
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 22 May 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1186455
 Nicolo Costantino Brembilla1*
Nicolo Costantino Brembilla1* Wolf-Henning Boehncke1,2
Wolf-Henning Boehncke1,2Psoriasis is a common chronic inflammatory skin disease, associated with substantial comorbidity. TH17 lymphocytes, differentiating under the influence of dendritic cell-derived IL-23, and mediating their effects via IL-17A, are believed to be central effector cells in psoriasis. This concept is underlined by the unprecedented efficacy of therapeutics targeting this pathogenetic axis. In recent years, numerous observations made it necessary to revisit and refine this simple “linear” pathogenetic model. It became evident that IL-23 independent cells exist that produce IL-17A, that IL-17 homologues may exhibit synergistic biological effects, and that the blockade of IL-17A alone is clinically less effective compared to the inhibition of several IL-17 homologues. In this review, we will summarize the current knowledge around IL-17A and its five currently known homologues, namely IL-17B, IL-17C, IL-17D, IL-17E (also known as IL-25) and IL-17F, in relation to skin inflammation in general and psoriasis in particular. We will also re-visit the above-mentioned observations and integrate them into a more comprehensive pathogenetic model. This may help to appreciate current as well as developing anti-psoriatic therapies and to prioritize the selection of future drugs’ mode(s) of action.
Psoriasis is a frequent, chronic, noncommunicable inflammatory skin disease, for which there is no clear cause or cure. Psoriasis affects all ethnicities and of all ages. The disease manifests as well-defined, red, scaly plaques, appearing with a chronic-recurrent course at preferential sites such as elbows, knees, and scalp (1). Individuals with psoriasis are at an increased risk of developing other chronic and serious diseases, including psoriatic arthritis, metabolic syndrome, cardiovascular diseases and depression (2, 3).
Psoriatic inflammation is triggered by environmental factors, such as infections (4). These initiate an innate immune response with dendritic cells playing a major role. A well-defined cascade starting with keratinocyte damage results in release of self-nucleic acids, which are subsequently complexed to antimicrobial peptides, and sensed by plasmacytoid dendritic cells (pDCs) (5). These produce IL-6 as well as transforming growth factor β, and induce differentiation of naïve T-lymphocytes towards a TH17 phenotype, which is subsequently maintained by IL-23, derived from the above-mentioned pDCs (6). TH17 cells are named after their key effector cytokines, namely different isoforms of IL-17 (7). These cytokines target keratinocytes, which respond by producing IL-17s themselves, but also neutrophil-attractant chemokines, thus constituting a positive feedback-loop (comprising dendritic cells and TH17-cells) and boosting inflammation further though recruitment of additional inflammatory cells, namely neutrophils (8). The presence of tissue-resident memory T-cells contributes to the chronicity of the disease and may explain the recurrent appearance of lesions at previously affected sites (9).
Taken together, the concept of a central pathogenetic axis evolved, starting with activated dendritic cells that produce IL-23 and thus favoring the development of TH17-cells, which exhibit their effector functions through IL-17s (10). Additional processes directly linked to this axis are leukocyte extravasation and migration as well as leukocyte activation, elements of which represent “drugable” targets (11). Indeed, all of these approaches have been or are currently being explored. However, most of the drugs currently used for targeted therapy of psoriasis block elements of the IL-23/TH17/IL-17 axis.
In this review, we will re-visit the above-mentioned observations and integrate them into a more comprehensive pathogenetic model (Figure 1). Subsequently, we will summarize the current knowledge around IL-17A and its five currently known homologues, namely IL-17B, IL-17C, IL-17D, IL-17E (also known as IL-25) and IL-17F, in relation to psoriasis with a focus on the most recent discoveries. In the last part of the review, we point towards the clinical implications of a more detailed understanding of IL-17 biology.
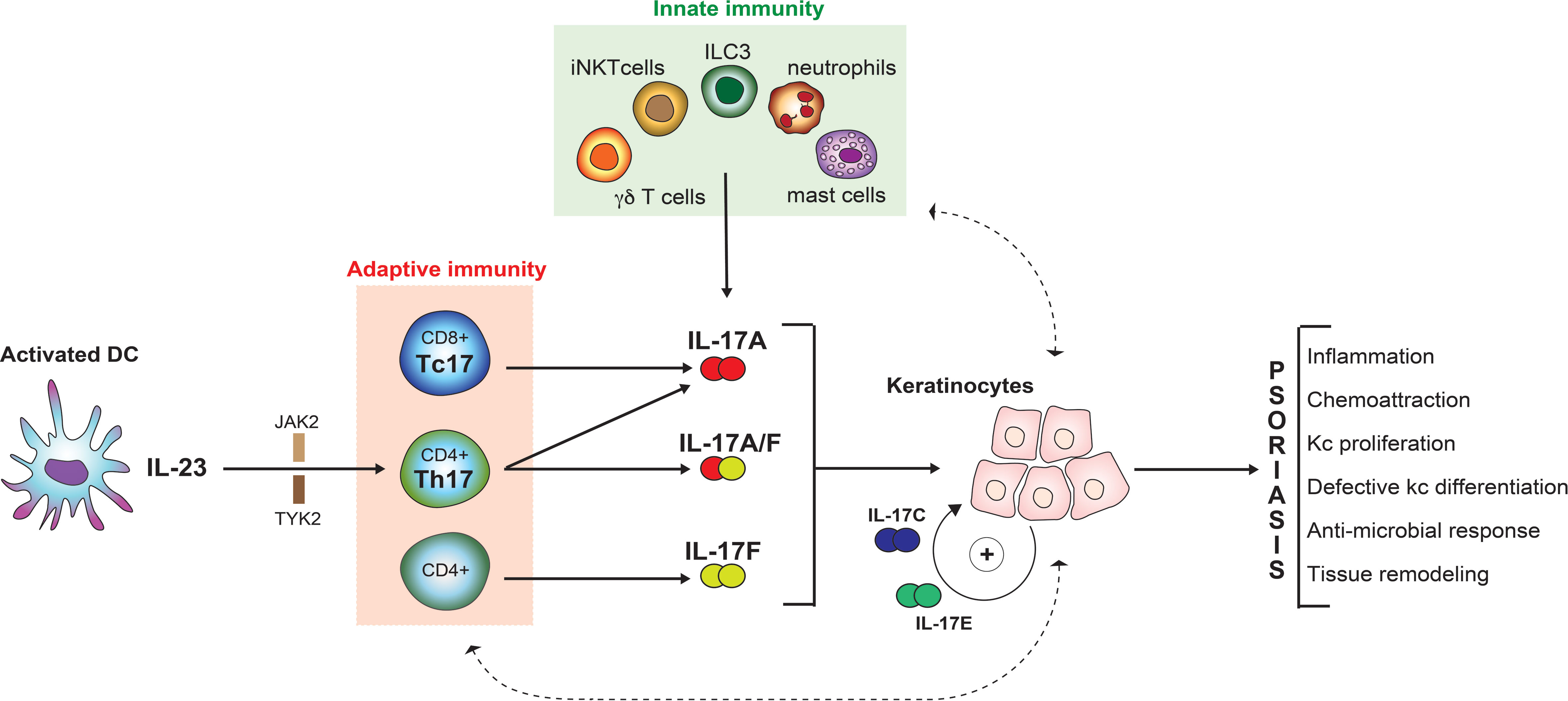
Figure 1 From linear to complex pathological model of psoriasis.
The above-mentioned “linear” model of psoriasis has proven most helpful when it comes to understanding the impressive progress in terms of therapeutic efficacy in the field:
● Biologics targeting elements of the central pathogenetic axis exhibit unprecedented efficacy: In direct comparator studies, the conventional disease-modifying anti-rheumatic drug (DMARD) methotrexate was inferior to the TNF-α inhibitor adalimumab (PASI75 after 12 weeks: 25 versus 76%) (12); TNF-α inhibition by etanercept was inferior to IL-23/IL-12 inhibition by ustekinumab (PASI75 at week 12: 57 versus 74%) (13), which was inferior to the IL-17A inhibitor secukinumab (PASI90 at week 16: 58 versus 79%) (14).
● IL-17A inhibition was shown to exhibit a faster mode of onset compared to IL-23 inhibition (15), which is in line with the idea that blocking a “downstream” effector cytokine such as IL-17A should reduce psoriasis symptoms faster than blocking an “upstream” regulatory cytokine such as IL-23.
● In contrast, IL-23 inhibition has a particularly long-lasting therapeutic effect, as evidenced in randomized-withdrawal studies (16). This observation was initially interpreted as being due to the longer time needed to produce TH17 cells again. More recent studies suggest that this may also be related to the reduction in resident memory T cells in the skin (17).
Despite these conformities between model-based predictions and clinical observations, more recent findings make it necessary to revisit this simple “linear” model. One such finding is that IL-23 inhibition is insufficient to completely suppress IL-17A production. This is due to the fact that IL-17A production is not restricted to conventional CD4+ T helper lymphocytes (TH17 cells). Innate-like lymphoid cells, namely γδ T-cells, invariant natural killer T cells (iNKTs), mucosal-associated invariant T-cells (MALTs), as well as innate lymphoid cells (ILCs) are often skewed to type-17 profiles and may substantially contribute to IL-17A production in an IL-23 independent manner [reviewed in (18)]. Besides, cells other than lymphoid cells are also capable of producing IL-17A (19). Moreover, IL-17 homologues other than IL-17A have long been overlooked. This is particularly true for IL-17F, which is actually more abundant in lesional psoriatic skin when compared to IL-17A (20). As IL-17 homologues exhibit partially overlapping biological effects (see below), molecules were developed that block IL-17A as well as IL-17F. This approach yields synergistic effects in-vitro (21) and proved superior to selective IL-17A inhibition in a direct comparator clinical trial (22).
The IL-17 cytokine family consists of six members, named IL-17A to IL-17F (Figure 2). All members form functionally active homodimers. IL-17A also forms heterodimers with IL-17F. IL-17 cytokines signal through the IL-17 receptor family (IL17Rs), exclusively using the adaptor Act1 as the primary intermediate to activate downstream signaling events. A functionally active receptor comprises a combination of two of five homologous receptor subunits, IL-17RA to IL17RE (7).
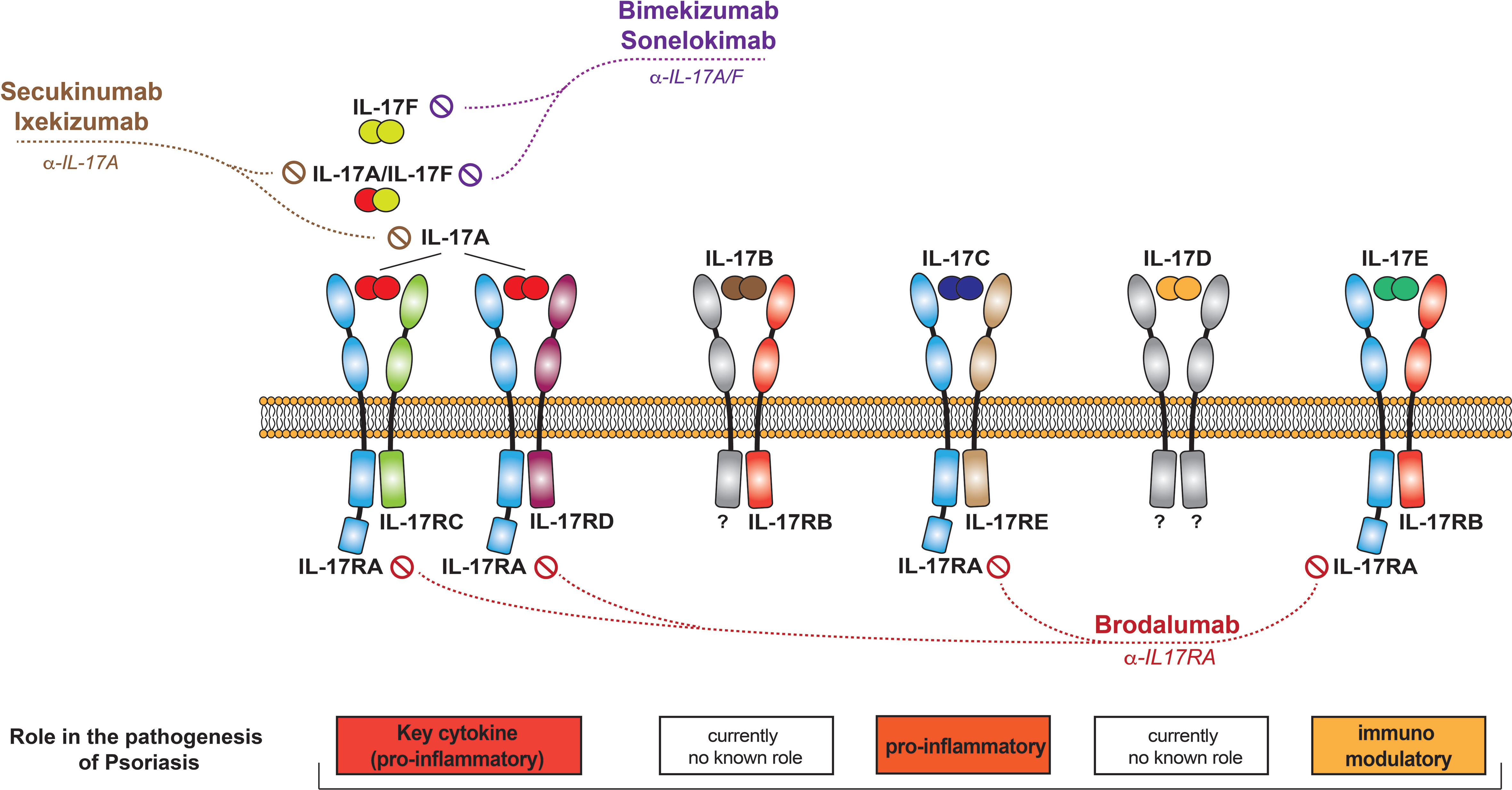
Figure 2 The IL-17 family of cytokines. Schematic representation of the different IL-17 cytokines, their receptors, and the currently available therapeutics that target them.
Described in 1993 and initially named CTLA-8, IL-17A is the most vigorously studied IL-17. It is produced by immune cells and targets primarily non-hematopoietic cells in barrier tissues, such as the skin, playing a central role in protective immune responses to extracellular pathogens and fungi.
IL-17A is classically considered to signal through the IL17RA-RC receptor, as do IL-17A/F heterodimers and IL-17F (7, 23). Recent data have however shown that these cytokines can also bind to a receptor composed of two IL17RC subunits, transducing signal in an IL-17RA-independent manner (24). The IL-17RA/RD heterodimer has also been reported as an alternative receptor for the IL-17A homodimer, but not for IL-17F/F and IL-17A/F (25, 26).
Activation of the IL17RA-RC heterodimer leads to the unique recruitment of the adaptor protein Act1 via homotypic SEFIR domain interaction. Act1 acts like a docking station for TRAF proteins, facilitating target gene transcription through the activation of NFκB, MAPK and C/EBP pathways. Additionally, activation of various RNA-binding proteins (such as HuR and Regnase-1) downstream the IL-17RA-RC receptors allows IL-17A to control post-transcriptional events, often resulting in increased target RNA stability (7, 27). Recent crystallography data point to the formation of a 2:2:2 hexameric signaling assembly composed of two units each of IL-17A, IL-17RA and IL-17RC. IL-17RA dimerization was shown to be functionally important, leading to the potentiation of IL-17-induced IL-36γ and CXCL1 mRNA expression in human keratinocytes (28).
IL-17A is secreted by IL-23-dependent Th17 cells in the skin. Additional cellular sources exist. 10 years ago, it was demonstrated that CD8+ T cells located in the epidermis of lesional psoriatic skin secrete IL-17A, possibly upon recognition of antigens presented by HLA-Cw6 (29). These so-called Tc17 cells exhibit a resident memory phenotype and express both PD-1 and IL-23R (30). Additionally, neutrophils, mast cells, and innate immune cells (MAILT, ILC3 and γδ T-cells) all are capable of producing IL-17A in an IL-23 independent manner (19, 31–33).
IL-17A has four major effects on mesenchymal and epithelial cells in the skin:
● establishing a pro-inflammatory loop geared towards activation of neutrophils and Th17 responses through induction of inflammatory mediators and chemoattractants such as IL-8, CCL20, and G-CSF
● promoting a cathelicidin- and defensin-dominated antimicrobial response
● de-regulating the keratinocyte differentiation program, leading to decreased expression of late differentiation molecules and loosening of the epidermal barrier
● promoting metalloproteinase-dependent tissue remodeling, facilitating the influx of newly recruited leukocytes
With regard to keratinocyte proliferation, recent publications show that IL-17A induces proliferation, either directly through activation of YAP-AREG signaling in keratinocytes, or indirectly via IL-19 and IL-24, produced in response to IL-17A in dermal stromal cells (34, 35).
Finally, new data suggest that IL-17A may alter immune regulatory responses. In psoriasis, regulatory T cells are dysfunctional, exhibiting reduced suppressive capacity and an exhausted phenotype (36, 37). The underlying mechanism comprises reduced TGF-β release and increased IFN-γ production. This skewing is reversed under therapy with IL-17A inhibitors (38). In addition, IL-17A (as well as IL17F and the IL-17A/F heterodimer) induces resistance to CD8-mediated suppression in CD4+ T cells primed with IL-17A, which is reversible via blocking IL-1β, IL-6, or STAT3 (39).
L-17F shows the highest homology to IL-17A amongst all IL-17 family members. Besides homodimers, there exist IL-17A/F heterodimers as well. IL-17F signals via the canonical IL-17A receptor, composed of an IL17RA and -RC subunit. It has functions similar to IL-17A, namely with regard to an effective defense against muco-epithelial bacterial and fungal infections. IL-17F is generally considered less “potent” than IL-17A, with the IL-17A/F heterodimer exhibiting an intermediate potency (40).
Similar to IL-17A, IL-17F is expressed by immune cells of both innate and adaptive lineages, including Th17 cells, γδ T cells, and ILC3. IL-17F is believed to be generally co-expressed with IL-17A (the two genes are at the same locus), but this concept is being challenged by more recent data, analyzing emigrating cells from psoriasis samples at a single cell level. Such analyses show that most T17 cells express either IL-17A or IL-17F, with <10% of cells co-expressing both cytokines (36). Interestingly, most T cells in lesional psoriatic skin express IL-17F and not IL-17A. These cells exhibit a cytokine profile which differs from “conventional”, i.e. IL-17A-producing T cells. This may explain the clinical observation that simultaneous blockade of IL-17A and IL-17F resulted in successful treatment of patients who failed to respond to selective anti IL17A therapy before (41). Besides, IL-17F may also play a role in the recurrent course of psoriasis (42).
While the contribution of IL-17F in psoriasis is now accepted, its role in psoriatic arthritis remains under debate. To this end, expression of both IL-17A as well as IL-17F has been shown to be increased in synovial tissue and entheses of patients with psoriatic arthritis. While data from head-to-head comparator trials are not yet available, indirect evidence suggests comparable efficacies of selective IL-17A inhibition and blockade of IL-17A along with -F (43).
IL-17C is only 23% homologous to IL-17A and, unlike the latter, is expressed by and acts on epithelial cells. Keratinocytes are the main producers of and responders to IL-17C in the skin. Signaling occurs via a receptor composed of the IL-17RA and IL-17RE subunits. IL-17Cs physiological role is to establish antimicrobial protective responses at barrier tissues.
It is now also widely accepted that IL-17C contributes to the psoriatic inflammation. It is increased in lesional skin, induces a gene expression pattern similar to that induced by IL-17A in keratinocytes in vitro, and is pathogenic in animal models in vivo (44–46).
Recent data, based on bioinformatic analysis of RNA seq experiments comparing non-lesional skin with the leading edge of evolving psoriatic plaques, identified IL-17C as a functional regulator of the initial psoriatic cytokine network, suggesting a role during the early stages of psoriatic inflammation, or the “priming” for plaque formation (47).
Of interest, the level of IL-17C in lesional skin was reduced to levels comparable to non-lesional skin upon therapy with an anti-IL17A/F inhibitor (48), and effective systemic therapy with methotrexate, ustekinumab, Secukinumab, or adalimumab were all associated with markedly decreased IL-17C concentrations in peripheral blood (49). These data suggest a potential role for IL-17C as a biomarker for therapeutic efficacy.
IL-17B signals through a receptor that contains the IL-17RB subunit. While IL-17B is recognized for its role in cancer pathophysiology, it may not play a major role in the context of inflammation (50). Its concentration in psoriatic plaques is low (51).
Little is known about IL-17D, and its receptor has yet to be identified. IL-17D induces the production of pro-inflammatory cytokines and appears to play a regulatory role in anti-tumor and anti-viral responses (52). IL-17D levels are decreased in chronic plaque-type (51), but increased in palmoplantar pustulosis (53). It inhibits the expression of DDX5 helicase RNA in keratinocytes, promoting the production of membrane-bound IL-36R, and leading to the amplification of IL-36R-mediated skin inflammation. This newly identified pathway suggests a novel role of IL-17D in the control of skin inflammatory disorders, which needs to be further elucidated and studied in the context of pathology (54).
L-17E, also known as IL-25, shares only 6% homology with IL-17A. It signals through a heterodimeric receptor composed of the IL-17RA and IL-17RB subunits. IL-17E is produced by and acts on several cell types, including those of epithelial, mesenchymal, and hematopoietic origin.
Although primarily recognized as a cytokine that participates in type 2 cell responses (55), IL-17E may also play a role in the maintenance of skin inflammation at large (56). Keratocyte-derived IL-17E promotes the expression of pro-inflammatory cytokines in an autocrine manner, participates in the amplification of the psoriatic inflammatory network (57–59), and promotes recruitment of neutrophils in neutrophilic dermatoses (60).
IL-17E exhibits effects on keratinocytes that are distinct from those of IL-17A in as much as it induces cell motility and proliferation rather than antimicrobial responses. IL-17Es functions clearly go beyond those of a “simple” alarmin; it seems to play an important role in epidermal homeostasis (61).
Interestingly, serum levels of IL-17E could perhaps be used as biomarkers of psoriasis activity or severity, as particularly high levels are detectable in erythrodermic psoriasis (62).
In recent years, IL-17 inhibition has become a mainstay of psoriasis treatment. Currently approved anti-psoriatic drugs interfering with IL-17 function comprise biologics and a JAK inhibitor, additional molecules are under development (11). These are (Table 1, Figure 2):
● Antibodies selectively blocking IL-17A (secukinumab, ixekizumab)
● Molecules blocking IL-17A and –F (bimekizumab, sonelokimab)
● Inhibitors of IL-17 signalling (brodalumab)
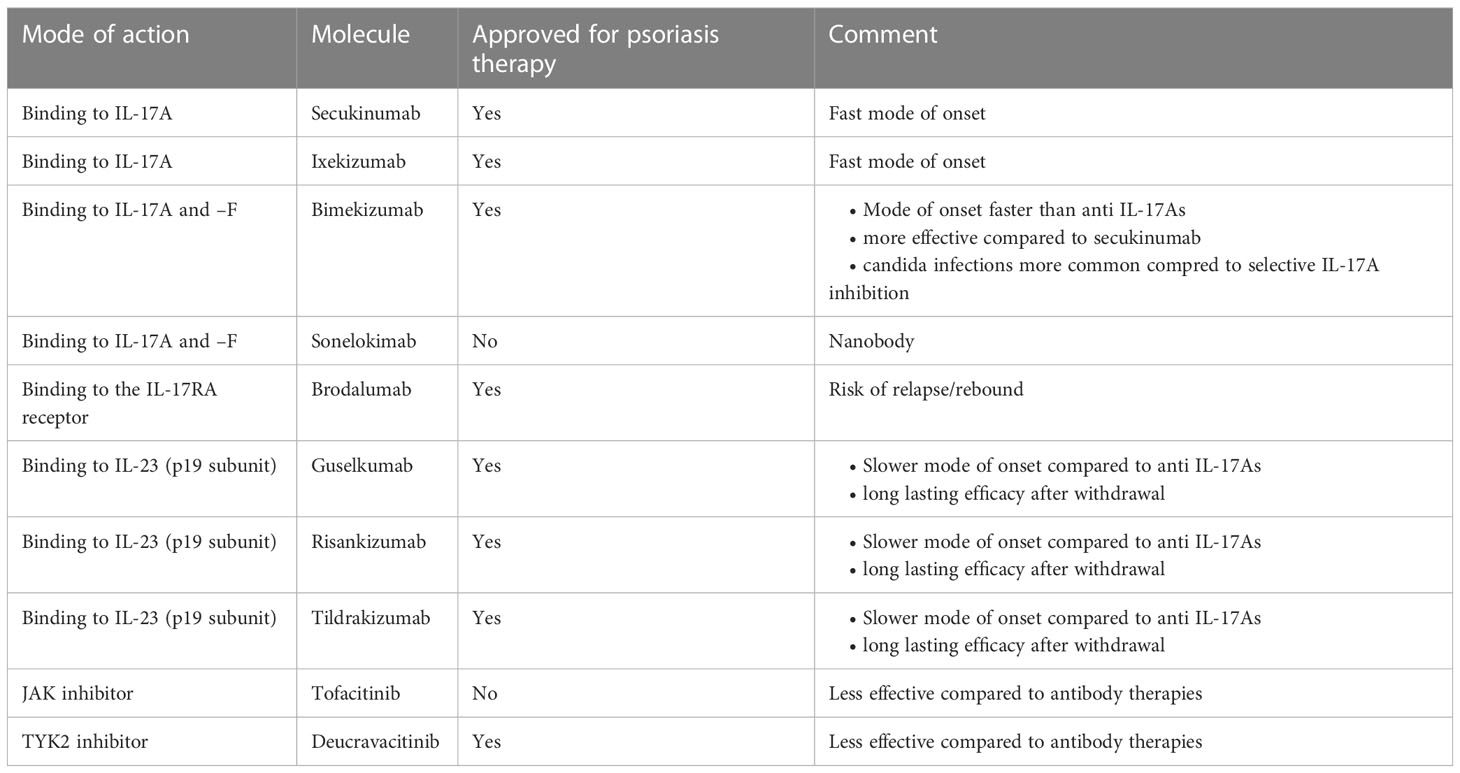
Table 1 Synopsis of anti-psoriatic drugs interfering with IL-17s. Only molecules with published positive phase III trial data are included.
Besides, IL-23 inhibitors can be regarded as indirect IL-17 inhibitors, as this mode of action results in reduced production of IL-17A. All of these molecules exhibit distinct efficacy and safety profiles reflecting their respective mode of action.
Secukinumab and ixekizumab are monoclonal antibodies binding IL-17A selectively and directly. They are both highly effective, and “clear skin” became a feasible treatment goal in psoriasis with the approval of these drugs, as most patients achieved treatment responses superior to the previous treatment goal of a 75% reduction in the Psoriasis Area and Severity Index (PASI) (63). They also exhibit a particularly fast mode of onset, as expected by the fact that they block a key “effector” cytokine directly (see above). This is evidenced by superior efficacy data in direct comparator studies against IL-23 inhibitors during the induction phase of treatment (15, 64). As IL-17A plays an important role in mucosal immunity in general and protection against infections (namely by candida) in particular, it is not surprising that candida infections are a common adverse event of these drugs (65). Nevertheless, the interest in novel strategies to block IL-17A remains unbroken, as exemplified by clinical development programs using innovative molecules such as izokibep, a fusion protein exhibiting a particularly high affinity to IL-17A (66) or the oral IL-17 inhibitor DC-806 (67).
IL-17F is an IL-17 homologue who’s relevance in the pathogenesis of psoriasis has long been overlooked. Once it was realized that IL-17F is actually more abundant in lesional psoriatic skin (20), with partially overlapping functions (21), it became an obvious target for novel anti-psoriatic therapies. The monoclonal antibody bimekizumab has recently been approved for this indication. It binds IL-17A and IL-17F homodimers as well as the IL-17A/F heterodimer. As it blocks two strongly pro-inflammatory cytokines involved in the pathogenesis of psoriasis, a greater depth of clinical response was hypothesized (68). This hypothesis proved right, as bimekizumab showed superior efficacy in a direct comparator trial against secukinumab. But as IL-17F does not only contribute to psoriasis, but also to mucosal immunity, the blockade of IL-17F along with IL-17A results in an increased risk for candida infections (22). Another molecule binding to both IL-17A and IL-17F is the nanobody sonelokimab. The clinical trial data published so far document its efficacy in the treatment of psoriasis (69).
Finally, the monoclonal antibody brodalumab blocks the IL-17RA receptor and thus signaling of IL-17A, -C –E, and –F. As expected, this strategy is highly effective (70). The brodalumab clinical development program was abruptly interrupted for safety reasons (several cases of major adverse cardiovascular events in the brodalumab arm but not the placebo arm in one clinical study, resulting in a statistically significant safety signal) and subsequently resumed (brodalumab’s safety profile with regard to such major adverse cardiovascular events has recently been documented by an integrated safety analysis based on 5 clinical trials (71). Discontinuation of brodalumab treatment resulted in numerous relapses and even rebounds (72). This observation may be explained by the fact that a mediator may still be present (or even increased in concentration due to blockade of potential feedback loops), ready to signal through the receptor once it becomes available for interaction again. Meanwhile, brodalumab is approved for the treatment of psoriasis.
IL-17 inhibition may also be achieved indirectly via blocking IL-23 and consequently a reduction of sources of IL-17. There are currently three anti IL-23 antibodies approved for the treatment of psoriasis, namely guselkumab, risankizumab, and tildrakizumab. Their efficacy is comparable to the one of the IL-17 blocking antibodies (63, 73). However, as expected when blocking an upstream “regulatory” cytokine, clinical trial data tend to document longer time to onset of action in studies assessing approved dosing ranges of IL-23 inhibitors compared with studies assessing IL-17 inhibitors (74). However, speed of onset does not depend on a drug’s mode of action alone, but is also influenced by affinity and potency, as well as the selected clinical end point. Therefore, individual comparisons between one specific IL-23 inhibitor and one specific IL-17 inhibitor may yield different results.
Blocking the intracellular signal transduction of IL-23 at the level of the JAKs follows a similar reasoning. The pan JAK inhibitor tofacitinib as well as the specific TYK2 inhibitor deucravacitinib, have both shown clinical efficacy in clinical trials (75, 76). Deucravacitinib has meanwhile been approved for the treatment of psoriasis, while the manufacturing company of tofacitinib decided not to file for approval.
In this review, we focused on psoriasis. From a physician’s perspective, it is also important to which extent psoriatic comorbidities can either also be treated or represent potential contraindications for a given anti-psoriatic drug (77). This is also highlighted in the current S3 treatment guidelines (78). Clinically, the single most important comorbidity of psoriasis is psoriatic arthritis, affecting up to 30% of psoriasis patients (79). In the context of this review, it is therefore important to stress that all of the above-mentioned anti-psoriatic drugs are also effective in the treatment of psoriatic arthritis. IL-17A inhibitors are by now readily accepted by rheumatologists and recommended by expert groups such as EULAR (80) or GRAPPA (81). JAK inhibition is an established treatment strategy for psoriatic arthritis as well (82). And there are positive data from a phase II trial with the above-mentioned TYK2 inhibitor deucravacitinib (83). Finally, guselkumab was the first IL-23 inhibitor approved for psoriatic arthritis (84), exhibiting efficacy data comparable to IL-17A inhibitors (85).
In summary, our growing understanding of the IL-17 cytokine family helps us to better understand the pathophysiology of psoriasis as well as the distinct profiles of numerous drugs either already approved or in advanced stages of clinical development for this indication with regard to efficacy, mode of onset, and safety. To this end, it seems as if direct blockade of more than one IL-17 homologue potentially increases efficacy, but also characteristic safety signals (candida infections), while modulating the inflammatory milieu via interference with signal transduction may result in a lower clinical efficacy compared to direct blockade of IL-17. This may open the door towards development of systemic therapies for moderate rather than severe psoriasis as well as – finally – innovative topical therapies, as small molecules such as JAK inhibitors may be used in topical formulations. It remains to be seen to which extent IL-17 homologues other than –A and -F are valid targets for anti-psoriatic therapies. One company’s Il-17C program has recently been shut down (WHB, personal communication). And IL-17E/IL-25 as an important player in epithelial homeostasis at large might be more relevant as therapeutic target in other pathologies (56).
W-HB and NB jointly selected the topic for the review, performed the literature review, wrote the manuscript, and designed the figure along with tables. All authors contributed to the article and approved the submitted version.
Open access funding by University of Geneva.
W-HB has received honoraria as a speaker and/or advisor from Abbvie, Almirall, BMS, Janssen, Leo, Lilly, Novartis, and UCB.
The remaining author declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Boehncke WH, Schon MP. Psoriasis. Lancet (2015). 386(9997):983–94. doi: 10.1016/S0140-6736(14)61909-7
2. Boehncke WH. Systemic inflammation and cardiovascular comorbidity in psoriasis patients: causes and consequences. Front Immunol (2018) 9:579. doi: 10.3389/fimmu.2018.00579
3. Dauden E, Castaneda S, Suarez C, Garcia-Campayo J, Blasco AJ, Aguilar MD, et al. Clinical practice guideline for an integrated approach to comorbidity in patients with psoriasis. J Eur Acad Dermatol Venereol (2013) 27(11):1387–404. doi: 10.1111/jdv.12024
4. Boehncke WH, Dressel D, Zollner Kaufmann TM R. Pulling the trigger on psoriasis. Nature (1996) 379(6568):777. doi: 10.1038/379777a0
5. Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature (2007) 449(7162):564–9. doi: 10.1038/nature06116
6. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24(2):179–89. doi: 10.1016/j.immuni.2006.01.001
7. Brembilla NC, Senra L, Boehncke WH. The IL-17 family of cytokines in psoriasis: IL-17A and beyond. Front Immunol (2018) 9:1682. doi: 10.3389/fimmu.2018.01682
8. Griffiths CEM, Armstrong AW, Gudjonsson JE, Barker J. Psoriasis. Lancet (2021) 397(10281):1301–15. doi: 10.1016/S0140-6736(20)32549-6
9. Clark RA. Resident memory T cells in human health and disease. Sci Transl Med (2015) 7(269):269rv1. doi: 10.1126/scitranslmed.3010641
10. Girolomoni G, Strohal R, Puig L, Bachelez H, Barker Boehncke WH, et al. The role of IL-23 and the IL-23/TH 17 immune axis in the pathogenesis and treatment of psoriasis. J Eur Acad Dermatol Venereol (2017) 31(10):1616–26. doi: 10.1111/jdv.14433
11. Boehncke WH, Brembilla NC. Pathogenesis-oriented therapy of psoriasis using biologics. Expert Opin Biol Ther (2022) 22(12):1463–73. doi: 10.1080/14712598.2022.2100219
12. Saurat JH, Stingl G, Dubertret L, Papp Langley K RG, Ortonne JP, et al. Efficacy and safety results from the randomized controlled comparative study of adalimumab vs. methotrexate vs. placebo in patients with psoriasis (CHAMPION). Br J Dermatol (2008) 158(3):558–66. doi: 10.1111/j.1365-2133.2007.08315.x
13. Griffiths CE, Strober BE, van de Kerkhof P, Ho V, Fidelus-Gort R, Yeilding N, et al. Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N Engl J Med (2010) 362(2):118–28. doi: 10.1056/NEJMoa0810652
14. Thaci D, Blauvelt A, Reich K, Tsai TF, Vanaclocha F, Kingo K, et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate to severe plaque psoriasis: CLEAR, a randomized controlled trial. J Am Acad Dermatol (2015) 73(3):400–9. doi: 10.1016/j.jaad.2015.05.013
15. Reich K, Armstrong AW, Langley RG, Flavin S, Randazzo B, Li S, et al. Guselkumab versus secukinumab for the treatment of moderate-to-severe psoriasis (ECLIPSE): results from a phase 3, randomised controlled trial. Lancet (2019) 394(10201):831–9. doi: 10.1016/S0140-6736(19)31773-8
16. Blauvelt A, Leonardi CL, Gooderham M, Papp KA, Philipp S, Wu JJ, et al. Efficacy and safety of continuous risankizumab therapy vs treatment withdrawal in patients with moderate to severe plaque psoriasis: a phase 3 randomized clinical trial. JAMA Dermatol (2020) 156(6):649–58. doi: 10.1001/jamadermatol.2020.0723
17. Mehta H, Mashiko S, Angsana J, Rubio M, Hsieh YM, Maari C, et al. Differential changes in inflammatory mononuclear phagocyte and T-cell profiles within psoriatic skin during treatment with guselkumab vs. secukinumab. J Invest Dermatol (2021) 141(7):1707–1718.e9. doi: 10.1016/j.jid.2021.01.005
18. O’Brien-Gore C, Gray EH, Durham LE, Taams LS, Kirkham BW. Drivers of inflammation in psoriatic arthritis: the old and the new. Curr Rheumatol Rep (2021) 23(6):40. doi: 10.1007/s11926-021-01005-x
19. Brembilla NC, Stalder R, Senra L, Boehncke WH. IL-17A localizes in the exocytic compartment of mast cells in psoriatic skin. Br J Dermatol (2017) 177(5):1458–60. doi: 10.1111/bjd.15358
20. Kolbinger F, Loesche C, Valentin MA, Jiang X, Cheng Y, Jarvis P, et al. Beta-defensin 2 is a responsive biomarker of IL-17A-driven skin pathology in patients with psoriasis. J Allergy Clin Immunol (2017) 139(3):923–932.e8. doi: 10.1016/j.jaci.2016.06.038
21. Glatt S, Baeten D, Baker T, Griffiths M, Ionescu L, Lawson ADG, et al. Dual IL-17A and IL-17F neutralisation by bimekizumab in psoriatic arthritis: evidence from preclinical experiments and a randomised placebo-controlled clinical trial that IL-17F contributes to human chronic tissue inflammation. Ann Rheum Dis (2018) 77(4):523–32. doi: 10.1136/annrheumdis-2017-212127
22. Reich K, Warren R.B, Lebwohl M, Gooderham M, Strober B, Langley RG, et al. Bimekizumab versus secukinumab in plaque psoriasis. N Engl J Med (2021) 385(2):142–52. doi: 10.1056/NEJMoa2102383
23. Chung SH, Ye XQ, Iwakura Y. Interleukin-17 family members in health and disease. Int Immunol (2021) 33(12):723–9. doi: 10.1093/intimm/dxab075
24. Goepfert A, Lehmann S, Blank J, Kolbinger F, Rondeau JM. Structural analysis reveals that the cytokine IL-17F forms a homodimeric complex with receptor IL-17RC to drive IL-17RA-Independent signaling. Immunity (2020) 52(3):499–512.e5. doi: 10.1016/j.immuni.2020.02.004
25. Billi AC, Gudjonsson JE. Interleukin-17 receptor d: an orphan receptor finds a home in the skin. Sci Immunol (2019) 4(36):eaax0687. doi: 10.1126/sciimmunol.aax0687
26. Su Y, Huang J, Zhao X, Lu H, Wang W, Yang XO, et al. Interleukin-17 receptor d constitutes an alternative receptor for interleukin-17A important in psoriasis-like skin inflammation. Sci Immunol (2019) 4(36):eaau9657. doi: 10.1126/sciimmunol.aau9657
27. Bechara R, McGeachy MJ, Gaffen SL. The metabolism-modulating activity of IL-17 signaling in health and disease. J Exp Med (2021) 218(5):e20202191. doi: 10.1084/jem.20202191
28. Goepfert A, Barske C, Lehmann S, Wirth E, Willemsen J, Gudjonsson JE, et al. IL-17-induced dimerization of IL-17RA drives the formation of the IL-17 signalosome to potentiate signaling. Cell Rep (2022) 41(3):111489. doi: 10.1016/j.celrep.2022.111489
29. Hijnen D, Knol EF, Gent YY, Giovannone B, Beijn SJ, Kupper TS, et al. CD8(+) T cells in the lesional skin of atopic dermatitis and psoriasis patients are an important source of IFN-gamma, IL-13, IL-17, and IL-22. J Invest Dermatol (2013) 133(4):973–9. doi: 10.1038/jid.2012.456
30. Phadungsaksawasdi P, Fujiyama T, Kurihara K, Ito T, Honda T, Tokura Y. PD-1 expression defines epidermal CD8(+)CD103(+) T cells preferentially producing IL-17A and using skewed TCR repertoire in psoriasis. J Invest Dermatol (2021) 141(10):2426–2435.e5. doi: 10.1016/j.jid.2021.03.011
31. Cole S, Murray J, Simpson C, Okoye R, Tyson K, Griffiths M, et al. Interleukin (IL)-12 and IL-18 synergize to promote MAIT cell IL-17A and IL-17F production independently of IL-23 signaling. Front Immunol (2020) 11:585134. doi: 10.3389/fimmu.2020.585134
32. Webster KE, Kim HO, Kyparissoudis K, Corpuz TM, Pinget GV, Uldrich AP, et al. IL-17-producing NKT cells depend exclusively on IL-7 for homeostasis and survival. Mucosal Immunol (2014) 7(5):1058–67. doi: 10.1038/mi.2013.122
33. Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Cayatte C, Chen Y, et al. Interleukin-23-Independent IL-17 production regulates intestinal epithelial permeability. Immunity (2015) 43(4):727–38. doi: 10.1016/j.immuni.2015.09.003
34. Xu X, Prens E, Florencia E, Leenen P, Boon L, Asmawidjaja P, et al. Interleukin-17A drives IL-19 and IL-24 expression in skin stromal cells regulating keratinocyte proliferation. Front Immunol (2021) 12:719562. doi: 10.3389/fimmu.2021.719562
35. Yu Z, Yu Q, Xu H, Dai X, Yu Y, Cui L, et al. IL-17A promotes psoriasis-associated keratinocyte proliferation through ACT1-dependent activation of YAP-AREG axis. J Invest Dermatol (2022) 142(9):2343–52. doi: 10.1016/j.jid.2022.02.016
36. Kim J, Lee J, Kim HJ, Kameyama N, Nazarian R, Der E, et al. Single-cell transcriptomics applied to emigrating cells from psoriasis elucidate pathogenic versus regulatory immune cell subsets. J Allergy Clin Immunol (2021) 148(5):1281–92. doi: 10.1016/j.jaci.2021.04.021
37. Sugiyama H, Gyulai R, Toichi E, Garaczi E, Shimada S, Stevens SR, et al. Dysfunctional blood and target tissue CD4+CD25high regulatory T cells in psoriasis: mechanism underlying unrestrained pathogenic effector T cell proliferation. J Immunol (2005) 174(1):164–73. doi: 10.4049/jimmunol.174.1.164
38. Liu Y, Zhang C, Li B, Yu C, Bai X, Xiao C, et al. A novel role of IL-17A in contributing to the impaired suppressive function of tregs in psoriasis. J Dermatol Sci (2021) 101(2):84–92. doi: 10.1016/j.jdermsci.2020.09.002
39. Crawford MP, Sinha S, Renavikar PS, Borcherding N, Karandikar NJ. CD4 T cell-intrinsic role for the T helper 17 signature cytokine IL-17: effector resistance to immune suppression. Proc Natl Acad Sci U.S.A. (2020) 117(32):19408–14. doi: 10.1073/pnas.2005010117
40. Noack M, Beringer A, Miossec P. Additive or synergistic interactions between IL-17A or IL-17F and TNF or IL-1beta depend on the cell type. Front Immunol (2019) 10:1726. doi: 10.3389/fimmu.2019.01726
41. Kokolakis G, Ghoreschi K. The clinical significance of simultaneous IL-17A and IL-17F blockade in psoriasis non-responding to anti-IL17A therapy. J Clin Med (2022) 12(1):35. doi: 10.3390/jcm12010035
42. Reich K, Cullen E, Weinberg M. Maintenance of response in moderate-to-severe psoriasis after withdrawal of the interleukin (IL)-17A and IL-17F nanobody sonelokimab: is there a role for IL-17F in disease reoccurrence? Br J Dermatol (2022) 187(4):591–3. doi: 10.1111/bjd.21617
43. Tam HKJ, Robinson PC, Nash P. Inhibiting IL-17A and IL-17F in rheumatic disease: therapeutics help to elucidate disease mechanisms. Curr Rheumatol Rep (2022) 24(10):310–20. doi: 10.1007/s11926-022-01084-4
44. Johnston A, Fritz Y, Dawes SM, Diaconu D, Al-Attar PM, Guzman AM, et al. Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J Immunol (2013) 190(5):2252–62. doi: 10.4049/jimmunol.1201505
45. Ramirez-Carrozzi V, Sambandam A, Luis E, Lin Z, Jeet S, Lesch J. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol (2011) 12(12):1159–66. doi: 10.1038/ni.2156
46. Vandeghinste N, Klattig J, Jagerschmidt C, Lavazais S, Marsais F, Haas JD, et al. Neutralization of IL-17C reduces skin inflammation in mouse models of psoriasis and atopic dermatitis. J Invest Dermatol (2018) 138(7):1555–63. doi: 10.1016/j.jid.2018.01.036
47. Boonpethkaew S, Meephansan J, Jumlongpim O, Tangtanatakul P, Soonthornchai W, Wongpiyabovorn J, et al. Transcriptomic profiling of peripheral edge of lesions to elucidate the pathogenesis of psoriasis vulgaris. Int J Mol Sci (2022) 23(9):4983. doi: 10.3390/ijms23094983
48. Oliver R, Krueger JG, Glatt S, Vajjah P, Mistry C, Page M, et al. Bimekizumab for the treatment of moderate-to-severe plaque psoriasis: efficacy, safety, pharmacokinetics, pharmacodynamics and transcriptomics from a phase IIa, randomized, double-blind multicentre study. Br J Dermatol (2022) 186(4):652–63. doi: 10.1111/bjd.20827
49. Wang X, Kaiser H, Kvist-Hansen A, McCauley BD, Skov L, Hansen PR, et al. IL-17 pathway members as potential biomarkers of effective systemic treatment and cardiovascular disease in patients with moderate-to-Severe psoriasis. Int J Mol Sci (2022) 23(1):555. doi: 10.3390/ijms23010555
50. Bastid J, Dejou C, Docquier A, Bonnefoy N. The emerging role of the IL-17B/IL-17RB pathway in cancer. Front Immunol (2020) 11:718. doi: 10.3389/fimmu.2020.00718
51. Johansen C, Usher PA, Kjellerup RB, Lundsgaard D, Iversen L, Kragballe K. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br J Dermatol (2009) 160(2):319–24. doi: 10.1111/j.1365-2133.2008.08902.x
52. Liu X, Sun S, Liu D. IL-17D: a less studied cytokine of IL-17 family. Int Arch Allergy Immunol (2020) 181(8):618–23. doi: 10.1159/000508255
53. Brunasso AMG, Massone C. Recent advances in palmoplantar pustulosis. Fac Rev (2021) 10:62. doi: 10.12703/r/10-62
54. Ni X, Xu Y, Wang W, Kong B, Ouyang J, Chen J, et al. IL-17D-induced inhibition of DDX5 expression in keratinocytes amplifies IL-36R-mediated skin inflammation. Nat Immunol (2022) 23(11):1577–87. doi: 10.1038/s41590-022-01339-3
55. Wu J, Zhang F, Tao H, Nawaz W, Chen D, Wu Z, et al. The potential roles of interleukin-25 in infectious diseases. Front Immunol (2022) 13:986118. doi: 10.3389/fimmu.2022.986118
56. Borowczyk J, Shutova M, Brembilla NC, Boehncke WH. IL-25 (IL-17E) in epithelial immunology and pathophysiology. J Allergy Clin Immunol (2021) 148(1):40–52. doi: 10.1016/j.jaci.2020.12.628
57. Senra L, Mylonas A, Kavanagh RD, Fallon PG, Conrad C, Borowczyk-Michalowska J, et al. IL-17E (IL-25) enhances innate immune responses during skin inflammation. J Invest Dermatol (2019) 139(8):1732–1742.e17. doi: 10.1016/j.jid.2019.01.021
58. Senra L, Stalder R, Alvarez Martinez D, Chizzolini C, Boehncke WH, Brembilla NC. Keratinocyte-derived IL-17E contributes to inflammation in psoriasis. J Invest Dermatol (2016) 136(10):1970–80. doi: 10.1016/j.jid.2016.06.009
59. Xu M, Lu H, Lee YH, Wu Y, Liu K, Shi Y, et al. An interleukin-25-Mediated autoregulatory circuit in keratinocytes plays a pivotal role in psoriatic skin inflammation. Immunity (2018) 48(4):787–798.e4. doi: 10.1016/j.immuni.2018.03.019
60. Stalder R, Brembilla N, Conrad C, Yawalkar N, Navarini A, Boehncke WH, et al. Interleukin-17E, inducible nitric oxide synthase and arginase1 as new biomarkers in the identification of neutrophilic dermatoses. Clin Exp Dermatol (2022) 47(4):675–83. doi: 10.1111/ced.14988
61. Borowczyk J, Buerger C, Tadjrischi N, Drukala J, Wolnicki M, Wnuk D, et al. IL-17E (IL-25) and IL-17A differentially affect the functions of human keratinocytes. J Invest Dermatol (2020) 140(7):1379–1389.e2. doi: 10.1016/j.jid.2019.12.013
62. Irie K, Yamamoto T. Serum levels and expression of IL-25 in patients with psoriatic erythroderma. Australas J Dermatol (2022) 63(3):e268–70. doi: 10.1111/ajd.13855
63. Sbidian E, Chaimani A, Garcia-Doval I, Do G, Hua C, Mazaud C, et al. Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis. Cochrane Database Syst Rev (2017) 12:CD011535. doi: 10.1002/14651858.CD011535.pub2
64. Warren RB, Blauvelt A, Poulin Y, Beeck S, Kelly M, Wu T, et al. Efficacy and safety of risankizumab vs. secukinumab in patients with moderate-to-severe plaque psoriasis (IMMerge): results from a phase III, randomized, open-label, efficacy-assessor-blinded clinical trial. Br J Dermatol (2021) 184(1):50–9. doi: 10.1111/bjd.19341
65. Saunte DM, Mrowietz U, Puig L, Zachariae C. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br J Dermatol (2017) 177(1):47–62. doi: 10.1111/bjd.15015
66. Behrens F, Taylor PC, Wetzel D, Brun NC, Brandt-Juergens J, Drescher E, et al. IZOKIBEP (ABY-035) in patients with active psoriatic arthritis - 16 week results from a phase 2 study. Ann Rheum Dis (2022) 81(1):OOP0258. doi: 10.1136/annrheumdis-2022-eular.536
67. DICE Therapeutics, I. DICE therapeutics announces positive topline data from phase 1 clinical trial of lead oral IL-17 antagonist, DC-806, for psoriasis. (South San Francisco, California: Globenewswire) (2022).
68. Adams R, Maroof A, Baker T, Lawson ADG, Oliver R, Paveley R, et al. Bimekizumab, a novel humanized IgG1 antibody that neutralizes both IL-17A and IL-17F. Front Immunol (2020) 11:1894. doi: 10.3389/fimmu.2020.01894
69. Papp KA, Weinberg MA, Morris A, Reich K. IL17A/F nanobody sonelokimab in patients with plaque psoriasis: a multicentre, randomised, placebo-controlled, phase 2b study. Lancet (2021) 397(10284):1564–75. doi: 10.1016/S0140-6736(21)00440-2
70. Papp KA, Leonardi C, Menter A, Ortonne JP, Krueger JG., Kricorian G, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med (2012) 366(13):1181–9. doi: 10.1056/NEJMoa1109017
71. Reich K, Thaci D, Stingl G, Andersen JS, Hiort LC, Lexner MO, et al. Safety of brodalumab in plaque psoriasis: integrated pooled data from five clinical trials. Acta Derm Venereol (2022) 102:adv00683. doi: 10.2340/actadv.v102.1993
72. Masson Regnault M, Konstantinou MP, Khemis A, Poulin Y, Bourcier M, Amelot F, et al. Early relapse of psoriasis after stopping brodalumab: a retrospective cohort study in 77 patients. J Eur Acad Dermatol Venereol (2017) 31(9):1491–6. doi: 10.1111/jdv.14387
73. Leonardi CL, See K, Burge R, Sun Z, Zhang Y, Mallbris L, et al. Number needed to treat network meta-analysis to compare biologic drugs for moderate-to-Severe psoriasis. Adv Ther (2022) 39(5):2256–69. doi: 10.1007/s12325-022-02065-w
74. Egeberg A, Andersen YMF, Halling-Overgaard AS, Alignahi F, Thyssen JP, Burge R, et al. Systematic review on rapidity of onset of action for interleukin-17 and interleukin-23 inhibitors for psoriasis. J Eur Acad Dermatol Venereol (2020) 34(1):39–46. doi: 10.1111/jdv.15920
75. Bachelez H, van de Kerkhof PC, Strohal R, Kubanov A, Valenzuela F, Lee JH, et al. Tofacitinib versus etanercept or placebo in moderate-to-severe chronic plaque psoriasis: a phase 3 randomised non-inferiority trial. Lancet (2015) 386(9993):552–61. doi: 10.1016/S0140-6736(14)62113-9
76. Papp K, Gordon K, Thaci D, Morita A, Gooderham M, Foley P, et al. Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N Engl J Med (2018) 379(14):1313–21. doi: 10.1056/NEJMoa1806382
77. Carrier Y, Ma HL, Ramon HE, Napierata L, Small C, O'Toole M, et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J Invest Dermatol (2011) 131(12):2428–37. doi: 10.1038/jid.2011.234
78. Nast A, Altenburg A, Augustin M, Boehncke WH, Harle P, Klaus J, et al. German S3-guideline on the treatment of psoriasis vulgaris, adapted from EuroGuiDerm - part 2: treatment monitoring and specific clinical or comorbid situations. J Dtsch Dermatol Ges (2021) 19(7):1092–115. doi: 10.1111/ddg.14507
79. Boehncke WH, Horvath R, Dalkilic E, Lima SAL, Okada M, Hojnik M, et al. Association between clinical specialty setting and disease management in patients with psoriatic arthritis: results from LOOP, a cross-sectional, multi-country, observational study. J Eur Acad Dermatol Venereol (2020) 34(9):2035–43. doi: 10.1111/jdv.16251
80. Gossec L, Baraliakos X, Kerschbaumer A, de Wit M, McInnes I, Dougados M, et al. EULAR recommendations for the management of psoriatic arthritis with pharmacological therapies: 2019 update. Ann Rheum Dis (2020) 79(6):700–12. doi: 10.1136/annrheumdis-2020-217159
81. Coates LC, Soriano ER, Corp N, Bertheussen H, Callis Duffin K, Campanholo CB, et al. Group for research and assessment of psoriasis and psoriatic arthritis (GRAPPA): updated treatment recommendations for psoriatic arthritis 2021. Nat Rev Rheumatol (2022) 18(8):465–79. doi: 10.1038/s41584-022-00798-0
82. Nash P, Kerschbaumer A, Dorner T, Dougados M, Fleischmann RM, Geissler K, et al. Points to consider for the treatment of immune-mediated inflammatory diseases with janus kinase inhibitors: a consensus statement. Ann Rheum Dis (2021) 80(1):71–87. doi: 10.1136/annrheumdis-2020-218398
83. Mease PJ, Deodhar AA, van der Heijde D, Behrens F, Kivitz AJ, Neal J, et al. Efficacy and safety of selective TYK2 inhibitor, deucravacitinib, in a phase II trial in psoriatic arthritis. Ann Rheum Dis (2022) 81(6):815–22. doi: 10.1136/annrheumdis-2021-221664
84. Boehncke WH, Brembilla NC, Nissen MJ. Guselkumab: the first selective IL-23 inhibitor for active psoriatic arthritis in adults. Expert Rev Clin Immunol (2021) 17(1):5–13. doi: 10.1080/1744666X.2020.1857733
Keywords: psoriasis, psoriatic arthritis, IL-17, IL-25, TH17 cells, TYK2
Citation: Brembilla NC and Boehncke WH (2023) Revisiting the interleukin 17 family of cytokines in psoriasis: pathogenesis and potential targets for innovative therapies. Front. Immunol. 14:1186455. doi: 10.3389/fimmu.2023.1186455
Received: 14 March 2023; Accepted: 09 May 2023;
Published: 22 May 2023.
Edited by:
Philippe Saas, Etablissement Français du Sang AuRA, FranceReviewed by:
Melinda Gooderham, SKiN Centre for Dermatology, CanadaCopyright © 2023 Brembilla and Boehncke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicolo Costantino Brembilla, bmljb2xvLmJyZW1iaWxsYUB1bmlnZS5jaA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.