
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 20 July 2023
Sec. Primary Immunodeficiencies
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1199727
This article is part of the Research TopicA year in review: Discussions in Primary ImmunodeficienciesView all 3 articles
 Silje F. Jorgensen1,2*
Silje F. Jorgensen1,2* Magnhild E. Macpherson1,2Tonje Skarpengland2
Magnhild E. Macpherson1,2Tonje Skarpengland2 Rolf K. Berge3,4
Rolf K. Berge3,4 Børre Fevang1,2
Børre Fevang1,2 Bente Halvorsen1,5
Bente Halvorsen1,5 Pål Aukrust1,2,5
Pål Aukrust1,2,5The relationship between metabolic and inflammatory pathways play a pathogenic role in various cardiometabolic disorders and is potentially also involved in the pathogenesis of other disorders such as cancer, autoimmunity and infectious diseases. Common variable immunodeficiency (CVID) is the most common primary immunodeficiency in adults, characterized by increased frequency of airway infections with capsulated bacteria. In addition, a large proportion of CVID patients have autoimmune and inflammatory complications associated with systemic inflammation. We summarize the evidence that support a role of a bidirectional pathogenic interaction between inflammation and metabolic disturbances in CVID. This include low levels and function of high-density lipoprotein (HDL), high levels of triglycerides (TG) and its major lipoprotein very low-density lipoprotein (VLDL), and an unfavorable fatty acid (FA) profile. The dysregulation of TG, VLDL and FA were linked to disturbed gut microbiota profile, and TG and VLDL levels were strongly associated with lipopolysaccharides (LPS), a marker of gut leakage in blood. Of note, the disturbed lipid profile in CVID did not include total cholesterol levels or high low-density lipoprotein levels. Furthermore, increased VLDL and TG levels in blood were not associated with diet, high body mass index and liver steatosis, suggesting a different phenotype than in patients with traditional cardiovascular risk such as metabolic syndrome. We hypothesize that these metabolic disturbances are linked to inflammation in a bidirectional manner with disturbed gut microbiota as a potential contributing factor.
Metabolic disturbances are thoroughly studied in classical metabolic disorders such as diabetes mellitus and various cardiometabolic diseases, uncovering strong links to inflammation which promotes disease progression (1–3). The relationship between metabolic and inflammatory pathways has also been studied in other disorders such as cancer, autoimmunity and infectious diseases, where it could potentially contribute to disease progression (4–8).
Common variable immunodeficiency (CVID) is the most common primary immunodeficiency in adults, affecting 1: 25 000 to 1: 50 000 Caucasians (9). CVID patients have a B-cell dysfunction leading to hypogammaglobulinemia and typically increased frequency of infections with capsulated bacteria in airways (10). In addition, 70-80% of CVID patients have non-infectious complication e.g., autoimmunity, lymphadenopathy, splenomegaly, granulomas, cytopenia and enteropathy, associated with increased mortality and morbidity (11). These autoimmune and inflammatory complications are linked to low-grade systemic inflammation and immune activation with abnormal T cell and monocyte/macrophage function (12–14). Moreover, during the recent years, some studies have shown that CVID patients have an altered composition of gut microbiota that could be related to systemic inflammation (15–17). So far, however, data on metabolic disturbances in CVID patients and their potential relationship to CVID phenotypes, systemic inflammation and gut microbiota composition are scarce. In this review article, we will present existing data on metabolic abnormalities in CVID and how these disturbances could potentially be an important bridge, linking gut microbiota to systemic inflammation and autoimmunity in these patients.
Cholesterol and its esterified form, cholesteryl esters, are essential to form and maintain cell membranes, and is also crucial for modulation of transmembrane signaling pathways, as well as for the synthesis of hormones, fat-soluble vitamins and production of bile acids. Although it is biosynthesized in all mammalian cells, humans mainly obtain cholesterol from endogenous production in the liver and intestines, as well as from the diet through intestinal absorption (18–20). Cholesterol absorption starts with micellar solubilization of nutrient cholesterol in the intestinal lumen, before transportation to the enterocyte brush border membrane where it interacts with the enterocyte and gains entry (20, 21). Once cholesterol has been absorbed, it undergoes intracellular esterification into cholesteryl esters. Within the enterocyte, diet-derived fatty acids (FAs) are re-esterified to form triglycerides (TGs), and packaged with apolipoproteins, cholesteryl esters, free cholesterol and phospholipids to form chylomicron particles for secretion at the basolateral surface of enterocytes (20, 22). Chylomicrons then enter the circulation via the lymphatic system, and transport its lipids to skeletal, muscle, cardiac and adipose tissues where their TG components are hydrolyzed by capillary lipoprotein lipase (LPL) to release free FAs for storage or oxidation (23) (Figure 1). Interesting, a recent study suggest that intestinal synthesized HDL particles containing ApoA-I, are generated and released in the portal vein (24). As the FA-depleted, cholesterol-rich chylomicron remnants reach the liver, they are cleared from the circulation by the hepatic low-density lipoprotein (LDL) receptor-related protein. From the liver, recycled and endogenously produced cholesterol can enter the metabolic pathway as very low-density lipoprotein (VLDL), of which the TG core is hydrolyzed to produce intermediate-density lipoprotein (IDL) (25). The IDL particles are taken up by the liver or further hydrolyzed to produce LDL. Oxidized LDL can enter macrophages via scavenger receptors (SR) CD36 and SR-A, that is not downregulated by intracellular cholesterol levels, and join the intracellular cholesterol pool in an unregulated way. These cells also contain LDL receptors that is regulated by intracellular cholesterol levels. Macrophages loaded with excess cholesterol become foam cells, a hallmark in the pathogenesis of disorders characterized by an interaction between lipids and inflammation such as atherosclerosis (26, 27). HDL cholesterol is transported from peripheral cells/tissues back to the liver and intestine, where cholesterol is recycled or eliminated through reverse cholesterol transport (28). HDL cholesterol can reach the liver either directly through binding to SR-B1, or indirectly after a lipid exchange with VLDL/LDL, which are cleared by hepatic LDL receptor (28). A simplified illustration of lipid metabolism is shown in Figure 1.
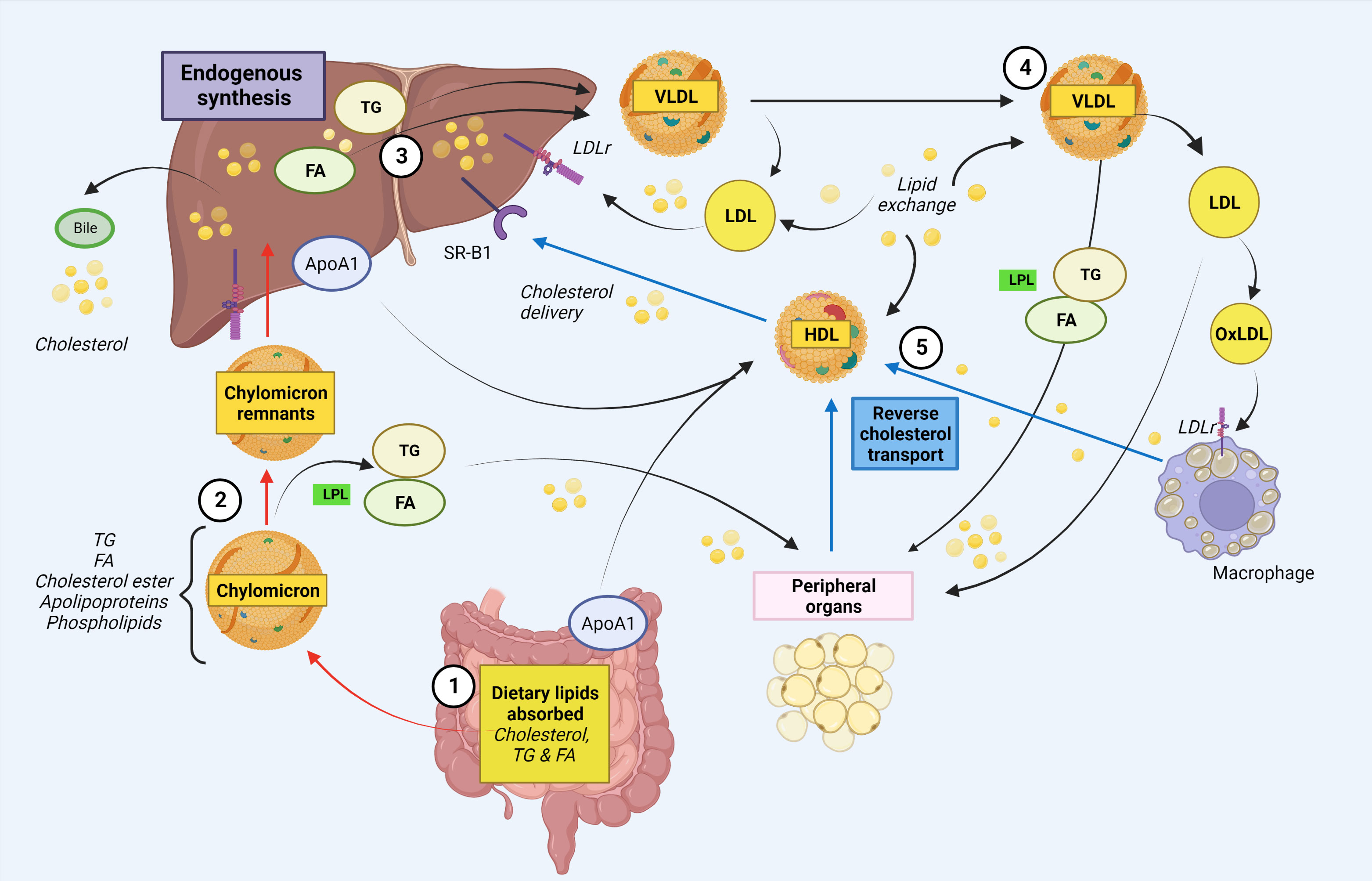
Figure 1 Simplified schematic illustration of cholesterol and fatty acid metabolism. 1. Intestinal absorption of dietary lipids. Cholesteryl esters, free fatty acids (FA) and triglycerides (TG) from the diet are absorbed in the intestinal lumen. In the enterocytes, chylomicrons are formed, which enters the circulation via the lymphatic system. In addition, ApoA1, the major protein component of HDL, is endogenous synthetized in enterocytes (and liver). 2. Chylomicrons deliver lipids to peripheral organs. Within the Chylomicrons, TGs are hydrolyzed by lipoprotein lipase (LPL) in peripheral organs, releasing FAs for storage. 3. The assembly of VLDL in the liver. The chylomicron remnants containing cholesterol are taken up by the liver. The liver delivers both endogenously synthesized and exogenously acquired FAs. FAs are re-esterified to form triglyceride and together with cholesterol form VLDL. VLDL enters the bloodstream as triglyceride rich lipoprotein VLDL. The liver also eliminate cholesterol via bile/intestine. 4. VLDLs transport triglycerides to peripheral tissues. Via the blood stream, VLDL can transport TGs to peripheral cells e.g. adipose tissue and muscle, where it is hydrolyzed by LPL to FAs for energy production or storage. VLDL can also generate circulating LDL which can either return to the liver or it can be oxidized and taken up by macrophages in peripheral organs. Macrophages loaded with excess cholesterol become foam cells. 5. Cholesterol is recycled or eliminated through reverse cholesterol transport. HDL cholesterol is transported from peripheral cells/tissues back to the liver and intestine, where cholesterol is recycled or eliminated. HDL cholesterol can reach the liver either directly through binding to SR-B1, or indirectly after a lipid exchange with VLDL/LDL, which are cleared by hepatic LDL receptor. Red arrows indicate exogenous pathways, black arrows indicate endogenous pathways and blue arrows indicate reverse cholesterol transport. ApoA-I, apolipoprotein A-I; FA, fatty acid; HDL, high-density lipoprotein, LDLr, low-density lipoprotein receptor; LPL, lipoprotein lipase; oxLDL, oxidized low-density lipoprotein; SR, scavenger receptor; TG, triglycerides; VLDL, very low-density lipoprotein. Created with BioRender.com.
Whereas normal lipid metabolism is essential for normal cellular function and homeostasis, abnormal regulation of these pathways may through a bidirectional interaction with immune cells have major implication for several disorders, not only atherosclerosis and related metabolic disorders, but also various disorders characterized by systemic inflammation and autoimmunity. A significant proportion of CVID patients develop inflammatory and autoimmune complications, and herein, we demonstrate support for our hypothesis that an unfavorable/dysregulated metabolic profile is linked to the inflammatory phenotype in CVID. The central role of the intestine in lipid metabolism, suggest that gut microbial composition may also influence lipid metabolism. Therefore, potential mechanisms linking gut microbiota to lipid metabolism and inflammation in CVID (29), will also be discussed in this review.
It is well known that HDL has anti-atherogenic properties that are primarily thought to be attributed to its key role in reverse cholesterol transport (30, 31). However, whereas most lipoproteins are thought to mediate inflammatory effects, several lines of evidence suggest that HDL has anti-inflammatory properties that could also contribute to its anti-atherogenic potential (32, 33). Thus, HDL have been shown to mediate inhibitory effects on several pathways relevant for immune activation and inflammation, at least partly through inhibitory effects on innate immunity, interacting both with toll-like receptors (TLR), such as TLR4 and TLR2, and NLRP inflammasomes, all central regulators of the innate immune responses in monocytes/macrophages (33–37).
Vieira et al. was the first to report deceased circulating levels of HDL cholesterol and Apolipoprotein A-I (ApoA-I), the major protein component in HDL, in a combined cohort of patients with CVID (n=18) and X-linked agammaglobulinemia (XLA, n=6), compared to healthy controls (38). More recently, Andrade et al. reported decreased levels of ApoA-I associated with increased oxidative stress in 32 CVID patients (39). MacPherson et al. went on to show decreased levels of HDL levels in a large cohort of 102 CVID patients (40). In a sub-analysis of 40 patients they also found decreased ApoA-1 levels compared to controls. In addition, they analyzed HDL subclasses, defined by density or size reflecting differences in relative content of proteins and lipids, and found decreased levels of extra-large, large and medium sized HDL, but not of the small sized HDL, compared to healthy controls (40). The small HDL subclass has been suggested to be of particular importance for reverse cholesterol transport (41), but how the actual HDL subclass pattern in CVID affects its function in these patients is at present unclear. Moreover, in the study by MacPherson et al., HDL levels were inversely correlated with tumor necrosis factor (TNF) and C-reactive protein (CRP), both markers of systemic inflammation (40).
While it is known that HDL can sequester lipopolysaccharide (LPS), and thereby prevent LPS-induced cellular activation through TLR4 (42), this mechanism alone does not explain why HDL can block a broad spectrum of TLR-mediated inflammatory responses in various cells such as macrophages. De Nardo E et al. showed that the induction of ATF3, a key transcriptional repressor of innate immune response genes, is the main mechanism by which HDL mediates its anti-inflammatory activities in macrophages (43). ATF3 can be induced by TLR activation as part of an important negative feedback loop to limit TLR-driven inflammatory responses in macrophages, and by inducing ATF3 (44), HDL utilizes an ancient regulatory feedback system to broadly and directly modify the inflammatory response of macrophages towards TLR stimulation (43). It seems that AFT3 is a more general regulator of inflammation and cell metabolisms with relevance to a wide range of diseases ranging from metabolic disorders to cancer (45). Interestingly, in the study by MacPherson et al. it was reported decreased expression of ATF3 in peripheral blood mononuclear cells (PBMC) from CVID patients, and PBMC from CVID patients had impaired anti-inflammatory effects of HDL in these cells, suggesting that attenuated HDL-AFT3 interaction could contribute to systemic inflammation in CVID (40) (Figure 2A).
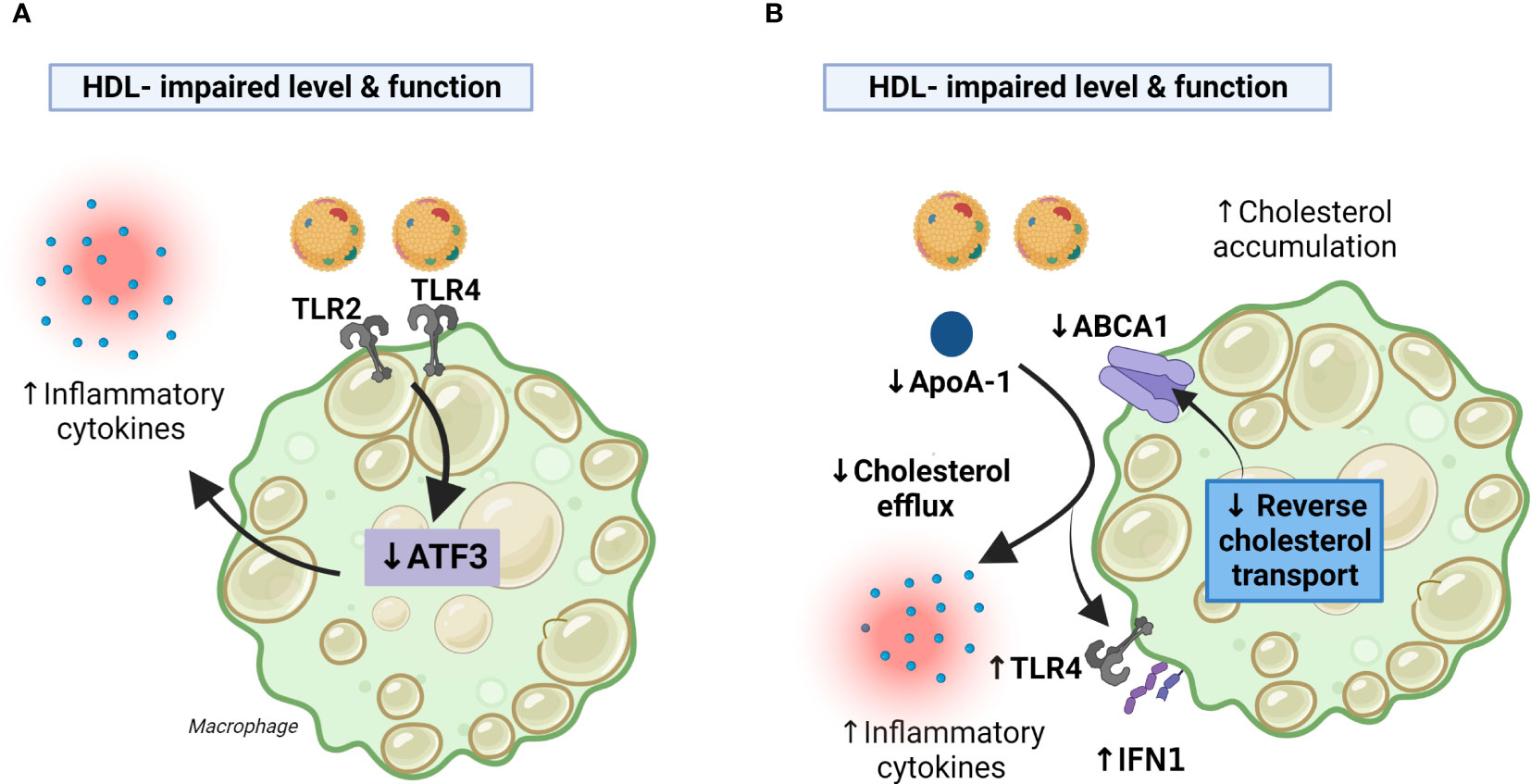
Figure 2 Decreased HDL levels and function in CVID may facilitate inflammatory responses in macrophages. Panel (A) HDL exert anti-inflammatory effects in macrophages by the induction of ATF3. CVID patients with systemic inflammation autoimmunity and inflammation have impaired HDL levels and function along with decreased AFT3 expression resulting in enhanced TLR2/4 signaling and increased production of inflammatory cytokines. Panel (B) CVID patients with autoimmunity and systemic inflammation had impaired cholesterol efflux capacity along with decreased expression of ABCA1 (PBMC) an ApoA-1 (serum), suggesting impaired RCT. This may result in (i) less inhibition of TLR4 and type I IFN signaling caused by attenuated RCT, (ii) less inhibition of TLR4 mediated release of inflammatory cytokines caused by down-regulation of ApoA-I and ABCA1. ABCA1, ATP-binding cassette transporter A1; ApoA-I, apolipoprotein A1; PBMC, peripheral blood mononuclear cells; RCT, reverse cholesterol transport; TLR, toll-like receptor. Created with BioRender.com.
In the study by Macpherson et al., it was shown that monocyte-derived macrophages from CVID patients had impaired cholesterol efflux capacity along with decreased expression ATP-binding cassette transporter A1 (ABCA1), a cell membrane protein that exports excess cholesterol from cells to ApoA-I, in their PBMC, suggesting impaired reverse cholesterol transport (40). Reverse cholesterol transport is an important part of the athero-protective properties of HDL (31), and it has also been shown that this process mediates anti-inflammatory effects. Thus, by counteracting cholesterol accumulation in macrophages, reverse cholesterol transport seems to inhibit TLR4 and type I interferon (IFN) signaling (37). Moreover, in addition to be of importance for cholesterol efflux mechanism, the interaction of apoA-I with ABCA1-expressing macrophages seems to suppress the ability of LPS to induce the inflammatory cytokines like interleukin (IL)-1β, IL-6, and TNF involving JAK2/STAT3 activation (46). Thus, it is plausible that the impaired cholesterol efflux mechanism, that involves impaired function and levels of HDL, decreased levels of ApoA-I (serum) and ABCA1 (macrophages) in CVID patients could contribute to the inflammatory phenotype in subgroups of these patients (Figure 2B). However, it has also been suggested that HDL could exert inflammatory effects in macrophages via passive cholesterol depletion and enhancement of TLR-induced signaling through a protein kinase C/NFkB/STAT1-IRF1 axis (38). These discrepancies could reflect differences in the experimental setup (e.g., in vivo versus in vitro experiments), but notably, this phenomenon could also reflect structural changes in the HDL particles during certain disease categories that include also chronic inflammatory disorders and the interaction between HDL and macrophages is still not fully elucidated (47, 48).
HDL and its main protein component ApoA-I can also exert anti-inflammatory effects by modulating T cell activation (49). Lipid rafts are regions of the plasma membrane which contain free cholesterol, sphingomyelin, and gangliosides that influence the functional capacity of both T and B cells (50). Enrichment of these lipid metabolites within cell membranes will enhance the cells autoimmune and inflammatory potential, and notably, HDL can attenuate these potential harmful properties through its enhancing effects on reverse cholesterol transport mechanisms (51). Of particular interest, regulatory T cells (Treg) are of major importance to ensure a balanced T cell response, interacting among others with dendritic cells (professional antigen-presenting cell type), to ensure a balanced immune response and notably (52–54), CVID patients have been shown to have a decreased proportion of this T cell subtype (55). In contrast to LDL, HDL seems to be taken up in Treg through scavenger receptor SR-BI/BII, and this appear to increase the survival of this important T cell subset (51, 56, 57). Thus, one could hypothesize that the decreased levels and impaired function of HDL in CVID could, at least partly, contribute to the decreased number of Treg and potentially also impaired function resulting in activated dendritic cells as well as enhanced production of effector T cells (Figure 3). In the largest HDL study on CVID, step-wise regression analysis identified soluble CD25, a reliable marker of T cell activation, as well as sex as the strongest predictor of HDL levels in CVID (40), further supporting a link between T cell pathology and low HDL levels in these patients.
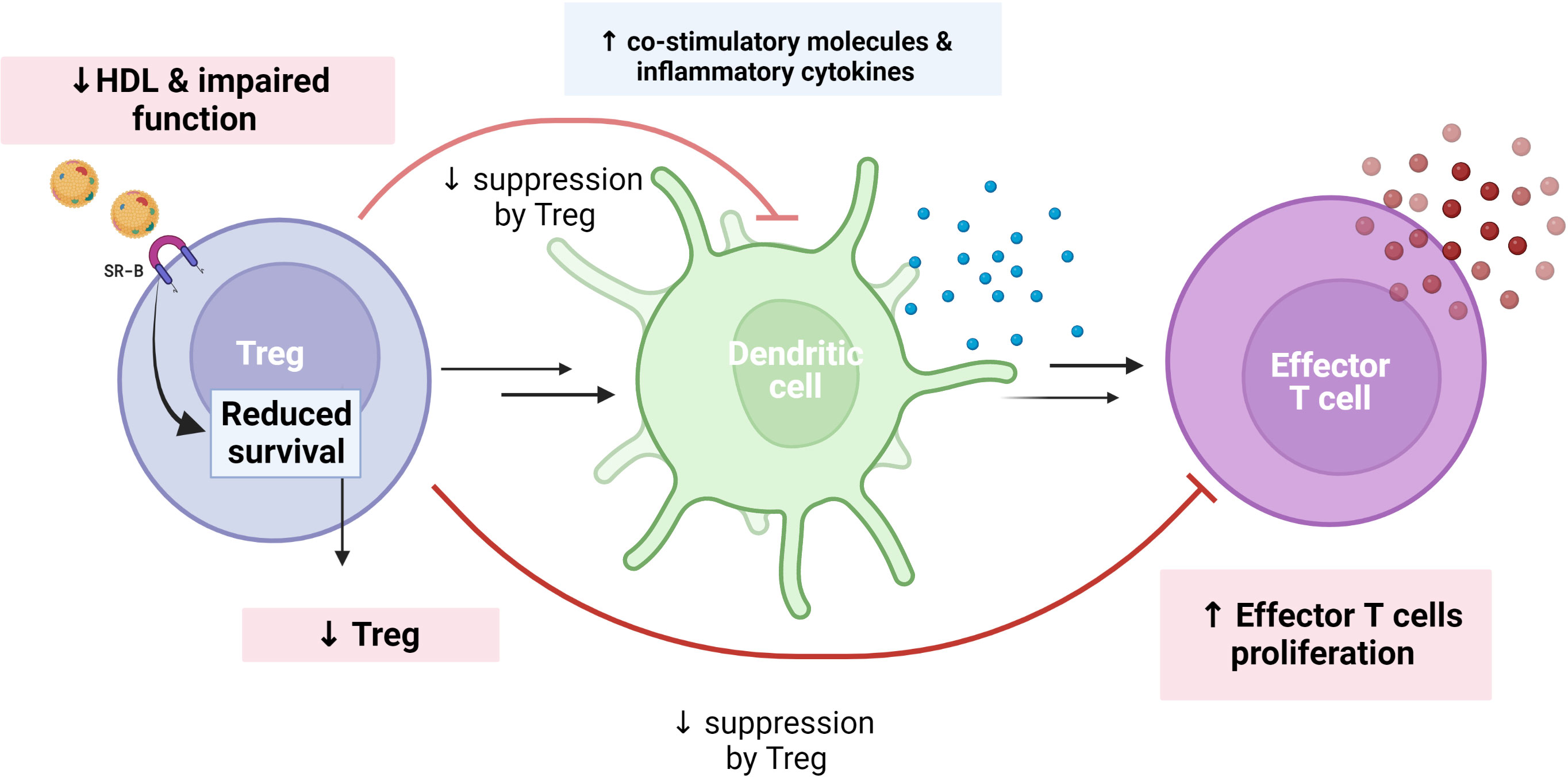
Figure 3 Theoretical figure showing how reduced HDL levels/function may explain reduce number/function of regulatory T cells (Treg) in CVID. In contrast to LDL, HDL is taken up in Tregs through the scavenger receptor SR-BI/BII, increasing the survival of this important T cell subset. Treg may attenuate dendritic cell response as well as proliferation of effector T cells to ensure a balanced immune response. This regulatory function of Tregs may be weakened in a situation with impaired HDL levels and function, as in CVID. We hypothesized that reduced HDL levels and function are associated with reduced Treg in CVID, which in turn decrease suppression of co-stimulatory molecules and increase effector T cell proliferation as well as inflammatory cytokines. Created with BioRender.com.
Data on the influence of HDL on B cell function are scarce. However, based on the ability of HDL to influence lipid rafts composition it is conceivable that this also will affect B cell function (58), and these issues will have to be clarified through further studies on the CVID population.
In contrast to HDL, which has both athero-protective and anti-inflammatory properties, TG and its major lipoprotein carrier, VLDL, are regarded as pro-atherogenic and related to inflammation (59–61). Although Vieira et al. found an inverse correlation between HDL and TG in CVID and XLA patients (n=18 and n=6, respectively), they found no differences in TG levels between these patients and healthy controls (38). More recently, however, Macpherson et al. reported significantly raised levels of TG in 95 CVID patients as compared with healthy controls (62). In a sub-study of 40 CVID patients, VLDL levels including all six sizes of this lipoprotein (extra-extra-large to extra-small), were also found to be increased as compared with healthy controls (62). It is well known that TG and VLDL are increased during acute infections (63, 64), but in the CVID study patients with ongoing acute infections were excluded (62). Moreover, TG and VLDL levels were not related to the occurrence of bronchiectasis, a condition that is characterized by low-grade and recurrent exacerbations of bacterial infections in the lower airways. In contrast, TG levels were significantly increased in the subgroup of CVID patients with autoimmunity and sterile inflammatory complications as compared to CVID patients without these complications (i.e., “infection only”) (62). TG levels in CVID patients were also correlated with increased levels of inflammatory markers (i.e., CRP, IL-6 and IL-12). Interestingly, there was a strong positive correlation between plasma levels of TG and LPS, linking disturbed VLDL/TG metabolism to gut leakage mechanisms in these patients (62). Thus, rather than being related to ongoing bacterial infections, it seems that raised levels of TG and VLDL in CVID is related to autoimmunity and non-infectious inflammation, which could also involve gut leakage mechanisms.
It is shown that inflammation could attenuate the effect and/or synthesis of enzymes that degrade TG such as lipoprotein lipase (LPL) and hepatic lipase (64, 65), and LPL has also been shown to enhance the levels of inflammatory cytokines like TNF (66). Moreover, the VLDL-associated lipoprotein ApoC-III has emerged as being particularly important as a regulator of TG transport involving mechanisms like impairment of LPL-mediated lipolysis, promotion of hepatic VLDL secretion, and suppression of TG clearance through the LDL receptor (67), and notably, ApoC-III may in itself promote inflammation (68). This displays the complexity of interactions between TG/VLDL metabolism and inflammation (Figure 4). Although it is tempting to hypothesize that persistent inflammation may downregulate LPL and hepatic lipase, whilst increasing ApoC-III in CVID, there is currently no data on these important regulators of TG and TG-rich lipoproteins (i.e., VLDL) in CVID.
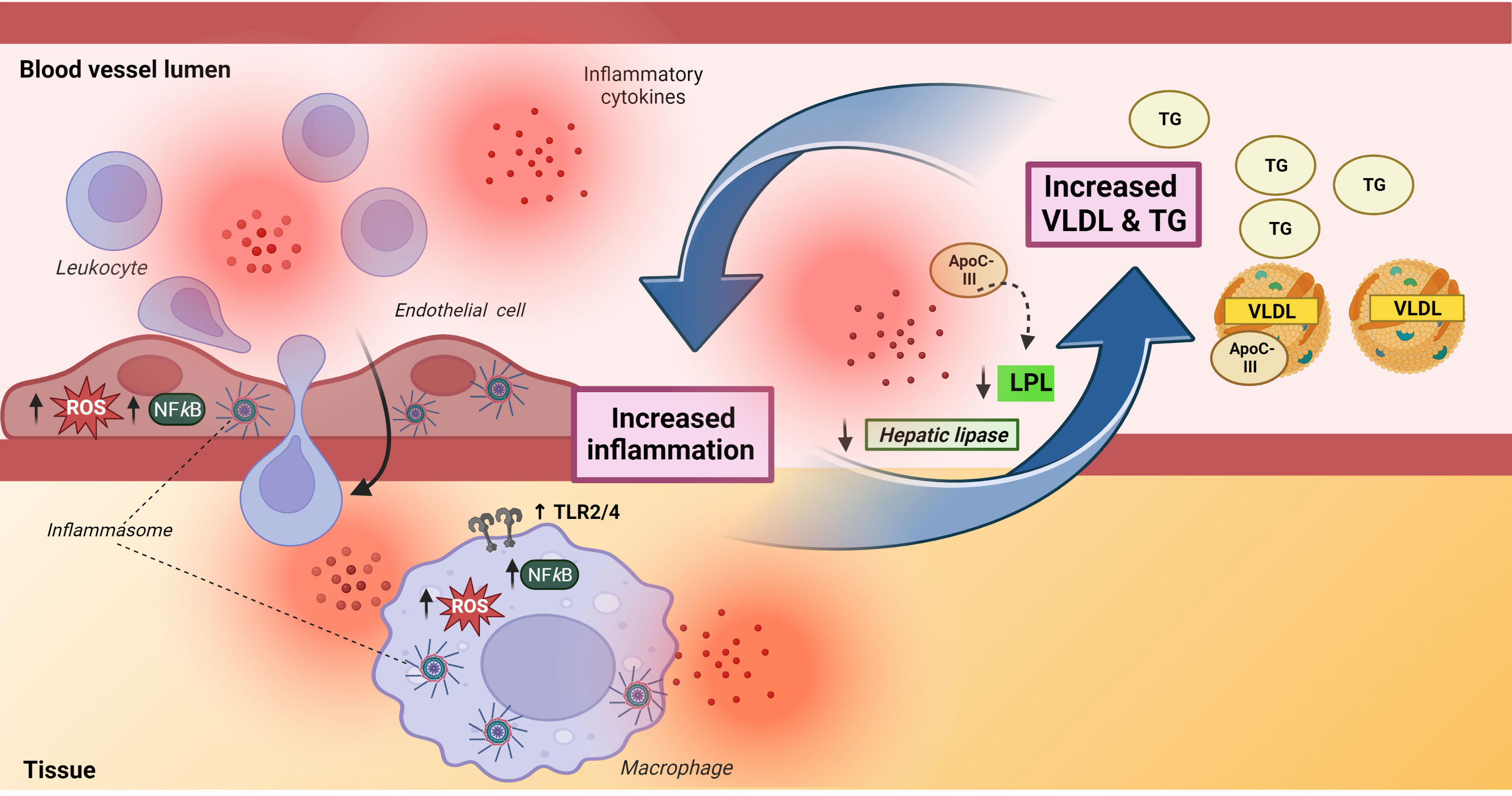
Figure 4 A bidirectional interaction between inflammation and triglycerides and very low-density lipoprotein. The left part of the Figure illustrates the complex effect of TG/VLDL on inflammation, inducing upregulation of TLR2/TLR4 in macrophages as well as promoting enhanced production of reactive oxygen species, activation of NFkB signaling pathways and activation of NLRP inflammasomes in both endothelial (NLRP1) and macrophages (NLRP3). The latter involves ApoC-III related mechanisms. The activation of endothelial cells facilitates leukocyte migration into inflamed tissue. The right part of the Figure illustrates that inflammation increases TG/VLDL levels by downregulating levels and attenuating the lipolytic activity of the enzymes, hepatic and lipoprotein lipase. Apoliprotein C-III, a major component of VLDL, could also attenuate the lipolytic activity of lipoprotein lipase. ApoC-III, apolipoprotein C-III; HL, hepatic lipase; LPL, lipoprotein lipase; ROS, reactive oxygen species, TG, triglycerides; VLDL, very low-density lipoprotein. Created with BioRender.com.
Whereas inflammation may promote hypertriglyceridemia and increased levels of VLDL, these lipid metabolites may themselves also promote inflammation, potentially representing a pathogenic loop in inflammatory disorders like CVID with autoimmune and inflammatory complications (61, 69), Figure 4. Thus, VLDL has been reported to induce inflammatory responses such as increased release of IL-1β and TNF in monocytes/macrophages through various mechanisms like activation of AP-1, ERK and NFκB pathways (70–72). Of particular interest, Zewinger and colleagues showed that ApoC-III, a major constituent of VLDL and a modulator of TG metabolism, activated the NLRP3 inflammasome in human monocytes in an alternative manner through caspase-8 and dimerization of TLR2 and TLR4 (73). This alternative inflammasome activation in human monocytes is mediated by the TLR adapter protein SCIMP, triggering Lyn/Syk-dependent calcium entry and the production of reactive oxygen species (ROS), leading to activation of caspase-8. More recently it has been reported that this effect of ApoC-III is not dependent of its binding of VLDL (74). However, VLDL is still a source of ApoC-III, but the exact molecular mechanisms for the ApoC-III-mediated activation of NLRP3 still need to be fully elucidated (75). TGs and in particular TG-rich lipoproteins (i.e., VLDL) can also promote inflammatory responses in endothelial cells, including induction of ROS and activation of various transcriptional factors like NFkB (59). VLDL has also been shown to promote expression of NLRP1 inflammasomes in human arterial endothelial cells (76), and in relevance to endothelial cell activation, it has been reported that fibrin interacts with VLDL and its receptor to promote leukocyte transmigration through inhibition of the src kinase Fyn (77). Thus, TG and in particular VLDL have several inflammatory properties with relevance for CVID such as monocyte activation, induction of inflammatory cytokines, enhanced ROS production and leukocyte/endothelial cell interaction (Figure 4), all features that have been demonstrated in subgroups of CVID patients (78–81).
FAs constitute a major component of TGs cholesteryl esters as well as phospholipids, serving among others as sources of energy and structural building blocks of cell membranes. Also, FAs and their derivatives take part in the regulation of several intracellular processes such as gene transcription, post-translational modification of proteins as well as direct modulation of enzyme activities as coactivators, thereby possessing a great potential to influence human health and disease (82). According to their degree of unsaturation, FAs have been classified into saturated FAs (SFAs), monounsaturated FAs and polyunsaturated FAs (PUFAs). In terms of inflammation, the most significant FA functions are attributed to PUFAs. These compounds contain more than one double bond in their carbon backbone and can be further categorized in various groups according to their chemical structure. The common classification of PUFAs as either n (omega)-3 PUFA or n-6 PUFA refers to the position of the first double bond relative to the methyl end of the carbon chain backbone (83).
Eicosanoids are a family of n-6 and n-3 PUFA metabolites synthesized by enzymatic oxygenation pathways, and includes prostaglandins, thromboxanes and leukotrienes. These classical eicosanoids, which can be biosynthesized in various cell types upon inflammatory signals, are well-documented regulators of pathological as well as physiological inflammation (84, 85). Of these, some of the best studied groups are eicosapentaenoic acid (EPA; 20:5 n-3), docosahexaenoate (DHA; 22:6 n-3), dihomo-γ-linolenic acid (DGLA; 20:3 n-6), and arachidonic acid (AA; 20:4 n-6). PUFA metabolites are involved in a variety of cellular functions involving modulation of several inflammatory pathways (85, 86). For instance, prostaglandins and leukotrienes execute their biological effects by activating their respective cell surface G protein-coupled receptors (84). Also, FA could modulate inflammation through “specific” FA receptors such as peroxisome proliferator-activated receptors (PPAR)y (82, 87)and affect signaling through established inflammatory receptors like TLRs (88). FAs could also modulate intracellular receptors/sensors that control inflammatory cell signaling such as NLRP3 inflammasomes (89) and NFkB (90). Thus, whereas the n-3 PUFAs DHA and EPA inhibit TLR4 signaling and NLRP3 activation (89), certain SFAs such as palmitate, the most abundant SFA in plasma, induces NLRP3 inflammasome and TLR4 activation (91, 92).
Data on FAs in CVID have been scarce or lacking. Recently, however, Skarpengland et al. reported an unfavorable FA profile in plasma with reduced proportions of EPA and DHA, as well as the n-6 PUFA Linoleic acid (LA) (93). Although still debated, several studies have proposed potential beneficial effects of increased n-3 PUFAs on autoimmunity, inflammation and related metabolic disorders (83, 94, 95), suggesting that the reduced proportion of EPA and DHA in CVID patients may represent a disadvantageous FA profile that may affect inflammation and the metabolic pathways in these patients. In contrast, the role of n-6 PUFA in health and disease appear less clear (96). Thus, whereas LA that was reduced in CVID (93) has been thought to promote inflammation, some studies have suggested beneficial and anti-inflammatory effects of this n-6 PUFA (97, 98). Also, de Pablo et al. showed that erythrocyte levels of LA were inversely associated with the risk of rheumatoid arthritis, whereas no associations were observed for other n-6 or n-3 PUFAs (99). At present, however, the clinical and immunological consequences of reduced LA (and EPA and DHA) in CVID are not clear. Thus, in contrast to the CVID study on HDL and TG (40, 62), Skarpengland et al. found no association between the potential unfavorable FA profile and any clinical subtypes including those with autoimmunity or any systemic markers of inflammation (93). This could potentially, at least partly, reflect that a lower number of patients were included in the FA study (n=39) as compared with the other (HDL and TG) (n=95-102). However, the authors reported that IgG levels correlated with plasma n-3 PUFA proportion (93) and notably, it has been reported that n-3 PUFA can boost B cell activation and antibody production potentially involving enhanced levels of Th2 cytokines (100). Although there was no difference in the proportion of AA between CVID patients and controls (93), this n-6 PUFA contributes to the formation of both leukotrienes and prostaglandins (101, 102). Interestingly, we have previously showed increased cAMP/protein kinase A type 1 activation in T cells from CVID contributing to their impaired T cell function, and this pathway interacts with AA and its derivate (103, 104).
More recently, eicosanoid-related studies have identified new classes of FA-derived bioactive metabolites with potentially anti-inflammatory and pro-resolving effects. These pro-resolving lipid mediators, including resolvins, lipoxines, protecines and maresines, are all derived from specific n-6 and n-3 PUFA precursors, and could represent novel preventive and therapeutic targets (105, 106). However, at present, data on these specialized pro-resolving mediators in CVID are, to the best of our knowledge, lacking.
Many inflammatory and metabolic diseases have been associated with gut microbial dysbiosis, but mechanistic studies are scarce in humans. For most diseases it is not known if the gut microbial dysbiosis is causing the disease or the result of the disease or a combination thereof. However, in mice-studies, the host-microbe interaction has been studied in more detail showing that microbes can regulate both immune and metabolic pathways (107–110). Due to the heterogeneity of CVID, no mouse model exists to study the host-microbe interaction in this disorder (111), and therefore all microbiota studies in CVID are in humans. A decade ago Shulzhenko et al. presented results from gut biopsies in three CVID patients, showing that the intestinal epithelium introduced its own protective mechanisms by upregulating INF-inducible immune response pathways and simultaneously repressing Gata4-related metabolic functions (112). More recently, a few studies have investigated the gut microbiota profile in CVID patients (15, 29, 113–118). Gut microbial imbalance in patients with CVID mainly includes changes in microbial diversity, a decrease in presumably beneficial bacteria, and an increase in pathobionts. Interestingly, in another study by Shulzhenko et al., CVID patients with enteropathy had reduced mucosal IgA expression in duodenal biopsies and more pathobionts bacteria such as A. baumannii (113). The largest CVID study on gut microbiota to date reported a CVID-specific dysbiosis index that captured the dysbiosis seen in CVID (29). This index consisted of ten taxa: Bacilli, Dorea, Roseburia, Gammaproteobacteria (increased in CVID), and Bifidobacterium, Odoribacteracea, Christensenellaceae, Blautia, Sutterella, Desulfovibrionacea (reduced in CVID). Subgroup analysis showed that gut microbial dysbiosis was significantly more pronounced in CVID patients with inflammatory and autoimmune complications than in CVID patients without. Furthermore, there was a clear positive association between markers of systemic inflammation in blood, representing activation of monocyte/macrophage (soluble CD14) and T cells (soluble CD25), and the degree of dysbiosis in stool samples (29). However, the role of gut microbiota in the pathogenesis of CVID is still not clear.
The relationship between gut microbiota and metabolic disturbances have been widely examined in particular in relation to cardiovascular and related metabolic disorders (119–124). A study by Fu et al. showed that gut microbiota contributed to lipid variation, independently of age, gender and genetics of the host (121). Notably, the association between gut microbial dysbiosis and lipids was particularly strong for HDL and TG, but weaker for LDL and total cholesterol. Moreover, whereas several taxa seemed to have an unfavorable effect on the lipid profile, certain orders (Bacteroidales [phylum Bacteroidetes] and family Clostridiaceae [phylum Firmicutes]), known to be involved in bile acid metabolism, were suggested to have a potential favorable effect. Both taxa are also involved in short chain FA acid (SCFA) metabolism and these microbiota-derived FAs seem to be protective against atherosclerosis and related metabolic complications like metabolic syndrome and type 2 diabetes mellitus (121, 125).
Data on the association between altered gut microbiota and lipid disturbances in CVID are scarce. However, Skarpengland et al. showed that the gut microbial alpha diversity was positively associated with plasma n-6 PUFAs, but not with n-3 PUFA, suggesting that plasma FA composition in blood could be related to gut microbial diversity (93). Interestingly, the FA composition was not related to the diet (93). Furthermore, a 2-week intervention with the non-absorbable antibiotic rifaximin, significantly altered FA composition in blood (29, 93, 126). These findings further support an important concept; that alteration of the gut microbial composition could affect FA levels in blood, at least in CVID.
In a recent study, Macpherson et al. reported a relationship between the CVID specific dysbiosis index in stools samples and TG levels in blood. Also, the major TG related lipoprotein VLDL was associated with the CVID specific dysbiosis index. However, there was no significant association between alpha diversity, reflecting a more general non-specific dysbiosis, and blood lipids in CVID (62). This may suggest that the association between TG and VLDL and gut microbiota is specific to the bacteria causing the dysbiosis in CVID. Moreover, CVID patients have increased plasma levels of LPS, a marker of gut leakage, and LPS was associated with gut dysbiosis and markers of systemic inflammation as well as T cell pathology in CVID (17, 29, 127, 128). In the study on TG and VLDL in CVID, Macpherson et al. reported a very strong correlation between LPS and TG, and LPS and VLDL (62). In the same cohort, there was no significant correlation between LPS and HDL (40), which is interesting since HDL has been shown to bind and neutralize LPS (129). However, more recent studies, suggest that LPS and related molecules are efficiently redistributed from HDL to other lipoprotein subclasses, in particular during pathological condition such as infections and inflammatory disorders (130, 131). As for TG, it is well known that bacterial endotoxins such as LPS elicits elevated plasma lipid levels due to increased synthesis and secretion of TG-rich lipoproteins by the liver and inhibition of LPL, often termed “the lipemia of sepsis” (132, 133). However, the association between TG and LPS may also reflect more indirect mechanisms. Thus, LPS is a potent and prototypical activator of TLR4, leading to increased production of inflammatory cytokines (134) that may also affect levels of TG and its major lipoprotein, VLDL. The mechanism causing the apparent shift in LPS affinity from HDL to VLDL must be further examined in more mechanistic studies and not only based on correlation analyses. Finally, and with relevance to CVID with both intestinal and hepatic pathology (15, 135), it has been suggested that intestinal synthesized HDL could be delivered into the portal circulation, thereby protecting the liver from injury in response to gut-derived LPS (24).
Another recent study showed that CVID patients had increased serum levels of bacterial DNA, belonging to gut commensals that were associated with parameters of systemic immune activation, increased serum IFN-γ and the low numbers of isotype-switched memory B cells (136). Whether these bacterial products also modulate lipid metabolisms has not been investigated. Except for one study of oropharyngeal microbial composition in CVID (137), there are to the best of our knowledge no data on dysbiosis in other organs. Forthcoming studies should address whether microbial dysbiosis outside the gut, e.g. the lungs could contribute to systemic inflammation and lipid dysregulation in CVID.
Macpherson et al. reported an association between increased plasma levels of the gut derived microbial metabolite, trimethylamine N-oxide (TMAO), and enhanced systemic inflammation, LPS, as well as the gut microbial abundance of Gammaproteobacteria in CVID patients (138). In terms of metabolic disturbances, these findings are interesting, since the TMAO-generating enzyme flavin monooxygenase 3 (FMO-3) may influence lipid metabolism and inflammation (139). Furthermore, a mice model has also suggested that TMAO may downregulate cholesterol 7α-hydroxylase, indicating a specific decline in the classical bile acid synthesis pathway with increased cholesterol levels as a consequence (140). The authors also identified the TMAO-generating enzyme FMO-3 as a powerful modifier of cholesterol metabolism and reverse cholesterol transport (140). More recently, phenylacetylglutamine has been identified as a gut microbiota-derived metabolite with impact on inflammation, thrombus formation and lipid metabolisms (141–143). In addition, SCFAs such as acetate, propionate and butyrate, mainly produced by anaerobic fermentation of gut microbes, has been associated with inflammation and lipid dysregulation in other disases (144, 145). Future studies should aim to analyze these important microbiota-derived metabolites and their relation to both metabolic and inflammatory phenotypes in CVID.
Recently, a multi-omics analysis was performed by Kaarbø et al., where duodenal biopsies with inflammation from CVID patients were compared to biopsies from healthy individuals. Pathway enrichment analyses suggested a different transcript signature for “cellular response to lipids” in CVID patients with duodenal inflammation compared to health controls (146). These findings further underscore a role for the intestine in mediating metabolic disturbances in CVID.
Based on the data presented above, CVID patients are characterized by low levels of HDL and high levels of TG and VLDL as well as an unfavorable FA profile in plasma, reflecting a lipid profile which could be suggestive of increased cardiovascular risk. Indeed, it has been proposed that accelerated atherosclerosis could be overlooked in CVID (147). However, LDL levels in the CVID patients were not different from those in healthy controls, including all subclasses of this pro-atherogenic lipoprotein (62). Moreover, in the TG/VLDL study, CVID patients had similar Body Mass Index (BMI) compared to healthy controls, and within the CVID group, there was no association between TG levels and gender, BMI, liver pathology or diet (including energy and fat intake), all known risk factors for atherosclerotic disease (62). Also, in the FA study, there were no differences in neither the dietary intake of fat, protein, carbohydrate, meat and egg, nor the dietary intake of saturated FA, mono-unsaturated FAs, and total PUFAs, between CVID patients and the general Norwegian population, suggesting that the lipid disturbances in CVID do not merely reflect differences in diet (93). Importantly, the CVID lipid profile does not fit with the classical lipid profile in metabolic syndrome where the lipid disturbances are associated with a high BMI, or the classical high risk patients in relation atherosclerosis-related disease where high levels of LDL is an important feature (148, 149). Moreover, whereas liver steatosis is an important feature of metabolic syndrome (150), this is not the case in CVID where nodular regenerative hyperplasia is the most common cause of liver pathology, a disorder that has been suggested to involve immune mediated mechanism (151). In the TG study it was estimated that 17% of the CVID patients had a diagnosis of cardiovascular disease, which does not appear to be increased compared to the general background population (62). Of note, CVID patients with cardiovascular disease showed no difference in TG levels as compared to CVID patients without cardiovascular disease (62). Interestingly, however, whereas age is risk factor for cardiovascular diseases (152), it was the younger CVID patients (<50 years of age) that had higher TG levels than age-matched healthy controls (62). Thus, it seems that the phenotype CVID is different from those with traditional cardiovascular risk including those with metabolic syndrome (Figure 5). However, several studies have shown that CVID patients, and in particular the large subgroup with autoimmune and inflammatory complications, are characterized by a state of persistent low-grade non-resolving inflammation that is a well-recognized risk factor for cardiovascular disease (153). Thus, before any conclusion can be drawn in relation to the cardiovascular risk in CVID, this will have to be investigated in much larger cohorts with clinical endpoints that due to the rarity of CVID will require international multi-centre studies.
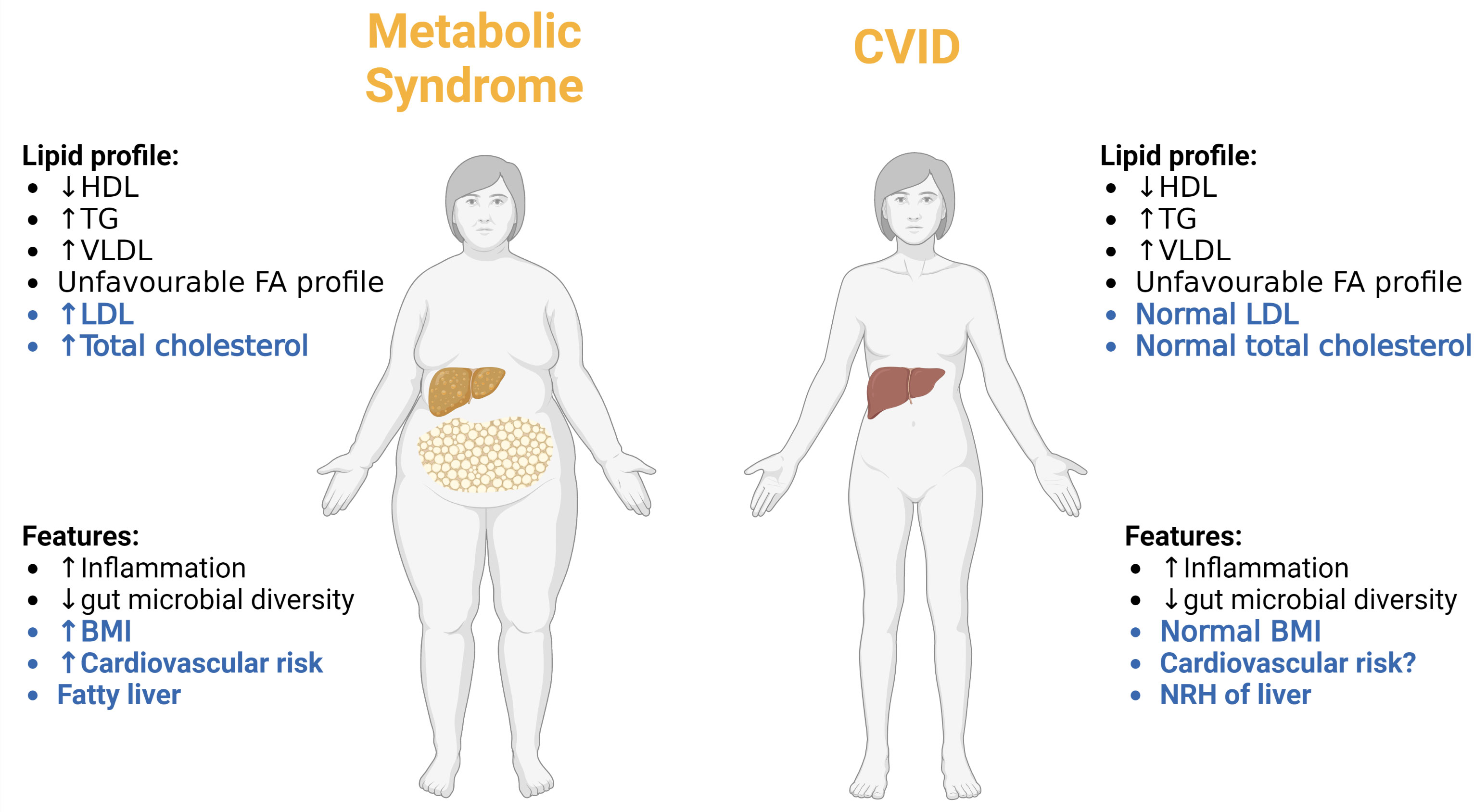
Figure 5 Comparing the metabolic phenotype of CVID patients to patients with classical metabolic syndrome. Differences are highlighted in blue and similarities in black. NRH, nodular regenerative hyperplasia. Created with BioRender.com.
CVID patients are characterized by persistent low-grade systemic inflammation and a large proportion of them have autoimmune and inflammatory complications. Recent data, as summarized in this review article, suggest that this inflammatory phenotype is associated with a disturbed lipid profile characterized by decrease levels and impaired function of HDL, increased levels of TG and its major lipoprotein VLDL as well as an unfavourable FA profile. We hypothesize that these metabolic disturbances are linked to inflammation in a bidirectional manner with disturbed gut microbiota as a potential missing link (Figure 6). These interactions between metabolic and inflammatory disturbances may also reflect interaction between environmental factors and genes, i.e., epigenetic and epitranscriptomic modifications (16). However, this hypothesis needs to be confirmed by in depth mechanistic studies as well as studies in lager CVID populations. Further studies should also explore potential therapeutic options that could attenuate the pathogenic loop between inflammation and metabolic disturbances, such as more targeted anti-inflammatory or immune modulating therapy like inhibition of the NLRP-3 inflammasome by IL-1 inhibition, lipid modulating drugs such as ApoA-1 mimetics, cholesteryl transfer protein inhibitors, statins or diet interventions targeting the gut microbial dysbiosis in CVID.
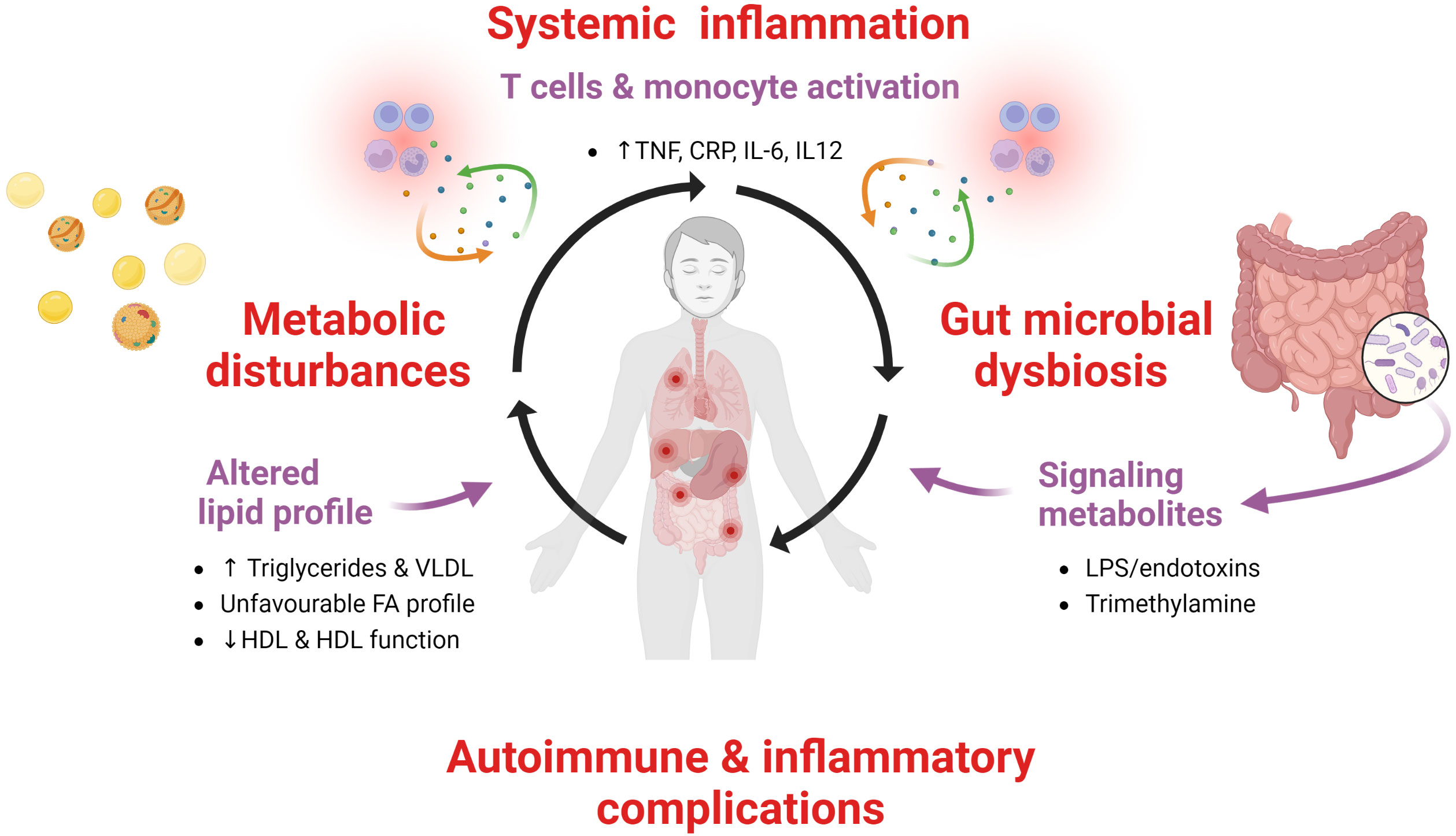
Figure 6 A pathogenic loop between persistent low-grade systemic inflammation, metabolic disturbances and gut microbiota in CVID. We hypothesize that in CVID patients with autoimmunity and inflammatory complications there is a pathogenic loop between persistent low-grade systemic inflammation and metabolic disturbances with altered gut microbiota as a contributing factor.
SJ and PA wrote the first draft of the paper. MM, TS, RB, BF, and BH provided intellectual input and contributed to the text. SJ made the figures in the manuscript. All authors critically revised the manuscript for important intellectual content. All authors contributed to the article and approved the submitted version.
SJ (project number 2019089) and MM (project number 2012/521) were funded by grants from the South-Eastern Norway Regional Health Authority.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. van Diepen JA, Berbée JF, Havekes LM, Rensen PC. Interactions between inflammation and lipid metabolism: relevance for efficacy of anti-inflammatory drugs in the treatment of atherosclerosis. Atherosclerosis (2013) 228:306–15. doi: 10.1016/j.atherosclerosis.2013.02.028
2. Wu H, Ballantyne CM. Metabolic inflammation and insulin resistance in obesity. Circ Res (2020) 126:1549–64. doi: 10.1161/CIRCRESAHA.119.315896
3. Esposito K, Giugliano D. The metabolic syndrome and inflammation: association or causation? Nutr Metab Cardiovasc Dis (2004) 14:228–32. doi: 10.1016/S0939-4753(04)80048-6
4. Yu JW, Lee MS. Mitochondria and the NLRP3 inflammasome: physiological and pathological relevance. Arch Pharm Res (2016) 39:1503–18. doi: 10.1007/s12272-016-0827-4
5. Piranavan P, Bhamra M, Perl A. Metabolic targets for treatment of autoimmune diseases. Immunometabolism (2020) 2. doi: 10.20900/immunometab20200012
6. Huang N, Perl A. Metabolism as a target for modulation in autoimmune diseases. Trends Immunol (2018) 39:562–76. doi: 10.1016/j.it.2018.04.006
7. Williams NC, O’Neill LAJ. A role for the Krebs cycle intermediate citrate in metabolic reprogramming in innate immunity and inflammation. Front Immunol (2018) 9:141. doi: 10.3389/fimmu.2018.00141
8. Zasłona Z, O’Neill LAJ. Cytokine-like roles for metabolites in immunity. Mol Cell (2020) 78:814–23. doi: 10.1016/j.molcel.2020.04.002
9. Cunningham-Rundles C. How I treat common variable immune deficiency. Blood (2010) 116:7–15. doi: 10.1182/blood-2010-01-254417
10. Oksenhendler E, Gérard L, Fieschi C, Malphettes M, Mouillot G, Jaussaud R, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis (2008) 46:1547–54. doi: 10.1086/587669
11. Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood (2008) 112:277–86. doi: 10.1182/blood-2007-11-124545
12. Ho HE, Cunningham-Rundles C. Seeking relevant biomarkers in common variable immunodeficiency. Front Immunol (2022) 13:857050. doi: 10.3389/fimmu.2022.857050
13. Peng XP, Caballero-Oteyza A, Grimbacher B. Common variable immunodeficiency: more pathways than roads to Rome. Annu Rev Pathol (2023) 18:283–310. doi: 10.1146/annurev-pathmechdis-031521-024229
14. Varzaneh FN, Keller B, Unger S, Aghamohammadi A, Warnatz K, Rezaei N. Cytokines in common variable immunodeficiency as signs of immune dysregulation and potential therapeutic targets - a review of the current knowledge. J Clin Immunol (2014) 34:524–43. doi: 10.1007/s10875-014-0053-0
15. Andersen IM, Jorgensen SF. Gut inflammation in CVID: causes and consequences. Expert Rev Clin Immunol (2022) 18:31–45. doi: 10.1080/1744666X.2021.2008241
16. Jorgensen SF, Fevang B, Aukrust P. Autoimmunity and inflammation in CVID: a possible crosstalk between immune activation, gut microbiota, and epigenetic modifications. J Clin Immunol (2019) 39:30–6. doi: 10.1007/s10875-018-0574-z
17. Varricchi G, Poto R, Ianiro G, Punziano A, Marone G, Gasbarrini A, et al. Gut microbiome and common variable immunodeficiency: few certainties and many outstanding questions. Front Immunol (2021) 12:712915. doi: 10.3389/fimmu.2021.712915
18. Cohn JS, Kamili A, Wat E, Chung RW, Tandy S. Dietary phospholipids and intestinal cholesterol absorption. Nutrients (2010) 2:116–27. doi: 10.3390/nu2020116
19. Luo J, Yang H, Song BL. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol (2020) 21:225–45. doi: 10.1038/s41580-019-0190-7
20. Afonso MS, Machado RM, Lavrador MS, Quintao ECR, Moore KJ, Lottenberg AM. Molecular pathways underlying cholesterol homeostasis. Nutrients (2018) 10:760. doi: 10.3390/nu10060760
21. Davis HR Jr., Altmann SW. Niemann–pick C1 like 1 (NPC1L1) an intestinal sterol transporter. Biochim Biophys Acta (BBA)-Molecular Cell Biol Lipids (2009) 1791:679–83. doi: 10.1016/j.bbalip.2009.01.002
22. Ko C-W, Qu J, Black DD, Tso P. Regulation of intestinal lipid metabolism: current concepts and relevance to disease. Nat Rev Gastroenterol Hepatol (2020) 17:169–83. doi: 10.1038/s41575-019-0250-7
23. Kumari A, Kristensen KK, Ploug M, Winther AL. The importance of lipoprotein lipase regulation in atherosclerosis. Biomedicines (2021) 9:782. doi: 10.3390/biomedicines9070782
24. Han Y-H, Onufer EJ, Huang L-H, Sprung RW, Davidson WS, Czepielewski RS, et al. Enterically derived high-density lipoprotein restrains liver injury through the portal vein. Science (2021) 373:eabe6729. doi: 10.1126/science.abe6729
25. Kwiterovich PO Jr. The metabolic pathways of high-density lipoprotein, low-density lipoprotein, and triglycerides: a current review Am J Cardiol (2000) 86:5–10 doi: 10.1016/S0002-9149(00)01461-2
26. Zhang K, Kaufman RJ. Unfolding the toxicity of cholesterol. Nat Cell Biol (2003) 5:769–70. doi: 10.1038/ncb0903-769
27. Poznyak AV, Nikiforov NG, Markin AM, Kashirskikh DA, Myasoedova VA, Gerasimova EV, et al. Overview of OxLDL and its impact on cardiovascular health: focus on atherosclerosis. Front Pharmacol (2021) 2248. doi: 10.3389/fphar.2020.613780
28. Ouimet M, Barrett TJ, Fisher EA. HDL and reverse cholesterol transport: basic mechanisms and their roles in vascular health and disease. Circ Res (2019) 124:1505–18. doi: 10.1161/CIRCRESAHA.119.312617
29. Jorgensen SF, Troseid M, Kummen M, Anmarkrud JA, Michelsen AE, Osnes LT, et al. Altered gut microbiota profile in common variable immunodeficiency associates with levels of lipopolysaccharide and markers of systemic immune activation. Mucosal Immunol (2016) 9:1455–65. doi: 10.1038/mi.2016.18
30. Marques LR, Diniz TA, Antunes BM, Rossi FE, Caperuto EC, Lira FS, et al. Reverse cholesterol transport: molecular mechanisms and the non-medical approach to enhance HDL cholesterol. Front Physiol (2018) 9:526. doi: 10.3389/fphys.2018.00526
31. Ouimet M, Barrett TJ, Fisher EA. HDL and reverse cholesterol transport. Circ Res (2019) 124:1505–18. doi: 10.1161/CIRCRESAHA.119.312617
32. Mineo C, Shaul PW. Novel biological functions of high-density lipoprotein cholesterol. Circ Res (2012) 111:1079–90. doi: 10.1161/CIRCRESAHA.111.258673
33. Taborda NA, Blanquiceth Y, Urcuqui-Inchima S, Latz E, Hernandez JC. High-density lipoproteins decrease proinflammatory activity and modulate the innate immune response. J Interferon Cytokine Res (2019) 39:760–70. doi: 10.1089/jir.2019.0029
34. Shridas P, De Beer MC, Webb NR. High-density lipoprotein inhibits serum amyloid a-mediated reactive oxygen species generation and NLRP3 inflammasome activation. J Biol Chem (2018) 293:13257–69. doi: 10.1074/jbc.RA118.002428
35. Thacker SG, Zarzour A, Chen Y, Alcicek MS, Freeman LA, Sviridov DO, et al. High-density lipoprotein reduces inflammation from cholesterol crystals by inhibiting inflammasome activation. Immunology (2016) 149:306–19. doi: 10.1111/imm.12638
36. Niyonzima N, Samstad EO, Aune MH, Ryan L, Bakke SS, Rokstad AM, et al. Reconstituted high-density lipoprotein attenuates cholesterol crystal-induced inflammatory responses by reducing complement activation. J Immunol (2015) 195:257–64. doi: 10.4049/jimmunol.1403044
37. Fotakis P, Kothari V, Thomas DG, Westerterp M, Molusky MM, Altin E, et al. Anti-inflammatory effects of HDL (High-density lipoprotein) in macrophages predominate over proinflammatory effects in atherosclerotic plaques. Arterioscler Thromb Vasc Biol (2019) 39:e253–72. doi: 10.1161/ATVBAHA.119.313253
38. Vieira DG, Costa-Carvalho BT, Hix S, da Silva R, Correia MSG, Sarni ROS. Higher cardiovascular risk in common variable immunodeficiency and X-linked agammaglobulinaemia patients. Ann Nutr Metab (2015) 66:237–41. doi: 10.1159/000435818
39. Andrade IGA, de Souza FIS, Fonseca FLA, Aranda CS, Sarni ROS. Selenium-related nutritional status in patients with common variable immunodeficiency: association with oxidative stress and atherosclerosis risk. BMC Immunol (2021) 22:31. doi: 10.1186/s12865-021-00425-9
40. Macpherson ME, Halvorsen B, Yndestad A, Ueland T, Mollnes TE, Berge RK, et al. Impaired HDL function amplifies systemic inflammation in common variable immunodeficiency. Sci Rep (2019) 9:9427. doi: 10.1038/s41598-019-45861-1
41. Heinecke JW. Small HDL promotes cholesterol efflux by the ABCA1 pathway in macrophages: implications for therapies targeted to HDL. Am Heart Assoc (2015) Volume 116:1101–3. doi: 10.1161/CIRCRESAHA.115.306052
42. Foit L, Thaxton CS. Synthetic high-density lipoprotein-like nanoparticles potently inhibit cell signaling and production of inflammatory mediators induced by lipopolysaccharide binding toll-like receptor 4. Biomaterials (2016) 100:67–75. doi: 10.1016/j.biomaterials.2016.05.021
43. De Nardo D, Labzin LI, Kono H, Seki R, Schmidt SV, Beyer M, et al. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat Immunol (2014) 15:152–60. doi: 10.1038/ni.2784
44. Zhang H, Reilly MP. Anti-inflammatory effects of high-density lipoprotein through activating transcription factor 3: benefit beyond cholesterol transport-dependent processes. Arterioscler Thromb Vasc Biol (2014) 34:e11–2. doi: 10.1161/ATVBAHA.114.303553
45. Ku HC, Cheng CF. Master regulator activating transcription factor 3 (ATF3) in metabolic homeostasis and cancer. Front Endocrinol (Lausanne) (2020) 11:556. doi: 10.3389/fendo.2020.00556
46. Tang C, Liu Y, Kessler PS, Vaughan AM, Oram JF. The macrophage cholesterol exporter ABCA1 functions as an anti-inflammatory receptor. J Biol Chem (2009) 284:32336–43. doi: 10.1074/jbc.M109.047472
47. van der Vorst EPC, Theodorou K, Wu Y, Hoeksema MA, Goossens P, Bursill CA, et al. High-density lipoproteins exert pro-inflammatory effects on macrophages via passive cholesterol depletion and PKC-NF-κB/STAT1-IRF1 signaling. Cell Metab (2017) 25:197–207. doi: 10.1016/j.cmet.2016.10.013
48. Kopecky C, Michlits G, Säemann MD, Weichhart T. Pro- versus anti-inflammatory actions of HDLs in innate immunity. Cell Metab (2017) 26:2–3. doi: 10.1016/j.cmet.2017.04.007
49. Grao-Cruces E, Lopez-Enriquez S, Martin ME, Montserrat-de la Paz S. High-density lipoproteins and immune response: a review. Int J Biol Macromol (2022) 195:117–23. doi: 10.1016/j.ijbiomac.2021.12.009
50. Wang SH, Yuan SG, Peng DQ, Zhao SP. HDL and ApoA-I inhibit antigen presentation-mediated T cell activation by disrupting lipid rafts in antigen presenting cells. Atherosclerosis (2012) 225:105–14. doi: 10.1016/j.atherosclerosis.2012.07.029
51. Wilhelm AJ, Zabalawi M, Owen JS, Shah D, Grayson JM, Major AS, et al. Apolipoprotein a-I modulates regulatory T cells in autoimmune LDLr-/-, ApoA-i-/- mice. J Biol Chem (2010) 285:36158–69. doi: 10.1074/jbc.M110.134130
52. Mahnke K, Enk AH. Dendritic cells: key cells for the induction of regulatory T cells? Curr Top Microbiol Immunol (2005) 293:133–50. doi: 10.1007/3-540-27702-1_7
53. Ness S, Lin S, Gordon JR. Regulatory dendritic cells, T cell tolerance, and dendritic cell therapy for immunologic disease. Front Immunol (2021) 12:633436. doi: 10.3389/fimmu.2021.633436
54. Mills KH. Regulatory T cells: friend or foe in immunity to infection? Nat Rev Immunol (2004) 4:841–55. doi: 10.1038/nri1485
55. Fevang B, Yndestad A, Sandberg WJ, Holm AM, Muller F, Aukrust P, et al. Low numbers of regulatory T cells in common variable immunodeficiency: association with chronic inflammation in vivo. Clin Exp Immunol (2007) 147:521–5. doi: 10.1111/j.1365-2249.2006.03314.x
56. Rueda CM, Rodríguez-Perea AL, Moreno-Fernandez M, Jackson CM, Melchior JT, Davidson W, et al. High density lipoproteins selectively promote the survival of human regulatory T cells. J Lipid Res (2017) 58:1514–23. doi: 10.1194/jlr.M072835
57. Sorci-Thomas MG, Thomas MJ. Anti-inflammatory liaisons: T regulatory cells and HDL. J Lipid Res (2017) 58:1491–2. doi: 10.1194/jlr.C078618
58. Catapano AL, Pirillo A, Bonacina F, Norata GD. HDL in innate and adaptive immunity. Cardiovasc Res (2014) 103:372–83. doi: 10.1093/cvr/cvu150
59. Öörni K, Lehti S, Sjövall P, Kovanen PT. Triglyceride-rich lipoproteins as a source of proinflammatory lipids in the arterial wall. Curr Med Chem (2019) 26:1701–10. doi: 10.2174/0929867325666180530094819
60. Wang L, Gill R, Pedersen TL, Higgins LJ, Newman JW, Rutledge JC. Triglyceride-rich lipoprotein lipolysis releases neutral and oxidized FFAs that induce endothelial cell inflammation. J Lipid Res (2009) 50:204–13. doi: 10.1194/jlr.M700505-JLR200
61. Huang JK, Lee HC. Emerging evidence of pathological roles of very-Low-Density lipoprotein (VLDL). Int J Mol Sci (2022) 23. doi: 10.3390/ijms23084300
62. Macpherson ME, Skarpengland T, Hov JR, Ranheim T, Vestad B, Dahl TB, et al. Increased plasma levels of triglyceride-enriched lipoproteins associate with systemic inflammation, lipopolysaccharides, and gut dysbiosis in common variable immunodeficiency. J Clin Immunol (2023), 1–12. doi: 10.1007/s10875-023-01475-x
63. Khovidhunkit W, Kim MS, Memon RA, Shigenaga JK, Moser AH, Feingold KR, et al. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res (2004) 45:1169–96. doi: 10.1194/jlr.R300019-JLR200
64. Khovidhunkit W, Memon RA, Feingold KR, Grunfeld C. Infection and inflammation-induced proatherogenic changes of lipoproteins. J Infect Dis (2000) 181 Suppl 3:S462–72. doi: 10.1086/315611
65. Semb H, Peterson J, Tavernier J, Olivecrona T. Multiple effects of tumor necrosis factor on lipoprotein lipase in vivo. J Biol Chem (1987) 262:8390–4. doi: 10.1016/S0021-9258(18)47576-X
66. Kota RS, Ramana CV, Tenorio FA, Enelow RI, Rutledge JC. Differential effects of lipoprotein lipase on tumor necrosis factor-alpha and interferon-gamma-mediated gene expression in human endothelial cells. J Biol Chem (2005) 280:31076–84. doi: 10.1074/jbc.M412189200
67. Gordts PL, Nock R, Son NH, Ramms B, Lew I, Gonzales JC, et al. ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J Clin Invest (2016) 126:2855–66. doi: 10.1172/JCI86610
68. Kohan AB. Apolipoprotein c-III: a potent modulator of hypertriglyceridemia and cardiovascular disease. Curr Opin Endocrinol Diabetes Obes (2015) 22:119–25. doi: 10.1097/MED.0000000000000136
69. Zhang BH, Yin F, Qiao YN, Guo SD. Triglyceride and triglyceride-rich lipoproteins in atherosclerosis. Front Mol Biosci (2022) 9:909151. doi: 10.3389/fmolb.2022.909151
70. Stollenwerk MM, Lindholm MW, Pörn-Ares MI, Larsson A, Nilsson J, Ares MP. Very low-density lipoprotein induces interleukin-1beta expression in macrophages. Biochem Biophys Res Commun (2005) 335:603–8. doi: 10.1016/j.bbrc.2005.07.123
71. Jinno Y, Nakakuki M, Kawano H, Notsu T, Mizuguchi K, Imada K, et al. Eicosapentaenoic acid administration attenuates the pro-inflammatory properties of VLDL by decreasing its susceptibility to lipoprotein lipase in macrophages. Atherosclerosis (2011) 219:566–72. doi: 10.1016/j.atherosclerosis.2011.09.046
72. Stollenwerk MM, Schiopu A, Fredrikson GN, Dichtl W, Nilsson J, Ares MP. Very low density lipoprotein potentiates tumor necrosis factor-alpha expression in macrophages. Atherosclerosis (2005) 179:247–54. doi: 10.1016/j.atherosclerosis.2004.12.002
73. Zewinger S, Reiser J, Jankowski V, Alansary D, Hahm E, Triem S, et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat Immunol (2020) 21:30–41. doi: 10.1038/s41590-019-0548-1
74. Hsu CC, Shao B, Kanter JE, He Y, Vaisar T, Witztum JL, et al. Apolipoprotein C3 induces inflammasome activation only in its delipidated form. Nat Immunol (2023) 24:408–11. doi: 10.1038/s41590-023-01423-2
75. Zewinger S, Reiser J, Jankowski V, Alansary D, Hahm E, Triem S, et al. Reply to: apolipoprotein C3 induces inflammasome activation only in its delipidated form. Nat Immunol (2023) 24:412–3. doi: 10.1038/s41590-023-01424-1
76. Bleda S, de Haro J, Varela C, Ferruelo A, Acin F. Elevated levels of triglycerides and vldl-cholesterol provoke activation of nlrp1 inflammasome in endothelial cells. Int J Cardiol (2016) 220:52–5. doi: 10.1016/j.ijcard.2016.06.193
77. Yakovlev S, Cao C, Galisteo R, Zhang L, Strickland DK, Medved L. Fibrin-VLDL receptor-dependent pathway promotes leukocyte transmigration by inhibiting src kinase fyn and is a target for fibrin β15-42 peptide. Thromb Haemost (2019) 119:1816–26. doi: 10.1055/s-0039-1695008
78. Aukrust P, Muller F, Froland SS. Enhanced generation of reactive oxygen species in monocytes from patients with common variable immunodeficiency. Clin Exp Immunol (1994) 97:232–8. doi: 10.1111/j.1365-2249.1994.tb06073.x
79. Aukrust P, Lien E, Kristoffersen AK, Muller F, Haug CJ, Espevik T, et al. Persistent activation of the tumor necrosis factor system in a subgroup of patients with common variable immunodeficiency–possible immunologic and clinical consequences. Blood (1996) 87:674–81. doi: 10.1182/blood.V87.2.674.bloodjournal872674
80. Cunningham-Rundles C. The many faces of common variable immunodeficiency. Hematol Am Soc Hematol Educ Program (2012) 2012:301–5. doi: 10.1182/asheducation.V2012.1.301.3798316
81. Baldovino S, Montin D, Martino S, Sciascia S, Menegatti E, Roccatello D. Common variable immunodeficiency: crossroads between infections, inflammation and autoimmunity. Autoimmun Rev (2013) 12:796–801. doi: 10.1016/j.autrev.2012.11.003
82. Papackova Z, Cahova M. Fatty acid signaling: the new function of intracellular lipases. Int J Mol Sci (2015) 16:3831–55. doi: 10.3390/ijms16023831
83. Sokoła-Wysoczańska E, Wysoczański T, Wagner J, Czyz K, Bodkowski R, Lochynski S, et al. Polyunsaturated fatty acids and their potential therapeutic role in cardiovascular system disorders-a review. Nutrients (2018) 10(10):1561. doi: 10.3390/nu10101561
84. Yamaguchi A, Botta E, Holinstat M. Eicosanoids in inflammation in the blood and the vessel. Front Pharmacol (2022) 3973. doi: 10.3389/fphar.2022.997403
85. Gutiérrez S, Svahn SL, Johansson ME. Effects of omega-3 fatty acids on immune cells. Int J Mol Sci (2019) 20. doi: 10.3390/ijms20205028
86. Djuricic I, Calder PC. Polyunsaturated fatty acids and metabolic health: novel insights. Curr Opin Clin Nutr Metab Care (2022) 25:436–42. doi: 10.1097/MCO.0000000000000865
87. Varga T, Czimmerer Z, Nagy L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim Biophys Acta (2011) 1812:1007–22. doi: 10.1016/j.bbadis.2011.02.014
88. Fessler MB, Rudel LL, Brown JM. Toll-like receptor signaling links dietary fatty acids to the metabolic syndrome. Curr Opin Lipidol (2009) 20:379–85. doi: 10.1097/MOL.0b013e32832fa5c4
89. Ralston JC, Lyons CL, Kennedy EB, Kirwan AM, Roche HM. Fatty acids and NLRP3 inflammasome-mediated inflammation in metabolic tissues. Annu Rev Nutr (2017) 37:77–102. doi: 10.1146/annurev-nutr-071816-064836
90. Hommelberg PP, Plat J, Langen RC, Schols AM, Mensink RP. Fatty acid-induced NF-kappaB activation and insulin resistance in skeletal muscle are chain length dependent. Am J Physiol Endocrinol Metab (2009) 296:E114–20. doi: 10.1152/ajpendo.00436.2007
91. Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest (2011) 121:2111–7. doi: 10.1172/JCI57132
92. Sokolova M, Vinge LE, Alfsnes K, Olsen MB, Eide L, Kaasboll OJ, et al. Palmitate promotes inflammatory responses and cellular senescence in cardiac fibroblasts. Biochim Biophys Acta Mol Cell Biol Lipids (2017) 1862:234–45. doi: 10.1016/j.bbalip.2016.11.003
93. Skarpengland T, Macpherson ME, Hov JR, Kong XY, Bohov P, Halvorsen B, et al. Altered plasma fatty acids associate with gut microbial composition in common variable immunodeficiency. J Clin Immunol (2022) 42:146–57. doi: 10.1007/s10875-021-01146-9
94. Djuricic I, Calder PC. Pros and cons of long-chain omega-3 polyunsaturated fatty acids in cardiovascular health. Annu Rev Pharmacol Toxicol (2023) 63:383–406. doi: 10.1146/annurev-pharmtox-051921-090208
95. Sherratt SCR, Libby P, Budoff MJ, Bhatt DL, Mason RP. Role of omega-3 fatty acids in cardiovascular disease: the debate continues. Curr Atheroscler Rep (2023) 25:1–17. doi: 10.1007/s11883-022-01075-x
96. Li J, Guasch-Ferré M, Li Y, Hu FB. Dietary intake and biomarkers of linoleic acid and mortality: systematic review and meta-analysis of prospective cohort studies. Am J Clin Nutr (2020) 112:150–67. doi: 10.1093/ajcn/nqz349
97. Wu JHY, Marklund M, Imamura F, Tintle N, Ardisson Korat AV, de Goede J, et al. Omega-6 fatty acid biomarkers and incident type 2 diabetes: pooled analysis of individual-level data for 39 740 adults from 20 prospective cohort studies. Lancet Diabetes Endocrinol (2017) 5:965–74. doi: 10.1016/S2213-8587(17)30307-8
98. Djuricic I, Calder PC. Beneficial outcomes of omega-6 and omega-3 polyunsaturated fatty acids on human health: an update for 2021. Nutrients (2021) 13:2421. doi: 10.3390/nu13072421
99. de Pablo P, Romaguera D, Fisk HL, Calder PC, Quirke AM, Cartwright AJ, et al. High erythrocyte levels of the n-6 polyunsaturated fatty acid linoleic acid are associated with lower risk of subsequent rheumatoid arthritis in a southern European nested case-control study. Ann Rheum Dis (2018) 77:981–7. doi: 10.1136/annrheumdis-2017-212274
100. Whelan J, Gowdy KM, Shaikh SR. N-3 polyunsaturated fatty acids modulate b cell activity in pre-clinical models: implications for the immune response to infections. Eur J Pharmacol (2016) 785:10–7. doi: 10.1016/j.ejphar.2015.03.100
101. Innes JK, Calder PC. Omega-6 fatty acids and inflammation. Prostaglandins Leukot Essent Fatty Acids (2018) 132:41–8. doi: 10.1016/j.plefa.2018.03.004
102. Wang B, Wu L, Chen J, Dong L, Chen C, Wen Z, et al. Metabolism pathways of arachidonic acids: mechanisms and potential therapeutic targets. Signal transduction targeted Ther (2021) 6:94. doi: 10.1038/s41392-020-00443-w
103. Holm AM, Aukrust P, Aandahl EM, Muller F, Tasken K, Froland SS. Impaired secretion of IL-10 by T cells from patients with common variable immunodeficiency–involvement of protein kinase a type I. J Immunol (2003) 170:5772–7. doi: 10.4049/jimmunol.170.11.5772
104. Aukrust P, Aandahl EM, Skålhegg BS, Nordoy I, Hansson V, Tasken K, et al. Increased activation of protein kinase a type I contributes to the T cell deficiency in common variable immunodeficiency. J Immunol (1999) 162:1178–85. doi: 10.4049/jimmunol.162.2.1178
105. Brouwers H, Jónasdóttir HS, Kuipers ME, Kwekkeboom JC, Auger JL, Gonzalez-Torres M, et al. Anti-inflammatory and proresolving effects of the omega-6 polyunsaturated fatty acid adrenic acid. J Immunol (2020) 205:2840–9. doi: 10.4049/jimmunol.1801653
106. Al-Shaer AE, Buddenbaum N, Shaikh SR. Polyunsaturated fatty acids, specialized pro-resolving mediators, and targeting inflammation resolution in the age of precision nutrition. Biochim Biophys Acta Mol Cell Biol Lipids (2021) 1866:158936. doi: 10.1016/j.bbalip.2021.158936
107. Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res (2020) 30:492–506. doi: 10.1038/s41422-020-0332-7
108. Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell (2014) 157:121–41. doi: 10.1016/j.cell.2014.03.011
109. Jiao Y, Wu L, Huntington ND, Zhang X. Crosstalk between gut microbiota and innate immunity and its implication in autoimmune diseases. Front Immunol (2020) 11:282. doi: 10.3389/fimmu.2020.00282
110. Sonnenburg JL, Bäckhed F. Diet-microbiota interactions as moderators of human metabolism. Nature (2016) 535:56–64. doi: 10.1038/nature18846
111. Jorgensen SF, Fevang B, Aukrust P. Commentary: gut antibody deficiency in a mouse model of CVID results in spontaneous development of a gluten-sensitive enteropathy. Front Immunol (2020) 11:1921. doi: 10.3389/fimmu.2020.01921
112. Shulzhenko N, Morgun A, Hsiao W, Battle M, Yao M, Gavrilova O, et al. Crosstalk between b lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nat Med (2011) 17:1585–93. doi: 10.1038/nm.2505
113. Shulzhenko N, Dong X, Vyshenska D, Greer RL, Gurung M, Vasquez-Perez S, et al. CVID enteropathy is characterized by exceeding low mucosal IgA levels and interferon-driven inflammation possibly related to the presence of a pathobiont. Clin Immunol (2018) 197:139–53. doi: 10.1016/j.clim.2018.09.008
114. Fadlallah J, Sterlin D, Fieschi C, Parizot C, Dorgham K, El Kafsi H, et al. Synergistic convergence of microbiota-specific systemic IgG and secretory IgA. J Allergy Clin Immunol (2019) 143:1575–1585 e4. doi: 10.1016/j.jaci.2018.09.036
115. Fiedorová K, Radvanský M, Bosák J, Grombirikova H, Nemcova E, Kralickova P, et al. Bacterial but not fungal gut microbiota alterations are associated with common variable immunodeficiency (CVID) phenotype. Front Immunol (2019) 10:1914. doi: 10.3389/fimmu.2019.01914
116. van Schewick CM, Nöltner C, Abel S, Burns SO, Workman S, Symes A, et al. Altered microbiota, impaired quality of life, malabsorption, infection, and inflammation in CVID patients with diarrhoea. Front Immunol (2020) 11:1654. doi: 10.3389/fimmu.2020.01654
117. Bosák J, Lexa M, Fiedorová K, Gadara DC, Micenkova L, Spacil Z, et al. Patients with common variable immunodeficiency (CVID) show higher gut bacterial diversity and levels of low-abundance genes than the healthy housemates. Front Immunol (2021) 12:671239. doi: 10.3389/fimmu.2021.671239
118. Jorgensen SF, Holm K, Macpherson ME, Storm-Larsen C, Kummen M, Fevang B, et al. Selective IgA deficiency in humans is associated with reduced gut microbial diversity. J Allergy Clin Immunol (2019) 143:1969–1971 e11. doi: 10.1016/j.jaci.2019.01.019
119. Jie Z, Xia H, Zhong SL, Feng Q, Li S, Liang S, et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat Commun (2017) 8:845. doi: 10.1038/s41467-017-00900-1
120. Xu Z, Jiang W, Huang W, Lin Y, Chan FKL, Ng SC. Gut microbiota in patients with obesity and metabolic disorders - a systematic review. Genes Nutr (2022) 17:2. doi: 10.1186/s12263-021-00703-6
121. Fu J, Bonder MJ, Cenit MC, Tigchelaar EF, Maatman A, Dekens JA, et al. The gut microbiome contributes to a substantial proportion of the variation in blood lipids. Circ Res (2015) 117:817–24. doi: 10.1161/CIRCRESAHA.115.306807
122. Makki K, Deehan EC, Walter J, Backhed F. The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe (2018) 23:705–15. doi: 10.1016/j.chom.2018.05.012
123. Wu H, Tremaroli V, Bäckhed F. Linking microbiota to human diseases: a systems biology perspective. Trends Endocrinol Metab (2015) 26:758–70. doi: 10.1016/j.tem.2015.09.011
124. Rahman MM, Islam F, Or-Rashid MH, Mamun AA, Rahaman MS, Islam MM, et al. The gut microbiota (Microbiome) in cardiovascular disease and its therapeutic regulation. Front Cell Infect Microbiol (2022) 12:903570. doi: 10.3389/fcimb.2022.903570
125. Vrieze A, Van Nood E, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology (2012) 143:913–6.e7. doi: 10.1053/j.gastro.2012.06.031
126. Jorgensen SF, Macpherson ME, Bjornetro T, Holm K, Kummen M, Rashidi A, et al. Rifaximin alters gut microbiota profile, but does not affect systemic inflammation - a randomized controlled trial in common variable immunodeficiency. Sci Rep (2019) 9:167. doi: 10.1038/s41598-018-35367-7
127. Le Coz C, Bengsch B, Khanna C, Trofa M, Ohtani T, Nolan BE, et al. Common variable immunodeficiency-associated endotoxemia promotes early commitment to the T follicular lineage. J Allergy Clin Immunol (2019) 144:1660–73. doi: 10.1016/j.jaci.2019.08.007
128. Perreau M, Vigano S, Bellanger F, Pellaton C, Buss G, Comte D, et al. Exhaustion of bacteria-specific CD4 T cells and microbial translocation in common variable immunodeficiency disorders. J Exp Med (2014) 211:2033–45. doi: 10.1084/jem.20140039
129. Feingold KR, Grunfeld C. The role of HDL in innate immunity. J Lipid Res (2011) 52:1–3. doi: 10.1194/jlr.E012138
130. Levels JH, Marquart JA, Abraham PR, van den Ende AE, Molhuizen HO, van Deventer SJ, et al. Lipopolysaccharide is transferred from high-density to low-density lipoproteins by lipopolysaccharide-binding protein and phospholipid transfer protein. Infect Immun (2005) 73:2321–6. doi: 10.1128/IAI.73.4.2321-2326.2005
131. Levels JH, Abraham PR, van den Ende A, van Deventer SJ. Distribution and kinetics of lipoprotein-bound endotoxin. Infect Immun (2001) 69:2821–8. doi: 10.1128/IAI.69.5.2821-2828.2001
132. Harris HW, Gosnell JE, Kumwenda ZL. The lipemia of sepsis: triglyceride-rich lipoproteins as agents of innate immunity. J Endotoxin Res (2000) 6:421–30.
133. Barker G, Leeuwenburgh C, Brusko T, Moldawer L, Reddy ST, Guirgis FW. Lipid and lipoprotein dysregulation in sepsis: clinical and mechanistic insights into chronic critical illness. J Clin Med (2021) 10. doi: 10.3390/jcm10081693
134. Miller SI, Ernst RK, Bader MW. LPS. TLR4 and infectious disease diversity. Nat Rev Microbiol (2005) 3:36–46. doi: 10.1038/nrmicro1068
135. Pecoraro A, Crescenzi L, Varricchi G, Marone G, Spadaro G. Heterogeneity of liver disease in common variable immunodeficiency disorders. Front Immunol (2020) 11:338. doi: 10.3389/fimmu.2020.00338
136. Ho HE, Radigan L, Bongers G, El-Shamy A, Cunningham-Rundles C. Circulating bioactive bacterial DNA is associated with immune activation and complications in common variable immunodeficiency. JCI Insight (2021) 6. doi: 10.1172/jci.insight.144777
137. Berbers RM, Mohamed Hoesein FAA, Ellerbroek PM, van Montfrans JM, Dalm V, van Hagen PM, et al. Low IgA associated with oropharyngeal microbiota changes and lung disease in primary antibody deficiency. Front Immunol (2020) 11:1245. doi: 10.3389/fimmu.2020.01245
138. Macpherson ME, Hov JR, Ueland T, Dahl TB, Kummen M, Otterdal K, et al. Gut microbiota-dependent trimethylamine n-oxide associates with inflammation in common variable immunodeficiency. Front Immunol (2020) 11:574500. doi: 10.3389/fimmu.2020.574500
139. Warrier M, Shih DM, Burrows AC, Ferguson D, Gromovsky AD, Brown AL, et al. The TMAO-generating enzyme flavin monooxygenase 3 is a central regulator of cholesterol balance. Cell Rep (2015) 10:326–38. doi: 10.1016/j.celrep.2014.12.036
140. Ding L, Chang M, Guo Y, Zhang L, Xue C, Yanagita T, et al. Trimethylamine-n-oxide (TMAO)-induced atherosclerosis is associated with bile acid metabolism. Lipids Health Dis (2018) 17:286. doi: 10.1186/s12944-018-0939-6
141. Witkowski M, Weeks TL, Hazen SL. Gut microbiota and cardiovascular disease. Circ Res (2020) 127:553–70. doi: 10.1161/CIRCRESAHA.120.316242
142. Zhu Y, Dwidar M, Nemet I, Buffa JA, Sangwan N, Li XS, et al. Two distinct gut microbial pathways contribute to meta-organismal production of phenylacetylglutamine with links to cardiovascular disease. Cell Host Microbe (2023) 31:18–32.e9. doi: 10.1016/j.chom.2022.11.015
143. Awoyemi A, Hov JR, Trøseid M. Phenylacetylglutamine from the gut microbiota: a future therapeutic target in heart failure? Circ Heart Fail (2023) 16:e010222. doi: 10.1161/CIRCHEARTFAILURE.122.010222
144. Nogal A, Valdes AM, Menni C. The role of short-chain fatty acids in the interplay between gut microbiota and diet in cardio-metabolic health. Gut Microbes (2021) 13:1–24. doi: 10.1080/19490976.2021.1897212
145. Gill PA, van Zelm MC, Muir JG, Gibson PR. Review article: short chain fatty acids as potential therapeutic agents in human gastrointestinal and inflammatory disorders. Aliment Pharmacol Ther (2018) 48:15–34. doi: 10.1111/apt.14689
146. Kaarbo M, Yang M, Hov JR, Holm K, de Sousa MML, Macpherson ME, et al. Duodenal inflammation in common variable immunodeficiency has altered transcriptional response to viruses. J Allergy Clin Immunol (2023) 151(3):767–77. doi: 10.1016/j.jaci.2022.09.029
147. Ucar R, Arslan S, Turkmen K, Caliskaner AZ. Accelerated atherosclerosis in patients with common variable immunodeficiency: is it overlooked or absent? Med Hypotheses (2015) 85:485–7. doi: 10.1016/j.mehy.2015.07.002
148. Shabana, Shahid SU, Sarwar S. The abnormal lipid profile in obesity and coronary heart disease (CHD) in Pakistani subjects. Lipids Health Dis (2020) 19:73. doi: 10.1186/s12944-020-01248-0
149. Hilvo M, Dhar I, Lääperi M, Lysne V, Sulo G, Tell GS, et al. Primary cardiovascular risk prediction by LDL-cholesterol in Caucasian middle-aged and older adults: a joint analysis of three cohorts. Eur J Prev Cardiol (2022) 29:e128–37. doi: 10.1093/eurjpc/zwab075
150. Yki-Järvinen H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol (2014) 2:901–10. doi: 10.1016/S2213-8587(14)70032-4
151. Fuss IJ, Friend J, Yang Z, He JP, Hooda L, Boyer J, et al. Nodular regenerative hyperplasia in common variable immunodeficiency. J Clin Immunol (2013) 33:748–58. doi: 10.1007/s10875-013-9873-6
152. Rodgers JL, Jones J, Bolleddu SI, Vanthenapalli S, Rodgers LE, Shah K, et al. Cardiovascular risks associated with gender and aging. J Cardiovasc Dev Dis (2019) 6. doi: 10.3390/jcdd6020019
Keywords: CVID - common variable immunodeficiency, antibody deficiencies, fatty acids, metabolism, inflammation, HDL - cholesterol, triglycerid
Citation: Jorgensen SF, Macpherson ME, Skarpengland T, Berge RK, Fevang B, Halvorsen B and Aukrust P (2023) Disturbed lipid profile in common variable immunodeficiency – a pathogenic loop of inflammation and metabolic disturbances. Front. Immunol. 14:1199727. doi: 10.3389/fimmu.2023.1199727
Received: 03 April 2023; Accepted: 03 July 2023;
Published: 20 July 2023.
Edited by:
Isabelle Meyts, KU Leuven, BelgiumReviewed by:
Charlotte Cunningham-Rundles, Icahn School of Medicine at Mount Sinai, United StatesCopyright © 2023 Jorgensen, Macpherson, Skarpengland, Berge, Fevang, Halvorsen and Aukrust. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silje F. Jorgensen, cy5mLmpvcmdlbnNlbkBtZWRpc2luLnVpby5ubw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.