
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 11 July 2023
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1198365
This article is part of the Research TopicThe Role of Negative Immune Checkpoints in the Treatment of Systemic Lupus Erythematosus and Rheumatoid ArthritisView all 5 articles
 Md Munnaf Hossen1,2,3†
Md Munnaf Hossen1,2,3† Yanmei Ma1,2,3†Zhihua Yin1,3
Yanmei Ma1,2,3†Zhihua Yin1,3 Yuhao Xia2,4
Yuhao Xia2,4 Jing Du4Jim Yi Huang5Jennifer Jin Huang6Linghua Zou1,7
Jing Du4Jim Yi Huang5Jennifer Jin Huang6Linghua Zou1,7 Zhizhong Ye1,3*
Zhizhong Ye1,3* Zhong Huang2,3*
Zhong Huang2,3*Autoimmune diseases (ADs) are characterized by the production of autoreactive lymphocytes, immune responses to self-antigens, and inflammation in related tissues and organs. Cytotoxic T-lymphocyte antigen 4 (CTLA-4) is majorly expressed in activated T cells and works as a critical regulator in the inflammatory response. In this review, we first describe the structure, expression, and how the signaling pathways of CTLA-4 participate in reducing effector T-cell activity and enhancing the immunomodulatory ability of regulatory T (Treg) cells to reduce immune response, maintain immune homeostasis, and maintain autoimmune silence. We then focused on the correlation between CTLA-4 and different ADs and how this molecule regulates the immune activity of the diseases and inhibits the onset, progression, and pathology of various ADs. Finally, we summarized the current progress of CTLA-4 as a therapeutic target for various ADs.
CTLA-4 is a T-cell co-receptor, also known as CD152. Compared to CD28, CTLA-4 has a superior binding affinity with B7 family molecules, including CD80 and CD86 on antigen-presenting cells (APCs). Although CTLA-4 binds with B7 co-stimulatory receptors, it plays a negative role in the activation of T cells (1). Following the T-cell receptor (TCR) recognizing the antigen presented by the major histocompatibility complex (MHC) of APC, the CD28 of T cells binds to B7 of APC, which initiates a signaling cascade and leads to T-cell activation. CTLA-4s express and move to the cell membranes after T-cell activation, take over B7 from CD28, and suppress T-cell activity (2, 3). The coordination of CTLA-4 and CD28 maintains the balance of T-cell immunity in the body, especially after infection and the onset and progression of autoimmune disease. However, the precise immune-regulating mechanism of CTLA-4 in T cells is debatable (4, 5) (Figure 1).

Figure 1 Signaling pathway of T-cell activation and CTLA-4 surface expression. Two signals are essential for T-cell activation. One is TCR-MHC signaling and another one is CD28-CD80/86 co-stimulation. After successful co-stimulation, naïve T cells become active and perform their function. After activation, T cells expresses CTLA-4 and CTLA-4 binds with CD80/86 molecules and functionally inactivated T cell (left panel). The newly synthesized CTLA-4 binds with TRIM in the TGN and comes out on the cell surface. PLD and ARF-1 are also responsible for externalization of CTLA-4. On the other side, CTLA-4 binds with CAP-1 and directs transport to lysosomal compartment. From the surface, dephosphorylated tyrosine 201 of CTLA-4 binds with clathrin adaptor protein-2 (CAP-2) and transports endosome to lysosome (right panel). APC, Antigen-presenting cell; TCR, T-cell receptor; MHC, Major histocompatibility complex; CTLA-4, Cytotoxic T lymphocyte antigen 4; CD28, TRIM, Transmembrane adaptor T-cell receptor interacting molecule; TGN, Trans Golgi network; PLD, Phospholipase D; ARF-1, GTPase ADP ribolysation factor-1; CAP-1, Clathrin adaptor protein-1; CAP-2, Clathrin adaptor protein-2.
In recent years, CTLA-4 has been shown to play a crucial role in immune checkpoint-based therapeutics, especially in cancer treatment, by using monoclonal antibodies against the molecule (6). Moreover, this regulatory molecule has been intimately involved in the treatment of autoimmune diseases (7). Elucidating the immunoregulatory mechanisms and roles of CTLA-4 in autoimmune diseases will provide potent immunotherapy targets for these diseases. In this review, we focused on the immunomodulatory role of CTLA-4 in T-cell immunity, discussed the key molecular signaling pathways mediated by CTLA-4, and summarized the latest immunoregulatory effects of CTLA-4 in various autoimmune diseases, especially its role in the progress and pathogenesis of the diseases and its clinical application in the diseases.
In humans and mice, the CTLA-4 gene consists of 4 exons encoded by chromosomes 2 and 1, respectively. Exon 1 provides the sequence for the leading peptide, while exon 2 has a CD80/CD86 binding site and a dimerization site. Exon 3 comprises a transmembrane region, while exon 4 contains the cytoplasmic tail (8). The CTLA-4 gene has distinct isoforms in humans and mice. In humans, there is a full-length CTLA-4 mRNA (flCTLA-4, containing exons 1 to 4) and a soluble cytotoxic T-lymphocyte antigen 4 (sCTLA-4) that is detectable in serum; however, it does not contain exon 3 (9, 10). The half-life of flCTLA-4 mRNA is longer than that of sCTLA-4 mRNA (11). In murine T cells, an extra CTLA-4 transcript dubbed ligand-independent CTLA-4 (liCTLA-4) is produced, which contains exons 1, 3, and 4 (12, 13).
In vitro studies reported that inhibiting NF-AT activity in T cells significantly reduced CTLA-4 transcription, suggesting that the activity of NF-AT was positively correlated with protein expression (14, 15). Upregulated CTLA-4 expression was also regulated by cyclic AMP (cAMP) (16). The mRNA of CTLA-4 was detected in T cells after 1 h of T-cell receptor (TCR) ligation and reached a peak approximately 24–36 h after T cells were activated by antigen (17, 18).
The level of CTLA-4 is closely regulated by numerous factors, such as ligand-inducing expression, cell surface translocation, fast internalization, recycling, and degradation. CTLA-4 is induced by TCR ligation and forms a complex with the T-cell receptor interacting molecule (TRIM) in the trans-Golgi network (TGN), which promotes protein transfer into the cell surface (19). Externalization of CTLA-4 is also facilitated by guanosine triphosphatases (GTPases), adenosine diphosphate ribosylation factor-1 (ARF-1), phospholipase D (PLD), calcium influx (20), and Rab11 (21). On the other hand, CTLA-4 internalization is regulated by both clathrin-dependent and clathrin-independent pathways. For the dependent pathway, the cell surface CTLA-4 associates with clathrin adaptor protein-1 (CAP-1) and clathrin adaptor protein-2 (CAP-2); for the independent pathway, the CTLA-4 binds to dynamin. After internalization, CTLA-4 is either delivered into lysosomes or endosomes (22, 23). To maintain the intracellular stable state of CTLA-4, it binds with CAP-1 in the TGN of T cells and is then transported to the lysosomal compartments for degradation (21). Phosphorylation of CTLA-4 by Lck and Fyn tyrosine kinases inhibits this interaction and blocks the trafficking of CTLA-4, thus prolonging the retention of the protein on the cell membrane and reducing degradation of the protein (24) (Figure 1).
The activation of T cells depends on the TCR binding to a specific antigen presented by the MHC of APCs. However, this recognition, the first T-cell activation signal, is not sufficient to cause T-cell activation (25, 26). To fully activate T cells, a second activation signal called co-stimulation is required. This signal is provided by a T cell’s inducible CD28 receptor, which binds to its ligand CD80 or CD86 on activated professional APCs (26). The activation of CD28 stimulates glucose absorption and cell cycle progression in T cells by increasing the expression of the anti-apoptotic proteins Bcl-X (Bcl-xL) and interleukin-2 (IL-2) to reduce apoptosis and increase proliferation of T cells (27–29). Without CD28 signaling, T cells will enter clonal anergy and apoptosis (30). When T cells receive the first and second activation signals, the cells become completely activated, and antigen-specific effector cells expand in peripheral lymphoid organs (31). If the antigen of the pathogen causes an expansion of particular T cells, they will assault contaminated cells and tissue at the site of infection. If the proliferated cells are due to self-antigen, they will move to target tissue and organs, cause inflammation, and damage the self-cell, tissue, and organ (Figure 2).
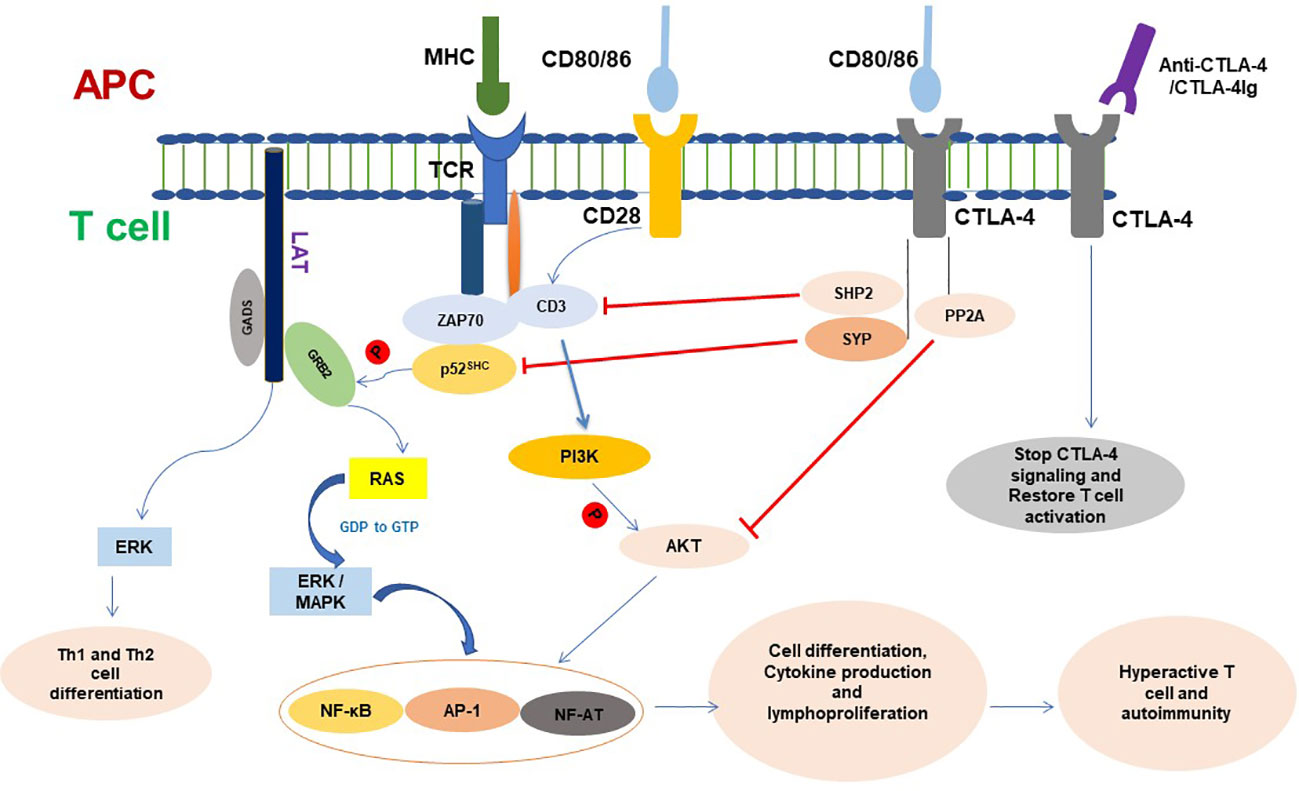
Figure 2 CTLA-4 signaling in T cell and functioning. CTLA-4 targets different molecules to inhibit T- cell activation and functioning. Once conjugated, T cell with APC initiates different signaling and T-cell proliferation and produces cytokines. CTLA-4 is expressed in cell surface of active T cells and binds with CD80/86. CTLA-4 signaling dephosphorylates TCR signaling and inhibits CD3 and ZAP70 signaling molecules and ultimately inhibits phosphorylation of GRB2 to RAS pathway. CTLA-4 inhibits Akt phosphorylation and activation and plays a negative role on the regulation of cell cycle and inhibits the transcription factors nuclear factor κB (NF-κB), AP-1, and NF-AT activation. Ligation of CTLA-4-CTLA-4Ig resumes T-cell signaling and functioning. APC, Antigen-presenting cell; AP-1, Activator protein 1; TCR, T-cell receptor; MHC, Major histocompatibility complex; CTLA-4, Cytotoxic T lymphocyte antigen 4; SYP, Tyrosine phosphatase synaptophysin; GRB2, Growth Factor Receptor-Bound 2; ZAP70, Zeta-chain associated protein kinase 70; NF-AF, Nuclear factor of activated T cells; ERK, Extracellular signal-regulated kinase; MAPK, Mitogen-activated protein kinase; PI3K, Phosphatidylinositol 3-kinase; PP2A, Protein phosphatase 2A.
CTLA-4 is expressed in activated T lymphocytes and is transported to the cell membrane. Despite the fact that CD80 and CD86 are ligands found on APCs that CTLA-4 and CD28 both share, CTLA-4 has a considerably higher affinity for these ligands than CD28; therefore, it preferentially binds to CD80/CD86 and transmits inhibitory signals to prevent CD28-mediated T-cell activation (1). On the cell membrane, CTLA-4 is phosphorylated by Fyn and Lck. The phosphorylated CTLA-4 recruits SHIP2, which dephosphorylates CD3 and linkers for activation of T cells (LAT). Thus, CTLA-4s disrupt TCR/CD3 and CD28/B7 signaling and result in downregulating T cells’ activity, immune response, and inflammatory cytokine production (32, 33), cell cycle progression, and the transcription factors nuclear factor B (NF-κB), activator protein 1 (AP-1), and NF-AT (34, 35) (Figure 2).
In regulatory T (Treg) cells, CTLA-4 is constitutively expressed and required for the cell’s immune suppressive activity (36). It interacts with CD80/CD86 on dendritic cells (DCs), which transmit the inhibitory signal to the APCs, downregulating the expression of CD80/CD86 and upregulating the expression of indoleamine-2,3-dioxygenase (IDO) in DCs, thus endowing DCs with immune tolerance properties (37). IDO released from DCs acts as an immune regulator for T cells by depleting tryptophan (38, 39). Deficiency of CTLA-4 in Treg cells impairs the immune suppressive function of the cells in vivo (40) and leads to abnormal activation and expansion of conventional T cells (41). On the other hand, CTLA-4 has been found to promote the generation of CD4+CD25+ regulatory T cells by increasing FoxP3 expression that is induced by transforming growth factor (TGF-β) (42). So far, the CTLA-4-mediated signaling in T cells and immune response in DCs still have not yet been completely elucidated; further study will not only help to explain T cells’ and DCs’ immune regulation but also help to understand the mechanism for other immune cell regulation.
The activation of TCR is initiated by binding the antigen presented by the MHC of DCs, which causes tyrosine phosphorylation of CD3. The adapter molecule p52SHC is recruited by activated CD3 and tyrosine phosphorylated by Lck. The phosphorylated p52SHC associates with SH2 and SH3 domains containing protein growth factor receptor bound protein (GRB2), and via the GRB2, with the guanine nucleotide exchange factor Son-of-Sevenless (SOS) and Ras form a complex (CD3/p52SHC/GRB2/SOS/Ras), which regulates the activity of the Ras signal (43, 44). It has been demonstrated that the tyrosine phosphatase synaptophysin (SYP) binds to the tyrosine phosphorylated YVKM motif in the cytoplasmic tail of CTLA-4. The CTLA-4-associated SYP exhibits tyrosine phosphatase activity towards p52SHC. The dephosphorylation of p52SHC interferes with the binding affinity of p52SHC to GRB2, thus disrupting the p52SHC/GRB2/SOS/Ras complex and resulting in Ras being unable to exchange GDP with GTP. This indicates that the regulatory effect of CTLA-4 on TCR-Ras signaling is via dephosphorylation of p52SHC by SYP (45) (Figure 2).
P52SHC has also been shown to be a component in C-X-C chemokine receptor type 4 (CXCR4) signaling. The binding of stromal-derived-factor-1α (SDF-1α) to CXCR4 results in activation of the receptor and association with Lck, which promotes phosphorylation of CD3 and p52SHC and assembly of the p52SHC/ZAP-70/Vav complex, implying that the TCR transactive pathway can be triggered by CXCR4 (46). On the other hand, given the tyrosine phosphatase SYP for p52SHC associated with CTLA-4, the complex of SYP/CTLA-4 may play a role in disrupting CXCR4-mediated chemotaxis and the activity of TCR signaling.
Phosphatidylinositol (3,4,5)-trisphosphates (PIP3), the product of PI3K, recruits the pleckstrin homology (PH) domain protein AKT to the membrane. The activation of AKT occurs through the phosphorylation of Thr-308 by PH domain kinase 1 (PDK1) and Ser-473 via mechanistic target of rapamycin complex 2 (mTORC2) (47, 48). The activated AKT is involved in many cellular functions such as immune regulation, cell proliferation, metabolism, survival, and anti-apoptosis by mediating the transcriptional factor activities of NF-κB, NFAT, and AP-1 (35, 49).
As early as 1995, PI3 kinase (PI3K) was reported to bind to the cytoplasmic pYVKM motif of CTLA-4, but the role of CTLA-4 in the PI3K/AKT pathway is still not fully understood (50, 51). The activation of PI3K by CTLA-4 signaling was demonstrated by its ligand stimulation increasing the phosphorylation of AKT (52). The function of CTLA-4 is closely related to that of its partners, serine/threonine phosphatase PP2A (PP2A) and tyrosine phosphatase SHIP2 (SHIP2). Phosphorylated CTLA-4 has been demonstrated to be associated with PP2A in its cytoplasmic tail (53) and SHIP2 in its pYVKM motif (54), but another study showed that CTLA-4 interaction with SHIP2 is indirect, possibly via PI3K (55). Differences with Schneider et al.’s study and Parry et al.’s study showed that CTLA-4 inhibited AKT activity through its partner PP2A, which was confirmed by the PP2A inhibitor okadaic acid or deleting its lysine-rich domain KLESS (a motif required for PP2A binding in CTLA-4) (56). This implies that the association of PP2A and its phosphatase activity is necessary for CTLA-4 to inhibit the activity of AKT. Interestingly, this inhibition preserved PI3K activity, which was further demonstrated by the PI3K inhibitor LY294002 reducing CTLA-4-induced Bcl-xL production (56). The function of SHIP2 in CTLA-4 signaling has not yet been elucidated, as their interaction pattern is still under debate, and SHIP2 has been shown to increase ERK activity (57). However, it is still possible for SHIP2 to dephosphorylate some TCR signaling proteins and reduce TCR-mediated T-cell activation (58, 59). On the other hand, Wu et al. showed that SHIP2 is required for PI3K/AKT activation via EGF, PDGF, and IGF signaling (60). AKT phosphorylates the apoptotic protein BL2 associated agonist of cell death (BAD), which releases the anti-apoptosis protein B-cell lymphoma 2 (BcL-2) or BcL-XL from the heterodimer of BAD-BcL-2 and BAD-BcL-XL (61).
Studies have shown that PI3K/AKT signaling activity enhanced by CTLA-4 plays a key role in maintaining the balance of T-cell survival, anergy, and apoptosis and in maintaining long-term immune tolerance (52). However, after combining with CD80/CD86, CTLA-4 recruits SHIP2 and PP2A to the membrane, the SHIP2 inhibits TCR signaling by dephosphorylation of CD3, as well as restrains PI3K/AKT signaling via dephosphorylation of PI3K, while PP2A directly dephosphorylates AKT to reduce its activity. The results indicate that the inhibitory activity of CTLA-4 depends on both PP2A and SHIP2, which coordinate the lower activity of NF-κB, mTOR, Bcl-xl, and the production of IL-2 in T cells (62), especially the reduced-AKT activity, which has been demonstrated to be required for the suppressive function of CD4+CD25+Foxp3+ regulatory T cells (63), and the differentiation and proliferation of natural Treg cells in the thymus are antagonized by PI3K/AKT signals via preventing FOXO factors translocating into the nucleus (50) (Figure 2). On the other hand, the study also reported that SHIP2 enhanced PI3K activity and was required for PDGF and IGF-induced AKT phosphorylation in mouse fibroblast (60). The effects of CTLA-4 on PI3K/AKT signaling are still debatable. The different results might come from different experimental conditions, cells, organs, and diseases, but nevertheless, the CTLA-4/PI3K/PKB pathway plays an important role in immune regulation, especially in the function of Treg cells.
CTLA-4 inhibits immune responses by mediating various immune-related signaling pathways. It competes with B7 and CD28 via SYP, dephosphorylates p52SHC in the CD3/p52SHC/GRB2/SOS complex to inhibit TCR signaling, and regulates TCR and the PI3K/AKT pathway via SHIP2 and PP2. Thus, CTLA-4 regulates T-cell signaling, modulates T-cell activity, maintains the balance of immunity, and protects the body from autoimmune diseases by downregulating the transcription factor activity of Fos, Jun, c-Myc, AP-1, NF-AT, and NF-κB (49), from which it protects the body from autoimmune diseases (Figure 2) (64). The mounting evidence regarding CTLA-4 provides an insight into the molecular underpinnings of T cells in their functions of immune suppression and immunological tolerance.
The mice lacking the CTLA-4 gene showed dysregulation of the T-cell immune response, which exhibited a dramatically increasing T-cell blast in the lymph nodes and spleens, and the mice died at 3 weeks of age (65). Mice with a specific deletion of CTLA-4 in Treg cells disrupted the suppressive activity of the cells, spontaneously developed lymphocyte proliferative disease, and increased CD80 and CD86 expression in DCs, suggesting that the deficiency of CTLA-4 in Treg cells may inhibit the immune function of DCs (5). Another study indicated that in the absence of CTLA-4 in mice, naïve CD4+ T cells spontaneously differentiated into T follicular helper cells (Tfh), the number of germinal centers increased, and high levels of cytokines (IL-2, IFN-γ, IL-4, and GM-CSF) were produced (66). Similar results appeared in the blockage of CTLA-4 with antibodies, which further confirms that Tfh differentiation is regulated by CTLA-4 (67). However, Paterson et al.’s study showed that CTLA-4-depleted Treg cells still had suppressive activity and were sufficient to protect mice from EAE, as well as upregulate the immune regulators IL-10, LAG-3, and PD-1 expression (68). The patients with the CTLA-4 mutation had dysfunction of FoxP3+ Treg cells, hyper-proliferation of lymphocytes, and activated effector T cells, which resulted in a large number of lymphocytes in the circulation and in lymphoid organs (69). Overall, the evidence has indicated that CTLA-4 plays an important role in controlling effector T-cell differentiation, proliferation, and apoptosis and endowing Treg cell with immune suppressive activity.
Patients with deficient CTLA-4 have lower B-cell death and BCR-induced proliferation (70). However, the study also showed that the patients with CTLA-4 mutations had lower levels of circulating B cells and were associated with hypogammaglobulinemia and lymphopenia (69, 71). In mice, CTLA-4 deficiency produced a much higher frequency of activated B cells and an increased amount of immunoglobulin in their serum. CTLA-4 deletion in both Tfh and Tfr increased B-cell responses, whereas CTLA-4 deletion in Tfr alone increased antigen-specific antibody production (72). It has been demonstrated that CTLA-4 controls the activity of follicular helper T cells (Tfh) and downregulates co-stimulatory molecules of B cells, hence suppressing B-cell activation and antibody production (72).
CTLA-4 expression was detected on activated natural killer (NK) cells, but the function of CTLA-4 in NK cells is under investigation. The patients with CTLA-4 haploidy significantly reduced degranulation activity and production of IFN-γ in NK cells (73). The patients with CTLA-4 deficiency exhibited a reduced number of NK cells and impaired the function of NK cells, including cytotoxicity and inflammatory cytokine generation; the upregulated expression of CTLA-4 in activated NK cells provided an inhibitory signal for controlling NK cell activity and cytokine generation, which were confirmed by studying CD28 or CTLA-4 gene knockout mice and showing that NK cell IFN-γ production was negatively correlated with the level of CTLA-4 and positively correlated with the level of CD28 (74).
Research has shown that CTLA-4’s extracellular domain is sufficient to exert its inhibitory effect on T cells. Recent studies have demonstrated that CTLA-4 may decrease T-cell immunity even in the absence of the whole extracellular domain and has a similar ability to inhibit T-cell activation and cytokine expression in vitro (75). Overexpression of the cytoplasmic domain of CTLA-4 (cdCTLA 4) promoted naive T-cell preferential differentiation into Foxp3+ T cells in Th17 differentiation conditions via the reduction of MAPK phosphorylation and increased nuclear localization of Smad2/3 (76). Contemporary studies also indicated that cdCTLA-4 raised the number of follicular regulatory T (Tfr) cells and lowered the number of follicular helper T (Tfh) cells and germinal center (GC) B cells in draining lymph nodes (77). It implies that not only the extracellular domain, but also the cytoplasmic domain of CTLA-4 also has immunoinhibiting activity.
To maintain the body’s immune homoeostasis and silence of autoimmunity, the immune system needs to be tightly regulated, especially the activity of T cells because they play an important role in autoimmune regulation (31). After TCRs receive the first activated signal, the binding of CD28 with B7 on activated APCs enhances TCR signaling and prevents non-responsiveness or anergy of T cells. Following the activation of T cells, CTLA-4 is motivated to move to the cell membrane and compete with CD28 to bind to CD80/86 on activated APCs. The signal of CTLA-4, directly and via DCs, inhibits T-cell activation, proliferation, and cytokine production, thus downregulating T-cell activity (2, 26).
Research has demonstrated that CTLA-4 is a critical negative regulator of autoimmune diseases (78). Polymorphisms in the CTLA-4 locus, such as CTLA-4 + 49 G/A, CT60, −1661A/G, and many more, have long been associated with autoimmunity (79–81). The heterozygous mutations of CTLA-4 in T cells have been reported to be linked to a variety of autoimmune diseases (65, 82). The animal deletion of CTLA-4 leads to unregulated proliferation and activation of CD4+T cells (83, 84). In mice, the CTLA-4 deficiency promoted the proliferation of T cells that infiltrated into targeted tissues and caused organ damage, which suggests that CTLA-4 dysfunction can induce activation of self-antigen-specific T cells (85). The functional integrity of the CD28 molecule was necessary for CTLA-4 knockout mice to cause autoimmune diseases, implying that CTLA-4 suppresses the autoimmunity caused by the CD28 signaling pathway (86). Taken together, dysfunctions of CTLA-4 in T cells can cause a breakdown of immunological self-tolerance and result in susceptibility to autoimmune diseases.
Deletion of CTLA-4 in B-1a cells led to higher production of autoantibodies, increased the number of Tfh cells and germinal centers, and promoted cell differentiation into APCs and greater self-replenishment in the mice, which caused disruption of immune homeostasis, loss of immune tolerance, and the development of autoimmune disease in the late life of the mice (87). In vitro studies reported that B cells isolated from healthy donors treated with CTLA-4Ig, a fusion protein of the extracellular domain of CTLA-4 and IgG1, inhibited Staphylococcus aureus-induced CD80/CD86 expression on B cells, especially on the surface of the cell membrane, and TNF-α and IL-6 secretion from B cells (88). Besides lymphocyte inactivation, CTLA-4Ig can also inhibit the differentiation of osteoclasts and therefore regulate osteogenesis, suggesting that CTLA-4Ig may have the function of preventing bone destruction in rheumatoid arthritis (RA) (89).
The CTLA-4 gene has previously been shown to be associated with RA, systemic lupus erythematosus (SLE), multiple sclerosis (MS), type 1 diabetes (T1D), and myasthenia gravis (MG). Patients with autoimmune diseases (e.g., RA, SLE, MS, and T1D) have lower levels of CTLA-4 mRNA and protein in their PBMC, spleen, and lymph nodes than healthy subjects (Table 1).
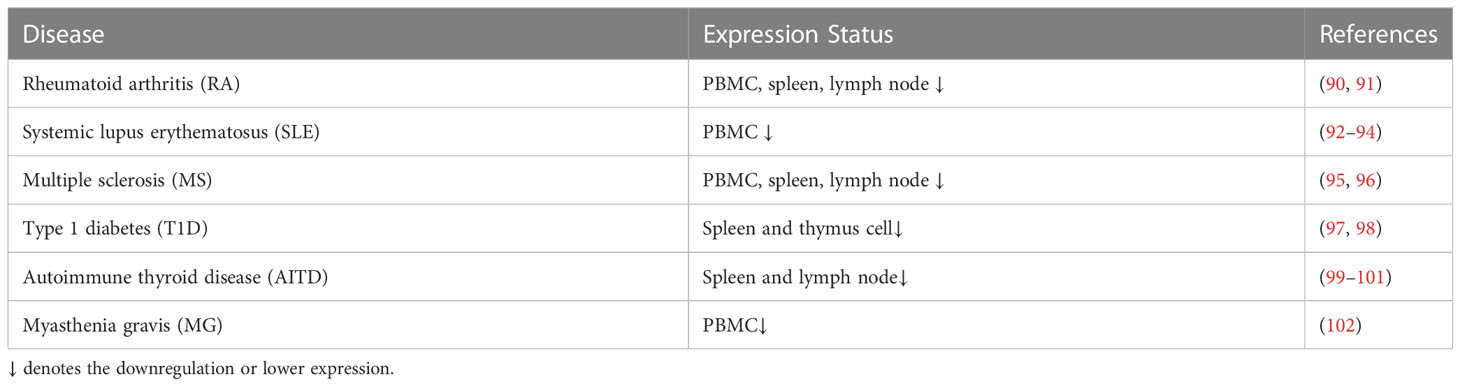
Table 1 Expression status of CTLA-4 in different autoimmune diseases.
RA is an autoimmune disease that is characterized by severe inflammation, hyperplasia of synovial lining cells, infiltration of mononuclear cells, and destruction of the articular joint. In addition, autoantibodies such as rheumatic factor (RF) and anti-citrullinated protein antibodies (ACPA) are also present in RA (103, 104).
CTLA-4 is a molecule that participates in the regulation of T-cell activity during autoimmune response, and multiple CTLA-4 single-nucleotide polymorphisms (SNPs) have been demonstrated to be closely associated with RA. Meta-analysis showed that polymorphisms of CTLA-4 (rs3087243, rs5742909, rs231775, and CTLA-4 + 49A/G) were significantly associated with the risk of RA (105–107). Klocke et al. (108) revealed that the expression of CTLA-4 by FoxP3+ regulatory T (Treg) cells attenuated the activity of disease and prevented tissue damage. They also found that overexpression of CTLA-4 in conventional T (Tcon) cells inhibited collagen-induced arthritis (CIA) by repressing the activity of T cells (Figure 3A). RA patients in the quiescent stage of the disease have lower levels of sCTLA-4 than the patients in the activating stage (90). CTLA-4 deficiency in Treg cells from RA patients significantly reduces their immune suppressing activity (91). Compared with the normal group, CTLA-4 expression in CD4+ Foxp3+ cells in rheumatoid arthritis patients was reduced, which was associated with an increased rate of CTLA-4 internalization; artificially driving CTLA-4 to the T cell surface with PMA restored the suppressive function of the cells, but this restoration can be reversed by CTLA-4 inhibition (91). The methylation of the CTLA-4 promotor’s DNA at the NF-AT binding site resulted in insufficient CTLA-4 expression in RA patients’ Treg cells, which, in turn, leads to the failure of the expression and activation of the tryptophan degrading enzyme indoleamine 2,3-dioxygenase (IDO); as a consequence, the Treg cells were unable to activate the kynurenine pathway, which exacerbates the development of the RA (109). RA clinical trials showed that the CTLA-4-Ig fusion protein Abatacept can reduce synovial inflammation and pathology by selectively modulating CD28, CD80, and CD86 co-stimulation signals in T cells (110, 111), which further confirmed that the functions of CTLA-4 in immune regulation, especially in Treg cells, play a very important role in controlling the onset and progression of RA (Figure 3A).
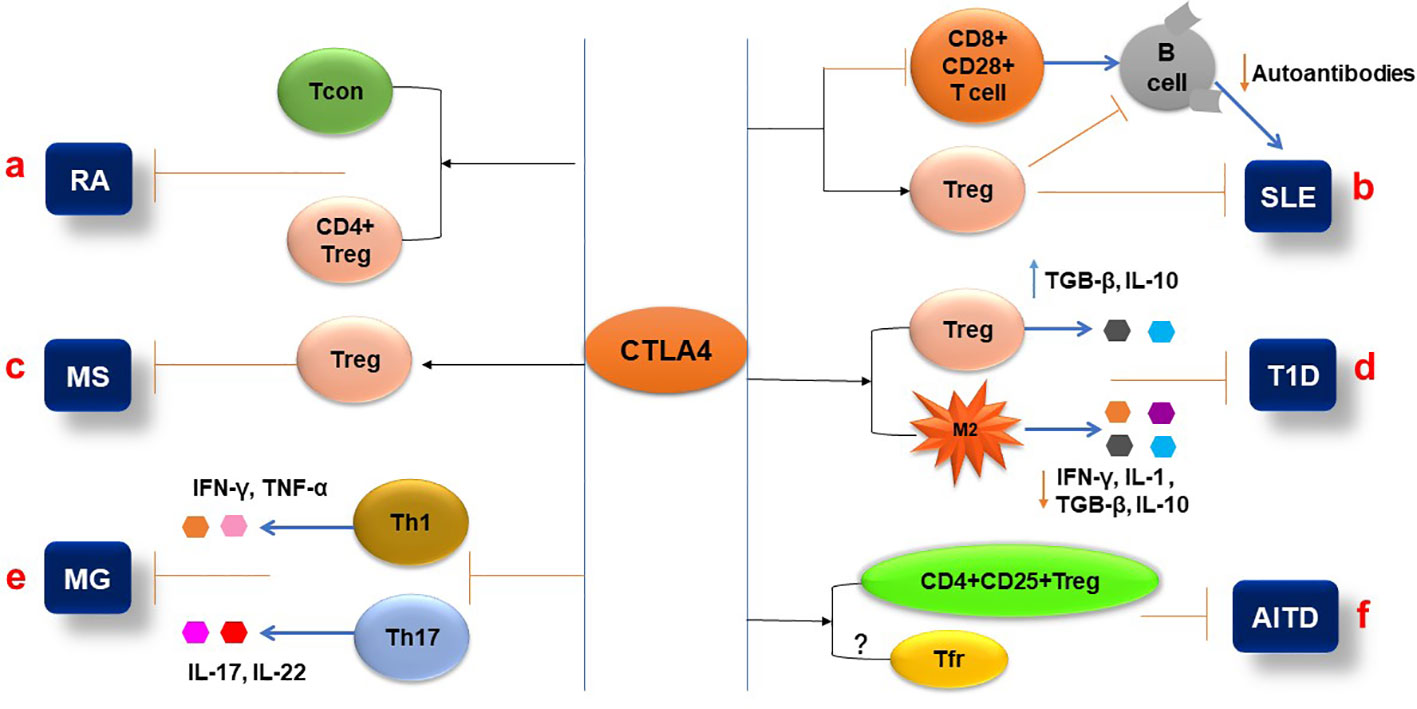
Figure 3 Pathophysiological aspects of autoimmune diseases are influenced by CTLA-4. (A) Expression of CTLA-4 in the T conventional and T regulatory cells (CD+FoxP3+) can inhibit the pathogenesis of RA. (B) CTLA-4 expression attenuates the Treg cell’s immune suppressive function and inhibits the CD8+ CD28+ T-cell functions that affect B-cell production of autoantibodies during SLE pathogenesis. (C) The CTLA4 signaling molecule can activate Treg cells and reduce MS activity. (D) CTLA-4 differentiates the CD4+CD25+ Treg cells and inhibits T1D pathogenesis by increasing the secretion of IL-10 and TGF-β. Additionally, CTLA-4 interaction with macrophages reduces pro-inflammatory cytokine (IL-1 and IFN-γ) secretion and inhibits T1D. (E) In MG, CTLA-4 expression decreases the frequency of Th1 and Th17 cells and their cytokine production. (F) In AITD, the CTLA-4 plays an inhibitory role by activating the CD4+CD25+ Treg cells and their immunosuppressive functions.
SLE is a systemic autoimmune disease resulting from autoimmune responses against nuclear autoantigens. In SLE, the body’s immune system attacks various self-tissues and damages a number of organs, including the kidney, brain, skin, and joints. Although the exact pathogeny of SLE is not clear, both genetic and environmental factors might be associated. Hyperactive B cells and the production of autoantibodies are common in SLE (112). The list of candidate genes related to SLE pathology is lengthy, especially MHC and CTLA-4. Several meta-analyses have reported that the CTLA-4 exon-1 + 49 (A/G) polymorphism is responsible for the development of SLE, especially in Asians (113, 114). Three other meta-analyses describe that CTLA-4 promoter -1722T/C, CT60A/G, and -318C/T polymorphisms also confer risk to SLE development in Asians and Iranians (115–117).
CTLA-4 is a concern for SLE-related studies due to its inhibitory role in immune responses and control of hyperactive T and B cells, the mechanism of which may be through the interaction between auto-reactive B cells and CD4+ T cells and the CD28/CD80-86/CTLA-4 axis. Simulating the effect of CTLA-4 competing with CD28 for B7, anti-CD28 was used to block CD28 signaling in NZB/NZW mice, which prevented lupus nephritis development, prolonged animal survival, reduced production of against double strand DNA (dsDNA) autoantibodies, and increased expression of IDO, receptor programmed cell-death-1 (PD-1), and ligand programmed death ligand-1 (PDL-1) (118). The CD8+CD28+ T cell subset of PBMCs from patients with active SLE expressed a lower level of CTLA-4, suggesting that the CD8+CD28+ T cells with higher activity may result from a lower expression of CTLA-4, which leads to the development of SLE (92) (Figure 3B). Further research showed that compared to the control group, the CTLA-4 levels of CD4+CD25+T cells and CD4+CD25+FoxP3+Treg cells isolated from the patients with SLE were significantly reduced and were negatively correlated with the SLE disease activity index and severity (93). Consistent with this result, a study indicated that the frequency of CTLA-4+ Treg and CD28+ Treg cells in peripheral blood mononuclear cells (PBMCs) of patients with SLE was also lower than that of healthy individuals (94). The low expression of CTLA-4 in patients with SLE leads to a decrease in the immune regulatory ability of Treg cells, an increase in autoimmune activity, and the deterioration of SLE. CTLA-4 has been reported to be essential for reducing Treg-inhibited effector T-cell proliferation, decreasing inflammatory cytokine release (119), and maturing of inducible Treg (iTreg) cells (120) (Figure 3B). Interestingly, another study showed a higher level of CTLA-4 in FOXP3- T cells from patients with SLE compared to other autoimmune diseases and healthy controls, but their study indicated that the FOXP3- T cells in SLE patients were unable to control activation and proliferation of effector T cells (121). The studies demonstrated that CD8+ CD28+ T cells secrete IL-2 and IFN-γ and stimulate B-cell proliferation and function to produce antibodies (122, 123), and that mechanism directly modulates SLE development. Further study highlighted that CD8+ T cells can modulate various cytokines, including CTLA-4, and play a role in the pathogenesis of autoimmune diseases such as SLE, MS, and T1D (124).
Substantial evidence has demonstrated that genetic changes, decreased expression, and function abnormalities of CTLA-4 increase the risk of developing SLE and contribute to the onset and progression of SLE (121, 125). The drugs targeting CTLA-4 are being investigated as potential treatments for SLE.
MS is a chronic autoimmune disease that affects the brain and central nervous system (CNS). In MS, the body’s immune cells usually attack the myelin sheath that covers nerve fibers and disrupts the connection between the brain and other parts of the body. In addition, auto-reactive T cells play a vital role in initiating the self-reactive immune response (126). The etiology of MS is unknown, but some studies suggest that either a viral infection or a direct autoimmune process is responsible for this disease. Currently, researchers are focusing on the role of inhibitory receptors in T cells, especially CTLA-4 and PD-1. It has been established that CTLA-4 is associated with MS genetically. The polymorphism of CTLA-4 has been reported to be associated with MS pathology. The G allele in the rs231775, A>G (+49 A>G) polymorphism of CTLA-4 contributes to the reduction of auto-reactive T-cell activation and leads to the development of MS (127). In contrast, two meta-analyses reported opposite results about CTLA-4 polymorphism and MS (128, 129).
Several studies have reported defective expression of various inhibitory receptors, such as CTLA-4, PD-1, and TIM-3, in MS patients. Freshly isolated PBMCs from MS patients showed lower levels of CTLA-4, PD-1, and TIM-3 than those from healthy people (95). Consistent with this finding, compared to healthy controls, lower surface and higher intracellular expression of CTLA-4 in CD4+CD25+ T cells were found in MS patients, and these were correlated with the levels of FoxP3 mRNA (96).
In experimental autoimmune encephalomyelitis (EAE) in mice, blocking CD80/CD86 molecules with CTLA-4Ig increased disease score with increased production of interleukin-17 (IL-17) and interferon-γ (IFN-γ). On the other hand, the CTLA-4Ig-treated EAE mouse model drastically reduced the number of CD4+FoxP3+ Treg cells and level of CTLA-4 compared to the untreated EAE mouse model (130). A study reported that CTLA-4 signaling peptide can induce Treg cells and inhibit the activity of MS (131) (Figure 3C). Furthermore, a cysteine-containing cell-penetrating peptide (AP)-conjugated CTLA-4 cytoplasmic domain (AP-ctCTLA-4) peptide attenuated the activity of EAE by inhibiting IL-17A expression and reducing the number of pathogenic IL-17A+GM-CSF+ CD4 T cells (132). The results suggest that CTLA-4 is a target for the treatment of MS.
T1D, once known as juvenile diabetes, is a chronic immune disorder in which the patients’ immune system destroys insulin-making pancreatic β cells, which are mediated by T cells, pro-inflammatory macrophages, and DCs (133). The underlying mechanism that causes T1D onset and progression is unknown. Genetic predisposition and environmental factors may play vital roles in this pathogenesis. A recent study reported that various genes have been associated with the pathogenesis of T1D. To date, research that shows that specific allele combinations like DRB1* and DQB1* in HLA are associated with T1D has been reported (134). Like other genes, the association between T1D and the polymorphism of CTLA-4 has been studied in several meta-analyses. One study described that the polymorphism of CTLA-4 (+49 A/G) is strongly associated with T1D in the south Indian population (134). A homogeneous combination, such as CTLA-4 + 49 GG/AA genotypes combined with HLA high risk alleles, confers a risk of T1D development than a heterogeneous gene combination. Further supporting this evidence is another meta-analysis that reported that the polymorphism of CTLA-4 + 49 G/A (rs231775) is associated with autoimmune diseases such as T1D, rheumatoid arthritis, and SLE in Asian and Caucasian populations (135). One study in Egypt revealed that the frequency of CTLA-4 polymorphism (+49 A/G) significantly increased in the T1D group than that in the control group, particularly in younger patients and female patients (136).
Regulatory T cells, including natural regulatory T cells (nTreg) and peripheral-induced regulatory T cells (iTreg), are coordinated to maintain immune homeostasis. A reduction in frequency and/or function of Treg cells is one of the main reasons for breaking the immune tolerance of the immune system to β cells and causing T1D (137, 138). The study showed that CTLA-4 plays a key role in controlling Treg cell-mediated immunological tolerance (5). According to a recent study, blocking CTLA-4 in non-obese diabetic (NOD) mice at 10 days of age induced mice to develop T1D more quickly than the control group. This result showed that CTLA-4 is essential for Treg cell differentiation and function in the NOD model (139). Wang and his colleagues described that the expression and membrane trafficking of CTLA-4 were significantly higher in Treg cells than in conventional T cells isolated from the pancreases of the DO11×RIP-mOVA diabetic mouse model, suggesting that the Treg cell CTLA-4 plays an important role in the regulation of diabetic immunity (97). In diabetic patients, Treg cells expressed lower CTLA-4 compared to the control group (98). The patients with melanoma were treated with anti-PD-1 or anti-CTLA-4, which increased part of the patients’ glycemia levels and caused T1D and type 2 diabetes (T2D) during the immunotherapy period. Based on a high level of C-reactive protein (CRP), they believe that the pathogeny of diabetes may be insulin resistance caused by inflammation (140) (Figure 3D).
The obesity-induced diabetic mouse model treated with CTLA-4Ig dramatically improved insulin sensitivity by promoting macrophage differentiation into M2 macrophages, which increased anti-inflammatory cytokine (IL-10 and TGF-β) and reduced proinflammatory cytokine (IL-1 and IFN-γ) production (141) (Figure D). One mechanism by which ethyl pyruvate (EP) reduced the incidences of streptozotocin-induced T1D was by increasing the level of CTLA-4 in CD4+CD25highFoxP3+ Treg cells, which also increased the expression of TGF-β and IL-10 (142) (Figure 3D). On the other hand, inhibition of CTLA-4 accelerated the development of T1D, such as miR-487a-3p, which promoted T1D development by suppressing CTLA-4 and FOXO3 through binding to their 3′UTR regions (143).
MG is defined as a long-term neuromuscular disease characterized by weakness and rapid fatigue of skeletal muscle. It is caused by interrupted communication between nerve and muscle cells at the neuromuscular junction (NMJ) (144). The destruction of the neuromuscular junction is due to the production a number of autoantibodies against acetylcholine receptor (AChR), muscle-specific kinase (MuSK), and LRP4 (145). Most of the autoantibodies (IgG1 and IgG3) are anti-AChR that inhibit the binding and degradation of muscle acetylcholine receptors. MuSK is a receptor tyrosine kinase that is activated by agrin and essential for NMJ formation. LRP4 is an agrin receptor that is required for agrin-induced activation of MuSK and AChR clustering. The effect of anti-MuSK is to block NMJ formation, and anti-LRP4 antibody aims to interfere with the activation of MuSK and AChR clustering. Together, these autoantibodies lead to the damage of NMJ formation, destruction of NMJ, and disruption of the signal transduction of NMJ (144, 145).
People who have rs733618, rs231775, and rs3087243*G polymorphisms in the CTLA-4 gene have increased susceptibility to MG (146). Functionally abnormal Treg cells were found in MG patients with low levels of CTLA-4 and CD25 (102). The hypermethylation at −658 and −793 CpGs of the CTLA-4 promoter has been indicated to be associated with MG by decreasing the frequency of Treg cells and CTLA-4+ Treg cells (147). The level of methylation was positively correlated with the level of anti-acetylcholine receptor (AChR) antibodies in MG patients. Th1 and Th17 CD4+ T cells and their cytokines IFN-γ and IL-17 showed that they drive anti-AChR and MuSK antibody production via B cells (148). Anti-CTLA-4 antibody increased the frequency of Th1 and Th17 cells and their cytokines IL-2, IFN-γ, and IL-17, respectively (149, 150). These imply that the abnormal CTLA-4 in Treg cells is associated with the generation of the antibodies (Figure 3E).
Autoimmune thyroid disease (AITD) is characterized by a loss of immunological tolerance for the thyroid tissue and damaged thyroid function. AITD includes Graves’ disease (GD) and Hashimoto’s thyroiditis (HT). In AITD, lymphocytic infiltration causes tissue damage and changes the function of the thyroid gland. It was shown that autoantibodies or autoreactive T cells are responsible for thyroid tissue injury or inflammation (151). The environmental factors and genetic associations are described in the multifactorial etiology of AITD.
Research has demonstrated a close relationship between CTLA-4 and AITD, including GD and HT (152). The studies demonstrated that the polymorphisms of CTLA-4 such as +49A/G and CT60, but not the -318C/T, were found to have a significant correlation with the risk of HT (153–155). According to recent studies, the +49A/G CTLA-4 polymorphism has been demonstrated to link to Down syndrome disorders in HT patients (156), correlate with antithyroid antibody production in children with HT (157), and increase susceptibility and relapse of GD (158, 159). Additionally, the results of using sodium iodide to induce AITD in NOD-H2h4 mice and of the treatment or non-treatment with anti-CTLA-4 antibody indicated that the amount of mononuclear cell infiltration in the thyroid as well as CD4+ effector T cells in the spleen and the level of thyroglobulin were significantly higher in the anti-CTLA-4-treated group than those in the control group (99). The study showed that CTLA-4 plays a key role in the immune suppressive function of naturally occurring CD4+CD25+ T (nTreg) cells that is essential for inducing immune tolerance in murine experimental Hashimoto’s thyroiditis (EHT) (100) (Figure 3F). Follicular T-helper (Tfh) cells promote the pathogenesis of AITD. The cell surface expression of CTLA-4 in T cells was higher in HT patients than in the control group; after phytohemagglutinin (PHA) stimulation for 48 h, the number of CD4 T cells expressing CTLA-4 increased in both HT patients and controls, but CTLA-4 expressed on the cell surface increased only in HT patients (101). Follicular helper T cells (Tfh) play a crucial role in the development and maintenance of lymphomatic germinal centers and provide key signals for germinal B cells to undergo somatic hypermutation, selection, and high-affinity maturation, which results in germinal B cells differentiating into plasma cells that produce high-affinity antibodies. Tfh cells have been found to facilitate the development of autoantibodies that target self-antigens in autoimmune disorders like HT (160). In patients with HT, Tfr cells are thought to inhibit the production of autoantibodies by suppressing the function of Tfh cells. This is achieved by producing anti-inflammatory cytokines such as IL-10 and TGF-β, which inhibit the proliferation and function of Tfh cells (161). In patients with GD, the number of circulated Tfh negatively correlated with serum concentrations of TSH receptor antibodies (162) (Figure 3F). Studies have demonstrated that expression of CTLA-4 in Tfr cells is essential for the immune suppressive activity of the cells, but paradoxically, Zhao et al. showed in their study that the percentage of CTLA-4 on Tfr cells was significantly reduced in patients with HT (161). APCs interact with CTLA-4 on T cells to suppress the activation and proliferation of the cells and promote the development of Treg cells (163). Therefore, it is thought that the interaction between Tfr cell CTLA-4 and APC CD28 is crucial for maintaining immunological tolerance and preventing the emergence of HT and GD. Tfh cells and B cells may become uncontrollably activated because of dysfunction or a lack of Tfr cells or CTLA-4, which leads to the production of autoantibodies and the autoimmune destruction of thyroid tissue (164, 165). This highlights the importance of Tfr cells and CTLA-4 in maintaining immune homeostasis and preventing AITD.
Although there is no reported association between CTLA-4 and sex, it is known that conditions such as SLE, AITD, RA, and MS are more prevalent in women than men. Genetic analysis of CTLA-4 (Table 2) has reported that co-stimulatory pathways are closely related to ADs. The study illustrated that deletion of CTLA-4 in adult mice leads to autoimmune disease (85). Nowadays, researchers have shown the CTLA-4 immunoglobulin (CTLA-4Ig) fusion protein has treatment effects for autoimmune diseases (Table 3). The first soluble CTLA-4Ig antibody, Abatacept, showed promising effects in RA clinical trials (166). The following improved variants of CTLA-4Ig, such as Belatacept, XPro95, and MEDI5256, especially MEDI5256, not only have 128-fold greater binding affinity to CD80 and CD86 than Abatacept but also have higher stability and longer pharmacokinetics (167, 168).
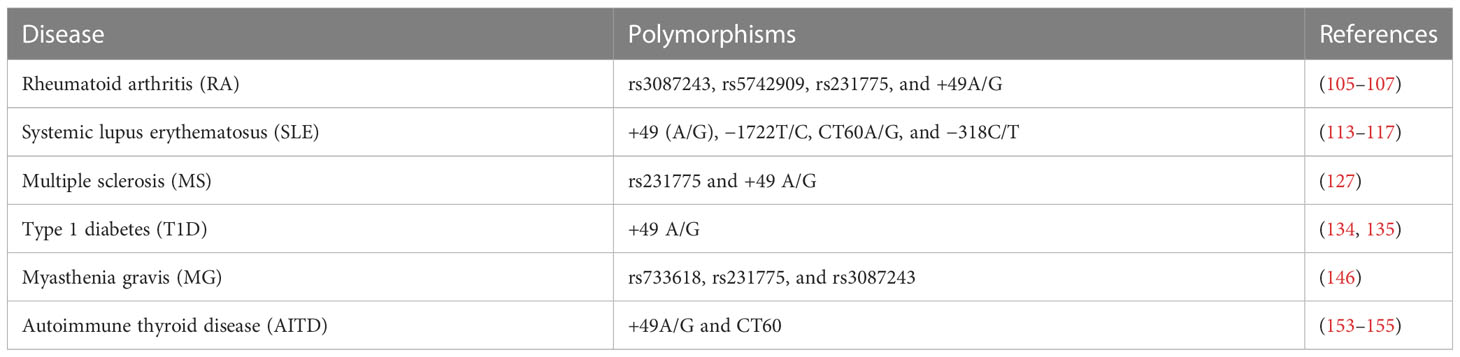
Table 2 The list of polymorphisms associated with ADs.

Table 3 Role of anti-CTLA-4/CTLA-4Ig in different autoimmune diseases.
Recently, abatacept (CTLA-4Ig) has become a new approach for RA immunotherapy (169). Treatment with abatacept (CTLA-4Ig) for rheumatoid arthritis considerably decreased disease severity by preventing T-cell proliferation, lowering the level of pro-inflammatory cytokines, and perhaps decreasing the quantity of autoreactive T cells (170–172). The study reported that CTLA-4Ig treatment reduces B-cell activity and also enhances the inhibitory capacity of Treg cells in RA patients (173, 174). The experiments suggested that CTLA-4Ig may govern humoral responses by interfering with the interaction between CD28 of T cells and CD80/CD86 of B cells, thus blocking the CD80/CD86 signal in B cells (88). Contradictorily, studies have shown that CTLA-4-Ig significantly increases the proportion of CD4+T and Treg cells by reducing the level of CD95 in the cells (175), but further analysis showed that Treg cell suppressive capacity and responsive T-cell proliferation ability were weakened in RA patients (176). There was a decrease in myelin basic protein proliferation and IFN-γ secretion in patients with MS treated with CTLA-4Ig (177). On the other hand, the study indicated that the patients who received anti-CTLA-4 antibody (ipilimumab) treatment showed clinical episodes of MS, which further confirms that CTLA-4 is a treatment target for MS (178). In individuals with T1D, treatment with Abatacept had a favorable safety profile (179). During the 2-year period of taking Abatacept, T1D patients continued to slow β cell damage and functional decline as well as maintain a low level of HbA1c, and these effects persisted for at least a year after the antibody cessation (180). In patients with newly diagnosed T1D, if they had been treated with Abatacept, the decreased rate of C-peptide significantly slowed down (181).
So far, AITD (182), SLE (183), diffuse cutaneous systemic sclerosis (184), MG (185), celiac disease (186), and allergic asthma (187) do not have the results of clinical trials for CTLA-4-Ig, but the clinical study indicated that a higher level of sCTLA-4 was found in the serum of the above autoimmune disease patients. In addition, studies showed that sCTLA-4 reduced the levels of proinflammatory cytokines such as IFN-γ, IL-2, IL-7, and IL-13 and increased the production of anti-inflammatory cytokines TGF-β and IL-10. These results imply that CTLA-4 can be a potential target for treatment of these autoimmune diseases.
Tissue damage or organ malfunction can result from the body’s immune system attacking its own self-antigens. Based on the T cell being located at the center of immune regulation, the functions and activities of Treg cells play a prominent role in the maintenance of body immune homeostasis. CTLA-4 and CD28 are two critical molecules that share a common ligand, CD80/CD86, on APCs required for T-cell regulation. CTLA-4 has the opposite effect of CD28 on T-cell immunity through competitive binding of CD80/CD86, thus blocking the second activating signal for T-cell activation and resulting in anergy and clonal tolerance of T cells, which also block DC differentiation and become immune tolerogenic cells. More and more data have suggested that CTLA-4 is crucial in the onset and progression of autoimmune diseases, and this is confirmed by the effects of CTLA-4Ig in the treatment of autoimmune diseases. However, it is worth noting that the mechanism by which CTLA-4 regulates the immunity of Treg cells, B cells, NK cells, DC, and macrophages needs to be further studied, as well as the effects of this molecule on endothelial, epithelial, and fibroblast immunoregulation, and its roles in the treatment of various autoimmune diseases. It is reasonable to believe that through the study of the CTLA-4 immune regulation signaling pathways and effects on various autoimmune diseases, there will be benefits for the treatment of autoimmune diseases.
MH and YM reviewed the literature, generated figures, and wrote the paper. ZHY, YX, and JD reviewed the literature, revised the paper, and offered feedback on the draft manuscript. JYH, JJH, and LZ reviewed the literature. ZZY and ZH designed the concept of the work, reviewed the literature, and wrote and edited the paper. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the Shenzhen Science and Technology Basic Research Project JCYJ20190808172005595 and JCYJ20190809151205630, Shenzhen Science and Technology Project JCYJ20180504170414637, Shenzhen Futian Public Welfare Scientific Research Project FTWS2021006 and FTWS2022021, and Sanming Project of Medicine in Shenzhen SZSM201602087.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Oosterwegel MA, Greenwald RJ, Mandelbrot DA, Lorsbach RB, Sharpe AH. CTLA-4 and T cell activation. Curr Opin Immunol (1999) 11(3):294–300. doi: 10.1016/S0952-7915(99)80047-8
2. Alegre ML, Frauwirth KA, Thompson CB. T-Cell regulation by CD28 and CTLA-4. Nat Rev Immunol (2001) 1(3):220–8. doi: 10.1038/35105024
3. Berg M, Zavazava N. Regulation of CD28 expression on CD8+ T cells by CTLA-4. J Leukoc Biol (2008) 83(4):853–63. doi: 10.1189/jlb.0107065
4. Walker LS. Treg and CTLA-4: two intertwining pathways to immune tolerance. J Autoimmun (2013) 45(100):49–57. doi: 10.1016/j.jaut.2013.06.006
5. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322(5899):271–5. doi: 10.1126/science.1160062
6. Abrams SI. Role of anti-CTLA-4 therapies in the treatment of cancer. Curr Opin Mol Ther (2004) 6(1):71–7.
7. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: a moving target in immunotherapy. Blood (2018) 131(1):58–67. doi: 10.1182/blood-2017-06-741033
8. Ling V, Wu PW, Finnerty HF, Sharpe AH, Gray GS, Collins M. Complete sequence determination of the mouse and human CTLA4 gene loci: cross-species DNA sequence similarity beyond exon borders. Genomics (1999) 60(3):341–55. doi: 10.1006/geno.1999.5930
9. Oaks MK, Hallett KM, Penwell RT, Stauber EC, Warren SJ, Tector AJ. A native soluble form of CTLA-4. Cell Immunol (2000) 201(2):144–53. doi: 10.1006/cimm.2000.1649
10. Iida T, Ohno H, Nakaseko C, Sakuma M, Takeda-Ezaki M, Arase H, et al. Regulation of cell surface expression of CTLA-4 by secretion of CTLA-4-containing lysosomes upon activation of CD4+ T cells. J Immunol (2000) 165(9):5062–8. doi: 10.4049/jimmunol.165.9.5062
11. Wang XB, Zheng CY, Giscombe R, Lefvert AK. Regulation of surface and intracellular expression of CTLA-4 on human peripheral T cells. Scand J Immunol (2001) 54(5):453–8. doi: 10.1046/j.1365-3083.2001.00985.x
12. Bednarczuk T, Hiromatsu Y, Fukutani T, Jazdzewski K, Miskiewicz P, Osikowska M, et al. Association of cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) gene polymorphism and non-genetic factors with graves' ophthalmopathy in European and Japanese populations. Eur J Endocrinol (2003) 148(1):13–8. doi: 10.1530/eje.0.1480013
13. Vijayakrishnan L, Slavik JM, Illés Z, Greenwald RJ, Rainbow D, Greve B, et al. An autoimmune disease-associated CTLA-4 splice variant lacking the B7 binding domain signals negatively in T cells. Immunity (2004) 20(5):563–75. doi: 10.1016/S1074-7613(04)00110-4
14. Gibson HM, Hedgcock CJ, Aufiero BM, Wilson AJ, Hafner MS, Tsokos GC, et al. Induction of the CTLA-4 gene in human lymphocytes is dependent on NFAT binding the proximal promoter. J Immunol (2007) 179(6):3831–40. doi: 10.4049/jimmunol.179.6.3831
15. Finn PW, He H, Wang Y, Wang Z, Guan G, Listman J, et al. Synergistic induction of CTLA-4 expression by costimulation with TCR plus CD28 signals mediated by increased transcription and messenger ribonucleic acid stability. J Immunol (1997) 158(9):4074–81. doi: 10.4049/jimmunol.158.9.4074
16. Teft WA, Kirchhof MG, Madrenas J. A molecular perspective of CTLA-4 function. Annu Rev Immunol (2006) 24:65–97. doi: 10.1146/annurev.immunol.24.021605.090535
17. Perkins D, Wang Z, Donovan C, He H, Mark D, Guan G, et al. Regulation of CTLA-4 expression during T cell activation. J Immunol (1996) 156(11):4154–9. doi: 10.4049/jimmunol.156.11.4154
18. Lindsten T, Lee KP, Harris ES, Petryniak B, Craighead N, Reynolds PJ, et al. Characterization of CTLA-4 structure and expression on human T cells. J Immunol (1993) 151(7):3489–99. doi: 10.4049/jimmunol.151.7.3489
19. Valk E, Leung R, Kang H, Kaneko K, Rudd CE, Schneider H. T Cell receptor-interacting molecule acts as a chaperone to modulate surface expression of the CTLA-4 coreceptor. Immunity (2006) 25(5):807–21. doi: 10.1016/j.immuni.2006.08.024
20. Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev (2009) 229(1):12–26. doi: 10.1111/j.1600-065X.2009.00770.x
21. Janman D, Hinze C, Kennedy A, Halliday N, Waters E, Williams C, et al. Regulation of CTLA-4 recycling by LRBA and Rab11. Immunology (2021) 164(1):106–19. doi: 10.1111/imm.13343
22. Schneider H, Martin M, Agarraberes FA, Yin L, Rapoport I, Kirchhausen T, et al. Cytolytic T lymphocyte-associated antigen-4 and the TCR zeta/CD3 complex, but not CD28, interact with clathrin adaptor complexes AP-1 and AP-2. J Immunol (1999) 163(4):1868–79. doi: 10.4049/jimmunol.163.4.1868
23. Qureshi OS, Kaur S, Hou TZ, Jeffery LE, Poulter NS, Briggs Z, et al. Constitutive clathrin-mediated endocytosis of CTLA-4 persists during T cell activation. J Biol Chem (2012) 287(12):9429–40. doi: 10.1074/jbc.M111.304329
24. Schneider H, Rudd CE. Diverse mechanisms regulate the surface expression of immunotherapeutic target ctla-4. Front Immunol (2014) 5:619. doi: 10.3389/fimmu.2014.00619
25. Smith-Garvin JE, Koretzky GA, Jordan MS. T Cell activation. Annu Rev Immunol (2009) 27:591–619. doi: 10.1146/annurev.immunol.021908.132706
26. Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 costimulation: from mechanism to therapy. Immunity (2016) 44(5):973–88. doi: 10.1016/j.immuni.2016.04.020
27. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16(6):769–77. doi: 10.1016/S1074-7613(02)00323-0
28. Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, et al. CD28 costimulation can promote T cell survival by enhancing the expression of bcl-XL. Immunity (1995) 3(1):87–98. doi: 10.1016/1074-7613(95)90161-2
29. June CH, Ledbetter JA, Gillespie MM, Lindsten T, Thompson CB. T-Cell proliferation involving the CD28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Mol Cell Biol (1987) 7(12):4472–81.
30. Schwartz RH. T Cell anergy. Annu Rev Immunol (2003) 21:305–34. doi: 10.1146/annurev.immunol.21.120601.141110
31. Rabb H. The T cell as a bridge between innate and adaptive immune systems: implications for the kidney. Kidney Int (2002) 61(6):1935–46. doi: 10.1046/j.1523-1755.2002.00378.x
32. Thompson CB, Allison JP. The emerging role of CTLA-4 as an immune attenuator. Immunity (1997) 7(4):445–50. doi: 10.1016/S1074-7613(00)80366-0
33. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity (1994) 1(5):405–13. doi: 10.1016/1074-7613(94)90071-X
34. Olsson C, Riesbeck K, Dohlsten M, Michaëlsson E. CTLA-4 ligation suppresses CD28-induced NF-kappaB and AP-1 activity in mouse T cell blasts. J Biol Chem (1999) 274(20):14400–5. doi: 10.1074/jbc.274.20.14400
35. Harlin H, Hwang KW, Palucki DA, Kim O, Thompson CB, Boothby M, et al. CTLA-4 engagement regulates NF-kappaB activation in vivo. Eur J Immunol (2002) 32(8):2095–104. doi: 10.1002/1521-4141(200208)32:8<2095::AID-IMMU2095>3.0.CO;2-E
36. Walker LSK. EFIS lecture: understanding the CTLA-4 checkpoint in the maintenance of immune homeostasis. Immunol Lett (2017) 184:43–50. doi: 10.1016/j.imlet.2017.02.007
37. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol (2004) 4(10):762–74. doi: 10.1038/nri1457
38. Boasso A, Herbeuval JP, Hardy AW, Winkler C, Shearer GM. Regulation of indoleamine 2,3-dioxygenase and tryptophanyl-tRNA-synthetase by CTLA-4-Fc in human CD4+ T cells. Blood (2005) 105(4):1574–81. doi: 10.1182/blood-2004-06-2089
39. Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol (2003) 4(12):1206–12. doi: 10.1038/ni1003
40. Read S, Greenwald R, Izcue A, Robinson N, Mandelbrot D, Francisco L, et al. Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. J Immunol (2006) 177(7):4376–83. doi: 10.4049/jimmunol.177.7.4376
41. Jain N, Nguyen H, Chambers C, Kang J. Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc Natl Acad Sci U.S.A. (2010) 107(4):1524–8. doi: 10.1073/pnas.0910341107
42. Zheng SG, Wang JH, Stohl W, Kim KS, Gray JD, Horwitz DA. TGF-beta requires CTLA-4 early after T cell activation to induce FoxP3 and generate adaptive CD4+CD25+ regulatory cells. J Immunol (2006) 176(6):3321–9. doi: 10.4049/jimmunol.176.6.3321
43. Patrussi L, Savino MT, Pellegrini M, Paccani SR, Migliaccio E, Plyte S, et al. Cooperation and selectivity of the two Grb2 binding sites of p52Shc in T-cell antigen receptor signaling to ras family GTPases and myc-dependent survival. Oncogene (2005) 24(13):2218–28. doi: 10.1038/sj.onc.1208384
44. Plyte S, Majolini MB, Pacini S, Scarpini F, Bianchini C, Lanfrancone L, et al. Constitutive activation of the Ras/MAP kinase pathway and enhanced TCR signaling by targeting the shc adaptor to membrane rafts. Oncogene (2000) 19(12):1529–37. doi: 10.1038/sj.onc.1203451
45. Marengère LE, Waterhouse P, Duncan GS, Mittrücker HW, Feng GS, Mak TW. Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science (1996) 272(5265):1170–3. doi: 10.1126/science.272.5265.1170
46. Patrussi L, Ulivieri C, Lucherini OM, Paccani SR, Gamberucci A, Lanfrancone L, et al. p52Shc is required for CXCR4-dependent signaling and chemotaxis in T cells. Blood (2007) 110(6):1730–8. doi: 10.1182/blood-2007-01-068411
47. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase balpha. Curr Biol (1997) 7(4):261–9. doi: 10.1016/S0960-9822(06)00122-9
48. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science (2005) 307(5712):1098–101. doi: 10.1126/science.1106148
49. Fraser JH, Rincón M, McCoy KD, Le Gros G. CTLA4 ligation attenuates AP-1, NFAT and NF-kappaB activity in activated T cells. Eur J Immunol (1999) 29(3):838–44. doi: 10.1002/(SICI)1521-4141(199903)29:03<838::AID-IMMU838>3.0.CO;2-P
50. Pompura SL, Dominguez-Villar M. The PI3K/AKT signaling pathway in regulatory T-cell development, stability, and function. J Leukoc Biol (2018). doi: 10.1002/JLB.2MIR0817-349R
51. Schneider H, Prasad KV, Shoelson SE, Rudd CE. CTLA-4 binding to the lipid kinase phosphatidylinositol 3-kinase in T cells. J Exp Med (1995) 181(1):351–5. doi: 10.1084/jem.181.1.351
52. Schneider H, Valk E, Leung R, Rudd CE. CTLA-4 activation of phosphatidylinositol 3-kinase (PI 3-K) and protein kinase b (PKB/AKT) sustains T-cell anergy without cell death. PloS One (2008) 3(12):e3842. doi: 10.1371/journal.pone.0003842
53. Teft WA, Chau TA, Madrenas J. Structure-function analysis of the CTLA-4 interaction with PP2A. BMC Immunol (2009) 10:23. doi: 10.1186/1471-2172-10-23
54. Miyatake S, Nakaseko C, Umemori H, Yamamoto T, Saito T. Src family tyrosine kinases associate with and phosphorylate CTLA-4 (CD152). Biochem Biophys Res Commun (1998) 249(2):444–8. doi: 10.1006/bbrc.1998.9191
55. Schneider H, Rudd CE. Tyrosine phosphatase SHP-2 binding to CTLA-4: absence of direct YVKM/YFIP motif recognition. Biochem Biophys Res Commun (2000) 269(1):279–83. doi: 10.1006/bbrc.2000.2234
56. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25(21):9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
57. Frearson JA, Alexander DR. The phosphotyrosine phosphatase SHP-2 participates in a multimeric signaling complex and regulates T cell receptor (TCR) coupling to the ras/mitogen-activated protein kinase (MAPK) pathway in jurkat T cells. J Exp Med (1998) 187(9):1417–26. doi: 10.1084/jem.187.9.1417
58. Rudd CE. The reverse stop-signal model for CTLA4 function. Nat Rev Immunol (2008) 8(2):153–60. doi: 10.1038/nri2253
59. Lee KM, Chuang E, Griffin M, Khattri R, Hong DK, Zhang W, et al. Molecular basis of T cell inactivation by CTLA-4. Science (1998) 282(5397):2263–6. doi: 10.1126/science.282.5397.2263
60. Wu CJ, O'Rourke DM, Feng GS, Johnson GR, Wang Q, Greene MI. The tyrosine phosphatase SHP-2 is required for mediating phosphatidylinositol 3-kinase/Akt activation by growth factors. Oncogene (2001) 20(42):6018–25. doi: 10.1038/sj.onc.1204699
61. Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell (1997) 91(2):231–41. doi: 10.1016/S0092-8674(00)80405-5
62. Nirschl CJ, Drake CG. Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clin Cancer Res (2013) 19(18):4917–24. doi: 10.1158/1078-0432.CCR-12-1972
63. Crellin NK, Garcia RV, Levings MK. Altered activation of AKT is required for the suppressive function of human CD4+CD25+ T regulatory cells. Blood (2007) 109(5):2014–22. doi: 10.1182/blood-2006-07-035279
64. Schneider H, Downey J, Smith A, Zinselmeyer BH, Rush C, Brewer JM, et al. Reversal of the TCR stop signal by CTLA-4. Science (2006) 313(5795):1972–5. doi: 10.1126/science.1131078
65. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in ctla-4. Science (1995) 270(5238):985–8. doi: 10.1126/science.270.5238.985
66. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3(5):541–7. doi: 10.1016/1074-7613(95)90125-6
67. Wang CJ, Heuts F, Ovcinnikovs V, Wardzinski L, Bowers C, Schmidt EM, et al. CTLA-4 controls follicular helper T-cell differentiation by regulating the strength of CD28 engagement. Proc Natl Acad Sci U.S.A. (2015) 112(2):524–9. doi: 10.1073/pnas.1414576112
68. Paterson AM, Lovitch SB, Sage PT, Juneja VR, Lee Y, Trombley JD, et al. Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J Exp Med (2015) 212(10):1603–21. doi: 10.1084/jem.20141030
69. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science (2014) 345(6204):1623–7. doi: 10.1126/science.1255904
70. Sic H, Speletas M, Cornacchione V, Seidl M, Beibel M, Linghu B, et al. An activating janus kinase-3 mutation is associated with cytotoxic T lymphocyte antigen-4-Dependent immune dysregulation syndrome. Front Immunol (2017) 8:1824. doi: 10.3389/fimmu.2017.01824
71. Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol (2018) 142(6):1932–46. doi: 10.1016/j.jaci.2018.02.055
72. Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor CTLA-4 controls b cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity (2014) 41(6):1026–39. doi: 10.1016/j.immuni.2014.12.005
73. Lougaris V, Tabellini G, Baronio M, Patrizi O, Gazzurelli L, Mitsuiki N, et al. CTLA-4 regulates human natural killer cell effector functions. Clin Immunol (2018) 194:43–5. doi: 10.1016/j.clim.2018.06.010
74. Stojanovic A, Fiegler N, Brunner-Weinzierl M, Cerwenka A. CTLA-4 is expressed by activated mouse NK cells and inhibits NK cell IFN-γ production in response to mature dendritic cells. J Immunol (2014) 192(9):4184–91. doi: 10.4049/jimmunol.1302091
75. Chikuma S, Abbas AK, Bluestone JA. B7-independent inhibition of T cells by CTLA-4. J Immunol (2005) 175(1):177–81. doi: 10.4049/jimmunol.175.1.177
76. Kim G-R, Lim S, Lee J-U, Choi J-M. The cytoplasmic domain of CTLA-4 control autoimmunity via inducing regulatory T cells. J Immunol (2019) 202(1 Supplement):193.4. doi: 10.4049/jimmunol.202.Supp.193.4
77. Lim S, Lee H-G, Kim G-R, Choi J-M. The cytoplasmic domain of CTLA-4 inhibits effector T cell responses and autoimmunity via increasing regulatory T cells in mice. J Immunol (2018) 200(1 Supplement):110.17. doi: 10.4049/jimmunol.200.Supp.110.17
78. Karandikar NJ, Vanderlugt CL, Walunas TL, Miller SD, Bluestone JA. CTLA-4: a negative regulator of autoimmune disease. J Exp Med (1996) 184(2):783–8. doi: 10.1084/jem.184.2.783
79. Butty V, Roy M, Sabeti P, Besse W, Benoist C, Mathis D. Signatures of strong population differentiation shape extended haplotypes across the human CD28, CTLA4, and ICOS costimulatory genes. Proc Natl Acad Sci U.S.A. (2007) 104(2):570–5. doi: 10.1073/pnas.0610124104
80. van Belzen MJ, Mulder CJ, Zhernakova A, Pearson PL, Houwen RH, Wijmenga C, et al. CTLA4 +49 A/G and CT60 polymorphisms in Dutch coeliac disease patients. Eur J Hum Genet (2004) 12(9):782–5. doi: 10.1038/sj.ejhg.5201165
81. Fang W, Zhang Z, Zhang J, Cai Z, Zeng H, Chen M, et al. Association of the CTLA4 gene CT60/rs3087243 single-nucleotide polymorphisms with graves' disease. BioMed Rep (2015) 3(5):691–6. doi: 10.3892/br.2015.493
82. Verma N, Burns SO, Walker LSK, Sansom DM. Immune deficiency and autoimmunity in patients with CTLA-4 (CD152) mutations. Clin Exp Immunol (2017) 190(1):1–7. doi: 10.1111/cei.12997
83. Gozalo-Sanmillan S, McNally JM, Lin MY, Chambers CA, Berg LJ. Cutting edge: two distinct mechanisms lead to impaired T cell homeostasis in janus kinase 3- and CTLA-4-deficient mice. J Immunol (2001) 166(2):727–30. doi: 10.4049/jimmunol.166.2.727
84. Chambers CA, Sullivan TJ, Allison JP. Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity (1997) 7(6):885–95. doi: 10.1016/S1074-7613(00)80406-9
85. Klocke K, Sakaguchi S, Holmdahl R, Wing K. Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood. Proc Natl Acad Sci U.S.A. (2016) 113(17):E2383–92. doi: 10.1073/pnas.1603892113
86. Tai X, Van Laethem F, Sharpe AH, Singer A. Induction of autoimmune disease in CTLA-4-/- mice depends on a specific CD28 motif that is required for in vivo costimulation. Proc Natl Acad Sci U.S.A. (2007) 104(34):13756–61. doi: 10.1073/pnas.0706509104
87. Yang Y, Li X, Ma Z, Wang C, Yang Q, Byrne-Steele M, et al. CTLA-4 expression by b-1a b cells is essential for immune tolerance. Nat Commun (2021) 12(1):525. doi: 10.1038/s41467-020-20874-x
88. Liu PC, Ssu CT, Tsao YP, Liou TL, Tsai CY, Chou CT, et al. Cytotoxic T lymphocyte-associated antigen-4-Ig (CTLA-4-Ig) suppresses staphylococcus aureus-induced CD80, CD86, and pro-inflammatory cytokine expression in human b cells. Arthritis Res Ther (2020) 22(1):64. doi: 10.1186/s13075-020-2138-x
89. Iwamoto N, Kawakami A. [The regulation of CTLA4-ig in bone and cartilage destruction of rheumatoid arthritis]. Clin Calcium (2015) 25(12):1817–23.
90. García-Chagollán M, Ledezma-Lozano IY, Hernández-Bello J, Sánchez-Hernández PE, Gutiérrez-Ureña SR, Muñoz-Valle JF. Expression patterns of CD28 and CTLA-4 in early, chronic, and untreated rheumatoid arthritis. J Clin Lab Anal (2020) 34(5):e23188.
91. Flores-Borja F, Jury EC, Mauri C, Ehrenstein MR. Defects in CTLA-4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc Natl Acad Sci U.S.A. (2008) 105(49):19396–401. doi: 10.1073/pnas.0806855105
92. Minning S, Xiaofan Y, Anqi X, Bingjie G, Dinglei S, Mingshun Z, et al. Imbalance between CD8(+)CD28(+) and CD8(+)CD28(-) T-cell subsets and its clinical significance in patients with systemic lupus erythematosus. Lupus (2019) 28(10):1214–23. doi: 10.1177/0961203319867130
93. Zhao L, Zhou X, Zhou X, Wang H, Gu L, Ke Y, et al. Low expressions of PD-L1 and CTLA-4 by induced CD4(+)CD25(+) Foxp3(+) tregs in patients with SLE and their correlation with the disease activity. Cytokine (2020) 133:155119. doi: 10.1016/j.cyto.2020.155119
94. Mesquita D Jr., Cruvinel WM, Araujo JA, Salmazi KC, Kallas EG, Andrade LE. Imbalanced expression of functional surface molecules in regulatory and effector T cells in systemic lupus erythematosus. Braz J Med Biol Res (2014) 47(8):662–9. doi: 10.1590/1414-431x20143483
95. Mohammadzadeh A, Rad IA, Ahmadi-Salmasi B. CTLA-4, PD-1 and TIM-3 expression predominantly downregulated in MS patients. J Neuroimmunol (2018) 323:105–8. doi: 10.1016/j.jneuroim.2018.08.004
96. Sellebjerg F, et al. FOXP3, CBLB and ITCH gene expression and cytotoxic T lymphocyte antigen 4 expression on CD4(+) CD25(high) T cells in multiple sclerosis. Clin Exp Immunol (2012) 170(2):149–55. doi: 10.1111/j.1365-2249.2012.04654.x
97. Wang CJ, et al. Immune regulation by CTLA-4–relevance to autoimmune diabetes in a transgenic mouse model. Diabetes Metab Res Rev (2011) 27(8):946–50. doi: 10.1002/dmrr.1277
98. Zóka A, et al. Extension of the CD4+Foxp3+CD25–/low regulatory T-cell subpopulation in type 1 diabetes mellitus. Autoimmunity (2015) 48(5):289–97. doi: 10.3109/08916934.2014.992518
99. Sharma R, Di Dalmazi G, Caturegli P. Exacerbation of autoimmune thyroiditis by CTLA-4 blockade: a role for IFNγ-induced indoleamine 2, 3-dioxygenase. Thyroid (2016) 26(8):1117–24. doi: 10.1089/thy.2016.0092
100. Morris GP, Brown NK, Kong YC. Naturally-existing CD4(+)CD25(+)Foxp3(+) regulatory T cells are required for tolerance to experimental autoimmune thyroiditis induced by either exogenous or endogenous autoantigen. J Autoimmun (2009) 33(1):68–76. doi: 10.1016/j.jaut.2009.03.010
101. Kucharska AM, Gorska E, Wasik M, Demkow U. Expression of cytotoxic T lymphocyte antigen-4 in T cells from children with hashimoto's thyroiditis. Adv Exp Med Biol (2013) 756:163–8. doi: 10.1007/978-94-007-4549-0_21
102. Danikowski KM, Jayaraman S, Prabhakar BS. Regulatory T cells in multiple sclerosis and myasthenia gravis. J Neuroinflamm (2017) 14(1):117. doi: 10.1186/s12974-017-0892-8
103. Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet (2010) 376(9746):1094–108. doi: 10.1016/S0140-6736(10)60826-4
104. Wasserman AM. Diagnosis and management of rheumatoid arthritis. Am Fam Physician (2011) 84(11):1245–52.
105. Mousavi MJ, Shayesteh MRH, Jamalzehi S, Alimohammadi R, Rahimi A, Aslani S, et al. Association of the genetic polymorphisms in inhibiting and activating molecules of immune system with rheumatoid arthritis: a systematic review and meta-analysis. J Res Med Sci (2021) 26:22. doi: 10.4103/jrms.JRMS_567_20
106. Zhou C, Gao S, Yuan X, Shu Z, Li S, Sun X, et al. Association between CTLA-4 gene polymorphism and risk of rheumatoid arthritis: a meta-analysis. Aging (Albany NY) (2021) 13(15):19397–414. doi: 10.18632/aging.203349
107. Lee YH, Bae SC, Choi SJ, Ji JD, Song GG. Association between the CTLA-4 +49 A/G polymorphism and susceptibility to rheumatoid arthritis: a meta-analysis. Mol Biol Rep (2012) 39(5):5599–605. doi: 10.1007/s11033-011-1364-3
108. Klocke K, Holmdahl R, Wing K. CTLA-4 expressed by FOXP3(+) regulatory T cells prevents inflammatory tissue attack and not T-cell priming in arthritis. Immunology (2017) 152(1):125–37. doi: 10.1111/imm.12754
109. Cribbs AP, Kennedy A, Penn H, Read JE, Amjadi P, Green P, et al. Treg cell function in rheumatoid arthritis is compromised by ctla-4 promoter methylation resulting in a failure to activate the indoleamine 2,3-dioxygenase pathway. Arthritis Rheumatol (2014) 66(9):2344–54. doi: 10.1002/art.38715
110. Cutolo M, Sulli A, Paolino S, Pizzorni C. CTLA-4 blockade in the treatment of rheumatoid arthritis: an update. Expert Rev Clin Immunol (2016) 12(4):417–25. doi: 10.1586/1744666X.2016.1133295
111. Fiocco U, Sfriso P, Oliviero F, Pagnin E, Scagliori E, Campana C, et al. Co-Stimulatory modulation in rheumatoid arthritis: the role of (CTLA4-ig) abatacept. Autoimmun Rev (2008) 8(1):76–82. doi: 10.1016/j.autrev.2008.07.035
112. Kiriakidou M, Ching CL. Systemic lupus erythematosus. Ann Intern Med (2020) 172(11):Itc81–itc96. doi: 10.7326/AITC202006020
113. Lee YH, Harley JB, Nath SK. CTLA-4 polymorphisms and systemic lupus erythematosus (SLE): a meta-analysis. Hum Genet (2005) 116(5):361–7. doi: 10.1007/s00439-004-1244-1
114. Zhai JX, Zou LW, Zhang ZX, Fan WJ, Wang HY, Liu T, et al. CTLA-4 polymorphisms and systemic lupus erythematosus (SLE): a meta-analysis. Mol Biol Rep (2013) 40(9):5213–23. doi: 10.1007/s11033-012-2125-7
115. Zhu JM, Li BK, Chen GM, Feng CC, Cen H, Fan YG, et al. CTLA-4 -1722T/C polymorphism and systemic lupus erythematosus susceptibility: a meta-analysis involving ten separate studies. Immunol Invest (2013) 42(2):91–105. doi: 10.3109/08820139.2012.724752
116. Zhu Y, Wang J, Feng X. CTLA-4 SNPs (CT60A/G, -1722T/C, -1661G/A, and -318C/T) and systemic lupus erythematosus: a meta-analysis. Crit Rev Eukaryot Gene Expr (2014) 24(2):89–100. doi: 10.1615/CritRevEukaryotGeneExpr.2014007884
117. Shojaa M, Aghaie M, Amoli M, Javid N, Shakeri F, Khashayar P, et al. Association between 318C/T polymorphism of the CTLA-4 gene and systemic lupus erythematosus in Iranian patients. Int J Rheum Dis (2017) 20(12):2040–4. doi: 10.1111/1756-185X.12275
118. Laurent L, Le Fur A, Bloas RL, Néel M, Mary C, Moreau A, et al. Prevention of lupus nephritis development in NZB/NZW mice by selective blockade of CD28. Eur J Immunol (2017) 47(8):1368–76. doi: 10.1002/eji.201746923
119. Verhagen J, Gabryšová L, Shepard ER, Wraith DC. Ctla-4 modulates the differentiation of inducible Foxp3+ treg cells but IL-10 mediates their function in experimental autoimmune encephalomyelitis. PloS One (2014) 9(9):e108023. doi: 10.1371/journal.pone.0108023
120. Sojka DK, Hughson A, Fowell DJ. CTLA-4 is required by CD4+CD25+ treg to control CD4+ T-cell lymphopenia-induced proliferation. Eur J Immunol (2009) 39(6):1544–51. doi: 10.1002/eji.200838603
121. Jury EC, Flores-Borja F, Kalsi HS, Lazarus M, Isenberg DA, Mauri C, et al. Abnormal CTLA-4 function in T cells from patients with systemic lupus erythematosus. Eur J Immunol (2010) 40(2):569–78. doi: 10.1002/eji.200939781
122. Valentine KM, et al. CD8 follicular T cells promote b cell antibody class switch in autoimmune disease. J Immunol (2018) 201(1):31–40. doi: 10.4049/jimmunol.1701079
123. Mohr E, et al. IFN-{gamma} produced by CD8 T cells induces T-bet-dependent and -independent class switching in b cells in responses to alum-precipitated protein vaccine. Proc Natl Acad Sci U.S.A. (2010) 107(40):17292–7. doi: 10.1073/pnas.1004879107
124. Deng Q, et al. The emerging epigenetic role of CD8+T cells in autoimmune diseases: a systematic review. Front Immunol (2019) 10:856. doi: 10.3389/fimmu.2019.00856
125. Ahmed S, Ihara K, Kanemitsu S, Nakashima H, Otsuka T, Tsuzaka K, et al. Association of CTLA-4 but not CD28 gene polymorphisms with systemic lupus erythematosus in the Japanese population. Rheumatology (2001) 40(6):662–7. doi: 10.1093/rheumatology/40.6.662
126. Cotsapas C, Mitrovic M, Hafler D. Multiple sclerosis. Handb Clin Neurol (2018) 148:723–30. doi: 10.1016/B978-0-444-64076-5.00046-6
127. Wagner M, Sobczynski M, Karabon L, Bilinska M, Pokryszko-Dragan A, Pawlak-Adamska E, et al. Polymorphisms in CD28, CTLA-4, CD80 and CD86 genes may influence the risk of multiple sclerosis and its age of onset. J Neuroimmunol (2015) 288:79–86. doi: 10.1016/j.jneuroim.2015.09.004
128. Gyu Song G, Ho Lee Y. CTLA-4 +49 A/G and -318 C/T polymorphisms and susceptibility to multiple sclerosis: a meta-analysis. Immunol Invest (2013) 42(5):409–22. doi: 10.3109/08820139.2013.803114
129. Haibing X, Xu C, Jifu C, Wenshuang Z, Ling L, Yuzhen C, et al. Correlation between CTLA-4 gene rs221775A>G single nucleotide polymorphism and multiple sclerosis susceptibility. A meta-analysis. Open Med (Wars) (2016) 11(1):264–9. doi: 10.1515/med-2016-0052
130. Vogel I, Kasran A, Cremer J, Kim YJ, Boon L, Van Gool SW, et al. CD28/CTLA-4/B7 costimulatory pathway blockade affects regulatory T-cell function in autoimmunity. Eur J Immunol (2015) 45(6):1832–41. doi: 10.1002/eji.201445190
131. Kim GR, Kim WJ, Lim S, Lee HG, Koo JH, Nam KH, et al. In vivo induction of regulatory T cells Via CTLA-4 signaling peptide to control autoimmune encephalomyelitis and prevent disease relapse. Adv Sci (Weinh) (2021) 8(14):2004973. doi: 10.1002/advs.202004973
132. Kim WJ, Kim GR, Cho HJ, Choi JM. The cysteine-containing cell-penetrating peptide AP enables efficient macromolecule delivery to T cells and controls autoimmune encephalomyelitis. Pharmaceutics (2021) 13(8):1839–50. doi: 10.3390/pharmaceutics13081134
133. Clark M, Kroger CJ, Tisch RM. Type 1 diabetes: a chronic anti-Self-Inflammatory response. Front Immunol (2017) 8:1898. doi: 10.3389/fimmu.2017.01898
134. Padma-Malini R, Rathika C, Ramgopal S, Murali V, Dharmarajan P, Pushkala S, et al. Associations of CTLA4 +49 A/G dimorphism and HLA-DRB1*/DQB1* alleles with type 1 diabetes from south India. Biochem Genet (2018) 56(5):489–505. doi: 10.1007/s10528-018-9856-7
135. Wang K, Zhu Q, Lu Y, Lu H, Zhang F, Wang X, et al. CTLA-4 +49 G/A polymorphism confers autoimmune disease risk: an updated meta-analysis. Genet testing Mol Biomarkers (2017) 21(4):222–7. doi: 10.1089/gtmb.2016.0335
136. Arafa RM, Desouky SM, Emam SM, Abed NT, Mohamed SY. Detection of cytotoxic T-lymphocyte associated antigen-4 gene polymorphism in type 1 diabetes mellitus. Egyptian J Immunol (2015) 22(1):49–57.
137. Hull CM, Peakman M, Tree TIM. Regulatory T cell dysfunction in type 1 diabetes: what's broken and how can we fix it? Diabetologia (2017) 60(10):1839–50. doi: 10.1007/s00125-017-4377-1
138. Zhang Y, Bandala-Sanchez E, Harrison LC. Revisiting regulatory T cells in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes (2012) 19(4):271–8. doi: 10.1097/MED.0b013e328355a2d5
139. You S, Alyanakian MA, Segovia B, Damotte D, Bluestone J, Bach JF, et al. Immunoregulatory pathways controlling progression of autoimmunity in NOD mice. Ann N Y Acad Sci (2008) 1150:300–10. doi: 10.1196/annals.1447.046
140. Gauci ML, Boudou P, Baroudjian B, Vidal-Trecan T, Da Meda L, Madelaine-Chambrin I, et al. Occurrence of type 1 and type 2 diabetes in patients treated with immunotherapy (anti-PD-1 and/or anti-CTLA-4) for metastatic melanoma: a retrospective study. Cancer Immunol Immunother (2018) 67(8):1197–208. doi: 10.1007/s00262-018-2178-0
141. Fujii M, Inoguchi T, Batchuluun B, Sugiyama N, Kobayashi K, Sonoda N, et al. CTLA-4Ig immunotherapy of obesity-induced insulin resistance by manipulation of macrophage polarization in adipose tissues. Biochem Biophys Res Commun (2013) 438(1):103–9. doi: 10.1016/j.bbrc.2013.07.034
142. Koprivica I, Vujičić M, Gajić D, Saksida T, Stojanović I. Ethyl pyruvate stimulates regulatory T cells and ameliorates type 1 diabetes development in mice. Front Immunol (2018) 9:3130. doi: 10.3389/fimmu.2018.03130
143. Zurawek M, Dzikiewicz-Krawczyk A, Izykowska K, Ziolkowska-Suchanek I, Skowronska B, Czainska M, et al. miR-487a-3p upregulated in type 1 diabetes targets CTLA4 and FOXO3. Diabetes Res Clin Pract (2018) 142:146–53. doi: 10.1016/j.diabres.2018.05.044
144. Lazaridis K, Tzartos SJ. Autoantibody specificities in myasthenia gravis; implications for improved diagnostics and therapeutics. Front Immunol (2020) 11:212–2. doi: 10.3389/fimmu.2020.00212
145. Barik A, Lu Y, Sathyamurthy A, Bowman A, Shen C, Li L, et al. LRP4 is critical for neuromuscular junction maintenance. J Neurosci (2014) 34(42):13892–905. doi: 10.1523/JNEUROSCI.1733-14.2014
146. Cai GM, Gao Z, Yue YX, Xie YC, Gao X, Hao HJ, et al. Association between CTLA-4 gene polymorphism and myasthenia gravis in a Chinese cohort. J Clin Neurosci (2019) 69:31–7. doi: 10.1016/j.jocn.2019.08.079
147. Xu W, Ren M, Ghosh S, Qian K, Luo Z, Zhang A, et al. Defects of CTLA-4 are associated with regulatory T cells in myasthenia gravis implicated by intravenous immunoglobulin therapy. Mediators Inflammation (2020) 2020:3645157. doi: 10.1155/2020/3645157
148. Yi JS, Guptill JT, Stathopoulos P, Nowak RJ, O'Connor KC. B cells in the pathophysiology of myasthenia gravis. Muscle Nerve (2018) 57(2):172–84. doi: 10.1002/mus.25973
149. Alegre ML, Shiels H, Thompson CB, Gajewski TF. Expression and function of CTLA-4 in Th1 and Th2 cells. J Immunol (1998) 161(7):3347–56. doi: 10.4049/jimmunol.161.7.3347
150. von Euw E, Chodon T, Attar N, Jalil J, Koya RC, Comin-Anduix B, et al. CTLA4 blockade increases Th17 cells in patients with metastatic melanoma. J Transl Med (2009) 7:35. doi: 10.1186/1479-5876-7-35
151. Kyritsi EM, Kanaka-Gantenbein C. Autoimmune thyroid disease in specific genetic syndromes in childhood and adolescence. Front Endocrinol (Lausanne) (2020) 11:543. doi: 10.3389/fendo.2020.00543
152. Chistiakov DA, Turakulov RI. CTLA-4 and its role in autoimmune thyroid disease. J Mol Endocrinol (2003) 31(1):21–36. doi: 10.1677/jme.0.0310021
153. Hu Y, Xu K, Jiang L, Zhang L, Shi H, Cui D. Associations between three CTLA-4 polymorphisms and hashimoto's thyroiditis risk: an updated meta-analysis with trial sequential analysis. Genet Test Mol Biomarkers (2018) 22(4):224–36. doi: 10.1089/gtmb.2017.0243
154. Qiu H, Tang W, Yin P, Cheng F, Wang L. Cytotoxic T-lymphocyte associated antigen 4 polymorphism and hashimoto's thyroiditis susceptibility: a meta-analysis. Endocrine (2014) 45(2):198–205. doi: 10.1007/s12020-013-9985-z
155. Ji R, Feng Y, Zhan WW. Updated analysis of studies on the cytotoxic T-lymphocyte-associated antigen-4 gene A49G polymorphism and hashimoto's thyroiditis risk. Genet Mol Res (2013) 12(2):1421–30. doi: 10.4238/2013.April.26.4
156. Faizi M, Rochmah N, Hisbiyah Y, Perwitasari RK, Putri QAN, Basuki S, et al. Cytotoxic T-lymphocyte-associated protein 4 +49A/G polymorphism in down syndrome children with hashimoto's thyroiditis. Biomol BioMed (2023) 18(3):324–32. doi: 10.17305/bb.2022.7869
157. Kucharska AM, Wisniewska A, Popko K, Demkow U. Association between the polymorphism A/G at position 49 of exon 1 of the CTLA-4 gene and antithyroid antibody production in children with hashimoto's thyroiditis. Horm Res Paediatr (2012) 78(2):67–72. doi: 10.1159/000338997
158. Jiang X, Hu H, Fu Z, Su Y, Long J. ASSOCIATION BETWEEN THE CTLA-4 EXON 1+49A/G POLYMORPHISM AND THE RELAPSE OF GRAVE'S DISEASE AFTER ATD WITHDRAWAL: a META-ANALYSIS. Acta Endocrinol (Buchar) (2022) 18(3):324–32. doi: 10.4183/aeb.2022.324
159. Huang F, He Q, Jiao X, Zhang H, Chang Q. Meta-analysis of CTLA-4 +49 gene polymorphism and susceptibility to graves' disease. Crit Rev Eukaryot Gene Expr (2020) 30(5):377–90. doi: 10.1615/CritRevEukaryotGeneExpr.2020034872
160. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med (2011) 17(8):975–82. doi: 10.1038/nm.2425
161. Zhao J, Chen Y, Xu Z, Yang W, Zhu Z, Song Y, et al. Increased circulating follicular regulatory T cells in hashimoto’s thyroiditis. Autoimmunity (2018) 51(7):345–51. doi: 10.1080/08916934.2018.1516759
162. Liu Y, Wang X, Luan W, Zou J, Xing J, Wang S, et al. MiR-29a-3p negatively regulates circulating tfh memory cells in patients with graves' disease by targeting ICOS. Immunol Res (2023) 71(2):173–84. doi: 10.1007/s12026-022-09333-5
163. Wing JB, Sakaguchi S. Multiple treg suppressive modules and their adaptability. Front Immunol (2012) 3:178. doi: 10.3389/fimmu.2012.00178
164. Sage PT, Sharpe AH. T Follicular regulatory cells in the regulation of b cell responses. Trends Immunol (2015) 36(7):410–8. doi: 10.1016/j.it.2015.05.005
165. Vasu C, Gorla SR, Prabhakar BS, Holterman MJ. Targeted engagement of CTLA-4 prevents autoimmune thyroiditis. Int Immunol (2003) 15(5):641–54. doi: 10.1093/intimm/dxg061
166. Brunner HI, Tzaribachev N, Vega-Cornejo G, Louw I, Berman A, Calvo Penadés I, et al. Subcutaneous abatacept in patients with polyarticular-course juvenile idiopathic arthritis: results from a phase III open-label study. Arthritis Rheumatol (2018) 70(7):1144–54. doi: 10.1002/art.40466
167. Douthwaite J, Moisan J, Privezentzev C, Soskic B, Sabbah S, Cohen S, et al. A CD80-biased CTLA4-ig fusion protein with superior In vivo efficacy by simultaneous engineering of affinity, selectivity, stability, and FcRn binding. J Immunol (2017) 198(1):528–37. doi: 10.4049/jimmunol.1600682
168. Bernett MJ, Chu SY, Leung I, Moore GL, Lee SH, Pong E, et al. Immune suppression in cynomolgus monkeys by XPro9523: an improved CTLA4-ig fusion with enhanced binding to CD80, CD86 and neonatal fc receptor FcRn. MAbs (2013) 5(3):384–96. doi: 10.4161/mabs.23976
169. Aldridge J, Andersson K, Gjertsson I, Ekwall AH, Hallström M, van Vollenhoven R, et al. Blood PD-1+TFh and CTLA-4+CD4+ T cells predict remission after CTLA-4Ig treatment in early rheumatoid arthritis. Rheumatol (Oxford) (2021). doi: 10.1093/rheumatology/keab454
170. Rubbert-Roth A, Enejosa J, Pangan AL, Haraoui B, Rischmueller M, Khan N, et al. Trial of upadacitinib or abatacept in rheumatoid arthritis. N Engl J Med (2020) 383(16):1511–21. doi: 10.1056/NEJMoa2008250
171. Judge TA, Tang A, Spain LM, Deans-Gratiot J, Sayegh MH, Turka LA. The in vivo mechanism of action of CTLA4Ig. J Immunol (1996) 156(6):2294–9. doi: 10.4049/jimmunol.156.6.2294
172. Weisman MH, Durez P, Hallegua D, Aranda R, Becker JC, Nuamah I, et al. Reduction of inflammatory biomarker response by abatacept in treatment of rheumatoid arthritis. J Rheumatol (2006) 33(11):2162–6.
173. Picchianti Diamanti A, Rosado MM, Scarsella M, Germano V, Giorda E, Cascioli S, et al. Abatacept (cytotoxic T lymphocyte antigen 4-immunoglobulin) improves b cell function and regulatory T cell inhibitory capacity in rheumatoid arthritis patients non-responding to anti-tumour necrosis factor-α agents. Clin Exp Immunol (2014) 177(3):630–40. doi: 10.1111/cei.12367
174. Lorenzetti R, Janowska I, Smulski CR, Frede N, Henneberger N, Walter L, et al. Abatacept modulates CD80 and CD86 expression and memory formation in human b-cells. J Autoimmun (2019) 101:145–52. doi: 10.1016/j.jaut.2019.04.016
175. Bonelli M, Göschl L, Blüml S, Karonitsch T, Hirahara K, Ferner E, et al. Abatacept (CTLA-4Ig) treatment reduces T cell apoptosis and regulatory T cell suppression in patients with rheumatoid arthritis. Rheumatol (Oxford) (2016) 55(4):710–20. doi: 10.1093/rheumatology/kev403
176. Blair HA, Deeks ED. Abatacept: a review in rheumatoid arthritis. Drugs (2017) 77(11):1221–33. doi: 10.1007/s40265-017-0775-4
177. Viglietta V, Bourcier K, Buckle GJ, Healy B, Weiner HL, Hafler DA, et al. CTLA4Ig treatment in patients with multiple sclerosis: an open-label, phase 1 clinical trial. Neurology (2008) 71(12):917–24. doi: 10.1212/01.wnl.0000325915.00112.61
178. Gerdes LA, Held K, Beltrán E, Berking C, Prinz JC, Junker A, et al. CTLA4 as immunological checkpoint in the development of multiple sclerosis. Ann Neurol (2016) 80(2):294–300. doi: 10.1002/ana.24715
179. Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Co-Stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet (2011) 378(9789):412–9. doi: 10.1016/S0140-6736(11)60886-6
180. Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Costimulation modulation with abatacept in patients with recent-onset type 1 diabetes: follow-up 1 year after cessation of treatment. Diabetes Care (2014) 37(4):1069–75. doi: 10.2337/dc13-0604
181. Orban T, Beam CA, Xu P, Moore K, Jiang Q, Deng J, et al. Reduction in CD4 central memory T-cell subset in costimulation modulator abatacept-treated patients with recent-onset type 1 diabetes is associated with slower c-peptide decline. Diabetes (2014) 63(10):3449–57. doi: 10.2337/db14-0047
182. Simone R, Pesce G, Antola P, Rumbullaku M, Bagnasco M, Bizzaro N, et al. The soluble form of CTLA-4 from serum of patients with autoimmune diseases regulates T-cell responses. BioMed Res Int (2014) 2014:215763. doi: 10.1155/2014/215763
183. Wong CK, Lit LC, Tam LS, Li EK, Lam CW. Aberrant production of soluble costimulatory molecules CTLA-4, CD28, CD80 and CD86 in patients with systemic lupus erythematosus. Rheumatol (Oxford) (2005) 44(8):989–94. doi: 10.1093/rheumatology/keh663
184. Sato S, Fujimoto M, Hasegawa M, Komura K, Yanaba K, Hayakawa I, et al. Serum soluble CTLA-4 levels are increased in diffuse cutaneous systemic sclerosis. Rheumatol (Oxford) (2004) 43(10):1261–6. doi: 10.1093/rheumatology/keh303
185. Wang XB, Kakoulidou M, Giscombe R, Qiu Q, Huang D, Pirskanen R, et al. Abnormal expression of CTLA-4 by T cells from patients with myasthenia gravis: effect of an AT-rich gene sequence. J Neuroimmunol (2002) 130(1-2):224–32. doi: 10.1016/S0165-5728(02)00228-X
186. Simone R, Brizzolara R, Chiappori A, Milintenda-Floriani F, Natale C, Greco L, et al. A functional soluble form of CTLA-4 is present in the serum of celiac patients and correlates with mucosal injury. Int Immunol (2009) 21(9):1037–45. doi: 10.1093/intimm/dxp069
Keywords: CTLA-4, autoimmunity, regulatory T cell, autoimmune disease, immune regulation
Citation: Hossen MM, Ma Y, Yin Z, Xia Y, Du J, Huang JY, Huang JJ, Zou L, Ye Z and Huang Z (2023) Current understanding of CTLA-4: from mechanism to autoimmune diseases. Front. Immunol. 14:1198365. doi: 10.3389/fimmu.2023.1198365
Received: 01 April 2023; Accepted: 19 June 2023;
Published: 11 July 2023.
Edited by:
Andrew W. Taylor, Boston University, United StatesReviewed by:
Mohammad Arif Rahman, National Institutes of Health (NIH), United StatesCopyright © 2023 Hossen, Ma, Yin, Xia, Du, Huang, Huang, Zou, Ye and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhizhong Ye, eWV6aGl6aG9uZzIwMDBAMTYzLmNvbQ==; Zhong Huang, emh1YW5nODA5QDEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.