 Victoria Navarro-Compán1†
Victoria Navarro-Compán1† Luis Puig2†
Luis Puig2† Silvia Vidal3
Silvia Vidal3 Julio Ramírez4
Julio Ramírez4 Mar Llamas-Velasco5
Mar Llamas-Velasco5 Cristina Fernández-Carballido6Raquel Almodóvar7
Cristina Fernández-Carballido6Raquel Almodóvar7 José Antonio Pinto8
José Antonio Pinto8 Eva Galíndez-Aguirregoikoa9Pedro Zarco7
Eva Galíndez-Aguirregoikoa9Pedro Zarco7 Beatriz Joven10Jordi Gratacós11Xavier Juanola12
Beatriz Joven10Jordi Gratacós11Xavier Juanola12 Ricardo Blanco13
Ricardo Blanco13 Salvador Arias-Santiago14,15,16
Salvador Arias-Santiago14,15,16 Jesús Sanz Sanz17
Jesús Sanz Sanz17 Rubén Queiro18*‡
Rubén Queiro18*‡ Juan D. Cañete4*‡
Juan D. Cañete4*‡- 1Department of Rheumatology, Hospital Universitario La Paz, IdiPaz, Madrid, Spain
- 2Department of Dermatology, Hospital de la Santa Creu i Sant Pau, Barcelona, Spain
- 3Immunology-Inflammatory Diseases, Institut de Recerca de l’Hospital de la Santa Creu i Sant Pau, Biomedical Research Institute Sant Pau (IIB Sant Pau), Barcelona, Spain
- 4Arthritis Unit, Department of Rheumatology, Hospital Clínic and Instituto de Investigaciones Biomédicas August Pi i Sunyer (IDIBAPS), Barcelona, Spain
- 5Department of Dermatology, Hospital Universitario La Princesa, Madrid, Spain
- 6Department of Rheumatology, Hospital Universitario San Juan de Alicante, Alicante, Spain
- 7Department of Rheumatology, Hospital Universitario Fundación Alcorcón, Alcorcón, Madrid, Spain
- 8Department of Rheumatology, Complejo Hospitalario Universitario de A Coruña, Instituto de Investigación Biomédica de A Coruña (INIBIC), A Coruña, Spain
- 9Department of Rheumatology, Hospital Universitario de Basurto, Bilbao, Spain
- 10Department of Rheumatology, Hospital Universitario 12 de Octubre, Madrid, Spain
- 11Department of Rheumatology, Medicine Department Autonomus University of Barcelona (UAB), I3PT, University Hospital Parc Taulí Sabadell, Barcelona, Spain
- 12Department of Rheumatology, University Hospital Bellvitge, Instituto de Investigación Biomédica de Bellvitge (IDIBELL), Barcelona, Spain
- 13Department of Rheumatology, Hospital Universitario Marqués de Valdecilla, Instituto de Investigación Marqués de Valdecilla (IDIVAL), Santander, Spain
- 14Department of Dermatology, Hospital Universitario Virgen de las Nieves, Granada, Spain
- 15Instituto de Investigación Biosanitaria ibs.GRANADA, Granada, Spain
- 16Department of Dermatology, Facultad de Medicina, Universidad de Granada, Granada, Spain
- 17Department of Rheumatology, Hospital Universitario Puerta del Hierro Majadahonda, Madrid, Spain
- 18Department of Rheumatology, Hospital Universitario Central de Asturias, Oviedo, Asturias, Spain
Interleukin-17 family (IL-17s) comprises six structurally related members (IL-17A to IL-17F); sequence homology is highest between IL-17A and IL-17F, displaying certain overlapping functions. In general, IL-17A and IL-17F play important roles in chronic inflammation and autoimmunity, controlling bacterial and fungal infections, and signaling mainly through activation of the nuclear factor-kappa B (NF-κB) pathway. The role of IL-17A and IL-17F has been established in chronic immune-mediated inflammatory diseases (IMIDs), such as psoriasis (PsO), psoriatic arthritis (PsA), axial spondylarthritis (axSpA), hidradenitis suppurativa (HS), inflammatory bowel disease (IBD), multiple sclerosis (MS), and asthma. CD4+ helper T cells (Th17) activated by IL-23 are well-studied sources of IL-17A and IL-17F. However, other cellular subtypes can also produce IL-17A and IL-17F, including gamma delta (γδ) T cells, alpha beta (αβ) T cells, type 3 innate lymphoid cells (ILC3), natural killer T cells (NKT), or mucosal associated invariant T cells (MAIT). Interestingly, the production of IL-17A and IL-17F by innate and innate-like lymphocytes can take place in an IL-23 independent manner in addition to IL-23 classical pathway. This would explain the limitations of the inhibition of IL-23 in the treatment of patients with certain rheumatic immune-mediated conditions such as axSpA. Despite their coincident functions, IL-17A and IL-17F contribute independently to chronic tissue inflammation having somehow non-redundant roles. Although IL-17A has been more widely studied, both IL-17A and IL-17F are overexpressed in PsO, PsA, axSpA and HS. Therefore, dual inhibition of IL-17A and IL-17F could provide better outcomes than IL-23 or IL-17A blockade.
1 Introduction
Interleukin (IL)-17A and IL-17F are a pair of rather newly described pro-inflammatory cytokines capable of bridging the adaptative and innate immune systems. Both cytokines have a role in maintaining epithelial barrier function (skin, intestinal epithelium, gingiva, vaginal mucosa, and conjunctiva) and providing protection against pathogens (1). However, alterations in the regulation and excess of these two cytokines have pathogenetic implications in chronic immune-mediated inflammatory diseases (IMIDs), including psoriasis (PsO), psoriatic arthritis (PsA), axial spondyloarthritis (axSpA), hidradenitis suppurativa (HS), and inflammatory bowel disease (IBD) (2).
IL-17A and IL-17F are produced by cells of the innate and adaptative immune system and activate the production of inflammatory mediators such as tumor necrosis factor α (TNFα), IL-1β, IL-6, granulocyte colony-stimulating factor (G-CSF), and granulocyte-macrophage colony-stimulating factor (GM-CSF). They also induce the production of chemokines, including CXCL1, CXCL5, CCL2, and CCL7, and the expression of antimicrobial peptides (AMPs), which mediate the activation and recruitment of inflammatory cells such as neutrophils (3).
IL-23 is a cytokine particularly important in maintaining the differentiation state of Th17 cells, the best-known producer of IL-17A and IL-17F. In this context, the discovery of the IL-23/IL-17 axis prompted the development of several therapeutic strategies for autoimmune disorders and chronic inflammation (4–6). To date, both IL-23p19 and IL-12/23 inhibitors (guselkumab, risankizumab, tildrakizumab, and ustekinumab), as well as IL-17RA and IL-17As inhibitors (brodalumab, ixekizumab, and secukinumab) have been tested in several IMIDs, including PsO, PsA, axSpA, IBD and HS (Table 1). However, their blockade did not yield the same clinical outcomes in all IMIDs (4, 6–10). A possible explanation is tissue specificity of these cytokines (11, 12). One example is the lack of efficacy of anti-IL-12/23 and anti-IL-23p19 monoclonal antibodies (mAb) therapy in axSpA and HS, unlike IL-17A inhibitors (13–19). Studies performed in animal models provided a plausible explanation for this differential behavior as IL-23 was found to be required for the initiation but not for the maintenance of the disease (20). Conversely, the inhibition of the IL-23 provides good results in IBD (21), bringing about the approval of biological treatments whereas neutralization of IL-17A did not (22, 23). However, IL-17F suppression was effective in a colitis mouse model, indicating that IL-17A and IL-17F could have differential roles in the gut (24). Indeed, recent studies suggest that IL-17A and IL-17F could have protective and pathogenic roles in the gut, respectively (24–27). Moreover, beyond Th17 cells, innate cells and innate-like lymphocytes also produce IL-17s in an IL-23-independent manner. Several studies have stressed that in certain contexts innate cells can be a major source of IL-17A and IL-17F and that they can produce these cytokines regardless of IL-23 stimulus, playing important roles in inflammation and autoimmunity (28, 29). Clinical and pre-clinical studies in PsO and SpA suggested that targeting IL-17F in addition to IL-17A could lead to increased suppression of proinflammatory genes (30, 31), reduced migration of both adaptive and innate immune cell types (31) and reduced periosteal stem cell bone formation (32), compared with the inhibition of IL-17A alone. Consequently, dual or bispecific inhibitors, such as bimekizumab or sonelokimab, have been developed with promising results in IMIDs (33). Altogether the evidence suggests a non-linear relationship between IL-23 and the IL-17s, as well as tissue-specific functions of IL-17A and IL-17F.
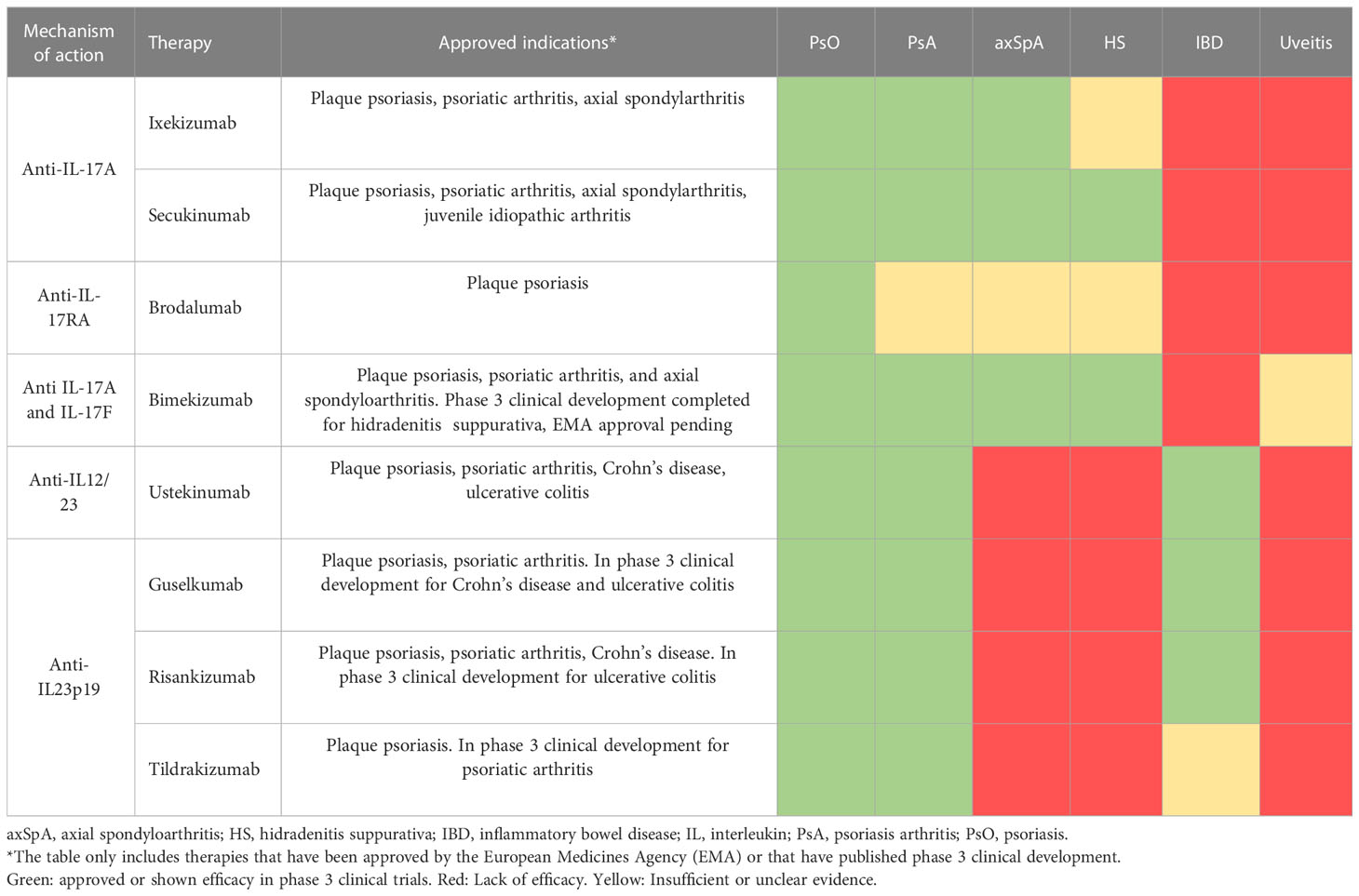
Table 1 Available therapies targeting IL-17 and IL-23 in IMIDs.
Thus, the role of IL-17A and IL-17F in IMIDs is complex, involving the adaptative and innate immune system, and cytokine signaling beyond the IL-23/IL-17 axis. The present narrative review aims to examine the evidence about the involvement of IL-17A and IL-17F in inflammatory diseases exploring alternative innate sources and IL-23-independent signaling, which represent alternative escape routes that perpetuate the inflammatory loop and can influence treatment choice.
2 Biology of IL-17s and their receptors
IL-17A is considered the founding member of the IL-17 family and thus is the most studied. However, the family includes five additional members, as revealed by sequence homology studies: IL-17B, IL-17C, IL-17D, IL-17E (also named IL-25), and IL-17F (34, 35). Among the IL-17 family members, the roles and functions of IL-17A and IL-17F are intertwined partly due to their high amino acid sequence homology (around 50%) and their shared common evolution, since their corresponding genes can be found in close proximity on chromosome 6 (36). The rest of the members have much lower homology (16-30%) and are located on different chromosomes (Supplementary Table 1) (37, 38). IL-17s function as dimeric cytokines, as revealed by structural analysis (39). In particular, IL-17A and IL-17F can form homodimers (IL-17A/A or IL-17F/F) or heterodimers (IL-17A/F) (40–42).
Regarding their function, IL-17A and IL-17F are modest activators in terms of their pro-inflammatory potency when working alone (43), but they can dramatically amplify their signal by synergizing with other pro-inflammatory molecules, such as TNF-α, IL-1β, and IL-22 (31). The synergistic effect of IL-17A and IL-17F with TNFα has been shown in animal and human models in many cell types (31). Interestingly, the synergistic effects of IL-17A and IL-17F with TNFα or IL-1β are cell type-specific (31, 44). Although IL-17A has a more potent pro-inflammatory effect, IL-17F is found at higher levels (up to 30-fold) in lesional skin and serum of patients with PsO (45). These higher serum levels of IL-17F have also been observed in other IMIDs, such as PsA, radiographic axSpA or HS (46–49). Furthermore, lower mRNA levels of IL-17A have been found in PsA synovial tissue than in paired PsO skin samples (50). Indeed, IL-17A mRNA levels were 2.7-fold lower than those of IL-17F in the skin and 17.3-fold higher in synovial tissue, but IL-17A protein levels were 37.4-fold higher than those of IL-17F in synovial fluid (50). In rheumatoid arthritis, screening analysis of synovial fluid by multiplex ELISA showed higher protein levels of interleukin IL-23 and IL-17F in ectopic lymphoid neogenesis (ELN), an aggregation of T and B lymphocytes in nonlymphoid tissues, compared to ELN-negative samples (51). Moreover, downstream of IL-23, expression of CD21L (a marker selectively expressed in germinal center-containing synovial tissues) was significantly associated with IL-17F, IL-21, and IL-22, but not IL-17A in two independent sample sets of synovial tissue, supporting differential expression, and maybe function, of IL-17F and IL-17A in synovial tissue with different T/B organization (51).
Signaling of IL-17s happens through a unique family of cell membrane receptors composed of five members (IL-17RA to IL-17RE, Supplementary Table 2, Figure 1). IL-17Rs can function either as homodimeric or heterodimeric proteins and are characterized by the presence of a conserved SEFIR (similar expression to fibroblast growth factor genes) domain in their intracellular region (53). Dimeric IL-17R complex induces binding of nuclear factor-kappa B (NF-κB) activator 1 (Act1) that functions as a linker with TRAF (TNF receptor-associated factor) family proteins. Recruitment of TRAF6 and transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1) by Act1 leads to the activation of the classical NF-κB pathway (34, 52). In addition, IL-17s activate the CCAAT/enhancer-binding protein (C/EBP) family of transcription factors C/EBPβ and C/EBPδ regulating the expression of inflammation-related genes (IL-6, chemokines or IL-23R encoding genes) (54). Thus, the binding of Act1 to the IL-17R along with the binding of TRAF6 mediates the canonical pathway where C/EBP, AP-1, and NF-kB activation results in the transcription of inflammatory genes (1, 2, 55, 56). Also, Act1 interacts with the TRAF2/TRAF5 complex regulating the noncanonical pathway controlled by several RNA-binding proteins (such as HuR and Arid5a). TRAF3 can bind to the IL-17R preventing the formation of the IL-17R-Act1-TRAF6 complex. Moreover, TRAF4 can compete with TRAF6 for the binding site on Act1 blocking its signaling (56).
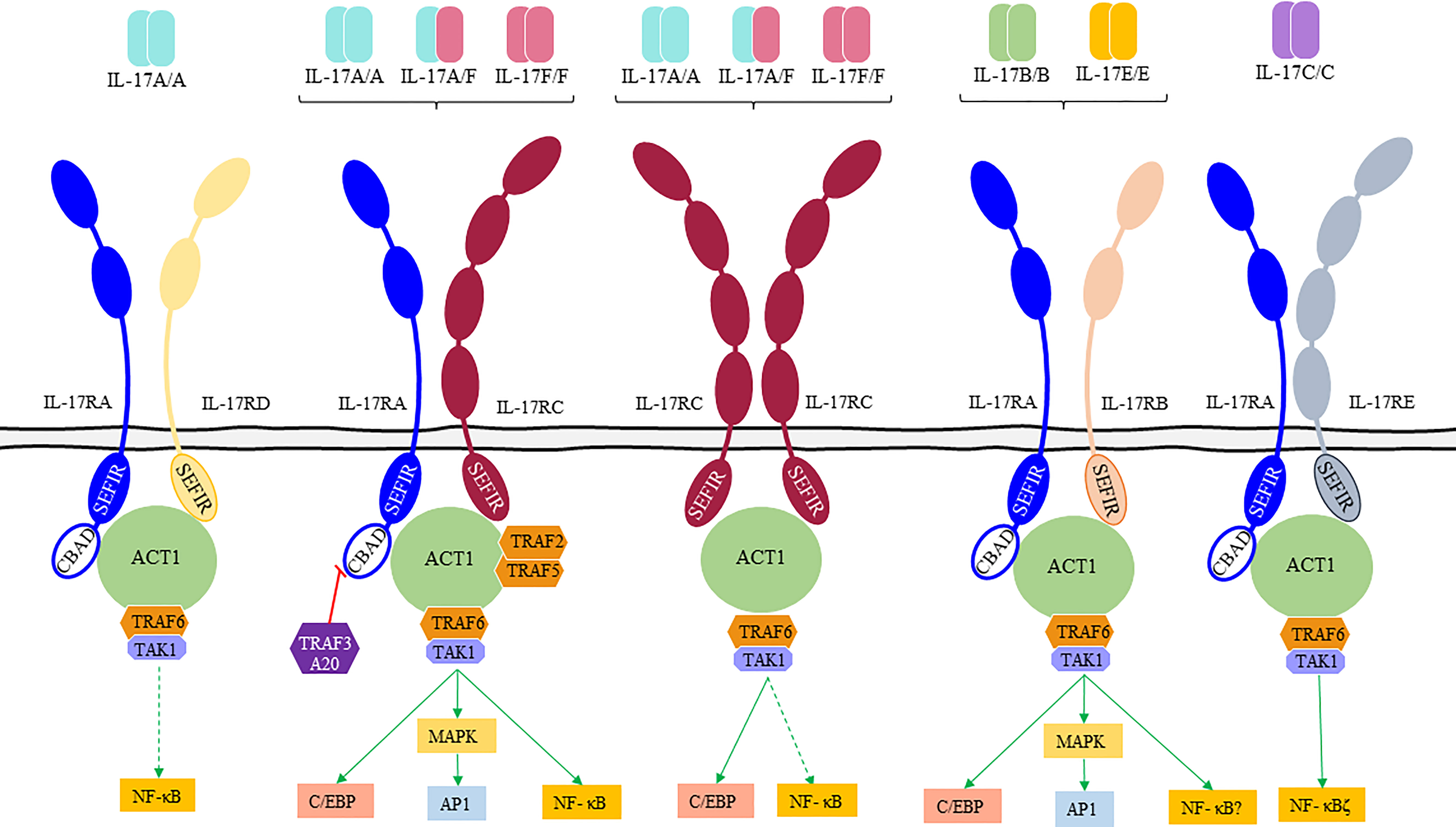
Figure 1 IL-17 receptor family (IL-17Rs), their ligands and downstream signaling pathways. IL-17Rs are classified in “short” and “tall” according to their extracellular domains (ECD). “Short” receptors (IL-17RA, IL-17RB, and IL-17RD) present a two-domains ECD and a disordered linker near the cytoplasmic membrane with a proline-rich motif, and “tall” receptors (IL-17RC and IL-17RE) have a larger ECD with two additional domains (52). IL-17A and IL-17F homodimers and IL-17A/IL-17F heterodimers bind to IL-17RA/C receptors leading to the recruitment of the nuclear factor-kappa B (NF-κB) activator 1 (Act1) by homotypic interactions between both SEFIR (similar expression to fibroblast growth factor genes) domains. Once the complex is formed other signaling molecules (TNF receptor-associated factor [TRAF]6, TRAF2, and TRAF5) are recruited to fully activate downstream signaling pathways. Bold arrows represent a preferential binding.
IL-17 receptors significantly differ in the composition of their extracellular domains, as revealed in the latest structural studies. IL-17RA is the only IL-17 chain known to contain a cytoplasmic domain named CBAD (C/EBP-β activation domain). This CBAD domain is part of a regulatory mechanism capable of coordinating the inhibition of IL-17s signaling through binding of TRAF3 to the ubiquitin-editing enzyme A20 (6). In addition to IL-17RA, both IL-17 A and F can signal as well through an IL-17RC:IL17RC homodimeric complex, debunking the long-standing concept that IL-17RA is a mandatory shared receptor (52). Mouse models have tested the functionality of the IL-17F/IL-17RC axis, showing that signaling through this axis can lead to a dysregulated inflammatory response (54, 57). This is mainly due to the inability of IL-17RC to bind to inhibitory regulatory partners such as A20 since the receptor lacks a CBAD domain (58, 59).
The functional patterns of IL-17RA and IL-17RC are not entirely overlapping (60), which might help to explain the tissue-specific roles observed of IL-17A and IL-17F. Blockade of IL-17RA could be potentiating an escape route via IL-17F/IL-17RC axis signaling (22, 57). Moreover, IL-17RA blockade leads to increase levels of circulating IL-17A in PsO that once the drug is withdrawn can readily signal through the IL-17RA/IL-17RC heterodimer inducing excessive IL-17A signaling (1).
Mechanistic studies have shown that immune cells, such as Th17 cells, are endowed with an autocrine regulatory feedback loop that tunes their pathogenicity (Figure 2) (61). IL-17A can bind in an autocrine manner to the IL-17RA : IL-17RC heterodimeric receptor, activates NF-κB and induces the secretion of IL-24. At the same time, IL-24 can also function in an autocrine manner and repress the expression of other Th17 signature cytokines including GM-CSF and IL-17F (61). Thus, blockade of IL-17A alone disrupts these autocrine pathways and unlocks the repressive role of IL-24, allowing for the release of GM-CSF and IL-17F and unlocking an unexpected pro-inflammatory escape route (Figure 2). This might explain the limitations of targeting IL-17A alone in some inflammatory conditions (62).
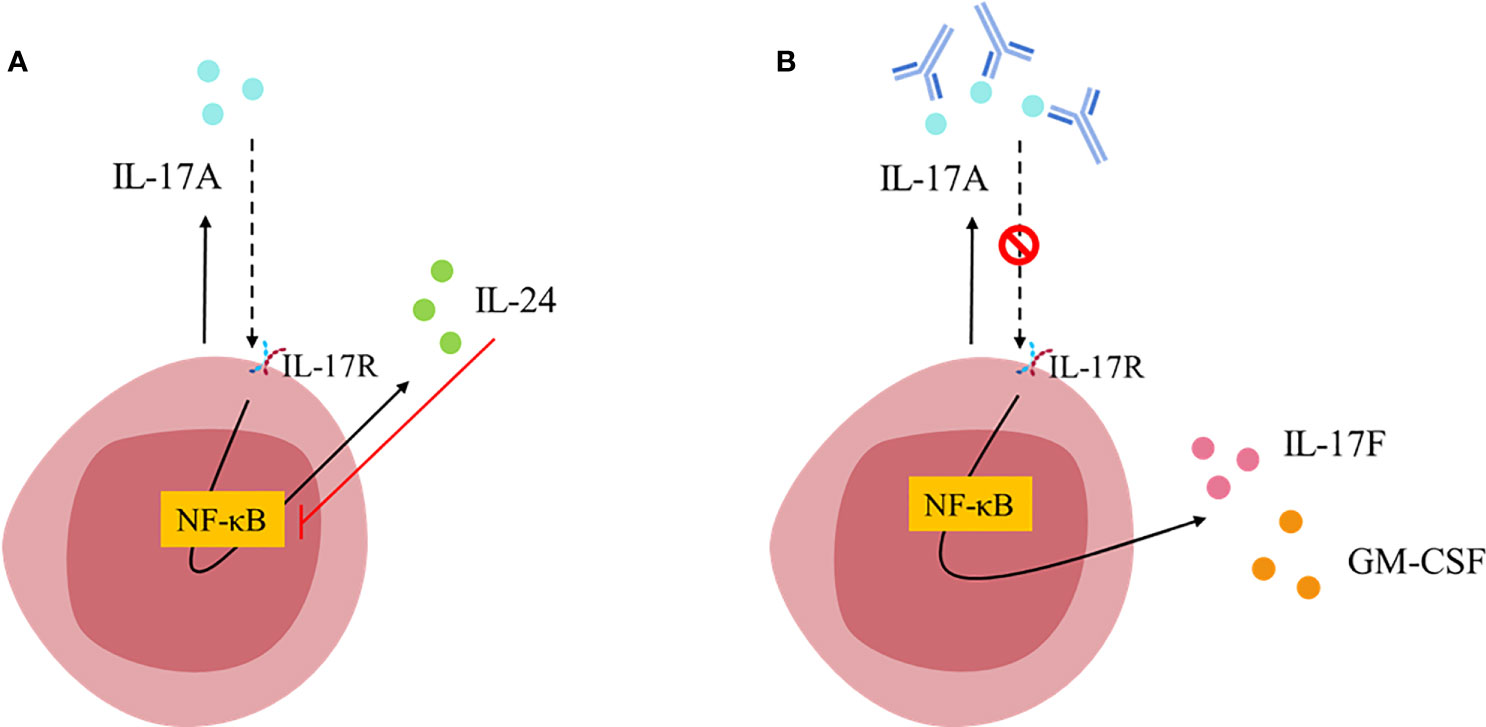
Figure 2 TH17 cell-intrinsic autocrine loop triggered by IL-17A [Figure adapted from Chong et al., 2020 (61)]. (A) IL-17A binds to IL-17RA/IL-17RC receptor and activates NF-κB inducing the expression of IL-24, which in turn inhibits NF-κB leading to the repression of other TH17 signature cytokines (such as IL-17F and GM-CSF). (B) Blockade of IL-17A breaks the autocrine loop allowing NF-κB signaling and therefore, favoring IL-17F and GM-CSF expression.
3 Cellular sources of IL-17A and IL-17F
As mentioned above, IL-17A and IL-17F are secreted by cells of the adaptative immune system such as Th17 cells and CD8+ cytotoxic T17 (Tc17) cells (63, 64), but also by innate immune cells such as group 3 innate lymphoid cells (ILC3s), and innate-like lymphocytes (ILLs) such as gamma delta (γδ) T, mucosal-associated invariant T (MAIT) cells, and natural killer T (NKT) cells (Figure 3, Table 2) (26, 64).
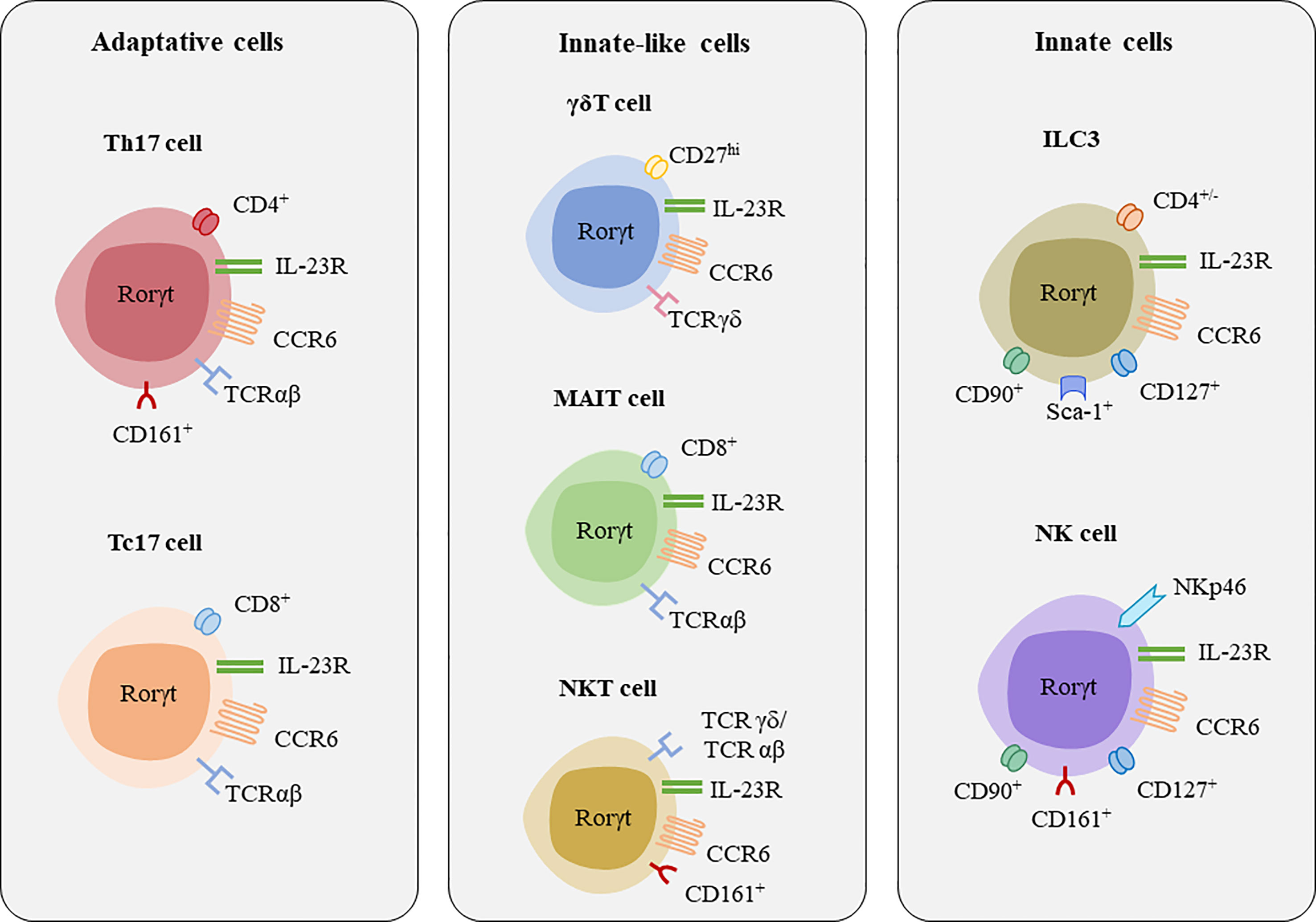
Figure 3 Major lymphocytes populations secreting IL-17A and IL-17F. [Figure adapted from Veldhoen, 2017 (60)]. Major transcription factor associated with these cells are RORγt, and some shared surface receptors include CCR6 and IL-23R.

Table 2 Main characteristics of the immune cell populations secreting IL-17A and IL-17F and their role in autoimmune diseases.
3.1 Th17 and Tc17 cells, the conventional IL-23-dependent producers of IL-17A and IL-17F
Th17 cells are the main responders against extracellular pathogens such as fungi and bacteria. They are a well-studied source of IL-17A and IL-17F, but they can also secrete other cytokines such as IL-21, IL-22, IL-24, and GM-CSF (114, 115). The differentiation of naïve CD4+ helper T cells (Th0) into effector subsets (Th1, Th2, or Th17) or regulatory T cells (Treg) depends on the local cytokine milieu (Figure 4) (117). T-bet (T-box expressed in T cells), GATA3 (GATA binding protein 3) and Foxp3 (forkhead box P3) are the main transcription factors expressed by Th1, Th2, and Treg cells, respectively, and are responsible for the lineage-specific cytokine profiles of each subset. Th1 are defined by their preferential production of IL-2 and IFNγ and respond mostly against intracellular pathogens (bacteria, virus, etc.); Th2 for IL-4, IL-5, and IL-13 and respond against parasites and allergens; and Treg for TGF-β and IL-10 and are involved in immune tolerance and lymphocyte homeostasis. Conversely, retinoid-related orphan receptor gamma t (RORγt) is the hallmark transcription factor that determines the differentiation of Th17 cells, inducing the production of IL-17A and IL-17F, and surface molecules such as the C-C chemokine receptor type 6 (CCR6) and IL-23R (119, 120). In general, it has been established that IL-1β and IL-6 are required for the differentiation of Th17 cells, whereas TGF-β and IL-23 favor lineage expansion, maintenance, and survival (121, 122). However, the balance between IL-23 and TGF-β prompts a different cytokine profile of Th17 cells and their pathogenicity (Figure 4) (123, 124). An increase in the TGF-β/IL-23 ratio can result in the generation of non-pathogenic Th17 cells, whereas a reduction in this ratio converts them to pathogenic Th17 cells (123, 125, 126). The lack of IL-10 expression and enhanced GM-CSF in Th17 cells has been also associated with a pathogenic phenotype (125). The pro-inflammatory mediator prostaglandin E2 (PEG2) is another potent activator of Th17 cells that induces a pathogenic phenotype (increasing RORγt and IL-17A and reducing IL-10) (127). Indeed, PEG2 and cyclooxygenase 2 (COX2) are increased in RA synovial tissue and, in mice models, PGE2 modulates the severity of the disease.
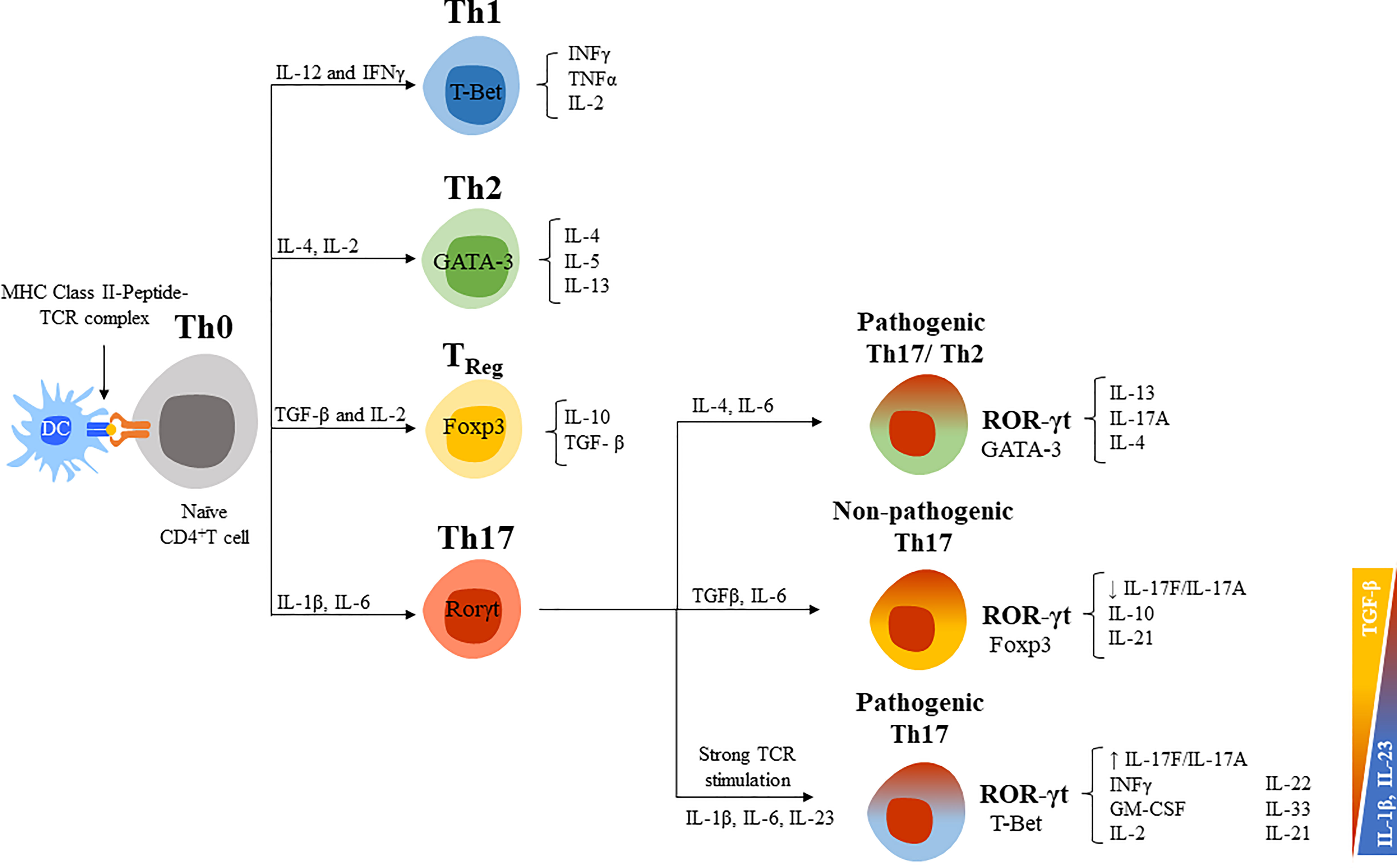
Figure 4 CD4+ helper T cells differentiation routes. [Figure adapted from Ruiz de Morales et al., 2020 (5)]. CD4+ helper T cells (Th0) differentiate into effector T cell subsets (Th1, Th2, or Th17) and regulatory T cells (TReg) with specific signal transduction mechanisms, transcription factors, and cytokine profiles for each cell lineage. IL-12 and IFNγ are critical cytokines initiating the downstream signaling cascade to develop Th1 cells, the T-box transcription factor (T-bet) is the master regulator for its differentiation, and mainly secrete IFNγ and IL-2 (116). For Th2 lineage development, IL-4 and IL-2 are crucial cytokines and GATA3 is the master regulator transcription factor. Among their key effector cytokines are IL-4, IL-5, and IL-13. TReg cells are generated after antigen stimulation under TGF-β and IL-2 presence, express the Foxp3 transcription factor and have a role in maintaining immune homeostasis through their main effector cytokines TGF-β and IL-10. IL-1β, IL-6, or IL-21 are required for the differentiation of Th17 cells, which express RORgt and secrete IL-17s (115, 117). TGF-β and IL-6 exposure sustained the differentiation of non-pathogenic Th17 cells (co-expression of T-Bet), whereas IL-23 induce a pathogenic profile (co-expression of Foxp3). Newly described Th17/Th2 cells express markers of both CD4+ T cells (CD161 and RORγt, and GATA3) and produce IL-4 and IL-17. This phenotype is also considered pathogenic (118).
Activated Th17 cells also differ in terms of the subtype of IL-17s that they preferentially express, differentiating between subpopulations that express IL-17A, IL-17A/F, or IL-17F. Three factors have been observed to control this differentiation, the cytokine environment, the strength or concentration of antigenic signaling through the T-cell receptor (TCR), and the duration of the stimulus (47, 128). Low-strength T cell activation preferentially promotes the induction of the IL-17A+ subpopulation of Th17 cells whereas high-strength stimulation favors the IL-17F+ subpopulation. Furthermore, IL-17A+ and IL-17F+ Th17 cells also differ in terms of the cytokine profiles that they produce. IL-17F+ cells have been associated with a more pathogenic phenotype in inflammatory diseases since they express reduced levels of IL-10 and GM-CSF and a higher level of IFN-γ compared to IL-17A+ cells (47). Moreover, the expression of IL-17A and IL-17F is differentially regulated over time. IL-17A is rapidly produced upon T-cells stimulation, whereas IL-17F expression shows a gradual increase with higher levels at later stages of activation. Conversely, unlike IL-17F, IL-17A expression is not sustained by continuous activation of T-cells (129). This might suggest that, whereas IL-17 A has an important role upon inflammation onset, IL-17F would acquire more relevance in its chronification. This would explain why in certain cases targeting IL-17A alone might not be enough for a long-term disease control (13–16, 61), but dual inhibition of IL-17 A and F might be of choice.
CD8+ cytotoxic T cells (Tc17) are another subset of cells from the adaptative immune system that produce IL-17A, IL-17F, IL-21, IL-22 and express RORγt (130). Like Th17 cells, cytokines IL-6 or IL-21 along with TGF-β determine the differentiation of Tc17 cells. In addition, IL-23 stabilizes their phenotype. Tc17 cells produce small amounts of IFNγ, granzyme and perforin, exerting a low cytotoxicity (131–133).
3.2 Innate lymphoid cells and innate-like lymphocytes: additional sources of IL-17A and IL-17F
Innate lymphoid cells (ILCs) and innate-like lymphocytes (ILLs such as ILC3, γδ T cells, MAIT cells, and NKT cells) are additional sources of IL-17A and IL-17F that have important roles in controlling homeostasis and protecting against infections (134–138). However, the dysregulation of these cells promotes inflammatory responses contributing to the pathogenesis of inflammatory diseases such as PsO, PsA, axSpA, and IBD (Table 1) (90, 116, 139–141). The innate nature of these cells allows them to be rapidly activated during early phases of inflammatory responses and become major sources of IL-17s through a restricted TCR engagement or in response to certain cytokine environments (91, 135). Several studies have highlighted the complexity and number of cytokines that can induce the production of IL-17A and IL-17F by innate and innate-like lymphocytes in response to other cytokines, different from the canonical IL-23, such as IL-7 (45, 141, 142), IL-9 (143), IL-12, IL-1β and IL-18 (28). This is consistent with data obtained in inflammatory diseases (PsO, PsA, axSpA, and HS) where IL-17F levels are higher than those of IL-17A (104, 144–146). Recent evidence has shown a trend in ILC3s, γδ T, and MAIT cells to produce predominantly IL-17F in a mode independent of IL-23 (28, 116). Production of IL-17s in ILCs and ILLs is also dependent on RORγt expression, and although these cells can present the cell surface marker IL-23R, they can also follow an IL-23-independent pathway (91, 142). One plausible explanation could be a molecular disconnection: IL-23 binds to the receptor but cannot activate IL-17s production due to a lower expression of tyrosine kinase 2 (TYK2) and signal transducer and activator of transcription 3 (STAT3), as some transcriptional studies seem to point out (91).
ILC3s, predominantly found in mucosal tissues and skin, are characterized for presenting an invariant TCR, expressing RORγt and therefore producing IL-17A, IL-17F and/or IL-22. Interestingly, in vitro studies performed with human samples have shown that ILC3s cells can be induced to produce IL-17A and IL-17F, in an IL-23 independent manner, upon exposure to a combination of IL-1β and IL-2 (28).
γδ T cells comprise 50% of the intraepithelial lymphocyte cells in mucosal and epithelial tissues and 3-5% of all blood lymphoid cells (135, 136). They are atypical T cells characterized by the expression of a semi-invariant γδ T cell receptor (TCR) that can recognize a broad range of microbial antigens. Most of the antigens and the recognition mechanism of γδ T cells are still unknown, binding to phosphorylated metabolites such as microbial (E)-4-Hydroxy-3-methyl-but-2-enyl pyrophosphate (HMB-PP) or eukaryotic isoprenoid precursor (IPP), or even to lipid antigens presented by cluster of differentiation 1 (CD1) molecules has been reported (135, 136). The γδ17 subset expresses RORγt and share many common features with Th17 cells (cell surface expression of IL-23R, CCR6, CCR2 and CXCR6) (147, 148). Secretion of IL-17A and IL-17F by γδ17 cells can take place upon IL-23 stimulation (138, 149), or in an IL-23-independent manner in the presence of other cytokines such as IL-7, or combined IL-12 and IL-18 stimulation (28, 83, 84, 135).
MAIT cells are predominantly CD8+ T cells and display rapid innate-like effector functions upon activation. These cells express an invariant TCR that recognizes small metabolites derived from the microbial vitamin B2 (riboflavin) biosynthesis and are restricted by MHC-related molecule-1 (MR1) (85). Overall, MAIT cells are abundant in humans and can be found in many tissues with varying frequency such as blood (up to 10% of T cells), liver (up to 20 to 50% of T cells), synovial tissue (~5%), intestine (1.5-4% CD3+ T cells), lungs (3%) and skin (0.5-2%) (72, 90, 92, 93, 150–152). Expression of RORγt polarises MAIT cells towards a Th17-like phenotype including the production of IL-17A and IL-17F cytokines, and receptors such as IL-1R, IL-7R, IL-12R, IL-18R and IL-23R (90, 94). In vitro studies of MAIT cells isolated from human blood samples have shown that exposure to a combination of IL-12 and IL-18 can induce the production of IL-17A and IL-17F independently of IL-23. Interestingly, strong TCR stimulation in the presence of IL-12 and IL-18 can influence the cytokine profile of MAIT cells with a bias towards IL-17F (28).
NKT cells have features of both NK cells and T cells (153). Three subpopulations can be distinguished based on the transcription factors and cytokine profiles that they express, namely analogous to Th1 (NKT1), Th2 (NKT2) and Th17 cells (NKT17) (152). NKT17 cells are mainly present in lymph nodes, skin, and lungs, and their survival and expression depend on IL-7 (98, 142, 154). NKT17 cells secrete Th17-related cytokines, such as IL-17A, IL-17F, IL-21 and IL-22 (155, 156) and express distinctive markers of Th17 cells such as IL-1R, IL-23R, CCR6, CD103, and CD138 (157–159). Apart from secreting IL-17A and IL-17F in an IL-23-dependent manner (160, 161), NKT can also produce IL-17s after stimulation with TGF-β and IL-1β (162). CD161 (or NK1.1 in murine models) is usually expressed on NK cells and associated with the inhibition of their function (163). The expression of CD161 is regulated by RORγt; it is a marker of IL-17 producing T cell subsets, including CD4+ and CD8+ T cells, and some populations of Treg cells besides NKT cells.
4 Conclusion
IL-17A and IL-17F have relevant physiological roles and their dysregulation can result in pathological conditions. Abnormal levels of these cytokines have been found in IMIDs, making them potential therapeutic targets. Adaptive Th17 cells are generally considered the main producers of IL-17s, and IL-23 was assumed to be indispensable to regulate their secretion. However, current evidence proves that other innate and innate-like cells can secrete IL-17A and IL-17F triggering different signaling pathways, which can be IL-23-independent. The crosstalk between IL-17s and IL-23 in autoimmune and inflammatory diseases is widely recognized, and the IL-23/IL-17 axis has been targeted in the development of therapeutic agents. Inhibitors of IL-17A, IL-17F, or IL-23 have promising results, although they do not yield the same clinical effect in all IMIDs. The evidence of a non-linear relationship between IL-23 and IL-17s can underly these tissue-specific functions of IL-17A and IL-17F.
Therefore, IL-23-independent signaling pathways and additional sources of IL-17A and IL-17F, apart from the adaptative immune cells, constitute alternative processes underpinning pathological conditions. The present review explores the recent literature regarding IL-17s’ alternative sources and signaling pathways independent of IL-23. The types of IL-17R are also important to modulate the response in different tissues. We hope this review may contribute to highlight the importance of considering all IL-17A and IL-17F cellular sources and alternative signaling pathways in designing new therapies and improving treatment selection for IMIDs.
Author contributions
All authors equally contributed to the literature review, drafting and critical revision of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The writing assistance has been sponsored by UCB Pharma.
Acknowledgments
Authors would express gratitude to Meisys (Madrid, Spain) for writing assistance.
Conflict of interest
Author VN-C has served as a speaker, consultant, and/or instructor for: AbbVie, Eli Lilly and Company, Galapagos, Janssen, Moonlake, Novartis, Pfizer, and UCB Pharma; and has received grant and/or research support from AbbVie and Novartis. Author LP has received consultancy and/or speaker’s honoraria from and/or participated in clinical trials and/or research projects sponsored by AbbVie, Almirall, Amgen, Biogen, Boehringer Ingelheim, Bristol Myers Squibb, Janssen, LEO Pharma, Lilly, Novartis, Pfizer, Sandoz, Sanofi, and UCB. Author SV has received speaker’s honoraria and participated in projects sponsored by Bayer, Janssen, LEO Pharma, Lilly, Novartis, Pfizer, Roche, Sanofi and UCB. Author JR has received consultancy and/or speaker’s honoraria from Abbvie, UCB, Janssen, Novartis, Pfizer, Amgen and Lilly and/or participated in clinical trials and/or research projects sponsored by Pfizer, Novartis and Janssen. Author ML-V has received consultancy and/or speaker’s honoraria from and/or participated in clinical trials and/or research projects sponsored by AbbVie, Almirall, Amgen, Boehringer Ingelheim, Celgene, Janssen, Kyowa Kirian, LEO Pharma, Lilly, Novartis, and UCB. Author CF-C has received consultancy and/or speaker’s honoraria and participated in clinical trials and/or research projects sponsored by AbbVie, Janssen, Lilly, MSD, Novartis, Pfizer, the Spanish Society of Rheumatology and UCB. Author RA has received consultancy and/or speaker’s honoraria from and/or participated in clinical trials and/or research projects sponsored by AbbVie, Almirall, Amgen, Galápagos, Gebro, Janssen, Lilly, MSD, Nordimet, Novartis, Pfizer and UCB. Author JP has received consultancy and/or speaker’s honoraria from and/or participated in clinical trials and/or research projects sponsored by Janssen, Novartis, Pfizer, MSD, Lilly, Amgen, BMS, AbbVie, and UCB. Author EG-A has received consultancy and/or speaker’s honoraria from and/or participated in clinical trials and/or research projects sponsored by AbbVie, MSD, Roche, Amgen, Janssen, Lilly, Novartis, Pfizer and UCB. Author PZ has received consultancy and/or speaker’s honoraria from and/or participated in clinical trials and/or research projects sponsored by AbbVie, Celgene, Galapagos, Janssen, Lilly, MSD, Novartis, Pfizer and UCB. Author BJ has received consultancy fees from Amgen, UCB and Janssen; has received speaker’s honoraria from Lilly, Abbvie and Janssen; has participated in clinical trials and/or research projects sponsored by Janssen, Lilly, Bristol Myers Squibb, Abbvie; has received support for attending congress from Novartis, Pfizer, UCB. Author JG has received consultancy and/or speaker’s honoraria from and/or participated in clinical trials and/or research projects sponsored by Novartis, UCB, Pfizer, BMS, MSD, AbbVie, Lilly, Janssen, AstraZeneca and Galápagos. Author XJ has received consultancy and/or speaker’s honoraria from and/or participated in clinical trials and/or research projects sponsored by AbbVie, Lilly, MSD, Nordic Pharma, Novartis, Pfizer and UCB. Author RB has received grants/research supports from Abbvie, MSD, and Roche, and had consultation fees/participation in company-sponsored speaker´s bureau from Abbvie, Pfizer, Roche, Bristol-Myers, Lilly, Janssen, and MSD. Author SA has received consultancy and/or speaker’s honoraria from and/or participated in clinical trials and/or research projects sponsored by AbbVie, Almirall, Janssen, LEO Pharma, Lilly, Novartis and UCB. Author JS has received consultancy and/or speaker’s honoraria from and/or participated in clinical trials and/or research projects sponsored by AbbVie, UCB, Novartis, Amgen, Pfizer and Janssen-Cilag. Author RQ has received consultancy and/or speaker’s honoraria from and/or participated in clinical trials and/or research projects sponsored by Novartis, Janssen, UCB, Pfizer, Amgen, MSD, Eli-Lilly and AbbVie. Author JC has received consultancy and/or speaker’s honoraria from and/or participated in clinical trials and/or research projects sponsored by AbbVie, Almirall, Biogen, Boehringer Ingelheim, Bristol Myers Squibb, Fresenius, Janssen, Lilly, Novartis, Pfizer, Sandoz and UCB.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1191782/full#supplementary-material
References
1. Monin L, Gaffen SL. Interleukin 17 family cytokines: signaling mechanisms, biological activities, and therapeutic implications. Cold Spring Harb Perspect Biol (2018) 10(4):a028522 . doi: 10.1101/cshperspect.a028522
2. McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity (2019) 50(4):892–906. doi: 10.1016/j.immuni.2019.03.021
3. Pappu R, Rutz S, Ouyang W. Regulation of epithelial immunity by IL-17 family cytokines. Trends Immunol (2012) 33(7):343–9. doi: 10.1016/j.it.2012.02.008
4. Fragoulis GE, Siebert S, McInnes IB. Therapeutic targeting of IL-17 and IL-23 cytokines in immune-mediated diseases. Annu Rev Med (2016) 67:337–53. doi: 10.1146/annurev-med-051914-021944
5. Ruiz de Morales JMG, Puig L, Dauden E, Canete JD, Pablos JL, Martin AO, et al. Critical role of interleukin (IL)-17 in inflammatory and immune disorders: An updated review of the evidence focusing in controversies. Autoimmun Rev (2020) 19(1):102429. doi: 10.1016/j.autrev.2019.102429
6. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol (2014) 14(9):585–600. doi: 10.1038/nri3707
7. Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov (2012) 11(10):763–76. doi: 10.1038/nrd3794
8. Moschen AR, Tilg H, Raine T. IL-12, IL-23 and IL-17 in IBD: immunobiology and therapeutic targeting. Nat Rev Gastroenterol Hepatol (2019) 16(3):185–96. doi: 10.1038/s41575-018-0084-8
9. Schinocca C, Rizzo C, Fasano S, Grasso G, La Barbera L, Ciccia F, et al. Role of the IL-23/IL-17 Pathway in Rheumatic Diseases: An Overview. Front Immunol (2021) 12:637829. doi: 10.3389/fimmu.2021.637829
10. Liu T, Li S, Ying S, Tang S, Ding Y, Li Y, et al. The IL-23/IL-17 Pathway in Inflammatory Skin Diseases: From Bench to Bedside. Front Immunol (2020) 11:594735. doi: 10.3389/fimmu.2020.594735
11. Brembilla NC, Senra L, Boehncke WH. The IL-17 Family of Cytokines in Psoriasis: IL-17A and Beyond. Front Immunol (2018) 9:1682. doi: 10.3389/fimmu.2018.01682
12. Yeremenko N. Out of the shadow of interleukin-17A: the role of interleukin-17F and other interleukin-17 family cytokines in spondyloarthritis. Curr Opin Rheumatol (2021) 33(4):333–40. doi: 10.1097/BOR.0000000000000805
13. Baeten D, Ostergaard M, Wei JC, Sieper J, Jarvinen P, Tam LS, et al. Risankizumab, an IL-23 inhibitor, for ankylosing spondylitis: results of a randomised, double-blind, placebo-controlled, proof-of-concept, dose-finding phase 2 study. Ann Rheum Dis (2018) 77(9):1295–302. doi: 10.1136/annrheumdis-2018-213328
14. van der Heijde D, Cheng-Chung Wei J, Dougados M, Mease P, Deodhar A, Maksymowych WP, et al. Ixekizumab, an interleukin-17A antagonist in the treatment of ankylosing spondylitis or radiographic axial spondyloarthritis in patients previously untreated with biological disease-modifying anti-rheumatic drugs (COAST-V): 16 week results of a phase 3 randomised, double-blind, active-controlled and placebo-controlled trial. Lancet (2018) 392(10163):2441–51. doi: 10.1016/S0140-6736(18)31946-9
15. Baeten D, Sieper J, Braun J, Baraliakos X, Dougados M, Emery P, et al. Secukinumab, an Interleukin-17A Inhibitor, in Ankylosing Spondylitis. N Engl J Med (2015) 373(26):2534–48. doi: 10.1056/NEJMoa1505066
16. Deodhar A, Gensler LS, Sieper J, Clark M, Calderon C, Wang Y, et al. Three Multicenter, Randomized, Double-Blind, Placebo-Controlled Studies Evaluating the Efficacy and Safety of Ustekinumab in Axial Spondyloarthritis. Arthritis Rheumatol (2019) 71(2):258–70. doi: 10.1016/S0140-6736(20)30265-8
17. Kimball AB, Jemec GBE, Alavi A, Reguiai Z, Gottlieb AB, Bechara FG, et al. Secukinumab in moderate-to-severe hidradenitis suppurativa (SUNSHINE and SUNRISE): week 16 and week 52 results of two identical, multicentre, randomised, placebo-controlled, double-blind phase 3 trials. Lancet. (2023) 401(10378):747–61. doi: 10.1016/S0140-6736(23)00022-3
18. Kimball AB, Prens EP, Passeron T, Maverakis E, Turchin I, Beeck S, et al. Efficacy and Safety of Risankizumab for the Treatment of Hidradenitis Suppurativa: A Phase 2, Randomized, Placebo-Controlled Trial. Dermatol Ther (Heidelb) (2023) 13(5):1099–111. doi: 10.1007/s13555-023-00913-3
19. Dudink K, Bouwman K, Chen Y, DePrimo SE, Munoz-Elias EJ, Aarts P, et al. Guselkumab for hidradenitis suppurativa: a phase II, open-label, mode-of-action study. Br J Dermatol (2023) 188(5):601–9. doi: 10.1093/bjd/ljad010
20. van Tok MN, Na S, Lao CR, Alvi M, Pots D, van de Sande MGH, et al. The Initiation, but Not the Persistence, of Experimental Spondyloarthritis Is Dependent on Interleukin-23 Signaling. Front Immunol (2018) 9:1550. doi: 10.3389/fimmu.2018.01550
21. Feagan BG, Panes J, Ferrante M, Kaser A, D'Haens GR, Sandborn WJ, et al. Risankizumab in patients with moderate to severe Crohn's disease: an open-label extension study. Lancet Gastroenterol Hepatol (2018) 3(10):671–80. doi: 10.1016/S2468-1253(18)30233-4
22. Targan SR, Feagan B, Vermeire S, Panaccione R, Melmed GY, Landers C, et al. A Randomized, Double-Blind, Placebo-Controlled Phase 2 Study of Brodalumab in Patients With Moderate-to-Severe Crohn's Disease. Am J Gastroenterol (2016) 111(11):1599–607. doi: 10.1038/ajg.2016.298
23. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. (2012) 61(12):1693–700. doi: 10.1136/gutjnl-2011-301668
24. Tang C, Kakuta S, Shimizu K, Kadoki M, Kamiya T, Shimazu T, et al. Suppression of IL-17F, but not of IL-17A, provides protection against colitis by inducing Treg cells through modification of the intestinal microbiota. Nat Immunol (2018) 19(7):755–65. doi: 10.1038/s41590-018-0134-y
25. Zhou C, Wu D, Jawale C, Li Y, Biswas PS, McGeachy MJ, et al. Divergent functions of IL-17-family cytokines in DSS colitis: Insights from a naturally-occurring human mutation in IL-17F. Cytokine. (2021) 148:155715. doi: 10.1016/j.cyto.2021.155715
26. Hall AO, Towne JE, Plevy SE. Get the IL-17F outta here! Nat Immunol (2018) 19(7):648–50. doi: 10.1038/s41590-018-0141-z
27. Maxwell JR, Zhang Y, Brown WA, Smith CL, Byrne FR, Fiorino M, et al. Differential Roles for Interleukin-23 and Interleukin-17 in Intestinal Immunoregulation. Immunity. (2015) 43(4):739–50. doi: 10.1016/j.immuni.2015.08.019
28. Cole S, Murray J, Simpson C, Okoye R, Tyson K, Griffiths M, et al. Interleukin (IL)-12 and IL-18 Synergize to Promote MAIT Cell IL-17A and IL-17F Production Independently of IL-23 Signaling. Front Immunol (2020) 11:585134. doi: 10.3389/fimmu.2020.585134
29. Cuthbert RJ, Watad A, Fragkakis EM, Dunsmuir R, Loughenbury P, Khan A, et al. Evidence that tissue resident human enthesis gammadeltaT-cells can produce IL-17A independently of IL-23R transcript expression. Ann Rheum Dis (2019) 78(11):1559–65. doi: 10.1136/annrheumdis-2019-215210
30. Oliver R, Krueger JG, Glatt S, Vajjah P, Mistry C, Page M, et al. Bimekizumab for the treatment of moderate-to-severe plaque psoriasis: efficacy, safety, pharmacokinetics, pharmacodynamics and transcriptomics from a phase IIa, randomized, double-blind multicentre study. Br J Dermatol (2022) 186(4):652–63. doi: 10.1111/bjd.20827
31. Glatt S, Baeten D, Baker T, Griffiths M, Ionescu L, Lawson ADG, et al. Dual IL-17A and IL-17F neutralisation by bimekizumab in psoriatic arthritis: evidence from preclinical experiments and a randomised placebo-controlled clinical trial that IL-17F contributes to human chronic tissue inflammation. Ann Rheum Dis (2018) 77(4):523–32. doi: 10.1136/annrheumdis-2017-212127
32. Shah M, Maroof A, Gikas P, Mittal G, Keen R, Baeten D, et al. Dual neutralisation of IL-17F and IL-17A with bimekizumab blocks inflammation-driven osteogenic differentiation of human periosteal cells. RMD Open (2020) 6(2):e001306. doi: 10.1136/rmdopen-2020-001306
33. Iznardo H, Puig L. Dual inhibition of IL-17A and IL-17F in psoriatic disease. Ther Adv Chronic Dis (2021) 12:20406223211037846. doi: 10.1177/2040622321103784634
34. Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol (2009) 9(8):556–67. doi: 10.1038/nri2586
35. Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol (2007) 25:821–52. doi: 10.1146/annurev.immunol.25.022106.141557
36. Loughlin J, Mustafa Z, Dowling B, Southam L, Marcelline L, Raina SS, et al. Finer linkage mapping of a primary hip osteoarthritis susceptibility locus on chromosome 6. Eur J Hum Genet (2002) 10(9):562–8. doi: 10.1038/sj.ejhg.5200848
37. Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity (2011) 34(2):149–62. doi: 10.1016/j.immuni.2011.02.012
38. Hymowitz SG, Filvaroff EH, Yin JP, Lee J, Cai L, Risser P, et al. IL-17s adopt a cystine knot fold: structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J (2001) 20(19):5332–41. doi: 10.1093/emboj/20.19.5332
39. Liu S. Structural Insights into the Interleukin-17 Family Cytokines and Their Receptors. Adv Exp Med Biol (2019) 1172:97–117. doi: 10.1007/978-981-13-9367-9_5
40. Wright JF, Bennett F, Li B, Brooks J, Luxenberg DP, Whitters MJ, et al. The human IL-17F/IL-17A heterodimeric cytokine signals through the IL-17RA/IL-17RC receptor complex. J Immunol (2008) 181(4):2799–805. doi: 10.4049/jimmunol.181.4.2799
41. Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, et al. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol (2006) 177(1):36–9. doi: 10.4049/jimmunol.177.1.36
42. Hu Y, Ota N, Peng I, Refino CJ, Danilenko DM, Caplazi P, et al. IL-17RC is required for IL-17A- and IL-17F-dependent signaling and the pathogenesis of experimental autoimmune encephalomyelitis. J Immunol (2010) 184(8):4307–16. doi: 10.4049/jimmunol.0903614
43. Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, et al. Regulation of inflammatory responses by IL-17F. J Exp Med (2008) 205(5):1063–75. doi: 10.1084/jem.20071978
44. Chuang SY, Lin CH, Sung CT, Fang JY. Murine models of psoriasis and their usefulness for drug discovery. Expert Opin Drug Discov (2018) 13(6):551–62. doi: 10.1080/17460441.2018.1463214
45. Kolbinger F, Loesche C, Valentin MA, Jiang X, Cheng Y, Jarvis P, et al. beta-Defensin 2 is a responsive biomarker of IL-17A-driven skin pathology in patients with psoriasis. J Allergy Clin Immunol (2017) 139(3):923–32 e8. doi: 10.1016/j.jaci.2016.06.038
46. Burns LA, Maroof A, Marshall D, Steel KJA, Lalnunhlimi S, Cole S, et al. Presence, function, and regulation of IL-17F-expressing human CD4(+) T cells. Eur J Immunol (2020) 50(4):568–80. doi: 10.1002/eji.201948138
47. Novatchkova M, Leibbrandt A, Werzowa J, Neubuser A, Eisenhaber F. The STIR-domain superfamily in signal transduction, development and immunity. Trends Biochem Sci (2003) 28(5):226–9. doi: 10.1016/S0968-0004(03)00067-7
48. Schlapbach C, Hanni T, Yawalkar N, Hunger RE. Expression of the IL-23/Th17 pathway in lesions of hidradenitis suppurativa. J Am Acad Dermatol (2011) 65(4):790–8. doi: 10.1016/j.jaad.2010.07.010
49. Rumberger BE, Boarder EL, Owens SL, Howell MD. Transcriptomic analysis of hidradenitis suppurativa skin suggests roles for multiple inflammatory pathways in disease pathogenesis. Inflamm Res (2020) 69(10):967–73. doi: 10.1007/s00011-020-01381-7
50. Chen S, Blijdorp IC, van Mens LJJ, Bowcutt R, Latuhihin TE, van de Sande MGH, et al. Interleukin 17A and IL-17F Expression and Functional Responses in Rheumatoid Arthritis and Peripheral Spondyloarthritis. J Rheumatol (2020) 47(11):1606–13. doi: 10.3899/jrheum.190571
51. Cañete JD, Celis R, Yeremenko N, Sanmarti R, van Duivenvoorde L, Ramirez J, et al. Ectopic lymphoid neogenesis is strongly associated with activation of the IL-23 pathway in rheumatoid synovitis. Arthritis Res Ther (2015) 17(1):173. doi: 10.1186/s13075-015-0688-0
52. Goepfert A, Lehmann S, Blank J, Kolbinger F, Rondeau JM. Structural analysis reveals that the cytokine IL-17F forms a homodimeric complex with receptor IL-17RC to drive IL-17RA-independent signaling. Immunity (2020) 52(3):499–512 e5. doi: 10.1016/j.immuni.2020.02.004
53. Shen F, Li N, Gade P, Kalvakolanu DV, Weibley T, Doble B, et al. IL-17 receptor signaling inhibits C/EBPbeta by sequential phosphorylation of the regulatory 2 domain. Sci Signal (2009) 2(59):ra8. doi: 10.1126/scisignal.2000066
54. De Luca A, Pariano M, Cellini B, Costantini C, Villella VR, Jose SS, et al. The IL-17F/IL-17RC Axis Promotes Respiratory Allergy in the Proximal Airways. Cell Rep (2017) 20(7):1667–80. doi: 10.1016/j.celrep.2017.07.063
55. Swaidani S, Liu C, Zhao J, Bulek K, Li X. TRAF Regulation of IL-17 Cytokine Signaling. Front Immunol (2019) 10:1293. doi: 10.3389/fimmu.2019.01293
56. Groen SS, Sinkeviciute D, Bay-Jensen AC, Thudium CS, Karsdal MA, Thomsen SF, et al. Exploring IL-17 in spondyloarthritis for development of novel treatments and biomarkers. Autoimmun Rev (2021) 20(3):102760. doi: 10.1016/j.autrev.2021.102760
57. Busse WW, Holgate S, Kerwin E, Chon Y, Feng J, Lin J, et al. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med (2013) 188(11):1294–302. doi: 10.1164/rccm.201212-2318OC
58. Kuestner RE, Taft DW, Haran A, Brandt CS, Brender T, Lum K, et al. Identification of the IL-17 receptor related molecule IL-17RC as the receptor for IL-17F. J Immunol (2007) 179(8):5462–73. doi: 10.4049/jimmunol.179.8.5462
59. Masson Regnault M, Konstantinou MP, Khemis A, Poulin Y, Bourcier M, Amelot F, et al. Early relapse of psoriasis after stopping brodalumab: a retrospective cohort study in 77 patients. J Eur Acad Dermatol Venereol (2017) 31(9):1491–6. doi: 10.1111/jdv.14387
60. Veldhoen M. Interleukin 17 is a chief orchestrator of immunity. Nat Immunol (2017) 18(6):612–21. doi: 10.1038/ni.3742
61. Chong WP, Mattapallil MJ, Raychaudhuri K, Bing SJ, Wu S, Zhong Y, et al. The Cytokine IL-17A Limits Th17 Pathogenicity via a Negative Feedback Loop Driven by Autocrine Induction of IL-24. Immunity (2020) 53(2):384–97 e5. doi: 10.1016/j.immuni.2020.06.022
62. Dick AD, Tugal-Tutkun I, Foster S, Zierhut M, Melissa Liew SH, Bezlyak V, et al. Secukinumab in the treatment of noninfectious uveitis: results of three randomized, controlled clinical trials. Ophthalmology (2013) 120(4):777–87. doi: 10.1016/j.ophtha.2012.09.040
63. Rosine N, Miceli-Richard C. Innate Cells: The Alternative Source of IL-17 in Axial and Peripheral Spondyloarthritis? Front Immunol (2020) 11:553742. doi: 10.3389/fimmu.2020.553742
64. Rosine N, Rowe H, Koturan S, Yahia-Cherbal H, Leloup C, Watad A, et al. Characterization of Blood Mucosal Associated Invariant T (MAIT) cells in Axial Spondyloarthritis and of resident MAITs from control axial enthesis. Arthritis Rheumatol (2022) 74(11):1786–95. doi: 10.1002/art.42090
65. Benham H, Norris P, Goodall J, Wechalekar MD, FitzGerald O, Szentpetery A, et al. Th17 and Th22 cells in psoriatic arthritis and psoriasis. Arthritis Res Ther (2013) 15(5):R136. doi: 10.1186/ar4317
66. Boehncke WH, Schon MP. Psoriasis. Lancet. (2015) 386(9997):983–94. doi: 10.1016/S0140-6736(14)61909-7
67. Martin DA, Towne JE, Kricorian G, Klekotka P, Gudjonsson JE, Krueger JG, et al. The emerging role of IL-17 in the pathogenesis of psoriasis: preclinical and clinical findings. J Invest Dermatol (2013) 133(1):17–26. doi: 10.1038/jid.2012.194
68. Mauro D, Simone D, Bucci L, Ciccia F. Novel immune cell phenotypes in spondyloarthritis pathogenesis. Semin Immunopathol (2021) 43(2):265–77. doi: 10.1007/s00281-021-00837-0
69. Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, et al. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat Med (2007) 13(6):711–8. doi: 10.1038/nm1585
70. Dagur PK, Biancotto A, Stansky E, Sen HN, Nussenblatt RB, McCoy JP. Secretion of interleukin-17 by CD8+ T cells expressing CD146 (MCAM). Clin Immunol (2014) 152(1-2):36–47. doi: 10.1016/j.clim.2014.01.009
71. Nakamura A, Haroon N. Recent Updates in the Immunopathology of Type 3 Immunity-Mediated Enthesitis. Curr Rheumatol Rep (2021) 23(5):31. doi: 10.1007/s11926-021-00995-y
72. Michel ML, Pang DJ, Haque SF, Potocnik AJ, Pennington DJ, Hayday AC. Interleukin 7 (IL-7) selectively promotes mouse and human IL-17-producing gammadelta cells. Proc Natl Acad Sci USA (2012) 109(43):17549–54. doi: 10.1073/pnas.1204327109
73. Leijten EF, van Kempen TS, Olde Nordkamp MA, Pouw JN, Kleinrensink NJ, Vincken NL, et al. Tissue-Resident Memory CD8+ T Cells From Skin Differentiate Psoriatic Arthritis From Psoriasis. Arthritis Rheumatol (2021) 73(7):1220–32. doi: 10.1002/art.41652
74. Bowes J, Budu-Aggrey A, Huffmeier U, Uebe S, Steel K, Hebert HL, et al. Dense genotyping of immune-related susceptibility loci reveals new insights into the genetics of psoriatic arthritis. Nat Commun (2015) 6:6046. doi: 10.1038/ncomms7046
75. Winchester R, Minevich G, Steshenko V, Kirby B, Kane D, Greenberg DA, et al. HLA associations reveal genetic heterogeneity in psoriatic arthritis and in the psoriasis phenotype. Arthritis Rheumatol (2012) 64(4):1134–44. doi: 10.1002/art.33415
76. Cortes A, Pulit SL, Leo PJ, Pointon JJ, Robinson PC, Weisman MH, et al. Major histocompatibility complex associations of ankylosing spondylitis are complex and involve further epistasis with ERAP1. Nat Commun (2015) 6:7146. doi: 10.1038/ncomms8146
77. Diani M, Casciano F, Marongiu L, Longhi M, Altomare A, Pigatto PD, et al. Increased frequency of activated CD8(+) T cell effectors in patients with psoriatic arthritis. Sci Rep (2019) 9(1):10870. doi: 10.1038/s41598-019-47310-5
78. Steel KJA, Srenathan U, Ridley M, Durham LE, Wu SY, Ryan SE, et al. Polyfunctional, Proinflammatory, Tissue-Resident Memory Phenotype and Function of Synovial Interleukin-17A+CD8+ T Cells in Psoriatic Arthritis. Arthritis Rheumatol (2020) 72(3):435–47. doi: 10.1002/art.41156
79. Keijsers RR, Joosten I, van Erp PE, Koenen HJ, van de Kerkhof PC. Cellular sources of IL-17 in psoriasis: a paradigm shift? Exp Dermatol (2014) 23(11):799–803. doi: 10.1111/exd.12487
80. Huang JC, Schleisman M, Choi D, Mitchell C, Watson L, Asquith M, et al. Preliminary Report on Interleukin-22, GM-CSF, and IL-17F in the Pathogenesis of Acute Anterior Uveitis. Ocul Immunol Inflamm (2021) 29(3):558–65. doi: 10.1080/09273948.2019.1686156
81. Globig AM, Hipp AV, Otto-Mora P, Heeg M, Mayer LS, Ehl S, et al. High-dimensional profiling reveals Tc17 cell enrichment in active Crohn's disease and identifies a potentially targetable signature. Nat Commun (2022) 13(1):3688. doi: 10.1038/s41467-022-31229-z
82. Srenathan U, Steel K, Taams LS. IL-17+ CD8+ T cells: Differentiation, phenotype and role in inflammatory disease. Immunol Lett (2016) 178:20–6. doi: 10.1016/j.imlet.2016.05.001
83. Guggino G, Ciccia F, Di Liberto D, Lo Pizzo M, Ruscitti P, Cipriani P, et al. Interleukin (IL)-9/IL-9R axis drives gammadelta T cells activation in psoriatic arthritis patients. Clin Exp Immunol (2016) 186(3):277–83. doi: 10.1111/cei.12853
84. Haas JD, Gonzalez FH, Schmitz S, Chennupati V, Fohse L, Kremmer E, et al. CCR6 and NK1.1 distinguish between IL-17A and IFN-gamma-producing gammadelta effector T cells. Eur J Immunol (2009) 39(12):3488–97. doi: 10.1002/eji.200939922
85. Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity (2009) 31(2):321–30. doi: 10.1016/j.immuni.2009.06.020
86. Blauvelt A, Chiricozzi A. The Immunologic Role of IL-17 in Psoriasis and Psoriatic Arthritis Pathogenesis. Clin Rev Allergy Immunol (2018) 55(3):379–90. doi: 10.1007/s12016-018-8702-3
87. Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Cayatte C, Chen Y, et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity. (2015) 43(4):727–38. doi: 10.1016/j.immuni.2015.09.003
88. Venken K, Jacques P, Mortier C, Labadia ME, Decruy T, Coudenys J, et al. RORgammat inhibition selectively targets IL-17 producing iNKT and gammadelta-T cells enriched in Spondyloarthritis patients. Nat Commun (2019) 10(1):9. doi: 10.1038/s41467-018-07911-6
89. Hysa E, Cutolo CA, Gotelli E, Pacini G, Schenone C, Kreps EO, et al. Immunopathophysiology and clinical impact of uveitis in inflammatory rheumatic diseases: An update. Eur J Clin Invest (2021) 51(8):e13572. doi: 10.1111/eci.13572
90. Raychaudhuri SK, Abria C, Mitra A, Raychaudhuri SP. Functional significance of MAIT cells in psoriatic arthritis. Cytokine. (2020) 125:154855. doi: 10.1016/j.cyto.2019.154855
91. Gracey E, Qaiyum Z, Almaghlouth I, Lawson D, Karki S, Avvaru N, et al. IL-7 primes IL-17 in mucosal-associated invariant T (MAIT) cells, which contribute to the Th17-axis in ankylosing spondylitis. Ann Rheum Dis (2016) 75(12):2124–32. doi: 10.1136/annrheumdis-2015-208902
92. Teunissen MBM, Yeremenko NG, Baeten DLP, Chielie S, Spuls PI, de Rie MA, et al. The IL-17A-producing CD8+ T-cell population in psoriatic lesional skin comprises mucosa-associated invariant T cells and conventional T cells. J Invest Dermatol (2014) 134(12):2898–907. doi: 10.1038/jid.2014.261
93. Nel I, Bertrand L, Toubal A, Lehuen A. MAIT cells, guardians of skin and mucosa? Mucosal Immunol (2021) 14(4):803–14. doi: 10.1038/s41385-021-00391-w
94. Dusseaux M, Martin E, Serriari N, Peguillet I, Premel V, Louis D, et al. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood (2011) 117(4):1250–9. doi: 10.1182/blood-2010-08-303339
95. Pisarska MM, Dunne MR, O'Shea D, Hogan AE. Interleukin-17 producing mucosal associated invariant T cells - emerging players in chronic inflammatory diseases? Eur J Immunol (2020) 50(8):1098–108. doi: 10.1002/eji.202048645
96. Toussirot E, Laheurte C, Gaugler B, Gabriel D, Saas P. Increased IL-22- and IL-17A-Producing Mucosal-Associated Invariant T Cells in the Peripheral Blood of Patients With Ankylosing Spondylitis. Front Immunol (2018) 9:1610. doi: 10.3389/fimmu.2018.01610
97. Haga K, Chiba A, Shibuya T, Osada T, Ishikawa D, Kodani T, et al. MAIT cells are activated and accumulated in the inflamed mucosa of ulcerative colitis. J Gastroenterol Hepatol (2016) 31(5):965–72. doi: 10.1111/jgh.13242
98. Menon B, Gullick NJ, Walter GJ, Rajasekhar M, Garrood T, Evans HG, et al. Interleukin-17+CD8+ T cells are enriched in the joints of patients with psoriatic arthritis and correlate with disease activity and joint damage progression. Arthritis Rheumatol (2014) 66(5):1272–81. doi: 10.1002/art.38376
99. Zhao M, Svensson MND, Venken K, Chawla A, Liang S, Engel I, et al. Altered thymic differentiation and modulation of arthritis by invariant NKT cells expressing mutant ZAP70. Nat Commun (2018) 9(1):2627. doi: 10.1038/s41467-018-05095-7
100. Grose RH, Thompson FM, Baxter AG, Pellicci DG, Cummins AG. Deficiency of invariant NK T cells in Crohn's disease and ulcerative colitis. Dig Dis Sci (2007) 52(6):1415–22. doi: 10.1007/s10620-006-9261-7
101. Grajewski RS, Hansen AM, Agarwal RK, Kronenberg M, Sidobre S, Su SB, et al. Activation of invariant NKT cells ameliorates experimental ocular autoimmunity by a mechanism involving innate IFN-gamma production and dampening of the adaptive Th1 and Th17 responses. J Immunol (2008) 181(7):4791–7. doi: 10.4049/jimmunol.181.7.4791
102. Hu Y, Chen Y, Chen Z, Zhang X, Guo C, Yu Z, et al. Dysregulated Peripheral Invariant Natural Killer T Cells in Plaque Psoriasis Patients. Front Cell Dev Biol (2021) 9:799560. doi: 10.3389/fcell.2021.799560
103. Yu HG, Lee DS, Seo JM, Ahn JK, Yu YS, Lee WJ, et al. The number of CD8+ T cells and NKT cells increases in the aqueous humor of patients with Behcet's uveitis. Clin Exp Immunol (2004) 137(2):437–43. doi: 10.1111/j.1365-2249.2004.02536.x
104. Dyring-Andersen B, Geisler C, Agerbeck C, Lauritsen JP, Gudjonsdottir SD, Skov L, et al. Increased number and frequency of group 3 innate lymphoid cells in nonlesional psoriatic skin. Br J Dermatol (2014) 170(3):609–16. doi: 10.1111/bjd.12658
105. Leijten EF, van Kempen TS, Boes M, Michels-van Amelsfort JM, Hijnen D, Hartgring SA, et al. Brief report: enrichment of activated group 3 innate lymphoid cells in psoriatic arthritis synovial fluid. Arthritis Rheumatol (2015) 67(10):2673–8. doi: 10.1002/art.39261
106. Soare A, Weber S, Maul L, Rauber S, Gheorghiu AM, Luber M, et al. Cutting Edge: Homeostasis of Innate Lymphoid Cells Is Imbalanced in Psoriatic Arthritis. J Immunol (2018) 200(4):1249–54. doi: 10.4049/jimmunol.1700596
107. Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, et al. Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J Invest Dermatol (2014) 134(4):984–91. doi: 10.1038/jid.2013.477
108. Ciccia F, Guggino G, Rizzo A, Saieva L, Peralta S, Giardina A, et al. Type 3 innate lymphoid cells producing IL-17 and IL-22 are expanded in the gut, in the peripheral blood, synovial fluid and bone marrow of patients with ankylosing spondylitis. Ann Rheum Dis (2015) 74(9):1739–47. doi: 10.1136/annrheumdis-2014-206323
109. Cuthbert RJ, Fragkakis EM, Dunsmuir R, Li Z, Coles M, Marzo-Ortega H, et al. Brief Report: Group 3 Innate Lymphoid Cells in Human Enthesis. Arthritis Rheumatol (2017) 69(9):1816–22. doi: 10.1002/art.40150
110. Chowdhury AC, Chaurasia S, Mishra SK, Aggarwal A, Misra R. IL-17 and IFN-gamma producing NK and gammadelta-T cells are preferentially expanded in synovial fluid of patients with reactive arthritis and undifferentiated spondyloarthritis. Clin Immunol (2017) 183:207–12. doi: 10.1016/j.clim.2017.03.016
111. Lin W, Man X, Li P, Song N, Yue Y, Li B, et al. NK cells are negatively regulated by sCD83 in experimental autoimmune uveitis. Sci Rep (2017) 7(1):12895. doi: 10.1038/s41598-017-13412-1
112. Kucuksezer UC, Aktas Cetin E, Esen F, Tahrali I, Akdeniz N, Gelmez MY, et al. The Role of Natural Killer Cells in Autoimmune Diseases. Front Immunol (2021) 12:622306. doi: 10.3389/fimmu.2021.622306
113. Dunphy S, Gardiner CM. NK cells and psoriasis. J BioMed Biotechnol (2011) 2011:248317. doi: 10.1155/2011/248317
114. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. (2006) 126(6):1121–33. doi: 10.1016/j.cell.2006.07.035
115. Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. (2008) 28(1):29–39. doi: 10.1016/j.immuni.2007.11.016
116. Luckheeram RV, Zhou R, Verma AD, Xia B. CD4(+)T cells: differentiation and functions. Clin Dev Immunol (2012) 2012:925135. doi: 10.1155/2012/925135
117. Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol (2007) 8(9):950–7. doi: 10.1038/ni1497
118. McCluskey D, Benzian-Olsson N, Mahil SK, Hassi NK, Wohnhaas CT, APRICOT and PLUM study team, et al. Single-cell analysis implicates TH17-to-TH2 cell plasticity in the pathogenesis of palmoplantar pustulosis. J Allergy Clin Immunol (2022) 150(4):882–93. doi: 10.1016/j.jaci.2022.04.027
119. Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. (2008) 454(7202):350–2. doi: 10.1038/nature07021
120. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. (2010) 467(7318):967–71. doi: 10.1038/nature09447
121. McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol (2007) 8(12):1390–7. doi: 10.1038/ni1539
122. Santarlasci V, Maggi L, Capone M, Frosali F, Querci V, De Palma R, et al. TGF-beta indirectly favors the development of human Th17 cells by inhibiting Th1 cells. Eur J Immunol (2009) 39(1):207–15. doi: 10.1002/eji.200838748
123. Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, et al. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol (2012) 13(10):991–9. doi: 10.1038/ni.2416
124. Purvis HA, Stoop JN, Mann J, Woods S, Kozijn AE, Hambleton S, et al. Low-strength T-cell activation promotes Th17 responses. Blood. (2010) 116(23):4829–37. doi: 10.1182/blood-2010-03-272153
125. Leng T, Akther HD, Hackstein CP, Powell K, King T, Friedrich M, et al. TCR and Inflammatory Signals Tune Human MAIT Cells to Exert Specific Tissue Repair and Effector Functions. Cell Rep (2019) 28(12):3077–91 e5. doi: 10.1016/j.celrep.2019.08.050
126. Van Kaer L, Postoak JL, Wang C, Yang G, Wu L. Innate, innate-like and adaptive lymphocytes in the pathogenesis of MS and EAE. Cell Mol Immunol (2019) 16(6):531–9. doi: 10.1038/s41423-019-0221-5
127. van Hamburg JP, Tas SW. Molecular mechanisms underpinning T helper 17 cell heterogeneity and functions in rheumatoid arthritis. J Autoimmun (2018) 87:69–81. doi: 10.1016/j.jaut.2017.12.006
128. Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol (2016) 17(7):765–74. doi: 10.1038/ni.3489
129. McDermott NL, Bridgewood C, MacLeod T, Khan A, Milner P, Loughenbury P, et al. P34. Co-expression of IL17F and IL17A secretion by entheseal resident T-cells and circulating T-cells in preferentially from the CD4 T-cell subset. Clin Exp Rheumatol (2022) 2022:41–2.
130. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol (2013) 13(2):145–9. doi: 10.1038/nri3365
131. Lawand M, Dechanet-Merville J, Dieu-Nosjean MC. Key Features of Gamma-Delta T-Cell Subsets in Human Diseases and Their Immunotherapeutic Implications. Front Immunol (2017) 8:761. doi: 10.3389/fimmu.2017.00761
132. Cai Y, Shen X, Ding C, Qi C, Li K, Li X, et al. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity. (2011) 35(4):596–610. doi: 10.1016/j.immuni.2011.08.001
133. Venken K, Elewaut D. IL-23 responsive innate-like T cells in spondyloarthritis: the less frequent they are, the more vital they appear. Curr Rheumatol Rep (2015) 17(5):30. doi: 10.1007/s11926-015-0507-2
134. Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol (2010) 10(7):479–89. doi: 10.1038/nri2800
135. Webster KE, Kim HO, Kyparissoudis K, Corpuz TM, Pinget GV, Uldrich AP, et al. IL-17-producing NKT cells depend exclusively on IL-7 for homeostasis and survival. Mucosal Immunol (2014) 7(5):1058–67. doi: 10.1038/mi.2013.122
136. Papotto PH, Ribot JC, Silva-Santos B. IL-17(+) gammadelta T cells as kick-starters of inflammation. Nat Immunol (2017) 18(6):604–11. doi: 10.1038/ni.3726
137. Cording S, Medvedovic J, Cherrier M, Eberl G. Development and regulation of RORgammat(+) innate lymphoid cells. FEBS Lett (2014) 588(22):4176–81. doi: 10.1016/j.febslet.2014.03.034
138. Sanati G, Aryan Z, Barbadi M, Rezaei N. Innate lymphoid cells are pivotal actors in allergic, inflammatory and autoimmune diseases. Expert Rev Clin Immunol (2015) 11(8):885–95. doi: 10.1586/1744666X.2015.1050382
139. Toubal A, Nel I, Lotersztajn S, Lehuen A. Mucosal-associated invariant T cells and disease. Nat Rev Immunol (2019) 19(10):643–57. doi: 10.1038/s41577-019-0191-y
140. Tupin E, Kinjo Y, Kronenberg M. The unique role of natural killer T cells in the response to microorganisms. Nat Rev Microbiol (2007) 5(6):405–17. doi: 10.1038/nrmicro1657
141. Zhang X, Angkasekwinai P, Dong C, Tang H. Structure and function of interleukin-17 family cytokines. Protein Cell (2011) 2(1):26–40. doi: 10.1007/s13238-011-1006-5
142. Hassane M, Jouan Y, Creusat F, Soulard D, Boisseau C, Gonzalez L, et al. Interleukin-7 protects against bacterial respiratory infection by promoting IL-17A-producing innate T-cell response. Mucosal Immunol (2020) 13(1):128–39. doi: 10.1038/s41385-019-0212-y
143. Yu HC, Lu MC, Huang KY, Huang HL, Liu SQ, Huang HB, et al. Sulfasalazine Treatment Suppresses the Formation of HLA-B27 Heavy Chain Homodimer in Patients with Ankylosing Spondylitis. Int J Mol Sci (2015) 17(1):46. doi: 10.3390/ijms17010046
144. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6(11):1133–41. doi: 10.1038/ni1261
145. Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med (2006) 203(10):2271–9. doi: 10.1084/jem.20061308
146. Maroof A. Presentation 3776. Translational data suggesting a pivotal role for IL-17A and IL-17F in hidradenitis suppurativa. SHSA 2022 (2022).
147. Torina A, Guggino G, La Manna MP, Sireci G. The Janus Face of NKT Cell Function in Autoimmunity and Infectious Diseases. Int J Mol Sci (2018) 19(2):440. doi: 10.3390/ijms19020440
148. Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol (2012) 12(4):239–52. doi: 10.1038/nri3174
149. Meyer EH, DeKruyff RH, Umetsu DT. iNKT cells in allergic disease. Curr Top Microbiol Immunol (2007) 314:269–91. doi: 10.1007/978-3-540-69511-0_11
150. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. (2009) 31(2):331–41. doi: 10.1016/j.immuni.2009.08.001
151. Lalor SJ, Dungan LS, Sutton CE, Basdeo SA, Fletcher JM, Mills KH. Caspase-1-processed cytokines IL-1beta and IL-18 promote IL-17 production by gammadelta and CD4 T cells that mediate autoimmunity. J Immunol (2011) 186(10):5738–48. doi: 10.4049/jimmunol.1003597
152. Godfrey DI, Koay HF, McCluskey J, Gherardin NA. The biology and functional importance of MAIT cells. Nat Immunol (2019) 20(9):1110–28. doi: 10.1038/s41590-019-0444-8
153. Tominaga K, Yamagiwa S, Setsu T, Kimura N, Honda H, Kamimura H, et al. Possible involvement of mucosal-associated invariant T cells in the progression of inflammatory bowel diseases. BioMed Res (2017) 38(2):111–21. doi: 10.2220/biomedres.38.111
154. Hinks TS. Mucosal-associated invariant T cells in autoimmunity, immune-mediated diseases and airways disease. Immunology. (2016) 148(1):1–12. doi: 10.1111/imm.12582
155. Godfrey DI, MacDonald HR, Kronenberg M, Smyth MJ, Van Kaer L. NKT cells: what's in a name? Nat Rev Immunol (2004) 4(3):231–7. doi: 10.1038/nri1309
156. Wang H, Hogquist KA. How Lipid-Specific T Cells Become Effectors: The Differentiation of iNKT Subsets. Front Immunol (2018) 9:1450. doi: 10.3389/fimmu.2018.01450
157. Dai H, Rahman A, Saxena A, Jaiswal AK, Mohamood A, Ramirez L, et al. Syndecan-1 identifies and controls the frequency of IL-17-producing naive natural killer T (NKT17) cells in mice. Eur J Immunol (2015) 45(11):3045–51. doi: 10.1002/eji.201545532
158. Lee YJ, Wang H, Starrett GJ, Phuong V, Jameson SC, Hogquist KA. Tissue-Specific Distribution of iNKT Cells Impacts Their Cytokine Response. Immunity. (2015) 43(3):566–78. doi: 10.1016/j.immuni.2015.06.025
159. Coquet JM, Chakravarti S, Kyparissoudis K, McNab FW, Pitt LA, McKenzie BS, et al. Diverse cytokine production by NKT cell subsets and identification of an IL-17-producing CD4-NK1.1- NKT cell population. Proc Natl Acad Sci USA (2008) 105(32):11287–92. doi: 10.1073/pnas.0801631105
160. Michel ML, Keller AC, Paget C, Fujio M, Trottein F, Savage PB, et al. Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J Exp Med (2007) 204(5):995–1001. doi: 10.1084/jem.20061551
161. Rachitskaya AV, Hansen AM, Horai R, Li Z, Villasmil R, Luger D, et al. Cutting edge: NKT cells constitutively express IL-23 receptor and RORgammat and rapidly produce IL-17 upon receptor ligation in an IL-6-independent fashion. J Immunol (2008) 180(8):5167–71. doi: 10.4049/jimmunol.180.8.5167
162. Doisne JM, Becourt C, Amniai L, Duarte N, Le Luduec JB, Eberl G, et al. Skin and peripheral lymph node invariant NKT cells are mainly retinoic acid receptor-related orphan receptor (gamma)t+ and respond preferentially under inflammatory conditions. J Immunol (2009) 183(3):2142–9. doi: 10.4049/jimmunol.0901059
Keywords: IL-17A, IL-17F, IL-23, spondyloarthritis, Th17 cells, MAIT cells, γδ T cells, psoriasis
Citation: Navarro-Compán V, Puig L, Vidal S, Ramírez J, Llamas-Velasco M, Fernández-Carballido C, Almodóvar R, Pinto JA, Galíndez-Aguirregoikoa E, Zarco P, Joven B, Gratacós J, Juanola X, Blanco R, Arias-Santiago S, Sanz Sanz J, Queiro R and Cañete JD (2023) The paradigm of IL-23-independent production of IL-17F and IL-17A and their role in chronic inflammatory diseases. Front. Immunol. 14:1191782. doi: 10.3389/fimmu.2023.1191782
Received: 22 March 2023; Accepted: 05 July 2023;
Published: 04 August 2023.
Edited by:
Gerald Nabozny, Boehringer Ingelheim, United StatesReviewed by:
Ganesan Ramamoorthi, Moffitt Cancer Center, United StatesNicolò Costantino Brembilla, University of Geneva, Switzerland
Copyright © 2023 Navarro-Compán, Puig, Vidal, Ramírez, Llamas-Velasco, Fernández-Carballido, Almodóvar, Pinto, Galíndez-Aguirregoikoa, Zarco, Joven, Gratacós, Juanola, Blanco, Arias-Santiago, Sanz Sanz, Queiro and Cañete. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan D. Cañete, amNhbmV0ZUBjbGluaWMuY2F0; Rubén Queiro, cnViZW5xdWU3QHlhaG9vLmVz
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship