 Abhinit Nagar
Abhinit Nagar Ravi Bharadwaj
Ravi Bharadwaj Mohammad Omar Faruk Shaikh
Mohammad Omar Faruk Shaikh Abhishek Roy
Abhishek Roy- 1Department of Flow Cytometry, Cytek Biosciences, Fremont, CA, United States
- 2MassBiologics of the University of Massachusetts Medical School, Boston, MA, United States
- 3Division of Critical Care Medicine, Department of Anesthesiology, Critical Care and Pain Medicine, Boston Children’s Hospital, Boston, MA, United States
- 4Department of Anesthesia, Harvard Medical School, Boston, MA, United States
- 5Department of Physiology and Biomedical Engineering, Mayo Clinic, Rochester, MN, United States
Speck assembly is the hallmark of NLRP3 inflammasome activation. The 1µm structure comprising of NLRP3 and ASC is the first observable phenotype of NLRP3 activation. While the common consensus is that the specks are the site of inflammasome activity, no direct experimental evidence exists to support this notion. In these 22 years, since the inflammasome discovery, several research studies have been published which directly or indirectly support or refute the idea of speck being the inflammasome. This review compiles the data from two decades of research to answer a long-standing question: “What are NLRP3-ASC specks?”
Introduction
Nod-like receptor protein containing pyrin 3 (NLRP3) is a cytosolic pathogen recognition receptor (PRR) predominantly expressed in myeloid cells (1, 2). It detects a wide range of chemically and structurally diverse stimuli and initiates an inflammatory response by forming multi-protein complexes called inflammasomes (1, 3, 4). Upon stimulation, NLRP3 recruits an adaptor protein called apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and pro-caspase-1. Caspase-1 activation leads to the processing of IL-1β, IL-18, and Gasdermin-D (GSDMD), which contribute to the inflammatory response against various threats (1, 3, 4). The inflammasome complexes, consisting of NLRs, ASC, and pro-caspase-1, are present in the high-molecular-weight (HMW) fraction of approximately 700 kDa (5–7). This suggests that the inflammasome may consist of 5-7 NLRP3-ASC-Casp1 units, similar to the apoptosome structure (8–10). Recent research by Xiao et al. revealed that NLRP3 forms a disk-like structure with 10 units, measuring around 32 nm (11). This configuration facilitates caspase-1 activation through proximity-induced interactions (10, 12–14).
Under normal conditions, NLRP3 and ASC are dispersed throughout the cytoplasm (15–17). However, upon NLRP3 activation, they rapidly relocate to form a perinuclear punctate structure known as the “speck” (15, 18). The speck, approximately 1 μm in diameter, can be visualized using fluorescence microscopy (9, 16, 17, 19). Speck formation concentrates the majority of inflammasome components and is crucial for the inflammasome response (20–23). Disruption of NLRP3:ASC interactions by mutations or small-molecule drugs impairs speck formation and inflammasome function (24–28). Colchicine, a drug that inhibits speck formation, is commonly used to treat inflammasome-related conditions like gout (4).
Although the speck is considered the site of inflammasome activity, its size is significantly larger than the observed size of an inflammasome (10, 17, 26). Additionally, some studies have shown caspase-1 activation in the absence of speck formation, challenging the notion that specks are inflammasomes (9, 17, 25, 29). Nonetheless, due to its rapid formation, recruitment of free NLRP3 and ASC, and its nature as a multi-protein complex, the speck is generally believed to represent the inflammasome.
This review aims to explore the research conducted in the field of inflammasomes, focusing on the nature and relevance of inflammasome-associated specks in the physiological functioning of the inflammasome. Recent studies have suggested that ASC specks exhibit prion-like polymerization and Supramolecular Organizing Center (SMOC)-like threshold properties, which enhance optimal inflammasome activation even with weaker stimuli. However, despite 22 years of inflammasome research, the direct experimental evidence supporting the speck as the inflammasome remains elusive, making it one of the biggest mysteries in inflammasome biology.
Inflammasome complex assembly
The relationship between speck assembly and NLRP3 activation remains complex, with multiple proposed models adding to the confusion (10, 14, 17, 20, 25–27, 30, 31). The precise mechanism by which NLRP3 and other inflammasome components assemble to form a functional multi-protein complex is still unclear. A comprehensive review by Elliot et al. has discussed the limitations of these models (8). Broadly, the proposed ideas can be categorized into two distinct models: the complex assembly model and the pre-assembled complex activation model.
The complex assembly model suggests that upon activation, NLRP3 recruits caspase-1 via ASC to form monomeric inflammasome units. These units then oligomerize through the Nucleotide Binding Domain (NBD) to create a functional inflammasome complex (Figure 1A). This model is supported by the observation that nearly all NLRP3 and ASC associate to form a singular speck. Furthermore, active NLRP3 co-immunoprecipitates with ASC and pro-caspase-1 (14, 27) and elutes with other components in complexes larger than 700 kDa (5, 6). In vitro studies have identified complexes smaller than a speck that contain caspase-1 (5, 6). However, whether these monomeric units further oligomerize to form a speck remains uncertain. Notably, these complexes exhibit different stoichiometry of monomeric units (11, 32), with mouse NLRP3 inflammasomes displaying a double-caged model of six units and human NLRP3 inflammasomes forming a disk shape composed of ten units (11). These species-specific differences raise questions about the unified activation of NLRP3 inflammasomes across different organisms and the function of complexes with varying monomeric units.
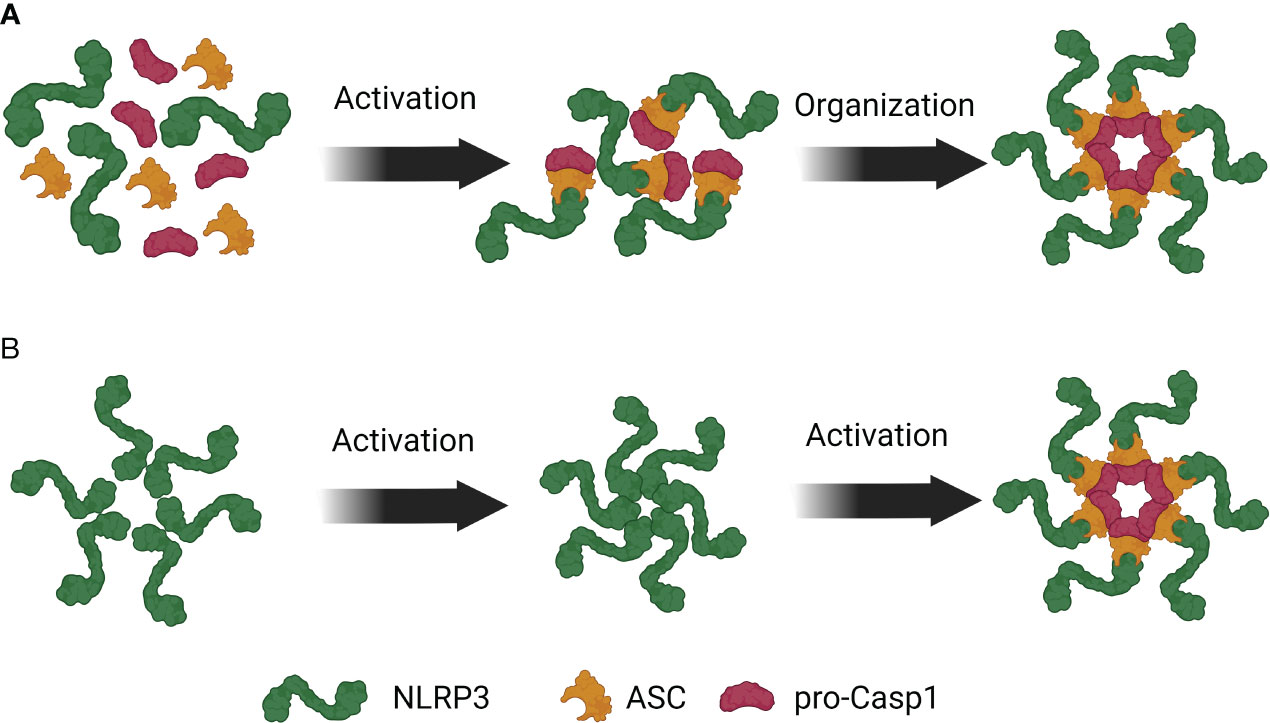
Figure 1 Competing models of NLRP3 inflammasome assembly. (A) Upon NLRP3 activation, ASC binds to NLRP3 and facilitates recruitment of pro-Caspase-1 to the complex. These trimeric complexes then oligomerize to form an inflammasome. (B) NLRP3 is present in the cytosol as a pre-formed inactive oligomeric complex where LRRs of NLRP3 are near the PYD. Activation induced structural changes in NLRP3 facilitate the recruitment of ASC and pro-Caspase-1 to the complex.
The counter-model proposes that a resting pre-assembled NLRP3 complex undergoes a conformational change upon activation and recruits ASC and pro-caspase-1 to the complex (Figure 1B). Under resting condition, NLRP1 and NLRP3 complexes elute in high-molecular-weight (HMW) complexes, indicating their presence in non-stimulated cells (5, 6). Bioluminescence Resonance Energy Transfer (BRET) experiments have shown that the leucine-rich repeats (LRRs) of NLRP3 are closely associated with the PYD domain, suggesting an interaction between them (33). This interaction maintains NLRP3 in an auto-repressed state that is disrupted upon ASC co-expression, indicating a basal level of NLRP3:ASC interaction (33). Furthermore, ASC PYD self-oligomerizes and can participate in pre-activation self-assembly (10, 18, 21, 26, 34).
The contrasting models highlight the complexity of the relationship between inflammasome activation and assembly. Importantly, these observations are made at different stages of inflammasome activation and in various cell types. Therefore, it is likely that both models hold some truth, and multiple states of inflammasome assembly and NLRP3 activation can coexist simultaneously.
Speck assembly
Speck assembly: structural dynamics and kinetics
Specks are perinuclear structures formed upon NLRP3 activation (5, 15, 35) and have recently been classified as supramolecular signaling complexes (SMOCs) (31, 36). SMOCs are higher order complexes that promote cooperative assembly of signaling components through weak allosteric interactions, enabling high local concentrations of these components (31, 36–38). SMOC assembly is tightly regulated and plays a role in threshold behavior, temporal and spatial control of signal transduction, and binary all-or-none responses (31, 36–38). However, the specific regulatory mechanisms involved in SMOC-mediated inflammasome activation remain poorly understood. The assembly of SMOCs involves prion-like polymerization, which lowers the Gibb’s free energy (ΔG) of a protein (21, 23, 26, 30), potentially altering protein conformation and kinetics (31, 36–38). Speck formation follows the SMOC assembly mechanism, indicating that specks enable inflammasome proteins to adopt a conformation that efficiently senses the activation signal (20, 22, 26, 39). This efficient signaling may contribute to the threshold and all-or-none nature of inflammasome responses.
While NLRP3 and ASC are unequivocally present within the speck, the presence of other components is still a subject of debate. In THP1 cells, NLRP3 and ASC oligomerizes under three minutes to form a speck making it an incredibly rapid process (35). Similarly, in HeLa cells, the concentration of cytosolic ASC drops from 20μM to around 0.1μM within 100 seconds (34). Similar observations have been made in other cell types like BMDMs and HEK293T cells (40, 41). Interestingly, the kinetics of speck formation differ in vivo. In Zebrafish, speck size stabilizes after a continuous growth period of 15 minutes (42). The advantages of such rapid protein oligomerization are not yet clear. Additionally, the morphology of the speck is a topic of discussion. Early studies suggested a hollow to fibrillar structure composed of small protein complexes (15, 18, 43), but recent studies using high-resolution microscopy and simulation models propose that the speck assembly consists of intertwined ASC filaments (37, 38).
These discrepancies may arise from various factors, including experimental conditions, cell types, and organisms. However, the differences highlight our current limitations in understanding speck formation, the regulation of speck assembly, the presence of a single speck per cell, and the mechanisms governing these processes. Some studies have reported the formation of multiple specks per cell (16, 44, 45), suggesting the existence of conditions and mechanisms that promote the formation of more than one speck. Investigating these conditions and mechanisms would be intriguing for further research.
Speck formation: the molecular view
Speck assembly is characterized traditionally as a microtubule-driven process (31, 36–38, 44, 46, 47). During infection and injury, PAMPs and DAMPs induce damage to the mitochondria which binds to ASC. Damaged-mitochondria laden ASC is transported towards the endoplasmic reticulum (ER) through microtubules, where NLRP3 is located (44). NLRP3 and ASC being in proximity interacts and colocalize to form one cytosolic perinuclear speck of 1 µm (35, 48). Microtubule depolymerization agents like colchicine and nocodazole block speck assembly further supporting the idea that microtubules facilitate speck assembly (35, 48). Consistent with speck’s proximity to centrosome, movement and activation of NLRP3 is dependent upon binding with two centrosomal proteins, Microtubule-affinity regulating kinase 4 (MARK4) (47) and NEK7, respectively (49–53). However, some studies suggest that NLRP3 is not localized exclusively on the centrosome (54) but also interacts with mitochondria (44, 55–62), Golgi apparatus (63, 64) and redox-associated proteins like TXNIP (62, 65). Thus, whether NLRP3 interaction with the centrosome is essential is still unclear. These differences can be the result of using different cell types and stimulation of NLRP3 by different agonists. It is likely that NLRP3 shuttles between different organelles or a sub-population of NLRP3 binds to one organelle while another sub-population binds to other organelle. However, it is still not clear what signals would determine shuttling from or the preference to bind one organelle to another. Recently few papers have shown the role of membrane association of NLRP3 in inflammasome formation. Membrane-bound NLRP3 promotes ASC oligomerization and inflammasome activation (63). Moreover, NLRP3 forms a double-ring cage structure facilitated by LRR interaction. Such LRR interactions are also required to disperse Trans-Golgi network (TGNs) into vesicles (63). In these studies, they have also revealed that a fraction of membrane unbound NLRP3 is also present in the cells which constitute the inactive form of NLRP3.
Specks as the site of inflammasome activity
NLRP3 activation results in rapid relocation of NLRP3 and ASC to form a speck, which is followed by caspase-1 activation suggesting that speck formation is required for inflammasome formation (1, 9, 16, 17, 20, 66). The idea that specks are needed for the inflammasomes stems from several observations. Firstly, speck formation is the first event which happens within 3 minutes of NLRP3 activation (19, 39, 40, 67, 68) and thus precedes inflammasome formation. Since caspase-1 is activated through proximity induced dimerization and only one speck is formed per cell, it is likely that caspase-1 activation occurs inside the speck (10, 12, 14). Secondly, small molecule drug, like MCC950, that inhibit specks also impair the NLRP3 inflammasome response (24, 28) suggesting that speck formation is necessary for inflammasome activation. MCC950 inhibit speck formation, specifically block NLRP3 inflammasomes in human and mouse macrophages and alleviate symptoms of Cryopyrin Associate Periodic Syndrome (CAPS) in a mouse model. Further, colchicine is used to treat gout (69) and apart from blocking speck formation (44), it also impairs IL-1β maturation after MSU stimulation (70). This finding suggests that speck assembly is required for inflammasome formation and/or possibly can be the site of inflammasome function, i.e., caspase-1 activation and subsequent IL-1β maturation. Thirdly, mutations that disrupt speck assembly also impair inflammasome function (25–27) further implicating that speck assembly is required for inflammasome function. Although indirect, but these observations strongly suggest that speck is the inflammasome. Conversely, these observations also suggest that preventing specks blocks the downstream events leading to inflammasome formation. Lastly, few studies have noted that active caspase-1 colocalizes with NLRP3 and ASC at the speck structure (2, 71) providing direct evidence of presence of caspase-1 at the speck. Several other tangential evidence exists that can be extrapolated to say that speck is the inflammasome, but none of such evidence directly establishes any such relationship.
Disparity between speck structure and inflammasomes
Contradictory to the idea that specks are inflammasomes, some observations suggests that specks may have some different roles. The reported size and stoichiometry of specks differ significantly from known inflammasome structures (10, 11, 20, 23, 26, 35, 39, 72, 73). Specks are approximately 100-1000-fold larger and display variations in the ratio of NLR, ASC, and Caspase-1 compared to other inflammasome complexes (10, 15, 71, 74). Discrepancies in stoichiometry (10, 26) can be attributed to the different proteins used in the studies, where full-sized NLRPs and ASC were used by Faustin et al. (10), while Lu et al. and Sborgi et al. used isolated PYD and CARD domains of ASC (26, 74). The absence of the CARD domain leads to uninterrupted filamentous PYD structures, as observed in these studies (26, 74). These differences in size and composition suggest that specks and inflammasomes are structurally distinct.
Interestingly, caspase-1 activity has also been observed in smaller death complexes that do not resemble specks (17, 25, 75). Moreover, Martinon et al., in their initial paper describing inflammasomes, passed cell lysates through a 0.45 µm filter, excluding the 1 µm specks (6). However, the filtered lysates still activated caspase-1 (6), suggesting that structures smaller than 0.45 µm can function as inflammasomes. Furthermore, specks are 500-1000-fold larger than the cryoelectron micrograph of inflammasomes (10, 26). Calculations based on size and mass relationships suggest that specks, assumed to be toroidal structures, would be approximately 150-15000 times larger than an inflammasome (76). These assumptions may vary depending on the torus’s major-to-minor radius ratio and the assumption of a perfect toroidal structure. While smaller structures resembling inflammasomes have been observed inside specks (26), their similarity to cryoelectron micrograph structures is yet to be established. Further investigations utilizing high-resolution microscopy and advanced biophysical techniques are needed to confirm the presence of these smaller structures in cells.
It is possible that specks are conglomerate structures composed of smaller inflammasomes, which are the sites of caspase-1 activity. Upon NLRP3 activation, these smaller structures may leave the speck, resulting in reduced speck size (2, 17). Interestingly, the gross-morphology of the specks is unaffected by the presence of caspase-1 suggesting that caspase-1 is either not an integral part of the speck or its recruitment is temporally regulated further complicating the relationship between these two structures (2, 17). The presence of caspase-1 within specks remains inconclusive, with some studies demonstrating caspase-1 colocalization while others suggesting only a fraction of active caspase-1 is localized within the speck (2, 71). Even in studies using super-resolution microscopy (71), point-spread function analysis suggests that the ring of the speck which is mostly ASC is in focus, whereas the center which is comprised of active caspase-1 is spread-out above and below the plane suggesting that only a fraction of active caspase-1 may be colocalized. Additionally, speck formation alone is not sufficient for inflammasome function, as specks lacking NLRP3 fail to activate caspase-1 or promote IL-1β maturation (9, 29). Studies have shown that higher doses of stimuli, even in the presence of colchicine, can still activate the inflammasome without inducing speck formation (17, 77). These phenotypes have been observed in in-vitro conditions and its physiological relevance are yet to be investigated. It is not clear how such high stimuli conditions manifest in a more relevant physiological condition. It is also possible that higher conditions doses alter the expression levels of inflammasome components which can account for different threshold behavior.
Abovementioned observations, in addition to the presence of multiple caspase-1 activating complexes within cells (17, 25, 75), highlight the complexity of inflammasome activation. Mutations disrupting speck formation without impairing IL-1β production (25), multiple smaller complexes instead of a single speck (44), and multiple caspase-1 activation sites (17) beyond the speck have been observed. Active caspase-1 was found to have low colocalization with specks (16, 17, 45), and live imaging showed caspase-1 activation throughout the cytoplasm rather than in a speck-like location (78). These findings suggest the presence of smaller death-complexes and non-speck locations for caspase-1 activation, challenging the notion that specks are the exclusive site of inflammasome activity.
In summary, conflicting evidence challenges the idea that specks are inflammasomes. The structural and functional distinctions between specks and inflammasomes, the presence of smaller inflammasome-like structures within specks, and the observation of caspase-1 activity outside the speck all contribute to the complexity and ambiguity surrounding the relationship between specks and inflammasome activation. However, it is crucial to conduct these studies under more physiological conditions to minimize potential variations resulting from in vitro stimulation. Further research using advanced techniques and physiological settings is required to unravel these intricate mechanisms.
Discussion
The role of specks in inflammasome function remains a subject of debate and requires further investigation. While some researchers propose that specks are the site of caspase-1 activation and inflammasome activity, others suggest that specks may serve as a critical node in the inflammasome activation pathway (Reviewed in Elliot et. al.) (8) (Figure 2). To answer the question of what NLRP3-ASC specks truly are, additional experiments utilizing advanced technologies such as imaging flow cytometry (16, 17, 79–81), cryo-electron microscopy, super-resolution microscopy (16, 17, 79–81), BRET and proximity ligation assays (33, 44) are needed.
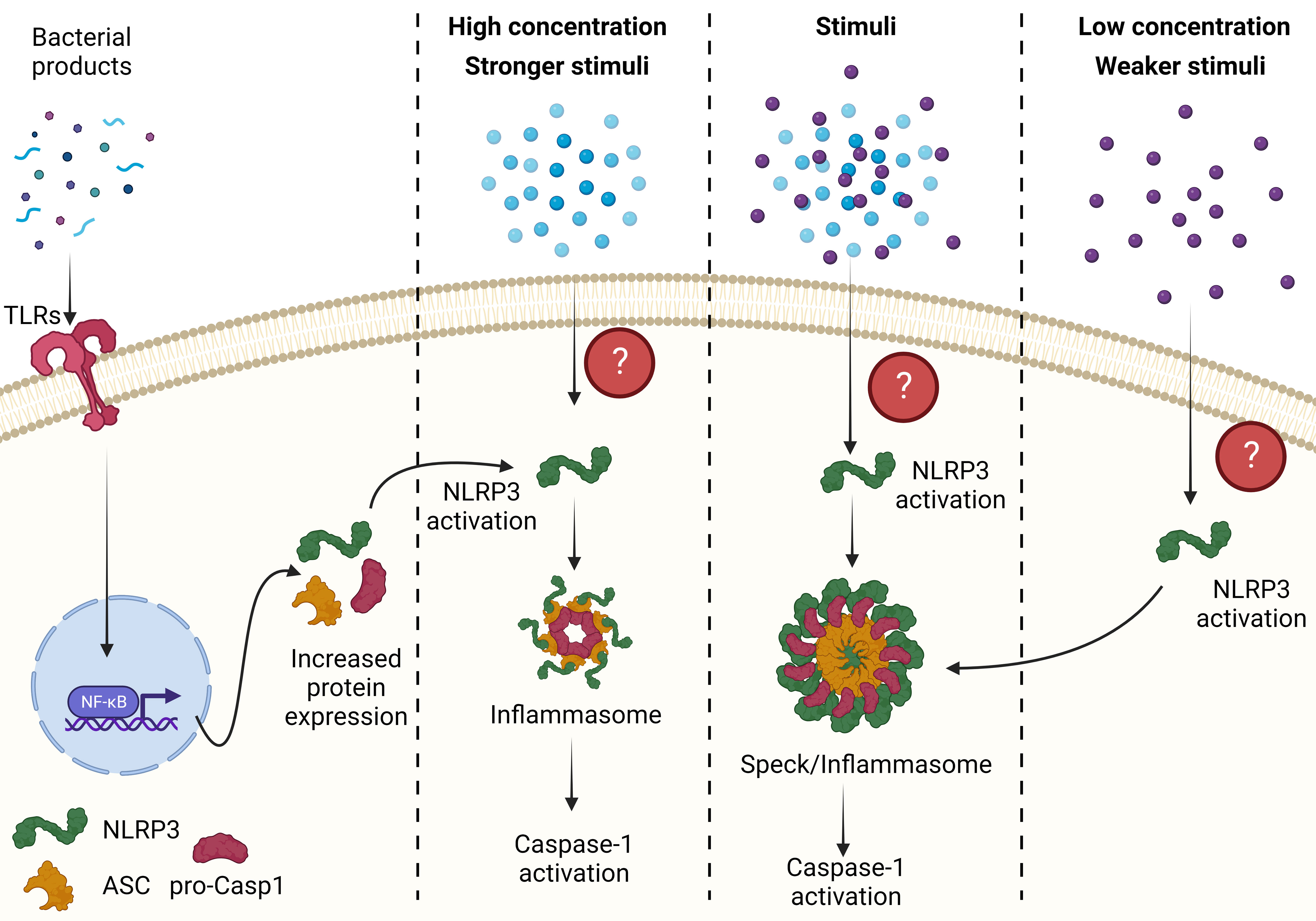
Figure 2 The relationship between an ASC speck and inflammasome. A weaker stimulus or a lower concentration of stimuli induces speck formation, which lower the threshold of NLRP3 activation possibly through prion-like folding of NLRP3, ASC and pro-caspase-1 and formation of smaller inflammasomes. These smaller inflammasome complex are the major activation site of caspase-1. However, in the presence of stronger stimulus or higher concentration of the stimuli, NLRP3 activation does not require prion-like folding and can assemble with ASC and caspase-1 to form inflammasomes. Conversely, if specks are the sole site of caspase-1 activation then any stimuli leading to NLRP3 activation forms a speck where caspase-1 is activated. All figures are drawn using BioRender.
Studying specks is essential despite caspase-1 activation occurring outside the speck and the presence of smaller death complexes. Investigating specks helps unravel their molecular composition, organization, and role in caspase-1 activation and regulation, shedding light on inflammasome signaling dynamics. Simultaneously, understanding alternative activation sites such as smaller death complexes provide insights into their composition, formation mechanisms, and regulatory roles. Examining the spatial and temporal regulation of caspase-1 activity under different cellular conditions enhances our comprehension of inflammasome signaling complexity. Re-evaluating existing literature in light of current knowledge can identify gaps in inflammasome assembly and ASC speck understanding. In addition to investigating the inflammasome-associated role of ASC specks, it is equally important to explore potential non-inflammasome functions of these structures. To truly comprehend NLRP3-ASC specks, advanced techniques are necessary to evaluate colocalization, modulate activation thresholds, and investigate non-inflammasome functions. These efforts will advance our understanding of specks and their significance in inflammasome biology.
Author contributions
AN conceptualized and wrote the manuscript. AN, RB, MS and AR edited the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
AN is employed at Cytek Biscience Inc. which have the rights to manufacture and sell AMNIS ISX and FlowSight cited in the paper.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol (2013) 13(6):397–411. doi: 10.1038/nri3452
2. Stein R, Kapplusch F, Heymann MC, Russ S, Staroske W, Hedrich CM, et al. Enzymatically Inactive Procaspase 1 stabilizes the ASC Pyroptosome and Supports Pyroptosome Spreading during Cell Division. J Biol Chem (2016) 291(35):18419–29. doi: 10.1074/jbc.M116.718668
3. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
4. Banerjee SK, Chatterjee A, Gupta S, Nagar A. Activation and regulation of NLRP3 by sterile and infectious insults. Front Immunol (2022) 13:896353. doi: 10.3389/fimmu.2022.896353
5. Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity (2004) 20(3):319–25. doi: 10.1016/S1074-7613(04)00046-9
6. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10(2):417–26. doi: 10.1016/S1097-2765(02)00599-3
7. Martinon F, Tschopp J. NLRs join TLRs as innate sensors of pathogens. Trends Immunol (2005) 26(8):447–54. doi: 10.1016/j.it.2005.06.004
8. Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev (2015) 265(1):35–52. doi: 10.1111/imr.12286
9. Rahman T, Nagar A, Duffy EB, Okuda K, Silverman N, Harton JA. NLRP3 sensing of diverse inflammatory stimuli requires distinct structural features. Front Immunol (2020) 11:1828. doi: 10.3389/fimmu.2020.01828
10. Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell (2007) 25(5):713–24. doi: 10.1016/j.molcel.2007.01.032
11. Xiao L, Magupalli VG, Wu H. Cryo-EM structures of the active NLRP3 inflammasome disc. Nature (2023) 613(7944):595–600. doi: 10.1038/s41586-022-05570-8
12. Salvesen GS, Dixit VM. Caspase activation: the induced-proximity model. Proc Natl Acad Sci U S A (1999) 96(20):10964–7. doi: 10.1073/pnas.96.20.10964
13. Shi Y. Caspase activation: revisiting the induced proximity model. Cell (2004) 117(7):855–8. doi: 10.1016/j.cell.2004.06.007
14. Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med (2018) 215(3):827–40. doi: 10.1084/jem.20172222
15. Masumoto J, Taniguchi S, Ayukawa K, Sarvotham H, Kishino T, Niikawa N, et al. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem (1999) 274(48):33835–8. doi: 10.1074/jbc.274.48.33835
16. Nagar A, DeMarco RA, Harton JA. Inflammasome and caspase-1 activity characterization and evaluation: an imaging flow cytometer-based detection and assessment of inflammasome specks and caspase-1 activation. J Immunol (2019) 202(3):1003–15. doi: 10.4049/jimmunol.1800973
17. Nagar A, Rahman T, Harton JA. The ASC speck and NLRP3 inflammasome function are spatially and temporally distinct. Front Immunol (2021) 12:752482. doi: 10.3389/fimmu.2021.752482
18. Richards N, Schaner P, Diaz A, Stuckey J, Shelden E, Wadhwa A, et al. Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. J Biol Chem (2001) 276(42):39320–9. doi: 10.1074/jbc.M104730200
19. Stutz A, Horvath GL, Monks BG, Latz E. ASC speck formation as a readout for inflammasome activation. Methods Mol Biol (2013) 1040:91–101. doi: 10.1007/978-1-62703-523-1_8
20. Hoss F, Rodriguez-Alcazar JF, Latz E. Assembly and regulation of ASC specks. Cell Mol Life Sci (2017) 74(7):1211–29. doi: 10.1007/s00018-016-2396-6
21. Sahillioglu AC, Sumbul F, Ozoren N, Haliloglu T. Structural and dynamics aspects of ASC speck assembly. Structure (2014) 22(12):1722–34. doi: 10.1016/j.str.2014.09.011
22. Gambin Y, Giles N, O'Carroll A, Polinkovsky ME, Johnston W, Hunter DJ, et al. Single-molecule fluorescence reveals the oligomerization and folding steps driving the prion-like behavior of ASC. J Mol Biol (2018) 430(4):491–508. doi: 10.1016/j.jmb.2017.12.013
23. Cai X, Chen J, Xu H, Liu S, Jiang QX, Halfmann R, et al. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell (2014) 156(6):1207–22. doi: 10.1016/j.cell.2014.01.063
24. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med (2015) 21(3):248–55. doi: 10.1038/nm.3806
25. Dick MS, Sborgi L, Ruhl S, Hiller S, Broz P. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat Commun (2016) 7:11929. doi: 10.1038/ncomms11929
26. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell (2014) 156(6):1193–206. doi: 10.1016/j.cell.2014.02.008
27. Vajjhala PR, Mirams RE, Hill JM. Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J Biol Chem (2012) 287(50):41732–43. doi: 10.1074/jbc.M112.381228
28. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med (2015) 21(3):263–9. doi: 10.1038/nm.3804
29. Compan V, Martin-Sanchez F, Baroja-Mazo A, Lopez-Castejon G, Gomez AI, Verkhratsky A, et al. Apoptosis-associated speck-like protein containing a CARD forms specks but does not activate caspase-1 in the absence of NLRP3 during macrophage swelling. J Immunol (2015) 194(3):1261–73. doi: 10.4049/jimmunol.1301676
30. Cai X, Chen ZJ. Prion-like polymerization as a signaling mechanism. Trends Immunol (2014) 35(12):622–30. doi: 10.1016/j.it.2014.10.003
31. Kagan JC, Magupalli VG, Wu H. SMOCs: supramolecular organizing centres that control innate immunity. Nat Rev Immunol (2014) 14(12):821–6. doi: 10.1038/nri3757
32. Andreeva L, David L, Rawson S, Shen C, Pasricha T, Pelegrin P, et al. NLRP3 cages revealed by full-length mouse NLRP3 structure control pathway activation. Cell (2021) 184(26):6299–312 e22. doi: 10.1016/j.cell.2021.11.011
33. Compan V, Baroja-Mazo A, Lopez-Castejon G, Gomez AI, Martinez CM, Angosto D, et al. Cell volume regulation modulates NLRP3 inflammasome activation. Immunity (2012) 37(3):487–500. doi: 10.1016/j.immuni.2012.06.013
34. Cheng J, Waite AL, Tkaczyk ER, Ke K, Richards N, Hunt AJ, et al. Kinetic properties of ASC protein aggregation in epithelial cells. J Cell Physiol (2010) 222(3):738–47. doi: 10.1002/jcp.22005
35. Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ (2007) 14(9):1590–604. doi: 10.1038/sj.cdd.4402194
36. Kagan JC. Signaling organelles of the innate immune system. Cell (2012) 151(6):1168–78. doi: 10.1016/j.cell.2012.11.011
37. Wu H. Higher-order assemblies in a new paradigm of signal transduction. Cell (2013) 153(2):287–92. doi: 10.1016/j.cell.2013.03.013
38. Wu H, Fuxreiter M. The structure and dynamics of higher-order assemblies: amyloids, signalosomes, and granules. Cell (2016) 165(5):1055–66. doi: 10.1016/j.cell.2016.05.004
39. Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The adaptor ASC has extracellular and 'prionoid' activities that propagate inflammation. Nat Immunol (2014) 15(8):727–37. doi: 10.1038/ni.2913
40. Sester DP, Thygesen SJ, Sagulenko V, Vajjhala PR, Cridland JA, Vitak N, et al. A novel flow cytometric method to assess inflammasome formation. J Immunol (2015) 194(1):455–62. doi: 10.4049/jimmunol.1401110
41. Loo YM, Gale M Jr. Immune signaling by RIG-I-like receptors. Immunity (2011) 34(5):680–92. doi: 10.1016/j.immuni.2011.05.003
42. Kuri P, Schieber NL, Thumberger T, Wittbrodt J, Schwab Y, Leptin M. Dynamics of in vivo ASC speck formation. J Cell Biol (2017) 216(9):2891–909. doi: 10.1083/jcb.201703103
43. McConnell BB, Vertino PM. Activation of a caspase-9-mediated apoptotic pathway by subcellular redistribution of the novel caspase recruitment domain protein TMS1. Cancer Res (2000) 60(22):6243–7.
44. Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol (2013) 14(5):454–60. doi: 10.1038/ni.2550
45. Nagar A, Harton JA. Flow imaging of the inflammasome: evaluating ASC speck characteristics and caspase-1 activity. Methods Mol Biol (2023) 2635:185–202. doi: 10.1007/978-1-0716-3020-4_11
46. Elliott JM, Rouge L, Wiesmann C, Scheer JM. Crystal structure of procaspase-1 zymogen domain reveals insight into inflammatory caspase autoactivation. J Biol Chem (2009) 284(10):6546–53. doi: 10.1074/jbc.M806121200
47. Li X, Thome S, Ma X, Amrute-Nayak M, Finigan A, Kitt L, et al. MARK4 regulates NLRP3 positioning and inflammasome activation through a microtubule-dependent mechanism. Nat Commun (2017) 8:15986. doi: 10.1038/ncomms15986
48. Bryan NB, Dorfleutner A, Rojanasakul Y, Stehlik C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol (2009) 182(5):3173–82. doi: 10.4049/jimmunol.0802367
49. He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci (2016) 41(12):1012–21. doi: 10.1016/j.tibs.2016.09.002
50. He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature (2016) 530(7590):354–7. doi: 10.1038/nature16959
51. Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, et al. A genome-wide CRISPR (Clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J Biol Chem (2016) 291(1):103–9. doi: 10.1074/jbc.C115.700492
52. Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature (2019) 570(7761):338–43. doi: 10.1038/s41586-019-1295-z
53. Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol (2016) 17(3):250–8. doi: 10.1038/ni.3333
54. Wang Y, Yang C, Mao K, Chen S, Meng G, Sun B. Cellular localization of NLRP3 inflammasome. Protein Cell (2013) 4(6):425–31. doi: 10.1007/s13238-013-2113-2
55. Adinolfi E, Callegari MG, Ferrari D, Bolognesi C, Minelli M, Wieckowski MR, et al. Basal activation of the P2X7 ATP receptor elevates mitochondrial calcium and potential, increases cellular ATP levels, and promotes serum-independent growth. Mol Biol Cell (2005) 16(7):3260–72. doi: 10.1091/mbc.e04-11-1025
56. Gross CJ, Mishra R, Schneider KS, Medard G, Wettmarshausen J, Dittlein DC, et al. K(+) efflux-independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity (2016) 45(4):761–73. doi: 10.1016/j.immuni.2016.08.010
57. Ichinohe T, Yamazaki T, Koshiba T, Yanagi Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc Natl Acad Sci U S A (2013) 110(44):17963–8. doi: 10.1073/pnas.1312571110
58. Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity (2013) 39(2):311–23. doi: 10.1016/j.immuni.2013.08.001
59. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity (2012) 36(3):401–14. doi: 10.1016/j.immuni.2012.01.009
60. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell (2013) 153(2):348–61. doi: 10.1016/j.cell.2013.02.054
61. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature (2018) 560(7717):198–203. doi: 10.1038/s41586-018-0372-z
62. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469(7329):221–5. doi: 10.1038/nature09663
63. Chen J, Chen ZJ. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature (2018) 564(7734):71–6. doi: 10.1038/s41586-018-0761-3
64. Zhang Z, Meszaros G, He WT, Xu Y, de Fatima Magliarelli H, Mailly L, et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J Exp Med (2017) 214(9):2671–93. doi: 10.1084/jem.20162040
65. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol (2010) 11(2):136–40. doi: 10.1038/ni.1831
66. Agrawal I, Jha S. Comprehensive review of ASC structure and function in immune homeostasis and disease. Mol Biol Rep (2020) 47(4):3077–96. doi: 10.1007/s11033-020-05345-2
67. Franklin BS, Latz E, Schmidt FI. The intra- and extracellular functions of ASC specks. Immunol Rev (2018) 281(1):74–87. doi: 10.1111/imr.12611
68. Baroja-Mazo A, Martin-Sanchez F, Gomez AI, Martinez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol (2014) 15(8):738–48. doi: 10.1038/ni.2919
69. Dalbeth N, Lauterio TJ, Wolfe HR. Mechanism of action of colchicine in the treatment of gout. Clin Ther (2014) 36(10):1465–79. doi: 10.1016/j.clinthera.2014.07.017
70. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature (2006) 440(7081):237–41. doi: 10.1038/nature04516
71. Man SM, Hopkins LJ, Nugent E, Cox S, Gluck IM, Tourlomousis P, et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U S A (2014) 111(20):7403–8. doi: 10.1073/pnas.1402911111
72. Hafner-Bratkovic I, Bencina M, Fitzgerald KA, Golenbock D, Jerala R. NLRP3 inflammasome activation in macrophage cell lines by prion protein fibrils as the source of IL-1beta and neuronal toxicity. Cell Mol Life Sci (2012) 69(24):4215–28. doi: 10.1007/s00018-012-1140-0
73. Shi F, Yang Y, Kouadir M, Fu Y, Yang L, Zhou X, et al. Inhibition of phagocytosis and lysosomal acidification suppresses neurotoxic prion peptide-induced NALP3 inflammasome activation in BV2 microglia. J Neuroimmunol (2013) 260(1-2):121–5. doi: 10.1016/j.jneuroim.2013.04.016
74. Sborgi L, Ravotti F, Dandey VP, Dick MS, Mazur A, Reckel S, et al. Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo-electron microscopy. Proc Natl Acad Sci U S A (2015) 112(43):13237–42. doi: 10.1073/pnas.1507579112
75. Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe (2010) 8(6):471–83. doi: 10.1016/j.chom.2010.11.007
76. Yoon S-H. Membrane Bioreactor Processes: Principles and Applications. Boca Raton, FL: Taylor and Francis/ CRC Press (2015).
77. Gao W, Yang J, Liu W, Wang Y, Shao F. Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc Natl Acad Sci U S A. (2016) 113(33):E4857–66. doi: 10.1073/pnas.1601700113
78. Liu T, Yamaguchi Y, Shirasaki Y, Shikada K, Yamagishi M, Hoshino K, et al. Single-cell imaging of caspase-1 dynamics reveals an all-or-none inflammasome signaling response. Cell Rep (2014) 8(4):974–82. doi: 10.1016/j.celrep.2014.07.012
79. La Rosa F, Saresella M, Marventano I, Piancone F, Ripamonti E, Al-Daghri N, et al. Stavudine reduces NLRP3 inflammasome activation and modulates amyloid-beta autophagy. J Alzheimers Dis (2019) 72(2):401–12. doi: 10.3233/JAD-181259
80. La Rosa F, Zoia CP, Bazzini C, Bolognini A, Saresella M, Conti E, et al. Modulation of MAPK- and PI3/AKT-dependent autophagy signaling by stavudine (D4T) in PBMC of alzheimer's disease patients. Cells (2022) 11(14). doi: 10.3390/cells11142180
Keywords: ASC specks, SMOCs, Casp-1, NLRP3, inflammasome
Citation: Nagar A, Bharadwaj R, Shaikh MOF and Roy A (2023) What are NLRP3-ASC specks? an experimental progress of 22 years of inflammasome research. Front. Immunol. 14:1188864. doi: 10.3389/fimmu.2023.1188864
Received: 17 March 2023; Accepted: 12 July 2023;
Published: 26 July 2023.
Edited by:
Haichao Wang, Feinstein Institute for Medical Research, United StatesReviewed by:
Francesca La Rosa, Fondazione Don Carlo Gnocchi Onlus (IRCCS), ItalyThirumala-Devi Kanneganti, St. Jude Children’s Research Hospital, United States
Copyright © 2023 Nagar, Bharadwaj, Shaikh and Roy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abhinit Nagar, YW5hZ2FyQGN5dGVrYmlvLmNvbQ==