 Betul Ibis1,2
Betul Ibis1,2 Konstantinos Aliazis1,2
Konstantinos Aliazis1,2 Carol Cao1,2,3
Carol Cao1,2,3 Sasitorn Yenyuwadee1,2†
Sasitorn Yenyuwadee1,2† Vassiliki A. Boussiotis1,2,4*
Vassiliki A. Boussiotis1,2,4*- 1Division of Hematology-Oncology Beth Israel Deaconess Medical Center, Boston, MA, United States
- 2Department of Medicine, Beth Israel Deaconess Medical Center, Boston, MA, United States
- 3Harvard College, Cambridge, MA, United States
- 4Cancer Center, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, United States
During the past decade, there has been a revolution in cancer therapeutics by the emergence of antibody-based immunotherapies that modulate immune responses against tumors. These therapies have offered treatment options to patients who are no longer responding to classic anti-cancer therapies. By blocking inhibitory signals mediated by surface receptors that are naturally upregulated during activation of antigen-presenting cells (APC) and T cells, predominantly PD-1 and its ligand PD-L1, as well as CTLA-4, such blocking agents have revolutionized cancer treatment. However, breaking these inhibitory signals cannot be selectively targeted to the tumor microenvironment (TME). Since the physiologic role of these inhibitory receptors, known as immune checkpoints (IC) is to maintain peripheral tolerance by preventing the activation of autoreactive immune cells, IC inhibitors (ICI) induce multiple types of immune-related adverse effects (irAEs). These irAEs, together with the natural properties of ICs as gatekeepers of self-tolerance, have precluded the use of ICI in patients with pre-existing autoimmune diseases (ADs). However, currently accumulating data indicates that ICI might be safely administered to such patients. In this review, we discuss mechanisms of well established and newly recognized irAEs and evolving knowledge from the application of ICI therapies in patients with cancer and pre-existing ADs.
Cancer immunosurveillance and immune escape in the tumor microenvironment
The tumor microenvironment (TME) is a complex multicellular space in which cancer can be subjected to immune-mediated control during the early stages of its evolution, such as the eradication and equilibrium phase, but ultimately favors the development of cancer immune escape by fostering cancer immunoediting. During that stage, under continuous immune pressure, cancer cells develop alterations to overcome the immune attack, resulting in tumors that are resistant to the physiological mechanisms utilized by innate immune cells to recognize and present antigens while shutting down the activation of helper and effector T cells (1).
Tumor-promoting factors in the TME include defective antigen presentation by antigen-presenting cells (APC), mainly dendritic cells (DC), which express inhibitory ligands, upregulation of multiple inhibitory receptors during persistent activation of T cells, such as PD-1, LAG-3, TIGIT, TIM-3, expansion of cell populations that mediate T cell inhibitory functions such as myeloid-derived suppressor cells (MDSCs) and T regulatory (Treg) cells, generation of tumor associated macrophages (TAMs) that lose the ability to phagocytose and present antigens but instead acquire protumorigenic functions, and soluble factors such as IDO, VEGF, and TGF-β, which support cancer cell survival and tumor growth while suppressing the function of immune cells (2).
Expression of immune checkpoints in the TME
T cells of the TME are critical determinants of tumor containment and progression. During the phase of cancer escape from immune control, T cells that can recognize tumor-associated antigens lose the ability to control tumor growth due to mechanisms of tumor-induced tolerance and immunosuppression. These dysfunctional T cells are characterized by features of T cell exhaustion (TEX) similar to those in chronic viral infections, including high expression of ICs including CTLA-4, PD-1, TIM-3, TIGIT and LAG-3, loss of expansion capacity, and impaired effector function as determined by the diminished production of cytokines such as IFN-γ and TNF-α (3). Conceptually, a central goal of novel immunotherapies is to achieve re-invigoration of tumor-specific TEX cells, for which the state of T cell exhaustion might be still reversible, and blockade of ICs has been the main approach to achieve this goal (4, 5).
The co-inhibitory molecules, such as CTLA-4 (CD152) and PD-1 (CD279), are induced during physiologic T cell activation. CTLA-4 being upregulated and acting early during T cell activation, is the high affinity receptor of CD80/CD86. CTLA-4 directly competes with CD28 for binding on CD80/CD86, but also induces depletion of CD80/CD86 by trans-endocytosis (6). Importantly, this interaction simultaneously releases free PD-L1 by eliminating availability of CD80 for PD-L1 engagement in PD-L1: CD80 interaction in cis (7). Thus, the interactions of CTLA4 interfere both with the key costimulatory pathway CD80/CD28 and the key co-inhibitory pathway PD-1/PD-L1.
PD-1 is physiologically induced upon TCR-mediated T cell activation. Engagement of PD-1 by its ligands PD-L1 (B7-H1 or CD274) and/or PD-L2 (B7-DC or B7-H3) counteracts TCR signaling and CD28-mediated co-stimulation (8–10). The expression of PD-L1 on tumor cells has served as a biomarker for patient stratification for anti-PD-1 ICI therapy. However, the relative contribution of PD-L1 on tumor cells and other cell types in limiting anti-tumor responses in the TME remains under investigation. By genetic deletion of PD-L1 in tumor cells or innate immune cells of host mice, it was found that PD-L1 expression on each of these compartments equally contributes to immune suppression (11). Subsequently, it was determined that PD-L1 expressed in APC, particularly DC, has the key and causative role in compromising anti-tumor T cell responses (12, 13).
Parallel studies revealed that PD-1 expression in myeloid cells has an important role in lineage fate commitment, effector differentiation and antigen presenting function (14). Specifically, it was found that PD-1 is expressed predominantly in myeloid progenitors, whereas ablation of PD-1 expression resulted their differentiation into mature myeloid cells with predominant features of monocyte and DC differentiation (14). RNAseq studies showed that PD-1 ablation in myeloid cells resulted in the differentiation of tumor infiltrating macrophages with features of potent immune function including activation, differentiation, phagocytosis and enhanced signaling and metabolic programs (15). Consistent with these findings, PD-1 expression in TAMs was associated with diminished phagocytosis and enhanced tumor growth (16). Thus, therapeutic targeting with PD-1/PD-L1 blocking compounds might lead to proinflammatory activation of both T cells and myeloid cells and release of multiple proinflammatory cytokines from both immune compartments.
PD-1 and CTLA-4 are the prototype ICs and the most extensively utilized therapeutic targets in cancer immunotherapy. However, other ICs, such as LAG-3, TIM-3, TIGIT, GITR, or VISTA are also exploited by tumor cells, contributing to the generation of an immunosuppressive TME and escape of immunosurveillance (17). Because clinical experience with ICIs for these receptors is limited, irAEs induced by therapies blocking these ICs have not been well-characterized. For these reasons, in the present review, we will focus on irAEs induced by blockade of CTLA-4 and the PD-1/PD-L1 pathway.
Role of ICs in central and peripheral tolerance and lessons from genetic models
CTLA-4
The importance of the CTLA-4 receptor in the establishment of peripheral tolerance was identified early by studies with CTLA-4-deficient mice. These mice developed splenomegaly and lymphadenopathy which led to death within 3-4 weeks of age (18–20). This extensive spontaneous lymphoproliferation resulted in massive infiltration of mononuclear cells in the heart, pancreas, liver, lungs, and other organs causing extensive damage to these tissues (18, 19). T cells from the spleen and lymph nodes showed an activated phenotype with upregulation of activation markers like CD69, CD25, and CD44 (18–20). Depletion of CD4+ but not CD8+ T cells led to inhibition of immune infiltration into non-lymphoid organs (20). Administration of CTLA-4-Ig to CTLA-4-deficient mice prevented the T cell expansion. When CTLA-4 was deleted in adulthood using a conditional knockout approach, these mice still developed lymphoproliferation and immune infiltration in various organs, including pancreatic β-islets, lungs, and stomach (21). Deletion of CTLA-4 in these adult mice resulted in expansion of Treg in the blood, spleen, and lymph nodes, although the levels of Treg decreased rapidly in the blood but not in the organs. CTLA-4 deletion also led to activation of T conventional cells (Tconv, non-Treg cells). In the collagen-induced arthritis (CIA) model, loss of CTLA-4 promoted a stark exacerbation of the disease with extended damage to the joints (21).
Specific deletion of CTLA-4 from Treg also led to systemic lymphoproliferation and death of the mice, albeit with slower kinetics (22). These mice developed immune infiltration of the myocardium and destruction of myocytes, which the authors speculated it caused heart failure due to myocarditis. When adoptively transferred into T cell-deficient mice, CTLA-4-/- Treg could provide tumor protection, in contrast to CTLA-4+/+ Treg, indicating that CTLA4-deficient Treg did not have suppressor function but rather acted as T effector cells. Similarly, in an autoimmune model for diabetes, adoptively transferred CTLA-4-deficient Treg were unable to prevent the destruction of the pancreas and the induction of diabetes (23). In contrast to previous reports which described the development of autoimmunity and exacerbation of autoimmune diseases by CTLA-4 deficiency, subsequent studies showed that CTLA-4 ablation in adult mice resulted in complete or transient resistance to experimental autoimmune encephalomyelitis (EAE), which was interpreted as a consequence of increased expansion of thymic Treg as a result of CTLA-4 deletion (24, 25).
B cells also showed an increased activation profile in CTLA-4-/- mice with upregulation of CD86, Fas, and CD5 but not CD80 (19). This correlated with a striking increase of all Ig subtypes in the serum of CTLA-4-deficient mice (19). In this model, CD4+ T cell depletion or administration of CTLA-4-Ig prevented the activation and expansion of B cells (20). CTLA-4 is expressed solely in B-1a B cells which are generated during fetal development (26). Deletion of CTLA-4 from B-1a B cells in CTLA-4f/fCD19Cre/+ mice led to activation and differentiation of these cells into antigen-presenting cells, and spontaneous germinal center formation in the spleens (26). These mice developed autoantibodies and late autoimmune characteristics which shared features with some human autoimmune diseases. Thus, abrogating CTLA-4 function induces autoimmunity by several mechanisms and by targeting several lymphocyte subsets.
The role of CTLA-4 in maintaining tolerance and preventing development of autoimmunity has also been documented in studies where treatment of mice with anti-CTLA-4 monoclonal antibody (mAb) exacerbated autoimmune diseases. When CTLA-4 was blocked in the mouse model of EAE, there was a marked increase in the proinflammatory cytokines TNF-α, IFN-γ, and IL-2, and symptoms were exacerbated (27, 28). Administration of anti-CTLA-4 blocking mAb also resulted in increased inflammatory foci in the brain and spinal cord, and increased demyelinating lesions (28). Similar results of disease exacerbation were observed using the nonobese diabetic (NOD) mouse model, in which administration of anti-CTLA-4 mAb induced rapid disease onset of about 15 days compared to the usual 5-6 months (29). CD4+ T cells from mAb-treated mice displayed a chronically activated phenotypic profile with increased expression levels of CD44 but low levels of CD62L, with a few T cells expressing early activation markers like CD69 and CD25 (29). Deletion of CTLA-4 on Treg led to spontaneous germinal center formation in the spleen and lymph nodes (30).
The CTLA4 locus has been involved in the regulation of several autoimmune diseases in humans (31). Polymorphisms on CTLA4 gene have been implicated in diabetes and thyroid disease (32, 33), rheumatoid arthritis (34), primary biliary cholangitis (35), and spontaneous abortion (36). Furthermore, a soluble form of CTLA-4 is present in patients with various autoimmune diseases, such as autoimmune thyroid diseases (37, 38), myasthenia gravis (39), and systemic lupus erythematosus (SLE) (40).
PD-1
Deletion of PD-1 in mice did not induce an immediate extreme phenotype, such as CTLA-4 deletion, however, in later stages of their lives, PD-1-deficient mice developed autoimmune symptoms. C57BL/6 PD-1-/- mice showed signs of mild splenomegaly early on but appeared to be healthy (41). The number of B cells and myeloid cells in the spleen increased, however, the number of T cells remained stable. An increase of IgA, IgG2b, and IgG3 was present in the serum. At 6 months of age, a few of these mice started showing signs of lupus-like glomerulonephritis and arthritis in the foot joints (42). At 14 months of age, the severity of glomerulonephritis had increased, whereas wild-type mice showed only mild symptoms (42). The severity of arthritis also progressed to an advanced stage in all the PD-1 KO mice.
Because C57BL/6/PD-1-/- mice showed a lupus-like phenotype, C57BL/6 PD-1-/- mice were crossed with the B6 lpr/lpr mouse strain, which is used as a model for SLE (43, 44). Glomerulonephritis was present much earlier in the C57BL/6-lpr/lpr-PD-1-/- mice compared to C57BL/6lpr/lpr mice (42). Depositions of IgG3 and C3 complement were present in the kidneys and histology showed arthritic lesions in the joints much earlier than in the control B6 lpr/lpr mice. Lymphadenopathy and extensive hyperplasia of bone marrow were also detected. The severity of symptoms in the C57BL/6lpr/lpr-PD-1-/- mice resembled that of the MRL-FASlpr/lpr mouse, which develops lupus-like symptoms much earlier than the C57BL/6lpr/lpr. PD-1 deletion in the MRL mouse resulted in the development of myocarditis and death of the mice by week 10 (45). Extensive immune infiltration was present in the hearts of the PD-1-deficient mice with both populations of CD4+ and CD8+ T cells showing an activated phenotype. Autoantibodies against cardiac myosin were also present despite the small number of B cells accumulated in the heart of the MRL PD-1-deficient mice. Notably, an increased accumulation of Mac1+Gr1+ cells was also present in the heart. These cells were able to strongly suppress infiltrating T cells and were considered similar to myeloid-derived suppressor cells (45).
Deletion of PD-1 in BALB/c mice resulted in a more severe phenotype than in the B57BL/6 mice, with development of multisystem autoimmunity. These mice suffered from splenomegaly, hepatomegaly, cardiomegaly, and immune infiltration of several organs such as the heart, lungs, liver, kidney, and developed skin lesions which resembled graft-versus-host disease (42, 46). Many mice died by the age of 5 weeks (42, 46). Histologic examination of the hearts revealed extensive damage, suggesting that the cause of death was heart failure (46). The BALB/c PD1-/- mice had an increased population of activated CD8+ T cells in comparison to WT mice. Interestingly, BALB/c-RAG-2-/-PD-1-/- mice did not die but remained healthy, highlighting the role of T cells and/or B cells in disease development (46). Indeed, the hearts of BALB/c PD-1-/- mice had increased depositions of IgG1, IgM, and C3 complement (46). Furthermore, serum from PD-1-deficient mice was enriched with autoantibodies against cardiac troponin I (46, 47).
Deletion of PD-1 from the NOD mouse presented a phenotype consistent with that observed to the two previous strains, with early and robust development of autoimmune activation. NOD-PD-1-/- mice developed diabetes much earlier than in NOD-PD-1+/+ mice with early insulitis and increased infiltration of CD4+ and CD8+ T cells (48). In vitro stimulation of T cells isolated from β-islets of NOD-PD-1-/- mice showed an increased proclivity for IFN-γ secretion.
These studies highlighted the importance of both CTLA-4 and PD-1 in immune homeostasis and their indispensable and non-redundant roles in preventing autoimmunity.
irAEs mediated by ICI therapies
Based on the physiological properties of CTLA-4 and PD-1 revealed by the extensive studies outlined above, it is anticipated that blockade of these ICs in humans for cancer therapy will inevitably lead to global immune activation leading to autoimmune manifestations of various organ systems (Figure 1). Immune checkpoint inhibitors (ICIs) employed for cancer therapy include PD-1 (pembrolizumab, nivolumab, cemiplimab, dostarlimab), PDL-1 (atezolizumab, avelumab, durvalumab), CTLA-4 (ipilimumab, tremelimumab), and the recently approved LAG-3 antibodies (relatlimab). These ICIs are used individually, in combination, or together with chemotherapy (49). ICI therapy differs from chemotherapy in mechanisms of action and side effect profile, which are intimately linked. ICIs aim to increase the activity of the immune system against cancer by breaking tolerance mediated by CTLA-4 and PD-1 in the TME. However, ICI therapy simultaneously activates non-cancer-specific T cell subsets, B cells and myeloid cells which are kept in check by ICs, thereby preventing autoimmunity as outlined in the previous section. Therefore, irAEs are typical side effects of ICIs. Several irAEs resemble features of ADs and may occur during or after discontinuation of ICI treatment. ICI may also trigger the clinical evolution and presentation of previously pre-clinical and unidentified ADs. In addition, irAEs may occur as paraneoplastic syndromes induced by autoantibody production like typical paraneoplastic syndromes in the context of several cancers.
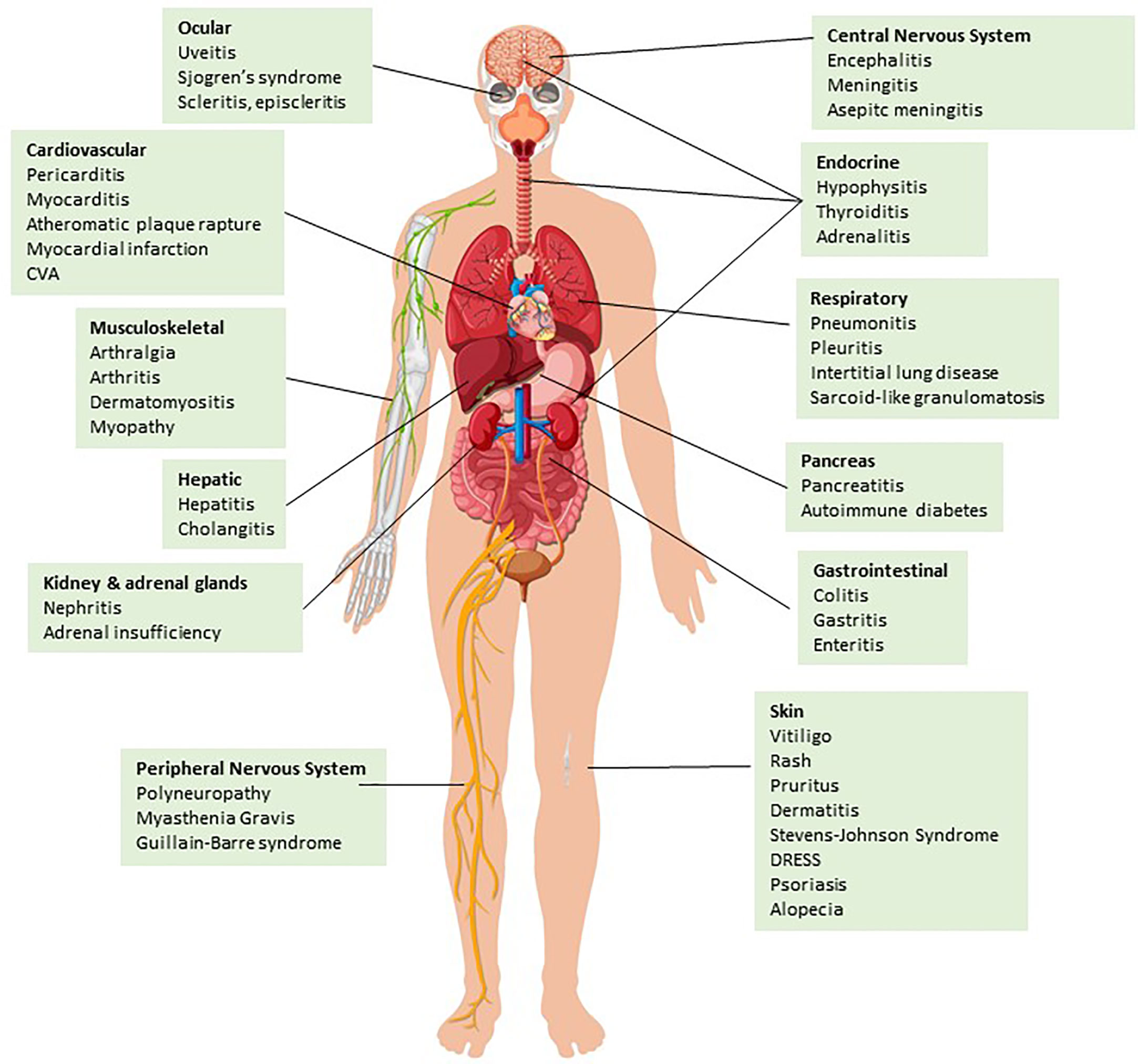
Figure 1 irAEs categorized by organ system. ICI treatment may result in inflammation of various tissues and organ systems leading to well-described irAEs (please see main text for details) Design by Brgfx-Freepik.com.
A cross-sectional study showed that 43.63% of patients with cancer in the US were eligible for ICI use (50). Hence, the use of ICIs is becoming increasingly common in various cancers, making the incidence of irAEs a critical problem, since up to 80% of patients treated with ICIs develop irAEs (51). Currently, there is no reliable biomarker predicting or correlating with irAEs, but several ones are under investigation including serum IL-6, IL-17, soluble CTLA-4 (sCTLA-4), absolute lymphocyte count, and tumor mutation burden (52–55). The most frequently reported irAEs include fatigue, pruritus, nausea, rash, diarrhea/colitis, and endocrinopathies (mainly hypophysitis and thyroiditis), the most common serious irAEs are pneumonitis and colitis, whereas myocarditis is the irAE with the highest fatality rate (Figure 1) (51, 56).
As the use of ICIs is currently increasing, previously unrecognized rare complications become clinically apparent. Importantly, there is increasing evidence that ICI immunotherapy induces cardiovascular complications to a previously unappreciated level. Myocarditis is a well recognized and extensively studied irAE occurring with higher incidence and severity among patients treated with ipilimumab and nivolumab combination compared to those treated with nivolumab alone (57, 58). Newer studies provide evidence the PD-1/PD-L1 blockade induces a prothrombotic environment leading to vein thromboembolism (59), and to the development or worsening of atherosclerotic cardiovascular disease (CVD) resulting in atherotic plaque rapture and presentation of various clinical pathologies of atherosclerotic CVD including coronary artery disease, myocardial infarction, and ischemic stroke (60). These findings are consistent with enhanced severity of atheromatous cardiac disease observed in experimental models of mice with genetic ablation of ICs or treated with ICIs (61–63). Importantly, recent studies provided evidence that pre-existing autoimmune conditions increase the incidence of CVD after ICI therapy (64). Given the immune-mediated nature of atheromatous disease (65, 66), these observations indicate that CVD is another form of ICI-mediated irAE.
irAEs generally improve with the discontinuation of ICIs with or without administration of immunosuppressive therapy. However, several case reports raise concerns about the potentially irreversible morbidity of irAEs, underlying the role of early diagnosis and proper management of these serious complications. Chronic irAEs may affect up to 40% of patients, with endocrine and rheumatologic manifestations being the more frequent forms of chronic irAEs (67, 68). Guidelines for diagnosis and treatment of irAEs are available and extensively reviewed elsewhere (69–71).
Pathogenesis of irAEs
Although the pathogenesis of irAEs is still not fully understood, several mechanisms have been identified including aberrant T-cell activation, autoantibody production, inflammatory monocyte activation, complement-mediated inflammation, inflammatory cytokines, host-specific factors including microbiome and genetics, and the type of ICI immunotherapy administered (Figure 2) (72). Briefly, irAEs can be mechanistically categorized as follows:
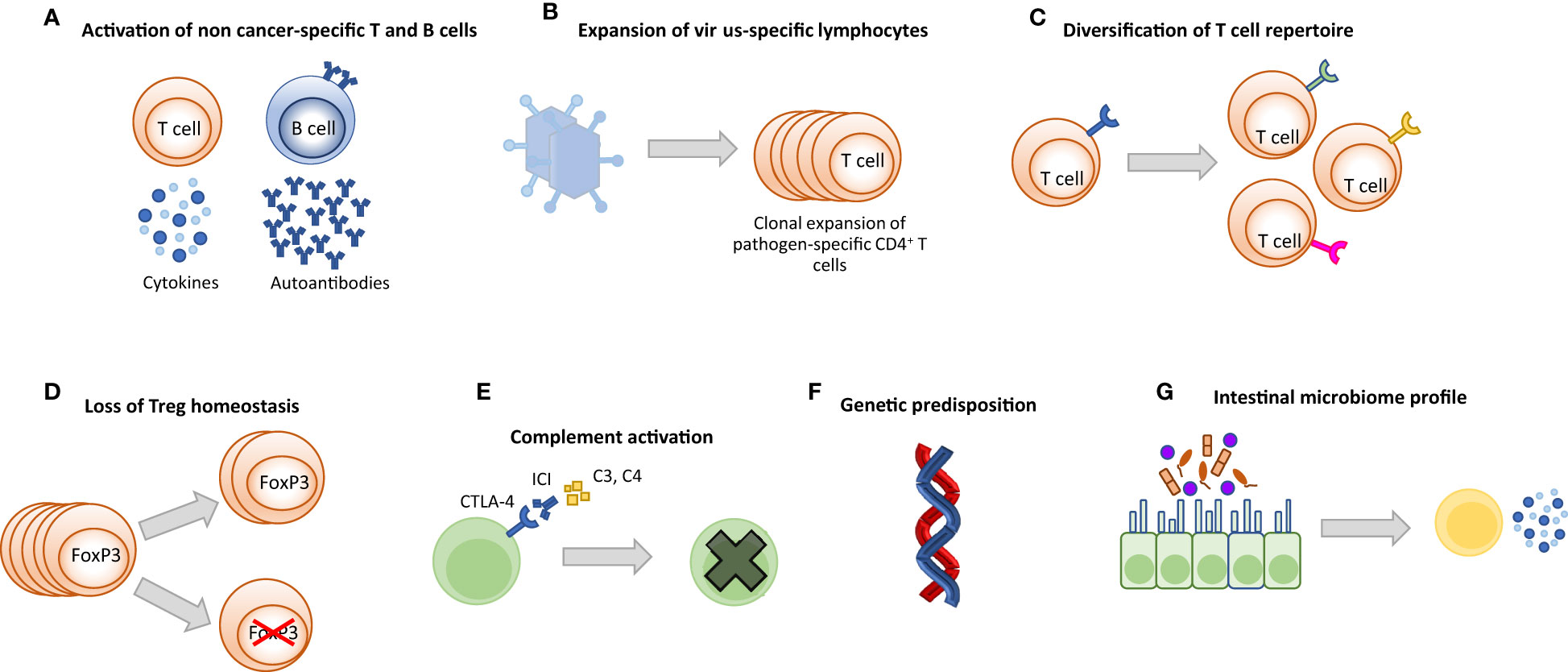
Figure 2 Potential mechanisms driving irAEs. (A) Activation of cytotoxic self-reactive T cells causes damage in off-target healthy tissues by extensive production of pro-inflammatory cytokines and/or direct attack. Activated autoreactive B cells may produce de novo auto-antibodies or increase the amount of pre-existing auto-antibodies. Antibodies bind to healthy tissues and cause inflammation and damage. (B) Clonal proliferation of virus-specific T cells may lead to excessive inflammation and destruction in the relevant organ and may be fatal. (C) Expansion of the T cell repertoire may cause T cells to attack off-target healthy tissues. (D) ICI treatment can lead to decreased number of FoxP3-expressing Treg cells and reprogramming of Treg cells, resulting in pro-inflammatory behavior. (E) Organ-specific expression of ICI targets can induce direct ICI binding followed by complement activation and antibody-mediated inflammation (type II hypersensitivity). (F) Genetic polymorphisms such as some HLA allele types, mutations of IC receptors and miRNAs are associated with the development of irAEs. (G) Microbiome composition (bacteria, metabolites, etc.) may cause aberrant activation of the immune system and increased production of inflammatory cytokines under ICI treatment.
T cell-mediated
ICI therapy directly causes activation of T cells outside of the TME as it inhibits inhibitory signals that prevent T cell activation against self antigens, leading to autoimmune manifestations from different organs. The increase in unique TCR V-beta CDR3 diversity, a marker of TCR richness, was significantly higher in patients who developed irAE compared to those who did not (73). Several case reports of severe and even lethal manifestations of irAEs provided evidence that clonally expanded self-reactive or virus-reactive T cells are accumulated in the affected tissues, linking self- and pathogen-recognizing T cell clones to lethal toxicity. For example, fulminant myocarditis was associated with infiltration of the myocardium by clonal T cells identical with those infiltrating the tumor and skeletal muscles (74); fatal ICI-associated encephalitis correlated with activation of EBV-specific memory CD4+ T cells, whereas ICI-associated hepatitis correlated with activation of CMV-specific T cells (75, 76). CD4+ and CD8+ T cell enrichment in target tissues was observed in irAEs leading to diabetes, colitis, and thyroiditis (77–79). Subsequent studies provided evidence that the TCR richness of activated CD4+ T effector memory cells underlies an overall increase in pretreatment TCR diversity in patients destined to develop severe irAEs. In this context, the magnitude of T cell clonal expansion correlated with the onset time of severe irAEs because patients with a greater magnitude of TCR clonal expansion developed irAEs sooner (80). In addition, Th17.1 cells, known for their role in chronic inflammation, were predominantly increased in bronchoalveolar lavage fluid of patients with ICI-related pneumonitis (81, 82). Lastly, a decrease in the number and an inflammatory reprogramming of Treg are also involved in the pathogenesis of irAEs (83).
B cell- and antibody-mediated
The presence of autoantibodies in the pathogenesis of ADs is well known. Although studies have shown that B cells play a role in response to immunotherapy, their role in the development of irAEs is still not fully understood (84). Inherited deficiencies in the B regulatory (Breg) cells, which produce IL-10, have been associated with the development of severe irAEs (85). An increase in CD21low B cell subset and plasmablasts has been identified in patients treated with combination ICIs., whereas circulating B cells were decreased (86). Patients experiencing organ-specific irAEs had a low baseline autoantibody level at baseline, which was significantly increased by 6 weeks after initiation of ICI therapy (87). This finding may suggest that the pathogenesis of irAEs is different from that of classic ADs, where baseline autoantibody levels positively correlate with AD development.
The role of disease-specific autoantibodies in the development of irAEs remains controversial. Sakakida et al. reported that positive anti-nuclear antibody (ANA) titers are associated with a higher risk of ICI-mediated colitis but not classic ANA-associated autoimmune diseases like SLE and scleroderma (88). However, other investigators reported a correlation between the occurrence of organ-specific irAEs and disease-specific autoantibody levels. Osorio et al. found that thyroid dysfunction is associated with anti-thyroid antibodies, whereas Suzuki et al. showed that ICI-related myasthenia gravis is associated with anti-acetylcholine receptor (AChR) antibodies (89, 90). In a retrospective study of patients with non-small cell lung cancer (NSCLC) treated with anti-PD-1, pre-existing ANA, rheumatoid factor (RF), anti-thyroiglobulin and anti-thyroid peroxidase antibodies positivity correlated with the development of irAEs but also with clinical benefit from ICI immunotherapy (91). Notably, these findings were confirmed by a smaller prospective study which determined that the levels of anti-thyroiglobulin and anti-thyroid peroxidase antibodies at baseline were higher in patients who developed thyroiditis after anti-PD-1 blocking immunotherapy (92). Other studies reported that pre-existing AChR antibodies are commonly found in patients who develop irAE-associated myositis and myasthenia gravis (93, 94).
Complement-mediated
Since ICIs are monoclonal antibodies, they can bind to off-target tissues and cause inflammation through classical complement cascade activation. This mechanism is thought to play a role in the pathogenesis of anti-CTLA4-mediated hypophysitis by binding abundantly expressed CTLA4 antigen in the pituitary, activating complement, and causing development of complement-dependent antibody-mediated cytotoxicity (CDC) against cells secreting thyrotropin, follicle-stimulating hormone or corticotropin. These cells became the site of deposition of C3d and C4d components and activation of an inflammatory cascade mimicking type II hypersensitivity (95). Autopsy results of patients who developed hypophysitis after treatment with ipilimumab showed that type II hypersensitivity was involved in the early stages of pathogenesis, whereas type IV hypersensitivity caused by infiltration of autoreactive T lymphocytes played a role in the later stages (96).
Cytokine-mediated
Since cytokines are mediators of inflammation, they can be used to assess the direction of the systemic immune response. ICI treatment causes a shift of cytokine balance toward an inflammatory profile. For example, elevated circulating IL-17, which is secreted especially by Th17 cells suppressing Treg activity, and IL-2 which enhances activity of cytotoxic CD8+ T cells, are associated with the development of irAEs (97, 98). Lim et al. defined a toxicity score (CYTOX) by integrating expression of 11 circulating pro-inflammatory cytokines (G-CSF, GM-CSF, Fractalkine, FGF-2, IFNα2, IL-12p70, IL-1a, IL-1b, IL-1RA, IL-2, IL-13) (98). CYTOX score were significantly higher in patients developed irAEs (98). Certain cytokines have been shown to be involved in the development of specific irAEs. For example, psoriasiform dermatitis has been correlated with increased IL-6 levels, pneumonitis with increased IL-1β, and colitis with increased IL-17 (99–101). In addition, chemokine ligands such as CXCL9, may also potentially contribute to the pathogenesis of irAEs (102).
Inflammatory monocyte-mediated
The inflammatory milieu generated by the production of effector cytokines by immune cells which are disinhibited by blockade of ICs during ICI immunotherapy, promotes the differentiation of M1-like monocytes which produce proinflammatory cytokines, such as TNF-α and IL-1β, and convert into macrophages in target tissues. Such inflammatory monocytes can also be generated by direct inhibition of PD-1 mediated signaling during differentiation of myeloid progenitors in response to hematopoietic growth factors produced by cancer and activated T cells (14, 15). Such inflammatory monocytes infiltrate target organs of irAEs such as the lung where they cause severe pneumonitis with granulomatosis (103) and the myocardium, causing life threatening myocarditis (104).
Host-specific factors
Factors that regulate immune responses by complex tissue-specific and systemic mechanisms, such as microbiota and gene polymorphisms, have been shown to play important roles in the development of irAEs (105–107). Polymorphism of microRNA146, which is associated with autoimmune diseases and is known to promote a proinflammatory Treg behavior, was shown to correlate with the development of severe irAEs (106, 107). Similarly to conventional ADs, several HLA types have been implicated in the pathogenesis of irAEs. Associations have been identified between HLA DRB1*04: 05 and ICI-induced inflammatory arthritis, HLA-DRB1*11:01 and pruritus, HLA-DQB1*03:01 and colitis (108, 109). However, a recent study reported no association between HLA type and irAE development (110). Thus, further work is required toward this direction.
The gut microbiome has been extensively studied regarding its tentative role in the therapeutic efficacy and toxicity of ICI therapy. Clostridiales, Ruminococcaceae or Faecalibacterium abundance and high diversity in the microbiota are associated with higher numbers of circulating T cells and responses to anti PD-1 immunotherapy in melanoma patients (111). In a recent prospective study of patients with advanced melanoma who were treated with a combination of anti-PD-1 and anti-CTLA-4 ICIs, the profiling of gut microbiota demonstrated a significantly higher pre-treatment fecal abundance of Bacteroides intestinalis in patients with any ≥ grade 3 toxicities, which correlated with upregulation of mucosal IL-1β in biopsy samples and a more diverse peripheral T cell repertoire (105). These findings contrasted with a previous report which had found that pre-treatment faucal abundance of Bacteroidetes phylum correlated with resistance to the development of colitis following ICI monotherapy with CTLA-4 blockade (112). It is evident that further work is required to understand the biological relevance and the potential exploitation of the microbiome to enhance the efficacy and limit the toxicity of ICI cancer immunotherapy (113).
Type of ICI immunotherapy
A systematic review of 35 randomized clinical trials consisting of 16,485 patients showed that the profile of irAEs depends on the type of immunotherapy. Colitis and hypophysitis were seen more often with CTLA-4 inhibitors, while hypothyroidism, hyperthyroidism, and pneumonitis were more common in PD-1 inhibitors (114). Furthermore, anti-CTLA-4-induced irAEs generally tend to be more frequent and severe (115). Although the differences in irAEs between ICI therapies have not been fully identified, the distinct prevalence of organ-specific irAEs might be related to the unique properties of the targeted receptors. CTLA-4 suppresses T cell responses in the early steps of the activation cascade in lymphoid organs, whereas PD-1 acts in the late stages of the immune response in both lymphoid organs and peripheral tissues (72). CTLA-4 is expressed by T cells and binds to its ligands CD80 or CD86, which are present on professional antigen-presenting cells, while PD-1 is expressed by T cells and many other immune cell types, and its ligand PD-L1 is present on several types of immune cells but also somatic cells and cancer (116–118). Due to the differential expression of CTLA-4 and PD-1, the combined use of PD-1 and CTLA-4 antibodies is associated with an increased risk of irAEs (119, 120), but also prolonged progression-free survival (121, 122).
irAEs and therapeutic response to ICI immunotherapy
Although the occurrence of irAEs indicates that the immune system has been successfully activated by ICI therapy suggesting that treatment has achieved its goal, there are conflicting data regarding the correlation between irAEs and therapeutic response. Recent studies showed that the development of irAEs in patients with various cancers receiving anti-PD-1 antibody is associated with a significantly higher response rate and increased progression-free survival rate (123, 124). These were consistent with earlier studies on patients treated with CTLA-4 inhibitors, which also showed that the development of irAEs correlated with a high response rate (125, 126). Subsequently, other reports indicated that the occurrence of irAEs in the early stages of treatment was associated with better outcomes, whereas other investigators found no correlation between irAEs and treatment outcome (70). Recently, it was noted that specific irAEs might be selectively associated with therapeutic response and might serve as biomarkers to predict clinical benefit. For example, thyroid, cutaneous and low-grade irAEs, as well as AD flares, are positively correlated with therapeutic outcome, whereas pneumonitis is associated with poor outcomes (127–130).
Diagnosis of iRAEs and therapeutic intervention
Early diagnosis and determination of the severity of irAEs are crucial to prevent morbidity and mortality. The grading system of irAEs is based on Common Terminology Criteria for Adverse Events (CTCAE) and grades are categorized from 1 to 5 according to the severity. Clinical criteria for diagnosis, grading, and treatment of irAEs have been reviewed extensively elsewhere (69, 70, 131).
Briefly, ICI discontinuation is not necessary in grade 1 irAEs when close monitoring is feasible, whereas holding ICIs should be considered for most grade 2 toxicities. In severe irAEs (grade 3-4), ICI should be discontinued, and steroid should be initiated. If symptoms worsen, steroid-sparing biologic immunosuppressants such as tumor necrosis factor-alpha (TNF-α) inhibitors or IL-6 antagonists should be considered (71). Corticosteroids are the first-line treatment modality and should be administered with dose adjustment according to the severity of symptoms for ≥ 2 grade irAEs (71). If symptoms do not improve with high-dose steroids, TNFα inhibitors can be an option (71). The management of endocrine and rheumatologic irAEs, which often cause chronic morbidity, may differ from therapy for other irAEs. A national multicenter study showed among 117 patients who developed rheumatologic irAEs, 44 patients required disease-modifying anti-rheumatic drugs (DMARD) (132). Hormone replacement is essential in the management of endocrine irAEs. Unlike other systemic irAEs, high-dose steroids are not required because it is unlikely to improve endocrine irAEs caused by damage to endocrine cells.
Cutaneous toxicities have been reported for 30% to 50% of all side effects of ICI therapies (133) and are classified separately. The ASCO committee divided dermatological irAEs into rash/inflammatory dermatitis, bullous dermatoses, and severe cutaneous adverse effects (SCARs) including Stevens-Johnson Syndrome (SJS), toxic epidermal necrolysis (TEN) and drug rash with eosinophilia and systemic symptoms (DRESS). Treatments for these skin manifestations depend on grading. Inflammatory reaction that affects the quality of life or is grade ≥ 2 should be considered as an indication to hold the ICI and monitor patients weekly for improvement. Blistering lesions that extend more than 10-30% of body surface area (BSA) or grade ≥ 2 should mandate withdrawing the ICI therapy and urge dermatological work up. Development of blistering lesions covering ≥ 30% of BSA mandates permanent discontinuation of ICI. SCARs, including SJS, TEN, acute generalized exanthematous pustulosis (AGEP), and DRESS/drug-induced hypersensitivity syndrome (DiHS) should mandate immediate discontinuation of ICI therapy, regardless of the grade, and close follow-up. Dermatology consultation is critical for appraising the risks and benefits of withdrawing or rechallenging with ICI immunotherapy. Systemic steroids are used in grade 3 of rash and inflammatory lesions. Blistering that affects the quality of life and meets the criteria for grade 2 toxicity with 10-30% BSA involvement should be managed with high potency topical steroid and consideration for systemic steroids therapy. For SCARs, prednisolone or equivalent agent should be initiated in grade 2, which is a morbilliform exanthem at 10-30% BSA together with a systemic symptom or lymphadenopathy.
A pigmented lesion which is a vitiligo-like depigmentation (VLD) has been reported exclusively in melanoma patients. To study the cumulative incidence of VLD, 137 studies conducted in patients with stage III or IV melanoma were recruited and showed an overall cumulative incidence of VLD at 3.4% (95% CI, 2.5% to 4.5%). The development of VLD significantly correlated with response to ICI therapy. Patients who developed VLD during ICI treatment for melanoma had a 2-fold lower risk of disease progression and a 4-fold lower risk of death, progression-free survival (hazard ratio [HR], 0.51; 95% CI, 0.32 to 0.82; p<0.005) and overall survival (HR, 0.25; 95% CI, 0.10 to 0.61; p<0.003) (134). Similarly, in a retrospective analysis of 148 patients who received nivolumab plus peptide vaccine or nivolumab alone, a significantly higher overall survival was observed in patients who developed rash (hazard ratio [HR], 0.423; 95% CI, 0.243 to 0.735; p=0.001) and VLD (HR, 0.184; 95% CI, 0.036 to 0.94; p=0.012 (31). VLD occurring after ICI therapy in melanoma illustrates that melanoma-specific T cells activated after ICI also recognize shared antigens of normal melanocytes and their presence might imply a better response to immunotherapy and a favorable prognosis (135).
The impact of high-dose steroid therapy on ICI efficacy and clinical outcome remains controversial. Some studies showed that high-dose corticosteroids are associated with poor outcomes (123, 136, 137) warning for cautious consideration of all parameters when the use of high-dose steroids is a tentative choice. Notably, a recent study revealed that only 47% of irAEs were managed according to guidelines, whereas 38.8% of irAEs had no documented management (138).
The role of ICIs in patients with cancer and pre-existing ADs
In the modern era of increasing incidence of cancer and ADs, up to a quarter of patients have both diseases at the same time (139). Thus, there are many cancer patients with pre-existing ADs who might be benefit from the use of ICI. Treatment of patients with pre-existing AD raises concerns for potential flair-ups due to their pre-activated immune system. As a result, clinical trials preclude enrollment of these patients. However, this raises important questions regarding the efficacy of ICI treatment in these patients and the potential complications such as flares or de novo irAEs post-ICI treatment if patients were administered immunosuppression for their AD. As currently there is increasing interest regarding eligibility of patients with pre-existing AD for ICI therapies, several retrospective studies and meta-analyses have begun assessing outcomes and toxicities in this cohort of patients (140–155). Key studies are summarized in Table 1.
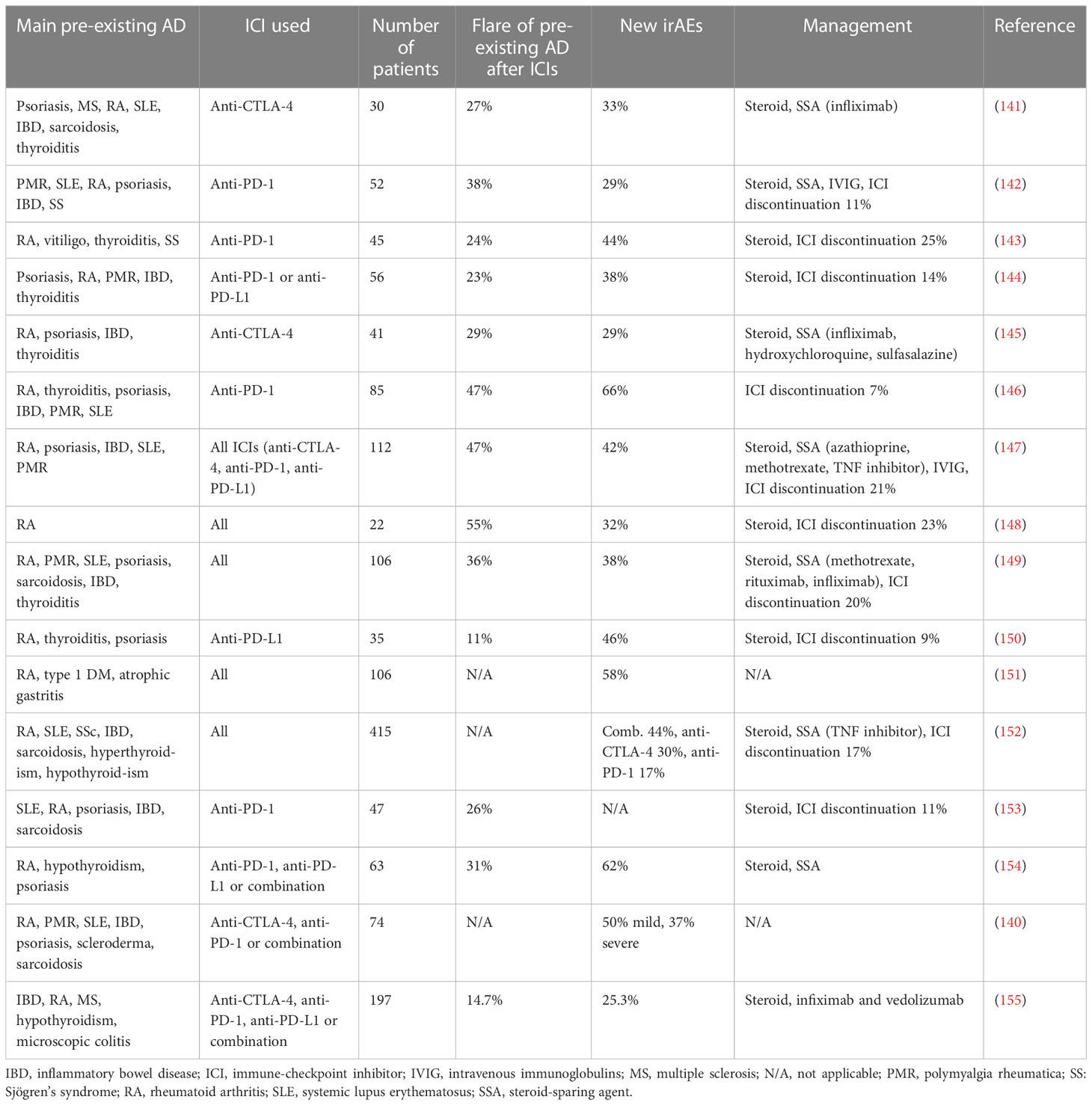
Table 1 Summary of key studies using ICI in patients with cancer and pre-existing autoimmune diseases.
Psoriasis and psoriatic arthritis patients have the highest risk of developing flare or de novo irAEs after ICI treatment (147, 156). Patients with rheumatoid arthritis (RA) experience flares of their disease at approximately 60% (147, 157). A retrospective study showed that there is no association between the presence of pre-existing AD and the development of irAEs, the number of irAEs, and their severity (154). In a study on melanoma and NSCLC patients, there was no association between overall survival and the presence of AD, however, a later study showed that patients with pre-existing AD had an increased overall survival compared to patients without AD (157). Other studies have shown that the development of irAEs is more prevalent in patients with a pre-existing AD (157, 158). Gulati et al. showed that melanoma patients with pre-existing AD had an increased progression-free survival and development of irAEs when treated with ICI (155). Patients with pre-existing AD who received ICI and survived for more than 1 year developed new-onset chronic kidney disease and experienced a rapid drop in glomerular filtration rate, which is used as a marker for kidney function (159).
Development of de novo irAEs is more common in patients with pre-existing AD after administration of ICI. Patients with RA have a higher frequency of all-grade irAEs and severe irAEs (157). A separate study found that only 14.7% of patients had an AD flare while 25.3% developed a new irAEs (140). Most of the patients in this study had quiescent AD and 9.6% were receiving steroid treatment at the time ICI therapy was initiated. Patients with gastrointestinal and rheumatologic AD had the most frequent flare-ups after ICI therapy, whereas patients with Hashimoto thyroiditis and neurologic AD developed mostly new irAEs. Studies with over 100 patients with pre-existing AD who received ICI treatment showed that 25-50% developed flares of their disease, whereas about 40% developed de novo irAEs (160). Abdel-Wahad et al. reported that no differences were observed in the presentation of irAEs between active and inactive AD, however, Wu et al. showed that patients with inactive AD presented high-grade irAEs more frequently compared to patients with active AD (156, 161). Moreover, patients on immunosuppression at the initiation of ICI showed lower rates for irAEs, especially high-grade, compared to those who were not on immunosuppression (156, 161).
Disease flares were increased in patients receiving anti-PD-1/PD-L1 treatment, whereas de novo irAEs were observed more often when patients received anti-CTLA-4 therapy (156). In a small study where anti-PD-L1 and anti-PD-1 treatment was administered to cancer patients with systemic sclerosis, 24% of patients experienced flares and almost 60% developed irAEs, with only 6% developing grade 3-4 irAEs (162). Patients with AD who received anti-PD-1 treatment developed irAEs of any grade at an increased level compared to those with no pre-existing AD (163). In a multicenter study, 33% of patients with advanced melanoma with AD which received a combination of anti-CTLA-4 and anti-PD-1 treatment developed a flare, most commonly rheumatic or gastrointestinal irAEs (164). Interestingly, patients without immunosuppression before the administration of ICI had an increased overall survival compared to those who were on immunosuppression. Furthermore, patients on immunosuppression had an increased risk of developing flares.
A novel retrospective study showed that patients who received ICI and had a pre-existing AD (most common being RA, psoriasis, and polymyalgia rheumatica) developed cardiovascular irAEs more often than patients without AD (64) indicating that such patients should be actively monitored for cardiovascular toxicities. However, more than two-thirds of patients receiving immunosuppression for their AD before ICI administration developed other non-cardiovascular-related adverse effects.
Patients without pre-existing AD had increased association of developing rheumatic-irAEs if they were positive for rheumatoid factor before getting administered ICI (165). Interestingly, other studies reported that the presence of rheumatic-irAEs and ICI-arthritis resulted in increased overall survival and only 35% of patients with pre-exisiting AD developed flare-ups. ICI-induced thyroiditis has been shown to be associated with improved overall survival and progression-free survival compared to patients who did not develop thyroiditis (166, 167). The association between overall survival and thyroiditis varied between different tumors, however, it was related strongly with lung cancer.
Conclusions and perspectives
There is a substantial number of patients with AD and cancer. Because such patients were excluded from clinical trials, only limited data are available regarding the efficacy and safety of ICIs in this large patient population. The limited number of studies available have shown that using immunotherapy in patients with ADs causes an increased risk of irAEs and AD flares, but irAEs are mostly transient and manageable and rarely mandate treatment discontinuation and life-threatening complications. The use of ICIs in patients with AD is often equally safe as in patients without AD. Hence, pre-existent AD is not an absolute contraindication for the use of ICIs. The lack of a validated predictive biomarker requires a careful evaluation for early recognition of irAEs.
The less frequent and milder adverse effect profile of PD-1 inhibitors compared to CTLA-4 inhibitors suggests that PD-1 inhibitors might be considered as the immunotherapy of choice for cancer patients with pre-existing ADs. In cases that develop irAEs with or without pre-existing ADs a multidisciplinary approach and close monitoring are mandatory. Since the pathogenesis of organ-specific irAEs is unique, approaches of personalized therapy will be required to avoid non-specific immunosuppression and preserve the therapeutic benefit of ICI immunotherapy.
Author contributions
BI wrote the main body of the manuscript and prepared figures. KA wrote main sections of the manuscript. CC wrote sections of the manuscript. SY wrote sections of the manuscript. VB wrote sections of the manuscript and supervised all the co-authors in the preparation of their contributions. All authors reviewed the manuscript and agree with its entire content. All authors contributed to the article.
Funding
This work was supported by NIH/NCI grants CA238263, CA229784 and CA257267 (VB).
Conflict of interest
VB has patents on the PD-1 pathway licensed by Bristol-Myers Squibb, Roche, Merck, EMD-Serono, Boehringer Ingelheim, AstraZeneca, Novartis and Dako.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science (2011) 331:1565–70. doi: 10.1126/science.1203486
2. Yenyuwadee S, Aliazis K, Wang Q, Christofides A, Shah R, Patsoukis N, et al. Immune cellular components and signaling pathways in the tumor microenvironment. Semin Cancer Biol (2022) 86:187–201. doi: 10.1016/j.semcancer.2022.08.004
3. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science (2016) 354:1160–5. doi: 10.1126/science.aaf2807
4. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature (2016) 537:417–21. doi: 10.1038/nature19330
5. Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity (2016) 45:389–401. doi: 10.1016/j.immuni.2016.07.011
6. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332:600–3. doi: 10.1126/science.1202947
7. Tekguc M, Wing JB, Osaki M, Long J, Sakaguchi S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc Natl Acad Sci USA (2021) 118:e2023739118. doi: 10.1073/pnas.2023739118
8. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T Cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science (2017) 355:1428–33. doi: 10.1126/science.aaf1292
9. Mizuno R, Sugiura D, Shimizu K, Maruhashi T, Watada M, Okazaki I-M, et al. PD-1 primarily targets TCR signal in the inhibition of functional T cell activation. Front Immunol (2019) 10:630. doi: 10.3389/fimmu.2019.00630
10. Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. Selective effects of PD-1 on akt and ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal (2012) 5:ra46. doi: 10.1126/scisignal.2002796
11. Lau J, Cheung J, Navarro A, Lianoglou S, Haley B, Totpal K, et al. Tumour and host cell PD-L1 is required to mediate suppression of anti-tumour immunity in mice. Nat Commun (2017) 8:14572. doi: 10.1038/ncomms14572
12. Oh SA, Wu D-C, Cheung J, Navarro A, Xiong H, Cubas R, et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat Cancer (2020) 1:681–91. doi: 10.1038/s43018-020-0075-x
13. Peng Q, Qiu X, Zhang Z, Zhang S, Zhang Y, Liang Y, et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat Commun (2020) 11:4835. doi: 10.1038/s41467-020-18570-x
14. Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol (2020) 5:eaay1863. doi: 10.1126/sciimmunol.aay1863
15. Christofides A, Katopodi X-L, Cao C, Karagkouni D, Aliazis K, Yenyuwadee S, et al. SHP-2 and PD-1-SHP-2 signaling regulate myeloid cell differentiation and antitumor responses. Nat Immunol (2023) 24:55–68. doi: 10.1038/s41590-022-01385-x
16. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature (2017) 545:495–9. doi: 10.1038/nature22396
17. Andrews LP, Yano H, Vignali DAA. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups. Nat Immunol (2019) 20:1425–34. doi: 10.1038/s41590-019-0512-0
18. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3:541–7. doi: 10.1016/1074-7613(95)90125-6
19. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in ctla-4. Science (1995) 270:985–8. doi: 10.1126/science.270.5238.985
20. Chambers CA, Sullivan TJ, Allison JP. Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity (1997) 7:885–95. doi: 10.1016/s1074-7613(00)80406-9
21. Klocke K, Sakaguchi S, Holmdahl R, Wing K. Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood. Proc Natl Acad Sci USA (2016) 113:E2383–92. doi: 10.1073/pnas.1603892113
22. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322:271–5. doi: 10.1126/science.1160062
23. Schmidt EM, Wang CJ, Ryan GA, Clough LE, Qureshi OS, Goodall M, et al. Ctla-4 controls regulatory T cell peripheral homeostasis and is required for suppression of pancreatic islet autoimmunity. J Immunol (2009) 182:274–82. doi: 10.4049/jimmunol.182.1.274
24. Verhagen J, Gabrysová L, Minaee S, Sabatos CA, Anderson G, Sharpe AH, et al. Enhanced selection of FoxP3+ T-regulatory cells protects CTLA-4-deficient mice from CNS autoimmune disease. Proc Natl Acad Sci USA (2009) 106:3306–11. doi: 10.1073/pnas.0803186106
25. Paterson AM, Lovitch SB, Sage PT, Juneja VR, Lee Y, Trombley JD, et al. Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J Exp Med (2015) 212:1603–21. doi: 10.1084/jem.20141030
26. Yang Y, Li X, Ma Z, Wang C, Yang Q, Byrne-Steele M, et al. CTLA-4 expression by b-1a b cells is essential for immune tolerance. Nat Commun (2021) 12:525. doi: 10.1038/s41467-020-20874-x
27. Perrin PJ, Maldonado JH, Davis TA, June CH, Racke MK. CTLA-4 blockade enhances clinical disease and cytokine production during experimental allergic encephalomyelitis. J Immunol (1996) 157:1333–6. doi: 10.4049/jimmunol.157.4.1333
28. Hurwitz AA, Sullivan TJ, Krummel MF, Sobel RA, Allison JP. Specific blockade of CTLA-4/B7 interactions results in exacerbated clinical and histologic disease in an actively-induced model of experimental allergic encephalomyelitis. J Neuroimmunol (1997) 73:57–62. doi: 10.1016/s0165-5728(96)00168-3
29. Lühder F, Höglund P, Allison JP, Benoist C, Mathis D. Cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) regulates the unfolding of autoimmune diabetes. J Exp Med (1998) 187:427–32. doi: 10.1084/jem.187.3.427
30. Wing JB, Ise W, Kurosaki T, Sakaguchi S. Regulatory T cells control antigen-specific expansion of tfh cell number and humoral immune responses via the coreceptor CTLA-4. Immunity (2014) 41:1013–25. doi: 10.1016/j.immuni.2014.12.006
31. Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature (2003) 423:506–11. doi: 10.1038/nature01621
32. Awata T, Kurihara S, Iitaka M, Takei S, Inoue I, Ishii C, et al. Association of CTLA-4 gene a-G polymorphism (IDDM12 locus) with acute-onset and insulin-depleted IDDM as well as autoimmune thyroid disease (Graves’ disease and hashimoto’s thyroiditis) in the Japanese population. Diabetes (1998) 47:128–9. doi: 10.2337/diab.47.1.128
33. Chen Y, Chen S, Gu Y, Feng Y, Shi Y, Fu Q, et al. CTLA-4 +49 G/A, a functional T1D risk SNP, affects CTLA-4 level in treg subsets and IA-2A positivity, but not beta-cell function. Sci Rep (2018) 8:10074. doi: 10.1038/s41598-018-28423-9
34. Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet (2010) 42:508–14. doi: 10.1038/ng.582
35. Agarwal K, Jones DE, Daly AK, James OF, Vaidya B, Pearce S, et al. CTLA-4 gene polymorphism confers susceptibility to primary biliary cirrhosis. J Hepatol (2000) 32:538–41. doi: 10.1016/s0168-8278(00)80213-5
36. Kaufman KA, Bowen JA, Tsai AF, Bluestone JA, Hunt JS, Ober C. The CTLA-4 gene is expressed in placental fibroblasts. Mol Hum Reprod (1999) 5:84–7. doi: 10.1093/molehr/5.1.84
37. Oaks MK, Hallett KM. Cutting edge: a soluble form of CTLA-4 in patients with autoimmune thyroid disease. J Immunol (2000) 164:5015–8. doi: 10.4049/jimmunol.164.10.5015
38. Saverino D, Brizzolara R, Simone R, Chiappori A, Milintenda-Floriani F, Pesce G, et al. Soluble CTLA-4 in autoimmune thyroid diseases: relationship with clinical status and possible role in the immune response dysregulation. Clin Immunol (2007) 123:190–8. doi: 10.1016/j.clim.2007.01.003
39. Wang XB, Kakoulidou M, Giscombe R, Qiu Q, Huang D, Pirskanen R, et al. Abnormal expression of CTLA-4 by T cells from patients with myasthenia gravis: effect of an AT-rich gene sequence. J Neuroimmunol (2002) 130:224–32. doi: 10.1016/s0165-5728(02)00228-x
40. Wong CK, Lit LC, Tam LS, Li EK, Lam CW. Aberrant production of soluble costimulatory molecules CTLA-4, CD28, CD80 and CD86 in patients with systemic lupus erythematosus. Rheumatol (Oxford) (2005) 44:989–94. doi: 10.1093/rheumatology/keh663
41. Nishimura H, Minato N, Nakano T, Honjo T. Immunological studies on PD-1 deficient mice: implication of PD-1 as a negative regulator for b cell responses. Int Immunol (1998) 10:1563–72. doi: 10.1093/intimm/10.10.1563
42. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity (1999) 11:141–51. doi: 10.1016/s1074-7613(00)80089-8
43. Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol (1985) 37:269–390. doi: 10.1016/s0065-2776(08)60342-9
44. Santiago-Raber ML, Laporte C, Reininger L, Izui S. Genetic basis of murine lupus. Autoimmun Rev (2004) 3:33–9. doi: 10.1016/S1568-9972(03)00062-4
45. Wang J, Okazaki IM, Yoshida T, Chikuma S, Kato Y, Nakaki F, et al. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int Immunol (2010) 22:443–52. doi: 10.1093/intimm/dxq026
46. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science (2001) 291:319–22. doi: 10.1126/science.291.5502.319
47. Okazaki T, Tanaka Y, Nishio R, Mitsuiye T, Mizoguchi A, Wang J, et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat Med (2003) 9:1477–83. doi: 10.1038/nm955
48. Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci USA (2005) 102:11823–8. doi: 10.1073/pnas.0505497102
49. Vilgelm AE, Johnson DB, Richmond A. Combinatorial approach to cancer immunotherapy: strength in numbers. J Leukocyte Biol (2016) 100:275–90. doi: 10.1189/jlb.5ri0116-013rr
50. Haslam A, Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw Open (2019) 2:e192535. doi: 10.1001/jamanetworkopen.2019.2535
51. Xing P, Zhang F, Wang G, Xu Y, Li C, Wang S, et al. Incidence rates of immune-related adverse events and their correlation with response in advanced solid tumours treated with NIVO or NIVO+IPI: a systematic review and meta-analysis. J immunotherapy Cancer (2019) 7:341. doi: 10.1186/s40425-019-0779-6
52. Bomze D, Hasan Ali O, Bate A, Flatz L. Association between immune-related adverse events during anti-PD-1 therapy and tumor mutational burden. JAMA Oncol (2019) 5:1633–5. doi: 10.1001/jamaoncol.2019.3221
53. Pistillo MP, Fontana V, Morabito A, Dozin B, Laurent S, Carosio R, et al. Soluble CTLA-4 as a favorable predictive biomarker in metastatic melanoma patients treated with ipilimumab: an Italian melanoma intergroup study. Cancer Immunology Immunotherapy (2019) 68:97–107. doi: 10.1007/s00262-018-2258-1
54. Tyan K, Baginska J, Brainard M, Giobbie-Hurder A, Severgnini M, Manos M, et al. Cytokine changes during immune-related adverse events and corticosteroid treatment in melanoma patients receiving immune checkpoint inhibitors. Cancer Immunology Immunotherapy (2021) 70:2209–21. doi: 10.1007/s00262-021-02855-1
55. Valpione S, Pasquali S, Campana LG, Piccin L, Mocellin S, Pigozzo J, et al. Sex and interleukin-6 are prognostic factors for autoimmune toxicity following treatment with anti-CTLA4 blockade. J Trans Med (2018) 16(1):94. doi: 10.1186/s12967-018-1467-x
56. Wang DY, Salem JE, Cohen JV, Chandra S, Menzer C, Ye F, et al. Fatal toxic effects associated with immune checkpoint inhibitors: a systematic review and meta-analysis. JAMA Oncol (2018) 4:1721–8. doi: 10.1001/jamaoncol.2018.3923
57. Moslehi JJ, Salem J-E, Sosman JA, Lebrun-Vignes B, Johnson DB. Increased reporting of fatal immune checkpoint inhibitor-associated myocarditis. Lancet (2018) 391:933. doi: 10.1016/S0140-6736(18)30533-6
58. Thavendiranathan P, Zhang L, Zafar A, Drobni ZD, Mahmood SS, Cabral M, et al. Myocardial T1 and T2 mapping by magnetic resonance in patients with immune checkpoint inhibitor-associated myocarditis. J Am Coll Cardiol (2021) 77:1503–16. doi: 10.1016/j.jacc.2021.01.050
59. Roopkumar J, Swaidani S, Kim AS, Thapa B, Gervaso L, Hobbs BP, et al. Increased incidence of venous thromboembolism with cancer immunotherapy. Med (N Y) (2021) 2:423–34. doi: 10.1016/j.medj.2021.02.002
60. Drobni ZD, Alvi RM, Taron J, Zafar A, Murphy SP, Rambarat PK, et al. Association between immune checkpoint inhibitors with cardiovascular events and atherosclerotic plaque. Circulation (2020) 142:2299–311. doi: 10.1161/CIRCULATIONAHA.120.049981
61. Gotsman I, Grabie N, Dacosta R, Sukhova G, Sharpe A, Lichtman AH. Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J Clin Invest (2007) 117:2974–82. doi: 10.1172/JCI31344
62. Koga N, Suzuki J, Kosuge H, Haraguchi G, Onai Y, Futamatsu H, et al. Blockade of the interaction between PD-1 and PD-L1 accelerates graft arterial disease in cardiac allografts. Arterioscler Thromb Vasc Biol (2004) 24:2057–62. doi: 10.1161/01.ATV.0000145015.23656.e4
63. Poels K, van Leent MMT, Boutros C, Tissot H, Roy S, Meerwaldt AE, et al. Immune checkpoint inhibitor therapy aggravates T cell-driven plaque inflammation in atherosclerosis. JACC CardioOncol (2020) 2:599–610. doi: 10.1016/j.jaccao.2020.08.007
64. Lee C, Drobni ZD, Zafar A, Gongora CA, Zlotoff DA, Alvi RM, et al. Pre-existing autoimmune disease increases the risk of cardiovascular and noncardiovascular events after immunotherapy. JACC CardioOncol (2022) 4:660–9. doi: 10.1016/j.jaccao.2022.11.008
65. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir E-AD, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med (2019) 25:1576–88. doi: 10.1038/s41591-019-0590-4
66. Roy P, Orecchioni M, Ley K. How the immune system shapes atherosclerosis: roles of innate and adaptive immunity. Nat Rev Immunol (2022) 22:251–65. doi: 10.1038/s41577-021-00584-1
67. Patrinely JR, Johnson R, Lawless AR, Bhave P, Sawyers A, Dimitrova M, et al. Chronic immune-related adverse events following adjuvant anti-PD-1 therapy for high-risk resected melanoma. JAMA Oncol (2021) 7:744–8. doi: 10.1001/jamaoncol.2021.0051
68. Johnson DB, Nebhan CA, Moslehi JJ, Balko JM. Immune-checkpoint inhibitors: long-term implications of toxicity. Nat Rev Clin Oncol (2022) 19:254–67. doi: 10.1038/s41571-022-00600-w
69. Brahmer JR, Abu-Sbeih H, Ascierto PA, Brufsky J, Cappelli LC, Cortazar FB, et al. Society for immunotherapy of cancer (SITC) clinical practice guideline on immune checkpoint inhibitor-related adverse events. J Immunother Cancer (2021) 9:e002435. doi: 10.1136/jitc-2021-002435
70. Horvat TZ, Adel NG, Dang TO, Momtaz P, Postow MA, Callahan MK, et al. Immune-related adverse events, need for systemic immunosuppression, and effects on survival and time to treatment failure in patients with melanoma treated with ipilimumab at memorial sloan kettering cancer center. J Clin Oncol (2015) 33:3193–8. doi: 10.1200/JCO.2015.60.8448
71. Schneider BJ, Naidoo J, Santomasso BD, Adkins LC, Anadkat SM, Atkins MB, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: ASCO guideline update. J Clin Oncol (2021) 39:4073–126. doi: 10.1200/JCO.21
72. Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. New Engl J Med (2018) 378:158–68. doi: 10.1056/nejmra1703481
73. Robert L, Tsoi J, Wang X, Emerson R, Homet B, Chodon T, et al. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin Cancer Res (2014) 20:2424–32. doi: 10.1158/1078-0432.CCR-13-2648
74. Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med (2016) 375:1749–55. doi: 10.1056/NEJMoa1609214
75. Hutchinson JA, Kronenberg K, Riquelme P, Wenzel JJ, Glehr G, Schilling HL, et al. Virus-specific memory T cell responses unmasked by immune checkpoint blockade cause hepatitis. Nat Commun (2021) 12(1):1439. doi: 10.1038/s41467-021-21572-y
76. Johnson DB, McDonnell WJ, Gonzalez-Ericsson PI, Al-Rohil RN, Mobley BC, Salem JE, et al. A case report of clonal EBV-like memory CD4+ T cell activation in fatal checkpoint inhibitor-induced encephalitis. Nat Med (2019) 25:1243–50. doi: 10.1038/s41591-019-0523-2
77. Kotwal A, Gustafson MP, Bornschlegl S, Kottschade L, Delivanis DA, Dietz AB, et al. Immune checkpoint inhibitor-induced thyroiditis is associated with increased intrathyroidal T lymphocyte subpopulations. Thyroid (2020) 30:1440–50. doi: 10.1089/thy.2020.0075
78. Luoma AM, Suo S, Williams HL, Sharova T, Sullivan K, Manos M, et al. Molecular pathways of colon inflammation induced by cancer immunotherapy. Cell (2020) 182:655–671.e22. doi: 10.1016/j.cell.2020.06.001
79. Yoneda S, Imagawa A, Hosokawa Y, Baden MY, Kimura T, Uno S, et al. T-Lymphocyte infiltration to islets in the pancreas of a patient who developed type 1 diabetes after administration of immune checkpoint inhibitors. Diabetes Care (2019) 42:e116–8. doi: 10.2337/dc18-2518
80. Lozano AX, Chaudhuri AA, Nene A, Bacchiocchi A, Earland N, Vesely MD, et al. T Cell characteristics associated with toxicity to immune checkpoint blockade in patients with melanoma. Nat Med (2022) 28:353–62. doi: 10.1038/s41591-021-01623-z
81. Fan NW, Wang S, Ortiz G, Chauhan SK, Chen Y, Dana R. Autoreactive memory Th17 cells are principally derived from T-bet+RORγt+ Th17/1 effectors. J Autoimmun (2022) 129:102816. doi: 10.1016/J.JAUT.2022.102816
82. Franken A, Mol PV, Vanmassenhove S, Donders E, Schepers R, Brussel TV, et al. Single-cell transcriptomics identifies pathogenic T-helper 17.1 cells and pro-inflammatory monocytes in immune checkpoint inhibitor-related pneumonitis. J ImmunoTherapy Cancer (2022) 10(9):e005323. doi: 10.1136/jitc-2022-005323
83. Grigoriou M, Banos A, Hatzioannou A, Kloetgen A, Kouzis P, Aggouraki D, et al. Regulatory T-cell transcriptomic reprogramming characterizes adverse events by checkpoint inhibitors in solid tumors. Cancer Immunol Res (2021) 9:726–34. doi: 10.1158/2326-6066.CIR-20-0969
84. Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature (2020) 577:549–55. doi: 10.1038/s41586-019-1922-8
85. Patel AJ, Willsmore ZN, Khan N, Richter A, Naidu B, Drayson MT, et al. Regulatory b cell repertoire defects predispose lung cancer patients to immune-related toxicity following checkpoint blockade. Nat Commun (2022) 13:3148. doi: 10.1038/s41467-022-30863-x
86. Das R, Bar N, Ferreira M, Newman AM, Zhang L, Bailur JK, et al. Early b cell changes predict autoimmunity following combination immune checkpoint blockade. J Clin Invest (2018) 128:715–20. doi: 10.1172/JCI96798
87. Ghosh N, Postow M, Zhu C, Jannat-Khah D, Li QZ, Vitone G, et al. Lower baseline autoantibody levels are associated with immune-related adverse events from immune checkpoint inhibition. J ImmunoTherapy Cancer (2022) 10(1):e004008. doi: 10.1136/jitc-2021-004008
88. Sakakida T, Ishikawa T, Chihara Y, Harita S, Uchino J, Tabuchi Y, et al. Safety and efficacy of PD-1/PD-L1 blockade in patients with preexisting antinuclear antibodies. Clin Trans Oncol (2020) 22:919–27. doi: 10.1007/s12094-019-02214-8
89. Osorio JC, Ni A, Chaft JE, Pollina R, Kasler MK, Stephens D, et al. Antibody-mediated thyroid dysfunction during T-cell checkpoint blockade in patients with non-small-cell lung cancer. Ann Oncol (2017) 28:583–9. doi: 10.1093/annonc/mdw640
90. Suzuki S, Ishikawa N, Konoeda F, Seki N, Fukushima S, Takahashi K, et al. Nivolumab-related myasthenia gravis with myositis and myocarditis in Japan. Neurology (2017) 89:1127–34. doi: 10.1212/WNL.0000000000004359
91. Toi Y, Sugawara S, Sugisaka J, Ono H, Kawashima Y, Aiba T, et al. Profiling preexisting antibodies in patients treated with anti-PD-1 therapy for advanced non-small cell lung cancer. JAMA Oncol (2019) 5:376–83. doi: 10.1001/jamaoncol.2018.5860
92. Kobayashi T, Iwama S, Yasuda Y, Okada N, Tsunekawa T, Onoue T, et al. Patients with antithyroid antibodies are prone to develop destructive thyroiditis by nivolumab: a prospective study. J Endocr Soc (2018) 2:241–51. doi: 10.1210/js.2017-00432
93. Mammen AL, Rajan A, Pak K, Lehky T, Casciola-Rosen L, Donahue RN, et al. Pre-existing antiacetylcholine receptor autoantibodies and b cell lymphopaenia are associated with the development of myositis in patients with thymoma treated with avelumab, an immune checkpoint inhibitor targeting programmed death-ligand 1. Ann Rheum Dis (2019) 78:150–2. doi: 10.1136/annrheumdis-2018-213777
94. Masi G, Pham MC, Karatz T, Oh S, Payne AS, Nowak RJ, et al. Clinicoserological insights into patients with immune checkpoint inhibitor-induced myasthenia gravis. Ann Clin Transl Neurol (2023) 10(5):825–31. doi: 10.1002/acn3.51761
95. Iwama S, Remigis AD, Callahan MK, Slovin SF, Wolchok JD, Caturegli P. Pituitary expression of CTLA-4 mediates hypophysitis secondary to administration of CTLA-4 blocking antibody. Sci Trans Med (2014) 6(230):230ra45. doi: 10.1126/scitranslmed.3008002
96. Caturegli P, Dalmazi GD, Lombardi M, Grosso F, Larman HB, Larman T, et al. Hypophysitis secondary to cytotoxic T-Lymphocyte–associated protein 4 blockade: insights into pathogenesis from an autopsy series. Am J Pathol (2016) 186:3225–35. doi: 10.1016/j.ajpath.2016.08.020
97. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi: 10.1038/nature04753
98. Lim SY, Lee JH, Gide TN, Menzies AM, Guminski A, Carlino MS, et al. Circulating cytokines predict immune-related toxicity in melanoma patients receiving Anti-PD-1–based immunotherapy. Clin Cancer Res (2019) 25:1557–63. doi: 10.1158/1078-0432.CCR-18-2795
99. Suresh K, Naidoo J, Zhong Q, Xiong Y, Mammen J, de Flores MV, et al. The alveolar immune cell landscape is dysregulated in checkpoint inhibitor pneumonitis. J Clin Invest (2019) 129:4305–15. doi: 10.1172/JCI128654
100. Tanaka R, Okiyama N, Okune M, Ishitsuka Y, Watanabe R, Furuta J, et al. Serum level of interleukin-6 is increased in nivolumab-associated psoriasiform dermatitis and tumor necrosis factor-α is a biomarker of nivolumab recativity. J Dermatol Sci (2017) 86:71–3. doi: 10.1016/j.jdermsci.2016.12.019
101. Tarhini AA, Zahoor H, Lin Y, Malhotra U, Sander C, Butterfield LH, et al. Baseline circulating IL-17 predicts toxicity while TGF-β1 and IL-10 are prognostic of relapse in ipilimumab neoadjuvant therapy of melanoma. J Immunother Cancer (2015) 3:39. doi: 10.1186/s40425-015-0081-1
102. Khan S, Khan SA, Luo X, Fattah FJ, Saltarski J, Gloria-McCutchen Y, et al. Immune dysregulation in cancer patients developing immune-related adverse events. Br J Cancer (2019) 120:63–8. doi: 10.1038/s41416-018-0155-1
103. Montaudié H, Pradelli J, Passeron T, Lacour J-P, Leroy S. Pulmonary sarcoid-like granulomatosis induced by nivolumab. Br J Dermatol (2017) 176:1060–3. doi: 10.1111/bjd.14808
104. Mahmood SS, Fradley MG, Cohen JV, Nohria A, Reynolds KL, Heinzerling LM, et al. Myocarditis in patients treated with immune checkpoint inhibitors. J Am Coll Cardiol (2018) 71:1755–64. doi: 10.1016/j.jacc.2018.02.037
105. Andrews MC, Duong CPM, Gopalakrishnan V, Iebba V, Chen WS, Derosa L, et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat Med (2021) 27:1432–41. doi: 10.1038/s41591-021-01406-6
106. Marschner D, Falk M, Javorniczky NR, Hanke-Müller K, Rawluk J, Schmitt-Graeff A, et al. MicroRNA-146a regulates immune-related adverse events caused by immune checkpoint inhibitors. JCI Insight (2020) 5(6):e132334. doi: 10.1172/jci.insight.132334
107. Zhou Q, Haupt S, Kreuzer JT, Hammitzsch A, Proft F, Neumann C, et al. Decreased expression of miR-146a and miR-155 contributes to an abnormal treg phenotype in patients with rheumatoid arthritis. Ann Rheumatic Dis (2015) 74:1265–74. doi: 10.1136/annrheumdis-2013-204377
108. Cappelli LC, Dorak MT, Bettinotti MP, Bingham CO, Shah AA. Association of HLA-DRB1 shared epitope alleles and immune checkpoint inhibitor-induced inflammatory arthritis. Rheumatol (Oxford) (2019) 58:476–80. doi: 10.1093/rheumatology/key358
109. Hasan Ali O, Berner F, Bomze D, Fässler M, Diem S, Cozzio A, et al. Human leukocyte antigen variation is associated with adverse events of checkpoint inhibitors. Eur J Cancer (2019) 107:8–14. doi: 10.1016/j.ejca.2018.11.009
110. Jiang N, Yu Y, Zhang M, Tang Y, Wu D, Wang S, et al. Association between germ-line HLA and immune-related adverse events. Front Immunol (2022) 13:952099. doi: 10.3389/fimmu.2022.952099
111. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science (2018) 359:97–103. doi: 10.1126/science.aan4236
112. Dubin K, Callahan MK, Ren B, Khanin R, Viale A, Ling L, et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun (2016) 7:10391. doi: 10.1038/ncomms10391
113. Soto Chervin C, Gajewski TF. Microbiome-based interventions: therapeutic strategies in cancer immunotherapy. Immunooncol Technol (2020) 8:12–20. doi: 10.1016/j.iotech.2020.11.001
114. Arnaud-Coffin P, Maillet D, Gan HK, Stelmes J-J, You B, Dalle S, et al. A systematic review of adverse events in randomized trials assessing immune checkpoint inhibitors: a systematic review of adverse events in randomized trials assessing immune checkpoint inhibitors. Int J Cancer (2019) 145:639–48. doi: 10.1002/ijc.32132
115. Khoja L, Day D, Wei-Wu Chen T, Siu LL, Hansen AR. Tumour- and class-specific patterns of immune-related adverse events of immune checkpoint inhibitors: a systematic review. Ann Oncol (2017) 28:2377–85. doi: 10.1093/annonc/mdx286
116. Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev (2008) 224:166–82. doi: 10.1111/j.1600-065X.2008.00662.x
117. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
118. Patsoukis N, Wang Q, Strauss L, Boussiotis VA. Revisiting the PD-1 pathway. Sci Adv (2020) 6:eabd2712. doi: 10.1126/sciadv.abd2712
119. Park R, Lopes L, Cristancho CR, Riano IM, Saeed A. Treatment-related adverse events of combination immune checkpoint inhibitors: systematic review and meta-analysis. Front Oncol (2020) 10:258. doi: 10.3389/fonc.2020.00258
120. Zhou X, Yao Z, Bai H, Duan J, Wang Z, Wang X, et al. Treatment-related adverse events of PD-1 and PD-L1 inhibitor-based combination therapies in clinical trials: a systematic review and meta-analysis. Lancet Oncol (2021) 22:1265–74. doi: 10.1016/S1470-2045(21)00333-8
121. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. New Engl J Med (2015) 372:2006–17. doi: 10.1056/nejmoa1414428
122. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob J-J, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. New Engl J Med (2017) 377:1345–56. doi: 10.1056/nejmoa1709684
123. Eggermont AMM, Kicinski M, Blank CU, Mandala M, Long GV, Atkinson V, et al. Association between immune-related adverse events and recurrence-free survival among patients with stage III melanoma randomized to receive pembrolizumab or placebo: a secondary analysis of a randomized clinical trial. JAMA Oncol (2020) 6:519–27. doi: 10.1001/jamaoncol.2019.5570
124. Maher VE, Fernandes LL, Weinstock C, Tang S, Agarwal S, Brave M, et al. Analysis of the association between adverse events and outcome in patients receiving a programmed death protein 1 or programmed death ligand 1 antibody. J Clin Oncol (2019) 37(30):2730–7. doi: 10.1200/JCO.19
125. Attia P, Phan GQ, Maker AV, Robinson MR, Quezado MM, Yang JC, et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J Clin Oncol (2005) 23:6043–53. doi: 10.1200/JCO.2005.06.205
126. Downey SG, Klapper JA, Smith FO, Yang JC, Sherry RM, Royal RE, et al. Prognostic factors related to clinical response in patients with metastatic melanoma treated by CTL-associated antigen-4 blockade. Clin Cancer Res (2007) 13:6681–8. doi: 10.1158/1078-0432.CCR-07-0187
127. Fukihara J, Sakamoto K, Koyama J, Ito T, Iwano S, Morise M, et al. Prognostic impact and risk factors of immune-related pneumonitis in patients with non–Small-Cell lung cancer who received programmed death 1 inhibitors. Clin Lung Cancer (2019) 20:442–450.e4. doi: 10.1016/j.cllc.2019.07.006
128. Hua C, Boussemart L, Mateus C, Routier E, Boutros C, Cazenave H, et al. Association of vitiligo with tumor response in patients with metastatic melanoma treated with pembrolizumab. JAMA Dermatol (2016) 152:45–51. doi: 10.1001/jamadermatol.2015.2707
129. Khan Z, Hammer C, Carroll J, Di Nucci F, Acosta SL, Maiya V, et al. Genetic variation associated with thyroid autoimmunity shapes the systemic immune response to PD-1 checkpoint blockade. Nat Commun (2021) 12:3355. doi: 10.1038/s41467-021-23661-4
130. Paderi A, Giorgione R, Giommoni E, Mela MM, Rossi V, Doni L, et al. Article association between immune related adverse events and outcome in patients with metastatic renal cell carcinoma treated with immune checkpoint inhibitors. Cancers (2021) 13:1–11. doi: 10.3390/cancers13040860
131. Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American society of clinical oncology clinical practice guideline. J Clin Oncol (2018) 36:1714–68. doi: 10.1200/JCO.2017.77.6385
132. Roberts J, Ennis D, Hudson M, Ye C, Saltman A, Himmel M, et al. Rheumatic immune-related adverse events associated with cancer immunotherapy: a nationwide multi-center cohort. Autoimmun Rev (2020) 19:102595. doi: 10.1016/j.autrev.2020.102595
133. Villadolid J, Amin A. Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities. Transl Lung Cancer Res (2015) 4:560–75. doi: 10.3978/j.issn.2218-6751.2015.06.06
134. Teulings H-E, Limpens J, Jansen SN, Zwinderman AH, Reitsma JB, Spuls PI, et al. Vitiligo-like depigmentation in patients with stage III-IV melanoma receiving immunotherapy and its association with survival: a systematic review and meta-analysis. J Clin Oncol (2015) 33:773–81. doi: 10.1200/JCO.2014.57.4756
135. Becker JC, Guldberg P, Zeuthen J, Bröcker EB, Straten PT. Accumulation of identical T cells in melanoma and vitiligo-like leukoderma. J Invest Dermatol (1999) 113:1033–8. doi: 10.1046/j.1523-1747.1999.00805.x
136. Bai X, Hu J, Warner AB, Quach HT, Cann CG, Zhang MZ, et al. Early use of high-dose glucocorticoid for the management of irAE is associated with poorer survival in patients with advanced melanoma treated with anti-PD-1 monotherapy. Clin Cancer Res (2021) 27:5993–6000. doi: 10.1158/1078-0432.CCR-21-1283
137. Arbour KC, Mezquita L, Long N, Rizvi H, Auclin E, Ni A, et al. Impact of baseline steroids on efficacy of programmed cell death-1 and programmed death-ligand 1 blockade in patients with non–Small-Cell lung cancer. JCO (2018) 36:2872–8. doi: 10.1200/JCO.2018.79.0006
138. Teimouri A, Minard LV, Scott SN, Daniels A, Snow S. Real-world adherence to toxicity management guidelines for immune-related adverse events. Curr Oncol (2022) 29:3104–17. doi: 10.3390/curroncol29050252
139. Khan SA, Pruitt SL, Xuan L, Gerber DE. Prevalence of autoimmune disease among patients with lung cancer: implications for immunotherapy treatment options. JAMA Oncol (2016) 2:1507. doi: 10.1001/jamaoncol.2016.2238
140. Gulati N, Celen A, Johannet P, Mehnert JM, Weber J, Krogsgaard M, et al. Preexisting immune-mediated inflammatory disease is associated with improved survival and increased toxicity in melanoma patients who receive immune checkpoint inhibitors. Cancer Med (2021) 10:7457–65. doi: 10.1002/cam4.4239
141. Johnson DB, Sullivan RJ, Ott PA, Carlino MS, Khushalani NI, Ye F, et al. Ipilimumab therapy in patients with advanced melanoma and preexisting autoimmune disorders. JAMA Oncol (2016) 2:234–40. doi: 10.1001/jamaoncol.2015.4368
142. Menzies AM, Johnson DB, Ramanujam S, Atkinson VG, Wong ANM, Park JJ, et al. Anti-PD-1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Ann Oncol (2017) 28:368–76. doi: 10.1093/annonc/mdw443
143. Danlos FX, Voisin AL, Dyevre V, Michot JM, Routier E, Taillade L, et al. Safety and efficacy of anti-programmed death 1 antibodies in patients with cancer and pre-existing autoimmune or inflammatory disease. Eur J Cancer (2018) 91:21–9. doi: 10.1016/j.ejca.2017.12.008
144. Leonardi GC, Gainor JF, Altan M, Kravets S, Dahlberg SE, Gedmintas L, et al. JOURNAL OF CLINICAL ONCOLOGY safety of programmed death-1 pathway inhibitors among patients with non-Small-Cell lung cancer and preexisting autoimmune disorders. J Clin Oncol (2018) 36:1905–12. doi: 10.1200/JCO
145. For the German Dermatologic Cooperative Oncology Group (DeCOG), Kähler KC, Eigentler TK, Gesierich A, Heinzerling L, Loquai C, et al. Ipilimumab in metastatic melanoma patients with pre-existing autoimmune disorders. Cancer Immunol Immunother (2018) 67:825–34. doi: 10.1007/s00262-018-2134-z
146. Cortellini A, Buti S, Santini D, Perrone F, Giusti R, Tiseo M, et al. Clinical outcomes of patients with advanced cancer and pre-existing autoimmune diseases treated with anti-programmed death-1 immunotherapy: a real-world transverse study. Oncologist (2019) 24:e327–37. doi: 10.1634/theoncologist.2018-0618
147. Tison A, Quéré G, Misery L, Funck-Brentano E, Danlos FX, Routier E, et al. Safety and efficacy of immune checkpoint inhibitors in patients with cancer and preexisting autoimmune disease: a nationwide, multicenter cohort study. Arthritis Rheumatol (2019) 71:2100–11. doi: 10.1002/art.41068
148. Efuni E, Cytryn S, Boland P, Niewold TB, Pavlick A, Weber J, et al. Risk of toxicity after initiating immune checkpoint inhibitor treatment in patients with rheumatoid arthritis. J Clin Rheumatol (2021) 27:267–71. doi: 10.1097/RHU.0000000000001314
149. Martinez Chanza N, Xie W, Issa M, Dzimitrowicz H, Tripathi A, Beuselinck B, et al. Safety and efficacy of immune checkpoint inhibitors in advanced urological cancers with pre-existing autoimmune disorders: a retrospective international multicenter study. J Immunother Cancer (2020) 8:e000538. doi: 10.1136/jitc-2020-000538
150. Loriot Y, Sternberg CN, Castellano D, Oosting SF, Dumez H, Huddart R, et al. Safety and efficacy of atezolizumab in patients with autoimmune disease: subgroup analysis of the SAUL study in locally advanced/metastatic urinary tract carcinoma. Eur J Cancer (2020) 138:202–11. doi: 10.1016/j.ejca.2020.07.023
151. Tully KH, Cone EB, Cole AP, Sun M, Chen X, Marchese M, et al. Risk of immune-related adverse events in melanoma patients with preexisting autoimmune disease treated with immune checkpoint inhibitors: a population-based study using SEER-Medicare data. Am J Clin Oncol (2021) 44:413–8. doi: 10.1097/COC.0000000000000840
152. van der Kooij MK, Suijkerbuijk KPM, Aarts MJB, van den Berkmortel FWPJ, Blank CU, Boers-Sonderen MJ, et al. Safety and efficacy of checkpoint inhibition in patients with melanoma and preexisting autoimmune Disease: a cohort study. Ann Intern Med (2021) 174:641–8. doi: 10.7326/M20-3419
153. Bhatlapenumarthi V, Patwari A, Harb AJ. Immune-related adverse events and immune checkpoint inhibitor tolerance on rechallenge in patients with irAEs: a single-center experience. J Cancer Res Clin Oncol (2021) 147:2789–800. doi: 10.1007/s00432-021-03610-w
154. Yeung C, Kartolo A, Holstead R, Moffat GT, Hanna L, Hopman W, et al. Safety and clinical outcomes of immune checkpoint inhibitors in patients with cancer and preexisting autoimmune diseases. J Immunother (2021) 44:362–70. doi: 10.1097/CJI.0000000000000377
155. Pizuorno Machado A, Shatila M, Liu C, Wang J, Altan M, Zhang HC, et al. Immune-related adverse events after immune checkpoint inhibitor exposure in adult cancer patients with pre-existing autoimmune diseases. J Cancer Res Clin Oncol (2023). doi: 10.1007/s00432-023-04582-9
156. Abdel-Wahab N, Shah M, Lopez-Olivo MA, Suarez-Almazor ME. Use of immune checkpoint inhibitors in the treatment of patients with cancer and preexisting autoimmune disease: a systematic review. Ann Intern Med (2018) 168:121. doi: 10.7326/M17-2073
157. Plaçais L, Dalle S, Dereure O, Trabelsi S, Dalac S, Legoupil D, et al. Risk of irAEs in patients with autoimmune diseases treated by immune checkpoint inhibitors for stage III or IV melanoma: results from a matched case-control study. Ann Rheum Dis (2022) 81:1445–52. doi: 10.1136/ard-2022-222186
158. Michailidou D, Khaki AR, Morelli MP, Diamantopoulos L, Singh N, Grivas P. Association of blood biomarkers and autoimmunity with immune related adverse events in patients with cancer treated with immune checkpoint inhibitors. Sci Rep (2021) 11:9029. doi: 10.1038/s41598-021-88307-3
159. Chute DF, Zhao S, Strohbehn IA, Rusibamayila N, Seethapathy H, Lee M, et al. Incidence and predictors of CKD and estimated GFR decline in patients receiving immune checkpoint inhibitors. Am J Kidney Dis (2022) 79(1):134–7. doi: 10.1053/j.ajkd.2021.05.012
160. Reid P, Cappelli LC. Treatment of rheumatic adverse events of cancer immunotherapy. Best Pract Res Clin Rheumatol (2022) 36(4), 101805. doi: 10.1016/j.berh.2022.101805
161. Wu C, Zhong L, Wu Q, Lin S, Xie X. The safety and efficacy of immune-checkpoint inhibitors in patients with cancer and pre-existing autoimmune diseases. Immunotherapy (2021) 13:527–39. doi: 10.2217/imt-2020-0230
162. Panhaleux M, Espitia O, Terrier B, Manson G, Maria A, Humbert S, et al. Anti-programmed death ligand 1 immunotherapies in cancer patients with pre-existing systemic sclerosis: a postmarketed phase IV safety assessment study. Eur J Cancer (2022) 160:134–9. doi: 10.1016/j.ejca.2021.10.018
163. Ansel S, Rulach R, Trotter N, Steele N. Pembrolizumab for advanced non-small cell lung cancer (NSCLC): impact of autoimmune comorbidity and outcomes following treatment completion. J Oncol Pharm Pract (2022), 10781552221079356. doi: 10.1177/10781552221079356
164. Brown LJ, Weppler A, Bhave P, Allayous C, Patrinely JR, Ott P, et al. Combination anti-PD1 and ipilimumab therapy in patients with advanced melanoma and pre-existing autoimmune disorders. J Immunother Cancer (2021) 9:e002121. doi: 10.1136/jitc-2020-002121
165. Mathias K, Rouhani S, Olson D, Bass AR, Gajewski TF, Reid P. Association between rheumatic autoantibodies and immune-related adverse events. Oncologist (2023) 28(5):440–8. doi: 10.1093/oncolo/oyac252
166. Grangeon M, Tomasini P, Chaleat S, Jeanson A, Souquet-Bressand M, Khobta N, et al. Association between immune-related adverse events and efficacy of immune checkpoint inhibitors in non-small-cell lung cancer. Clin Lung Cancer (2019) 20:201–7. doi: 10.1016/j.cllc.2018.10.002
167. Street S, Chute D, Strohbehn I, Zhao S, Rengarajan M, Faje A, et al. The positive effect of immune checkpoint inhibitor-induced thyroiditis on overall survival accounting for immortal time bias: a retrospective cohort study of 6596 patients. Ann Oncol (2021) 32(8):1050–1. doi: 10.1016/j.annonc.2021.05.357
Keywords: Immune checkpoint inhibitors (ICIs), immune-related adverse events (irAEs), immunotherapy, autoimmune disease, cancer
Citation: Ibis B, Aliazis K, Cao C, Yenyuwadee S and Boussiotis VA (2023) Immune-related adverse effects of checkpoint immunotherapy and implications for the treatment of patients with cancer and autoimmune diseases. Front. Immunol. 14:1197364. doi: 10.3389/fimmu.2023.1197364
Received: 30 March 2023; Accepted: 15 May 2023;
Published: 05 June 2023.
Edited by:
Evelyn Ullrich, Goethe University Frankfurt, GermanyReviewed by:
Zhida Liu, Shanxi Academy of Advanced Research and Innovation, ChinaStefania Canova, San Gerardo Hospital, Italy
Copyright © 2023 Ibis, Aliazis, Cao, Yenyuwadee and Boussiotis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vassiliki A. Boussiotis, dmJvdXNzaW9AYmlkbWMuaGFydmFyZC5lZHU=
†Present address: Sasitorn Yenyuwadee, Department of Dermatology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand