 Thivya Balendran
Thivya Balendran Keith Lim2
Keith Lim2 John A. Hamilton
John A. Hamilton Adrian A. Achuthan
Adrian A. Achuthan- 1Department of Medicine, Royal Melbourne Hospital, The University of Melbourne, Parkville, VIC, Australia
- 2Department of Medicine, Western Health, The University of Melbourne, St Albans, VIC, Australia
Rheumatoid arthritis (RA) is a destructive inflammatory autoimmune disease that causes pain and disability. Many of the currently available drugs for treating RA patients are aimed at halting the progression of the disease and alleviating inflammation. Further, some of these treatment options have drawbacks, including disease recurrence and adverse effects due to long-term use. These inefficiencies have created a need for a different approach to treating RA. Recently, the focus has shifted to direct targeting of transcription factors (TFs), as they play a vital role in the pathogenesis of RA, activating key cytokines, chemokines, adhesion molecules, and enzymes. In light of this, synthetic drugs and natural compounds are being explored to target key TFs or their signaling pathways in RA. This review discusses the role of four key TFs in inflammation, namely NF-κB, STATs, AP-1 and IRFs, and their potential for being targeted to treat RA.
1 Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease that mostly affects joints. Joint inflammation is initiated and maintained by a complex interaction between many cells, including T cells, dendritic cells, B cells, macrophages, neutrophils, osteoclasts, and fibroblast-like synoviocytes (FLS) (1). These cells can release pro-inflammatory cytokines, chemokines, reactive oxidative species, matrix metalloproteinases (MMPs) and autoantibodies into synovial joints and thus contribute inflammation, cartilage damage, osteoclast activation, and bone destruction (2–4).
Many pro-inflammatory mediators have been implicated in the pathogenesis of RA (5). For example, tumor necrosis factor (TNF), interferons (IFNs), interleukin (IL) -1β, IL-2, IL-4, IL-6, IL-8, IL-17, IL-18, IL-21, IL-22, IL-23 and granulocyte macrophage-colony stimulating factor (GM-CSF) have been suggested to play a central role in RA pathogenesis (6–8). These cytokines activate key transcription factors (TFs), such as nuclear factor-κB (NF-κB), activator protein-1 (AP-1), interferon regulatory factors (IRFs), and signal transducer and activator of transcription (STAT) proteins, which can further promote the production of pro-inflammatory mediators (9). Therefore, targeting these key TFs or the signaling pathways associated with these TFs is a feasible strategy for treating RA. While several synthetic drugs are currently being trialed aimed at targeting key TFs in RA, several natural compounds have also been explored as potential alternative treatment options with a focus on targeting TFs. In this review, we summarize the role of four families of TFs, namely NF-κB, STATs, AP-1 and IRFs, in the pathogenesis of RA, and provide an update on the latest preclinical and clinical trials targeting them.
2 NF-κB
2.1 NF-κB signaling pathway
The NF-κB signaling pathway controls many biological processes, but its dysregulation is often associated with inflammation, for example, that associated with RA. Activated NF-κB is observed in RA synovium in early and late stages of joint inflammation and initiation of inflammation is triggered by NF-κB activation in both T cells and antigen presenting cells (10). Different extracellular and/or intracellular stimuli (e.g., TNF, IL-1β, IL-6, MMPs and RANKL) can activate the NF-κB signaling pathway, either directly or indirectly (11). The NF-κB family is composed of five structurally related members that include NF-κB1/p50 (precursor p105), NF-κB2/p52 (precursor p100), RelA/p65, RelB, and c-Rel which bind to specific DNA and κB enhancer elements that mediate the transcription of target genes (12, 13). NF-κB is activated via two different pathways. Direct activation includes canonical and non-canonical pathways, mediated by inhibitor of kappa B (IκB) kinase (IKK) and NF-κB-inducing kinase (NIK), respectively. The indirect activation of NF-κB is interconnected with other cellular pathways, including mitogen-activated protein kinase (MAPK), Rho, and phosphoinositide 3-kinase (PI3-K) (11).
2.1.1 Canonical pathway
In inflammatory conditions, such as in RA, cytokines, chemokines and free radicals provide signals that lead to degradation of IκB protein resulting in the disassociation of NF-κB (12, 13) (Figure 1). Activation of the canonical pathway occurs through stimulation of the TNF receptors, Toll-like receptors (TLRs), interleukin receptors, pattern recognition receptors (PRRs), T cell receptors (TCRs) and B cell receptors (BCRs) (14, 15). The canonical pathway has an IKK complex, comprising IKKα and IKKβ, the homologous catalytic subunits, and IKKγ, a regulatory subunit of the complex that activates IKKβ (14). Receptor activation stimulates numerous kinases, such as transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1), receptor-interacting protein kinase 1 (RIP1), MAPK kinase ERK1 (MEKK1) and TANK-binding kinase (TBK1), which phosphorylate IKKβ and activate the IKK complex (16). Activated IKKβ then phosphorylates IκBα and activates its downstream TFs, RelA/p50 and p50/c-Rel (13). The liberated RelA/p50 and p50/c-Rel translocate to the nucleus and activate the transcription of NF-κB-dependent inflammatory genes (14, 17).
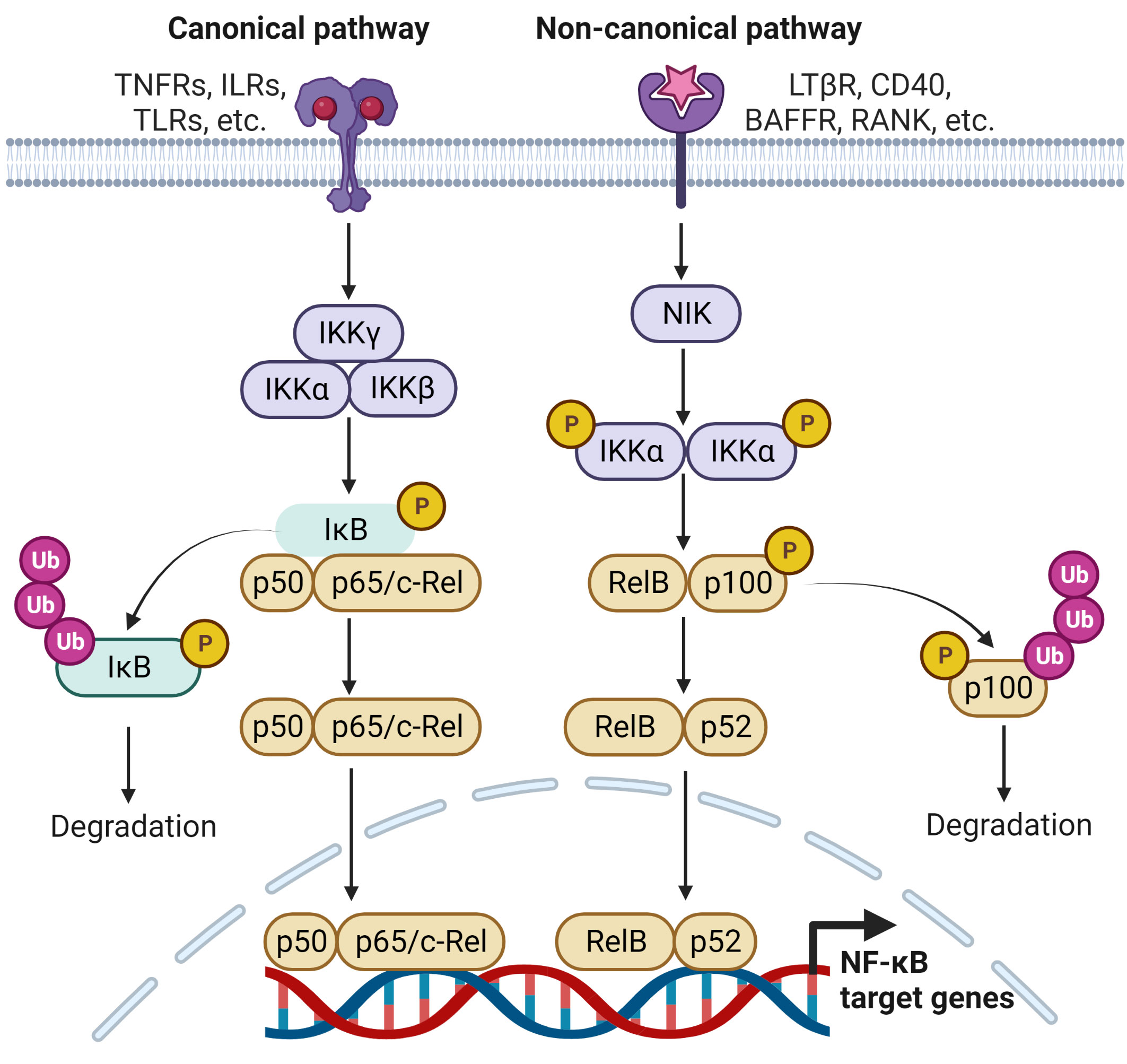
Figure 1 Signaling pathways leading to the regulation of NF-κB target genes. The activation of NF-κB involves two signaling pathways, the canonical and non-canonical pathways. Both are activated through engagement with distinct receptors, leading to transcriptional regulation several NF-κB target genes that are responsible for immune and inflammatory responses.
2.1.2 Non-canonical pathway
Noncanonical NF-κB pathway respond to a certain type of stimulus, such as the lymphotoxin β receptor (LTβR), CD40, the B-cell activating factor receptor (BAFFR) and receptor activator of NF-κB (RANK) (18) (Figure 1). NIK is essential for the activation of this pathway and is central to the signaling that activates IKKα and forms a functional cooperation with IKKα to phosphorylate p100. Phosphorylation of p100 stimulates the partial proteasomal processing of p52 (11). This generates NF-κB2/p52 through the degradation of the p100 C-terminal IκB-like structure, and leads to the nuclear translocation of p52/RelB occurs (19).
2.2 NF-κB-regulated genes
NF-κB regulates more than 150 genes involved in anti-apoptosis, cell proliferation, immunity, and inflammation. It plays a key role in regulating the activation, survival, and differentiation of innate and adaptive immune cells (20). In RA, a deregulated NF-κB signaling pathway contributes to the pathogenic process and activates both immune and non-immune cells (e.g., FLS) through transcriptional regulation of inflammatory mediators, including TNF, IL-1, IL-2, IL-6, IL-8, IL-9, IL-12, IL-18, IL-23, GM-CSF, VEGF, RANKL, MCP-1, MIP-2, CXCL1, CXCL10, RANTES, ICAM-1, VCAM-1, MMPs, and COX-2. (10, 13, 20, 21). These NF-κB-regulated inflammatory mediators have been reported to play a crucial role in the pathogenesis of RA by activating both immune and non-immune cells.
T cells and macrophages are key responders to the NF-κB signaling pathway. Deregulated NF-κB signaling causes aberrant activation of T cells and each member of the NF-κB family is responsible in activating different types of T cells in RA. RelA and c-Rel activate naïve T cells by inducing TCR activation. c-Rel promotes the transcription of Foxp3, a key regulator of Tregs (22). NF-κB differentiates Th1 and Th17 cells by inducing IL-12 production and promotes IL-17 synthesis in Th17 cells, and thereby recruiting neutrophils and monocytes to sites of inflammation. Th17 cells contribute to inflammation by regulating expression of TNF, IL-1β, IL17, IL-21, and IL-22 (23). Noncanonical NF-κB regulates Th17 to induce GM-CSF. On the other hand, Th2 responses are regulated by NF-κB1/p50. In macrophages, NF-κB induces a range of inflammatory mediators, including TNF, IL-1β, IL-6, IL-12, and COX-2. Activated c-Rel is essential for IL-12B expression and also for NF-κB-ATF3-CEBPδ transcriptional circuit, which enables macrophages to analyze the responses received from persistent and transient TLR4 stimulation (24). In FLS, NF-κB p50/p52 and NFATc1 respond to RANKL and exhibit an inflammatory response along with osteoclast activation and osteoclast genesis (18). NFATC1 is a major TF that regulates osteoclast differentiation (25). Together with NFATC1, RelB regulates osteoclast formation (26). Given the broad range of inflammatory roles of NF-B, its targeting might be beneficial for treating RA.
2.3 Current treatments targeting NF-κB
2.3.1 Synthetic drugs
Conventional disease-modifying antirheumatic drugs (cDMARDs) and biological DMARDs (bDMARDs) have been used to treat RA for many decades. Methotrexate (MTX) is a first-line drug widely used to treat RA, while bDMARDs, such as TNF inhibitors, have been used since 1980. Currently, five main classes of TNF-inhibiting bDMARDs are available: etanercept, adalimumab, certolizumab pegol, golimumab, and infliximab. A recent clinical trial suggests switching from TNF inhibitors to tacrolimus (TAC) after acquiring low disease activity. TAC is an immunosuppressant that can block the calcineurin pathway in T cells by inhibiting cytokine production and T cell proliferation (27). Artemisinin-type compounds inhibit several receptor-coupled signaling pathways that include IL-1, TNF, RANKL, growth factor receptors, and TLRs (4). Terfenadine and Fexofenadine have recently been identified as more cost-effective and safer TNF inhibitors (28). Regulation of RANKL levels is maintained by bDMARDs (e.g., Denosumab) (29). All the above-mentioned drugs target cytokines that can activate NF-κB, thereby indirectly suppressing its activity. The long-term use of these drugs and the need to increase the dosage for an effective result can lead to adverse effects, such as osteoporosis, hyperlipidemia, hepatitis, tuberculosis, malignancies, and adrenal insufficiency (30). Furthermore, there is an increase in resistance to these drug in 30% of cases of RA (31).
Since there is a need for a different approach to reduce side effects, recent studies focus directly on targeting NF-κB, thus potentially achieving more precision in treating RA (Table 1). Tetrandrine, a bisbenzylisoquinoline, blocks NF-κB/RelA (32). Iguratimod is a new synthetic targeted DMARD (stDMARD) that inhibits the translocation of NF-κB to the nucleus and is approved only in China and Japan for RA treatment (33). Small-interfering RNA (siRNA) targeting NF-κB, delivered in combination with MTX inside a liposome capsule, prevents its release in the circulation, avoiding possible adverse effects of MTX (17). Chen et al., have demonstrated that low molecular weight polyethyleneimine cholesterol polyethylene glycol encapsulates siRNA as an efficient way to silence NF-κB/p65 to restore an anti-inflammatory microenvironment in RA (34). Drug delivery via nanocarriers is now being explored to deliver controlled doses of drug of interest to promote cell/tissue specific treatment, thus minimizing the potential side effects (43).
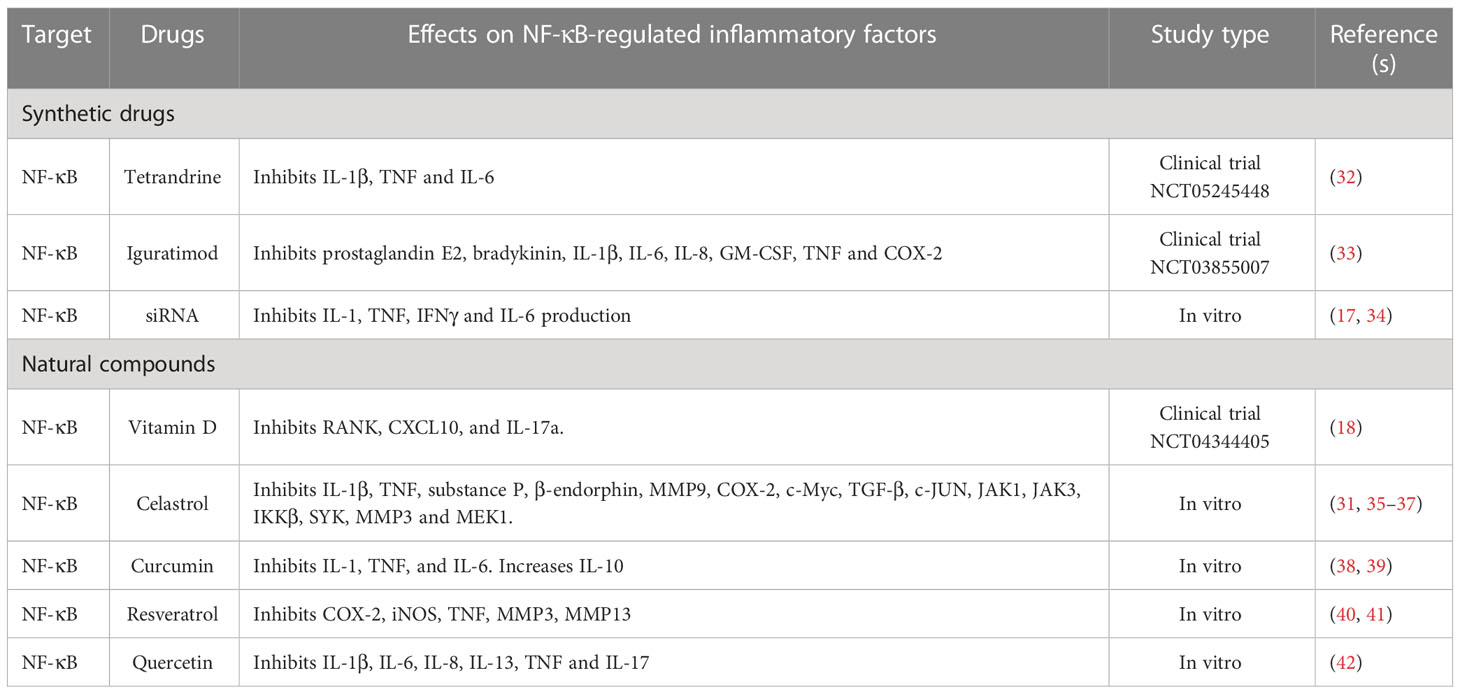
Table 1 Synthetic drugs and natural compounds targeting NF-κB either directly or indirectly.
2.3.2 Natural compounds
To minimize side effects caused by synthetic drugs, many studies are now focusing on natural compounds that can alleviate RA disease (Table 1). Celastrol, triptolide, resveratrol, curcumin, myricetin, fisetin and quercetin have been identified to hopefully reduce RA severity by targeting numerous cytokines, signaling pathways and proteases (41, 44). Numerous studies have shown the effect of celastrol on actively improving RA severity through suppression of the following: ROS-NF-κB-NLRP3 signaling (37), HIF expression and ROS release (36), the PI3-K/AKT/mTOR axis (45, 46) and NF-κB by degrading IκB (44, 47). Resveratrol, a polyphenol, activates sirt1, which suppresses the transcriptional activity of NF-κB/p65 by deacetylation and inhibits the COX/MMP pathway and the production of IL-1β, IL-6, and TNF (48). Curcumin suppresses the expression of NF-κB by upregulating that of miR-124 (39). Emerging findings suggest that treating RA patients with vitamin D supplementation can lower RANKL and CXCL10 levels, and suppress activation of NF-κB (18). Glucosamine prevents the demethylation of particular CpG sites in the promotor region of IL-1β, thereby preventing NF-κB from binding to the promotor region and suppressing the expression of IL-1β (49). These studies indicate the potential of natural compounds to not only target NF-κB, but also to suppress inflammation in RA.
3 JAK/STAT
3.1 JAK/STAT signaling pathway
The Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway is a key signaling pathway important in governing many biological processes, including cell differentiation, proliferation, and immune functions. Several studies have identified that the JAK/STAT signaling pathway is deregulated in RA (50, 51). Many of the proinflammatory cytokines, including TNF, IL-β, IL-6, IL-7, IL-8, IL-12, IL-15, IL-17, IL-23, IL-32, IFN and GM-CSF, that are highly expressed in RA are known to be regulated by JAK/STAT signaling pathway (50, 52). The JAK family has four members, JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2), while the STAT family of TFs consists of seven members, namely STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. Upon receptor ligation, JAKs are autophosphorylated, and recruit and phosphorylate members of the STAT family (Figure 2). Phosphorylated STATs dissociate from the receptor and form homo or heterodimers before translocating to the nucleus to activate the transcription of STAT-regulated genes (53). STATs bound to gene promoters can be dephosphorylated by nuclear protein tyrosine phosphatases (N-PTPs) and subsequently exit the nucleus to the cytoplasm for further activation cycles (54). Negative regulators of the JAK/STAT pathway, such as PTPs, protein inhibitors of activated STAT (PIAS), and suppressors of cytokine signaling proteins (SOCS), play crucial roles in controlling STAT-regulated gene expression (55–57).
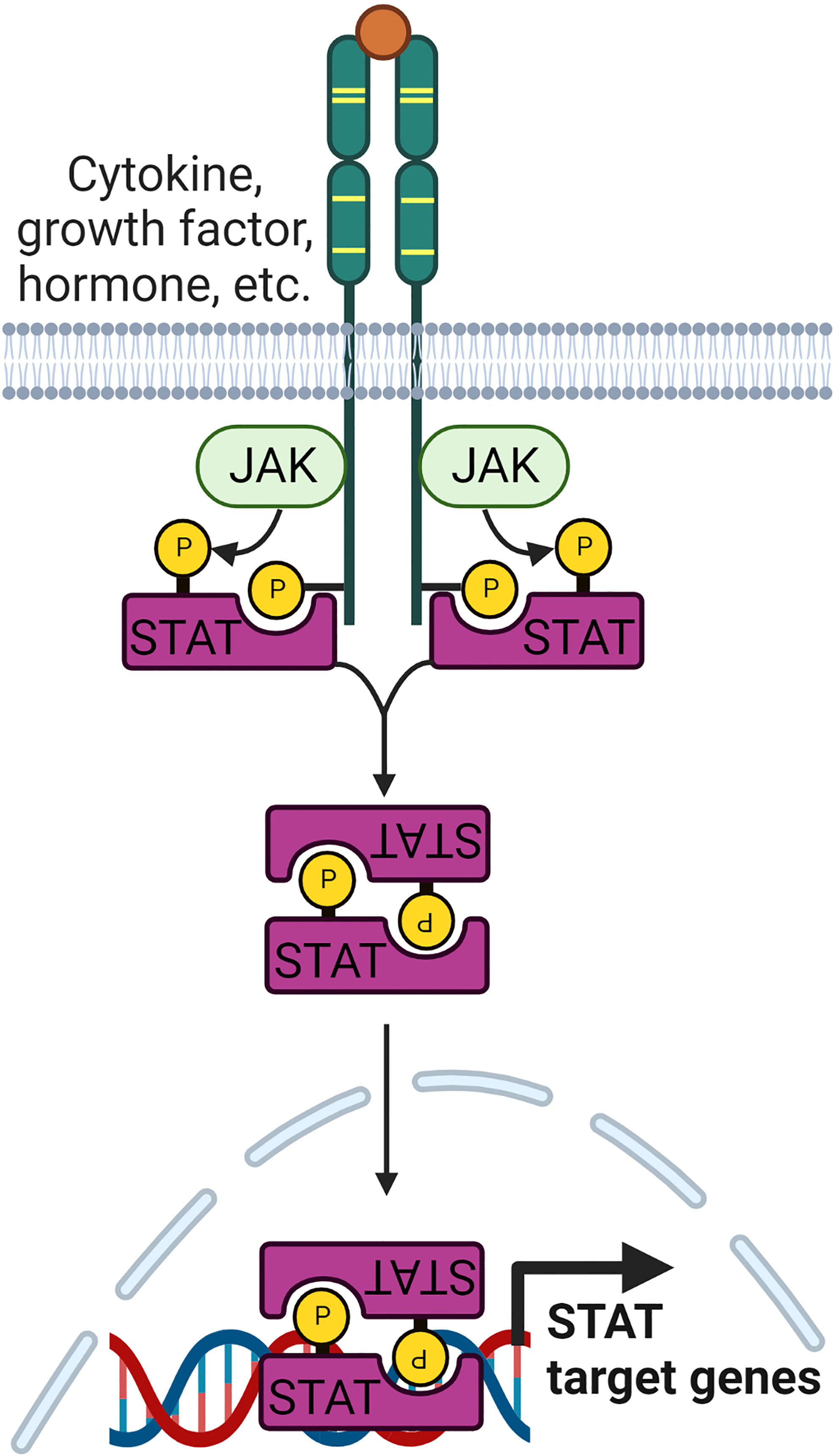
Figure 2 Signaling pathway leading to the regulation of JAK/STAT target genes. The family of JAK kinases are activated upon receptor ligation. Subsequently, they recruit STAT family proteins and phosphorylate them. Phosphorylated STATs form homo or heterodimers before being translocated to the nucleus to regulate transcription of STAT target genes.
JAKs and STATs are activated by stimulation with various cytokines (51, 58). JAK1 is phosphorylated by four types of cytokine receptor families: (i) cytokine receptor with γc (IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 receptors); (ii) receptors with gp130 subunits (IL-6, IL-11, IL-27, oncostatin M (OSM), cardiotrophin-1 (CT-1), leukemia inhibitory factor (LIF), cardiotrophin-like cytokine (CLC), and ciliary neurotrophic factor (CNTF) receptors; (59) (iii) class 2 cytokine receptors (IL-10 family, type 1 and 2 IFN receptors); and (iv) IL-3, IL-5, and GM-CSF receptors. As for JAK1, JAK2 is activated by (i) the gp130 receptor family, (ii) the IL-3R family (IL-3, IL-5R and GM-CSF receptors), (iii) the class 2 cytokine receptor family, and (iv) single chain receptors, such as growth hormone, thrombopoietin, prolactin and erythropoietin receptors. JAK3 is activated by IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21 receptors and TYK2 is activated by IFN type 1, IL-6 family, IL-10 family, IL-12, IL-13, and IL-23 receptors (60).
STAT1 is activated by IFNγ, IL-2, IL-6, IL-7, IL-21, epidermal growth factor (EGF), platelet-derived growth factor (PDGF), TNF, hepatocyte growth factor (HGF) and angiotensin 2. It has been found that IFNα/β are the only cytokines that can activate STAT2. STAT3 is activated by the IL-6 cytokine family (IL-6, IL-11, IL-27, IL-31, CNTF, OSM, and LIF), the IL-10 cytokine family (IL-10, IL-19, IL-20, IL-22, IL-24, IL-26, IL-28A, IL-28B and IL-29), GM-CSF, IL-2, IL-7, IL-21, IFNα/β, and leptin. STAT4 is activated by IL-12, IL-23 and IFNα/β whereas STAT5 is activated by the IL-3, the IL-2 cytokine family (IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21), prolactin, EGF, GM-CSF, PDGF and GH. STAT6 is activated only by IL-4 and IL-13 (58, 61, 62).
3.2 STAT-regulated genes
STATs bind directly to DNA regulatory elements and regulate gene transcription. STAT1, STAT3, STAT4, STAT5, and STAT6 have been shown to be highly expressed in RA (63). STATs often interact with other TFs, which assemble in the promotor or enhancer regions of target genes. Examples of such interactions are STAT1-STAT2 with IRF9, STAT1 with NF-κB, STAT3 with Jun, STAT3 with IRF4, RORγt, and BAFT in T cells, and STAT1 with IRF1, IRF8 and PU.1 in macrophages. These complex transcription networks highlight the fact that multiple TFs can be involved in regulating gene expression in various cell types.
3.2.1 STAT1
Various STAT proteins play a different role in different cell types that lead to RA pathogenesis. In synovial macrophages, STAT1 activates CXCL9 and CXCL10, which recruit T cells, induce Th1 differentiation, and upregulate IFNγ production (64, 65). STAT1 activation is essential for activated IRF1 and TLR3 in macrophages (66). STAT1 can induce iNOS expression and produce NO, which can reduce cell migration, while suppressing STAT3 activity (67). Most importantly, STAT1 regulates MMP3 and MMP13, thereby inducing cartilage degradation in the knee joint (68). A study by Kuuliala et al. has suggested that the activation of STAT1 and STAT6 in circulating leukocytes helps predict the response to treatment in RA (69).
3.2.2 STAT3
STAT3 is another major TF involved in inflammation. STAT3 induces angiogenesis, transcription of B cell lymphoma protein 2 (BCL-2), several MMPs, including MMP1, 3, and 13, and cyclins (70, 71). STAT3 activates RORγt, which induces IL-6 production and leads to Th17 polarization and stabilization (72). It can inhibit fibroblast apoptosis (73), the function of STAT1, and the expression of IFNα (74).
3.2.3 STAT4, STAT5, and STAT6
The production of the Th1-driven cytokine, IL-12, is mediated through STAT4. STAT5 promotes the production of CD4+ T cells (75). A few studies have reported the role of STAT5 in GM-CSF-induced CCL17 production (76, 77), a chemokine found to be important in inflammatory arthritis (78). IL-4 transduces signal through STAT6, which regulates the Treg cell response (55, 69).
3.3 Current treatments targeting JAK/STAT
3.3.1 Synthetic drugs
JAK/STATs are key regulators of cytokines produced in RA pathogenesis and therefore, are considered as feasible drug targets (60). Currently, JAK inhibitors (JAKi) are used as third-line therapy for RA patients with disease recurrence after using MTX and bDMARDs. JAKi are tsDMARDs, and they competitively inhibit by binding to the ATP binding site of the kinase domain present in JAKs, thereby inhibiting the JAK phosphorylation and preventing STAT activation (60). Among the currently available JAKi, baricitinib and tofacitinib are pan JAKi (Table 2). Baricitinib, tofacitinib and upadacitinib are approved by the FDA for the treatment of RA (87), while filgotinib and peficitinib are being evaluated (82). Tofacitinib is highly selective for JAK1 and JAK3, with less selectivity for JAK2 and TYK2 (64). Baricitinib inhibits JAK1 and JAK2, while moderately inhibits TYK2 and JAK3 (88). One study showed a similar safety profile for baricitinib and tofacitinib, but a better clinical outcome with baricitinib (89). The introduction of a nanostructure-based drug delivery system enables site-specific delivery of tofacitinib and the JNK inhibitor SP600125 (90). Upadacitinib and filgotinib selectively inhibit JAK1 and have been proven to be efficient in phase 2 and 3 studies (91). Another study demonstrated that baricitinib combined with MTX and upadacitinib with MTX can effectively inhibit the JAK/STAT signaling pathway (82).
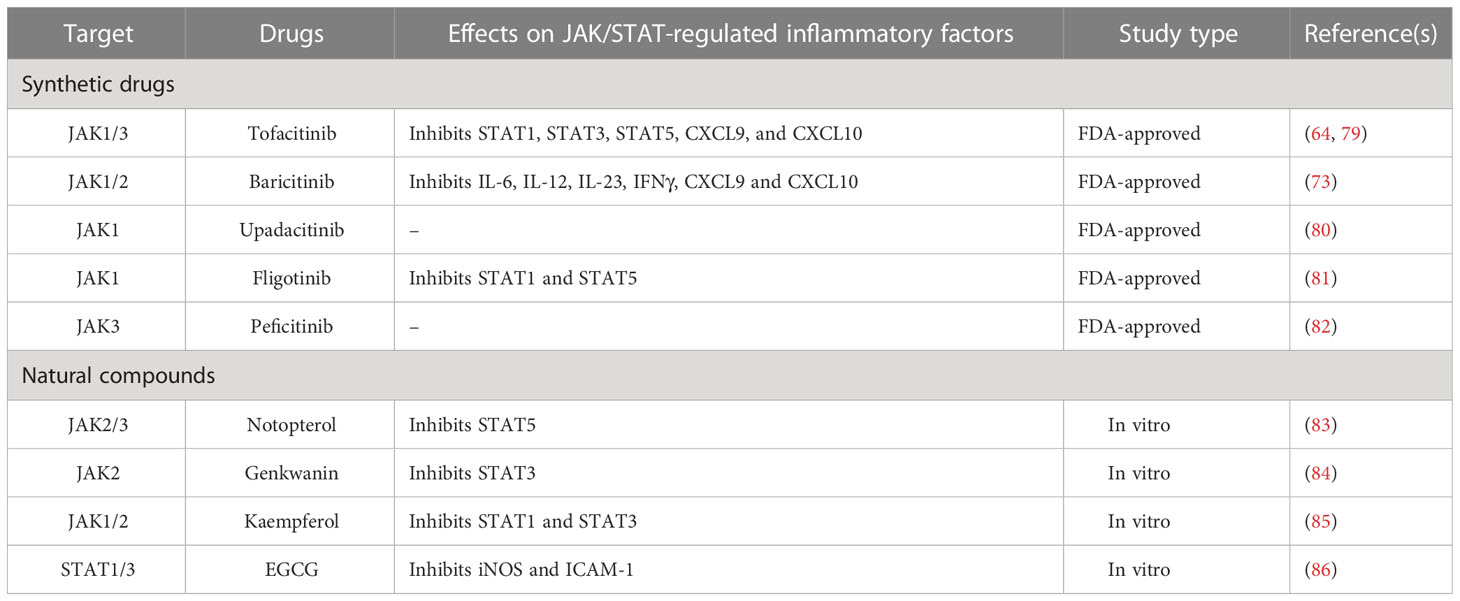
Table 2 Synthetic drugs and natural compounds targeting JAK/STAT either directly or indirectly.
3.3.2 Natural compounds
Although JAKi function effectively in RA patients, they are expensive for broader application and demonstrate adverse effects, including hepatotoxicity, gastrointestinal perforations, thromboembolism, herpes zoster, and tuberculosis; therefore, some studies are focusing on exploring natural compounds that can inhibit JAK/STAT signaling (87) (Table 2). Notopterol is a natural compound that effectively inhibits JAK2/JAK3 and suppresses the production of CXCL2, CXCL9, CXCL10, CXCL12, CCL5, IL-1β, IL-6, and TNF levels in bone marrow-derived macrophages. Genkwanin, a flavone, inhibits the JAK/STAT pathway by binding to JAK2 and NF-κB, reducing TNF, NO and IL-6 levels, while increasing IL-10 production (84, 92). Quercetin, epigallocatechin-3-gallate (EGCG), resveratrol, curcumin, genistein, chlorogenic acid, swertiamarin, cyanidin, ferulic acid, baicalein, falcarindiol, cinnamaldehyde and cryptotanshinone have been found to be effective in inhibiting JAK/STATs.
4 AP-1
4.1 Activation of AP-1
The activator protein-1 (AP-1) is proposed to play an important role in inflammation and pathogenesis of RA (93). Increased levels of c-Fos and c-Jun in RA synovium are correlated with disease severity (94). In the initial phase of RA, ROS activated AP-1, but in the late phase, proinflammatory cytokines can upregulate AP-1 (95). It is a leucine zipper TF composed of Fos, Jun, and ATF families of proteins (96). Fos proteins (FosB, Fra-1, Fra-2, c-Fos) heterodimerize with members of the Jun family, whereas Jun proteins (c-Jun, JunB and JunD) can heterodimerize and/or homodimerize with members of the Fos family to form transcriptionally functional complexes that bind to the promotor region of AP-1 sites (97) (Figure 3). The dimer composition of AP-1 and the active state of the Jun and Fos components determine the target of AP-1 (98). Jun : Jun and Fos : Jun dimers selectively bind to AP-1 motifs, known as the 12-O-tetra-decanoylphorbol-13-acetate (TPA) responsive element (TRE) and the cAMP-responsive element (CRE) (99).
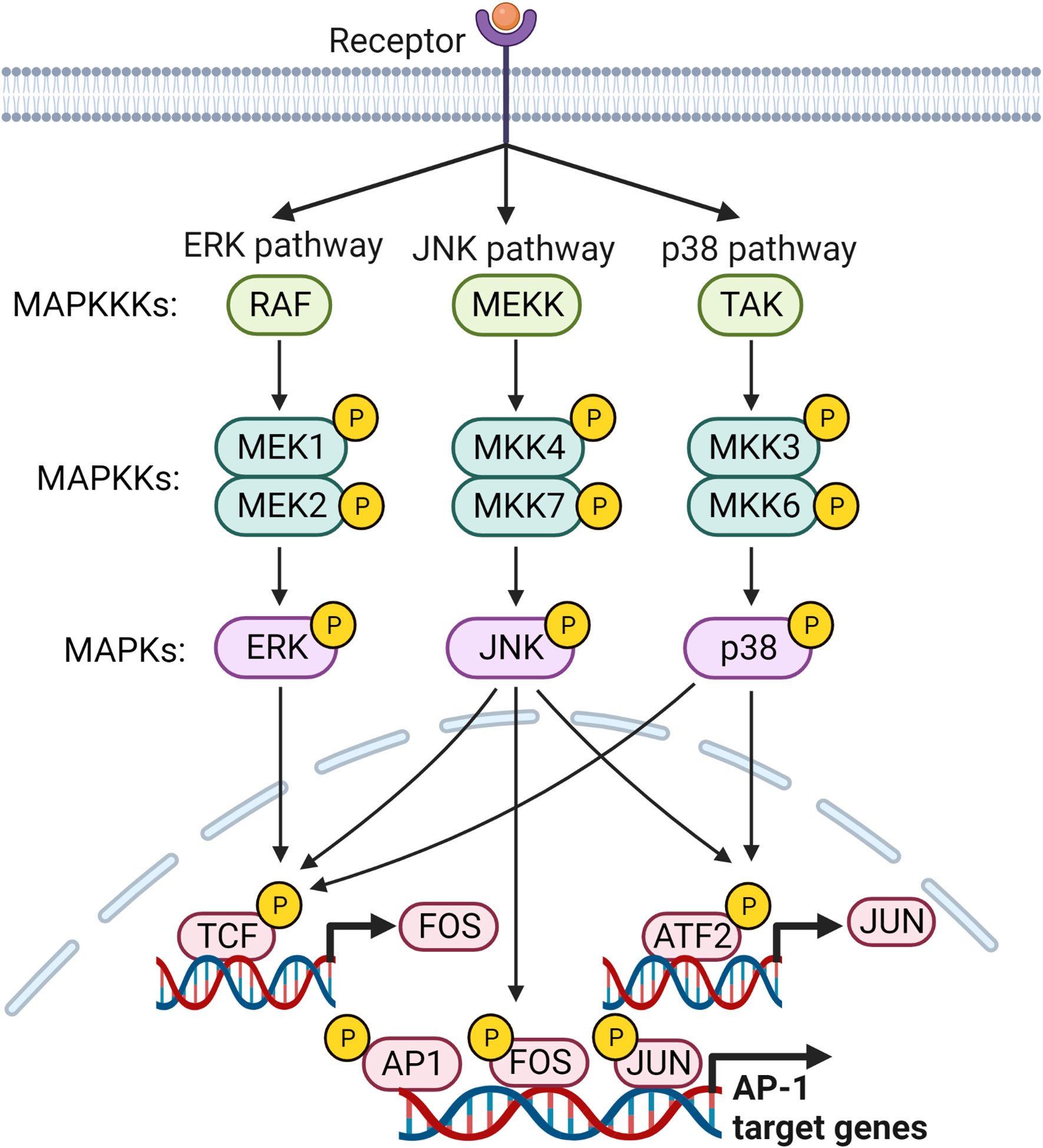
Figure 3 MAP kinase signaling pathways leading to the regulation of AP-1 target genes. Members of MAP kinases, ERK, JNK and p38, are responsible for activating transcription factors that regulate transcription of FOS and JUN genes and their subsequent activation. Subsequently, activated AP-1 binds to the promoter regions of its target genes and regulates their expression.
AP-1 is activated primarily by MAPK signaling. The three main subfamilies in the MAPK signaling pathway, extracellular signal-regulated kinases (ERKs), p38, and c-Jun N-terminal kinase (JNK), are essential for activation of AP-1 (96). MAPKs are activated by a cascade of phosphorylation events, wherein activated mitogen-activated protein kinase kinase kinase (MAPKKK) phosphorylates mitogen-activated protein kinase kinase (MAPKK), which finally phosphorylates MAPK (100). IL-1β, IL-6, TNF, TGF-β, and TPA up-regulates AP-1 through the MAPK pathway (101, 102). ERK1/2 is activated through a signaling cascade via phosphorylation of Ras, Raf, and MEK 1/2 (98). Stimulation of TLR4, IL-1R, and TNFR activates MyD88 and TAK1, which activates MKK4/7 or MKK3/6, thereby activating c-Fos/c-Jun of AP-1 by JNK and p38, respectively (103, 104). CXCL1 induces c-Jun phosphorylation in RA synovial fibroblasts (RASFs), and increased activation of AP-1 is observed in CXCL1 treated cells (93). AP-1 is activated by enhanced PI3K/AKT activation through stimulation of TNF- and thrombin-induced EGFR transactivation in chondrocytes (105, 106). Another study in RASFs showed that myostatin-induced TNF expression through the PIK3-AKT-AP-1 signaling pathway by activating the c-Jun binding site found in the TNF gene promoter region (107). Activating transcription factor 2 (ATF2), a member of the AP-1 TF family, is highly expressed in RA FLS activated via ERK and MAPK. Sprouty2 can inhibit ATF2 overexpression by inhibiting the phosphorylation of ERK and MAPK (108).
4.2 AP-1-regulated genes
4.2.1 Fos
AP-1 selectively regulates a range of cytokines, chemokines, proteinases, and TFs. Each member of the AP-1 family differentially regulates genes. c-Fos/AP-1 induce the expression of MMPs (e.g., MMP1, 2, 3, 8, 9, and 13) and cytokines (e.g., IL-23) (109, 110). MMPs are mainly regulated by IL-1β-induced c-Fos/AP-1; most genes in the MMP family have an AP-1 binding site in the promoter regions near the TATA box and a mutation in the AP-1 binding site completely suppresses MMP expression (111). MMPs are essential for cartilage joint matrix breakdown and MMP13 predominantly degrades cartilage by cleaving type 2 collagen (111). IL-1β can induce osteoclast genesis directly and/or indirectly through RANKL signaling. Integration of RANKL and M-CSF signaling requires Fos/AP-1 (112).
4.2.2 Jun
c-Jun differentially regulates cyclooxygenase-2 (COX-2) and arginase-1 (ARG-1) and promotes macrophage activation, thus contributing to arthritis progression (113). JunB can control Th17 differentiation by inducing the expression of RORγt and RORα, while suppressing the expression of Foxp3 (72). JunB synergizes with c-MAF and GATA3 and induces activation of IL-4, which induces Th2 cell differentiation (114). cJun and JunB together activate AKT1 by binding directly to its promoter region (115).
4.2.3 NFAT
The nuclear factor of activated T cells (NFAT) is suggested to play a role in the pathogenesis of RA (116). AP-1 interacts with NFAT and cooperatively forms an AP-1/NFAT complex, which enhances transcriptional activity compared to Fos-Jun or NFAT binding and regulates most cytokines. It regulates IL-2, which is required for Treg proliferation (114). As AP-1 regulates some important inflammatory mediators that promote RA, it serves as a treatment target to alleviate RA. Synthetic drugs and natural compounds targeting AP-1 are being studied at present.
4.3 Current treatments targeting AP-1
4.3.1 Synthetic drugs
Given the role of AP-1 in the regulation of key inflammatory mediators known to promote RA, targeting it is a potential treatment solution; however, there are no FDA-approved AP-1 inhibitors available in the clinic. Many in vitro and in vivo studies are currently focusing on drugs that can inhibit AP-1 (Table 3). CKD-506 is an orally administered hydroxamate that blocks the activation of AP-1 and NF-κB transcription in peripheral blood mononuclear cells isolated from RA patients (117). T-5224, a molecular inhibitor of c-Fos/AP-1, inhibits the DNA binding of c-Fos/c-Jun, thus inhibiting IL-1β, IL-6, TNF, MMP1, 3, and 13. N-(3-acetamidophenyl)-2-[5-(1H-benzimidazol-2-yl) pyridin2-yl] sulfanylacetamide can disrupt the interaction between AP-1 and NFAT and blocks the transcription of IL-2 and some cyclosporin A-sensitive cytokines (126). A cyclin-dependent kinase 4/6 (CDK) inhibitor (CDKi) blocks AP-1 transcription by decreasing Jun stability, thus blocking the production of MMP3 and attenuating cartilage destruction in the collagen-induced arthritis model (119). A novel JNK inhibitor, 11H-indeno[1,2-b] quinoxaline-11-one oxime, has been shown to not only inhibit JNK phosphorylation but also block the transcriptional activity of AP-1 and NF-κB (120). Roflumilast, a selective phosphodiester-4 inhibitor, inhibits the production of IL-1β, IL-6, TNF, CCL5, CXCL9, CXCL10, MMP3, and MMP13 by blocking the transcriptional activity of AP-1 and NF-κB (118). Many of these synthetic drugs targeting AP-1 show promise in preclinical studies, but further research and clinical trials are needed before obtaining FDA approval.

Table 3 Synthetic drugs and natural compounds targeting AP-1 either directly or indirectly.
4.3.2 Natural compounds
While there are no FDA-approved AP-1 inhibitors available, many studies have explored natural compounds that can potentially block components in the AP-1 pathway (Table 3). Antcin K is an extract taken from a medicinal mushroom, Antrodia cinnamomea, that inhibits vascular cell adhesion molecule 1 (VCAM-1) and monocyte adhesion to RASFs by inhibiting the phosphorylation of p38 and MEK1/2-ERK (122). Apigenin-4´-O-α-L-rhamnoside, a natural flavonoid, exhibits inhibitory mechanisms against MMP1, MMP3, TNF, and RNAKL in RA FLS by inhibiting the MAPK/JNK/p38 pathway (102). Anticin K and Apigenin-4´-O-α-L-rhamnoside inhibit the inflammatory mediators of AP-1 and indirectly suppress the activation of AP-1. Treatment with resveratrol directly suppresses bradykinin-mediated AP-1 and NF-κB activities and inhibits COX-2 production in RASFs (40). Melittin, the primary component of bee venom, exhibits inhibitory properties by suppressing MMP1 and MMP8 by blocking the phosphorylation of PI3-K/AKT and ERK/JNK and the translocation of c-fos (106). Thymoquinone is another natural compound found in Nigella sative, which shows anti-inflammatory properties in preclinical arthritis models, blocking multiple pathways that include AP-1 and NF-κB (127). The ethanolic extract of Sigesbeckia orientalis inhibits pannus formation and reduces cartilage damage and bone erosion in the collagen-induced arthritic model, while it leads to decreased expression of IL-1β, IL-6, IL-8, COX-2, MMP9, and NLRP3 by inhibiting MAPKs, AP-1, and NF-κB in in vitro studies carried out in synovial cells (125).
5 IRFs
5.1 Activation of IRFs
In humans, the interferon regulatory factor (IRF) family of transcription factors consists of nine members, IRF1 to IRF9. They share a homology region found in the N-terminal DNA binding domain, which binds to the interferon stimulated response element (ISRE). The diverse C-terminal domain is unique to each member and binds to a wide range the proteins outside the IRF family. They are activated via signals received by activation of TLR and BCR (128). Upon activation, IRFs can form homo- or hetero dimers, which are translocated to the nucleus. Hyperactivated IRFs mainly produce IFNs and thus contribute to inflammatory diseases. Each member of the IRF family is regulated by a range of inflammatory mediators present in RA synovium and their mode of activation is discussed below (Figure 4).
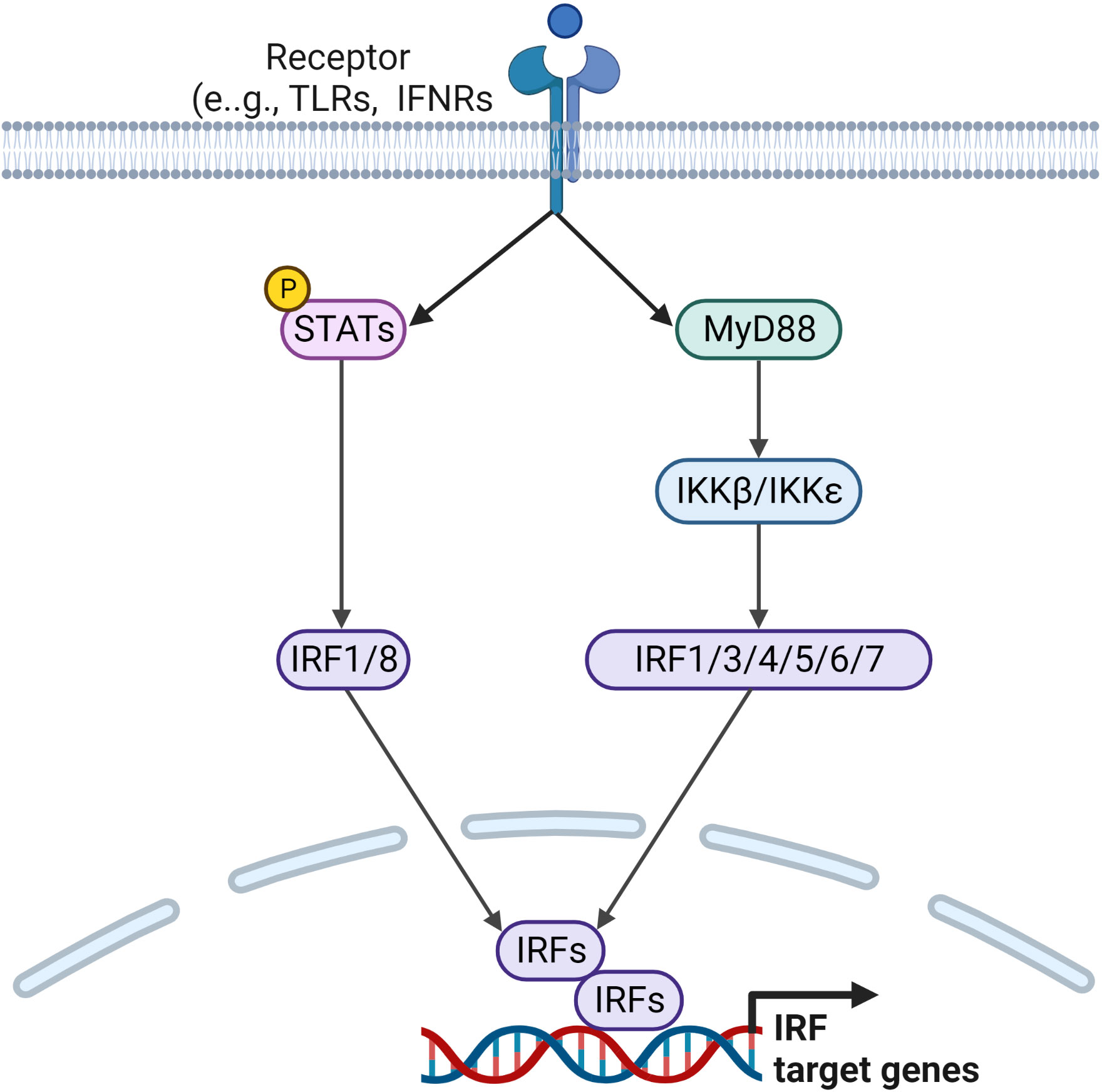
Figure 4 Signaling pathways leading to the regulation of IRF target genes. Ligands binding to TLRs and IFNRs initiate downstream signaling pathways via downstream activators and adaptor molecules, such as STATs and MyD88, which lead to the activation of IRF family transcription factors. Activated IRFs form homo or heterodimers before being translocated to the nucleus, where they regulate transcription of IRF target genes.
5.1.1 IRF1
TLR-activated TAK1 can induce IRF1 transcription via the RelA/p50 complex (129). Furthermore, the JAK/STAT signaling cascade has also been shown to induce the expression of IRF1 (130). The following studies have also demonstrated a similar pathway of IRF1 activation. Pristane induces autophagy in macrophages and can induce IRF1 activation by activating STAT1 (66), and IKKβ regulates IRF1 transcription in conventional type 1 dendritic cells (129).
5.1.2 IRF3
IRF3 is activated by various signals received from intracellular receptors such as RIG-1, MDA5, TLR3, TLR4, and cytosolic double-stranded DNA (dsDNA) sensors (131). Activated TLR3 signals via TRIF to activate TBK1 and IKKε via TRAF3. dsDNA in the cytosol can trigger type 1 IFN through STING, which can activate IRF3 by stimulating its phosphorylation by activating TBK1. It is suggested that the C-terminal tail of the STING oligomer can recruit both TBK1 and IRF3 by binding to the IRF3 motif and delivers IRF3 to TBK1 (132). Finally, TBK1, along with IKKϵ, phosphorylate IRF3 to form dimers and then translocate to the nucleus (133).
5.1.3 IRF4
IRF4 is induced by activation of BCR and CD40 in B cells, TCR in T cells, and TLR in macrophages. However, it is not activated by type 1 or type 2 IFNs. The receptor activation leads to activation of c-Rel, which binds to the promotor region of IRF4 to induce transcription. The promotor region contains Foxp3, STAT4, STAT6, and IRF4 binding sites, suggesting that IRF4 is capable of autoregulating its expression (134).
5.1.4 IRF5
IRF5 activation is initiated upon binding of ligands to TLR7/8/9, wherein MyD88, IRAK1/4, and TRAF6 are recruited. Then autophosphorylation of IRAK4 activates TAK1 to phosphorylate IKKβ. Meanwhile, TRAF6 ubiquitinates IRF5, which is subsequently phosphorylated by IKKβ, thus forming homodimers. IRF5 homodimers are translocated to the nucleus to activate target genes (128).
5.1.5 IRF6, IRF7 and IRF8
IRF6 is regulated via TLR2 in epithelial cells and TLR3 in keratinocytes (135). In keratinocytes, RIPK4 phosphorylates and activates IRF6 (136). IRF7 is activated through TLR3 and TLR7 via two different pathways. TLR3 is activated by dsRNA and phosphorylates IRF7 via TRIF, while TLR7 signals via MyD88-TRAF6 signaling, which is induced by ssRNA (137). IRF8 is activated in macrophages, dendritic cells, T cells, and NK cells. Binding of IFNγ to its receptor activates IRF8 by activating STAT1. IRF8 is also activated following stimulation with IFNα/β and LPS (138).
5.2 IRF-regulated genes
IRFs play a major role in autoinflammation and autoimmunity (139). IRF1, IRF3, IRF5 and IRF7 are important in the induction of type 1 IFN, where IRF4, IRF5 and IRF8 regulate the development of myeloid cells and play a crucial role in inflammatory responses (140). A wide range of studies have suggested a role for each IRF in inflammatory diseases, including RA.
5.2.1 IRF1
IRF1 regulates several IFN-regulated genes (e.g., CXCL9, CXCL10, and CXCL1)1 in rheumatoid synovium and activates B cell activating factor (BAFF), which is highly expressed in RA (141). IRF1 regulates TNF-induced IFNβ expression and subsequently activates STAT1 to activate IFN-regulated genes (141). IRF1 can induce TLR3 expression in pristane-induced arthritis (66). A recent study demonstrated that the invasiveness of synovial fibroblasts is regulated by the expression of follistalin-like protein 1 induced by IRF1 (142).
5.2.2 IRF2 and IRF3
IRF2 negatively regulates IFN type 1 signaling and counterbalances the activity of IRF1. IRF2 activates IL-12p40 and VCAM-1, leading to the development of NK cells and Th1 cells, respectively (143). IRF2 stimulates inflammatory ROS levels, TNF, IL-1β, and IL-6 expression, and suppresses superoxide dismutase. The knockdown of IRF2 gene is shown to inhibit the JAK/STAT signaling pathway (144). IRF3 regulates the expression of IL-6, IL-8, MMP3, and MMP9 in RA FLS by activating c-Jun/AP-1 (145).
5.2.3 IRF4
IRF4 plays a diverse role in inflammation and arthritis. It is mainly involved in T cell differentiation. IRF4 responds to IL-4 and regulates Th1 and Th2 differentiation through interaction with T-bet and GATA3, respectively. IRF4 binds directly to RORγt and mediates the differentiation of Th17 cells (146) and regulates Glut1, IL-17 and IL-21 levels (147, 148). Furthermore, it interacts with BCL-6 and Foxp3 to produce T follicular helper cells and Tregs, respectively (146). In macrophages, IRF4 distorts macrophages into the M2 phenotype through JMJD3 competing for MyD88 with IRF5 while suppressing M1 polarization of M1 and inducing IL-4 and IL-10 secretion. Previous studies have shown that a pro-inflammatory cytokine GM-CSF can regulate CCL17 formation in monocytes/macrophages through JMJD3 and IRF4 (78). In addition to these functions, IRF4 binds to STAT3, STAT6, and NFATs to carry out transcription. IRF4 functions as a transcriptional repressor by forming a homodimer or heterodimer with IRF8, suppressing the expression of IFN-inducible genes and inhibiting IRF1 activity in macrophages and T cells (134).
5.2.4 IRF5
Studies show that IRF5 acts in T cells, monocytes, and macrophages. Increased expression of IRF5 induces M1 polarization while suppressing M2 polarization (128). IRF5 increases the expression of IL-12 in circulating monocytes in samples from OA patients without treatment and promotes Th1-related genes in resting T cells (149). Furthermore, IRF5 induces a wide range of pro-inflammatory cytokines such as IL-17, monocyte chemotactic proteins (MCP-1), TNF-α, RANTES, IL-6, IL-12p40, and IL-23p40 (150, 151). IRF5 up-regulates MMP3 production mediated via NF-κB (151).
5.2.5 IRF6 and IRF7
TLR3-induced activation of IRF6 leads to enhanced expression of IL-23p19, while negatively regulating IFNβ expression by competing with IRF3 in the IFNβ promotor region or by forming a heterodimer complex with IRF3 (135). In addition, IRF6 induces the expression of IL-8, CCL5, and CXCL11 (136, 152) and IRF7 mediates RANKL production in RA FLS (137).
5.2.6 IRF8
IRF8 is crucial for the development and maturation of myeloid cells. At the transcription level, IRF8 is co-recruited to form ternary complexes with other TFs. It forms a heterodimer with IRF1, STATs, AP-1, and PU.1 (138, 143) and induce the production of IL-6, IL-12p40 and TNF. On the other hand, IRF8 negatively regulates osteoclastogenesis by inducing IFNγ (153). Recently, it was found that IRF8 can promote the expression of MMP13 in OA (154).
5.3 Current treatments targeting IRFs
Since the IRF family of TFs is involved in the regulation of a wide range of inflammatory mediators, they can be potential treatment targets for RA. Currently a type 1 IFN inhibitor, anifrolumab, is subjected to phase 2 clinical trials (155). A study in RA patients by Juge et al. shows a IRF5 response to rituximab within 24 weeks (156). Certain JAKi, such as tofacitinib and baricitinib, are documented to suppress the activity of certain IRFs by inhibiting STAT1 activity (141).
6 Other TFs
In addition to the TFs discussed above, other TFs such as, hypoxia-inducible factor (HIF) and nuclear factor-erythroid 2-related factor-2 (Nrf2), are also implicated in the pathogenesis of RA. In RA synovium, HIF is activated during hypoxia, which aggravates angiogenesis, synovial hyperplasia, and pannus formation (157–159). TNF, IL-1β, and IL-33 can induce the expression of HIF in RASFs and resident macrophages (157). Production of HIF primarily induces the expression of VEGF, which promotes the synthesis of proteolytic enzymes in endothelial cells (160). Furthermore, it promotes the generation of M1-type macrophages and Th17 cells (161, 162). Knockdown of HIF-α in collagen-induced arthritis (CIA) mouse model has been shown to inhibit multiple inflammatory pathways and thereby, ameliorating arthritis (163). In recent years, several studies are focusing on HIF inhibitors as potential therapeutics for treating arthritis. Pharmacological HIF inhibitor, PT2977 has been shown to ameliorate arthritis in the CIA mouse model (164). Moreover, natural compounds, that include andrographolide, geniposide, dihydroarteannuin, and tylophorine-based compounds, can inhibit HIF and be effective in attenuating RA progression (163, 165–167).
Nrf2 is a redox regulator, which plays a protective role by exerting anti-inflammatory and antioxidant effects (168). Significantly, the protective role of Nrf2 has been linked to relieving severe symptoms in RA via detoxification, regulation of redox balance, and metabolism (34, 169). TNF and increased ROS levels can induce the expression of Nrf2 in RA synovium, which in return suppresses the proliferation and MMPs production in RAFLS via inhibition of inflammatory mediators activated in RA (169). Due to this protective role of Nrf2, studies are focusing on synthetic drugs, including, dihydroartemisinin, and dimethyl fumarate, as well as natural compounds, including sinomenine, licochalcone, 7-deacetyl-gedunin, calycosin and resveratrol, that increase the expression of Nrf2 to treat RA (170–174; 90). While both HIF and Nrf2 have been identified as potential treatment targets for RA, further studies, utilizing the above-mentioned synthetic and natural compounds, are required to explore their therapeutic potential.
7 Network of TFs
In a complex disease, such as in RA, all the aforementioned TFs can form a network to cross-regulate each other or function cooperatively to activate or antagonize downstream target genes (175, 176). Examples of such interactions are STAT1-STAT2 with IRF9, STAT1 with NF-κB, STAT3 with Jun, STAT3 with IRF4, RORγt, and BAFT in T cells, and STAT1 with IRF1, IRF8 and PU.1 in macrophages (146, 177). Further, c-Fos/AP-1 and NFATc1 together control the osteoclast differentiation (110). IRF4 can bind to STAT3, STAT6, and NFATs to facilitate transcription of their downstream genes but it can also function as a transcriptional repressor by forming a heterodimer with IRF8, suppressing the expression of IFN-inducible genes and inhibiting IRF1 activity in macrophages and T cells (134). AP-1, NF-κB, and IRFs together are known to activate MMPs, (178). NF-κB, IRF4/8, PU.1, AP-1, and STAT1 induce the expression of IL-1β (179). STAT3 can activate HIF in RA synovium (180). While these studies highlight the complex network of TFs and their regulation of downstream inflammatory mediators, a careful approach is warranted when targeting them for therapeutic benefits. Since these TFs function both individually and cooperatively, targeting one or more TFs can effectively ameliorate RA. However, the key TFs involved in RA pathogenesis are also associated with biological processes involving homeostasis, and therefore inhibiting these TFs may lead to undesirable side effects. This challenge is currently being addressed by tissue-specific/joint-specific drug delivery via nanocarriers, which increase the specificity and efficacy, while minimizing potential adverse effects (17, 30, 31, 35, 121, 181–183).
8 Conclusion and prospect
RA is a chronic inflammatory autoimmune disease, causing pain and disability. Several drugs that are currently used for RA treatment are effective only delaying the progression of the disease or alleviating inflammatory symptoms. Many of these drugs have drawbacks, including disease recurrence and adverse effects due to long-term use. Therefore, there is a need to develop novel therapeutic strategies to address these shortcomings.
TFs play important roles in immune and nonimmune cells through regulation of gene expression. Studies emphasize the importance of the forementioned TFs in RA disease initiation and progression of RA disease. With the approval of JAK inhibitors in the treatment of RA, the pursuit of TFs or their signaling pathways as potential treatment targets has gained momentum. Currently, several inhibitors of TFs are being investigated, and they block TF function by inhibiting protein-protein interaction, translocation of TFs from the cytosol to the nucleus, or protein-DNA binding.
In summary, this review highlights key TFs and their signaling pathways that may become targets for future RA therapies; it also provides an update on several synthetic drugs and natural compounds that are in consideration for targeting such TFs or the signaling pathways that activate TFs.
Author contributions
All authors contributed to the writing and revision of the manuscript. TB and AA developed the concept, structure, and prepared tables and figures. TB drafted the original manuscript. AA, JH, and KL reviewed and edited.
Funding
The project was funded by grants to AA from the National Health and Medical Research Council (NHMRC) of Australia (1159901) and the University of Melbourne (UoM) Research Grant Support Scheme.
Acknowledgments
Figures were drawn with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lin YJ, Anzaghe M, Schülke S. Update on the pathomechanism, diagnosis, and treatment options for rheumatoid arthritis. Cells (2020) 9(4):880. doi: 10.3390/cells9040880
2. Chang MR, Rosen H, Griffin PR. RORs in autoimmune disease. Curr Top Microbiol Immunol (2014) 378:171–82. doi: 10.1007/978-3-319-05879-5_8
3. Chen SJ, Lin GJ, Chen JW, Wang KC, Tien CH, Hu CF, et al. Immunopathogenic mechanisms and novel immune-modulated therapies in rheumatoid arthritis. Int J Mol Sci (2019) 20(6):1332. doi: 10.3390/ijms20061332
4. Efferth T, Oesch F. The immunosuppressive activity of artemisinin-type drugs towards inflammatory and autoimmune diseases. Med Res Rev (2021) 41(6):3023–61. doi: 10.1002/med.21842
5. Huang J, Fu X, Chen X, Li Z, Huang Y, Liang C. Promising therapeutic targets for treatment of rheumatoid arthritis. Front Immunol (2021) 12:686155. doi: 10.3389/fimmu.2021.686155
6. Hamilton JA. GM-CSF-Dependent inflammatory pathways. Front Immunol (2019) 10:2055. doi: 10.3389/fimmu.2019.02055
7. Achuthan AA, Lee KMC, Hamilton JA. Targeting GM-CSF in inflammatory and autoimmune disorders. Semin Immunol (2021) 54:101523. doi: 10.1016/j.smim.2021.101523
8. Kondo N, Kuroda T, Kobayashi D. Cytokine networks in the pathogenesis of rheumatoid arthritis. Int J Mol Sci (2021) 22(20):10922. doi: 10.3390/ijms222010922
9. Papavassiliou KA, Papavassiliou AG. Transcription factor drug targets. J Cell Biochem (2016) 117(12):2693–6. doi: 10.1002/jcb.25605
10. Makarov SS. NF-kappa b in rheumatoid arthritis: a pivotal regulator of inflammation, hyperplasia, and tissue destruction. Arthritis Res (2001) 3(4):200–6. doi: 10.1186/ar300
11. Li J, Tang RS, Shi Z, Li JQ. Nuclear factor-κB in rheumatoid arthritis. Int J Rheum Dis (2020) 23(12):1627–35. doi: 10.1111/1756-185x.13958
12. Roman-Blas JA, Jimenez SA. Targeting NF-kappaB: a promising molecular therapy in inflammatory arthritis. Int Rev Immunol (2008) 27(5):351–74. doi: 10.1080/08830180802295740
13. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal transduction targeted Ther (2017) 2:17023–. doi: 10.1038/sigtrans.2017.23
14. Pflug KM, Sitcheran R. Targeting NF-κB-Inducing kinase (NIK) in immunity, inflammation, and cancer. Int J Mol Sci (2020) 21(22):8470. doi: 10.3390/ijms21228470
15. Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal transduction targeted Ther (2020) 5(1):209. doi: 10.1038/s41392-020-00312-6
16. Choi MC, Jo J, Park J, Kang HK, Park Y. NF-κB signaling pathways in osteoarthritic cartilage destruction. Cells (2019) 8(7):734. doi: 10.3390/cells8070734
17. Duan W, Li H. Combination of NF-kB targeted siRNA and methotrexate in a hybrid nanocarrier towards the effective treatment in rheumatoid arthritis. J Nanobiotechnology (2018) 16(1):58. doi: 10.1186/s12951-018-0382-x
18. Ilchovska DD, Barrow DM. An overview of the NF-kB mechanism of pathophysiology in rheumatoid arthritis, investigation of the NF-kB ligand RANKL and related nutritional interventions. Autoimmun Rev (2021) 20(2):102741. doi: 10.1016/j.autrev.2020.102741
19. Koch PD, Pittet MJ, Weissleder R. The chemical biology of IL-12 production via the non-canonical NFkB pathway. RSC Chem Biol (2020) 1(4):166–76. doi: 10.1039/d0cb00022a
20. Nejatbakhsh Samimi L, Farhadi E, Tahmasebi MN, Jamshidi A, Sharafat Vaziri A, Mahmoudi M. NF-κB signaling in rheumatoid arthritis with focus on fibroblast-like synoviocytes. Autoimmun Highlights (2020) 11(1):11. doi: 10.1186/s13317-020-00135-z
21. Kusiak A, Brady G. Bifurcation of signalling in human innate immune pathways to NF-kB and IRF family activation. Biochem Pharmacol (2022) 205:115246. doi: 10.1016/j.bcp.2022.115246
22. Dong Y, Yang C, Pan F. Post-translational regulations of Foxp3 in treg cells and their therapeutic applications. Front Immunol (2021) 12:626172. doi: 10.3389/fimmu.2021.626172
23. Pandolfi F, Franza L, Carusi V, Altamura S, Andriollo G, Nucera E. Interleukin-6 in rheumatoid arthritis. Int J Mol Sci (2020) 21(15):5238. doi: 10.3390/ijms21155238
24. Dorrington MG, Fraser IDC. NF-κB signaling in macrophages: dynamics, crosstalk, and signal integration. Front Immunol (2019) 10:705. doi: 10.3389/fimmu.2019.00705
25. Yao Z, Getting SJ, Locke IC. Regulation of TNF-induced osteoclast differentiation. Cells (2021) 11(1):132. doi: 10.3390/cells11010132
26. Zhao Z, Hou X, Yin X, Li Y, Duan R, Boyce BF, et al. TNF induction of NF-κB RelB enhances RANKL-induced osteoclastogenesis by promoting inflammatory macrophage differentiation but also limits it through suppression of NFATc1 expression. PloS One (2015) 10(8):e0135728. doi: 10.1371/journal.pone.0135728
27. Jung SY, Koh JH, Kim KJ, Park YW, Yang HI, Choi SJ, et al. Switching from TNFα inhibitor to tacrolimus as maintenance therapy in rheumatoid arthritis after achieving low disease activity with TNFα inhibitors and methotrexate: 24-week result from a non-randomized, prospective, active-controlled trial. Arthritis Res Ther (2021) 23(1):182. doi: 10.1186/s13075-021-02566-z
28. Liu R, Chen Y, Fu W, Wang S, Cui Y, Zhao X, et al. Fexofenadine inhibits TNF signaling through targeting to cytosolic phospholipase A2 and is therapeutic against inflammatory arthritis. Ann Rheum Dis (2019) 78(11):1524–35. doi: 10.1136/annrheumdis-2019-215543
29. Chiu YG, Ritchlin CT. Denosumab: targeting the RANKL pathway to treat rheumatoid arthritis. Expert Opin Biol Ther (2017) 17(1):119–28. doi: 10.1080/14712598.2017.1263614
30. Li J, Hao J. Treatment of neurodegenerative diseases with bioactive components of tripterygium wilfordii. Am J Chin Med (2019) 47(4):769–85. doi: 10.1142/s0192415x1950040x
31. An L, Li Z, Shi L, Wang L, Wang Y, Jin L, et al. Inflammation-targeted celastrol nanodrug attenuates collagen-induced arthritis through NF-κB and Notch1 pathways. Nano Lett (2020) 20(10):7728–36. doi: 10.1021/acs.nanolett.0c03279
32. Gao LN, Feng QS, Zhang XF, Wang QS, Cui YL. Tetrandrine suppresses articular inflammatory response by inhibiting pro-inflammatory factors via NF-κB inactivation. J Orthop Res (2016) 34(9):1557–68. doi: 10.1002/jor.23155
33. Xie S, Li S, Tian J, Li F. Iguratimod as a new drug for rheumatoid arthritis: current landscape. Front Pharmacol (2020) 11:73. doi: 10.3389/fphar.2020.00073
34. Chen J, Zhu G, Sun Y, Wu Y, Wu B, Zheng W, et al. 7-deacetyl-gedunin suppresses proliferation of human rheumatoid arthritis synovial fibroblast through activation of Nrf2/ARE signaling. Int Immunopharmacol (2022) 107:108557. doi: 10.1016/j.intimp.2022.108557
35. Kang Q, Liu J, Zhao Y, Liu X, Liu XY, Wang YJ, et al. Transdermal delivery system of nanostructured lipid carriers loaded with celastrol and indomethacin: optimization, characterization and efficacy evaluation for rheumatoid arthritis. Artif Cells Nanomed Biotechnol (2018) 46(sup3):S585–s597. doi: 10.1080/21691401.2018.1503599
36. Xinqiang S, Erqin D, Yu Z, Hongtao D, Lei W, Ningning Y. Potential mechanisms of action of celastrol against rheumatoid arthritis: transcriptomic and proteomic analysis. PloS One (2020) 15(7):e0233814. doi: 10.1371/journal.pone.0233814
37. Jing M, Yang J, Zhang L, Liu J, Xu S, Wang M, et al. Celastrol inhibits rheumatoid arthritis through the ROS-NF-κB-NLRP3 inflammasome axis. Int Immunopharmacol (2021) 98:107879. doi: 10.1016/j.intimp.2021.107879
38. Yan F, Li H, Zhong Z, Zhou M, Lin Y, Tang C, et al. Co-Delivery of prednisolone and curcumin in human serum albumin nanoparticles for effective treatment of rheumatoid arthritis. Int J Nanomedicine (2019) 14:9113–25. doi: 10.2147/ijn.S219413
39. Qiu B, Xu X, Yi P, Hao Y. Curcumin reinforces MSC-derived exosomes in attenuating osteoarthritis via modulating the miR-124/NF-kB and miR-143/ROCK1/TLR9 signalling pathways. J Cell Mol Med (2020) 24(18):10855–65. doi: 10.1111/jcmm.15714
40. Yang CM, Chen YW, Chi PL, Lin CC, Hsiao LD. Resveratrol inhibits BK-induced COX-2 transcription by suppressing acetylation of AP-1 and NF-κB in human rheumatoid arthritis synovial fibroblasts. Biochem Pharmacol (2017) 132:77–91. doi: 10.1016/j.bcp.2017.03.003
41. Khan H, Sureda A, Belwal T, Çetinkaya S, Süntar İ., Tejada S, et al. Polyphenols in the treatment of autoimmune diseases. Autoimmun Rev (2019) 18(7):647–57. doi: 10.1016/j.autrev.2019.05.001
42. Shen P, Lin W, Ba X, Huang Y, Chen Z, Han L, et al. Quercetin-mediated SIRT1 activation attenuates collagen-induced mice arthritis. J Ethnopharmacol (2021) 279:114213. doi: 10.1016/j.jep.2021.114213
43. Rahimizadeh P, Rezaieyazdi Z, Behzadi F, Hajizade A, Lim SI. Nanotechnology as a promising platform for rheumatoid arthritis management: diagnosis, treatment, and treatment monitoring. Int J Pharm (2021) 609:121137. doi: 10.1016/j.ijpharm.2021.121137
44. Song X, Zhang Y, Dai E. Therapeutic targets of thunder god vine (Tripterygium wilfordii hook) in rheumatoid arthritis (Review). Mol Med Rep (2020) 21(6):2303–10. doi: 10.3892/mmr.2020.11052
45. Xi J, Li Q, Luo X, Wang Y, Li J, Guo L, et al. Celastrol inhibits glucocorticoid−induced osteoporosis in rat via the PI3K/AKT and wnt signaling pathways. Mol Med Rep (2018) 18(5):4753–9. doi: 10.3892/mmr.2018.9436
46. Yang J, Liu J, Li J, Jing M, Zhang L, Sun M, et al. Celastrol inhibits rheumatoid arthritis by inducing autophagy via inhibition of the PI3K/AKT/mTOR signaling pathway. Int Immunopharmacol (2022) 112:109241. doi: 10.1016/j.intimp.2022.109241
47. Ng SW, Chan Y, Chellappan DK, Madheswaran T, Zeeshan F, Chan YL, et al. Molecular modulators of celastrol as the keystones for its diverse pharmacological activities. BioMed Pharmacother (2019) 109:1785–92. doi: 10.1016/j.biopha.2018.11.051
48. Moon MH, Jeong JK, Lee YJ, Seol JW, Jackson CJ, Park SY. SIRT1, a class III histone deacetylase, regulates TNF-α-induced inflammation in human chondrocytes. Osteoarthritis Cartilage (2013) 21(3):470–80. doi: 10.1016/j.joca.2012.11.017
49. Imagawa K, de Andrés MC, Hashimoto K, Pitt D, Itoi E, Goldring MB, et al. The epigenetic effect of glucosamine and a nuclear factor-kappa b (NF-kB) inhibitor on primary human chondrocytes–implications for osteoarthritis. Biochem Biophys Res Commun (2011) 405(3):362–7. doi: 10.1016/j.bbrc.2011.01.007
50. Malemud CJ. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther Adv Musculoskelet Dis (2018) 10(5-6):117–27. doi: 10.1177/1759720X18776224
51. Simon LS, Taylor PC, Choy EH, Sebba A, Quebe A, Knopp KL, et al. The Jak/STAT pathway: a focus on pain in rheumatoid arthritis. Semin Arthritis Rheum (2021) 51(1):278–84. doi: 10.1016/j.semarthrit.2020.10.008
52. Ivashkiv LB, Hu X. The JAK/STAT pathway in rheumatoid arthritis: pathogenic or protective? Arthritis Rheum (2003) 48(8):2092–6. doi: 10.1002/art.11095
53. Moura RA, Fonseca JE. JAK inhibitors and modulation of b cell immune responses in rheumatoid arthritis. Front Med (Lausanne) (2020) 7:607725. doi: 10.3389/fmed.2020.607725
54. Zare F, Dehghan-Manshadi M, Mirshafiey A. The signal transducer and activator of transcription factors lodge in immunopathogenesis of rheumatoid arthritis. Reumatismo (2015) 67(4):127–37. doi: 10.4081/reumatismo.2015.851
55. Seif F, Khoshmirsafa M, Aazami H, Mohsenzadegan M, Sedighi G, Bahar M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun Signal (2017) 15(1):23. doi: 10.1186/s12964-017-0177-y
56. Liau NPD, Laktyushin A, Lucet IS, Murphy JM, Yao S, Whitlock E, et al. The molecular basis of JAK/STAT inhibition by SOCS1. Nat Commun (2018) 9(1):1558. doi: 10.1038/s41467-018-04013-1
57. Niu GJ, Xu JD, Yuan WJ, Sun JJ, Yang MC, He ZH, et al. Protein inhibitor of activated STAT (PIAS) negatively regulates the JAK/STAT pathway by inhibiting STAT phosphorylation and translocation. Front Immunol (2018) 9:2392. doi: 10.3389/fimmu.2018.02392
58. Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal transduction targeted Ther (2021) 6(1):402. doi: 10.1038/s41392-021-00791-1
59. Rose-John S. Interleukin-6 family cytokines. Cold Spring Harbor Perspect Biol (2018) 10(2):a028415. doi: 10.1101/cshperspect.a028415
60. Hodge JA, Kawabata TT, Krishnaswami S, Clark JD, Telliez JB, Dowty ME, et al. The mechanism of action of tofacitinib - an oral janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol (2016) 34(2):318–28.
61. Hubbard SR, Miller WT. Receptor tyrosine kinases: mechanisms of activation and signaling. Curr Opin Cell Biol (2007) 19(2):117–23. doi: 10.1016/j.ceb.2007.02.010
62. Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol (2011) 29:71–109. doi: 10.1146/annurev-immunol-031210-101312
63. Świerkot J, Nowak B, Czarny A, Zaczyńska E, Sokolik R, Madej M, et al. The activity of JAK/STAT and NF-κB in patients with rheumatoid arthritis. Adv Clin Exp Med (2016) 25(4):709–17. doi: 10.17219/acem/61034
64. Paik JJ, Casciola-Rosen L, Shin JY, Albayda J, Tiniakou E, Leung DG, et al. Study of tofacitinib in refractory dermatomyositis: an open-label pilot study of ten patients. Arthritis Rheumatol (2021) 73(5):858–65. doi: 10.1002/art.41602
65. Sharma V, Pope BJ, Santiago NV, Boland MT, Sun D, Reynolds RJ, et al. Decreased levels of STAT1 and interferon-γ-Induced STAT1 phosphorylation in rheumatoid arthritis CD4 and CD8 T cells. ACR Open Rheumatol (2021) 3(4):277–83. doi: 10.1002/acr2.11244
66. Zhu W, Xu J, Jiang C, Wang B, Geng M, Wu X, et al. Pristane induces autophagy in macrophages, promoting a STAT1-IRF1-TLR3 pathway and arthritis. Clin Immunol (2017) 175:56–68. doi: 10.1016/j.clim.2016.11.017
67. Kongdang P, Jaitham R, Thonghoi S, Kuensaen C, Pradit W, Ongchai S. Ethanolic extract of kaempferia parviflora interrupts the mechanisms-associated rheumatoid arthritis in SW982 culture model via p38/STAT1 and STAT3 pathways. Phytomedicine (2019) 59:152755. doi: 10.1016/j.phymed.2018.11.015
68. Cui SJ, Zhang T, Fu Y, Liu Y, Gan YH, Zhou YH, et al. DPSCs attenuate experimental progressive TMJ arthritis by inhibiting the STAT1 pathway. J Dent Res (2020) 99(4):446–55. doi: 10.1177/0022034520901710
69. Kuuliala K, Kuuliala A, Koivuniemi R, Kautiainen H, Repo H, Leirisalo-Repo M. STAT6 and STAT1 pathway activation in circulating lymphocytes and monocytes as predictor of treatment response in rheumatoid arthritis. PloS One (2016) 11(12):e0167975. doi: 10.1371/journal.pone.0167975
70. Araki Y, Tsuzuki Wada T, Aizaki Y, Sato K, Yokota K, Fujimoto K, et al. Histone methylation and STAT-3 differentially regulate interleukin-6-Induced matrix metalloproteinase gene activation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol (2016) 68(5):1111–23. doi: 10.1002/art.39563
71. Maioli G, Caporali R, Favalli EG. Lessons learned from the preclinical discovery and development of sarilumab for the treatment of rheumatoid arthritis. Expert Opin Drug Discovery (2022) 17(8):799–813. doi: 10.1080/17460441.2022.2093852
72. Yamazaki S, Tanaka Y, Araki H, Kohda A, Sanematsu F, Arasaki T, et al. The AP-1 transcription factor JunB is required for Th17 cell differentiation. Sci Rep (2017) 7(1):17402. doi: 10.1038/s41598-017-17597-3
73. Ciobanu DA, Poenariu IS, Crînguș LI, Vreju FA, Turcu-Stiolica A, Tica AA, et al. JAK/STAT pathway in pathology of rheumatoid arthritis (Review). Exp Ther Med (2020) 20(4):3498–503. doi: 10.3892/etm.2020.8982
74. Ho HH, Ivashkiv LB. Role of STAT3 in type I interferon responses. negative regulation of STAT1-dependent inflammatory gene activation. J Biol Chem (2006) 281(20):14111–8. doi: 10.1074/jbc.M511797200
75. Sheng W, Yang F, Zhou Y, Yang H, Low PY, Kemeny DM, et al. STAT5 programs a distinct subset of GM-CSF-producing T helper cells that is essential for autoimmune neuroinflammation. Cell Res (2014) 24(12):1387–402. doi: 10.1038/cr.2014.154
76. Monaghan KL, Aesoph D, Ammer AG, Zheng W, Rahimpour S, Farris BY, et al. Tetramerization of STAT5 promotes autoimmune-mediated neuroinflammation. Proc Natl Acad Sci U.S.A. (2021) 118(52)e2116256118880. doi: 10.1073/pnas.2116256118
77. Feng G, Bajpai G, Ma P, Koenig A, Bredemeyer A, Lokshina I, et al. CCL17 aggravates myocardial injury by suppressing recruitment of regulatory T cells. Circulation (2022) 145(10):765–82. doi: 10.1161/circulationaha.121.055888
78. Achuthan A, Cook AD, Lee MC, Saleh R, Khiew HW, Chang MW, et al. Granulocyte macrophage colony-stimulating factor induces CCL17 production via IRF4 to mediate inflammation. J Clin Invest (2016) 126(9):3453–66. doi: 10.1172/jci87828
79. Palmroth M, Kuuliala K, Peltomaa R, Virtanen A, Kuuliala A, Kurttila A, et al. Tofacitinib suppresses several JAK-STAT pathways in rheumatoid arthritis In vivo and baseline signaling profile associates with treatment response. Front Immunol (2021) 12:738481. doi: 10.3389/fimmu.2021.738481
80. Rubbert-Roth A, Enejosa J, Pangan AL, Haraoui B, Rischmueller M, Khan N, et al. Trial of upadacitinib or abatacept in rheumatoid arthritis. N Engl J Med (2020) 383(16):1511–21. doi: 10.1056/NEJMoa2008250
81. Kim ES, Keam SJ. Filgotinib in rheumatoid arthritis: a profile of its use. Clin Drug Investig (2021) 41(8):741–9. doi: 10.1007/s40261-021-01055-0
82. Lee YH, Song GG. Relative efficacy and safety of tofacitinib, baricitinib, upadacitinib, and filgotinib in comparison to adalimumab in patients with active rheumatoid arthritis. Z Rheumatol (2020) 79(8):785–96. doi: 10.1007/s00393-020-00750-1
83. Wang Q, Zhou X, Yang L, Zhao Y, Chew Z, Xiao J, et al. The natural compound notopterol binds and targets JAK2/3 to ameliorate inflammation and arthritis. Cell Rep (2020) 32(11):108158. doi: 10.1016/j.celrep.2020.108158
84. Bao Y, Sun YW, Ji J, Gan L, Zhang CF, Wang CZ, et al. Genkwanin ameliorates adjuvant-induced arthritis in rats through inhibiting JAK/STAT and NF-κB signaling pathways. Phytomedicine (2019) 63:153036. doi: 10.1016/j.phymed.2019.153036
85. Yin Q, Wang L, Yu H, Chen D, Zhu W, Sun C. Pharmacological effects of polyphenol phytochemicals on the JAK-STAT signaling pathway. Front Pharmacol (2021) 12:716672. doi: 10.3389/fphar.2021.716672
86. Senggunprai L, Kukongviriyapan V, Prawan A, Kukongviriyapan U. Quercetin and EGCG exhibit chemopreventive effects in cholangiocarcinoma cells via suppression of JAK/STAT signaling pathway. Phytother Res (2014) 28(6):841–8. doi: 10.1002/ptr.5061
87. Kour G, Choudhary R, Anjum S, Bhagat A, Bajaj BK, Ahmed Z. Phytochemicals targeting JAK/STAT pathway in the treatment of rheumatoid arthritis: is there a future? Biochem Pharmacol (2022) 197:114929. doi: 10.1016/j.bcp.2022.114929
88. Honda S, Harigai M. The safety of baricitinib in patients with rheumatoid arthritis. Expert Opin Drug Saf (2020) 19(5):545–51. doi: 10.1080/14740338.2020.1743263
89. Miyazaki Y, Nakano K, Nakayamada S, Kubo S, Inoue Y, Fujino Y, et al. Efficacy and safety of tofacitinib versus baricitinib in patients with rheumatoid arthritis in real clinical practice: analyses with propensity score-based inverse probability of treatment weighting. Ann Rheum Dis (2021) 80(9):1130–6. doi: 10.1136/annrheumdis-2020-219699
90. Wang G, Xie X, Yuan L, Qiu J, Duan W, Xu B, et al. Resveratrol ameliorates rheumatoid arthritis via activation of SIRT1-Nrf2 signaling pathway. Biofactors (2020) 46(3):441–53. doi: 10.1002/biof.1599
91. Jamilloux Y, El Jammal T, Vuitton L, Gerfaud-Valentin M, Kerever S, Sève P. JAK inhibitors for the treatment of autoimmune and inflammatory diseases. Autoimmun Rev (2019) 18(11):102390. doi: 10.1016/j.autrev.2019.102390
92. Sun YW, Bao Y, Yu H, Chen QJ, Lu F, Zhai S, et al. Anti-rheumatoid arthritis effects of flavonoids from Daphne genkwa. Int Immunopharmacol (2020) 83:106384. doi: 10.1016/j.intimp.2020.106384
93. Hou SM, Chen PC, Lin CM, Fang ML, Chi MC, Liu JF. CXCL1 contributes to IL-6 expression in osteoarthritis and rheumatoid arthritis synovial fibroblasts by CXCR2, c-raf, MAPK, and AP-1 pathway. Arthritis Res Ther (2020) 22(1):251. doi: 10.1186/s13075-020-02331-8
94. Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev (2010) 233(1):233–55. doi: 10.1111/j.0105-2896.2009.00859.x
95. Chi PL, Chen YW, Hsiao LD, Chen YL, Yang CM. Heme oxygenase 1 attenuates interleukin-1β-induced cytosolic phospholipase A2 expression via a decrease in NADPH oxidase/reactive oxygen species/activator protein 1 activation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum (2012) 64(7):2114–25. doi: 10.1002/art.34371
96. Cahill CM, Zhu W, Oziolor E, Yang YJ, Tam B, Rajanala S, et al. Differential expression of the activator protein 1 transcription factor regulates interleukin-1ß induction of interleukin 6 in the developing enterocyte. PloS One (2016) 11(1):e0145184. doi: 10.1371/journal.pone.0145184
97. Chen TK, Smith LM, Gebhardt DK, Birrer MJ, Brown PH. Activation and inhibition of the AP-1 complex in human breast cancer cells. Mol Carcinog (1996) 15(3):215–26. doi: 10.1002/(SICI)1098-2744(199603)15:3<215::AID-MC7>3.0.CO;2-G
98. Young MR, Nair R, Bucheimer N, Tulsian P, Brown N, Chapp C, et al. Transactivation of fra-1 and consequent activation of AP-1 occur extracellular signal-regulated kinase dependently. Mol Cell Biol (2002) 22(2):587–98. doi: 10.1128/mcb.22.2.587-598.2002
99. Bejjani F, Evanno E, Zibara K, Piechaczyk M, Jariel-Encontre I. The AP-1 transcriptional complex: local switch or remote command? Biochim Biophys Acta Rev Cancer (2019) 1872(1):11–23. doi: 10.1016/j.bbcan.2019.04.003
100. Liu S, Ma H, Zhang H, Deng C, Xin P. Recent advances on signaling pathways and their inhibitors in rheumatoid arthritis. Clin Immunol (2021) 230:108793. doi: 10.1016/j.clim.2021.108793
101. Noh EM, Kim JS, Hur H, Park BH, Song EK, Han MK, et al. Cordycepin inhibits IL-1beta-induced MMP-1 and MMP-3 expression in rheumatoid arthritis synovial fibroblasts. Rheumatol (Oxford) (2009) 48(1):45–8. doi: 10.1093/rheumatology/ken417
102. Cao D, Fan Q, Li Z, Chen M, Jiang Y, Lin R, et al. Transcriptomic profiling revealed the role of apigenin-4'-O-α-L-rhamnoside in inhibiting the activation of rheumatoid arthritis fibroblast-like synoviocytes via MAPK signaling pathway. Phytomedicine (2022) 102:154201. doi: 10.1016/j.phymed.2022.154201
103. Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res (2002) 12(1):9–18. doi: 10.1038/sj.cr.7290105
104. Khan A, Khan SU, Khan A, Shal B, Rehman SU, Rehman SU, et al. Anti-inflammatory and anti-rheumatic potential of selective plant compounds by targeting TLR-4/AP-1 signaling: a comprehensive molecular docking and simulation approaches. Molecules (Basel Switzerland) (2022) 27(13):4319. doi: 10.3390/molecules27134319
105. Huang CY, Lin HJ, Chen HS, Cheng SY, Hsu HC, Tang CH. Thrombin promotes matrix metalloproteinase-13 expression through the PKCδ c-Src/EGFR/PI3K/Akt/AP-1 signaling pathway in human chondrocytes. Mediators Inflammation (2013) 2013:326041. doi: 10.1155/2013/326041
106. Jeong YJ, Shin JM, Bae YS, Cho HJ, Park KK, Choe JY, et al. Melittin has a chondroprotective effect by inhibiting MMP-1 and MMP-8 expressions via blocking NF-κB and AP-1 signaling pathway in chondrocytes. Int Immunopharmacol (2015) 25(2):400–5. doi: 10.1016/j.intimp.2015.02.021
107. Su CM, Hu SL, Sun Y, Zhao J, Dai C, Wang L, et al. Myostatin induces tumor necrosis factor-α expression in rheumatoid arthritis synovial fibroblasts through the PI3K-akt signaling pathway. J Cell Physiol (2019) 234(6):9793–801. doi: 10.1002/jcp.27665
108. Zhang X, Zhang D, Wang Q, Guo X, Chen J, Jiang J, et al. Sprouty2 inhibits migration and invasion of fibroblast-like synoviocytes in rheumatoid arthritis by down-regulating ATF2 expression and phosphorylation. Inflammation (2021) 44(1):91–103. doi: 10.1007/s10753-020-01311-z
109. Liu FL, Chen CH, Chu SJ, Chen JH, Lai JH, Sytwu HK, et al. Interleukin (IL)-23 p19 expression induced by IL-1beta in human fibroblast-like synoviocytes with rheumatoid arthritis via active nuclear factor-kappaB and AP-1 dependent pathway. Rheumatol (Oxford) (2007) 46(8):1266–73. doi: 10.1093/rheumatology/kem055
110. Shiozawa S, Tsumiyama K, Yoshida K, Hashiramoto A. Pathogenesis of joint destruction in rheumatoid arthritis. Arch Immunol Ther Exp (Warsz) (2011) 59(2):89–95. doi: 10.1007/s00005-011-0116-3
111. Motomura H, Seki S, Shiozawa S, Aikawa Y, Nogami M, Kimura T. A selective c-Fos/AP-1 inhibitor prevents cartilage destruction and subsequent osteophyte formation. Biochem Biophys Res Commun (2018) 497(2):756–61. doi: 10.1016/j.bbrc.2018.02.147
112. Okamoto H, Cujec TP, Yamanaka H, Kamatani N. Molecular aspects of rheumatoid arthritis: role of transcription factors. FEBS J (2008) 275(18):4463–70. doi: 10.1111/j.1742-4658.2008.06582.x
113. Hannemann N, Jordan J, Paul S, Reid S, Baenkler HW, Sonnewald S, et al. The AP-1 transcription factor c-jun promotes arthritis by regulating cyclooxygenase-2 and arginase-1 expression in macrophages. J Immunol (Baltimore Md. 1950) (2017) 198(9):3605–14. doi: 10.4049/jimmunol.1601330
114. Katagiri T, Kameda H, Nakano H, Yamazaki S. Regulation of T cell differentiation by the AP-1 transcription factor JunB. Immunol Med (2021) 44(3):197–203. doi: 10.1080/25785826.2021.1872838
115. Atsaves V, Zhang R, Ruder D, Pan Y, Leventaki V, Rassidakis GZ, et al. Constitutive control of AKT1 gene expression by JUNB/CJUN in ALK+ anaplastic large-cell lymphoma: a novel crosstalk mechanism. Leukemia (2015) 29(11):2162–72. doi: 10.1038/leu.2015.127
116. Horsley V, Pavlath GK. NFAT: ubiquitous regulator of cell differentiation and adaptation. J Cell Biol (2002) 156(5):771–4. doi: 10.1083/jcb.200111073
117. Park JK, Jang YJ, Oh BR, Shin J, Bae D, Ha N, et al. Therapeutic potential of CKD-506, a novel selective histone deacetylase 6 inhibitor, in a murine model of rheumatoid arthritis. Arthritis Res Ther (2020) 22(1):176. doi: 10.1186/s13075-020-02258-0
118. Zhong B, Guo S, Yang Z, Han L, Du J, Chen J, et al. Roflumilast reduced the IL-18-Induced inflammatory response in fibroblast-like synoviocytes (FLS). ACS Omega (2021) 6(3):2149–55. doi: 10.1021/acsomega.0c05281
119. Hosoya T, Saito T, Baba H, Tanaka N, Noda S, Komiya Y, et al. Chondroprotective effects of CDK4/6 inhibition via enhanced ubiquitin-dependent degradation of JUN in synovial fibroblasts. Rheumatol (Oxford) (2022) 61(8):3427–38. doi: 10.1093/rheumatology/keab874
120. Liakhov SA, Schepetkin IA, Karpenko OS, Duma HI, Haidarzhy NM, Kirpotina LN, et al. Novel c-jun n-terminal kinase (JNK) inhibitors with an 11H-Indeno[1,2-b]quinoxalin-11-one scaffold. Molecules (Basel Switzerland) (2021) 26(18):5688. doi: 10.3390/molecules26185688
121. Pal RR, Rajpal V, Singh N, Singh S, Mishra N, Singh P, et al. Downregulation of pro-inflammatory markers IL-6 and TNF-α in rheumatoid arthritis using nano-lipidic carriers of a quinone-based phenolic: an in vitro and in vivo study. Drug Delivery Transl Res (2023) 13(2):627–41. doi: 10.1007/s13346-022-01221-7
122. Achudhan D, Li-Yun Chang S, Liu SC, Lin YY, Huang WC, Wu YC, et al. Antcin K inhibits VCAM-1-dependent monocyte adhesion in human rheumatoid arthritis synovial fibroblasts. Food Nutr Res (2022) 66. doi: 10.29219/fnr.v66.8645
123. Thummuri D, Jeengar MK, Shrivastava S, Nemani H, Ramavat RN, Chaudhari P, et al. Thymoquinone prevents RANKL-induced osteoclastogenesis activation and osteolysis in an in vivo model of inflammation by suppressing NF-KB and MAPK signalling. Pharmacol Res (2015) 99:63–73. doi: 10.1016/j.phrs.2015.05.006
124. Umar S, Hedaya O, Singh AK, Ahmed S. Thymoquinone inhibits TNF-α-induced inflammation and cell adhesion in rheumatoid arthritis synovial fibroblasts by ASK1 regulation. Toxicol Appl Pharmacol (2015) 287(3):299–305. doi: 10.1016/j.taap.2015.06.017
125. Linghu KG, Xiong SH, Zhao GD, Zhang T, Xiong W, Zhao M, et al. Sigesbeckia orientalis l. extract alleviated the collagen type II-induced arthritis through inhibiting multi-Target-Mediated synovial hyperplasia and inflammation. Front Pharmacol (2020) 11:547913. doi: 10.3389/fphar.2020.547913
126. Mognol GP, González-Avalos E, Ghosh S, Spreafico R, Gudlur A, Rao A, et al. Targeting the NFAT:AP-1 transcriptional complex on DNA with a small-molecule inhibitor. Proc Natl Acad Sci U.S.A. (2019) 116(20):9959–68. doi: 10.1073/pnas.1820604116
127. Pal RR, Rajpal V, Singh P, Saraf SA. Recent findings on thymoquinone and its applications as a nanocarrier for the treatment of cancer and rheumatoid arthritis. Pharmaceutics (2021) 13(6):775. doi: 10.3390/pharmaceutics13060775
128. Thompson CD, Matta B, Barnes BJ. Therapeutic targeting of IRFs: pathway-dependence or structure-based? Front Immunol (2018) 9:2622. doi: 10.3389/fimmu.2018.02622
129. Ghislat G, Cheema AS, Baudoin E, Verthuy C, Ballester PJ, Crozat K, et al. NF-κB-dependent IRF1 activation programs cDC1 dendritic cells to drive antitumor immunity. Sci Immunol (2021) 6(61):eabg3570. doi: 10.1126/sciimmunol.abg3570
130. Feng H, Zhang YB, Gui JF, Lemon SM, Yamane D. Interferon regulatory factor 1 (IRF1) and anti-pathogen innate immune responses. PloS Pathog (2021) 17(1):e1009220. doi: 10.1371/journal.ppat.1009220
131. Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signaling (2012) 5(214):ra20. doi: 10.1126/scisignal.2002521
132. Zhang C, Shang G, Gui X, Zhang X, Bai XC, Chen ZJ. Structural basis of STING binding with and phosphorylation by TBK1. Nature (2019) 567(7748):394–8. doi: 10.1038/s41586-019-1000-2
133. Al Hamrashdi M, Brady G. Regulation of IRF3 activation in human antiviral signaling pathways. Biochem Pharmacol (2022) 200:115026. doi: 10.1016/j.bcp.2022.115026
134. Wong RWJ, Ong JZL, Theardy MS, Sanda T. IRF4 as an oncogenic master transcription factor. Cancers (Basel) (2022) 14(17):4314. doi: 10.3390/cancers14174314
135. Ramnath D, Tunny K, Hohenhaus DM, Pitts CM, Bergot AS, Hogarth PM, et al. TLR3 drives IRF6-dependent IL-23p19 expression and p19/EBI3 heterodimer formation in keratinocytes. Immunol Cell Biol (2015) 93(9):771–9. doi: 10.1038/icb.2015.77
136. Kwa MQ, Scholz GM, Reynolds EC. RIPK4 activates an IRF6-mediated proinflammatory cytokine response in keratinocytes. Cytokine (2016) 83:19–26. doi: 10.1016/j.cyto.2016.03.005
137. Kim KW, Kim BM, Won JY, Lee KA, Kim HR, Lee SH. Toll-like receptor 7 regulates osteoclastogenesis in rheumatoid arthritis. J Biochem (2019) 166(3):259–70. doi: 10.1093/jb/mvz033
138. Salem S, Salem D, Gros P. Role of IRF8 in immune cells functions, protection against infections, and susceptibility to inflammatory diseases. Hum Genet (2020) 139(6-7):707–21. doi: 10.1007/s00439-020-02154-2
139. Jefferies CA. Regulating IRFs in IFN driven disease. Front Immunol (2019) 10:325. doi: 10.3389/fimmu.2019.00325
140. Wang F, Qiao L, Lv X, Trivett A, Yang R, Oppenheim JJ, et al. Alarmin human α defensin HNP1 activates plasmacytoid dendritic cells by triggering NF-κB and IRF1 signaling pathways. Cytokine (2016) 83:53–60. doi: 10.1016/j.cyto.2016.03.015
141. Bonelli M, Dalwigk K, Platzer A, Olmos Calvo I, Hayer S, Niederreiter B, et al. IRF1 is critical for the TNF-driven interferon response in rheumatoid fibroblast-like synoviocytes : JAKinibs suppress the interferon response in RA-FLSs. Exp Mol Med (2019) 51(7):1–11. doi: 10.1038/s12276-019-0267-6
142. Wang Q, Yu X, Gong M. Single-cell transcriptome analysis reveals the importance of IRF1/FSTL1 in synovial fibroblast subsets for the development of rheumatoid arthritis. Comput Math Methods Med (2022) 2022:1169614. doi: 10.1155/2022/1169614
143. Li JY, Xiao J, Gao M, Zhou HF, Fan H, Sun F, et al. IRF/Type I IFN signaling serves as a valuable therapeutic target in the pathogenesis of inflammatory bowel disease. Int Immunopharmacol (2021) 92:107350. doi: 10.1016/j.intimp.2020.107350
144. He T, Yang L, Wu D. Effect of interferon regulatory factor 2 on inflammatory response and oxidative stress in lipopolysaccharide-induced acute kidney injury. Drug Dev Res (2022) 83(4):940–51. doi: 10.1002/ddr.21919
145. Sweeney SE, Corr M, Kimbler TB. Role of interferon regulatory factor 7 in serum-transfer arthritis: regulation of interferon-β production. Arthritis Rheum (2012) 64(4):1046–56. doi: 10.1002/art.33454
146. Nalbant A, Eskier D. Genes associated with T helper 17 cell differentiation and function. Front Biosci (Elite Ed) (2016) 8(3):427–35. doi: 10.2741/e777
147. Lopez-Isac E, Martin JE, Assassi S, Simeon CP, Carreira P, Ortego-Centeno N, et al. Brief report: IRF4 newly identified as a common susceptibility locus for systemic sclerosis and rheumatoid arthritis in a cross-disease meta-analysis of genome-wide association studies. Arthritis Rheumatol (2016) 68(9):2338–44. doi: 10.1002/art.39730
148. Xiao F, Rui K, Han M, Zou L, Huang E, Tian J, et al. Artesunate suppresses Th17 response via inhibiting IRF4-mediated glycolysis and ameliorates Sjog̈ren's syndrome. Signal transduction targeted Ther (2022) 7(1):274. doi: 10.1038/s41392-022-01103-x
149. Ni Z, Zhao X, Dai X, Zhao L, Xia J. The role of interferon regulatory factor 5 in macrophage inflammation during osteoarthritis. Inflammation (2019) 42(5):1821–9. doi: 10.1007/s10753-019-01044-8
150. Duffau P, Menn-Josephy H, Cuda CM, Dominguez S, Aprahamian TR, Watkins AA, et al. Promotion of inflammatory arthritis by interferon regulatory factor 5 in a mouse model. Arthritis Rheumatol (2015) 67(12):3146–57. doi: 10.1002/art.39321
151. Guo L, Hao R, Tian F, An N, Wang K. Interferon regulatory factor 5 (IRF5) regulates the expression of matrix metalloproteinase-3 (MMP-3) in human chondrocytes. Int Immunopharmacol (2018) 55:231–6. doi: 10.1016/j.intimp.2017.11.035
152. Kwa MQ, Nguyen T, Huynh J, Ramnath D, De Nardo D, Lam PY, et al. Interferon regulatory factor 6 differentially regulates toll-like receptor 2-dependent chemokine gene expression in epithelial cells. J Biol Chem (2014) 289(28):19758–68. doi: 10.1074/jbc.M114.584540
153. Ivashkiv LB, Zhao B, Park-Min KH, Takami M. Feedback inhibition of osteoclastogenesis during inflammation by IL-10, m-CSF receptor shedding, and induction of IRF8. Ann N Y Acad Sci (2011) 1237:88–94. doi: 10.1111/j.1749-6632.2011.06217.x
154. Yang Q, Ding W, Cao Y, Zhou Y, Ni S, Shi T, et al. Interferonregulatoryfactor-8(IRF-8) regulates the expression of matrix metalloproteinase-13 (MMP-13) in chondrocytes. Cell Stress Chaperones (2018) 23(3):393–8. doi: 10.1007/s12192-017-0849-y
155. Merrill JT, Furie R, Werth VP, Khamashta M, Drappa J, Wang L, et al. Anifrolumab effects on rash and arthritis: impact of the type I interferon gene signature in the phase IIb MUSE study in patients with systemic lupus erythematosus. Lupus Sci Med (2022) 5(1):e000284. doi: 10.1136/lupus-2018-000284
156. Juge PA, Gazal S, Constantin A, Mariette X, Combe B, Tebib J, et al. Variants of genes implicated in type 1 interferon pathway and b-cell activation modulate the EULAR response to rituximab at 24 weeks in rheumatoid arthritis. RMD Open (2017) 3(2):e000448. doi: 10.1136/rmdopen-2017-000448
157. Westra J, Molema G, Kallenberg CG. Hypoxia-inducible factor-1 as regulator of angiogenesis in rheumatoid arthritis - therapeutic implications. Curr Med Chem (2010) 17(3):254–63. doi: 10.2174/092986710790149783
158. Guo X, Chen G. Hypoxia-inducible factor is critical for pathogenesis and regulation of immune cell functions in rheumatoid arthritis. Front Immunol (2020) 11:1668. doi: 10.3389/fimmu.2020.01668
159. Hong Z, Tie Q, Zhang L. Targeted inhibition of the GRK2/HIF-1α pathway is an effective strategy to alleviate synovial hypoxia and inflammation. Int Immunopharmacol (2022) 113(Pt A):109271. doi: 10.1016/j.intimp.2022.109271
160. Ba X, Huang Y, Shen P, Huang Y, Wang H, Han L, et al. WTD attenuating rheumatoid arthritis via suppressing angiogenesis and modulating the PI3K/AKT/mTOR/HIF-1α pathway. Front Pharmacol (2021) 12:696802. doi: 10.3389/fphar.2021.696802
161. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi: 10.1016/j.cell.2011.07.033
162. Liu X, Hu Z, Zhou H. N-acetylcysteine improves inflammatory response in COPD patients by regulating Th17/Treg balance through hypoxia inducible factor-1α pathway. BioMed Res Int (2021) 2021:6372128. doi: 10.1155/2021/6372128
163. Gan P, Sun M, Wu H, Ke J, Dong X, Chen F. A novel mechanism for inhibiting proliferation of rheumatoid arthritis fibroblast-like synoviocytes: geniposide suppresses HIF-1α accumulation in the hypoxic microenvironment of synovium. Inflammation Res (2022) 71(10-11):1375–88. doi: 10.1007/s00011-022-01636-5
164. Wen J, Lyu P, Stolzer I, Xu J, Gießl A, Lin Z, et al. Epithelial HIF2α expression induces intestinal barrier dysfunction and exacerbation of arthritis. Ann Rheum Dis (2022). doi: 10.1136/annrheumdis-2021-222035
165. Li GF, Qin YH, Du PQ. Andrographolide inhibits the migration, invasion and matrix metalloproteinase expression of rheumatoid arthritis fibroblast-like synoviocytes via inhibition of HIF-1α signaling. Life Sci (2015) 136:67–72. doi: 10.1016/j.lfs.2015.06.019
166. Lee YZ, Guo HC, Zhao GH, Yang CW, Chang HY, Yang RB, et al. Tylophorine-based compounds are therapeutic in rheumatoid arthritis by targeting the caprin-1 ribonucleoprotein complex and inhibiting expression of associated c-myc and HIF-1α. Pharmacol Res (2020) 152:104581. doi: 10.1016/j.phrs.2019.104581
167. Zhang M, Wu D, Xu J, Liu L, Jiao W, Yu J, et al. Suppression of NLRP3 inflammasome by dihydroarteannuin via the HIF-1α and JAK3/STAT3 signaling pathway contributes to attenuation of collagen-induced arthritis in mice. Front Pharmacol (2022) 13:884881. doi: 10.3389/fphar.2022.884881
168. Manda G, Milanesi E, Genc S, Niculite CM, Neagoe IV, Tastan B, et al. Pros and cons of NRF2 activation as adjunctive therapy in rheumatoid arthritis. Free Radic Biol Med (2022) 190:179–201. doi: 10.1016/j.freeradbiomed.2022.08.012
169. Du Y, Wang Q, Tian N, Lu M, Zhang XL, Dai SM. Knockdown of nrf2 exacerbates TNF-α-Induced proliferation and invasion of rheumatoid arthritis fibroblast-like synoviocytes through activating JNK pathway. J Immunol Res (2020) 2020:6670464. doi: 10.1155/2020/6670464
170. Su X, Huang Q, Chen J, Wang M, Pan H, Wang R, et al. Calycosin suppresses expression of pro-inflammatory cytokines via the activation of p62/Nrf2-linked heme oxygenase 1 in rheumatoid arthritis synovial fibroblasts. Pharmacol Res (2016) 113(Pt A):695–704. doi: 10.1016/j.phrs.2016.09.031
171. Fan M, Li Y, Yao C, Liu X, Liu J, Yu B. DC32, a dihydroartemisinin derivative, ameliorates collagen-induced arthritis through an Nrf2-p62-Keap1 feedback loop. Front Immunol (2018) 9:2762. doi: 10.3389/fimmu.2018.02762
172. Su X, Li T, Liu Z, Huang Q, Liao K, Ren R, et al. Licochalcone a activates Keap1-Nrf2 signaling to suppress arthritis via phosphorylation of p62 at serine 349. Free Radic Biol Med (2018) 115:471–83. doi: 10.1016/j.freeradbiomed.2017.12.004
173. Lal R, Dhaliwal J, Dhaliwal N, Dharavath RN, Chopra K. Activation of the Nrf2/HO-1 signaling pathway by dimethyl fumarate ameliorates complete freund's adjuvant-induced arthritis in rats. Eur J Pharmacol (2021) 899:174044. doi: 10.1016/j.ejphar.2021.174044
174. Liao K, Su X, Lei K, Liu Z, Lu L, Wu Q, et al. Sinomenine protects bone from destruction to ameliorate arthritis via activating p62(Thr269/Ser272)-Keap1-Nrf2 feedback loop. BioMed Pharmacother (2021) 135:111195. doi: 10.1016/j.biopha.2020.111195
175. Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The human transcription factors. Cell (2018) 172(4):650–65. doi: 10.1016/j.cell.2018.01.029
176. Platanitis E, Decker T. Regulatory networks involving STATs, IRFs, and NFκB in inflammation. Front Immunol (2018) 9:2542. doi: 10.3389/fimmu.2018.02542
177. Mogensen TH. IRF and STAT transcription factors - from basic biology to roles in infection, protective immunity, and primary immunodeficiencies. Front Immunol (2018) 9:3047. doi: 10.3389/fimmu.2018.03047
178. Chao PZ, Hsieh MS, Cheng CW, Lin YF, Chen CH. Regulation of MMP-3 expression and secretion by the chemokine eotaxin-1 in human chondrocytes. J BioMed Sci (2011) 18(1):86. doi: 10.1186/1423-0127-18-86
179. Rébé C, Ghiringhelli F. Interleukin-1β and cancer. Cancers (Basel) (2020) 12(7):1791. doi: 10.3390/cancers12071791
180. Gao W, McCormick J, Connolly M, Balogh E, Veale DJ, Fearon U. Hypoxia and STAT3 signalling interactions regulate pro-inflammatory pathways in rheumatoid arthritis. Ann Rheum Dis (2015) 74(6):1275–83. doi: 10.1136/annrheumdis-2013-204105
181. Li X, Qu J, Zhang T, He X, Jiang Y, Chen J. Nuclear factor kappa b (NF-kappaB) targeted self-assembled nanoparticles loaded with methotrexate for treatment of rheumatoid arthritis. Med Sci Monit (2019) 25:8204–12. doi: 10.12659/MSM.917396
182. Chen X, Zhou B, Gao Y, Wang K, Wu J, Shuai M, et al. Efficient treatment of rheumatoid arthritis by degradable LPCE nano-Conjugate-Delivered p65 siRNA. Pharmaceutics (2022) 14(1):162. doi: 10.3390/pharmaceutics14010162
Keywords: transcription factors, rheumatoid arthritis, cytokines, NF-κB, AP-1, STAT and IRF
Citation: Balendran T, Lim K, Hamilton JA and Achuthan AA (2023) Targeting transcription factors for therapeutic benefit in rheumatoid arthritis. Front. Immunol. 14:1196931. doi: 10.3389/fimmu.2023.1196931
Received: 30 March 2023; Accepted: 16 June 2023;
Published: 29 June 2023.
Edited by:
Li Wu, Tsinghua University, ChinaReviewed by:
Fengyang Chen, Hangzhou Medical College, ChinaGanesan Ramamoorthi, Moffitt Cancer Center, United States
Copyright © 2023 Balendran, Lim, Hamilton and Achuthan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adrian A. Achuthan, YWFhQHVuaW1lbGIuZWR1LmF1