mutation presenting with nonimmune hydrops fetalis in two siblings. Pediatrics (2022) 149;e2021052411. doi: 10.1542/peds.2021-052411
54. Zhang X, Kang X, Yang M, Cai Z, Song Y, Zhou X, et al. A variant of RAG1 gene identified in severe combined immunodeficiency: a case report. BMC Pediatr (2023) 23:56. doi: 10.1186/s12887-022-03822-0
55. Hromas R, Williamson E, Lee SH, Nickoloff J. Preventing the chromosomal translocations that cause cancer. Trans Am Clin Climatol Assoc (2016) 127:176–95.
56. Rommel PC, Oliveira TY, Nussenzweig MC, Robbiani DF. RAG1/2 induces genomic insertions by mobilizing DNA into RAG1/2-independent breaks. J Exp Med (2017) 214:815–31. doi: 10.1084/jem.20161638
57. Kirkham CM, Scott JNF, Wang X, Smith AL, Kupinski AP, Ford AM, et al. Cut-and-Run: a distinct mechanism by which V(D)J recombination causes genome instability. Mol Cell (2019) 74:584–597.e589. doi: 10.1016/j.molcel.2019.02.025
58. Thomson DW, Shahrin NH, Wang PPS, Wadham C, Shanmuganathan N, Scott HS, et al. Aberrant RAG-mediated recombination contributes to multiple structural rearrangements in lymphoid blast crisis of chronic myeloid leukemia. Leukemia (2020) 34:2051–63. doi: 10.1038/s41375-020-0751-y
59. Jakobczyk H, Jiang Y, Debaize L, Soubise B, Avner S, Serandour AA, et al. ETV6-RUNX1 and RUNX1 directly regulate RAG1 expression: one more step in the understanding of childhood b-cell acute lymphoblastic leukemia leukemogenesis. Leukemia (2022) 36:549–54. doi: 10.1038/s41375-021-01409-9
60. Paranjape AM, Desai SS, Nishana M, Roy U, Nilavar NM, Mondal A, et al. Nonamer dependent RAG cleavage at CpGs can explain mechanism of chromosomal translocations associated to lymphoid cancers. PLoS Genet (2022) 18:e1010421. doi: 10.1371/journal.pgen.1010421
61. Sterrenberg JN, Folkerts ML, Rangel V, Lee SE, Pannunzio NR. Diversity upon diversity: linking DNA double-strand break repair to blood cancer health disparities. Trends Cancer (2022) 8:328–43. doi: 10.1016/j.trecan.2022.01.003
62. Miyazaki K, Miyazaki M. The interplay between chromatin architecture and lineage-specific transcription factors and the regulation of rag gene expression. Front Immunol (2021) 12:659761. doi: 10.3389/fimmu.2021.659761
63. Nezlin R. Combinatorial events in generation of antibody diversity. Comb Chem High Throughput Screen (2001) 4:377–83. doi: 10.2174/1386207013330977
64. Elhanati Y, Sethna Z, Marcou Q, Callan CG Jr., Mora T, Walczak AM. Inferring processes underlying b-cell repertoire diversity. Philos Trans R Soc Lond B Biol Sci (2015) 370:20140243. doi: 10.1098/rstb.2014.0243
65. Imkeller K, Wardemann H. Assessing human b cell repertoire diversity and convergence. Immunol Rev (2018) 284:51–66. doi: 10.1111/imr.12670
66. Boyd SD, Joshi SA. High-throughput DNA sequencing analysis of antibody repertoires. Microbiol Spectr (2014) 2. doi: 10.1128/microbiolspec.AID-0017-2014
67. Rees AR. Understanding the human antibody repertoire. MAbs (2020) 12:1729683. doi: 10.1080/19420862.2020.1729683
68. Rothstein TL. Polyreactive low-affinity IgM antibodies produced by CD5+ b cells. Immunol Today (1990) 11:152. doi: 10.1016/0167-5699(90)90062-E
69. Jones DD, Deiulio GA, Winslow GM. Antigen-driven induction of polyreactive IgM during intracellular bacterial infection. J Immunol (2012) 189:1440–7. doi: 10.4049/jimmunol.1200878
70. Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem (2007) 76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740
71. Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, et al. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center b cells. J Biol Chem (1999) 274:18470–6. doi: 10.1074/jbc.274.26.18470
72. Casali P, Pal Z, Xu Z, Zan H. DNA Repair in antibody somatic hypermutation. Trends Immunol (2006) 27:313–21. doi: 10.1016/j.it.2006.05.001
73. Franklin A, Steele EJ. RNA-Directed DNA repair and antibody somatic hypermutation. Trends Genet (2022) 38:426–36. doi: 10.1016/j.tig.2021.10.005
74. Cogne M. Activation-induced deaminase in b lymphocyte maturation and beyond. BioMed J (2013) 36:259–68. doi: 10.4103/2319-4170.113191
75. Wei L, Chahwan R, Wang S, Wang X, Pham PT, Goodman MF, et al. Overlapping hotspots in CDRs are critical sites for V region diversification. Proc Natl Acad Sci U. S .A. (2015) 112:E728–737. doi: 10.1073/pnas.1500788112
76. Hershberg U, Shlomchik MJ. Differences in potential for amino acid change after mutation reveals distinct strategies for kappa and lambda light-chain variation. Proc Natl Acad Sci USA (2006) 103:15963–8.
77. Del Pozo-Yauner L, Becerril B, Ochoa-Leyva L, Rodríguez-Ambriz SL, Pérez Carrión JI, Zavala-Padilla G, et al. The structural determinants of the immunoglobulin light chain amyloid aggregation. In: Olivares-Quiroz L. G-LO, Jardón-Valadez H, editors. Physical biology of proteins and peptides. Switzerland: Springer, Cham (2015). p. 1–28.
78. Del Pozo-Yauner L, Turbat-Herrera EA, Pérez-Carreón JI, Herrera GA. From the light chain sequence to the tissue microenvironment: contribution of the mesangial cells to glomerular amyloidosis. Hemato (2022) 3:232–67. doi: 10.3390/hemato3010019
79. Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese LS. Structure of the human MutSalpha DNA lesion recognition complex. Mol Cell (2007) 26:579–92. doi: 10.1016/j.molcel.2007.04.018
80. Shang Y, Meng FL. Repair of programmed DNA lesions in antibody class switch recombination: common and unique features. Genome Instab Dis (2021) 2:115–25. doi: 10.1007/s42764-021-00035-0
81. Sale JE. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb Perspect Biol (2013) 5:a012708. doi: 10.1101/cshperspect.a012708
82. Matsuda T, Bebenek K, Masutani C, Rogozin IB, Hanaoka F, Kunkel TA. Error rate and specificity of human and murine DNA polymerase eta. J Mol Biol (2001) 312:335–46. doi: 10.1006/jmbi.2001.4937
83. Alexeeva M, Moen MN, Xu XM, Rasmussen A, Leiros I, Kirpekar F, et al. Intrinsic strand-incision activity of human UNG: implications for nick generation in immunoglobulin gene diversification. Front Immunol (2021) 12:762032. doi: 10.3389/fimmu.2021.762032
84. Stratigopoulou M, Van Dam TP, Guikema JEJ. Base excision repair in the immune system: small DNA lesions with big consequences. Front Immunol (2020) 11:1084. doi: 10.3389/fimmu.2020.01084
85. Tang Q, Caglayan M. The scaffold protein XRCC1 stabilizes the formation of polbeta/gap DNA and ligase IIIalpha/nick DNA complexes in base excision repair. J Biol Chem (2021) 297:101025. doi: 10.1016/j.jbc.2021.101025
86. Harriman W, Volk H, Defranoux N, Wabl M. Immunoglobulin class switch recombination. Annu Rev Immunol (1993) 11:361–84. doi: 10.1146/annurev.iy.11.040193.002045
87. Viret C, Gurr W. The origin of the "one cell-one antibody" rule. J Immunol (2009) 182:1229–30. doi: 10.4049/jimmunol.182.3.1229
88. Mills FC, Brooker JS, Camerini-Otero RD. Sequences of human immunoglobulin switch regions: implications for recombination and transcription. Nucleic Acids Res (1990) 18:7305–16. doi: 10.1093/nar/18.24.7305
89. Gritzmacher CA. Molecular aspects of heavy-chain class switching. Crit Rev Immunol (1989) 9:173–200.
90. Schmitz J, Radbruch A. An interleukin 4-induced DNase I hypersensitive site indicates opening of the gamma 1 switch region prior to switch recombination. Int Immunol (1989) 1:570–5. doi: 10.1093/intimm/1.6.570
91. Li SC, Rothman PB, Zhang J, Chan C, Hirsh D, Alt FW. Expression of I mu-c gamma hybrid germline transcripts subsequent to immunoglobulin heavy chain class switching. Int Immunol (1994) 6:491–7. doi: 10.1093/intimm/6.4.491
92. Qiao Q, Wang L, Meng FL, Hwang JK, Alt FW, Wu H. AID recognizes structured DNA for class switch recombination. Mol Cell (2017) 67:361–373.e364. doi: 10.1016/j.molcel.2017.06.034
93. Spiegel J, Adhikari S, Balasubramanian S. The structure and function of DNA G-quadruplexes. Trends Chem (2020) 2:123–36. doi: 10.1016/j.trechm.2019.07.002
94. Deze O, Laffleur B, Cogne M. Roles of G4-DNA and G4-RNA in class switch recombination and additional regulations in b-lymphocytes. Molecules (2023) 28;1159. doi: 10.3390/molecules28031159
95. Xu Z, Fulop Z, Wu G, Pone EJ, Zhang J, Mai T, et al. 14-3-3 adaptor proteins recruit AID to 5'-AGCT-3'-rich switch regions for class switch recombination. Nat Struct Mol Biol (2010) 17:1124–35. doi: 10.1038/nsmb.1884
96. Pasqualucci L, Kitaura Y, Gu H, Dalla-Favera R. PKA-mediated phosphorylation regulates the function of activation-induced deaminase (AID) in b cells. Proc Natl Acad Sci USA (2006) 103:395–400. doi: 10.1073/pnas.0509969103
97. Chaudhuri J, Khuong C, Alt FW. Replication protein a interacts with AID to promote deamination of somatic hypermutation targets. Nature (2004) 430:992–8. doi: 10.1038/nature02821
98. Matthews AJ, Zheng S, Dimenna LJ, Chaudhuri J. Regulation of immunoglobulin class-switch recombination: choreography of noncoding transcription, targeted DNA deamination, and long-range DNA repair. Adv Immunol (2014) 122:1–57. doi: 10.1016/B978-0-12-800267-4.00001-8
99. Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol (2002) 12:1748–55. doi: 10.1016/S0960-9822(02)01215-0
100. Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both ig switching and the A/T-focused phase of somatic mutation. Mol Cell (2004) 16:163–71. doi: 10.1016/j.molcel.2004.10.011
101. Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol (2008) 26:261–92. doi: 10.1146/annurev.immunol.26.021607.090248
102. Igarashi K, Ochiai K, Itoh-Nakadai A, Muto A. Orchestration of plasma cell differentiation by Bach2 and its gene regulatory network. Immunol Rev (2014) 261:116–25. doi: 10.1111/imr.12201
103. Budzynska PM, Kylaniemi MK, Kallonen T, Soikkeli AI, Nera KP, Lassila O, et al. Bach2 regulates AID-mediated immunoglobulin gene conversion and somatic hypermutation in DT40 b cells. Eur J Immunol (2017) 47:993–1001. doi: 10.1002/eji.201646895
104. Ochiai K, Igarashi K. Exploring novel functions of BACH2 in the acquisition of antigen-specific antibodies. Int Immunol (2022) 35(6):257–65. doi: 10.1093/intimm/dxac065
105. De Yebenes VG, Belver L, Pisano DG, Gonzalez S, Villasante A, Croce C, et al. miR-181b negatively regulates activation-induced cytidine deaminase in b cells. J Exp Med (2008) 205:2199–206. doi: 10.1084/jem.20080579
106. Elton TS, Selemon H, Elton SM, Parinandi NL. Regulation of the MIR155 host gene in physiological and pathological processes. Gene (2013) 532:1–12. doi: 10.1016/j.gene.2012.12.009
107. Recaldin T, Hobson PS, Mann EH, Ramadani F, Cousins DJ, Lavender P, et al. miR-29b directly targets activation-induced cytidine deaminase in human b cells and can limit its inappropriate expression in naive b cells. Mol Immunol (2018) 101:419–28. doi: 10.1016/j.molimm.2018.07.028
108. Mu Y, Mcbride KM. Targeting mutagenesis in b cells: phosphorylation goes beyond AID association. Mol Cell Oncol (2018) 5:e1432259. doi: 10.1080/23723556.2018.1432259
109. Mcbride KM, Gazumyan A, Woo EM, Schwickert TA, Chait BT, Nussenzweig MC. Regulation of class switch recombination and somatic mutation by AID phosphorylation. J Exp Med (2008) 205:2585–94. doi: 10.1084/jem.20081319
110. Gazumyan A, Timachova K, Yuen G, Siden E, Di Virgilio M, Woo EM, et al. Amino-terminal phosphorylation of activation-induced cytidine deaminase suppresses c-myc/IgH translocation. Mol Cell Biol (2011) 31:442–9. doi: 10.1128/MCB.00349-10
111. Hu Y, Ericsson I, Doseth B, Liabakk NB, Krokan HE, Kavli B. Activation-induced cytidine deaminase (AID) is localized to subnuclear domains enriched in splicing factors. Exp Cell Res (2014) 322:178–92. doi: 10.1016/j.yexcr.2014.01.004
112. Chao J, Rothschild G, Basu U. Ubiquitination events that regulate recombination of immunoglobulin loci gene segments. Front Immunol (2014) 5:100. doi: 10.3389/fimmu.2014.00100
113. Bello A, Muller A, Hirth G, Giebeler LN, Bottcher K, Voigt S, et al. Cell cycle-mediated regulation of secondary ig diversification. J Immunol (2023) 210(10):1508–18. doi: 10.4049/jimmunol.2100880
114. Endo Y, Marusawa H, Chiba T. Involvement of activation-induced cytidine deaminase in the development of colitis-associated colorectal cancers. J Gastroenterol (2011) 46 Suppl 1:6–10. doi: 10.1007/s00535-010-0326-1
115. Mechtcheriakova D, Svoboda M, Meshcheryakova A, Jensen-Jarolim E. Activation-induced cytidine deaminase (AID) linking immunity, chronic inflammation, and cancer. Cancer Immunol Immunother (2012) 61:1591–8. doi: 10.1007/s00262-012-1255-z
116. Shimizu T, Marusawa H, Endo Y, Chiba T. Inflammation-mediated genomic instability: roles of activation-induced cytidine deaminase in carcinogenesis. Cancer Sci (2012) 103:1201–6. doi: 10.1111/j.1349-7006.2012.02293.x
117. Machida K, Cheng KT, Pavio N, Sung VM, Lai MM. Hepatitis c virus E2-CD81 interaction induces hypermutation of the immunoglobulin gene in b cells. J Virol (2005) 79:8079–89. doi: 10.1128/JVI.79.13.8079-8089.2005
118. Ito M, Murakami K, Suzuki T, Mochida K, Suzuki M, Ikebuchi K, et al. Enhanced expression of lymphomagenesis-related genes in peripheral blood b cells of chronic hepatitis c patients. Clin Immunol (2010) 135:459–65. doi: 10.1016/j.clim.2010.02.002
119. Pauklin S, Sernandez IV, Bachmann G, Ramiro AR, Petersen-Mahrt SK. Estrogen directly activates AID transcription and function. J Exp Med (2009) 206:99–111. doi: 10.1084/jem.20080521
120. Incorvaia E, Sicouri L, Petersen-Mahrt SK, Schmitz KM. Hormones and AID: balancing immunity and autoimmunity. Autoimmunity (2013) 46:128–37. doi: 10.3109/08916934.2012.748752
121. Frasca D, Andrisani G, Diaz A, Felice C, Guidi L, Blomberg BB. AID in aging and autoimmune diseases. Autoimmunity (2013) 46:168–75. doi: 10.3109/08916934.2012.750300
122. Takai A, Marusawa H, Chiba T. Acquisition of genetic aberrations by activation-induced cytidine deaminase (AID) during inflammation-associated carcinogenesis. Cancers (Basel) (2011) 3:2750–66. doi: 10.3390/cancers3022750
123. Gushima M, Hirahashi M, Matsumoto T, Fujita K, Ohuchida K, Oda Y, et al. Expression of activation-induced cytidine deaminase in ulcerative colitis-associated carcinogenesis. Histopathology (2011) 59:460–9. doi: 10.1111/j.1365-2559.2011.03965.x
124. Takai A, Marusawa H, Minaki Y, Watanabe T, Nakase H, Kinoshita K, et al. Targeting activation-induced cytidine deaminase prevents colon cancer development despite persistent colonic inflammation. Oncogene (2012) 31:1733–42. doi: 10.1038/onc.2011.352
125. Kou T, Marusawa H, Kinoshita K, Endo Y, Okazaki IM, Ueda Y, et al. Expression of activation-induced cytidine deaminase in human hepatocytes during hepatocarcinogenesis. Int J Cancer (2007) 120:469–76. doi: 10.1002/ijc.22292
126. Matsumoto T, Shimizu T, Nishijima N, Ikeda A, Eso Y, Matsumoto Y, et al. Hepatic inflammation facilitates transcription-associated mutagenesis via AID activity and enhances liver tumorigenesis. Carcinogenesis (2015) 36:904–13. doi: 10.1093/carcin/bgv065
127. Nonaka T, Toda Y, Hiai H, Uemura M, Nakamura M, Yamamoto N, et al. Involvement of activation-induced cytidine deaminase in skin cancer development. J Clin Invest (2016) 126:1367–82. doi: 10.1172/JCI81522
128. Matsushita M, Iwasaki T, Nonaka D, Kuwamoto S, Nagata K, Kato M, et al. Higher expression of activation-induced cytidine deaminase is significantly associated with merkel cell polyomavirus-negative merkel cell carcinomas. Yonago Acta Med (2017) 60:145–53. doi: 10.33160/yam.2017.09.002
129. Nakanishi Y, Kondo S, Wakisaka N, Tsuji A, Endo K, Murono S, et al. Role of activation-induced cytidine deaminase in the development of oral squamous cell carcinoma. PLoS One (2013) 8:e62066. doi: 10.1371/journal.pone.0062066
130. Koduru S, Wong E, Strowig T, Sundaram R, Zhang L, Strout MP, et al. Dendritic cell-mediated activation-induced cytidine deaminase (AID)-dependent induction of genomic instability in human myeloma. Blood (2012) 119:2302–9. doi: 10.1182/blood-2011-08-376236
131. Bolli N, Maura F, Minvielle S, Gloznik D, Szalat R, Fullam A, et al. Genomic patterns of progression in smoldering multiple myeloma. Nat Commun (2018) 9:3363. doi: 10.1038/s41467-018-05058-y
132. Maura F, Rustad EH, Yellapantula V, Luksza M, Hoyos D, Maclachlan KH, et al. Role of AID in the temporal pattern of acquisition of driver mutations in multiple myeloma. Leukemia (2020) 34:1476–80. doi: 10.1038/s41375-019-0689-0
133. Rustad EH, Yellapantula V, Leongamornlert D, Bolli N, Ledergor G, Nadeu F, et al. Timing the initiation of multiple myeloma. Nat Commun (2020) 11:1917. doi: 10.1038/s41467-020-15740-9
134. Greisman HA, Lu Z, Tsai AG, Greiner TC, Yi HS, Lieber MR. IgH partner breakpoint sequences provide evidence that AID initiates t(11;14) and t(8;14) chromosomal breaks in mantle cell and burkitt lymphomas. Blood (2012) 120:2864–7. doi: 10.1182/blood-2012-02-412791
135. Bal S, Kumar SK, Fonseca R, Gay F, Hungria VT, Dogan A, et al. Multiple myeloma with t(11;14): unique biology and evolving landscape. Am J Cancer Res (2022) 12:2950–65.
136. Hayman SR, Bailey RJ, Jalal SM, Ahmann GJ, Dispenzieri A, Gertz MA, et al. Translocations involving the immunoglobulin heavy-chain locus are possible early genetic events in patients with primary systemic amyloidosis. Blood (2001) 98:2266–8. doi: 10.1182/blood.V98.7.2266
137. Bryce AH, Ketterling RP, Gertz MA, Lacy M, Knudson RA, Zeldenrust S, et al. Translocation t(11;14) and survival of patients with light chain (AL) amyloidosis. Haematologica (2009) 94:380–6. doi: 10.3324/haematol.13369
138. Xu L, Su Y. Genetic pathogenesis of immunoglobulin light chain amyloidosis: basic characteristics and clinical applications. Exp Hematol Oncol (2021) 10:43. doi: 10.1186/s40164-021-00236-z
139. Muchtar E, Dispenzieri A, Kumar SK, Ketterling RP, Dingli D, Lacy MQ, et al. Interphase fluorescence in situ hybridization in untreated AL amyloidosis has an independent prognostic impact by abnormality type and treatment category. Leukemia (2017) 31:1562–9. doi: 10.1038/leu.2016.369
140. Tandon N, Sidana S, Rajkumar SV, Dispenzieri A, Gertz MA, Lacy MQ, et al. Predictors of early treatment failure following initial therapy for systemic immunoglobulin light-chain amyloidosis. Amyloid (2017) 24:183–8. doi: 10.1080/13506129.2017.1351354
141. Dumas B, Yameen H, Sarosiek S, Sloan JM, Sanchorawala V. Presence of t(11;14) in AL amyloidosis as a marker of response when treated with a bortezomib-based regimen. Amyloid (2020) 27:244–9. doi: 10.1080/13506129.2020.1778461
142. Worn A, Pluckthun A. Mutual stabilization of VL and VH in single-chain antibody fragments, investigated with mutants engineered for stability. Biochemistry (1998) 37:13120–7. doi: 10.1021/bi980712q
143. Bellotti V, Mangione P, Merlini G. Review: immunoglobulin light chain amyloidosis–the archetype of structural and pathogenic variability. J Struct Biol (2000) 130:280–9. doi: 10.1006/jsbi.2000.4248
144. Luo J, Obmolova G, Huang A, Strake B, Teplyakov A, Malia T, et al. Coevolution of antibody stability and vkappa CDR-L3 canonical structure. J Mol Biol (2010) 402:708–19. doi: 10.1016/j.jmb.2010.08.009
145. Richardson JS, Richardson DC. Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci USA (2002) 99:2754–9. doi: 10.1073/pnas.052706099
146. Cheng PN, Pham JD, Nowick JS. The supramolecular chemistry of beta-sheets. J Am Chem Soc (2013) 135:5477–92. doi: 10.1021/ja3088407
147. Holding S, Spradbery D, Hoole R, Wilmot R, Shields ML, Levoguer AM, et al. Use of serum free light chain analysis and urine protein electrophoresis for detection of monoclonal gammopathies. Clin Chem Lab Med (2011) 49:83–8. doi: 10.1515/CCLM.2011.010
148. Messiaen T, Deret S, Mougenot B, Bridoux F, Dequiedt P, Dion JJ, et al. Adult fanconi syndrome secondary to light chain gammopathy. clinicopathologic heterogeneity and unusual features in 11 patients. Med (Baltimore) (2000) 79:135–54. doi: 10.1097/00005792-200005000-00002
149. Sathick IJ, Drosou ME, Leung N. Myeloma light chain cast nephropathy, a review. J Nephrol (2019) 32:189–98. doi: 10.1007/s40620-018-0492-4
150. Palladini G, Milani P. Diagnosis and treatment of AL amyloidosis. Drugs (2023) 83:203–16. doi: 10.1007/s40265-022-01830-z
151. Sabinot A, Ghetti G, Pradelli L, Bellucci S, Lausi A, Palladini G. State-of-the-art review on AL amyloidosis in Western countries: epidemiology, health economics, risk assessment and therapeutic management of a rare disease. Blood Rev (2023) 101040. doi: 10.1016/j.blre.2023.101040
152. Helms LR, Wetzel R. Specificity of abnormal assembly in immunoglobulin light chain deposition disease and amyloidosis. J Mol Biol (1996) 257:77–86. doi: 10.1006/jmbi.1996.0148
153. Stevens FJ, Pokkuluri PR, Schiffer M. Protein conformation and disease: pathological consequences of analogous mutations in homologous proteins. Biochemistry (2000) 39:15291–6. doi: 10.1021/bi001017x
154. Sikkink LA, Ramirez-Alvarado M. Biochemical and aggregation analysis of bence Jones proteins from different light chain diseases. Amyloid (2008) 15:29–39. doi: 10.1080/13506120701815324
155. Hwa YL, Fogaren T, Sams A, Faller DV, Stull DM, Thuenemann S, et al. Immunoglobulin light-chain amyloidosis: clinical presentations and diagnostic approach. J Adv Pract Oncol (2019) 10:470–81. doi: 10.6004/jadpro.2019.10.5.5
156. Li G, Han D, Wei S, Wang H, Chen L. Multiorgan involvement by amyloid light chain amyloidosis. J Int Med Res (2019) 47:1778–86. doi: 10.1177/0300060518814337
157. Kyle RA, Maldonado JE, Bayrd ED. Idiopathic bence Jones proteinuria–a distinct entity? Am J Med (1973) 55:222–6. doi: 10.1016/0002-9343(73)90172-1
158. Kyle RA, Greipp PR. "Idiopathic" bence Jones proteinuria: long-term follow-up in seven patients. N Engl J Med (1982) 306:564–7. doi: 10.1056/NEJM198203113061002
159. Kanoh T, Ohnaka T, Uchino H, Fujii H. The outcome of idiopathic bence Jones proteinuria. Tohoku J Exp Med (1987) 151:121–6. doi: 10.1620/tjem.151.121
160. Stevens PW, Raffen R, Hanson DK, Deng YL, Berrios-Hammond M, Westholm FA, et al. Recombinant immunoglobulin variable domains generated from synthetic genes provide a system for in vitro characterization of light-chain amyloid proteins. Protein Sci (1995) 4:421–32. doi: 10.1002/pro.5560040309
161. Mian M, Franz I, Wasle I, Herold M, Griesmacher A, Prokop W, et al. "Idiopathic bence-Jones proteinuria": a new characterization of an old entity. Ann Hematol (2013) 92:1263–70. doi: 10.1007/s00277-013-1739-8
162. Kyle RA, Larson DR, Therneau TM, Dispenzieri A, Melton LJ 3rd, Benson JT, et al. Clinical course of light-chain smouldering multiple myeloma (idiopathic bence Jones proteinuria): a retrospective cohort study. Lancet Haematol (2014) 1:e28–36. doi: 10.1016/S2352-3026(14)70001-8
163. Jiang X, Myatt E, Lykos P, Stevens FJ. Interaction between glycosaminoglycans and immunoglobulin light chains. Biochemistry (1997) 36:13187–94. doi: 10.1021/bi970408h
164. Davis DP, Gallo G, Vogen SM, Dul JL, Sciarretta KL, Kumar A, et al. Both the environment and somatic mutations govern the aggregation pathway of pathogenic immunoglobulin light chain. J Mol Biol (2001) 313:1021–34. doi: 10.1006/jmbi.2001.5092
165. Lorenz EC, Sethi S, Poshusta TL, Ramirez-Alvarado M, Kumar S, Lager DJ, et al. Renal failure due to combined cast nephropathy, amyloidosis and light-chain deposition disease. Nephrol Dial Transplant (2010) 25:1340–3. doi: 10.1093/ndt/gfp735
166. Ami D, Lavatelli F, Rognoni P, Palladini G, Raimondi S, Giorgetti S, et al. In situ characterization of protein aggregates in human tissues affected by light chain amyloidosis: a FTIR microspectroscopy study. Sci Rep (2016) 6:29096. doi: 10.1038/srep29096
167. Comenzo RL, Zhang Y, Martinez C, Osman K, Herrera GA. The tropism of organ involvement in primary systemic amyloidosis: contributions of ig V(L) germ line gene use and clonal plasma cell burden. Blood (2001) 98:714–20. doi: 10.1182/blood.V98.3.714
168. Perfetti V, Casarini S, Palladini G, Vignarelli MC, Klersy C, Diegoli M, et al. Analysis of v(lambda)-j(lambda) expression in plasma cells from primary (AL) amyloidosis and normal bone marrow identifies 3r (lambdaIII) as a new amyloid-associated germline gene segment. Blood (2002) 100:948–53. doi: 10.1182/blood-2002-01-0114
169. Del Pozo Yauner L, Ortiz E, Sanchez R, Sanchez-Lopez R, Guereca L, Murphy CL, et al. Influence of the germline sequence on the thermodynamic stability and fibrillogenicity of human lambda 6 light chains. Proteins (2008) 72:684–92. doi: 10.1002/prot.21934
170. Kourelis TV, Dasari S, Theis JD, Ramirez-Alvarado M, Kurtin PJ, Gertz MA, et al. Clarifying immunoglobulin gene usage in systemic and localized immunoglobulin light-chain amyloidosis by mass spectrometry. Blood (2017) 129:299–306. doi: 10.1182/blood-2016-10-743997
171. Sidana S, Dasari S, Kourelis TV, Dispenzieri A, Murray DL, King RL, et al. IGVL gene region usage correlates with distinct clinical presentation in IgM vs non-IgM light chain amyloidosis. Blood Adv (2021) 5:2101–5. doi: 10.1182/bloodadvances.2020003671
172. Hurle MR, Helms LR, Li L, Chan W, Wetzel R. A role for destabilizing amino acid replacements in light-chain amyloidosis. Proc Natl Acad Sci USA (1994) 91:5446–50. doi: 10.1073/pnas.91.12.5446
173. Wetzel R. Domain stability in immunoglobulin light chain deposition disorders. Adv Protein Chem (1997) 50:183–242. doi: 10.1016/S0065-3233(08)60322-8
174. Poshusta TL, Sikkink LA, Leung N, Clark RJ, Dispenzieri A, Ramirez-Alvarado M. Mutations in specific structural regions of immunoglobulin light chains are associated with free light chain levels in patients with AL amyloidosis. PLoS One (2009) 4:e5169. doi: 10.1371/journal.pone.0005169
175. Blancas-Mejia LM, Hammernik J, Marin-Argany M, Ramirez-Alvarado M. Differential effects on light chain amyloid formation depend on mutations and type of glycosaminoglycans. J Biol Chem (2015) 290:4953–65. doi: 10.1074/jbc.M114.615401
176. Maritan M, Ambrosetti A, Oberti L, Barbiroli A, Diomede L, Romeo M, et al. Modulating the cardiotoxic behaviour of immunoglobulin light chain dimers through point mutations. Amyloid (2019) 26:105–6. doi: 10.1080/13506129.2019.1583185
177. Solomon A, Frangione B, Franklin EC. Bence Jones proteins and light chains of immunoglobulins. preferential association of the V lambda VI subgroup of human light chains with amyloidosis AL (lambda). J Clin Invest (1982) 70:453–60. doi: 10.1172/JCI110635
178. Ozaki S, Abe M, Wolfenbarger D, Weiss DT, Solomon A. Preferential expression of human lambda-light-chain variable-region subgroups in multiple myeloma, AL amyloidosis, and waldenstrom's macroglobulinemia. Clin Immunol Immunopathol (1994) 71:183–9. doi: 10.1006/clin.1994.1070
179. Comenzo RL, Wally J, Kica G, Murray J, Ericsson T, Skinner M, et al. Clonal immunoglobulin light chain variable region germline gene use in AL amyloidosis: association with dominant amyloid-related organ involvement and survival after stem cell transplantation. Br J Haematol (1999) 106:744–51. doi: 10.1046/j.1365-2141.1999.01591.x
180. Abraham RS, Geyer SM, Price-Troska TL, Allmer C, Kyle RA, Gertz MA, et al. Immunoglobulin light chain variable (V) region genes influence clinical presentation and outcome in light chain-associated amyloidosis (AL). Blood (2003) 101:3801–8. doi: 10.1182/blood-2002-09-2707
181. Garay Sanchez SA, Rodriguez Alvarez FJ, Zavala-Padilla G, Mejia-Cristobal LM, Cruz-Rangel A, Costas M, et al. Stability and aggregation propensity do not fully account for the association of various germline variable domain gene segments with light chain amyloidosis. Biol Chem (2017) 398:477–89. doi: 10.1515/hsz-2016-0178
182. Pokkuluri PR, Solomon A, Weiss DT, Stevens FJ, Schiffer M. Tertiary structure of human lambda 6 light chains. Amyloid (1999) 6:165–71. doi: 10.3109/13506129909007322
183. Wall J, Schell M, Murphy C, Hrncic R, Stevens FJ, Solomon A. Thermodynamic instability of human lambda 6 light chains: correlation with fibrillogenicity. Biochemistry (1999) 38:14101–8. doi: 10.1021/bi991131j
184. Takahashi N, Takayasu T, Isobe T, Shinoda T, Okuyama T, Shimizu A. Comparative study on the structure of the light chains of human immunoglobulins. II. assignment of a new subgroup. J Biochem (1979) 86:1523–35. doi: 10.1093/oxfordjournals.jbchem.a132670
185. Nau A, Shen Y, Sanchorawala V, Prokaeva T, Morgan GJ. Complete variable domain sequences of monoclonal antibody light chains identified from untargeted RNA sequencing data. Front Immunol (2023) 14:1167235. doi: 10.3389/fimmu.2023.1167235
186. Desikan KR, Dhodapkar MV, Hough A, Waldron T, Jagannath S, Siegel D, et al. Incidence and impact of light chain associated (AL) amyloidosis on the prognosis of patients with multiple myeloma treated with autologous transplantation. Leuk Lymphoma (1997) 27:315–9. doi: 10.3109/10428199709059685
187. Bahlis NJ, Lazarus HM. Multiple myeloma-associated AL amyloidosis: is a distinctive therapeutic approach warranted? Bone Marrow Transplant (2006) 38:7–15. doi: 10.1038/sj.bmt.1705395
188. Usnarska-Zubkiewicz L, Holojda J, Jelen M, Zubkiewicz-Zarebska A, Debski J, Kuliczkowski K. The occurrence of Al amyloidosis (light-chain amyloidosis) in patients with multiple myeloma in lower Silesia region, Poland. Adv Clin Exp Med (2014) 23:235–44. doi: 10.17219/acem/37068
189. Ballegaard M, Nelson LM, Gimsing P. Comparing neuropathy in multiple myeloma and AL amyloidosis. J Peripher Nerv Syst (2021) 26:75–82. doi: 10.1111/jns.12428
190. Villalba MI, Canul-Tec JC, Luna-Martinez OD, Sanchez-Alcala R, Olamendi-Portugal T, Rudino-Pinera E, et al. Site-directed mutagenesis reveals regions implicated in the stability and fiber formation of human lambda3r light chains. J Biol Chem (2015) 290:13039. doi: 10.1074/jbc.A114.629550
191. Jaffe DB, Shahi P, Adams BA, Chrisman AM, Finnegan PM, Raman N, et al. Functional antibodies exhibit light chain coherence. Nature (2022) 611:352–7. doi: 10.1038/s41586-022-05371-z
192. Denoroy L, Deret S, Aucouturier P. Overrepresentation of the V kappa IV subgroup in light chain deposition disease. Immunol Lett (1994) 42:63–6. doi: 10.1016/0165-2478(94)90036-1
193. Decourt C, Touchard G, Preud'homme JL, Vidal R, Beaufils H, Diemert MC, et al. Complete primary sequences of two lambda immunoglobulin light chains in myelomas with nonamyloid (Randall-type) light chain deposition disease. Am J Pathol (1998) 153:313–8. doi: 10.1016/S0002-9440(10)65573-3
194. Sirac C, Bridoux F, Carrion C, Devuyst O, Fernandez B, Goujon JM, et al. Role of the monoclonal kappa chain V domain and reversibility of renal damage in a transgenic model of acquired fanconi syndrome. Blood (2006) 108:536–43. doi: 10.1182/blood-2005-11-4419
195. El Hamel C, Thierry A, Trouillas P, Bridoux F, Carrion C, Quellard N, et al. Crystal-storing histiocytosis with renal fanconi syndrome: pathological and molecular characteristics compared with classical myeloma-associated fanconi syndrome. Nephrol Dial Transplant (2010) 25:2982–90. doi: 10.1093/ndt/gfq129
196. Bender S, Javaugue V, Saintamand A, Ayala MV, Alizadeh M, Filloux M, et al. Immunoglobulin variable domain high-throughput sequencing reveals specific novel mutational patterns in POEMS syndrome. Blood (2020) 135:1750–8. doi: 10.1182/blood.2019004197
197. Bellotti V, Merlini G. Toward understanding the molecular pathogenesis of monoclonal immunoglobulin light-chain deposition. Nephrol Dial Transplant (1996) 11:1708–11. doi: 10.1093/oxfordjournals.ndt.a027651
198. Raffen R, Dieckman LJ, Szpunar M, Wunschl C, Pokkuluri PR, Dave P, et al. Physicochemical consequences of amino acid variations that contribute to fibril formation by immunoglobulin light chains. Protein Sci (1999) 8:509–17. doi: 10.1110/ps.8.3.509
199. Kim Y, Wall JS, Meyer J, Murphy C, Randolph TW, Manning MC, et al. Thermodynamic modulation of light chain amyloid fibril formation. J Biol Chem (2000) 275:1570–4. doi: 10.1074/jbc.275.3.1570
200. Kim YS, Cape SP, Chi E, Raffen R, Wilkins-Stevens P, Stevens FJ, et al. Counteracting effects of renal solutes on amyloid fibril formation by immunoglobulin light chains. J Biol Chem (2001) 276:1626–33. doi: 10.1074/jbc.M007766200
201. Khurana R, Gillespie JR, Talapatra A, Minert LJ, Ionescu-Zanetti C, Millett I, et al. Partially folded intermediates as critical precursors of light chain amyloid fibrils and amorphous aggregates. Biochemistry (2001) 40:3525–35. doi: 10.1021/bi001782b
202. Kim YS, Randolph TW, Stevens FJ, Carpenter JF. Kinetics and energetics of assembly, nucleation, and growth of aggregates and fibrils for an amyloidogenic protein. insights into transition states from pressure, temperature, and co-solute studies. J Biol Chem (2002) 277:27240–6. doi: 10.1074/jbc.M202492200
203. Souillac PO, Uversky VN, Millett IS, Khurana R, Doniach S, Fink AL. Effect of association state and conformational stability on the kinetics of immunoglobulin light chain amyloid fibril formation at physiological pH. J Biol Chem (2002) 277:12657–65. doi: 10.1074/jbc.M109230200
204. Blancas-Mejia LM, Tellez LA, Del Pozo-Yauner L, Becerril B, Sanchez-Ruiz JM, Fernandez-Velasco DA. Thermodynamic and kinetic characterization of a germ line human lambda6 light-chain protein: the relation between unfolding and fibrillogenesis. J Mol Biol (2009) 386:1153–66. doi: 10.1016/j.jmb.2008.12.069
205. Gonzalez-Andrade M, Becerril-Lujan B, Sanchez-Lopez R, Cecena-Alvarez H, Perez-Carreon JI, Ortiz E, et al. Mutational and genetic determinants of lambda6 light chain amyloidogenesis. FEBS J (2013) 280:6173–83. doi: 10.1111/febs.12538
206. Poshusta TL, Katoh N, Gertz MA, Dispenzieri A, Ramirez-Alvarado M. Thermal stability threshold for amyloid formation in light chain amyloidosis. Int J Mol Sci (2013) 14:22604–17. doi: 10.3390/ijms141122604
207. Marin-Argany M, Guell-Bosch J, Blancas-Mejia LM, Villegas S, Ramirez-Alvarado M. Mutations can cause light chains to be too stable or too unstable to form amyloid fibrils. Protein Sci (2015) 24:1829–40. doi: 10.1002/pro.2790
208. Qin Z, Hu D, Zhu M, Fink AL. Structural characterization of the partially folded intermediates of an immunoglobulin light chain leading to amyloid fibrillation and amorphous aggregation. Biochemistry (2007) 46:3521–31. doi: 10.1021/bi061716v
209. Misra P, Ramirez-Alvarado M. Early events in light chain aggregation at physiological pH reveal new insights on assembly, stability, and aggregate dissociation. Amyloid (2021) 28:113–24. doi: 10.1080/13506129.2021.1877129
210. Mann MJ, Flory AR, Oikonomou C, Hayes CA, Melendez-Suchi C, Hendershot LM. Identification of two rate-limiting steps in the degradation of partially folded immunoglobulin light chains. Front Cell Dev Biol (2022) 10:924848. doi: 10.3389/fcell.2022.924848
211. Luna-Martinez OD, Hernandez-Santoyo A, Villalba-Velazquez MI, Sanchez-Alcala R, Fernandez-Velasco DA, Becerril B. Stabilizing an amyloidogenic lambda6 light chain variable domain. FEBS J (2017) 284:3702–17. doi: 10.1111/febs.14265
212. Sletten K, Natvig JB, Husby G, Juul J. The complete amino acid sequence of a prototype immunoglobulin-lambda light-chain-type amyloid-fibril protein AR. Biochem J (1981) 195:561–72. doi: 10.1042/bj1950561
213. Wall J, Murphy CL, Solomon A. In vitro immunoglobulin light chain fibrillogenesis. Methods Enzymol (1999) 309:204–17. doi: 10.1016/S0076-6879(99)09016-3
214. Mohan S, Kourentzi K, Schick KA, Uehara C, Lipschultz CA, Acchione M, et al. Association energetics of cross-reactive and specific antibodies. Biochemistry (2009) 48:1390–8. doi: 10.1021/bi801901d
215. Adhikary R, Yu W, Oda M, Zimmermann J, Romesberg FE. Protein dynamics and the diversity of an antibody response. J Biol Chem (2012) 287:27139–47. doi: 10.1074/jbc.M112.372698
216. Wu NR, Nicely NI, Lee EM, Reed RK, Watts BE, Cai F, et al. Cooperation between somatic mutation and germline-encoded residues enables antibody recognition of HIV-1 envelope glycans. PLoS Pathog (2019) 15:e1008165. doi: 10.1371/journal.ppat.1008165
217. Otzen DE. Driving forces in amyloidosis: how does a light chain make a heavy heart? J Biol Chem (2021) 296:100785. doi: 10.1016/j.jbc.2021.100785
218. Uversky VN, Van Regenmortel MHV. Mobility and disorder in antibody and antigen binding sites do not prevent immunochemical recognition. Crit Rev Biochem Mol Biol (2021) 56:149–56. doi: 10.1080/10409238.2020.1869683
219. Baden EM, Owen BA, Peterson FC, Volkman BF, Ramirez-Alvarado M, Thompson JR. Altered dimer interface decreases stability in an amyloidogenic protein. J Biol Chem (2008) 283:15853–60. doi: 10.1074/jbc.M705347200
220. Rennella E, Morgan GJ, Kelly JW, Kay LE. Role of domain interactions in the aggregation of full-length immunoglobulin light chains. Proc Natl Acad Sci USA (2019) 116:854–63. doi: 10.1073/pnas.1817538116
221. Rottenaicher GJ, Absmeier RM, Meier L, Zacharias M, Buchner J. A constant domain mutation in a patient-derived antibody light chain reveals principles of AL amyloidosis. Commun Biol (2023) 6:209. doi: 10.1038/s42003-023-04574-y
222. Brumshtein B, Esswein SR, Salwinski L, Phillips ML, Ly AT, Cascio D, et al. Inhibition by small-molecule ligands of formation of amyloid fibrils of an immunoglobulin light chain variable domain. Elife (2015) 4:e10935. doi: 10.7554/eLife.10935.014
223. Wolwertz ML, Nguyen PT, Quittot N, Bourgault S. Probing the role of lambda6 immunoglobulin light chain dimerization in amyloid formation. Biochim Biophys Acta (2016) 1864:409–18. doi: 10.1016/j.bbapap.2016.01.009
224. Morgan GJ, Buxbaum JN, Kelly JW. Light chain stabilization: a therapeutic approach to ameliorate AL amyloidosis. Hemato (2021) 2:645–59. doi: 10.3390/hemato2040042
225. Yan NL, Nair R, Chu A, Wilson IA, Johnson KA, Morgan GJ, et al. Amyloidogenic immunoglobulin light chain kinetic stabilizers comprising a simple urea linker module reveal a novel binding sub-site. Bioorg Med Chem Lett (2022) 60:128571. doi: 10.1016/j.bmcl.2022.128571
226. Blancas-Mejia LM, Tischer A, Thompson JR, Tai J, Wang L, Auton M, et al. Kinetic control in protein folding for light chain amyloidosis and the differential effects of somatic mutations. J Mol Biol (2014) 426:347–61. doi: 10.1016/j.jmb.2013.10.016
227. Oberti L, Rognoni P, Barbiroli A, Lavatelli F, Russo R, Maritan M, et al. Concurrent structural and biophysical traits link with immunoglobulin light chains amyloid propensity. Sci Rep (2017) 7:16809. doi: 10.1038/s41598-017-16953-7
228. Peterle D, Klimtchuk ES, Wales TE, Georgescauld F, Connors LH, Engen JR, et al. A conservative point mutation in a dynamic antigen-binding loop of human immunoglobulin lambda6 light chain promotes pathologic amyloid formation. J Mol Biol (2021) 433:167310. doi: 10.1016/j.jmb.2021.167310
229. Ruiz-Zamora RA, Guillaume S, Al-Hilaly YK, Al-Garawi Z, Rodriguez-Alvarez FJ, Zavala-Padilla G, et al. The CDR1 and other regions of immunoglobulin light chains are hot spots for amyloid aggregation. Sci Rep (2019) 9:3123. doi: 10.1038/s41598-019-39781-3
230. Tzotzos S, Doig AJ. Amyloidogenic sequences in native protein structures. Protein Sci (2010) 19:327–48. doi: 10.1002/pro.314
231. Lecoq L, Wiegand T, Rodriguez-Alvarez FJ, Cadalbert R, Herrera GA, Del Pozo-Yauner L, et al. A substantial structural conversion of the native monomer leads to in-register parallel amyloid fibril formation in light-chain amyloidosis. Chembiochem (2019) 20:1027–31. doi: 10.1002/cbic.201800732
232. Swuec P, Lavatelli F, Tasaki M, Paissoni C, Rognoni P, Maritan M, et al. Cryo-EM structure of cardiac amyloid fibrils from an immunoglobulin light chain AL amyloidosis patient. Nat Commun (2019) 10:1269. doi: 10.1038/s41467-019-09133-w
233. Radamaker L, Lin YH, Annamalai K, Huhn S, Hegenbart U, Schonland SO, et al. Cryo-EM structure of a light chain-derived amyloid fibril from a patient with systemic AL amyloidosis. Nat Commun (2019) 10:1103. doi: 10.1038/s41467-019-09032-0
234. Radamaker L, Karimi-Farsijani S, Andreotti G, Baur J, Neumann M, Schreiner S, et al. Role of mutations and post-translational modifications in systemic AL amyloidosis studied by cryo-EM. Nat Commun (2021) 12:6434. doi: 10.1038/s41467-021-26553-9
235. Radamaker L, Baur J, Huhn S, Haupt C, Hegenbart U, Schonland S, et al. Cryo-EM reveals structural breaks in a patient-derived amyloid fibril from systemic AL amyloidosis. Nat Commun (2021) 12:875. doi: 10.1038/s41467-021-21126-2
236. Burnett DL, Schofield P, Langley DB, Jackson J, Bourne K, Wilson E, et al. Conformational diversity facilitates antibody mutation trajectories and discrimination between foreign and self-antigens. Proc Natl Acad Sci USA (2020) 117:22341–50. doi: 10.1073/pnas.2005102117
237. Saini S, Agarwal M, Pradhan A, Pareek S, Singh AK, Dhawan G, et al. Exploring the role of framework mutations in enabling breadth of a cross-reactive antibody (CR3022) against the SARS-CoV-2 RBD and its variants of concern. J Biomol Struct Dyn (2023) 41:2341–54. doi: 10.1080/07391102.2022.2030800
238. Klimtchuk ES, Gursky O, Patel RS, Laporte KL, Connors LH, Skinner M, et al. The critical role of the constant region in thermal stability and aggregation of amyloidogenic immunoglobulin light chain. Biochemistry (2010) 49:9848–57. doi: 10.1021/bi101351c
239. Blancas-Mejia LM, Martin EB, Williams A, Wall JS, Ramirez-Alvarado M. Kinetic stability and sequence/structure studies of urine-derived bence-Jones proteins from multiple myeloma and light chain amyloidosis patients. Biophys Chem (2017) 230:89–98. doi: 10.1016/j.bpc.2017.08.011
240. Souillac PO, Uversky VN, Millett IS, Khurana R, Doniach S, Fink AL. Elucidation of the molecular mechanism during the early events in immunoglobulin light chain amyloid fibrillation. evidence for an off-pathway oligomer at acidic pH. J Biol Chem (2002) 277:12666–79. doi: 10.1074/jbc.M109229200
241. Mukherjee S, Pondaven SP, Jaroniec CP. Conformational flexibility of a human immunoglobulin light chain variable domain by relaxation dispersion nuclear magnetic resonance spectroscopy: implications for protein misfolding and amyloid assembly. Biochemistry (2011) 50:5845–57. doi: 10.1021/bi200410c
242. Arosio P, Owczarz M, Muller-Spath T, Rognoni P, Beeg M, Wu H, et al. In vitro aggregation behavior of a non-amyloidogenic lambda light chain dimer deriving from U266 multiple myeloma cells. PLoS One (2012) 7:e33372. doi: 10.1371/journal.pone.0033372
243. Perfetti V, Ubbiali P, Vignarelli MC, Diegoli M, Fasani R, Stoppini M, et al. Evidence that amyloidogenic light chains undergo antigen-driven selection. Blood (1998) 91:2948–54. doi: 10.1182/blood.V91.8.2948.2948_2948_2954
244. Vitagliano L, Stanzione F, De Simone A, Esposito L. Dynamics and stability of amyloid-like steric zipper assemblies with hydrophobic dry interfaces. Biopolymers (2009) 91:1161–71. doi: 10.1002/bip.21182
245. Schmidt A, Annamalai K, Schmidt M, Grigorieff N, Fandrich M. Cryo-EM reveals the steric zipper structure of a light chain-derived amyloid fibril. Proc Natl Acad Sci USA (2016) 113:6200–5. doi: 10.1073/pnas.1522282113
246. Bemporad F, Taddei N, Stefani M, Chiti F. Assessing the role of aromatic residues in the amyloid aggregation of human muscle acylphosphatase. Protein Sci (2006) 15:862–70. doi: 10.1110/ps.051915806
247. Hee JS, Mitchell SM, Liu X, Leonhardt RM. Melanosomal formation of PMEL core amyloid is driven by aromatic residues. Sci Rep (2017) 7:44064. doi: 10.1038/srep44064
248. Pradhan T, Annamalai K, Sarkar R, Huhn S, Hegenbart U, Schonland S, et al. Seeded fibrils of the germline variant of human lambda-III immunoglobulin light chain FOR005 have a similar core as patient fibrils with reduced stability. J Biol Chem (2020) 295:18474–84. doi: 10.1074/jbc.RA120.016006
249. Tveteraas T, Sletten K, Westermark P. The amino acid sequence of a carbohydrate-containing immunoglobulin-light-chain-type amyloid-fibril protein. Biochem J (1985) 232:183–90. doi: 10.1042/bj2320183
250. Fykse EM, Sletten K, Husby G, Cornwell GG 3rd. The primary structure of the variable region of an immunoglobin IV light-chain amyloid-fibril protein (AL GIL). Biochem J (1988) 256:973–80. doi: 10.1042/bj2560973
251. Aucouturier P, Khamlichi AA, Preud'homme JL, Bauwens M, Touchard G, Cogne M. Complementary DNA sequence of human amyloidogenic immunoglobulin light-chain precursors. Biochem J (1992) 285(Pt 1):149–52. doi: 10.1042/bj2850149
252. Kagimoto T, Nakakuma H, Hata H, Hidaka M, Horikawa K, Kawaguti T, et al. Differential glycosylation of bence Jones protein and kidney impairment in patients with plasma cell dyscrasia. J Lab Clin Med (1997) 129:217–23. doi: 10.1016/S0022-2143(97)90142-6
253. Foss GS, Nilsen R, Cornwell GC 3rd, Husby G, Sletten K. A glycosylated bence Jones protein and its autologous amyloid light chain containing potentially amyloidogenic residues. Scand J Immunol (1998) 47:348–54. doi: 10.1046/j.1365-3083.1998.00311.x
254. Omtvedt LA, Bailey D, Renouf DV, Davies MJ, Paramonov NA, Haavik S, et al. Glycosylation of immunoglobulin light chains associated with amyloidosis. Amyloid (2000) 7:227–44. doi: 10.3109/13506120009146437
255. Stevens FJ. Four structural risk factors identify most fibril-forming kappa light chains. Amyloid (2000) 7:200–11. doi: 10.3109/13506120009146835
256. Kumar S, Murray D, Dasari S, Milani P, Barnidge D, Madden B, et al. Assay to rapidly screen for immunoglobulin light chain glycosylation: a potential path to earlier AL diagnosis for a subset of patients. Leukemia (2019) 33:254–7. doi: 10.1038/s41375-018-0194-x
257. Mellors PW, Dasari S, Kohlhagen MC, Kourelis T, Go RS, Muchtar E, et al. MASS-FIX for the detection of monoclonal proteins and light chain n-glycosylation in routine clinical practice: a cross-sectional study of 6315 patients. Blood Cancer J (2021) 11:50. doi: 10.1038/s41408-021-00444-0
258. Dispenzieri A, Larson DR, Rajkumar SV, Kyle RA, Kumar SK, Kourelis T, et al. N-glycosylation of monoclonal light chains on routine MASS-FIX testing is a risk factor for MGUS progression. Leukemia (2020) 34:2749–53. doi: 10.1038/s41375-020-0940-8
259. Nevone A, Girelli M, Mangiacavalli S, Paiva B, Milani P, Cascino P, et al. An n-glycosylation hotspot in immunoglobulin kappa light chains is associated with AL amyloidosis. Leukemia (2022) 36:2076–85. doi: 10.1038/s41375-022-01599-w
260. Abe Y, Shibata H, Oyama K, Ueda T. Effect of O-glycosylation on amyloid fibril formation of the variable domain in the Vlambda6 light chain mutant wil. Int J Biol Macromol (2021) 166:342–51. doi: 10.1016/j.ijbiomac.2020.10.194
261. Dasari S, Theis JD, Vrana JA, Meureta OM, Quint PS, Muppa P, et al. Proteomic detection of immunoglobulin light chain variable region peptides from amyloidosis patient biopsies. J Proteome Res (2015) 14:1957–67. doi: 10.1021/acs.jproteome.5b00015
262. Ricagno S, Puri S, Schulte T, Chaves-Sanjuan A, Mazzini G, Caminito S, et al. The cryo-EM structure of renal amyloid fibril suggests structurally homogeneous multiorgan aggregation in AL amyloidosis. Res Square (2023). doi: 10.21203/rs.3.rs-2759584/v1
263. Bhutani D, Leng S, Lentzsch S. Fibril-directed therapies in systemic light chain AL amyloidosis. Clin Lymphoma Myeloma Leuk (2019) 19:555–9. doi: 10.1016/j.clml.2019.03.029
264. Jambhekar K, Pandey T, Kaushik C, Viswamitra S. Protean manifestations of amyloidosis. Contemp Diagn Radiol (2009) 32:1–6. doi: 10.1097/01.CDR.0000362207.95849.95
265. Wechalekar AD, Fontana M, Quarta CC, Liedtke M. AL amyloidosis for cardiologists: awareness, diagnosis, and future prospects: JACC: CardioOncology state-of-the-Art review. JACC CardioOncol (2022) 4:427–41. doi: 10.1016/j.jaccao.2022.08.009
266. Oubari S, Naser E, Papathanasiou M, Luedike P, Hagenacker T, Thimm A, et al. Impact of time to diagnosis on Mayo stages, treatment outcome, and survival in patients with AL amyloidosis and cardiac involvement. Eur J Haematol (2021) 107:449–57. doi: 10.1111/ejh.13681
267. Bellavia D, Pellikka PA, Al-Zahrani GB, Abraham TP, Dispenzieri A, Miyazaki C, et al. Independent predictors of survival in primary systemic (Al) amyloidosis, including cardiac biomarkers and left ventricular strain imaging: an observational cohort study. J Am Soc Echocardiogr (2010) 23:643–52. doi: 10.1016/j.echo.2010.03.027
268. Palladini G, Barassi A, Klersy C, Pacciolla R, Milani P, Sarais G, et al. The combination of high-sensitivity cardiac troponin T (hs-cTnT) at presentation and changes in n-terminal natriuretic peptide type b (NT-proBNP) after chemotherapy best predicts survival in AL amyloidosis. Blood (2010) 116:3426–30. doi: 10.1182/blood-2010-05-286567
269. Palladini G, Milani P, Basset M, Russo F, Lavatelli F, Nuvolone M, et al. Severity and reversibility of cardiac dysfunction and residual concentration of amyloidogenic light chain predict overall survival of patients with AL amyloidosis who attain complete response. Amyloid (2017) 24:54–5. doi: 10.1080/13506129.2017.1289916
270. Brenner DA, Jain M, Pimentel DR, Wang B, Connors LH, Skinner M, et al. Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ Res (2004) 94:1008–10. doi: 10.1161/01.RES.0000126569.75419.74
271. Shi J, Guan J, Jiang B, Brenner DA, Del Monte F, Ward JE, et al. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38alpha MAPK pathway. Proc Natl Acad Sci USA (2010) 107:4188–93. doi: 10.1073/pnas.0912263107
272. Mcwilliams-Koeppen HP, Foster JS, Hackenbrack N, Ramirez-Alvarado M, Donohoe D, Williams A, et al. Light chain amyloid fibrils cause metabolic dysfunction in human cardiomyocytes. PLoS One (2015) 10:e0137716. doi: 10.1371/journal.pone.0137716
273. Jordan TL, Maar K, Redhage KR, Misra P, Blancas-Mejia LM, Dick CJ, et al. Light chain amyloidosis induced inflammatory changes in cardiomyocytes and adipose-derived mesenchymal stromal cells. Leukemia (2020) 34:1383–93. doi: 10.1038/s41375-019-0640-4
274. Marin-Argany M, Lin Y, Misra P, Williams A, Wall JS, Howell KG, et al. Cell damage in light chain amyloidosis: FIBRIL INTERNALIZATION, TOXICITY AND CELL-MEDIATED SEEDING. J Biol Chem (2016) 291:19813–25. doi: 10.1074/jbc.M116.736736
275. Guan J, Mishra S, Shi J, Plovie E, Qiu Y, Cao X, et al. Stanniocalcin1 is a key mediator of amyloidogenic light chain induced cardiotoxicity. Basic Res Cardiol (2013) 108:378. doi: 10.1007/s00395-013-0378-5
276. Mishra S, Guan J, Plovie E, Seldin DC, Connors LH, Merlini G, et al. Human amyloidogenic light chain proteins result in cardiac dysfunction, cell death, and early mortality in zebrafish. Am J Physiol Heart Circ Physiol (2013) 305:H95–103. doi: 10.1152/ajpheart.00186.2013
277. Lavatelli F, Imperlini E, Orru S, Rognoni P, Sarnataro D, Palladini G, et al. Novel mitochondrial protein interactors of immunoglobulin light chains causing heart amyloidosis. FASEB J (2015) 29:4614–28. doi: 10.1096/fj.15-272179
278. Solomon A, Weiss DT, Pepys MB. Induction in mice of human light-chain-associated amyloidosis. Am J Pathol (1992) 140:629–37.
279. Martinez-Rivas G, Bender S, Sirac C. Understanding AL amyloidosis with a little help from in vivo models. Front Immunol (2022) 13:1008449. doi: 10.3389/fimmu.2022.1008449
280. Sirac C, Bender S, Jaccard A, Bridoux F, Lacombe C, Touchard G, et al. Strategies to model AL amyloidosis in mice. Amyloid (2011) 18 Suppl 1:45–7. doi: 10.3109/13506129.2011.574354016
281. Ward JE, Soohoo P, Toraldo G, Jasuja R, Connors LH, O'hara C, et al. Metabolic phenotype in an AL amyloidosis transgenic mouse model. Amyloid (2011) 18 Suppl 1:40–1. doi: 10.3109/13506129.2011.574354014
282. Sirac C. (2023). Pre-clinical models in MGCS, in: Fifth International Meeting of International Kidney & Monoclonal Gammopathy (IKMG), Hilton Milan Hotel, Milan, Italy.
283. Mishra S, Joshi S, Ward JE, Buys EP, Mishra D, Mishra D, et al. Zebrafish model of amyloid light chain cardiotoxicity: regeneration versus degeneration. Am J Physiol Heart Circ Physiol (2019) 316:H1158–66. doi: 10.1152/ajpheart.00788.2018
284. Diomede L, Rognoni P, Lavatelli F, Romeo M, Di Fonzo A, Foray C, et al. Investigating heart-specific toxicity of amyloidogenic immunoglobulin light chains: a lesson from c. elegans. Worm (2014) 3:e965590. doi: 10.4161/21624046.2014.965590
285. Liao R. Worming along in amyloid cardiotoxicity. Blood (2014) 123:3525–6. doi: 10.1182/blood-2014-04-566703
286. Guan J, Mishra S, Qiu Y, Shi J, Trudeau K, Las G, et al. Lysosomal dysfunction and impaired autophagy underlie the pathogenesis of amyloidogenic light chain-mediated cardiotoxicity. EMBO Mol Med (2014) 6:1493–507. doi: 10.15252/emmm.201404190
287. Maritan M, Romeo M, Oberti L, Sormanni P, Tasaki M, Russo R, et al. Inherent biophysical properties modulate the toxicity of soluble amyloidogenic light chains. J Mol Biol (2020) 432:845–60. doi: 10.1016/j.jmb.2019.12.015
288. Roland BP, Kodali R, Mishra R, Wetzel R. A serendipitous survey of prediction algorithms for amyloidogenicity. Biopolymers (2013) 100:780–9. doi: 10.1002/bip.22305
289. Rawat P, Prabakaran R, Kumar S, Gromiha MM. Exploring the sequence features determining amyloidosis in human antibody light chains. Sci Rep (2021) 11:13785. doi: 10.1038/s41598-021-93019-9
290. Garofalo M, Piccoli L, Romeo M, Barzago MM, Ravasio S, Foglierini M, et al. Machine learning analyses of antibody somatic mutations predict immunoglobulin light chain toxicity. Nat Commun (2021) 12:3532. doi: 10.1038/s41467-021-23880-9
291. Allegra A, Mirabile G, Tonacci A, Genovese S, Pioggia G, Gangemi S. Machine learning approaches in diagnosis, prognosis and treatment selection of cardiac amyloidosis. Int J Mol Sci (2023) 24:5680. doi: 10.3390/ijms24065680
292. Bloom MW, Gorevic PD. Cardiac amyloidosis. Ann Intern Med (2023) 176:ITC33–48. doi: 10.7326/AITC202303210
293. Isaac J, Kerby JD, Russell WJ, Dempsey SC, Sanders PW, Herrera GA. In vitro modulation of AL-amyloid formation by human mesangial cells exposed to amyloidogenic light chains. Amyloid (1998) 5:238–46. doi: 10.3109/13506129809007296
294. Keeling J, Teng J, Herrera GA. AL-amyloidosis and light-chain deposition disease light chains induce divergent phenotypic transformations of human mesangial cells. Lab Invest (2004) 84:1322–38. doi: 10.1038/labinvest.3700161
295. Vora M, Kevil CG, Herrera GA. Contribution of human smooth muscle cells to amyloid angiopathy in AL (light-chain) amyloidosis. Ultrastruct Pathol (2017) 41:358–68. doi: 10.1080/01913123.2017.1349852
296. Herrera GA, Teng J, Zeng C, Xu H, Liang M, Alexander JS, et al. Phenotypic plasticity of mesenchymal stem cells is crucial for mesangial repair in a model of immunoglobulin light chain-associated mesangial damage. Ultrastruct Pathol (2018) 42:262–88. doi: 10.1080/01913123.2018.1449772
297. Herrera GA, Teng J, Turbat-Herrera EA, Zeng C, Del Pozo-Yauner L. Understanding mesangial pathobiology in AL-amyloidosis and monoclonal ig light chain deposition disease. Kidney Int Rep (2020) 5:1870–93. doi: 10.1016/j.ekir.2020.07.013
298. Herrera GA, Del Pozo-Yauner L, Teng J, Zeng C, Shen X, Moriyama T, et al. Glomerulopathic light chain-mesangial cell interactions: sortilin-related receptor (SORL1) and signaling. Kidney Int Rep (2021) 6:1379–96. doi: 10.1016/j.ekir.2021.02.014
299. Wochner RD, Strober W, Waldmann TA. The role of the kidney in the catabolism of bence Jones proteins and immunoglobulin fragments. J Exp Med (1967) 126:207–21. doi: 10.1084/jem.126.2.207
300. Nakano T, Matsui M, Inoue I, Awata T, Katayama S, Murakoshi T. Free immunoglobulin light chain: its biology and implications in diseases. Clin Chim Acta (2011) 412:843–9. doi: 10.1016/j.cca.2011.03.007
301. Herrera GA. Low molecular weight proteins and the kidney: physiologic and pathologic considerations. Ultrastruct Pathol (1994) 18:89–98. doi: 10.3109/01913129409016277
302. Batuman V, Verroust PJ, Navar GL, Kaysen JH, Goda FO, Campbell WC, et al. Myeloma light chains are ligands for cubilin (gp280). Am J Physiol (1998) 275:F246–254. doi: 10.1152/ajprenal.1998.275.2.F246
303. Christensen EI, Birn H. Megalin and cubilin: synergistic endocytic receptors in renal proximal tubule. Am J Physiol Renal Physiol (2001) 280:F562–573. doi: 10.1152/ajprenal.2001.280.4.F562
304. Parasuraman R, Wolforth SC, Wiesend WN, Dumler F, Rooney MT, Li W, et al. Contribution of polyclonal free light chain deposition to tubular injury. Am J Nephrol (2013) 38:465–74. doi: 10.1159/000356557
305. Gozzetti A, Guarnieri A, Zamagni E, Zakharova E, Coriu D, Bittrich M, et al. Monoclonal gammopathy of renal significance (MGRS): real-world data on outcomes and prognostic factors. Am J Hematol (2022) 97:877–84. doi: 10.1002/ajh.26566
306. Herrera GA. Renal amyloidosis with emphasis on the diagnostic role of electron microscopy. Ultrastruct Pathol (2020) 44:325–41. doi: 10.1080/01913123.2020.1844355
307. Herrera GA. The contributions of electron microscopy to the understanding and diagnosis of plasma cell dyscrasia-related renal lesions. Med Electron Microsc (2001) 34:1–18. doi: 10.1007/s007950100000
308. Herrera GA, J.T. C, Del Pozo-Yauner L, Liu B, Turbat- Herrera EA. AL(light chain)-amyloidogenesis by mesangial cells involves active participation of lysosomes: an ultrastructural study. Heliyon. (2023) e15190. doi: 10.1016/j.heliyon.2023.e15190
Keywords: light chain (AL) amyloidosis, somatic hypermutation, V(D)J rearrangement, protein aggregation, amyloid, immune system, antibodies
Citation: Del Pozo-Yauner L, Herrera GA, Perez Carreon JI, Turbat-Herrera EA, Rodriguez-Alvarez FJ and Ruiz Zamora RA (2023) Role of the mechanisms for antibody repertoire diversification in monoclonal light chain deposition disorders: when a friend becomes foe. Front. Immunol. 14:1203425. doi: 10.3389/fimmu.2023.1203425
Received: 10 April 2023; Accepted: 20 June 2023;
Published: 13 July 2023.
Edited by:
Francesca Lavatelli, University of Pavia, ItalyReviewed by:
Gareth J. Morgan, Boston University, United StatesRobert W. Maul, National Institute on Aging (NIH), United States
Stefano Ricagno, University of Milan, Italy
Copyright © 2023 Del Pozo-Yauner, Herrera, Perez Carreon, Turbat-Herrera, Rodriguez-Alvarez and Ruiz Zamora. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis Del Pozo-Yauner, bGRlbHBvem95YXVuZXJAaGVhbHRoLnNvdXRoYWxhYmFtYS5lZHU=