 David P. Maison
David P. Maison Youping Deng
Youping Deng Mariana Gerschenson
Mariana Gerschenson
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 19 June 2023
Sec. Viral Immunology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1195871
This article is part of the Research TopicRegulation of the Host Immune Activation in Respiratory Virus InfectionsView all 16 articles
The SARS-CoV-2 pandemic and the COVID-19 disease have affected everyone globally, leading to one of recorded history’s most significant research surges. As our knowledge evolves, our approaches to the virus and treatments must also evolve. The evaluation of future research approaches to SARS-CoV-2 will necessitate reviewing the host immune response and viral antagonism of that response. This review provides an overview of the current knowledge on SARS-CoV-2 by summarizing the virus and human response. The focuses are on the viral genome, replication cycle, host immune activation, response, signaling, and antagonism. To effectively fight the pandemic, efforts must focus on the current state of research to help develop treatments and prepare for future outbreaks.
SARS-CoV-2 emerged in November 2019 in Wuhan, China, from multiple zoonotic cross-species transmission events in humans. The subsequent COVID-19 pandemic - declared by the WHO on March 11, 2020 - is ongoing, and has seen substantial viral evolution and new variants (1–9). Between November 2019 and November 2022, over 632 million cases of COVID-19 were confirmed globally, with more than 6.6 million confirmed deaths and models predicting nearly 20 million deaths (10, 11).
Our understanding of the host immune response to SARS-CoV-2 is constantly growing, as are the discoveries related to immune modulatory effects related to the virus and viral proteins. This review will cover the SARS-CoV-2 genome, virion structure, viral entry and replication, the host immune response, and SARS-CoV-2 immune antagonism. Understanding the host immune response and viral immune antagonism is crucial - as the current state of research - as this knowledge can guide novel treatment strategies and inform public health measures.
SARS-CoV-2 is an enveloped, positive-sense RNA virus of the family coronaviridae (12). Within the coronaviridae family, SARS-CoV-2 is part of the betacoronavirus genera, also known as genus sarbecoviruses (13). The 29.903 kilobase SARS-CoV-2 genome was published in January 2020 (3, 5, 14). The ancestral lineage A and B genomes are ~80% similar to SARS-CoV and 96.2% similar to the Bat RaTG13 virus (4, 14, 15). From 5’ to 3’, the genome is organized as 5’-cap structure, 5’ UTR, ORF1ab, S, ORF3a-d, E, M, ORF6, ORF7a-b, ORF8, ORF9b-c, N, ORF10, and 3’ UTR (Figure 1A) (13, 16–19). The genome contains coding for four structural proteins, 11 accessory proteins, and 16 nsps (12). The four structural genes are S, E, M, and N. The structural proteins and the genome form an enveloped virion able to infect cells (15). The 11 accessory genes and proteins are ORF3a-d, ORF6, ORF7a and b, ORF8, ORF9b and c, and ORF10, and serve various functions from host interaction to immune modulation (18–20). The 16 nsps are identified numerically as nsp1-16 (12). All the nsps are synthesized from ORF1a and ORF1ab (12, 15–17). The change from ORF1a to ORF1ab is facilitated by the ribosomal frameshift of a stop codon at the end of ORF1a (16, 17).
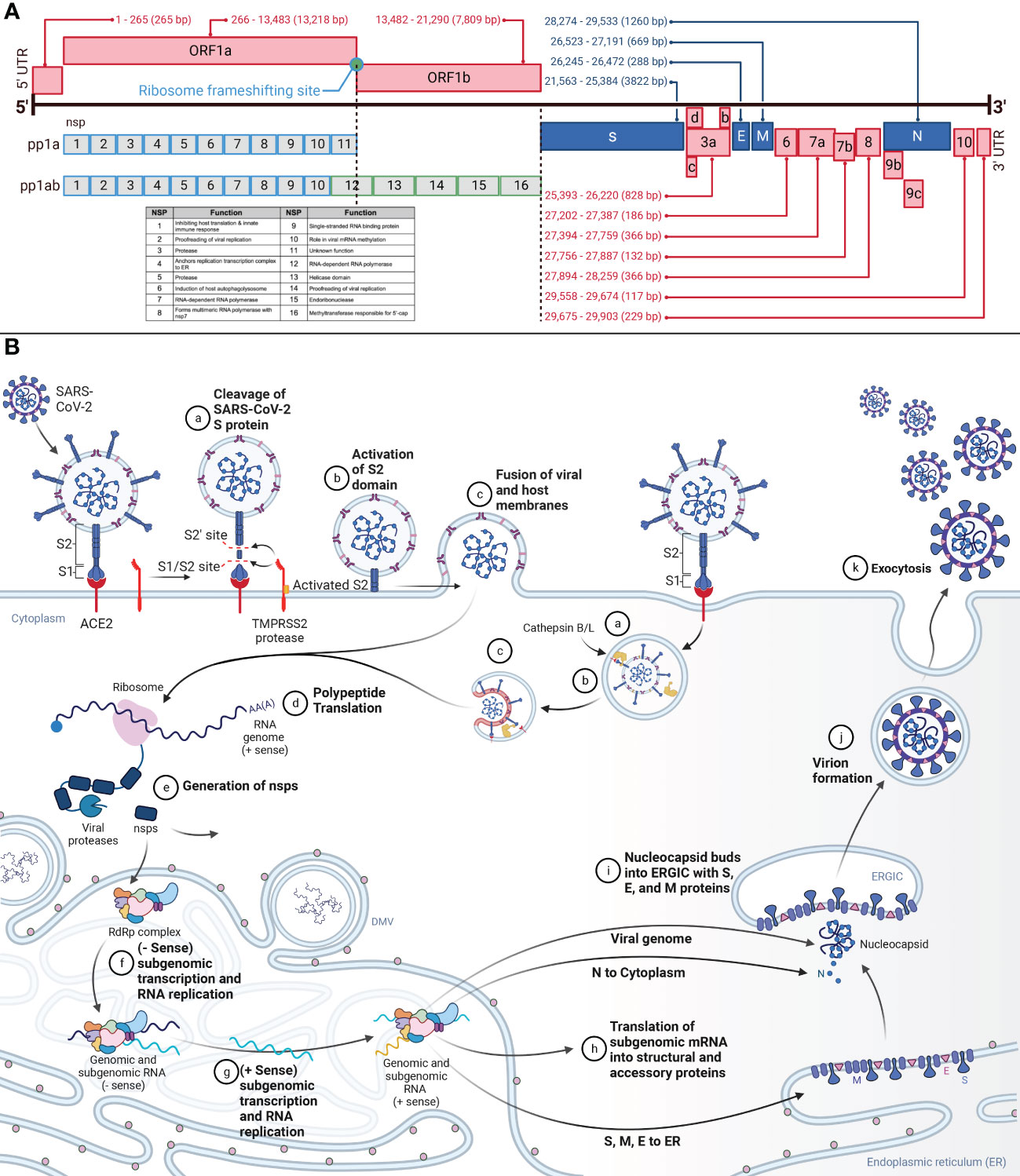
Figure 1 (A) Genomic Organization of SARS-CoV-2 and SARS-CoV-2 Viral Entry and Replication Cycle. This figure demonstrates the genome of SARS-CoV-2, a positive-sense RNA virus, and the proteins produced from each genome segment. The 29.903 kilobase genome was first identified in Wuhan, China, in December 2019. From 5’ to 3’, the genome is organized as 5’-cap structure, 5’ UTR, ORF1ab, spike (S), ORF3a-d, envelope (E), membrane (M), ORF6, ORF7a-b, ORF8, ORF9b-c, nucleocapsid (N), ORF10, 3’ UTR, and 3’-poly-A tail. Nsps 1-16 are translated from ORF1ab. Structural and accessory proteins are translated from their corresponding segment. Nucleotide annotations acquired from GenBank Accession NC_045512. (B) This figure represents the overview of SARS-CoV-2 viral entry and the replication cycle. aSARS-CoV-2 entry begins after binding the S protein S1 domain to the ACE2. The S1/S2 site on the S protein will then be cleaved by TMPRSS2, resulting in the removal of S1. bFollowing, the S2’ site is cleaved by TMPRSS2, furin, and cathepsins, priming the S protein by allowing S2 to form a pre-hairpin in the cellular membrane. cThe pre-hairpin forms a six-hairpin bundle that pulls the viral membrane and cellular membranes together, fusing them and allowing the viral RNA to release into the cytosol. dUpon release into the cytosol, translation of the viral RNA will begin to be translated. eThe non-structural proteins (nsps) polyproteins pp1a and pp1ab will be translated from ORF1ab. Autoproteolysis and post-translational processing will form the replication transcription complex (RTC) and the RNA-dependent-RNA-polymerase (RdRp) within the endoplasmic reticulum (ER). fWithin the ER, in specialized double-membrane vesicles (DMV), the RTC will synthesize the complementary negative-strand genomes and negative-strand subgenomic RNAs. gThe negative strands will serve as the template for progeny genomes and subgenomic RNAs. hThe subgenomic RNAs are translated as structural and accessory proteins. The structural proteins S, E, and M are translated into the ER membrane and the N into the cytoplasm to encapsulate the viral genome. iThe N-coated viral genome will bud into the ERGIC complex containing S, E, and M proteins. jThe virion will be budded into a vesicle bound to the cellular membrane. kThe vesicle will fuse with the cellular membrane, and exocytosis will release the fully-formed virus into extracellular space.
The structural proteins form the virus’s envelope and hold the RNA genome. The N protein coats the genome, whereas S, M, and E are embedded in the lipid bilayer membrane (16). The N protein is composed of positively charged amino acids and binds the negatively charged backbone of genomic RNA (12). The S protein is a 1273 amino acid protein divided into S1 and S2 portions (21), and domains (17, 22). S1 contains the NTD and the RBD (22). Juxtaposed is the S1/S2 furin cleavage site. S2 includes the S2’ cleavage site, FP, HR1, CH, HR2, TM, and the cytoplasmic tail (22, 23). Spike proteins complex into trimers that protrude from the viral envelope surface. On the viral envelope is an average of 15-30 of these S protein trimers (12, 24). Cellular entry by SARS-CoV-2 relies on S protein binding to ACE2 and S protein priming by TMPRSS2 (25). Cathepsin B and L can also serve the same function as TMPRSS2 but within the endosome (15, 21, 26). The E protein, a single-span transmembrane protein, facilitates viral assembly and release, and is an important component in pathogenesis (27). In addition to being transmembrane, the E protein contains channel activity, forming pores and allowing ion transport (28). The M protein is the most abundant structural protein, and spans the membrane three times and interacts with both the E and the S proteins to facilitate the structure of the lipid envelope (12, 27).
The replication cycle (Figure 1B) begins after SARS-CoV-2 binding to ACE2 via the S1 subunit of the S protein (16, 25). The spike protein S1 ectodomain changes conformation from open to closed, and in the open conformation, the RBD interacts with ACE2 in humans (12, 14, 29). The S2 subunit of the S protein then facilitates membrane fusion (16). Two cleavage sites in the S protein are responsible for the pre-fusion to post-fusion conformational change (30, 31). The S1/S2 site, consisting of the polybasic furin motif, separates the S1 and S2 domains (4, 26, 32, 33). The S2’ cleavage site drives the fusion of the virus with the cell membrane (16, 34). The S1/S2 site is cleaved by TMPRSS2 and results in the removal of the S1 subunit (16, 35). The S2’ site is cleaved by TMPRSS2, furin, and cathepsins, inducing an irreversible conformational change in the S protein (16, 36). Removing the S1 subunit and activating S2 allows S2 to form a pre-hairpin that embeds into the target cellular membrane. The pre-hairpin folds back and creates a six-hairpin bundle that pulls the viral and cellular membranes together.
The genomic vRNA will release into the cytosol and uncoat (21). Immediately, the polyproteins pp1a and pp1ab will be translated from ORF1ab (21). These proteins are required for replication and viral survival. The majority of the replication machinery is in the ORF1b portion of the genome and are produced with the ORF1a to ORF1ab -1 frameshift, which occurs at a ~20-50% rate (16). These polyproteins are co- and post-translationally processed into the RTC by nsp3 (PLPro) and nsp5 (3CLpro) (13, 16, 21, 37). The RTC is responsible for replication, transcription, RNA processing, and formation of within the ER to serve as replication factories (15, 16). The viral RTC will replicate the viral genome and subgenomic mRNA for the assembly of new viruses (21).
The RTC will synthesize negative-strand full-length genomes from the positive-strand RNA genome for the subsequent generation of positive-strand progeny genomes (16). Additionally, the RTC will generate subgenomic mRNA via discontinuous transcription utilizing transcription regulatory sequences located at the 5’ end of each ORF (16). The negative-strand subgenomic mRNAs then facilitate the generation of positive-strand subgenomic mRNA. These positive-strand subgenomic mRNA then serve as translation templates for viral protein production. Replication occurs in specialized ER structures known as DMV (16, 38). Structural proteins are processed through the endoplasmic reticulum to the Golgi apparatus. Here the S, E, and M structural proteins are retained at the budding ERGIC, where interaction with N-encapsidated genomic vRNA results in the formation of secretory vesicles (21). The M protein incorporates viral components into the virions. The N protein interacts with both the genome and the E protein to enable packaging into the virion. The E protein participates in viral assembly by functioning as an ion channel and participating in membrane curvature (16). Virions are released from the cell via exocytosis.
Upon infection with SARS-CoV-2, a cell and host will begin a robust innate immune response (Figure 2). PRRs such as RIG-I, MDA-5, and TLRs will recognize SARS-CoV-2 within the infected cell (28, 39–42). Beginning with TLR2, it senses the SARS-CoV-2 envelope protein to initiate an immune response before viral entry and replication, causing the release of TNF-α and IFN-γ (28). TLR2 also activates the assembly of the NLRP3 inflammasome (28). TLR1 is predicted to form a heterodimer with TLR2 and has significantly higher RNA counts in severe and critical COVID-19 (28). Shifting our focus to TLR3, this receptor interacts with viral PAMPs and dsRNA (43). The TLR3 response induces both the IRF3 and NF-κB response via the TRIF-dependent pathway and increases NLRP3 expression, enabling recognition of SARS-CoV-2 and formation of an inflammasome alongside other proteins. The inflammasome results in the maturation and release of IL-1β and IL-18, triggering pyroptosis (40, 41, 44, 45). In the case of TLR4, its response is to DAMPs and PAMPs resulting from SARS-CoV-2 infection and upregulates IL-6 production via NF-κB and MAPK (44, 46, 47). TLR1, TLR4, and TLR5 have been proposed - and in-silico predicted - to respond to the SARS-CoV-2 spike glycoprotein (44, 45, 47), which activates the MyD88 and TRIF innate immunity signaling pathways (48). Turning to TLR7, TLR8, and TLR9, these endosomal receptors play distinct roles. TLR7 and TLR8 sense ssRNA and causes a release of IL-6, TNFα, and type I and III IFNs (42, 45). TLR9 detects viral RNA and DNA with unmethylated CpG, and mtDNA released due to SARS-CoV-2-induced cellular damage. TLR9 causes the release of cytokines, including IL-1β, IL-6, IL-10, IL-17, TNFα, and type I IFN (45).
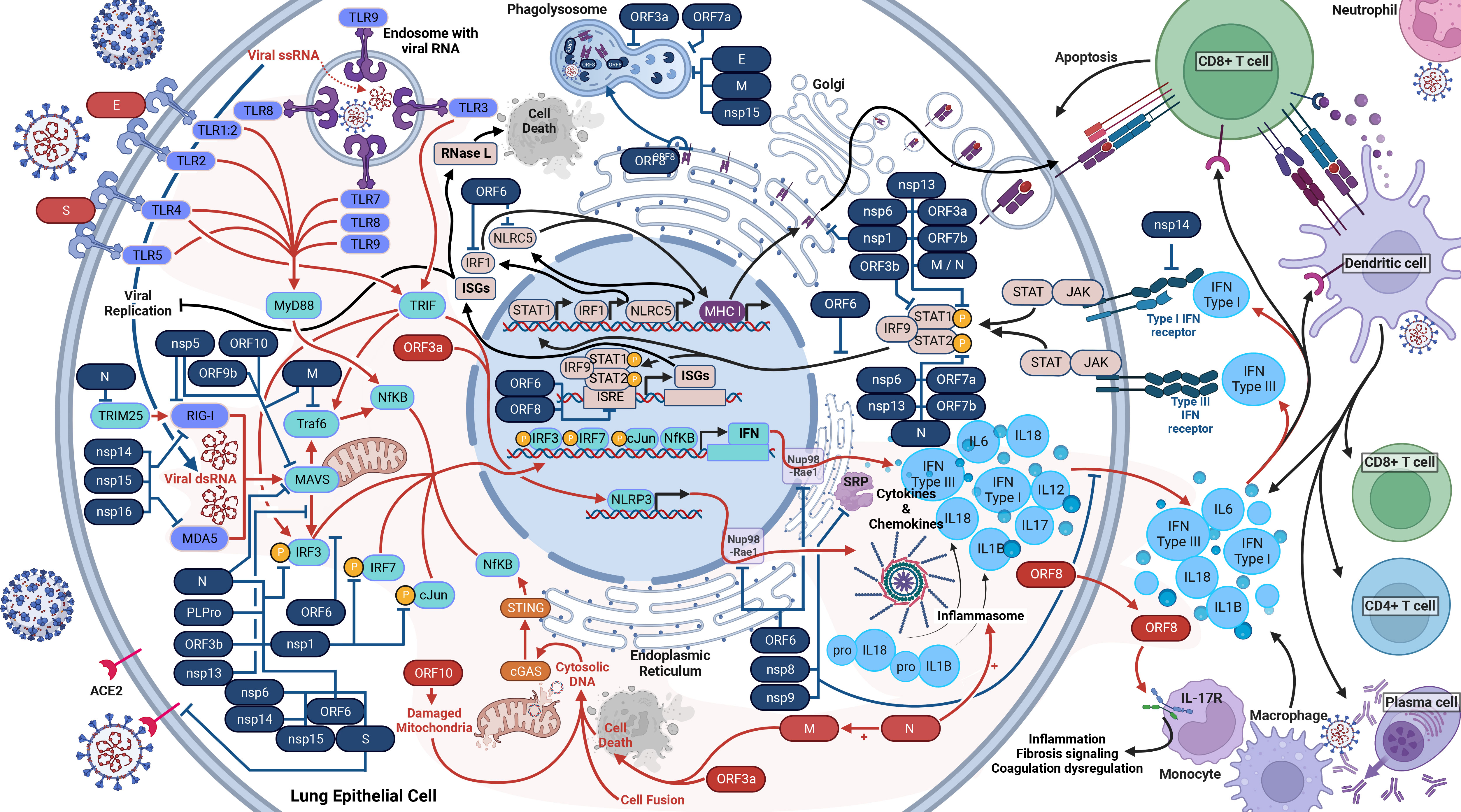
Figure 2 Host Immune Response to SARS-CoV-2 Infection. SARS-CoV-2 activates and antagonizes several arms of the immune response to infection. In total, SARS-CoV-2 will activate and proliferate an immune response (red arrows) through cellular damage (dysfunctional mitochondria), single-stranded RNA (ssRNA), double-stranded RNA (dsRNA), cytosolic DNA, open-reading frame 8 (ORF8), the envelope (E) protein, the spike (S) glycoprotein, and the nucleocapsid (N) protein (shown in red). These parts of the virus initiate the response to induce the production of type I interferons (IFN), type III IFN, and inflammasomes (red lines). These cytokine responses will act in autocrine and paracrine manners to activate IFN receptors (IFNAR, IFNLR) to induce interferon-stimulated genes (ISGs), interferon regulatory factor 1 (IRF1), and NOD-like receptor family CARD domain-containing five (NLRC5) (black lines). IRF1 and NLRC5 induce major histocompatibility complex (MHC) class I expression. ISGs will inhibit viral replication as well as activate RNase L, which will cleave all cellular and viral ssRNA and induce cell death (black lines). The cytokine responses will also recruit dendritic cells, CD8+ killer T cells, and initiate the adaptive immune response. The adaptive immune response to SARS-CoV-2 includes CD8+ T cells, CD4+ T cells, and B cells (plasma cells) to produce antibodies. At least 26 SARS-CoV-2 proteins (shown in blue with blue lines) antagonize some part of the immune response.
As the immune response develops, RIG-I and MDA5, upon activation by dsRNA, interact with MAVS, which initiates the phosphorylation of IRF3 (38, 49, 50). In parallel, TLRs activate MyD88, which initiates NF-κB (16, 41). These activated responses stimulate the production of many cytokines, including types I (IFNα and IFNβ) and III IFN (IFNλ), via several pathways (39–42). As such, the IFN antiviral pathway is one of the most important innate mechanisms against viral infections (38, 51). Notably, the suppressed IFN response is a major determinant of COVID-19 clinical severity (52). Meanwhile, the host cell also produces cytokines and chemokines such as IL-6, IP-10, and TNF, resulting in the cytokine storm (28, 39, 40, 53).
In the next stage of the immune response, in autocrine and paracrine manners, type I IFNs bind to IFNAR, and type III IFNs bind to IFNLR. The binding of IFNs to IFNAR and IFNLR activates the JAK/STAT pathway, inducing the expression of MHC class I and ISG via the ISRE (16, 41, 42). ISGs repress viral replication and activate RNase L, which degrades both viral and host ssRNA, leading to cell death without allowing the virus to spread (38).
Moreover, SARS-CoV-2 infection also leads to activation of cGAS-STING signaling via cell death, cell fusion, mitochondrial stress, and DNA damage, which mediates recognition of the genome DNA from fused cells and the activation of NF-κB and subsequent upregulation of TNF and IL-6 (39, 54). cGAS-STING also stimulates the IRF3 signaling pathway (39). Lastly for the innate response, proteasomes will degrade viral proteins and present them on MHC class I proteins to facilitate cytotoxic T-cell destruction of the infected cell. The dysregulation caused by the many avenues of cytokine release ultimately damages tissues and organs (28). Inflammatory cells are also activated and migrate to the lungs, producing a compounded cytokine response to add to the cytokine storm (40).
The pro-inflammatory cytokine response recruits immune cells and begins the adaptive response (39). There are several essential facets of the adaptive immune response against SARS-CoV-2, which include plasma cells, CD4+ T helper cells, and CD8+ Killer T cells (55, 56). This section will discuss the adaptive response specific to SARS-CoV-2 infection. All three of these arms (antibodies, CD4+, and CD8+) work together to combat infection with SARS-CoV-2, and follow the recognition of the virus by dendritic cells, and migration of those activated dendritic cells to lymph nodes (57). nAbs are the most common route to immunity against SARS-CoV-2 progression to COVID-19 and are therefore essential, and a focus for vaccines elicited responses (56, 58, 59). nAbs bind the virus to prevent the virus from entering cells. Long-term protection from antibodies is facilitated by memory B cells. Memory B cells, both circulating and in bone marrow, are detectable up to 6 months after SARS-CoV-2 mRNA vaccination and a year following SARS-CoV-2 infection (24, 60). B cells differentiate into plasma cells, which produce nAbs (IgG, IgA, and IgM) within severals days after infection or vaccination (24). This differentiation can occur either in the extrafollicular region during the EF phase or in the germinal centers during the GC phase. Following the EF phase, the B cells undergo somatic hypermutation and selection in germinal centers during the GC phase and compartmentalize in the bone marrow (24). However, a unique finding in acute COVID-19 is the lack of germinal centers in lymph nodes. Germinal centers are essential for developing and differentiating memory B cells and plasma cells with high-affinity antibodies (61). During COVID-19 disease, there is an absence of Bcl-6 transcription factor expressing B cells, a critical transcription factor for B cell development in germinal centers (62). This absence correlates with the lack of germinal centers in acute COVID-19, which results in “disease-related” extrafollicular B cells. The “disease-related” B cells result from class-switching and not selection in germinal centers. This class of B cells does not impart long-lasting protection. Thus, the development of B cells underscores the importance of vaccination in generating a high-affinity nAb response and protecting against COVID-19 (62).
CD4+ T helper cells are vital to the antibody responses through interactions with B cells, and are part of almost all infections with SARS-CoV-2 (55, 56). CD4+ T cell responses are more prominent than CD8+ in SARS-CoV-2 infection, are strongly associated with lessened disease severity, and have been demonstrated against 21 SARS-CoV-2 proteins; prominently S, M, N, nsp3, nsp4, nsp12, ORF3a, ORF7a, and ORF8 (55, 63–65). Memory CD4+ T cells persist and can generate a response on secondary challenge, circulate by 30 days post-symptom onset and have a half-life of 94 days (64). There are several cell functions for CD4+ seen in COVID-19 (55). The specialized subset of CD4+ T cells known as TFH are important for helping generate nAbs and help B cells. TFH are detectable and durable for more than six months post-symptom onset (24, 64). However, Bcl-6+ GC-TFH, a requirement for inducing germinal centers, are decreased in COVID-19. Instead, there is a robust TH1 response in COVID-19 (62). The TH1 response is involved in cytokine secretion (IFNγ) and innate cell recruitment (55). Even so, the host humoral immune response evolves continuously via affinity maturation with CD4+ T cells in germinal centers after viral resolution (66). Additionally, slower decay rates of antibodies and a higher-affinity antibody response correlate with a higher frequency of CD4+ T cells (66). TRM are those memory T cells that are non-recirculating and persist in tissues, and serve to limit re-infection (67). TRM in the respiratory tract exists for ten months post-infection (67). IL-22 secreting CD4+ T-cells play a role in mucosal wound healing (55).
CD8+ T cells play an essential role in many viral infections and are responsible for destroying infected cells (56). CD8+ T cell responses have been recognized against SARS-CoV-2 S protein, M protein, N protein, nsp6, and ORF3a (63–65). Memory CD8+ T cells circulate by 20-50 days post-symptom onset and have a half-life of 225 days. The preponderance of these circulating CD8+ T cells are TEMRA, with lesser amounts of TEM and TCM. TEMRA plays a role in protection against severe disease, as shown in other viral infections (64).
Upon infection with SARS-CoV-2, those able to generate immune responses using B cells, CD4+ cells, and CD8+ cells are able to limit disease severity (56). Further, developing a robust response with these three arms was inversely correlated with age (56). The broad spectrum of all three responses defines the adaptive immune response to SARS-CoV-2 infection and lessens the disease severity (56). An important consideration and implication for vaccine design is the inclusion of M and N to better mimic natural SARS-CoV-2 CD4+ T cell and CD8+ T cell responses (65). In vaccinated individuals, regardless of vaccine platform, memory CD4+ T cells and memory CD8+ T cells are preserved and not impacted by evolving variants. Whereas memory B cells recognition of spike proteins, and antibody reactivity, are significantly reduced against variants in both vaccinated and naturally infected persons (68, 69).
We are now delving into the role of SARS-CoV-2 modulation or antagonism of the immune system, which SARS-CoV-2 is very effective at (55). Firstly, we will cover the structural proteins. Inside the cell, the Spike protein will antagonize the immune response by interacting with IRF3 (70). In the case of the M protein, it antagonizes innate immunity by inhibiting the TRAF complex, which is involved in the promoter activation of NF-κB and subsequent IFN transcription (16, 71). Additionally, the M protein interacts with MAVS, impairing the IFN downstream response, and further, blocks phosphorylation of STAT1, an element responsible for inducing ISG (72, 73). Lastly, the M protein triggers cell apoptosis with the N protein as a cofactor (74). Switching to the N protein, it targets the initiation of RIG-1 pathway via blocking TRIM25, and additionally prevents phosphorylation of STAT1, STAT2, and IRF3, blocking the entry of all three into the nucleus and inhibiting the IFN and ISG responses (16, 51). The N protein is also shown to prevent the aggregation of MAVS, as well as promote the activation and assembly of the inflammasome (75, 76).
Now moving on to the non-structural proteins. Nsp1 blocks the phosphorylation of IRF3, IRF7, STAT1, and cJun. Nsp1 additionally directly inhibits the IFN and NF-κB promoters and interacts with host 40S ribosomal subunits via 18S rRNA to inhibit host protein translation. Further, nsp1 degrades transcripts lacking a 5’ viral leader sequence (16, 38, 52, 77–80). Regarding nsp3, it blocks cytokine production (81). Nsp3, like N protein, also block the phosphorylation of IRF3, preventing nuclear translocation, and antagonize type I IFN activity (16, 81, 82). Transitioning to nsp5, it proteolytically cleaves RIG-I and induces the degradation of MAVS, thus preventing detection of viral dsRNA and inhibiting the IFN pathway (83). In the case of nsp8 and nsp9, they bind to the SRP and disrupt protein trafficking, suppressing the type I IFN response (52). As for nsp10, it impairs the activity of IRF3 and NF-κB binding sites (81). Exploring nsp6 and nsp13, they bind to an intermediary between MAVS and IRF3 signaling; thus limiting IRF3 activation (63, 73, 77, 84). Nsp6 and nsp13 also inhibit phosphorylation of STAT1 and STAT2 (73). Additionally, nsp13 limits nuclear translocation of NF-κB (77). Nsp13, nsp14, nsp15, and ORF6 prevent nuclear translocation of IRF3 (85). Turning to nsp14, nsp15, and nsp16, they modify the viral RNA and prevent recognition by RIG-1 and MDA-5 (38). Nsp14 additionally targets the IFNAR receptor for lysosomal degradation (81). Lastly, nsp16 additionally binds to the spliceosome and interrupts mRNA splicing, further suppressing the IFN response (52).
Now we move on to the ORFs. ORF3a blocks the phosphorylation of STAT1, activates the NLRP3 inflammasome, prevents phagosome and lysosome fusion, and induces cell death via the extrinsic apoptosis pathway (18, 20, 73, 81). ORF3b antagonizes type I IFN activity (86). Regarding ORF6, it blocks the translocation of IRF3 and the STAT1 complex into the nucleus and inhibits the MHC class I pathway (16, 73, 87). ORF6 also binds to the interferon-inducible nuclear export complex of Nup98 and Rae1, preventing the nuclear release of mRNA (63, 84). Moreover, ORF6, ORF3b and ORF8 inhibit the ISRE to type I IFN production (38, 88). Focusing on ORF7a, it inhibits the translocation of STAT2 to the nucleus, reduces phagolysosome acidity, and binds to monocytes, decreasing their ability to present antigens (18, 73, 81, 89). In the case of ORF7b, it suppresses STAT1 and STAT2 phosphorylation (73). Turning to ORF8, it directly interacts with MHC class I proteins on the ER membrane and facilitates their degradation via autophagosome degradation (90). Additionally, ORF8 is a secreted protein that mimics IL17A and interacts with IL17 receptors on monocytes. This interaction of monocytes upregulates gene expression in fibrosis signaling, coagulation dysregulation, and inflammation (91). Examining ORF9b, it interacts with Tom70, thereby interfering with MAVS and type I IFN expression (84, 92). As for ORF9c, it upregulates IL-6 signaling while impairing IFN signaling (93). Lastly, ORF10 induces mitophagy and thereby causes the degradation of MAVS (94).
The rapid emergence and spread of SARS-CoV-2 worldwide has vastly changed research over the past years. This dynamic has included more than 10,000 new SARS-CoV-2 articles released per month (95). This review aims to summarize some of the most relevant and referenced immune response resources into a comprehensive summary of the current state of the host immune response to SARS-CoV-2. This amalgamation of information represents the background and current state of understanding in an ever-evolving and dynamic field. Understanding the virus-host immune interactions can provide valuable insights for developing targeted therapeutics, such as antivirals and immunomodulatory drugs, which can mitigate the severity of the infection and limit viral spread. Furthermore, this knowledge can inform pandemic preparedness by helping to identify potential therapeutic targets and designing effective pre-emptive interventions.
The topics covered herein require constant reevaluation as more evidence is published worldwide. Such reflection will allow for refining the selection of drug targets and elucidating and understanding the molecular basis of pathogenesis. Understanding the molecular pathway of SARS-CoV-2 viral inhibition of IFN production and signaling for drug targets and pathogenesis can point to other prospective treatments, such as IFN mimicry, small molecules, solubilizable ACE2 in place of monoclonal antibodies (96), prophylactic RIG-I agonists (97), or nanobody-fusions targeting specific ORFs. There are many currently available and approved IFN receptor agonists that clinical trials could further explore (ropeginterferon alfa-2b, peginterferon alfa-2b, etc). These points of continuous assessment allow for the most effective management of SARS-CoV-2, COVID-19, and future pandemics.
DM prepared the initial manuscript. DM, YD, and MG collectively produced the final version. All authors contributed to the article and approved the submitted version.
This work was partially supported by the National Institutes of Health, Department of Health and Human Services, USA [grant numbers 5P20GM103466, 5U54MD007601, 5P30GM114737, 1P20GM139753, 1U54GM138062, and 5P20GM113134].
Figures 1, 2 created with BioRender.com. Figure 1 adapted from “Genome Organization of SARS-CoV,” “Life Cycle of Coronavirus,” and “Mechanism of SARS-CoV-2 Viral Entry” by BioRender.com (2022). Retrieved from https://apps.biorender.com/biorender-templates.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
2. Worobey M, Levy JI, Serrano LM, Crits-Christoph A, Pekar JE, Goldstein SA, et al. The huanan seafood wholesale market in wuhan was the early epicenter of the COVID-19 pandemic. Science (2022) 377(6609):951–9. doi: 10.1126/science.abp8715
3. Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al. A new coronavirus associated with human respiratory disease in China. Nature (2020) 579(7798):265–9. doi: 10.1038/s41586-020-2008-3
4. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med (2020) 26(4):450–2. doi: 10.1038/s41591-020-0820-9
5. CDC. CDC Museum COVID-19 timeline. Centers for Disease Control and Prevention (2021). Available at: https://www.cdc.gov/museum/timeline/covid19.html.
6. Rambaut A, Holmes EC, O’Toole Á, Hill V, McCrone JT, Ruis C, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol (2020) 5(11):1403–7. doi: 10.1038/s41564-020-0770-5
7. O’Toole Á, Scher E, Underwood A, Jackson B, Hill V, McCrone J, et al. Pangolin: lineage assignment in an emerging pandemic as an epidemiological tool. PANGO lineages (2021). Available at: https://github.com/cov-lineages/pangolin
8. Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, Callender C, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics (2018) 34(23):4121–3. doi: 10.1093/bioinformatics/bty407
9. Maison DP, Ching LL, Cleveland SB, Tseng AC, Nakano E, Shikuma CM, et al. Dynamic SARS-CoV-2 emergence algorithm for rationally-designed logical next-generation vaccines. Commun Biol (2022) 5(1):1–12. doi: 10.1038/s42003-022-04030-3
10. Johns Hopkins university of medicine coronavirus resource center. Johns Hopkins Coronavirus Resource Center (2021). Available at: https://coronavirus.jhu.edu/.
11. Adam D. The pandemic’s true death toll: millions more than official counts. Nature (2022) 601(7893):312–5. doi: 10.1038/d41586-022-00104-8
12. Kadam SB, Sukhramani GS, Bishnoi P, Pable AA, Barvkar VT. SARS-CoV-2, the pandemic coronavirus: molecular and structural insights. J Basic Microbiol (2021) 61(3):180–202. doi: 10.1002/jobm.202000537
13. Cascella M, Rajnik M, Aleem A, Dulebohn SC, Di Napoli R. StatPearls. Treasure Island (FL: StatPearls Publishing (2022). Available at: http://www.ncbi.nlm.nih.gov/books/NBK554776/.
14. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature (2020) 579(7798):270–3. doi: 10.1038/s41586-020-2012-7
15. Lamers MM, Haagmans BL. SARS-CoV-2 pathogenesis. Nat Rev Microbiol (2022) 20(5):270–84. doi: 10.1038/s41579-022-00713-0
16. Hartenian E, Nandakumar D, Lari A, Ly M, Tucker JM, Glaunsinger BA. The molecular virology of coronaviruses. J Biol Chem (2020) 295(37):12910–34. doi: 10.1074/jbc.REV120.013930
17. Naqvi AAT, Fatima K, Mohammad T, Fatima U, Singh IK, Singh A, et al. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. Biochim Biophys Acta (BBA) - Mol Basis Disease. (2020) 1866(10):165878. doi: 10.1016/j.bbadis.2020.165878
18. Redondo N, Zaldívar-López S, Garrido JJ, Montoya M. SARS-CoV-2 accessory proteins in viral pathogenesis: knowns and unknowns. Front Immunol (2021) 12:708264. doi: 10.3389/fimmu.2021.708264
19. Shang J, Han N, Chen Z, Peng Y, Li L, Zhou H, et al. Compositional diversity and evolutionary pattern of coronavirus accessory proteins. Briefings Bioinf (2020) 22(2):1267–78. doi: 10.1093/bib/bbaa262
20. Ren Y, Shu T, Wu D, Mu J, Wang C, Huang M, et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol Immunol (2020) 17(8):881–3. doi: 10.1038/s41423-020-0485-9
21. V’kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol (2021) 19(3):155–70. doi: 10.1038/s41579-020-00468-6
22. Huang Y, Yang C, Xu XF, Xu W, Liu SW. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta pharmacologica Sinica. (2020) 41(9):1141. doi: 10.1038/s41401-020-0485-4
23. Duan L, Zheng Q, Zhang H, Niu Y, Lou Y, Wang H. The SARS-CoV-2 spike glycoprotein biosynthesis, structure, function, and antigenicity: implications for the design of spike-based vaccine immunogens. Front Immunol (2020) 11:576622. doi: 10.3389/fimmu.2020.576622
24. Qi H, Liu B, Wang X, Zhang L. The humoral response and antibodies against SARS-CoV-2 infection. Nat Immunol (2022) 23(7):1008–20. doi: 10.1038/s41590-022-01248-5
25. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell (2020) 181(2):271–280.e8. doi: 10.1016/j.cell.2020.02.052
26. Peacock TP, Goldhill DH, Zhou J, Baillon L, Frise R, Swann OC, et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat Microbiol (2021) 6(7):899–909. doi: 10.1038/s41564-021-00908-w
27. Zhou S, Lv P, Li M, Chen Z, Xin H, Reilly S, et al. SARS-CoV-2 e protein: pathogenesis and potential therapeutic development. Biomedicine Pharmacotherapy. (2023) 159:114242. doi: 10.1016/j.biopha.2023.114242
28. Zheng M, Karki R, Williams EP, Yang D, Fitzpatrick E, Vogel P, et al. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat Immunol (2021) 22(7):829–38. doi: 10.1038/s41590-021-00937-x
29. Shah SM, Alsaab HO, Rawas-Qalaji MM, Uddin MN. A review on current COVID-19 vaccines and evaluation of particulate vaccine delivery systems. Vaccines (2021) 9(10):1086. doi: 10.3390/vaccines9101086
30. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science (2020) 367(6483):1260–3. doi: 10.1126/science.abb2507
31. Fan X, Cao D, Kong L, Zhang X. Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nat Commun (2020) 11(1):3618. doi: 10.1038/s41467-020-17371-6
32. Mlcochova P, Kemp SA, Dhar MS, Papa G, Meng B, Ferreira IATM, et al. SARS-CoV-2 B.1.617.2 delta variant replication and immune evasion. Nature (2021) 599(7883):114–9. doi: 10.1038/s41586-021-03944-y
33. Maison DP, Ching LL, Shikuma CM, Nerurkar VR. Genetic characteristics and phylogeny of 969-bp s gene sequence of SARS-CoV-2 from Hawaii reveals the worldwide emerging P681H mutation. Hawaii J Health Soc Welf (2021) 80(3):52–61.
34. Hoffmann M, Kleine-Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell (2020) 78(4):779–84. doi: 10.1016/j.molcel.2020.04.022
35. Corbett KS, Edwards DK, Leist SR, Abiona OM, Boyoglu-Barnum S, Gillespie RA, et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature (2020) 586(7830):567–71. doi: 10.1038/s41586-020-2622-0
36. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell (2020) 181(2):281–292.e6. doi: 10.1016/j.cell.2020.02.058
37. Raj R. Analysis of non-structural proteins, NSPs of SARS-CoV-2 as targets for computational drug designing. Biochem Biophysics Rep (2021) 25:100847. doi: 10.1016/j.bbrep.2020.100847
38. Kim YM, Shin EC. Type I and III interferon responses in SARS-CoV-2 infection. Exp Mol Med (2021) 53(5):750–60. doi: 10.1038/s12276-021-00592-0
39. Neufeldt CJ, Cerikan B, Cortese M, Frankish J, Lee JY, Plociennikowska A, et al. SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-κB. Commun Biol (2022) 5(1):1–15.
40. Brodin P. Immune determinants of COVID-19 disease presentation and severity. Nat Med (2021) 27(1):28–33. doi: 10.1038/s41591-020-01202-8
41. Sampaio NG, Chauveau L, Hertzog J, Bridgeman A, Fowler G, Moonen JP, et al. The RNA sensor MDA5 detects SARS-CoV-2 infection. Sci Rep (2021) 11(1):13638. doi: 10.1038/s41598-021-92940-3
42. Park A, Iwasaki A. Type I and type III interferons – induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe (2020) 27(6):870–8. doi: 10.1016/j.chom.2020.05.008
43. Bortolotti D, Gentili V, Rizzo S, Schiuma G, Beltrami S, Strazzabosco G, et al. TLR3 and TLR7 RNA sensor activation during SARS-CoV-2 infection. Microorganisms (2021) 9(9):1820. doi: 10.3390/microorganisms9091820
44. Gadanec LK, McSweeney KR, Qaradakhi T, Ali B, Zulli A, Apostolopoulos V. Can SARS-CoV-2 virus use multiple receptors to enter host cells? Int J Mol Sci (2021) 22(3):992. doi: 10.3390/ijms22030992
45. Manik M, Singh RK. Role of toll-like receptors in modulation of cytokine storm signaling in SARS-CoV-2-induced COVID-19. J Med Virology (2021) 94(3):869–77. doi: 10.1002/jmv.27405
46. Taefehshokr N, Taefehshokr S, Hemmat N, Heit B. Covid-19: perspectives on innate immune evasion. Front Immunol (2020) 11:580641. doi: 10.3389/fimmu.2020.580641
47. Aboudounya MM, Heads RJ. COVID-19 and toll-like receptor 4 (TLR4): SARS-CoV-2 may bind and activate TLR4 to increase ACE2 expression, facilitating entry and causing hyperinflammation. Mediators Inflammation. (2021) 2021:e8874339. doi: 10.1155/2021/8874339
48. Kayesh MEH, Kohara M, Tsukiyama-Kohara K. An overview of recent insights into the response of TLR to SARS-CoV-2 infection and the potential of TLR agonists as SARS-CoV-2 vaccine adjuvants. Viruses (2021) 13(11):2302. doi: 10.3390/v13112302
49. Kouwaki T, Nishimura T, Wang G, Oshiumi H. RIG-I-Like receptor-mediated recognition of viral genomic RNA of severe acute respiratory syndrome coronavirus-2 and viral escape from the host innate immune responses. Front Immunol (2021) 12:700926. doi: 10.3389/fimmu.2021.700926
50. Thorne LG, Reuschl AK, Zuliani-Alvarez L, Whelan MVX, Turner J, Noursadeghi M, et al. SARS-CoV-2 sensing by RIG-I and MDA5 links epithelial infection to macrophage inflammation. EMBO J (2021) 40(15):e107826. doi: 10.15252/embj.2021107826
51. Mu J, Fang Y, Yang Q, Shu T, Wang A, Huang M, et al. SARS-CoV-2 n protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discovery (2020) 6(1):1–4. doi: 10.1038/s41421-020-00208-3
52. Banerjee AK, Blanco MR, Bruce EA, Honson DD, Chen LM, Chow A, et al. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell (2020) 183(5):1325–39. doi: 10.1016/j.cell.2020.10.004
53. Ravindra NG, Alfajaro MM, Gasque V, Huston NC, Wan H, Szigeti-Buck K, et al. Single-cell longitudinal analysis of SARS-CoV-2 infection in human airway epithelium identifies target cells, alterations in gene expression, and cell state changes. PloS Biol (2021) 19(3):e3001143. doi: 10.1371/journal.pbio.3001143
54. Liu X, Wei L, Xu F, Zhao F, Huang Y, Fan Z, et al. SARS-CoV-2 spike protein-induced cell fusion activates the cGAS-STING pathway and the interferon response. Sci Signal (2022) 15(729):eabg8744. doi: 10.1126/scisignal.abg8744
55. Sette A, Crotty S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell (2021) 184(4):861–80. doi: 10.1016/j.cell.2021.01.007
56. Rydyznski Moderbacher C, Ramirez SI, Dan JM, Grifoni A, Hastie KM, Weiskopf D, et al. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell (2020) 183(4):996–1012.e19. doi: 10.1016/j.cell.2020.09.038
57. Borges RC, Hohmann MS, Borghi SM. Dendritic cells in COVID-19 immunopathogenesis: insights for a possible role in determining disease outcome. Int Rev Immunol (2021) 40(1–2):108–25. doi: 10.1080/08830185.2020.1844195
58. Corbett KS, Flynn B, Foulds KE, Francica JR, Boyoglu-Barnum S, Werner AP, et al. Evaluation of the mRNA-1273 vaccine against SARS-CoV-2 in nonhuman primates. New Engl J Med (2020) 383(16):1544–55. doi: 10.1056/NEJMoa2024671
59. Kalnin KV, Plitnik T, Kishko M, Zhang J, Zhang D, Beauvais A, et al. Immunogenicity and efficacy of mRNA COVID-19 vaccine MRT5500 in preclinical animal models. NPJ Vaccines (2021) 6(1):61. doi: 10.1038/s41541-021-00324-5
60. Kim W, Zhou JQ, Horvath SC, Schmitz AJ, Sturtz AJ, Lei T, et al. Germinal centre-driven maturation of b cell response to mRNA vaccination. Nature (2022) 604(7904):141–5. doi: 10.1038/s41586-022-04527-1
61. De Silva NS, Klein U. Dynamics of b cells in germinal centres. Nat Rev Immunol (2015) 15(3):137–48. doi: 10.1038/nri3804
62. Kaneko N, Kuo HH, Boucau J, Farmer JR, Allard-Chamard H, Mahajan VS, et al. Loss of bcl-6-Expressing T follicular helper cells and germinal centers in COVID-19. Cell (2020) 183(1):143–157.e13. doi: 10.1016/j.cell.2020.08.025
63. Dong Y, Dai T, Wei Y, Zhang L, Zheng M, Zhou F. A systematic review of SARS-CoV-2 vaccine candidates. Sig Transduct Target Ther (2020) 5(1):1–14. doi: 10.1038/s41392-020-00352-y
64. Dan JM, Mateus J, Kato Y, Hastie KM, Yu ED, Faliti CE, et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science (2021) 371(6529):eabf4063. doi: 10.1126/science.abf4063
65. Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, et al. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell (2020) 181(7):1489–1501.e15. doi: 10.1016/j.cell.2020.05.015
66. Wang Z, Yang X, Mei X, Zhou Y, Tang Z, Li G, et al. SARS-CoV-2-specific CD4+ T cells are associated with long-term persistence of neutralizing antibodies. Sig Transduct Target Ther (2022) 7(1):1–10. doi: 10.1038/s41392-022-00978-0
67. Grau-Expósito J, Sánchez-Gaona N, Massana N, Suppi M, Astorga-Gamaza A, Perea D, et al. Peripheral and lung resident memory T cell responses against SARS-CoV-2. Nat Commun (2021) 12(1):3010. doi: 10.1038/s41467-021-23333-3
68. Tarke A, Coelho CH, Zhang Z, Dan JM, Yu ED, Methot N, et al. SARS-CoV-2 vaccination induces immunological T cell memory able to cross-recognize variants from alpha to omicron. Cell (2022) 185(5):847–59. doi: 10.1016/j.cell.2022.01.015
69. Liu L, Iketani S, Guo Y, Chan JFW, Wang M, Liu L, et al. Striking antibody evasion manifested by the omicron variant of SARS-CoV-2. Nature (2022) 602(7898):676–81. doi: 10.1038/s41586-021-04388-0
70. Freitas RS, Crum TF, Parvatiyar K. SARS-CoV-2 spike antagonizes innate antiviral immunity by targeting interferon regulatory factor 3. Front Cell Infection Microbiol (2022) 11:789462. doi: 10.3389/fcimb.2021.789462
71. Sa Ribero M, Jouvenet N, Dreux M, Nisole S. Interplay between SARS-CoV-2 and the type I interferon response. PloS Pathog (2020) 16(7):e1008737. doi: 10.1371/journal.ppat.1008737
72. Fu YZ, Wang SY, Zheng ZQ, Huang Y, Li WW, Xu ZS, et al. SARS-CoV-2 membrane glycoprotein m antagonizes the MAVS-mediated innate antiviral response. Cell Mol Immunol (2021) 18(3):613–20. doi: 10.1038/s41423-020-00571-x
73. Xia H, Cao Z, Xie X, Zhang X, Chen JYC, Wang H, et al. Evasion of type I interferon by SARS-CoV-2. Cell Rep (2020) 33(1):108234. doi: 10.1016/j.celrep.2020.108234
74. Ren Y, Wang A, Fang Y, Shu T, Wu D, Wang C, et al. SARS-CoV-2 membrane glycoprotein m triggers apoptosis with the assistance of nucleocapsid protein n in cells. Front Cell Infection Microbiol (2021) 11:706252. doi: 10.3389/fcimb.2021.706252
75. Pan P, Shen M, Yu Z, Ge W, Chen K, Tian M, et al. SARS-CoV-2 n protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat Commun (2021) 12(1):4664. doi: 10.1038/s41467-021-25015-6
76. Zotta A, Hooftman A, O’Neill LAJ. SARS-CoV-2 targets MAVS for immune evasion. Nat Cell Biol (2021) 23(7):682–3. doi: 10.1038/s41556-021-00712-y
77. Vazquez C, Swanson SE, Negatu SG, Dittmar M, Miller J, Ramage HR, et al. SARS-CoV-2 viral proteins NSP1 and NSP13 inhibit interferon activation through distinct mechanisms. PloS One (2021) 16(6):e0253089. doi: 10.1371/journal.pone.0253089
78. Thoms M, Buschauer R, Ameismeier M, Koepke L, Denk T, Hirschenberger M, et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science (2020) 369(6508):1249–55. doi: 10.1126/science.abc8665
79. Sunshine S, Puschnik AS, Replogle JM, Laurie MT, Liu J, Zha BS, et al. Systematic functional interrogation of SARS-CoV-2 host factors using perturb-seq. bioRxiv (2022). Available at: https://www.biorxiv.org/content/10.1101/2022.07.15.500120v1.
80. Finkel Y, Gluck A, Nachshon A, Winkler R, Fisher T, Rozman B, et al. SARS-CoV-2 uses a multipronged strategy to impede host protein synthesis. Nature (2021) 594(7862):240–5. doi: 10.1038/s41586-021-03610-3
81. Hayn M, Hirschenberger M, Koepke L, Nchioua R, Straub JH, Klute S, et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep (2021) 35(7):109126. doi: 10.1016/j.celrep.2021.109126
82. Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A, et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature (2020) 587(7835):657–62. doi: 10.1038/s41586-020-2601-5
83. Liu Y, Qin C, Rao Y, Ngo C, Feng JJ, Zhao J, et al. SARS-CoV-2 Nsp5 demonstrates two distinct mechanisms targeting RIG-I and MAVS to evade the innate immune response. mBio (2021) 12(5):e02335–21. doi: 10.1128/mBio.02335-21
84. Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature (2020) 583(7816):459–68. doi: 10.1038/s41586-020-2286-9
85. Yuen CK, Lam JY, Wong WM, Mak LF, Wang X, Chu H, et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerging Microbes Infections. (2020) 9(1):1418–28. doi: 10.1080/22221751.2020.1780953
86. Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y, et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep (2020) 32(12):108185. doi: 10.1016/j.celrep.2020.108185
87. Yoo JS, Sasaki M, Cho SX, Kasuga Y, Zhu B, Ouda R, et al. SARS-CoV-2 inhibits induction of the MHC class I pathway by targeting the STAT1-IRF1-NLRC5 axis. Nat Commun (2021) 12:6602. doi: 10.1038/s41467-021-26910-8
88. Li JY, Liao CH, Wang Q, Tan YJ, Luo R, Qiu Y, et al. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res (2020) 286:198074. doi: 10.1016/j.virusres.2020.198074
89. Cao Z, Xia H, Rajsbaum R, Xia X, Wang H, Shi PY. Ubiquitination of SARS-CoV-2 ORF7a promotes antagonism of interferon response. Cell Mol Immunol (2021) 18(3):746–8. doi: 10.1038/s41423-020-00603-6
90. Zhang Y, Chen Y, Li Y, Huang F, Luo B, Yuan Y, et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-I. Proc Natl Acad Sci (2021) 118(23):e2024202118. doi: 10.1073/pnas.2024202118
91. Wu X, Xia T, Shin WJ, Yu KM, Jung W, Herrmann A, et al. Viral mimicry of interleukin-17A by SARS-CoV-2 ORF8. mBio (2022) 13(2):e0040222. doi: 10.1128/mbio.00402-22
92. Jiang Hw, Zhang Hn, Meng Qf, Xie J, Li Y, Chen H, et al. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell Mol Immunol (2020) 17(9):998–1000. doi: 10.1038/s41423-020-0514-8
93. Dominguez Andres A, Feng Y, Rosa Campos A, Yin J, Yang CC, James B, et al. SARS-CoV-2 ORF9c is a membrane-associated protein that suppresses antiviral responses in cells. bioRxiv (2020). Available at: https://www.biorxiv.org/content/10.1101/2020.08.18.256776v1.full
94. Li X, Hou P, Ma W, Wang X, Wang H, Yu Z, et al. SARS-CoV-2 ORF10 suppresses the antiviral innate immune response by degrading MAVS through mitophagy. Cell Mol Immunol (2022) 19(1):67–78. doi: 10.1038/s41423-021-00807-4
95. Chen Q, Allot A, Lu Z. LitCovid: an open database of COVID-19 literature. Nucleic Acids Res (2021) 49(D1):D1534–40. doi: 10.1093/nar/gkaa952
96. Krishnamurthy S, Lockey RF, Kolliputi N. Soluble ACE2 as a potential therapy for COVID-19. Am J Physiol Cell Physiol (2021) 320(3):C279–81. doi: 10.1152/ajpcell.00478.2020
Keywords: COVID-19, immune response, innate immunity, adaptive immunity, viral immune evasion
Citation: Maison DP, Deng Y and Gerschenson M (2023) SARS-CoV-2 and the host-immune response. Front. Immunol. 14:1195871. doi: 10.3389/fimmu.2023.1195871
Received: 29 March 2023; Accepted: 05 June 2023;
Published: 19 June 2023.
Edited by:
Avijit Dutta, Chang Gung University, TaiwanReviewed by:
Feng-Liang Liu, Kunming Institute of Zoology (CAS), ChinaCopyright © 2023 Maison, Deng and Gerschenson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David P. Maison, ZGF2aWRtMjJAaGF3YWlpLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.