
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 16 June 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1198551
 Weihong Tian1,2
Weihong Tian1,2 Gaofeng Qin2Miaomiao Jia3Wuhao Li1
Gaofeng Qin2Miaomiao Jia3Wuhao Li1 Weili Cai4
Weili Cai4 Hui Wang1Yangjing Zhao1
Hui Wang1Yangjing Zhao1 Xuanwen Bao5
Xuanwen Bao5 Wangzhi Wei2
Wangzhi Wei2 Yu Zhang2*
Yu Zhang2* Qixiang Shao1,4*
Qixiang Shao1,4*The fundamental principle of immune checkpoint blockade (ICB) is to protect tumor-infiltrating T cells from being exhausted. Despite the remarkable success achieved by ICB treatment, only a small group of patients benefit from it. Characterized by a hypofunctional state with the expression of multiple inhibitory receptors, exhausted T (Tex) cells are a major obstacle in improving ICB. T cell exhaustion is a progressive process which adapts to persistent antigen stimulation in chronic infections and cancers. In this review, we elucidate the heterogeneity of Tex cells and offer new insights into the hierarchical transcriptional regulation of T cell exhaustion. Factors and signaling pathways that induce and promote exhaustion are also summarized. Moreover, we review the epigenetic and metabolic alterations of Tex cells and discuss how PD-1 signaling affects the balance between T cell activation and exhaustion, aiming to provide more therapeutic targets for applications of combinational immunotherapies.
Despite the tremendous clinical success of ICB therapy, the challenge brought by T cell exhaustion, which sustains immune tolerance, and limits the prolonged benefits of cancer immunotherapy, has raised great concerns about its linkage with poor clinical outcomes. Defined with decreased cytotoxic efficacy, diminished inflammatory cytokines secretion (e.g. IL-2, IFN-γ, and TNF), and elevated expression of suppressive immune checkpoints (such as PD-1, CTLA-4, LAG-3, TIM-3, TIGIT, and CD39), exhausted T (Tex) cells are dysfunctional clusters in hierarchical heterogeneity (1). Ranging from the stem-like stage (precursor Tex cells, Tpex) to the entirely function-lost stage (terminal Tex cells, Texterm), Tex cells are gradually developed with significant alterations in transcriptome and epigenome (2, 3).
The Tpex cells are capable of self-renewal and maintaining a chronically stimulated Tex cell pool (4). They are the main responder in mediating the therapeutic response of PD-1 blockade while the Texterm cells are totally function-restrained (4–7). T cell exhaustion is an inevitable outcome initiated by sustained TCR signaling under long-lasting inflammation (8). Nevertheless, this program protects Tpex against excessive differentiation or over-stimulation-induced apoptosis (9). This mechanism partly explains why serious autoimmunity is a side effect of ICB treatments even though interfering with exhaustion contributes to therapeutic benefits.
Tex cells exhibit altered transcriptomes compared to effector T (Teff) cells and memory T (Tmem) cells. Transcription factor (TF) NFAT and NR4A, downstream of persistent TCR signaling, play vital roles in initiating T cell exhaustion (10). A transcriptional hierarchy of coordinated TF networks regulates the differentiation of distinct Tex clusters. The feedback loop of T cell factor-1 (TCF-1)-thymocyte selection-associated high mobility group box protein (TOX) and T-box family members is critical to control the development of T cell exhaustion (10–12). Instead of causing a temporary reduction of T cell function, exhaustion is a unique and epigenetically fixed state which is irreversible during T cell development. Tex cells show decreased chromatin accessibility of effector-related genes but increased accessibility of genes encoding inhibitory receptors (IRs). Observations indicate that Tex cells could only be transiently rejuvenated by anti-PD-1 and they will ultimately tend to terminal exhaustion (13). Targeting epigenetic modifications of Tex cells are supposed to be taken into consideration sufficiently to improve immunotherapies.
Efforts to overcome the limitations of ICB have focused on better understanding the molecular mechanisms underlying T cell exhaustion, as well as the transcriptional interactions between Tex cells and immune checkpoint receptors and ligands. In this review, we elucidate developmental pathways and transcriptional regulation of Tex cells. We aim to examine the cellular and molecular machinery that governs Tex formation, differentiation, and response to PD-1 blockade. Additionally, we discuss therapeutic strategies aimed at restoring the effector functions of Tex, which may ultimately improve the clinical response to immune checkpoint blockade.
Exhausted T cells exhibit a hierarchical cell differentiation trajectory (Figure 1). Naïve CD8+ T cells differentiate into cytotoxic effector cells and memory cell precursors after cell activation (14, 15). While in chronic infections and tumors, an early Tpex cell cluster develops and preferentially localizes in lymphoid organs (4, 7). This Tpex cluster is proliferative and function-reserved. Tpex cells subsequently generate additional subsets of Tex cells containing the transitory Tex clusters and the terminal Tex cell clusters (2, 16, 17) in chronic infections and tumor microenvironments (TME), reflecting a cell differentiation hierarchy necessary for maintaining the functional T cell pool. This progressive development of T cell exhaustion results in a high heterogeneity of Tex cells. We discuss the heterogeneity of Tex cells and the epigenetic modifications underlining this stepwise differentiation in the following.
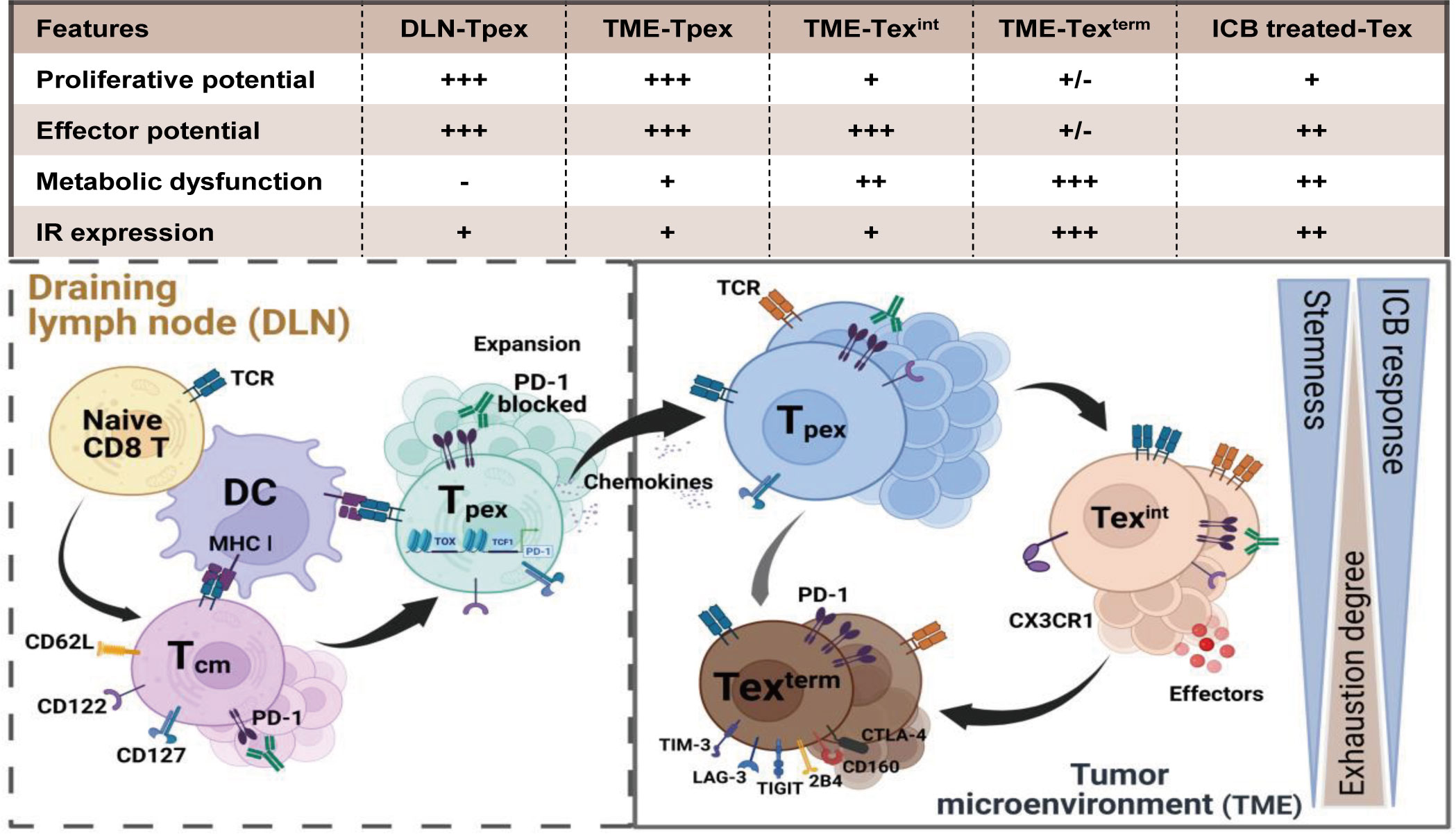
Figure 1 The differentiation hierarchy of exhausted T cells. The exhaustion program of Tex cells follows a hierarchical differentiation process with progressive loss of effector function and proliferative capability. Activated by tumor antigen, naïve CD8+ T cells differentiate into central memory T cells (Tcm). However, persistent stimulation of tumor antigens triggers Tcm cells to form the function-restrained progenitor Tex cells (Tpex). Tpex cell cluster preferentially localizes in lymphoid organs and is responsive to PD-1 blockade. Once migrating into the TME, the Tpex cluster subsequently differentiates spanning from the effector-like intermediate Tex cells (Texint) to the terminal Tex cells (Texterm) for maintaining the anti-tumor T cell pool. Persistent TCR signaling and suppressive co-stimulatory signals in the TME force the development of T cell exhaustion. Tpex cells gradually lose the proliferative potential while converting to progeny Tex cells. The expression of inhibitory receptors (e.g. PD-1, LAG3, TIM3 and TIGIT) elevates in terminal exhausted T cells. Accompanied by metabolic dysfunctions and epigenetic modifications, the exhaustion state of Texterm cells is irreversible and they can minorly respond after PD-1 blockade. Created with BioRender.com.
The heterogeneity of Tex cells mainly depends on their distinguished phenotypes, effector molecules, key transcription factors and inhibitory receptors. Four heterogenous Tex subpopulations with developmental relationships were identified during chronic viral infections and tumors (18). There existed two progenitor Tex (Texprog) subpopulations, Texprog1 and Texprog2, which shared related open-chromatin landscapes but differed in phenotypes and functions. Texprog1 upregulated genes associated with stem-cell biologies, such as Tcf7, Myb, Il7r and Sell. While genes preferentially up-regulated in Texprog2 were related to cell cycle (Cdk and Mki67) and cell migration (Anxa2, Itgb7 and Alcam). Texprog1 was resident and capable of transforming into Texprog2, which could re-localize from blood-inaccessible lymphoid niches to blood-accessible locales. The Texprog2 subpopulation underwent a proliferation-driven transition to the intermediate Tex (Texint) cells, which presented as a transitional population with some cytolytic and migratory capacities. The transcriptional characteristic of the Texint population contained genes linked to cytotoxic functions, such as Gzma, Gzmb, Prf1, Klrk1 and Cx3cr1. Texint also expressed higher TF genes like Tbx1, Zeb2 and Id2. The Texterm subpopulation exhibited the most severe exhausted profile and the weakest cytotoxicity. Texterm up-regulated multiple inhibitory receptor genes (e.g., Pdcd1, Lag3, Tigit and Cd244) and molecule genes related to terminal depletion (Entpd1, Cd101 and Cd38). Meanwhile, the Texterm showed elevated TCR signalings with highly expressed Zap70 and Nfatc1. Moreover, although the key exhaustion regulator TOX (19, 20) was expressed by all Tex cells, the Texterm population exhibited the highest expression.
The differentiation hierarchy of Tex cells confirms the progressive development of T cell exhaustion. Within the spectrum of T cell exhaustion, numerous Tex cells are identified. Zhang et al. identified five main pan-cancer Tex clusters across 30 bulk solid tumor types (21). These five Tex clusters in the TME showed Tex-specific profiles in glycolysis, chemokinesis and cytotoxic functions. The transcriptional factor TCF-1 and T-bet gradually decreased among the five clusters from Texprog to Texterm. TOX had a decreasing trend from Texprog to Texint but was highest in the Texterm. Besides, the Texterm subset displayed reduced gene mutations and DNA methylations when comparing the genomic alterations. Similarly, Bengsch et al. also uncovered nine distinct Tex clusters in human HIV infection and lung cancer by using a comprehensive mass cytometry approach (22). Validated by a functional exhaustion score, the Tex clusters with higher scores had elevated co-expression of IRs and TF Eomesodermin (Eomes). Conversely, a lower score owned Tex clusters expressed higher levels of functional markers like CD127 and TCF-1 but lower levels of IRs like PD-1, CD38 and 2B4. Additional observations in patients with B-ALL also illustrated that Tex cells are remarkably heterogeneous in cell cycle and expression patterns of cytotoxic molecules (23).
Totally, the expression of IRs like PD-1, CTLA-4, LAG-3 and TIM-3 is considered a crucial factor for categorizing Tex cells. The PD-1lo subset expressed fewer or no IRs such as TIM-3 and 2B4, and can differentiate into the PD-1hi subset (24). TCF-1 plays a crucial role as a central transcription factor in the PD-1lo Tex cluster (12, 25–27). The TCF-1+PD-1lo Tex cluster exhibits a self-renewing capacity and maintains persistent T cell responses (18, 21, 22, 28, 29) in chronic infections and cancers. Based on the expression intensity of IRs and TCF-1, Tex cells are generally divided into three stages (1): the Tpex, which is the stem-like PD-1loTCF-1+ Tex precursors (2); the Texterm, which is the terminally differentiated PD-1hiTCF-1- Tex cluster with the highest exhaustion profile; and (3) the Texint, which is at a transitional stage between the Tpex and Texterm, characterized by intermediate PD-1 expression but without TCF-1 and has the highest effector functions.
Epigenetic studies have shown that exhaustion is a unique and enduring stage in T cell differentiation rather than merely a momentary reduction of T cell function. Approximately 6,000 open chromatin regions distinguish CD8+ Tex cells from effector and memory T cells (30, 31). The exhaustion program is firmly established in the epigenome of Tex cells. Although the inhibitory receptor has been blocked, the epigenetic remodelings of Tex cells are minorly changed (30). This explains why reinvigorating PD-1loCD8+ Tex cells can merely cause a transient cytotoxic activity in response to ICB therapy.
Tex cells exist within a range of steady and constant epigenetic landscapes. In a general model, exhaustion-regulated genes are accessible in Tex cells and are transcriptionally controlled by exhaustion-related transcription factors. Memory-related genes are inaccessible to the transcriptional machinery, whereas several effector-related genes are still available in exhausted T cells but are not effectively bound by transcription factors. For example, the chromatin-accessible regions at the Ifng and Gzmb locus both emerged in CD8+ Teff cells and Tex cells, but the gene expressions were significantly distinct on the basis of functional pathway enrichment (31). Tex cells exhibited significant alterations in the regulation of IFN-γ production and immune effector process pathways (31). However, in reinvigorated Tex cells by PD-L1 blockade, the exhaustion relevant genes like Pdcd1 and Ctla4 are still expressed for DNA demethylation of these gene loci. Although inhibition of PD-1 signaling activated the expression of many effector genes like Gzmb and Ifng, the revived Tex cells eventually returned to be exhausted under consistent high antigen stimulation in chronic LCMV infection. The anti-PD-L1 treated Tex cells were unable to differentiate into Tmem cells even if the antigen was eliminated (30). This suggests that the powerful reinvigoration of checkpoint blockade may not be long-lasting without any epigenetic alterations.
Via transposase-accessible chromatin sequencing (ATAC-Seq), CD8+ Tex cells reveal numerous distinctive genomic features. A Nr4a1 binding motif at -22.4 kb of the murine Pdcd1 locus becomes accessible in Tex cells (32), and a de novo enhancer at approximately 23.8 kb upstream the transcription start site of Pdcd1 is also identified (30, 31). Functional analysis of this enhancer has shown that it is necessary for the persistent and integrated expression of PD-1 in Tex cells (31). Epigenetic modifications at the Pdcd1 locus reflect the regulatory differences between Tex and Teff cells. During the early phase of chronic infection, the differentiation of CD8+ Teff cells is followed by a temporal loss of DNA methylation at the Pdcd1 locus (33). Following durable TCR signaling, histone acetylation facilitates the opening of Pdcd1 enhancer elements. Some of these gene loci are found to be recognized by T-bet, directly supporting that T-bet also functions to regulate PD-1 expression (34). In exhausted CD8+ T cells, the Pdcd1 region is entirely demethylated and it remains in this status even when inflammation is eliminated. The absence of DNA methylation leaves the Pdcd1 locus primed for accelerated expression (35).
It is crucial to identify gene loci that are epigenetically modified and related to severe T cell exhaustion for improving immunotherapies. Zhang et al. demonstrated that treating TILs with valproic acid, a histone deacetylase inhibitor, ex vivo reduced the exhaustion degree of Tex cells and improved the therapeutic effect of ICB treatment (36). Additionally, Ghoneim et al. found that Dnmt3a mediates the DNA methylation reprogramming in the development of terminal T cell exhaustion and targeting Dnmt3a synergizes with PD-1 blockade to improve ICB therapy (13). To develop new therapeutic options, an in-depth understanding of the molecular mechanisms underlying epigenetic modification involved in T cell exhaustion is needed.
Exhausted T cells exhibit altered transcriptional profiles. Key TFs in regulating heterogeneous Tex clusters display different expression patterns of concentrations and subcellular localizations, and they also show distinct downstream interactions with specific co-factors and accessible chromatins (37). Importantly, a transcriptional hierarchy of coordinated TF networks exists to establish and regulate the exhaustion program (38). Under continuous TCR signaling, Tex cells undergo a hierarchical differentiation trajectory from the Tpex cells to the transitory Tex cells, eventually to the Texterm cells. The critical transcription factors TCF-1, TOX, and T-box family members T-bet and Eomes involved in varying regulation networks take turns to modulate the exhaustion program of different Tex clusters (Figure 2).
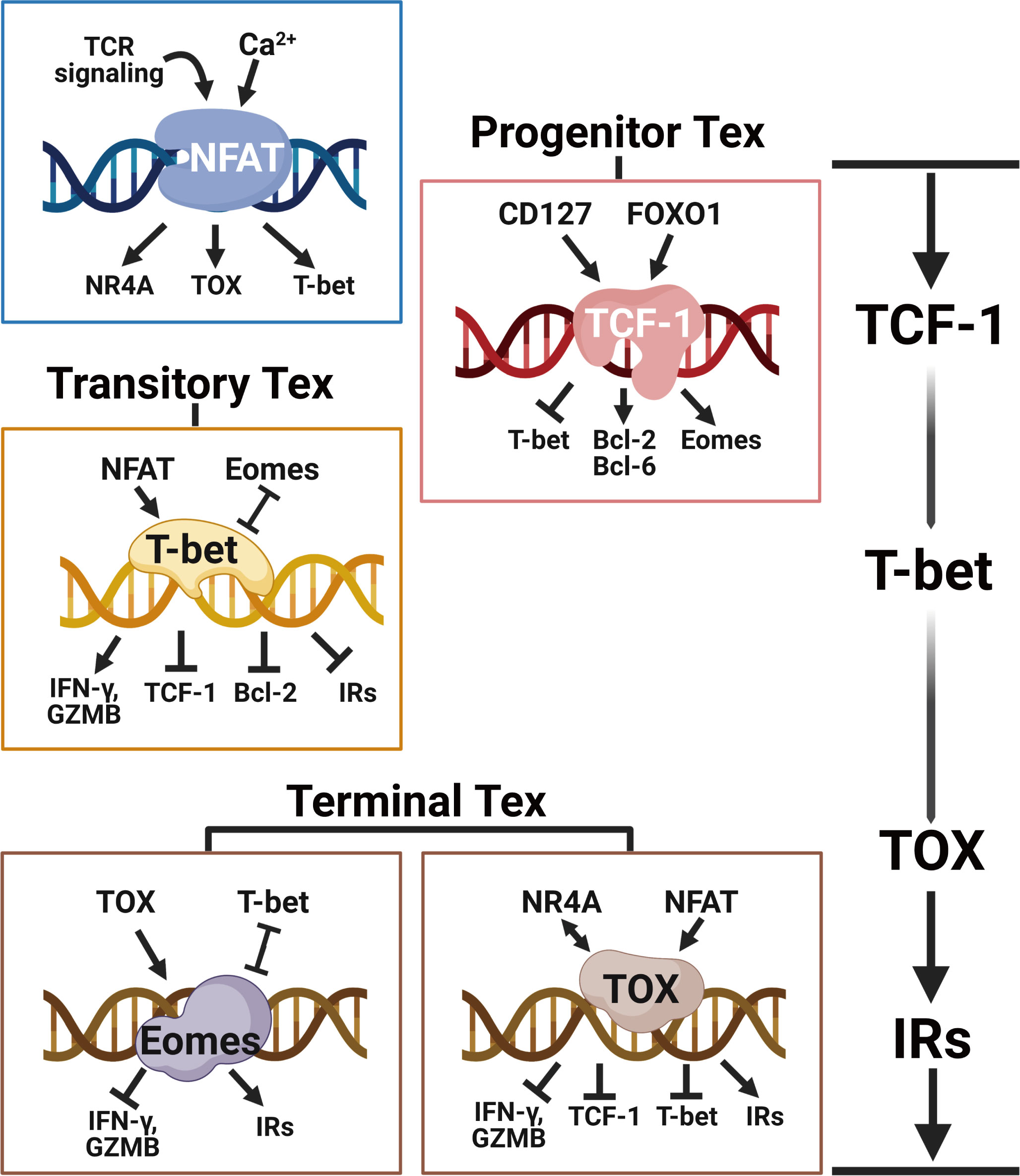
Figure 2 The transcriptional hierarchy of regulating T cell exhaustion. Following continued TCR stimulation and downstream Ca2+ signaling, the transcriptional network orchestrated by NFAT directly induces the expression of TOX and NR4A to initiate the T cell exhaustion program. TCF-1 is central for maintaining the stemness of Tpex cells by enhancing Eomes and Bcl-2 expressions. With the ongoing NFAT signaling, T-bet counterbalances TCF-1 and Eomes to facilitate the expression of effector molecules like IFN-γ and granzyme B in transitory Tex cells. The eventually upregulated TOX and Eomes prevent T-bet-mediated effector programming and induce the expression of multiple inhibitory receptors (e.g. PD-1, LAG-3, TIM-3, 2B4 and TIGIT), epigenetically imposing the terminal T cell exhaustion program. Created with BioRender.com.
T cell exhaustion is initially driven by persistent and high amounts of TCR stimulation in chronic infections or tumors (39). The calcium influx from TCR signaling promotes the activation of TFs NFAT, BATF and IRF4 (40). This TCR-responsive TF network is crucial for maintaining T cell expansion and function (41–43). Associated with the antigen loads, T cells express higher levels of these TFs in chronic infections than in acutely solved infections (44). At the early stage of T cell activation, NFAT translocates into the nucleus to form heterodimers with AP-1 for fostering the expression of T cell effector genes (45, 46). However, at the late effector stage, the ratio of nuclear NFAT : AP-1 was abnormally risen (46, 47). On the one hand, the consistent TCR signaling recruits superfluous NFAT to relocate into the nucleus, resulting in a higher ratio of NFAT : AP-1 and leaving many NFATs free. On the other hand, the increased BATF signaling antagonizes the formation of classic AP-1 complex, leading to less AP-1 and a much higher NFAT : AP-1 ratio (42, 48). NFAT is reported to function as an effective PD-1 promoter (46) and elevate the expression of NR4A and TOX (49). As a result, the excessively free NFATs primarily promote the expression of inhibitory receptor genes of T cells, and gradually drive the acquisition of exhausted phenotypes (11, 49, 50)
What’s more, the TCR signal strength determines the differentiation of Tex cells. It has been observed that the T cell cluster specific for antigen with lower affinity, compared with another cluster recognizing antigen with higher affinity, is more abundant in Tpex cells in the same tumor (51). Besides, Daniel et al. found that high TCR signaling correlates with more terminal Tex cells, whereas low TCR signal strength generates the effector-like Tex cell cluster with a killer cell lectin-like receptor (KLR)-expressed cytotoxic profile (52). Shakiba et al. also demonstrated that tumor-specific CD8+ T cells, whether undergoing low or high TCR signal strength, both upregulated inhibitory receptors and couldn’t mediate tumor control. However, high TCR signaling leads to an effector function loss and a dysfunctional state of CD8+ T cells while the low TCR signal restrains them in a functional state (53). These findings all illustrate that TCR signal strength is a key determinant of T cell response and Tex cell formation.
Progenitor Tex cell cluster generates early during chronic infections and they are able to proliferate and self-renew for their expression of TCF-1 (38). TCF-1 signaling plays a crucial role in generating Tpex cells during persistent inflammation by antagonizing the Teff cell-driving TFs like T-bet and ID-2 (4, 12). In early bifurcation of T cell differentiation, TCF-1 represses the developmental path to the terminal KLRG1hi Teff cells and promotes the formation of the Tpex cluster (12, 40). Studies have shown that ectopic TCF-1 expression strongly promotes the formation of Tpex in both chronic infection and pre-clinical tumor models (26). Adoptive transfer experiments have similarly demonstrated that TCF-1+ Tex cells are capable of proliferating and maintaining T cell responses. In contrast, Tex cells that have lost TCF-1 expression are unable to proliferate and maintain T cell responses (4, 7). Furthermore, TCF-1 overexpression diminishes the expression of co-inhibitory receptors and enhances cytokine production capacity (54). TCF-1+ Tpex population is the main cluster of the proliferative burst after PD-1 blockade, indicating their retained functions in comparison to other Tex clusters (4).
TOX is a pivotal regulator of T cell exhaustion (19, 20). In chronic infections and tumors, TOX can be induced by TCR signaling quickly and keeps high expression specifically in the Tex cell lineage. But in acute infections, TOX is transiently induced and expressed at low levels with no persistence (11, 49, 55). NFAT signaling downstream of TCR stimulation activates Tox gene expression directly (55). The expression of TOX increases the production of PD-1 and other inhibitory receptors (55, 56), establishing the exhaustion phenotype of early exhausted T cells. The deletion of Tox abrogated T cell exhaustion program in chronic viral infection. In Tox-deficient tumor-infiltrating T cells, a large fraction of gene loci for inhibitory receptors like Pdcd1, Havcr2 and Tigit are revealed to be inaccessible (11). However, the Tox-deficient T cells preserved high expression of TCF-1 (11). Indeed, at the early stage of LCMV infection, the TOX+ T cell subset positively connected with TCF-1 expression (55). It was reported that KLRG1+ terminal Teff was unable to generate TOX+ T cells, perhaps due to their lack of TCF-1 (57, 58).
The T-box transcription factors Eomes and T-bet are both indispensable for the development of T cell exhaustion. During early chronic infections, TCF-1 antagonizes T-bet and promotes a T-bet to Eomes conversion in precursor Tex cells by increasing Eomes expression, which controls a Bcl-2 mediated Tex cell survival (12). Eomes promotes IL-15Rβ expression and T cell memory formation during acute infections (59, 60), while T-bet is crucial for effector function and drives KLRG-1+ Teff cells formation. Eomes is straightforwardly connected with the TCF-1 expression in naive and memory CD8+ T cells. Actually, enhanced expression of Eomes partly saves TCF-1-deficient memory CD8+ T cells from progressively losing (27). The acquisition of memory features in Tpex is then promoted by the activation of Eomes gene transcription, which is necessary to maintain the Tex cell pool (26).
In conclusion, at the early stage of chronic infections and tumors, probably within the first seven days of chronic antigen stimulation (61), TCF-1, under control of TCR and PD-1 signaling, fosters Eomes expression in Tpex to blunt the activity of T-bet, antagonizing terminal Teff cell differentiation. TCF-1 signaling allows adequate time for TOX-dependent epigenetic remodeling in stepwise fixing the exhaustion program of Tpex cells (20). Importantly, the TCF-1+ Tpex cells are responsible for the proliferative boost after immune checkpoint blockade (4, 62), and the proportion of this cluster in tumors correlates positively with advanced clinical outcomes (7, 63, 64). In this way, the Tpex cell cluster is potential to perform as a therapeutic marker for ICB therapy clinically.
The transitory Tex cell cluster shows enrichments of T-bet and RUNX motifs in ATAC-seq analysis of TF motif accessibility, while the Texterm cluster shows enrichments of NR4A and Eomes motifs (18). For one thing, it implies a transcriptional signaling shift of T-box family members whose activity may emphasize distinct Tex clusters when differentiation. For another thing, it indicates that T-bet and Eomes have separate functions in chronic infections and cancers from their roles in Teff and Tmem cells during acute infections (65).
The transitory Tex cell cluster is effector-like and exists at an intermediate cell state between Tpex cells and Texterm cells. T-bet counterbalances TOX to stabilize the transitional exhaustion state and prevent terminal exhaustion. T-bet inhibits the transcription of Pdcd1 and possibly other immune checkpoint genes (34) while Eomes expression positively correlates with the expression of PD-1 and other IRs in Tex cells (22, 66). Differential nuclear localization of T-bet and Eomes regulates the T cell exhaustion program. T-bet and Eomes compete for binding to the same DNA motifs that contain the Pdcd1 locus. A decreased nuclear Eomes in Tex cells allows T-bet to play a dominant role as a stronger inhibitor of Pdcd1. Silencing PD-1 signaling in Tex cells increases T-bet nuclear localization, which initiates the expression of T-bet-related functional genes for chemotaxis, homing, and activation (67).
Dynamically, Eomes is higher in the Tpex cluster for TCF-1 activation whereas it declines in the transient Texint cluster. T-bet reaches its highest level in the Texint cluster before dropping at the Texterm stage (18). Most notably, although researches have shown that transient Tex cells give rise to Texterm cells, there still has evidence that transitory Tex cells may serve as the endpoint of Tex cell differentiation based on the TCR lineage tracing analysis (16, 68). One possible explanation of this finding could be the heterogeneity of transitory Tex cells. The Texint cells are also heterogeneous and could progress through a CX3CR1+ KLR+Tex cell state which represents an alternative terminus of Tex cell differentiation (52).
Terminal exhausted T cells derived from transitory Tex cells are defined by their high expression of TOX and multiple IRs like PD-1, LAG-3, TIM-3, CD38 and CD101 (2, 18). Persistent TCR and NFAT signalings induce Tox expression adequately. The endurable TOX signal interacts directly with histone acetyltransferase and indirectly with DNA methyltransferases to epigenetically fix CD8+ Tpex cells towards terminal exhaustion (20, 69). Eomes expression is higher in the Texterm cluster (67). Eomes synergizes with TOX at the late stage of chronic infection to promote more severe T cell exhaustion (18). The higher expression of Eomes in the Texterm subset is distinct from its expression in the quiescent self-renewing Tmem cells during acutely resolved infections. Eomes partially antagonizes T-bet and determines the Tex profile through greater nuclear localization and higher affinity to Pdcd1 binding sites. Eomes is higher in the Tpex cluster for TCF-1 activation and it declines in the transient Texint cluster while rebounding at the Texterm stage.
With consecutive antigen exposure, Tex cells eventually proceed to the Texterm cluster with elevated expression of TOX and Eomes. These two key transcriptional factors orchestrate other exhaustion regulators like NR4A to promote the terminal exhaustion program that includes highly expressed inhibitory receptors, decreased effector functions and elevated metabolic alterations.
Exhausted T cells display metabolic dysregulations in diminished glycolysis, impaired mitochondrial fitness and enhanced endoplasmic reticulum (ER)-related stress (Figure 3). The dysregulated metabolism profile also contributes to T cell exhaustion in long-lasting inflammations. Hypoxia, low pH and insufficient nutrients are representative conditions of TME to play active roles in T cell exhaustion. It was found that the cytolytic activity of CD8+ T cells was regulated partly by the hypoxia-inducible factors (HIFs) and the von Hippel-Lindau (VHL) tumor suppressor protein during chronic infections (70, 71). The VHL-deficient cytotoxic T lymphocytes exhibited improved control of persistent viral infection and tumor growth for their enhanced response to hypoxia (70). Impaired aerobic glycolysis strongly dampens T cell activation and cytokine secretions (72). Limited glucose and downregulated transporter GLUT1 result in poor glucose usage and accelerate the exhaustion program of Tex cells (73, 74).
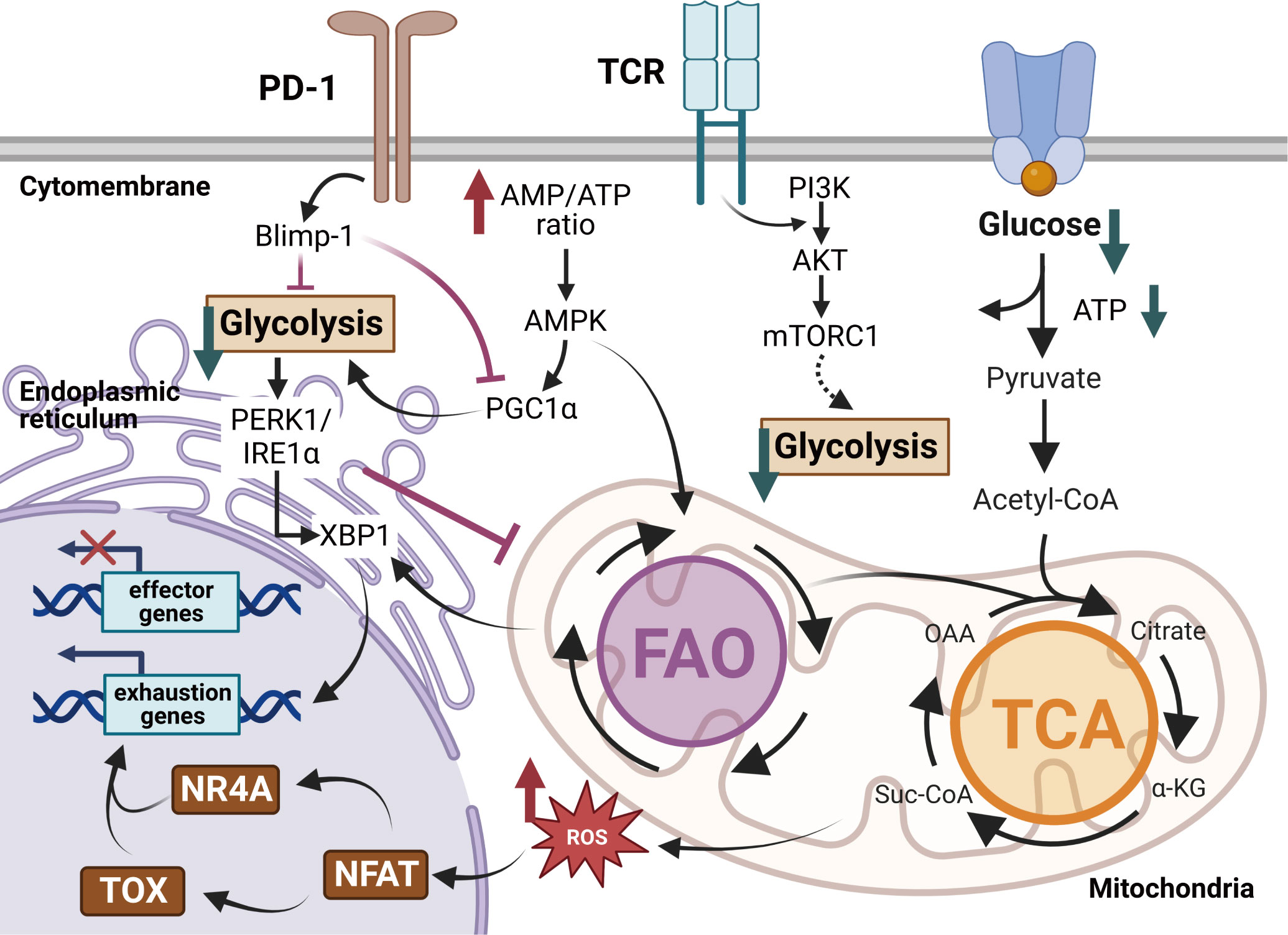
Figure 3 Metabolic regulations of exhausted T cells in the TME. Exhausted T cells exhibit metabolic insufficiency with inhibited glycolysis, mitochondrial respiration and endoplasmic reticulum (ER) function. Enhanced PD-1 signaling in Tex cells dampens the transcriptional coactivator PGC1α, decreasing glycolysis but promoting fatty acid oxidation (FAO). The overloaded cholesterol elicits more ER stress via the XBP1 cascades and fosters the expression of inhibitory receptors. Moreover, impaired glycolysis activates the IRE1-XBP1 signaling pathway, resulting in elevated ER stress and dysregulations of mitochondrial function. The dysfunctional mitochondria produce robust ROS, ultimately promoting TOX expression and repressing effector functions in Tex cells. Created with BioRender.com.
Faulty metabolic reprogramming and altered mitochondrial responses lock T cells in the exhausted state (73, 75). Signalings of inhibitory receptors trigger metabolic insufficiency, mitochondrial dysfunctions and endoplasmic reticulum (ER)-related stress in Tex cells (76). The PD-1+ Tex cells exhibit abnormal metabolic programs in reduced glycolysis, protein glycosylation and oxidative phosphorylation (OXPHOS) (77, 78). The PD-1-related mitochondrial dysfunction follows the downregulation of the transcriptional coactivator PGC1α, which is essential for maintaining mitochondrial activity (79). Overexpression of PGC1α promotes mitochondrial fitness and maintains effector functions of tumor-infiltrating CD8+ T cells. Dysfunctional mitochondria result in a strong production of mitochondrial reactive oxygen species (mtROS), which facilitates T cell exhaustion by enhancing inhibitory receptors expression and impairing effector function genes (80). ER-related stress upregulates IRE1-XBP1 signaling, leading to the over-expression of inhibitory receptor genes like Pdcd1 and Cd244 (81). XBP1 signaling cascades repress the expression of Tbx21 (82), which impairs T cell function. The mitochondria-ER contacts play an important role in T cell exhaustion. The metabolic interactions between mitochondria and ER are controlled by Ca2+ flux, mitochondria ROS (mtROS) and mitochondria-associated membranes (MAMs). MAMs have been demonstrated to act as critical immunometablic molecules that participate in metabolic reprogramming and secretion of IFN-γ in CD8+ T cells. Abnormal Ca2+-NFAT signaling coupled with the accumulation of mtROS under persistent TCR stimulation disrupts mitochondrial metabolism homeostasis and damages T cell survival.
The epigenetic exhaustion alterations are the major regulatory mechanisms linked with mitochondrial disorders and nuclear reprogramming (83). Metabolic insufficiency and dysfunctional mitochondrial activity increase epigenetic modifications in T cell exhaustion. Limited availability of glucose catabolism acetyl-CoA alters the chromatin accessibility to histone acetylation at Ifng and Tbx21 gene loci. Under glucose restriction, acetate treatment speeds up histone acetylation and reduces Ifng gene accessibility, restoring IFN-γ secretion in CD8+ T cells (84). Moreover, NAD-dependent histone deacetylation accelerates T cell exhaustion in reconstructing chromatin structure by suppressing deacetylase sirtuins (SIRTs) activity. The activity of SIRT1 involves affecting IFN-γ production and the expression of Tbx21 genes in CD8+ Teff cells (85). It is also reported that S-adenosylmethionine (SAM) contributes to the exhausted epigenetic remodeling during Tex cell differentiation (73). SAM metabolism is inferred to modulate DNA methylation programs on Tcf7, Tbx21, and Ifng gene loci (13) that are engaged in the differentiation of progenitor Tex cells.
Fine-tuning the metabolic activity of exhausted T cells improves T cell effects and responds to ICB treatment. PD-1 blocking during chronic LCMV infection reinstates mitochondrial fitness and boosts glucose absorption in Tex cells (77). The blockades of glutamine and acetate relieve certain metabolic dysfunctions and prevent T cell exhaustion in the TME (86). Promoting fatty acid oxidation with chemokine treatments and peroxisome-proliferator-activated receptor agonists improves CD8+ T cell tumoricidal effects (87). In addition, the IRE1 and PERK inhibitors effectively reduce ER stress and regain T cell functions. Closely monitoring the mitochondrial activity and controlling the metabolic demands seem to be efficient ways to overcome T cell exhaustion.
Factors and signaling pathways involved in the development of T cell exhaustion are diversified and complex. Besides the TCR signal strength, key transcriptional pathways and metabolic alterations mentioned above, the co-stimulatory signals and soluble mediators also widely participate in the progress of T cell exhaustion. We summarize the intrinsic and extrinsic factors contributing to T cell exhaustion in four general signal models in Table 1. And in the following, we discuss the co-stimulatory molecules, cytokines and kinases in T cell exhaustion.

Table 1 Summary of signal models in development of T cell exhaustion.
Costimulatory molecules play pivotal roles in regulating T cell proliferation, activation and differentiation. However, inappropriately dampened or enhanced co-stimulatory signals contribute to T cell exhaustion. The activation of T cells relies on both TCR signaling and co-stimulatory signaling from CD28. The effects of CD28 signal are compromised during persistent inflammation, where a significant level of CTLA-4 is expressed. Moreover, the engagement of PD-1 directly inhibits CD28 signaling and prevents T cell activation (88). Critically, CD28 is necessary for successful PD-1 blockade during chronic LCMV infection. Conditional deletion of CD28 has been found to diminish the efficacy of anti-PD-1 therapy in mouse tumor models (89). Continual CD27 signaling has also been reported to result in more severe T cell exhaustion due to excessive T cell stimulation (90). However, another member in the TNF receptor family, 4-1BB, may synergize with anti-PD-L1 to rejuvenate Tex cells (91). The immunoglobulin superfamily member CD226 also plays a role in T cell exhaustion. As a co-stimulatory receptor that mediates T cell adhesion and execution of cytotoxicity (92), CD226 competes with immune checkpoints like TIGIT for the same ligands expressed by cancer cells to execute the anti-tumor effects (93–95). Chronic stimulations on tumor-infiltrating Tex cells induce high expression of Eomes, which downregulates CD226 (67, 96, 97). The Eomes-dependent loss of CD226 limits TCR signaling and anti-tumor effects (98–101). Moreover, the loss of CD226 has been found to impair T cell response to PD-1 blockade (102–104). In a mouse melanoma model, anti-PD-1 therapy was unable to rescue effector functions of Tex cells lacking CD226, implying that the CD226- Tex group displayed a more terminally exhausted profile (105).
Both proinflammatory and immunosuppressive cytokines have regulatory roles in T cell exhaustion. Specifically, high levels of proinflammatory factors trigger the production of unfavorable cytokines which promote T cell exhaustion during long-term infections and malignancies. IL-10 is typically responsive-elevated in inflammatory microenvironments, and blocking IL-10 can prevent and reverse T cell exhaustion (106). The mechanism of reversing exhaustion by blockade of IL-10 may be directly through activating T cells by STAT-3, or indirectly via modulation of suppressive antigen-presenting cells (APCs) (107). In addition, IL-6 and IL-27 are also important cytokines that upregulate co-inhibitory signals on tumor-infiltrating T cells, correlating with T cell exhaustion in both human and mouse tumor models (108–110). The transforming growth factor-β (TGF-β) is another kind of immunosuppressive cytokines that seemingly participated in terminal T cell exhaustion. The expression of TGF-β and the activation of downstream SMAD2 are hallmarks of Tex cells (111, 112). Inhibiting TGF-β1 signaling synergizes with the agonist of bone morphogenetic protein in CD8+ T cells in vitro enhanced the efficacy of Tex cells (113). However, it was also reported that therapeutic inhibition of TGF-β alone failed to control the persistent LCMV infection and scarcely enhanced the efficacy of antiviral T cells (114, 115).
In contrast, IL-2 responsiveness is crucial for maintaining the Tex cell pool, as IL-2R deficient Tex rapidly disappear during chronic antigen stimulation (116). In vivo IL-2 therapy after LCMV chronic infection has been shown to increase the number of antigen-specific Tex and achieve excellent viral control (117). Besides IL-2, recent studies have also indicated that supplying adequate amounts of IL-21 may allow TCF-1+ Tpex to escape from exhaustion and differentiate into a CX3CR1+ Teff subpopulation that exhibits lower PD-1 and displays improved cytotoxicity compared to Tex (16). What’s more, ambiguous in promoting or preventing T cell exhaustion, type I interferons (IFN-I) play complex roles in controlling T lymphocytes. IFN-I is critical for activating and inducing antiviral functions of CD8+ T cells (118), which seems beneficial to avoid T cell exhaustion. However, elevated IFN-I in the tumor microenvironment may also induce high levels of IL-10 and PD-L1 (119). What’s more, it has been reported that IFN-I signaling can promote terminal exhaustion by suppressing Tpex cell sustainment through antagonizing TCF-1 (25). On-going IFN-I exposure has also been linked to the promotion of co-inhibitory receptors (including PD-1, TIM-3, LAG-3, and TIGIT) expression in chronic viral infections (120–124).
In addition to the co-stimulatory molecules and cytokines that regulate T cell exhaustion, the TCR-linked phosphoinositide 3 kinase (PI3K)-Akt-mTOR pathway also plays an important role in regulating CD8+ T cell exhaustion. Upon T cell activation, PI3Kδ inactivates the downstream transcriptional targets FOXO1 and BACH2 to suppress Tcf7 expression (125–127). This results in the repression of TCF-1-mediated central memorization and the up-regulation of effector genes such as Tbx21 and Gzmb, which execute cytotoxicity functions (128–130). Inhibition of PI3Kδ has been reported to promote the expansion of the TCF-1+ Tpex subpopulation and improve the response to ICB therapy (131). Tex cells with reduced PI3Kδ activity preserve TCF-1 and promote the generation of a self-renewing memory Tpex population as well (125, 129, 130). Pre-inhibition of Akt or mTOR is also proven to helpfully maintain the stem-like CD8+ T cell pool and enhance the antitumor efficacy (132–134). However, it is noticeable that with the exhaustion progressing, mTOR inhibition may no longer limit the chronic viral infection (134), partly because PD-1 signaling weakens the PI3K-Akt-mTOR signaling and causes metabolic reprogramming including impaired glycolysis and mitochondrial activities in late-stage Tex cells (135). So, a more careful modification of this kinase signaling pathway is necessary to balance the stimulation of TCF-1+ Tpex cluster properly.
PD-1 plays an important role in regulating the threshold of antigen response and limiting cytotoxic damage to maintain self-tolerance in T lymphocytes (136). Hence, interrupting the exhaustion program, such as PD-1 blockade, not only can result in enhanced viral control, but also may cause serious immunopathology (137). In fact, immune-mediated pathology is a common side effect of checkpoint blockade in anti-tumor immunity (138, 139). Upon binding with PD-L1/2, PD-1 undergoes a conformational change that brings phosphorylated SHP-2 close to the TCR and CD28 signaling complex (88, 89, 140), resulting in less activation of LCK-mediated ZAP70 and attenuation of downstream RAS-MEK-ERK and PI3K-Akt-mTOR signaling pathways (88). These transduction events ultimately lead to the suppression of T cell proliferation, differentiation, and function execution. This is why higher expression of PD-1 is correlated with more severe T cell exhaustion. Nevertheless, T cell exhaustion should also be considered a physiological adaptation to chronic infections and cancers rather than a deficit in the function of the immune system.
Since chronic infections have existed throughout evolution, T cell exhaustion is a process that avoids immune-mediated pathology while maintaining a degree of functional activity (141). T cells with exhaustion phenotypes can still exert effector functions and mediate the pathogen and tumor control (142–144). CD8+ T lymphocytes in established chronic simian immunodeficiency virus (SIV) infections were demonstrated to control viremia efficiently. Virus replication burst rapidly if depleting CD8+ T cells in SIV-infected macaques (145, 146). Moreover, a favorable clinical outcome is positively connected with the quantity of tumor-infiltrating CD8+ T cells (147). The immune efficacy of CD8+ PD-1hi T cells was similar to or even higher than those with less exhausted profiles in secreting cytotoxic molecules (136, 142). Similar phenomena indicate that the effector function of Tex cells is reserved by immune suppressive regulators rather than attenuated intrinsically and irreversibly.
According to this theory, PD-1 stabilizes the TCF-1+ Tex precursor cell pool and maintains the survival of this early TCF-1+ subset (12). PD-1 activation prevents the loss of TCF-1 expression, and one possible mechanism of PD-1 and TCF-1 interaction is the suppressive effect on TCR and CD28 signaling induced by PD-1 (148). Another theory suggests that PD-1 induces the expression of the transcriptional factor BATF, which positively regulates TCF-1 expression and represses effector-related genes (149). Moreover, BATF has also been predicted as a downstream responder of TCF-1 in the Tpex subpopulation (150). Thus, it is conceivable that a PD-1-BATF-TCF-1 feedback circuit occurs in the Tpex cluster to maintain the early survival of tumor-infiltrating Tex. The Tpex cell cluster is required for an efficient response to ICB therapy because the PD-1hi subpopulation is terminally exhausted and unable to respond to PD-1 pathway inhibition.
PD-1 also functions as an adaptive immune molecule in tumor-specific memory T (Ttsm) cells derived from tumor draining lymph nodes (TdLN). PD-1+TCF-1+TdLN-residing Ttsm cells do not activate TOX signaling and significantly differ from Tpex and Tex on a transcriptional and epigenetic level (151). Investigations have shown that the TdLN-Ttsm cluster is the primary responder to PD-(L)1 blockade treatment. Blocking PD-L1 effectively amplifies TdLN-Ttsm clusters, resulting in the enlargement of Tpex and Tex in the tumor microenvironment (151). It implies that PD-1 is dispensable for the initiation of T cell exhaustion but it is critical for maintaining the restrained functional state of anti-tumor T cells. To fully understand the impact of anti-PD-1 on various immune cell types, such as B cells, myeloid cells, Treg, and NKs, further investigations are needed. In addition, removing extra immune checkpoints, such as CTLA-4, LAG3, TIM3, and TIGIT) on CD8+ T cells might synergistically advance the success of anti-PD-(L)1 therapy (152). However, it is still unclear if there is a mechanism by which inhibitory receptors cooperate, nor is it clear how to optimize interactions between various inhibitory receptors.
T cell exhaustion is an inevitable outcome of repeated antigen stimulation in chronic infections and tumors. The quantity of Tex cells in the TME is not only a signature for conducting immunotherapy but also a long-lasting “biomarker” for predicting the prognosis. Exploring the heterogeneity of Tex cells reveals advanced therapeutic approaches for reinvigorating T cell exhaustion and improves the efficacy of current ICB therapy. The degree of T cell exhaustion is regulated directly by the intensity and longevity of TCR signaling. Critically, a differentiation hierarchy of T cell exhaustion exists. The progressive transcriptional regulation mechanically orchestrates epigenetic modifications and metabolic reprogrammings to shape distinct Tex clusters. Multiple signal models synergistically contribute to T cell exhaustion, providing many opportunities for combinational treatments to overcome the limitations and achieve success of immunotherapies. Inhibitory receptors play a crucial role in keeping the delicate balance between T cell exhaustion and activation. The best approach to reverse T cell exhaustion still has to be investigated with a considerable understanding of this dysfunctional cell state.
WT designed the conceptualization. QS, GQ, and XB assisted in the validation. WT and GQ wrote the original draft. QS, WW, and YZ supervised this work. WT performed the visualization. YZ, QS, WC, WW, XB, MJ, YJZ, WL, and HW provided the theoretical support. QS and YZ provided financial support. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Grant No. 82071738), and the Medical Leadership Program of Jiangsu College of Nursing (Grant No. 2021001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Chow A, Perica K, Klebanoff CA, Wolchok JD. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat Rev Clin Oncol (2022) 19(12):775–90. doi: 10.1038/s41571-022-00689-z
2. Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P, et al. Proliferating transitory T cells with an effector-like transcriptional signature emerge from pd-1(+) stem-like Cd8(+) T cells during chronic infection. Immunity (2019) 51(6):1043–58.e4. doi: 10.1016/j.immuni.2019.11.002
3. Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell (2018) 175(4):998–1013.e20. doi: 10.1016/j.cell.2018.10.038
4. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining Cd8+ T cells that provide the proliferative burst after pd-1 therapy. Nature (2016) 537(7620):417–21. doi: 10.1038/nature19330
5. Kallies A, Zehn D, Utzschneider DT. Precursor exhausted T cells: key to successful immunotherapy? Nat Rev Immunol (2020) 20(2):128–36. doi: 10.1038/s41577-019-0223-7
6. Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following pd-1 blockade. Nat Med (2019) 25(8):1251–9. doi: 10.1038/s41591-019-0522-3
7. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted Cd8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol (2019) 20(3):326–36. doi: 10.1038/s41590-019-0312-6
8. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862
9. van der Heide V, Humblin E, Vaidya A, Kamphorst AO. Advancing beyond the twists and turns of T cell exhaustion in cancer. Sci Transl Med (2022) 14(670):eabo4997. doi: 10.1126/scitranslmed.abo4997
10. Seo W, Jerin C, Nishikawa H. Transcriptional regulatory network for the establishment of Cd8(+) T cell exhaustion. Exp Mol Med (2021) 53(2):202–9. doi: 10.1038/s12276-021-00568-0
11. Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, et al. Tox is a critical regulator of tumour-specific T cell differentiation. Nature (2019) 571(7764):270–4. doi: 10.1038/s41586-019-1324-y
12. Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, et al. Tcf-1-Centered transcriptional network drives an effector versus exhausted Cd8 t cell-fate decision. Immunity (2019) 51(5):840–55.e5. doi: 10.1016/j.immuni.2019.09.013
13. Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De novo epigenetic programs inhibit pd-1 blockade-mediated T cell rejuvenation. Cell (2017) 170(1):142–57.e19. doi: 10.1016/j.cell.2017.06.007
14. Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector Cd8 T cells that give rise to long-lived memory cells. Nat Immunol (2003) 4(12):1191–8. doi: 10.1038/ni1009
15. Zhang N, Bevan MJ. Cd8(+) T cells: foot soldiers of the immune system. Immunity (2011) 35(2):161–8. doi: 10.1016/j.immuni.2011.07.010
16. Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, et al. Cd4(+) T cell help is required for the formation of a cytolytic Cd8(+) T cell subset that protects against chronic infection and cancer. Immunity (2019) 51(6):1028–42.e4. doi: 10.1016/j.immuni.2019.10.009
17. Raju S, Xia Y, Daniel B, Yost KE, Bradshaw E, Tonc E, et al. Identification of a T-Bet(Hi) quiescent exhausted Cd8 T cell subpopulation that can differentiate into Tim3(+)Cx3cr1(+) effectors and memory-like cells. J Immunol (2021) 206(12):2924–36. doi: 10.4049/jimmunol.2001348
18. Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, et al. Developmental relationships of four exhausted Cd8(+) T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity (2020) 52(5):825–41.e8. doi: 10.1016/j.immuni.2020.04.014
19. Yao C, Sun HW, Lacey NE, Ji Y, Moseman EA, Shih HY, et al. Single-cell rna-seq reveals tox as a key regulator of Cd8(+) T cell persistence in chronic infection. Nat Immunol (2019) 20(7):890–901. doi: 10.1038/s41590-019-0403-4
20. Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, et al. Tox reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature (2019) 571(7764):265–9. doi: 10.1038/s41586-019-1326-9
21. Zhang Z, Chen L, Chen H, Zhao J, Li K, Sun J, et al. Pan-cancer landscape of T-cell exhaustion heterogeneity within the tumor microenvironment revealed a progressive roadmap of hierarchical dysfunction associated with prognosis and therapeutic efficacy. EBioMedicine (2022) 83:104207. doi: 10.1016/j.ebiom.2022.104207
22. Bengsch B, Ohtani T, Khan O, Setty M, Manne S, O’Brien S, et al. Epigenomic-guided mass cytometry profiling reveals disease-specific features of exhausted Cd8 t cells. Immunity (2018) 48(5):1029–45.e5. doi: 10.1016/j.immuni.2018.04.026
23. Wang X, Chen Y, Li Z, Huang B, Xu L, Lai J, et al. Single-cell rna-seq of T cells in b-all patients reveals an exhausted subset with remarkable heterogeneity. Adv Sci (Weinh) (2021) 8(19):e2101447:eaai8593. doi: 10.1002/advs.202101447
24. Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted Cd8 T cells by alphapd-L1 blockade. Proc Natl Acad Sci U.S.A. (2008) 105(39):15016–21. doi: 10.1073/pnas.0801497105
25. Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, et al. The Tcf1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol (2016) 1(6):eaai8593. doi: 10.1126/sciimmunol.aai8593
26. Wang Y, Hu J, Li Y, Xiao M, Wang H, Tian Q, et al. The transcription factor Tcf1 preserves the effector function of exhausted Cd8 T cells during chronic viral infection. Front Immunol (2019) 10:169. doi: 10.3389/fimmu.2019.00169
27. Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH. Differentiation and persistence of memory Cd8(+) T cells depend on T cell factor 1. Immunity (2010) 33(2):229–40. doi: 10.1016/j.immuni.2010.08.002
28. Milner JJ, Toma C, He Z, Kurd NS, Nguyen QP, McDonald B, et al. Heterogenous populations of tissue-resident Cd8(+) T cells are generated in response to infection and malignancy. Immunity (2020) 52(5):808–24.e7. doi: 10.1016/j.immuni.2020.04.007
29. Bassez A, Vos H, Van Dyck L, Floris G, Arijs I, Desmedt C, et al. A single-cell map of intratumoral changes during anti-Pd1 treatment of patients with breast cancer. Nat Med (2021) 27(5):820–32. doi: 10.1038/s41591-021-01323-8
30. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by pd-1 blockade. Science (2016) 354(6316):1160–5. doi: 10.1126/science.aaf2807
31. Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The epigenetic landscape of T cell exhaustion. Science (2016) 354(6316):1165–9. doi: 10.1126/science.aae0491
32. Mognol GP, Spreafico R, Wong V, Scott-Browne JP, Togher S, Hoffmann A, et al. Exhaustion-associated regulatory regions in Cd8(+) tumor-infiltrating T cells. Proc Natl Acad Sci U.S.A. (2017) 114(13):E2776–e85. doi: 10.1073/pnas.1620498114
33. Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, et al. Chronic virus infection enforces demethylation of the locus that encodes pd-1 in antigen-specific Cd8(+) T cells. Immunity (2011) 35(3):400–12. doi: 10.1016/j.immuni.2011.06.015
34. Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, et al. Transcription factor T-bet represses expression of the inhibitory receptor pd-1 and sustains virus-specific Cd8+ T cell responses during chronic infection. Nat Immunol (2011) 12(7):663–71. doi: 10.1038/ni.2046
35. Ahn E, Youngblood B, Lee J, Lee J, Sarkar S, Ahmed R. Demethylation of the pd-1 promoter is imprinted during the effector phase of Cd8 T cell exhaustion. J Virol (2016) 90(19):8934–46. doi: 10.1128/jvi.00798-16
36. Zhang F, Zhou X, DiSpirito JR, Wang C, Wang Y, Shen H. Epigenetic manipulation restores functions of defective Cd8+ T cells from chronic viral infection. Mol Ther (2014) 22(9):1698–706. doi: 10.1038/mt.2014.91
37. McLane LM, Abdel-Hakeem MS, Wherry EJ. Cd8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol (2019) 37:457–95. doi: 10.1146/annurev-immunol-041015-055318
38. Belk JA, Daniel B, Satpathy AT. Epigenetic regulation of T cell exhaustion. Nat Immunol (2022) 23(6):848–60. doi: 10.1038/s41590-022-01224-z
39. Utzschneider DT, Alfei F, Roelli P, Barras D, Chennupati V, Darbre S, et al. High antigen levels induce an exhausted phenotype in a chronic infection without impairing T cell expansion and survival. J Exp Med (2016) 213(9):1819–34. doi: 10.1084/jem.20150598
40. Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, et al. Transcription factor Irf4 promotes Cd8(+) T cell exhaustion and limits the development of memory-like T cells during chronic infection. Immunity (2017) 47(6):1129–41.e5. doi: 10.1016/j.immuni.2017.11.021
41. Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S, et al. The transcription factor Irf4 is essential for tcr affinity-mediated metabolic programming and clonal expansion of T cells. Nat Immunol (2013) 14(11):1155–65. doi: 10.1038/ni.2710
42. Kurachi M, Barnitz RA, Yosef N, Odorizzi PM, DiIorio MA, Lemieux ME, et al. The transcription factor batf operates as an essential differentiation checkpoint in early effector Cd8+ T cells. Nat Immunol (2014) 15(4):373–83. doi: 10.1038/ni.2834
43. Klein-Hessling S, Muhammad K, Klein M, Pusch T, Rudolf R, Flöter J, et al. Nfatc1 controls the cytotoxicity of Cd8(+) T cells. Nat Commun (2017) 8(1):511. doi: 10.1038/s41467-017-00612-6
44. Scott-Browne JP, López-Moyado IF, Trifari S, Wong V, Chavez L, Rao A, et al. Dynamic changes in chromatin accessibility occur in Cd8(+) T cells responding to viral infection. Immunity (2016) 45(6):1327–40. doi: 10.1016/j.immuni.2016.10.028
45. Macián F, García-Cózar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell (2002) 109(6):719–31. doi: 10.1016/s0092-8674(02)00767-5
46. Martinez GJ, Pereira RM, Äijö T, Kim EY, Marangoni F, Pipkin ME, et al. The transcription factor nfat promotes exhaustion of activated Cd8+ T cells. Immunity (2015) 42(2):265–78. doi: 10.1016/j.immuni.2015.01.006
48. Karin M, Liu Z, Zandi E. Ap-1 function and regulation. Curr Opin Cell Biol (1997) 9(2):240–6. doi: 10.1016/s0955-0674(97)80068-3
49. Seo H, Chen J, González-Avalos E, Samaniego-Castruita D, Das A, Wang YH, et al. Tox and Tox2 transcription factors cooperate with Nr4a transcription factors to impose Cd8(+) T cell exhaustion. Proc Natl Acad Sci U.S.A. (2019) 116(25):12410–5. doi: 10.1073/pnas.1905675116
50. Liu X, Wang Y, Lu H, Li J, Yan X, Xiao M, et al. Genome-wide analysis identifies Nr4a1 as a key mediator of T cell dysfunction. Nature (2019) 567(7749):525–9. doi: 10.1038/s41586-019-0979-8
51. Burger ML, Cruz AM, Crossland GE, Gaglia G, Ritch CC, Blatt SE, et al. Antigen dominance hierarchies shape Tcf1(+) progenitor Cd8 T cell phenotypes in tumors. Cell (2021) 184(19):4996–5014.e26. doi: 10.1016/j.cell.2021.08.020
52. Daniel B, Yost KE, Hsiung S, Sandor K, Xia Y, Qi Y, et al. Divergent clonal differentiation trajectories of T cell exhaustion. Nat Immunol (2022) 23(11):1614–27. doi: 10.1038/s41590-022-01337-5
53. Shakiba M, Zumbo P, Espinosa-Carrasco G, Menocal L, Dündar F, Carson SE, et al. Tcr signal strength defines distinct mechanisms of T cell dysfunction and cancer evasion. J Exp Med (2022) 219(2):e20201966. doi: 10.1084/jem.20201966
54. Shan Q, Hu S, Chen X, Danahy DB, Badovinac VP, Zang C, et al. Ectopic Tcf1 expression instills a stem-like program in exhausted Cd8(+) T cells to enhance viral and tumor immunity. Cell Mol Immunol (2021) 18(5):1262–77. doi: 10.1038/s41423-020-0436-5
55. Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, et al. Tox transcriptionally and epigenetically programs Cd8(+) T cell exhaustion. Nature (2019) 571(7764):211–8. doi: 10.1038/s41586-019-1325-x
56. Wang X, He Q, Shen H, Xia A, Tian W, Yu W, et al. Tox promotes the exhaustion of antitumor Cd8(+) T cells by preventing Pd1 degradation in hepatocellular carcinoma. J Hepatol (2019) 71(4):731–41. doi: 10.1016/j.jhep.2019.05.015
57. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, et al. Inflammation directs memory precursor and short-lived effector Cd8(+) T cell fates Via the graded expression of T-bet transcription factor. Immunity (2007) 27(2):281–95. doi: 10.1016/j.immuni.2007.07.010
58. Angelosanto JM, Blackburn SD, Crawford A, Wherry EJ. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J Virol (2012) 86(15):8161–70. doi: 10.1128/jvi.00889-12
59. Banerjee A, Gordon SM, Intlekofer AM, Paley MA, Mooney EC, Lindsten T, et al. Cutting edge: the transcription factor eomesodermin enables Cd8+ T cells to compete for the memory cell niche. J Immunol (2010) 185(9):4988–92. doi: 10.4049/jimmunol.1002042
60. Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and memory Cd8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol (2005) 6(12):1236–44. doi: 10.1038/ni1268
61. Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature (2017) 545(7655):452–6. doi: 10.1038/nature22367
62. Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, et al. T Cell factor 1-expressing memory-like Cd8(+) T cells sustain the immune response to chronic viral infections. Immunity (2016) 45(2):415–27. doi: 10.1016/j.immuni.2016.07.021
63. Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, et al. Intratumoral Tcf1(+)Pd-1(+)Cd8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity (2019) 50(1):195–211.e10. doi: 10.1016/j.immuni.2018.12.021
64. Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell (2019) 176(1-2):404. doi: 10.1016/j.cell.2018.12.034
65. Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, Wherry EJ. Network analysis reveals centrally connected genes and pathways involved in Cd8+ T cell exhaustion versus memory. Immunity (2012) 37(6):1130–44. doi: 10.1016/j.immuni.2012.08.021
66. Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, et al. Progenitor and terminal subsets of Cd8+ T cells cooperate to contain chronic viral infection. Science (2012) 338(6111):1220–5. doi: 10.1126/science.1229620
67. McLane LM, Ngiow SF, Chen Z, Attanasio J, Manne S, Ruthel G, et al. Role of nuclear localization in the regulation and function of T-bet and eomes in exhausted Cd8 T cells. Cell Rep (2021) 35(6):109120. doi: 10.1016/j.celrep.2021.109120
68. Gabriel SS, Tsui C, Chisanga D, Weber F, Llano-León M, Gubser PM, et al. Transforming growth factor-B-Regulated mtor activity preserves cellular metabolism to maintain long-term T cell responses in chronic infection. Immunity (2021) 54(8):1698–714.e5. doi: 10.1016/j.immuni.2021.06.007
69. Zeng Z, Wei F, Ren X. Exhausted T cells and epigenetic status. Cancer Biol Med (2020) 17(4):923–36. doi: 10.20892/j.issn.2095-3941.2020.0338
70. Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-inducible factors enhance the effector responses of Cd8(+) T cells to persistent antigen. Nat Immunol (2013) 14(11):1173–82. doi: 10.1038/ni.2714
71. Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, et al. Pdk1 regulation of mtor and hypoxia-inducible factor 1 integrate metabolism and migration of Cd8+ T cells. J Exp Med (2012) 209(13):2441–53. doi: 10.1084/jem.20112607
72. Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153(6):1239–51. doi: 10.1016/j.cell.2013.05.016
73. Franco F, Jaccard A, Romero P, Yu YR, Ho PC. Metabolic and epigenetic regulation of T-cell exhaustion. Nat Metab (2020) 2(10):1001–12. doi: 10.1038/s42255-020-00280-9
74. Saka D, Gökalp M, Piyade B, Cevik NC, Arik Sever E, Unutmaz D, et al. Mechanisms of T-cell exhaustion in pancreatic cancer. Cancers (Basel) (2020) 12(8):2274. doi: 10.3390/cancers12082274
75. Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL, et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol (2021) 22(2):205–15. doi: 10.1038/s41590-020-00834-9
76. Li X, Wenes M, Romero P, Huang SC, Fendt SM, Ho PC. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat Rev Clin Oncol (2019) 16(7):425–41. doi: 10.1038/s41571-019-0203-7
77. Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor pd-1 are an early driver of Cd8(+) T cell exhaustion. Immunity (2016) 45(2):358–73. doi: 10.1016/j.immuni.2016.07.008
78. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. Pd-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6:6692. doi: 10.1038/ncomms7692
79. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity (2016) 45(2):374–88. doi: 10.1016/j.immuni.2016.07.009
80. Siska PJ, Beckermann KE, Mason FM, Andrejeva G, Greenplate AR, Sendor AB, et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair Cd8 T cells infiltrating human renal cell carcinoma. JCI Insight (2017) 2(12):e93411. doi: 10.1172/jci.insight.93411
81. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces Cd8(+) T cell exhaustion in the tumor microenvironment. Cell Metab (2019) 30(1):143–56.e5. doi: 10.1016/j.cmet.2019.04.002
82. Cao Y, Trillo-Tinoco J, Sierra RA, Anadon C, Dai W, Mohamed E, et al. Er stress-induced mediator C/Ebp homologous protein thwarts effector T cell activity in tumors through T-bet repression. Nat Commun (2019) 10(1):1280. doi: 10.1038/s41467-019-09263-1
83. Matilainen O, Quirós PM, Auwerx J. Mitochondria and epigenetics - crosstalk in homeostasis and stress. Trends Cell Biol (2017) 27(6):453–63. doi: 10.1016/j.tcb.2017.02.004
84. Qiu J, Villa M, Sanin DE, Buck MD, O’Sullivan D, Ching R, et al. Acetate promotes T cell effector function during glucose restriction. Cell Rep (2019) 27(7):2063–74.e5. doi: 10.1016/j.celrep.2019.04.022
85. Kuroda S, Yamazaki M, Abe M, Sakimura K, Takayanagi H, Iwai Y. Basic leucine zipper transcription factor, atf-like (Batf) regulates epigenetically and energetically effector Cd8 T-cell differentiation Via Sirt1 expression. Proc Natl Acad Sci U.S.A. (2011) 108(36):14885–9. doi: 10.1073/pnas.1105133108
86. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell (2015) 162(6):1217–28. doi: 10.1016/j.cell.2015.08.012
87. Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing Cd8(+) T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell (2017) 32(3):377–91.e9. doi: 10.1016/j.ccell.2017.08.004
88. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T Cell costimulatory receptor Cd28 is a primary target for pd-1-Mediated inhibition. Science (2017) 355(6332):1428–33. doi: 10.1126/science.aaf1292
89. Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted Cd8 T cells by pd-1-Targeted therapies is Cd28-dependent. Science (2017) 355(6332):1423–7. doi: 10.1126/science.aaf0683
90. Penaloza-MacMaster P, Ur Rasheed A, Iyer SS, Yagita H, Blazar BR, Ahmed R. Opposing effects of Cd70 costimulation during acute and chronic lymphocytic choriomeningitis virus infection of mice. J Virol (2011) 85(13):6168–74. doi: 10.1128/jvi.02205-10
91. Vezys V, Penaloza-MacMaster P, Barber DL, Ha SJ, Konieczny B, Freeman GJ, et al. 4-1bb signaling synergizes with programmed death ligand 1 blockade to augment Cd8 T cell responses during chronic viral infection. J Immunol (2011) 187(4):1634–42. doi: 10.4049/jimmunol.1100077
92. Martinet L, Smyth MJ. Balancing natural killer cell activation through paired receptors. Nat Rev Immunol (2015) 15(4):243–54. doi: 10.1038/nri3799
93. Guillerey C, Harjunpää H, Carrié N, Kassem S, Teo T, Miles K, et al. Tigit immune checkpoint blockade restores Cd8(+) T-cell immunity against multiple myeloma. Blood (2018) 132(16):1689–94. doi: 10.1182/blood-2018-01-825265
94. Blake SJ, Dougall WC, Miles JJ, Teng MW, Smyth MJ. Molecular pathways: targeting Cd96 and tigit for cancer immunotherapy. Clin Cancer Res (2016) 22(21):5183–8. doi: 10.1158/1078-0432.Ccr-16-0933
95. Blake SJ, Stannard K, Liu J, Allen S, Yong MC, Mittal D, et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discovery (2016) 6(4):446–59. doi: 10.1158/2159-8290.Cd-15-0944
96. Buggert M, Tauriainen J, Yamamoto T, Frederiksen J, Ivarsson MA, Michaëlsson J, et al. T-Bet and eomes are differentially linked to the exhausted phenotype of Cd8+ T cells in hiv infection. PloS Pathog (2014) 10(7):e1004251. doi: 10.1371/journal.ppat.1004251
97. Li J, He Y, Hao J, Ni L, Dong C. High levels of eomes promote exhaustion of anti-tumor Cd8(+) T cells. Front Immunol (2018) 9:2981. doi: 10.3389/fimmu.2018.02981
98. Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, et al. The receptors Cd96 and Cd226 oppose each other in the regulation of natural killer cell functions. Nat Immunol (2014) 15(5):431–8. doi: 10.1038/ni.2850
99. Guillerey C, Ferrari de Andrade L, Vuckovic S, Miles K, Ngiow SF, Yong MC, et al. Immunosurveillance and therapy of multiple myeloma are Cd226 dependent. J Clin Invest (2015) 125(7):2904. doi: 10.1172/jci82646
100. Cella M, Presti R, Vermi W, Lavender K, Turnbull E, Ochsenbauer-Jambor C, et al. Loss of dnam-1 contributes to Cd8+ T-cell exhaustion in chronic hiv-1 infection. Eur J Immunol (2010) 40(4):949–54. doi: 10.1002/eji.200940234
101. Scharf L, Pedersen CB, Johansson E, Lindman J, Olsen LR, Buggert M, et al. Inverted Cd8 T-cell exhaustion and Co-stimulation marker balance differentiate aviremic hiv-2-Infected from seronegative individuals. Front Immunol (2021) 12:744530. doi: 10.3389/fimmu.2021.744530
102. Braun M, Aguilera AR, Sundarrajan A, Corvino D, Stannard K, Krumeich S, et al. Cd155 on tumor cells drives resistance to immunotherapy by inducing the degradation of the activating receptor Cd226 in Cd8(+) T cells. Immunity (2020) 53(4):805–23.e15. doi: 10.1016/j.immuni.2020.09.010
103. Weulersse M, Asrir A, Pichler AC, Lemaitre L, Braun M, Carrié N, et al. Eomes-dependent loss of the Co-activating receptor Cd226 restrains Cd8(+) T cell anti-tumor functions and limits the efficacy of cancer immunotherapy. Immunity (2020) 53(4):824–39.e10. doi: 10.1016/j.immuni.2020.09.006
104. Wang B, Zhang W, Jankovic V, Golubov J, Poon P, Oswald EM, et al. Combination cancer immunotherapy targeting pd-1 and gitr can rescue Cd8(+) T cell dysfunction and maintain memory phenotype. Sci Immunol (2018) 3(29):eaat7061. doi: 10.1126/sciimmunol.aat7061
105. Banta KL, Xu X, Chitre AS, Au-Yeung A, Takahashi C, O’Gorman WE, et al. Mechanistic convergence of the tigit and pd-1 inhibitory pathways necessitates Co-blockade to optimize anti-tumor Cd8(+) T cell responses. Immunity (2022) 55(3):512–26.e9. doi: 10.1016/j.immuni.2022.02.005
106. Zarour HM. Reversing T-cell dysfunction and exhaustion in cancer. Clin Cancer Res (2016) 22(8):1856–64. doi: 10.1158/1078-0432.Ccr-15-1849
107. Couper KN, Blount DG, Riley EM. Il-10: the master regulator of immunity to infection. J Immunol (2008) 180(9):5771–7. doi: 10.4049/jimmunol.180.9.5771
108. Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, et al. Induction and transcriptional regulation of the Co-inhibitory gene module in T cells. Nature (2018) 558(7710):454–9. doi: 10.1038/s41586-018-0206-z
109. DeLong JH, O’Hara Hall A, Rausch M, Moodley D, Perry J, Park J, et al. Il-27 and tcr stimulation promote T cell expression of multiple inhibitory receptors. Immunohorizons (2019) 3(1):13–25. doi: 10.4049/immunohorizons.1800083
110. Zhang W, Liu Y, Yan Z, Yang H, Sun W, Yao Y, et al. Il-6 promotes pd-L1 expression in monocytes and macrophages by decreasing protein tyrosine phosphatase receptor type O expression in human hepatocellular carcinoma. J Immunother Cancer (2020) 8(1):e000285. doi: 10.1136/jitc-2019-000285
111. Tinoco R, Alcalde V, Yang Y, Sauer K, Zuniga EI. Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific Cd8+ T cell deletion and viral persistence in vivo. Immunity (2009) 31(1):145–57. doi: 10.1016/j.immuni.2009.06.015
112. Nelson DR, Gonzalez-Peralta RP, Qian K, Xu Y, Marousis CG, Davis GL, et al. Transforming growth factor-beta 1 in chronic hepatitis c. J Viral Hepat (1997) 4(1):29–35. doi: 10.1046/j.1365-2893.1997.00124.x
113. Saadey AA, Yousif A, Osborne N, Shahinfar R, Chen YL, Laster B, et al. Rebalancing Tgfβ1/Bmp signals in exhausted T cells unlocks responsiveness to immune checkpoint blockade therapy. Nat Immunol (2023) 24(2):280–94. doi: 10.1038/s41590-022-01384-y
114. Garidou L, Heydari S, Gossa S, McGavern DB. Therapeutic blockade of transforming growth factor beta fails to promote clearance of a persistent viral infection. J Virol (2012) 86(13):7060–71. doi: 10.1128/jvi.00164-12
115. Boettler T, Cheng Y, Ehrhardt K, von Herrath M. Tgf-B blockade does not improve control of an established persistent viral infection. Viral Immunol (2012) 25(3):232–8. doi: 10.1089/vim.2011.0079
116. Bachmann MF, Wolint P, Walton S, Schwarz K, Oxenius A. Differential role of il-2r signaling for Cd8+ T cell responses in acute and chronic viral infections. Eur J Immunol (2007) 37(6):1502–12. doi: 10.1002/eji.200637023
117. West EE, Jin HT, Rasheed AU, Penaloza-Macmaster P, Ha SJ, Tan WG, et al. Pd-L1 blockade synergizes with il-2 therapy in reinvigorating exhausted T cells. J Clin Invest (2013) 123(6):2604–15. doi: 10.1172/jci67008
118. Crouse J, Kalinke U, Oxenius A. Regulation of antiviral T cell responses by type I interferons. Nat Rev Immunol (2015) 15(4):231–42. doi: 10.1038/nri3806
119. Papatriantafyllou M. Regulatory T cells: distilling regulatory T cell inducers. Nat Rev Immunol (2013) 13(8):546. doi: 10.1038/nri3506
120. Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, et al. Persistent lcmv infection is controlled by blockade of type I interferon signaling. Science (2013) 340(6129):207–11. doi: 10.1126/science.1235214
121. Han Y, Liu D, Li L. Pd-1/Pd-L1 pathway: current researches in cancer. Am J Cancer Res (2020) 10(3):727–42.
122. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and tigit: Co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44(5):989–1004. doi: 10.1016/j.immuni.2016.05.001
123. Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BE, et al. Molecular and transcriptional basis of Cd4+ T cell dysfunction during chronic infection. Immunity (2014) 40(2):289–302. doi: 10.1016/j.immuni.2014.01.005
124. Zhen A, Rezek V, Youn C, Lam B, Chang N, Rick J, et al. Targeting type I interferon-mediated activation restores immune function in chronic hiv infection. J Clin Invest (2017) 127(1):260–8. doi: 10.1172/jci89488
125. Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, et al. The transcription factor Foxo1 sustains expression of the inhibitory receptor pd-1 and survival of antiviral Cd8(+) T cells during chronic infection. Immunity (2014) 41(5):802–14. doi: 10.1016/j.immuni.2014.10.013
126. Ando R, Shima H, Tamahara T, Sato Y, Watanabe-Matsui M, Kato H, et al. The transcription factor Bach2 is phosphorylated at multiple sites in murine b cells but a single site prevents its nuclear localization. J Biol Chem (2016) 291(4):1826–40. doi: 10.1074/jbc.M115.661702
127. Yao C, Lou G, Sun HW, Zhu Z, Sun Y, Chen Z, et al. Bach2 enforces the transcriptional and epigenetic programs of stem-like Cd8(+) T cells. Nat Immunol (2021) 22(3):370–80. doi: 10.1038/s41590-021-00868-7
128. Cannons JL, Villarino AV, Kapnick SM, Preite S, Shih HY, Gomez-Rodriguez J, et al. Pi3kδ coordinates transcriptional, chromatin, and metabolic changes to promote effector Cd8(+) T cells at the expense of central memory. Cell Rep (2021) 37(2):109804. doi: 10.1016/j.celrep.2021.109804
129. Pichler AC, Cannons JL, Schwartzberg PL. The road less taken: less appreciated pathways for manipulating Cd8(+) T cell exhaustion. Front Immunol (2022) 13:926714. doi: 10.3389/fimmu.2022.926714
130. Lin WH, Adams WC, Nish SA, Chen YH, Yen B, Rothman NJ, et al. Asymmetric Pi3k signaling driving developmental and regenerative cell fate bifurcation. Cell Rep (2015) 13(10):2203–18. doi: 10.1016/j.celrep.2015.10.072
131. Chen YH, Kratchmarov R, Lin WW, Rothman NJ, Yen B, Adams WC, et al. Asymmetric Pi3k activity in lymphocytes organized by a Pi3k-mediated polarity pathway. Cell Rep (2018) 22(4):860–8. doi: 10.1016/j.celrep.2017.12.087
132. Zhang L, Tschumi BO, Lopez-Mejia IC, Oberle SG, Meyer M, Samson G, et al. Mammalian target of rapamycin complex 2 controls Cd8 T cell memory differentiation in a Foxo1-dependent manner. Cell Rep (2016) 14(5):1206–17. doi: 10.1016/j.celrep.2015.12.095
133. Mousset CM, Hobo W, Ji Y, Fredrix H, De Giorgi V, Allison RD, et al. Ex vivo akt-inhibition facilitates generation of polyfunctional stem cell memory-like Cd8(+) T cells for adoptive immunotherapy. Oncoimmunology (2018) 7(10):e1488565. doi: 10.1080/2162402x.2018.1488565
134. Ando S, Perkins CM, Sajiki Y, Chastain C, Valanparambil RM, Wieland A, et al. Mtor regulates T cell exhaustion and pd-1-Targeted immunotherapy response during chronic viral infection. J Clin Invest (2023) 133(2):e160025. doi: 10.1172/jci160025
135. Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. Selective effects of pd-1 on akt and ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal (2012) 5(230):ra46. doi: 10.1126/scisignal.2002796
136. Speiser DE, Utzschneider DT, Oberle SG, Münz C, Romero P, Zehn D. T Cell differentiation in chronic infection and cancer: functional adaptation or exhaustion? Nat Rev Immunol (2014) 14(11):768–74. doi: 10.1038/nri3740
137. Frebel H, Nindl V, Schuepbach RA, Braunschweiler T, Richter K, Vogel J, et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J Exp Med (2012) 209(13):2485–99. doi: 10.1084/jem.20121015
138. Hasan Ali O, Diem S, Markert E, Jochum W, Kerl K, French LE, et al. Characterization of nivolumab-associated skin reactions in patients with metastatic non-small cell lung cancer. Oncoimmunology (2016) 5(11):e1231292. doi: 10.1080/2162402x.2016.1231292
139. Berner F, Bomze D, Diem S, Ali OH, Fässler M, Ring S, et al. Association of checkpoint inhibitor-induced toxic effects with shared cancer and tissue antigens in non-small cell lung cancer. JAMA Oncol (2019) 5(7):1043–7. doi: 10.1001/jamaoncol.2019.0402
140. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase Shp2. J Exp Med (2012) 209(6):1201–17. doi: 10.1084/jem.20112741
141. Zehn D, Utzschneider DT, Thimme R. Immune-surveillance through exhausted effector T-cells. Curr Opin Virol (2016) 16:49–54. doi: 10.1016/j.coviro.2016.01.002
142. Baitsch L, Baumgaertner P, Devêvre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific Cd8+ T cells in metastases from melanoma patients. J Clin Invest (2011) 121(6):2350–60. doi: 10.1172/jci46102
143. Zelinskyy G, Myers L, Dietze KK, Gibbert K, Roggendorf M, Liu J, et al. Virus-specific Cd8+ T cells upregulate programmed death-1 expression during acute friend retrovirus infection but are highly cytotoxic and control virus replication. J Immunol (2011) 187(7):3730–7. doi: 10.4049/jimmunol.1101612
144. Larsen M, Sauce D, Deback C, Arnaud L, Mathian A, Miyara M, et al. Exhausted cytotoxic control of Epstein-Barr virus in human lupus. PloS Pathog (2011) 7(10):e1002328. doi: 10.1371/journal.ppat.1002328
145. Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, et al. Control of viremia in simian immunodeficiency virus infection by Cd8+ lymphocytes. Science (1999) 283(5403):857–60. doi: 10.1126/science.283.5403.857
146. Jin X, Bauer DE, Tuttleton SE, Lewin S, Gettie A, Blanchard J, et al. Dramatic rise in plasma viremia after Cd8(+) T cell depletion in simian immunodeficiency virus-infected macaques. J Exp Med (1999) 189(6):991–8. doi: 10.1084/jem.189.6.991
147. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313(5795):1960–4. doi: 10.1126/science.1129139
148. Li K, Yuan Z, Lyu J, Ahn E, Davis SJ, Ahmed R, et al. Pd-1 suppresses tcr-Cd8 cooperativity during T-cell antigen recognition. Nat Commun (2021) 12(1):2746. doi: 10.1038/s41467-021-22965-9
149. Boi SK, Lan X, Youngblood B. Batf targets T cell exhaustion for termination. Nat Immunol (2021) 22(8):936–8. doi: 10.1038/s41590-021-00978-2
150. Chen Y, Zander RA, Wu X, Schauder DM, Kasmani MY, Shen J, et al. Batf regulates progenitor to cytolytic effector Cd8(+) T cell transition during chronic viral infection. Nat Immunol (2021) 22(8):996–1007. doi: 10.1038/s41590-021-00965-7
151. Huang Q, Wu X, Wang Z, Chen X, Wang L, Lu Y, et al. The primordial differentiation of tumor-specific memory Cd8(+) T cells as bona fide responders to pd-1/Pd-L1 blockade in draining lymph nodes. Cell (2022) 185(22):4049–66.e25. doi: 10.1016/j.cell.2022.09.020
Keywords: T cell exhaustion, heterogeneous, T cell factor 1, chronic TCR stimulation, TOX, PD-1, immune checkpoint blockade
Citation: Tian W, Qin G, Jia M, Li W, Cai W, Wang H, Zhao Y, Bao X, Wei W, Zhang Y and Shao Q (2023) Hierarchical transcriptional network governing heterogeneous T cell exhaustion and its implications for immune checkpoint blockade. Front. Immunol. 14:1198551. doi: 10.3389/fimmu.2023.1198551
Received: 01 April 2023; Accepted: 06 June 2023;
Published: 16 June 2023.
Edited by:
Chih-Hao Chang, Jackson Laboratory, United StatesReviewed by:
Lily Wang, Cleveland Clinic, United StatesCopyright © 2023 Tian, Qin, Jia, Li, Cai, Wang, Zhao, Bao, Wei, Zhang and Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Zhang, emhhbmd5dTAwN0Biam11LmVkdS5jbg==; Qixiang Shao, c2hhb19xeEB1anMuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.