 Zihan Deng
Zihan Deng Lisen Lu2†
Lisen Lu2† Binghui Li
Binghui Li Xiujuan Shi
Xiujuan Shi Honglin Jin
Honglin Jin Weidong Hu
Weidong Hu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 11 July 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1195572
This article is part of the Research TopicPolarization of cellular immune response in the context of inflammation and cancerView all 8 articles
Inflammation is a key characteristic of all stages of tumor development, including tumor initiation, progression, malignant transformation, invasion, and metastasis. Inflammasomes are an important component of the inflammatory response and an indispensable part of the innate immune system. Inflammasomes regulate the nature of infiltrating immune cells by signaling the secretion of different cytokines and chemokines, thus regulating the anti-tumor immunity of the body. Inflammasome expression patterns vary across different tumor types and stages, playing different roles during tumor progression. The complex diversity of the inflammasomes is determined by both internal and external factors relating to tumor establishment and progression. Therefore, elucidating the specific effects of different inflammasomes in anti-tumor immunity is critical for promoting the discovery of inflammasome-targeting drugs. This review focuses on the structure, activation pathway, and identification methods of the NLRP3, NLRC4, NLRP1 and AIM2 inflammasomes. Herein, we also explore the role of inflammasomes in different cancers and their complex regulatory mechanisms, and discuss current and future directions for targeting inflammasomes in cancer therapy. A detailed knowledge of inflammasome function and regulation may lead to novel therapies that target the activation of inflammasomes as well as the discovery of new drug targets.
The occurrence and progression of cancer is characterized by several hallmarks, including uncontrolled proliferation, autocrine growth signals, anti-apoptotic signaling, neovascularization, increased tissue invasion and metastasis, and an inflammatory environment (1). The correlation between inflammation and cancer was first discovered in the 19th century by Rudolf Virchow, who observed the infiltration of leukocytes in tumor tissue and suggested that cancer could occur at the site of chronic inflammation (2). The innate immune system can modulate cancer-associated inflammation, either by triggering the malignant development of tumors, or by providing an inflammatory microenvironment that promotes tumorigenesis (3). Inflammasomes are involved in innate immunity and mediate the release of inflammatory cytokines, trigger inflammatory cascades and promote the formation of an inflammatory microenvironment that is conducive to tumor cell growth. Therefore, a detailed study of the molecular mechanisms that govern inflammation and cancer may lead to the development of novel therapeutic approaches to cancer treatment.
An inflammasome is a kind of multiprotein complex that is assembled in cells in response to pathogen associated molecular patterns (PAMPs) and damage associated molecular patterns (DAMPs). In response to external stimulation, such as the presence of bacterial pathogens, procaspase-1 is activated and the precursors of IL-1β and IL-18 are cleaved to release the active forms of these cytokines, which result in the death of phagocytes (4, 5). Inflammasome complexes affect a variety of pathogens and their homeostatic signaling, while also modulating the host response, especially in the context of bacterial infection, autoimmune diseases, and tumors (6). Members of the nucleotide-binding domain and leucine-rich repeat containing receptor (NLR) family, NLRP1, NLRP3, NLRC4 and absent in melanoma 2 (AIM2), have been confirmed to be involved in the assembly of inflammasomes (7). Inflammasomes play an important role in the different stages of development of numerous inflammatory and tumor-like diseases in humans. They not only promote tumorigenesis and metastasis, but also exert tumor-suppressive effects, reflecting the complexity of inflammasome action during the process of tumorigenesis (8). Herein, we review the composition and activation process of different inflammasomes, and analyze the molecular mechanisms by which they may mediate tumor pathogenesis, to obtain a deeper understanding of the molecular mechanisms of tumor development. In addition, we also discuss possible therapeutic directions and potential targets for cancer therapies directed at inflammasomes. Overall, understanding the mechanisms of inflammasome formation and action in different cancers and different stages of tumor formation, development, progression, and invasion will provide new targets for clinical drugs and immunotherapies.
The notion of the inflammasome was first proposed by Tschopp et al. in 2002 (9). The inflammasome consists of three parts. The first is the pattern recognition receptor (PRR), which acts as an intracellular receptor. The second is the cytokine activation system, that is, ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD)], which is an adaptor protein consisting of a pyrin domain (PYD) and a CARD. One end of ASC is linked to the PRR, and pro-caspase-1 is recruited through its CARD domain at the other end. Once activated, pro-caspase-1 becomes caspase-1, which is the third important component of the inflammasome. Caspase-1 can mediate the inflammatory response, induce pyroptosis and activate caspase enzymes to cause cell lysis and apoptosis (10, 11). Because the structure of adaptor protein ASC and effector protein caspase-1 are rarely changed, inflammasomes are generally classified according to the different receptor proteins, which are more varied. Inflammasomes may be considered classical or non-classical. Classical inflammasomes require the participation of caspase-1, and include NLRP1, NLRP3, NLRC4, and AIM2. Non-classical inflammasomes are usually dependent on caspase-4 or caspase-5, and are less studied and need to be further explored (12). Classical pathways recruit pro-caspase-1 through ASC and transform it into active caspase-1. Activated caspase-1 promotes the conversion of pro-IL-1β and pro-IL-18 into the pro-inflammatory cytokines IL-1β and IL-18, resulting in apoptosis and pathogen clearance. This process plays an important role in innate immune defense (13) (Figure 1). IL-1β secreted extracellularly facilitates the pro-inflammatory effects of IL-6 and TNF-α, while IL-18 can mediate neutrophil maturation and local infiltration and induce the transformation of T-cells to the Th2 state. Th2 cells secrete IL-4 and IL-13, which can induce macrophages to differentiate into the M2 state and mediate anti-inflammatory effects (14).
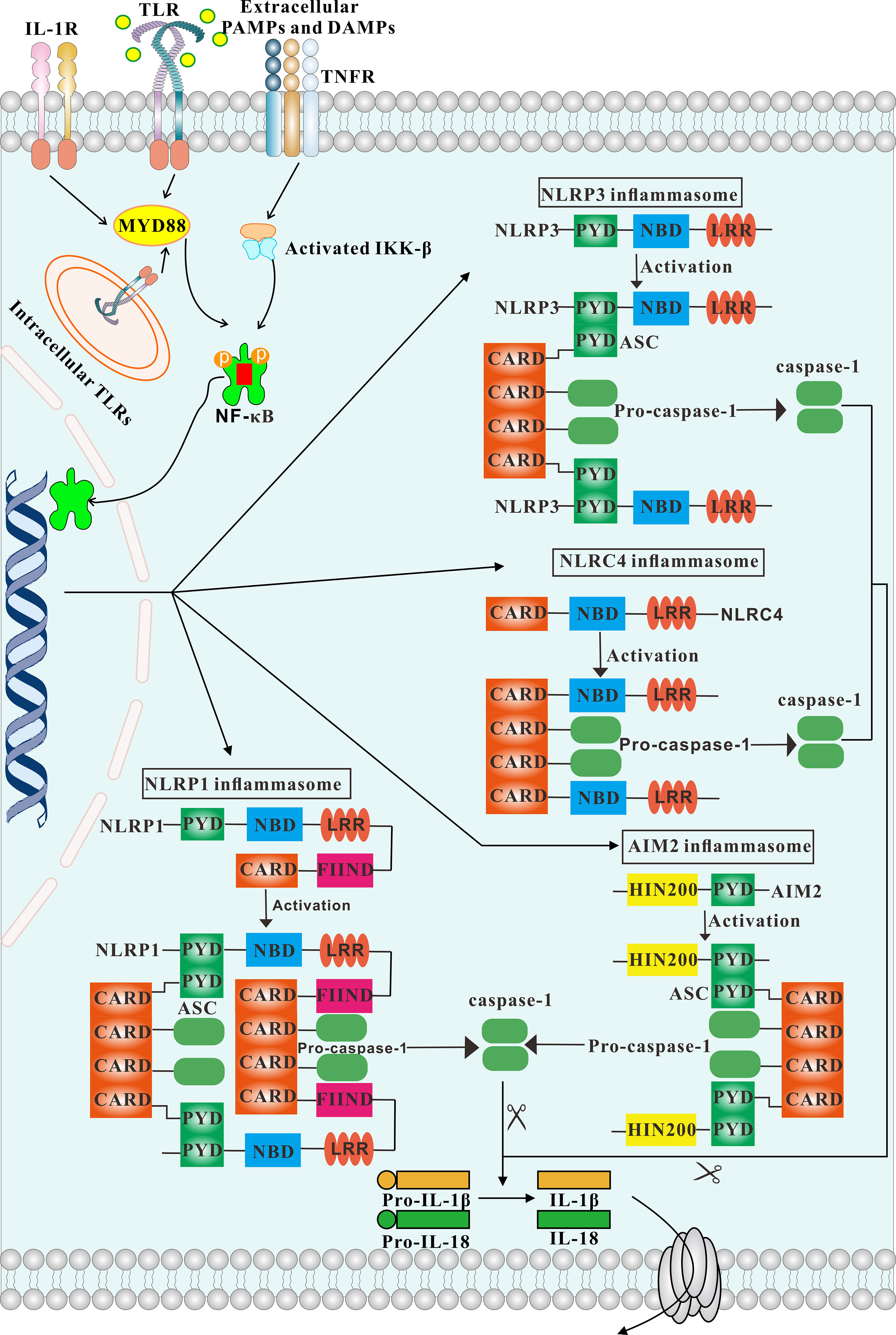
Figure 1 Overview of different inflammasomes. The structure and activation process of inflammasomes are depicted. Engagement of TLR, IL-1 and TNF receptors lead to activation of IKKs, which in turn results in activation of NF-κB signaling. Consequently, NF-κB activates inflammasomes. The NLRP3, AIM2 inflammasomes is strictly dependent on the adaptor ASC. In contrast, NLRP1 and NLRC4 possess a CARD domain and can recruit caspase-1 directly.
NLRP3, the most thoroughly studied inflammasome, consists of the sensor NLRP3, the adaptor protein ASC, and the effector protein caspase-1. The NLRP3 inflammasome is widely present in immune cells, including granulocytes, dendritic cells, macrophages, epithelial cells and osteoblasts (15). NLRP3 belongs to the family of NLR proteins and consists of three structural domains: a C-terminal leucine-rich repeat (LRR), a central nucleotide-binding domain (NBD), and an N-terminal PYD. The LRR is the key regulator of NLRP3 activity and senses endogenous damage and microbial presence. The NBD has ATPase activity and promotes self-oligomerization. The PYD is primarily involved in mediating downstream protein interactions. Normally, the NBD of NLRP3 binds to the LRR, inducing a state of self-inhibition. In the presence of PAMPs or DAMPs, NLRP3 exposes the NBD domain and oligomerizes. The PYD at the N-terminus of NLRP3 recruits PYD-bearing ASC splice proteins, and the CARD of ASC recruits pro-CARD-bearing caspase-1 to complete the assembly of the inflammasome (16).
The activation of the NLRP3 inflammasome requires two steps. In the first step (initiation), microbial or endogenous molecules such as TLR ligands activate NF-κB, which in turn induces the transcription and production of pro-IL-1β, pro-IL-18, and NLRP3. In the second step (activation), the NLRP3 inflammasome recognizes various PAMPs and DAMPs from diverse stimuli, such as fungi (10), bacterial (17), viruses (18, 19), fibrillar amyloid-β (Aβ) peptide (20), ATP (20), glucose (21), and so on. These stimuli promote the activation of the inflammasome and the maturation of pro-IL-1β and pro-IL-18. There are four main activation mechanisms of the NLRP3 inflammasome, including ionic flux, ROS production, mitochondrial dysfunction and lysosomal damage. In addition, atypical inflammasome activation can occur through a pathway stimulated by lipopolysaccharide. In mice, this process is predominantly controlled by caspase-11, while in humans, caspase-4 and caspase-5 are the main players. After the activation of caspase-4, caspase-5 or caspase-11, the intact gasdermin D (GSDMD) is cleaved to generate an N-terminal fragment, which binds to the cell membrane to form a pore, thus inducing pyroptosis (22). In a study aimed at identifying additional sensors for intracellular LPS by biochemical screening, the authors identified the orphan nuclear receptor Nur77 as an LPS-binding protein in macrophage lysates, which could bind mitochondrial DNA and LPS to activate the non-classical NLRP3 inflammasome (23). During microbial infections, the NLRP3 inflammasome can support the host immune system to resist infection, thus maintaining homeostasis. However, abnormal activation of the NLRP3 inflammasome causes excessive IL-1β and IL-18 production, which can trigger certain genetic or acquired inflammatory diseases.
NLRC4, also known as IPAF (IL-1β converting enzyme protease activating factor), is similar to other NLRs and consists of three structural domains. The C-terminal LRR domain, consisting of 15 repetitive units with a total of 440 amino acids, with each unit connected to a helix structure containing 8-15 amino acids, recognizes ligands such as PAMP. The N-terminal CARD is usually composed of the first 94 amino acids of NLRC4, and folds into six inversely parallel α-helices wrapped around the hydrophobic core; the N-terminal CARD can connect the binding protein ASC and the effector molecule caspase-1, and mediate the downstream signaling. Finally, the central NBD, which includes a central NTPase domain, two helical domains (HD1, HD2) and the winged-helical domain (WHD). These protein domains occur in the order of NBD-HD1-WHD-HD2 from N-terminus to C-terminus, and can mediate the oligomerization of NLR molecules, thus changing their conformation (24).
The activation of the NLRC4 inflammasome is primarily regulated at the translational and post-translational levels by two main mechanisms: ligand binding and phosphorylation. In the event of bacterial infection, the LRR recognizes the ligands and undergoes a conformational change, facilitating the conversion of ADP to ATP. Consequently, the inhibition of NBD oligomerization by the LRR is removed, exposing the CARD and activating the NLRC4 inflammasome. Gram-negative pathogenic bacteria, such as Salmonella typhimurium and Pseudomonas aeruginosa, can activate the NLRC4 inflammasome by introducing flagellin or type three secretion system (T3SS) rod protein (PrgJ) into host cells through the transmembrane needle-like structure of the T3SS (25). Some NLRC4 proteins can also directly recruit and activate pro-caspase-1 without ASC, but require interaction with the NLR family apoptosis inhibitory protein (NAIP), which acts as a ligand to activate the NLRC4 inflammasome (26). After NLRC4 inflammasome activation, pro-caspase-1 can be recruited through the CARD of ASC or NLRC proteins to become the active caspase-1, which cleaves pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18, resulting in further inflammation. In addition, caspase-1 can also activate GSDMD to cause pyroptosis. Pyroptosis not only induces host inflammation, but also captures pathogenic bacteria through cell membrane pores, thus promoting the host to initiate immune defense to eliminate the pathogenic bacteria (27).
The NLRP1 inflammasome is a multi-protein complex composed of the receptor NLRP1, the adaptor protein ASC and the effector protein caspase-1. Humans have only one NLRP1 gene, which is similar to other NLRs but also has its own unique domains, consisting of an N-terminal PYD, a central NBD, an LRR, a function to find domain (FIIND) and a C-terminal CARD. In contrast, mice have three homologous NLRP genes: NLRP1a, NLRP1b, and NLRP1c, which are different from human NLRP1 in that they lack the N-terminal PYD and contain a truncated LRR, instead possessing an NR100 domain at the N-terminus, which activates the inflammasome and triggers subsequent pyroptosis when cleavage occurs (28). In the absence of no external stimulation, the NBD in NLRP1 binds to the LRR and inhibits self-oligomerization, resulting in an inactive state. However, when cells are exposed to activating substances that bind to the LRR in NLRP1, a conformational change in NLRP1 is induced, exposing the PYD and CARD. In general, PYD and CARD domains mediate other downstream proteins containing PYD and CARD, such as homotypic interactions with ASC and caspase-1, respectively. The NBD domain can interact with the LRR domain, and the FIIND domain enables NLRP1 to perceive the stability of its own proteins to detect and respond to pathogen-related activities. The unique CARD domain of NLRP1 can directly bind to caspase-1 through CARD-CARD interaction to complete the assembly of the NLRP1 inflammasome, so although ASC can promote NLRP1-mediated activation of caspase-1, it is not necessary for NLRP1 inflammasome activation (29). Oligomerized pro-caspase-1 is cleaved by auto-phosphorylation to form active caspase-1, which in turn promotes the cleavage of pro-IL-18 and pro-IL-1β to generate IL-18 and IL-1β, thereby inducing inflammation (30).
At present, several substances that activate the NLRP1 inflammasome have been identified, including bacterial cytosolic acyl dipeptide (MDP), anthrax lethal toxin (LT), and parasites (31). Recent studies have also shown that the expression of NLRP1 inflammasome in human lung epithelial cells is important for the detection of and response to SARS-CoV-2. However, SARS-CoV-2 infection can inhibit the classical caspase-1-GSDMD inflammasome pathway through its NSP5 protease (3CL protease) activity, while triggering epithelial cell pyroptosis via GSDME. Therefore, the NLRP1 inflammasome can sense SARS-CoV-2, but the NSP5 protease is capable of inhibiting innate immunity (32).
The AIM2 protein is a member of the interferon-inducible HIN-200 protein family, and contains a PYD at its N-terminus that binds to the adaptor protein ASC, as well as two adjacent oligonucleotide/oligosaccharide binding domains (OB) at the C-terminus, which are associated with dsDNA recognition and binding (33). During the assembly of the AIM2 inflammasome, the PYD is responsible for the recruitment of the adaptor protein ASC. In the resting state, the PYD of AIM2 binds to the molecular complex within the HIN domain, thus maintaining the inhibitory state. However, when dsDNA binds to the HIN domain, a conformational change occurs, causing the PYD of AIM2 to bind directly and interact with the PYD of ASC. In turn, the CARD of ASC binds to the CARD of pro-caspase-1, promoting caspase-1 activation and the maturation of the downstream inflammatory cytokines IL-1β and IL-18 (34).
As an intracellular DNA receptor, AIM2 can recognize both viral DNA that invades host cells and bacterial DNA that exerts an escape mechanism to enter the cytoplasm by punching holes in the phagosomal membrane; both sources of dsDNA act as ligands to activate AIM2. By recognizing dsDNA, the AIM2 inflammasome plays an indispensable role in host innate immunity against pathogenic microorganisms. AIM2 not only recognizes bacterial and viral dsDNA in the cytoplasm, but also detects damaged and mislocalized DNA molecules (35). However, AIM2 activation can be inhibited by repeated TTAGGG sequences, which are often seen in mammalian telomeric DNA and function to maintain the inhibitory state of AIM2 by competing with dsDNA. Synthetic DNA containing this sequence also inhibits the activation of the AIM2 inflammasome (36). In addition, POP1 and POP3 proteins of the PYD can also inhibit ASC-dependent inflammasome assembly by preventing inflammasome nucleation, thus inhibiting AIM2 inflammasome activation and interfering with caspase-l activation and the release of IL-1β and IL-18 (37).
The inflammasome is a molecular platform that accumulates in response to various intracellular stimuli and may influence tumorigenesis by regulating both innate and adaptive immune responses. Aberrant inflammasome activation is associated with inflammation, metabolic diseases, neurodegeneration and malignant tumors. Inflammasomes play a dual role in the occurrence and development of different tumors (Table 1). On the one hand, the activation of inflammasomes can trigger an effective immune response to limit the invasion of pathogens, while also inducing cell death under stress and inflammatory pathological conditions by regulating caspase-1-dependent cell pyroptosis (56). On the other hand, dysregulated activation of inflammasomes could lead to a hyperinflammatory state, creating an environment that encourages tumor growth and promotes metastasis (57). The specific role of inflammasomes is influenced by their expression level, downstream effector molecules, tumor types and stages of tumor growth.
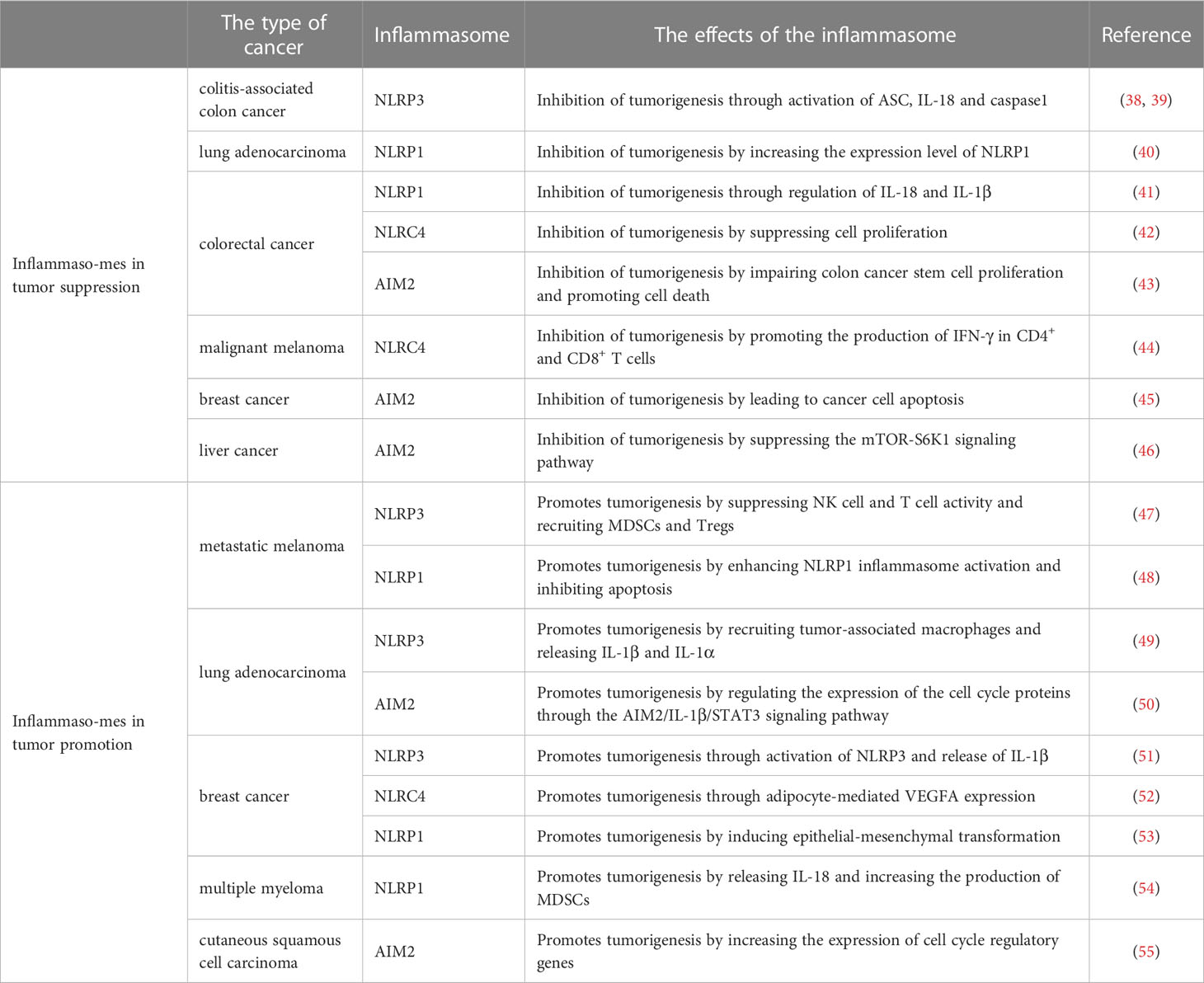
Table 1 The dual role of inflammasomes in different types of cancers.
NLRP3 is the most widely studied inflammasome that not only regulates the tumor itself, but also influences the composition of the tumor microenvironment. The biological importance of the NLRP3 inflammasome in many diseases, including colorectal cancer, melanoma and metastases, is dependent on its ability to respond to multiple signals. In a mouse model of azoxymethane (AOM)-dextran sulfate sodium (DSS)-induced colon cancer, NLRP3 knockout mice were more likely to develop colon polyps and were highly sensitive to AOM-DSS-induced colitis-associated colon cancer. Moreover, tumor growth was observed to be accelerated in NLRP3 knockout mice, concomitant with decreased IL-18 levels in the tissue. IL-18 helps repair the epithelial barrier to counteract damage, and after AOM-DSS induction, IL-18-/- mice were more likely to develop tumors than control mice, indicating that IL-18 has potential anti-tumor effects (38). In addition, ASC and caspase-1 knockout mice are also susceptible to DSS-induced colitis and colitis-associated colon cancer, suggesting that the activation of the NLRP3 inflammasome may have an inhibitory effect on colorectal carcinogenesis (39) (Figures 2A–C). Furthermore, IL-18 secretion mediated by the NLRP3 inflammasome can indirectly inhibit the tumor progression of colitis-associated colorectal cancer by inducing regulatory T (Treg) cells to produce IFN-γ and enhancing the cytotoxicity of T cells and NK cells (58). Hence, the presence of the NLRP3 inflammasome is also necessary in the anti-tumor adaptive immune response. Moreover, chemotherapy treatment of tumor cells (e.g., with oxaliplatin and anthracyclines) can cause tumor cells to release ATP, which is an activation signal for the NLRP3 inflammasome and the IL-1β/IL-1 receptor (IL1R) signaling axis in dendritic cells (DCs) (59). Subsequent P2X7 receptor binding drives the anti-tumor immune response of CD8+ T cells against transplanted tumors (41). In addition, the activation of inflammasomes may enhance the therapeutic effect of anti-PD-1 antibody immunotherapy by activating CD8+ T cells. Activated CD8+ T cells may further promote the activation of NLRP3 inflammasomes of antigen-presenting cells through perforin-dependent mechanisms and achieve anti-tumor effects through a positive feedback mechanism (60).
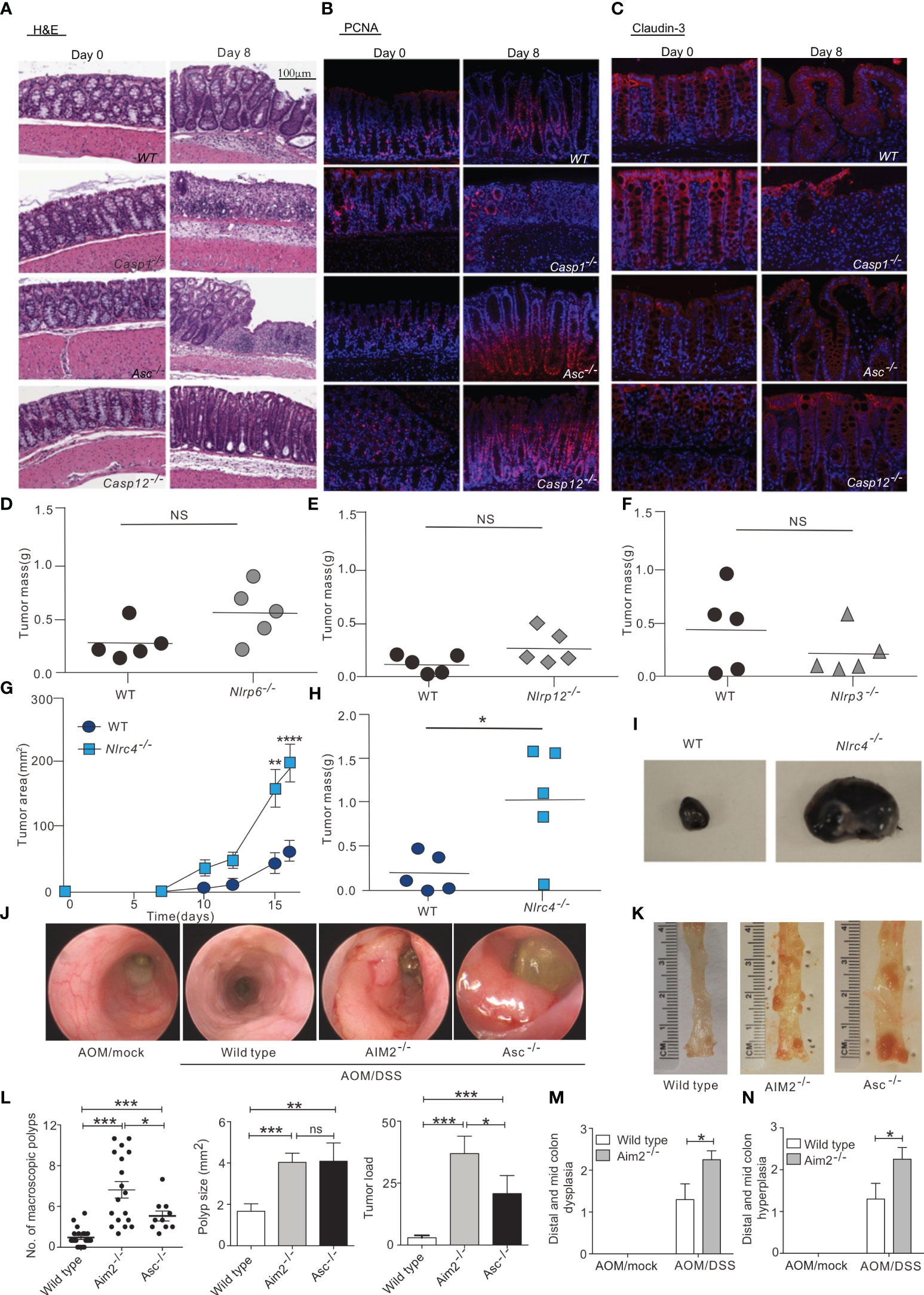
Figure 2 Inflammasome in tumor suppression. (A) Hematoxylin and eosin (H&E) staining of colon sections derived from WT, Casp1-/-, Asc-/-and Casp12 -/- mice on days 0 and 8 after the start of DSS treatment (magnification 100×). (B, C), Immunofluorescence was performed on colon sections derived from WT, Casp1-/-, Asc-/-and Casp12 -/- mice on days 0 and 8 for examination of proliferating cells (PCNA) (B) and tight junction (Claudin-3) (C); (magnification 100×). , WT and Nlrp6–/– (D), Nlrp12–/– (E), Nlrp3–/– (F), and Nlrc4–/– (G–I) mice were injected s.c. with 1 × 105 B16F10 cells. (D-F, H) Tumor mass was determined at 16 to 20 days after inoculation. (G) WT and Nlrc4–/– tumor areas (length × width) were measured every 2 to 3 days. (I) Representative images of excised WT and Nlrc4–/– B16F10 tumors. (G, H) Data are representative of 3 independent experiments with n = 5 mice per group. (J, K) Mini-endoscopy (J) and representative images (K) of colons from AOM/DSS-treated wild-type (n = 5), Asc−/− (n = 3) and Aim2−/− mice (n = 5). (L) Macroscopic polyp counts, average polyp size per mouse and tumor load (n = 21 for WT, n = 18 for Aim2−/− and n = 10 for Asc−/−). (M, N) Semiquantitative scoring of colon hyperplasia (M) and dysplasia (N) in AOM/DSS-treated wild-type and Aim2−/− mice (n = 6 for WT AOM/mock and Aim2−/− AOM/DSS, and n = 5 for WT AOM/DSS and Aim2−/− AOM/mock). The figures A–C are reprinted with permission from Ref (39). Copyright Cell Press. The figures D–I are reprinted with permission from Ref (44). Copyright American Society for Clinical. The figures (J–N) are reprinted with permission from Ref (43). Copyright Nature Publishing Company. *P<0.05, **P<0.01, ***P<0.001, ns: no significance.
Similarly, NLRP1 inflammasome activity can also influence cancer growth. In inflammatory bowel disease and colorectal cancer, the NLRP1 inflammasome exerts effects through IL-1β and IL-18. NLRP1 expression levels were downregulated in biopsied tissues of colon cancer patients compared with healthy controls, suggesting that NLRP1 can act as a key regulator of colon homeostasis (41). Additionally, a biological analysis of lung adenocarcinoma (LUAD) data from the TCGA and GEO databases found that the levels of NLRP1 mRNA were significantly lower in LUAD tissues than in non-cancerous lung tissues, and that LUAD progression was negatively associated with NLRP1 expression, in terms of clinical features such as tumor pathological stage, lymph node metastasis, and primary tumor status. In addition, NLRP1 has been positively correlated with the degree of infiltration of tumor-infiltrating immune cells (40). Similarly, the NLRC4 inflammasome can also inhibit the occurrence of colorectal cancer by suppressing cell proliferation and promoting cell death (42). The NLRC4 inflammasome has also been found to enhance inflammatory signaling pathways in macrophages via non-inflammasome pathways and promote the production of IFN-γ in CD4+ and CD8+ T cells, thereby inhibiting the growth of melanoma in mice (44) (Figures 2D–I).
In addition to the NLR family, the AIM2 inflammasome, which recognizes DNA, was also shown to inhibit AOM-DSS-induced colorectal cancer and spontaneous colorectal carcinogenesis by impairing colon cancer stem cell proliferation and promoting cell death (43) (Figures 2J–N). AIM2 is an interferon-inducible protein and contains HIN-200, which can inhibit the cell cycle and hinder tumor growth. Studies have shown that AIM2 can inhibit cell proliferation and also promote apoptosis when expressed in large amounts by inducing the release of cytochrome c from the mitochondria to the cytoplasm. In breast cancer, overexpression of AIM2 can decrease the expression of the anti-apoptotic protein Bcl-xl, increase the expression of the pro-apoptotic Bax, and induce the cleavage of the DNA repair protein PARP, thus leading to breast cancer cell apoptosis and the inhibition of breast cancer progression (45). Compared with the terminally differentiated, normal, cancer-free tissues, the expression of AIM2 in hepatocellular carcinoma tissues was significantly reduced, and AIM2 expression was negatively correlated with tumor burden. AIM2 may inhibit hepatocarcinogenesis in immunocompromised nude mice by suppressing the mTOR-S6K1 signaling pathway. Studies involving AIM2 silencing and overexpression in hepatocellular carcinoma cells further revealed that AIM2 exerts anti-tumor effects through inflammasome activity leading to pyroptosis (46).
Hence, inflammasomes can inhibit tumor occurrence and development in certain types of cancer. These tumor suppressive activities depend on the ability of the inflammasome to produce and regulate cytokines, especially those that affect T-cell activity and cell proliferation and maturation.
However, despite their tumor-suppressive effects in some contexts, it has also been shown that inflammasomes can contribute to tumor development by influencing host tumor immunity, promoting tumor cell proliferation and differentiation, and regulating the tumor microenvironment.
The tumor microenvironment includes proliferating tumor parenchymal cells, vascular endothelial cells, inflammatory infiltrating cells, and associated stromal cells, as well as their secreted proteins and cytokines. Among these, pro-inflammatory immune cells are a key component and play an important role in tumorigenesis, proliferation, progression, invasion, and metastasis (61). Myeloid-derived suppressor cells (MDSCs), a key component of the tumor microenvironment (TME), produce anti-inflammatory cytokines and encourage Treg proliferation, both of which have strong immunosuppressive activity. In mouse models of sarcoma and metastatic melanoma, activated NLRP3 inflammasome promotes tumorigenesis by suppressing NK cell and T cell activity and recruiting MDSCs and Tregs. Notably, in NLRP3-/- mice, a 5-fold reduction in the number and activity of MDSCs was found, suggesting that NLRP3 affects tumorigenesis by modulating host immunity (47). Tumor-associated macrophages (TAMs) are among the immune cells that infiltrate into tumors and play an important role in tumor lymphangiogenesis and proliferation, TAMs are highly aggregated in the tumor microenvironment of non-small cell lung cancer (NSCLC). The NLRP3 inflammasome in the cytoplasm of lung TAMs initiates TLR4/caspase-1 and TLR4/caspase-11 signaling pathways through TLR4, which are involved in the release of IL-1β and IL-1α, and promote lung tumor formation (49). Ershaid et al. found that cancer-associated fibroblasts can sense DAMPs and activate the NLRP3 inflammasome, which in turn triggers IL-1β secretion and further promotes the growth and metastasis of mouse and human breast cancer cells. However, when NLRP3 or IL-1β is silenced, the tumor-promoting effect is impaired. These results suggest that NLRP3 can induce the growth, metastasis and invasion of breast cancer and increase its incidence and development (51) (Figures 3A–D).
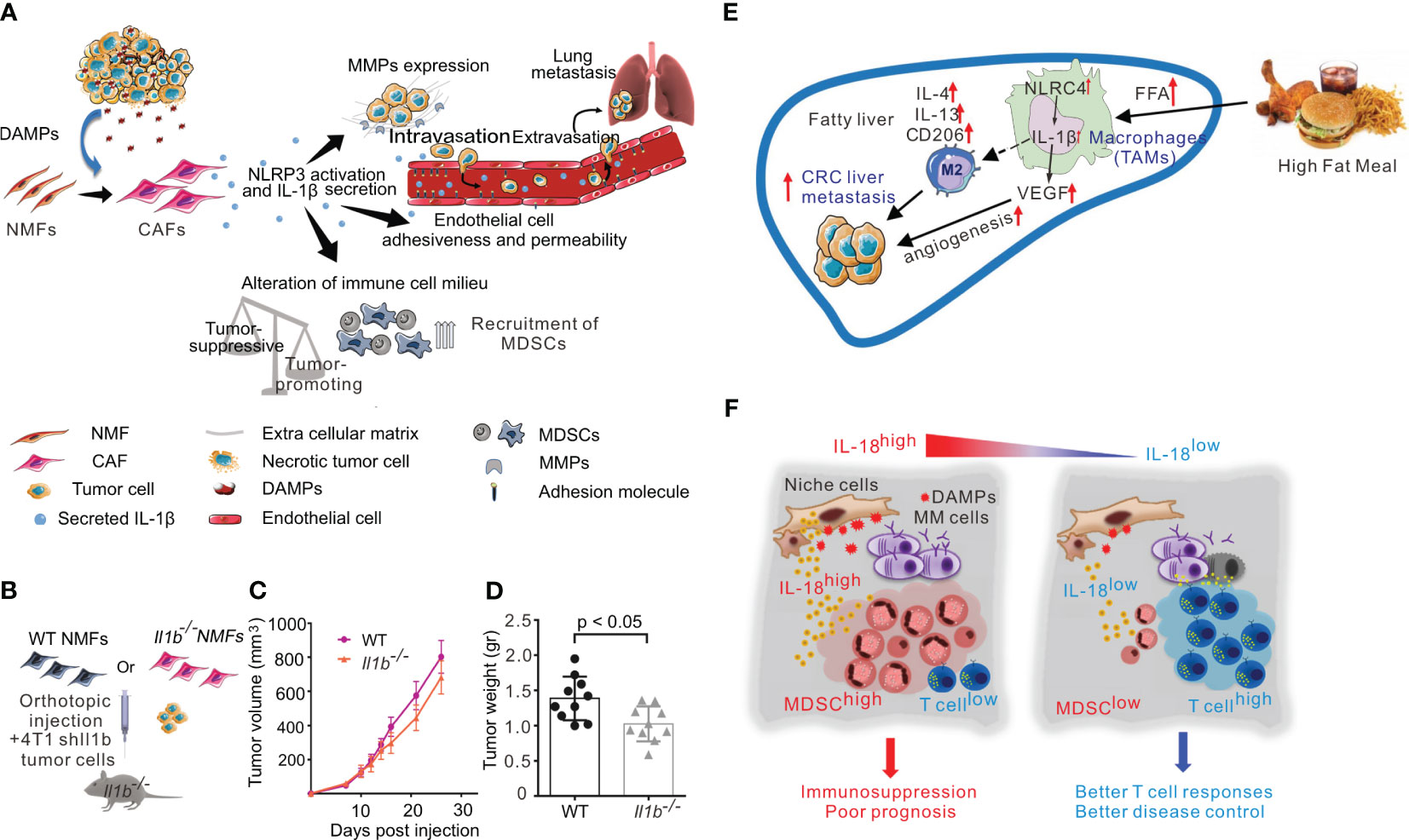
Figure 3 Inflammasome in tumor promotion. (A) Activation of the NLRP3 inflammasome in cancer-associated fibroblasts links tissue damage with tumor-promoting inflammation in breast cancer progression and metastasis. (B–D): CAF-derived IL-1β facilitates lung metastasis. (B)Scheme of experiments analyzed in B-D. shIl1b 4T1 mammary carcinoma cells were orthotopically coinjected with WT NMFs (Il1b+/+) or with Il1b−/− NMFs into the right inguinal mammary glands of Il1b−/− female mice. (C) Growth curves of 4T1 tumors injected with WT NMFs (Il1b+/+) or with Il1b−/− NMFs. n = 10 mice per group. (D)Tumor weight at termination of experiment. n = 10 mice per group. (E) In NAFLD, NLRC4 contributes to M2 polarization, IL-1β, and VEGF production in TAMs, which promote metastatic liver tumor growth. (F) The pro-inflammatory cytokine IL-18 was critically involved in MM-associated inflammation and immunosuppression, and dysregulated IL-18 was a potential therapeutic target in the MM niche. The figures A-D are reprinted with permission from Ref (51). Copyright Nature Publishing Group. The figure E is reprinted with permission from Ref (62). Copyright WILEY. The figure F is reprinted with permission from Ref (55). Copyright Cell Press.
Obesity is known to increase the risk of cancer, and inflammasome activity may partially account for this observation. Kolb et al. found that NLRC4 inflammasome/IL-1β signaling promotes the progression of breast cancer. In obesity, the TME causes NLRC4 inflammasome activation in TAMs, leading the production of the pro-inflammatory cytokine IL-1β, which then promotes the angiogenesis of trophoblast tumor cells through adipocyte-mediated vascular endothelial growth factor A (VEGFA) expression (52). In a mouse model of non-alcoholic steatohepatitis (NASH), NLRC4 knockout promoted the growth and recurrence of liver metastases of colorectal cancer, but significantly restricted hepatic tumorigenesis. NLRC4-/- mice with NAFLD also had decreased TAMs and decreased IL-1β and VEGF expression, suggesting that NLRC4 plays a key role in the growth and recurrence of liver metastases in the context of both NAFLD and colorectal cancer (62) (Figure 3E).
NRLP1 has been implicated in melanoma, with one study examining 216 melanoma tissue samples and 13 human melanoma cell lines to report that NLRP1 expression was elevated in melanoma. Meanwhile, knockdown of NLRP1 in human metastatic melanoma cells decreased caspase-1 activity, IL-1β secretion and NF-κB activity. Futhermore, the activity of caspase-2 and caspase-9 were enhanced and apoptosis was promoted. Therefore, there is evidence to suggest that NLRP1 promotes melanoma growth by enhancing inflammasome activation and inhibiting apoptosis in melanoma cells (48). The NLRP1 inflammasome has also been studied in the TME of multiple myeloma, where NLRP1 increases the production of MDSCs by sensing disturbances in hematopoietic stress or cellular homeostasis and responding by secreting IL-18, which leads to accelerated tumor progression (54). High expression levels of NLRP1 have also been found in human breast cancer mcf-7 cells and breast cancer tissues, and were correlated with lymph node metastasis, tumor lymph node metastasis stage and Ki-67 levels. In nude mouse transplantation models of breast cancer, overexpression of NLRP1 was found to promote breast cancer migration, invasion and growth by inducing epithelial-mesenchymal transformation (53).
In addition to the NLR family inflammasomes, AIM2 has also been implicated in cancer growth. Dysregulation of pro-inflammatory cytokines in the lung is one of the most important causes of inflammatory diseases and NSCLC. In NSCLC cells, AIM2 overexpression has been shown to enhance cell viability and migration. AIM2 also played an oncogenic role to regulate the expression of the cell cycle proteins cyclinB1 and cDC2 through the AIM2/IL-1β/STAT3 signaling pathway, thereby regulating NSCLC cell cycle progression (50). In the context of skin cancer, primary and metastatic cutaneous squamous cell carcinoma (cSCC) were found to exhibit higher levels of AIM2 expression than normal skin. AIM2 inflammasome activity can cause cSCC cells to release indirect effector cytokines and increase the expression of cell cycle regulatory genes, such as CDK1, CDC7, and CCNA1. Consequently, cell death is inhibited and the proliferation rate is increased, leading to rapid tumor development. Conversely, AIM2 knockdown decreased CDK1 expression, tumor cell invasion, and metastatic potential, and inhibited the growth and angiogenesis of metastatic tumor cells. Therefore, AIM2 is likely to promote the occurrence and metastasis of cSCC and can be used as a therapeutic target for cSCC (55) (Figure 3F).
To sum up, the role of inflammasomes in tumor growth metastasis is bidirectional. The precise function of any one inflammasome in a specific context depends on a variety of factors, including the type of cancer and the route of inoculation.
In recent years, there have been major advances in cancer immunotherapy, which now involves multiple modes of action. Therapeutic strategies include targeting specific cells, regulating the TME, systemic administration of immunosuppressive cells, and influencing immune checkpoint inhibitors, among others (63). T cells and NK cells remain the primary targets for tumor treatment, as T cells can exhibit antigen-specific tumor cell-targeting and NK cells can kill both tumor cells and virally infected cells. Additionally, antigen-presenting cells (APCs) remain central to enhancing the anti-tumor response (64). Inflammasomes play an important role in the regulation of tumor immunity, as the inflammatory response can frequently stimulate the maturation and antigen presentation of APCs such as DCs. Hence, inflammasomes can be targeted or modulated to achieve tumor immunotherapy.
Chemotherapeutic drugs (such as anthracyclines and platinum drugs) selectively kill immunosuppressive cells such as Tregs, MDSCs and TAMs, similar to inflammasome activity, which also regulates the death of immunosuppressive cells (65). The NLRP3 inflammasome has been observed to be activated in DCs during chemotherapy and enhanced the anthracycline-induced anti-tumor immune response by secreting caspase-1 and IL-1β. However, the release of various DAMPs, including ATP, HMGB1 and IL-1α, from dead tumor cells activates the NLRP3 inflammasome in DCs, which can lead to resistance to chemotherapy and promote tumor growth (66). In addition, gemcitabine and 5-fluorouracil (5-FU) can trigger the release of cathepsin B in MDSCs and activate the NLRP3 inflammasome. Although the level of IL-1β that is subsequently produced cannot activate CD8+ T cells, they can induce CD4+ T cells to produce IL-17, thus promoting tumor growth and angiogenesis (67).
Recent studies have shown that the expression of NLRP3, AIM2 and caspase-1 was significantly increased in tumor specimens from melanoma patients who had been effectively treated with PD-1. Although this study cannot show a causal relationship between inflammasome-secreted IL-1 β or IL-18 and the efficacy of PD-1 treatment, it nevertheless provides observational evidence that NLRP3 expression was associated with increased numbers of CD8+ T cells and memory CD4+ T cells (68). After treatment with PD-1 inhibitors, in vivo and in vitro studies have shown that the activation of CD8+ T cells can cause a signaling cascade mediated by endogenous PD-L1/NLRP3 inflammasomes, resulting in the recruitment of polymorphonuclear-MDSCs into tumor tissues, thereby suppressing the anti-tumor immune response (69). TIM-3 is another emerging immune checkpoint molecule similar to PD-1/PD-L1 and CTLA-4. Specific knockdown of TIM-3 in DCs resulted in significant inhibition of tumor growth and improved the antigen-presenting ability of DCs. In addition, single-cell sequencing data showed that inflammasome-related genes such as AIM2, NLRP3, IL-1β and caspase1 were significantly enriched in TIM-3 knockout DCs. Changes in cellular function in the TME and altered molecular pathways in DCs suggested that TIM-3 deficiency activates inflammasomes in DCs, which maintains the proliferative activity of CD8+ T cells and increases their tumor-killing capacity (70).
The NLRP3 inflammasome can also impair the efficacy of anti-tumor vaccines. In a mouse model of melanoma, NLRP3-/- mice that were vaccinated with DC-based vaccines had significantly increased survival compared to wild-type controls, possibly due to the presence of fewer MDSCs in NLRP3-/- mice, which may have contributed to impaired tumorigenesis (47). Knockout of the NLRP3 inflammasome also induced NK cell infiltration, increased production of chemokines CCL5 and CXCL9, and enhanced anti-metastatic activity of NK cells. Therefore, targeted inhibition of inflammasome activation can reduce the number of immunosuppressive cells and thus inhibit tumor growth and metastasis.
Tumor progression is not solely dependent on the behavior of cancer cells, but regulated by a complex plethora of cellular and non-cellular signals from the inflammatory microenvironment. As discussed above, inflammasomes can promote carcinogenesis, but they are also an ideal target for the prevention and treatment of tumors. There are several approaches to target inflammasome activity, including blocking upstream signaling pathways, inhibiting inflammasome components and antagonizing the end products of inflammasome activation (71) (Table 2). However, the use of synthetic compounds or small molecule inhibitors that target inflammasomes and their components is limited by poor specificity and low efficiency. In clinical studies, inhibition of cytokine signaling has been the most successful approach. For example, monoclonal antibodies that block IL-1R (e.g., anakinra, rilonacept, canakinumab, and gevokizumab) have been used to impair IL-1α- and IL-1β-mediated signaling via IL-1R, for the prevention or treatment of multiple myeloma through inhibition of IL-1β-induced production of IL-6 (72).

Table 2 Inflammasomes inhibitors.
Avastin (bevacizumab) is a specific inhibitor of the P2X7 receptor, a trimeric ATP-gated channel essential for the NLRP3/caspase-1 cascade. By blocking NLRP3 signaling, Avastin effectively inhibits tumor growth. Avastin has been approved by the FDA for the treatment of breast, colon, and lung cancers. Avastin inhibits proliferation and anti-apoptotic gene expression in tumor cells, while also preventing the activation of the transcription factor NFATc1 and suppressing the production of vascular endothelial growth factor (VEGF) (73). Glibenclamide is another chemotherapy drug that influences NLRP3. Glibenclamide inhibits K+ influx and thereby suppresses the maturation of caspase-1 and pro-IL-1β in macrophages. In human trophoblast cells, glibenclamide acts downstream of the P2X7 receptor and upstream of NLRP3, effectively blocking the activation of the NLRP3 inflammasome (74).
Other inflammasome-targeting therapies in development include MCC950, a small molecule NLRP3 inflammasome inhibitor. MCC950 has been shown to prevent both typical and atypical NLRP3 inflammasome activation and the secretion of IL-1β in a mouse model of experimental autoimmune encephalitis (EAE). However, MCC950 not only alleviates the symptoms of EAE, but also inhibits tumor development, indicating that it may have potential use for the treatment of NLRP3 inflammasome-related cancer (75). For instance, Chen et al. showed that the application of MCC950 in a mouse model of head and neck squamous cell carcinoma (HNSCC) significantly reduced IL-1β production in tumors. MCC950 also reduced the number of MDSCs, Tregs, and TAMs, depleted PD-1+ and Tim3+ T cells, and increased the number of CD4+ and CD8+ T cells. These results suggest that the NLRP3/IL-1β pathway promotes the development of HNSCC and that modulating of the tumor microenvironment by targeting the NLRP3/IL-1β pathway is expected to be a new therapeutic approach for HNSCC (76). In addition, in mouse macrophages and DCs, microRNA-223 inhibits IL-1β secretion and NLRP3 expression by directly binding to the 3’ untranslated region (3’UTR) of the NLRP3 mRNA, thus suppressing NLRP3 inflammasome activation (77). Some other microRNAs, including microRNA-155, microRNA-377, and microRNA-133a-1, are also involved in NLRP3 inflammasome activation and are expected to exert inhibitory effects. Another small molecule inhibitor, andrographolide, slows down the development of colitis-associated colon cancer (CAC) in mice by mediating mitochondrial phagocytosis and inhibiting NLRP3 inflammasome activation in macrophages (78). Therefore, novel treatments that target inflammasome activation may have a role in tumor therapy.
To optimize drug delivery, advances in nanomaterials have demonstrated promising results in the field of biomedicine. Nano-drug delivery systems have been widely used in oncology and immunotherapy, as encapsulating drugs in nanoparticles can improve the stability of hydrophobic drugs and reduce toxicity (79). However, the size, shape, surface charge and other physicochemical properties of nanoparticles can affect the activation of inflammasomes. Lunov et al. found that amine-modified polystyrene nanoparticles (PS-NH2) can trigger the assembly of NLRP3 inflammasomes and promote the downstream release of pro-inflammatory factor IL-1β from human macrophages (80). Similarly, silica nanoparticles (SiNPs) have also been shown to activate NLRP3 inflammasomes in vitro, resulting in the production of pro-inflammatory cytokines IL-1β and IL-18 (81). Moreover, studies have shown that chiral adjuvants can activate the NLRP3 inflammasome pathway by specifically binding to G protein-coupled receptors on the surfaces of antigen-presenting cells. Consequently, cytokines such as IL-2, IL-4, IL-12, and IFN-γ are released, simultaneously inducing cellular and humoral immune responses to achieve lasting vaccine protection (82) (Figures 4C, D). Other researchers have proposed an anti-tumor vaccine based on a proton-driven deformable nano-delivery system, which not only efficiently delivers antigen peptides into the cytoplasm of immune cells, but also enhances the innate immune system by acting as an adjuvant to activate the NLRP3. Deformable nano-tumor vaccine delivery systems can effectively inhibit the growth of mouse melanoma (B16) and human papillomavirus (HPV) tumors. In addition, the combination of this deformable nano-vaccine and anti-PD-L1 antibody therapy could significantly inhibit tumor growth in tumor-bearing mice, with about half of the mice exhibiting complete tumor regression in vivo (83) (Figures 4A, B). However, despite the beneficial effects of NLRP3 inflammasome activation in anti-tumor vaccine therapy, the activation of NLRP3 inflammasomes by nanoparticles may also have adverse effects for cancer treatment due to the release of large quantities of pro-inflammatory cytokines (e.g., TGF-β). Cytokine treatment of human cervical cancer cells (Hela), alveolar epithelial adenocarcinoma cells (A549) and ovarian epithelial adenocarcinoma cells (SKOV3) has been shown to result in significantly increased levels of epithelial-mesenchymal transformation. In addition, spontaneous metastasis of subcutaneously implanted mouse lung squamous carcinoma cells (KLN 205) was observed after intravenous injection of nanoparticles into tumor-bearing mice, whereas no spontaneous metastasis occurred in the control group (84). To sum up, nanomaterials play an important role in the activation of inflammasomes. While multi-functional small molecular nanomedicines show strong potential for targeted therapy of malignant tumors, their application in clinical practice should be undertaken with caution.
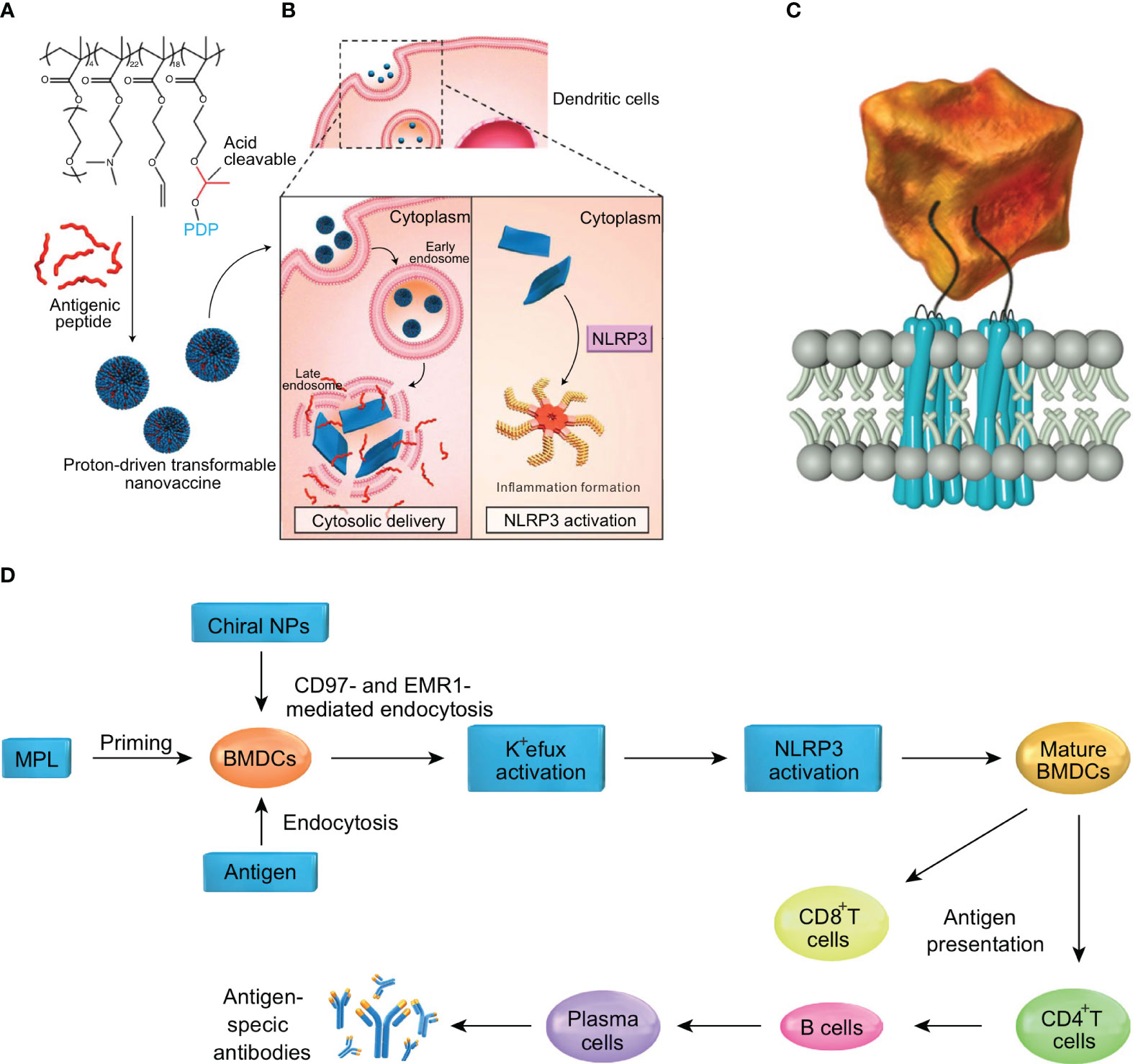
Figure 4 Nanoparticles by activating inflammasomes enhance antitumor immunity. (A) The nanotransformer-based vaccine (NTV) is composed of a polymer–peptide conjugate-based nanotransformer (NT) loaded with antigenic peptide (AP). The NTV has a spherical morphology with a diameter of about 100 nm at pH 7.4. (B) After the NTV is internalized by DCs, the acidic endosomal environment (pH 5.6) will trigger fast cleavage of the pyrene-conjugated D-peptide (PDP), which will then re-assemble into nanosheets with sizes in the range 5–8 μm. The morphological change leads to disruption of the endosomal membrane and delivery of AP into the cytosol. Moreover, the cytoplasmic nanosheets activate the NLRP3 inflammasome pathway, which promotes DC maturation and antigen processing. These two features contribute to the enhanced cross-presentation of AP to CD8+ T-cells and efficient antitumor immunity. (C) Diagram showing the interaction of chiral NPs with extracellular chiral chains of EGF-like domains on cellular AGPCR receptors. (D) The mechanism of induction of immune responses by chiral NPs. The figures A and B are reprinted with permission from Ref (83). Copyright John Wiley & Sons Inc. The figures C and D are reprinted with permission from Ref (82). Copyright Nature Publishing Group.
Inflammasomes can be divided into classical and non-classical categories. Classical inflammasomes require the participation of caspase-1, and include NLRP1, NLRP3, NLRC4, and AIM2, which are widely studied at present, among which the NLRP3 inflammasome is the most thoroughly studied. Non-classical inflammasomes are mainly dependent on caspase-4 or caspase-5, and there are few studies in this area, so further exploration is required. NLRP11 has been found to be an important component of the NLRP3 inflammasomes in human macrophages, which may have been overlooked in the past as mouse cells do not contain NLRP11 and the NBD structural domain between NLRP11 and NLRP3 does not bind specifically in other human cells such as HEK293 cells. NLRP11 interacts with NLRP3 and ASC to prevent the assembly of NLRP3 inflammasomes, the polymerization of NLRP3 and ASC, the activation of caspase-1, pyroptosis and the release of cytokines. NLRP11 knockout can specifically influence the activation of NLRP3 inflammasomes without affecting the activation of other inflammasomes (85), further indicating the complexity of the regulation of different inflammasome complexes. Hence, although inflammasomes are highly conserved in evolution, attention should be paid to their species-specificity.
The numerous pathways regulated by inflammasome activation are complex and diverse, pertaining to several aspects of cell function. First, multiple modalities of regulation are present simultaneously, such as post-transcriptional regulation and post-translational modification, including phosphorylation and ubiquitination. Second, the same mode of regulation may mediate completely opposite results. Furthermore, different regulatory proteins or small molecules may act on the same site of the inflammasome and exert the same or opposite effects. Finally, the same regulatory mechanism may act on different inflammasomes to produce different outcomes.
Future research will undoubtedly shed further light on the molecular and cellular mechanisms by which inflammasomes and immune cell function, which may provide new therapeutic strategies for cancer. Based on the close relationship between inflammasomes and tumors, targeting inflammasomes is likely to be a key strategy for improving cancer treatment. Inflammation may be targeted in cancer therapy by activating anti-cancer immune cells, such as DCs, NK cells, NKT cells, cytotoxic T cells, Th1 cells and B cells to enhance their anti-tumor activities. At the same time, immunotherapies should aim to suppress cancer-promoting immune cells such as mast cells, TAMs, MDSCs, eosinophils, Th2 cells, Th17 cells, Treg cells and Breg cells, or seek to polarize them into anti-tumor types by targeting key signaling pathways to prevent immunosuppressive effects and cancer progression. Further in-depth research is needed to determine the specific roles of DC subsets, macrophages and Treg cells in the activation of inflammasomes.
Considering the complex role of inflammasomes in cancer, it is necessary to study in more detail the role of different inflammasomes in different tissues, different types of cells and different stages of cancer, and explore the mechanism behind these different therapeutic effects. At the same time, to support more effective, personalized cancer treatment, future studies should seek to determine the best application timeline for each treatment model, in order to more precisely develop and prescribe drugs to individual cancer patients. Translational efforts may focus on small molecule drugs, as small molecule inhibitors that directly target inflammasomes are more potent and cost-effective compared to larger molecular biological agents, affording them greater application prospects due to their low concentration and low toxicity. Already, some drugs that target inflammasomes and their downstream effectors, such as MCC950, OLT1177, anakinra and CY-09, have achieved significant efficacy in the treatment of inflammatory diseases such as osteoarthritis, rheumatoid arthritis and inflammatory bowel disease. However, there are still few studies on the application of such drugs for tumors. Therefore, there is also a need to validate key molecular targets of inflammasome activation to design more selective inhibitors for tumor therapy. In addition, activated inflammasomes regulate tumor immunity by secreting the cytokines IL-1β and IL-18, and have shown good therapeutic effect in preclinical models. However, monotherapy strategies that aim to decrease inflammation are subject to the risk of promoting tumor growth. Because of the heterogeneity and plasticity of the TME, tumors can quickly evolve to evade monoclonal antibodies that target a single immune regulatory factor, leading to drug resistance and even promoting tumor progression. In future, the comprehensive use of high-resolution technology, such as multi-omics analysis and single-cell technology, will assist in the identification of more specific inflammasome targets and the exploration of the cellular and molecular levels of the local treatment response. Detailed data on inflammasome activity will help to translate anti-inflammatory therapies into clinical practice, to better improve the quality of life and prolong the survival of cancer patients. Ideal future oncology treatments may include individualized, multi-drug combinations of anti-inflammasome therapies.
Overall, inflammasomes play a nuanced and central role in regulating the tumor microenvironment, with effects on both tumor and immune cells. Given this, inflammasomes and inflammasome-related molecules are potential drug targets for cancer therapy. However, as inflammasomes partake in diverse and complex effects in response to a plethora of different signals, further research is required to identify effective, safe, and appropriate targets for therapeutic development.
WH and ZD selected the topic. ZD and LL reviewed all published articles and posters, and wrote the manuscript. HJ and XS provided some revising suggestions on the Discussion section. WH, BL, and HJ revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the Zhongnan Hospital of Wuhan University Medical Science and Technology Innovation Platform Construction Support Project (Grant No. PTXM2021019), Translational Medicine and Interdisciplinary Research Joint Fund of Zhongnan Hospital of Wuhan University (Grant No. ZNJC202015), the National Natural Science Foundation of China (No. 82102900).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
2. Balkwill F, Mantovani A. Inflammation and cancer: back to virchow? Lancet (9255) 2001:539–45:357. doi: 10.1016/S0140-6736(00)04046-0
3. Place DE, Kanneganti TD. Recent advances in inflammasome biology. Curr Opin Immunol (2018) 50:32–8. doi: 10.1016/j.coi.2017.10.011
4. Franchi L, Warner N, Viani K, Nuñez G. Function of nod-like receptors in microbial recognition and host defense. Immunol Rev (2009) 227(1):106–28. doi: 10.1111/j.1600-065X.2008.00734.x
5. Franchi L, Muñoz-Planillo R, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol (2012) 13(4):325–32. doi: 10.1038/ni.2231
6. Cao X, Xu J. Insights into inflammasome and its research advances in cancer. Tumori (2019) 105(6):456–64. doi: 10.1177/0300891619868007
7. Thi HTH, Hong S. Inflammasome as a therapeutic target for cancer prevention and treatment. J Cancer Prev (2017) 22(2):62–73. doi: 10.15430/JCP.2017.22.2.62
8. Karki R, Kanneganti TD. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat Rev Cancer (2019) 19(4):197–214. doi: 10.1038/s41568-019-0123-y
9. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10(2):417–26. doi: 10.1016/S1097-2765(02)00599-3
10. Gross O, Poeck H, Bscheider M, Dostert C, Hannesschläger N, Endres S, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature (2009) 459(7245):433–6. doi: 10.1038/nature07965
11. Bae JY, Lee SW, Shin YH, Lee JH, Jahng JW, Park K. P2X7 receptor and NLRP3 inflammasome activation in head and neck cancer. Oncotarget (2017) 8(30):48972–82. doi: 10.18632/oncotarget.16903
12. Li Y, Huang H, Liu B, Zhang Y, Pan X, Yu XY, et al. Inflammasomes as therapeutic targets in human diseases. Signal Transduct Target Ther (2021) 6(1):247. doi: 10.1038/s41392-021-00650-z
13. Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature (2012) 481(7381):278–86. doi: 10.1038/nature10759
14. Kaplanski G. Interleukin-18: biological properties and role in disease pathogenesis. Immunol Rev (2018) 281(1):138–53. doi: 10.1111/imr.12616
15. Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in muckle-wells autoinflammatory disorder. Immunity (2004) 20(3):319–25. doi: 10.1016/S1074-7613(04)00046-9
16. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell (2014) 156(6):1193–206. doi: 10.1016/j.cell.2014.02.008
17. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature (2006) 440(7081):228–32. doi: 10.1038/nature04515
18. Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem (2006) 281(48):36560–8. doi: 10.1074/jbc.M607594200
19. Muruve DA, Pétrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature (2008) 452(7183):103–7. doi: 10.1038/nature06664
20. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol (2008) 9(8):857–65. doi: 10.1038/ni.1636
21. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol (2010) 11(2):136–40. doi: 10.1038/ni.1831
22. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin d for non-canonical inflammasome signalling. Nature (2015) 526(7575):666–71. doi: 10.1038/nature15541
23. Zhu F, Ma J, Li W, Liu Q, Qin X, Qian Y, et al. The orphan receptor Nur77 binds cytoplasmic LPS to activate the non-canonical NLRP3 inflammasome. Immunity (2023) 56(4):753–67.e8. doi: 10.1016/j.immuni.2023.03.003
24. Li Y, Fu TM, Lu A, Witt K, Ruan J, Shen C, et al. Cryo-EM structures of ASC and NLRC4 CARD filaments reveal a unified mechanism of nucleation and activation of caspase-1. Proc Natl Acad Sci U S A (2018) 115(43):10845–52. doi: 10.1073/pnas.1810524115
25. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140(6):821–32. doi: 10.1016/j.cell.2010.01.040
26. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature (2011) 477(7366):592–5. doi: 10.1038/nature10394
27. Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med (2016) 213(10):2113–28. doi: 10.1084/jem.20151613
28. Bauernfried S, Scherr MJ, Pichlmair A, Duderstadt KE, Hornung V. Human NLRP1 is a sensor for double-stranded RNA. Science (2021) 371(6528). doi: 10.1126/science.abd0811
29. Gong Q, Robinson K, Xu C, Huynh PT, Chong KHC, Tan EYJ, et al. Structural basis for distinct inflammasome complex assembly by human NLRP1 and CARD8. Nat Commun (2021) 12(1):188. doi: 10.1038/s41467-020-20319-5
30. Chavarría-Smith J, Vance RE. The NLRP1 inflammasomes. Immunol Rev (2015) 265(1):22–34. doi: 10.1111/imr.12283
31. Nozaki K, Li L, Miao EA. Innate sensors trigger regulated cell death to combat intracellular infection. Annu Rev Immunol (2022) 40:469–98. doi: 10.1146/annurev-immunol-101320-011235
32. Planès R, Pinilla M, Santoni K, Hessel A, Passemar C, Lay K, et al. Human NLRP1 is a sensor of pathogenic coronavirus 3CL proteases in lung epithelial cells. Mol Cell (2022) 82(13):2385–400.e9. doi: 10.1016/j.molcel.2022.04.033
33. Wang B, Tian Y, Yin Q. AIM2 inflammasome assembly and signaling. Adv Exp Med Biol 2019 (1172) 143–55. doi: 10.1007/978-981-13-9367-9_7
34. Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L, et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity (2012) 36(4):561–71. doi: 10.1016/j.immuni.2012.02.014
35. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458(7237):514–8. doi: 10.1038/nature07725
36. Lee S, Karki R, Wang Y, Nguyen LN, Kalathur RC, Kanneganti TD. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature (2021) 597(7876):415–9. doi: 10.1038/s41586-021-03875-8
37. Ratsimandresy RA, Chu LH, Khare S, Almeida L, Gangopadhyay A, Indramohan M, et al. The PYRIN domain-only protein POP2 inhibits inflammasome priming and activation. Nat Commun (2017) 8:15556. doi: 10.1038/ncomms15556
38. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity (2010) 32(3):379–91. doi: 10.1016/j.immuni.2010.03.003
39. Dupaul-Chicoine J, Yeretssian G, Doiron K, Bergstrom KS, McIntire CR, LeBlanc PM, et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity (2010) 32(3):367–78. doi: 10.1016/j.immuni.2010.02.012
40. Shen E, Han Y, Cai C, Liu P, Chen Y, Gao L, et al. Low expression of NLRP1 is associated with a poor prognosis and immune infiltration in lung adenocarcinoma patients. Aging (Albany NY) (2021) 13(5):7570–88. doi: 10.18632/aging.202620
41. Williams TM, Leeth RA, Rothschild DE, Coutermarsh-Ott SL, McDaniel DK, Simmons AE, et al. The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis. J Immunol (2015) 194(7):3369–80. doi: 10.4049/jimmunol.1402098
42. Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci U S A (2010) 107(50):21635–40. doi: 10.1073/pnas.1016814108
43. Wilson JE, Petrucelli AS, Chen L, Koblansky AA, Truax AD, Oyama Y, et al. Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and akt. Nat Med (2015) 21(8):906–13. doi: 10.1038/nm.3908
44. Janowski AM, Colegio OR, Hornick EE, McNiff JM, Martin MD, Badovinac VP, et al. NLRC4 suppresses melanoma tumor progression independently of inflammasome activation. J Clin Invest (2016) 126(10):3917–28. doi: 10.1172/JCI86953
45. Liu ZY, Yi J, Liu FE. The molecular mechanism of breast cancer cell apoptosis induction by absent in melanoma (AIM2). Int J Clin Exp Med (2015) 8(9):14750–8.
46. Schneider KS, Thomas CJ, Groß O. Inflammasome activation and inhibition in primary murine bone marrow-derived cells, and assays for IL-1α, IL-1β, and caspase-1. Methods Mol Biol (2013) (1040) 117–35. doi: 10.1007/978-1-62703-523-1_10
47. van Deventer HW, Burgents JE, Wu QP, Woodford RM, Brickey WJ, Allen IC, et al. The inflammasome component NLRP3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer Res (2010) 70(24):10161–9. doi: 10.1158/0008-5472.CAN-10-1921
48. Zhai Z, Liu W, Kaur M, Luo Y, Domenico J, Samson JM, et al. NLRP1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene (2017) 36(27):3820–30. doi: 10.1038/onc.2017.26
49. Terlizzi M, Colarusso C, Popolo A, Pinto A, Sorrentino R. IL-1α and IL-1β-producing macrophages populate lung tumor lesions in mice. Oncotarget (2016) 7(36):58181–92. doi: 10.18632/oncotarget.11276
50. Zhang M, Jin C, Yang Y, Wang K, Zhou Y, Zhou Y, et al. AIM2 promotes non-small-cell lung cancer cell growth through inflammasome-dependent pathway. J Cell Physiol (2019) 234(11):20161–73. doi: 10.1002/jcp.28617
51. Ershaid N, Sharon Y, Doron H, Raz Y, Shani O, Cohen N, et al. NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat Commun (2019) 10(1):4375. doi: 10.1038/s41467-019-12370-8
52. Kolb R, Phan L, Borcherding N, Liu Y, Yuan F, Janowski AM, et al. Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat Commun (2016) 7:13007. doi: 10.1038/ncomms13007
53. Kesavardhana S, Kanneganti TD. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int Immunol (2017) 29(5):201–10. doi: 10.1093/intimm/dxx018
54. Nakamura K, Kassem S, Cleynen A, Chrétien ML, Guillerey C, Putz EM, et al. Dysregulated IL-18 is a key driver of immunosuppression and a possible therapeutic target in the multiple myeloma microenvironment. Cancer Cell (2018) 33(4):634–48.e5. doi: 10.1016/j.ccell.2018.02.007
55. Farshchian M, Nissinen L, Siljamäki E, Riihilä P, Piipponen M, Kivisaari A, et al. Tumor cell-specific AIM2 regulates growth and invasion of cutaneous squamous cell carcinoma. Oncotarget (2017) 8(28):45825–36. doi: 10.18632/oncotarget.17573
56. Zitvogel L, Kepp O, Galluzzi L, Kroemer G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol (2012) 13(4):343–51. doi: 10.1038/ni.2224
57. Moossavi M, Parsamanesh N, Bahrami A, Atkin SL, Sahebkar A. Role of the NLRP3 inflammasome in cancer. Mol Cancer (2018) 17(1):158. doi: 10.1186/s12943-018-0900-3
58. Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature (2012) 491(7423):259–63. doi: 10.1038/nature11535
59. Lee PH, Yamamoto TN, Gurusamy D, Sukumar M, Yu Z, Hu-Li J, et al. Host conditioning with IL-1β improves the antitumor function of adoptively transferred T cells. J Exp Med (2019) 216(11):2619–34. doi: 10.1084/jem.20181218
60. Jiao S, Subudhi SK, Aparicio A, Ge Z, Guan B, Miura Y, et al. Differences in tumor microenvironment dictate T helper lineage polarization and response to immune checkpoint therapy. Cell (2019) 179(5):1177–90.e13. doi: 10.1016/j.cell.2019.10.029
61. Terme M, Ullrich E, Aymeric L, Meinhardt K, Desbois M, Delahaye N, et al. IL-18 induces PD-1-dependent immunosuppression in cancer. Cancer Res (2011) 71(16):5393–9. doi: 10.1158/0008-5472.CAN-11-0993
62. Ohashi K, Wang Z, Yang YM, Billet S, Tu W, Pimienta M, et al. NOD-like receptor C4 inflammasome regulates the growth of colon cancer liver metastasis in NAFLD. Hepatology (2019) 70(5):1582–99. doi: 10.1002/hep.30693
63. Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol (2020) 17(8):807–21. doi: 10.1038/s41423-020-0488-6
64. Dong Y, Wan Z, Gao X, Yang G, Liu L. Reprogramming immune cells for enhanced cancer immunotherapy: targets and strategies. Front Immunol (2021) 12:609762. doi: 10.3389/fimmu.2021.609762
65. Locher C, Conforti R, Aymeric L, Ma Y, Yamazaki T, Rusakiewicz S, et al. Desirable cell death during anticancer chemotherapy. Ann N Y Acad Sci 2010. (1209) 99–108. doi: 10.1111/j.1749-6632.2010.05763.x
66. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med (2009) 15(10):1170–8. doi: 10.1038/nm.2028
67. Bruchard M, Ghiringhelli F. [Impact of chemotherapies on immunosuppression and discovery of new therapeutic targets]. Bull Cancer (2014) 101(6):605–7. doi: 10.1684/bdc.2014.1936
68. Segovia M, Russo S, Jeldres M, Mahmoud YD, Perez V, Duhalde M, et al. Targeting TMEM176B enhances antitumor immunity and augments the efficacy of immune checkpoint blockers by unleashing inflammasome activation. Cancer Cell (2019) 35(5):767–81.e6. doi: 10.1016/j.ccell.2019.04.003
69. Theivanthiran B, Haykal T, Cao L, Holtzhausen A, Plebanek M, DeVito NC, et al. Overcoming immunotherapy resistance by targeting the tumor-intrinsic NLRP3-HSP70 signaling axis. Cancers (Basel) (2021) 13(19):4753. doi: 10.3390/cancers13194753
70. Dixon KO, Tabaka M, Schramm MA, Xiao S, Tang R, Dionne D, et al. TIM-3 restrains anti-tumour immunity by regulating inflammasome activation. Nature (2021) 595(7865):101–6. doi: 10.1038/s41586-021-03626-9
71. Lee C, Do HTT, Her J, Kim Y, Seo D, Rhee I. Inflammasome as a promising therapeutic target for cancer. Life Sci (2019) 231:116593. doi: 10.1016/j.lfs.2019.116593
72. Lust JA, Lacy MQ, Zeldenrust SR, Witzig TE, Moon-Tasson LL, Dinarello CA, et al. Reduction in c-reactive protein indicates successful targeting of the IL-1/IL-6 axis resulting in improved survival in early stage multiple myeloma. Am J Hematol (2016) 91(6):571–4. doi: 10.1002/ajh.24352
73. Adinolfi E, Raffaghello L, Giuliani AL, Cavazzini L, Capece M, Chiozzi P, et al. Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res (2012) 72(12):2957–69. doi: 10.1158/0008-5472.CAN-11-1947
74. Tamura K, Ishikawa G, Yoshie M, Ohneda W, Nakai A, Takeshita T, et al. Glibenclamide inhibits NLRP3 inflammasome-mediated IL-1β secretion in human trophoblasts. J Pharmacol Sci (2017) 135(2):89–95. doi: 10.1016/j.jphs.2017.09.032
75. Perera AP, Fernando R, Shinde T, Gundamaraju R, Southam B, Sohal SS, et al. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci Rep (2018) 8(1):8618. doi: 10.1038/s41598-018-26775-w
76. Chen L, Huang CF, Li YC, Deng WW, Mao L, Wu L, et al. Blockage of the NLRP3 inflammasome by MCC950 improves anti-tumor immune responses in head and neck squamous cell carcinoma. Cell Mol Life Sci (2018) 75(11):2045–58. doi: 10.1007/s00018-017-2720-9
77. Bauernfeind F, Rieger A, Schildberg FA, Knolle PA, Schmid-Burgk JL, Hornung V. NLRP3 inflammasome activity is negatively controlled by miR-223. J Immunol (2012) 189(8):4175–81. doi: 10.4049/jimmunol.1201516
78. Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol (2015) 6:262. doi: 10.3389/fphar.2015.00262
79. Ali ES, Sharker SM, Islam MT, Khan IN, Shaw S, Rahman MA, et al. Targeting cancer cells with nanotherapeutics and nanodiagnostics: current status and future perspectives. Semin Cancer Biol (2021) 69:52–68. doi: 10.1016/j.semcancer.2020.01.011
80. Lunov O, Syrovets T, Loos C, Nienhaus GU, Mailänder V, Landfester K, et al. Amino-functionalized polystyrene nanoparticles activate the NLRP3 inflammasome in human macrophages. ACS Nano (2011) 5(12):9648–57. doi: 10.1021/nn203596e
81. Gómez DM, Urcuqui-Inchima S, Hernandez JC. Silica nanoparticles induce NLRP3 inflammasome activation in human primary immune cells. Innate Immun (2017) 23(8):697–708. doi: 10.1177/1753425917738331
82. Xu L, Wang X, Wang W, Sun M, Choi WJ, Kim JY, et al. Enantiomer-dependent immunological response to chiral nanoparticles. Nature (2022) 601(7893):366–73. doi: 10.1038/s41586-021-04243-2
83. Gong N, Zhang Y, Teng X, Wang Y, Huo S, Qing G, et al. Proton-driven transformable nanovaccine for cancer immunotherapy. Nat Nanotechnol (2020) 15(12):1053–64. doi: 10.1038/s41565-020-00782-3
84. Manshian BB, Poelmans J, Saini S, Pokhrel S, Grez JJ, Himmelreich U, et al. Nanoparticle-induced inflammation can increase tumor malignancy. Acta Biomater (2018) 68:99–112. doi: 10.1016/j.actbio.2017.12.020
Keywords: inflammasome, cancer, immunology, immunotherapy, nanoparticle agents
Citation: Deng Z, Lu L, Li B, Shi X, Jin H and Hu W (2023) The roles of inflammasomes in cancer. Front. Immunol. 14:1195572. doi: 10.3389/fimmu.2023.1195572
Received: 28 March 2023; Accepted: 26 June 2023;
Published: 11 July 2023.
Edited by:
Elisa Wirthgen, University Hospital Rostock, GermanyReviewed by:
Iman Mamdouh Talaat, University of Sharjah, United Arab EmiratesCopyright © 2023 Deng, Lu, Li, Shi, Jin and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weidong Hu, aHV3ZEB3aHUuZWR1LmNu; Honglin Jin, amluQG1haWwuaHphdS5lZHUuY24=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.