 Qian Lei
Qian Lei Jie Yang
Jie Yang Li Li
Li Li Ning Zhao
Ning Zhao Cheng Lu
Cheng Lu Aiping Lu
Aiping Lu Xiaojuan He
Xiaojuan He- 1Institute of Basic Research in Clinical Medicine, China Academy of Chinese Medical Sciences, Beijing, China
- 2Law Sau Fai Institute for Advancing Translational Medicine in Bone and Joint Diseases, School of Chinese Medicine, Hong Kong Baptist University, Hong Kong, Hong Kong SAR, China
- 3Shanghai GuangHua Hospital of Integrated Traditional Chinese and Western Medicine, Institute of Arthritis Research, Shanghai Academy of Chinese Medical Sciences, Shanghai, China
- 4Guangdong-Hong Kong-Macau Joint Lab on Chinese Medicine and Immune Disease Research, Guangzhou, China
As a chronic progressive autoimmune disease, rheumatoid arthritis (RA) is characterized by mainly damaging the synovium of peripheral joints and causing joint destruction and early disability. RA is also associated with a high incidence rate and mortality of cardiovascular disease. Recently, the relationship between lipid metabolism and RA has gradually attracted attention. Plasma lipid changes in RA patients are often detected in clinical tests, the systemic inflammatory status and drug treatment of RA patients can interact with the metabolic level of the body. With the development of lipid metabolomics, the changes of lipid small molecules and potential metabolic pathways have been gradually discovered, which makes the lipid metabolism of RA patients or the systemic changes of lipid metabolism after treatment more and more comprehensive. This article reviews the lipid level of RA patients, as well as the relationship between inflammation, joint destruction, cardiovascular disease, and lipid level. In addition, this review describes the effect of anti-rheumatic drugs or dietary intervention on the lipid profile of RA patients to better understand RA.
1 Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease, which is prevalent in about 0.5–1% of the population. The pathological basis of RA is synovial inflammation, which then erodes the articular cartilage and bone tissue from the synovium, resulting in the destruction of joints and affecting the normal joint function of patients (1–3). Extraarticular manifestations, such as cardiovascular diseases (CVD) and comorbidities are common manifestations of RA, which increase the incidence rate and mortality of RA patients. Although the exact etiology is still unclear, it is currently believed that genetic, autoimmunity, environmental, and intestinal microorganisms are important causes of the disease (4, 5). In addition to these generally acknowledged factors, recent studies suggest that there is also a profound relationship between lipid metabolism and RA.
As the most abundant type of cellular metabolites, lipids are important for energy supply and storage, cell membrane construction, and signal transduction (6). Lipid metabolism is a process of digestion, absorption, decomposition, and metabolism of lipids, such as triglyceride (TG), cholesterol, and fatty acids (FAs) (7), which is an important and complex biological process (Figure 1). Under normal conditions, the cholesterol that the body absorbs from food and synthesizes in the liver will be converted into steroid hormones or become a component of the cell membrane, keeping the concentration of cholesterol in the blood constant. The fat in food is absorbed and synthesized into TG through the intestine, stored in the liver or fat, or the blood in a free form. TG is decomposed into glycerol and FAs under the catalysis of enzymes. FAs usually exist in the blood in free form or the body as the main component of some high-order lipids (such as phospholipids and glycolipids). Lipid reprogramming within cells further promotes membrane remodeling, affecting membrane fluidity, lipid raft formation, and proinflammatory signaling cascades (9). In addition, lipids are also the precursors of various molecules with important biological roles. For example, polyunsaturated fatty acids (PUFA) generate oxidized lipids through the action of specific enzymes. Among them, arachidonic acid (AA) in n-6 PUFA and its derivatives, such as prostaglandin (PG), thromboxane, and leukotriene, are mainly considered substrates of pro-inflammatory mediators. Eicosapentaenoic acid (EPA), docosahexaenoic acid (10) in n-3 PUFA, and their derivatives are considered substrates of mediators inducing inflammation regression (11). Lipoxins from AA, E-series resolvins from EPA, D-series resolvins, protectins, and maresins from DHA are called specialized pro-resolving mediators (SPMs), which play an important role in the regression of inflammation (12).
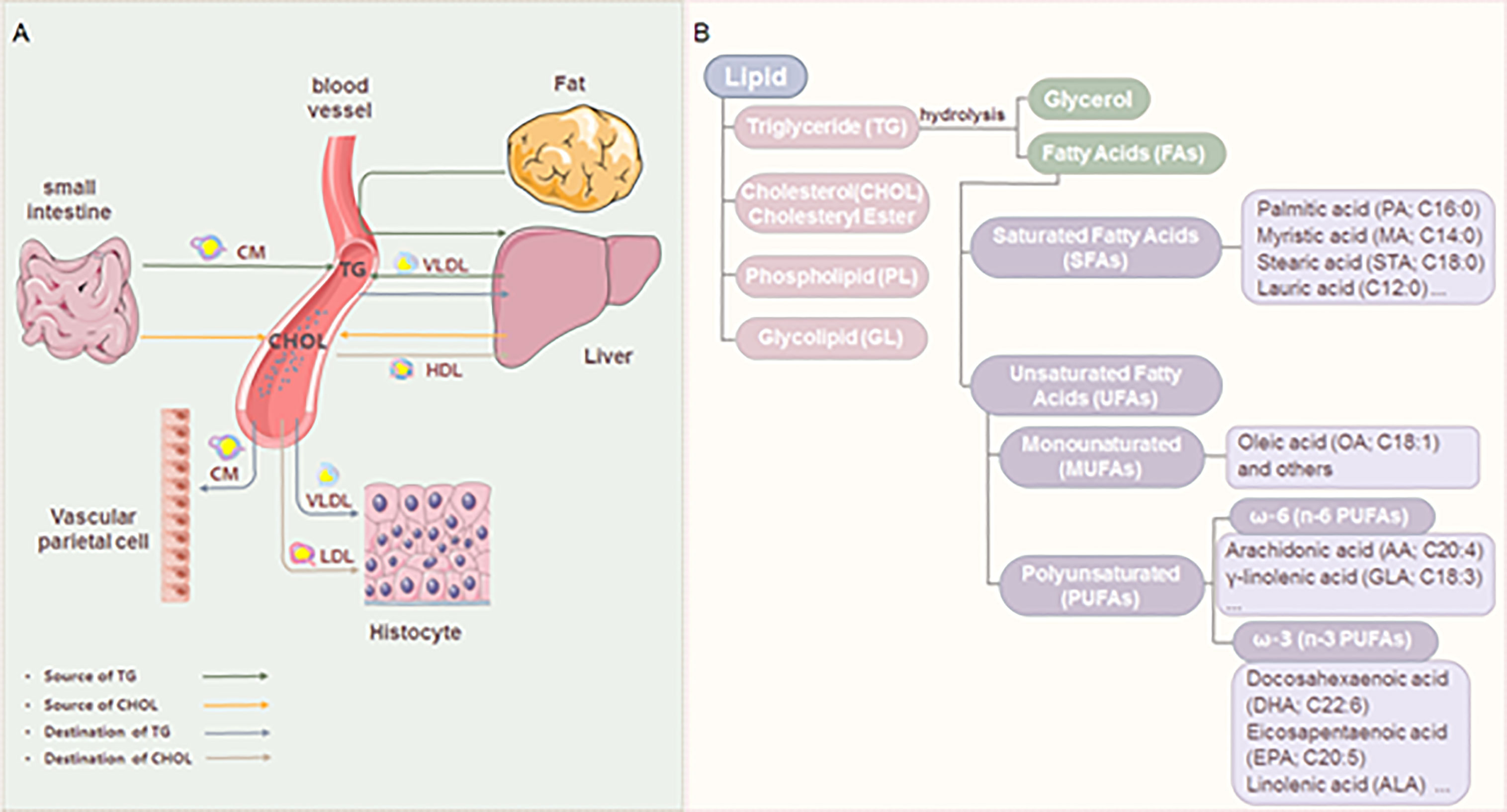
Figure 1 Lipid metabolism and classification. (A) The source and destination of triglyceride and cholesterol. Triglyceride (TG): The fat in the food is digested to form chylomicron (8) in the small intestine. TG is carried by CM through blood circulation to adipose tissue for storage. A part of TG in adipose tissue is decomposed into glycerol and FAs, which are transported to the liver. The liver reconstitutes them into TG for storage and can also transport blood in the form of VLDL. Cholesterol (CHOL): absorbed by food, and synthesized by the liver or small intestine; It is carried by LDL and transported to the whole body to synthesize lipid hormones and form cell membranes. The excess CHOL in the tissue is absorbed by HDL, transported to the liver, and then discharged from the body. (B) Classification of lipids and FAs.
Under normal circumstances, the balance of lipids is maintained by strictly controlling the levels of cholesterol and FAs. But the imbalance of these regulatory mechanisms becomes the foundation of human diseases. Recent studies have shown that RA patients have abnormal lipid metabolism, and these abnormal lipid metabolites affect the progress of RA (13, 14). During the treatment of RA, the lipid level of RA patients is regulated under the effect of drugs. In addition, dietary intervention of lipid intake can also play a role in regulating the lipid level of RA patients. Combined with the complex process of lipid metabolism and the disease characteristics of RA, therefore, this review discusses the lipid level of RA patients, the relationship between disease progression and lipid level, and the effect of anti-rheumatic drugs or dietary intervention on the lipid mass spectrum of RA patients. We hope this review can deepen the understanding of RA from the perspective of lipid metabolism by providing updated evidence.
2 Changes in lipid metabolism in RA
2.1 Blood lipid profile of RA patients
Blood lipids refer to TG, cholesterol, and lipids (such as phospholipids and glycolipids) in plasma. Plasma proteins generally include apolipoproteins in blood and molecular complexes composed of a variety of lipids. In clinical practice, the parameters generally concerned with measuring blood lipid metabolism in RA patients are TG, total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), phospholipids, FAs, lipoproteins, and apolipoproteins, et al. Previously, it was generally believed that the TC, LDL-C, and HDL-C of untreated patients with RA before and during the active period were reduced (15–17). Consistent with this, recent studies also found that HDL-C (16, 18–20) and LDL-C (17) decreased, and TC/HDL-C (16, 18) increased in RA patients. Studies on TC have both down-regulated results consistent with the previous report (19), and conflicting views, such as the level of TC in RA patients remained unchanged (18), or the level was up-regulated (20, 21). The serum lipoprotein (a) (Lp(a)) concentration of RA patients increased significantly (16, 19), and the apolipoprotein B (apo B)/apolipoprotein A-I (apo A-I) ratio was also significantly higher than that of the control group (16). In addition, the levels of circulating FAs in RA patients have also been detected. Compared with the control group, the percentage or concentration of palmitic acid, palmitoleic acid, oleic acid, linoleic acid, erucic acid, AA, EPA, 20-22C monounsaturated fatty acid (MUFA), and/or total n-6 PUFA decreased (22). Lipid derivatives, such as leukotriene B4 (LTB4) derived from AA, are elevated in RA due to their pro-inflammatory effects (23). About SPMs, resolvin D3 (RvD3), RvD4 and resolvin E3 (RvE3) decreased in the serum of RA patients (24). Although a single detection of plasma lipid profiles may display lipid levels in RA patients to some extent, there are always conflicting results in clinical detection due to differences in detection populations or methods. If new discoveries are to be made from this perspective, it is necessary to link lipid levels more widely with disease markers or related complications such as CVD, which will be beneficial for the clinical diagnosis and treatment of RA.
Actually, a few studies have begun to explore the relationship between plasm lipid change and inflammation related markers in RA patients (20). The level and duration of C-reactive protein (CRP) are closely related to the severity and activity of inflammation, so CRP is often used as an inflammatory marker of RA (25). In addition, erythrocyte sedimentation rate (ESR) is also commonly used to diagnose the degree of RA inflammation. Clinical diagnosis of RA should consider the influence of concentration of plasma lipids to make a reasonable judgment on the diagnosis of the disease. Compared with the control group, the average level of inflammation marker high-sensitivity CRP (hsCRP) in RA patients was higher. HDL-C was negatively correlated with hsCRP (20, 26), and higher TC/HDL-C and higher LDL/HDL ratios positively correlate with hsCRP (20). Furthermore, in patients in the increased inflammation cohort, the increase in hsCRP levels was associated with a significant decrease in LDL-C, TG, TC, apoB, and apoA-I levels (27). ESR and RA also showed gender correlation. ESR level was negatively correlated with HDL-C concentration in RA patients, and in male RA patients, ESR concentration increased with the increase of LDL-C (26). These results showed that the lipid levels exhibited a correlation with hsCRP and ESR to some extent.
To further understand the change of serum lipids in RA, different RA animal models were established for related research. Compared with clinical specimens, animal models can dynamically simulate the occurrence and progression of RA and provide theoretical support for clinical data. In the rat model of pristane-induced arthritis, it was found that the serum TC and TG were low in the acute phase and chronic phase after induction. In addition, adiponectin levels were low in the acute phase, but not in the chronic phase (28). In a more comprehensive collagen-induced arthritis (CIA) model, joint tissue and plasma samples were taken as research objects to study the possible ways of lipid changes in RA. Compared with the control group, TG, TC, and phospholipids in plasma lipids of CIA group rats decreased significantly, TC and phospholipids in joint tissue increased significantly, while TG slightly decreased without significant difference. TG, phospholipids, and TC in the joints of CIA rats were negatively correlated with TG, phospholipids, and TC in plasma (29). Moreover, the levels of AA and its metabolites, phospholipid metabolites, and EPA metabolites in the RA rat model were significantly increased, such as PGE2, PGF2α, PGI2, LTB4, and thromboxane B2 (30–32). There are also differences in the levels of oxylipins between CIA mice and healthy mice. Among them, the oxylipins derived from AA, DHA, and EPA were up-regulated, while the oxylipins derived from linoleic acid were down-regulated (33).
2.2 Lipid levels in synovial fluid of RA
Synovial fluid (SF) provides nutrition and lubrication for articular cartilage. Normal human SF contains very low concentrations of lipoproteins and apolipoproteins, and there is a significant difference between them and the lipid content in blood (34). In RA, SF accumulates at the synovial junction of the joint (35). SF of RA is rich in inflammatory cytokines and immune cells, which can further enhance synovial inflammation. Therefore, the detection results of SF in RA patients are usually compared with other bone diseases, such as osteoarthritis (OA). In terms of FA content, the percentage of palmitic acid, total saturated FA, long chain MUFA and/or total MUFA increased in SF of RA compared with SF of trauma control or OA patients (36). The myristic acid and palmitoleic acid levels were lower than those in patients with non-RA inflammatory arthritis (37). Interestingly, the lipid level in SF of RA was different in location (38). The proportion of AA and DHA in the SF of RA shoulder increased, and the proportion of oleic acid decreased. The proportion of linoleic acid, DHA, and total n-6 PUFAs in SF of the RA knee joint was low. These changes may affect joint lubrication, synovitis, pannus formation, cartilage, and bone degeneration, and lead to the pathogenesis of inflammatory joint disease (30). In terms of the content of SPMs, maresins 1 ll, 5S-hydroxy eicosapentaenoic acid (HETE), 12S-diHETE, RvD1, RvD3, RvD5, and lipoxins A4 (LXA4) were detected in SF of RA (39–41), and it was confirmed that the levels of PGE 2, 15-HETE and LTB4 increased in SF of RA (40, 42). Although there are currently few comparative studies on lipids in the joint synovial fluid between RA and normal individuals, some oxidized lipids have specificity in RA compared to OA patients, such as LXA4, 5-HETE, 12-HETE, etc. These specific oxidized lipids may serve as biomarkers for diagnosis.
3 Effect of lipids on RA
3.1 Effect of lipids on inflammation of RA
The pathophysiology of RA inflammation involves numerous cell types, including T cells, B cells, dendritic cells (DCs), neutrophils, macrophages, and fibroblast-like synoviocytes (FLS). Lipid metabolites can affect the progress of RA by regulating the activity and function of these cells (Table 1).
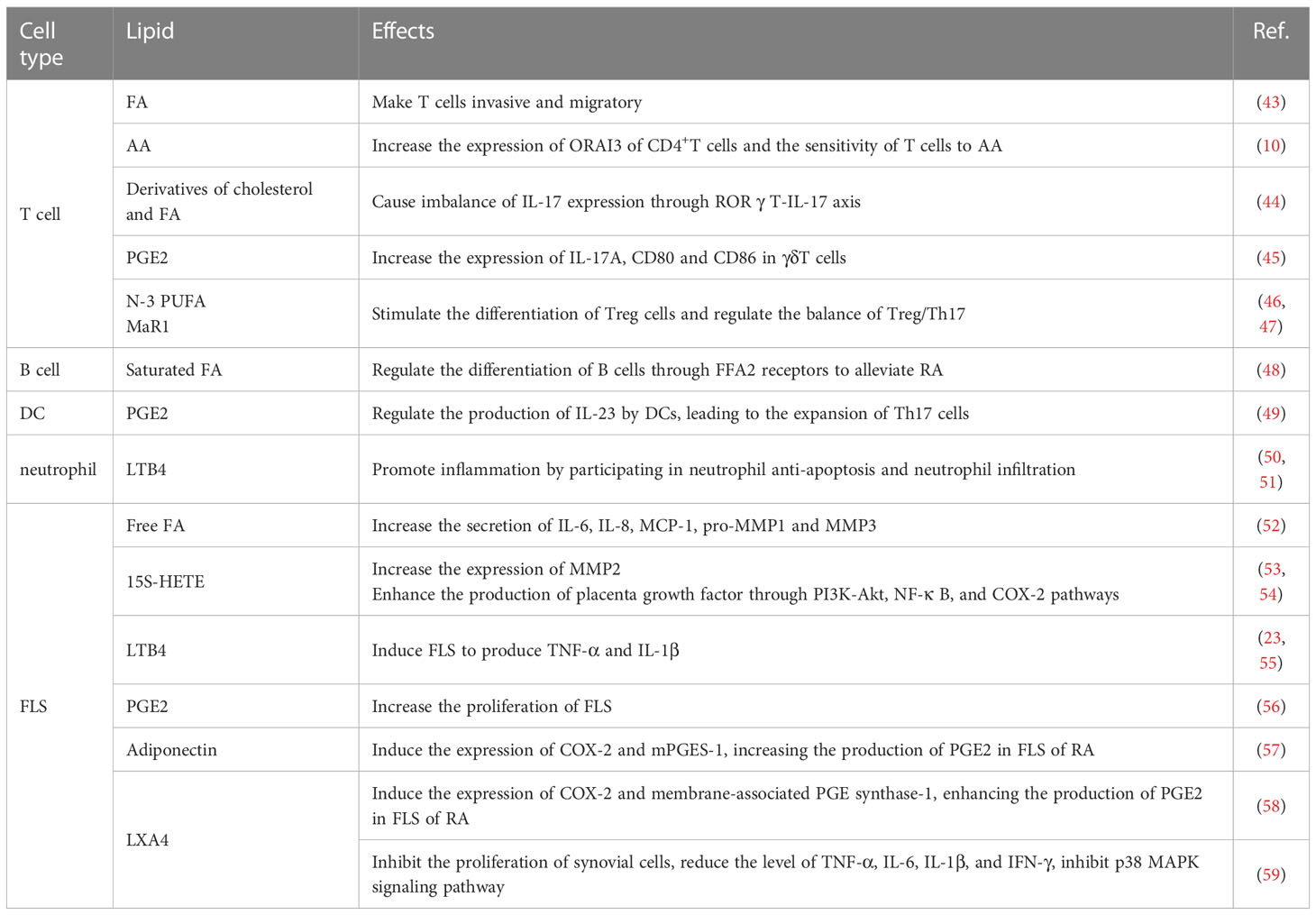
Table 1 Effect of lipid on Inflammation of RA.
3.1.1 Effect of lipids on the immune cell of RA
3.1.1.1 Effect of lipid on T cells and B cells of RA
As an autoimmune disease, RA is associated with immune cell dysfunction at different developmental stages. In RA, lipid abnormalities in T cells contribute to tissue invasion and migration, promoting synovial inflammation and eroding cartilage and bones (60, 61). At present, it is believed that lipid abnormalities in T cells are mainly caused by mitochondrial metabolic defects (60, 62, 63). Mitochondrial metabolic defects in T cells of RA are not conducive to glycolysis, thus shunting to pentose phosphate pathway, producing NADPH and biosynthetic precursors, and finally producing FAs. T cells adapt to the formation of excessive FAs by depositing lipid droplets in the cytoplasm. Subsequently, the T cell membrane containing lipid droplets was then arranged into invasive folds, which promoted the migration of T cells in the extracellular matrix and tissues (43).
Regarding other aspects of lipids and RA T cells, AA-regulated calcium signals in T cells of RA promote the development of synovitis. The expression of ORAI3, the pore-forming calcium channel component of CD4+T cells from RA patients, increased, thus enhancing the activity of AA-regulated calcium selective channels, making T cells sensitive to AA (10). Retinoid-related orphan nuclear receptor γt (RORγt) is expressed in mouse and human T cells, RORγt is the main transcription factor of interleukin (IL) -17, and the derivatives of cholesterol and FAs are also the natural ligand of RORγt (64). The imbalance of IL-17 expression is closely related to autoimmune diseases and inflammatory diseases, therefore RORγt-IL-17 axis is an interesting target for studying T cell function in RA (44). The number of γδT cells and the level of IL-17A increased in RA patients. In addition, after being treated with PGE2, the level of IL-17A in γδT cells increased correspondingly, and the expression of costimulatory molecules CD80 and CD86 in these cells also increased (45). Th17 and Treg subset dysregulations were present in RA patients (65). Fat-1 transgenic mice expressed the fat-1 gene, which encodes an n-3 FA desaturase that can convert n-6 FAs into n-3 FAs, producing abundant n-3 FAs that do not require dietary supply. This allows for the exploration of the role of n-3 PUFA by a collagen antibody-induced arthritis model. It found that n-3 PUFA decreased the expression of IL-6, IL-23, and IL-17, and stimulated the expression of FoxP3, thereby stimulating the differentiation of Treg cells (46). In the CIA model, MaR1 increased the proportion of Treg cells and decreased the proportion of Th17 cells, regulating the balance of Treg/Th17, thereby improving RA progress (47).
Compared with T cells, the effect of lipids on B cells in RA is less studied at present. Although B cells participate in the disease process of RA through antigen presentation, cytokine secretion, and autoantibody production, not all B cells can promote the pathogenesis of RA, and some antibodies produced by B cells can prevent and protect RA (66). It is reported that short-chain FAs regulated the differentiation of B cells through its receptor FFA2 to alleviate RA (48).
3.1.1.2 Effect of lipids on dendritic cells, neutrophils, and macrophages of RA
In addition to adaptive immune cells, some innate immune cells including DCs, neutrophils, and macrophages also play important roles in the progression of RA. In DCs, in addition to the role of energy storage and structural components of the cell membrane, lipids act as second messengers and effectors in the steps of cell differentiation and regulate important functions of DCs (67). The chronic inflammatory environment in RA seriously affects the distribution and function of DCs, resulting in tolerance deficiency and aggravation of inflammation. The synovial fluid of RA patients is rich in DCs subpopulations derived from monocytes, which can promote the harmful Th17 response (68). Synovial cells release PGE2 to produce IL-23, IL-6, IL-1β, and TNF-α through the EP2/EP4 (membrane receptors) signal of DCs, which leads T cells in arthritis to differentiate into Th17 cells. Subsequently, Th17 induces the production of IL-1β, IL-6, matrix metallopeptidase-1 (MMP-1), and MMP-3 through synovial cells, further promoting the release of PGE2 in synovial cells. This process establishes a pro-inflammatory cycle in RA through DCs (49).
Neutrophils account for over 90% of the cells found in the synovial fluid of RA patients. Synovial fluid contains various immune complexes and complement components. Previous studies have shown that inflammatory mediators (PGE2, TXA2, LTB4) released by neutrophils after ingesting immune complexes and complement components in synovial fluid are one of the causes of RA inflammation. These inflammatory mediators can inhibit the functions of neutrophils, platelets, macrophages, and mast cells. As a product of AA, LTB4 is considered the most potent proinflammatory agent (69, 70). LTB4 promotes inflammation in RA by participating in neutrophil anti-apoptosis and neutrophil infiltration (50, 51).
Lipids and macrophages in inflammation are extensively studied. On the one hand, lipids serve as membrane components, and the lipid composition of cell membranes can alter membrane fluidity or lipid raft structure, affecting cellular signaling pathways (71). Changes in cholesterol content and lipid raft composition reduce macrophage inflammation in the absence of FA synthase (72). On the other hand, exogenous lipids, long-chain saturated FAs, such as palmitate or stearate, can act directly as pro-inflammatory signals (73). Derivatives of certain FAs, such as PGs, thromboxanes, leukotrienes, resolvins, and maresins, can act as immunomodulators, mediating the effects of macrophages (74). Although there has been extensive research on the role of macrophages with lipids in inflammation, in RA, apart from the metabolic features of lipid-scavenging macrophages that will be described in the 3.3 section, there are currently few studies that combine the immune effects of macrophages with lipids, and there is still a long way to go in the future.
3.1.2 Effect of lipid on fibroblast-like synoviocytes of RA
Synovitis is the pathological basis of RA. In RA, FLS, also known as synovial fibroblast, is the most common cell type at the pannus cartilage junction. It invades and degrades the cartilage matrix by producing cytokines, chemokines, and matrix degradation molecules, leading to joint destruction (75). Compared with FLS of OA, the levels of 12-22C saturated FA, palmitoleic acid, oleic acid, and linoleic acid in FLS of RA are higher (76).
In RA, lipids affect FLS function in many ways. Free FA contributes to the pro-inflammatory environment of FLS in RA. In FLS, free FA dose-dependently increases the secretion of pro-inflammatory cytokines, chemokines, and matrix degrading enzymes (such as IL-6, IL-8, monocyte chemoattractant protein-1 (MCP-1), pro-MMP1 and MMP3). And the effects of saturated and unsaturated free FA on FLS in promoting inflammation are similar (52). TLR4 is involved in FA-induced signal transduction (77). Palmitic acid can induce the secretion of IL-6 in FLS. However, after the use of inhibitors to inhibit extracellular and intracellular TLR4, PA-induced IL-6 secretion was inhibited through FA transporter CD36/FA translocase (52). Bioactive lipids have various regulatory effects on FLS. A kind of AA metabolite, the downstream product of 15-LOX of 15S-HETE, can increase the mRNA and protein levels of MMP2 in FLS from RA patients (53). 15S-HETE can also enhance the production of placenta growth factor in FLS through PI3K-Akt, nuclear factor κ-B (NF-κ B), and cyclooxygenase-2 (COX-2) pathways (54). Another AA metabolite, LTB4, is an effective pro-inflammatory lipid mediator. Its expression in active RA patients is higher than that in inactive RA patients and healthy donors. The main LTB4 receptor expressed on FLS is LTB4 receptor 2. LTB4 induces FLS of RA to produce TNF-α and IL-1β through LTB4 receptor 2 (23, 55). PGs are beneficial to leukocyte infiltration, synovial hyperplasia, and angiogenesis, thus promoting synovitis (78). Compared with OA and joint trauma patients, PGE2 and its processing enzymes COX-2 and membrane-associated PGE synthase-1 have the highest content in synovial tissue of RA patients, mainly in FLS and plasma cells (79). FLS promotes the expression of triggering receptor expressed on myeloid cells-1 in monocytes through the COX-2/PGE2 pathway (80). Adiponectin induced the expression of COX-2 and membrane-associated PGE synthase-1, thereby enhancing the production of PGE2 in the FLS of RA (57). Other bioactive lipids may protect FLS through their anti-inflammatory effects. For example, 15-LOX induced by IL-13 may regulate the production of LXA4, thus having an anti-inflammatory effect (81). In vitro experiments confirmed that LXA4 could inhibit IL-1β induced production of IL-6, IL-8, and MMP3 in human FLS, and enhance the synthesis of tissue inhibitors of metalloproteinases (58). In vivo experiments demonstrated that LXA4 could inhibit the proliferation of synovial cells of CIA mice, reduce the level of TNF-α, IL-6, IL-1β, and IFN-γ, and inhibit the p38 MAPK signaling pathway (59).
3.2 Effect of lipids on cartilage and bone of RA
Lipids play an important role in bone remodeling in the microenvironment of bone. On the one hand, the interaction between osteoblasts and cholesterol contributes to bone homeostasis, and adipogenic molecules contribute to the differentiation balance of osteoblasts. On the other hand, osteoclasts can respond to changes in metabolic dysfunction (82). RA patients often suffer from progressive injury of joint bone and cartilage, bone loss around joints and the whole body increases the risk of fracture in RA patients. Bone loss in RA patients is the result of excessive osteoclasts (OCs) mediated absorption and limited osteoblasts (OBs) mediated formation (83). OBs are the main functional cells of bone formation, responsible for the synthesis, secretion, and mineralization of bone matrix. On the contrary, OCs mediate bone resorption during bone transformation and OCs over-absorb bone in RA (84). Cartilage is the key component of the synovial joint. In addition to various matrix proteins, chondrocytes are the only cells in cartilage. There are many inflammatory mediators in the synovial joints of RA patients. Chondrocytes can not only act as target cells of these inflammatory mediators but also lead to cell dysfunction. Chondrocytes can also act as effector cells to directly or indirectly promote joint injury of RA (85). The role of lipid regulation in the bone cells and chondrocytes of RA is complex (Table 2).

Table 2 Effect of lipid on bone cells and chondrocytes of RA.
3.2.1 Effect of lipid on osteoclasts of RA
OCs are derived from monocytes/macrophage precursors and developed into OCs after the activation of macrophage colony stimulating factor and receptor activator of nuclear factor-κB receptor ligand (RANKL) (95). Among them, the RANKL-RANK signal plays a key role in the generation and function of OCs (96). TNF receptor-associated factor 6 activated by RANKL signal transduction leads to the activation of NF-κ B and mitogen-activated protein kinase (MAPK), including c-Jun N-terminal kinase and p38 (97). Through the examination of the bone erosion site at the bone pannus interface of RA patients, multinuclear giant cells could be found, which were tartrate-resistant acid phosphatase positive, calcitonin receptor-positive, and cathepsin K positive (98).
Osteoclast precursors (OCPs) show monocyte phenotype after being stimulated by macrophage colony stimulating factor and RANKL, and circulating OCPs play a crucial role in bone erosion of RA. However, increasing FA oxidation can induce OCPs fusion and promote RA joint destruction (86). The differentiation of OCs induced by free FA, PA, and linoleic acid enhanced the secretion of IL-8 compared with OA, especially at the earliest point of differentiation, indicating that the sensitivity of RA to free FA increased (87). LTB4 can promote OCs formation and induce bone injury in many ways. On the one hand, LTB4 can indirectly stimulate human OCs differentiation by increasing RANKL expression of FLS (88). On the other hand, as a receptor of LTB4, BLT1 regulates OCs differentiation negatively through NF-κ B and Ca2+ signals, thus linking the dual role of the LTB4 receptor in OCs formation (89). Lysophosphatidic acid is a bioactive lipid that can bind to LPA receptors. LPA1 is highly expressed in the synovium of RA patients, and LPA/LPA1 signal promotes the development of arthritis through OCs generation and cell infiltration (90).
Although some lipids and their derivatives can promote bone resorption of OCs, other lipids can inhibit OCs formation by promoting apoptosis or other signaling pathways, thus playing a role in bone protection in RA. DHA inhibited c-Jun N-terminal kinase, ERK, and p38 MAPK (91). In addition, DHA can inhibit NF-κ B signal cascade, which weakens the induction of c-Fos and nuclear factor of activated T cells c1 (91). DHA plays an anti-osteoclastogenic role by inhibiting the proliferation and differentiation of bone marrow-derived macrophages and enhancing the apoptosis of mature OCs (91). DHA accelerates the apoptosis of mature OCs by inducing the expression of Bim, which is a key regulator of OCs apoptosis (91). RvE1 inhibits the expression of nuclear factor of activated T cells c1 and c-fos in OCs induced by RANKL, thus inhibiting the formation of OCs and bone resorption (92).
3.2.2 Effect of lipid on osteoblasts of RA
Although OBs play an important role in RA bone loss, the effect of lipid metabolism on OBs is less concerning. Current research mainly focuses on free FA. The effect of free FA on OBs of RA is multifaceted (87). When stimulated with free FA, such as PA or LA, OBs of RA secretes more pro-inflammatory cytokines IL-6, chemokine IL-8, and growth-related oncogenes α and MCP-1 (87). The mineralization activity of OBs is negatively correlated with the level of IL-6 secretion induced by free FA (87). TLR4 is considered the receptor of free FA and is involved in arthritis-dependent joint destruction. After specifically blocking the expression of LTR4, the IL-8 secreted by PA-induced OBs decreased (87).
3.2.3 Effect of lipid on chondrocytes of RA
In addition to bone destruction, cartilage damage is also a typical feature of RA. The stimulation of PA induced human chondrocytes to secrete a large amount of IL-6. However, the stimulation of unsaturated FAs, especially PUFAs, has a weak effect on the secretion of IL-6 by human chondrocytes (52). PGE2 is a regulator of chondrocyte maturation rate, which can activate the response reporter genes in cAMP response element/cyclic AMP response element binding protein and AP-1, and play a role through protein kinase A and protein kinase C signaling pathways. The activation of the cAMP response element/cyclic AMP response element binding protein signal of PGE2 could be completely blocked by inhibition of protein kinase A/cyclic AMP response element binding protein and partially blocked by inhibition of c-Fos/PKC, thus suppressing chondrocyte differentiation (93). RvD1 often shows cartilage protection in RA. RvD1 can significantly reduce the serum marker of cartilage renewal (99). The stable epimer 17R-RvD1 can prevent inflammatory arthritis in mice by significantly reducing the biosynthesis of PGE2 and increasing the level of protective SPMs (39). RvD1 can also reduce the cartilage erosion of CIA mice through miRNA-146a-5p (94).
3.3 Effect of lipid on CVD in RA
In addition to systemic inflammation and joint destruction, RA has other extraarticular manifestations, of which CVD is the most serious (100, 101). Lipid abnormalities help accelerate atherosclerosis and increase the risk of CVD, which is the main cause of increased mortality in RA patients (102). Although the lipoprotein level of RA patients is lower than that of the general population, the risk of death caused by CVD is significantly increased, which is known as the “lipid paradox”. Patients with RA had elevated TC, LDL-C, LDL/HDL-C, TC/HDL-C ratio, and decreased HDL-C (103, 104). CVD risk in RA patients is associated with elevated levels of LDL-C, TG, and hsCRP (105). In the follow-up cases of RA, it was found that the CVD incidence rate of male RA patients was higher than that of female patients because the TC of male patients was higher than that of female patients (8). A retrospective study of RA patients in China found that the incidence rate of CVD was significantly higher than those of patients with HDL ≤ 1.04 mmol/L (106). In RA patients, myeloperoxidase can mediate an increase in HDL oxidation, and it is more prominent in patients with CVD (107). Paraoxonase 1 (PON1) can metabolize oxidized lipids and participate in maintaining oxidative balance, and the activity of PON1 is lower in RA patients (108). Compared with the healthy control group, the myeloperoxidase/PON1 ratio of RA patients with a history of CVD was higher, indicating that HDL dysfunction determined by myeloperoxidase/PON1 ratio may be related to the increase of CVD in RA (109).
The increase of LDL level is considered as a strong predictor of CVD, the more LDL-C is reduced by drugs, the greater the subsequent reduction of CVD risk (105). In RA, compared with the control group, the level of small dense LDL granules was higher, while the level of small HDL granules was lower (110). Because small dense LDL particles are easier to penetrate endothelial cells than their larger counterparts and are more susceptible to oxidation (111). LDL transports cholesterol from the liver to tissues, and excessive accumulation of cholesterol in tissues will lead to atherosclerosis. When the endothelial cells of the artery wall change, LDL enters, and the small dense LDL is converted into oxidized LDL, which is eventually swallowed by macrophages to form foam cells through innate and adaptive immunity, causing plaque lesions (111–113). L5 is the most negatively charged subcomponent of LDL, which is related to atherosclerosis. In RA, L5 may promote atherosclerosis by enhancing the formation of macrophage foam cells, up-regulating the expression of M1 macrophage-related markers in vascular plaque formation and atherosclerosis, and improving the expression level of atherosclerosis-related mediators (such as IL-6, IL-8 and TNF-α) (111). RA plasma adversely affects the ability of monocytes/macrophages in the arterial wall to metabolize cholesterol and maintain lipid homeostasis, leading to premature atherosclerosis (114). Lp(a) is rich in cholesterol and consists of LDL particles attached to apoA. The increase of serum Lp(a) is related to the increased risk of atherosclerosis. As a positive acute phase protein, the mechanism of Lp(a) increase during inflammation may be due to the increased synthesis and up-regulation of Lp(a) expression (115).
In healthy individuals, in the absence of oxidative stress and systemic inflammation, HDL has an anti-inflammatory effect due to its function of reverse cholesterol transport and thus has cardioprotective properties (116). While in RA patients characterized by oxidative stress and systemic inflammation, the normal protective anti-inflammatory HDL changes to pro-inflammatory HDL due to changes in their structure and enzymatic content. During the acute phase reaction, serum amyloid A, apolipoprotein J, and pancreatic phospholipase A2 are present in the serum at high concentrations and mixed with HDL to replace their common components, such as cholesteryl ester transfer protein. Cholesteryl ester transfer protein enzyme transfers cholesterol esters to lipoproteins containing apo B, which are then absorbed by the liver to assist HDL to transport cholesterol to the liver and be absorbed. In addition, PON1 decreased and platelet-activating factor acetylhydrolase levels increased in HDL particles. PON1 has anti-inflammatory properties while platelet-activating factor acetylhydrolase has pro-inflammatory properties, these changes make HDL have the characteristics of promoting atherosclerosis (111, 117).
4 Strategies for regulating abnormal lipid metabolism in RA
4.1 Drugs affecting the blood lipid profile of RA
Common RA drugs include nonsteroidal anti-inflammatory drugs, statins, glucocorticoids, traditional disease-modifying antirheumatic drugs (DMARDs), and biological agents. Because of the different targets of pathophysiological pathways in the treatment of RA, they have different effects on the remission of RA. For example, nonsteroidal anti-inflammatory drugs are used to control the symptoms of RA. The therapeutic effect of statins is mainly attributed to their anti-inflammatory and immunomodulatory properties (118). In addition, statins can reduce the risk of CVD in RA patients through a lipid-lowering effect (119, 120). We will mainly review the effects of traditional DMARDs and biological agents on lipid and lipoprotein parameters in RA patients.
4.1.1 DMARDs
Methotrexate (MTX) is the commonly used DMARDs in RA. The changes in plasma metabolome in patients with RA before and after MTX treatment were evaluated by semi-targeted metabolomics. It was found that the changes in multiple metabolites were related to the efficacy of MTX, including TC, FAs, and metabolites related to FA/lipid and energy metabolism (121, 122). As anti-atherosclerotic proteins, ATP binding cassette transporter-A1 and 27 hydroxylase are known to promote cell cholesterol efflux. Compared with other subjects taking DMARDs, there was no difference in blood lipid levels in patients taking MTX, while the expressions of ATP binding cassette transporter-A1 and 27 hydroxylase were significantly increased (121). However, using serum lipidomics to predict the response of RA patients to MTX, it was found that the detection of serum lipid profile before or at the early stage of treatment has no supporting effect in routine clinical practice (123). As an antimalarial drug, hydroxychloroquine (HCQ) has the characteristics of DMARDs and is commonly used in the treatment of mild RA (121). It can improve the blood lipid level of RA patients. Patients taking HCQ showed significantly lower TC, LDL, TC/HDL, and LDL/HDL and higher HDL (124, 125). Consistent with these results, the oral administration of 400mg/day HCQ within three months reduced the levels of serum TC and TG in a clinical trial of 15 RA patients (126).
4.1.2 Biological agents
Anti-TNF-α agents and anti-IL-6 agents are common biological agents for the treatment of RA, which can effectively alleviate inflammation (127). TNF-α, a key inflammatory cytokine, can induce atherogenic changes in lipid mass spectrometry and may increase the cardiovascular risk of RA patients. The use of anti-TNF-α agents is related to the changes in serum lipid profile in RA patients, but some research results are contradictory. At present, the common biological agents used to treat RA are infliximab (IFX), etanercept (ETN), adalimumab, certolizumab pegol, and golimumab (128). Whether compared with baseline or non-TNF-α inhibitor users, TNF-α inhibitors were not related to the difference in blood lipid level in RA patients, including TC, TG, LDL-C, HDL-C, LDL/HDL, TC/HDL, apo A-I, apo B and apo B/apo A-I (129, 130). Another study found no change in the levels of TC, HDL-C, LDL-C, and atherogenic index, while TG levels increased significantly (131). ETN treatment resulted in a significant and sustained decrease in the apo B/apo A-I ratio in patients with good or moderate EULAR response (132). Neutralizing TNF with monoclonal anti-TNF antibody (one kind of adalimumab) increased HDL-C levels and decreased CRP and IL-6 levels after 2 weeks (133). The effect of IFX treatment on blood lipid levels seems to be neutral because there is no significant change in LDL-C level and TC/HDL-C and TG/HDL-C ratio during treatment (134). Another similar study suggests that IFX can change the blood lipid level of patients with active RA, TC, TG, HDL-C, LDL-C, and apoB levels increased significantly from baseline, while ATI, LDL-C/HDL-C ratio, and LDL-C/apoB ratio remained unchanged (135). In patients who successfully received IFX or MTX treatment, although the serum TC level increased and normalized, there was a difference in the regulation of lipid components between IFX and MTX, for example, IFX treatment preferentially induced ultra-high levels of VLDL-TG. Therefore, it is necessary to pay attention to the level of TG during IFX treatment (136). In addition, IFX was beneficial to lipids by changing the antioxidant capacity of HDL and contributing to the protective effect of anti-TNF agents on CVD in RA (137). However, some studies believed that the beneficial effect of anti-TNF-α treatment on CVD was not related to the effect on lipid metabolism (129, 134). TNF-α blocking therapy had no significant overall effect on the atherosclerosis index in RA patients (138, 139). JAK inhibitor (JAKi) can target and block cytokine signal transduction mediated by Janus kinase signal transduction and transcriptional activator pathway, thus regulating immune response and cell growth. Targeted inhibition of JAK in autoimmune diseases such as RA can effectively control diseases (140). A study reported that JAKi could reduce pain reported by RA patients. In JAKi treated patients, there is a significant association between pain reduction and DHA changes, and the analgesic effect may be related to n-3 FAs and increased DHA levels (141).
In short, studies on changes in lipid levels after drug treatment, to further identify potential metabolic biomarkers of response to drug treatment, or to combine lipid levels with other pathological symptoms accompanying RA, such as diabetes and CVD, can not only guide drug treatment of RA through lipid metabolism but also explore the therapeutic effects of drugs on complications such as CVD from multiple perspectives, such as metabolism and immunity.
4.2 Dietary intervention
In addition to drug treatment, diet also has beneficial or harmful effects on RA. Overall, energy intake, total FA and saturated FA, the unbalanced ratio of n-3 to n-6 FA, and the western type diet characterized by a high sugar diet, low fiber, and low antioxidant content can increase obesity, thereby increasing the risk of RA (142, 143). Therefore, dietary intervention strategy in RA is also a hot spot in RA research. The anti-inflammatory diet has a positive effect on the disease activity of RA (144). It has been found in different systematic reviews of randomized controlled trials that a Mediterranean diet can reduce the disease activity and drug treatment failure rate of RA. Although vitamin D supplementation does not affect disease activity score 28 (DAS28), it is beneficial to the outcome of RA (142, 145). 1.7-3 g/d of n-3 FA did not affect DAS28, but 3-6 g/d of n-3 FA could effectively reduce RA-related pain (145, 146). Accumulation of toxic lipid mediators in skeletal muscle can induce mitochondrial dysfunction and insulin resistance, which are key determinants of CVD and sarcopenia. Most clinical trials have shown that n-3 PUFA supplements are beneficial to RA patients (147–150). High n-3 PUFA intake can prevent the occurrence of RA (147, 151, 152) and improve the prognosis of RA (41, 148). N-3 FA can improve muscle metabolism and limit muscle atrophy in obese and insulin-resistant subjects, thereby preventing CVD, poor activity, and death risk in RA patients (153). In a longitudinal study, compared to the group with lower n-3 FAs intake, RA patients with high levels of n-3 FAs, palmitoleic acid, and AA in their diet showed a decrease in arterial stiffness, which helps to slow the progression of arterial aging (154). In addition, through the meta-analysis of RA disease severity markers, it was found that oral n-3 FA was beneficial to RA disease activity and improved the disease severity markers (155). Dietary regulation also has a certain effect on the SPMs level of RA patients. Marine oils are a rich source of n-3 FAs, especially the essential FAs, DHA, and EPA (156). By supplementing enriched marine oil supplements, the concentration of SPMs in the peripheral blood of RA patients increased and the peripheral blood cells were reprogrammed. SPMs have a guiding role in mediating the immune response of this supplement (157). After supplementation with n-3 PUFA, LTB4 levels were decreased in RA subjects (155, 158). In conclusion, a reasonable diet or dietary supplementation, such as a Mediterranean diet, supplemented with enriched marine oil supplements or n-3 FA, can suppress the clinical symptoms of RA to a certain extent. The current difficulty is that a broad and applicable dietary standard cannot be proposed, but in clinical treatment, doctors can provide reasonable dietary recommendations to assist patients.
Compared with only receiving RA drugs, proper dietary supplementation has an effective effect on delaying the progression of RA disease. Data from a population-based prospective study in Sweden found that a higher intake of dietary vitamin D and n-3 FA can enhance the therapeutic effect of DMARDs (159). Supplementation of n-3 FA with indomethacin may improve disease activity compared to only receiving indomethacin, or supplementation with n-3 FA (160). In the CIA mouse model, the combination of curcumin and n-3 FA delayed disease progression, reduced disease severity, and significantly inhibited the factors related to RA occurrence, such as TNF, IFN-γ, and MCP-1 (161). Long-term combined supplementation of coenzyme Q10 and n-3 FA can restore the damaged mitochondrial bioenergy and antioxidant status in RA animal models (162). Higher plasma EPA is associated with a greater reduction of DAS28, and EPA is associated with clinical improvement of anti-TNF therapy in vivo. ETN increases Th17 frequency in vitro, and EPA prevents the effect of ETN on Th17 cells (163). Clinically, in addition to exploring the therapeutic effect of drugs on RA, it is also important to explore the early intervention of diet in RA progression or the therapeutic effect of synergistic drugs. There is a long way to go for systematic drug therapy and food therapy.
5 Conclusion and perspectives
Although RA is still incurable, the development of DMARDs and the improvement of treatment schemes make it an almost controllable disease. Like the pathological mechanism of RA, lipid metabolism in RA is also complex. At present, more and more attention has been paid to the relationship between lipid metabolism and the progress of RA and the changes in plasma lipid levels after different drug treatments, drug combinations, or dietary interventions. As the most abundant type of cellular metabolites, lipids play an undeniable role in energy supply and storage, cell membrane construction, transportation, and signal transduction. Secondly, using lipids and lipid mediators in plasma as diagnostic and therapeutic targets not only facilitates clinical detection but also enables more targeted treatment based on individual differences. However, there are still many shortcomings in related research. Firstly, there are often conflicting results regarding lipid levels due to differences in population, experimental duration, or analytical methods. Secondly, it is currently unclear whether changes in lipid metabolism are the cause, consequence, or both of RA. In addition, at the cellular immune level, some cells, such as B cells, neutrophils, and macrophages, have been extensively studied in lipid metabolism, but there is still a lack of relevant evidence in RA. Finally, although some lipids have the effect of promoting RA progression, there are still various types of lipids and their metabolites, such as DHA, EPA, and the oxylipins derived from them (resolvins, maresins, and protectins), which have been proven to have protective effects on RA. How to guide the treatment or intervention of RA from the perspective of lipids and provide corresponding explanations from a molecular perspective remains a challenging task. Therefore, we need further research to address existing issues to better determine the pathophysiology of lipid abnormalities and the potential impact of drug therapy on RA lipid metabolism. To better integrate the procession of lipid metabolism with the prevention, occurrence, development, and treatment of RA and even related complications will undoubtedly enable us to have a more comprehensive understanding of RA.
Author contributions
QL wrote the manuscript. JY participated in the writing. LL, NZ, and CL helped in revising the manuscript. AL and XH conceptualized the idea and revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Science Foundation of China (No. 81873053, 82274342), the Fundamental Research Funds for the Central Public Welfare Research Institutes (No. Z0736), CACMS Innovation Fund (CI2021A01509) and the 2020 Guangdong Provincial Science and Technology Innovation Strategy Special Fund (Guangdong-Hong Kong-Macau Joint Lab, No: 2020B1212030006).
Acknowledgments
Parts of the figure were drawn using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Gibofsky A. Epidemiology, pathophysiology, and diagnosis of rheumatoid arthritis: a synopsis. Am J Manag Care (2014) 20(7 Suppl):S128–35.
2. Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ, Xu J. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res (2018) 6:15. doi: 10.1038/s41413-018-0016-9
3. Symmons D, Turner G, Webb R, Asten P, Barrett E, Lunt M, et al. The prevalence of rheumatoid arthritis in the united kingdom: new estimates for a new century. Rheumatol (Oxford) (2002) 41(7):793–800. doi: 10.1093/rheumatology/41.7.793
4. Deane KD, Demoruelle MK, Kelmenson LB, Kuhn KA, Norris JM, Holers VM. Genetic and environmental risk factors for rheumatoid arthritis. Best Pract Res Clin Rheumatol (2017) 31(1):3–18. doi: 10.1016/j.berh.2017.08.003
5. Kang Y, Cai Y, Zhang X, Kong X, Su J. Altered gut microbiota in RA: implications for treatment. Z Rheumatol (2017) 76(5):451–7. doi: 10.1007/s00393-016-0237-5
6. DeBose-Boyd RA. Significance and regulation of lipid metabolism. Semin Cell Dev Biol (2018) 81:97. doi: 10.1016/j.semcdb.2017.12.003
7. Salciccia S, Capriotti AL, Laganà A, Fais S, Logozzi M, De Berardinis E, et al. Biomarkers in prostate cancer diagnosis: from current knowledge to the role of metabolomics and exosomes. Int J Mol Sci (2021) 22(9):48–54. doi: 10.3390/ijms22094367
8. Crowson CS, Rollefstad S, Ikdahl E, Kitas GD, van Riel PLCM, Gabriel SE, et al. Impact of risk factors associated with cardiovascular outcomes in patients with rheumatoid arthritis. Ann Rheum Dis (2018) 77(1):48–54. doi: 10.1136/annrheumdis-2017-211735
9. Chiapparino A, Maeda K, Turei D, Saez-Rodriguez J, Gavin AC. The orchestra of lipid-transfer proteins at the crossroads between metabolism and signaling. Prog Lipid Res (2016) 61:30–9. doi: 10.1016/j.plipres.2015.10.004
10. Ye Z, Shen Y, Jin K, Qiu J, Hu B, Jadhav RR, et al. Arachidonic acid-regulated calcium signaling in T cells from patients with rheumatoid arthritis promotes synovial inflammation. Nat Commun (2021) 12(1):907. doi: 10.1038/s41467-021-21242-z
11. Navarini L, Afeltra A, Gallo Afflitto G, Margiotta DPE. Polyunsaturated fatty acids: any role in rheumatoid arthritis? Lipids Health Dis (2017) 16(1):197. doi: 10.1186/s12944-017-0586-3
12. Basil MC, Levy BD. Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat Rev Immunol (2016) 16(1):51–67. doi: 10.1038/nri.2015.4
13. Pašková U. Lipid profile and risks of cardiovascular diseases in conditions of rheumatoid arthritis. Ceska Slov Farm (2019) 68(6):219–28.
14. McGrath CM, Young SP. Lipid and metabolic changes in rheumatoid arthritis. Curr Rheumatol Rep (2015) 17(9):57. doi: 10.1007/s11926-015-0534-z
15. Myasoedova E, Crowson CS, Kremers HM, Roger VL, Fitz-Gibbon PD, Therneau TM, et al. Lipid paradox in rheumatoid arthritis: the impact of serum lipid measures and systemic inflammation on the risk of cardiovascular disease. Ann Rheum Dis (2011) 70(3):482–7. doi: 10.1136/ard.2010.135871
16. Park YB, Lee SK, Lee WK, Suh CH, Lee CW, Lee CH, et al. Lipid profiles in untreated patients with rheumatoid arthritis. J Rheumatol (1999) 26(8):1701–4. doi: 10.1097/00124743-199908000-00019
17. Myasoedova E, Crowson CS, Kremers HM, Fitz-Gibbon PD, Therneau TM, Gabriel SE. Total cholesterol and LDL levels decrease before rheumatoid arthritis. Ann Rheum Dis (2010) 69(7):1310–4. doi: 10.1136/ard.2009.122374
18. Kim JY, Lee EY, Park JK, Song YW, Kim JR, Cho KH, et al. Patients with rheumatoid arthritis show altered lipoprotein profiles with dysfunctional high-density lipoproteins that can exacerbate inflammatory and atherogenic process. PloS One (2016) 11(10):e0164564. doi:?10.1371/journal.pone.0164564
19. Govindan KP, Basha S, Ramesh V, Kumar CN, Swathi S. A comparative study on serum lipoprotein (a) and lipid profile between rheumatoid arthritis patients and normal subjects. J Pharm Bioallied Sci (2015) 7(Suppl 1):S22–5. doi: 10.4103/0975-7406.155767
20. Dessie G, Tadesse Y, Demelash B, Genet S. Assessment of serum lipid profiles and high-sensitivity c-reactive protein among patients suffering from rheumatoid arthritis at tikur anbessa specialized hospital, Addis Ababa, Ethiopia: a cross-sectional study. Open Access Rheumatol (2020) 12:223–32. doi: 10.2147/OARRR.S264466
21. Salem HR, Zahran ES. Vascular cell adhesion molecule-1 in rheumatoid arthritis patients: relation to disease activity, oxidative stress, and systemic inflammation. Saudi Med J (2021) 42(6):620–8. doi: 10.15537/smj.2021.42.6.20200753
22. Mustonen AM, Nieminen P. Fatty acids and oxylipins in osteoarthritis and rheumatoid arthritis-a complex field with significant potential for future treatments. Curr Rheumatol Rep (2021) 23(6):41. doi: 10.1007/s11926-021-01007-9
23. Miyabe Y, Miyabe C, Luster AD. LTB(4) and BLT1 in inflammatory arthritis. Semin Immunol (2017) 33:52–7. doi: 10.1016/j.smim.2017.09.009
24. Arnardottir HH, Dalli J, Norling LV, Colas RA, Perretti M, Serhan CN. Resolvin D3 is dysregulated in arthritis and reduces arthritic inflammation. J Immunol (2016) 197(6):2362–8. doi: 10.4049/jimmunol.1502268
25. Hensor EMA, McKeigue P, Ling SF, Colombo M, Barrett JH, Nam JL, et al. Validity of a two-component imaging-derived disease activity score for improved assessment of synovitis in early rheumatoid arthritis. Rheumatol (Oxford) (2019) 58(8):1400–9. doi: 10.1093/rheumatology/kez049
26. Gan L, He Y, Liu L, Ou Q, Lin J. Association of serum lipids with autoantibodies and inflammatory markers in rheumatoid arthritis patients. Clin Chim Acta (2018) 486:282–90. doi: 10.1016/j.cca.2018.08.028
27. Weber B, He Z, Yang N, Playford MP, Weisenfeld D, Iannaccone C, et al. Divergence of cardiovascular biomarkers of lipids and subclinical myocardial injury among rheumatoid arthritis patients with increased inflammation. Arthritis Rheumatol (2021) 73(6):970–9. doi: 10.1002/art.41613
28. Chouk M, Bordy R, Moretto J, Wendling D, Totoson P, Demougeot C. Pristane-induced arthritis in dark agouti rat is a relevant model for mimicking vascular dysfunction and lipid paradox in rheumatoid arthritis. Joint Bone Spine (2019) 86(4):483–90. doi: 10.1016/j.jbspin.2018.12.001
29. Srivastava NK, Sharma S, Sinha N, Mandal SK, Sharma D. Abnormal lipid metabolism in a rat model of arthritis: one possible pathway. Mol Cell Biochem (2018) 448(1-2):107–24. doi: 10.1007/s11010-018-3318-8
30. Wang N, Zhao X, Huai J, Li Y, Cheng C, Bi K, et al. Arachidonic acid metabonomics study for understanding therapeutic mechanism of huo luo xiao ling Dan on rat model of rheumatoid arthritis. J Ethnopharmacol (2018) 217:205–11. doi: 10.1016/j.jep.2018.02.027
31. Ding X, Hu J, Li J, Zhang Y, Shui B, Ding Z, et al. Metabolomics analysis of collagen-induced arthritis in rats and interventional effects of oral tolerance. Anal Biochem (2014) 458:49–57. doi: 10.1016/j.ab.2014.04.035
32. Wang N, Zhao X, Wang W, Peng Y, Bi K, Dai R. Targeted profiling of arachidonic acid and eicosanoids in rat tissue by UFLC-MS/MS: application to identify potential markers for rheumatoid arthritis. Talanta (2017) 162:479–87. doi: 10.1016/j.talanta.2016.10.065
33. He M, van Wijk E, Berger R, Wang M, Strassburg K, Schoeman JC, et al. Collagen induced arthritis in DBA/1J mice associates with oxylipin changes in plasma. Mediators Inflamm (2015) 2015:543541. doi: 10.1155/2015/543541
34. Prete PE, Gurakar-Osborne A, Kashyap ML. Synovial fluid lipids and apolipoproteins: a contemporary perspective. Biorheology (1995) 32(1):1–16. doi: 10.3233/BIR-1995-32101
35. Brouwers H, von Hegedus J, Toes R, Kloppenburg M, Ioan-Facsinay A. Lipid mediators of inflammation in rheumatoid arthritis and osteoarthritis. Best Pract Res Clin Rheumatol (2015) 29(6):741–55. doi: 10.1016/j.berh.2016.02.003
36. Mustonen AM, Käkelä R, Lehenkari P, Huhtakangas J, Turunen S, Joukainen A, et al. Distinct fatty acid signatures in infrapatellar fat pad and synovial fluid of patients with osteoarthritis versus rheumatoid arthritis. Arthritis Res Ther (2019) 21(1):124. doi: 10.1186/s13075-019-1914-y
37. Kim S, Hwang J, Xuan J, Jung YH, Cha HS, Kim KH. Global metabolite profiling of synovial fluid for the specific diagnosis of rheumatoid arthritis from other inflammatory arthritis. PloS One (2014) 9(6):e97501. doi: 10.1371/journal.pone.0097501
38. Mustonen AM, Käkelä R, Joukainen A, Lehenkari P, Jaroma A, Kääriäinen T, et al. Synovial fluid fatty acid profiles are differently altered by inflammatory joint pathologies in the shoulder and knee joints. Biol (Basel) (2021) 10(5):401. doi: 10.3390/biology10050401
39. Norling LV, Headland SE, Dalli J, Arnardottir HH, Haworth O, Jones HR, et al. Proresolving and cartilage-protective actions of resolvin D1 in inflammatory arthritis. JCI Insight (2016) 1(5):e85922. doi: 10.1172/jci.insight.85922
40. Giera M, Ioan-Facsinay A, Toes R, Gao F, Dalli J, Deelder AM, et al. Lipid and lipid mediator profiling of human synovial fluid in rheumatoid arthritis patients by means of LC-MS/MS. Biochim Biophys Acta (2012) 1821(11):1415–24. doi: 10.1016/j.bbalip.2012.07.011
41. Lee YH, Bae SC, Song GG. Omega-3 polyunsaturated fatty acids and the treatment of rheumatoid arthritis: a meta-analysis. Arch Med Res (2012) 43(5):356–62. doi: 10.1016/j.arcmed.2012.06.011
42. Charles-Schoeman C, Meriwether D, Lee YY, Shahbazian A, Reddy ST. High levels of oxidized fatty acids in HDL are associated with impaired HDL function in patients with active rheumatoid arthritis. Clin Rheumatol (2018) 37(3):615–22. doi: 10.1007/s10067-017-3896-y
43. Shen Y, Wen Z, Li Y, Matteson EL, Hong J, Goronzy JJ, et al. Metabolic control of the scaffold protein TKS5 in tissue-invasive, proinflammatory T cells. Nat Immunol (2017) 18(9):1025–34. doi: 10.1038/ni.3808
44. Kumar R, Theiss AL, Venuprasad K. RORγt protein modifications and IL-17-mediated inflammation. Trends Immunol (2021) 42(11):1037–50. doi: 10.1016/j.it.2021.09.005
45. Du B, Zhu M, Li Y, Li G, Xi X. The prostaglandin E2 increases the production of IL-17 and the expression of costimulatory molecules on γδ T cells in rheumatoid arthritis. Scand J Immunol (2020) 91(5):e12872. doi: 10.1111/sji.12872
46. Kim JY, Lim K, Kim KH, Kim JH, Choi JS, Shim SC. N-3 polyunsaturated fatty acids restore Th17 and treg balance in collagen antibody-induced arthritis. PloS One (2018) 13(3):e0194331. doi: 10.1371/journal.pone.0194331
47. Jin S, Chen H, Li Y, Zhong H, Sun W, Wang J, et al. Maresin 1 improves the Treg/Th17 imbalance in rheumatoid arthritis through miR-21. Ann Rheum Dis (2018) 77(11):1644–52. doi: 10.1136/annrheumdis-2018-213511
48. Yao Y, Cai X, Zheng Y, Zhang M, Fei W, Sun D, et al. Short-chain fatty acids regulate b cells differentiation via the FFA2 receptor to alleviate rheumatoid arthritis. Br J Pharmacol (2022) 179(17):4315–29. doi: 10.1111/bph.15852
49. Jia XY, Chang Y, Sun XJ, Dai X, Wei W. The role of prostaglandin E2 receptor signaling of dendritic cells in rheumatoid arthritis. Int Immunopharmacol (2014) 23(1):163–9. doi: 10.1016/j.intimp.2014.08.024
50. Li KX, Zheng LX, Guo HQ, Hong FF, Yang SL. LTB4-induced anti-apoptosis and infiltration of neutrophils in rheumatoid arthritis. Clin Exp Rheumatol (2020) 38(3):543–51.
51. Canetti CA, Leung BP, Culshaw S, McInnes IB, Cunha FQ, Liew FY. IL-18 enhances collagen-induced arthritis by recruiting neutrophils via TNF-alpha and leukotriene B4. J Immunol (2003) 171(2):1009–15. doi: 10.4049/jimmunol.171.2.1009
52. Frommer KW, Schäffler A, Rehart S, Lehr A, Müller-Ladner U, Neumann E. Free fatty acids: potential proinflammatory mediators in rheumatic diseases. Ann Rheum Dis (2015) 74(1):303–10. doi: 10.1136/annrheumdis-2013-203755
53. Wu MY, Lin TH, Chiu YC, Liou HC, Yang RS, Fu WM. Involvement of 15-lipoxygenase in the inflammatory arthritis. J Cell Biochem (2012) 113(7):2279–89. doi: 10.1002/jcb.24098
54. Wu MY, Yang RS, Lin TH, Tang CH, Chiu YC, Liou HC, et al. Enhancement of PLGF production by 15-(S)-HETE via PI3K-akt, NF-κB and COX-2 pathways in rheumatoid arthritis synovial fibroblast. Eur J Pharmacol (2013) 714(1-3):388–96. doi: 10.1016/j.ejphar.2013.07.010
55. Xu S, Lu H, Lin J, Chen Z, Jiang D. Regulation of TNFalpha and IL1beta in rheumatoid arthritis synovial fibroblasts by leukotriene B4. Rheumatol Int (2010) 30(9):1183–9. doi: 10.1007/s00296-009-1125-y
56. Yan Y, Singh GK, Zhang F, Wang P, Liu W, Zhong L, et al. Comparative study of normal and rheumatoid arthritis fibroblast-like synoviocytes proliferation under cyclic mechanical stretch: role of prostaglandin E2. Connect Tissue Res (2012) 53(3):246–54. doi: 10.3109/03008207.2011.632828
57. Kusunoki N, Kitahara K, Kojima F, Tanaka N, Kaneko K, Endo H, et al. Adiponectin stimulates prostaglandin E(2) production in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum (2010) 62(6):1641–9. doi: 10.1002/art.27450
58. Sodin-Semrl S, Taddeo B, Tseng D, Varga J, Fiore S. Lipoxin A4 inhibits IL-1 beta-induced IL-6, IL-8, and matrix metalloproteinase-3 production in human synovial fibroblasts and enhances synthesis of tissue inhibitors of metalloproteinases. J Immunol (2000) 164(5):2660–6. doi: 10.4049/jimmunol.164.5.2660
59. Li J, Sun Q, Zheng C, Bai C, Liu C, Zhao X, et al. Lipoxin A4-mediated p38 MAPK signaling pathway protects mice against collagen-induced arthritis. Biochem Genet (2021) 59(1):346–65. doi: 10.1007/s10528-020-10016-9
60. Qiu J, Wu B, Goodman SB, Berry GJ, Goronzy JJ, Weyand CM. Metabolic control of autoimmunity and tissue inflammation in rheumatoid arthritis. Front Immunol (2021) 12:652771. doi: 10.3389/fimmu.2021.652771
61. Panayi GS, Corrigall VM, Pitzalis C. Pathogenesis of rheumatoid arthritis. the role of T cells and other beasts. Rheum Dis Clin North Am (2001) 27(2):317–34. doi: 10.1016/S0889-857X(05)70204-0
62. Weyand CM, Wu B, Goronzy JJ. The metabolic signature of T cells in rheumatoid arthritis. Curr Opin Rheumatol (2020) 32(2):159–67. doi: 10.1097/BOR.0000000000000683
63. Masoumi M, Alesaeidi S, Khorramdelazad H, Behzadi M, Baharlou R, Alizadeh-Fanalou S, et al. Role of T cells in the pathogenesis of rheumatoid arthritis: focus on immunometabolism dysfunctions. Inflammation (2022) 46(1):88–102. doi: 10.1007/s10753-022-01751-9
64. Sladek FM. What are nuclear receptor ligands? Mol Cell Endocrinol (2011) 334(1-2):3–13. doi: 10.1016/j.mce.2010.06.018
65. Wang T, Rui J, Shan W, Xue F, Feng D, Dong L, et al. Imbalance of Th17, treg, and helper innate lymphoid cell in the peripheral blood of patients with rheumatoid arthritis. Clin Rheumatol (2022) 41(12):3837–49. doi: 10.1007/s10067-022-06315-8
66. Wu F, Gao J, Kang J, Wang X, Niu Q, Liu J, et al. B cells in rheumatoid Arthritis: Pathogenic mechanisms and treatment prospects. Front Immunol (2021) 12:750753. doi: 10.3389/fimmu.2021.750753
67. Thurnher M. Lipids in dendritic cell biology: messengers, effectors, and antigens. J Leukoc Biol (2007) 81(1):154–60. doi: 10.1189/jlb.0706438
68. Coutant F. Shaping of monocyte-derived dendritic cell development and function by environmental factors in rheumatoid arthritis. Int J Mol Sci (2021) 22(24):13670. doi: 10.3390/ijms222413670
69. Weissmann G, Korchak H. Rheumatoid arthritis. the role of neutrophil activation. Inflammation (1984) 8 Suppl:S3–14. doi: 10.1007/BF00915708
70. Elmgreen J, Nielsen OH, Ahnfelt-Rønne I. Enhanced capacity for release of leucotriene B4 by neutrophils in rheumatoid arthritis. Ann Rheum Dis (1987) 46(7):501–5. doi: 10.1136/ard.46.7.501
71. Schumann J. It is all about fluidity: fatty acids and macrophage phagocytosis. Eur J Pharmacol (2016) 785:18–23. doi: 10.1016/j.ejphar.2015.04.057
72. Wei X, Song H, Yin L, Rizzo MG, Sidhu R, Covey DF, et al. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature (2016) 539(7628):294–8. doi: 10.1038/nature20117
73. Hernandez ED, Lee SJ, Kim JY, Duran A, Linares JF, Yajima T, et al. A macrophage NBR1-MEKK3 complex triggers JNK-mediated adipose tissue inflammation in obesity. Cell Metab (2014) 20(3):499–511. doi: 10.1016/j.cmet.2014.06.008
74. Castoldi A, Monteiro LB, van Teijlingen Bakker N, Sanin DE, Rana N, Corrado M, et al. Triacylglycerol synthesis enhances macrophage inflammatory function. Nat Commun (2020) 11(1):4107. doi: 10.1038/s41467-020-17881-3
75. Bustamante MF, Garcia-Carbonell R, Whisenant KD, Guma M. Fibroblast-like synoviocyte metabolism in the pathogenesis of rheumatoid arthritis. Arthritis Res Ther (2017) 19(1):110. doi: 10.1186/s13075-017-1303-3
76. Ahn JK, Kim S, Hwang J, Kim J, Kim KH, Cha HS. GC/TOF-MS-based metabolomic profiling in cultured fibroblast-like synoviocytes from rheumatoid arthritis. Joint Bone Spine (2016) 83(6):707–13. doi: 10.1016/j.jbspin.2015.11.009
77. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest (2006) 116(11):3015–25. doi: 10.1172/JCI28898
78. Ma Y, Hong FF, Yang SL. Role of prostaglandins in rheumatoid arthritis. Clin Exp Rheumatol (2021) 39(1):162–72. doi: 10.55563/clinexprheumatol/1jlh15
79. Al-Madol MA, Shaqura M, John T, Likar R, Ebied RS, Salih MM, et al. Prostanoid receptor subtypes and its endogenous ligands with processing enzymes within various types of inflammatory joint diseases. Mediators Inflamm (2020) 2020:4301072. doi: 10.1155/2020/4301072
80. Peng A, Lu X, Huang J, He M, Xu J, Huang H, et al. Rheumatoid arthritis synovial fibroblasts promote TREM-1 expression in monocytes via COX-2/PGE(2) pathway. Arthritis Res Ther (2019) 21(1):169. doi: 10.1186/s13075-019-1954-3
81. Hashimoto A, Hayashi I, Murakami Y, Sato Y, Kitasato H, Matsushita R, et al. Antiinflammatory mediator lipoxin A4 and its receptor in synovitis of patients with rheumatoid arthritis. J Rheumatol (2007) 34(11):2144–53.
82. Wang B, Wang H, Li Y, Song L. Lipid metabolism within the bone micro-environment is closely associated with bone metabolism in physiological and pathophysiological stages. Lipids Health Dis (2022) 21(1):5. doi: 10.1186/s12944-021-01615-5
83. Shim JH, Stavre Z, Gravallese EM. Bone loss in rheumatoid arthritis: basic mechanisms and clinical implications. Calcif Tissue Int (2018) 102(5):533–46. doi: 10.1007/s00223-017-0373-1
84. Hu L, Liu R, Zhang L. Advance in bone destruction participated by JAK/STAT in rheumatoid arthritis and therapeutic effect of JAK/STAT inhibitors. Int Immunopharmacol (2022) 111:109095. doi: 10.1016/j.intimp.2022.109095
85. Tseng CC, Chen YJ, Chang WA, Tsai WC, Ou TT, Wu C, et al. Dual role of chondrocytes in rheumatoid arthritis: the chicken and the egg. Int J Mol Sci (2020) 21(3):1071. doi: 10.3390/ijms21031071
86. Huang Z, Luo R, Yang L, Chen H, Zhang X, Han J, et al. CPT1A-mediated fatty acid oxidation promotes precursor osteoclast fusion in rheumatoid arthritis. Front Immunol (2022) 13:838664. doi: 10.3389/fimmu.2022.838664
87. Frommer KW, Hasseli R, Schäffler A, Lange U, Rehart S, Steinmeyer J, et al. Free fatty acids in bone pathophysiology of rheumatic diseases. Front Immunol (2019) 10:2757. doi: 10.3389/fimmu.2019.02757
88. Chen ZK, Lv HS, Jiang J. LTB4 can stimulate human osteoclast differentiation dependent of RANKL. Artif Cells Blood Substit Immobil Biotechnol (2010) 38(1):52–6. doi: 10.3109/10731190903495785
89. Bouchareychas L, Grössinger EM, Kang M, Qiu H, Adamopoulos IE. Critical role of LTB4/BLT1 in IL-23-Induced synovial inflammation and osteoclastogenesis via NF-κB. J Immunol (2017) 198(1):452–60. doi: 10.4049/jimmunol.1601346
90. Miyabe Y, Miyabe C, Iwai Y, Takayasu A, Fukuda S, Yokoyama W, et al. Necessity of lysophosphatidic acid receptor 1 for development of arthritis. Arthritis Rheum (2013) 65(8):2037–47. doi: 10.1002/art.37991
91. Kim HJ, Ohk B, Yoon HJ, Kang WY, Seong SJ, Kim SY, et al. Docosahexaenoic acid signaling attenuates the proliferation and differentiation of bone marrow-derived osteoclast precursors and promotes apoptosis in mature osteoclasts. Cell Signal (2017) 29:226–32. doi: 10.1016/j.cellsig.2016.11.007
92. Funaki Y, Hasegawa Y, Okazaki R, Yamasaki A, Sueda Y, Yamamoto A, et al. Resolvin E1 inhibits osteoclastogenesis and bone resorption by suppressing IL-17-induced RANKL expression in osteoblasts and RANKL-induced osteoclast differentiation. Yonago Acta Med (2018) 61(1):8–18. doi: 10.33160/yam.2018.03.002
93. Li TF, Zuscik MJ, Ionescu AM, Zhang X, Rosier RN, Schwarz EM, et al. PGE2 inhibits chondrocyte differentiation through PKA and PKC signaling. Exp Cell Res (2004) 300(1):159–69. doi: 10.1016/j.yexcr.2004.06.019
94. Sun W, Ma J, Zhao H, Xiao C, Zhong H, Ling H, et al. Resolvin D1 suppresses pannus formation via decreasing connective tissue growth factor caused by upregulation of miRNA-146a-5p in rheumatoid arthritis. Arthritis Res Ther (2020) 22(1):61. doi: 10.1186/s13075-020-2133-2
95. Arai F, Miyamoto T, Ohneda O, Inada T, Sudo T, Brasel K, et al. Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-fms and receptor activator of nuclear factor kappaB (RANK) receptors. J Exp Med (1999) 190(12):1741–54. doi: 10.1084/jem.190.12.1741
96. Li J, Sarosi I, Yan XQ, Morony S, Capparelli C, Tan HL, et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci U.S.A. (2000) 97(4):1566–71. doi: 10.1073/pnas.97.4.1566
97. Kobayashi N, Kadono Y, Naito A, Matsumoto K, Yamamoto T, Tanaka S, et al. Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J (2001) 20(6):1271–80. doi: 10.1093/emboj/20.6.1271
98. Gravallese EM, Harada Y, Wang JT, Gorn AH, Thornhill TS, Goldring SR. Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. Am J Pathol (1998) 152(4):943–51.
99. Benabdoun HA, Kulbay M, Rondon EP, Vallières F, Shi Q, Fernandes J, et al. In vitro and in vivo assessment of the proresolutive and antiresorptive actions of resolvin D1: relevance to arthritis. Arthritis Res Ther (2019) 21(1):72. doi: 10.1186/s13075-019-1852-8
100. Crowson CS, Liao KP, Davis JM 3rd, Solomon DH, Matteson EL, Knutson K, et al. Rheumatoid arthritis and cardiovascular disease. Am Heart J (2013) 166(4):622–628.e1. doi: 10.1016/j.ahj.2013.07.010
101. Zhang M, Wang M, Tai Y, Tao J, Zhou W, Han Y, et al. Triggers of cardiovascular diseases in rheumatoid arthritis. Curr Probl Cardiol (2022) 47(6):100853. doi: 10.1016/j.cpcardiol.2021.100853
102. Boekholdt SM, Arsenault BJ, Mora S, Pedersen TR, LaRosa JC, Nestel PJ, et al. Association of LDL cholesterol, non-HDL cholesterol, and apolipoprotein b levels with risk of cardiovascular events among patients treated with statins: a meta-analysis. Jama (2012) 307(12):1302–9. doi: 10.1001/jama.2012.366
103. Chavan VU, Ramavataram D, Patel PA, Rupani MP. Evaluation of serum magnesium, lipid profile and various biochemical parameters as risk factors of cardiovascular diseases in patients with rheumatoid arthritis. J Clin Diagn Res (2015) 9(4):Bc01–5. doi: 10.7860/JCDR/2015/12206.5740
104. Dessie G. Association of atherogenic indices with c-reactive protein and risk factors to assess cardiovascular risk in rheumatoid arthritis patient at tikur anbessa specialized hospital, Addis Ababa. PloS One (2022) 17(6):e0269431. doi: 10.1371/journal.pone.0269431
105. Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American college of Cardiology/American heart association task force on clinical practice guidelines. J Am Coll Cardiol (2019) 73(24):e285–350. doi: 10.1016/j.jacc.2018.11.003
106. Zeng T, Tan L, Yu J, Wu Y. High density lipoprotein in rheumatoid arthritis: emerging role in predicting inflammation level and osteoporosis occurrence. Scand J Clin Lab Invest (2020) 80(5):375–80. doi: 10.1080/00365513.2020.1747109
107. Vivekanandan-Giri A, Slocum JL, Byun J, Tang C, Sands RL, Gillespie BW, et al. High density lipoprotein is targeted for oxidation by myeloperoxidase in rheumatoid arthritis. Ann Rheum Dis (2013) 72(10):1725–31. doi: 10.1136/annrheumdis-2012-202033
108. Bae SC, Lee YH. Associations between paraoxonase 1 (PON1) polymorphisms and susceptibility and PON1 activity in rheumatoid arthritis patients, and comparison of PON1 activity in patients and controls: a meta-analysis. Clin Rheumatol (2019) 38(8):2141–9. doi: 10.1007/s10067-019-04499-0
109. Alisik T, Alisik M, Nacir B, Ayhan FF, Genc H, Erel O. Evaluation of dysfunctional high-density lipoprotein levels with myeloperoxidase/paraoxonase-1 ratio in rheumatoid arthritis. Int J Clin Pract (2021) 75(7):e14172. doi: 10.1111/ijcp.14172
110. Toms TE, Panoulas VF, Kitas GD. Dyslipidaemia in rheumatological autoimmune diseases. Open Cardiovasc Med J (2011) 5:64–75. doi: 10.2174/1874192401105010064
111. García-Gómez C, Bianchi M, de la Fuente D, Badimon L, Padró T, Corbella E, et al. Inflammation, lipid metabolism and cardiovascular risk in rheumatoid arthritis: a qualitative relationship? World J Orthop (2014) 5(3):304–11. doi: 10.5312/wjo.v5.i3.304
112. Puig N, Montolio L, Camps-Renom P, Navarra L, Jiménez-Altayó F, Jiménez-Xarrié E, et al. Electronegative LDL promotes inflammation and triglyceride accumulation in macrophages. Cells (2020) 9(3):583. doi: 10.3390/cells9030583
113. Rhoads JP, Major AS. How oxidized low-density lipoprotein activates inflammatory responses. Crit Rev Immunol (2018) 38(4):333–42. doi: 10.1615/CritRevImmunol.2018026483
114. Voloshyna I, odayil S, Littlefield MJ, Belilos E, Belostocki K, Bonetti L, et al. Plasma from rheumatoid arthritis patients promotes pro-atherogenic cholesterol transport gene expression in THP-1 human macrophages. Exp Biol Med (Maywood) (2013) 238(10):1192–7. doi: 10.1177/1535370213503262
115. Ramharack R, Barkalow D, Spahr MA. Dominant negative effect of TGF-beta1 and TNF-alpha on basal and IL-6-induced lipoprotein(a) and apolipoprotein(a) mRNA expression in primary monkey hepatocyte cultures. Arterioscler Thromb Vasc Biol (1998) 18(6):984–90. doi: 10.1161/01.ATV.18.6.984
116. Rosenson RS, Brewer HB Jr, Davidson WS, Fayad ZA, Fuster V, Goldstein J, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation (2012) 125(15):1905–19. doi: 10.1161/CIRCULATIONAHA.111.066589
117. Hima Bindu G, Rao VS, Kakkar VV. Friend turns foe: transformation of anti-inflammatory HDL to proinflammatory HDL during acute-phase response. Cholesterol (2011) 2011:274629. doi: 10.1155/2011/274629
118. Soulaidopoulos S, Nikiphorou E, Dimitroulas T, Kitas GD. The role of statins in disease modification and cardiovascular risk in rheumatoid arthritis. Front Med (Lausanne) (2018) 5:24. doi: 10.3389/fmed.2018.00024
119. Mihaylova B, Emberson J, Blackwell L, Keech A, Simes J, Barnes EH, et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet (2012) 380(9841):581–90. doi: 10.1016/S0140-6736(12)60367-5
120. Akiyama M, Mawatari T, Nakashima Y, Miyahara H, Yamada H, Okazaki K, et al. Prevalence of dyslipidemia in Japanese patients with rheumatoid arthritis and effects of atorvastatin treatment. Clin Rheumatol (2015) 34(11):1867–75. doi: 10.1007/s10067-015-3049-0
121. Chen DY, Chih HM, Lan JL, Chang HY, Chen WW, Chiang EP. Blood lipid profiles and peripheral blood mononuclear cell cholesterol metabolism gene expression in patients with and without methotrexate treatment. BMC Med (2011) 9:4. doi: 10.1186/1741-7015-9-4
122. Medcalf MR, Bhadbhade P, Mikuls TR, O'Dell JR, Gundry RL, Funk RS. Plasma metabolome normalization in rheumatoid arthritis following initiation of methotrexate and the identification of metabolic biomarkers of efficacy. Metabolites (2021) 11(12):824. doi: 10.3390/metabo11120824
123. Maciejewski M, Sands C, Nair N, Ling S, Verstappen S, Hyrich K, et al. Prediction of response of methotrexate in patients with rheumatoid arthritis using serum lipidomics. Sci Rep (2021) 11(1):7266. doi: 10.1038/s41598-021-86729-7
124. Restrepo JF, Del Rincon I, Molina E, Battafarano DF, Escalante A. Use of hydroxychloroquine is associated with improved lipid profile in rheumatoid arthritis patients. J Clin Rheumatol (2017) 23(3):144–8. doi: 10.1097/RHU.0000000000000502
125. Morris SJ, Wasko MC, Antohe JL, Sartorius JA, Kirchner HL, Dancea S, et al. Hydroxychloroquine use associated with improvement in lipid profiles in rheumatoid arthritis patients. Arthritis Care Res (Hoboken) (2011) 63(4):530–4. doi: 10.1002/acr.20393
126. Araiza-Casillas R, Díaz-Molina R, González-Ortiz M, Robinson-Navarro OM. [Effects of hydroxychloroquine on insulin sensitivity and lipid profile in patients with rheumatoid arthritis]. Rev Med Chil (2013) 141(8):1019–25. doi: 10.4067/S0034-98872013000800008
127. Lin YJ, Anzaghe M, Schülke S. Update on the pathomechanism, diagnosis, and treatment options for rheumatoid arthritis. Cells (2020) 9(4):880. doi: 10.3390/cells9040880
128. Nam JL, Winthrop KL, van Vollenhoven RF, Pavelka K, Valesini G, Hensor EM, et al. Current evidence for the management of rheumatoid arthritis with biological disease-modifying antirheumatic drugs: a systematic literature review informing the EULAR recommendations for the management of RA. Ann Rheum Dis (2010) 69(6):976–86. doi: 10.1136/ard.2009.126573
129. Soubrier M, Jouanel P, Mathieu S, Poujol D, Claus D, Dubost JJ, et al. Effects of anti-tumor necrosis factor therapy on lipid profile in patients with rheumatoid arthritis. Joint Bone Spine (2008) 75(1):22–4. doi: 10.1016/j.jbspin.2007.04.014
130. Bili A, Morris SJ, Sartorius JA, Kirchner HL, Antohe JL, Dancea S, et al. Tumor necrosis factor-α inhibitor use is not associated with lipid changes in rheumatoid arthritis. J Rheumatol (2012) 39(5):946–8. doi: 10.3899/jrheum.111093
131. Ferraz Filho AC, dos Santos LP, Silva MB, Skare TL. Lipid profile and anti-TNF-α use. Rev Bras Reumatol (2013) 53(5):444–7. doi: 10.1590/S0482-50042013000500013
132. Jamnitski A, Visman IM, Peters MJ, Dijkmans BA, Voskuyl AE, Nurmohamed MT. Beneficial effect of 1-year etanercept treatment on the lipid profile in responding patients with rheumatoid arthritis: the ETRA study. Ann Rheum Dis (2010) 69(11):1929–33. doi: 10.1136/ard.2009.127597
133. Popa C, Netea MG, Radstake T, Van der Meer JW, Stalenhoef AF, van Riel PL, et al. Influence of anti-tumour necrosis factor therapy on cardiovascular risk factors in patients with active rheumatoid arthritis. Ann Rheum Dis (2005) 64(2):303–5. doi: 10.1136/ard.2004.023119
134. Kiortsis DN, Mavridis AK, Filippatos TD, Vasakos S, Nikas SN, Drosos AA. Effects of infliximab treatment on lipoprotein profile in patients with rheumatoid arthritis and ankylosing spondylitis. J Rheumatol (2006) 33(5):921–3.
135. Tam LS, Tomlinson B, Chu TT, Li TK, Li EK. Impact of TNF inhibition on insulin resistance and lipids levels in patients with rheumatoid arthritis. Clin Rheumatol (2007) 26(9):1495–8. doi: 10.1007/s10067-007-0539-8
136. Saiki O, Takao R, Naruse Y, Kuhara M, Imai S, Uda H. Infliximab but not methotrexate induces extra-high levels of VLDL-triglyceride in patients with rheumatoid arthritis. J Rheumatol (2007) 34(10):1997–2004.
137. Popa C, van Tits LJ, Barrera P, Lemmers HL, van den Hoogen FH, van Riel PL, et al. Anti-inflammatory therapy with tumour necrosis factor alpha inhibitors improves high-density lipoprotein cholesterol antioxidative capacity in rheumatoid arthritis patients. Ann Rheum Dis (2009) 68(6):868–72. doi: 10.1136/ard.2008.092171
138. van Sijl AM, Peters MJ, Knol DL, de Vet RH, Sattar N, Dijkmans BA, et al. The effect of TNF-alpha blocking therapy on lipid levels in rheumatoid arthritis: a meta-analysis. Semin Arthritis Rheum (2011) 41(3):393–400. doi: 10.1016/j.semarthrit.2011.04.003
139. Kirkham BW, Wasko MC, Hsia EC, Fleischmann RM, Genovese MC, Matteson EL, et al. Effects of golimumab, an anti-tumour necrosis factor-α human monoclonal antibody, on lipids and markers of inflammation. Ann Rheum Dis (2014) 73(1):161–9. doi: 10.1136/annrheumdis-2012-202089
140. Tanaka Y, Luo Y, O'Shea JJ, Nakayamada S. Janus kinase-targeting therapies in rheumatology: a mechanisms-based approach. Nat Rev Rheumatol (2022) 18(3):133–45. doi: 10.1038/s41584-021-00726-8
141. Chang CK, Chen PK, Chen CC, Chang SH, Chen CH, Chen DY. Increased levels of omega-3 fatty acids and DHA are linked to pain reduction in rheumatoid arthritis patients treated with janus kinase inhibitors. Nutrients (2021) 13(9):3050. doi: 10.3390/nu13093050
142. Philippou E, Nikiphorou E. Are we really what we eat? nutrition and its role in the onset of rheumatoid arthritis. Autoimmun Rev (2018) 17(11):1074–7. doi: 10.1016/j.autrev.2018.05.009
143. Sigaux J, Bellicha A, Buscail C, Julia C, Flipo RM, Cantagrel A, et al. Serum fatty acid profiles are associated with disease activity in early rheumatoid arthritis: results from the ESPOIR cohort. Nutrients (2022) 14(14):2947. doi: 10.3390/nu14142947
144. Vadell AKE, Bärebring L, Hulander E, Gjertsson I, Lindqvist HM, Winkvist A. Anti-inflammatory diet in rheumatoid arthritis (ADIRA)-a randomized, controlled crossover trial indicating effects on disease activity. Am J Clin Nutr (2020) 111(6):1203–13. doi: 10.1093/ajcn/nqaa019
145. Nelson J, Sjöblom H, Gjertsson I, Ulven SM, Lindqvist HM, Bärebring L. Do interventions with diet or dietary supplements reduce the disease activity score in rheumatoid arthritis? a systematic review of randomized controlled trials. Nutrients (2020) 12(10):2991. doi: 10.3390/nu12102991
146. Abdulrazaq M, Innes JK, Calder PC. Effect of ω-3 polyunsaturated fatty acids on arthritic pain: a systematic review. Nutrition (2017) 39-40:57–66. doi: 10.1016/j.nut.2016.12.003
147. Gan RW, Young KA, Zerbe GO, Demoruelle MK, Weisman MH, Buckner JH, et al. Lower omega-3 fatty acids are associated with the presence of anti-cyclic citrullinated peptide autoantibodies in a population at risk for future rheumatoid arthritis: a nested case-control study. Rheumatol (Oxford) (2016) 55(2):367–76. doi: 10.1093/rheumatology/kev266
148. Proudman SM, James MJ, Spargo LD, Metcalf RG, Sullivan TR, Rischmueller M, et al. Fish oil in recent onset rheumatoid arthritis: a randomised, double-blind controlled trial within algorithm-based drug use. Ann Rheum Dis (2015) 74(1):89–95. doi: 10.1136/annrheumdis-2013-204145
149. Kristensen S, Schmidt EB, Schlemmer A, Rasmussen C, Johansen MB, Christensen JH. Beneficial effect of n-3 polyunsaturated fatty acids on inflammation and analgesic use in psoriatic arthritis: a randomized, double blind, placebo-controlled trial. Scand J Rheumatol (2018) 47(1):27–36. doi: 10.1080/03009742.2017.1287304
150. Lourdudoss C, Di Giuseppe D, Wolk A, Westerlind H, Klareskog L, Alfredsson L, et al. Dietary intake of polyunsaturated fatty acids and pain in spite of inflammatory control among methotrexate-treated early rheumatoid arthritis patients. Arthritis Care Res (Hoboken) (2018) 70(2):205–12. doi: 10.1002/acr.23245
151. Di Giuseppe D, Wallin A, Bottai M, Askling J, Wolk A. Long-term intake of dietary long-chain n-3 polyunsaturated fatty acids and risk of rheumatoid arthritis: a prospective cohort study of women. Ann Rheum Dis (2014) 73(11):1949–53. doi: 10.1136/annrheumdis-2013-203338
152. Gan RW, Demoruelle MK, Deane KD, Weisman MH, Buckner JH, Gregersen PK, et al. Omega-3 fatty acids are associated with a lower prevalence of autoantibodies in shared epitope-positive subjects at risk for rheumatoid arthritis. Ann Rheum Dis (2017) 76(1):147–52. doi: 10.1136/annrheumdis-2016-209154
153. Lanchais K, Capel F, Tournadre A. Could omega 3 fatty acids preserve muscle health in rheumatoid arthritis? Nutrients (2020) 12(1):223. doi: 10.3390/nu12010223
154. Woodman RJ, Baghdadi LR, Shanahan EM, de Silva I, Hodgson JM, Mangoni AA. Diets high in n-3 fatty acids are associated with lower arterial stiffness in patients with rheumatoid arthritis: a latent profile analysis. Br J Nutr (2019) 121(2):182–94. doi: 10.1017/S0007114518003100
155. Gioxari A, Kaliora AC, Marantidou F, Panagiotakos DP. Intake of ω-3 polyunsaturated fatty acids in patients with rheumatoid arthritis: a systematic review and meta-analysis. Nutrition (2018) 45:114–124.e4. doi: 10.1016/j.nut.2017.06.023
156. Uauy R, Valenzuela A. Marine oils, in drugs and lactation database (LactMed®). Bethesda (MD: National Institute of Child Health and Human Development (2006).
157. Souza PR, Marques RM, Gomez EA, Colas RA, De Matteis R, Zak A, et al. Enriched marine oil supplements increase peripheral blood specialized pro-resolving mediators concentrations and reprogram host immune responses: a randomized double-blind placebo-controlled study. Circ Res (2020) 126(1):75–90. doi: 10.1161/CIRCRESAHA.119.315506
158. Jiang J, Li K, Wang F, Yang B, Fu Y, Zheng J, et al. Effect of marine-derived n-3 polyunsaturated fatty acids on major eicosanoids: a systematic review and meta-analysis from 18 randomized controlled trials. PloS One (2016) 11(1):e0147351. doi: 10.1371/journal.pone.0147351
159. Lourdudoss C, Wolk A, Nise L, Alfredsson L, Vollenhoven RV. Are dietary vitamin d, omega-3 fatty acids and folate associated with treatment results in patients with early rheumatoid arthritis? data from a Swedish population-based prospective study. BMJ Open (2017) 7(6):e016154. doi: 10.1136/bmjopen-2017-016154
160. Das Gupta AB, Hossain AK, Islam MH, Dey SR, Khan AL. Role of omega-3 fatty acid supplementation with indomethacin in suppression of disease activity in rheumatoid arthritis. Bangladesh Med Res Counc Bull (2009) 35(2):63–8. doi: 10.3329/bmrcb.v35i2.3020
161. Hemshekhar M, Anaparti V, El-Gabalawy H, Mookherjee N. A bioavailable form of curcumin, in combination with vitamin-d- and omega-3-enriched diet, modifies disease onset and outcomes in a murine model of collagen-induced arthritis. Arthritis Res Ther (2021) 23(1):39. doi: 10.1186/s13075-021-02423-z
162. Kucharská J, Poništ S, Vančová O, Gvozdjáková A, Uličná O, Slovák L, et al. Treatment with coenzyme Q10, omega-3-polyunsaturated fatty acids and their combination improved bioenergetics and levels of coenzyme Q9 and Q10 in skeletal muscle mitochondria in experimental model of arthritis. Physiol Res (2021) 70(5):723–33. doi: 10.33549/physiolres.934664
163. Jeffery L, Fisk HL, Calder PC, Filer A, Raza K, Buckley CD, et al. Plasma levels of eicosapentaenoic acid are associated with anti-TNF responsiveness in rheumatoid arthritis and inhibit the etanercept-driven rise in Th17 cell differentiation in vitro. J Rheumatol (2017) 44(6):748–56. doi: 10.3899/jrheum.161068
Keywords: rheumatoid arthritis, lipid metabolism, inflammation, joint destruction, treatment
Citation: Lei Q, Yang J, Li L, Zhao N, Lu C, Lu A and He X (2023) Lipid metabolism and rheumatoid arthritis. Front. Immunol. 14:1190607. doi: 10.3389/fimmu.2023.1190607
Received: 28 March 2023; Accepted: 17 May 2023;
Published: 31 May 2023.
Edited by:
Kerstin Klein, University Hospital Bern, SwitzerlandReviewed by:
Stanislav Kotlyarov, Ryazan State Medical University, RussiaKristina Zec, University of Oxford, United Kingdom
Copyright © 2023 Lei, Yang, Li, Zhao, Lu, Lu and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaojuan He, aHhqMTlAMTI2LmNvbQ==; Aiping Lu, YWlwaW5nbHVAaGtidS5lZHUuaGs=