
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 30 May 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1188365
This article is part of the Research Topic Community Series in Combinational Immunotherapy of Cancer: Novel Targets, Mechanisms, and Strategies, volume II View all 9 articles
 Desheng Qi1,2
Desheng Qi1,2 Milin Peng2,3*
Milin Peng2,3*Cell death is a universal biological process in almost every physiological and pathological condition, including development, degeneration, inflammation, and cancer. In addition to apoptosis, increasing numbers of cell death types have been discovered in recent years. The biological significance of cell death has long been a subject of interest and exploration and meaningful discoveries continue to be made. Ferroptosis is a newfound form of programmed cell death and has been implicated intensively in various pathological conditions and cancer therapy. A few studies show that ferroptosis has the direct capacity to kill cancer cells and has a potential antitumor effect. As the rising role of immune cells function in the tumor microenvironment (TME), ferroptosis may have additional impact on the immune cells, though this remains unclear. In this study we focus on the ferroptosis molecular network and the ferroptosis-mediated immune response, mainly in the TME, and put forward novel insights and directions for cancer research in the near future.
The homeostasis of living entities is delicately regulated by the cell death of damaged cells and survival of properly working cells in a spatio-temporal manner. Cell death can take the form of either non-programmed cell death or programmed cell death (PCD). Necrosis is a non-programmed form of cell death that is induced by repeated freezing and thawing or other forms of physical tissue damage, and was identified as early as 1951 (1). Studies on cell death have increased in the past 50 years following the discovery of apoptosis (2). It has been found that apoptosis is closely involved in numerous biological conditions (3, 4), which is the first category of programmed cell death and is regulated by caspases. Previous researchers have generally considered apoptosis and necrosis to be the only cell death types of interest. However, this understanding has been challenged and many scientific discoveries in last two decades have brought to light new forms for cell death (5–8). Necroptosis is another fundamental programmed cell death and is found in the universal biological conditions of development, inflammation, and cancer (9–11), and pyroptosis is a Gasdermin D (GSDMD)-mediated PCD that is implicated in inflammation, immunity, and sepsis and other diseases (12).
Ferroptosis is a newly identified type of iron-dependent non-apoptotic cell death that was discovered in the last 10 years by Stockwell (7, 13, 14). By reviewing its discovery history, we can obtain a comprehensive understanding of ferroptosis. First, a novel compound, erastin, was identified to selectively kill engineered cancer cells expressing oncogenic RAS and small T oncoprotein in a non-apoptotic manner through compounds screening for engineered human tumor cells (13). This first discovery led to much more comprehensive studies. Interestingly, using the synthetic lethal screening modality in vitro, Stockwell found that the introduction of oncogenic RAS into cancer cells leads to increased levels of cellular labile iron pool by upregulating transferrin receptor gene TfR1 and downregulating ferritin genes FTH and FTL. This non-apoptotic cell death is inhibited by iron chelator, which indicates that it is iron-dependent. In addition, two other compounds, RSL3 and RSL5, were identified as inducers of this novel form of cell death. RSL5 and erastin both lead to this cell death by targeting voltage-dependent anion channel 3 (VDAC3), whereas RSL3 induces death in a VDAC3-independent manner (14). VDACs are mitochondrial pores that transport small metabolites and ions. Furthermore, ferroptosis is named to represent this iron-dependent non-apoptotic cell death after one more key study (7), in which ferroptosis was proven to have unique morphological, bioenergetic, and genetic traits that are distinct from previous identified types of cell death. Cells undergoing ferroptosis have the morphological features of small and shrunken mitochondria under electron microscope scan. In terms of bioenergetics, ferroptosis does not consume much intracellular adenosine triphosphate (ATP) nor rely on mitochondrial electron transport chain (ETC) to generate reactive oxygen species (ROS). Lipid peroxidation was found to be another characteristic of ferroptosis. As for genetic features, six high-confidence genes were found to significantly change in cells undergoing ferroptosis. These were IREB2 (iron response element binding protein 2), RPL8 (ribosomal protein L8), CS (citrate synthase), ATP5G3 (ATP synthase F0 complex subunit C3), ACSF2 (acyl-CoA synthetase family member 2), and TTC35 (tetratricopeptide repeat domain 35) (7). There are other landmarks in the history of ferroptosis. Glutathione peroxidases 4 (GPX4), a key component of keeping intracellular reducing state, plays a crucial role in the regulation of ferroptosis (15). P53, a canonical tumor suppressor, sensitizes ferroptotic cancer cell death (16). BAP1, another tumor suppressor, also sensitizes ferroptotic tumor cell death by repressing the amino acid antiporter system Xc- expression, which is important for synthesizing cellular reduced glutathione (GSH) to protect lipids from peroxidation and ferroptosis (17). Thus, ferroptosis is closely associated with conventional tumor suppressors’ activities and is an endogenous mechanism that represses the survival of tumor cells. Inversely, tumor cells utilize various mechanisms to evade ferroptosis, such as upregulating ferroptosis suppressor protein 1 (FSP1) (18). In addition, prominin 2 promotes ferritin-containing exosomes to transport iron out of cells and protects cancer cells from ferroptosis stress (19). Primary tumor growth relies on the inhibition of endogenous ferroptosis. Drug-resistant and highly mesenchymal cancer cells depend on GPX4 activity to repress ferroptosis and survive (20, 21). Furthermore, immunotherapy-activated CD8+ T cells induces the ferroptosis of tumor cells by secreting interferon gamma (IFN-γ) and inhibiting system Xc- on the cell membrane (22). Radiotherapy also directly induces the ferroptosis of tumor cells, which is also a novel junction of synergy between radiotherapy and immunotherapy (23). Recently, it was also found that dihydroorotate dehydrogenase (DHODH) is an alternative way to protect against ferroptosis aside from GPX4 and FSP1 (24). All the above milestones comprise a sketch of the discovery history of ferroptosis (Figure 1).
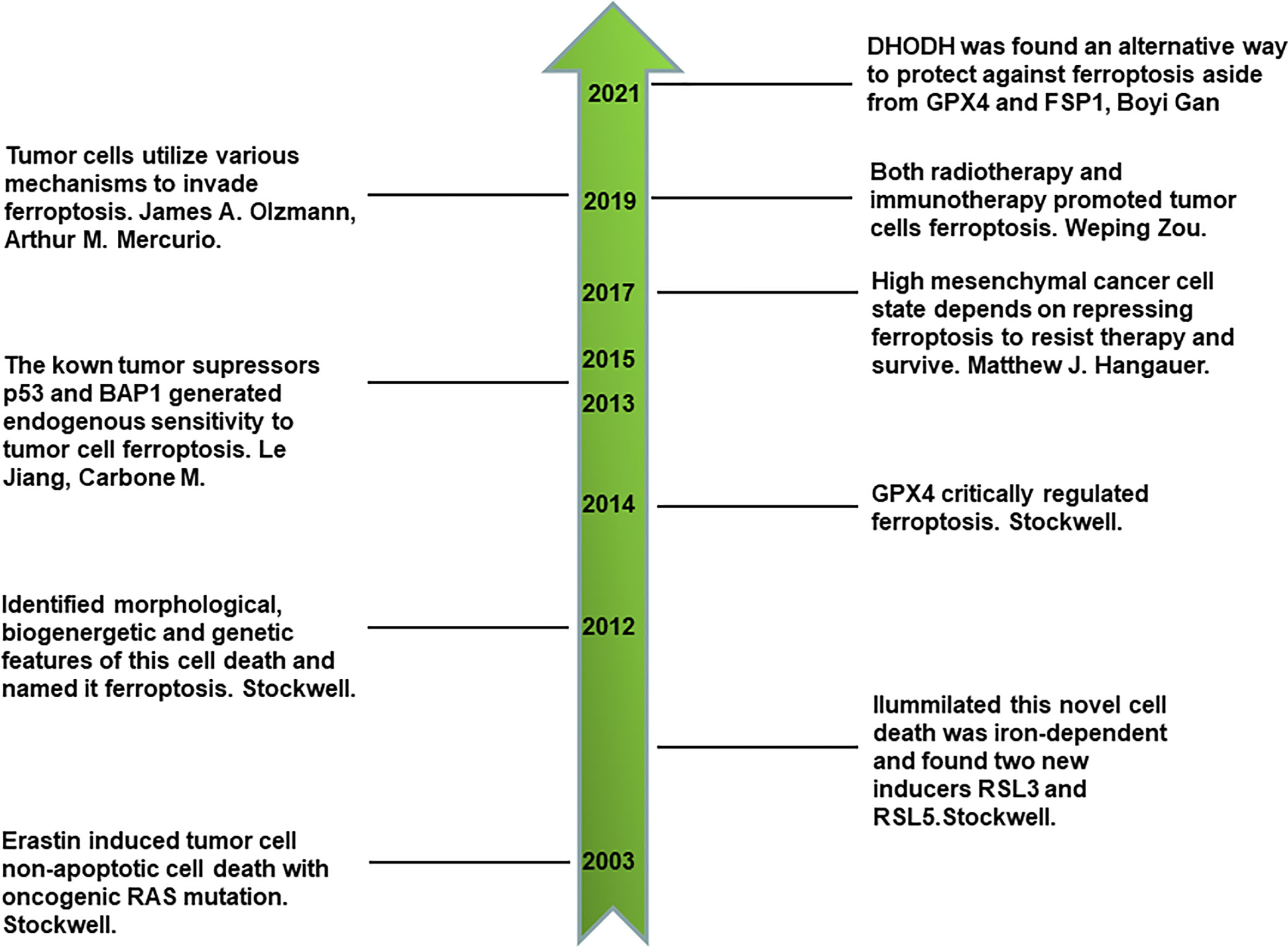
Figure 1 Milestones in the discovery history of ferroptosis.
From the above introduction to the discovery history of ferroptosis, we can conclude that ferroptosis is an iron-dependent programmed form of cell death that is induced by lipid peroxidation and the breaking of the intracellular reducing state maintained by amino acid and antioxidation metabolism. Thus, ferroptosis is by nature a form of metabolic cell death mainly involved in iron metabolism, lipid metabolism, and amino acid and antioxidation metabolism. In general, iron overload can induce the Fenton reaction and lead to the significant accumulation of oxygen free radicals in cells. When the oxidizability induced by oxygen free radical overpowers the reducing state that is mainly maintained by GPX4 and system Xc- activity, arachidonate lipoxygenases (ALOXs) are activated and turn polyunsaturated fatty acids (PUFAs) into peroxidative lipids; this is known as lipid peroxidation. Lipid peroxidation can lead to cell plasma membrane remodeling and perforation, after which cell normal structure is destroyed. Finally, ferroptosis occurs (Figure 2).
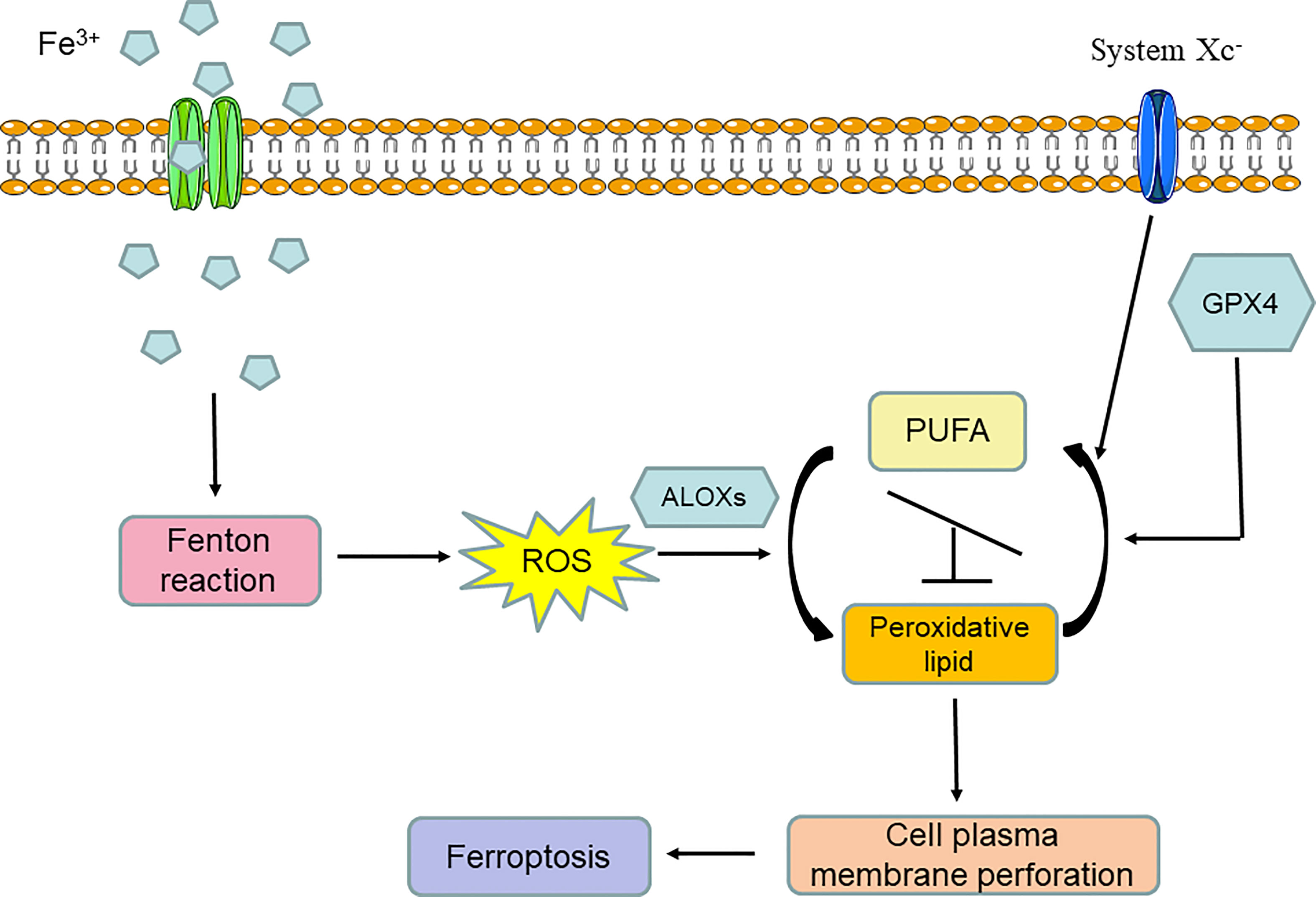
Figure 2 Molecular network of ferroptosis.
As shown above, iron overload is the critical initial procedure for ferroptosis, and iron metabolism is heavily involved in the cancer microenvironment. Malignant cells rely on high levels of intracellular iron to fulfill the biosynthesis of DNA and proliferation (25). However, accumulated iron in tumor cells is a double-edged sword, as excessive labile iron in cytoplasm can also elicit the Fenton reaction, which induces lipid peroxidation and lipid toxicity (26). Iron-dependent lipid peroxidation can generate an overload in the levels of oxygen free radicals, which eventually leads to ferroptosis. A number of genes and proteins implicated in iron metabolism have been reported to regulate ferroptosis. When inducing ferroptosis, the iron-starvation response is activated and results in the downregulation of ferritin (genes: FTH1 and FTL; function: store intracellular iron) and ferroportin (function: export iron out of the cell), and the upregulation of transferrin receptor (gene: TfR1; function: import iron into the cell) (14), which leads to the accumulation of intracellular free iron pool. The iron–sulfur cluster biosynthetic enzyme NFS1 maintains the iron–sulfur co-factors level and serves as an upstream regulator to control the iron-starvation response and limit the effect of high levels of oxygen tension in a lung adenocarcinoma context. NFS1 suppression sensitizes lung adenocarcinoma cells to ferroptosis by stimulating the iron-starvation response, stabilizing TfR1 mRNA and inhibiting FTH1 expression (27). Another critical mediator is nuclear receptor coactivator 4 (NCOA4), which is required for transporting ferritin to lysosomes to degrade, resulting in the release of ferritin-bound iron to increase intracellular iron levels (28). Heme oxygenase-1 (HO-1), a main source of intracellular iron, has been proven to play a decisive role in erastin-induced ferroptosis, and the inhibition of HO-1 prevents erastin-triggered ferroptosis in HT-1080 fibrosarcoma cells (29). However, in hepatocellular carcinoma cells (HCCs), the knockdown of HO-1 by small interfering RNA facilitates ferroptosis induced by sorafenib and erastin (30). Thus, the role of HO-1 in ferroptosis is still in debate and is specific to different cells. In addition, the p62-Keap1-nuclear factor erythroid 2-related factor 2 (NRF2) pathway serves as an upstream modulator regulating HO-1 and FTH1 stability and protecting HCCs from ferroptosis (30). Fanconi anemia complementation group D2 (FANCD2) also protects bone marrow stromal cells from ferroptosis by regulating the genes or proteins in iron metabolism (31). Divalent metal transporter 1 (DMT1) is another key regulator involved in iron metabolism and it has four isoforms. DMT1A1, one of the isoforms, is localized on the apical cell membrane and has the function of non-transferritin-bound ferrous iron uptake. DMT1A/B-II, localized at the endosomal membrane, imports ferrous iron from endosome to cytosol (32). Importantly, human poly (rC)–binding protein 1 (PCBP1) acts as an iron chaperone that delivers ferrous iron to ferritin for storage in the cytoplasm (33), which can protect cells from labile ferrous iron-related oxidative damage. However, the relationship between DMT1, PCBPs, and ferroptosis has not yet been explored and needs further study. Phosphorylase kinase G2 (PHKG2) is another mediator of cellular labile iron pool tha facilitates ferroptosis (34). In a word, these markers in iron metabolism probably have a close relationship with ferroptosis and can be utilized for further investigation.
Iron-associated Fenton reaction is the catalyzer for ALOXs activity, resulting in lipid peroxidation and ferroptosis. In addition, PUFAs are the specific substrates for this biochemical reaction. The bis-allylic position in PUFAs is the critical site for the production of peroxidation and determines the radical chain reaction of lipid peroxidation (34). Acyl-CoA synthetase long chain family member 4 (ACSL4) and lyso-phosphatidylcholine acyltransferase (LPCAT) play important roles in the esterification of PUFAs into membrane phospholipids, which are the key signals for the stimulation of ferroptosis (35, 36). PUFAs insert into cell membrane phospholipids and formulate PUFAs-containing membrane phospholipids, such as phosphatidylethanolamines (PEs) containing arachidonic acid (C20:4) and adrenic acid (C22:4) (36). Recently, polyunsaturated ether phospholipid (PUFA-ePL) was found to be another important substrate for lipid peroxidation reaction, eventually leading to ferroptosis (37). PUFAs-containing PE phospholipids on cell membranes are oxidated by the ALOXs family (34, 36, 38). There are six ALOX genes in the human ALOXs family: ALOXE3, ALOX5, ALOX12, ALOX12B, ALOX15, and ALOX15B. These six genes have different expression levels in various cell lines and tissues. The human renal carcinoma cell line G-401 expresses all six ALOX genes. The silencing of all six ALOX genes drives G-401 cell resistance to ferroptosis inducer imidazole ketone erastin (IKE) treatment rather than (1S, 3R)-RSL3 stimulation (34). IKE inhibits system Xc- on cell membrane to exhaust cellular GSH and inhibits GPX4 indirectly (like erastin function), whereas (1S, 3R)-RSL3 suppresses GPX4 activity directly in the absence of cellular GSH exhaustion (39). BJeLR or HT-1080 cell line does not express the ALOX5, ALOX12, ALOX12B, or ALOX15 genes but does express ALOXE3 and ALOX15B. The silencing of ALOXE3 and ALOX15B in the BJeLR or HT-1080 cell lines rescues erastin-induced ferroptosis, which also confirms that ferroptosis that occurs after GSH exhaustion requires ALOXs (34). In addition, ALOX12 and ALOX15 are specific downstream markers of ferroptosis that work by activating apoptosis-inducing factor (AIF). Cell death is completely prevented by inhibiting ALOX12 and ALOX15, or silencing AIF (38). The detailed role of AIF in ferroptosis needs further study. Supplement of PUFAs together with transforming growth factor-β1 (TGF-β1) synergistically inhibit B16 cell growth, and this effect can be completely reversed by the antioxidant vitamin E, but adding PUFAs alone does not inhibit B16 cell growth (40). As vitamin E is a potential anti-ferroptosis agent (41, 42), further study is needed to confirm whether TGF-β1-mediated B16 cell growth inhibition and enhancement by PUFAs is due to the inducement of ferroptosis. In addition, mevalonate metabolism is implicated in ferroptosis pathway (43). However, the total net effect of mevalonate pathway on ferroptosis is still undetermined and requires further study (43, 44).
The intracellular reducing state is an aspect of homeostasis that is crucial to the maintenance of normal function in living beings. Superoxide anion radical (O2•–) and hydrogen peroxide (H2O2) are the two main types of reactive oxygen species (ROS) (45). Oxidized lipid is another important kind of ROS. The influences of ROS on inflammation, cancer, senescence, and other pathologic conditions are prevailing and vital (46–48). In cells, there are certain mechanisms that regulate ROS levels. Amino acid metabolism, especially that of cystine and glutathione, has the key function of controlling ROS and maintaining an antioxidant state in normal cells. The cystine and glutathione metabolic pathway under normal circumstances is as follows. System xc– on the cell membrane is made up of the disulfide-linked heterodimers SLC3A2 and xCT and serves as the bidirectional channel to transfer cystine into cells and export intracellular glutamate out at a 1:1 ratio. Subsequently, intracellular cystine and glutamate are synthesized into GSH by glutamate-cysteine ligase (GCL) and glutathione synthetase (GSS). Some cells take advantage of the transsulfuration pathway to produce cysteine from methionine, thus bypassing the system xc– function of importing cysteine. GSH is the cofactor of GPX4 in its reduction of phospholipid hydroperoxides to phospholipids, which is the unique function of GPX4 (49). GPX4, a member of the glutathione peroxidases family (GPXs), is the core mediator to maintaining an antioxidant state in cells. The disruption in the above antioxidant mechanism by inhibiting key targets such as system xc–, GCL, and GPX4 generates the accumulation of ROS and lipid peroxides, leading to ferroptosis (7, 15). GPX4 is the key regulator in the ferroptosis pathway (15). By using 60 different human cells in the NCI-60 cell line panel, it was found that intracellular nicotinamide adenine dinucleotide phosphate (NADPH) abundance is negatively correlated with ferroptosis sensitivity (50). All ROS are generated by mitochondrial electron transport chain (ETC) complexes and NADPH oxidases. While in ferroptosis, cells eliminated from the mitochondria still retain their sensitivity to ferroptosis, indicating that ROS generated by mitochondria are dispensable for inducing ferroptosis (7). However, inhibiting ETC complexes in mitochondria blocks ferroptosis induced by system xc– inhibition, but not by GPX4 suppression (51). The true underlying mechanism remains obscure, and the role of mitochondria in ferroptosis is still undetermined and, furthermore, more comprehensive studies are needed. In addition, the inhibition of thioredoxin reductase (TXNRD) activity induces ferroptosis through upregulating Ptgs2 expression (52). In a word, amino acid and antioxidant metabolism, and associated key regulators, are closely related to ferroptosis.
Glucose metabolism has a close correlation with ferroptosis. The tricarboxylic acid (TCA) cycle, the fundamental form of glucose metabolism, has close links with ferroptosis. The mitochondrial TCA cycle and ETC complexes promote cysteine-deprivation-induced (CDI) ferroptosis by inducing oxidative phosphorylation (OXPHOS) and producing cellular lipid peroxides (51). Glutaminolysis is also deeply implicated in the regulation of ferroptosis through its replenishing of TCA cycle intermediates such as α-ketoglutaric acid (α-KG). Glutamine is absorbed in cells by SLC1A5 transporters on the cell membrane. Subsequently, glutamine is transformed to glutamate by glutaminase (GLS1) in the cytoplasm. Glutamine can also enter mitochondria and form glutamate by glutaminase (GLS2). Glutamate in mitochondria can be turned into and subsequently supply α-ketoglutaric acid (α-KG) for TCA cycle through its reaction with glutamic-oxaloacetic transaminase 1 (GOT1). Suppressing glutamine uptake by the SLC1A5 transporter, or inhibiting glutamine transformation to glutamate by GLS2, or restraining glutamate conversion to a-KG in mitochondria, all inhibit ferroptosis (53). In addition, as we know, fumarate hydratase (FH) is an enzyme in the TCA cycle that catalyzes the transformation of fumarate into malate and has the function of suppressing cancer and the DNA damage response pathway (DDR). Intriguingly, FH-inactivated renal cell carcinoma (HLRCC) exhibits oxidative stress promotion, and metabolic alteration, and increased sensitivity to ferroptosis through the mechanisms of inducing fumarate accumulation, the succination of GPX4 proteins at the C94 site, and the inhibition of GPX4 function (54). On the other hand, FH-inactivated HLRCC also demonstrates the fumarate accumulation-related attenuation of NRF2 degradation (54), which is the notable protective factor in the inhibition of ferroptosis (30, 55, 56). The net effect of ferroptosis on FH-inactivated HLRCC is still unknown, and further, more comprehensive studies are needed to determine the impact of fumarate hydratase on ferroptosis in other cancers. This entry point is interesting and reflects the in-depth correlation between glycometabolism and ferroptosis through post-translation modulation in the form of succination, which is a promising future research direction. In addition to, FH as cancer suppressor gene involved in cancer cell ferroptosis, two other cancer suppressor genes, p53 and BAP1, have the direct function of repressing the expression of system xc-, inhibiting cystine uptake, and sensitizing cancer cells to ferroptosis (16, 57, 58). In a word, cancer suppressor genes utilize ferroptosis, by means of glycometabolism modification, as a critical endogenous way of inhibiting tumor growth.
Ferroptosis has not only a complex molecular network but also vital functions in many pathologic conditions, including acute kidney injury, cardiomyopathy, traumatic nerve injury, and neurodegenerative diseases (59–62). In addition, as ferroptosis was firstly discovered using high-throughput compounds screening for killing tumor cells (13), it has been intensely studied by those in the tumor research field over the years. Some intractable cancer cells that are resistant to apoptosis, necroptosis, pyroptosis, and other cell death types, have been found to be susceptible to ferroptosis. A therapy-resistant state, depending on the lipid peroxidase pathway, can be induced in cancer cells by ferroptosis (21), meaning that ferroptosis is potentially a viable way to cure cancer. This topic can be dissected into two main aspects; one is the effect of ferroptosis on tumor cells themselves, and the other is the role of ferroptosis in the tumor microenvironment (TME). The first aspect has been continuously explored since the discovery of ferroptosis. In subcutaneous xenograft mouse tumor models, Stockwell found that the ferroptosis inducer (1S, 3R)-RSL3 can suppress tumor growth in athymic nude mice (15). Another study showed that IKE, a ferroptosis inducer, exerted an antitumor effect in an immunodeficient mouse lymphoma model (63). However, recent research has demonstrated that the compound of ferroptocide can induce ferroptosis in an immunocompetent murine model of breast cancer, but not in immunodeficient mice (64). Zou reported that cyst(e)inase alone, an ferroptosis inductive agent, could not suppress ovarian cancer ID8 growth, but that the combination of cyst(e)inase and anti-PD-L1 could inhibit ID8 development (22). These studies indicate that ferroptosis has the direct effect of killing tumors and also of activating an antitumor immune microenvironment. On the other hand, many kinds of tumor cells are equipped with various defense mechanisms to withstand ferroptosis, such as transporting iron out of cells by way of exosomes, or halting lipid peroxidation by way of ferroptosis suppressor protein 1 (FSP1) (18, 19). However, the metabolic vulnerability of tumors provides therapy opportunities (65), and drug-tolerant persistent cancer cells rely on GPX4 activity to resist treatment (20). Thus, inhibiting GPX4 to induce ferroptosis is considered to be a useful means of taking advantage of this “Achilles heel” to inhibit cancer progression. Although there is increasing interest in utilizing ferroptosis to kill tumor cells directly and suppress tumor growth, the role of ferroptosis in the TME remains obscure. In consideration of the pivotal role of TME in cancer progression, the aim of this review is to investigate the potential impact of ferroptosis on TME, in order to highlight a crucial and valuable field for future investigation.
The role of ferroptosis in the TME was first explored in cell death-related immunological studies. From the first and second Cold Spring Harbor Symposium and the notable work of immunologist CA Janeway, Jr., a more comprehensive and profound conception of immunity from an evolutionary perspective was put forward, in particular Burnet’s crucial clonal selection theory, which posited that the first signal of class I major histocompatibility complex molecules (MHC-I) was their interaction with T-cell receptors, that the second signal was a co-stimulator to activate T cells, and that the initial signal of pattern recognition receptors (PRRs) to recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) by antigen-presenting cells (APCs) was also vitally important for immune system function (66). Cross-priming is the prevailing and critical model used by APCs, mainly dendritic cells (DCs), to process extrinsic protein antigens and present to T cells through MHC-I (67). The self/non-self model of immunity comprises the microbial non-self, missing self, and altered self (68). The microbial non-self contains extrinsic invading pathogens, and in the missing self contains cells with decreased expressions of MHC-1, CD47, or sialic acid, which are “do not eat me” signals. Dying cells belong to the altered self and release DAMPs, which are “find me” and “eat me” signals that either stimulate or suppress immune responses. In addition, there is an emerging theory in the field of cancer research regarding the association of dying cancer cells with immune responses through their mimicry of pathogen defense responses. Immunogenic dying cancer cells exhibit behavior that is similar to pathogen-infected dying cells, on the one hand releasing chemokines such as CXCL1, CCL2, and CXCL10 (pathogen response-like chemokine signature, PARC) that recruit neutrophils as first responders to phagocytose dying cells. On the other hand, dying cancer cells release DAMPs, including ATP, nucleic acids that can be recognized by APCs, activates neutrophils and eventually facilitates nitric oxide-based respiratory burst and hydrogen peroxide release, which causes the death of residual viable cancer cells (68, 69). In this way, the altered self-mimicry of dying cells can be perceived by the innate immune cells to clear residual dying cells. Numerous clinical studies have shown that T cell-infiltrated tumors respond sensitively to immunotherapy (70, 71), whereas in non-T cell-infiltrated tumors, other means of maximally expediting innate immune activation in the TME are required (72). Immunogenic cell death (ICD) was found to have an antitumor capacity by activating immune response (73–76) and is an emerging determinant in various clinical cancer therapeutic modalities, including radiation therapy, chemotherapy, molecular targeted therapy, and immunotherapy (77–81). Whether or not ferroptosis has an immunoregulatory effect in the TME is still being explored. The immune system is composed of two main parts: innate immunity and adaptive immunity. The components of innate immunity consist of physical/chemical barriers, humoral innate immunity, and inherent immune cells (82). External protective structures, external mucous secretions, and specialized skin and mucosa form physical/chemical barriers. Lysozymes, antimicrobial peptides, acute phase proteins, complements, cytokines, and natural antibodies make up humoral innate immunity. Intraepithelial T lymphocytes, myeloid phagocytic cells, innate lymphoid cells, phagocytic B cells, non-specific cytotoxic cells, and mucosa-associated invariant T cells are types of inherent immune cells (82). Innate immunity works as the first guard to kill invading pathogens and mutant cancer cells by phagocytosis, extracellular traps, and other mechanisms, and is therefore critical to the maintenance of homeostasis. The innate immune system is also contains sentry cells that recognize the “not me” signal, and processing and presenting the alien antigens to adaptive immune cells. Efforts have been made to explore the effect of ferroptosis on innate immunity. Keratinocytes, the protective shelters of skin, show features of cell death including ferroptosis in vitro and in vivo by specifically knocking out the glutamate-cysteine ligase gene in keratinocytes (83). Macrophages are the main drivers of innate immunity. An in vitro study shows that when human leukemic Jurkat T cells were induced to ferroptosis and then co-cultured with macrophages, this led to the exposure of phosphatidylserine being detected on the surface of dying cells, and enhanced phagocytosis by macrophages (84). Ferroptosis can promote the release of KRASG12D protein from pancreatic ductal adenocarcinoma cells (PDAC). KRASG12D protein is then delivered to macrophages by exosomes and stimulates macrophage polarization to the M2 pro-tumor type (85). A recent study discovered that proteoglycan decorin (DCN), as the novel DAMP released by cells dying from ferroptosis, can trigger the production of inflammatory macrophage cytokines and elicit an anticancer immune response (86). In addition, the knockdown of MXRA8 can elicit ferroptosis in glioma cells and decrease the infiltration of M2 macrophages (87). One study indirectly proved that the inhibition of ferroptosis might prevent macrophage polarization to the M1 pro-inflammatory type and limit the release of TNF-α and IL-1β in folic acid-induced kidney injury conditions (88). In addition, one study found that M1 type macrophages exert high resistance to pharmacologically induced ferroptosis, whereas M2 type macrophages are sensitive to ferroptosis in TME conditions and brain trauma under regulation by inducible nitric oxide synthase (iNOS)/NO• (89). In general, ferroptosis may result in enhanced phagocytosis and clearance abilities of macrophages, and the increase of the M1/M2 type macrophage ratio in some conditions, which is a desirable outcome for improving the TME packed with immunosuppressive M2 type macrophages.
As for other types of innate immune cells, one study demonstrates that photosensor photodynamic therapy (PS-PDT) induced the immunogenic cell death of glioma GL261 and fibrosarcoma MCA205 cells. This process was inhibited by ferrostatin-1 and DFO (ferroptosis inhibitors), which indicated that ferroptosis was involved in PS-PDT-induced cell death. After co-culturing, PS-PDT-induced dying cells were more effectively engulfed by bone marrow-derived DCs (BMDCs) than live cells, and BMDCs were efficiently engulfed by BMDCs. And BMDCs were then matured and activated of surface co-stimulatory molecule CD86, MHCII expression, and releasing higher interleukin-6 (IL-6) (90). The results indicate that cells undergoing ferroptosis promote the production of BMDCs to gain a higher phagocytotic capacity, maturation, and activation, and that ferroptosis is a type of ICD. Another study found that ferroptosis was the cell death type for cells in an injured myocardium, and orchestrated the recruitment of neutrophils to the injured myocardium after heart transplantation through a toll-like receptor 4 (TLR4)/TIR-domain-containing adapter-inducing interferon-β (TRIF)/type I interferon (IFN) signaling pathway (91).
When discussing the interaction between cancer cells and innate immune cells in the TME in depth, there is a series of steps that we followed in our attempt to shed light on this process. The first step is how immune cells distinguish altered self ferroptosis cancer cells from normal self cells in the early stages before rupture. We hypothesize that cell membrane epitopes on the outside surfaces of tumor cells are reprogramming when undergoing ferroptosis and serve as “find me” and “eat me” signals. What is the initial signal in the interaction between ferroptosis cells and innate immune cells? In a study comparing three types of cell death, that is apoptosis, necroptosis, and ferroptosis, Katharina detected phosphatidylserine exposure on the outside membranes of all three types of dying cells, subsequently recognized and engulfed by macrophages in vitro (84). As phosphatidylserine exposure is the notable characteristic of apoptotic cells to be recognized by macrophages and induces the clearance of residual apoptotic cells and immune tolerance, the subsequent effect of phosphatidylserine exposure on ferroptosis cells in the TME remains unclear. In another study using the dynamic lipid membranes model, Stockwell simulated ferroptosis cell membrane by combining a high concentration of PUFAs, lipid peroxides, and longer acyl tails, and found that a higher composition of oxidized phosphatidylethanolamine (PE-AA-OOH) on the membrane was the main characteristic of cell membrane change in ferroptosis cells, which was probably the main cause of ferroptosis cell membrane rupture and final death (92). Another study found that the oxidized phospholipid of 1-steaoryl-2-15-HpETE-sn-glycero-3-phosphatidylethanolamine (SAPE-OOH) was a key “eat me” signal on the ferroptosis cell surface that could be recognized by TLR2 on macrophages (93). The oxidation of arachidonic acid via ACSL4, acyl-CoA:lysophosphatidylcholine acyltransferase (LPCAT), and ALOX enzymes generated PE-AA-OOH production. Other types of eicosanoids are also produced in the context of ferroptosis, including prostaglandin E2 (PGE2), oxidized phospholipids (oxPLs), 5-Hydroxyeicosatetraenoic (5-HETE), 11-HETE, 12-HETE, and 15-HETE (94). Multiple studies have extensively explored the crucial immunometabolic modulatory function of eicosanoids interaction with inflammatory cells by specific receptors (95). However, in the TME, a selective inhibitor of 20-HETE synthesis, HET0016, was reported to be able to decrease breast tumor volume and lung metastasis and reduce the population of granulocytic myeloid-derived suppressive cells (MDSCs) (g-MDSCs: CD11b+Ly6G+) and the expression of pro-inflammatory cytokines (96). As it has been established that MDSCs are capable of constructing an immunosuppressive niche in favor of tumor growth, it was expected that 20-HETE would have the ability. Furthermore, we learn from previous studies that the immunometabolic modulatory function of eicosanoids has a specificity that is time- and space-dependent (97). The evolution process of specific types of eicosanoids on the surface of ferroptosis cells, varying with time and space, may exert diverse effects on associated immune responses in tumor immune niche in different stages of ferroptosis, and this needs future study. Except for cell membrane lipid metabolism reprogramming on the surface of ferroptosis cells determining immune response, the second step of interaction between ferroptosis cells and immune cells may closely correlate to DAMPs, which are endogenous messages that deliver signals to immune cells or non-immune cells by binding specific receptors. It has been reported that certain DAMPs have the capacity to trigger the recruitment of monocytes and neutrophils to activate inflammatory responses (98) and serve as the initial signal to induce autophagy and immunity (99). The release of DAMPs is the hallmark of ICD. The immunogenicity of DAMPs, as “find me” or “eat me” signals that emitted from dying cells, determines their ability to activate immune responses, which has a beneficial function in antitumor therapy (100). The common types of DAMPs include high mobility group protein B1 (HMGB1), secreted ATP, and surface-exposed calreticulin (CRT). It was reported that ferroptosis cancer cells and non-cancer cells could release HMGB1, and then activate co-cultured BMDMs (bone marrow-derived macrophages) to secrete proinflammatory cytokine tumor necrosis factor alpha (TNF-α) via the HMGB1-AGER signaling pathway. This process can be blocked by adding anti-HMGB1 antibodies (101). Therefore, ferroptosis may have the ability to drive macrophage polarization to the M1 type by a HMGB1-mediated immune response. Another study showed that the depletion of Arntl, a circadian transcription factor, contributed to increased HMGB1 release and neutrophils recruitment and induced acute pancreatitis, which could be attenuated by anti-HMGB1 neutralizing antibodies and ferroptosis inhibitors (102). This study also suggested that ferroptosis might be involved in HMGB1-induced inflammatory and immune responses, and this thesis was supported by the results of another study (90). Paradoxically, it has been reported that HMGB1 was associated with various cancer progression and poor outcomes (103, 104). However, with there being scarce evidence regarding the impact of ferroptosis-released HMGB1 on tumor growth in vivo, it is not enough to conclude that HMGB1 plays a beneficial role in the ferroptosis-induced immune response for antitumor therapy, and further investigations are still required. Calreticulin is another type of DAMPs that has been implicated in eliciting an anticancer effect by initiating a cancer cell death-associated immune response and promoting the increased production of DCs and phagocytotic tumor cells (100). One study has reported the increase of calreticulin exposure on the surface of ferroptotic cancer cells, which, combined with higher emissions of HMGB1 and ATP, induced the activation and maturation of BMDCs to exert an antitumor effect (90). Another important type of DAMPs, ATP, can trigger immunogenic signaling after cancer cell death; and was used as a candidiate for an effective anticancer vaccine (105). In the ferroptosis microenvironment, ferroptosis not only causes mitochondrial fragmentation and reduces ATP levels in neuronal cell (106), but elicits ATP release from ferroptosis cancer cells, and triggers an antitumor immune response (90). Furthermore, in the context of kidney cell death, increasing evidence shows that ferroptosis plays a crucial role in renal cell death, and that lipid peroxides are unique types of ferroptosis-released DAMPs and predominate immunogenic effects (107). Arachidonic acid (AA) peroxidation products and other lipid mediators, including PGE2, PE-AA-OOH, oxPLs, and lyso-phospholipids, are emitted from ferroptosis cells. There is no direct evidence that shows that these lipid mediators released by ferroptosis cells have an effect on antitumor immunity in the TME. However, indirect evidence has demonstrated that ALOX12/15-produced phospholipid oxidation products limits the maturation of DCs and the differentiation of fine-tuning T helper 17 (TH17) cells (108). On the other hand, oxidative lipids-equipped lipid bodies are absorbed by DCs and dampen its antigen cross-presentation function and silent subsequent CD8+ T cells response in the TME (109). The effect of oxidative lipid mediators on immune response may vary among different disease conditions, cell lines, and animal models. In a word, although there is little direct evidence to support this, ferroptosis-released DAMPs and their associated immune reactions are the potential target to achieve optimal antitumor immunity effect and need to be analyzed further in the future.
In conclusion, ferroptosis may have an effect on innate immunity reprogramming to favor pro-inflammatory and antitumor response (Figure 3), and rely on cell membrane lipid metabolism reprogramming on the surface of ferroptosis cells or releasing soluble antigens as the initial signals to activate innate immune cells (Figure 4). Due to a shortage of comprehensive studies and evidence, more studies are required to further explore the impact of ferroptosis on innate immune cells’ reprogramming in diverse disease conditions and TME in the future.

Figure 3 Ferroptosis mediated immune response.
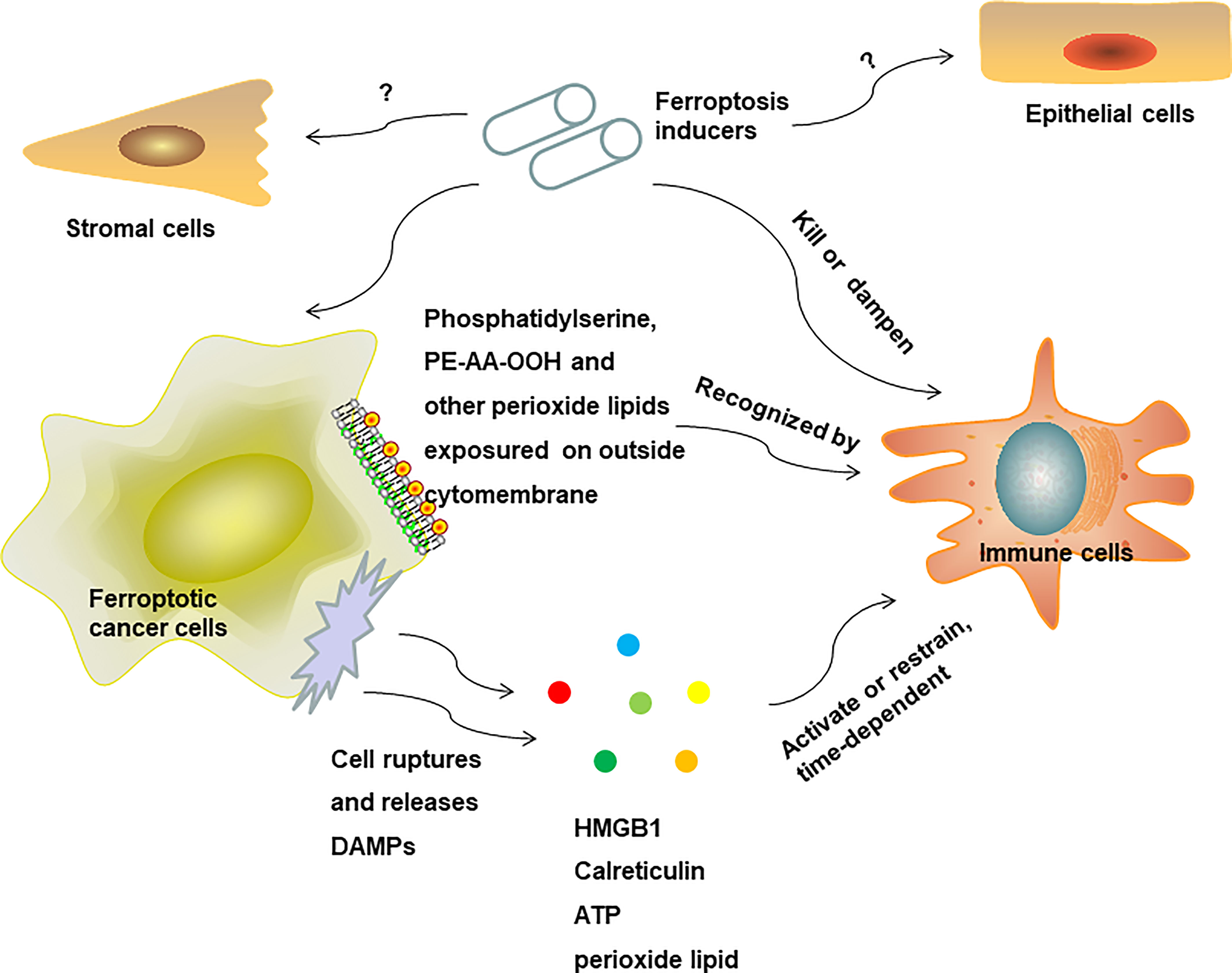
Figure 4 Potential mechanisms for ferroptosis mediated immune response in the TME.
Having explored adaptive immunity responses to the activation of innate immune cells, we move forward to explore the influence of ferroptosis on adaptive immunity. The activation of adaptive immune system by innate immune cells depends on the first and second signals (66). MHC I or MHC II interaction with T-cell receptors is the first signal. DCs then activate and prime T cells, with the second signal as co-stimulator, which predetermines the eventual immune effect cooperating with the first signal. The second signals are often chemokines like IL-1 released by APCs to stimulate T-cell activity (110). Only the first signal without the second signal leads to T cell anergy and immune tolerance (66). How ferroptosis cells prime APCs and further activate adaptive immune cells, and what the first signal and the second signal are during this process, remains obscure. Recently, several studies have focused on ferroptosis associated adaptive immune response. One study showed that ferroptosis was a kind of ICD and promoted maturation of BMDCs, then continuously activated adaptive immune system and inhibited tumor growth in vivo (90). Ferroptosis can activate resting memory CD4+ T cells and M0 macrophages for igniting anticancer immune response via ferroptosis-related lncRNA in hepatocellular carcinoma (111). Later, but still in the early stages of ferroptosis, ferroptosis cells generate ATP and HMGB1 as immunogenic signals to induce the adaptive immune system and maturation of BMDCs in vitro and in vivo (112). The time-dependent immunogenicity may be because of early-dying cells being more immunogenic than late-dying cells, meaning that they tend to possess more intact membranes and replicate their intracellular material before it leaks out. In another recent study, ferroptosis cancer cells are divided into three phases in the process: “initial”, related to lipid peroxidation; “intermediate”, associated with ATP release; and “terminal”, defined as a loss of plasma membrane integrity and inceased release of HMGB1s. “Initial” ferroptotic cells reduce the maturation of DCs and impede antigen cross-presentation (113). The contradictory results may arise from specific experimental condition in each study, but whether ferroptosis cancer cells have time-dependent immunogenicity and can be used in vaccines for antitumor immunity still needs to be clarified in studies carried out in the near future.
However, ferroptosis also exerts a direct impact on T cells themselves. One study demonstrated that T-cell clearance was mediated by lipid peroxidation and ferroptosis and that ferroptosis was the vital backup scavenger system that maintained T-cell homeostasis (114). With regard to T-cell immunity, another study demonstrated that T cell-specific GPX4 deficient mice had defective CD8+ T cell homeostasis in the periphery and a compromised defense ability against viral and parasitic infection. In ex vivo GPX4 deficient T cells accumulated membrane lipid peroxides rapidly and undergone ferroptosis (115). Another study showed that GPX4-deficient CD8+ T cells absorbed oxidized lipids by the receptor CD36 in the TME were susceptible to ferroptosis and lost their antitumor immunity functionality (116). Furthermore, ferroptosis is also involved in antitumor immunotherapy. The combination of the ferroptosis inducer cyst(e)inase and immune checkpoint inhibitor PD-L1 synergistically facilitates T cell-regulated antitumor immunity, induces ferroptosis, and inhibits tumor growth in vivo (22). This work sheds light on the critical role of ferroptosis in immunotherapy in killing tumor cells. Therefore, ferroptosis exerts an effect on T-cell immunity by maintaining the homeostatic balance of T cells, activating the secondary immune response of T cells, and enhancing T cell-mediated anti-tumor immunity. In addition, the deletion of GPX4 in regulatory T cells (Tregs)-facilitated ferroptosis that can enhance the antitumor immune response without inducing overt autoimmunity (117). The other important members in the adaptive immune system are B cells. GPX4 was also found to have the crucial function of maintaining B1 and marginal zone (MZ) B-cell homeostasis, but not that for follicular (Fo) B2 cells (118). The regulation of redox systems plays an essential role in carcinogenesis and cancer development. A clinical and experimental study showed that patients with GPX4-positive diffuse large B-cell lymphoma had significantly poor progression-free survival and overall survival compared with those of the GPX4-negative group by immunohistochemistry results (119). This result might be due to GPX4-positive cancer cells having a powerful reducing capacity that makes them impervious to the effect of ferroptosis triggered by oxidative stress. Hence, targeting GPX4 function, or cysteine levels, or other redox factors may have significant application value for B-cell leukemia treatment. GPX4, a selenium-containing enzyme, is pivotal to regulating the ferroptosis pathway, and the underpinning rationale is that GPX4 is the only enzyme to reduce phospholipid hydroperoxides (49). The depletion of GPX4 renders cancer cells vulnerable to ferroptosis (15) and the therapy-resistant state of cancer cells is dependent on GPX4 being able to escape multiple therapy challenges (20, 21). In addition, GPX4 plays an important role in mediating immune cells themselves function. GPX4 deficient CD8+ and CD4+T cells fail to withstand virus and parasite infections (115), and in the chicken bursa of the Fabricius model, selenium deprivation elicits a decrease in GPX4, the downregulation of IL-2, IL-6, IL-8, IL-10, IL17, IL-1β, IFN-α, IFN-β, and IFN-γ compared with the controls, and in the bursa having a higher TNF-α level than the controls (120). GPX4 is a potential target that can reshape the TME; its use is thus beneficial in cancer therapy and points to a direction future research can take. System xc– is also crucial to the regulation of ferroptosis and reshaping of the TME. The function of system xc– is to transport cystine into cells at the expense of exporting glutamate out to fulfill cells’ requirement of maintaining a reducing state. Tumor cells utilize this mechanism to evade oxidative stress and ferroptosis. Meanwhile, the exported glutamate can also exert an effect on immune cells in the TME. It was reported that elevated extracellular glutamate contributed to Tregs’ proliferation and activation, thus enhancing immunosuppression in the TME of glioblastoma (121). Another study showed that the loss of system xc– enhanced the anticancer capacity of the immunotherapeutic agent anti-CTLA-4 (122). GPX4 and system Xc- are critical regulators of intracellular metabolic pathways and help to maintain a reducing-state homeostasis. As increasing evidence shows that metabolic environment modification builds up the bridge for crosstalk between cancer cells and immune cells, exploring the role of metabolic reprogramming involved in ferroptosis by targeting GPX4 and system xc– in both tumor cells and immune cells in the TME is a very promising direction for future research.
In conclusion, ferroptosis is the primary mechanism that maintains adaptive immune cells’ homeostasis. However, the overwhelmingly reducing capacity of immune cells may facilitate their development into B cells, or the progressionof leukemia T-cells. More work should be done to clarify how to maintain the proper and delicate balance of redox levels in tumor and immune cells in the TME, and also the optimal way to utilize ferroptosis to inhibit cancer progression in a specific context in the future. GPX4 and system xc– are not only critical regulators of ferroptosis, but also exert a crucial effect on the TME, making them potential targets for cancer therapy.
In the following text we introduce some new applications for ferroptosis-associated immune response in cancer treatment. In this way, we analyze the close relationship between ferroptosis and immune system and present promising combination therapy strategies that could cure cancer in the near future.
As certain ferroptosis inducers have short half-lives and potentially systematic adverse effects, the approach of combining ferroptosis inducers with nanomaterials has been intensively implicated in strengthening anticancer immunity in recent years. Zanganeh found that iron oxide nanoparticles (SPIONs) limited tumor growth by regulating macrophages to polarize to pro-inflammatory M1 types in tumor niches (123). Although ferroptosis was not mentioned in this study, abundant iron input probably elevated the labile iron pool burden and elicited the Fenton reaction that leads to ferroptosis. Moreover, SPIONs could directly promote macrophages to undergo classical autophagy and stimulate the expression of inflammatory cytokines at higher levels through the activation of TLR4 signaling via the TLR4-p38-Nrf2-p62 signal axis (124). Furthermore, iron chelated melanin-like nanoparticles (Fe@PDA-PEG) were shown to induce the M2-to-M1 repolarization of tumor-associated macrophages (TAMs), and, in combination with photothermal therapy, synergistically inhibited tumor growth (PTT) (125). The reason for this was that the amplified antigens, stimulated by ferroptosis, acquired the ability of macrophages, and exerted a synergistic effect with the release of PTTinduced tumor-associated antigens (TAAS). Another study demonstrated that αMSH-PEG-C′ nanoparticles could induce tumors to ferroptosis in vivo by accelerating iron absorption into cancer cells (126). However, an immunodeficient animal model was used in the study and the ferroptosis-related immune response was not explored. In addition, the engineering magnetosomes, constructed with the core of Fe3O4 magnetic nanocluster, the cloak of leukocyte membranes with inner face implanted with TGF-β inhibitor and outer face loaded with PD-1 antibody, showed optimal antitumor therapy capacity (127). Biomimetic magnetic nanoparticles Fe3O4-SAS @ PLT can induce ferroptosis and sensitize the activity of PD-1 immune checkpoint blockade treatment (128). The nanoplatform combining sorafenib (ferroptosis promoter) with photosensitized hemoglobin can strengthen ferroptosis and stimulate an antitumor immune response by recruiting immune cells to secrete IFN-γ (129). There have been plenty of studies conducted in recent years that investigate the activity of nanocomposites, comprising ferroptosis inducers and other components that exhibit amplifying anticancer immunity (130–134). Thus, the synergism of ferroptosis and immunomodulation gives rise to potent therapeutic effects, and exhibits more advantages by uniting characteristics of modified nanomaterial, which is a promising approach for strengthening antitumor immunity for cancer therapy in the future (135).
Last but not least, we focus on whether ferroptosis-involving combined therapy can induce antitumor immunity. Zou found that uniting reagents that induce ferroptosis with immune checkpoint inhibitors displayed the optimal therapeutic effect for suppressing tumor progression (22). This research also showed that anti-PD-L1 immunotherapy could elicit tumor cell ferroptosis via the CD8+ T cell-IFN-γ-system xc– signal pathway. Another study also confirmed the superior synergy of combining ferroptosis with immunotherapy (122, 127). In addition, ferroptosis was found to create bond of synergy between radiotherapy and immunotherapy (23). In addition, the Krebs cycle enzyme fumarate hydratase deprivation (FH-/-) demonstrated synergy with ferroptosis inducers for the limiting of hereditary leiomyomatosis and renal cell cancer (HLRCC) progression (136). Nanocomposite containing Fe3+ with cancer cell membrane was absorbed by breast cancer cells and induced ferroptosis that could reinforce the synergistic therapeutic effect of sequential PDT and chemotherapy (137). The chemotherapeutics drugs sorafenib was also shown to induce ferroptosis via a ferritinophagy cascade, thus triggering an antitumor immune response (138). In addition, incorporating ferroptosis and ultrasound-triggered sonodynamic therapy (SDT) synergistically elicited strong antitumor immunity by increasing the numbers of mature DCs and activated CD8+ cells and decreasing the number of MDSCs in the TME (139). These findings indicate that ferroptosis is capable of synergistic action with chemotherapy, PDT, or SDT, in turn meaning that it can serve as the critical therapeutic target to enhance chemotherapy and PDT treatment outcomes. The underlying synergistic effect mechanism for ferroptosis and chemotherapy may be owing to th ability of these two therapy methods to greatly increase the ROS level in the TME (140). PDT directly leads to ferroptosis in tumor cells, and this would explain the cooperative effect between ferroptosis and PDT (90, 141). We believe that the more intricate and core mechanism of the synergy between ferroptosis and other treatment manners for cancer lies in the potential capacity of ferroptosis to induce a strong antitumor immune reaction, which would augment the therapeutic effect of other treatment methods. In depth, ferroptosis would change tumor cells into “altered self” or “missing self” with the downregulation of the “do not eat me signals” such as CD47 on the surfaces of tumor cells, or through mimicking microbial antigens by releasing DAMPs like oxidized lipids. All these transformations can be recognized by innate immune cells and lead to anticancer immune responses, and these potential mechanisms could be explored and verified in the future. Thus, as the role of ferroptosis in multiple metabolic pathways and in classical cancer treatment approaches of immunotherapy, radiotherapy, chemotherapy, PDT and SDT, has been extensively and thoroughly covered, moving to develop ferroptosis-involving combination therapies for antitumor treatment in the near future is promising (142).
This review focuses on ferroptosis molecular network and associated immune response in tumor immune niche, but there are some undeniable challenge associated with inducing ferroptosis in the TME. As shown above, although ferroptosis may exert antitumor capacity by killing cancer cells and boosting the immune response, it also has a direct impact on immune cells. In the TME, ferroptosis may also kill immune cells, and disarm the defense force monitoring cancer cells. In addition, normal cells such as vascular endothelial cells and stromal cells would be affected by ferroptosis inducers, which may facilitate metastasis, and the likelihood of this needs further study and confirmation. Therefore, as ferroptosis has a comprehensive influence on all units in the TME, it is crucial to clarify the unique effect on each unit and grasp its overall effect on various cancer types (Figure 4). By utilizing novel biological materials and through other physical and chemical means, we could amplify the beneficial role of ferroptosis in cancer therapy and eradicate its potential adverse effect on immune cells and other normal cells in the meantime.
In conclusion, although there is a great number of gaps and dilemmas in this area that needs to be addressed before clinical applications are feasible, we have confidence that working on this subject will lead to a promising future, wherein cancer could be eliminated by the reshaping of tumor immune niches via ferroptosis.
DQ performed the majority of the research and wrote the manuscript, MP checked and revised the manuscript. All authors contributed to the article and approved the submitted version.
The funding of the study was supported by the National Natural Science Foundation of China (grant numbers 82102282); Natural Science Foundation of Hunan Province (grant numbers 2022JJ30953); China Postdoctoral Science Foundation (grant numbers 2022M713528) and the Natural Science Foundation of Changsha City (grant numbers kq2202381).
The authors declare that the research was conducted in the absence of any financial or commercial relationships that could be construed as potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ellenberg M, Osserman KE. The role of shock in the production of central liver cell necrosis. Am J Med (1951) 11(2):170–8. doi: 10.1016/0002-9343(51)90102-7
2. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. (1972) 26(4):239–57. doi: 10.1038/bjc.1972.33
3. Searle J, Collins DJ, Harmon B, Kerr JF. The spontaneous occurrence of apoptosis in squamous carcinomas of the uterine cervix. Pathology (1973) 5(2):163–9. doi: 10.3109/00313027309060831
4. Weedon D, Searle J, Kerr JF. Apoptosis. its nature and implications for dermatopathology. Am J Dermatopathol. (1979) 1(2):133–44. doi: 10.1097/00000372-197900120-00003
5. Hu X, Han W, Li L. Targeting the weak point of cancer by induction of necroptosis. Autophagy (2007) 3(5):490–2. doi: 10.4161/auto.4592
6. Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol (2006) 8(11):1812–25. doi: 10.1111/j.1462-5822.2006.00751.x
7. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
8. Gudipaty SA, Conner CM, Rosenblatt J, Montell DJ. Unconventional ways to live and die: cell death and survival in development, homeostasis, and disease. Annu Rev Cell Dev Biol (2018) 34:311–32. doi: 10.1146/annurev-cellbio-100616-060748
9. Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, Pham VC, et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature (2016) 540(7631):129–33. doi: 10.1038/nature20559
10. Wang R, Li H, Wu J, Cai ZY, Li B, Ni H, et al. Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature (2020) 580(7803):386–90. doi: 10.1038/s41586-020-2127-x
11. Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and mincle-induced immune suppression. Nature (2016) 532(7598):245–9. doi: 10.1038/nature17403
12. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci (2017) 42(4):245–54. doi: 10.1016/j.tibs.2016.10.004
13. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell (2003) 3(3):285–96. doi: 10.1016/s1535-6108(03)00050-3
14. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol (2008) 15(3):234–45. doi: 10.1016/j.chembiol.2008.02.010
15. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2014) 156(1-2):317–31. doi: 10.1016/j.cell.2013.12.010
16. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature (2015) 520(7545):57–62. doi: 10.1038/nature14344
17. Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G. BAP1 and cancer. Nat Rev Cancer. (2013) 13(3):153–9. doi: 10.1038/nrc3459
18. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature (2019) 575(7784):688–92. doi: 10.1038/s41586-019-1705-2
19. Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell (2019) 51(5):575–86 e4. doi: 10.1016/j.devcel.2019.10.007
20. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature (2017) 551(7679):247–50. doi: 10.1038/nature24297
21. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature (2017) 547(7664):453–7. doi: 10.1038/nature23007
22. Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature (2019) 569(7755):270–4. doi: 10.1038/s41586-019-1170-y
23. Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discovery (2019) 9(12):1673–85. doi: 10.1158/2159-8290.CD-19-0338
24. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature (2021) 593(7860):586–90. doi: 10.1038/s41586-021-03539-7
25. Pfeifhofer-Obermair C, Tymoszuk P, Petzer V, Weiss G, Nairz M. Iron in the tumor microenvironment-connecting the dots. Front Oncol (2018) 8:549. doi: 10.3389/fonc.2018.00549
26. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell (2019) 35(6):830–49. doi: 10.1016/j.ccell.2019.04.002
27. Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature (2017) 551(7682):639–43. doi: 10.1038/nature24637
28. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature (2014) 509(7498):105–9. doi: 10.1038/nature13148
29. Kwon MY, Park E, Lee SJ, Chung SW. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget (2015) 6(27):24393–403. doi: 10.18632/oncotarget.5162
30. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology (2016) 63(1):173–84. doi: 10.1002/hep.28251
31. Song X, Xie Y, Kang R, Hou W, Sun X, Epperly MW, et al. FANCD2 protects against bone marrow injury from ferroptosis. Biochem Biophys Res Commun (2016) 480(3):443–9. doi: 10.1016/j.bbrc.2016.10.068
32. Yanatori I, Kishi F. DMT1 and iron transport. Free Radic Biol Med (2019) 133:55–63. doi: 10.1016/j.freeradbiomed.2018.07.020
33. Shi H, Bencze KZ, Stemmler TL, Philpott CC. A cytosolic iron chaperone that delivers iron to ferritin. Science (2008) 320(5880):1207–10. doi: 10.1126/science.1157643
34. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. (2016) 113(34):E4966–75. doi: 10.1073/pnas.1603244113
35. Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol (2015) 10(7):1604–9. doi: 10.1021/acschembio.5b00245
36. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol (2017) 13(1):81–90. doi: 10.1038/nchembio.2238
37. Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature (2020) 585(7826):603–8. doi: 10.1038/s41586-020-2732-8
38. Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab (2008) 8(3):237–48. doi: 10.1016/j.cmet.2008.07.005
39. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
40. Newman MJ. Inhibition of carcinoma and melanoma cell growth by type 1 transforming growth factor beta is dependent on the presence of polyunsaturated fatty acids. Proc Natl Acad Sci U S A. (1990) 87(14):5543–7. doi: 10.1073/pnas.87.14.5543
41. Khanna S, Roy S, Ryu H, Bahadduri P, Swaan PW, Ratan RR, et al. Molecular basis of vitamin e action: tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. J Biol Chem (2003) 278(44):43508–15. doi: 10.1074/jbc.M307075200
42. Dysken MW, Sano M, Asthana S, Vertrees JE, Pallaki M, Llorente M, et al. Effect of vitamin e and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA (2014) 311(1):33–44. doi: 10.1001/jama.2013.282834
43. Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol (2016) 12(7):497–503. doi: 10.1038/nchembio.2079
44. Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol (2016) 26(3):165–76. doi: 10.1016/j.tcb.2015.10.014
45. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol (2020) 21(7):363–83. doi: 10.1038/s41580-020-0230-3
46. Qian X, Nie X, Yao W, Klinghammer K, Sudhoff H, Kaufmann AM, et al. Reactive oxygen species in cancer stem cells of head and neck squamous cancer. Semin Cancer Biol (2018) 53:248–57. doi: 10.1016/j.semcancer.2018.06.001
47. Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol (2010) 10(3):210–5. doi: 10.1038/nri2725
48. Davalli P, Mitic T, Caporali A, Lauriola A, D'Arca DROS, Senescence C. And novel molecular mechanisms in aging and age-related diseases. Oxid Med Cell Longev (2016) 2016:3565127. doi: 10.1155/2016/3565127
49. Ursini F, Maiorino M, Valente M, Ferri L, Gregolin C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim Biophys Acta (1982) 710(2):197–211. doi: 10.1016/0005-2760(82)90150-3
50. Shimada K, Hayano M, Pagano NC, Stockwell BR. Cell-line selectivity improves the predictive power of pharmacogenomic analyses and helps identify NADPH as biomarker for ferroptosis sensitivity. Cell Chem Biol (2016) 23(2):225–35. doi: 10.1016/j.chembiol.2015.11.016
51. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell (2019) 73(2):354–63 e3. doi: 10.1016/j.molcel.2018.10.042
52. Yang L, Wang H, Yang X, Wu Q, An P, Jin X, et al. Auranofin mitigates systemic iron overload and induces ferroptosis via distinct mechanisms. Signal Transduct Target Ther (2020) 5(1):138. doi: 10.1038/s41392-020-00253-0
53. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell (2015) 59(2):298–308. doi: 10.1016/j.molcel.2015.06.011
54. Ooi A. Advances in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) research. Semin Cancer Biol (2020) 61:158–66. doi: 10.1016/j.semcancer.2019.10.016
55. Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M, Buchfelder M, et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis (2017) 6(8):e371. doi: 10.1038/oncsis.2017.65
56. Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol (2019) 23:101107. doi: 10.1016/j.redox.2019.101107
57. Zhang Y, Zhuang L, Gan B. BAP1 suppresses tumor development by inducing ferroptosis upon SLC7A11 repression. Mol Cell Oncol (2019) 6(1):1536845. doi: 10.1080/23723556.2018.1536845
58. Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol (2018) 20(10):1181–92. doi: 10.1038/s41556-018-0178-0
59. Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. (2019) 116(7):2672–80. doi: 10.1073/pnas.1821022116
60. Zhang Y, Sun C, Zhao C, Hao J, Zhang Y, Fan B, et al. Ferroptosis inhibitor SRS 16-86 attenuates ferroptosis and promotes functional recovery in contusion spinal cord injury. Brain Res (2019) 1706:48–57. doi: 10.1016/j.brainres.2018.10.023
61. Hu Z, Zhang H, Yi B, Yang S, Liu J, Hu J, et al. VDR activation attenuate cisplatin induced AKI by inhibiting ferroptosis. Cell Death Dis (2020) 11(1):73. doi: 10.1038/s41419-020-2256-z
62. Hambright WS, Fonseca RS, Chen L, Na R, Ran Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol (2017) 12:8–17. doi: 10.1016/j.redox.2017.01.021
63. Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem Biol (2019) 26(5):623–33 e9. doi: 10.1016/j.chembiol.2019.01.008
64. Llabani E, Hicklin RW, Lee HY, Motika SE, Crawford LA, Weerapana E, et al. Diverse compounds from pleuromutilin lead to a thioredoxin inhibitor and inducer of ferroptosis. Nat Chem (2019) 11(6):521–32. doi: 10.1038/s41557-019-0261-6
65. Kim J, DeBerardinis RJ. Mechanisms and implications of metabolic heterogeneity in cancer. Cell Metab (2019) 30(3):434–46. doi: 10.1016/j.cmet.2019.08.013
66. Janeway CA Jr. Approaching the asymptote? evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol (1989) 54 Pt 1:1–13. doi: 10.1101/sqb.1989.054.01.003
67. Bevan MJ. Antigen recognition. Class discrimination World Immunol Nature. (1987) 325(6101):192–4. doi: 10.1038/325192b0
68. Garg AD, Agostinis P. Cell death and immunity in cancer: from danger signals to mimicry of pathogen defense responses. Immunol Rev (2017) 280(1):126–48. doi: 10.1111/imr.12574
69. Garg AD, Vandenberk L, Fang S, Fasche T, Van Eygen S, Maes J, et al. Pathogen response-like recruitment and activation of neutrophils by sterile immunogenic dying cells drives neutrophil-mediated residual cell killing. Cell Death Differ (2017) 24(5):832–43. doi: 10.1038/cdd.2017.15
70. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med (2007) 13(9):1050–9. doi: 10.1038/nm1622
71. Azimi F, Scolyer RA, Rumcheva P, Moncrieff M, Murali R, McCarthy SW, et al. Tumor-infiltrating lymphocyte grade is an independent predictor of sentinel lymph node status and survival in patients with cutaneous melanoma. J Clin Oncol (2012) 30(21):2678–83. doi: 10.1200/JCO.2011.37.8539
72. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol (2013) 14(10):1014–22. doi: 10.1038/ni.2703
73. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol (2013) 31:51–72. doi: 10.1146/annurev-immunol-032712-100008
74. Birge RB, Ucker DS. Innate apoptotic immunity: the calming touch of death. Cell Death Differ (2008) 15(7):1096–102. doi: 10.1038/cdd.2008.58
75. Sancho D, Joffre OP, Keller AM, Rogers NC, Martinez D, Hernanz-Falcon P, et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature (2009) 458(7240):899–903. doi: 10.1038/nature07750
76. Snyder AG, Hubbard NW, Messmer MN, Kofman SB, Hagan CE, Orozco SL, et al. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci Immunol (2019) 4(36):eaaw2004. doi: 10.1126/sciimmunol.aaw2004
77. Kasmann L, Eze C, Manapov F. Stereotactic body radiation therapy (SBRT) combined with immune check-point inhibition (ICI) in advanced lung cancer: which metastatic site should be irradiated to induce immunogenic cell death? Int J Radiat Oncol Biol Phys (2020) 108(1):225–226. doi: 10.1016/j.ijrobp.2020.04.002
78. Galluzzi L, Kepp O, Kroemer G. Immunogenic cell death in radiation therapy. Oncoimmunology (2013) 2(10):e26536. doi: 10.4161/onci.26536
79. Wang Q, Ju X, Wang J, Fan Y, Ren M, Zhang H. Immunogenic cell death in anticancer chemotherapy and its impact on clinical studies. Cancer Lett (2018) 438:17–23. doi: 10.1016/j.canlet.2018.08.028
80. Pozzi C, Cuomo A, Spadoni I, Magni E, Silvola A, Conte A, et al. The EGFR-specific antibody cetuximab combined with chemotherapy triggers immunogenic cell death. Nat Med (2016) 22(6):624–31. doi: 10.1038/nm.4078
81. Gebremeskel S, Lobert L, Tanner K, Walker B, Oliphant T, Clarke LE, et al. Natural killer T-cell immunotherapy in combination with chemotherapy-induced immunogenic cell death targets metastatic breast cancer. Cancer Immunol Res (2017) 5(12):1086–97. doi: 10.1158/2326-6066.CIR-17-0229
82. Riera Romo M, Perez-Martinez D, Castillo Ferrer C. Innate immunity in vertebrates: an overview. Immunology (2016) 148(2):125–39. doi: 10.1111/imm.12597
83. Telorack M, Meyer M, Ingold I, Conrad M, Bloch W, Werner S. A glutathione-Nrf2-Thioredoxin cross-talk ensures keratinocyte survival and efficient wound repair. PloS Genet (2016) 12(1):e1005800. doi: 10.1371/journal.pgen.1005800
84. Kloditz K, Fadeel B. Three cell deaths and a funeral: macrophage clearance of cells undergoing distinct modes of cell death. Cell Death Discovery (2019) 5:65. doi: 10.1038/s41420-019-0146-x
85. Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy (2020) 2020:1–15. doi: 10.1080/15548627.2020.1714209
86. Liu J, Zhu S, Zeng L, Li J, Klionsky DJ, Kroemer G, et al. DCN released from ferroptotic cells ignites AGER-dependent immune responses. Autophagy (2022) 18(9):2036–49. doi: 10.1080/15548627.2021.2008692
87. Xu Z, Chen X, Song L, Yuan F, Yan Y. Matrix remodeling-associated protein 8 as a novel indicator contributing to glioma immune response by regulating ferroptosis. Front Immunol (2022) 13:834595. doi: 10.3389/fimmu.2022.834595
88. Li X, Zou Y, Xing J, Fu YY, Wang KY, Wan PZ, et al. Pretreatment with roxadustat (FG-4592) attenuates folic acid-induced kidney injury through antiferroptosis via Akt/GSK-3beta/Nrf2 pathway. Oxid Med Cell Longev (2020) 2020:6286984. doi: 10.1155/2020/6286984
89. Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol (2020) 16(3):278–90. doi: 10.1038/s41589-019-0462-8
90. Turubanova VD, Balalaeva IV, Mishchenko TA, Catanzaro E, Alzeibak R, Peskova NN, et al. Immunogenic cell death induced by a new photodynamic therapy based on photosens and photodithazine. J Immunother Cancer. (2019) 7(1):350. doi: 10.1186/s40425-019-0826-3
91. Li W, Feng G, Gauthier JM, Lokshina I, Higashikubo R, Evans S, et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Invest. (2019) 129(6):2293–304. doi: 10.1172/JCI126428
92. Agmon E, Solon J, Bassereau P, Stockwell BR. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep (2018) 8(1):5155. doi: 10.1038/s41598-018-23408-0
93. Luo X, Gong HB, Gao HY, Wu YP, Sun WY, Li ZQ, et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ (2021) 28(6):1971–89. doi: 10.1038/s41418-020-00719-2
94. Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. (2019) 19(7):405–14. doi: 10.1038/s41568-019-0149-1
95. Araujo AC, Wheelock CE, Haeggstrom JZ. The eicosanoids, redox-regulated lipid mediators in immunometabolic disorders. Antioxid Redox Signal (2018) 29(3):275–96. doi: 10.1089/ars.2017.7332
96. Borin TF, Shankar A, Angara K, Rashid MH, Jain M, Iskander A, et al. HET0016 decreases lung metastasis from breast cancer in immune-competent mouse model. PloS One (2017) 12(6):e0178830. doi: 10.1371/journal.pone.0178830
97. Piper K, Garelnabi M. Eicosanoids: atherosclerosis and cardiometabolic health. J Clin Transl Endocrinol (2020) 19:100216. doi: 10.1016/j.jcte.2020.100216
98. Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol (2020) 15:493–518. doi: 10.1146/annurev-pathmechdis-012419-032847
99. Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev (2012) 249(1):158–75. doi: 10.1111/j.1600-065X.2012.01146.x
100. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med (2007) 13(1):54–61. doi: 10.1038/nm1523
101. Wen Q, Liu J, Kang R, Zhou B, Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun (2019) 510(2):278–83. doi: 10.1016/j.bbrc.2019.01.090
102. Liu Y, Wang Y, Liu J, Kang R, Tang D. The circadian clock protects against ferroptosis-induced sterile inflammation. Biochem Biophys Res Commun (2020) 525(3):620–625. doi: 10.1016/j.bbrc.2020.02.142
103. Lv DJ, Song XL, Huang B, Yu YZ, Shu FP, Wang C, et al. HMGB1 promotes prostate cancer development and metastasis by interacting with Brahma-related gene 1 and activating the akt signaling pathway. Theranostics (2019) 9(18):5166–82. doi: 10.7150/thno.33972
104. Cheng KJ, Alshawsh MA, Mejia Mohamed EH, Thavagnanam S, Sinniah A, Ibrahim ZA. HMGB1: an overview of its versatile roles in the pathogenesis of colorectal cancer. Cell Oncol (Dordr). (2019) 43(2):177–193. doi: 10.1007/s13402-019-00477-5
105. Garg AD, Krysko DV, Vandenabeele P, Agostinis P. Extracellular ATP and P(2)X(7) receptor exert context-specific immunogenic effects after immunogenic cancer cell death. Cell Death Dis (2016) 7:e2097. doi: 10.1038/cddis.2015.411
106. Neitemeier S, Jelinek A, Laino V, Hoffmann L, Eisenbach I, Eying R, et al. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol (2017) 12:558–70. doi: 10.1016/j.redox.2017.03.007
107. Sarhan M, von Massenhausen A, Hugo C, Oberbauer R, Linkermann A. Immunological consequences of kidney cell death. Cell Death Dis (2018) 9(2):114. doi: 10.1038/s41419-017-0057-9
108. Rothe T, Gruber F, Uderhardt S, Ipseiz N, Rossner S, Oskolkova O, et al. 12/15-lipoxygenase-mediated enzymatic lipid oxidation regulates DC maturation and function. J Clin Invest. (2015) 125(5):1944–54. doi: 10.1172/JCI78490
109. Veglia F, Tyurin VA, Mohammadyani D, Blasi M, Duperret EK, Donthireddy L, et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat Commun (2017) 8(1):2122. doi: 10.1038/s41467-017-02186-9
110. Sokol CL, Luster AD. The chemokine system in innate immunity. Cold Spring Harb Perspect Biol (2015) 7(5):a016303. doi: 10.1101/cshperspect.a016303
111. Xu Z, Peng B, Liang Q, Chen X, Cai Y, Zeng S, et al. Construction of a ferroptosis-related nine-lncRNA signature for predicting prognosis and immune response in hepatocellular carcinoma. Front Immunol (2021) 12:719175. doi: 10.3389/fimmu.2021.719175
112. Efimova I, Catanzaro E, van der Meeren L, Turubanova VD, Hammad H, Mishchenko TA, et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J Immunother Cancer. (2020) 8(2):e001369. doi: 10.1136/jitc-2020-001369
113. Wiernicki B, Maschalidi S, Pinney J, Adjemian S, Vanden Berghe T, Ravichandran KS, et al. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat Commun (2022) 13(1):3676. doi: 10.1038/s41467-022-31218-2
114. Dash B, Belmonte PJ, Fine SR, Shapiro MJ, Chung JY, Schwab AD, et al. Murine T cell maturation entails protection from MBL2, but complement proteins do not drive clearance of cells that fail maturation in the absence of NKAP. J Immunol (2019) 203(2):408–17. doi: 10.4049/jimmunol.1801443
115. Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T Cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med (2015) 212(4):555–68. doi: 10.1084/jem.20140857
116. Xu S, Chaudhary O, Rodriguez-Morales P, Sun X, Chen D, Zappasodi R, et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8+ T cells in tumors. Immunity (2021) 54(7):1561–77 e7. doi: 10.1016/j.immuni.2021.05.003
117. Xu C, Sun S, Johnson T, Qi R, Zhang S, Zhang J, et al. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain treg cell activation and suppression of antitumor immunity. Cell Rep (2021) 35(11):109235. doi: 10.1016/j.celrep.2021.109235
118. Muri J, Thut H, Bornkamm GW, Kopf M. B1 and marginal zone b cells but not follicular B2 cells require Gpx4 to prevent lipid peroxidation and ferroptosis. Cell Rep (2019) 29(9):2731–44 e4. doi: 10.1016/j.celrep.2019.10.070
119. Kinowaki Y, Kurata M, Ishibashi S, Ikeda M, Tatsuzawa A, Yamamoto M, et al. Glutathione peroxidase 4 overexpression inhibits ROS-induced cell death in diffuse large b-cell lymphoma. Lab Invest. (2018) 98(5):609–19. doi: 10.1038/s41374-017-0008-1
120. Khoso PA, Yang Z, Liu C, Li S. Selenoproteins and heat shock proteins play important roles in immunosuppression in the bursa of fabricius of chickens with selenium deficiency. Cell Stress Chaperones. (2015) 20(6):967–78. doi: 10.1007/s12192-015-0625-9
121. Long Y, Tao H, Karachi A, Grippin AJ, Jin L, Chang YE, et al. Dysregulation of glutamate transport enhances treg function that promotes VEGF blockade resistance in glioblastoma. Cancer Res (2020) 80(3):499–509. doi: 10.1158/0008-5472.CAN-19-1577
122. Arensman MD, Yang XS, Leahy DM, Toral-Barza L, Mileski M, Rosfjord EC, et al. Cystine-glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity. Proc Natl Acad Sci U S A. (2019) 116(19):9533–42. doi: 10.1073/pnas.1814932116
123. Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. (2016) 11(11):986–94. doi: 10.1038/nnano.2016.168
124. Jin R, Liu L, Zhu W, Li D, Yang L, Duan J, et al. Iron oxide nanoparticles promote macrophage autophagy and inflammatory response through activation of toll-like receptor-4 signaling. Biomaterials (2019) 203:23–30. doi: 10.1016/j.biomaterials.2019.02.026
125. Rong L, Zhang Y, Li WS, Su Z, Fadhil JI, Zhang C. Iron chelated melanin-like nanoparticles for tumor-associated macrophage repolarization and cancer therapy. Biomaterials (2019) 225:119515. doi: 10.1016/j.biomaterials.2019.119515
126. Kim SE, Zhang L, Ma K, Riegman M, Chen F, Ingold I, et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol. (2016) 11(11):977–85. doi: 10.1038/nnano.2016.164
127. Zhang F, Li F, Lu GH, Nie W, Zhang L, Lv Y, et al. Engineering magnetosomes for Ferroptosis/Immunomodulation synergism in cancer. ACS Nano. (2019) 13(5):5662–73. doi: 10.1021/acsnano.9b00892
128. Jiang Q, Wang K, Zhang X, Ouyang B, Liu H, Pang Z, et al. Platelet membrane-camouflaged magnetic nanoparticles for ferroptosis-enhanced cancer immunotherapy. Small (2020) 16(22):e2001704. doi: 10.1002/smll.202001704
129. Xu T, Ma Y, Yuan Q, Hu H, Hu X, Qian Z, et al. Enhanced ferroptosis by oxygen-boosted phototherapy based on a 2-in-1 nanoplatform of ferrous hemoglobin for tumor synergistic therapy. ACS Nano. (2020) 14(3):3414–25. doi: 10.1021/acsnano.9b09426
130. Deng H, Zhang J, Yang Y, Yang J, Wei Y, Ma S, et al. Chemodynamic and photothermal combination therapy based on dual-modified metal-organic framework for inducing tumor Ferroptosis/Pyroptosis. ACS Appl Mater Interfaces. (2022) 14(21):24089–101. doi: 10.1021/acsami.2c00574
131. Yun T, Liu Z, Wang J, Wang R, Zhu L, Zhu Z, et al. Microenvironment immune response induced by tumor ferroptosis-the application of nanomedicine. Front Oncol (2022) 12:1019654. doi: 10.3389/fonc.2022.1019654
132. Chen C, Wang Z, Jia S, Zhang Y, Ji S, Zhao Z, et al. Evoking highly immunogenic ferroptosis aided by intramolecular motion-induced photo-hyperthermia for cancer therapy. Adv Sci (Weinh). (2022) 9(10):e2104885. doi: 10.1002/advs.202104885
133. Wang Y, Chen Q, Song H, Zhang Y, Chen H, Liu P, et al. A triple therapeutic strategy with antiexosomal iron efflux for enhanced ferroptosis therapy and immunotherapy. Small (2022) 18(41):e2201704. doi: 10.1002/smll.202201704
134. Li Z, Rong L. Cascade reaction-mediated efficient ferroptosis synergizes with immunomodulation for high-performance cancer therapy. Biomater Sci (2020) 8(22):6272–85. doi: 10.1039/d0bm01168a
135. Zhang X, Ma Y, Wan J, Yuan J, Wang D, Wang W, et al. Biomimetic nanomaterials triggered ferroptosis for cancer theranostics. Front Chem (2021) 9:768248. doi: 10.3389/fchem.2021.768248
136. K MJ, M J, W JA. Fumarate hydratase inactivation in hereditary leiomyomatosis and renal cell cancer is synthetic lethal with ferroptosis induction. Cancer Sci (2018) 109(9):2757–66. doi: 10.1111/cas.13701
137. Pan WL, Tan Y, Meng W, Huang NH, Zhao YB, Yu ZQ, et al. Microenvironment-driven sequential ferroptosis, photodynamic therapy, and chemotherapy for targeted breast cancer therapy by a cancer-cell-membrane-coated nanoscale metal-organic framework. Biomaterials (2022) 283:121449. doi: 10.1016/j.biomaterials.2022.121449
138. Zuo T, Fang T, Zhang J, Yang J, Xu R, Wang Z, et al. pH-sensitive molecular-Switch-Containing polymer nanoparticle for breast cancer therapy with ferritinophagy-cascade ferroptosis and tumor immune activation. Adv Healthc Mater (2021) 10(21):e2100683. doi: 10.1002/adhm.202100683
139. Xu Q, Zhan G, Zhang Z, Yong T, Yang X, Gan L. Manganese porphyrin-based metal-organic framework for synergistic sonodynamic therapy and ferroptosis in hypoxic tumors. Theranostics (2021) 11(4):1937–52. doi: 10.7150/thno.45511
140. Zhang J, Yang J, Zuo T, Ma S, Xokrat N, Hu Z, et al. Heparanase-driven sequential released nanoparticles for ferroptosis and tumor microenvironment modulations synergism in breast cancer therapy. Biomaterials (2021) 266:120429. doi: 10.1016/j.biomaterials.2020.120429
141. Mishchenko T, Balalaeva I, Gorokhova A, Vedunova M, Krysko DV. Which cell death modality wins the contest for photodynamic therapy of cancer? Cell Death Dis (2022) 13(5):455. doi: 10.1038/s41419-022-04851-4
Keywords: cell death, ferroptosis, immunity, tumor microenvironment (TME), cancer immunotherapy
Citation: Qi D and Peng M (2023) Ferroptosis-mediated immune responses in cancer. Front. Immunol. 14:1188365. doi: 10.3389/fimmu.2023.1188365
Received: 17 March 2023; Accepted: 08 May 2023;
Published: 30 May 2023.
Edited by:
Xian Zeng, Fudan University, ChinaReviewed by:
Wan Zhuo, Air Force Medical University, ChinaCopyright © 2023 Qi and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Milin Peng, cGVuZ21pbGluQGNzdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.