 Markus Zeisbrich
Markus Zeisbrich Jens Thiel
Jens Thiel Nils Venhoff
Nils Venhoff- 1Department of Rheumatology and Clinical Immunology, Medical Center – University of Freiburg, Freiburg, Germany
- 2Division of Rheumatology and Clinical Immunology, Medical University Graz, Graz, Austria
The network of IL-17 cytokines is considered a key component of autoimmune and inflammatory processes. Blocking IL-17 showed great success in psoriasis as well as psoriatic arthritis, and in patients with axial spondyloarthritis. Secukinumab is one of the approved IL-17A inhibitors for these diseases and is now routinely used. In giant cell arteritis, a large vessel vasculitis, there is accumulating evidence for a pathogenic role of IL-17 and Th17 cells, which are part of the CD4+ T-cell subset. Giant cell arteritis occurs in individuals over 50 years of age and many have relative contraindications to glucocorticoid therapy, which today still represents the mainstay therapy. Despite the approval of tocilizumab, which targets the IL-6 receptor, a high demand for glucocorticoid-sparing agents remains that combine the effective suppression of the acute inflammation observed in giant cell arteritis with a safety profile that matches the needs of an older patient population. The first results from a phase II proof-of-principle study (TitAIN) support an optimistic outlook on a potential new treatment option with secukinumab in giant cell arteritis.
Introduction
Interleukin (IL)-17 was discovered 30 years ago (1, 2) and is now recognized as one of the main cytokines for barrier protection on epithelial and mucosal surfaces with an essential function in immunity to mucosal fungal infections (3). Moreover, its pro-inflammatory function is relevant in immune-mediated diseases, first demonstrated in a mouse model for multiple sclerosis (4). The identification of the immuno-pathological role of IL-17 subsequently led to the development of IL-17 blocking agents. While the use of monoclonal anti-IL-17A antibodies was disappointing in rheumatoid arthritis and multiple sclerosis (5, 6), these therapeutics are now used with great success in patients with psoriasis, psoriatic arthritis, and axial spondyloarthritis (7, 8).
Giant cell arteritis (GCA) leads to vascular inflammation in large arteries as well as medium-sized arteries with a tissue tropism preferentially affecting the thoracic aorta, its proximal branches (e.g., A. subclavia, A. carotidea, and A. axillaris), and cranial arteries (e.g., A. temporalis superficiales and occipital arteries) (9). Vascular inflammation drives circular vessel wall thickening and luminal occlusion, which may cause ischemic symptoms like headaches, visual symptoms or blindness, scalp tenderness, or jaw claudication when branches of the carotid arteries are affected (10). GCA generally occurs over 50 years of age with the highest incidence among persons who are 75 to 85 years old (11). Glucocorticoids are still the standard treatment although their long-term use entails grave side effects in this older patient population (12, 13).
This review focuses on IL-17 and Th17 cells, and presents available evidence for the involvement of the IL-17 pathway in GCA. Additionally, the results of the phase II proof-of-concept study TitAIN with an IL-17A blocking agent in GCA patients with active disease are depicted and discussed.
Signaling and effector functions of the IL-17 cytokine family members
Six isoforms of IL-17 (IL-17A to IL-17F) are identified with IL-17A and IL-17F having the greatest homology and some overlapping functions (14, 15). IL-17A is the most intensively studied isoform and more potent than its closest sibling IL-17F (16). IL-17A and IL-17F can exist as homodimers or form IL-17A/F heterodimers and they are co-expressed by linked genes (17). Monoclonal antibodies such as secukinumab and ixekizumab directly bind IL-17A, while bimekizumab neutralizes both IL-17A and IL-17F.
IL-17 signaling is mediated through five receptors (IL-17RA to IL-17RE) (18). The IL-17 receptors RA, RC, and RD are required for the signaling of IL-17A and IL-17F (19). Receptor ligation activates common downstream intracellular signaling via nuclear factor-kappa B (NF-κB), mitogen-activated protein kinases (MAPK), CCAAT/enhancer-binding protein (C/EBP), Janus kinase (JAK), PI3K (phosphatidylinositol 3-kinase), and the JAK/STAT pathway (20) to promote gene transcription. In homeostasis, large numbers of IL-17-producing cells reside on mucocutaneus surfaces (19) and IL-17 is a key cytokine in promoting barrier functions in order to limit fungal and bacterial invasion (3). Defects in IL-17 production lead to impaired protection against candida and other fungal pathogens (21, 22). Thus, patients under treatment with anti-IL-17 mAbs are at risk to suffer from mostly mild and localized mucocutaneous candidiasis (23).
Another important aspect of IL-17-mediated host protection is executed by the recruitment of neutrophils. IL-17 increases the release of IL-8 (also called CXCL8 or neutrophil chemotactic factor) in target cells (24), which is a chemokine-inducing chemotaxis of neutrophils towards the side of its production, and the production of G-CSF, a major cytokine for proliferation, differentiation, and function of neutrophils (25, 26).
Another characteristic of IL-17 is its cooperative effect with other inflammatory mediators. Not only do the two isoforms IL-17A and IL-17F act synergistically with each other (27), but also other cytokines like granulocyte-macrophage colony-stimulating factor (GM-CSF), IFN-, IL-22, IL-1β, and TNF-α cooperate with IL-17 to exert pro-inflammatory effects (28). The synergy between IL-17 and TNF-α becomes evident on the post-transcriptional level. IL-17 or TNF-α alone are both rather weak inducers of messenger RNA for CXCL1, CXCL2, IL-6, I-κBζ, and CXCL5, which is then subject to rapid degradation. In contrast, when IL-17 and TNF-α act synergistically, the stability of those messenger RNAs is enhanced significantly (29). In essence, despite being a faint inducer of target genes, synergistic effects of IL-17 together with other cytokines potently shape inflammatory gene expression patterns.
In 2005, CD4+ T helper 17 (Th17) cells were described as the major source of IL-17 (30, 31). Since then, many other cell types with the ability to produce IL-17 have been identified: innate lymphoid cells and natural killer cells, γδ T cells and CD8+ T cells, NK T cells and mucosal-associated T cells, Paneth cells, and mast cells (3, 32–35). Owing to their relevance in GCA, this mini-review focuses on Th17 cells.
The differentiation and the developmental pathway of Th17 cells
The CD4+ T-cell lineage is considered to have four major subsets with Th1 cells, Th2 cells, Th17 cells, and T regulatory cells (Tregs). Of those, Th17 cells characteristically secrete high levels of IL-17A, IL-17F, and also IL-22 and GM-CSF. Interestingly, in vitro differentiated Th17 cells produce more IL-17F than IL-17A, while the majority of pathogenic Th17 cells in inflammatory diseases produce both cytokines at a similar level (36).
Differentiation of Th17 cells is a rather complex process compared to differentiation of other cellular lineages, e.g., Th1 or Th2 cells. Induction of Th17 cells is orchestrated by multiple cytokines with transforming growth factor-beta (TGF-β) and IL-6 being the primary cytokines initiating Th17 differentiation (37). In addition, co-signaling with IL-1β and IL-23 further guides Th17 survival, proliferation, and development (38, 39). On the transcriptional level, RORγt and STAT3 cooperatively induce expression of IL-17. RORγt is acknowledged as the key transcription factor in this process; its overexpression alone induces IL17 transcription in the absence of other cytokines, while STAT3 overexpression fails to do so in the absence of RORγt (40).
The developmental pathway of Th17 cells is interconnected with that of Tregs as TGF-β is crucial for both subsets to induce differentiation. Adding IL-6 then shifts CD4+ T cells towards the Th17 lineage by inhibiting FoxP3, the key transcription factor of Tregs, and upregulating RORγt (41, 42). The association of a reduced expression of FoxP3 in T cells together with the loss of immunological tolerance and autoimmunity is reported repeatedly (43–45). Thus, the homeostasis between Th17 cells on one side and Tregs on the other side is an important gatekeeper in the prevention of uncontrolled inflammation and autoimmunity (46).
Because of their plasticity, the phenotypical fate of Th17 cells and Tregs depends on the stimulation they receive from their immunological microenvironment. In this context, cytokines from innate immune cells are particularly able to push these cells into a pro-inflammatory and pathogenic direction. IL-6 inhibits the key transcription factor of Tregs while, at the same time, enhancing the key transcription factor of Th17 cells. IL-1β boosts Th17 cell differentiation, promotes expression of IL-17, and suppresses the production of anti-inflammatory IL-10 in Th17 cells (47), enhancing their pathogenic potential in autoimmune disease (48). IL-23 is required for the stabilization and expansion of the Th17 lineage (39). Therefore, these cytokines are sometimes called Th17-related cytokines. On the other hand, IL-2 is indispensable for the differentiation and proliferation of Tregs (49) and low-dose IL-2 enables a shift from the Th17 population toward Treg cells (50).
Evidence for the involvement of the IL-17 pathway in giant cell arteritis
In 2008, an in vivo model by Chen et al. (51) studied the impact of interferon regulatory factor 4 (IRF-4) and IRF-4-binding protein (IBP) on the Th17 lineage. Transgenic IBP-deficient mice were characterized by hyperresponsive CD4+ T cells with increased production of IL-17 and IL-21, both signature cytokines of Th17 cells. To the investigators’ surprise, IBP-deficient mice started dying from an age of 3 months or older and histologic analysis revealed that all mice suffered from severe inflammation of the aortic roof resembling granulomatous large vessel vasculitis with the presence of multinucleated giant cells, a typical finding in GCA. This is the first in vivo model pointing to a role of IL-17 in large vessel vasculitis.
In GCA, increased production of IL-6 was first reported in 1993 by Roche et al. (52). Based on findings on the Th17 lineage published almost 20 years later, one could conclude that IL-6 tips the balance from Tregs towards Th17 cells in vasculitis patients. Indeed, in 2012, two independent studies reported an expansion of the Th17 population in the peripheral blood of GCA patients while Treg counts were decreased at the same time (53, 54).
Another study showed Treg dysfunction in GCA patients and downregulation of FoxP3 in GCA Tregs. Interestingly, treatment with tocilizumab partially normalized Treg dysfunction but not FoxP3 expression (55). Miyabe et al. demonstrate that GCA Tregs from active disease possessed impaired suppressive capacity and a hypofunctional isoform of FoxP3 with missing exon 2. These cells displayed a Th17-like phenotype, which, again, illustrates the plasticity of Tregs. In this study, tocilizumab but not corticosteroids were able to correct Treg abnormalities (56).
An important study by Deng et al. illustrated that in patients with active GCA, the subsets of Th1 and Th17 cells were both expanded, but only the presence of relevant Th17 cell numbers was characteristic of new-onset disease. While Th1 cells already accounted for 11.8% of CD4 T cells in age-matched healthy controls with an increase to 20.6% in GCA patients, Th17 cells in controls were only marginal with 0.03% to 0.59% and increased in GCA patients 8-fold, making up to 5.3% of CD4 T cells in individual cases. In temporal artery biopsies, IL-17-producing cells were abundant in all wall layers and around adventitial vasa vasorum. This study also delivered evidence that the underlying immune defect might emerge from the innate immune system as monocytes from GCA patients were capable of inducing Th17 differentiation by producing IL-1β, IL-6, and IL-23. Glucocorticoid treatment normalized Th17 cell counts within 10 weeks, while Th1 cell frequency was unaffected (57).
In line with this, patients with high IL-17A expression in temporal artery specimens had fewer relapses and required shorter duration of glucocorticoid treatment in another study (58), indicating that the IL-17 pathway is involved in the acute phase of the disease and is suppressed by glucocorticoids.
Work from Wen et al. shed light on the interaction of the GCA vasculature with T cells. Adventitial microvascular endothelial cells control the access of inflammatory cells to the inner vascular wall, and infiltration of immune cells from the adventitia to the media and intima is a key step in the pathophysiology of GCA. High levels of vascular endothelial growth factor (VEGF) in GCA blood induce the expression of the Notch ligand Jagged1 on adventitial endothelial cells. This induces differentiation of CD4+ T that express the Notch1 receptor towards pathogenic Th1 and Th17 cells (59).
Investigating the histological pattern in GCA-affected arteries, Ciccia et al. reported IL-17 expression mainly in arteries displaying granulomatous transmural inflammation—with IL-17-positive giant cells—and vasa vasorum vasculitis. Furthermore, the expression of IL-17 in arteries was directly correlated with the intensity of the systemic inflammatory response in GCA patients (60). To expand the complex histological picture of vasculitis in GCA, a recent study reported neutrophil extracellular traps (NETs) in temporal arteries from patients with GCA—found primarily in the adventitia close to vasa vasorum. In all of the 10 patients, these NETs were decorated with IL-17A (61).
Epigenetic investigations of temporal arteries revealed hypomethylated genes that indicate strong activation of Th1 and Th17 pathways (62). Last, a meta-analysis including over 1,000 GCA patients and more than 3,000 healthy individuals from Spain, Italy, Germany, and Norway revealed that polymorphisms (SNPs rs4711998, rs2275913, and rs7747909) within the IL-17A locus confer a risk to GCA (63).
IL-17A inhibition in giant cell arteritis
The first case report of successful GCA treatment with a monoclonal antibody against IL-17A (secukinumab) appeared in 2018 in a patient with additional psoriatic arthritis, in which secukinumab is approved for therapy (64). In addition to psoriatic arthritis, secukinumab is approved for the treatment of plaque psoriasis and active non-radiographic and radiographic axial spondyloarthritis (65).
The phase II proof-of-concept trial TitAIN (66) was the first randomized controlled trial investigating the efficacy and safety of secukinumab in patients with active GCA. This randomized, parallel-grouped, double-blinded, placebo-controlled multi-center study was designed to compare secukinumab 300 mg to placebo in patients with active GCA with a 26-week prednisolone taper regimen (67). Fifty-two patients with relapsing (19.2%) or newly diagnosed (80.8%) GCA were included in the 52-week trial (66). A total of 37 patients (71.2%) completed the study and the proportion of GCA patients in sustained remission at week 28 was higher with secukinumab (70.1%) than with placebo (20.3%). At the end of the study in week 52, these numbers were 59.3% in the secukinumab arm and 8.0% in the placebo arm. In addition, secukinumab treatment led to a longer time to first GCA relapse compared to placebo treatment. Importantly, no unexpected or new safety signals compared to the use of secukinumab in other indications were detected (66).
For this reason, two phase III studies (NCT04930094 and NCT05380453) are currently recruiting to comprehensively investigate secukinumab in the treatment of active relapsing or newly diagnosed GCA. In addition, the treatment of patients with active polymyalgia rheumatica (PMR) without concomitant GCA is being investigated in a phase III study (NCT05767034).
Discussion
There is robust evidence for the involvement of the IL-17 pathway in the disease process of GCA (Table 1). Th17 cell populations are expanded in active GCA and IL-17-producing cells are abundant in all vascular wall layers of affected temporal artery specimens. As the developmental pathway of Th17 cells is interconnected with that of Tregs, it is interesting that, at the same time, Treg counts are lower in GCA patients and that these Tregs are dysfunctional. This leads to the suggestion that the dysbalance between these important T-cell subtypes is of relevance in the vasculitic process in GCA (Figure 1).

Table 1 Supporting findings for the contribution of IL-17 to the pathogenesis of GCA.
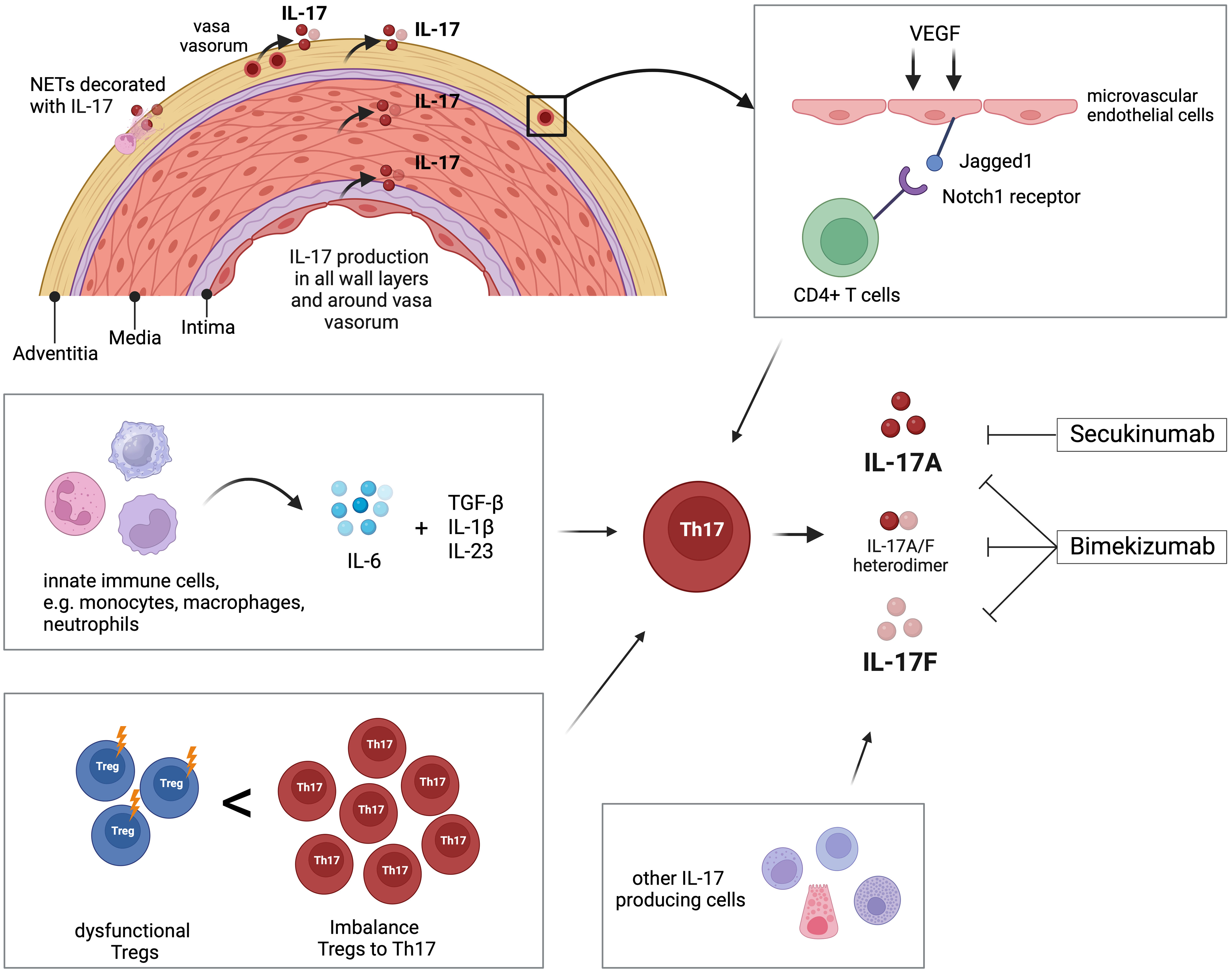
Figure 1 Overview of the role of IL-17 and Th17 cells in giant cell arteritis (GCA). Interleukin-17 (IL-17) is detectable in all layers of temporal arteries, around adventitial vasa vasorum, and in neutrophil extracellular traps (NETs) located in the adventitia. Microvascular endothelial cells in the adventitia are stimulated by vascular endothelial growth factor (VEGF) to present the Notch-ligand Jagged1 to bind CD4+ T cells, which then differentiate into Th17 cells. Innate immune cells like monocytes, macrophages, and neutrophils produce IL-6, which, in combination with TGF-β, IL-1β, and IL23, promotes differentiation of Th17 cells. The peripheral blood of GCA patients is characterized by increased frequency of Th17 compared to decreased number of T regulatory cells (Tregs). Additionally, those Tregs are dysfunctional regarding their regulatory function. Th17 cells and other immune and non-immune cells produce IL-17 that is linked to the pathology of GCA. The pharmaceutical agent secukinumab blocks IL-17A, while bimekizumab blocks IL-17A and IL-17F. Created with BioRender.com.
In contrast to many other rheumatic diseases, in which new therapies have emerged over the last decades, the standard of care in GCA is still defined by long-term treatment with high-dose glucocorticoids. Although this treatment approach effectively reduces systemic inflammation, histopathological studies of temporal artery biopsies to multiple time points in steroid-treated patients show that clinical symptoms were suppressed, but inflammatory alterations in the vascular wall often persisted (68). Furthermore, treatment with glucocorticoids entails high numbers of serious adverse effects (12, 69), which is especially relevant in GCA patients. They are, by definition, of older age, and many have relative contraindications to glucocorticoid therapy, e.g., diabetes or osteoporosis. This problem gets amplified by long-term intake of glucocorticoids when prednisolone cannot be reduced on time. In this context, another study showed that after 2 years, only 55% of patients have a daily dose of prednisolone lower than 5 mg (70). The only glucocorticoid-sparing agent approved for GCA so far is the IL-6 receptor antagonist tocilizumab. Yet, tocilizumab suppresses acute-phase proteins that are routinely used for GCA relapse detection, and MRI studies in tocilizumab-treated patients showed normalization of vessel wall enhancement only in one-third of the patients (71), leaving the remaining question of whether these findings have an impact on long-term prognosis.
In other words, there is a high need for other glucocorticoid-sparing agents for the treatment of GCA that combine effective suppression of acute inflammation with a safety profile that matches the needs of an elderly patient population. Considering these expectations, it was timely to investigate a pharmaceutical agent in GCA patients that has already been shown to be effective and safe in other rheumatic diseases. Secukinumab fulfilled these requirements and first results from the TitAIN study led to an optimistic outlook on a new potential treatment option in GCA.
Author contributions
MZ, JT, and NV wrote the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
NV and JT received honoraria from Novartis for lecturing and consulting activities. In addition, research projects from NV and JT were financially supported by Novartis. Novartis was sponsor of the TitAIN study in which NV and JT were investigators. JT and NV are named on the Novartis patent application for secukinumab in the treatment of giant cell arteritis.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rouvier E, Luciani MF, Mattei MG, Denizot F, Golstein P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J Immunol (1993) 150(12):5445–56. doi: 10.4049/jimmunol.150.12.5445
2. Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, et al. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity (1995) 3(6):811–21. doi: 10.1016/1074-7613(95)90070-5
3. Mills KHG. IL-17 and IL-17-producing cells in protection versus pathology. Nat Rev Immunol (2023) 23(1):38–54. doi: 10.1038/s41577-022-00746-9
4. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med (2005) 201(2):233–40. doi: 10.1084/jem.20041257
5. Genovese MC, Durez P, Richards HB, Supronik J, Dokoupilova E, Mazurov V, et al. Efficacy and safety of secukinumab in patients with rheumatoid arthritis: a phase II, dose-finding, double-blind, randomised, placebo controlled study. Ann Rheum Dis (2013) 72(6):863–9. doi: 10.1136/annrheumdis-2012-201601
6. Kunwar S, Dahal K, Sharma S. Anti-IL-17 therapy in treatment of rheumatoid arthritis: a systematic literature review and meta-analysis of randomized controlled trials. Rheumatol Int (2016) 36(8):1065–75. doi: 10.1007/s00296-016-3480-9
7. Bilal J, Berlinberg A, Bhattacharjee S, Trost J, Riaz IB, Kurtzman DJB. A systematic review and meta-analysis of the efficacy and safety of the interleukin (IL)-12/23 and IL-17 inhibitors ustekinumab, secukinumab, ixekizumab, brodalumab, guselkumab and tildrakizumab for the treatment of moderate to severe plaque psoriasis. J Dermatolog Treat (2018) 29(6):569–78. doi: 10.1080/09546634.2017.1422591
8. Dubash S, Bridgewood C, McGonagle D, Marzo-Ortega H. The advent of IL-17A blockade in ankylosing spondylitis: secukinumab, ixekizumab and beyond. Expert Rev Clin Immunol (2019) 15(2):123–34. doi: 10.1080/1744666X.2019.1561281
9. Weyand CM, Goronzy JJ. Clinical practice. Giant-cell arteritis and polymyalgia rheumatica. N Engl J Med (2014) 371(1):50–7.
10. Dejaco C, Duftner C, Buttgereit F, Matteson EL, Dasgupta B. The spectrum of giant cell arteritis and polymyalgia rheumatica: revisiting the concept of the disease. Rheumatol (Oxford) (2017) 56(4):506–15.
11. Hunder GG. Epidemiology of giant-cell arteritis. Cleve Clin J Med (2002) 69 Suppl 2:SII79–82. doi: 10.3949/ccjm.69.Suppl_2.SII79
12. Petri H, Nevitt A, Sarsour K, Napalkov P, Collinson N. Incidence of giant cell arteritis and characteristics of patients: data-driven analysis of comorbidities. Arthritis Care Res (Hoboken) (2015) 67(3):390–5. doi: 10.1002/acr.22429
13. Proven A, Gabriel SE, Orces C, O'Fallon WM, Hunder GG. Glucocorticoid therapy in giant cell arteritis: duration and adverse outcomes. Arthritis Rheumatol (2003) 49(5):703–8. doi: 10.1002/art.11388
14. Hymowitz SG, Filvaroff EH, Yin JP, Lee J, Cai L, Risser P, et al. IL-17s adopt a cystine knot fold: structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J (2001) 20(19):5332–41. doi: 10.1093/emboj/20.19.5332
15. Wright JF, Guo Y, Quazi A, Luxenberg DP, Bennett F, Ross JF, et al. Identification of an interleukin 17F/17A heterodimer in activated human CD4+ T cells. J Biol Chem (2007) 282(18):13447–55. doi: 10.1074/jbc.M700499200
16. Li X, Bechara R, Zhao J, McGeachy MJ, Gaffen SL. IL-17 receptor-based signaling and implications for disease. Nat Immunol (2019) 20(12):1594–602. doi: 10.1038/s41590-019-0514-y
17. Goepfert A, Lehmann S, Wirth E, Rondeau JM. The human IL-17A/F heterodimer: a two-faced cytokine with unique receptor recognition properties. Sci Rep (2017) 7(1):8906. doi: 10.1038/s41598-017-08360-9
18. Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol (2009) 9(8):556–67. doi: 10.1038/nri2586
19. Adamopoulos IE, Kuchroo V. IL-17A and IL-17F in tissue homeostasis, inflammation and regeneration. Nat Rev Rheumatol (2023) 19(9):535–6. doi: 10.1038/s41584-023-01004-5
20. Ge Y, Huang M, Yao YM. Biology of interleukin-17 and its pathophysiological significance in sepsis. Front Immunol (2020) 11:1558. doi: 10.3389/fimmu.2020.01558
21. Levy R, Okada S, Beziat V, Moriya K, Liu C, Chai LY, et al. Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc Natl Acad Sci USA (2016) 113(51):E8277–E85. doi: 10.1073/pnas.1618300114
22. Whibley N, Tritto E, Traggiai E, Kolbinger F, Moulin P, Brees D, et al. Antibody blockade of IL-17 family cytokines in immunity to acute murine oral mucosal candidiasis. J Leukoc Biol (2016) 99(6):1153–64. doi: 10.1189/jlb.4A0915-428R
23. Davidson L, van den Reek J, Bruno M, van Hunsel F, Herings RMC, Matzaraki V, et al. Risk of candidiasis associated with interleukin-17 inhibitors: A real-world observational study of multiple independent sources. Lancet Reg Health Eur (2022) 13:100266. doi: 10.1016/j.lanepe.2021.100266
24. Luzza F, Parrello T, Monteleone G, Sebkova L, Romano M, Zarrilli R, et al. Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J Immunol (2000) 165(9):5332–7. doi: 10.4049/jimmunol.165.9.5332
25. Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med (2008) 14(3):275–81. doi: 10.1038/nm1710
26. Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med (2001) 194(4):519–27. doi: 10.1084/jem.194.4.519
27. Hot A, Miossec P. Effects of interleukin (IL)-17A and IL-17F in human rheumatoid arthritis synoviocytes. Ann Rheum Dis (2011) 70(5):727–32. doi: 10.1136/ard.2010.143768
28. Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol (2010) 10(7):479–89. doi: 10.1038/nri2800
29. Hartupee J, Liu C, Novotny M, Li X, Hamilton T. IL-17 enhances chemokine gene expression through mRNA stabilization. J Immunol (2007) 179(6):4135–41. doi: 10.4049/jimmunol.179.6.4135
30. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol (2005) 6(11):1123–32. doi: 10.1038/ni1254
31. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6(11):1133–41. doi: 10.1038/ni1261
32. Chen L, He Z, Slinger E, Bongers G, Lapenda TLS, Pacer ME, et al. IL-23 activates innate lymphoid cells to promote neonatal intestinal pathology. Mucosal Immunol (2015) 8(2):390–402. doi: 10.1038/mi.2014.77
33. Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med (2014) 20(1):54–61. doi: 10.1038/nm.3423
34. Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity (2009) 31(2):321–30. doi: 10.1016/j.immuni.2009.06.020
35. Michel ML, Keller AC, Paget C, Fujio M, Trottein F, Savage PB, et al. Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J Exp Med (2007) 204(5):995–1001.
36. Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell (2015) 163(6):1400–12. doi: 10.1016/j.cell.2015.11.009
37. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441(7090):235–8. doi: 10.1038/nature04753
38. Bhaumik S, Basu R. Cellular and molecular dynamics of Th17 differentiation and its developmental plasticity in the intestinal immune response. Front Immunol (2017) 8:254. doi: 10.3389/fimmu.2017.00254
39. Stritesky GL, Yeh N, Kaplan MH. IL-23 promotes maintenance but not commitment to the Th17 lineage. J Immunol (2008) 181(9):5948–55. doi: 10.4049/jimmunol.181.9.5948
40. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol (2007) 8(9):967–74. doi: 10.1038/ni1488
41. Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med (2009) 361(9):888–98. doi: 10.1056/NEJMra0707449
42. Robert M, Miossec P, Hot A. The Th17 pathway in vascular inflammation: culprit or consort? Front Immunol (2022) 13:888763. doi: 10.3389/fimmu.2022.888763
43. Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, et al. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity (2013) 39(5):949–62. doi: 10.1016/j.immuni.2013.10.016
44. Hua J, Inomata T, Chen Y, Foulsham W, Stevenson W, Shiang T, et al. Pathological conversion of regulatory T cells is associated with loss of allotolerance. Sci Rep (2018) 8(1):7059. doi: 10.1038/s41598-018-25384-x
45. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med (2014) 20(1):62–8. doi: 10.1038/nm.3432
46. Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev (2014) 13(6):668–77. doi: 10.1016/j.autrev.2013.12.004
47. Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, et al. Pathogen-induced human TH17 cells produce IFN-gamma or IL-10 and are regulated by IL-1beta. Nature (2012) 484(7395):514–8. doi: 10.1038/nature10957
48. Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, et al. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol (2012) 13(10):991–9. doi: 10.1038/ni.2416
49. Thornton AM, Donovan EE, Piccirillo CA, Shevach EM. Cutting edge: IL-2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J Immunol (2004) 172(11):6519–23. doi: 10.4049/jimmunol.172.11.6519
50. Rosenzwajg M, Lorenzon R, Cacoub P, Pham HP, Pitoiset F, El Soufi K, et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Ann Rheum Dis (2019) 78(2):209–17. doi: 10.1136/annrheumdis-2018-214229
51. Chen Q, Yang W, Gupta S, Biswas P, Smith P, Bhagat G, et al. IRF-4-binding protein inhibits interleukin-17 and interleukin-21 production by controlling the activity of IRF-4 transcription factor. Immunity (2008) 29(6):899–911. doi: 10.1016/j.immuni.2008.10.011
52. Roche NE, Fulbright JW, Wagner AD, Hunder GG, Goronzy JJ, Weyand CM. Correlation of interleukin-6 production and disease activity in polymyalgia rheumatica and giant cell arteritis. Arthritis Rheumatol (1993) 36(9):1286–94. doi: 10.1002/art.1780360913
53. Samson M, Audia S, Fraszczak J, Trad M, Ornetti P, Lakomy D, et al. Th1 and Th17 lymphocytes expressing CD161 are implicated in giant cell arteritis and polymyalgia rheumatica pathogenesis. Arthritis Rheumatol (2012) 64(11):3788–98. doi: 10.1002/art.34647
54. Terrier B, Geri G, Chaara W, Allenbach Y, Rosenzwajg M, Costedoat-Chalumeau N, et al. Interleukin-21 modulates Th1 and Th17 responses in giant cell arteritis. Arthritis Rheumatol (2012) 64(6):2001–11. doi: 10.1002/art.34327
55. Adriawan IR, Atschekzei F, Dittrich-Breiholz O, Garantziotis P, Hirsch S, Risser LM, et al. Novel aspects of regulatory T cell dysfunction as a therapeutic target in giant cell arteritis. Ann Rheum Dis (2022) 81(1):124–31. doi: 10.1136/annrheumdis-2021-220955
56. Miyabe C, Miyabe Y, Strle K, Kim ND, Stone JH, Luster AD, et al. An expanded population of pathogenic regulatory T cells in giant cell arteritis is abrogated by IL-6 blockade therapy. Ann Rheum Dis (2017) 76(5):898–905. doi: 10.1136/annrheumdis-2016-210070
57. Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation (2010) 121(7):906–15. doi: 10.1161/CIRCULATIONAHA.109.872903
58. Espigol-Frigole G, Corbera-Bellalta M, Planas-Rigol E, Lozano E, Segarra M, Garcia-Martinez A, et al. Increased IL-17A expression in temporal artery lesions is a predictor of sustained response to glucocorticoid treatment in patients with giant-cell arteritis. Ann Rheum Dis (2013) 72(9):1481–7. doi: 10.1136/annrheumdis-2012-201836
59. Wen Z, Shen Y, Berry G, Shahram F, Li Y, Watanabe R, et al. The microvascular niche instructs T cells in large vessel vasculitis via the VEGF-Jagged1-Notch pathway. Sci Transl Med (2017) 9(399). doi: 10.1126/scitranslmed.aal3322
60. Ciccia F, Rizzo A, Guggino G, Cavazza A, Alessandro R, Maugeri R, et al. Difference in the expression of IL-9 and IL-17 correlates with different histological pattern of vascular wall injury in giant cell arteritis. Rheumatol (Oxford) (2015) 54(9):1596–604. doi: 10.1093/rheumatology/kev102
61. Palamidas DA, Argyropoulou OD, Georgantzoglou N, Karatza E, Xingi E, Kapsogeorgou EK, et al. Neutrophil extracellular traps in giant cell arteritis biopsies: presentation, localization and co-expression with inflammatory cytokines. Rheumatol (Oxford) (2022) 61(4):1639–44. doi: 10.1093/rheumatology/keab505
62. Coit P, De Lott LB, Nan B, Elner VM, Sawalha AH. DNA methylation analysis of the temporal artery microenvironment in giant cell arteritis. Ann Rheum Dis (2016) 75(6):1196–202. doi: 10.1136/annrheumdis-2014-207116
63. Marquez A, Hernandez-Rodriguez J, Cid MC, Solans R, Castaneda S, Fernandez-Contreras ME, et al. Influence of the IL-17A locus in giant cell arteritis susceptibility. Ann Rheum Dis (2014) 73(9):1742–5. doi: 10.1136/annrheumdis-2014-205261
64. Rotar Z, Tomsic M, Hocevar A. Secukinumab for the maintenance of glucocorticoid-free remission in a patient with giant cell arteritis and psoriatic arthritis. Rheumatol (Oxford) (2018) 57(5):934–6. doi: 10.1093/rheumatology/kex507
65. Blair HA. Secukinumab: A review in psoriatic arthritis. Drugs (2021) 81(4):483–94. doi: 10.1007/s40265-021-01476-3
66. Venhoff N, Schmidt WA, Bergner R, Rech J, Unger L, Tony H-P, et al. Safety and efficacy of secukinumab in patients with giant cell arteritis (TitAIN): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Rheumatol (2023) 5(6):e341–e50. doi: 10.1016/S2665-9913(23)00101-7
67. Venhoff N, Schmidt WA, Lamprecht P, Tony HP, App C, Sieder C, et al. Efficacy and safety of secukinumab in patients with giant cell arteritis: study protocol for a randomized, parallel group, double-blind, placebo-controlled phase II trial. Trials (2021) 22(1):543. doi: 10.1186/s13063-021-05520-1
68. Maleszewski JJ, Younge BR, Fritzlen JT, Hunder GG, Goronzy JJ, Warrington KJ, et al. Clinical and pathological evolution of giant cell arteritis: a prospective study of follow-up temporal artery biopsies in 40 treated patients. Mod Pathol (2017) 30(6):788–96. doi: 10.1038/modpathol.2017.10
69. Salvarani C, Cantini F, Boiardi L, Hunder GG. Polymyalgia rheumatica. Best Pract Res Clin Rheumatol (2004) 18(5):705–22. doi: 10.1016/j.berh.2004.06.003
70. Paskins Z, Whittle R, Sultan AA, Muller S, Blagojevic-Bucknall M, Helliwell T, et al. Risk of fracture among patients with polymyalgia rheumatica and giant cell arteritis: a population-based study. BMC Med (2018) 16(1):4. doi: 10.1186/s12916-017-0987-1
Keywords: vasculitis, giant cell arteriitis (GCA), IL-17, secukinumab, Th-17
Citation: Zeisbrich M, Thiel J and Venhoff N (2024) The IL-17 pathway as a target in giant cell arteritis. Front. Immunol. 14:1199059. doi: 10.3389/fimmu.2023.1199059
Received: 02 April 2023; Accepted: 15 December 2023;
Published: 17 January 2024.
Edited by:
Valentin Sebastian Schäfer, University Hospital Bonn, GermanyReviewed by:
Arash Pourgholaminejad, Gilan University of Medical Sciences, IranClaus-Juergen Bauer, University Hospital Bonn, Germany
Copyright © 2024 Zeisbrich, Thiel and Venhoff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Zeisbrich, bWFya3VzLnplaXNicmljaEB1bmlrbGluaWstZnJlaWJ1cmcuZGU=