 Kotaro Matsumoto
Kotaro Matsumoto Katsuya Suzuki
Katsuya Suzuki Masaru Takeshita
Masaru Takeshita Tsutomu Takeuchi
Tsutomu Takeuchi Yuko Kaneko
Yuko Kaneko- Division of Rheumatology, Department of Internal Medicine, Keio University School of Medicine, Shinjuku-ku, Tokyo, Japan
Giant cell arteritis and Takayasu arteritis are two types of primary large-vessel vasculitis (LVV). Although glucocorticoids (GC) are the standard treatment for LVV, the disease relapse rates are high. Recent clinical trials on biological disease-modifying anti-rheumatic drugs (bDMARDs) and Janus kinase (JAK) inhibitors have demonstrated their efficacy in reducing LVV relapse rates and GC dosages. However, the control of residual inflammation and degenerative alterations in the vessel wall remains an outstanding requirement in the clinical management of LVV. The analysis of immune cell phenotypes in patients with LVV may predict their response to treatment with bDMARDs and JAK inhibitors and guide their optimal use. In this mini-review, we focused on molecular markers, including the immune cell proportions and gene expression, in patients with LVV and in mouse models of LVV treated with bDMARDs and JAK inhibitors.
1 Introduction
Giant cell arteritis (GCA) and Takayasu arteritis (TAK) are two types of primary large-vessel vasculitis (LVV) characterized by a predominantly granulomatous infiltration of T cells, macrophages, and multinucleated giant cells (1). The pathophysiology of LVV is not sufficiently elucidated; however, the involvement of Th1 and Th17 immune-mediated responses and an imbalance between Th17 and regulatory T (Treg) cells have been demonstrated in LVV (2–12). Glucocorticoids (GC) are used as a standard treatment for LVV and are effective in inducing remission. However, relapse during the maintenance phase is common, and long-term GC use may be associated with substantial negative effects (13–15). GC therapy ameliorates vasculitis symptoms, and the persistence of Th1, Th17, and myeloid cells during treatment can lead to disease relapse (2–4, 16–20).
Recent clinical trials have demonstrated promising results for biological disease-modifying anti-rheumatic drugs (bDMARDs) and Janus kinase (JAK) inhibitors in the reduction of LVV relapse and tapering of GC dose. However, the response to these therapies is variable. Therefore, it is essential to identify molecular signatures that can predict treatment response and guide treatment optimization. In patients with LVV and in mouse models of LVV, analysis of immune cell phenotypes can provide insight into the molecular mechanisms of bDMARDs and JAK inhibitors and predict treatment response. In this mini-review, we investigated the longitudinal changes in immune cell phenotypes in patients with LVV and in mouse models of LVV treated with bDMARDs and JAK inhibitors.
2 Molecular profile alterations in LVV under treatment
The molecular profiles of patients with LVV and mouse models of LVV treated with bDMARDs and JAK inhibitors are presented in Tables 1, 2, respectively. We reviewed the evidence for the following treatments: interleukin (IL)-6 receptor antagonists, tumor necrosis factor (TNF)-α receptor antagonists, IL-1 receptor antagonists, granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor antagonists, and JAK inhibitors. Although there are no established animal models of LVV, studies that used IL-1Ra-deficient mice and human artery engrafted mice as LVV models were reviewed. IL-1Ra-deficient mice have autoimmune diseases similar to those in humans, including aortitis, arthritis, and skin manifestations (36, 37). Aortitis involves aortic valve wall thickening and regurgitation and resembles TAK. The human artery engrafted mouse is a model of GCA in which human temporal arteries are grafted into severe combined immunodeficiency mice (38).
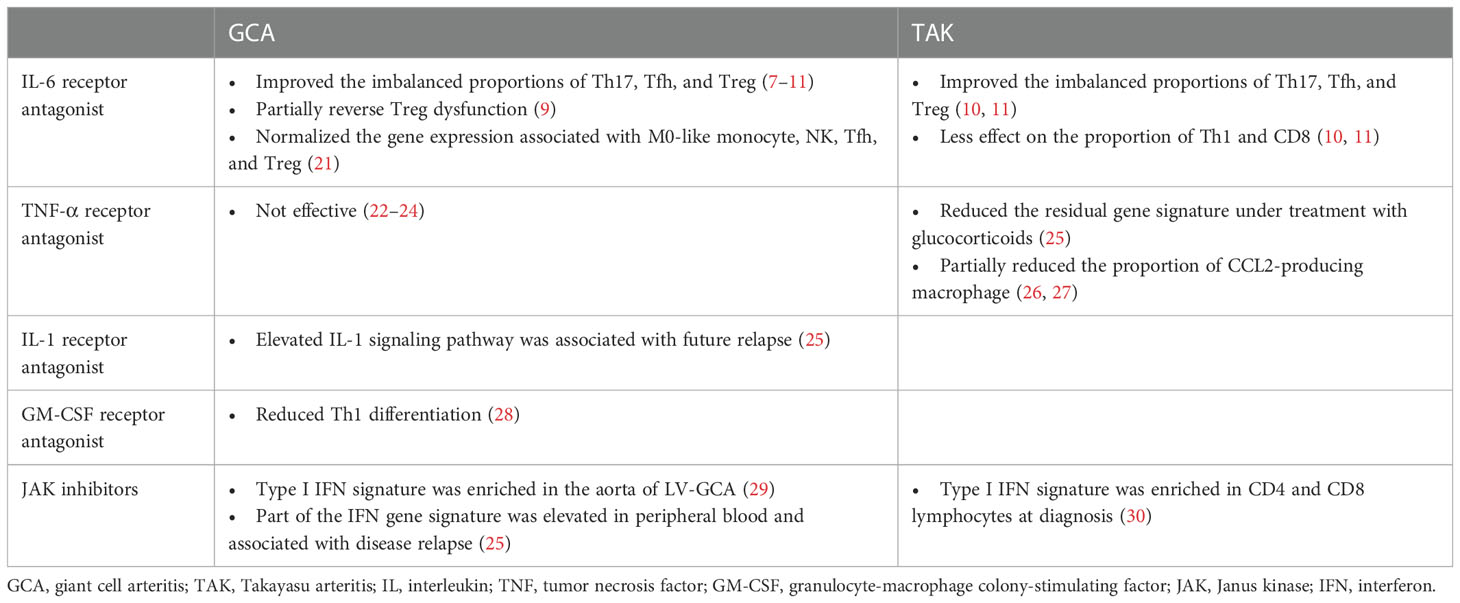
Table 1 Molecular profiles in patients with large vessel vasculitis associated with treatments.

Table 2 Molecular profiles in mouse models of large vessel vasculitis associated with treatments.
2.1 IL-6 receptor antagonist
In LVV, IL-6 is produced by lymphocytes, macrophages, and neutrophils and plays a critical role in the pathogenesis of the disease (4, 39–41). Clinical trials in GCA and TAK have revealed that tocilizumab (TCZ), an IL-6 receptor antagonist, induces sustained remission, prolongs relapse time, and reduces the rate of new vascular events compared to a placebo. At the same time, the effect was more pronounced in patients with GCA than in those with TAK (42–47). TCZ inhibits antibody production by B cells in general (48), and normalized the imbalanced proportion of M0-like monocytes, activated/resting NK, Treg, Tfh, and Th17 cells in LVV (7, 21). In addition, TCZ reverses the glycolysis and calcium signaling abnormalities observed in Treg dysfunction in patients with GCA (8). However, it is important to note that TCZ has less effect on the proportion of Th1 and CD8 T cells in the circulation, and that Th1- and CD8-mediated inflammation may persist under TCZ treatment, particularly in TAK (10, 11). Th1- and CD8-mediated interferon (IFN)-γ responses are observed in TAK rather than in GCA (2–5). IFN-γ promotes proinflammatory response by exploring Th1 differentiation, cytotoxic T cell proliferation, and M1 macrophage polarization (49, 50). The incomplete response of TCZ in TAK pathogenesis is highlighted in IL-1Ra-deficient mice, where inflammation persists in IL-6 -/- mice and in those treated with TCZ, but is suppressed in Tnfsf1a (TNF-α) -/- mice (31–33).
2.2 TNF-α receptor antagonist
Available evidence suggests that both TNF-α and IL-6 play important roles in LVV pathogenesis, but their contributions may differ in GCA and TAK. TNF-α inhibition-based treatments such as infliximab, adalimumab, etanercept, and certolizumab have been shown to be effective in the treatment of TAK (51–53), suggesting that TNF-α is a substantial mediator in the pathogenesis of this disease. Conversely, clinical trials of TNF-α antagonists have failed to demonstrate efficacy in GCA treatment (22–24). Studies in IL-1Ra-deficient mice also support the association between TNF-α and TAK progression, as the mice exhibited significantly reduced aortitis and arthritis in the absence of TNF-α (32, 33). TNF-α acts in conjunction with IFN-γ to stimulate macrophages and induce the production of monocyte chemotactic proteins, particularly CCL2, which recruits monocytes expressing CCR2 to form multinucleated giant cells that are characteristic of LVV (26, 27, 54–56). Elevated CCL2 serum levels after treatment with GC suggest that CCL2 may contribute to treatment failure in LVV (57). At the transcriptome level, TNF-α inhibition, like TCZ, could improve the residual gene signature compared to GC monotherapy (25, 58). Methylome profiling of LVV patients has also revealed novel pathways in the disease pathogenesis (59); however, there is limited data associated with TNF-α inhibition. Further secondary analyses of immunocellular dynamics are required as evidence for the efficacy of TNF-α inhibition in TAK accumulates.
2.3 IL-1 receptor antagonist
IL-1 β, like IL-6 and TNF-α, is highly expressed in the inflamed arterial walls of patients with LVV, and may play a role in the disease pathogenesis (39, 40). Whole blood transcriptome gene expression analysis revealed that the molecular pathway related to IL-1 was significantly upregulated in patients with LVV compared to healthy controls. This correlated with the positron emission tomography (PET) vascular activity score, a disease extent score based on the distribution of affected arteries (25). Increased transduction of IL-1 signaling activates CD4+ T cells. Specifically, activated CD4+ T cells migrate CCR2+ γ delta T cells toward CCL2+ inflammatory tissues (34). Considering that IL-1Ra deficient mice develop TAK-like aortitis, inhibition of IL-1 signaling may be a promising approach for the treatment of LVV. While clinical trials evaluating IL-1 receptor antagonists in the LVV are ongoing, previous reports have shown that anakinra is effective in patients with refractory GCA, with improvement in inflammatory biomarker levels and symptoms, and resolution of arterial inflammation on PET-CT images (60, 61).
2.4 GM-CSF receptor antagonist
CD4+ T cells, macrophages, myofibroblasts, and endothelial cells produce GM-CSF. The GM-CSF receptor α is highly expressed in GCA-affected arteries, resulting in an autocrine amplification loop (28, 62). GM-CSF is a macrophage differentiation factor fundamentally involved in vascular inflammation. Recent data have shown that GM-CSF induces the differentiation of CD206+ MMP9+ macrophage, that play a role in arterial wall destruction during neo-angiogenesis or intimal hyperplasia (63–65). Using an ex vivo temporal artery culture model, GM-CSF increased macrophage activation, Th1 cell polarization, neo-angiogenesis, and tissue injury. Treatment with the GM-CSF receptor antagonist, mavrilimumab, reduced CD16- and CD3ϵ- positive cell infiltration and downregulated key molecules involved in T cell activation and differentiation (28). Among T cells, mavrilimumab decreased Th1 differentiation by reducing TNF-α and IFN-γ, while a direct effect on Th17 differentiation was not assessed (28, 55). In a recent phase 2 clinical trial, mavrilimumab demonstrated superiority over placebo in analyses of the time to flare and sustained remission in patients with GCA (66).
2.5 JAK inhibitors
As described previously, the pathogenesis of LVV involves multiple cytokines, including IL-6, TNF-α, IL-1, GM-CSF, and IFN-γ. Inhibition of these cytokines or agents that inhibit subsequent cellular signaling pathways may effectively treat LVV. Although JAK/STAT signaling and type I IFN signatures were not identified as distinct pathways using whole blood RNA sequencing from patients with GCA-dominant LVV, one of the IFN signature genes, APOBEC3A, was elevated at diagnosis and was associated with disease relapse (25). Pathways associated with JAK/STAT signaling and the type I IFN signature are enriched in CD4 and CD8 lymphocytes in the TAK and the aorta of LV-GCA (29, 30). Type I IFN transcripts were also shown to increase in the vessel walls of immunodeficient mice trans-engrafted with inflamed temporal arteries from patients with GCA (67). In an experimental animal model of LVV, inhibition of JAK1/3 effectively suppressed arterial wall lesioned T cells and inhibited macrophage infiltration and growth factor production, resulting in reduced neo-angiogenesis and intimal hyperplasia (35). JAK inhibitors, including baricitinib (JAK1/2 inhibitor) and tofacitinib (JAK1/2/3 inhibitor), have recently shown efficacy in pilot studies in patients with LVV (68, 69). Considering that blocking JAK1/2 may promote upregulation of Th17- and CD206+ macrophage-mediated inflammatory response (50, 70), investigation of the clinical efficacy depend on targeted JAK isoform selectivity is anticipated.
3 Discussion
We investigated alterations in the molecular profiles of patients with LVV and in mouse models of LVV associated with the following treatments: IL-6 receptor antagonist, TNF-α receptor antagonist, IL-1 receptor antagonist, GM-CSF receptor antagonist, and JAK inhibitors. Other potential therapies, including CTLA4-Ig (abatacept), anti-IL-17A antibody (secukinumab), anti-IL-12/23p40 antibody (ustekinumab), and anti-CD20 antibody (rituximab), were outside the scope of this study. Our study demonstrated that bDMARDs and JAK inhibitors improved the levels of dysregulated molecular profiles compared to GC monotherapy. These results are consistent with a study in patients with rheumatoid arthritis, which revealed that treatment with bDMARDs normalized the molecular signature to a greater extent than methotrexate monotherapy (71). Using bDMARDs and JAK inhibitors may induce deep molecular remission in various inflammatory connective tissue diseases (72–74).
CD4+ and CD8+ T cells play central roles in the pathogenesis of LVV (75, 76). Recent evidence suggests that Th1- and CD8-mediated inflammation may be less responsive to TCZ treatment, whereas the molecular signatures of Th17, Tfh, and Treg cells are improved. The incomplete response of IL-1Ra deficient mice to TCZ suggests a limitation of this treatment. Based on clinical evidence which demonstrated that the effect of TCZ is modest in TAK but dramatic in GCA, it is inferred that Th1-and CD8-mediated inflammation may be dominant in TAK. Therefore, targeting the Th1- and CD8-IFN-γ axis may be an important therapeutic strategy in TAK. Based on this evidence, TNF-α receptor antagonists, IL-1 receptor antagonists, GM-CSF receptor antagonists, and JAK inhibitors are potential therapies that may ameliorate residual inflammation in the LVV.
In this study, we reviewed the alterations in molecular profile of LVV. Evidence from human and murine studies has revealed changes in the immune profiles of the LVV following treatment with bDMARDs and JAK inhibitors. Despite treatment with bDMARDs and JAK inhibitors, residual immune cell activation was observed, which contributed to immune cell infiltration and damage to large arteries, resulting in arterial stenosis, aneurysm, and potentially life-threatening complications. Further studies are required to elucidate the molecular mechanisms underlying LVV.
Author contributions
KM drafted the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from Keio University School of Medicine and JSPS KAKENHI (grant number JP21K16306).
Acknowledgments
We would like to thank Editage (www.editage.com) for English language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. (2013). 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum (2013) 65:1–11. doi: 10.1002/art.37715
2. Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation (2010) 121:906–15. doi: 10.1161/CIRCULATIONAHA.109.872903
3. Samson M, Audia S, Fraszczak J, Trad M, Ornetti P, Lakomy D, et al. Th1 and Th17 lymphocytes expressing CD161 are implicated in giant cell arteritis and polymyalgia rheumatica pathogenesis. Arthritis Rheum (2012) 64:3788–98. doi: 10.1002/art.34647
4. Saadoun D, Garrido M, Comarmond C, Desbois AC, Domont F, Savey L, et al. Th1 and Th17 cytokines drive inflammation in takayasu arteritis. Arthritis Rheumatol (2015) 67:1353–60. doi: 10.1002/art.39037
5. Watanabe R, Berry GJ, Liang DH, Goronzy JJ, Weyand CM. Pathogenesis of giant cell arteritis and takayasu arteritis-similarities and differences. Curr Rheumatol Rep (2020) 22:68. doi: 10.1007/s11926-020-00948-x
6. Wen Z, Shen Y, Berry G, Shahram F, Li Y, Watanabe R, et al. The microvascular niche instructs T cells in large vessel vasculitis via the VEGF-Jagged1-notch pathway. Sci Transl Med (2017) 9:eaal3322. doi: 10.1126/scitranslmed.aal3322
7. Miyabe C, Miyabe Y, Strle K, Kim ND, Stone JH, Luster AD, et al. An expanded population of pathogenic regulatory T cells in giant cell arteritis is abrogated by IL-6 blockade therapy. Ann Rheum Dis (2017) 76:898–905. doi: 10.1136/annrheumdis-2016-210070
8. Samson M, Greigert H, Ciudad M, Gerard C, Ghesquière T, Trad M, et al. Improvement of treg immune response after treatment with tocilizumab in giant cell arteritis. Clin Transl Immunol (2021) 10:e1332. doi: 10.1002/cti2.1332
9. Adriawan IR, Atschekzei F, Dittrich-Breiholz O, Garantziotis P, Hirsch S, Risser LM, et al. Novel aspects of regulatory T cell dysfunction as a therapeutic target in giant cell arteritis. Ann Rheum Dis (2022) 81:124–31. doi: 10.1136/annrheumdis-2021-220955
10. Matsumoto K, Suzuki K, Yoshimoto K, Seki N, Tsujimoto H, Chiba K, et al. Significant association between clinical characteristics and changes in peripheral immuno-phenotype in large vessel vasculitis. Arthritis Res Ther (2019) 21:304. doi: 10.1186/s13075-019-2068-7
11. Matsumoto K, Suzuki K, Yoshida H, Magi M, Kaneko Y, Takeuchi T. Longitudinal monitoring of circulating immune cell phenotypes in large vessel vasculitis. Autoimmun Rev (2022) 21:103160. doi: 10.1016/j.autrev.2022.103160
12. Weyand CM, Goronzy JJ. Immune mechanisms in medium and large-vessel vasculitis. Nat Rev Rheumatol (2013) 9:731–40. doi: 10.1038/nrrheum.2013.161
13. Marie I, Proux A, Duhaut P, Primard E, Lahaxe L, Girszyn N, et al. Long-term follow-up of aortic involvement in giant cell arteritis: a series of 48 patients. Med (Baltim) (2009) 88:182–92. doi: 10.1097/MD.0b013e3181a68ae2
14. Kermani TA, Warrington KJ, Crowson CS, Ytterberg SR, Hunder GG, Gabriel SE, et al. Large-Vessel involvement in giant cell arteritis: a population-based cohort study of the incidence-trends and prognosis. Ann Rheum Dis (2013) 72:1989–94. doi: 10.1136/annrheumdis-2012-202408
15. Dejaco C, Ramiro S, Touma Z, Bond M, Soowamber M, Sanchez-Alvarez C, et al. What is a response in randomised controlled trials in giant cell arteritis? Ann Rheum Dis (2023) In Press. doi: 10.1136/ard-2022-223751. ard-2022-223751.
16. van Sleen Y, Graver JC, Abdulahad WH, van der Geest KSM, Boots AMH, Sandovici M, et al. Leukocyte dynamics reveal a persistent myeloid dominance in giant cell arteritis and polymyalgia rheumatica. Front Immunol (2019) 10:1981. doi: 10.3389/fimmu.2019.01981
17. Quinn KA, Ahlman MA, Malayeri AA, Marko J, Civelek AC, Rosenblum JS, et al. Comparison of magnetic resonance angiography and 18F-fluorodeoxyglucose positron emission tomography in large-vessel vasculitis. Ann Rheum Dis (2018) 77:1165–71. doi: 10.1136/annrheumdis-2018-213102
18. Saadoun D, Vautier M, Cacoub P. Medium- and large-vessel vasculitis. Circulation (2021) 143:267–82. doi: 10.1161/CIRCULATIONAHA.120.046657
19. Camellino D, Matteson EL, Buttgereit F, Dejaco C. Monitoring and long-term management of giant cell arteritis and polymyalgia rheumatica. Nat Rev Rheumatol (2020) 16:481–95. doi: 10.1038/s41584-020-0458-5
20. Weyand CM, Kaiser M, Yang H, Younge B, Goronzy JJ. Therapeutic effects of acetylsalicylic acid in giant cell arteritis. Arthritis Rheum (2002) 46:457–66. doi: 10.1002/art.10071
21. Matsumoto K, Suzuki K, Yoshida H, Magi M, Matsumoto Y, Noguchi-Sasaki M, et al. Distinct gene signatures of monocytes and b cells in patients with giant cell arteritis: a longitudinal transcriptome analysis. Arthritis Res Ther (2023) 25:1. doi: 10.1186/s13075-022-02982-9
22. Hoffman GS, Cid MC, Rendt-Zagar KE, Merkel PA, Weyand CM, Stone JH, et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med (2007) 146:621–30. doi: 10.7326/0003-4819-146-9-200705010-00004
23. Martínez-Taboada VM, Rodríguez-Valverde V, Carreño L, López-Longo J, Figueroa M, Belzunegui J, et al. A double-blind placebo controlled trial of etanercept in patients with giant cell arteritis and corticosteroid side effects. Ann Rheum Dis (2008) 67:625–30. doi: 10.1136/ard.2007.082115
24. Seror R, Baron G, Hachulla E, Debandt M, Larroche C, Puéchal X, et al. Adalimumab for steroid sparing in patients with giant-cell arteritis: results of a multicentre randomized controlled trial. Ann Rheum Dis (2014) 73:2074–81. doi: 10.1136/annrheumdis-2013-203586
25. Matsumoto K, Suzuki K, Yoshimoto K, Ishigaki S, Yoshida H, Magi M, et al. Interleukin-1 pathway in active large vessel vasculitis patients with a poor prognosis: a longitudinal transcriptome analysis. Clin Transl Immunol (2021) 10:e1307. doi: 10.1002/cti2.1307
26. Cid MC, Hoffman MP, Hernández-Rodríguez J, Segarra M, Elkin M, Sánchez M, et al. Association between increased CCL2 (MCP-1) expression in lesions and persistence of disease activity in giant-cell arteritis. Rheumatol (Oxford) (2006) 45:1356–63. doi: 10.1093/rheumatology/kel128
27. Kong X, Xu M, Cui X, Ma L, Cheng H, Hou J, et al. Potential role of macrophage phenotypes and CCL2 in the pathogenesis of takayasu arteritis. Front Immunol (2021) 12:646516. doi: 10.3389/fimmu.2021.646516
28. Corbera-Bellalta M, Alba-Rovira R, Muralidharan S, Espígol-Frigolé G, Ríos-Garcés R, Marco-Hernández J, et al. Blocking GM-CSF receptor alpha with mavrilimumab reduces infiltrating cells, pro-inflammatory markers and neoangiogenesis in ex vivo cultured arteries from patients with giant cell arteritis. Ann Rheum Dis (2022) 81:524–36. doi: 10.1136/annrheumdis-2021-220873
29. Vieira M, Régnier P, Maciejewski-Duval A, Le Joncour AL, Darasse-Jèze G, Rosenzwajg M, et al. Interferon signature in giant cell arteritis aortitis. J Autoimmun (2022) 127:102796. doi: 10.1016/j.jaut.2022.102796
30. Régnier P, Le Joncour A, Maciejewski-Duval A, Desbois AC, Comarmond C, Rosenzwajg M, et al. Targeting JAK/STAT pathway in takayasu’s arteritis. Ann Rheum Dis (2020) 79:951–9. doi: 10.1136/annrheumdis-2019-216900
31. Hada Y, Uchida HA, Mukai T, Kojima F, Yoshida M, Takeuchi H, et al. Inhibition of interleukin-6 signaling attenuates aortitis, left ventricular hypertrophy and arthritis in interleukin-1 receptor antagonist deficient mice. Clin Sci (Lond) (2020) 134:2771–87. doi: 10.1042/CS20201036
32. Nakajima A, Matsuki T, Komine M, Asahina A, Horai R, Nakae S, et al. TNF, but not IL-6 and IL-17, is crucial for the development of T cell-independent psoriasis-like dermatitis in Il1rn–/– mice. J Immunol (2010) 185:1887–93. doi: 10.4049/jimmunol.1001227
33. Matsuki T, Isoda K, Horai R, Nakajima A, Aizawa Y, Suzuki K, et al. Involvement of tumor necrosis factor-alpha in the development of T cell–dependent aortitis in interleukin-1 receptor antagonist–deficient mice. Circulation (2005) 112:1323–31. doi: 10.1161/CIRCULATIONAHA.105.564658
34. Akitsu A, Ishigame H, Kakuta S, Chung SH, Ikeda S, Shimizu K, et al. IL-1 receptor antagonist-deficient mice develop autoimmune arthritis due to intrinsic activation of IL-17-producing CCR2(+)Vγ6(+)γδ T cells. Nat Commun (2015) 6:7464. doi: 10.1038/ncomms8464
35. Zhang H, Watanabe R, Berry GJ, Tian L, Goronzy JJ, Weyand CM. Inhibition of JAK-STAT signaling suppresses pathogenic immune responses in medium and large vessel vasculitis. Circulation (2018) 137:1934–48. doi: 10.1161/CIRCULATIONAHA.117.030423
36. Horai R, Saijo S, Tanioka H, Nakae S, Sudo K, Okahara A, et al. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist–deficient mice. J Exp Med (2000) 191:313–20. doi: 10.1084/jem.191.2.313
37. Isoda K, Matsuki T, Kondo H, Iwakura Y, Ohsuzu F. Deficiency of interleukin-1 receptor antagonist induces aortic valve disease in BALB/c mice. Arterioscler Thromb Vasc Biol (2010) 30:708–15. doi: 10.1161/ATVBAHA.109.201749
38. Ma-Krupa W, Jeon MS, Spoerl S, Tedder TF, Goronzy JJ, Weyand CM. Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis. J Exp Med (2003) 199:173–83. doi: 10.1084/jem.20030850
39. Wagner AD, Goronzy JJ, Weyand CM. Functional profile of tissue-infiltrating and circulating CD68+ cells in giant cell arteritis. evidence for two components of the disease. J Clin Invest (1994) 94:1134–40. doi: 10.1172/JCI117428
40. Herńandez-Rodríguez J, Segarra M, Vilardell C, Sánchez M, García-Martínez A, Esteban MJ, et al. Tissue production of pro-inflammatory cytokines (IL-1beta, TNFalpha and IL-6) correlates with the intensity of the systemic inflammatory response and with corticosteroid requirements in giant-cell arteritis. Rheumatol (Oxf Engl) (2004) 43:294–301. doi: 10.1093/rheumatology/keh058
41. Palamidas DA, Argyropoulou OD, Georgantzoglou N, Karatza E, Xingi E, Kapsogeorgou EK, et al. Neutrophil extracellular traps in giant cell arteritis biopsies: presentation, localization and co-expression with inflammatory cytokines. Rheumatol (Oxford) (2022) 61:1639–44. doi: 10.1093/rheumatology/keab505
42. Villiger PM, Adler S, Kuchen S, Wermelinger F, Dan D, Fiege V, et al. Tocilizumab for induction and maintenance of remission in giant cell arteritis: a phase 2, randomized, double-blind, placebo-controlled trial. Lancet (2016) 387:1921–7. doi: 10.1016/S0140-6736(16)00560-2
43. Stone JH, Tuckwell K, Dimonaco S, Klearman M, Aringer M, Blockmans D, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med (2017) 377:317–28. doi: 10.1056/NEJMoa1613849
44. Nakaoka Y, Isobe M, Takei S, Tanaka Y, Ishii T, Yokota S, et al. Efficacy and safety of tocilizumab in patients with refractory takayasu arteritis: results from a randomized, double-blind, placebo-controlled, phase 3 trial in Japan (the TAKT study). Ann Rheum Dis (2018) 77:348–54. doi: 10.1136/annrheumdis-2017-211878
45. Nakaoka Y, Isobe M, Tanaka Y, Ishii T, Ooka S, Niiro H, et al. Long-term efficacy and safety of tocilizumab in refractory takayasu arteritis: final results of the randomized controlled phase 3 TAKT study. Rheumatol (Oxford) (2020) 59:2427–34. doi: 10.1093/rheumatology/kez630
46. Mekinian A, Saadoun D, Vicaut E, Thietart S, Lioger B, Jego P, et al. Tocilizumab in treatment-naïve patients with takayasu arteritis: TOCITAKA French prospective multicenter open-labeled trial. Arthritis Res Ther (2020) 22:218. doi: 10.1186/s13075-020-02311-y
47. Kang L, Liu Y, Luo Z, Zhou Y, Chen B, Yin G, et al. Systematic review and meta-analysis of the current literature on tocilizumab in patients with refractory takayasu arteritis. Front Immunol (2023) 14:1084558. doi: 10.3389/fimmu.2023.1084558
48. Choy EH, De Benedetti FD, Takeuchi T, Hashizume M, John MR, Kishimoto T. Translating IL-6 biology into effective treatments. Nat Rev Rheumatol (2020) 16:335–45. doi: 10.1038/s41584-020-0419-z
49. Ren YL, Li TT, Cui W, Zhao LM, Gao N, Liao H, et al. CD8+ T lymphocyte is a main source of interferon-gamma production in takayasu’s arteritis. Sci Rep (2021) 11:17111. doi: 10.1038/s41598-021-96632-w
50. Chen R, Wang J, Dai X, Wu S, Huang Q, Jiang L, et al. Augmented PFKFB3-mediated glycolysis by interferon-γ promotes inflammatory M1 polarization through the JAK2/STAT1 pathway in local vascular inflammation in takayasu arteritis. Arthritis Res Ther (2022) 24:266. doi: 10.1186/s13075-022-02960-1
51. Alibaz-Oner F, Kaymaz-Tahra S, Bayındır Ö, Yazici A, Ince B, Kalkan K, et al. Biologic treatments in takayasu’s arteritis: a comparative study of tumor necrosis factor inhibitors and tocilizumab. Semin Arthritis Rheum (2021) 51:1224–9. doi: 10.1016/j.semarthrit.2021.09.010
52. Molloy ES, Langford CA, Clark TM, Gota CE, Hoffman GS. Anti-tumour necrosis factor therapy in patients with refractory takayasu arteritis: long-term follow-up. Ann Rheum Dis (2008) 67:1567–9. doi: 10.1136/ard.2008.093260
53. Mekinian A, Comarmond C, Resche-Rigon M, Mirault T, Kahn JE, Lambert M, et al. Efficacy of biological-target treatments in takayasu arteritis: multicenter, retrospective study of 49 patients. Circulation (2015) 132:1963–700. doi: 10.1161/CIRCULATIONAHA.114.014321
54. Shepherd J, Nicklin MJH. Elastic-vessel arteritis in interleukin-1 receptor antagonist–deficient mice involves effector Th1 cells and requires interleukin-1 receptor. Circulation (2005) 111:3135–40. doi: 10.1161/CIRCULATIONAHA.104.519132
55. Corbera-Bellalta M, Planas-Rigol E, Lozano E, Terrades-García N, Alba MA, Prieto-González S, et al. Blocking interferon gamma reduces expression of chemokines CXCL9, CXCL10 and CXCL11 and decreases macrophage infiltration in ex vivo cultured arteries from patients with giant cell arteritis. Ann Rheum Dis (2016) 75:1177–86. doi: 10.1136/annrheumdis-2015-208371
56. Watanabe R, Hashimoto M. Pathogenic role of monocytes/macrophages in large vessel vasculitis. Front Immunol (2022) 13:859502. doi: 10.3389/fimmu.2022.859502
57. Abe N, Kono M, Kono M, Katsuyama T, Ohmura K, Sato T, et al. Cytokine and chemokine multiplex analysis-based exploration for potential treatment and prognostic prediction in large-vessel vasculitis: a preliminary observational study. Front Immunol (2022) 13:1066916. doi: 10.3389/fimmu.2022.1066916
58. Matsumoto K, Kurasawa T, Yoshimoto K, Suzuki K, Takeuchi T. Identification of neutrophil β2-integrin LFA-1 as a potential mechanistic biomarker in ANCA-associated vasculitis via microarray and validation analyses. Arthritis Res Ther (2021) 23:136. doi: 10.1186/s13075-021-02510-1
59. Estupiñán-Moreno E, Ortiz-Fernández L, Li T, Hernández-Rodríguez J, Ciudad L, Andrés-León E, et al. Methylome and transcriptome profiling of giant cell arteritis monocytes reveals novel pathways involved in disease pathogenesis and molecular response to glucocorticoids. Ann Rheum Dis (2022) 81:1290–300. doi: 10.1136/annrheumdis-2022-222156
60. Ly KH, Stirnemann J, Liozon E, Michel M, Fain O, Fauchais AL. Interleukin-1 blockade in refractory giant cell arteritis. Joint Bone Spine (2014) 81:76–8. doi: 10.1016/j.jbspin.2013.06.004
61. Deshayes S, Ly KH, Rieu V, Maigné G, Martin Silva N, Manrique A, et al. Steroid-sparing effect of anakinra in giant-cell arteritis: a case series with clinical, biological and iconographic long-term assessments. Rheumatol (Oxf Engl) (2021) 61:400–6. doi: 10.1093/rheumatology/keab280
62. Greigert H, Genet C, Ramon A, Bonnotte B, Samson M. New insights into the pathogenesis of giant cell arteritis: mechanisms involved in maintaining vascular inflammation. J Clin Med (2022) 11:2905. doi: 10.3390/jcm11102905
63. Jiemy WF, van Sleen Y, van der Geest KS, Ten Berge HA, Abdulahad WH, Sandovici M, et al. Distinct macrophage phenotypes skewed by local granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colonystimulating factor (M-CSF) are associated with tissue destruction and intimal hyperplasia in giant cell arteritis. Clin Transl Immunol (2020) 9:e1164. doi: 10.1002/cti2.1164
64. van Sleen Y, Jiemy WF, Pringle S, van der Geest KSM, Abdulahad WH, Sandovici M, et al. A distinct macrophage subset mediating tissue destruction and neovascularization in giant cell arteritis: implication of the YKL-40/interleukin-13 receptor α2 axis. Arthritis Rheumatol (2021) 73:2327–37. doi: 10.1002/art.41887
65. Esen I, Jiemy WF, van Sleen Y, van der Geest KSM, Sandovici M, Heeringa P, et al. Functionally heterogenous macrophage subsets in the pathogenesis of giant cell arteritis: novel targets for disease monitoring and treatment. J Clin Med (2021) 10:4958. doi: 10.3390/jcm10214958
66. Cid MC, Unizony SH, Blockmans D, Brouwer E, Dagna L, Dasgupta B, et al. Efficacy and safety of mavrilimumab in giant cell arteritis: a phase 2, randomised, double-blind, placebo-controlled trial. Ann Rheum Dis (2022) 81:653–61. doi: 10.1136/annrheumdis-2021-221865
67. Watanabe R, Hashimoto M. Perspectives of JAK inhibitors for large vessel vasculitis. Front Immunol (2022) 13:881705. doi: 10.3389/fimmu.2022.881705
68. Koster MJ, Crowson CS, Giblon RE, Jaquith JM, Duarte-García A, Matteson EL, et al. Baricitinib for relapsing giant cell arteritis: a prospective open-label 52-week pilot study. Ann Rheum Dis (2022) 81:861–7. doi: 10.1136/annrheumdis-2021-221961
69. Kong X, Sun Y, Dai X, Wang L, Ji Z, Chen H, et al. Treatment efficacy and safety of tofacitinib versus methotrexate in takayasu arteritis: a prospective observational study. Ann Rheum Dis (2022) 81:117–23. doi: 10.1136/annrheumdis-2021-220832
70. Kelchtermans H, Schurgers E, Geboes L, Mitera T, Van Damme J, Van Snick J, et al. Effector mechanisms of interleukin-17 in collagen-induced arthritis in the absence of interferon-gamma and counteraction by interferon-gamma. Arthritis Res Ther (2009) 11:R122. doi: 10.1186/ar2787
71. Tasaki S, Suzuki K, Kassai Y, Takeshita M, Murota A, Kondo Y, et al. Multi-omics monitoring of drug response in rheumatoid arthritis in pursuit of molecular remission. Nat Commun (2018) 9:2755. doi: 10.1038/s41467-018-05044-4
72. Matsumoto K, Suzuki K, Yoshimoto K, Seki N, Tsujimoto H, Chiba K, et al. Significant association between clinical characteristics and immuno-phenotypes in patients with ANCA-associated vasculitis. Rheumatol (Oxf Engl) (2020) 59:545–53. doi: 10.1093/rheumatology/kez327
73. Matsumoto K, Suzuki K, Yoshimoto K, Seki N, Tsujimoto H, Chiba K, et al. Longitudinal immune cell monitoring identified CD14++ CD16+ intermediate monocyte as a marker of relapse in patients with ANCA-associated vasculitis. Arthritis Res Ther (2020) 22:145. doi: 10.1186/s13075-020-02234-8
74. Matsumoto K, Suzuki K, Yasuoka H, Hirahashi J, Yoshida H, Magi M, et al. Longitudinal monitoring of circulating immune cell phenotypes in anti-neutrophil cytoplasmic antibody-associated vasculitis. Autoimmun Rev (2023) 22:103271. doi: 10.1016/j.autrev.2023.103271
75. Watanabe R, Berry GJ, Liang DH, Goronzy JJ, Weyand CM. Cellular signaling pathways in medium and large vessel vasculitis. Front Immunol (2020) 11:587089. doi: 10.3389/fimmu.2020.587089
Keywords: giant cell arteritis, takayasu arteritis, biological disease-modifying anti-rheumatic drugs, janus kinase inhibitors, molecular remission
Citation: Matsumoto K, Suzuki K, Takeshita M, Takeuchi T and Kaneko Y (2023) Changes in the molecular profiles of large-vessel vasculitis treated with biological disease-modifying anti-rheumatic drugs and Janus kinase inhibitors. Front. Immunol. 14:1197342. doi: 10.3389/fimmu.2023.1197342
Received: 30 March 2023; Accepted: 19 April 2023;
Published: 01 May 2023.
Edited by:
Ryu Watanabe, Osaka Metropolitan University, JapanReviewed by:
Chie Miyabe, St. Marianna University School of Medicine, JapanCopyright © 2023 Matsumoto, Suzuki, Takeshita, Takeuchi and Kaneko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kotaro Matsumoto, YWE2MTUxMTlAa2Vpby5qcA==
†ORCID: Kotaro Matsumoto, orcid.org/0000-0003-2047-7626