 Ming Zhao
Ming Zhao Hui Huang
Hui Huang Feng He
Feng He Xiangsheng Fu
Xiangsheng Fu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 18 May 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1188277
Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer and shows high global incidence and mortality rates. The liver is an immune-tolerated organ with a specific immune microenvironment that causes traditional therapeutic approaches to HCC, such as chemotherapy, radiotherapy, and molecular targeted therapy, to have limited efficacy. The dramatic advances in immuno-oncology in the past few decades have modified the paradigm of cancer therapy, ushering in the era of immunotherapy. Currently, despite the rapid integration of cancer immunotherapy into clinical practice, some patients still show no response to treatment. Therefore, a rational approach is to target the tumor microenvironment when developing the next generation of immunotherapy. This review aims to provide insights into the hepatic immune microenvironment in HCC and summarize the mechanisms of action and clinical usage of immunotherapeutic options for HCC, including immune checkpoint blockade, adoptive therapy, cytokine therapy, vaccine therapy, and oncolytic virus-based therapy.
Hepatocellular carcinoma (HCC), the major type of hepatic malignancy, is the third leading cause of cancer-related death worldwide (1). Surgery is the best choice of intervention for early-stage HCC. However, most patients are not able to undergo surgery at the time of diagnosis due to the advanced disease stage. Other strategies, including chemotherapy, radiotherapy, molecular targeted medications, radiofrequency ablation (RFA), and transarterial chemoembolization (TACE), have limited efficacy against advanced HCC (2).
In the past decade, dramatic advances in immunology have modified the paradigm of cancer therapy, ushering in the era of immunotherapy. In cancer immunotherapy, the immune system is activated and attacks malignant cells through natural mechanisms, therefore avoiding damage to normal tissues, which is one of the most serious side effects of chemotherapy and radiotherapy. This attractive feature has supported the rapid application of immunotherapy in clinical practice. Currently, the most prevalent cancer immunotherapies include checkpoint inhibitors, engineered T cells, lymphocyte-promoting cytokines, and cancer vaccines. Immune checkpoint blockade (ICB) by monoclonal antibodies targeting cytotoxic T-lymphocyte associated protein 4 (CTLA-4) and programmed cell death 1 (PD-1) has been approved for several types of tumors and substantially benefited a subset of patients. However, most patients still show resistance to ICB (3). Therefore, targeting the tumor microenvironment is a rational approach for the development of the next generation of immunotherapy. This review will summarize the components of the immune microenvironment in HCC and the development of immunotherapy.
The liver is an organ that is nourished by a dual blood supply from both the hepatic artery and the portal vein. Blood converges in the hepatic sinusoids, which slow the blood flow and allow interactions between immune cells and antigens from the gut microbiota and circulation (4). The liver has a strong ability to remove deleterious compounds because of its abundance of innate and adaptive immune cells, including resident macrophages (Kupffer cells [KCs]), natural killer (NK) cells, and lymphocytes, with a higher CD8+/CD4+ T cells than in the periphery (4, 5). Accordingly, the liver is an immune-modulating organ with a controlled T-cell response that is maintained by resident cells, including KCs, hepatic stellate cells (HSCs), dendritic cells (DCs), and regulatory T cells (Tregs) (6). The intrinsic immune tolerogenicity prevents the liver from mounting a hyperactive response to deleterious stimuli. On the other hand, it impedes immune surveillance and facilitates oncogenesis and progression (7).
Up to 80% of HCC cases arise from chronic inflammation, which is driven by immune cell infiltration and resident cells, such as KCs, HSCs, and liver sinusoidal cells (8). Chronic inflammation creates an oxidative microenvironment and induces DNA damage and genetic alterations, favoring neoplasia initiation and progression (8).
The cellular components in the immune microenvironment in HCC include several types of cells, such as tumor-infiltrating lymphocytes (TILs), tumor-associated macrophages (TAMs), dendritic cells (DCs), tumor-associated neutrophils (TANs), myeloid-derived suppressor cells (MDSCs), mast cells (MCs), HSCs, and cancer-associated fibroblasts (CAFs), as well as noncellular components, such as cytokines and the extracellular matrix (Figure 1).
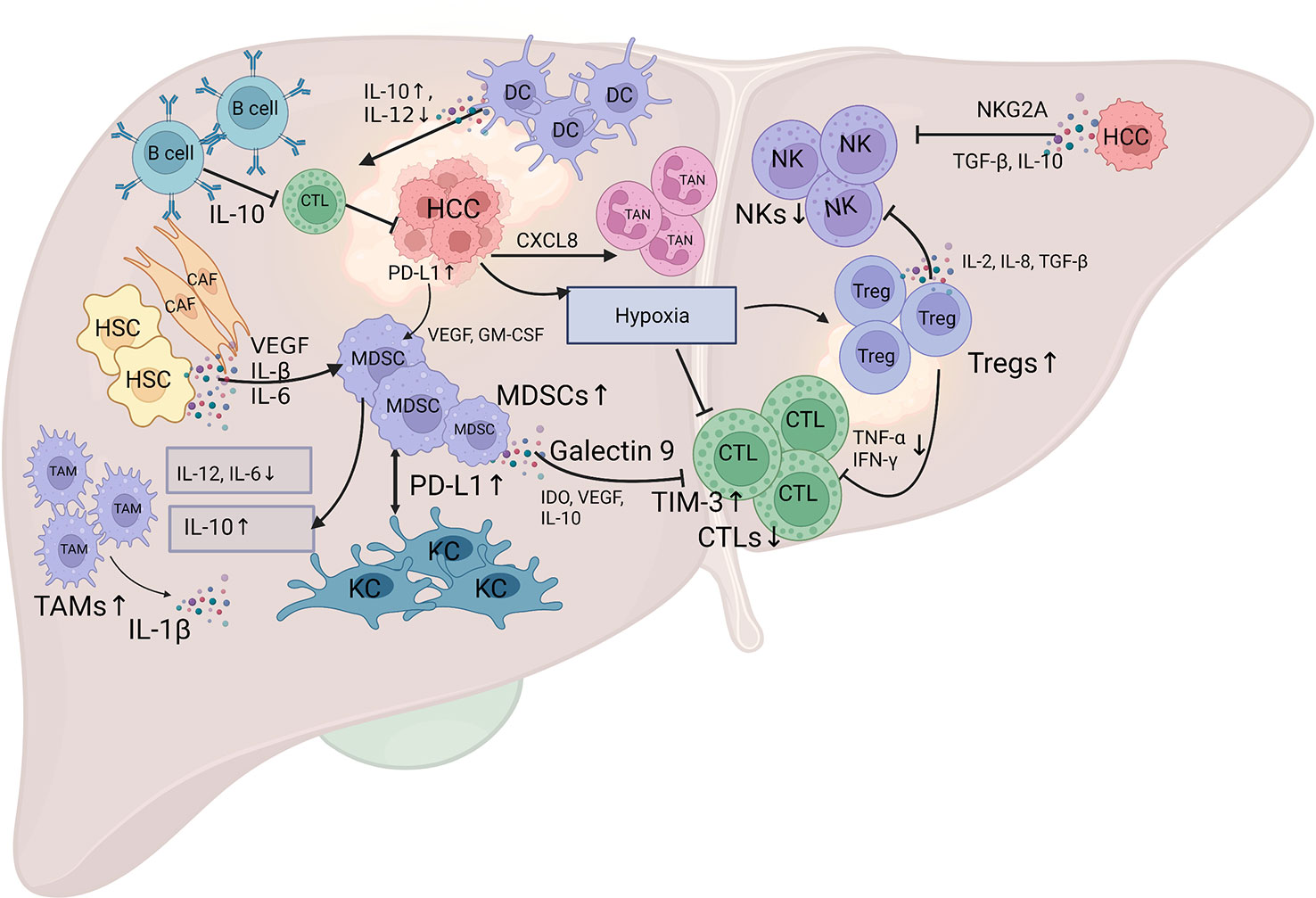
Figure 1 Schematic diagram of the landscape of the immune microenvironment in HCC. The infiltration of various subpopulations of immune cells, regulatory cytokines, and certain inhibitory signals mediate the specific immune response in HCC. The HCC tumor cells produce various factors, such as IDO, VEGF, IL-10, and hypoxia, to suppress the tumoricidal ability of CTL, and TGF-β, IL-10 and inhibitory receptors NKG2A, to suppress the tumoricidal ability of NK cells. Additionally, the HCC tumor cells secrete CXC chemokines, especially CXCL8, to attract TANs into the tumor stroma. The activation of PD-1 signaling in B cells promotes the production of IL-10, which inhibits the anti-tumor immunity of effector T cells. The crosstalk between MDSCs and TAMs results in a decreased secretion of IL-6 and IL-12, and an increased secretion of IL-10, which impair the cytotoxicity of CTL and NK cells. The aggregation of MDSCs in the tumor stroma is mediated by various cytokines, such as VEGF, IL-β, and IL-6 that produced by CAFs and HSCs, and VEGF and GM-CSF that produced by HCC tumor cells. MDSCs interact with KCs to induce PD-L1 expression on KCs, which interact with PD-1 on T cells to mediate immune evasion. Galectin-9 expression on MDSCs can bind to TIM-3 on T cells to induce T cell apoptosis. Tregs can inhibit CTL activation that mediated by reduced production of TNF-α and IFN-γ, and weaken the anti-tumor ability of NK cells through the production of IL-2, IL-8, and TGF-β. DCs can mediate IL-10 production and IL-12 reduction to suppress the antitumor response of CTL. CTL, cytotoxic T lymphocyte; NK, natural killer cells; TAM, tumor-associated macrophage; MDSC, myeloid-derived suppressor cell; TAN, tumor-associated neutrophil; CAF, cancer-associated fibroblast; HSC, hepatic stellate cell; KC, Kupffer cell; DC, dendritic cell; IDO, indoleamine 2,3-dioxygenase; VEGF, vascular endothelial growth factor; IL-10, interleukin-10; NKG2A, inhibitory receptors natural killer group 2A; TGF-β, transforming growth factor β; GM-CSF, granulocyte-macrophage colony-stimulating factor; ICI, immune checkpoint inhibitor. The figure was drawn at BioRender.com.
TILs are a heterogeneous cell population comprising T cells, NK cells, and B cells. HCC patients with T-cell accumulation after surgery experience lower recurrence and longer survival (34). The accumulation of CD8+ T cells, the major cytotoxic T lymphocytes (CTLs) in the liver, is associated with reduced tumor progression and improved survival of HCC patients (35). However, their tumor-killing ability is counteracted by various factors, such as indoleamine 2,3-dioxygenase (IDO), vascular endothelial growth factor (VEGF), interleukin 10 (IL-10), hypoxia, and CD4+ T-cell deprivation (35). CD4+ T cells secrete IFN-γ and TNF-α to remove senescent hepatocytes in a CXCR6-dependent manner, thus controlling hepatocarcinogenesis (36). Accordingly, the ROS-mediated apoptosis of CD4+ T cells promotes HCC in patients with NAFLD (37). Conversely, regulatory T cells (Tregs), a subtype of CD4+ T cells characterized by the expression of CD25 and forkhead box P3 (FoxP3), are capable of suppressing the tumor immune response and are associated with the metastasis and recurrence of HCC (38). Transforming growth factor β (TGF-β) induces HCC progression by promoting Treg polarization and the consequent repression of cytotoxic CD8+ T cells (39). This phenotype was reversed by the specific TGF-β inhibitor SM-16, which induced attenuated Treg infiltration and HCC regression (39). The CTL/Treg ratio represents the immune ecosystem. A high level of CTLs might overcome Treg-mediated immune suppression, thus suppressing HCC tumor growth (40).
NK cells are innate lymphocytes that mediate cancer immune surveillance and eradicate tumor cells without prior sensitization (41). They account for 30%-50% of hepatic lymphocytes and play an important role in preventing fibrosis and resisting cancer and viral infections through strong cytotoxicity and the production of IFN-γ (42). With the progression of HCC, the tumor killing activity of NK cells decreases, with the characteristics of reduced expression of the NK cytotoxic factors granzyme and perforin, as well as reduced secretion of the tumor killing-related cytokines TNF-α and IFN-γ (43). The infiltration of activated NK cells with high natural killer group 2 member D (NKG2D) expression correlates with prolonged survival of patients with cholangiocarcinoma (44). In contrast, the accumulation of immature and inactivated NK cells with poor catalytic activity is associated with HCC progression (45). The catalytic activity of NK cells is counteracted by several factors, such as the inhibitory receptor NKG2A and the immunosuppressive cytokines TGF-β and IL-10 (46, 47). Specifically, a subgroup of NK cells expressing CD49a was discovered to reside in HCC tissues, with high expression of the inhibitory receptors PD1 and CD96, and was correlated with a poor prognosis of HCC (48).
B cells directly present tumor-associated antigens to CD4+ T cells and CD8+ T cells, produce antibodies to promote the uptake of tumor antigens by TAMs and DCs, and secrete cytokines to promote antitumor immunity or directly kill tumor cells. In HCC, IgG+ memory B cells that accumulate at the edge of the invading tumor generate granzyme B, TRAIL, and IFN-γ, express surface markers of APCs, and cooperate with CD8+ T cells, resulting in a good prognosis (49). B-cell infiltration is positively correlated with survival and the response to immunotherapy (50). However, in patients with advanced HCC, activation of PD-1 signaling in B cells promotes IL-10 production and inhibits effector T-cell-mediated antitumor immunity, leading to early recurrence after tumor resection (51).
DCs are the major type of antigen-presenting cells that capture antigens, present them to T cells, and activate the adaptive immune response. In the healthy liver, most DCs are immature and activate effector T cells by inducing the production of antigen-specific CD8+ Tregs (52). This response might be attributed to the low level of IL-12 and high level of L-10 in the hepatic microenvironment (53). The liver contains two major groups of DCs, plasma cell-like DCs (pDCs) and conventional DCs (cDCs), which are further divided into cDC1s and cDC2s (54). cDC1s are essential for antitumor immune responses, and their presence in the tumor microenvironment is associated with a better prognosis (55), whereas cDC2s act as effective stimulators of nascent T helper (Th) cells (56). In contrast, immature pDCs in the tumor microenvironment are mainly associated with a poor prognosis for some types of human cancers (57). Compared to healthy controls, both circulating pDCs and cDCs showed a lower quantity and expression of human leukocyte antigen-DR (HLA-DR) and molecules CD80 and CD86 in the peripheral blood of HCC patients (58). Intratumorally infiltrated pDCs predict a poor prognosis among patients with primary HCC who have undergone resection, which might be caused by the induction of Treg cell and IL-17 production (59).
TAMs in HCC are derived from resident KCs in the liver and CD14+/CD16+ monocytes in the peripheral blood. They infiltrate into the tumor following stimulation with cytokines and chemokines, such as M-CSF, VEGF and CCL chemokines, secreted by tumor cells or mesenchymal cells (60). KCs are the major population of liver-resident macrophages with high expression of PD-L1 and low expression of CD80 and CD86, and are crucial for maintaining the immunosuppressive hepatic microenvironment (61). Antigen presentation by KCs induces the arrest of CD4+ T cells, expansion of antigen-specific Tregs and tolerogenic immunity. However, KCs lose tolerogenic markers in the injured liver. The deletion of KCs reduces hepatic tolerance to particulate antigens (61). Myeloid-derived monocytes can be attracted to infiltrate HCC, differentiate into TAMs, and acquire a phenotype similar to that of KCs. Monocyte-derived macrophages help CD4+ T cells remove senescent premalignant hepatocytes, thus inhibiting hepatocarcinogenesis (62). Conversely, senescent hepatocytes secrete CCL2 to attract CCR2+ monocytes into the liver, differentiate into macrophages and remove senescent premalignant hepatocytes (63). CD68 is a widely used marker to identify TAMs; however, it is not specific to distinguish liver-resident KCs and monocyte-derived macrophages. A recent study identified that Clecf4 and Tim4 are specifically expressed on the surface of KCs instead of monocyte-derived macrophages in HCC and thus might be used as markers for the TAM origin (64).
Similar to most cancers, TAMs in HCC are trained by specific tumor microenvironments and polarized to the M1 type and M2 type (65). M1 polarization is attributed to typical Th1 cytokines, such as interferon-γ (IFN-γ), and microbial components, such as lipopolysaccharide (LPS). M1 macrophages are classic active macrophages associated with pathogen clearance, pro-inflammatory cytokine liberation and matrix component lysis (66). M1 macrophages suppress tumor growth by directly killing tumor cells or presenting antigens to T cells. They efficiently produce pro-inflammatory cytokines, such as IL-1β, IL-6, and TNF-α, and are characterized by high expression of IL-12 and low expression of IL-10 (65). M1 macrophages secrete cytokines, such as CXCL9 and CXCL10, to attract Th1 cells, which in turn promote polarization to M1 macrophages (67). On the other hand, M2 polarization is induced by IL-4 and IL-13 (68) and is associated with parasite engulfment, anti-inflammatory cytokine release, matrix deposition and liver injury repair (66). M2 macrophages are characterized by low expression of IL-12 and high expression of IL-10 (65). Additionally, tumor-derived cytokines, such as CCL2 and CSF1, attract peripheral blood-derived macrophages to tumors and induce their differentiation to the M2 type (69). M2 macrophages are alternatively activated macrophages that secrete large amounts of anti-inflammatory cytokines, such as IL-8, IL-10, and TGF-β, to promote tumor vasculature development, CD8+ T-cell apoptosis, and the Th1 immune response, thus favoring tumor growth and metastasis (68). Notably, macrophages in peritumoral liver tissue also promote HCC progression and predict a poor prognosis (70).
TANs are tumor-infiltrating neutrophils that directly influence tumor development and progression. Similar to TAMs, TANs are classified into the N1 and N2 subtypes, which have antitumor and protumor roles, respectively (71). TGF-β is the key molecule driving polarization to the N2 type with an immunosuppressive response, while TGF-β inhibition promotes N1-type polarization characterized by enhanced cytotoxicity and inflammation (71, 72). In HCC patients, tumor cells secrete CXC chemokines, especially CXCL8, to attract neutrophil accumulation in the peritumor stroma (73). CXCL5 overexpression was also reported to be correlated with neutrophil infiltration in HCC tumors and predicted a poor prognosis (74). TANs may enhance the stemness of HCC cells through the miR-301b-3p/LSAMP/CYLD axis. Conversely, stem-like HCC cells secrete CXCL5 to facilitate TAN infiltration into the tumor. The positive feedback loop between TANs and HCC stem-like cells promotes HCC progression and metastasis (75). TANs also recruit TAMs and Tregs, thus creating an immunosuppressive microenvironment to facilitate HCC carcinogenesis (76). Moreover, TANs produce an increased number of neutrophil extracellular traps (NETs) to provoke an inflammatory response and sequester circulating cancer cells (CSCs) to promote HCC metastasis (77, 78).
MDSCs represent a heterogeneous population of immature myeloid cells that are mainly located in the bone marrow, spleen, peripheral blood, and tumors (79). Normally, myeloid cells differentiate into mature granulocytes, macrophages, or DCs to clear pathogens. However, myeloid cells cannot differentiate and become immature under conditions of inflammation or cancer. They are classified into M-MDSCs and PMN-MDSCs based on their phenotype and function (79). MDSCs create an immunosuppressive microenvironment through various mechanisms, including Treg differentiation, DC inhibition, macrophage M2 polarization, and oxidative stress (80–82). Compared with healthy volunteers or hepatitis patients, peripheral blood from HCC patients contained more MDSCs, which were subsequently revealed to indicate a poor prognosis (83). Additionally, the number of MDSCs increases with HCC progression, and these cells are closely associated with tumor size, tumor node metastasis and tumor multiplicity (84). MDSCs were reported to reduce the antigen presentation activity of KCs. In a coculture system containing MDSCs and KCs, KCs showed increased expression of CD86, CD274 and MHCII, increased secretion of IL-1β and IL-10, and reduced secretion of CCL2 and IL-18 (85). MDSC differentiation and infiltration in HCC are induced by activated hepatic stellate cells (HSCs) through the COX2-PGE2-EP4 pathway (86). HCC cells produce cytokines, such as VEGF, GM-CSF, and IL-1β, to attract MDSCs, and treatments targeting MDSCs might increase the sensitivity of immune checkpoint inhibitors (ICIs) promoting the antitumor response (87). MDSCs also reduce NK cell activity through a direct interaction to reduce NKp30 receptor expression in NK cells, which is independent of inducible nitric oxide synthase (iNOS) or arginase expression in MDSCs (88).
MCs are innate tissue-resident immune cells in the bone marrow lineage that were previously presumed to be central to the development of allergy and inflammation (89). However, MCs have recently been reported to affect tumor cells and the surrounding tumor-associated stroma, thereby altering tumor cell behavior and the tumor microenvironment (90). Previous studies have shown that total MC release, including effluent granules and histamine, reduces the viability and proliferation of HuH-6 cells (91). In addition, MCs actively participate in tumor cell clearance and tumor rejection through the release of IL-1, IL-4, IL-6, and TNF-α. Conversely, MCs release mediators, such as IL-8, FGF-2, VEGF, NGF, and PDGF, to favor tumorigenesis and angiogenesis (92). Based on their key role in orchestrating the cancer stroma, MCs have been recognized as a promising target for cancer immunotherapy.
HSCs account for 13% of all liver cells and are located in the Disse space between hepatocytes and endothelial cells, where substance exchange between hepatocytes and blood occurs (93). HSCs are mesenchymal cells with the characteristics of adipocytes, fibroblasts, and myocytes; thus, they function to maintain vitamin A homeostasis, induce hepatofibrosis, and modulate the hepatic bloodstream (94). Normally, HSCs remain quiet to maintain vitamin A homeostasis, with low activity to synthesize the extracellular matrix (ECM) (94). Once stimulated with liver-damaging factors, such as hepatic viruses and toxic chemicals, damaged hepatocytes secrete a number of cytokines, such as TGF-β, PDGF, and TNF, to induce lipid loss and morphological changes and are transdifferentiated into myofibroblasts (MFBs) that synthesize large amounts of ECM, leading to hepatofibrosis (94). TGF-β, which is derived from various types of cells, including macrophages and even HSCs themselves, is the key stimulator of HSC activation and MFBs (95). Activated HSCs produce angiogenic growth factors, stimulating angiogenesis and establishing a new vascular system in the tumor (96). Additionally, activated HSCs recruit MDSCs and Tregs and reduce the activation of CD8+ T cells (97). HSC-mediated inflammation/MFBs are well recognized as risk factors for HCC.
CAFs are detected in almost all types of solid tumors. CAFs in tumors produce a wide variety of factors to create an immunosuppressive microenvironment that favors tumor development (98, 99). A high level of CAF-specific biomarkers, such as α-smooth muscle cell (α-SMC) actin and fibroblast-activating protein (FAP), predicts a poor prognosis for HCC patients (100). CAFs have been reported to promote cancer progression and metastasis by producing cytokines (101). Activated HCC cells produce more CCL26 to promote higher levels of CAF infiltration that facilitate HCC progression (102). In turn, CAFs induce the epithelial-mesenchymal transition (EMT) process in HCC cells, thus promoting metastasis (103). Additionally, CAFs create an immunosuppressive microenvironment by inducing MDSCs and TANs, thus facilitating immune evasion (104, 105).
T-cell activation is a critical event in tumor immune therapy. Upon activation, negative costimulatory molecules, also called immune checkpoints, are expressed on the T-cell surface. The binding of these molecules to their ligands mediates coinhibitory signaling, resulting in the suppression of the proliferation and differentiation of T cells, thus promoting immune evasion (106). ICIs block ligand−receptor interactions, thus reactivating exhausted T cells to enhance the antitumor immune response (Figure 2).
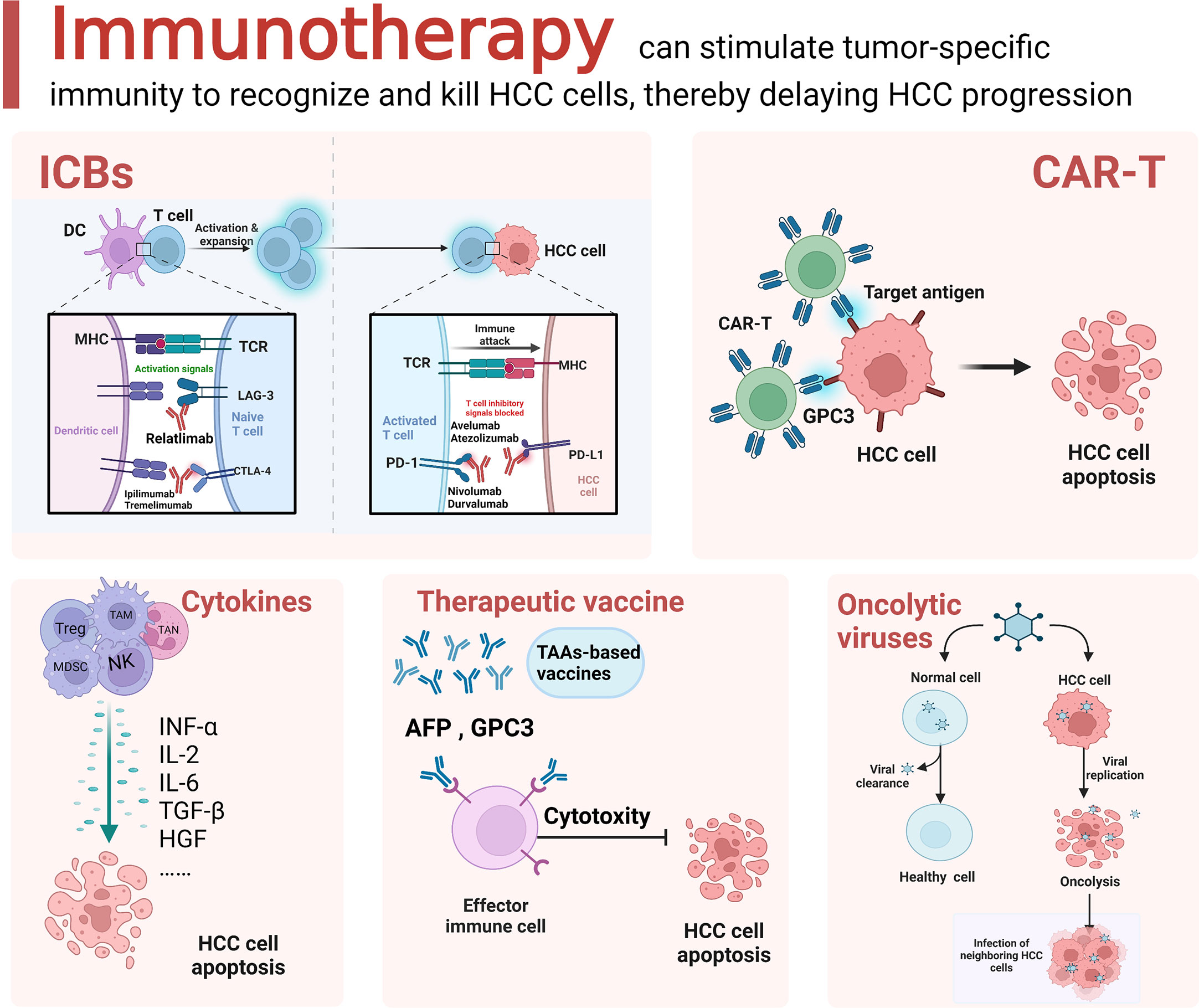
Figure 2 Schematic diagram of the current immunotherapeutic options for HCC. Immunotherapeutic approaches for HCC mainly include immune checkpoint blockade (ICB), adoptive cell therapy (ACT), cytokine based immune regimens, therapeutic vaccine and oncolytic virus. The figure was drawn at BioRender.com.
CTLA-4 is expressed primarily by Tregs and activated T cells and transmits inhibitory signals to effector T cells upon interacting with CD80/86 molecules expressed on antigen-presenting cell membranes (107). CTLA-4 outperforms CD28 in binding to B7 family costimulatory receptors (CD80/CD86) because of its 10-fold higher affinity (108), thus blocking B7/CD28-mediated costimulatory signaling (109). Tremelimumab and ipilimumab are CTLA-4 inhibitors that have shown anticancer effects on HCC patients. Tremelimumab has been shown to block signaling from the costimulatory receptor (CD80/CD86), preventing T-cell deactivation and subsequently avoiding the induction of tumor immune escape (110). In a phase II multicenter clinical trial (NCT01008358), patients with advanced HCC who were administered tremelimumab showed a disease control rate (DCR) of 76.4%, partial response (PR) rate of 17.6%, and time to progression (TTP) of 6.48 months, with good tolerance (13). Moreover, this treatment was also able to reduce the HCV load in the patient’s body (13). The most common adverse event of tremelimumab was pruritic rash, which was alleviated by antihistamine medication in most cases. These adverse events reached grade 3 or higher in 45% of cases but were unrelated to a decline in liver function (13).
PD-1 is the most important immune checkpoint molecule that belongs to the CD28 family. PD-L1 is the major ligand of PD-1 and is mainly expressed on the surface of tumor cells and DCs (111). The interaction between PD-1 and PD-L1 suppresses T-cell activity, induces apoptosis, and promotes Treg differentiation, thus creating an immunosuppressive microenvironment and leading to immune evasion (112). A higher level of the PD-L1 protein is detected in HCC tissues than in normal liver tissues and is closely associated with progression, recurrence, and the prognosis (113). Inhibitors of PD-1 and PD-L1 can block the interaction between these proteins and thus reverse T-cell activity for tumor killing. Following the clinical trials CheckMate-040 (9), KEYNOTE-224 (11), and IMbravel50 (114), the PD-1 inhibitors nivolumab and pembrolizumab and the PD-L1 inhibitor atezolizumab were approved for HCC treatment. In addition, other PD-1 inhibitors, such as tirelizumab and carrelizumab, and PD-L1 inhibitors, such as atirizumab, valileumab, and avelumab, have shown encouraging efficacy in clinical trials as HCC therapies (115).
Nivolumab (anti-PD-1) is a second-line ICI approved by the FDA in September 2017 for HCC patients who were treated with or intolerant to sorafenib. In a phase I/II clinical trial (CheckMate-040) with a cohort of 262 patients with advanced HCC, 48 patients in the dose-escalation cohort received 0.1–10 mg/kg nivolumab every 2 weeks for up to 2 years, while the other 214 patients in the dose-expansion cohort received 3 mg/kg nivolumab (9). The objective remission rates (ORRs) were 15% and 20% during the dose escalation and dose expansion periods, respectively. In the dose escalation cohort, the median OS was 15 months, and the median duration of efficacy was 17 months; in the dose expansion cohort, although the median OS could not be calculated, the median duration of efficacy was 9.9 months. This trial provides a rationale for the use of nivolumab as first-line therapy for patients with advanced HCC. A phase III trial (CheckMate-459) evaluated the efficacy of nivolumab versus sorafenib as a first-line treatment for patients with advanced HCC. The median OS was 16.4 months in the nivolumab group and 14.7 months in the sorafenib group (116).
Pembrolizumab (anti-PD-1) is a second-line ICI that was approved by the FDA in November 2018 for HCC patients who experienced treatment failure with sorafenib. A phase II clinical trial (KEYNOTE-224) with a cohort of 104 HCC patients reported an ORR of 17% (95% CI 11–26), median OS of 12.9 months (95% CI 9.7–15.5), and median progression-free survival (mPFS) of 4.9 months (11). In a randomized controlled phase III trial (KEYNOTErate-240), a median OS and PFS of 13.9 months and 3 months, respectively, were reported in the pembrolizumab group compared with 10.6 months and 2.8 months, respectively, in the placebo group. The ORR of the pembrolizumab group was 18.3%, which was significantly higher than the ORR of 4.4% for the placebo group (117).
Tislelizumab is a humanized antibody against PD-1 that was approved for HCC by the China National Medical Products Administration (NMPA) based on the results of the randomized phase II/III clinical trial RATIONALE-301 (NCT03412773). Although the OS of patients receiving the combination therapy was not superior to that of patients treated with sorafenib, the noninferiority median OS was 15.9 months for patients with combined therapy compared with 14.1 months for patients with sorafenib (P = 0.0398) (118). An international multicenter phase IA/IB clinical study (NCT02407990) indicated that tislelizumab monotherapy produced durable responses in patients with advanced tumors. The overall response rate (ORR) was 12.2%, and the disease control rate (DCR) was 51% in a cohort of 50 patients with HCC (119).
TIM-3 modulates the immune microenvironment of HCC by mediating T-cell depletion and apoptosis and enhancing Treg-mediated immune suppression (120). Preclinical studies have documented the antitumor efficacy of TIM-3 blockade alone or in combination with PD-1 inhibitors (121–123). Currently, a phase I clinical study of TSR-022, an anti-TIM-3 antibody, is ongoing in patients with advanced cancers, including HCC, as a first-in-man study (NCT02817633).
Although LAG-3 is not expressed on naive T cells, its expression can be induced on CD4+ and CD8+ T cells with sustained antigen stimulation (124, 125). LAG-3 blockade was shown to restore the cytotoxic activity of T cells for tumor killing (126, 127). Evidence suggests that LAG-3 signal transduction exerts a negative regulatory effect on Th1 cell proliferation and activation and cytokine secretion (128). In the tumor microenvironment, tumor cells use this pathway to avoid immune surveillance. Relatlimab was the first LAG-3 blocking antibody in clinical development (129). It was evaluated in clinical trials against solid tumors and hematologic malignancies as a monotherapy or in combination with nivolumab (anti-PD-1 mAb), and preliminary data showed that it was relatively well tolerated and exhibited clinical efficacy (130, 131).
TIGIT is expressed on activated T cells, NK cells, and B cells and inhibits immune cell-mediated antitumor responses (132, 133). TIGIT and the immunoactivating receptor CD226 trigger a signaling pathway similar to CD28/CTLA-4 to regulate tumor immune responses (134). In preclinical studies, anti-TIGIT antibodies combined with anti-PD-1/PD-L1 drugs had a synergistic effect (135, 136). According to related studies, TIGIT expression is elevated on lymphocytes infiltrating HCC tissues and may be involved in tumorigenesis and progression. At present, several exciting new drugs targeting TIGIT, such as tiragolumab, vibostolimab, etigilimab, ociperlimab, and domvanalimab, are in preclinical and clinical trials.
Based on the temporospatial expression patterns of CTLA-4 and PD-1/PD-L1, dual ICI therapy further improves therapeutic efficacy (15, 137). In the CheckMate-040 trial (Cohort 4), a combination of ipilimumab (anti-CTLA-4) and nivolumab (anti-PD-1) was administered to HCC patients with sorafenib treatment failure (15). The recommended regimen was ipilimumab (3 mg/kg) plus nivolumab (1 mg/kg) every 3 weeks for 4 cycles, followed by nivolumab (240 mg) biweekly, which yielded an ORR of 32% and a medium duration of response (DOR) of 17 months. This impressive result encouraged the approval of the ipilimumab plus nivolumab strategy after sorafenib by the FDA in March 2020 (137). In a phase I/II clinical trial of patients with advanced HCC who were previously treated with sorafenib, the regimen of tremelimumab (anti-CTLA4, 1500 mg) plus durvalumab (anti-PD-L1, 300 mg) every 4 weeks produced an ORR of 24% with a median OS of 18.7 months (17) (Table 1).
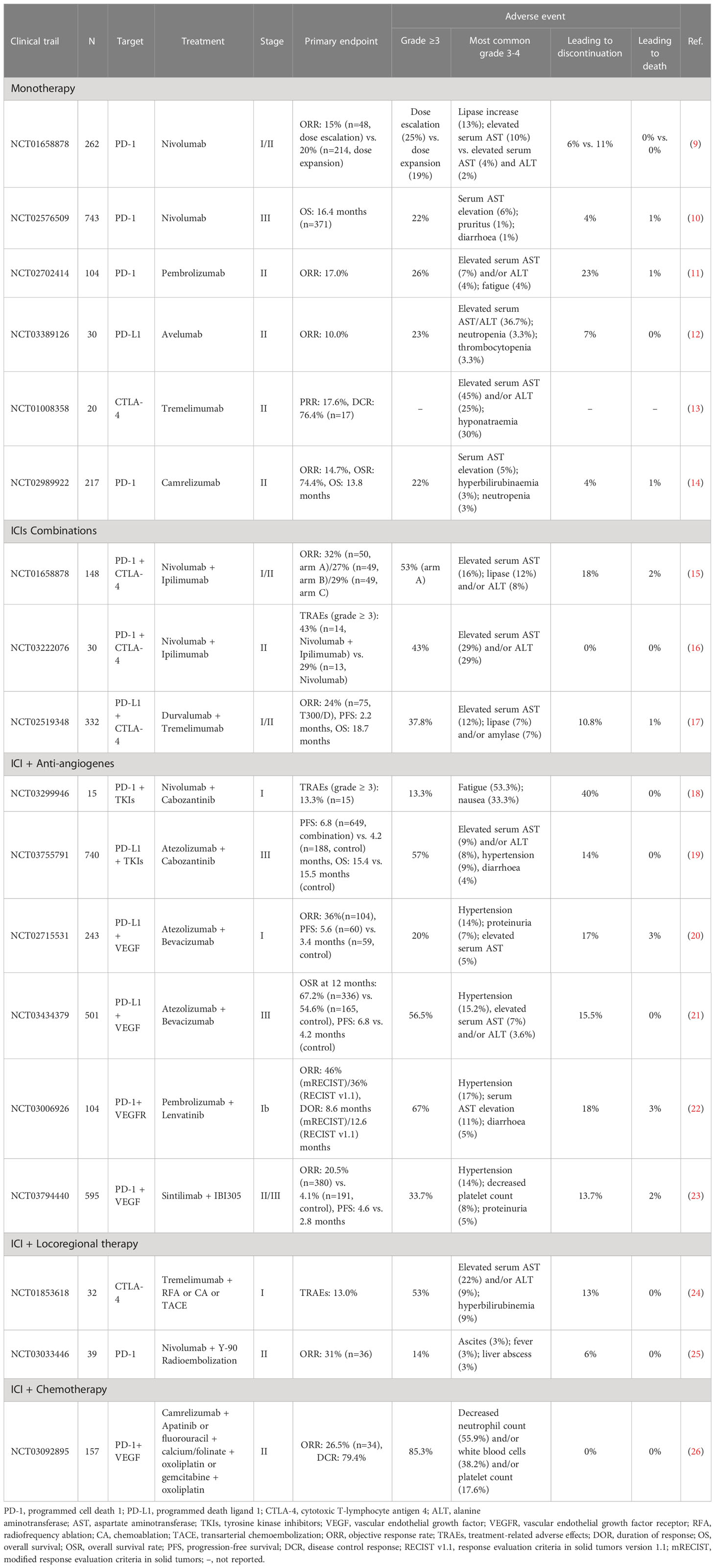
Table 1 Summary of clinical trials investigating ICIs combination therapy in HCC.
Molecular targeted drugs, such as sorafenib, regorafenib, and lenvatinib, promote tumor antigen presentation, thus activating T cells to improve the efficacy of immunotherapy (138). VEGF is a chemokine that induces angiogenesis and contributes to tumor invasion and metastasis. In addition, it helps recruit and induce the activation of Tregs, TAMs, and MDSCs to shape an immunosuppressive microenvironment (139). Although anti-VEGF therapy alone reduces the activity of Tregs and MDSCs and induces cytotoxic T-cell infiltration, it shows only modest efficacy against HCC (140, 141).
Based on a phase Ib clinical trial of patients with unresectable HCC, the PD-L1 inhibitor atezolizumab plus the VEGF inhibitor bevacizumab significantly suppressed HCC progression and improved PFS compared to atezolizumab monotherapy (20). In 2020, a randomized phase III clinical trial, IMbrave150 (NCT03434379), was approved by the FDA to compare the efficacy of atezolizumab plus bevacizumab (n = 336) with sorafenib (n = 165) in 501 treatment-naive patients with unresectable Child–Pugh A HCC (21, 142). The atezolizumab plus bevacizumab regimen mediated dual immune modulation by inhibiting PD-L1 and VEGF signaling and showed an OS benefit compared with sorafenib. The median survival time (MST) was 19.2 months (HR 0.66; 95% CI 17.0–23.7) in the atezolizumab plus bevacizumab group and 13.4 months in the sorafenib group. Currently, the atezolizumab plus bevacizumab combination is the standard regimen for patients with advanced Child–Pugh A HCC.
ORIENT-32 (NCT03794440) is a phase II/III clinical trial comparing the PD-1 inhibitor sintilimab plus a bevacizumab biosimilar, IBI305, with sorafenib in Chinese patients with unresectable or metastatic HCC (23). Sintilimab plus IBI305 produced both PFS and OS benefits compared with sorafenib, and had an acceptable safety profile. Another PD-1 inhibitor, camrelizumab, plus a tyrosine kinase inhibitor (TKI), apatinib, was reported to effectively prolong the survival of patients with mid- and advanced-stage HCC with good tolerance (143).
Lenvatinib is a multi-TKI that targets FGFRs, VEGFRs, PDGFRα, and KIT that was approved by the FDA in August 2018 as the first-line drug for patients with unresectable HCC (144). The combination of pembrolizumab and lenvatinib was evaluated in a phase Ib trial of untreated HCC patients, generating an ORR of 46%, a median OS of 22 months, and a median PFS of 9.3 months (22). Although grade ≥ 3 treatment-related adverse events (TRAEs) occurred in up to 67% of the patients, these toxicities were controlled by dose modifications and interruptions (22).
The combination of chemotherapy and immunotherapy produces multiple complex effects. While killing cancer cells, chemotherapy stimulates the release of antigens from tumor cells, reduces the number of immunomodulatory cells, and induces an increase in the number of antitumor effector T cells (145–147). A phase II study (NCT03434379) evaluated camrelizumab plus the FOLFOX4 or GEMOX regimens as a first-line strategy for patients with advanced primary HCC or cholangiocarcinoma (26). The ORR was 26.5%, and the disease control rate (DCR) was 79.4% in 34 patients. In terms of safety, 85.5% of HCC patients experienced TRAEs of grade ≥ 3, with myelosuppression and allergic reactions reported as the most common TRAEs.
Radiation therapy (RT) induces tumor cell apoptosis and the release of tumor-specific antigens to activate the immune system, suggesting that radiotherapy may exert a synergistic effect on tumor killing with immunotherapy. Numerous preclinical studies have shown that the combination of RT and ICIs produces synergistic effects and significant tumor suppression (148–151). A study reported results from five patients with large unresectable HCC who were treated with nivolumab after SBRT. They all achieved an ORR of 100%, with two patients in complete remission and three patients in partial remission (152). Another combination of Y-90-RE with nivolumab resulted in an ORR of 31%, and the combination proved to be safe and tolerable, with only a minority (11%) of treated patients with advanced HCC experiencing grade 3 or higher TRAEs (153).
TACE is one of the main treatments for patients with inoperable HCC. It causes ischemic necrosis of the tumor tissue by embolizing the artery with chemotherapeutic agents delivered to the hepatic artery. TACE therapy for patients with unresectable HCC has been proven effective, but the long-term survival of patients is not satisfactory. In a clinical trial evaluating the safety of TACE plus pembrolizumab for HCC, TACE plus pembrolizumab had a tolerable safety profile with no synergistic toxicity (154).
RFA is the first-line therapy for early local ablation and plays a leading role in the treatment of HCC. A large body of evidence suggests that ablative therapy not only physically eliminates tumors but also activates systemic antitumor immune responses and suppresses immunosuppressive effects (155–157). In a clinical study evaluating the efficacy of tremelimumab plus RFA for advanced HCC, it showed that 5 of the 19 evaluable patients achieved partial remission, 12 of 14 HCV+ patients had a significantly reduced viral load, and patients who experienced a clinical benefit had significantly increased CD8+ T-cell numbers, with a median OS of 12.3 months and median time to tumor progression of 7.4 months (24).
Although ICIs activate antitumor immunity, they inevitably alter the original immune homeostasis, resulting in a hyperactive immune response and a series of adverse reactions similar to autoimmune diseases. irAEs involve multiple organs but mainly affect the skin, liver, lung, gastrointestinal tract and endocrine organs (158). HCC usually develops from cirrhosis and has systemic manifestations (159). Therefore, the symptoms caused by multiple organs overlap with irAEs and increase their severity. Overall, the PD1 inhibitors pembrolizumab and nivolumab result in severe irAEs in 10-20% of patients with advanced HCC and over 30% of patients with other cancer types (160). However, the incidence of liver-related irAEs is higher in patients with HCC than in patients with other tumor types. Anti-PD1 monotherapy induces hepatitis-related enzymes in 14% of HCC patients but in only 3% of patients with other cancer types (161, 162).
Principally, irAEs are attributed to the hyperactive immune system. The role of the CTLA-4 and PD-1 checkpoints in autoimmunity and immune homeostasis is fundamental to our understanding of the mechanisms of irAEs. Although the precise mechanism of irAEs is not fully understood, the incidence of irAEs involves complex dynamic changes, including the regulation of effector T-cell and Treg activity, toxic effects of macrophages and granulocytes, cytokine release, and antibody production by B cells (160). The life-threatening nature of irAEs limits the application of ICIs. Therefore, biomarkers predicting the severity of irAEs are key points to improve patient survival and quality of life. The possible biomarkers for irAEs are summarized below. (1) Autoimmune diseases are caused by hyperactive or suppressive immune-related cells, and the immunosuppressive microenvironment is a characteristic of tumor initiation and progression. The investigation of the heterogeneity of immune-related cells may promote a better understanding of immune disorders, thus improving the prediction and management of irAEs. (2) Cytokines, such as interleukin, TNF-α, and IFN-γ, are indispensable for cancer immunotherapy. Their secretion changes remarkably with ICI treatment and might be involved in irAEs. Previous studies examining immunotherapy revealed that the levels of cytokines are closely correlated with the incidence of irAEs, suggesting the possibility that cytokines may serve as biomarkers for irAEs. (3) Tumorigenesis is acknowledged as a consequence of genetic mutations, which may interfere with communication between immune cells and tumor cells. NGS and single-cell RNA sequencing technology may enable the extensive screening of genetic mutations as potential biomarkers for irAEs. (4) Serum biomarkers reflecting the tumor burden were revealed to be associated with the response to ICIs. Therefore, studies investigating the prediction of irAEs are worthwhile.
Adoptive cell therapy (ACT) is a host cell-based immunotherapy that endows lymphocytes with long-lasting antitumor immunity and produces low cytotoxic effects on normal cells. The basic principle involves isolating host lymphocytes, followed by their enrichment, activation, and amplification in vitro via stimulation with cytokines or peptides or via genetic modification. The modified cells are then infused into the host to exert cytotoxic effects on autologous tumor cells (163). The most commonly used ACT-based therapies for HCC include chimeric antigen receptor (CAR)-modified T cells, cytokine-induced killing (CIK) cells, and T-cell receptor (TCR)-engineered T cells (Table 2).

Table 2 Summary of clinical trials investigating ACT in HCC.
CAR is a fusion protein composed of a tumor-associated antigen (TAA) binding domain, an extracellular hinge region, a transmembrane domain, and an intracellular signal domain (164, 165). Following CAR modifications, T lymphocytes recognize a broader range of target antigens than the natural TCR (164) and participate in the identification of cancer cells independently of the major histocompatibility complex (MHC). Glypican-3 (GPC3) is specifically expressed on the surface of HCC cells and was used as the TAA for CAR-T cell construction (166, 167). CAR-T cells targeting GPC3 have proven effective in vitro and in animal models, with a clinically significant prolongation of survival (168). In recent phase I trials (NCT02395250 and NCT03146234) comprising 13 patients with advanced HCC receiving CAR-T-cell infusions, two patients showed partial responses, and one patient showed persistent stable disease (SD) (27). In another ongoing phase I clinical trial (NCT03198546), complete tumor disappearance was observed in patients with GPC3+ advanced HCC after 30 days of treatment with intratumor injections of 19.9 × 108 CAR-GPC3 T cells (169).
Although CAR-T cell therapy has made breakthroughs in the treatment of hematologic malignancies through its targeting specificity, CAR-T cell therapy for solid tumors, including HCC, still faces many challenges. These challenges are mainly attributed to the intricate immune microenvironment within solid tumors, with specific histopathological features leading to hypoxia, a low pH, immunosuppressive cells, an increased number of inhibitory checkpoints, and higher levels tumor-derived cytokines (170, 171). These characteristics make T-cell infiltration in local tumor tissues more challenging (172), thus affecting the cytotoxic antitumor effect of T cells. A number of studies have been conducted to overcome the detrimental effects of the tumor microenvironment in solid tumors, including the modification of CAR-T cells by knocking down PD-1 expression and the use of CAR-T cells in combination with ICIs (173–175).
CIK cells are heterogeneous and non-MHC-restricted lymphocytes consisting of T cells (CD3+CD56−), NK cells (CD3−CD56+), and NKT cells (CD3+CD56+) (176). CD3+CD56+ NKT cells are the main effector cells exerting antitumor activity due to the higher proportion of CD8+ cells, more differentiated effector cells, and higher granzyme A content (177). CIK cells are produced by the expansion and activation of peripheral blood mononuclear cells (PBMCs) in response to stimulators such as an anti-CD3 antibody, IFN-γ, and IL-2 (178). With the advantages of non-MHC-restricted cytotoxicity, dual function of T cells and NK cells, high proliferation rate, high specificity for tumors, and low impact on normal cells (179), CIK cells are applied to both hematological malignancies and solid tumors.
A randomized phase II trial of CIK cells for HCC revealed that CIK cell therapy significantly prolonged OS and recurrence-free survival (RFS) compared with the control group (31). In a multicenter phase III clinical trial consisting of 230 HCC patients, adjuvant immunotherapy with activated CIK cells was administered to HCC patients that treated with surgical resection, RFA, or a percutaneous ethanol injection. The median PFS was 44 months in the CIK group and 30 months in the control group. The CIK group showed lower HRs for all-cause death (0.21; 95% CI 0.06–0.75; P = 0.008) and cancer-related death (0.19; 95% CI 0.04–0.87; P = 0.02). Although the overall frequency of adverse events (AEs) was higher in the CIK group than in the control group (62% vs. 41%; P = 0.002), the severe AE incidence did not differ significantly (7.8% vs. 3.5%; P = 0.15) (30). A 5-year follow-up study (NCT01890291) revealed the sustained therapeutic efficacy of autologous CIK cell immunotherapy against HCC. The study enrolled 162 patients, with 89 in the CIK group and 73 in the control group. After an average follow-up of 68.5 months, the recurrence-free survival (RFS) rate in the CIK group was 44.8% compared to 33.1% in the control group. Furthermore, the CIK group showed a significant reduction in the risk of all-cause death with a hazard ratio of 0.33 (95% CI: 0.15-0.76, P = 0.006) (180). To date, several clinical trials have evaluated the role of CIK cells in the treatment of HCC and have obtained encouraging results, suggesting that CIK cells are a very promising immunotherapy for the treatment of HCC (29, 181, 182).
TCR-T cell therapy is performed by extracting the α and β chain genes encoding the TCR from tumor antigen-induced effector T cells, introducing them into mature T cells using genetic engineering techniques, and then infusing the cells back into patients who lack tumor antigen-specific responsive T cells. TCR-T cells perform antigen recognition in an MHC-dependent manner (183) and have a wider range of applications than CAR-T cells. Hepatitis B virus (HBV) infection is the leading cause of HCC in Asia, accounting for 80% of cases (184). HBV infection stimulates the production of high-affinity T-cell receptors (TCRs), which may provide a potential therapeutic strategy for targeting HBV-infected cells. However, a major concern is that viral antigens may also be expressed on nonmalignant liver tissue in patients with HBV-associated HCC, posing a risk of severe liver injury (185). Current candidate TAAs for HCC TCR-T therapy include New York esophageal squamous cell carcinoma 1 (NY-ESO-1) (186), alpha-fetoprotein (AFP) (187), GPC3 (188), and human telomerase reverse transcriptase (hTERT) (189). NY-ESO-1 is the most commonly used target antigen in clinical trials of TCR-T cells. NY-ESO-1 is expressed in approximately 45% of HCC tumors (190); therefore, NY-ESO-1-specific TCR-T cells have been used in clinical therapeutic trials for HCC (NCT01967823, NCT02869217, and NCT03159585). Recently, Docta et al. obtained an HLA-A2+/AFP-specific TCR (191), and a clinical trial of this TCR-T cell population for HCC is underway to confirm its effectiveness in treating HCC by examining AFP expression and T-cell infiltration in biopsied tissues (NCT03132792). In contrast to AFP, fewer studies have been conducted on GPC3-specific TCRs. A study obtained HLA-A2/GPC3367-specific TCRs by transfecting HLA-A2 with GPC3 into HLA-A2-negative donor DCs and coculturing them with host T cells; the expression of this receptor on T cells enabled them to recognize and kill GPC3-positive HCC cells (188).
Cytokines are an important component of the immune system and promising therapeutic targets in liver diseases due to their importance in modulating immune and inflammatory responses (192). In recent years, many cytokines, such as IL-2, IL-15, IL-21, GM-CSF, and IFN-α, have proven effective in preclinical tumor models and are used as potential options for cancer immunotherapy (193). With the ability to mediate antiviral, antitumor, and other immune responses, IFN-α reduces the mortality and early relapse rates of HCC patients after curative treatment (194). In addition, adjuvant IFN therapeutic efficacy on postoperative recurrence differed between patients with HBV-related HCC and patients with HCV-related HCC; therefore, the adjuvant IFN strategy should be used according to the hepatitis background (195). A preclinical study suggested that hepatocyte growth factor (HGF) may be involved in the progression of HCC (196). Two studies have documented that pretreatment serum HGF levels are potential independent predictors of OS in prospective cohorts of HCC patients (197, 198). Notably, lower HGF levels at the initiation of treatment are often associated with longer OS and PFS benefits from sorafenib treatment (198). IL-2 is capable of promoting T-cell proliferation and activation, which is essential for tumor-killing activity. In a clinical trial of patients with inoperable HCC, IL-2 administration prolonged OS (199). A study of patients with HCC treated with sorafenib therapy evaluated the prognostic value of pretreatment serum IL-6 levels. In both the discovery and validation cohorts, higher pretreatment serum IL-6 levels (threshold: 4.28 pg/mL) were an independent predictor of shorter OS. However, this result was unrelated to the efficacy of sorafenib, as PFS and TTP were similar, regardless of pretreatment IL-6 levels. Additionally, pretreatment IL-6 levels were not associated with macrovascular invasion or extrahepatic metastasis (200). A recent study using cellular models showed that inhibition of IL-6-related pathways may reduce resistance to sorafenib (201). TGF-β serves as a tumor suppressor at early stages of tumorigenesis. However, with tumor progression, TGF-β loses its growth-inhibitory ability and initiates the EMT and cell migration (202). Galunisertib (LY2157299), a novel TGF-β inhibitor, was evaluated in a phase II trial of HCC patients with disease progression on sorafenib therapy. The median OS for the cohort with AFP levels < 200 ng/mL was 17 months compared to 8.4 months for patients with AFP levels > 200 ng/mL. Notably, an obvious improvement in OS was noted in patients with TGF-β1 levels that were reduced by more than 20% (203, 204). In addition, preclinical studies have shown that galunisertib modulates the inhibition of T-cell proliferation and shows potential synergy with PD-1/PD-L1 inhibitors (205). Galunisertib is currently being evaluated for efficacy and safety in the treatment of HCC patients in several ongoing clinical trials (NCT02240433, NCT01246986, NCT02178358, NCT02906397, and NCT02423343). However, only high concentrations of cytokines are able to reach the tumor microenvironment, and parenterally administered cytokines have difficulty reaching effective concentrations and exerting their effects on the tumor microenvironment. The combination of cytokines with other immunotherapies is currently being clinically investigated to enhance their antitumor capacity and avoid these obstacles.
Therapeutic vaccines are active immunotherapies in which TAAs (including peptides, tumor cells, and viruses) are introduced into patients to overcome the immunosuppressive tumor microenvironment and further activate the patient’s immune system to produce a tumor-specific response with enhanced potency (206). The identification of target TAAs is the most critical step in exerting a specific antitumor response. Several HCC peptide vaccines targeting TAAs, such as GPC3, AFP, and hTERT, have been developed for use as immunotherapeutic targets for vaccines (207).
The GPC3 peptide is considered an ideal vaccine for HCC treatment due to its overexpression on HCC tumor cells and low level on normal cells (208). A phase I clinical study of patients with advanced HCC showed that the GPC3 vaccine was well tolerated and induced a GPC3-specific CTL response in 30/33 patients (91%) (209). In a phase II study of 41 HCC patients who underwent surgical resection, adjuvant GPC3 vaccine administration significantly reduced the recurrence rate (1- and 2-year recurrence rates of 24% vs. 48% and 52.4% vs. 61.9%, respectively; P = 0.047, 0.387). In addition, the postoperative administration of the GPC3 peptide vaccine was shown to prolong RFS (210).
AFP is a classic diagnostic marker for HCC due to its specific expression on HCC cells (211). AFP blockade reduces the proliferation and promotes apoptosis of liver cancer cells (212). In a clinical trial, two AFP-positive HCC patients received an AFP vaccine, which showed good tolerance and safety without clinically significant adverse events (213).
Oncolytic viruses (OVs) are a group of genetically modified viruses that have acquired the ability to kill tumor cells in a targeted manner without damaging normal cells. During the killing of tumor cells by oncolytic viruses, the antitumor immune response is activated in the body, and the inflammatory response generated by viral infection further promotes the immune response (214). OVs are usually divided into two categories, namely, natural viruses (including Newcastle disease virus and eutherian virus) and genetically modified viruses (including adenovirus and herpes simplex virus) (215). JX-594 is an engineered oncolytic virus that selectively targets tumors by inactivating viral thymidine kinase (vTK) (216). In a phase II clinical trial of the cowpox vaccine JX-594 for the treatment of HCC, JX-594 was well tolerated even at high doses. The study showed a positive correlation between the dose of JX-594 and survival benefit, with a longer median OS for the high-dose group than the low-dose group (14.1 months and 6.7 months for the high- and low-dose groups, respectively) (216). Currently, two clinical studies of OVs in combination with anti-PD-1 antibodies for the treatment of HCC (NCT04612504 and NCT05061537) are ongoing. A recent study showed that treatment with the cancer-favoring vaccinia virus (CVV) resulted in a significantly lower incidence of metastasis than treatment with sorafenib alone in an animal model (217). Although OVs for HCC show promising clinical applications, more clinical trials are needed to further validate their efficacy and safety.
The tumor microenvironment and cancer immunotherapy have been at the forefront of cancer research in recent decades. Immune cells in the immune microenvironment of HCC tumors provide new targets for the next generation of immunotherapy. Immune checkpoint therapies targeting CTLA-4 and PD-1/PD-L1 are the main drugs used in the treatment of HCC, and other immunotherapies are rapidly evolving, including CAR-T cells, TCR-T cells, therapeutic vaccines, and OVs. Although response rates to ICIs in HCC patients are modest, more new treatment modalities will emerge as our understanding of the interaction of ICIs with adaptive and innate immune responses improves. Many strategies have been developed that combine ICIs with other first-line therapies (including chemotherapy, local therapies, and targeted therapies) in ideal combinations, and these new therapies have provided palliation and prolonged the survival of patients with HCC.
MZ and HH wrote the manuscript. FH and XF revised and edited the manuscript. All authors read and approved the final version of the manuscript.
This work is supported by the Startup Research Grant (CYFY-GQ50), the Scientific Research Grant of the First Affiliated Hospital of Chengdu Medical College (CYFY2021ZD02), the Scientific Research Grant of Chengdu Medical College (CYZZD22-03), the Scientific Research Grant of Chengdu Municipal Health Commission (2022099) granted to MZ.
We thank members of the Fu laboratory for critical reading and discussion.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Vogel A, Meyer T, Sapisochin G, Salem R, Saborowski A. Hepatocellular carcinoma. Lancet (2022) 400(10360):1345–62. doi: 10.1016/S0140-6736(22)01200-4
2. Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. nature reviews. Dis Primers (2021) 7(1):6. doi: 10.1038/s41572-020-00240-3
3. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell (2015) 27(4):450–61. doi: 10.1016/j.ccell.2015.03.001
4. Rizvi S, Wang J, El-Khoueiry AB. Liver cancer immunity. Hepatology (2021) 73(Suppl 1):86–103. doi: 10.1002/hep.31416
5. Ding P, Peng B, Ming YZ. Cross-talk between hepatic stellate cells and T lymphocytes in liver fibrosis. Hepatobil Pancreat Dis Int (2021) 20(3):207–14. doi: 10.1016/j.hbpd.2021.04.007
6. Protzer U, Maini MK, Knolle PA. Living in the liver: hepatic infections. Nat Rev Immunol (2012) 12(3):201–13. doi: 10.1038/nri3169
7. Flecken T, Schmidt N, Hild S, Gostick E, Drognitz O, Zeiser R, et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8(+) T-cell responses in hepatocellular carcinoma. Hepatol (Baltimore Md.) (2014) 59:1415–26. doi: 10.1002/hep.26731
8. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
9. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet (2017) 389(10088):2492–502. doi: 10.1016/S0140-6736(17)31046-2
10. Yau T, Park JW, Finn RS, Cheng AL, Mathurin P, Edeline J, et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol (2022) 23(1):77–90. doi: 10.1016/S1470-2045(21)00604-5
11. Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol (2018) 19(7):940–52. doi: 10.1016/S1470-2045(18)30351-6
12. Lee DW, Cho EJ, Lee JH, Yu SJ, Kim YJ, Yoon JH, et al. Phase II study of avelumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. Clin Cancer Res (2021) 27(3):713–8. doi: 10.1158/1078-0432.CCR-20-3094
13. Sangro B, Gomez-Martin C, de la Mata M, Iñarrairaegui M, Garralda E, Barrera P, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis c. J Hepatol (2013) 59(1):81–8. doi: 10.1016/j.jhep.2013.02.022
14. Qin S, Ren Z, Meng Z, Chen Z, Chai X, Xiong J, et al. Camrelizumab in patients with previously treated advanced hepatocellular carcinoma: a multicentre, open-label, parallel-group, randomised, phase 2 trial. Lancet Oncol (2020) 21(4):571–80. doi: 10.1016/S1470-2045(20)30011-5
15. Yau T, Kang YK, Kim TY, El-Khoueiry AB, Santoro A, Sangro B, et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: the CheckMate 040 randomized clinical trial. JAMA Oncol (2020) 6(11):e204564. doi: 10.1001/jamaoncol.2020.4564
16. Kaseb AO, Hasanov E, Cao HST, Xiao L, Vauthey JN, Lee SS, et al. Perioperative nivolumab monotherapy versus nivolumab plus ipilimumab in resectable hepatocellular carcinoma: a randomised, open-label, phase 2 trial. Lancet Gastroenterol Hepatol (2022) 7(3):208–18. doi: 10.1016/S2468-1253(21)00427-1
17. Kelley RK, Sangro B, Harris W, Ikeda M, Okusaka T, Kang YK, et al. Safety, efficacy, and pharmacodynamics of tremelimumab plus durvalumab for patients with unresectable hepatocellular carcinoma: randomized expansion of a phase I/II study. J Clin Oncol (2021) 39(27):2991–3001. doi: 10.1200/JCO.20.03555
18. Ho WJ, Zhu Q, Durham J, Popovic A, Xavier S, Leatherman J, et al. Neoadjuvant cabozantinib and nivolumab converts locally advanced HCC into resectable disease with enhanced antitumor immunity. Nat Cancer (2021) 2(9):891–903. doi: 10.1038/s43018-021-00234-4
19. Kelley RK, Rimassa L, Cheng AL, Kaseb A, Qin S, Zhu AX, et al. Cabozantinib plus atezolizumab versus sorafenib for advanced hepatocellular carcinoma (COSMIC-312): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol (2022) 23(8):995–1008. doi: 10.1016/S1470-2045(22)00326-6
20. Lee MS, Ryoo BY, Hsu CH, Numata K, Stein S, Verret W, et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): an open-label, multicentre, phase 1b study. Lancet Oncol (2020) 21(6):808–20. doi: 10.1016/S1470-2045(20)30156-X
21. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med (2020) 382(20):1894–905. doi: 10.1056/NEJMoa1915745
22. Finn RS, Ikeda M, Zhu AX, Sung MW, Baron AD, Kudo M, et al. Phase ib study of lenvatinib plus pembrolizumab in patients with unresectable hepatocellular carcinoma. J Clin Oncol (2020) 38(26):2960–70. doi: 10.1200/JCO.20.00808
23. Ren Z, Xu J, Bai Y, Xu A, Cang S, Du C, et al. Sintilimab plus a bevacizumab biosimilar (IBI305) versus sorafenib in unresectable hepatocellular carcinoma (ORIENT-32): a randomised, open-label, phase 2-3 study. Lancet Oncol (2021) 22(7):977–90. doi: 10.1016/S1470-2045(21)00252-7
24. Duffy AG, Ulahannan SV, Makorova-Rusher O, Rahma O, Wedemeyer H, Pratt D, et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J Hepatol (2017) 66(3):545–51. doi: 10.1016/j.jhep.2016.10.029
25. Tai D, Loke K, Gogna A, Kaya NA, Tan SH, Hennedige T, et al. Radioembolisation with Y90-resin microspheres followed by nivolumab for advanced hepatocellular carcinoma (CA 209-678): a single arm, single centre, phase 2 trial. Lancet Gastroenterol Hepatol (2021) 6(12):1025–35. doi: 10.1016/S2468-1253(21)00305-8
26. Qin S, Chen Z, Liu Y, Xiong J, Zou J. A phase II study of anti–PD-1 antibody camrelizumab plus FOLFOX4 or GEMOX systemic chemotherapy as first-line therapy for advanced hepatocellular carcinoma or biliary tract cancer. J Clin Oncol (2019) 37(15_suppl):4074–4. doi: 10.1200/JCO.2019.37.15_suppl.4074
27. Shi D, Shi Y, Kaseb AO, Qi X, Zhang Y, Chi J, et al. Chimeric antigen receptor-Glypican-3 T-cell therapy for advanced hepat ocellular carcinoma: results of phase I trials. Clin Cancer Res (2020) 26(15):3979–89. doi: 10.1158/1078-0432.CCR-19-3259
28. Fang W, Fu Q, Zhao Q, Zheng Y, Liu L, Li Z, et al. Phase I trial of fourth-generation chimeric antigen receptor T-cells targeting glypican-3 for advanced hepatocellular carcinoma. J Clin Oncol (2021) 39(15_suppl):4088–8. doi: 10.1200/JCO.2021.39.15_suppl.4088
29. Xu L, Wang J, Kim Y, Shuang Z-y, Zhang Y-j, Lao X-m, et al. A randomized controlled trial on patients with or without adjuvant autologous cytokine-induced killer cells after curative resection for hepatocellular carcinoma. OncoImmunology (2016) 5(3):e1083671. doi: 10.1080/2162402X.2015.1083671
30. Lee JH. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology (2015) 148(7):1383–1391.e6. doi: 10.1053/j.gastro.2015.02.055
31. Yu X, Zhao H, Liu L, Cao S, Ren B, Zhang N, et al. A randomized phase II study of autologous cytokine-induced killer cells in treatment of hepatocelluar carcinoma. J Clin Immunol (2014) 34(2):194–203. doi: 10.1007/s10875-013-9976-0
32. Meng F, Zhao J, Tan AT, Hu W, Wang SY, Jin J, et al. Immunotherapy of HBV-related advanced hepatocellular carcinoma with short-term HBV-specific TCR expressed T cells: results of dose escalation, phase I trial. Hepatol Int (2021) 15(6):1402–12. doi: 10.1007/s12072-021-10250-2
33. Chen W, Cheng J, Zheng X, Yang F, Fam R, Koh S, et al. 273 phase i study of LioCyx-m, autologous hepatitis b virus (HBV)-specific T cell receptor (TCR) T-cells, in recurrent HBV-related hepatocellular carcinoma (HCC) post-liver transplantation. J ImmunoTher Cancer (2020) 8(Suppl 3):A167. doi: 10.1136/jitc-2020-SITC2020.0273
34. Unitt E, Marshall A, Gelson W, Rushbrook SM, Davies S, Vowler SL, et al. Tumour lymphocytic infiltrate and recurrence of hepatocellular carcinoma following liver transplantation. J Hepatol (2006) 45(2):246–53. doi: 10.1016/j.jhep.2005.12.027
35. Garnelo M, Tan A, Her Z, Yeong J, Lim CJ, Chen J, et al. Interaction between tumour-infiltrating b cells and T cells controls the progression of hepatocellular carcinoma. Gut (2017) 66(2):342–51. doi: 10.1136/gutjnl-2015-310814
36. Mossanen JC, Kohlhepp M, Wehr A, Krenkel O, Liepelt A, Roeth AA, et al. CXCR6 inhibits hepatocarcinogenesis by promoting natural killer T- and CD4(+) T-Cell-Dependent control of senescence. Gastroenterology (2019) 156(6):1877–1889.e4. doi: 10.1053/j.gastro.2019.01.247
37. Brown ZJ, Fu Q, Ma C, Kruhlak M, Zhang H, Luo J, et al. Carnitine palmitoyltransferase gene upregulation by linoleic acid induces CD4(+) T cell apoptosis promoting HCC development. Cell Death Dis (2018) 9(6):620. doi: 10.1038/s41419-018-0687-6
38. Huang Y, Wang FM, Wang T, Wang YJ, Zhu ZY, Gao YT, et al. Tumor-infiltrating FoxP3+ tregs and CD8+ T cells affect the prognosis of hepatocellular carcinoma patients. Digestion (2012) 86(4):329–37. doi: 10.1159/000342801
39. Shen Y, Wei Y, Wang Z, Jing Y, He H, Yuan J, et al. TGF-β regulates hepatocellular carcinoma progression by inducing treg cell polarization. Cell Physiol Biochem (2015) 35(4):1623–32. doi: 10.1159/000373976
40. Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, et al. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol (2007) 25(18):2586–93. doi: 10.1200/JCO.2006.09.4565
41. Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer (2016) 16(1):7–19. doi: 10.1038/nrc.2015.5
42. Notas G, Kisseleva T, Brenner D. NK and NKT cells in liver injury and fibrosis. Clin Immunol (2009) 130(1):16–26. doi: 10.1016/j.clim.2008.08.008
43. Hosseinzadeh F, Ai J, Ebrahimi-Barough S, Seyhoun I, Hajifathali A, Muhammadnejad S, et al. Natural killer cell expansion with autologous feeder layer and anti-CD3 antibody for immune cell therapy of hepatocellular carcinoma. Asian Pac J Cancer Prev (2019) 20(12):3797–803. doi: 10.31557/APJCP.2019.20.12.3797
44. Tsukagoshi M, Wada S, Yokobori T, Altan B, Ishii N, Watanabe A, et al. Overexpression of natural killer group 2 member d ligands predicts favorable prognosis in cholangiocarcinoma. Cancer Sci (2016) 107(2):116–22. doi: 10.1111/cas.12853
45. Zhang QF, Yin WW, Xia Y, Yi YY, He QF, Wang X, et al. Liver-infiltrating CD11b(-)CD27(-) NK subsets account for NK-cell dysfunction in patients with hepatocellular carcinoma and are associated with tumor progression. Cell Mol Immunol (2017) 14(10):819–29. doi: 10.1038/cmi.2016.28
46. Sui Q, Zhang J, Sun X, Zhang C, Han Q, Tian Z. NK cells are the crucial antitumor mediators when STAT3-mediated immunosuppression is blocked in hepatocellular carcinoma. J Immunol (2014) 193(4):2016–23. doi: 10.4049/jimmunol.1302389
47. Sun C, Xu J, Huang Q, Huang M, Wen H, Zhang C, et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology (2017) 6(1):2:e126456. doi: 10.1080/2162402X.2016.1264562
48. Sun H, Liu L, Huang Q, Liu H, Huang M, Wang J, et al. Accumulation of tumor-infiltrating CD49a(+) NK cells correlates with poor prognosis for human hepatocellular carcinoma. Cancer Immunol Res (2019) 7(9):1535–46. doi: 10.1158/2326-6066.CIR-18-0757
49. Shi JY, Gao Q, Wang ZC, Zhou J, Wang XY, Min ZH, et al. Margin-infiltrating CD20(+) b cells display an atypical memory phenotype and correlate with favorable prognosis in hepatocellular carcinoma. Clin Cancer Res (2013) 19(21):5994–6005. doi: 10.1158/1078-0432.CCR-12-3497
50. Petitprez F, de Reyniès A, Keung EZ, Chen TW, Sun CM, Calderaro J, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature (2020) 577(7791):556–60. doi: 10.1038/s41586-019-1906-8
51. Xiao X, Lao XM, Chen MM, Liu RX, Wei Y, Ouyang FZ, et al. PD-1hi identifies a novel regulatory b-cell population in human hepatoma that promotes disease progression. Cancer Discovery (2016) 6(5):546–59. doi: 10.1158/2159-8290.CD-15-1408
52. Lurje I, Hammerich L, Tacke F. Dendritic cell and T cell crosstalk in liver fibrogenesis and hepatocarcinogenesis: implications for prevention and therapy of liver cancer. Int J Mol Sci (2020) 21(19):7378. doi: 10.3390/ijms21197378
53. Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol (2013) 14(10):996–1006. doi: 10.1038/ni.2691
54. Huber A, Dammeijer F, Aerts J, Vroman H. Current state of dendritic cell-based immunotherapy: opportunities for in vitro antigen loading of different DC subsets? Front Immunol (2018) 9:2804. doi: 10.3389/fimmu.2018.02804
55. Böttcher JP, Reis e Sousa C. The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer (2018) 4(11):784–92. doi: 10.1016/j.trecan.2018.09.001
56. Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology (2018) 154(1):3–20. doi: 10.1111/imm.12888
57. Li S, Wu J, Zhu S, Liu YJ, Chen J. Disease-associated plasmacytoid dendritic cells. Front Immunol (2017) 8:1268. doi: 10.3389/fimmu.2017.01268
58. Beckebaum S, Zhang X, Chen X, Yu Z, Frilling A, Dworacki G, et al. Increased levels of interleukin-10 in serum from patients with hepatocellular carcinoma correlate with profound numerical deficiencies and immature phenotype of circulating dendritic cell subsets. Clin Cancer Res (2004) 10(21):7260–9. doi: 10.1158/1078-0432.CCR-04-0872
59. Zhou Z-J, Xin H-Y, Li J, Hu Z-Q, Luo C-B, Zhou S-L. Intratumoral plasmacytoid dendritic cells as a poor prognostic factor for hepatocellular carcinoma following curative resection. Cancer Immunol Immunother (2019) 68(8):1223–33. doi: 10.1007/s00262-019-02355-3
60. Sawa-Wejksza K, Kandefer-Szerszeń M. Tumor-associated macrophages as target for antitumor therapy. Arch Immunol Ther Exp (Warsz) (2018) 66(2):97–111. doi: 10.1007/s00005-017-0480-8
61. Heymann F, Peusquens J, Ludwig-Portugall I, Kohlhepp M, Ergen C, Niemietz P, et al. Liver inflammation abrogates immunological tolerance induced by kupffer cells. Hepatology (2015) 62(1):279–91. doi: 10.1002/hep.27793
62. Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature (2011) 479(7374):547–51. doi: 10.1038/nature10599
63. Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell (2016) 30(4):533–47. doi: 10.1016/j.ccell.2016.09.003
64. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated kupffer cells. Nat Commun (2016) 7:10321. doi: 10.1038/ncomms10321
65. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122(3):787–95. doi: 10.1172/JCI59643
66. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest (2005) 115(1):56–65. doi: 10.1172/JCI200522675
67. Mills CD. Anatomy of a discovery: m1 and m2 macrophages. Front Immunol (2015) 6:212. doi: 10.3389/fimmu.2015.00212
68. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23(11):549–55. doi: 10.1016/S1471-4906(02)02302-5
69. Huang C, Ou R, Chen X, Zhang Y, Li J, Liang Y, et al. Tumor cell-derived SPON2 promotes M2-polarized tumor-associated macrophage infiltration and cancer progression by activating PYK2 in CRC. J Exp Clin Cancer Res (2021) 40(1):304. doi: 10.1186/s13046-021-02108-0
70. Zhu XD, Zhang JB, Zhuang PY, Zhu HG, Zhang W, Xiong YQ, et al. High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. J Clin Oncol (2008) 26(16):2707–16. doi: 10.1200/JCO.2007.15.6521
71. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN. Cancer Cell (2009) 16(3):183–94. doi: 10.1016/j.ccr.2009.06.017
72. Shaul ME, Fridlender ZG. Neutrophils as active regulators of the immune system in the tumor microenvironment. J Leukoc Biol (2017) 102(2):343–9. doi: 10.1189/jlb.5MR1216-508R
73. Kuang DM, Zhao Q, Wu Y, Peng C, Wang J, Xu Z, et al. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J Hepatol (2011) 54(5):948–55. doi: 10.1016/j.jhep.2010.08.041
74. Zhou SL, Dai Z, Zhou ZJ, Wang XY, Yang GH, Wang Z, et al. Overexpression of CXCL5 mediates neutrophil infiltration and indicates poor prognosis for hepatocellular carcinoma. Hepatology (2012) 56(6):2242–54. doi: 10.1002/hep.25907
75. Zhou SL, Yin D, Hu ZQ, Luo CB, Zhou ZJ, Xin HY, et al. A positive feedback loop between cancer stem-like cells and tumor-associated neutrophils controls hepatocellular carcinoma progression. Hepatology (2019) 70(4):1214–30. doi: 10.1002/hep.30630
76. Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, et al. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology (2016) 150(7):1646–1658.e17. doi: 10.1053/j.gastro.2016.02.040
77. Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest (2013) 123(8):3446–58. doi: 10.1172/JCI67484
78. Yang LY, Luo Q, Lu L, Zhu WW, Sun HT, Wei R, et al. Increased neutrophil extracellular traps promote metastasis potential of hepatocellular carcinoma via provoking tumorous inflammatory response. J Hematol Oncol (2020) 13(1):3. doi: 10.1186/s13045-019-0836-0
79. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res (2017) 5(1):3–8. doi: 10.1158/2326-6066.CIR-16-0297
80. Consonni FM, Porta C, Marino A, Pandolfo C, Mola S, Bleve A, et al. Myeloid-derived suppressor cells: ductile targets in disease. Front Immunol (2019) 10:949. doi: 10.3389/fimmu.2019.00949
81. Qu P, Wang LZ, Lin PC. Expansion and functions of myeloid-derived suppressor cells in the tumor microenvironment. Cancer Lett (2016) 380(1):253–6. doi: 10.1016/j.canlet.2015.10.022
82. Tcyganov E, Mastio J, Chen E, Gabrilovich DI. Plasticity of myeloid-derived suppressor cells in cancer. Curr Opin Immunol (2018) 51:76–82. doi: 10.1016/j.coi.2018.03.009
83. Gao XH, Tian L, Wu J, Ma XL, Zhang CY, Zhou Y, et al. Circulating CD14(+) HLA-DR(-/low) myeloid-derived suppressor cells predicted early recurrence of hepatocellular carcinoma after surgery. Hepatol Res (2017) 47(10):1061–71. doi: 10.1111/hepr.12831
84. Wang D, An G, Xie S, Yao Y, Feng G. The clinical and prognostic significance of CD14(+)HLA-DR(-/low) myeloid-derived suppressor cells in hepatocellular carcinoma patients receiving radiotherapy. Tumour Biol (2016) 37(8):10427–33. doi: 10.1007/s13277-016-4916-2
85. Lacotte S, Slits F, Orci LA, Meyer J, Oldani G, Delaune V, et al. Impact of myeloid-derived suppressor cell on kupffer cells from mouse livers with hepatocellular carcinoma. Oncoimmunology (2016) 5(11):e1234565. doi: 10.1080/2162402X.2016.1234565
86. Xu Y, Zhao W, Xu J, Li J, Hong Z, Yin Z, et al. Activated hepatic stellate cells promote liver cancer by induction of myeloid-derived suppressor cells through cyclooxygenase-2. Oncotarget (2016) 7(8):8866–78. doi: 10.18632/oncotarget.6839
87. Zhou J, Liu M, Sun H, Feng Y, Xu L, Chan AWH, et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut (2018) 67(5):931–44. doi: 10.1136/gutjnl-2017-314032
88. Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology (2009) 50(3):799–807. doi: 10.1002/hep.23054
89. González-de-Olano D, Álvarez-Twose I. Mast cells as key players in allergy and inflammation. J Investig Allergol Clin Immunol (2018) 28(6):365–78. doi: 10.18176/jiaci.0327
90. Aponte-López A, Muñoz-Cruz S. Mast cells in the tumor microenvironment. Adv Exp Med Biol (2020) 1273:159–73. doi: 10.1007/978-3-030-49270-0_9
91. Lampiasi N, Azzolina A, Montalto G, Cervello M. Histamine and spontaneously released mast cell granules affect the cell growth of human hepatocellular carcinoma cells. Exp Mol Med (2007) 39(3):284–94. doi: 10.1038/emm.2007.32
92. Ribatti D. Mast cells and macrophages exert beneficial and detrimental effects on tumor progression and angiogenesis. Immunol Lett (2013) 152(2):83–8. doi: 10.1016/j.imlet.2013.05.003
93. Freitas-Lopes M, Mafra K, David B, Carvalho-Gontijo R, Menezes G. Differential location and distribution of hepatic immune cells. Cells (2017) 6(4):48. doi: 10.3390/cells6040048
94. Luo N, Li J, Wei Y, Lu J, Dong R. Hepatic stellate cell: a double-edged sword in the liver. Physiol Res (2021) 70(6):821–9. doi: 10.33549/physiolres.934755
95. Gressner AM, Weiskirchen R, Breitkopf K, Dooley S. Roles of TGF-beta in hepatic fibrosis. Front Biosci (2002) 7:d793–807. doi: 10.2741/gressner
96. Barry AE, Baldeosingh R, Lamm R, Patel K, Zhang K, Dominguez DA, et al. Hepatic stellate cells and hepatocarcinogenesis. Front Cell Dev Biol (2020) 8:709. doi: 10.3389/fcell.2020.00709
97. Pinato DJ, Guerra N, Fessas P, Murphy R, Mineo T, Mauri FA, et al. Immune-based therapies for hepatocellular carcinoma. Oncogene (2020) 39(18):3620–37. doi: 10.1038/s41388-020-1249-9
98. Pein M, Insua-Rodríguez J, Hongu T, Riedel A, Meier J, Wiedmann L, et al. Metastasis-initiating cells induce and exploit a fibroblast niche to fuel malignant colonization of the lungs. Nat Commun (2020) 11(1):1494. doi: 10.1038/s41467-020-15188-x
99. Multhaupt HA, Leitinger B, Gullberg D, Couchman JR. Extracellular matrix component signaling in cancer. Adv Drug Delivery Rev (2016) 97:28–40. doi: 10.1016/j.addr.2015.10.013
100. Lau EY, Lo J, Cheng BY, Ma MK, Lee JM, Ng JK, et al. Cancer-associated fibroblasts regulate tumor-initiating cell plasticity in hepatocellular carcinoma through c-Met/FRA1/HEY1 signaling. Cell Rep (2016) 15(6):1175–89. doi: 10.1016/j.celrep.2016.04.019
101. Liu J, Chen S, Wang W, Ning BF, Chen F, Shen W, et al. Cancer-associated fibroblasts promote hepatocellular carcinoma metastasis through chemokine-activated hedgehog and TGF-β pathways. Cancer Lett (2016) 379(1):49–59. doi: 10.1016/j.canlet.2016.05.022
102. Luo Q, Wang CQ, Yang LY, Gao XM, Sun HT, Zhang Y, et al. FOXQ1/NDRG1 axis exacerbates hepatocellular carcinoma initiation via enhancing crosstalk between fibroblasts and tumor cells. Cancer Lett (2018) 417:21–34. doi: 10.1016/j.canlet.2017.12.021
103. Sun L, Wang Y, Wang L, Yao B, Chen T, Li Q, et al. Resolvin D1 prevents epithelial-mesenchymal transition and reduces the stemness features of hepatocellular carcinoma by inhibiting paracrine of cancer-associated fibroblast-derived COMP. J Exp Clin Cancer Res (2019) 38(1):170. doi: 10.1186/s13046-019-1163-6
104. Deng Y, Cheng J, Fu B, Liu W, Chen G, Zhang Q, et al. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene (2017) 36(8):1090–101. doi: 10.1038/onc.2016.273
105. Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, et al. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis (2018) 9(4):422. doi: 10.1038/s41419-018-0458-4
106. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med (2016) 375(18):1767–78. doi: 10.1056/NEJMra1514296
107. Chan LL, Chan SL. Emerging immune checkpoint inhibitors for the treatment of hepatocellular carcinoma. Expert Opin Emerg Drugs (2021) 26(1):39–52. doi: 10.1080/14728214.2021.1902503
108. Kudo M. Scientific rationale for combination immunotherapy of hepatocellular carcinoma with anti-PD-1/PD-L1 and anti-CTLA-4 antibodies. Liver Cancer (2019) 8(6):413–26. doi: 10.1159/000503254
109. Fulgenzi CAM, Talbot T, Murray SM, Silletta M, Vincenzi B, Cortellini A, et al. Immunotherapy in hepatocellular carcinoma. Curr Treat Opt Oncol (2021) 22(10):87. doi: 10.1007/s11864-021-00886-5
110. Agdashian D, ElGindi M, Xie C, Sandhu M, Pratt D, Kleiner DE, et al. The effect of anti-CTLA4 treatment on peripheral and intra-tumoral T cells in patients with hepatocellular carcinoma. Cancer Immunol Immunother (2019) 68(4):599–608. doi: 10.1007/s00262-019-02299-8
111. Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory pathways in the B7-CD28 ligand-receptor family. Immunity (2016) 44(5):955–72. doi: 10.1016/j.immuni.2016.05.002
112. Fan J, Shen X, Wang Y, Zhou HL, Liu G, Li YL, et al. Biomarkers for immune checkpoint therapy targeting programmed death 1 and programmed death ligand 1. BioMed Pharmacother (2020) 130:110621. doi: 10.1016/j.biopha.2020.110621
113. Gao Q, Wang XY, Qiu SJ, Yamato I, Sho M, Nakajima Y, et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res (2009) 15(3):971–9. doi: 10.1158/1078-0432.CCR-08-1608
114. Galle PR, Finn RS, Qin S, Ikeda M, Zhu AX, Kim T-Y, et al. Patient-reported outcomes with atezolizumab plus bevacizumab versus sorafenib in patients with unresectable hepatocellular carcinoma (IMbrave150): an open-label, randomised, phase 3 trial. Lancet Oncol (2021) 22(7):991–1001. doi: 10.1016/S1470-2045(21)00151-0
115. Xu W, Liu K, Chen M, Sun JY, McCaughan GW, Lu XJ, et al. Immunotherapy for hepatocellular carcinoma: recent advances and future perspectives. Ther Adv Med Oncol (2019) 11:1758835919862692. doi: 10.1177/1758835919862692
116. Yau T, Park JW, Finn RS, Cheng AL, Mathurin P, Edeline J, et al. CheckMate 459: a randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann Oncol (2019) 30:v874–5. doi: 10.1093/annonc/mdz394.029
117. Finn RS, Ryoo BY, Merle P, Kudo M, Bouattour M, Lim HY, et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: a randomized, double-blind, phase III trial. J Clin Oncol (2020) 38(3):193–202. doi: 10.1200/JCO.19.01307
118. Qin S, Finn RS, Kudo M, Meyer T, Vogel A, Ducreux M, et al. RATIONALE 301 study: tislelizumab versus sorafenib as first-line treatment for unresectable hepatocellular carcinoma. Future Oncol (2019) 15(16):1811–22. doi: 10.2217/fon-2019-0097
119. Desai J, Deva S, Lee JS, Lin CC, Millward M. Phase IA/IB study of single-agent tislelizumab, an investigational anti-PD-1 antibody, in solid tumors. J ImmunoTher Cancer (2020) 8(1):e000453. doi: 10.1136/jitc-2019-000453
120. Liu F, Liu Y, Chen Z. Tim-3 expression and its role in hepatocellular carcinoma. J Hematol Oncol (2018) 11(1):126. doi: 10.1186/s13045-018-0667-4
121. Liu F, Zeng G, Zhou S, He X, Sun N, Zhu X, et al. Blocking Tim-3 or/and PD-1 reverses dysfunction of tumor-infiltrating lymphocytes in HBV-related hepatocellular carcinoma. Bull Cancer (2018) 105(5):493–501. doi: 10.1016/j.bulcan.2018.01.018
122. Zhou G, Sprengers D, Boor PPC, Doukas M, Schutz H, Mancham S, et al. Antibodies against immune checkpoint molecules restore functions of tumor-infiltrating T cells in hepatocellular carcinomas. Gastroenterology (2017) 153(4):1107–1119.e10. doi: 10.1053/j.gastro.2017.06.017
123. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med (2010) 207(10):2187–94. doi: 10.1084/jem.20100643
124. Okazaki T, Okazaki IM, Wang J, Sugiura D, Nakaki F, Yoshida T, et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J Exp Med (2011) 208(2):395–407. doi: 10.1084/jem.20100466
125. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med (1990) 171(5):1393–405. doi: 10.1084/jem.171.5.1393
126. Puhr HC, Ilhan-Mutlu A. New emerging targets in cancer immunotherapy: the role of LAG3. ESMO Open (2019) 4(2):e000482. doi: 10.1136/esmoopen-2018-000482
127. Ruffo E, Wu RC, Bruno TC, Workman CJ, Vignali DAA. Lymphocyte-activation gene 3 (LAG3): the next immune checkpoint receptor. Semin Immunol (2019) 42:101305. doi: 10.1016/j.smim.2019.101305
128. Maçon-Lemaître L, Triebel F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology (2005) 115(2):170–8. doi: 10.1111/j.1365-2567.2005.02145.x
129. Yu X, Huang X, Chen X, Liu J, Wu C, Pu Q, et al. Characterization of a novel anti-human lymphocyte activation gene 3 (LAG-3) antibody for cancer immunotherapy. MAbs (2019) 11(6):1139–48. doi: 10.1080/19420862.2019.1629239
130. Long L, Zhang X, Chen F, Pan Q, Phiphatwatchara P, Zeng Y, et al. The promising immune checkpoint LAG-3: from tumor microenvironment to cancer immunotherapy. Genes Cancer (2018) 9(5-6):176–89. doi: 10.18632/genesandcancer.180
131. Davar D, Zarour HM. Immunological targets for immunotherapy: inhibitory T cell receptors. Methods Mol Biol (2020) 2055:23–60. doi: 10.1007/978-1-4939-9773-2_2
132. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44(5):989–1004. doi: 10.1016/j.immuni.2016.05.001
133. Hasan MM, Nair SS, O'Leary JG, Thompson-Snipes L, Nyarige V, Wang J, et al. Implication of TIGIT(+) human memory b cells in immune regulation. Nat Commun (2021) 12(1):1534. doi: 10.1038/s41467-021-21413-y
134. Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev (2017) 276(1):112–20. doi: 10.1111/imr.12518
135. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell (2014) 26(6):923–37. doi: 10.1016/j.ccell.2014.10.018
136. Srivastava MK, Rui Y, Mayes E, Yu J, Park AI. Abstract 2612: anti-tigit induces T cell mediated anti-tumor immune response and combines with immune checkpoint inhibitors to enhance strong and long term anti-tumor immunity. Cancer Res (2017) 77(13 Supplement):2612–2. doi: 10.1158/1538-7445.AM2017-2612
137. Saung MT, Pelosof L, Casak S, Donoghue M, Lemery S, Yuan M, et al. FDA Approval summary: nivolumab plus ipilimumab for the treatment of patients with hepatocellular carcinoma previously treated with sorafenib. Oncologist (2021) 26(9):797–806. doi: 10.1002/onco.13819
138. Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity (2016) 44(3):609–21. doi: 10.1016/j.immuni.2016.01.024
139. Zhang Y, Brekken RA. Direct and indirect regulation of the tumor immune microenvironment by VEGF. J Leukoc Biol (2022) 111(6):1269–86. doi: 10.1002/JLB.5RU0222-082R
140. Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front Immunol (2018) 9:978. doi: 10.3389/fimmu.2018.00978
141. Siegel AB, Cohen EI, Ocean A, Lehrer D, Goldenberg A, Knox JJ, et al. Phase II trial evaluating the clinical and biologic effects of bevacizumab in unresectable hepatocellular carcinoma. J Clin Oncol (2008) 26(18):2992–8. doi: 10.1200/JCO.2007.15.9947
142. Cheng AL, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Updated efficacy and safety data from IMbrave150: atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J Hepatol (2022) 76(4):862–73. doi: 10.1016/j.jhep.2021.11.030
143. Yuan GS, He WM, Hu XY, Li Q, Zang MY, Cheng X, et al. [Clinical efficacy and safety analysis of camrelizumab combined with apatinib as a second-line therapy for unresectable hepatocellular carcinoma: a multicenter retrospective study]. Zhonghua Gan Zang Bing Za Zhi (2021) 29(4):326–31. doi: 10.3760/cma.j.cn501113-20210329-00148
144. Mossenta M, Busato D, Baboci L, Cintio FD, Toffoli G, Bo MD. New insight into therapies targeting angiogenesis in hepatocellular carcinoma. Cancers (Basel) (2019) 11(8):1086. doi: 10.3390/cancers11081086
145. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol (2017) 17(2):97–111. doi: 10.1038/nri.2016.107
146. McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science (2016) 351(6280):1463–9. doi: 10.1126/science.aaf1490
147. Mpekris F, Baish JW, Stylianopoulos T, Jain RK. Role of vascular normalization in benefit from metronomic chemotherapy. Proc Natl Acad Sci (2017) 114(8):1994–9. doi: 10.1073/pnas.1700340114
148. Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest (2014) 124(2):687–95. doi: 10.1172/JCI67313
149. Dovedi SJ, Adlard AL, Lipowska-Bhalla G, Mckenna C, Jones S, Cheadle EJ, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res (2014) 74(19):5458–68. doi: 10.1158/0008-5472.CAN-14-1258
150. Kim KJ, Kim JH, Lee SJ, Lee EJ, Shin EC, Seong J. Radiation improves antitumor effect of immune checkpoint inhibitor in murine hepatocellular carcinoma model. Oncotarget (2017) 8(25):41242–55. doi: 10.18632/oncotarget.17168
151. Young KH, Baird JR, Savage T, Cottam B, Crittenden MR, et al. Optimizing timing of immunotherapy improves control of tumors by hypofractionated radiation therapy. PloS One (2016) 11(6):e0157164. doi: 10.1371/journal.pone.0157164
152. Chiang CL, Chan A, Chiu K, Kong F. Combined stereotactic body radiotherapy and checkpoint inhibition in unresectable hepatocellular carcinoma: a potential synergistic treatment strategy. Front Oncol (2019) 9:1157. doi: 10.3389/fonc.2019.01157
153. Tai WMD, Loke KSH, Gogna A, Tan SH, Ng DCE, Hennedige TP, et al. A phase II open-label, single-center, nonrandomized trial of Y90-radioembolization in combination with nivolumab in Asian patients with advanced hepatocellular carcinoma: CA 209-678. J Clin Oncol (2020) 38(15_suppl):4590–0. doi: 10.1200/JCO.2020.38.15_suppl.4590
154. Pinato DJ, Cole T, Bengsch B, Tait P, Sayed AA, Abomeli F, et al. 750P - a phase ib study of pembrolizumab following trans-arterial chemoembolization (TACE) in hepatocellular carcinoma (HCC): PETAL. Ann Oncol (2019) 30:v288. doi: 10.1093/annonc/mdz247.076
155. Ali MY, Grimm CF, Ritter M, Mohr L, Allgaier HP, Weth R, et al. Activation of dendritic cells by local ablation of hepatocellular carcinoma. J Hepatol (2005) 43(5):817–22. doi: 10.1016/j.jhep.2005.04.016
156. Rochigneux P, Nault JC, Mallet F, Chretien AS, Barget N, Garcia AJ, et al. Dynamic of systemic immunity and its impact on tumor recurrence after radiofrequency ablation of hepatocellular carcinoma. Oncoimmunology (2019) 8(8):1615818. doi: 10.1080/2162402X.2019.1615818
157. Zerbini A, Pilli M, Penna A, Pelosi G, Schianchi C, Molinari A, et al. Radiofrequency thermal ablation of hepatocellular carcinoma liver nodules can activate and enhance tumor-specific T-cell responses. Cancer Res (2006) 66(2):1139–46. doi: 10.1158/0008-5472.CAN-05-2244
158. Fessas P, Possamai LA, Clark J, Daniels E, Gudd C, Mullish BH, et al. Immunotoxicity from checkpoint inhibitor therapy: clinical features and underlying mechanisms. Immunology (2020) 159(2):167–77. doi: 10.1111/imm.13141
159. Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, et al. Hepatocellular carcinoma. Nat Rev Dis Primers (2016) 2:16018. doi: 10.1038/nrdp.2016.18
160. Sangro B, Chan SL, Meyer T, Reig M, El-Khoueiry A, Galle PR. Diagnosis and management of toxicities of immune checkpoint inhibitors in hepatocellular carcinoma. J Hepatol (2020) 72(2):320–41. doi: 10.1016/j.jhep.2019.10.021
161. Voutsadakis IA. PD-1 inhibitors monotherapy in hepatocellular carcinoma: meta-analysis and systematic review. Hepatobil Pancreat Dis Int (2019) 18(6):505–10. doi: 10.1016/j.hbpd.2019.09.007
162. Wang Y, Zhou S, Yang F, Qi X, Wang X, Guan X, et al. Treatment-related adverse events of PD-1 and PD-L1 inhibitors in clinical trials: a systematic review and meta-analysis. JAMA Oncol (2019) 5(7):1008–19. doi: 10.1001/jamaoncol.2019.0393
163. Perica K, Varela JC, Oelke M, Schneck J. Adoptive T cell immunotherapy for cancer. Rambam Maimonides Med J (2015) 6(1):e0004. doi: 10.5041/RMMJ.10179
164. Sadelain M, Rivière I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer (2003) 3(1):35–45. doi: 10.1038/nrc971
165. Bridgeman J S, Hawkins R E, Hombach A A, Abken H E, Gilham D. Building better chimeric antigen receptors for adoptive T cell therapy. Curr Gene Ther (2010) 10(2):77–90. doi: 10.2174/156652310791111001
166. Li W, Guo L, Rathi P, Marinova E, Gao X, Wu MF, et al. Redirecting T cells to glypican-3 with 4-1BB zeta chimeric antigen receptors results in Th1 polarization and potent antitumor activity. Hum Gene Ther (2017) 28(5):437–48. doi: 10.1089/hum.2016.025
167. Gao H, Li K, Tu H, Pan X, Jiang H, Shi B, et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin Cancer Res (2014) 20(24):6418–28. doi: 10.1158/1078-0432.CCR-14-1170
168. Batra SA, Rathi P, Guo L, Courtney AN, Fleurence J, Balzeau J, et al. Glypican-3-Specific CAR T cells coexpressing IL15 and IL21 have superior expansion and antitumor activity against hepatocellular carcinoma. Cancer Immunol Res (2020) 8(3):309–20. doi: 10.1158/2326-6066.CIR-19-0293
169. Pang N, Shi J, Qin L, Chen A, Tang Y, Yang H, et al. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. J Hematol Oncol (2021) 14:1–6. doi: 10.1186/s13045-021-01128-9
170. Beatty GL, Moon EK. Chimeric antigen receptor T cells are vulnerable to immunosuppressive mechanisms present within the tumor microenvironment. OncoImmunology (2014) 3(11):e970027. doi: 10.4161/21624011.2014.970027
171. Anderson KG, Stromnes IM, Greenberg PD. Obstacles posed by the tumor microenvironment to T cell activity: a case for synergistic therapies. Cancer Cell (2017) 31(3):311–25. doi: 10.1016/j.ccell.2017.02.008
172. Xia A-L, Wang X-C, Lu Y-J, Lu X-J, Sun B. Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: challenges and opportunities. Oncotarget (2017) 8(52):90521–31. doi: 10.18632/oncotarget.19361
173. Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep (2017) 7(1):737. doi: 10.1038/s41598-017-00462-8
174. Hegde UP, Mukherji B. Current status of chimeric antigen receptor engineered T cell-based and immune checkpoint blockade-based cancer immunotherapies. Cancer Immunol Immunother (2017) 66(9):1113–21. doi: 10.1007/s00262-017-2007-x
175. Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther (2017) 25(9):2214–24. doi: 10.1016/j.ymthe.2017.05.012
176. Zhang Y, Sharma A, Weiher H, Schmid M, Kristiansen G, Schmidt-Wolf IGH. Clinical studies on cytokine-induced killer cells: lessons from lymphoma trials. Cancers (Basel) (2021) 13(23):6007. doi: 10.3390/cancers13236007
177. Introna M. CIK as therapeutic agents against tumors. J Autoimmun (2017) 85:32–44. doi: 10.1016/j.jaut.2017.06.008
178. Wang F-S. Antitumor activities of human autologous cytokine-induced killer (CIK) cells against hepatocellular carcinoma cellsin vitroandin vivo. World J Gastroenterol (2002) 8(3):464. doi: 10.3748/wjg.v8.i3.464
179. Wang H, Liu A, Bo W, Feng X, Hu Y, Tian L, et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma patients after curative resection, a systematic review and meta-analysis. Digest Liver Dis (2016) 48(11):1275–82. doi: 10.1016/j.dld.2016.07.010
180. Lee JH, Lee JH, Lim YS, Yeon JE, Song TJ, Yu SJ, et al. Sustained efficacy of adjuvant immunotherapy with cytokine-induced killer cells for hepatocellular carcinoma: an extended 5-year follow-up. Cancer Immunol Immunother (2019) 68(1):23–32. doi: 10.1007/s00262-018-2247-4
181. Weng D-S, Zhou J, Zhou Q-M, Zhao M, Wang Q-J, Huang L-X, et al. Minimally invasive treatment combined with cytokine-induced killer cells therapy lower the short-term recurrence rates of hepatocellular carcinomas. J Immunother (2008) 31(1):63–71. doi: 10.1097/CJI.0b013e31815a121b
182. Pan C-C, Huang Z-L, Li W, Zhao M, Zhou Q-M, Xia J-C, et al. Serum alpha-fetoprotein measurement in predicting clinical outcome related to autologous cytokine-induced killer cells in patients with hepatocellular carcinoma undergone minimally invasive therapy. Chin J Cancer (2010) 29(6):596–602. doi: 10.5732/cjc.009.10580
183. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (2015) 348(6230):62–8. doi: 10.1126/science.aaa4967
184. Wang SH, Yeh SH, Chen PJ. Unique features of hepatitis b virus-related hepatocellular carcinoma in pathogenesis and clinical significance. Cancers (Basel) (2021) 13(10):2454. doi: 10.3390/cancers13102454
185. Qasim W, Brunetto M, Gehring AJ, Xue SA, Schurich A, Khakpoor A, et al. Immunotherapy of HCC metastases with autologous T cell receptor redirected T cells, targeting HBsAg in a liver transplant patient. J Hepatol (2015) 62(2):486–91. doi: 10.1016/j.jhep.2014.10.001
186. Remy T, Ghaneya AK, Jessica R, Wouter H, Said D, Davide B, et al. NY-ESO-1 based immunotherapy of cancer: current perspectives. Front Immunol (2018) 9:947. doi: 10.3389/fimmu.2018.00947
187. Zhu W, Peng Y, Wang L, Hong Y, Jiang X, Li Q, et al. Identification of α-fetoprotein-specific T-cell receptors for hepatocellular carcinoma immunotherapy. Hepatology (2018) 68(2):574–89. doi: 10.1002/hep.29844
188. Dargel C, Bassani-Sternberg M, Hasreiter J, Zani F, Bockmann JH, Thiele F, et al. T Cells engineered to express a T-cell receptor specific for glypican-3 to recognize and kill hepatoma cells In vitro and in mice. Gastroenterology (2015) 149(4):1042–52. doi: 10.1053/j.gastro.2015.05.055
189. Mizukoshi E, Nakamoto Y, Marukawa Y, Arai K, Yamashita T, Tsuji H, et al. Cytotoxic T cell responses to human telomerase reverse transcriptase in patients with hepatocellular carcinoma. Hepatology (2006) 43(6):1284–94. doi: 10.1002/hep.21203
190. Li K, Lan Y, Wang J, Liu L. Chimeric antigen receptor–engineered T cells for liver cancers, progress and obstacles. Tumor Biol (2017) 39(3):1010428317692229. doi: 10.1177/1010428317692229
191. Docta RY, Ferronha T, Sanderson JP, Weissensteiner T, Pope GR, Bennett AD, et al. Tuning T-cell receptor affinity to optimize clinical risk-benefit when targeting alpha-Fetoprotein-Positive liver cancer. Hepatology (2019) 69(5):2061–75. doi: 10.1002/hep.30477
192. Del Campo JA, Gallego P, Grande L. Role of inflammatory response in liver diseases: therapeutic strategies. World J Hepatol (2018) 10(1):1–7. doi: 10.4254/wjh.v10.i1.1
193. Waldmann TA. Cytokines in cancer immunotherapy. Cold Spring Harbor Perspect Biol (2017) 10(12):a028472. doi: 10.1101/cshperspect.a028472
194. Qu LS, Jin F, Huang XW, Shen XZ. Interferon-α therapy after curative resection prevents early recurrence and improves survival in patients with hepatitis b virus-related hepatocellular carcinoma. J Surg Oncol (2010) 102(7):796–801. doi: 10.1002/jso.21741
195. Wei Z, Song T, Zhang TI, Qiang WU, Kong D, Qiang LI, et al. Adjuvant interferon for early or late recurrence of hepatocellular carcinoma and mortality from hepatocellular carcinoma following curative treatment: a meta-analysis with comparison of different types of hepatitis. Mol Clin Oncol (2014) 2(6):1125. doi: 10.3892/mco.2014.386
196. Giordano S, Columbano A. Met as a therapeutic target in HCC: facts and hopes. J Hepatol (2014) 60(2):442–52. doi: 10.1016/j.jhep.2013.09.009
197. Llovet JM, Peña CE, Lathia CD, Shan M, Meinhardt G, Bruix J. Plasma biomarkers as predictors of outcome in patients with advanced hepatocellular carcinoma. Clin Cancer Res (2012) 18(8):2290–300. doi: 10.1158/1078-0432.CCR-11-2175
198. Miyahara K, Nouso K, Morimoto Y, Takeuchi Y, Hagihara H, Kuwaki K, et al. Pro-angiogenic cytokines for prediction of outcomes in patients with advanced hepatocellular carcinoma. Br J Cancer (2013) 109(8):2072–8. doi: 10.1038/bjc.2013.554
199. Bertelli R, Neri F, Tsivian M, Ruhrman N, Cavallari G, Beltempo P, et al. Endolymphatic immunotherapy in inoperable hepatocellular carcinoma. Transplant Proc (2008) 40(6):1913–5. doi: 10.1016/j.transproceed.2008.05.049
200. Shao Y-Y, Lin H, Li Y-S, Lee Y-H, Chen H-M, Cheng A-L, et al. High plasma interleukin-6 levels associated with poor prognosis of patients with advanced hepatocellular carcinoma. Japan J Clin Oncol (2017) 47(10):949–53. doi: 10.1093/jjco/hyx103
201. Li Y, Chen G, Han Z, Cheng H, Qiao L, Li Y. IL-6/STAT3 signaling contributes to sorafenib resistance in hepatocellular carcinoma through targeting cancer stem cells. OncoTargets Ther (2020) 13:9721. doi: 10.2147/OTT.S262089
202. Pickup M, Novitskiy S, Moses HL. The roles of TGFβ in the tumour microenvironment. Nat Rev Cancer (2013) 13(11):788–99. doi: 10.1038/nrc3603
203. Faivre SJ, Santoro A, Kelley RK, Merle P, Gane E, Douillard J-Y, et al. A phase 2 study of a novel transforming growth factor-beta (TGF-β1) receptor I kinase inhibitor, LY2157299 monohydrate (LY), in patients with advanced hepatocellular carcinoma (HCC). J Clin Oncol (2014) 32(3_suppl):LBA173–3. doi: 10.1200/jco.2014.32.3_suppl.lba173
204. Faivre SJ, Santoro A, Gane E, Kelley RK, Hourmand IO, Assenat E, et al. A phase 2 study of galunisertib, a novel transforming growth factor-beta (TGF-β) receptor I kinase inhibitor, in patients with advanced hepatocellular carcinoma (HCC) and low serum alpha fetoprotein (AFP). J Clin Oncol (2016) 34(15_suppl):4070–0. doi: 10.1200/JCO.2016.34.15_suppl.4070
205. Holmgaard RB, Schaer DA, Li Y, Castaneda SP, Murphy MY, Xu X, et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J Immunother Cancer (2018) 6(1):47. doi: 10.1186/s40425-018-0356-4
206. Hu Z, Ott PA, Wu CJ. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol (2018) 18(3):168–82. doi: 10.1038/nri.2017.131
207. Liu JKH, Irvine AF, Jones RL, Samson A. Immunotherapies for hepatocellular carcinoma. Cancer Med (2022) 11(3):571–91. doi: 10.1002/cam4.4468
208. Nishida T, Kataoka H. Glypican 3-targeted therapy in hepatocellular carcinoma. Cancers (2019) 11(9):1339. doi: 10.3390/cancers11091339
209. Sawada Y, Yoshikawa T, Nobuoka D, Shirakawa H, Kuronuma T, Motomura Y, et al. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: immunologic evidence and potential for improving overall survival. Clin Cancer Res (2012) 18(13):3686–96. doi: 10.1158/1078-0432.CCR-11-3044
210. Sawada Y, Yoshikawa T, Ofuji K, Yoshimura M, Tsuchiya N, Takahashi M, et al. Phase II study of the GPC3-derived peptide vaccine as an adjuvant therapy for hepatocellular carcinoma patients. OncoImmunology (2016) 5(5):e1129483. doi: 10.1080/2162402X.2015.1129483
211. Wang X, Wang Q. Alpha-fetoprotein and hepatocellular carcinoma immunity. Can J Gastroenterol Hepatol (2018) 2018:9049252. doi: 10.1155/2018/9049252
212. Hung T-M, Hu R-H, Ho C-M, Chiu Y-L, Lee J-L, Jeng Y-M, et al. Downregulation of alpha-fetoprotein expression by LHX4: a critical role in hepatocarcinogenesis. Carcinogenesis (2011) 32(12):1815–23. doi: 10.1093/carcin/bgr219
213. Butterfield LH, Economou JS, Gamblin T, Geller DA. Alpha fetoprotein DNA prime and adenovirus boost immunization of two hepatocellular cancer patients. J Trans Med (2014) 12(1):86. doi: 10.1186/1479-5876-12-86
214. Kooti W, and Cancer OV. Do you know the main mechanism? Front Oncol (2021) 11:761015. doi: 10.3389/fonc.2021.761015
215. Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res (2014) 2(4):295–300. doi: 10.1158/2326-6066.CIR-14-0015
216. Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med (2013) 19(3):329–36. doi: 10.1038/nm.3089
217. Yoo SY, Jeong SN, Kang DH, Heo J. Evolutionary cancer-favoring engineered vaccinia virus for metastatic hepatocellular carcinoma. Oncotarget (2017) 8(42):71489–99. doi: 10.18632/oncotarget.17288

Keywords: hepatocellular carcinoma, tumor microenvironment, cancer immunotherapy, immunocheck point inhibitors, adoptive cell therapy
Citation: Zhao M, Huang H, He F and Fu X (2023) Current insights into the hepatic microenvironment and advances in immunotherapy for hepatocellular carcinoma. Front. Immunol. 14:1188277. doi: 10.3389/fimmu.2023.1188277
Received: 20 March 2023; Accepted: 04 May 2023;
Published: 18 May 2023.
Edited by:
Zong Sheng Guo, University at Buffalo, United StatesReviewed by:
Wantong Song, Chinese Academy of Sciences (CAS), ChinaCopyright © 2023 Zhao, Huang, He and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng He, ODU0NTMwMDFAcXEuY29t; Xiangsheng Fu, ZnV4aWFuZ3NoZW5nQGNtYy5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.