
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 20 June 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1190943
This article is part of the Research TopicStructural Immunology of Molecular Innate ImmunityView all 14 articles
 Sergio Navas-Yuste1
Sergio Navas-Yuste1 Karla de la Paz1,2Javier Querol-García1,2
Karla de la Paz1,2Javier Querol-García1,2 Sara Gómez-Quevedo1,3
Sara Gómez-Quevedo1,3 Santiago Rodríguez de Córdoba1,4
Santiago Rodríguez de Córdoba1,4 Francisco J. Fernández1,2*
Francisco J. Fernández1,2* M. Cristina Vega1*
M. Cristina Vega1*Leptospirosis is a neglected worldwide zoonosis involving farm animals and domestic pets caused by the Gram-negative spirochete Leptospira interrogans. This bacterium deploys a variety of immune evasive mechanisms, some of them targeted at the complement system of the host’s innate immunity. In this work, we have solved the X-ray crystallographic structure of L. interrogans glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to 2.37-Å resolution, a glycolytic enzyme that has been shown to exhibit moonlighting functions that potentiate infectivity and immune evasion in various pathogenic organisms. Besides, we have characterized the enzyme’s kinetic parameters toward the cognate substrates and have proven that the two natural products anacardic acid and curcumin are able to inhibit L. interrogans GAPDH at micromolar concentration through a noncompetitive inhibition modality. Furthermore, we have established that L. interrogans GAPDH can interact with the anaphylatoxin C5a of human innate immunity in vitro using bio-layer interferometry and a short-range cross-linking reagent that tethers free thiol groups in protein complexes. To shed light into the interaction between L. interrogans GAPDH and C5a, we have also carried out cross-link guided protein-protein docking. These results suggest that L. interrogans could be placed in the growing list of bacterial pathogens that exploit glycolytic enzymes as extracellular immune evasive factors. Analysis of the docking results indicates a low affinity interaction that is consistent with previous evidence, including known binding modes of other α-helical proteins with GAPDH. These findings allow us to propose L. interrogans GAPDH as a potential immune evasive factor targeting the complement system.
The complement system is a central part of the innate immune defense against pathogens (1). It comprises about forty soluble and membrane-associated proteins, which survey the blood and interstitial fluids for pathogens, immune complexes, and apoptotic cell debris. Those stimuli can activate the complement system very swiftly through three main activation pathways: the alternative (AP), classical (CP), and lectin (LP) pathways. Normal complement activation on surfaces involves a self-amplification cascade where the so-called C3 convertases proteolytically cleave C3, the most abundant complement factor, to yield C3b, which remains attached to the activating surface, and C3a. Surface-attached C3b can assemble C3-convertase enzyme complexes, propagating C3b deposition in a process known as opsonization. C3b can be quickly cleaved by factor I into iC3b (2). On densely opsonized surfaces like those of pathogens and other foreign or damaged surfaces, C3b-containing enzyme complexes can cleave C5 into C5b and soluble C5a, a 74-amino-acid anaphylatoxin (3). The former remains bound to surfaces and nucleate the assembly of the so-called membrane attack complex (MAC) (4), which can lyse targeted cells directly through osmotic shock in a process known as terminal pathway. Like C3a, C5a is a soluble factor that diffuses away from the site of activation and acts as one of the most powerful chemoattractants of innate immunity. Once liganded to its cognate receptor (C5aR1/CD88), C5a stimulates proinflammatory responses like chemotaxis and vascular permeability, which result in the recruitment of inflammatory neutrophils and macrophages to the sites of activation (5). On self-cells, however, complement activation is strongly suppressed by self-protective fluid-phase regulators such as factor H and C4b binding protein (C4BP), both involved in the inactivation of iC3b on opsonized cell surfaces, and membrane regulators such as MCP and DAF, which disassemble C3 convertases to prevent further deposition of C3b (6).
Leptospirosis is a widespread zoonotic disease caused by the highly motile Gram-negative spirochete Leptospira (7, 8). Leptospira colonizes a range of hosts including humans, domestic and farm animals, and some wild animal species such as mice, rats, and bats, which typically serve as reservoirs of infection (9). In humans, leptospirosis typically presents with mild fever and flu-like symptoms, but in its more severe forms it can lead to fatal multi-organ failure. Leptospirosis causes about 1 million severe cases in humans every year with 60,000 fatalities (10). In cattle and swine, leptospirosis causes veterinary and economic damage through reproductive failure, abortion, still-births, fetal mummification, weak calves/piglets, and agalactia (8, 11). Its prevalence has surged in recent years due to global warming, intensive farming, and other geographic and socioeconomic factors (12). Globally, leptospirosis represents an increasing public and veterinary health threat, as evidenced by growing incidence rates and multiple outbreaks around the world, compounded by frequent misdiagnosis.
Pathogenic Leptospira produces the activation of all three complement activation pathways and, not surprisingly, it has evolved sophisticated immune evasion mechanisms to escape it (13). The deployment of Leptospira’s complement-targeting molecular weaponry accelerates the decay of the three complement activation pathways and inhibits the terminal pathway, thereby promoting the pathogen’s dissemination and infection. Examples of immunoevasion strategies deployed by Leptospira include: 1) acquisition via surface evasion molecules of host’s soluble complement regulators like factor H and C4BP (molecular mimicry) (14); 2) terminal pathway inhibition either through the direct interaction of surface pathogenic proteins with C9 or through the indirect interaction with vitronectin, an inhibitor of C5b7 complex formation and C9 polymerization; 3) plasminogen binding to and cleavage of C3b, C4b, and C5 (mixed molecular mimicry/proteolytic cleavage) (15); and 4) direct proteolytic degradation of complement proteins C2, C3, C4, and factor B. Several Leptospira virulence factors comprising extracellular enzymes and cell-surface proteins have been demonstrated to play key roles in host-cell adherence and immunoevasion. To date, two Leptospira proteins displaying moonlighting functions have been found: elongation factor-thermal unstable (EF-Tu), shown to interact with host extracellular membrane (ECM) molecules, plasminogen, and factor H, and the glycolytic enzyme α-enolase, described to interact with plasminogen, factor H, and C4BP.
We and others have proposed that the ubiquitous glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH; E.C. 1.2.1.12) from pathogenic bacteria may double as an innate immune evasive factor when it is found in the extracellular environment (16). To perform these moonlighting functions, GAPDH must be relocated to the extracellular space by cell lysis (e.g., via streptococcal lysins) (16), secretion (e.g., type-3 secretion systems in enteropathogenic Escherichia coli) (17), or outer membrane shedding (e.g., Francisella tularensis, Mycobacterium tuberculosis, Staphylococcus aureus, Atopobium vaginae, and Leptospira interrogans). Some of the infectivity enhancing functions attributed to moonlighting GAPDH are mostly targeted at the innate immunity and, specifically, the complement system. Examples of these mechanisms include sequestering nascent C5a as it is being generated by C5 cleavage, a mechanism described for Gram-positive bacteria such as Streptococcus pneumoniae, S. pyogenes, A. vaginae, and Clostridium perfringens (16, 18, 19); binding complement factors like C3 and C1q (20, 21); and increasing pathogen dissemination by binding to ECM components as plasminogen to help to degrade tissue barriers, basement membranes, and fibrin clots (13, 22).
In this work, we set out to characterize the structure and function of GAPDH from the Gram-negative spirochete L. interrogans and investigate whether it could operate as a virulence factor by binding to C5a. We have characterized LiGAPDH’s enzymatic activity and inhibition by curcumin and anacardic acid, two natural products, and we have solved its crystal structure at 2.37-Å resolution complete with its NAD+ cofactor. Furthermore, we have shown by bio-layer interferometry and controlled cross-linking experiments that LiGAPDH can bind C5a, a property shared by GAPDH enzymes from other pathogenic bacteria that might contribute to immune evasion in the mammalian host. To shed light into the C5a recognition mechanism, we have performed cross-link guided protein-protein docking.
The gene encoding full-length LiGAPDH (UniProt Accession No. Q72QM3_LEPIC) was amplified by PCR from L. interrogans serovar Copenhageni strain Fiocruz L1-130 genomic DNA (ATCC) and cloned into the pETM-11 expression vector by restriction-ligation after digesting the PCR fragment with BsaI-XhoI and the pETM-11 expression vector with NcoI-XhoI, conferring an N-terminal hexahistidine tag and a tobacco etch virus cleavage (TEV) site in frame with the LiGAPDH gene. The expression plasmid was verified by sequencing the entire ORF. For protein expression, the LiGAPDH construct was transformed into Rosetta(DE3) chemically competent cells. An overnight starter culture was used to inoculate a 2.5-L expression culture at 37 °C. The culture was allowed to grow at 37 °C in Luria-Bertani medium supplemented with 50 µg/ml kanamycin and 34 μg/ml chloramphenicol to an absorbance of 0.6 at 590 nm and then induced with 1 mM isopropyl β-D-thiogalactopyranoside (IPTG) for 20 h. Cell pellet was lysed by sonication in IMAC-A buffer (50 mM Tris-HCl (pH 8.0), 500 mM NaCl, 20 mM imidazole) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF) and one tablet of EDTA-free protease inhibitor cocktail. Supernatant was collected upon centrifugation for 20 min at 4 °C. The sample was clarified further by filtration through a 0.22 µm membrane and loaded on a HisTrap column (Cytiva) pre-equilibrated in IMAC-A buffer and eluted in IMAC-A buffer with 250 mM imidazole. Peak fractions were analyzed by SDS-PAGE and fractions containing LiGAPDH were pooled and dialyzed against a buffer containing 10 mM HEPES-NaOH (pH 7.4), 150 mM NaCl, 3.4 mM EDTA. Then, LiGAPDH was subjected to size exclusion chromatography on a HiLoad 16/60 Superdex 200 pg (Cytiva) pre-equilibrated in the same buffer. Comparison of the elution volume of LiGAPDH with a calibration curve constructed using high and low molecular weight calibration kits (Cytiva) revealed that the quaternary structure of LiGAPDH corresponds to a tetrameric oligomeric state. Finally, LiGAPDH was concentrated to 10 mg/ml, dispensed in 50-µl aliquots, snap-frozen in liquid nitrogen and stored at −80 °C until use. The yield was ~2 mg LiGAPDH/l of culture, showing a >95% purity on a Coomassie brilliant blue-stained SDS-PAGE gel.
The LiGAPDH enzyme activity was followed spectrophotometrically by the change in absorbance at 340 nm due to NADH formation (ϵ = 6220 M−1 cm−1), adapted from a previously described method (23). Temperature controlled assays were performed in an Eppendorf BioSpectrometer spectrophotometer at 25 °C. One unit of enzyme activity was defined as the amount of GAPDH that converts 1 µmol/min of NAD+ to NADH at 25 °C. A standard assay was carried out in a final volume of 0.15 ml using 40 mM Tris-HCl (pH 8.5), 2 mM EDTA, 10 nM LiGAPDH, and indicated concentrations of the different substrates: nicotinamide adenine dinucleotide (NAD+), glyceraldehyde 3-phosphate (G3P), and inorganic phosphate (Pi). NAD+ concentration was varied between 0.02 and 1.62 mM while keeping fixed G3P at 2 mM and Pi at 5 mM; G3P concentration between 0.14 and 11.7 mM at 2 mM NAD+ and 5 mM Pi; and Pi concentration between 0.38 and 31.5 mM at 2 mM NAD+ and 2 mM G3P. The reaction was initiated by adding 0.56 μg of enzyme. Michaelis-Menten parameters were obtained by non-linear regression fitting of the kinetic data using SigmaPlot 14.5 (Systat Software Inc.).
Inhibition assays were performed with two natural compounds, anacardic acid and curcumin. Four inhibitor concentrations each were tested for anacardic acid (10, 24, 64, and 160 μM) and curcumin (10, 25, 62.5, and 150 μM). To ascertain the inhibition modality with respect to G3P and NAD+, initial velocity measurements at each inhibitor concentration were carried out varying G3P concentration (0.06-1.56 mM G3P for both inhibitors) while maintaining a saturating concentration of 2 mM NAD+; or varying NAD+ concentration (0.02-1.1 mM NAD+ for both inhibitors) while maintaining a saturating concentration of 2 mM G3P. In either case, potassium phosphate was kept at a saturating concentration of 5 mM. The software SigmaPlot 14.5 (Systat Software Inc.) was used to analyze the data.
To find the optimal crystallization conditions, we performed extensive scans of crystallization conditions from commercial screenings by Hampton Research and Molecular Dimensions (Crystal Screening 1 and 2, Salt Screening 1 and 2, JSCG+ 1 and 2). The best results were obtained with Bis-Tris buffer at different concentrations of PEG 3350 as precipitant. The crystals presented a form of elongated prisms with a dimension of 150-300 μm in their longest axis. For freezing, 20% (v/v) glycerol was used as cryoprotectant. Crystals were mounted on Micromount loops (MiTeGen) and frozen in liquid nitrogen. The crystals were diffracted at the BL13-XALOC beamline of the ALBA synchrotron (Barcelona) (24). The maximum observable resolution was 2.37 Å with unit cell dimensions of a = 79.8 Å, b = 82.0 Å, c = 123.2 Å, α = 94.0°, β = 95.1°, and γ = 112.5° and P1 space group. The data was processed with XDS (25) and scaled and merged with Aimless (26).
The crystallographic structure of LiGAPDH was solved by molecular replacement using PHASER (27) in the PHENIX suite (28) with the structure of AvGAPDH as a model (PDB ID 5LD5; http://doi.org/10.2210/pdb5LD5/pdb) (18). The crystal structure contained two tetramers in the asymmetric unit. The difference map (Fo-Fc) showed a clear position and conformation for an NAD+ cofactor in the active site of all the monomers from the two tetramers. Refinement cycles with phenix.refine (29) of the PHENIX suite were interspersed with cycles of manual construction (placing first NAD+ and then solvent molecules) and validation cycles with Coot (30). Non-crystallographic symmetry was applied as a constraint on the main-chain dihedral angles during the initial refinement, but they were removed during the later stages of refinement. At the end of the refinement, the LiGAPDH model obtained a Rwork/Rfree of 0.19/0.23 with an r.m.s.d. of 0.011 Å and 1.31° for distance and bond angles, respectively. The final model consists of 2674 amino acid residues, eight NAD+ molecules, 380 water molecules, 4 phosphate anions, and 33 glycerol molecules from the cryoprotectant solution. The crystallographic refinement statistics are summarized in Table 1.
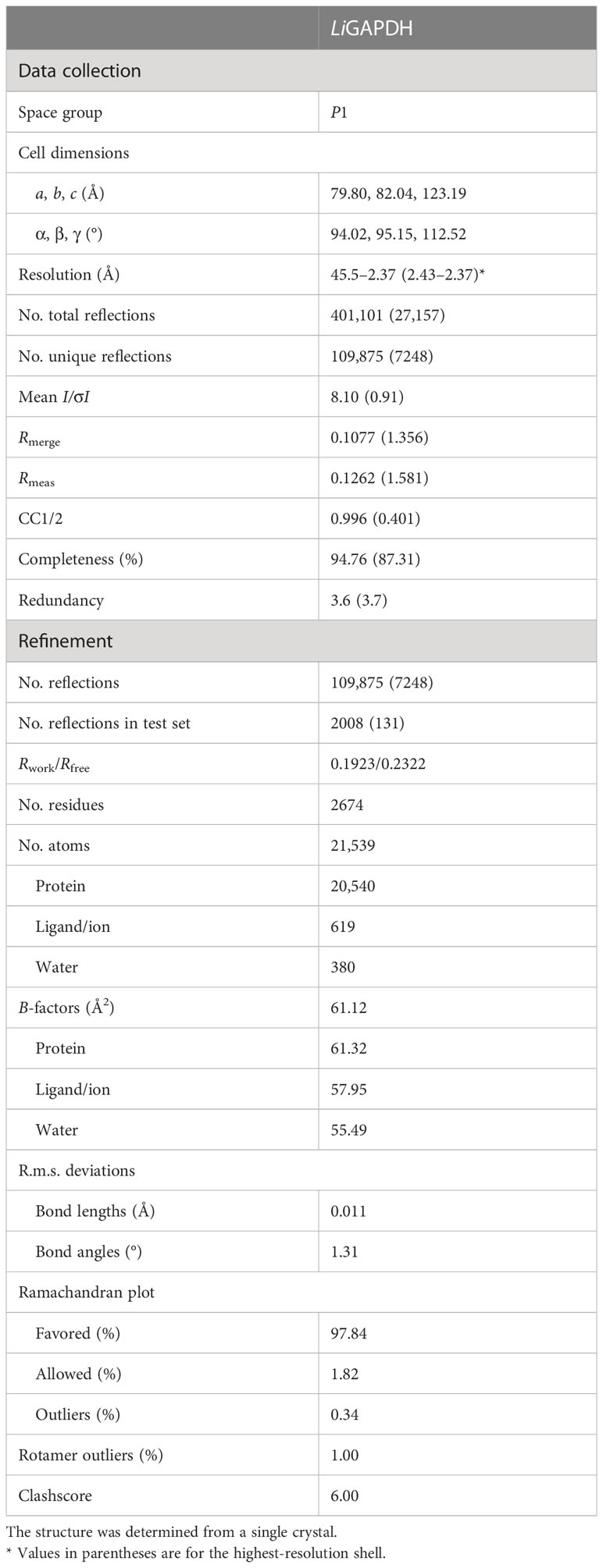
Table 1 Data collection and refinement statistics (molecular replacement).
The coordinate and structure factors files have been deposited with the Protein Data Bank (PDB) with PDB ID 8OHA (http://doi.org/10.2210/pdb8OHA/pdb).
SAXS experiments were carried out at the B21 beamline (31) from the Diamond Light Source synchrotron (DLS, UK). To improve sample purity and monodispersity, we collected SAXS data using continuous flow in HPLC-SAXS mode (620 images/3 s) at 9 °C. Sample LiGAPDH at 7 mg/ml in 10 mM HEPES-NaOH (pH 7.4), 150 mM NaCl, 3.4 mM EDTA, 2 mM TCEP, 3% (v/v) glycerol were injected on a Shodex KW-403 size-exclusion column (theoretical separation range 10-700 kDa, 4.6-ml column volume), previously equilibrated in the same buffer. Individual 2D data images were radially averaged to produce 1D diffraction profiles I(q) vs. q without subtracting buffer. For the final data reduction process, statistical checks were performed to rule out images affected by radiation damage or systematic scaling errors (32). The data were averaged, buffer subtracted, and combined to produce the final SAXS profile covering the transfer momentum range 0.0026 to 0.3400 Å-1. The ATSAS 3.0 software package (33) was used to extract structural information and perform an ab initio shape restoration of LiGAPDH. Firstly, the number of Shannon channels and the maximum usable q were estimated with SHANUM (34). Next, the direct diffraction extrapolated to zero angle I(0) and the Rg were evaluated using the Guinier approximation (35) and the pairwise distance distribution function in real space (P(r) vs. r) computed with GNOM (36). From the P(r) profile it was possible to evaluate the maximum dimension (Dmax) of the particle. Additionally, two different concentration-independent methods were used to estimate the molecular mass of LiGAPDH: the correlation volume (VC) and the empirical Porod volume (VP) correction (37), implemented in the ATSAS toolsets called DATMOW, DATVC, and DATPOROD. With DATCLASS, the shape of LiGAPDH derived from the SAXS data was classified as compact and potentially unique (38). Ab initio shape restoration was performed using a dummy bead model of 50 independent runs with DAMMIF (39), which were superimposed, averaged, and clustered with DAMAVER (40), and further refined with DAMMIN (41) to create the final ab initio shape. The most representative cluster contained >90% of all bead models with a normalized spatial discrepancy (NSD) threshold of 0.55 (42). The resolution was estimated from the Fourier shell correlation (FSC) at FSC = 0.5 (43). The fit of the crystal structure of LiGAPDH to the SAXS data was evaluated using CRYSOL (44).
Bio-layer interferometry (BLI) studies were performed on a BLItz instrument (ForteBio) at 25 °C with shaking at 2200 rpm. BLI assay buffer consisted of 10 mM HEPES-NaOH (pH 7.4), 50 mM NaCl, 0.34 mM EDTA, 0.02% (v/v) polysorbate 20 (P20), which was 0.22-µm filtered. Before use, (anti-biotin) streptavidin (SA) biosensors (ForteBio 18-5019) were hydrated in BLI assay buffer for 10 min. All samples for BLI measurements were prepared in 4.5 µl. The BLI assay was as follows: baseline (30 s) (Equilibration), loading (300 s) (BLI assay buffer for nonspecific binding or biotinylated-C5a (34 µg/ml) (Abvance Biotech ABVC5ARBIO1), stabilization (300 s) (BLI assay buffer), baseline (30 s) (Equilibration), association (300 s) (230 µM LiGAPDH), and dissociation (300 s) (BLI assay buffer). Loading of biotinylated-C5a for 300 s onto SA sensor tips resulted in a wavelength shift signal of ~2.75 nm. Loading of LiGAPDH for 300 s onto either the mock SA sensor tips or the biotinylated-C5a SA sensor tips resulted in wavelength shift signals of 0.5 and 4.5 nm, respectively.
Specific cross-linking assays were performed with bis(maleimido)ethane (BMOE; Pierce 22322), a 7-atom, 8.0-Å short spacer arm cross-linking reagent that generates non-cleavable cross-links between sulfhydryl groups that are in proximity (< 8 Å apart). Cross-linking reactions were carried out by mixing 10 µg LiGAPDH with 0.5, 1.0, 2.0, 4.0, or 5.0 µg human recombinant C5a (ABVC5A, Abvance Biotech) in assay buffer (10 mM HEPES-NaOH (pH 7.4), 150 mM NaCl, 3.4 mM EDTA) containing 0.3 mM BMOE, and incubating the reaction mixtures for 2 h at 4 °C. The C5a:LiGAPDH molar ratio for these assays was 0.27, 0.53, 1.07, 2.13, or 2.67; and the BMOE:LiGAPDH molar ratio was 1.44. To control for nonspecific cross-linking, we treated identical amounts of LiGAPDH (10 µg) and C5a (5 µg) with 0.3 mM BMOE and without BMOE. We followed the appearance of cross-linked products by 12% SDS-PAGE gel electrophoresis and Coomassie-Brilliant Blue (CBB) staining. A second SDS-PAGE gel was run with one-fourth of the cross-linking reactions under otherwise identical conditions and transferred onto a nitrocellulose membrane (1 h at 80 V) for Western blotting. The membrane was incubated 1 h at RT with blocking solution (5% (w/v) BSA in TBST), probed with an anti-C5a primary antibody (1:6000, 1 h at 37 °C) and a Goat anti-Rabbit HRP secondary antibody (1:2000, 30 min at 37 °C), developed with luminol and water peroxide, and imaged on a ChemiDoc Imaging System (Bio-Rad). Afterwards, the same blot was treated with Restore Stripping buffer (ThermoFisher Scientific 21059) for 30 min at 37 °C, reblocked, reprobed with an anti-His HRP antibody (1:3000, 1 h at RT), developed with luminol and water peroxide, and imaged.
We applied a cross-link guided protein-protein docking protocol to predict LiGAPDH-C5a complexes using the standard ROSETTA docking protocol (45), with modifications. Receptor (LiGAPDH) and ligand (C5a) were first relaxed and then subjected to cross-link guided docking using the known length of the BMOE cross-link as restraint. The protocol filters and ranks the protein-protein docking poses by the sequential application of Xwalk (46) to simulate cross-links on protein surfaces, FreeSASA (47) and PISA (48) to calculate the size of predicted binding interfaces, and an energy-based clustering approach implemented in ROSETTA (49).
Electrostatic potential surfaces of LiGAPDH and C5a were calculated with the APBS (Adaptive Poisson-Boltzmann Solver) (50) software as a plugin in PyMOL (51) using default parameters.
We have determined the first crystal structure of LiGAPDH at 2.37-Å resolution (Figure 1). The crystal structure corresponds to the holoenzyme with an NAD+ molecule tightly bound into the active site (Figure 1). We solved the structure by molecular replacement using Atopobium vaginae GAPDH as a model (AvGAPDH; PDB ID 5LD5; http://doi.org/10.2210/pdb5LD5/pdb) (18). There are two independent tetramers in the asymmetric unit that are nearly identical, with an r.m.s.d. of 0.30 Å. This remarkable similarity is mirrored by the structure of the individual subunits, which can be superimposed with an r.m.s.d. of 0.26 Å on average. Crystallographic data processing and refinement and validation statistics are reported in Table 1.
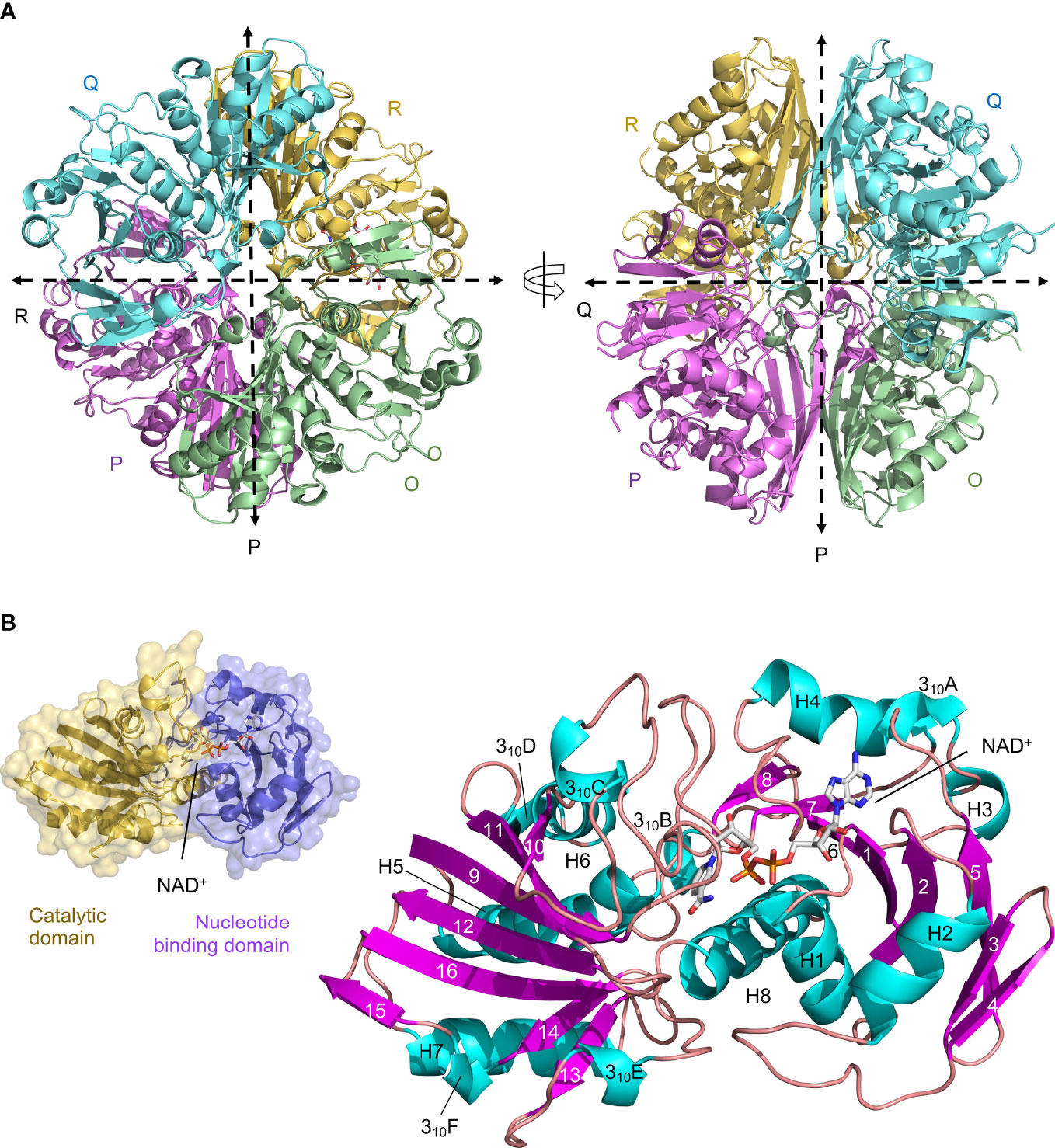
Figure 1 Crystal structure of LiGAPDH. (A) Overall tetrameric structure of LiGAPDH in cartoon representation with chain colors (O in green, P in violet, Q in cyan, and R in yellow). Crossing dashed lines indicate the directions of the two molecular symmetry axes on the plane of the figure. The NAD+ cofactor is shown in sticks and CPK colors. Two views of the LiGAPDH tetramer are shown down the Q-axis or the R-axis, which are perpendicular to the plane of the figure. (B) Cartoon representation of the LiGAPDH subunit structure color coded according to secondary structure: helices in cyan, β-strands in violet, and irregular segments and loops in salmon. Secondary structural elements and the NAD+ are annotated. In the inset (top left corner) we show the same monomer in molecular surface representation, color according to the domain: the N-terminal nucleotide-binding domain is in violet and the C-terminal catalytic domain in gold.
The quaternary structure of LiGAPDH consists in a homotetramer with O, P, Q, and R subunits related by a 222/D2 molecular symmetry, which gives rise to three non-equivalent interfaces related by three mutually perpendicular axes referred to as P, Q, and R (Figure 1A). The monomers are composed of two domains: an N-terminal domain that contains the NAD+ binding pocket (residues 1-152), and a catalytic C-terminal domain spanning residues 153-335 (Figure 1B). The N-terminal domain adopts an α/β/α Rossmann fold characterized by the classic α/β nucleotide binding pocket, which typically contains a central 7-stranded β-sheet and a tightly bound NAD+ cofactor occupying the active site. In LiGAPDH there are 8 β-strands (β1 to β8) because the canonical seventh β-strand is split into two smaller β-strands (β7 and β8) by a small irregular segment of extended conformation. Small helices are inserted between consecutive β-strands in this domain, which is further stabilized by packing against the C-terminal H8 helix. The C-terminal domain contains an 8-stranded β-sheet (β9 to β16), with helices inserted between β9-β10 (310C), β10-β11 (H6 and 310D), β12-β13 (H7), and β13-β14 (310E). Helix H8 is the last secondary structural motif of LiGAPDH.
The main interfaces through which each LiGAPDH monomer interacts with its two neighboring chains within the tetramer are not equivalent and have different surface areas (Figure 1A and Supplementary Figures 1, 2). First, the interface between the O-P subunits is the most extensive, with an average surface area of 1901 Å2 (Supplementary Figure 1). Nine H-bonds and 19 salt bridges stabilize the O-P interface. Second, the O-R interface, with an average surface area of 1412 Å2, has up to 13 H-bonds (Supplementary Figure 2A). Finally, the smallest intersubunit interface lies between the O-Q subunits, with an average surface area of 492 Å2, 10 H-bonds, and 2 salt bridges (Supplementary Figure 2B).
Another key structural feature of LiGAPDH is the S loop, an extended and irregular segment comprising residues Ala180-Ile207 that inserts itself between the NAD+-binding site and the adjacent subunit (Figure 1B and Supplementary Figure 2). The S loop contains residues lying between two of the catalytic triad residues His179 and Arg234 that are important for catalysis, cofactor-binding, and dimerization.
We analyzed the size and other hydrodynamic properties of LiGAPDH by solution small-angle X-ray scattering (SAXS) at the B21 beamline of the Diamond Light Synchrotron (DLS) (31). SAXS parameters are reported in Table 2 and Supplementary Table 1. Results indicated that LiGAPDH is a fairly spherical homotetramer with a well-folded structure (Figures 2A, B), with a radius of gyration Rg of 34.8 Å and a maximum dimension Dmax of 90.9 Å (Figure 2C). These hydrodynamic parameters match well those obtained from the crystallographic structure (Rg 32.5 Å, Dmax 99.6 Å). Indeed, direct comparison of the theoretical scattering calculated with CRYSOL, and the experimental scattering confirmed the excellent agreement with a χ2 = 1.5 (Figure 2A). Ab initio shape reconstruction of LiGAPDH using dummy-bead models as implemented in DAMMIF resulted in a family of volumes with a consistent shape and a calculated resolution for the consensus reconstruction of 28.3 Å (Supplementary Figure 3). Attempts to rigid-body fit the crystallographic model of LiGAPDH into the ab-initio SAXS shape showed a close agreement between the crystal and solution structures (Figure 2D). From these observations, we concluded that the overall organization of LiGAPDH in solution is preserved in the crystal lattice.
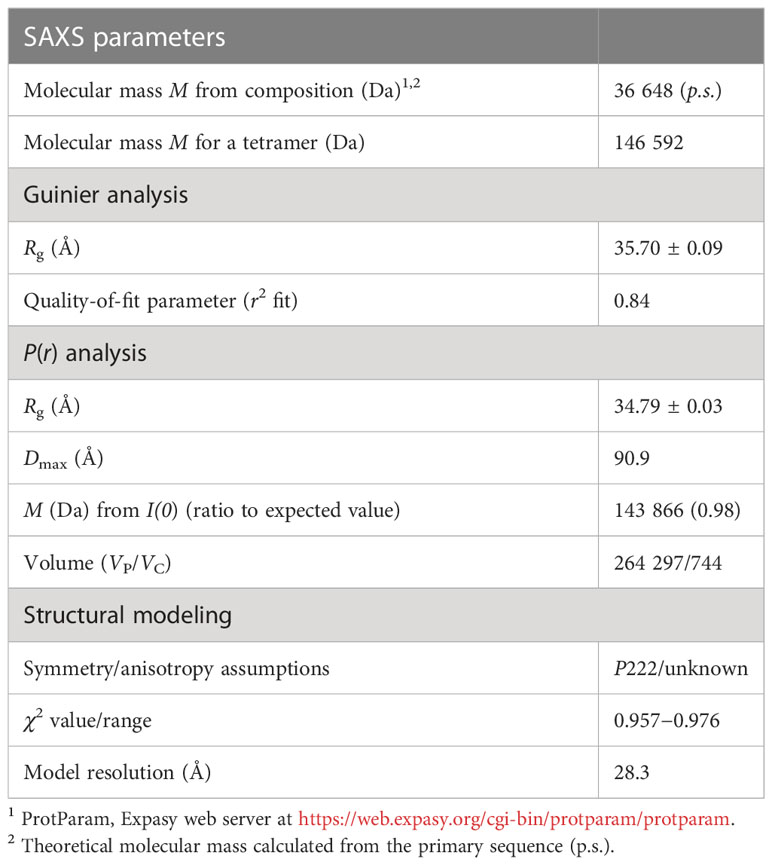
Table 2 Small-angle X-ray scattering (SAXS) parameters.
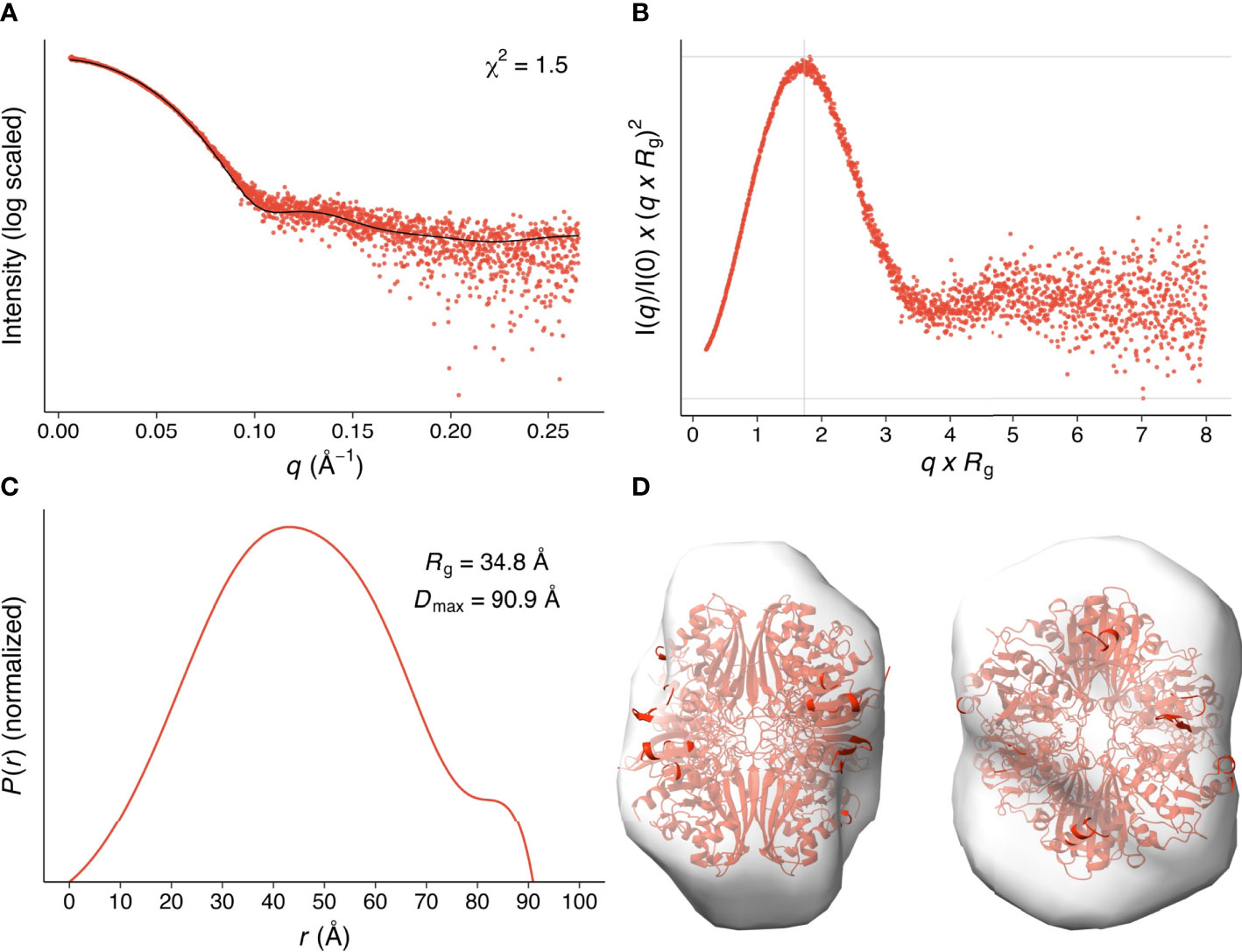
Figure 2 SAXS hydrodynamic properties and shape restoration for LiGAPDH. (A) 1D diffraction intensity of LiGAPDH plotted as a function of the diffraction momentum transfer q. Experimental data shown as a scatter plot (red circles). The black line corresponds to the average of the theoretical scattering profile of the two LiGAPDH tetramers in the asymmetric unit (χ2 = 1.5). (B) Dimensionless Kratky representation showing the degree of protein folding in solution. The experimental pattern is shown as a scatter plot (red circles). The cross (gray lines) marks the so-called Guinier-Kratky point (1.732, 1.1), i.e., where the position of the main peak for globular proteins would be located. (C) Pair distance distribution function P(r) plotted as a function of r. The experimental pattern is shown as a solid line (red color). The value of Dmax is the largest non-negative value that the distribution function supports. (D) Cartoon representation of LiGAPDH (in red) fitted inside the ab initio shape calculated with DAMMIF. Two orientations 90° apart are shown.
The active site of LiGAPDH is a large cavity covered by a lid spanning about 50 amino acid residues (residues 114-164). At the bottom of the groove, the NAD+ cofactor occupies an elongated binding site between the central β-sheet of the N-terminal domain and helices H1-H5, where it makes contacts with the main-chain atoms of Asn32 (from β2), Glu76, and Arg77 (from the β5-310A loop) (Figure 3). The catalytic Cys152 residue is located at the intersection between the N-terminal and C-terminal domains, where it interacts with the side chains of His179 and Arg234, the two other catalytic triad residues, responsible for lowering the pKa of the Cys152 thiol nucleophile. Arg234, in turn, interacts with Thr182 and Gln185. LiGAPDH lacks an aspartic acid residue between Thr182 and Gln185, unlike other GAPDH sequences like those of C. perfringens and S. pyogenes (19).
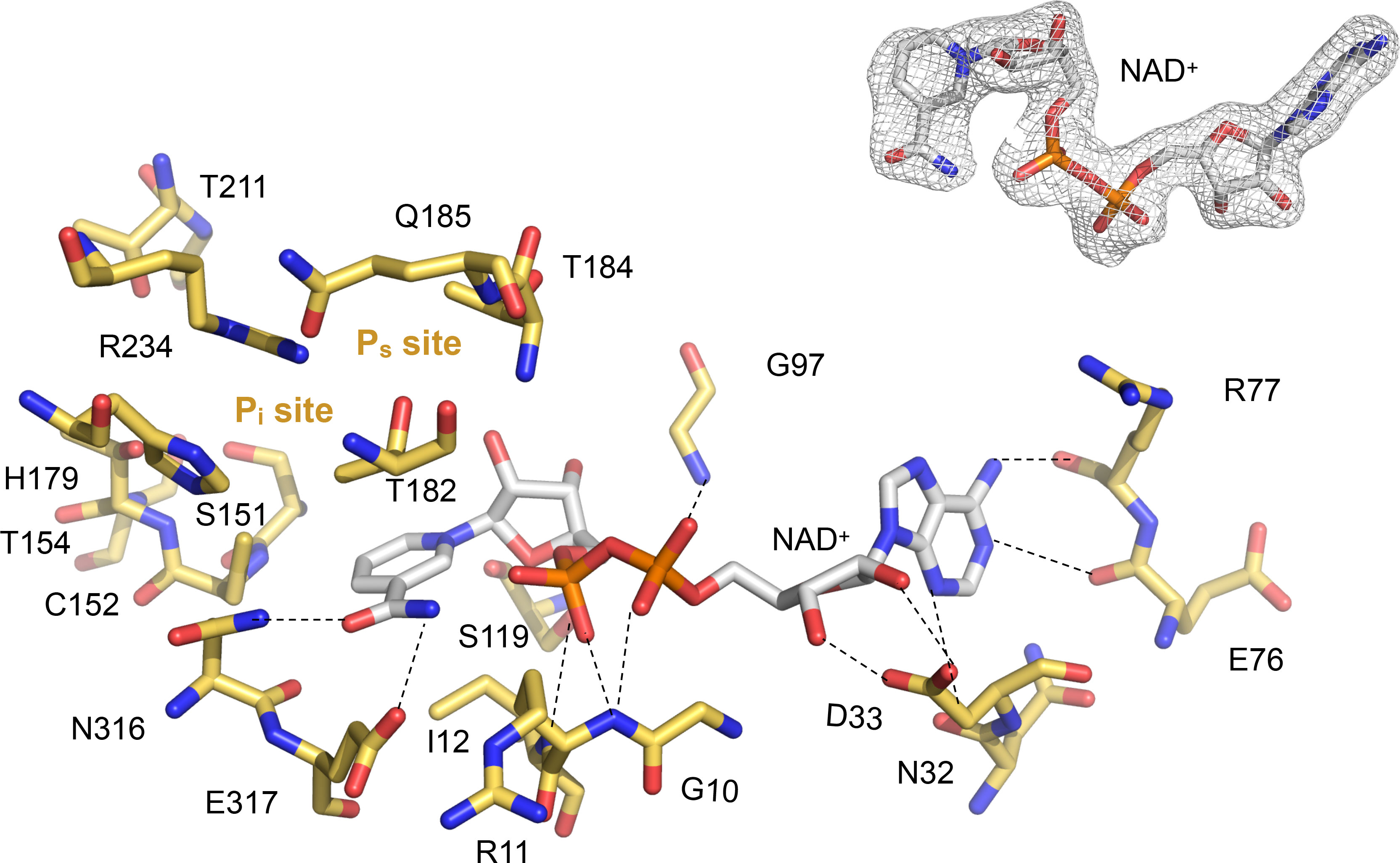
Figure 3 Close-up into the active site of LiGAPDH. Catalytic residues are shown in sticks in CPK colors (non-C atoms) or gold (C atoms). The NAD+ cofactor is shown in sticks in CPK colors. Dashed lines represent polar interactions (H-bonds). The Ps and Pi sites are annotated. Inset (top right) shows the quality of the experimental electron density surrounding the cofactor.
In contrast to other GAPDH structures, in LiGAPDH, well-ordered electron density was found for the residues responsible for binding the inorganic phosphate moieties of substrates and products, the so-called Ps and Pi binding sites (Figure 3). Inspection of the eight active sites in the crystal structure revealed that some of them had the Ps and Pi sites occupied by phosphate anions from the purification buffers or glycerol molecules from the cryoprotectant solution, which acted as substrates or substrate analogs. These ligands corroborated the relevance of the observed active-site configuration, which remained unchanged with or without substrate analogs across all subunits. The Ps site is formed by residues Thr182, Thr184, and Gln185, and the Pi site by residues Ser151, Thr153, His179, Thr211, and Gly212. The catalytic triad residue Arg234 interacts with both phosphate sites, thereby it belongs to the Ps and Pi sites.
Besides carrying out moonlighting functions in the extracellular space, LiGAPDH is a glycolytic enzyme located in the cytoplasm. We have measured its enzymatic activity using a well-established assay that reports on the reduction of the NAD+ cofactor to NADH at 25 °C and pH 8.5 (18, 19). In these assays, the kinetic parameters for the direct reaction catalyzed by LiGAPDH were determined by systematically varying NAD+, G3P, and Pi concentrations. While we observed a classic hyperbolic dependence of the catalytic activity for G3P/Pi at low substrate concentration (Michaelis-Menten model), with excess of either G3P or Pi the enzyme exhibited significant substrate inhibition with inhibitory constants KSS larger though within one order of magnitude of the corresponding Km values (Supplementary Figure 4). Kinetic parameters are reported in Table 3.

Table 3 LiGAPDH kinetic parameters.
Next, we examined the inhibitory effect of two natural compounds, anacardic acid and curcumin, which have previously shown efficacy against GAPDH from the Gram-positive bacterial pathogens A. vaginae (18) and S. pyogenes (19), and, in the case of anacardic acid, Trypanosoma cruzi (52). The safety of these compounds for use in humans makes them attractive lead compounds in repositioning campaigns for leptospirosis.
We obtained comparable results after testing the two natural compounds in the range 10-160 μM (anacardic acid) and 10-150 μM (curcumin) while varying the concentrations of either the G3P substrate or the NAD+ cofactor (Table 4; Figure 4; Supplementary Figures 5, 6). Both anacardic acid and curcumin behaved as micromolar non-competitive inhibitors of LiGAPDH with respect to G3P and NAD+. Not only was the inhibitory modality the same for both natural products, but also the magnitude of the inhibition constants was comparable: Kiana/G3P = 135 μM vs. Kicur/G3P = 148 μM and Kiana/NAD = 41 μM vs. Kicur/NAD = 59 μM.
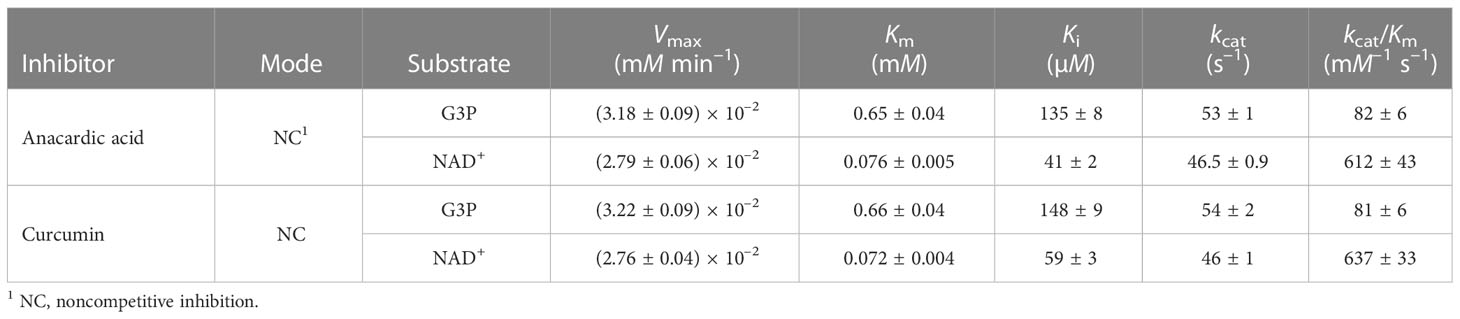
Table 4 Inhibition of LiGAPDH by anacardic acid and curcumin.
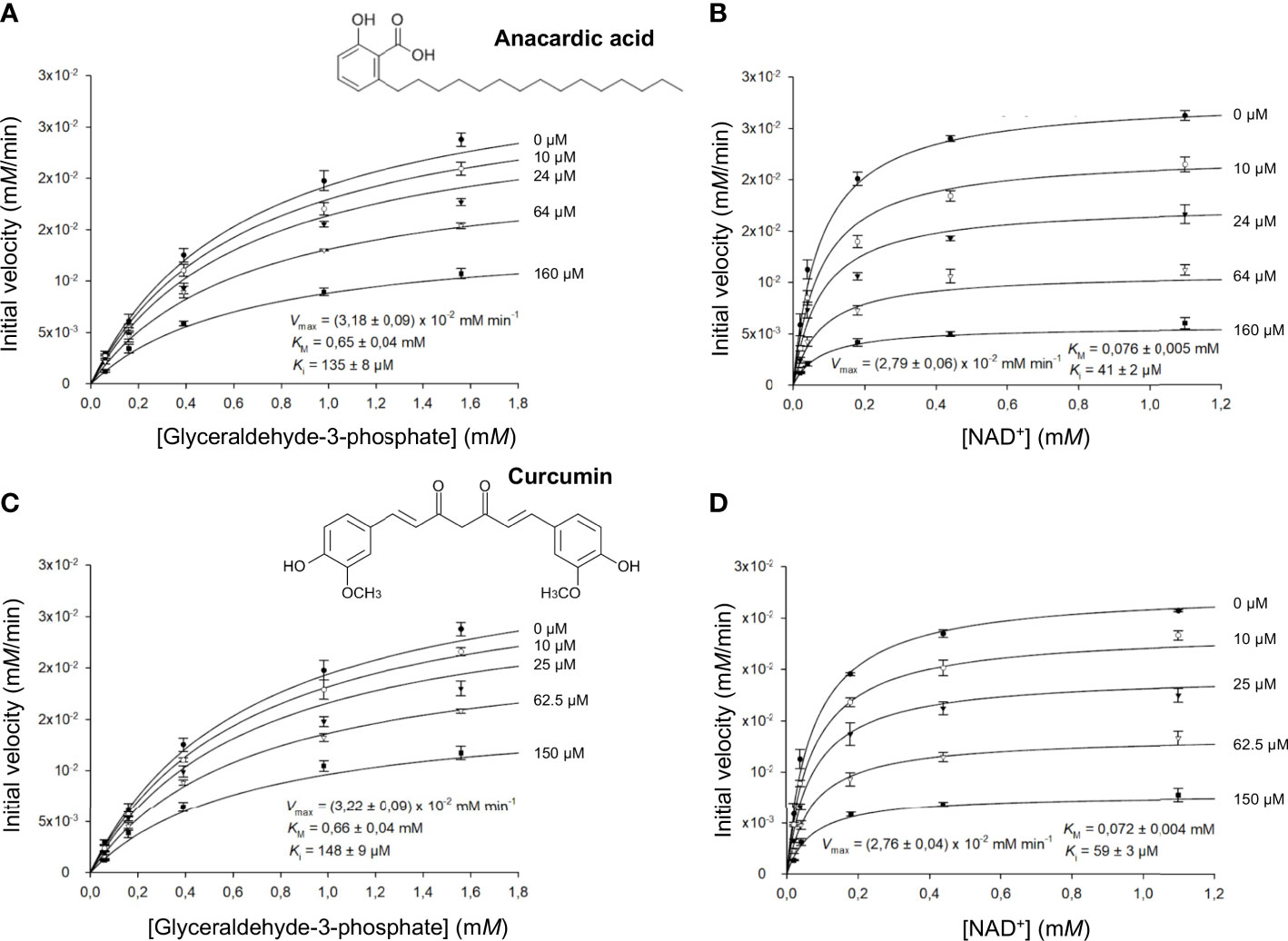
Figure 4 LiGAPDH can be inhibited by natural products. Initial rate of reaction plotted in the presence of various concentrations of anacardic acid (10-160 µM) against the concentration of (A) glyceraldehyde-3-phosphate (G3P) or (B) NAD+, and in the presence of curcumin (10-150 µM) against the concentration of (C) glyceraldehyde-3-phosphate (G3P) or (D) NAD+. Each experimental data point represents the mean and the errors are standard deviations of the mean (SEM) from three independent experiments. Nonlinear regression to a Michaelis-Menten hyperbolic model with noncompetitive inhibition was carried out with SigmaPlot 14.5 (R2 = 0.98).
It has been reported that C5a can interact with immune evasive factors like GAPDH on the pathogen’s surface through weak, transient interactions, which might be enhanced by the high local concentration of surface associated GAPDH (16, 18, 19). To reveal a potential interaction between natively folded LiGAPDH and C5a in vitro, we performed bio-layer interferometry experiments with streptavidin (SA) biosensors loaded with biotinylated-C5a. At a high LiGAPDH concentration (230 µM, ~9 mg/mL), we observed a clear binding event with immobilized C5a characterized by a low kinetic association constant (kon estimated at 37.3 ± 0.2 M–1 s–1) and a slow dissociation constant, which could not be accurately determined (Figure 5A). Binding to the unmodified SA biosensors was far lower at the same concentration of LiGAPDH, ruling out strong nonspecific interactions between the biosensor and the analyte.
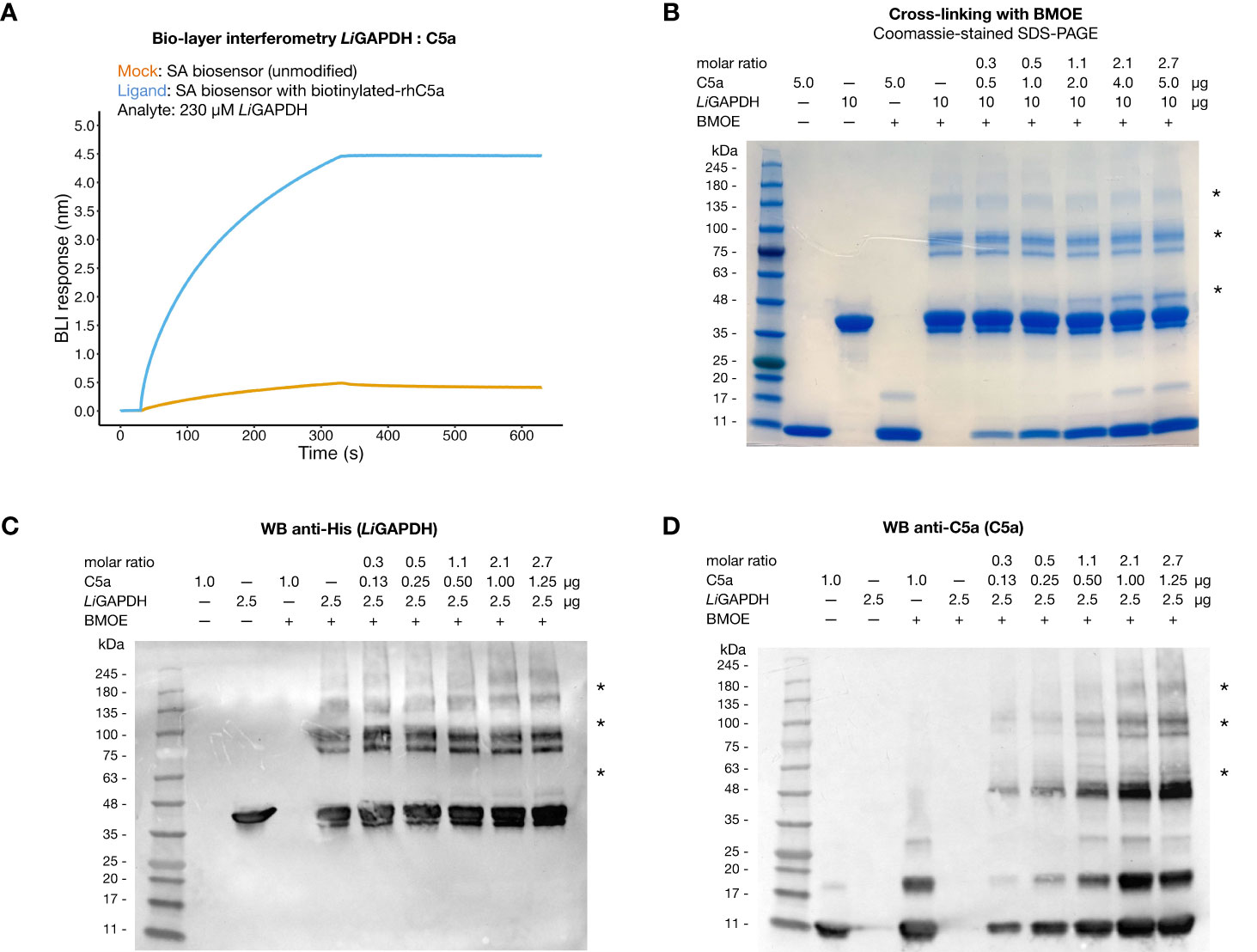
Figure 5 Interaction of LiGAPDH-C5a. (A) Bio-layer interferometry sensorgrams showing the wavelength shift length or BLI response (nm) obtained by incubating streptavidin (SA) biosensors previously loaded with biotinylated-C5a (blue line) or unmodified (orange line) with 230 µM LiGAPDH. (B) SDS-PAGE electrophoretic separation of cross-linking reactions of LiGAPDH and C5a with BMOE. Gel loaded with mock-treated control samples and BMOE-treated samples (increasing concentrations of C5a for a fixed concentration of LiGAPDH). The first two lanes with added BMOE represent internal controls for C5a (maximum load) and LiGAPDH without added C5a. (C) Western blotting of the same samples in (A) revealed with an anti-His HRP antibody. (D) Like (C) with an anti-C5a primary antibody (D). Asterisks indicate protein bands containing cross-linked LiGAPDH:C5a complexes.
To provide independent evidence for the interaction, we resorted to the highly specific cross-linker reagent BMOE. BMOE covalently tethers the free sulfhydryl groups of Cys residues that lie in close proximity, typically at a distance of 8 Å apart. This stringent condition discriminates nonspecific interactions from meaningful though weak interactions, at the risk of missing some authentic but little populated complexes or those that would have required a longer tether. As shown in Figure 5B, SDS-PAGE electrophoresis of BMOE-treated mixtures of LiGAPDH and C5a revealed the appearance of a band corresponding to cross-linked LiGAPDH-C5a complexes, clearly discernable already at substoichiometric C5a:LiGAPDH molar ratios (between 0.53-1.07). The molecular mass of this cross-linked band (~10 kDa greater than monomeric LiGAPDH) coincided with the expected mass increment due to C5a (molecular mass ~9 kDa) (Figure 5B, indicated by an asterisk). The intensity of this band depended on the amount of C5a in the cross-linking reaction, thus proving that a native, though weak interaction is likely to exist between LiGAPDH and C5a. We could identify proteins bands simultaneously containing C5a and hexahistidine-tagged LiGAPDH by Western blotting with anti-C5a and anti-His antibodies (Figures 5C, D, indicated by asterisks), demonstrating the formation of LiGAPDH:C5a cross-linked complexes. This interaction necessarily involves protein surfaces containing a free Cys residue. The only free Cys in C5a (Cys704 in C5 numbering, Cys27 in C5a numbering) must therefore mediate this interaction. Based on previous evidence (16) we hypothesize that the catalytic Cys residue (Cys152 in LiGAPDH) participates in this interaction. Cys152 is the most reactive Cys residue in LiGAPDH since the nucleophilicity of its thiol moiety is enhanced by the catalytic environment. The specificity of the reaction was corroborated by the observation that no cross-linked bands developed in samples containing only C5a until later than the LiGAPDH-C5a cross-linked complexes, nor did they spontaneously appear on samples not treated with BMOE (Figure 5B).
To shed light on the structure of the LiGAPDH-C5a binary complexes, we carried out cross-link guided protein-protein docking with ROSETTA, as distance restraints from chemical cross-linking experiments can guide protein-protein docking calculations and significantly improve the accuracy of the simulations (45). All the calculated docking poses were clustered and filtered according to binding energy, compliance with distance restraints derived from the cross-linker, and the buried surface area. After analyzing the 10 most populated clusters of binary complexes (Supplementary Figure 7), the most likely docking pose was identified (Figure 6A). In this pose, C5a approximates the LiGAPDH deep groove at the interface between chains Q and P, making polar interactions with residues from both chains (shown in aquamarine and violet, respectively, in Figures 6A, B). Residues from C5a implicated in binding include residues near Cys27 (residues 23-36) and also from the N- and the C-terminal helices (Figure 6C, color coded as in Figures 6A, B). As far as the electrostatic potential is concerned, the face of C5a that comes closest to LiGAPDH bears a slightly negative charge, which is complementary to the long and positively charged cavity of the active site and neighboring residues (Figures 6D, E). The C-terminal helix of C5a tilts at an angle that allows it to slide out of the binding site. This orientation is consistent with known facts about the interaction of GAPDH from various organisms with mostly α-helical proteins of small size (53–55).
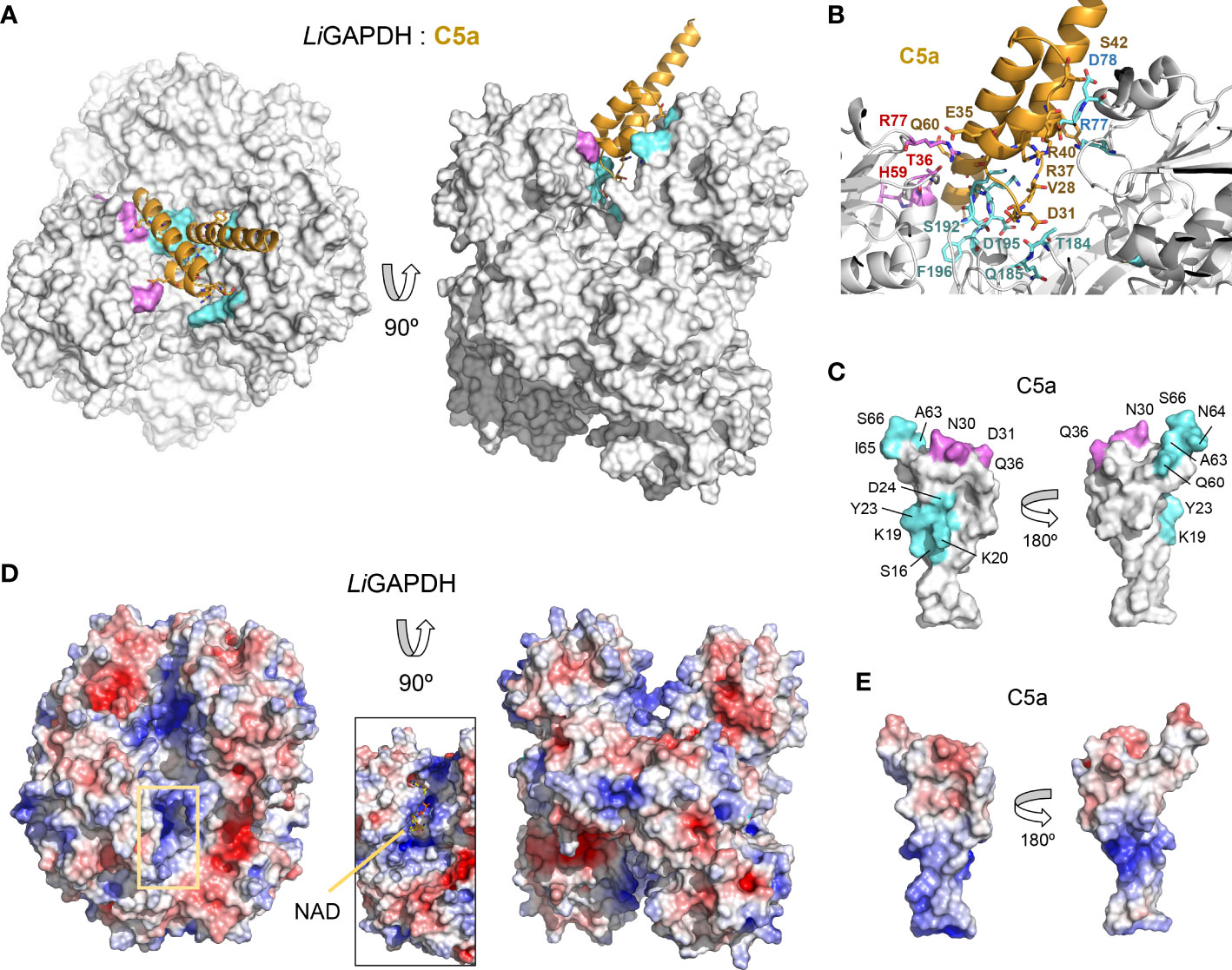
Figure 6 LiGAPDH-C5a complex by guided docking. (A) Two views 90° apart of the top-ranking LiGAPDH-C5a docking pose, shown in molecular surface representation (LiGAPDH, in white) and in cartoon (C5a, in orange). LiGAPDH residues engaged in polar interactions with C5a are shown in aquamarine (Q subunit) or violet (P subunit). (B) Zoom into the binding interface with interacting residues shown in sticks and annotated, color coded as in (A). (C) Two orientations 180° apart of C5a shown in molecular surface representation, with residues engaged in interactions with the Q and P subunits of LiGAPDH colored in aquamarine and violet, respectively. (D) Electrostatic potential surface of LiGAPDH mapped onto the same two orientations shown in (A). The inset zooms into the NAD+ binding site, highlighting the positively charged residues in the neighboring area. (E) Electrostatic potential surface of C5a mapped onto the same two orientations shown in (C). It can be noted that the region around Cys27 is slightly negatively charged.
Other high-ranking docking poses exploited the same or very similar docking surfaces as the top-ranking pose, differing mainly in the angle with which C5a approximated LiGAPDH (Supplementary Figure 7). Using the most representative poses, which differ minimally in the binding site, we can describe a consensus binding interface (Figure 7). Concerning LiGAPDH, the surface residues that mediate most contacts with C5a belong to the N- and C-terminal domains and to two chains, Q and P. On subunit Q, the three identified patches comprise residues 75-81 (patch 1Q), 183-185 (patch 2Q), and 192-196 (patch 3Q); and, on subunit P, residues 36-38 (patch 1P), 7 and 96-97 (patch 2P), and 182-186 and 191-196 (patch 3P) (Figure 7A). Reciprocally, the C5a residues that consistently contributed to the docking interface in the top-ranking docking solutions comprised residues surrounding Cys27 and from the two long helices (Figure 7B); the end of the C-terminal helix was pointing out of the docking site in most poses. The generally polar nature of the interface of most docking poses and the relatively wide range of compatible tilts for C5a (within the same small docking area) are compatible with a weak interaction between C5a and LiGAPDH, which could be enhanced in the biological context through electrostatic and avidity effects as previously described for GAPDH from Gram-positive pathogens (18, 19).
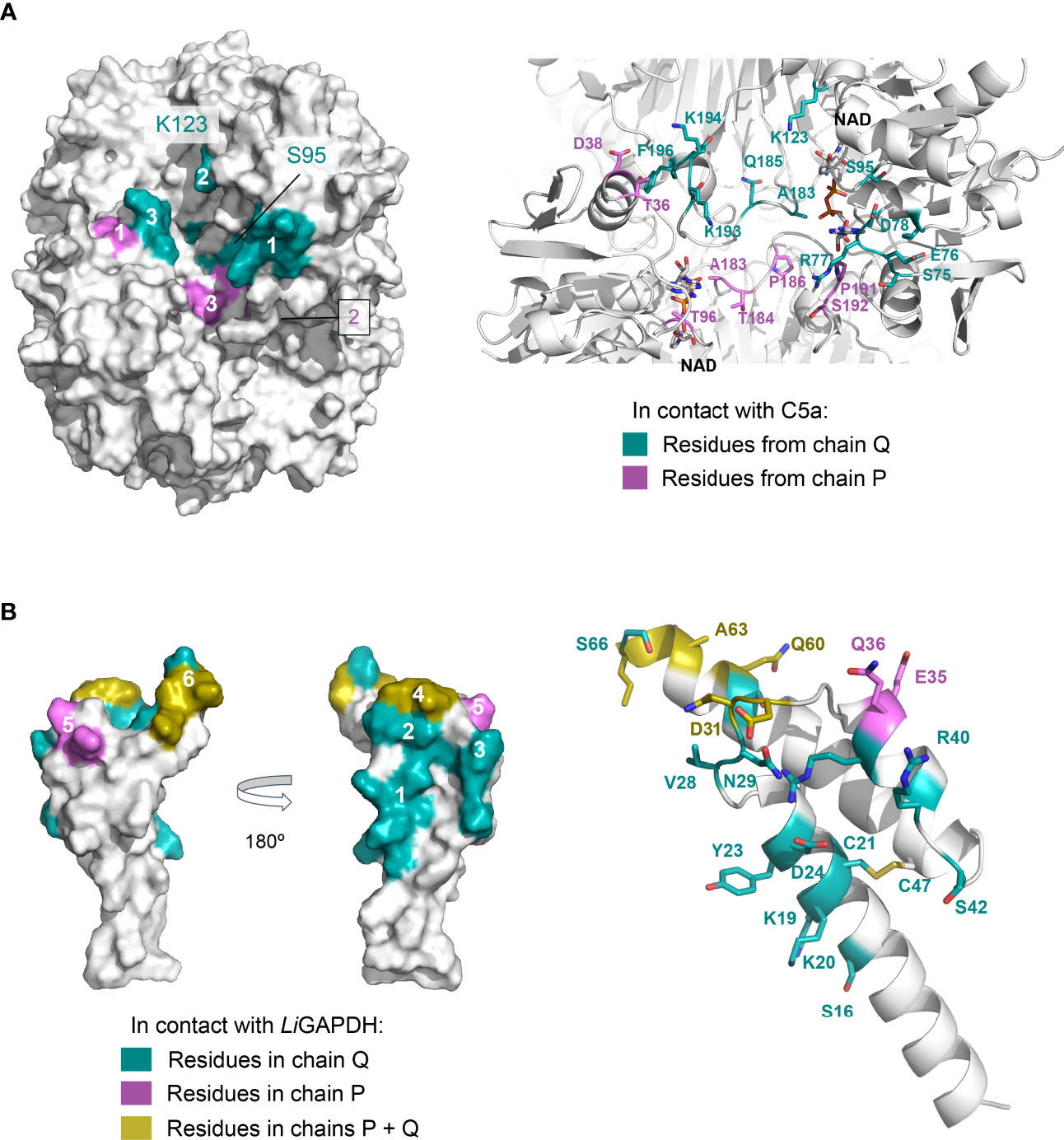
Figure 7 Consensus docking surfaces for LiGAPDH-C5a. (A) Molecular surface representation of LiGAPDH (in white). Surface areas that interact with C5a in most of the top-ranking docking poses (consensus docking surfaces) are highlighted in aquamarine if the residues come from the Q subunit, and violet if they come from the P subunit. Spatially close residues define up to three interaction patches on chains Q and P. The inset shows a zoom into the area containing the consensus docking residues (shown in sticks with the same color coding); LiGAPDH is shown in cartoon (in white). (B) Likewise, for C5a. Two orientations 180° apart are shown for the molecular surface representation. Residues in aquamarine interact with the Q subunit and in violet with the P subunit of LiGAPDH; the few residues that interact with both the Q and P subunits are colored yellow. Patches are numbered 1 through 6. Patches 4 and 6 (in yellow) pivot between binding the Q or the P subunits depending on the specific pose. In addition, C5a is shown in cartoons with the residues predicted to interact with LiGAPDH shown in sticks and with the same color coding.
Vertebrate innate immunity has evolved to prevent and fight infections by recognizing features of pathogens that are broadly shared such as plasma membrane lipids and cell-wall composition, called pathogen-associated molecular patterns (PAMPs). The complement system is one of the oldest and most efficient branches of our innate immunity. It therefore makes evolutionary sense that many pathogens have evolved sophisticated molecular weaponry to circumvent, inactivate, or mimic components of the complement system. Bacteria (and many eukaryotic parasites) rely on multiple complement-evasive strategies, often deployed simultaneously. One of these complement-targeting strategies consists in the neutralization of the C5a anaphylatoxin, which bacteria have learned to do in two separate but complementary ways: by proteolytic inactivation and by direct binding (sequestration). Proteases that can cleave C5a are deployed by pathogenic bacteria (56) as diverse as Pseudomonas aeruginosa (the alkaline protease ArpA and elastase B LasB) (57) and all sequenced serotypes of Group B streptococci (streptococcal cell-wall C5a peptidase) (58, 59).
Besides the direct proteolytic cleavage of C5a, some pathogens have also evolved the capacity to bind and retain C5a close to the site of infection, thereby precluding the anaphylatoxin (or, at least, slowing it down) from recruiting neighboring phagocytes. One of the virulence factors is the moonlighting protein GAPDH. Intracellularly, GAPDH is the well-known glycolytic enzyme; however, outside the cell, GAPDH can bind C5a (and, in some microorganisms, C3). In S. pneumoniae, GAPDH remains associated with the cell wall, indicating that bound C5a remains attached to the pathogen cells (16). Reciprocally, in clinical isolates of S. pyogenes, added C5a binds the cells in a dose-dependent fashion (19). These two complementary views show that streptococcal cells have the ability to “soak in” C5a, effectively shielding it from macrophages. Other cell-wall components might help to retain C5a besides GAPDH, further enhancing the immune evasive effect.
L. interrogans is a Gram-negative pathogen with an impressive array of immune evasive mechanisms, including many targeted at complement factors (13, 60). Several of the best characterized immune evasion mechanisms of L. interrogans include recruiting endogenous complement regulators such as FH and C4BP (13, 61, 62). However, complement-targeting immune evasion mechanisms directly interfering with C5a had not been demonstrated. In this work, we have shown that C5a binding and sequestration through LiGAPDH can provide an additional immune evasive mechanism to an already impressive weaponry. In support of this view, other metabolic and glycolytic enzymes have been shown to perform similar functions, such as enolase (22), EF-Tu (63), and the chaperonin GroEL (64). The in vivo relevance of these interactions for the pathogen’s survival in the host is still a matter of debate, further complicated by the essential nature of the bacterial genes encoding most moonlighting proteins, which precludes the analysis of gene deletion phenotypes, and the high concentrations found for these proteins in both the bacterial cytosol and exoproteome. Another difficulty for dissecting the relevance of specific moonlighting/virulence factors arises from the multiplicity of redundant and nonredundant immune evasive strategies that appear to contribute to the adaptation of L. interrogans and other bacterial pathogens to their hosts.
To better understand the unconventional roles of LiGAPDH, we have solved the crystal structure of the holoenzyme to 2.37-Å resolution. The crystallographic structure agrees with the solution SAXS data, suggesting that the crystal lattice has trapped the native conformation. The structural information it provides contributes to a significant pool of GAPDH structures available at the PDB. This information can be used for purposes such as drug discovery and repurposing campaigns in cases where GAPDH has a role in completely expressing pathogen’s virulence. This unconventional role appears to be rather prevalent as phylogenetically diverse Gram-negative and Gram-positive bacteria (and at least one eukaryotic parasite too) exhibit it (16, 18–21, 56). In this light, the kinetic characterization that we have performed on LiGAPDH shows that the enzyme is susceptible to inhibition by natural products such as anacardic acid and curcumin. Given the favorable safety profile of these natural products, they represent promising starting points for further drug development.
In the context of leptospirosis, extracellular LiGAPDH may play a virulence role by binding to C5a generated by the activation of complement’s terminal pathway. Indeed, LiGAPDH has been shown to be one of the twenty most abundant proteins in the extracellular proteome of pathogenic L. interrogans strains (11). Although not yet known, LiGAPDH could be exported to the cellular exterior by type I or II secretion systems or via extracellular vesicles, two of the most common secretion mechanisms characterized in L. interrogans.
In this work, we have shown by bio-layer interferometry and cross-linking experiments that LiGAPDH and C5a can form a specific complex at a sufficiently high concentration to overcome an intrinsically slow kinetic association constant. Initially, a weak interaction between nascent C5a (generated in situ on the surface of opsonized bacteria) and LiGAPDH could delay C5a diffusion long enough to be proteolytically degraded by nonspecific proteases from the pathogen or dearginated by serum carboxypeptidases; in fact, deargination of anaphylatoxins C5a and C3a in vivo is a fast and irreversible process that dampens the chemotactic response. In either scenario, neutrophil recruitment to the site of infection would be much reduced. The cross-link guided docking protocol that we have explored in this work produces LiGAPDH-C5a complexes that are compatible with the known facts about the interaction: proximity between C5a Cys27, the only free Cys residue in C5a, and the highly reactive catalytic Cys152 residue; structural and electrostatic complementarity at the docking site; a considerable buried interface (>2000 Å2); a predominantly electrostatic nature; and a variety of compatible poses differing in the overall tilt of C5a inside the interfacial groove between LiGAPDH Q-P subunits.
As our structural knowledge of the molecular machinery of the host’s innate immunity and the pathogens’ immune evasion factors expands and refines, our tools to fight recalcitrant infections will likely become more efficient and sophisticated. In the face of the dwindling efficacy of antibiotics and the looming medical and humanitarian crisis unleashed by global warming, further research is sorely needed to generate new approaches to curb infectious diseases through the combination of structural information from the host’s innate immune system and the pathogens’ virulence factors.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
FJF and MCV contributed to the conception of the study and the design of all the experiments. SNY and KdlP expressed and purified the protein samples for biochemical and structural analysis. SNY, KdlP, and SGQ performed kinetics experiments. SNY performed the BLI experiments. JQG carried out the cross-linking experiments, FJF and MCV ran the cross-link guide docking calculations and analyses. FJF and MCV collected the diffraction data. SNY, FJF and MCV solved and refined the crystal structure and the SAXS shape. FJF and MCV wrote the manuscript. SRdC read the manuscript critically. All authors contributed to the article and approved the submitted version.
This work was funded by Spanish Ministerio de Ciencia, Innovación y Universidades-FEDER grants RTI2018-102242-B-I00 (to MCV) and PID2019-104912RB-I00 (to SRdC); and the Spanish Ministerio de Ciencia e Innovación-Recovery, Transformation and Resilience Plan (PRTR) grant PDC2022-133713-I00. It was also funded by Grants S2017/BMD-3673 and S2022/BMD-7278 of the Regional Government of Madrid and the European Commission – NextGenerationEU through CSIC’s Global Health Platform (“PTI Salud Global”) (SGL2103020) (to SRdC and MCV), and the CSIC Special Intramural Grant PIE-201620E064 (to MCV). It was additionally supported by the Research Network on Complement in Health and Disease (RED2022-134750-T). SRdC was also supported by the CIBER de Enfermedades Raras. KdlP was supported by an Industrial PhD grant (IND2018-010094) awarded by the Spanish Ministerio de Economía y Competitividad.
The authors acknowledge the ALBA synchrotron light source (Barcelona, Spain) for the provision of synchrotron radiation facilities at the BL13-XALOC beamline and the Diamond Light Source (Leeds, UK) for access to the BioSAXS B21 beamline under Proposal SM28015. SNY and KdlP acknowledge the support of the PhD program in Biochemistry, Molecular Biology and Biomedicine of the Universidad Complutense de Madrid (UCM).
Abvance Biotech slr provided salaries for KdlP, JQG and FJF.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1190943/full#supplementary-material
1. Walport MJ. Complement. first of two parts. N Engl J Med (2001) 344:1058–66. doi: 10.1056/NEJM200104053441406
2. Medicus RG, Melamed J, Arnaout MA. Role of human factor I and C3b receptor in the cleavage of surface-bound C3bi molecules. Eur J Immunol (1983) 13:465–70. doi: 10.1002/eji.1830130607
3. Rawal N, Pangburn MK. Structure/function of C5 convertases of complement. Int Immunopharmacol (2001) 1:415–22. doi: 10.1016/s1567-5769(00)00039-4
4. Xie CB, Jane-Wit D, Pober JS. Complement membrane attack complex: new roles, mechanisms of action, and therapeutic targets. Am J Pathol (2020) 190:1138–50. doi: 10.1016/j.ajpath.2020.02.006
5. Guo R-F, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol (2005) 23:821–52. doi: 10.1146/annurev.immunol.23.021704.115835
6. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I – molecular mechanisms of activation and regulation. Front Immunol (2015) 6:262. doi: 10.3389/fimmu.2015.00262
7. Bharti AR, Nally JE, Ricaldi JN, Matthias MA, Diaz MM, Lovett MA, et al. Leptospirosis: a zoonotic disease of global importance. Lancet Infect Dis (2003) 3:757–71. doi: 10.1016/s1473-3099(03)00830-2
8. Adler B, de la Peña Moctezuma A. Leptospira and leptospirosis. Vet Microbiol (2010) 140:287–96. doi: 10.1016/j.vetmic.2009.03.012
9. Putz EJ, Nally JE. Investigating the immunological and biological equilibrium of reservoir hosts and pathogenic leptospira: balancing the solution to an acute problem? Front Microbiol (2020) 11:2005. doi: 10.3389/fmicb.2020.02005
10. Costa F, Hagan JE, Calcagno J, Kane M, Torgerson P, Martinez-Silveira MS, et al. Global morbidity and mortality of leptospirosis: a systematic review. PloS Negl Trop Dis (2015) 9:e0003898. doi: 10.1371/journal.pntd.0003898
11. Eshghi A, Pappalardo E, Hester S, Thomas B, Pretre G, Picardeau M. Pathogenic leptospira interrogans exoproteins are primarily involved in heterotrophic processes. Infect Immun (2015) 83:3061–73. doi: 10.1128/IAI.00427-15
12. Goarant C. Leptospirosis: risk factors and management challenges in developing countries. Res Rep Trop Med (2016) 7:49–62. doi: 10.2147/RRTM.S102543
13. Fraga TR, Isaac L, Barbosa AS. Complement evasion by pathogenic leptospira. Front Immunol (2016) 7:623. doi: 10.3389/fimmu.2016.00623
14. Barbosa AS, Abreu PAE, Vasconcellos SA, Morais ZM, Gonçales AP, Silva AS, et al. Immune evasion of leptospira species by acquisition of human complement regulator C4BP. Infect Immun (2009) 77:1137–43. doi: 10.1128/IAI.01310-08
15. Cosate MR, Siqueira GH, de Souza GO, Vasconcellos SA, Nascimento ALTO. Mammalian cell entry (Mce) protein of leptospira interrogans binds extracellular matrix components, plasminogen and beta2 integrin. Microbiol Immunol (2016) 60:586–98. doi: 10.1111/1348-0421.12406
16. Terao Y, Yamaguchi M, Hamada S, Kawabata S. Multifunctional glyceraldehyde-3-phosphate dehydrogenase of streptococcus pyogenes is essential for evasion from neutrophils. J Biol Chem (2006) 281:14215–23. doi: 10.1074/jbc.M513408200
17. Aguilera L, Ferreira E, Giménez R, Fernández FJ, Taulés M, Aguilar J, et al. Secretion of the housekeeping protein glyceraldehyde-3-phosphate dehydrogenase by the LEE-encoded type III secretion system in enteropathogenic escherichia coli. Int J Biochem Cell Biol (2012) 44:955–62. doi: 10.1016/j.biocel.2012.03.002
18. Querol-García J, Fernández FJ, Marin AV, Gómez S, Fullà D, Melchor-Tafur C, et al. Crystal structure of glyceraldehyde-3-Phosphate dehydrogenase from the gram-positive bacterial pathogen a. vaginae, an immunoevasive factor that interacts with the human C5a anaphylatoxin. Front Microbiol (2017) 8:541. doi: 10.3389/fmicb.2017.00541
19. Gómez S, Querol-García J, Sánchez-Barrón G, Subias M, González-Alsina À, Franco-Hidalgo V, et al. The antimicrobials anacardic acid and curcumin are not-competitive inhibitors of gram-positive bacterial pathogenic glyceraldehyde-3-Phosphate dehydrogenase by a mechanism unrelated to human C5a anaphylatoxin binding. Front Microbiol (2019) 10:326. doi: 10.3389/fmicb.2019.00326
20. Terrasse R, Tacnet-Delorme P, Moriscot C, Pérard J, Schoehn G, Vernet T, et al. Human and pneumococcal cell surface glyceraldehyde-3-phosphate dehydrogenase (GAPDH) proteins are both ligands of human C1q protein. J Biol Chem (2012) 287:42620–33. doi: 10.1074/jbc.M112.423731
21. Sahoo S, Murugavel S, Devi IK, Vedamurthy GV, Gupta SC, Singh BP, et al. Glyceraldehyde-3-phosphate dehydrogenase of the parasitic nematode haemonchus contortus binds to complement C3 and inhibits its activity. Parasite Immunol (2013) 35:457–67. doi: 10.1111/pim.12058
22. Nogueira SV, Backstedt BT, Smith AA, Qin J-H, Wunder EA, Ko A, et al. Leptospira interrogans enolase is secreted extracellularly and interacts with plasminogen. PloS One (2013) 8:e78150. doi: 10.1371/journal.pone.0078150
23. Ferdinand W. The isolation and specific activity of rabbit-muscle glyceraldehyde phosphate dehydrogenase. Biochem J (1964) 92:578–85. doi: 10.1042/bj0920578
24. Juanhuix J, Gil-Ortiz F, Cuní G, Colldelram C, Nicolás J, Lidón J, et al. Developments in optics and performance at BL13-XALOC, the macromolecular crystallography beamline at the alba synchrotron. J Synchrotron Radiat (2014) 21:679–89. doi: 10.1107/S160057751400825X
25. Kabsch W. XDS. In: International tables for crystallography (2012). (International Union of Crystallography). 11.6, 304–10. doi: 10.1107/97809553602060000835
26. Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr (2013) 69:1204–14. doi: 10.1107/S0907444913000061
27. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr (2007) 40:658–74. doi: 10.1107/S0021889807021206
28. Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr (2010) 66:213–21. doi: 10.1107/S0907444909052925
29. Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr Sect Biol Crystallogr (2012) 68:352–67. doi: 10.1107/S0907444912001308
30. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr Sect Biol Crystallogr (2010) 66:486–501. doi: 10.1107/S0907444910007493
31. Cowieson NP, Edwards-Gayle CJC, Inoue K, Khunti NS, Doutch J, Williams E, et al. Beamline B21: high-throughput small-angle X-ray scattering at diamond light source. J Synchrotron Radiat (2020) 27:1438–46. doi: 10.1107/S1600577520009960
32. Franke D, Jeffries CM, Svergun DI. Correlation map, a goodness-of-fit test for one-dimensional X-ray scattering spectra. Nat Methods (2015) 12:419–22. doi: 10.1038/nmeth.335
33. Manalastas-Cantos K, Konarev PV, Hajizadeh NR, Kikhney AG, Petoukhov MV, Molodenskiy DS, et al. ATSAS 3.0: expanded functionality and new tools for small-angle scattering data analysis. J Appl Crystallogr (2021) 54:343–55. doi: 10.1107/S1600576720013412
34. Konarev PV, Svergun DI. A posteriori determination of the useful data range for small-angle scattering experiments on dilute monodisperse systems. IUCrJ (2015) 2:352–60. doi: 10.1107/S2052252515005163
35. Guinier A. La diffraction des rayons X aux très petits angles: application à l’étude de phénomènes ultramicroscopiques. Ann Phys (1939) 11:161–237. doi: 10.1051/anphys/193911120161
36. Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr (1992) 25:495–503. doi: 10.1107/S0021889892001663
37. Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, et al. New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Crystallogr (2012) 45:342–50. doi: 10.1107/S0021889812007662
38. Petoukhov MV, Svergun DI. Ambiguity assessment of small-angle scattering curves from monodisperse systems. Acta Crystallogr Sect D (2015) 71:1051–8. doi: 10.1107/S1399004715002576
39. Franke D, Svergun DI. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J Appl Crystallogr (2009) 42:342–6. doi: 10.1107/S0021889809000338
40. Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr (2003) 36:860–4. doi: 10.1107/S0021889803000268
41. Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J (1999) 76:2879–86. doi: 10.1016/S0006-3495(99)77443-6
42. Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. J Appl Crystallogr (2001) 34:33–41. doi: 10.1107/S0021889800014126
43. Tuukkanen AT, Kleywegt GJ, Svergun DI. Resolution of ab initio shapes determined from small-angle scattering. IUCrJ (2016) 3:440–7. doi: 10.1107/S2052252516016018
44. Svergun D, Barberato C, Koch MHJ. CRYSOL – a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr (1995) 28:768–73. doi: 10.1107/S0021889895007047
45. Kahraman A, Herzog F, Leitner A, Rosenberger G, Aebersold R, Malmström L. Cross-link guided molecular modeling with ROSETTA. PloS One (2013) 8:e73411. doi: 10.1371/journal.pone.0073411
46. Kahraman A, Malmström L, Aebersold R. Xwalk: computing and visualizing distances in cross-linking experiments. Bioinformatics (2011) 27:2163–4. doi: 10.1093/bioinformatics/btr348
47. Mitternacht S. FreeSASA: an open source c library for solvent accessible surface area calculations. F1000Research (2016) 5:189. doi: 10.12688/f1000research.7931.1
48. Krissinel E. Macromolecular complexes in crystals and solutions. Acta Crystallogr Sect Biol Crystallogr (2011) 67:376–85. doi: 10.1107/S0907444911007232
49. Hosseinzadeh P, Bhardwaj G, Mulligan VK, Shortridge MD, Craven TW, Pardo-Avila F, et al. Comprehensive computational design of ordered peptide macrocycles. Science (2017) 358:1461–6. doi: 10.1126/science.aap7577
50. Jurrus E, Engel D, Star K, Monson K, Brandi J, Felberg LE, et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci (2018) 27:112–28. doi: 10.1002/pro.3280
51. The PyMOL molecular graphics system, version 2.0. Available at: https://pymol.org/2/support.html?#citing (Accessed December 9, 2022).
52. Pereira JM, Severino RP, Vieira PC, Fernandes JB, da Silva MFGF, Zottis A, et al. Anacardic acid derivatives as inhibitors of glyceraldehyde-3-phosphate dehydrogenase from trypanosoma cruzi. Bioorg Med Chem (2008) 16:8889–95. doi: 10.1016/j.bmc.2008.08.057
53. Matsumura H, Kai A, Maeda T, Tamoi M, Satoh A, Tamura H, et al. Structure basis for the regulation of glyceraldehyde-3-Phosphate dehydrogenase activity via the intrinsically disordered protein CP12. Structure (2011) 19:1846–54. doi: 10.1016/j.str.2011.08.016
54. McFarlane CR, Shah NR, Kabasakal BV, Echeverria B, Cotton CAR, Bubeck D, et al. Structural basis of light-induced redox regulation in the Calvin–Benson cycle in cyanobacteria. Proc Natl Acad Sci (2019) 116:20984–90. doi: 10.1073/pnas.1906722116
55. Yu A, Xie Y, Pan X, Zhang H, Cao P, Su X, et al. Photosynthetic phosphoribulokinase structures: enzymatic mechanisms and the redox regulation of the Calvin-Benson-Bassham cycle. Plant Cell (2020) 32:1556–73. doi: 10.1105/tpc.19.00642
56. Fernández FJ, Gómez S, Vega MC. Pathogens’ toolbox to manipulate human complement. Semin Cell Dev Biol (2019) 85:98–109. doi: 10.1016/j.semcdb.2017.12.001
57. Mateu-Borrás M, González-Alsina A, Doménech-Sánchez A, Querol-García J, Fernández FJ, Vega MC, et al. Pseudomonas aeruginosa adaptation in cystic fibrosis patients increases C5a levels and promotes neutrophil recruitment. Virulence (2022) 13:215–24. doi: 10.1080/21505594.2022.2028484
58. Cheng Q, Stafslien D, Purushothaman SS, Cleary P. The group b streptococcal C5a peptidase is both a specific protease and an invasin. Infect Immun (2002) 70:2408–13. doi: 10.1128/IAI.70.5.2408-2413.2002
59. Lynskey NN, Reglinski M, Calay D, Siggins MK, Mason JC, Botto M, et al. Multi-functional mechanisms of immune evasion by the streptococcal complement inhibitor C5a peptidase. PloS Pathog (2017) 13:e1006493. doi: 10.1371/journal.ppat.1006493
60. Alves da Silva PYO, Midon LM, Heinemann MB, de Moraes Vasconcelos D, Barbosa AS, Isaac L. Contribution of complement system pathways to the killing of leptospira spp. Microbes Infect (2020) 22:550–7. doi: 10.1016/j.micinf.2020.07.005
61. Castiblanco-Valencia MM, Fraga TR, da SLB, Monaris D, Abreu PAE, Strobel S, et al. Leptospiral immunoglobulin-like proteins interact with human complement regulators factor h, FHL-1, FHR-1, and C4BP. J Infect Dis (2012) 205:995–1004. doi: 10.1093/infdis/jir875
62. Meri T, Murgia R, Stefanel P, Meri S, Cinco M. Regulation of complement activation at the C3-level by serum resistant leptospires. Microb Pathog (2005) 39:139–47. doi: 10.1016/j.micpath.2005.07.003
63. Wolff DG, Castiblanco-Valencia MM, Abe CM, Monaris D, Morais ZM, Souza GO, et al. Interaction of leptospira elongation factor tu with plasminogen and complement factor h: a metabolic leptospiral protein with moonlighting activities. PloS One (2013) 8:e81818. doi: 10.1371/journal.pone.0081818
Keywords: structural biology, innate immunity, complement system, C5a anaphylatoxin, GAPDH – glyceraldehyde 3-phospate dehydrogenase, leptospirosis, moonlighting proteins
Citation: Navas-Yuste S, de la Paz K, Querol-García J, Gómez-Quevedo S, Rodríguez de Córdoba S, Fernández FJ and Vega MC (2023) The structure of Leptospira interrogans GAPDH sheds light into an immunoevasion factor that can target the anaphylatoxin C5a of innate immunity. Front. Immunol. 14:1190943. doi: 10.3389/fimmu.2023.1190943
Received: 21 March 2023; Accepted: 30 May 2023;
Published: 20 June 2023.
Edited by:
Hyun Ho Park, Chung-Ang University, Republic of KoreaReviewed by:
Jinghua Lu, National Institute of Allergy and Infectious Diseases (NIH), United StatesCopyright © 2023 Navas-Yuste, de la Paz, Querol-García, Gómez-Quevedo, Rodríguez de Córdoba, Fernández and Vega. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Cristina Vega, Y3ZlZ2FAY2liLmNzaWMuZXM=; Francisco J. Fernández, ZmpmZXJuYW5kZXpAYWJ2YW5jZS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.