 Xiurong Cai
Xiurong Cai Frank Tacke
Frank Tacke Adrien Guillot
Adrien Guillot Hanyang Liu
Hanyang Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 16 May 2023
Sec. Cytokines and Soluble Mediators in Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1192840
This article is part of the Research Topic Community Series in Hepatic Immune Response underlying Liver Cirrhosis and Portal Hypertension, volume II View all 14 articles
The biliary epithelial cells, also known as cholangiocytes, line the intra- and extrahepatic bile ducts, forming a barrier between intra- and extra-ductal environments. Cholangiocytes are mostly known to modulate bile composition and transportation. In hepatobiliary diseases, bile duct injury leads to drastic alterations in cholangiocyte phenotypes and their release of soluble mediators, which can vary depending on the original insult and cellular states (quiescence, senescence, or proliferation). The cholangiocyte-secreted cytokines (also termed cholangiokines) drive ductular cell proliferation, portal inflammation and fibrosis, and carcinogenesis. Hence, despite the previous consensus that cholangiocytes are bystanders in liver diseases, their diverse secretome plays critical roles in modulating the intrahepatic microenvironment. This review summarizes recent insights into the cholangiokines under both physiological and pathological conditions, especially as they occur during liver injury-regeneration, inflammation, fibrosis and malignant transformation processes.
Cholangiocytes, also known as biliary epithelial cells (BECs), are specialized epithelial cells forming the biliary epithelium and lining the bile ducts (1). In general, cholangiocytes are polarized with apical and basal membranes corresponding to different functions: 1) maintain bile flow via the cilium system and intraductal homeostasis via active biomolecule transport; 2) modify bile via secreting bicarbonate (HCO3−) through the plasma membrane domain; 3) maintain cross-ductal interaction in the liver, depending on their tight junctions and immunoglobulin A (IgA) secretion; 4) reabsorb different molecules, including bile salts, bile acids, glucose, amino acids and ions (2–4). Cholangiocytes represent a heterogeneous population in terms of morphological characteristics, classically described as small or large cholangiocytes (5). Accordingly, cholangiocyte transcriptome is highly variable, so as their structural and metabolic functions. Large cholangiocytes typically line the larger branches of the biliary tree and form more complex structures than those small cholangiocytes. Simultaneously, large cholangiocytes engage in hormone-modulated bile secretion, while small cholangiocytes are able to proliferate and exhibit functional plasticity in diseases (6–8). Small cholangiocytes appear more capable of self-replication during liver injury, implying their potential in the liver regeneration and ductular reaction (DR) (9). DR is described as a complex of dynamic interactions among liver parenchymal cells, stromal cells, and immune cells, which serves a crucial machinery during liver injury-regeneration, fibrogenesis, and malignant transformation processes. Though not affirmatively being recognized as the origins of DR, cholangiocytes participate in DR as both initiators and executors (10).
To date, cholangiocyte biology has been merely studied in liver diseases, due to their relatively small population in the liver. However, a rising number of studies unveiled crucial functions of cholangiocytes in liver pathobiology. Interacting with both intra- and extrahepatic ductal environments, cholangiocytes are exposed to both hepatic molecules and gut-derived stimuli [pathogen-associated molecular patterns (PAMPs), danger-associated molecular patterns (DAMPs) and microorganisms] (11, 12). Cholangiocytes have been identified as collateral targets of various liver diseases such as fatty liver disease [nonalcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH)] and alcohol-related liver disease (ALD). BECs are also directly injured in chronic cholestatic liver diseases including primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), biliary atresia (BA) and cholangiocarcinoma (13, 14). Furthermore, stimulated cholangiocytes can adopt varying secretory phenotypes. Importantly, bile duct-derived ductular cells are considered to play active roles in liver regeneration, although contradictory results suggest the necessity of a more comprehensive analysis of their function. More interestingly, activated cholangiocytes exhibit a peculiar secretory phenotype that dramatically shapes their surrounding microenvironment by modulating immune cell recruitment and mesenchymal cell migration and activation (15). From our current understanding, the release of cholangiokines (cholangiocyte-secreted cytokines, including chemokines, growth factors, ect.) is associated with cell statuses, which are affected by tissue inflammation, infection, and metabolic dysregulations. Both acute and chronic liver disorders have been shown to alter the BEC secretory profiles (16–18). Consequently, elevating attention has been given to cholangiokines in the liver, which inspires more work on obtaining in-depth and systematic understandings.
More than constituents of bile ducts, cholangiocytes play a critical role in maintaining liver homeostasis, which refers to the balance of various physiological processes in the liver. One of the essential functions of cholangiocytes is to regulate biliary composition and bile flow by secreting and absorbing electrolytes, water, and other solutes. Standing to reason, cholangiocytes are vulnerable targets in cholangiopathies, which is a complex umbrella term encompassing inherited disorders, autoimmune or other poorly understood diseases (e.g., PSC, PBC and autoimmune cholangitis), exogenous stimuli-induced injury (e.g., infection and drug), ischemic injury and other undefined types of insults. Under such injury conditions, cholangiocytes assuasively secrete cholangiokines to sustain the microenvironment of the portal area. Moreover, cholangiocytes participate in the immune response and inflammation regulation through cytokine and chemokine production. Therefore, circulating immune cells are attracted and activated to promote portal inflammation (1, 19–22).
According to variable chronic liver injury mouse models, BECs actively interact with hepatocytes and liver progenitor cells (HPCs) to promote the DR, which ultimately constitutes an alternative liver regeneration process (23, 24). Synchronously, BECs have been determined to fuel DR by several cholangiokines (25, 26). Hence, this section will demonstrate intriguing secretory phenotypes that occur in cholangiocytes, triggered by a complex portal niche (Figure 1).
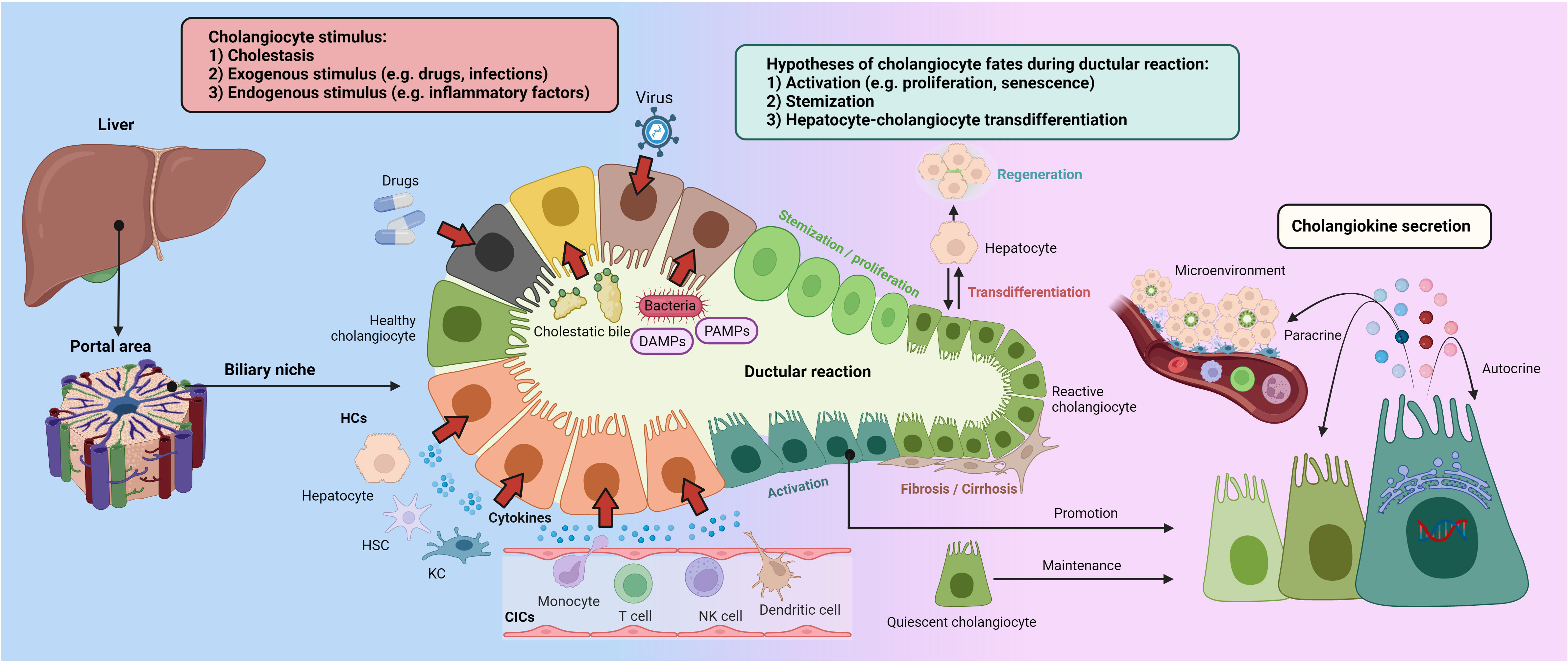
Figure 1 Ductular reaction and cholangiokine secretion Cholangiocytes stand as a crucial component in the hepatic portal areas, maintaining liver homeostasis. Cholangiocellular phenotypes can be altered by cholestasis, exogenous stimulus, and inflammatory factors. Besides evidence and debates on cholangiocyte stemization and hepatocyte-cholangiocyte trans-differentiation, activated cholangiocytes drive the DR by producing variable cytokines and chemokines, termed cholangiokines. Furthermore, cholangiokines are responsible for autocrine and paracrine effects in the portal microenvironment. CIC, circulating immune cell; DAMPs, damage-associated molecular patterns; HC, hepatic cell; HSC, hepatic stellate cell; KC, Kupffer cell; PAMPs, pathogen-associated molecular patterns.
Bile flow perturbation generally leads to an impaired bile efflux to the intestines, resulting in a pathogenic accumulation of bile acids in the intra-hepatic environment. Gradually concentrated and thickened bile exerts detrimental effects on the gut-liver axis, thus referred to as ‘toxic bile’ (27). Due to their anatomical location along the biliary tree, cholangiocytes are amongst the first cells to be affected by cholestasis. Studies have showed that higher levels of interleukin-8 (IL-8), a potent chemoattractant for neutrophils, were detected in the bile of PSC patients as compared to non-PSC patients (28, 29), which suggests that bile duct injury induces the secretion of inflammatory cytokines into the bile (30). Of note, the effects of the main bile salts, namely tauroursodeoxycholate (TUDC), taurocholate (TC), taurodeoxycholate (TDC), taurochenodeoxycholate (TCDC) and taurolithocholate (TLC), also remain to be defined. Results from Lamireau et al. indicated that TC effectively stimulated murine BECs to release monocyte chemoattractant protein-1/C-C motif chemokine ligand-2 (MCP-1/CCL-2) and IL-6 (31). In addition, oxysterols were revealed to insult cholangiocytes and induce malignancy transformation, which can be taken as a destructive part of ‘toxic bile’ (32–34).
Neuroendocrine hormones including secretin (Sct), are released by proliferating cholangiocytes (35, 36). Other than inducing biliary bicarbonate secretion by binding with its basolateral receptor (SR) (7, 37, 38), the Sct/SR axis plays a key role in the modulation of biliary proliferation and hepatic fibrosis by influencing the BEC secretome (35, 36, 39). The SR gene expression was shown to be elevated in the biliary obstruction animal model after bile duct ligation (BDL) (40). Furthermore, studies have shown that increased expression of vascular endothelial growth factor-A (VEGF-A) and transforming growth factor-beta 1 (TGF-β1) occurs when the Sct/SR axis is activated, leading to enhanced proliferation of ductular cells and fibrogenesis. Moreover, DR and liver fibrosis can be ameliorated when the SR expression was genetically disrupted in BDL and Mdr2−/− (Abcb4−/−) mouse models (35, 39). Additionally, increased activity of the Sct/SR/TGF-β1 axis was observed in the liver of PSC patients compared to healthy livers (39).
Furthermore, fibroblast growth factor 19 (FGF19) was found in the liver samples from patients with cholestasis (41). Exposure to FGF19 has been associated with the proliferation and IL-6 release of cholangiocytes (42). More importantly, human gallbladder cells secrete FGF19 into the bile, which is assumed to participate in cholangiopathies (43). Even though many other cell populations have been identified as sources of FGF19, it would be interesting to elucidate the functions of cholangiocyte-secreted FGF19, particularly in the hepatoportal regions (44).
It has been reported that BDL-induced bile duct obstruction in mice triggers cholangiocytes to secrete osteopontin (OPN) (45, 46). Additionally, nerve growth factor (NGF) was found to be secreted by cholangiocytes in an experimental mouse cholestasis model (47). TGR5, well-known as a G-protein-coupled bile acid receptor, is highly expressed on cholangiocytes and hepatic macrophages. It is postulated that TGR5 can participate in bile production, proliferation regulation and inflammation modulation. The beneficial secretion of bicarbonate and chloride was known attributing to the TGR5-mediated cholangiocyte activation (48). Furthermore, it is hypothesized that TGR5 might impede hepatic cell-cell communication, which either directly or indirectly affects the cholangiocyte-associated secretory characteristics. Nonetheless, the effects of TGR5 on the cholangiocyte secretome have already been discussed elsewhere (49).
Environmental factors (microorganisms, drugs, ischemia, etc.) serve a pivotal role in the cholangiocyte activation and pathogenesis of cholangiopathies. Notably, the microbiota has emerged as a crucial mediator of BEC functions (50). Although hepatocytes and Kupffer cells are generally responsible for the clearance of bacterial products in the liver (51, 52), cholangiocytes can also play auxiliary roles in this process, especially regarding the response to intraductal stimuli. In terms of physiology, the gut barrier stands as the first line of defense preventing external insults from entering the organism via the venous system, while bile duct acts as the front-line defending bile-derived insults (12). The portal channel, however, may allow certain bacteria, PAMPs, and DAMPs to enter the liver, affecting biliary inflammation or possibly inducing inflammation in the biliary tree. Paik et al. recently reported that gut-resident bacteria can inhibit Th-17 cell functions by producing bile acids (3-oxoLCA), which evidences a bacteria-induced immune turbulencein the gut-liver inflammatory modulation (53). Additionally, a growing number of studies have revealed the critical functions that gut microbiota plays in influencing the progression of liver disease, particularly in PSC and PBC (54–56). Indeed, PAMPs refluxed into the bile duct can be sensed by cholangiocytes via pattern recognition receptors, which can provoke a variety of inflammatory signaling pathways and cytokine secretion.
Fundamentally, cholangiocytes express the Toll-like receptor (TLR) family proteins, which are well-known as mediators in innate immune responses (57, 58). TLRs can recognize microbial and other exogenous molecules, PAMPs and DAMPs. Furthermore, the TLR activation induced by PAMPs and DAMPs triggering their conrrespondingsignaling pathways, results in the recruitment of toll/IL-1-domain containing adaptor molecules [e.g., myeloid differentiation protein 88 (MyD88)], and the activation of protein kinases [e.g., IL-1 receptor associated kinase (IRAK)]. The activation of these specific intracellular pathways leads to a nuclear factor kappa-B (NF-κB)-dependent secretion of proinflammatory cytokines/chemokines (59). Furthermore, cholangiocellular autocrine and paracrine signals are robustly enhanced by several cytokines including IL-1, IL-6, IL-8 and interferon-γ (IFN-γ) (60). Moreover, emerging studies have elucidated the mechanisms of microbiota implication on cholangiopathies. For instance, PBC patients were found to tolerate autoantibodies that can cross-react with bacterial antigens from E. coli and N. aromaticivorans (61). E. coli infection is known as a key factor in breaking immunological tolerance against the mitochondria, resulting in the production of PBC-specific autoantibodies (termed anti-mitochondrial autoantibodies) (62). These findings lend credence to prospective mechanisms underlying the secretome changes in association with cholangiocytes.
In the past three years, Coronavirus disease 2019 (COVID-19) has swept the world and brought new challenges to human diseases, leading to investigations and discussions on the COVID-19-interfered cholangiopathies (63, 64). A case report of COVID-19 patients discovered unique histologic features, including severe cholangiocyte injury and intrahepatic microangiopathy in their liver samples, suggesting a SARS-CoV-2-induced hepatic injury (64). Additionally, SARS-CoV-2 can infect cholangiocytes via the angiotensin-converting enzyme 2 (ACE2), which can be reduced by ursodeoxycholic acid while being induced by farnesoid X receptor (FXR) signaling in cholangiocytes (65).
Drug-induced cholangiopathies [also known as drug-induced vanishing bile duct syndrome (VBDS)] were first described in rare clinical cases (66). Certain medications, including carbamazepine and amoxicillin/clavulanic acid, have been shown to cause biliary damage (67). Additionally, fluorodeoxyuridines and 5-fluorouracil were revealed to selectively induce injuries in large bile ducts (68). Interestingly, cholangiocytes are implicated in drug metabolism as they were shown to express cytochrome P450 (CYP450) superfamily members (69, 70). Therefore, functional investigations linking drug metabolism or drug-induced liver damage to the secretory characteristics of cholangiocytes are highly anticipated.
Similar cholangiopathies with vanishing bile ducts, biliary strictures and protein casts also occur after ischemic insults, including ischemic-type biliary lesions (ITBL) after liver transplantation, secondary sclerosing cholangitis of critically ill patients (SC-CIP) after acute respiratory distress syndrome, COVID-19, shock and sepsis (71). Regrettably, most studies only investigated cellular injury or histological manifestations of such cholangiopathies without a detailed description of cholangiocyte-associated secretory phenotypes.
In comparison to injuries, endogenous stimulus, mainly inflammatory factors, play pivotal roles in modulating a variety of cholangiocyte phenotypes. In this context, cholangiocytes act as a major sensor rather than an initiator of inflammation, which possibly explains the general notion of cholangiopathies in most acute and chronic liver diseases (14).
When there is a disturbance in homeostasis, cholangiocytes are more susceptible to immunological responses, which enhances their secretion of cytokines including chemokines, and angiogenic growth factors. For instance, IL-1 and tumor necrosis factor-α (TNF-α) trigger cholangiocytes to release epithelial cell-derived neutrophil-activating protein (ENA-78) and growth-related gene products (72). Moreover, primary human cholangiocytes treated with cytokines (IL-1β, TNF-α and IL-17) or TLRs-related PAMPs [Pam3CSK4, poly(I:C) and LPS] can attract periductal Langerhans cells (Langerin+ periductal cells) via secreting the chemokine macrophage inflammatory protein-3α (MIP-3α) to activate PAMPs-sensing TLRs, thereby regulating biliary innate immune response in PBC (73). Poly (I:C)-treated primary cholangiocytes, mimicking biliary damage in BA, also trigger a stronger release of chemokine (C-X3-C motif) ligand 1 (CX3CL1) and the subsequent attraction of malfunctional natural killer (NK) cells (74). Other cytokines (IL-1β, IL-6, and IL-23p19) and chemokines [chemokine (C-XC-C motif) ligand (CXCL)-1/2/3/6/8, CCL-2 and CCL-20) were found enriched in the interlobular bile ducts from PBC patients, which was also confirmed in the in vitro stimulation of primary cholangiocytes with PAMPs [Pam3CSK4, poly(I:C) and LPS] and IL-17 (75).
During persistent liver injury, cholangiocytes synthesize and release TGF-β, especially TGF-β2, which was significantly increased in reactive bile ducts of fibrotic livers. In turn, TGF-β further promotes cholangiocytes to secrete endothelin-1 and regulates, in a paracrine manner, the deposition of extracellular matrix in the adjacent mesenchymal cells (76). Cholangiocytes appear to be responsive to IFN-γ, is mainly secreted by CD8+ T cells (77). IFN-γ was revealed to ameliorate fibrosis and cholestasis in carbon tetrachloride-treated mice (78). On the other hand, IFN-γ induces a shift of cytokine secretion in cholangiocytes from an acute inflammation pattern to a chronic inflammation feature, serving an important driver of persistent inflammation in cholangiopathies. Specifically, IFN-γ represses IL-8 secretion while enhancing the secretion of several cytokines including MCP-1 (79), monokine (80), interferon-inducible T cell alpha chemoattractant (ITAC) (81) and interferon-γ-inducible protein 10 (IP10) (82). Furthermore, IFN-γ and IL-6 stimulate nitric oxide (NO) production in cholangiocytes by inducing nitric oxide synthase-2 (NOS-2) expression (83). Besides, BECs exposed to IFN-γ exhibit a phenotypic flip between the acute and chronic inflammatory processes in terms of their release of inflammatory components (84).
IL-6, HGF and epidermal growth factors (EGF) can promote the proliferation of cholangiocytes in vitro, while the secretion of IL-6 can be further enhanced by IL-1β and phorbol myristate acetate (85). Exogenous IL-6 addition can also rescue the activin-A-induced growth inhibition of primary cholangiocytes in vitro (86). With the assistance of NO, IL-6 is involved in the LPS-induced sepsis-related systemic inflammatory response and is one of the most powerful mitogens for cholangiocytes (87, 88). Moreover, LPS and IFN-activated liver-derived macrophages (LDM) express high level of CD154 (also known as CD40 ligand, CD40L), which triggers the CD40-dependent changes of secreting proinflammatory cytokines with increased IL-3, IL-12p70, IL-10 and GM-CSF but reduced CXCL10, IL-6 and CCL2 in human cholangiocytes (89). Studies have showed that TNF-α and IFN-γ could disrupt the barrier function of cholangiocytes (90–92). In addition, inflammatory macrophages secrete TNF-α in the earlier phases of liver diseases, causing an upregulation of integrin αvβ6 on the membrane of epithelial cells and leading to the binding and activation of latent TGF-β1 (93). Furthermore, BECs can produce MIP-3α/CCL-20 in response to cytokines (IL-1β, TNF-α and IL-17) and PAMPs (73). Such evidence suggests that mutual influence exists between macrophages and cholangiocytes during inflammatory hepatic processes.
Furthermore, the inflammatory milieu directly drives the alterations of the cholangiocyte secretory profile, leading to the recruitment of activated liver mesenchymal cells, thereby participating in the positive feedback loop of the inflammatory response as part of the DR. As a hallmark of epithelial–mesenchymal crosstalk, alterations in the reactive cholangiocyte secretome include the upregulation of TGF-β1, TGF-β2, IL-6, platelet-derived growth factor-B (PDGF-B) and CCL-2 (76, 94, 95). Hypothetically, endogenous stimulus derived by neighboring cells and circulating immune cells play constant roles in cholangiocyte activation and cholangiokine secretion, which eventually stimulate cholangiocytes to be a remarkable mediator in liver diseases.
Under physiological conditions, cholangiocytes stay quiescent, maintaining both local and systemic bile homeostasis (38, 96). Even though the replication rate is limited in the quiescent state, cholangiocytes are able to re-enter cell-cycle and proliferate upon various exogenous or endogenous insults (97), and even compensate for the proliferation-incapable hepatocytes to regenerate liver parenchyma (98). The secretory dynamics of cholangiocytes act in autocrine, paracrine, and endocrine manners to maintain biliary homeostasis and regulate other cell types including hepatocytes, HSCs, portal fibroblasts (PFs) and immune cells (99). Cholangiocytes detect pathogens via the TLRs and then secrete antimicrobial IgA into the bile, which serves a vital barrier against germs from both the duodenum and portal vein (100, 101), as well as a variety of cytokines (IL-6 and MCP-1) (102), chemokines (103) and other active anti-microbial peptides (e.g., human beta-defensin-1, hBD-1) into the portal microenvironment (104). Accordingly, these secreted substances have a variable composition depending on the cellular state of cholangiocytes, which creates a complicated secretory network that distinguishes and maintains various cholangiopathies. In general, quiescent cholangiocytes in the biliary system become activated as a result of ongoing distress (e.g., targeted BEC injury and/or inflammatory response caused by broader liver insults). Active cholangiocytes have different cell cycle fates depending on the nature and duration of the injury, primarily cell death, growth and senescence (97).
Following an acute insult, injured cholangiocytes undergo cell death, either programmed (e.g., apoptosis) or non-programmed (e.g., necrosis). The release of apoptotic bodies or DAMPs can trigger a local inflammatory response, which aids in the clearance of cell debris, leading to a time-constrained immune response. However, when a moderate injury occurs or persists, cholangiocytes may re-enter the cell cycle, or engage into an irreversible cell cycle arrest (termed cellular senescence), both of which are accompanied by unique secretory patterns. Nevertheless, several factors, including IL-1β, IL-6, MCP-1, stem cell factor (SCF), TGF-β1, and PDGF, can be secreted by both proliferative and senescent cholangiocytes (105). The similarities and differences of secreted factors from cholangiocytes in proliferative and senescent states are described in the following sections. To understand the complexity of cell status-cholangiokine association, we summarize relevant evidence in Table 1.

Table 1 Cell status-associated cholangiokines.
The anatomic location of the biliary system makes biliary epithelium a fundamental barrier against microorganisms mainly ascending from the duodenum and partially from the portal vein, or as suggested by recent studies present in the bile (1, 124–126). Thereby, under physiological conditions, cholangiocytes establish an intricate cooperative machinery with other hepatocytes and resident immune cells through direct or paracrine factor-mediated intercellular communication. This is supported by a recent study using single-cell RNA-sequencing data of human liver samples, revealing an up-regulation of genes involved in the secretion- and inflammation-related pathway in a subset of cholangiocytes, thereby indicating a crucial role of quiescent cholangiocyte secretome in maintaining homeostatic liver-biliary microenvironment. Additionally, it shows the heterogeneity of quiescent cholangiocyte populations in the liver, suggesting that cholangiocytes may be in a dynamic physiological state as they respond to occasional microbial assaults (127).
The majority of the immunoglobulins (Igs) in human bile are secretory Igs, which significantly maintain liver homeostasis. Hepatocytes effectively secrete most of the IgA in rodents, whereas cholangiocytes represent the main source of IgA secretion in human liver (101). Biliary immunoglobulins, especially IgA, are crucial innate defenders against microorganisms in the biliary tract and upper intestine. Quiescent cholangiocytes also secrete alternative antimicrobial peptides [such as defensins (104), mucins and mucin-associated trefoil peptides (TFF) (120), lactoferrin (121) and cathelicidin (122)], contributing to the basic defense of microorganisms in the biliary tract (128).
In addition to direct immunological defense through the bile, quiescent cholangiocytes can recruit and/or maintain different immune cells by expressing immune-modulating proteins on their surface or by secreting chemokines and cytokines (2). For instance, the cholangiocytes’ surface protein CD1d, which resembles the MHC class I molecule, can activate NKT cells by presenting lipid antigens (129). Cholangiocytes can also activate mucosal-associated invariant T (MAIT) cells, which are prevalent in the human liver and locate near bile ducts, by presenting bacterial antigens via MHC class I-related protein (130). Under normal circumstances, quiescent cholangiocytes secrete TGF-β2, which is involved in maintaining periductular connective tissues and is markedly up-regulated in the proliferating bile ducts of fibrotic livers (76). Last but not least, unstimulated primary human intrahepatic BECs secrete a panel of cytokines/chemokines in vitro, including IL-8, IL-4 and MCP-1 (102, 106), as well as insulin-like growth factor-1 (IGF-1) from healthy cholangiocytes (123).
Conclusively, in the healthy scenario, cholangiocytes mostly remain quiescent but retain baseline secretion of immunoglobulins, antimicrobial peptides, TGF-β2, IGF-1, etc., thereby dynamically maintaining the homeostatic hepatic microenvironment.
When a moderate injury occurs, cholangiocytes can re-proliferate to compensate for cell loss and repair the injury, which is aided by the acute inflammatory response. Cholangiocellular proliferation can be triggered by multiple pathways and stimulus, including IL-6, hepatocyte growth factor (HGF), estrogen, acetylcholine, and bile acids, all of which function through binding to their specific receptors (8). One fundamental feature of proliferating cholangiocytes is their enhanced secretion of variable pro-inflammatory cytokines, chemokines, growth factors, defensin, and other bioactive factors (99). With the timely repair of injury, inflammation would also resolve, and this scenario represents an acute inflammatory response without inducing aberrant hyperproliferation of cholangiocytes (131). However, if the damage persists to prevails over repair processes, cholangiocytes abnormally proliferate and induce chronic inflammation through interaction with various infiltrated immune cells, causing angiogenesis and fibrotic response in the liver, termed DR (14).
In response to a variety of insults, including infections, cholestasis and ischemia, quiescent cholangiocytes can be activated (97), and acquire a hyperproliferative and neuroendocrine-like phenotype with pro-fibrotic and pro-inflammatory secretome (57). Acting in an autocrine/paracrine fashion, these released bioactive factors, including pro-inflammatory cytokines and chemokines (e.g., IL-6, IL-8, TNF-α and various growth factors), modulate cholangiocyte biology and direct the prognosis of biliary damage (108, 132–134). For example, IL-8 and TNF-α levels are substantially elevated in cholangiocytes from individuals with advanced PSC compared to those at the early disease stage (107). In the infection scenario, Helicobacter bilis or fluke products (Opisthorchis viverrini excretory/secretory products) can activate cholangiocytes to proliferate and massively secrete IL-6 and IL-8, thereby initiating innate mucosal immunity against microorganisms (135, 136).
Other chemokines secreted by reactive proliferating cholangiocytes including fractalkine from injured small bile ducts of PBC. Fractalkine possesses chemoattractant and cell-adhesive functions in recruiting intraepithelial monocytes/lymphocytes by binding to its receptor CX3CR1 (109). Moreover, MCP-1 expression was intensively but not exclusively up-regulated in the epithelial cells of regenerating bile ducts (106), which contributes to myofibroblastic trans-differentiation of portal fibroblasts, resulting in biliary fibrosis and cirrhosis (94).
In a chronic damage scenario or under a susceptible genetic background, injury-induced inflammation persists and causes cellular senescence in the biliary epithelium. Senescence can be induced by various factors, including repetitive replication-related telomere shortening, oncogene activation or inactivation of tumor suppressor genes, DNA-damaging interventions, and oxygen radicals (137). The first unveiled feature of senescence is the irreversible cell cycle arrest, leading to the limitation of cell division in vitro (138). With the deepened investigation of senescence in organisms, more hallmarks of senescence have been revealed, including intracellular accumulation of dysfunctional mitochondria, epigenetic alteration, apoptosis resistance, metabolism changes and secretion of multiple bioactive factors, so-called senescence-associated secretory phenotypes (SASP) (139). The initiation of senescence is triggered by DNA damage response (DDR), resulting in the activation of the p53 and the ERK/ETS1/2 pathways, which ultimately up-regulate the expression of p21CIP1 (also known as CDKN1A) and p16INK4a (also known as CDKN2A), respectively (140, 141). As cell cycle blockers, the overexpressed p21CIP1 and p16INK4a prevent cells from entering S phase from the G1 phase. Moreover, unsolvable DDR activates the retinoblastoma (Rb) and p53 pathways and promotes the formation of promyelocytic leukemia nuclear bodies, which ultimately leads to senescence-associated heterochromatin foci (SAHF) through the ASF1A and HIRA chaperones (142, 143).
Senescent cholangiocytes are accumulated in patients with PSC and alcoholic steatohepatitis and are associated with disease exacerbation (117, 144, 145). Entering senescence enhances metabolic levels, allowing cholangiocytes to resist apoptosis. Furthermore, immune cells are responsible to eliminate these apoptosis-resistant cells to prevent abnormal growth and oncogenesis (142). Nonetheless, when senescent cells persist due to an unsuccessful immunologic clearance, these senescent cells may promote aggressiveness of their neighboring malignant cells (146) or even acquire a stem cell-like phenotype themselves once being released from cell cycle withdrawal (147). The significant role of senescent cells root from not only their cell-autonomous changes but also their non-cell autonomous traits for inducing neighboring cells into senescence through their secreted TGF-β, as a bystander effect (113). The bystander effect was also unveiled in the in vitro N-Ras-induced senescent cholangiocytes, which promoted cell cycle arrest and SASP secretion in their surrounding cholangiocytes (117).
Senescent cholangiocytes can be found in mouse PBC samples at the early disease stage, resulting from the over-activation of the Sct/SR pathway and its induced TGF-β1 secretion, which triggered cytokine-induced senescence in an autocrine manner. SASP from these senescent cholangiocytes activates Kupffer cells and HSCs in a paracrine manner, leading to local inflammation and liver fibrosis (114). Moreover, clinical evidence also indicated an unneglected role of SASP from cholangiocytes in various cholangiopathies. For instance, C-X-C motif chemokine ligand-11 (CXCL-11) and CCL-20 from senescent cholangiocytes showed predictive value in detecting ursodeoxycholic acid (UDCA) non-responsive PBC patients (115). Further in vitro study revealed that oxidative stress- and DNA damage-induced senescent BECs exhibited stronger secretion of chemokines (CCL-2/3/4/5, CX3CL-1, CXCL-1, CXCL-2, CXCL-10, and CXCL-16), thereby attracting monocyte/macrophage-like RAW264.7 cells, which suggested that the influence of senescent cholangiocytes on the pathogenesis of PBC was likely achieved by their environmental modulation (116). Furthermore, elevated secretion of pro-inflammatory factors [IL-6, IL-8, CCL-2, plasminogen activator inhibitor-1 (PAI-1)] was evident in senescent cholangiocytes in PSC (117). More evidence showed that CCL-2, CCL-20, IL-3, IL-11 and IL-15 were upregulated in senescent BECs as SASP (118).Even though senescent cholangiocytes are not well understood in BA, intrahepatic bile duct-derived organoids exhibited reduced cholangiocyte proliferation after receiving acetaminophen treatment, while enhancing the secretion of TGF-β1 and PDGF-BB, which indicated a possible role of senescence in this regard (110). The pro-inflammatory factors from SASP label senescent cholangiocytes as harmful actors involving in disease progression, which opens the door for senescence-targeted therapy, such as TGF-inhibition and senolytics, in the treatment of senescent cholangiocytes-related bile duct disorders. For example, genetic or pharmacological (Fisetin) elimination of cholangiocyte senescence reduced the release of inflammatory markers (TNF-α, IL-1 and MCP-1) and alleviated fibrosis in the progression of PSC (119).
Besides the canonical secretion of cholangiokines, cholangiocytes were reported to possibly release extracellular vesicles (EVs) containing IL-13Ra1 into the serum of PSC patients (148). Higher protein levels of Cystatin-S, IL-13Ra1, CD83, IL-1β and EMAP-2 were found in these serum EVs. However, whether and how these EVs are released by cholangiocytes are unclear due to the lack of EVs-tracing evidence. Furthermore, another study revealed that LPS-induced or PSC patient-derived senescent cholangiocytes can also release EVs, which contain multiple growth factors, including EGF, while containing low levels of cytokine/chemokine (149).
In summary, the secretory phenotypes of cholangiocytes are dynamically modified by intrinsic evolutionary factors during the life course of cholangiocytes, and by extrinsic microenvironmental factors engaging with cholangiocytes. Regarding the complexity of the cholangiocyte secretome, temporospatial regulation and cellular context must be taken into account when deciphering the role of cholangiocytes and other cell types in cholangiopathies.
During liver injuries, biliary cells are susceptibly disturbed by both exogenous and endogenous stimulus, leading to cell damage. Thus, persistent damage and dysfunction in cholangiocytes trigger immune cell accumulation and inflammatory reaction, which cause variable pathological consequences, including excessive deposition of scar tissue in portal areas and biliary cirrhosis. This complex response triggered by immune cells, mesenchymal cells, and activated cholangiocytes is termed as DR (14). DR is orchestrated by a finely tuned interplay between proliferation, differentiation and trans-differentiation of cholangiocytes, hepatocytes and HPCs, ulteriorly fueling fibrogenesis and inflammation. Generally, in hepatobiliary diseases, DR refers to similar manifestations, including cholestasis, proliferation, inflammation, fibrosis, and eventually carcinogenesis (150). Nonetheless, the nature of DR remains obscure. As discussed in previous sections, cholangiocyte phenotype alterations (e.g., SASP, proliferation) during DR can drive cholangiocellular proliferation and inflammation by secreting cholangiokines, which further favors DR progression. Coinciding with current opinions, cholangiocytes are considered as not only reactors but also potential initiators in DR (14, 151, 152). In this context, cholangiokines may play crucial roles in different liver/bile duct pathological models by modulating complex cellular interactions, which will be discussed in detail in the following sections (Figure 2).
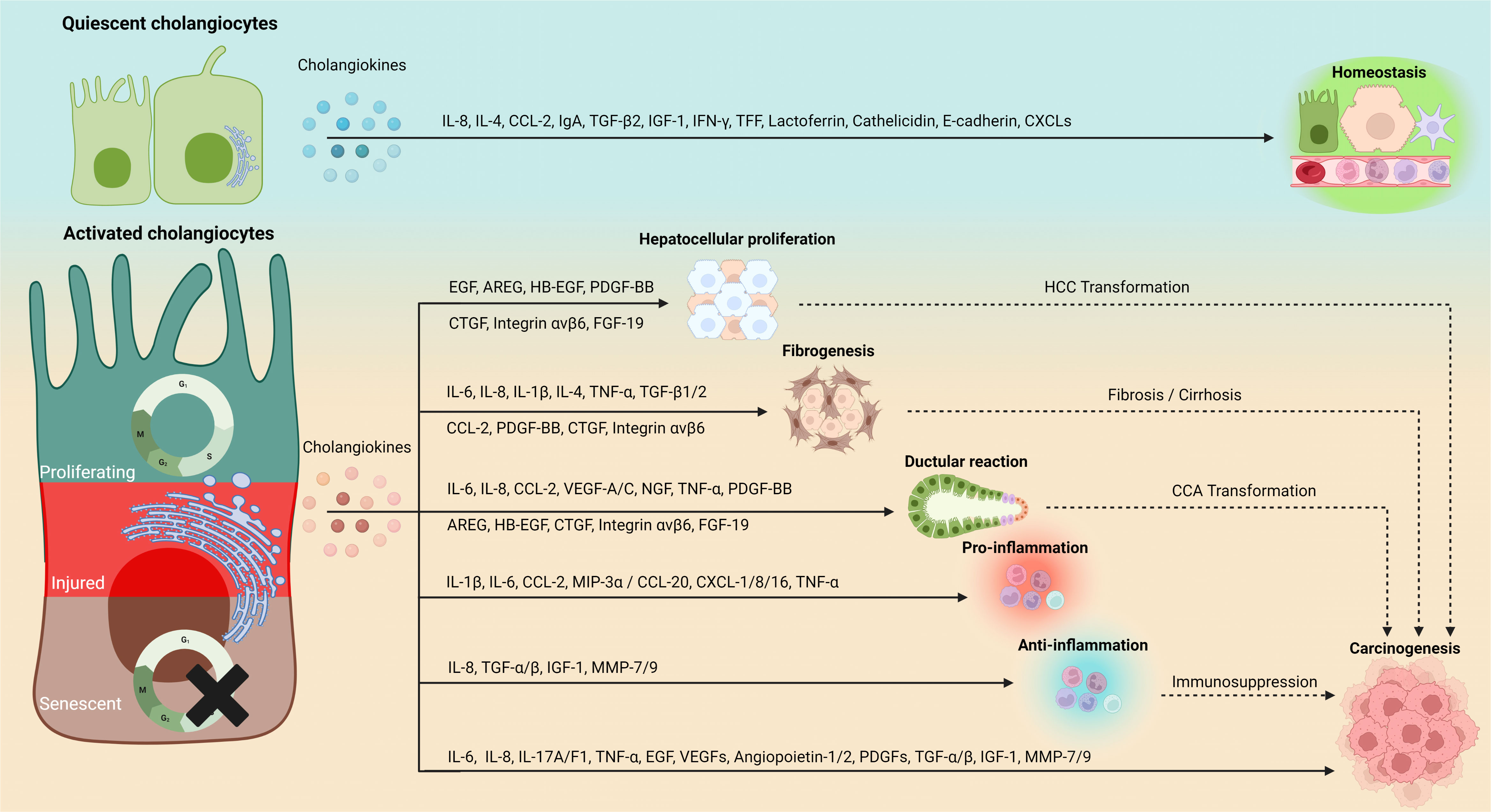
Figure 2 Phenotype-depending secretion and functionality of cholangiokines. Cholangiocytes convert to a major source of functional cytokines in addition to hepatocytes and immune cells. Cholangiokines exert perpetual influences on the hepatic environment. Quiescence-associated cholangiokines maintain liver homeostasis, whereas cholangiokines released by cholangiocytes at their activated statuses (e.g., proliferation, senescence and injury) mediate hepatocellular proliferation, fibrogenesis, DR and inflammation, which eventually cause hepatic carcinogenesis. IL, interleukin; CCL, chemokine (C-C motif) ligand; EGF, epidermal growth factor; IgA, immunoglobulin A; TGF-β, transforming growth factor-β; IGF-1, insulin-like growth factor 1; IFN-γ, Interferon gamma; TFF, trefoil factor; CXCL, chemokine (C-X-C motif) ligand; AREG, amphiregulin; HB-EGF, heparin-binding-EGF; FGF-19, fibroblast growth factor-19; TNF-α, tumor necrosis factor-α; VEGF, vascular endothelial growth factor, PDGF-BB, platelet-derived growth factor; CTGF, connective tissue growth factor; NGF, nerve growth factor; MIP-3α, macrophage inflammatory protein-3α; MMP, matrix metallopeptidase.
The liver has a remarkable capacity to regenerate due to the persistent occurrence of hepatocyte self-renewal. While the facultative stemization of hepatocytes has been assumed as the main origin of liver regeneration for centuries, cholangiocyte proliferation and trans-differentiation appear to be a recently recognized mechanism to enhance the liver regenerative capacity (22).
Fundamentally, HGF and ligands of epidermal growth factor receptor (EGFR), viewed as ‘complete mitogens’, can induce hepatocyte proliferation, even in cultures without serum supplement (153). In terms of liver regeneration, EGF, amphiregulin (AREG), TGF-α and heparin-binding EGF-like growth factor (HB-EGF) are more relevant to hepatocellular proliferation by binding to EGFR (22). Uriarte et al. discovered an increased secretion of HGF from HGF-19-treated murine cholangiocytes (154). In the Mdr2-/- mouse model, senescent cholangiocytes were found enriched with multiple growth factors, including EGF (149). Zhao et al. used cholangiocytes with Cul3 (known as a tumor suppressor) gene deficiency to show that cancerous cholangiocytes are prone to secrete AREG (155). Moreover, another study indicated that cancerous cholangiocytes upregulated HSC-based HB-EGF upon TGF-β secretion (156). In addition, TNFs and IL-6 are known as ‘auxiliary mitogens’. A delayed liver regeneration was recorded in mice with genetic TNF receptor 1/2 (TNFR1/2)-deficiency (157) Simultaneously, IL-6-deficient mice showed reduced activation of hepatocellular STAT3, which is a determinant in promoting proliferation (158). Interestingly, cytokines discussed above (e.g., TNF-α, IL-6) have been known as a fundamental part of cholangiocyte SASP (105).
Other than supportive functions in hepatocellular proliferation, activated cholangiocytes conduct a ‘self-rescuing’ program to sustain their own proliferation and survival. During this ‘self-rescuing’ procedure, cholangiocellular proliferation is initiated not only by genetic/epigenetic alterations but also by autocrine/paracrine cytokines. As described previously, IL-8 levels increase in PSC patients’ bile. In addition, IL-8 caused cell proliferation when added to primary human cholangiocyte cultures (28). The bile component TC protects cholangiocytes against injury. In mouse BDL models, TC administration can enhance VEGF-A and VEGF-C, which are key regulators of biliary proliferation during cholestasis (159, 160). Gigliozzi et al. demonstrated that cholangiocytes secrete NGF, which stimulated the proliferation of cholangiocytes via protein kinase B (AKT)- and ERK1/2-dependent mechanisms. In vivo, NGF neutralization decreased the proliferative capacity of BECs in post-BDL rats (47). More interestingly, we reported that the secretion of CCL-2 by injured cholangiocytes attracts monocytes, which in turn upregulate integrin-β6 and favor cholangiocyte proliferation. This study proposed a novel concept regarding cholangiocyte-associated cellular crosstalk during liver injury (112, 161). Taken together, bile duct repair is driven by stimulatory and inhibitory, autocrine or paracrine secretory factors originating from cholangiocytes. Promisingly, variable cells may be involved in the complex regulation of such regenerative processes.
Inflammation is a fundamental orchestrator of BEC response to liver injury. As discussed above, inflammatory factors effectively influence the cholangiocyte secretory programs. In turn, cholangiocytes with active secretory phenotypes regulate immune cell accumulation and polarization.
Cholangiocytes are capable of sensing exogenous stimuli, including PAMPs and DAMPs, via TLRs and the downstream signal pathways. Upon sensing these stimuli, signaling cascades mainly involving NF-κB, mitogen-activated protein kinase (MAPK) and inflammasome, are rapidly activated (102, 111). Consequently, a broad spectrum of proinflammatory cytokines (e.g., IL-1β, IL-8, IL-6, MCP-1, TNF-α, INF-γ and TGF-β) and chemokines (e.g., CXCL-1, -8 and -16), is released by cholangiocytes (2, 162, 163). Investigations of the liver immune landscape revealed that, the recruited leukocytes are the leading responders in the immune response towards bile duct alterations (2, 164). The first single-cell analysis of liver samples from PSC patients indicates a strong dynamism of T cells, among which naive CD4+ T cells are prone to develop into T-helper (Th) 17 cells (165). Th17 cell accumulation has also been observed in the liver biopsies of PBC patients, specifically around the activated or injured intrahepatic BECs (75, 166). Reactive cholangiocytes regulate Th17 cell differentiation by IL-6 and IL-1β (167). In addition, fractalkine/CX3CL1 and CXCL1 are released by reactive cholangiocytes, which further recruit monocytes and T cells (109, 131, 168, 169). In response to biliary injury, injured or senescent cholangiocytes dramatically release TNF-α and IL-6 (105, 134). TNF-α not only activates naïve and effector T cells, but also induces apoptosis of highly activated effector T cells, further determining the scale of the pathogenic or protective conventional T-cell pool (170). Meanwhile, IL-6 is not only a key player in regulating the Th17/Treg balance, but also exerts paracrine functions to promote terminal differentiation of B cells and their subsequent secretion of immunoglobulins (171, 172).
Another important part of the liver’s innate immunity is the hepatic myeloid cells, which execute crucial roles in either driving liver injury or repairing hepatic malfunction in liver diseases, such as cholangiopathies (161, 173, 174). We have revealed the cholangiocyte-monocyte crosstalk using an acute biliary cell injury mouse model. We found that the injured cholangiocytes can promote the accumulation of CCR2+ monocyte-derived macrophages (MoMFs) and alter bile acid metabolism, while the MoMFs provide important factors for cholangiocytes to proliferate and restore biliary function (112). Furthermore, we have learned from the liver samples of PSC patients and the ex vivo experiments that, secretion of CCL-20 and CCL-2 from human primary cholangiocytes favors monocyte infiltration (175). Additionally, Mip-3a/CCL-20 can be released by the activated cholangiocytes to induce the chemoattraction of immature dendritic cells by its binding to CC chemokine receptor 6 (CCR6) (73). To sum up, cholangiokines play a crucial role in hepatic immunomodulation. However, a more precise understanding of cholangiocyte-driven inflammation is necessary.
In response to an injury, DR is driven by cholangiocyte proliferation and their secretome, participating in the complex regulation of portal inflammation and fibrogenesis (14). Inflammation generates signals that attract liver mesenchymal cells to bile ducts and portal areas. This process is considered to be the primary stage of biliary or portal fibrosis. In this context, interaction between reactive ductular cells and myofibroblast cells, so-called epithelial–mesenchymal crosstalk, is a constant key modulator in liver fibrogenesis, the process of which also involves several profibrogenic factors (e.g., IL-6, TGF-β1/2, CCL-2 and PDGF-B) (76, 94, 95).
During biliary fibrosis, proliferating BECs represent a predominant source of the profibrogenic connective tissue growth factor (CTGF) besides HSCs (176, 177). According to a recent study, reactive cholangiocytes secrete TGF-β depending on the Mothers against decapentaplegic homolog 3 (SMAD3) and lysine Acetyltransferases 2A (KAT2A). Pharmacological inhibition of Kat2a protein or cholangiocyte-selective deletion of Kat2a gene was protective in mouse models of biliary fibrosis (178). BECs can regulate the proliferation and myofibroblastic trans-differentiation of HSCs to provoke the portal fibrosis by the CCL-2-based paracrine (94). TGF-β1 and TGF-β2 were found upregulated in cholangiocytes during chronic liver diseases, suggesting their implication in biliary hyperplasia and fibrogenesis (76). Likewise, IL-8 secreted by the activated cholangiocytes can stimulate the production of profibrotic genes, suggesting that IL-8 may be involved in the pathogenesis of cholangiopathies (28). Grappone et al. suggested that PDGF-B chains can be produced by cholangiocytes during chronic cholestasis (179). Recently, Moncsek et al. disclosed that senescent cholangiocytes promoted the activation of quiescent mesenchymal cells in a PDGF-dependent manner (180). Another latest study has demonstrated that biliary NF-κB-inducing kinase (NIK) could trigger DR. While the ablation of NIK significantly decreased the expression of Il-1β, Il-4, Il-6, iNos, Tnfα, Mcp1 and Tgfb1, thereby attenuating liver fibrosis (25). What’s more, Liu et al. reported that cholangiocyte-derived exosomal H19 plays a critical role in the progression of cholestatic liver fibrosis by promoting the differentiation and activation of HSCs (181). Integrin αvβ6 acts as not only a crucial mediator but also a therapeutic target in liver fibrosis (182, 183). Moreover, genetic suppression of Itgb6 (a gene encoding integrin αvβ6) in the mouse models of biliary injury is therapeutically relevant to the attenuation of DR and biliary fibrosis (184, 185). Pi et al. have revealed that CTGF and integrin αvβ6 regulate biliary cell activation and fibrosis, probably through the secretion of fibronectin and TGF-β1 (176). In conclusion, activated cholangiocytes and their cholangiokines might be promising therapeutic targets for ameliorating liver fibrosis.
Primary liver cancers, including hepatocellular carcinoma (HCC) and CCA are a tremendous burden to global health, but their pathomechanisms are only partially understood (137, 186). From a short-term perspective, cholangiokines contribute to hyperplasia, inflammation and fibrogenesis of the hepatic portal areas. In the long run, cholangiokines may eventually fuel the malignant transformation of hepatic cells through continuous autocrine and paracrine stimulation.
IL-6 has been determined by several studies as not only a key driver but also a promising therapeutic target for liver cancers (187–189). IL-6 levels are highly presented in the serum and bile of CCA patients and culture medium of CCA cell lines (190). Recent studies concluded that HCC and intrahepatic CCA (iCCA) are significantly driven by IL-6 and its associated inflammatory processes (191, 192). IL-6 promotes the survival of transformed cholangiocytes through different pathways. In particular, the IL-6-activated p38 pathway determines cell proliferation by mediating p21WAF1/CIP1 and p44/p42 MAPK (193). Even more intriguingly, single-cell analysis of iCCA patient specimens showed that CCA-derived exosomal miR-9-5p elicited a high secretory possibility of IL-6 in cancer-associated fibroblasts to promote tumor progression, suggesting broader roles of cholangiocyte- derived IL-6 in the tumor microenvironment (TME) (194).
EGF administration can provoke CCA progression by triggering epithelial-mesenchymal transition (EMT). In addition, the upregulation of TGF-α favors the proliferative levels of HCC cells (195). EGF and TGF-α regulate cell proliferation and differentiation by binding EGFR (196). Earlier studies also revealed a positive correlation between EGFR inhibition and HCC suppression (197, 198). Inoue et al. characterized that blocking EGFR by vandetanib in liver cancer models yielded a significantly reduced tumor vessel density and tumor growth, while enhancing tumor cell apoptosis and survival prolongation with reduced number of intrahepatic metastases (199). Moreover, it has been well elucidated that hepatic myofibroblasts promote malignancy progression in CCA patients through their HB-EGF-induced activation (156, 200), which is consistent with the fact that myofibroblasts are also prone to trigger the cholangiocyte-secreted PDGF-B (201).
TGF-β and its related signaling cascades play a central role in inflammation, fibrogenesis and immunomodulation in the TME of liver cancers (202, 203). A recent study indicated a positive feedback loop of TGF-β and LIN28B in CCA metastasis (204). TGF-β has also been found to promote the progression of CCA and HCC by interacting with non-coding RNAs (205–208). More strikingly, TGF-β exerts immunoregulatory functions in HCC, mainly via suppressing T cells (202, 209). Interestingly, the blockade of TGF-β-induced activated dendritic cells enhances the lethal effects of T cells in CCA (210). Thus, TGF-β potentially disturbs immunotherapies in liver cancers, which makes it a promising target to attenuate immunotherapy resistance. Besides, TGF-β was also found to regulate monocyte/macrophages in liver cancers. Yan et al. reported that TGF-β fosters the expression of T cell immunoglobulin domain and mucin domain-3 (TIM-3/CD366) on monocytes, which augments the infiltration of tumor-associated macrophages in HCC (211). Ning et al. demonstrated that the induction of imbalanced TGF-β1/BMP-7 pathways in HCC cells could significantly reinforce the aggressiveness and stemness of HCC cells (212).
Novel observations indicate that VEGF is a master factor in lymphangiogenesis and the immune response to cholangiocarcinoma (84). The secretion of VEGFs, angiopoietin-1/2, PDGF and TGF-β from tumor cells or other cell types robustly modulate the TME, which is a critical component of tumor biology (213). The VEGF-A secretion by CCA cells can be mediated by other factors including IGF-1, its receptor IGFR as well as the estrogen receptor (ER) family (214, 215). Furthermore, estrogens induce the proliferation of CCA cells by VEGF/VEGFR2 mediation (216). VEGF-A, on the other hand, induces cholangiokines, including matrix metalloproteinase (MMP)-7 and -9, from CCA cells, which contribute to the significant remodeling of extracellular matrix (ECM) and the extensive tumor metastasis (217).
Notably, TNF-α plays contradictory roles in liver cancers. Commonly known as a participant in maintaining homeostasis of cancer immunobiology, TNF-α unveils its ‘dark side’ to provoke chronic inflammation, EMT and angiogenesis, which may fuel the aggressiveness of cancers (218). Interestingly, high-dose administration of TNF-α inhibits neovascularization in mice, whereas low –dosed TNF-α promotes angiogenesis by increasing the expression of VEGF, VEGFR, IL-8 and basic FGF (219). Another study underlined that TNF-α strengthened the migration behaviors of CCA cells by upregulating their EMT markers, including ZEB2, vimentin and S100A4. Moreover, TNF-α has been shown to induce TGFB overexpression, which eventually promotes cancer cells to migrate (220). Yuan et al. described a novel phenomenon that TNFs favor cholangiocellular proliferation, differentiation and transformation due to the induced chronic mitochondrial dysfunction and the accumulation of reactive oxygen species (ROS). This finding enriches the research directions of TNF-α meditation in CCA (221). Even though cholangiokines can hardly be concluded as a robust oncogenic secretome based on our current knowledge, various tumor-promoting cytokines secreted by cholangiocytes have been evidenced to regulate TME.
Although the quantitative contribution of cholangiocytes to the total liver mass and the hepatic secretome appears modest, cholangiocytes play essential roles in a vast array of disease-related mechanisms and shape the portal area microenvironment. Sensitized by various injuries, stimuli or immune disturbances, cholangiocytes release cholangiokines, which broadly participate in liver immunology, inflammation, fibrogenesis and malignant transformation. Particularly, cholangiokines are gaining recognition for their involvement in cholangiopathies and primary liver cancers. Of note, better characterization of the cholangiokines may provide an in-depth understanding of cholangiocyte-driven pathophysiological processes. Nonetheless, the paracrine and autocrine nature of cholangiokines poses some technical challenges, as their functions need to be interpreted in the spatiotemporal context of the hepatic microenvironment. Even though the practicability of cholangiokines as diagnostic/prognostic markers is still hidden in fog, emerging biotechnics can incarnate wind to achieve it. Recently, several novel approaches, such as multiplex immunostaining, imaging mass cytometry and spatially resolved single-cell sequencing, have emerged for in situ liver studies, which shed light on differential spatial heterogeneity of the hepatic parenchymal and immune cells (164, 222–224). Furthermore, by tying up the single-cell spatial or newly developed single-cell Stereo-sequencing methods (225), pathomechanisms of cholangiokines associated with time phases, zonation and functionality are anticipated to be soon and decently determined.
HL conceived the topic. XC and HL drafted the manuscript and prepared the figures. XC, FT, AG, and HL revised the manuscript. All authors have approved the published version of the manuscript. All authors contributed to the article and approved the submitted version.
This study is funded by the German Research Foundation (DFG SFB/TRR 296 and CRC1382, 403224013) and the German Ministry of Education and Research (BMBF DEEP-HCC consortium), Young talent project of Changzhou (China) Health Commission (CZQM2022007), Youth fund of Changzhou (China) Health Commission (QN202121), Basic application project of Changzhou (China) Science and Technology Bureau (CJ20220142) and General project of Changzhou Medical Center, Nanjing Medical University (CZKYCMCB202221). XC and HL are funded by China Scholarship Council Foundation.
We acknowledge financial support from the Open Access Publication Fund of Charité – Universitätsmedizin Berlin and the German Research Foundation (DFG). The figures were created with BioRender.com.
Author FT’s lab received research grants from Gilead, Allergan, Bristol-Myers Squibb and Inventiva.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Banales JM, Huebert RC, Karlsen T, Strazzabosco M, LaRusso NF, Gores GJ. Cholangiocyte pathobiology. Nat Rev Gastroenterol Hepatol (2019) 16(5):269–81. doi: 10.1038/s41575-019-0125-y
2. Bjorkstrom NK. Immunobiology of the biliary tract system. J Hepatol (2022) 77(6):1657–69. doi: 10.1016/j.jhep.2022.08.018
3. Wang W, Chen D, Wang J, Wen L. Cellular homeostasis and repair in the biliary tree. Semin Liver Dis (2022) 42(3):271–82. doi: 10.1055/a-1869-7714
4. Lanzoni G, Cardinale V, Carpino G. The hepatic, biliary, and pancreatic network of stem/progenitor cell niches in humans: a new reference frame for disease and regeneration. Hepatology. (2016) 64(1):277–86. doi: 10.1002/hep.28326
5. Alpini G, McGill JM, Larusso NF. The pathobiology of biliary epithelia. Hepatology. (2002) 35(5):1256–68. doi: 10.1053/jhep.2002.33541
6. Marzioni M, Glaser SS, Francis H, Phinizy JL, LeSage G, Alpini G. Functional heterogeneity of cholangiocytes. Semin Liver Dis (2002) 22(3):227–40. doi: 10.1055/s-2002-34501
7. Alpini G, Roberts S, Kuntz SM, Ueno Y, Gubba S, Podila PV, et al. Morphological, molecular, and functional heterogeneity of cholangiocytes from normal rat liver. Gastroenterology. (1996) 110(5):1636–43. doi: 10.1053/gast.1996.v110.pm8613073
8. Yoo KS, Lim WT, Choi HS. Biology of cholangiocytes: from bench to bedside. Gut Liver (2016) 10(5):687–98. doi: 10.5009/gnl16033
9. Mancinelli R, Franchitto A, Gaudio E, Onori P, Glaser S, Francis H, et al. After damage of large bile ducts by gamma-aminobutyric acid, small ducts replenish the biliary tree by amplification of calcium-dependent signaling and de novo acquisition of large cholangiocyte phenotypes. Am J Pathol (2010) 176(4):1790–800. doi: 10.2353/ajpath.2010.090677
10. Gouw AS, Clouston AD, Theise ND. Ductular reactions in human liver: diversity at the interface. Hepatology. (2011) 54(5):1853–63. doi: 10.1002/hep.24613
11. Maroni L, Ninfole E, Pinto C, Benedetti A, Marzioni M. Gut-liver axis and inflammasome activation in cholangiocyte pathophysiology. Cells. (2020) 9(3):736. doi: 10.3390/cells9030736
12. Giordano DM, Pinto C, Maroni L, Benedetti A, Marzioni M. Inflammation and the gut-liver axis in the pathophysiology of cholangiopathies. Int J Mol Sci (2018) 19(10):3003. doi: 10.3390/ijms19103003
13. Chen Y, Gao WK, Shu YY, Ye J. Mechanisms of ductular reaction in non-alcoholic steatohepatitis. World J Gastroenterol (2022) 28(19):2088–99. doi: 10.3748/wjg.v28.i19.2088
14. Sato K, Marzioni M, Meng F, Francis H, Glaser S, Alpini G. Ductular reaction in liver diseases: pathological mechanisms and translational significances. Hepatology. (2019) 69(1):420–30. doi: 10.1002/hep.30150
15. Boyer JL, Soroka CJ. Bile formation and secretion: an update. J Hepatol (2021) 75(1):190–201. doi: 10.1016/j.jhep.2021.02.011
16. Pinzani M, Luong TV. Pathogenesis of biliary fibrosis. Biochim Biophys Acta Mol Basis Dis (2018) 1864(4 Pt B):1279–83. doi: 10.1016/j.bbadis.2017.07.026
17. Fabris L, Spirli C, Cadamuro M, Fiorotto R, Strazzabosco M. Emerging concepts in biliary repair and fibrosis. Am J Physiol Gastrointest Liver Physiol (2017) 313(2):G102–G16. doi: 10.1152/ajpgi.00452.2016
18. Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. Lancet. (2015) 386(10003):1565–75. doi: 10.1016/S0140-6736(15)00154-3
19. Trussoni CE, O'Hara SP, LaRusso NF. Cellular senescence in the cholangiopathies: a driver of immunopathology and a novel therapeutic target. Semin Immunopathol (2022) 44(4):527–44. doi: 10.1007/s00281-022-00909-9
20. Tam PKH, Yiu RS, Lendahl U, Andersson ER. Cholangiopathies - towards a molecular understanding. EBioMedicine. (2018) 35:381–93. doi: 10.1016/j.ebiom.2018.08.024
21. Lazaridis KN, LaRusso NF. The cholangiopathies. Mayo Clin Proc (2015) 90(6):791–800. doi: 10.1016/j.mayocp.2015.03.017
22. Michalopoulos GK, Bhushan B. Liver regeneration: biological and pathological mechanisms and implications. Nat Rev Gastroenterol Hepatol (2021) 18(1):40–55. doi: 10.1038/s41575-020-0342-4
23. Russell JO, Lu WY, Okabe H, Abrams M, Oertel M, Poddar M, et al. Hepatocyte-specific beta-catenin deletion during severe liver injury provokes cholangiocytes to differentiate into hepatocytes. Hepatology. (2019) 69(2):742–59. doi: 10.1002/hep.30270
24. Rodrigo-Torres D, Affo S, Coll M, Morales-Ibanez O, Millan C, Blaya D, et al. The biliary epithelium gives rise to liver progenitor cells. Hepatology. (2014) 60(4):1367–77. doi: 10.1002/hep.27078
25. Zhang Z, Zhong X, Shen H, Sheng L, Liangpunsakul S, Lok AS, et al. Biliary NIK promotes ductular reaction and liver injury and fibrosis in mice. Nat Commun (2022) 13(1):5111. doi: 10.1038/s41467-022-32575-8
26. Coll M, Arino S, Martinez-Sanchez C, Garcia-Pras E, Gallego J, Moles A, et al. Ductular reaction promotes intrahepatic angiogenesis through Slit2-roundabout 1 signaling. Hepatology. (2022) 75(2):353–68. doi: 10.1002/hep.32140
27. Kullak-Ublick GA, Beuers U, Paumgartner G. Hepatobiliary transport. J Hepatol (2000) 32(1 Suppl):3–18. doi: 10.1016/S0168-8278(00)80411-0
28. Zweers SJ, Shiryaev A, Komuta M, Vesterhus M, Hov JR, Perugorria MJ, et al. Elevated interleukin-8 in bile of patients with primary sclerosing cholangitis. Liver Int (2016) 36(9):1370–7. doi: 10.1111/liv.13092
29. Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol (2016) 13(2):88–110. doi: 10.1038/nrgastro.2015.200
31. Lamireau T, Zoltowska M, Levy E, Yousef I, Rosenbaum J, Tuchweber B, et al. Effects of bile acids on biliary epithelial cells: proliferation, cytotoxicity, and cytokine secretion. Life Sci (2003) 72(12):1401–11. doi: 10.1016/S0024-3205(02)02408-6
32. Jusakul A, Loilome W, Namwat N, Haigh WG, Kuver R, Dechakhamphu S, et al. Liver fluke-induced hepatic oxysterols stimulate DNA damage and apoptosis in cultured human cholangiocytes. Mutat Res (2012) 731(1-2):48–57. doi: 10.1016/j.mrfmmm.2011.10.009
33. Seo DW, Choi HS, Lee SP, Kuver R. Oxysterols from human bile induce apoptosis of canine gallbladder epithelial cells in monolayer culture. Am J Physiol Gastrointest Liver Physiol (2004) 287(6):G1247–56. doi: 10.1152/ajpgi.00013.2004
34. Yoon JH, Canbay AE, Werneburg NW, Lee SP, Gores GJ. Oxysterols induce cyclooxygenase-2 expression in cholangiocytes: implications for biliary tract carcinogenesis. Hepatology. (2004) 39(3):732–8. doi: 10.1002/hep.20125
35. Glaser S, Meng F, Han Y, Onori P, Chow BK, Francis H, et al. Secretin stimulates biliary cell proliferation by regulating expression of microRNA 125b and microRNA let7a in mice. Gastroenterology. (2014) 146(7):1795–808 e12. doi: 10.1053/j.gastro.2014.02.030
36. Glaser S, Lam IP, Franchitto A, Gaudio E, Onori P, Chow BK, et al. Knockout of secretin receptor reduces large cholangiocyte hyperplasia in mice with extrahepatic cholestasis induced by bile duct ligation. Hepatology. (2010) 52(1):204–14. doi: 10.1002/hep.23657
37. Yao Y, Eshun JK, Lu S, Berschneider HM, Black DD. Regulation of triacylglycerol and phospholipid trafficking by fatty acids in newborn swine enterocytes. Am J Physiol Gastrointest Liver Physiol (2002) 282(5):G817–24. doi: 10.1152/ajpgi.00397.2001
38. Alpini G, Lenzi R, Sarkozi L, Tavoloni N. Biliary physiology in rats with bile ductular cell hyperplasia. evidence for a secretory function of proliferated bile ductules. J Clin Invest (1988) 81(2):569–78. doi: 10.1172/JCI113355
39. Wu N, Meng F, Invernizzi P, Bernuzzi F, Venter J, Standeford H, et al. The secretin/secretin receptor axis modulates liver fibrosis through changes in transforming growth factor-beta1 biliary secretion in mice. Hepatology. (2016) 64(3):865–79. doi: 10.1002/hep.28622
40. Alpini G, Ulrich CD 2nd, Phillips JO, Pham LD, Miller LJ, LaRusso NF. Upregulation of secretin receptor gene expression in rat cholangiocytes after bile duct ligation. Am J Physiol (1994) 266(5 Pt 1):G922–8. doi: 10.1152/ajpgi.1994.266.5.G922
41. Schaap FG, van der Gaag NA, Gouma DJ, Jansen PL. High expression of the bile salt-homeostatic hormone fibroblast growth factor 19 in the liver of patients with extrahepatic cholestasis. Hepatology. (2009) 49(4):1228–35. doi: 10.1002/hep.22771
42. Yang J, Sontag D, Kung S, Minuk GY. Fibroblast growth factor 19 induced changes in non-malignant cholangiocytes. J Clin Transl Hepatol (2021) 9(6):909–16. doi: 10.14218/JCTH.2021.00087
43. Zweers SJ, Booij KA, Komuta M, Roskams T, Gouma DJ, Jansen PL, et al. The human gallbladder secretes fibroblast growth factor 19 into bile: towards defining the role of fibroblast growth factor 19 in the enterobiliary tract. Hepatology. (2012) 55(2):575–83. doi: 10.1002/hep.24702
44. Alvarez-Sola G, Uriarte I, Latasa MU, Jimenez M, Barcena-Varela M, Santamaria E, et al. Bile acids, FGF15/19 and liver regeneration: from mechanisms to clinical applications. Biochim Biophys Acta Mol Basis Dis (2018) 1864(4 Pt B):1326–34. doi: 10.1016/j.bbadis.2017.06.025
45. Wen Y, Jeong S, Xia Q, Kong X. Role of osteopontin in liver diseases. Int J Biol Sci (2016) 12(9):1121–8. doi: 10.7150/ijbs.16445
46. Ramaiah SK, Rittling S. Pathophysiological role of osteopontin in hepatic inflammation, toxicity, and cancer. Toxicol Sci (2008) 103(1):4–13. doi: 10.1093/toxsci/kfm246
47. Gigliozzi A, Alpini G, Baroni GS, Marucci L, Metalli VD, Glaser SS, et al. Nerve growth factor modulates the proliferative capacity of the intrahepatic biliary epithelium in experimental cholestasis. Gastroenterology. (2004) 127(4):1198–209. doi: 10.1053/j.gastro.2004.06.023
48. Reich M, Deutschmann K, Sommerfeld A, Klindt C, Kluge S, Kubitz R, et al. TGR5 is essential for bile acid-dependent cholangiocyte proliferation in vivo and in vitro. Gut (2016) 65(3):487–501. doi: 10.1136/gutjnl-2015-309458
49. Merlen G, Ursic-Bedoya J, Jourdainne V, Kahale N, Glenisson M, Doignon I, et al. Bile acids and their receptors during liver regeneration: "Dangerous protectors". Mol Aspects Med (2017) 56:25–33. doi: 10.1016/j.mam.2017.03.002
50. Ozdirik B, Muller T, Wree A, Tacke F, Sigal M. The role of microbiota in primary sclerosing cholangitis and related biliary malignancies. Int J Mol Sci (2021) 22(13):6975. doi: 10.3390/ijms22136975
51. Etienne-Mesmin L, Vijay-Kumar M, Gewirtz AT, Chassaing B. Hepatocyte toll-like receptor 5 promotes bacterial clearance and protects mice against high-fat diet-induced liver disease. Cell Mol Gastroenterol Hepatol (2016) 2(5):584–604. doi: 10.1016/j.jcmgh.2016.04.007
52. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol (2017) 17(5):306–21. doi: 10.1038/nri.2017.11
53. Paik D, Yao L, Zhang Y, Bae S, D'Agostino GD, Zhang M, et al. Human gut bacteria produce Tau(Eta)17-modulating bile acid metabolites. Nature. (2022) 603(7903):907–12. doi: 10.1038/s41586-022-04480-z
54. Ji J, Wu L, Wei J, Wu J, Guo C. The gut microbiome and ferroptosis in MAFLD. J Clin Transl Hepatol (2023) 11(1):174–87. doi: 10.14218/JCTH.2022.00136
55. Tilg H, Adolph TE, Trauner M. Gut-liver axis: pathophysiological concepts and clinical implications. Cell Metab (2022) 34(11):1700–18. doi: 10.1016/j.cmet.2022.09.017
56. Zhang X, Liu H, Hashimoto K, Yuan S, Zhang J. The gut-liver axis in sepsis: interaction mechanisms and therapeutic potential. Crit Care (2022) 26(1):213. doi: 10.1186/s13054-022-04090-1
57. Pinto C, Giordano DM, Maroni L, Marzioni M. Role of inflammation and proinflammatory cytokines in cholangiocyte pathophysiology. Biochim Biophys Acta Mol Basis Dis (2018) 1864(4 Pt B):1270–8. doi: 10.1016/j.bbadis.2017.07.024
58. Syal G, Fausther M, Dranoff JA. Advances in cholangiocyte immunobiology. Am J Physiol Gastrointest Liver Physiol (2012) 303(10):G1077–86. doi: 10.1152/ajpgi.00227.2012
59. Kumar S, Duan Q, Wu R, Harris EN, Su Q. Pathophysiological communication between hepatocytes and non-parenchymal cells in liver injury from NAFLD to liver fibrosis. Adv Drug Delivery Rev (2021) 176:113869. doi: 10.1016/j.addr.2021.113869
60. Kemp DR. Inappropriate diagnosis of necrotizing arachnidism. watch out miss muffet–but don't get paranoid. Med J Aust (1990) 152(12):669–71. doi: 10.5694/j.1326-5377.1990.tb125430.x
61. Sasatomi K, Noguchi K, Sakisaka S, Sata M, Tanikawa K. Abnormal accumulation of endotoxin in biliary epithelial cells in primary biliary cirrhosis and primary sclerosing cholangitis. J Hepatol (1998) 29(3):409–16. doi: 10.1016/S0168-8278(98)80058-5
62. Tanaka A, Leung PSC, Gershwin ME. Pathogen infections and primary biliary cholangitis. Clin Exp Immunol (2019) 195(1):25–34. doi: 10.1111/cei.13198
63. Faruqui S, Okoli FC, Olsen SK, Feldman DM, Kalia HS, Park JS, et al. Cholangiopathy after severe COVID-19: clinical features and prognostic implications. Am J Gastroenterol (2021) 116(7):1414–25. doi: 10.14309/ajg.0000000000001264
64. Roth NC, Kim A, Vitkovski T, Xia J, Ramirez G, Bernstein D, et al. Post-COVID-19 cholangiopathy: a novel entity. Am J Gastroenterol (2021) 116(5):1077–82. doi: 10.14309/ajg.0000000000001154
65. Brevini T, Maes M, Webb GJ, John BV, Fuchs CD, Buescher G, et al. FXR inhibition may protect from SARS-CoV-2 infection by reducing ACE2. Nature (2022) 615:134–42. doi: 10.1038/s41586-022-05594-0
66. Xia X, Demorrow S, Francis H, Glaser S, Alpini G, Marzioni M, et al. Cholangiocyte injury and ductopenic syndromes. Semin Liver Dis (2007) 27(4):401–12. doi: 10.1055/s-2007-991516
67. Visentin M, Lenggenhager D, Gai Z, Kullak-Ublick GA. Drug-induced bile duct injury. Biochim Biophys Acta Mol Basis Dis (2018) 1864(4Pt B):1498–506. doi: 10.1016/j.bbadis.2017.08.033
68. Geubel AP, Sempoux CL. Drug and toxin-induced bile duct disorders. J Gastroenterol Hepatol (2000) 15(11):1232–8. doi: 10.1046/j.1440-1746.2000.2369.x
69. Almazroo OA, Miah MK, Venkataramanan R. Drug metabolism in the liver. Clin Liver Dis (2017) 21(1):1–20. doi: 10.1016/j.cld.2016.08.001
70. Lakehal F, Wendum D, Barbu V, Becquemont L, Poupon R, Balladur P, et al. Phase I and phase II drug-metabolizing enzymes are expressed and heterogeneously distributed in the biliary epithelium. Hepatology. (1999) 30(6):1498–506. doi: 10.1002/hep.510300619
71. Strnad P, Tacke F, Koch A, Trautwein C. Liver - guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol (2017) 14(1):55–66. doi: 10.1038/nrgastro.2016.168
72. Adams DH, Afford SC. The role of cholangiocytes in the development of chronic inflammatory liver disease. Front Biosci (2002) 7:e276–85. doi: 10.2741/a923
73. Harada K, Shimoda S, Ikeda H, Chiba M, Hsu M, Sato Y, et al. Significance of periductal langerhans cells and biliary epithelial cell-derived macrophage inflammatory protein-3alpha in the pathogenesis of primary biliary cirrhosis. Liver Int (2011) 31(2):245–53. doi: 10.1111/j.1478-3231.2010.02367.x
74. Okamura A, Harada K, Nio M, Nakanuma Y. Participation of natural killer cells in the pathogenesis of bile duct lesions in biliary atresia. J Clin Pathol (2013) 66(2):99–108. doi: 10.1136/jclinpath-2012-201097
75. Harada K, Shimoda S, Sato Y, Isse K, Ikeda H, Nakanuma Y. Periductal interleukin-17 production in association with biliary innate immunity contributes to the pathogenesis of cholangiopathy in primary biliary cirrhosis. Clin Exp Immunol (2009) 157(2):261–70. doi: 10.1111/j.1365-2249.2009.03947.x
76. Milani S, Herbst H, Schuppan D, Stein H, Surrenti C. Transforming growth factors beta 1 and beta 2 are differentially expressed in fibrotic liver disease. Am J Pathol (1991) 139(6):1221–9.
77. St Paul M, Ohashi PS. The roles of CD8(+) T cell subsets in antitumor immunity. Trends Cell Biol (2020) 30(9):695–704. doi: 10.1016/j.tcb.2020.06.003
78. Alpini G, Elias I, Glaser SS, Rodgers RE, Phinizy JL, Robertson WE, et al. Gamma-interferon inhibits secretin-induced choleresis and cholangiocyte proliferation in a murine model of cirrhosis. J Hepatol (1997) 27(2):371–80. doi: 10.1016/S0168-8278(97)80184-5
79. Morland CM, Fear J, McNab G, Joplin R, Adams DH. Promotion of leukocyte transendothelial cell migration by chemokines derived from human biliary epithelial cells in vitro. Proc Assoc Am Physicians (1997) 109(4):372–82.
80. Mazzi V. Mig chemokine in primary biliary cirrhosis. Clin Ter (2019) 170(3):e211–e5. doi: 10.7417/CT.2019.2135
81. Morland CM, Fear J, Joplin R, Adams DH. Inflammatory cytokines stimulate human biliary epithelial cells to express interleukin-8 and monocyte chemotactic protein-1. Biochem Soc Trans (1997) 25(2):232S. doi: 10.1042/bst025232s
82. Nishikawa H, Enomoto H, Iwata Y, Hasegawa K, Nakano C, Takata R, et al. Impact of serum wisteria floribunda agglutinin positive mac-2-binding protein and serum interferon-gamma-inducible protein-10 in primary biliary cirrhosis. Hepatol Res (2016) 46(6):575–83. doi: 10.1111/hepr.12595
83. Spirli C, Fabris L, Duner E, Fiorotto R, Ballardini G, Roskams T, et al. Cytokine-stimulated nitric oxide production inhibits adenylyl cyclase and cAMP-dependent secretion in cholangiocytes. Gastroenterology. (2003) 124(3):737–53. doi: 10.1053/gast.2003.50100
84. Mariotti V, Fiorotto R, Cadamuro M, Fabris L, Strazzabosco M. New insights on the role of vascular endothelial growth factor in biliary pathophysiology. JHEP Rep (2021) 3(3):100251. doi: 10.1016/j.jhepr.2021.100251
85. Matsumoto K, Fujii H, Michalopoulos G, Fung JJ, Demetris AJ. Human biliary epithelial cells secrete and respond to cytokines and hepatocyte growth factors in vitro: interleukin-6, hepatocyte growth factor and epidermal growth factor promote DNA synthesis in vitro. Hepatology (1994) 20(2):376–82. doi: 10.1002/hep.1840200217
86. Yokomuro S, Tsuji H, Lunz JG 3rd, Sakamoto T, Ezure T, Murase N, et al. Growth control of human biliary epithelial cells by interleukin 6, hepatocyte growth factor, transforming growth factor beta1, and activin a: comparison of a cholangiocarcinoma cell line with primary cultures of non-neoplastic biliary epithelial cells. Hepatology. (2000) 32(1):26–35. doi: 10.1053/jhep.2000.8535
87. Liu Z, Sakamoto T, Ezure T, Yokomuro S, Murase N, Michalopoulos G, et al. Interleukin-6, hepatocyte growth factor, and their receptors in biliary epithelial cells during a type I ductular reaction in mice: interactions between the periductal inflammatory and stromal cells and the biliary epithelium. Hepatology. (1998) 28(5):1260–8. doi: 10.1002/hep.510280514
88. Hung YL, Fang SH, Wang SC, Cheng WC, Liu PL, Su CC, et al. Corylin protects LPS-induced sepsis and attenuates LPS-induced inflammatory response. Sci Rep (2017) 7:46299. doi: 10.1038/srep46299
89. Alabraba EB, Lai V, Boon L, Wigmore SJ, Adams DH, Afford SC. Coculture of human liver macrophages and cholangiocytes leads to CD40-dependent apoptosis and cytokine secretion. Hepatology. (2008) 47(2):552–62. doi: 10.1002/hep.22011
90. Hu G, Gong AY, Liu J, Zhou R, Deng C, Chen XM. miR-221 suppresses ICAM-1 translation and regulates interferon-gamma-induced ICAM-1 expression in human cholangiocytes. Am J Physiol Gastrointest Liver Physiol (2010) 298(4):G542–50. doi: 10.1152/ajpgi.00490.2009
91. Gong AY, Zhou R, Hu G, Li X, Splinter PL, O'Hara SP, et al. MicroRNA-513 regulates B7-H1 translation and is involved in IFN-gamma-induced B7-H1 expression in cholangiocytes. J Immunol (2009) 182(3):1325–33. doi: 10.4049/jimmunol.182.3.1325
92. Hanada S, Harada M, Koga H, Kawaguchi T, Taniguchi E, Kumashiro R, et al. Tumor necrosis factor-alpha and interferon-gamma directly impair epithelial barrier function in cultured mouse cholangiocytes. Liver Int (2003) 23(1):3–11. doi: 10.1034/j.1600-0676.2003.01707.x
93. Locatelli L, Cadamuro M, Spirli C, Fiorotto R, Lecchi S, Morell CM, et al. Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in congenital hepatic fibrosis. Hepatology. (2016) 63(3):965–82. doi: 10.1002/hep.28382
94. Kruglov EA, Nathanson RA, Nguyen T, Dranoff JA. Secretion of MCP-1/CCL2 by bile duct epithelia induces myofibroblastic transdifferentiation of portal fibroblasts. Am J Physiol Gastrointest Liver Physiol (2006) 290(4):G765–71. doi: 10.1152/ajpgi.00308.2005
95. Kinnman N, Francoz C, Barbu V, Wendum D, Rey C, Hultcrantz R, et al. The myofibroblastic conversion of peribiliary fibrogenic cells distinct from hepatic stellate cells is stimulated by platelet-derived growth factor during liver fibrogenesis. Lab Invest (2003) 83(2):163–73. doi: 10.1097/01.LAB.0000054178.01162.E4
96. Han Y, Glaser S, Meng F, Francis H, Marzioni M, McDaniel K, et al. Recent advances in the morphological and functional heterogeneity of the biliary epithelium. Exp Biol Med (Maywood) (2013) 238(5):549–65. doi: 10.1177/1535370213489926
97. O'Hara SP, Tabibian JH, Splinter PL, LaRusso NF. The dynamic biliary epithelia: molecules, pathways, and disease. J Hepatol (2013) 58(3):575–82. doi: 10.1016/j.jhep.2012.10.011
98. Raven A, Lu WY, Man TY, Ferreira-Gonzalez S, O'Duibhir E, Dwyer BJ, et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature. (2017) 547(7663):350–4. doi: 10.1038/nature23015
99. Guicciardi ME, Trussoni CE, LaRusso NF, Gores GJ. The spectrum of reactive cholangiocytes in primary sclerosing cholangitis. Hepatology. (2020) 71(2):741–8. doi: 10.1002/hep.31067
100. Sakisaka S, Gondo K, Yoshitake M, Harada M, Sata M, Kobayashi K, et al. Functional differences between hepatocytes and biliary epithelial cells in handling polymeric immunoglobulin A2 in humans, rats, and guinea pigs. Hepatology. (1996) 24(2):398–406. doi: 10.1002/hep.510240218
101. Brown WR, Kloppel TM. The role of the liver in translocation of IgA into the gastrointestinal tract. Immunol Invest (1989) 18(1-4):269–85. doi: 10.3109/08820138909112242
102. Yokoyama T, Komori A, Nakamura M, Takii Y, Kamihira T, Shimoda S, et al. Human intrahepatic biliary epithelial cells function in innate immunity by producing IL-6 and IL-8 via the TLR4-NF-kappaB and -MAPK signaling pathways. Liver Int (2006) 26(4):467–76. doi: 10.1111/j.1478-3231.2006.01254.x
103. Rupp C, Bode KA, Leopold Y, Sauer P, Gotthardt DN. Pathological features of primary sclerosing cholangitis identified by bile proteomic analysis. Biochim Biophys Acta Mol Basis Dis (2018) 1864(4 Pt B):1380–9. doi: 10.1016/j.bbadis.2017.09.012
104. Harada K, Ohba K, Ozaki S, Isse K, Hirayama T, Wada A, et al. Peptide antibiotic human beta-defensin-1 and -2 contribute to antimicrobial defense of the intrahepatic biliary tree. Hepatology. (2004) 40(4):925–32. doi: 10.1002/hep.20379
105. Meadows V, Baiocchi L, Kundu D, Sato K, Fuentes Y, Wu C, et al. Biliary epithelial senescence in liver disease: there will be SASP. Front Mol Biosci (2021) 8:803098. doi: 10.3389/fmolb.2021.803098
106. Marra F, DeFranco R, Grappone C, Milani S, Pastacaldi S, Pinzani M, et al. Increased expression of monocyte chemotactic protein-1 during active hepatic fibrogenesis: correlation with monocyte infiltration. Am J Pathol (1998) 152(2):423–30.
107. Mueller T, Beutler C, Pico AH, Shibolet O, Pratt DS, Pascher A, et al. Enhanced innate immune responsiveness and intolerance to intestinal endotoxins in human biliary epithelial cells contributes to chronic cholangitis. Liver Int (2011) 31(10):1574–88. doi: 10.1111/j.1478-3231.2011.02635.x
108. Isse K, Harada K, Nakanuma Y. IL-8 expression by biliary epithelial cells is associated with neutrophilic infiltration and reactive bile ductules. Liver Int (2007) 27(5):672–80. doi: 10.1111/j.1478-3231.2007.01465.x
109. Isse K, Harada K, Zen Y, Kamihira T, Shimoda S, Harada M, et al. Fractalkine and CX3CR1 are involved in the recruitment of intraepithelial lymphocytes of intrahepatic bile ducts. Hepatology. (2005) 41(3):506–16. doi: 10.1002/hep.20582
110. Chusilp S, Lee C, Li B, Lee D, Yamoto M, Ganji N, et al. A novel model of injured liver ductal organoids to investigate cholangiocyte apoptosis with relevance to biliary atresia. Pediatr Surg Int (2020) 36(12):1471–9. doi: 10.1007/s00383-020-04765-2
111. Maroni L, Agostinelli L, Saccomanno S, Pinto C, Giordano DM, Rychlicki C, et al. Nlrp3 activation induces il-18 synthesis and affects the epithelial barrier function in reactive cholangiocytes. Am J Pathol (2017) 187(2):366–76. doi: 10.1016/j.ajpath.2016.10.010
112. Guillot A, Guerri L, Feng D, Kim SJ, Ahmed YA, Paloczi J, et al. Bile acid-activated macrophages promote biliary epithelial cell proliferation through integrin alphavbeta6 upregulation following liver injury. J Clin Invest (2021) 131(9):e132305. doi: 10.1172/JCI132305
113. Ferreira-Gonzalez S, Lu WY, Raven A, Dwyer B, Man TY, O'Duibhir E, et al. Paracrine cellular senescence exacerbates biliary injury and impairs regeneration. Nat Commun (2018) 9(1):1020. doi: 10.1038/s41467-018-03299-5
114. Kennedy L, Francis H, Invernizzi P, Venter J, Wu N, Carbone M, et al. Secretin/secretin receptor signaling mediates biliary damage and liver fibrosis in early-stage primary biliary cholangitis. FASEB J (2019) 33(9):10269–79. doi: 10.1096/fj.201802606R
115. Barron-Millar B, Ogle L, Mells G, Flack S, Badrock J, Sandford R, et al. The serum proteome and ursodeoxycholic acid response in primary biliary cholangitis. Hepatology. (2021) 74(6):3269–83. doi: 10.1002/hep.32011
116. Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Modulation of the microenvironment by senescent biliary epithelial cells may be involved in the pathogenesis of primary biliary cirrhosis. J Hepatol (2010) 53(2):318–25. doi: 10.1016/j.jhep.2010.03.008
117. Tabibian JH, O'Hara SP, Splinter PL, Trussoni CE, LaRusso NF. Cholangiocyte senescence by way of n-ras activation is a characteristic of primary sclerosing cholangitis. Hepatology. (2014) 59(6):2263–75. doi: 10.1002/hep.26993
118. Sasaki M, Sato Y, Nakanuma Y. Interferon-induced protein with tetratricopeptide repeats 3 may be a key factor in primary biliary cholangitis. Sci Rep (2021) 11(1):11413. doi: 10.1038/s41598-021-91016-6
119. Alsuraih M, O'Hara SP, Woodrum JE, Pirius NE, LaRusso NF. Genetic or pharmacological reduction of cholangiocyte senescence improves inflammation and fibrosis in the Mdr2 (-/-) mouse. JHEP Rep (2021) 3(3):100250. doi: 10.1016/j.jhepr.2021.100250
120. Srivatsa G, Giraud AS, Ulaganathan M, Yeomans ND, Dow C, Nicoll AJ. Biliary epithelial trefoil peptide expression is increased in biliary diseases. Histopathology. (2002) 40(3):261–8. doi: 10.1046/j.1365-2559.2002.01347.x
121. Saito K, Nakanuma Y. Lactoferrin and lysozyme in the intrahepatic bile duct of normal livers and hepatolithiasis. an immunohistochemical study. J Hepatol (1992) 15(1-2):147–53. doi: 10.1016/0168-8278(92)90028-N
122. D'Aldebert E, Biyeyeme Bi Mve MJ, Mergey M, Wendum D, Firrincieli D, Coilly A, et al. Bile salts control the antimicrobial peptide cathelicidin through nuclear receptors in the human biliary epithelium. Gastroenterology. (2009) 136(4):1435–43. doi: 10.1053/j.gastro.2008.12.040
123. Alvaro D, Metalli VD, Alpini G, Onori P, Franchitto A, Barbaro B, et al. The intrahepatic biliary epithelium is a target of the growth hormone/insulin-like growth factor 1 axis. J Hepatol (2005) 43(5):875–83. doi: 10.1016/j.jhep.2005.04.011
124. Hay AJ, Zhu J. In sickness and in health: the relationships between bacteria and bile in the human gut. Adv Appl Microbiol (2016) 96:43–64. doi: 10.1016/bs.aambs.2016.07.019
125. Ye F, Shen H, Li Z, Meng F, Li L, Yang J, et al. Influence of the biliary system on biliary bacteria revealed by bacterial communities of the human biliary and upper digestive tracts. PloS One (2016) 11(3):e0150519. doi: 10.1371/journal.pone.0150519
126. Sung JY, Costerton JW, Shaffer EA. Defense system in the biliary tract against bacterial infection. Dig Dis Sci (1992) 37(5):689–96. doi: 10.1007/BF01296423
127. MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun (2018) 9(1):4383. doi: 10.1038/s41467-018-06318-7
128. Farina A, Dumonceau JM, Delhaye M, Frossard JL, Hadengue A, Hochstrasser DF, et al. A step further in the analysis of human bile proteome. J Proteome Res (2011) 10(4):2047–63. doi: 10.1021/pr200011b
129. Schrumpf E, Tan C, Karlsen TH, Sponheim J, Bjorkstrom NK, Sundnes O, et al. The biliary epithelium presents antigens to and activates natural killer T cells. Hepatology. (2015) 62(4):1249–59. doi: 10.1002/hep.27840
130. Jeffery HC, van Wilgenburg B, Kurioka A, Parekh K, Stirling K, Roberts S, et al. Biliary epithelium and liver b cells exposed to bacteria activate intrahepatic MAIT cells through MR1. J Hepatol (2016) 64(5):1118–27. doi: 10.1016/j.jhep.2015.12.017
131. Mohamad Zaki NH, Shiota J, Calder AN, Keeley TM, Allen BL, Nakao K, et al. C-X-C motif chemokine ligand 1 induced by hedgehog signaling promotes mouse extrahepatic bile duct repair after acute injury. Hepatology. (2022) 76(4):936–50. doi: 10.1002/hep.32492
132. Ehrlich L, Scrushy M, Meng F, Lairmore TC, Alpini G, Glaser S. Biliary epithelium: a neuroendocrine compartment in cholestatic liver disease. Clin Res Hepatol Gastroenterol (2018) 42(4):296–305. doi: 10.1016/j.clinre.2018.03.009
133. Strazzabosco M, Fiorotto R, Cadamuro M, Spirli C, Mariotti V, Kaffe E, et al. Pathophysiologic implications of innate immunity and autoinflammation in the biliary epithelium. Biochim Biophys Acta Mol Basis Dis (2018) 1864(4 Pt B):1374–9. doi: 10.1016/j.bbadis.2017.07.023
134. Yasoshima M, Kono N, Sugawara H, Katayanagi K, Harada K, Nakanuma Y. Increased expression of interleukin-6 and tumor necrosis factor-alpha in pathologic biliary epithelial cells: in situ and culture study. Lab Invest (1998) 78(1):89–100.
135. Yamashita M, Adachi T, Ono S, Yoshino K, Imamura H, Matsushima H, et al. Helicobacter bilis infection induces oxidative stress in and enhances the proliferation of human cholangiocytes. Helicobacter. (2022) 27(4):e12908. doi: 10.1111/hel.12908
136. Ninlawan K, O'Hara SP, Splinter PL, Yongvanit P, Kaewkes S, Surapaitoon A, et al. Opisthorchis viverrini excretory/secretory products induce toll-like receptor 4 upregulation and production of interleukin 6 and 8 in cholangiocyte. Parasitol Int (2010) 59(4):616–21. doi: 10.1016/j.parint.2010.09.008
137. Cai X, Guillot A, Liu H. Cellular senescence in hepatocellular carcinoma: the passenger or the driver? Cells. (2022) 12(1):132. doi: 10.3390/cells12010132
138. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res (1961) 25:585–621. doi: 10.1016/0014-4827(61)90192-6
139. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol (2018) 28(6):436–53. doi: 10.1016/j.tcb.2018.02.001
140. Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev (1998) 12(19):3008–19. doi: 10.1101/gad.12.19.3008
141. Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. (2006) 444(7119):633–7. doi: 10.1038/nature05268
142. Rai TS, Cole JJ, Nelson DM, Dikovskaya D, Faller WJ, Vizioli MG, et al. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev (2014) 28(24):2712–25. doi: 10.1101/gad.247528.114
143. Chandra T, Kirschner K, Thuret JY, Pope BD, Ryba T, Newman S, et al. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol Cell (2012) 47(2):203–14. doi: 10.1016/j.molcel.2012.06.010
144. Wan Y, McDaniel K, Wu N, Ramos-Lorenzo S, Glaser T, Venter J, et al. Regulation of cellular senescence by miR-34a in alcoholic liver injury. Am J Pathol (2017) 187(12):2788–98. doi: 10.1016/j.ajpath.2017.08.027
145. Tabibian JH, Trussoni CE, O'Hara SP, Splinter PL, Heimbach JK, LaRusso NF. Characterization of cultured cholangiocytes isolated from livers of patients with primary sclerosing cholangitis. Lab Invest (2014) 94(10):1126–33. doi: 10.1038/labinvest.2014.94
146. Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell (2016) 30(4):533–47. doi: 10.1016/j.ccell.2016.09.003
147. Milanovic M, Fan DNY, Belenki D, Dabritz JHM, Zhao Z, Yu Y, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. (2018) 553(7686):96–100. doi: 10.1038/nature25167
148. Povero D, Tameda M, Eguchi A, Ren W, Kim J, Myers R, et al. Protein and miRNA profile of circulating extracellular vesicles in patients with primary sclerosing cholangitis. Sci Rep (2022) 12(1):3027. doi: 10.1038/s41598-022-06809-0
149. Al Suraih MS, Trussoni CE, Splinter PL, LaRusso NF, O'Hara SP. Senescent cholangiocytes release extracellular vesicles that alter target cell phenotype via the epidermal growth factor receptor. Liver Int (2020) 40(10):2455–68. doi: 10.1111/liv.14569
150. Zhou T, Kundu D, Robles-Linares J, Meadows V, Sato K, Baiocchi L, et al. Feedback signaling between cholangiopathies, ductular reaction, and non-alcoholic fatty liver disease. Cells (2021) 10(8):2072. doi: 10.3390/cells10082072
151. Campana L, Esser H, Huch M, Forbes S. Liver regeneration and inflammation: from fundamental science to clinical applications. Nat Rev Mol Cell Biol (2021) 22(9):608–24. doi: 10.1038/s41580-021-00373-7
152. De Assuncao TM, Jalan-Sakrikar N, Huebert RC. Regenerative medicine and the biliary tree. Semin Liver Dis (2017) 37(1):17–27. doi: 10.1055/s-0036-1597818
153. Michalopoulos GK. Liver regeneration. J Cell Physiol (2007) 213(2):286–300. doi: 10.1002/jcp.21172
154. Uriarte I, Fernandez-Barrena MG, Monte MJ, Latasa MU, Chang HC, Carotti S, et al. Identification of fibroblast growth factor 15 as a novel mediator of liver regeneration and its application in the prevention of post-resection liver failure in mice. Gut. (2013) 62(6):899–910. doi: 10.1136/gutjnl-2012-302945
155. Zhao M, Quan Y, Zeng J, Lyu X, Wang H, Lei JH, et al. Cullin3 deficiency shapes tumor microenvironment and promotes cholangiocarcinoma in liver-specific Smad4/Pten mutant mice. Int J Biol Sci (2021) 17(15):4176–91. doi: 10.7150/ijbs.67379
156. Claperon A, Mergey M, Aoudjehane L, Ho-Bouldoires TH, Wendum D, Prignon A, et al. Hepatic myofibroblasts promote the progression of human cholangiocarcinoma through activation of epidermal growth factor receptor. Hepatology. (2013) 58(6):2001–11. doi: 10.1002/hep.26585
157. Kirillova I, Chaisson M, Fausto N. Tumor necrosis factor induces DNA replication in hepatic cells through nuclear factor kappaB activation. Cell Growth Differ (1999) 10(12):819–28.
158. Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol (2004) 5(10):836–47. doi: 10.1038/nrm1489
159. Mancinelli R, Onori P, Gaudio E, Franchitto A, Carpino G, Ueno Y, et al. Taurocholate feeding to bile duct ligated rats prevents caffeic acid-induced bile duct damage by changes in cholangiocyte VEGF expression. Exp Biol Med (Maywood) (2009) 234(4):462–74. doi: 10.3181/0808-RM-255
160. Gaudio E, Onori P, Pannarale L, Alvaro D. Hepatic microcirculation and peribiliary plexus in experimental biliary cirrhosis: a morphological study. Gastroenterology. (1996) 111(4):1118–24. doi: 10.1016/S0016-5085(96)70081-1
161. Bruneau A, Guillot A, Tacke F. Macrophages in cholangiopathies. Curr Opin Gastroenterol (2022) 38(2):114–20. doi: 10.1097/MOG.0000000000000814
162. Govaere O, Cockell S, Van Haele M, Wouters J, Van Delm W, Van den Eynde K, et al. High-throughput sequencing identifies aetiology-dependent differences in ductular reaction in human chronic liver disease. J Pathol (2019) 248(1):66–76. doi: 10.1002/path.5228
163. Vesterhus M, Holm A, Hov JR, Nygard S, Schrumpf E, Melum E, et al. Novel serum and bile protein markers predict primary sclerosing cholangitis disease severity and prognosis. J Hepatol (2017) 66(6):1214–22. doi: 10.1016/j.jhep.2017.01.019
164. Guillot A, Winkler M, Silva Afonso M, Aggarwal A, Lopez D, Berger H, et al. Mapping the hepatic immune landscape identifies monocytic macrophages as key drivers of steatohepatitis and cholangiopathy progression. Hepatology (2023). Online ahead of print. doi: 10.1097/HEP.0000000000000270
165. Poch T, Krause J, Casar C, Liwinski T, Glau L, Kaufmann M, et al. Single-cell atlas of hepatic T cells reveals expansion of liver-resident naive-like CD4(+) T cells in primary sclerosing cholangitis. J Hepatol (2021) 75(2):414–23. doi: 10.1016/j.jhep.2021.03.016
166. Guillot A, Gasmi I, Brouillet A, Ait-Ahmed Y, Calderaro J, Ruiz I, et al. Interleukins-17 and 27 promote liver regeneration by sequentially inducing progenitor cell expansion and differentiation. Hepatol Commun (2018) 2(3):329–43. doi: 10.1002/hep4.1145
167. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med (2005) 201(2):233–40. doi: 10.1084/jem.20041257
168. Shimoyama S, Kawata K, Ohta K, Chida T, Suzuki T, Tsuneyama K, et al. Ursodeoxycholic acid impairs liver-infiltrating T-cell chemotaxis through IFN-gamma and CX3CL1 production in primary biliary cholangitis. Eur J Immunol (2021) 51(6):1519–30. doi: 10.1002/eji.202048589
169. Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. (1997) 91(4):521–30. doi: 10.1016/S0092-8674(00)80438-9
170. Mehta AK, Gracias DT, Croft M. TNF activity and T cells. Cytokine. (2018) 101:14–8. doi: 10.1016/j.cyto.2016.08.003
171. Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol (2010) 40(7):1830–5. doi: 10.1002/eji.201040391
172. Wu CT, Davis PA, Luketic VA, Gershwin ME. A review of the physiological and immunological functions of biliary epithelial cells: targets for primary biliary cirrhosis, primary sclerosing cholangitis and drug-induced ductopenias. Clin Dev Immunol (2004) 11(3-4):205–13. doi: 10.1080/17402520400004177
173. Roohani S, Tacke F. Liver injury and the macrophage issue: molecular and mechanistic facts and their clinical relevance. Int J Mol Sci (2021) 22(14):7249. doi: 10.3390/ijms22147249
174. Guillot A, Tacke F. Liver macrophages: old dogmas and new insights. Hepatol Commun (2019) 3(6):730–43. doi: 10.1002/hep4.1356
175. Kunzmann LK, Schoknecht T, Poch T, Henze L, Stein S, Kriz M, et al. Monocytes as potential mediators of pathogen-induced T-helper 17 differentiation in patients with primary sclerosing cholangitis (PSC). Hepatology. (2020) 72(4):1310–26. doi: 10.1002/hep.31140
176. Pi L, Robinson PM, Jorgensen M, Oh SH, Brown AR, Weinreb PH, et al. Connective tissue growth factor and integrin alphavbeta6: a new pair of regulators critical for ductular reaction and biliary fibrosis in mice. Hepatology. (2015) 61(2):678–91. doi: 10.1002/hep.27425
177. Sedlaczek N, Jia JD, Bauer M, Herbst H, Ruehl M, Hahn EG, et al. Proliferating bile duct epithelial cells are a major source of connective tissue growth factor in rat biliary fibrosis. Am J Pathol (2001) 158(4):1239–44. doi: 10.1016/S0002-9440(10)64074-6
178. Aseem SO, Jalan-Sakrikar N, Chi C, Navarro-Corcuera A, De Assuncao TM, Hamdan FH, et al. Epigenomic evaluation of cholangiocyte transforming growth factor-beta signaling identifies a selective role for histone 3 lysine 9 acetylation in biliary fibrosis. Gastroenterology. (2021) 160(3):889–905 e10. doi: 10.1053/j.gastro.2020.10.008
179. Grappone C, Pinzani M, Parola M, Pellegrini G, Caligiuri A, DeFranco R, et al. Expression of platelet-derived growth factor in newly formed cholangiocytes during experimental biliary fibrosis in rats. J Hepatol (1999) 31(1):100–9. doi: 10.1016/S0168-8278(99)80169-X
180. Moncsek A, Al-Suraih MS, Trussoni CE, O'Hara SP, Splinter PL, Zuber C, et al. Targeting senescent cholangiocytes and activated fibroblasts with b-cell lymphoma-extra large inhibitors ameliorates fibrosis in multidrug resistance 2 gene knockout (Mdr2(-/-) ) mice. Hepatology. (2018) 67(1):247–59. doi: 10.1002/hep.29464
181. Liu R, Li X, Zhu W, Wang Y, Zhao D, Wang X, et al. Cholangiocyte-derived exosomal long noncoding RNA H19 promotes hepatic stellate cell activation and cholestatic liver fibrosis. Hepatology. (2019) 70(4):1317–35. doi: 10.1002/hep.30662
182. Slack RJ, Macdonald SJF, Roper JA, Jenkins RG, Hatley RJD. Emerging therapeutic opportunities for integrin inhibitors. Nat Rev Drug Discovery (2022) 21(1):60–78. doi: 10.1038/s41573-021-00284-4
183. Koivisto L, Bi J, Hakkinen L, Larjava H. Integrin alphavbeta6: structure, function and role in health and disease. Int J Biochem Cell Biol (2018) 99:186–96. doi: 10.1016/j.biocel.2018.04.013
184. Peng ZW, Ikenaga N, Liu SB, Sverdlov DY, Vaid KA, Dixit R, et al. Integrin alphavbeta6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology. (2016) 63(1):217–32. doi: 10.1002/hep.28274
185. Popov Y, Patsenker E, Stickel F, Zaks J, Bhaskar KR, Niedobitek G, et al. Integrin alphavbeta6 is a marker of the progression of biliary and portal liver fibrosis and a novel target for antifibrotic therapies. J Hepatol (2008) 48(3):453–64. doi: 10.1016/j.jhep.2007.11.021
186. Beaufrere A, Calderaro J, Paradis V. Combined hepatocellular-cholangiocarcinoma: an update. J Hepatol (2021) 74(5):1212–24. doi: 10.1016/j.jhep.2021.01.035
187. Xu J, Lin H, Wu G, Zhu M, Li M. IL-6/STAT3 is a promising therapeutic target for hepatocellular carcinoma. Front Oncol (2021) 11:760971. doi: 10.3389/fonc.2021.760971
188. Zhou YF, Song SS, Tian MX, Tang Z, Wang H, Fang Y, et al. Cystathionine beta-synthase mediated PRRX2/IL-6/STAT3 inactivation suppresses tregs infiltration and induces apoptosis to inhibit HCC carcinogenesis. J Immunother Cancer (2021) 9(8):e003031. doi: 10.1136/jitc-2021-003031
189. Isomoto H, Kobayashi S, Werneburg NW, Bronk SF, Guicciardi ME, Frank DA, et al. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology. (2005) 42(6):1329–38. doi: 10.1002/hep.20966
190. Goydos JS, Brumfield AM, Frezza E, Booth A, Lotze MT, Carty SE. Marked elevation of serum interleukin-6 in patients with cholangiocarcinoma: validation of utility as a clinical marker. Ann Surg (1998) 227(3):398–404. doi: 10.1097/00000658-199803000-00012
191. Rosenberg N, Van Haele M, Lanton T, Brashi N, Bromberg Z, Adler H, et al. Combined hepatocellular-cholangiocarcinoma derives from liver progenitor cells and depends on senescence and IL-6 trans-signaling. J Hepatol (2022) 77(6):1631–41. doi: 10.1016/j.jhep.2022.07.029
192. Schmidt-Arras D, Rose-John S. IL-6 pathway in the liver: from physiopathology to therapy. J Hepatol (2016) 64(6):1403–15. doi: 10.1016/j.jhep.2016.02.004
193. Tadlock L, Patel T. Involvement of p38 mitogen-activated protein kinase signaling in transformed growth of a cholangiocarcinoma cell line. Hepatology. (2001) 33(1):43–51. doi: 10.1053/jhep.2001.20676
194. Zhang M, Yang H, Wan L, Wang Z, Wang H, Ge C, et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J Hepatol (2020) 73(5):1118–30. doi: 10.1016/j.jhep.2020.05.039
195. Hennig M, Yip-Schneider MT, Klein P, Wentz S, Matos JM, Doyle C, et al. Ethanol-TGFalpha-MEK signaling promotes growth of human hepatocellular carcinoma. J Surg Res (2009) 154(2):187–95. doi: 10.1016/j.jss.2008.11.836
196. Kadry YA, Lee JY, Witze ES. Regulation of EGFR signalling by palmitoylation and its role in tumorigenesis. Open Biol (2021) 11(10):210033. doi: 10.1098/rsob.210033
197. Claperon A, Mergey M, Nguyen Ho-Bouldoires TH, Vignjevic D, Wendum D, Chretien Y, et al. EGF/EGFR axis contributes to the progression of cholangiocarcinoma through the induction of an epithelial-mesenchymal transition. J Hepatol (2014) 61(2):325–32. doi: 10.1016/j.jhep.2014.03.033
198. Fuchs BC, Hoshida Y, Fujii T, Wei L, Yamada S, Lauwers GY, et al. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology. (2014) 59(4):1577–90. doi: 10.1002/hep.26898
199. Inoue K, Torimura T, Nakamura T, Iwamoto H, Masuda H, Abe M, et al. Vandetanib, an inhibitor of VEGF receptor-2 and EGF receptor, suppresses tumor development and improves prognosis of liver cancer in mice. Clin Cancer Res (2012) 18(14):3924–33. doi: 10.1158/1078-0432.CCR-11-2041
200. Dong ZR, Sun D, Yang YF, Zhou W, Wu R, Wang XW, et al. TMPRSS4 drives angiogenesis in hepatocellular carcinoma by promoting HB-EGF expression and proteolytic cleavage. Hepatology. (2020) 72(3):923–39. doi: 10.1002/hep.31076
201. Kandhi R, Bobbala D, Yeganeh M, Mayhue M, Menendez A, Ilangumaran S. Negative regulation of the hepatic fibrogenic response by suppressor of cytokine signaling 1. Cytokine. (2016) 82:58–69. doi: 10.1016/j.cyto.2015.12.007
202. Chen J, Gingold JA, Su X. Immunomodulatory TGF-beta signaling in hepatocellular carcinoma. Trends Mol Med (2019) 25(11):1010–23. doi: 10.1016/j.molmed.2019.06.007
203. Sirica AE, Gores GJ, Groopman JD, Selaru FM, Strazzabosco M, Wei Wang X, et al. Intrahepatic cholangiocarcinoma: continuing challenges and translational advances. Hepatology. (2019) 69(4):1803–15. doi: 10.1002/hep.30289
204. Puthdee N, Sriswasdi S, Pisitkun T, Ratanasirintrawoot S, Israsena N, Tangkijvanich P. The LIN28B/TGF-beta/TGFBI feedback loop promotes cell migration and tumour initiation potential in cholangiocarcinoma. Cancer Gene Ther (2022) 29(5):445–55. doi: 10.1038/s41417-021-00387-5
205. Wu MZ, Yuan YC, Huang BY, Chen JX, Li BK, Fang JH, et al. Identification of a TGF-beta/SMAD/lnc-UTGF positive feedback loop and its role in hepatoma metastasis. Signal Transduct Target Ther (2021) 6(1):395. doi: 10.1038/s41392-021-00781-3
206. Mahpour A, Mullen AC. Our emerging understanding of the roles of long non-coding RNAs in normal liver function, disease, and malignancy. JHEP Rep (2021) 3(1):100177. doi: 10.1016/j.jhepr.2020.100177
207. Zhang D, Li H, Jiang X, Cao L, Wen Z, Yang X, et al. Role of AP-2alpha and MAPK7 in the regulation of autocrine TGF-beta/miR-200b signals to maintain epithelial-mesenchymal transition in cholangiocarcinoma. J Hematol Oncol (2017) 10(1):170. doi: 10.1186/s13045-017-0528-6
208. Maemura K, Natsugoe S, Takao S. Molecular mechanism of cholangiocarcinoma carcinogenesis. J Hepatobiliary Pancreat Sci (2014) 21(10):754–60. doi: 10.1002/jhbp.126
209. Chen W, Ten Dijke P. Immunoregulation by members of the TGFbeta superfamily. Nat Rev Immunol (2016) 16(12):723–40. doi: 10.1038/nri.2016.112
210. Thepmalee C, Panya A, Junking M, Chieochansin T, Yenchitsomanus PT. Inhibition of IL-10 and TGF-beta receptors on dendritic cells enhances activation of effector T-cells to kill cholangiocarcinoma cells. Hum Vaccin Immunother (2018) 14(6):1423–31. doi: 10.1080/21645515.2018.1431598
211. Yan W, Liu X, Ma H, Zhang H, Song X, Gao L, et al. Tim-3 fosters HCC development by enhancing TGF-beta-mediated alternative activation of macrophages. Gut. (2015) 64(10):1593–604. doi: 10.1136/gutjnl-2014-307671
212. Ning J, Ye Y, Bu D, Zhao G, Song T, Liu P, et al. Imbalance of TGF-beta1/BMP-7 pathways induced by M2-polarized macrophages promotes hepatocellular carcinoma aggressiveness. Mol Ther (2021) 29(6):2067–87. doi: 10.1016/j.ymthe.2021.02.016
213. Cadamuro M, Stecca T, Brivio S, Mariotti V, Fiorotto R, Spirli C, et al. The deleterious interplay between tumor epithelia and stroma in cholangiocarcinoma. Biochim Biophys Acta Mol Basis Dis (2018) 1864(4 Pt B):1435–43. doi: 10.1016/j.bbadis.2017.07.028
214. Mancino A, Mancino MG, Glaser SS, Alpini G, Bolognese A, Izzo L, et al. Estrogens stimulate the proliferation of human cholangiocarcinoma by inducing the expression and secretion of vascular endothelial growth factor. Dig Liver Dis (2009) 41(2):156–63. doi: 10.1016/j.dld.2008.02.015
215. Alvaro D, Barbaro B, Franchitto A, Onori P, Glaser SS, Alpini G, et al. Estrogens and insulin-like growth factor 1 modulate neoplastic cell growth in human cholangiocarcinoma. Am J Pathol (2006) 169(3):877–88. doi: 10.2353/ajpath.2006.050464
216. Lorent K, Yeo SY, Oda T, Chandrasekharappa S, Chitnis A, Matthews RP, et al. Inhibition of jagged-mediated notch signaling disrupts zebrafish biliary development and generates multi-organ defects compatible with an alagille syndrome phenocopy. Development. (2004) 131(22):5753–66. doi: 10.1242/dev.01411
217. Fabris L, Perugorria MJ, Mertens J, Bjorkstrom NK, Cramer T, Lleo A, et al. The tumour microenvironment and immune milieu of cholangiocarcinoma. Liver Int (2019) 39 Suppl 1:63–78. doi: 10.1111/liv.14098
218. Balkwill F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev (2006) 25(3):409–16. doi: 10.1007/s10555-006-9005-3
219. Yao X, Wang X, Wang Z, Dai L, Zhang G, Yan Q, et al. Clinicopathological and prognostic significance of epithelial mesenchymal transition-related protein expression in intrahepatic cholangiocarcinoma. Onco Targets Ther (2012) 5:255–61. doi: 10.2147/OTT.S36213
220. Techasen A, Namwat N, Loilome W, Duangkumpha K, Puapairoj A, Saya H, et al. Tumor necrosis factor-alpha modulates epithelial mesenchymal transition mediators ZEB2 and S100A4 to promote cholangiocarcinoma progression. J Hepatobiliary Pancreat Sci (2014) 21(9):703–11. doi: 10.1002/jhbp.125
221. Yuan D, Huang S, Berger E, Liu L, Gross N, Heinzmann F, et al. Kupffer cell-derived tnf triggers cholangiocellular tumorigenesis through JNK due to chronic mitochondrial dysfunction and ROS. Cancer Cell (2017) 31(6):771–89 e6. doi: 10.1016/j.ccell.2017.05.006
222. Guillot A, Tacke F. Location, location, location - spatial insight into hepatic macrophage populations. Nat Rev Gastroenterol Hepatol (2022) 19(5):281–2. doi: 10.1038/s41575-022-00600-2
223. Sheng J, Zhang J, Wang L, Tano V, Tang J, Wang X, et al. Topological analysis of hepatocellular carcinoma tumour microenvironment based on imaging mass cytometry reveals cellular neighbourhood regulated reversely by macrophages with different ontogeny. Gut. (2022) 71(6):1176–91. doi: 10.1136/gutjnl-2021-324339
224. Guillot A, Kohlhepp MS, Bruneau A, Heymann F, Tacke F. Deciphering the immune microenvironment on a single archival formalin-fixed paraffin-embedded tissue section by an immediately implementable multiplex fluorescence immunostaining protocol. Cancers (Basel) (2020) 12(9):2449. doi: 10.3390/cancers12092449
Keywords: biliary epithelial cells, cholangiocyte secretome, cholangiopathies, ductular reaction, cellular senescence, inflammation, fibrosis, hepatic carcinogenesis
Citation: Cai X, Tacke F, Guillot A and Liu H (2023) Cholangiokines: undervalued modulators in the hepatic microenvironment. Front. Immunol. 14:1192840. doi: 10.3389/fimmu.2023.1192840
Received: 24 March 2023; Accepted: 02 May 2023;
Published: 16 May 2023.
Edited by:
Jinhang Gao, Sichuan University, ChinaReviewed by:
Christy Trussoni, Mayo Clinic, United StatesCopyright © 2023 Cai, Tacke, Guillot and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hanyang Liu, aGFueWFuZy5saXVAY2hhcml0ZS5kZQ==; Adrien Guillot, YWRyaWVuLmd1aWxsb3RAY2hhcml0ZS5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.