 Chiara Capelli1†Carolina Cuofano1†Chiara Pavoni1Simona Frigerio2
Chiara Capelli1†Carolina Cuofano1†Chiara Pavoni1Simona Frigerio2 Daniela Lisini2Sara Nava2Michele Quaroni3Valentina Colombo3
Daniela Lisini2Sara Nava2Michele Quaroni3Valentina Colombo3 Francesco Galli4
Francesco Galli4 Svetlana Bezukladova5,6
Svetlana Bezukladova5,6 Paola Panina-Bordignon5,6
Paola Panina-Bordignon5,6 Giuseppe Gaipa3Patrizia Comoli7
Giuseppe Gaipa3Patrizia Comoli7 Giulio Cossu4,5
Giulio Cossu4,5 Gianvito Martino6,7
Gianvito Martino6,7 Andrea Biondi8,9
Andrea Biondi8,9 Martino Introna1*
Martino Introna1* Josée Golay1*
Josée Golay1*- 1Center of Cellular Therapy “G. Lanzani”, ASST Papa Giovanni XXIII, Bergamo, Italy
- 2Cell Therapy Production Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy
- 3Laboratory of Cell and Gene Therapy Stefano Verri, ASST Monza Ospedale San Gerardo, Monza, Italy
- 4Division of Cell Matrix Biology & Regenerative Medicine, Faculty of Biology, Medicine and Health (FBMH), University of Manchester, Manchester, United Kingdom
- 5Università Vita-Salute San Raffaele, Milan, Italy
- 6IRCCS San Raffaele Hospital, Neuroimmunology Unit, Division of Neuroscience, Milan, Italy
- 7Pediatric Hematology/Oncology, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy
- 8Department of Pediatrics, Fondazione IRCCS San Gerardo dei Tintori, Monza, Italy
- 9Department of Medicine and Surgery, University of Milano-Bicocca, Monza, Italy
Advanced Therapy Medicinal Products (ATMPs) based on somatic cells expanded in vitro, with or without genetic modification, is a rapidly growing area of drug development, even more so following the marketing approval of several such products. ATMPs are produced according to Good Manufacturing Practice (GMP) in authorized laboratories. Potency assays are a fundamental aspect of the quality control of the end cell products and ideally could become useful biomarkers of efficacy in vivo. Here we summarize the state of the art with regard to potency assays used for the assessment of the quality of the major ATMPs used clinic settings. We also review the data available on biomarkers that may substitute more complex functional potency tests and predict the efficacy in vivo of these cell-based drugs.
1 Introduction
The use of extensively manipulated, eventually genetically modified cells, is now well established in the clinic. Among these advanced therapy medicinal products (ATMPs), perhaps the most widely used due to their clear clinical efficacy, are epithelial cells (keratinocytes) for skin or cornea repair, but others have seen important breakthroughs in the last 10 years, such as chimeric antigen receptor (CAR)-modified lymphocytes. Other cell types that have shown activity in vivo and reached marketing approval are mesenchymal stromal cells (MSCs), fibroblasts, chondrocytes, dendritic cells (DCs), genetically modified CD34+ hematopoietic stem cells and virus-specific cytotoxic lymphocytes (CTL) (1). Finally, tumor- or virus- specific T cells or NK cells, neuronal stem cells, mesoangioblasts (MABs) as well as cells derived from induced pluripotent stem cells (iPSCs) have reached the clinic and are in active development. Table 1 lists the major cell-based ATMPs.
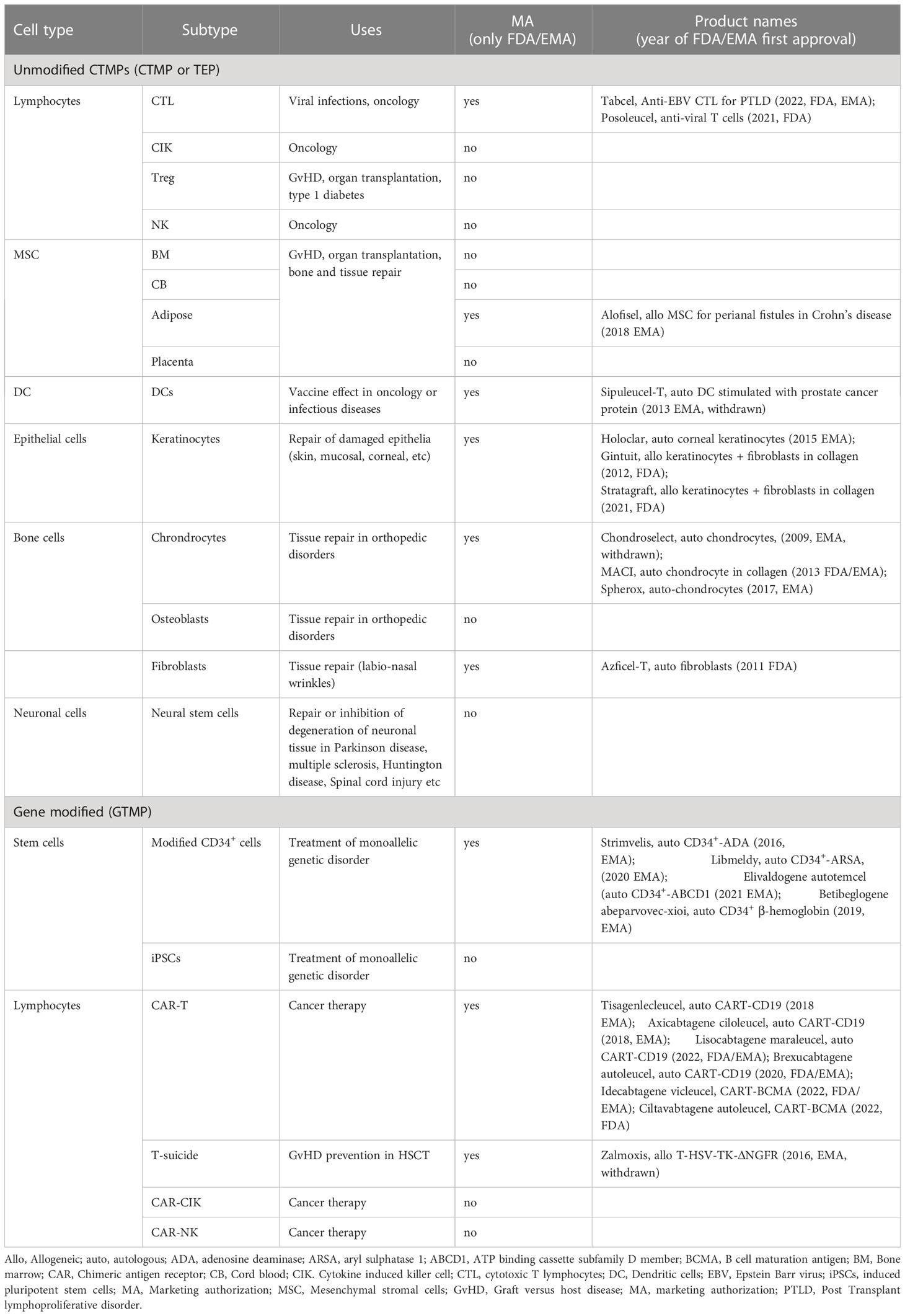
Table 1 Major cell based ATMPs that have reached the clinic or marketing authorization.
ATMPs are drugs and, as such, need to be produced in specialized laboratories (i.e. Cell Factories) according to Good Manufacturing Practices (GMPs), as defined in Europe by Regulation (EC) n. 1394/2007 and subsequent EU Directives and GMP guidelines, and in the US by FDA regulations for cellular and gene therapy products (CGTs) (2, 3). These legislations aim to guarantee the safety and efficacy of the cell products for the patients. Many ATMPs used in experimental clinical trials are produced by academic Cell Factories such as ours, which have to face the challenge of complying with stringent GMP standards for novel and complex drugs, also considering the limited human and economic resources of these laboratories (2).
One important problem with ATMPs, whether produced for established use or in the context of experimental clinical trials, is the fact that these cells derive from different individuals or pools of individuals and therefore may show intrinsic phenotypic and functional variability. Potency assays are tests that are recommended to be carried out in GMP conditions on final batches of ATMPs, before their formal release for clinical use, to guarantee the effectiveness, functional quality and consistency of the cell products that will be administered to the patients (4). These potency assays may be particularly important in the context of experimental clinical trials, which aim to establish the efficacy of a specific treatment for a defined condition.
This review aims to summarize the state of the art with respect to potency assays, performed in GMP on ATMPs produced for established or experimental clinical use, and pinpoint the specific problems still present for different types of products. Whether alternative markers can be employed in lieu of more complex potency assays will be discussed, as well as possible improvements that could be applied to this field. Whether potency assays predict in vivo efficacy will also be reviewed. Some results obtained from our cell factory network, composed of 5 approved cell factories located in and financed by the Lombardy Region of Italy and collaborating towards the development of novel cell-based drugs for treatment of severe clinical conditions, will be summarized, to illustrate some specific observations.
2 Unmodified or CAR-modified cytotoxic T or NK cells or antigen-specific T-cell lines
A common type of ATMP is based on cytotoxic T lymphocytes (CTL) that are selected or induced to have specific MHC-restricted or -unrestricted cytotoxicity against specific targets. Targets can be virus-infected or neoplastic cells. MHC-restricted cytotoxicity is mediated by T cells selected for or induced in vitro to have anti-tumor or anti-viral activity. In some cases specificity for the target is also increased by genetic modification of T cells with defined T cells receptors (TCRs) (5). MHC-unrestricted cytotoxicity can be mediated by cytokine induced killer cells (CIKs), γδ T cells, NK, NKT or all the above cytotoxic cells, genetically modified with chimeric antigen receptors (CARs) (Table 1). Several of these products have been approved by FDA/EMA as marketed drugs (Table 1). T cells are expanded in vitro using IL2 and/or IL7/IL15 after different stimulation protocols. NK cells can also be expanded in vitro using IL2, IL15, IL12, IL18, IL21 or combinations thereof, and have been tested in clinical trials as anti-cancer agents, with limited success, in part due to limited in vivo persistence (6). CAR-modified NK cells are therefore an option to increase efficacy, with the advantage over T cells-based products that NK cells do not express TCRs and do not induce graft versus host disease (GvHD). Several CAR-NK products are being tested in clinical trials with some encouraging results, but none has yet received marketing approval (Table 1).
A common mechanism of action of T/NK cells is cytotoxicity, which is therefore commonly used as a potency assay for both cell types. Standard cytotoxicity assays include the release of measurable, cell-associated molecules, either endogenous proteins (e.g. LDH) or dyes loaded into the target cells before the test (e.g. 51chromium, calcein, etc) and released by the dying cells, or measuring target cell death by flow cytometry with dead/live cell dyes. In some cases surrogate markers of cytotoxic activity, expressed by the effector cells, are employed, such as the induction of degranulation markers (CD107a or granzyme B) or of inflammatory and pro-apoptotic cytokines, most commonly IFNγ, TNFα or IL2, following contact of the effector with the target cells (7). The potency tests most commonly used for T/NK cells are summarized in Table 2.
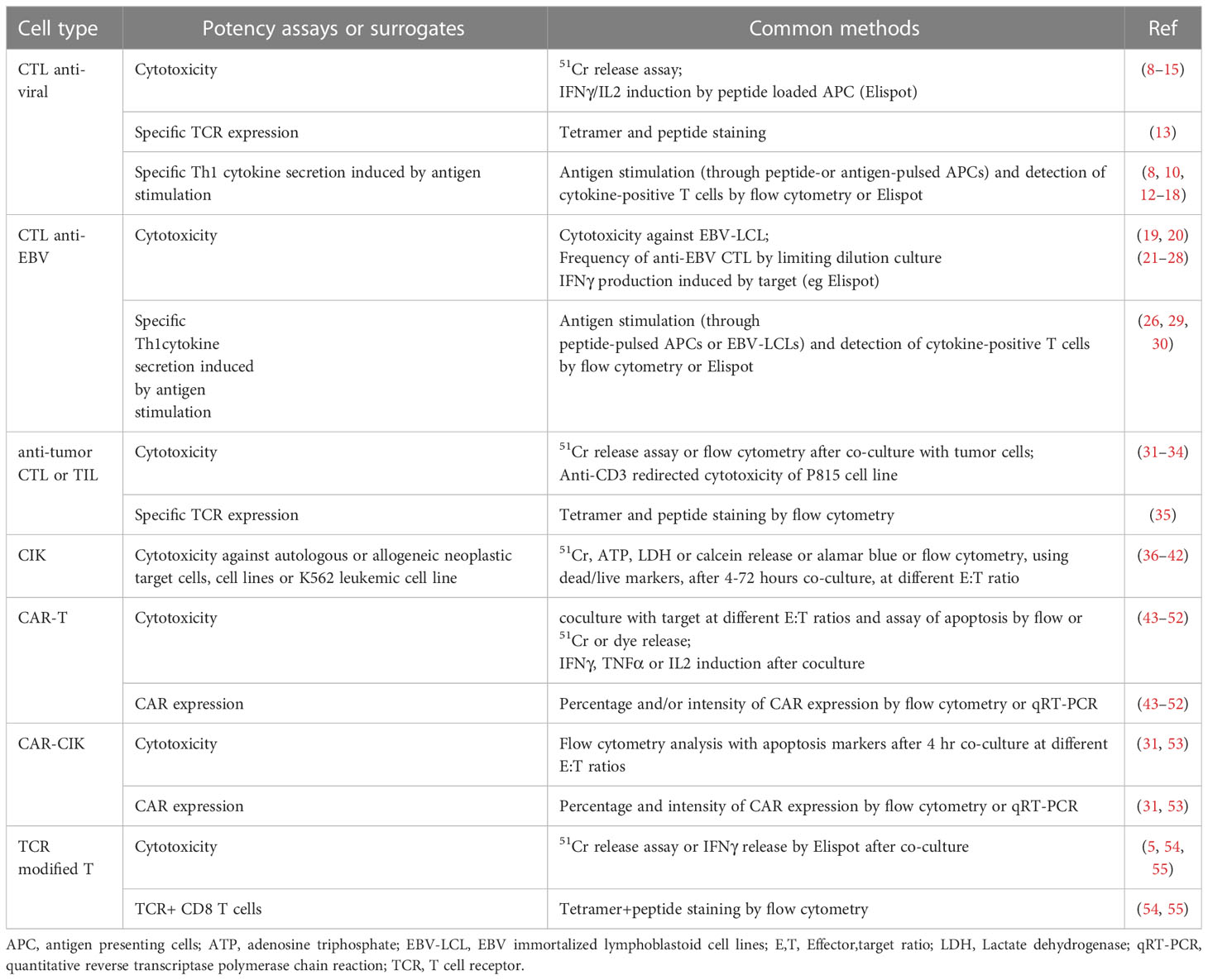
Table 2 Potency assays commonly used for cytotoxic T lymphocytes.
Cytotoxicity assay protocols are relatively complex and quite variable between different laboratories. For example, co-culture more commonly is carried out for 4 hours in the case of dye release assays or 4-24 hours for assays based on flow cytometry. Effector target ratios used are also variable (40:1 to 1:1). Targets may be peptide-primed APCs, such as autologous PHA stimulated peripheral blood mononuclear cells (PBMCs) or DCs, or autologous tumor cells, depending on the specific T cell product, or allogeneic cell lines as surrogate targets (Table 2) (5, 8–14, 19–27, 29, 31–33, 36–55). Another, non-specific cytotoxicity assay indicating the presence of functional cytotoxic T cells within the product, irrespective of antigen specificity, is the anti-CD3 redirected lysis of the NK-resistant, FcγR+ P815 cell line (34, 56).
Surrogate markers of cytotoxicity include, for expanded antigen-specific T cells, tetramer staining and flow cytometry which measures the frequency of cells expressing TCR against a specific peptide presented on an appropriate MHC (13, 31, 35). For polyclonal T cells it does not therefore identify all the effector cells that may mediate cytotoxicity against a specific target. For gene modified (TCR or CAR) cells, the TCR and CAR can be quantified by flow cytometry or quantitative PCR (pRT-PCR) and are therefore more accurate surrogate markers of potency, as they should detect all target-specific T cells within the product (Table 2) (31, 43–53). Regarding virus specific ATMPs, another surrogate marker of potency is the detection by flow cytometry or by Elispot of Th1 cytokines, such as IFNγ, secreted by the T cells upon stimulation with the specific antigen. The Th1 cytokine-secreting function is considered a marker of cell activation and specific activity, and, although it does not directly reflect the presence of cytotoxic T cells, it is likely to be associated to immune protective activities, such as lytic function (8, 12, 15–18, 30) (Table 2).
Whether surrogate markers correlate with functional potency in vitro is an important issue in the context of clinical trials and with the view of defining predictive markers of response in vivo. Regarding gene-modified T cells, whether the quantitative assay of CAR expression reflects the functional potency in vitro of CAR-CIK cells has been investigated by our groups. The percentages CAR-CD19 positive cells was demonstrated to correlate significantly with cytotoxicity in vitro, performed against a standard CD19+ B-acute lymphoblastic leukemia (ALL) cell line, in the context of our two clinical studies using 60 batches of CIK cells modified with a CAR anti-CD19 (NCT03389035 and NCT05252403 and product validation data for these trials) (Figure 1). These data suggest that quantitative analysis of CAR expression can indeed be used as a rapid and surrogate potency assay for GMP release, instead of more laborious cytotoxicity assays.
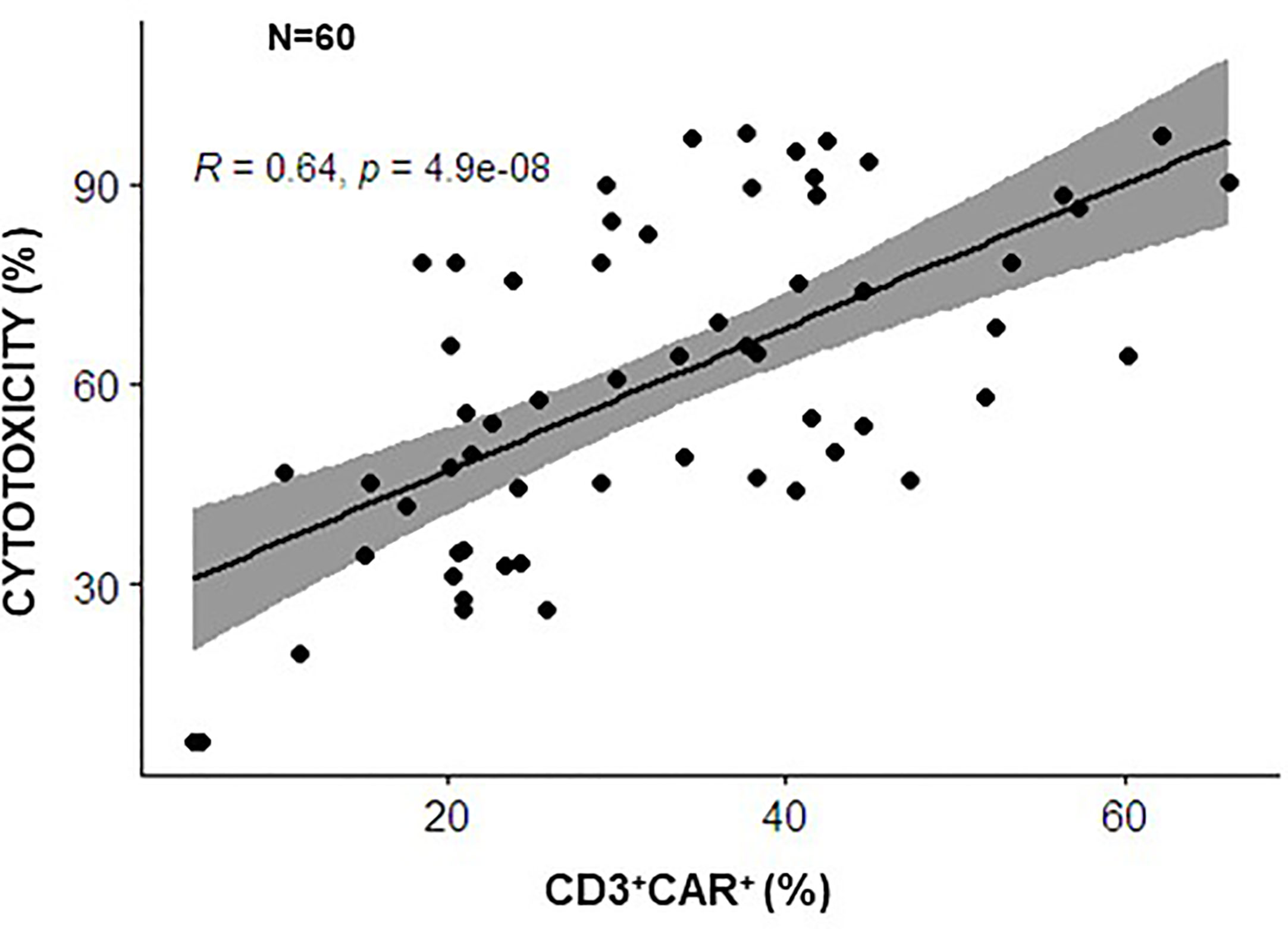
Figure 1 Correlation between CAR expression and the cytotoxic potential of CARCIK-CD19 cells. CARCIK-CD19 cells expanded for clinical use of validation purposes, using the same GMP standard operating procedure, in 2 different Cell Factories (Center of Cellular Therapy “G. Lanzani, Bergamo and Laboratory of Cell and Gene Therapy Stefano Verri, Monza, Italy), in the context of common clinical trials (NCT03389035 and NCT05252403), were tested for percentage of CAR-CD19 expression and cytotoxic activity against the REH leukemic cell line, using the same assays conditions (N=60). Percentage CAR expression correlated significantly with percentage cytotoxicity (potency) (The Pearson correlation coefficient R=0,64, p<0.0001).
Given the fact that functional potency assays are quite laborious and may show a relatively high level of variability, as detailed above, they are sometimes employed only in the development and validation phase of ATMPs and not used as a routine GMP release test during clinical experimentation. In this latter context, they may be used “for information only” (FIO), for example to evaluate retrospectively whether in vitro cytotoxicity correlates with the clinical response or to perform statistical analyses of production efficiency. Indeed, presently, there are no standardized and universally recognized potency methods or parameters for GMP-compliant batch release of T- or NK- cell products. Specifications may also vary between laboratories and depend upon the results of analyses made during development. In contrast, surrogate tests of the cells overall fitness are universally applied. For example, T cell overall fitness is measured by testing the cell product viability, as well as the immunophenotype that defines the ATMP product identity, detecting as a minimum the CD3, CD4 and CD8; defined TCR or CAR expression are also measured in case of genetically modified T cells.
More extensive characterization of the T cell based ATMPs, including the quantification of naive-memory subsets, the expression of co-stimulatory molecules or checkpoint inhibitors and other makers that may indirectly correlate with potency are usually performed only as ancillary tests. In particular these assays are used to gain knowledge of the product characteristics and to allow retrospective correlative studies that may explain the clinical response of patients (45, 47, 48, 57, 58). There are relatively few data reporting whether potency assays or surrogate markers of functionality do correlate or not with the efficacy of T- or NK-cell based ATMPs in vivo in the clinic. The data available on CAR-T cells, mostly directed against CD19 and used for the treatment of acute lymphoblastic leukemia (ALL) and B-non Hodgkin’s lymphoma (B-NHL), are however informative in this regard. The published results show that in vitro cytotoxicity, CAR expression, CAR-T dosage or CAR-T CD4/CD8 ratio in the product do not usually correlate significantly with the clinical response (59, 60). Rather CAR-T cell expansion in vivo following cell infusion, and the persistence of a less exhausted phenotype of the cells recovered at different time points after infusion, predicted response in vivo (60, 61). Also patients and tumor characteristics such as tumor burden, state and frequency of circulating T cells upon infusion, tumour microenvironment parameters, including expression of checkpoint inhibitors or presence of immune suppressive cells (62–64), have been observed to be predictive factors of the clinical response (19, 57, 58, 65, 66).
However, in the last 5 years, quite extensive analyses of the characteristics of the infused CAR-T cell products have been conducted in the context of clinical trials, probing more complex phenotypes, in some cases using single cell RNA sequencing (scRNAseq) or epigenetic analyses, in order to identify characteristics of the cell products that may predict in vivo cell expansion, persistence and clinical response. These investigations have demonstrated that an early memory and less exhausted phenotype of the infused cell product (60, 67–69), a high frequency of CD8+CD27+PD1- cells showing an upregulated STAT3/IL6 pathway (60), polyfunctionality (70, 71), shorter effector-target contact and higher out-of-contact migratory capacity (71), IFNγ secretion (70), a Th2 profile following activation in vitro (67), as well as a specific epigenetic pattern of DNA methylation (72), are all biomarkers that may predict in part the persistence of the infused cells and/or the clinical response to CAR-T therapy. Homing receptors such as CCR7 and CXCR3 have also been suggested to correlate with clinical response but results are still controversial (60, 67, 70). Indeed despite the extensive characterization of chemokine receptors, such as CXCR1, CXCR4, XCL, CCL3/4 and 5 for the recruitment of cells to different tissues in vivo (73), there is still a lack of clear understanding of their effective role in the context of passively administered T/NK cells (unmodified or gene modified) for their effective intratumoral recruitment. Altogether, the present data on product characteristics confirm that batch variability does exist and that parameters other than CAR expression and in vitro cytotoxicity need to be applied to better characterize the efficacy of this class of ATMPs. Furthermore the more precise characterization of an effective CAR-T product will allow to define improved expansion conditions to more reproducibly generate less exhausted and more effective cell products (69, 74). Indeed the quality of the starting cell material used to expand CAR-T cells, as well as specific CAR and expansion conditions, may influence the quality and efficacy of the final CAR-T cells (60, 74, 75). Clearly, further work and larger studies will be required to confirm all these data and define appropriate GMP compliant markers of CAR-T potency that may be applied to clinical grade CAR-T cell products, to improve their reproducibility and quality. Similar work is warranted for MHC-restricted T cells and NK/NKT cells used in clinical trials. New technologies and in vivo models may also be useful to better understand the dynamics of T cell function and define predictive parameters of efficacy in vivo (76–78).
NK and T cells have also been generated in vitro from iPSCs for immunotherapeutic purposes (79, 80). The expansion potential of iPSCs eliminates the need for multiple donors, increasing cell product reproducibility, and epigenetic rejuvenation during iPSC should produce biologically younger cells (81). These may therefore also become “off-the-shelf” products. Nonetheless, despite impressive progress to obtain meaningful number of cells using simplified steps applicable to GMP conditions (82), very few clinical reports have yet been published so far with these ATMPs (59, 60, 83, 84). Furthermore, the frequent chromosomal alterations as well as the teratomas observed in mouse experiments may represent a severe limitation to current clinical translation (79, 80), suggesting the necessity of adding suicide genes for safety issues.
iPSCs can be established from T-cell clones and re-differentiated into functional T cells. CD8+ T lymphocytes derived from iPSC with specificity for tumor antigens, such as MART-1, or viruses, such as HIV-1, have been generated using this technique (85). These rejuvenated CD8+ cells have shown IFN-ɣ production and cytotoxicity against the relevant targets, improved proliferation capacity and a less differentiated profile associated with prolonged persistency with respect with unmodified T cells (85–89). Potency assays for iPSCs generated T or NK cells are the same as those for standard cells.
Suicide systems are available for both unmodified or CAR-T cells and include the introduction of proteins (CD20, HSV-TK) that can be targeted by drugs (rituximab, gancyclovir) or inducible pro-apoptotic protein such as iCasp9 (90). Clearly potency assays also need to verify gene copy number, level of cell transduction, the effective expression and/or function of the suicide gene, depending on the system used. Few such suicide systems for ATMPs have yet reached the clinic (91, 92).
3 Dendritic cells
Different subsets of DCs are present in man, such as conventional DCs, plasmacytoid DCs and Langheran cells, each with specific phenotypes, localization and functions. Conventional DCs are very efficient antigen presenting cells (APCs) that can be relatively easily generated in vitro by culturing monocytes (usually derived from peripheral blood), in presence of appropriate cytokines, most often GM-CSF and IL4 for the differentiation phase to generate immature DCs (iDCs), followed by a 24-48 hour culture in presence of TNFα, IL1β, IL6 and Prostaglandin E2 (PGE2), to generate mature DCs (mDCs) (93, 94). These in vitro generated cells are CD1a/CD80/CD83/CD86/CD40/CD209+, HLA-DR+, express MHC-class I cells and are called monocyte-derived DCs. Mature DCs process and present peptides from intracellularly processed proteins on MHC class I/II complexes, in cooperation with the costimulatory molecules (CD80/CD86/CD40) and pro-inflammatory cytokines (IL12), and thus prime T cells. These cells, once loaded with antigens, peptides or cells (for example viral antigens or tumor cells), can induce Th1 responses as well as activation and proliferation of naive or memory CD8+ cells to induce CTLs directed against the presented antigens. Antigen processing, co-stimulatory signals, in particular CD40, CD80/CD86 and cytokines production (mostly IL12) are essential for effective T cell stimulation and polarization toward an effective immune function.
Unmodified DCs have been extensively used in clinical trials as vaccines with the scope of inducing or boosting T cells response against tumors or infectious agents (95, 96). However, regulatory or tolerogenic DCs can be induced by a tolerogenic tumor environment which may negatively influence the efficacy of autologous mDCs, including DCs generated ex vivo in GMP (95, 97). Due to the still relatively limited success of unmodified DC-based vaccines (98), more recently, gene-modified DCs are being generated with the hope of enhancing their efficacy in vivo. Genetic modifications include the introduction by viral infection or transfection of the desired target antigens (viral or tumor antigens), with or without a co-stimulatory molecule, or of the cDNAs encoding the GM-CSF/IL4 cytokines that will permit the efficient generation of “self-generated” iDCs from immature precursors (SmartDCs) (99–103).
Potency assays for DCs generally measure allogeneic mixed lymphocyte reaction (MLR) in vitro and assay conditions and detection methods vary quite significantly between laboratories. Furthermore, such assays, which are based on proliferation measured over several days and employ different donors, show intrinsic variability. Specifications also vary between laboratories and are based on the validation data (31, 93, 94, 104, 105) (Table 3). Thus, no standardized assays are defined for DCs. Potency assays for genetically modified DCs are similar to those used in the context of unmodified DCs, since the desired function of these ATMPs is the same, and the same caveats apply, i.e. variability and lack of accuracy in the assays; they may additionally include quantitative evaluation of the introduced genes by quantitative PCR (99, 100, 102). Potency assays may in some cases be used as a GMP release assay but are more likely employed in the validation phase and/or used for information only in the context of clinical trials. They may be performed before cryopreservation or on cryopreserved cells (31, 93, 94, 104, 105). Immunophenotyping to define the identity of the clinical grade DC products is therefore often used as surrogate for potency assays (positivity for CD11c, CD80/83/86, HLA-DR, CD209 and negativity for CD14, CD3 CD19), but the set of markers used and the specifications somewhat vary between laboratories (31, 93, 94, 104, 105).

Table 3 Most common potency assays for DCs.
Whether the potency of a DC vaccine may be predictive of in vivo efficacy is not clear. Several groups have shown increased T cell responses and cytokines, or decreased Tregs post DC-based vaccination, which may or may not correlate with the clinical response (98, 106–109). There is also some indication that the presence in vivo of lymphocytes that can be activated by pulsed DCs to become cytotoxic against the autologous tumor correlates with the clinical outcome of ALL patients, but these patients were not treated with a DC vaccine (110). Other groups have shown a correlation between in vitro potency or surrogate markers thereof and clinical response (101, 111). These data need to be confirmed on larger cohorts of patients.
To summarize, potency assays are performed for DCs intended in a vaccine mode to induce T cell mediated immunity against tumor cells or virus infections. These are performed in the validation phase and in some cases also for release of the GMP grade ATMPs. Assays are quite variable between laboratories and are not standardized. Furthermore, few data are yet available to demonstrate that potency in vitro predicts efficacy in vivo. More extensive characterization of the cell products, similar to what has been done for T cell-based products, is warranted to define better biomarkers predictive of efficacy.
4 Mesenchymal stromal cells
MSCs have been extensively expanded in vitro for clinical use, due to the ready availability of the tissues from which they can be derived: mostly bone marrow (BM), adipose tissue (AT) and newer sources such umbilical cord (UC), cord blood (CB), placenta (PL), amniotic fluid or amniotic membrane (112–115). In addition, MSCs show multiple functions in vitro and in vivo. In particular, they display immunosuppressive properties acting on different cell types, as well as a capacity to differentiate to adipose or bone tissue, making these cells potential drugs in many different clinical contexts. MSCs, expanded in vitro according to a variety of different protocols and starting from different tissues have therefore been extensively used in the clinic to treat a variety of conditions. Three MSC products have been approved worldwide: Alofisel, an allogeneic adipose derived product from Takeda and TiGenix approved by EMA to treat enterocutaneous fistulae (Table 1). In addition, two MSC products have been approved in Korea: Cartistem, based on human CB derived MSCs for the treatment of knee cartilage defects, as a result of degenerative osteoarthritis or repeated trauma and Cupistem, which is an autologous adipose tissue derived MSC product for Crohn’s Fistula. None have yet been approved by FDA (112, 116–120). Unfortunately, most of the clinical applications of MSCs have not yet produced very significant and/or reproducible clinical results (113, 121).
Potency assays performed on MSCs are different according to the expected in vivo function of these ATMPs, i.e. whether their pharmacological use is based on their immunosuppressive (122–124) or tissue repair properties (31, 124–128). However, MSCs’ mechanism of action in vivo may involve the effect of many effector pathways with synergistic and overlapping functionalities. For example, dominant MSC functionalities in immune modulation may overlap with those relevant in tissue injury response, with some unique components for each. Considering this, it has been difficult to define and identify a cellular universal reference standard that would contemporaneously display all of the potential functions of MSCs identified to date (129–131).
Table 4 summarizes the potency tests that are most commonly applied to MSCs in different clinical contexts. Some are routinely used for release assays, others have been described and employed only in the development and characterization phase of the cell product and are not performed on clinical batches for GMP release, since the development of an appropriate potency assay is challenging (132). Indeed induction of IDO, PGE2, adhesion molecules or the transcription factor TWIST as well as others by inflammatory mediators have been demonstrated for MSCs, but their detection relies on complex assays so far described only in the preclinical development phase (133). Furthermore, the methodologies and findings may vary markedly between different laboratories (134). Other tests are qualitative more than quantitative (e.g. the differentiation potential to different cell types; Table 4) (31, 124, 126–128). One approach to reduce the observed batch to batch variability when testing for functional assays of MSCs is to generate batches from pooled donors. This approach has been suggested to result in more reproducible products (119). These data will need to be confirmed in larger studies.
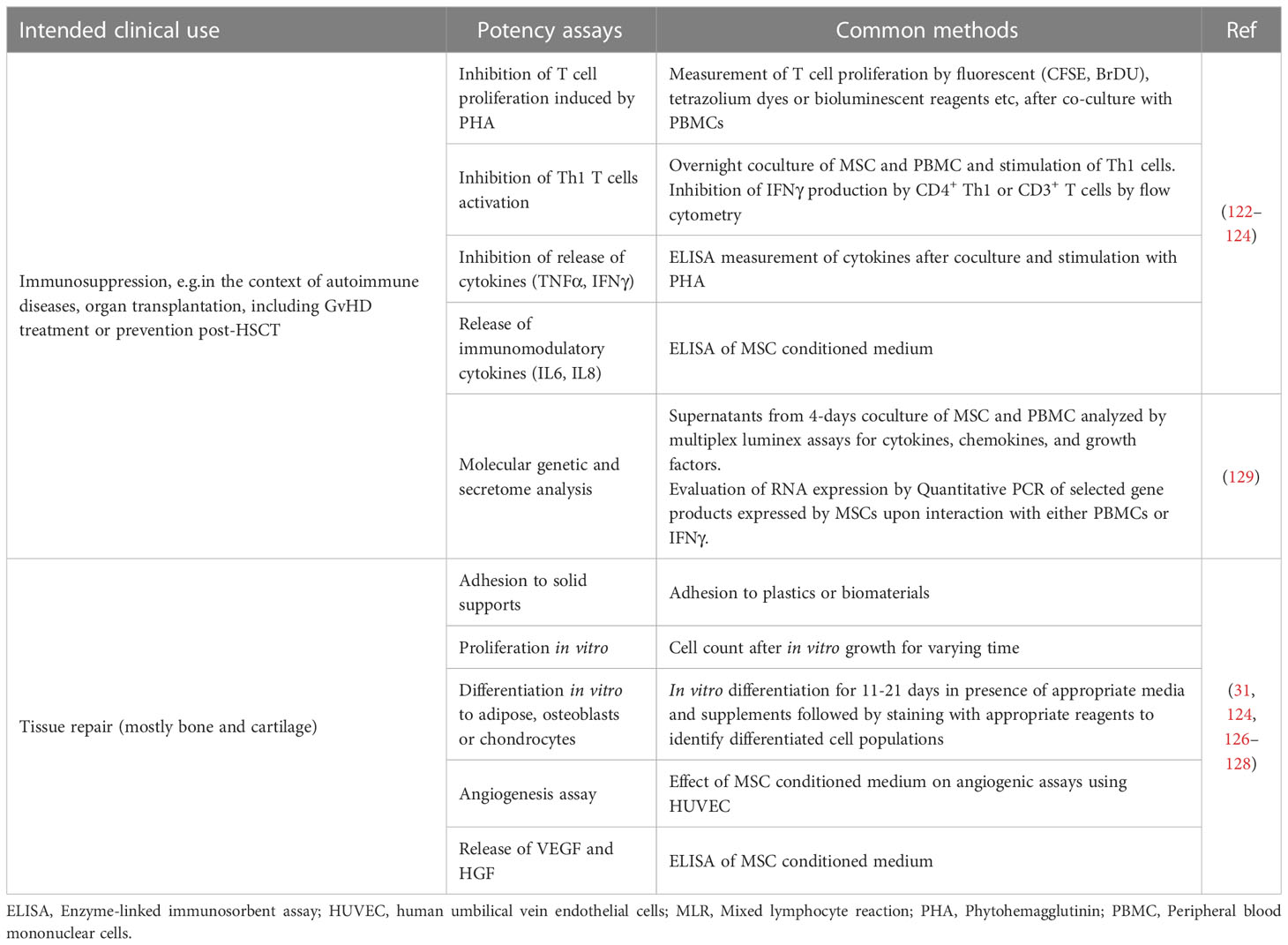
Table 4 Most commonly used potency assays to functionally qualify MSCs.
With regard to the tissue regenerating function of MSCs, potency assays are usually performed only to demonstrate the capacity of the expanded MSCs to differentiate into the expected cell types, depending on the specific clinical application (e.g. bone or cartilage repair) (31, 124, 126–128, 135).
Whether the GMP release assays correlate with the clinical response to the MSC infusion is still unclear. Indeed little correlation has been observed between inhibition of proliferation in vitro and immunosuppressive function in vivo, or between differentiation potential in the test tube and tissue repair (summarized in (4, 133)). Similarly, reliable in vivo markers of disease and response to MSCs are still lacking (133, 136). With regard to the immune suppressing function of MSCs, this is in part due to a still incomplete understanding of the MSCs mechanisms of action in vivo in different disease contexts. Chinnadurai and colleagues have tested an in vitro assay matrix approach combining molecular, genetic and secretome analysis, elements of which could be deployed to define MSC immune modulatory potency (129). Indeed MSCs may need to respond to in vivo clues to become activated and unleash their full potential (113, 125). Moreover, recent data suggest that apoptosis of infused MSCs and their phagocytosis by macrophages, called efferocytosis, may trigger immunosuppressive cascades of events and may be more important than the direct immunosuppression by the MSCs themselves (133, 137, 138). Indeed the apoptosis of MSCs induced by the PBMCs of patients has been suggested to be a better predictor of in vivo MSC activity (138, 139). Finally, evidence indicates that another mechanism of MSC function is via the release of extracellular vesicles by these cells (EVs, see below) (139).
MSC products are heterogeneous cell populations which are known to gradually lose their multilineage and proliferation potential during culture in adherent monolayers (140–142). Indeed the ISCT has recommended the term mesenchymal stromal cells rather than mesenchymal stem cells, due to lack of evidence of real stemness of the cells, i.e. the capacity to self-replicate and differentiate into different cell progeny (143). In addition, no marker of stemness or of differentiation is available to define the MSC precursors present in culture and their capacity to proliferate and differentiate to the desired tissue. Such lack of marker makes the further development of MSC-based therapeutics more difficult at present (136, 144, 145).
In this context, the lack of clear knowledge of relevant potency assays to define MSCs activity in vivo, as well as their intrinsic variability and complexity, has led many groups to propose as release assays the sole immunophenotypic and viability analyses, i.e. the definition of a minimum phenotype and vitality of MSCs, which are part of the ISCT recommendations for this ATMP (31, 146). The consensus ISCT criteria for MSC identification is positivity (≥ 95%) for CD105, CD73 and CD90 and negativity (≤ 2%) for CD45, CD34, CD14 or CD11b, CD70a or CD19, and HLA-DR. Some groups have used less stringent immunophenotypic criteria for GMP purposes and do not apply all recommended markers in this context (e.g. ≥80%, ≥85%, ≥90%, positivity for CD73, CD90 and CD105) (31, 126).
The identification of markers of stemness and/or lineage differentiation potential, similar to what has been identified in epithelial stem cells (see below) should continue to be pursued also for MSC products.
EVs have been produced from MSCs for therapeutic use and in GMP conditions. given the reported functional activity of these products (139, 147). EVs are purified and used in lieu of live cells and are not therefore strictly ATMPs, but ATMP-derived, and are still in search of classification (148). They are however presently extensively tested in clinical studies across different therapeutic areas (149–151). Their qualification and potency evaluation should follow the recently published guidelines, that include expression of at least 3 markers as well as physical characterization (152). Indeed MSC derived EVs express markers such as CD9, CD63, CD81, TSG101 and do not express calnexin and cytochrome C (147, 151). Physical characterization of particle size and concentration should be evaluated by two different but complementary techniques. These EVs are thought to act via paracrine intercellular communication effects, inducing tissue repair and immunomodulation by different mechanisms. Functional potency assays for EVs show similar limitations to that described for MSCs and their relevance to the in vivo efficacy is still a little understood (139).
5 Epithelial stem cells
Autologous epithelial cells expanded in vitro from different tissues and organs have been used extensively for the treatment of severe burns and were the first success of extensively manipulated cell-based drugs (136, 153–156). These autologous epithelial stem cells are marketed for tissue repair as Holoclar, whereas allogeneic epithelial cells are marketed as Gintuit and Stratagraft (Table 1).
Successful engraftment of keratinocytes in culture depends upon the presence and amplification of epithelial stem cells capable of forming holoclones (156). In vivo, stem cells are usually slowly cycling and can give rise, after isolation, to distinct epithelial lineages characteristic of the different tissues from which they were derived, even after in vitro culture (157). Stem cells for corneal epithelium reside in the limbus whereas stem cells for conjunctival epithelium reside in the bulbar and forniceal conjunctiva and generate both conjunctival keratinocytes and goblet cells, both of which are important for the integrity of the ocular surface (158). Stem cells for skin epithelium are distributed in the basal layer of interfollicular epidermis and various niches of the hair follicles. Stem cells are also found in mucosal epithelia (136, 158–160).
Epithelial stem cells can be expanded in vitro according to a variety of culture conditions and show a high proliferative capacity (up to 80-160 cell divisions in vitro). Final products are characterized for research purposes by cell number, clonogenic assays (to measure the frequency of holoclones as well as of more differentiated meroclones and paraclones) and analyses of the proliferative and differentiative capacity of these clones (136). Stem cells (holoclone forming cells) also express high levels of stem cell markers, most specifically ΔNp63α, but also other less specific markers of stemness such as vimentin, ABCB5 and keratins CK14 and CK15 (see below) (161–163). Differentiation in vitro to different epithelial types can be defined by staining for other keratins specific of different tissues (158). More recent technology includes the generation and transplantation of cellularized scaffold products such as StrataGraft or Gintuit (sheets of allogeneic human keratinocytes and fibroblasts in collagen), the investigation of pluripotent embryonic (hPSCs) or induced pluripotent stem cells (iPSCs) to generate epithelial cells in vitro and on improved culture conditions, substrates and co-cultures to expand epithelial sheets containing stem cells able to differentiate into different epithelia (155, 161, 164–167). Allogeneic material, generally containing also fibroblasts, is used for temporary skin replacement until autologous cells can be generated and administered (168). Finally other developments are the genetic modification of epithelial stem cells in vitro to correct monosomic genetic diseases affecting the skin (136).
A fundamental aspect of epithelial stem cell use for tissue regenerative purposes in the clinic is to define the content and frequency of stem cells (holoclones) within the cell graft, i.e. a measurement of potency of the product (136, 156). Although colony assays can be used to functionally characterize the cell-based drug, these are long and laborious and are not always suitable for GMP release purposes. They are therefore usually performed in the product development/validation phase or for information only during clinical trials. A number of phenotypic markers have therefore been identified to try and define epithelial stem cells as well as their differentiated progeny. The p63 transcription factor is a regulator of epithelial proliferation and has been shown to be the best marker of stemness in epithelial cell cultures. Indeed the ΔNp63α isoform is a marker of corneal, epidermal, oral and conjunctival epithelial stem cells, i.e. holoclones (136, 163, 169, 170). Furthermore this marker is the only marker so far that correlates with the clinical response to limbal stem cells (171, 172). It is therefore routinely used to measure the number of stem cells in epithelial cell products for clinical use. Other, far less clear-cut, parameters for stem cells are small cell size, positivity for ABCG2, cytokeratins CK14 and CK15 and/or integrin α6 and negativity for differentiated epithelial cells (CK1, CK10, CK3, CK12 and CK19) (163, 173–177). Some groups have also attempted to standardize these assays, adding reference standards, and define specifications values (170). Recent evidence suggests that proliferative capacity in vitro, as well as the presence of stem cells strongly expressing ΔNp63α (p63bright), may both be important factors to predict epithelium regeneration in vivo. Therefore such parameters should be investigated in clinical studies to further improve the characterization of the cell products and identify factors that determine successful engraftment (136, 163, 178). Finally, additional work is being conducted to further improve safe and more effective culture methods for generation of epithelial stem cells for clinical use and potency assays are important in this context as well (136, 178).
Also in the case of genetically modified epithelial stem cells to correct monosomic diseases, the evidence is compelling that long-term reconstitution of corrected keratinocytes depends upon the presence of holoclones (179). In the case of genetically modified cells, clonal analyses of potency are usually performed on the product in parallel with product administration (in fresh). The correct expression of the introduced gene is additionally performed by qRT-PCR or protein analyses.
We conclude that it would be useful to standardize the detection assay for ΔNp63α and routinely add such potency test marker for all epithelial products used in clinical trials, in order to allow correlative studies to be made and establish whether this marker can predict the engraftment and duration of the graft (136, 157).
6 Neuronal stem cells
In the last 30 years, the generation in vitro of large numbers of neuronal stem cells (NSCs) from fetal brain tissue has been developed in order to treat few degenerative or post-trauma neural diseases (180, 181). Ethical and regulatory issues, as well as difficulties in reproducibly expanding standardized cell products have delayed the clinical application of these products, but in recent years Phase I or II studies have been performed with non-genetically modified, in vitro expanded, GMP-grade NSCs in different clinical contexts, in particular Parkinson disease (PD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS) and spinal cord injury (SCI) (182–185). Human NSCs are generated by extensive culture expansion in presence of EGF and bFGF, which lasts from few weeks to several months (186). NSCs can differentiate into neural and glial cells (astrocytes and oligodendrocytes) in vivo, and can produce neurotrophic factors, cytokines and extracellular vesicles (EVs), promoting neuronal tissue repair, immunomodulation, or neuroprotection (187). Defining potency assays for such products is therefore particularly difficult, given the multiple and not yet fully defined mechanisms of action of these cells, which may vary in different disease contexts and in the presence of multiple factors in the host environment. Thus, similarly to MSCs, potency assays in vitro and in vivo relevant to the intended target disease should be performed (Potency Tests for Cellular and Gene Therapy Products | FDA. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/potency-tests-cellular-and-gene-therapy-products).
This is obviously a difficult task, specifically for assays performed in vivo in small animals, due to its labor-intensive nature. The in vivo models are often preferred over in vitro systems due to their highest level of biological complexity, being the most relevant to the clinical settings. Potency assays in vivo differ according to disease model, but usually include verification of infiltration of human NSCs or graft survival into different brain regions of treated immunodeficient mice or rat models, 3-6 months after NSC transplantation (186, 188). As clinical outcomes, partial reversal of neurodegeneration or neuronal repair are investigated using various functional tests at different time points following NSC administration. These tests are model-specific, thus difficult to standardize across different laboratories.
On the other hand, tissue- or cell-based potency assays help to reduce the use of animal models according to the 3R principle (replacement, reduction and refinement) and can directly evaluate the functionality of the cellular product. In vitro potency assays include growth rate and capacity to form embryoid bodies or colonies at low density (186); however culture conditions and growth rate evaluations can substantially vary between laboratories. Additional in vitro biological potency assays may include the evaluation of surface markers expression, release of soluble mediators and changes in gene transcription. Current evidence suggests that performing potency assays with clinical grade NSC cell lines, in parallel with reference standard cell lines, should be mandatory before performing clinical trials, as nicely described by Anderson and colleagues (188). Furthermore, transparency and full functional description of the biological functions tested for the cell products used in clinical trials should be included in the report of clinical efficacy or lack thereof, in order to allow a correct interpretation of positive and negative results and further advance the field (188). Nonetheless, progress has been made in the last 10 years to better define the origin of the NSC starting material used for expansion, and more standardized culture protocols have been proposed by some groups (186). Importantly, sufficient NSC numbers can be obtained from a single donor, which are then expanded to create the master cell bank and cryopreserved, allowing to perform larger clinical trials and to treat hundreds of patients with rigorously quality-controlled cell products. The number of cells generated in large batches for each expansion (each cell line) is sufficient to perform more extensive in vitro and in vivo potency assays before clinical use (183, 186). Nonetheless, cellular products even obtained from a single donor typically show a large degree of lot-to-lot variability, due in part to inherent variability of the starting material, donor genetic factors, epigenetic changes and genetic polymorphisms. Therefore, human iPSCs differentiated in vitro to neural precursor cells (NPCs) can be employed as an alternative to NSCs of fetal brain origin (180, 189).
In the past years, the number of iPSC-based clinical trials have increased dramatically (190), including the first case report of iPSC-derived dopaminergic precursors transplantation in Parkinson’s disease patients (191). The majority of clinical trials using iPSC-derived products are being conducted in an allogeneic setting, since the generation of iPSC under GMP conditions (required for clinical use) for a single patient is time-consuming, laborious and not cost-effective, despite the fact that initially iPSCs were intended to be used in an autologous setting. Therefore, iPSC banks with common HLA haplotypes have been established in several countries, to advance the development of cell therapies using iPSC-based products (192, 193).
Before clinical use, iPSCs and their products are rigorously controlled for the absence of adventitious viruses and mycoplasma. Chromosome analysis (karyotyping), DNA fingerprinting, and whole genome sequencing performed to control for absence of cancer-related and neurodegenerative-associated mutations. Detailed genetic integrity analyses of iPSCs are described by Popp and colleagues (194). Furthermore, the final iPSC-derived products are quality controlled for absence of undifferentiated iPSCs, accurately characterized in vitro by immunocytochemistry (NESTIN, SOX2 neural progenitor markers expression) and RT-PCR (absence of OCT4, SSEA4 expression) and functionally assessed with electrophysiology analyses. Human NPCs can be generated with a two-step approach from iPSCs, however the presence of even few undifferentiated iPSCs in the final cell product could lead to tumor formation upon transplantation. To address this issue, genetically modified NSCs are being proposed for clinical use, including more stable NSC cell lines (136, 189). Genetically engineered NSC show an enhanced capacity to produce the specific therapeutic molecules (e.g. LINGO-1-Fc, IL10, NT-3 or TGFβ1) to counteract neuroinflammation and promote tissue repair (195). The direct differentiation of human fibroblasts towards neural progenitors for clinical use, via viral delivery of transcription factors to generate iPSCs, have intrinsic potential safety risks. For this reason, novel methods of direct differentiation via non-genetic chemical modulation or by transgene-free delivery of transcription factors using mRNA or proteins have been developed (196, 197). The induced NSCs (iNSCs) obtained have showed efficacy in several preclinical studies, including stroke, Parkinson’s, and Alzheimer’s disease, multiple sclerosis and brain cancer, however no clinical data of these cells have been reported up to date (198).
The therapeutic potential of neural stem cells is undoubtedly encouraging but further development of GMP-grade standardized protocols for NSC differentiation and characterization are urgent. Better understanding of NSC mechanisms of action will allow to develop more efficient potency assays to ensure cellular product safety and efficacy in the clinical phases.
7 Mesoangioblasts
A successful therapy for patients suffering from different muscular dystrophies is still missing. In the case of cell therapy, initial studies attempted to inject satellite cells (muscle stem cells)-derived myogenic progenitors from a parent into a single muscle of patients affected by Duchenne Muscular Dystrophy (DMD). However, the limited survival and migration of the injected cells into the tissues led to disappointing results. More recently however, injection of autologous myogenic progenitors (isolated from non-affected muscles) in the pharyngeal muscles of patients affected by Facio-Scapulo Muscular Dystrophy (FSMD) resulted in a significant amelioration of swallowing, the main problem of these patients (199). Other muscle stem/progenitor cells were subsequently isolated and tested mainly intra-muscularly in dystrophic, immune deficient rodents (200). Mesoangioblasts (MABs) are a subset of muscle pericytes (201) that can be expanded extensively in culture and maintain the ability to differentiate into skeletal and smooth muscle cells. Of relevance, they express some of the proteins that leukocytes use to bind and cross endothelium in the presence of inflammation, which suggested the possibility of a systemic, intra-arterial delivery, not possible with other myogenic cells. Preclinical work in dystrophic mice (202) and dogs (203) led to a first in-man trial, that proved safe but of very limited efficacy (204, 205).
In particular a current promising approach investigates the clinical use of muscle pericyte-derived MABs (206). Indeed, one of us (GC) has developed culture conditions to generate sufficient number of genetically modified autologous MABs for a clinical trial in patients with DMD (Acronym: DMD06-Mab). The cells are genetically modified with a lentiviral vector expressing the U7 small nuclear RNA, engineered to skip exon 51 of the dystrophin gene. The MABs can be expanded in vitro up to approximately 20 population doublings (PD), maintain an euploid karyotype up to senescence and do not form tumors when injected subcutaneously into nude or SCID mice (201).
MABs express several stem cell surface markers but these are not specific for MABs (200). Adult MABs express PW1/Peg3 which is one of the drivers of muscle differentiation. Nonetheless, potency assays for gene-modified MABs rely presently on rather complex and long functional assays carried out on an aliquot of ATMP product to be administered to the patient (206). Potency assays include in vitro differentiation for 5-10 days in appropriate media and measurement of dystrophin production (207, 208). Dystrophin expression can be tested at the level of the RNA or protein level. The first is more quantitative but may detect abnormal transcripts that will not be translated (depending on the specific mutation), while western blot detects the protein, but is semi-quantitative. The differentiation potency can also be revealed by immunofluorescence with antibodies specific for myosin heavy chain or other sarcomeric proteins (208). Differentiation potential varies significantly among different individuals, as demonstrated by the example of myosin staining shown in Figure 2 (204).
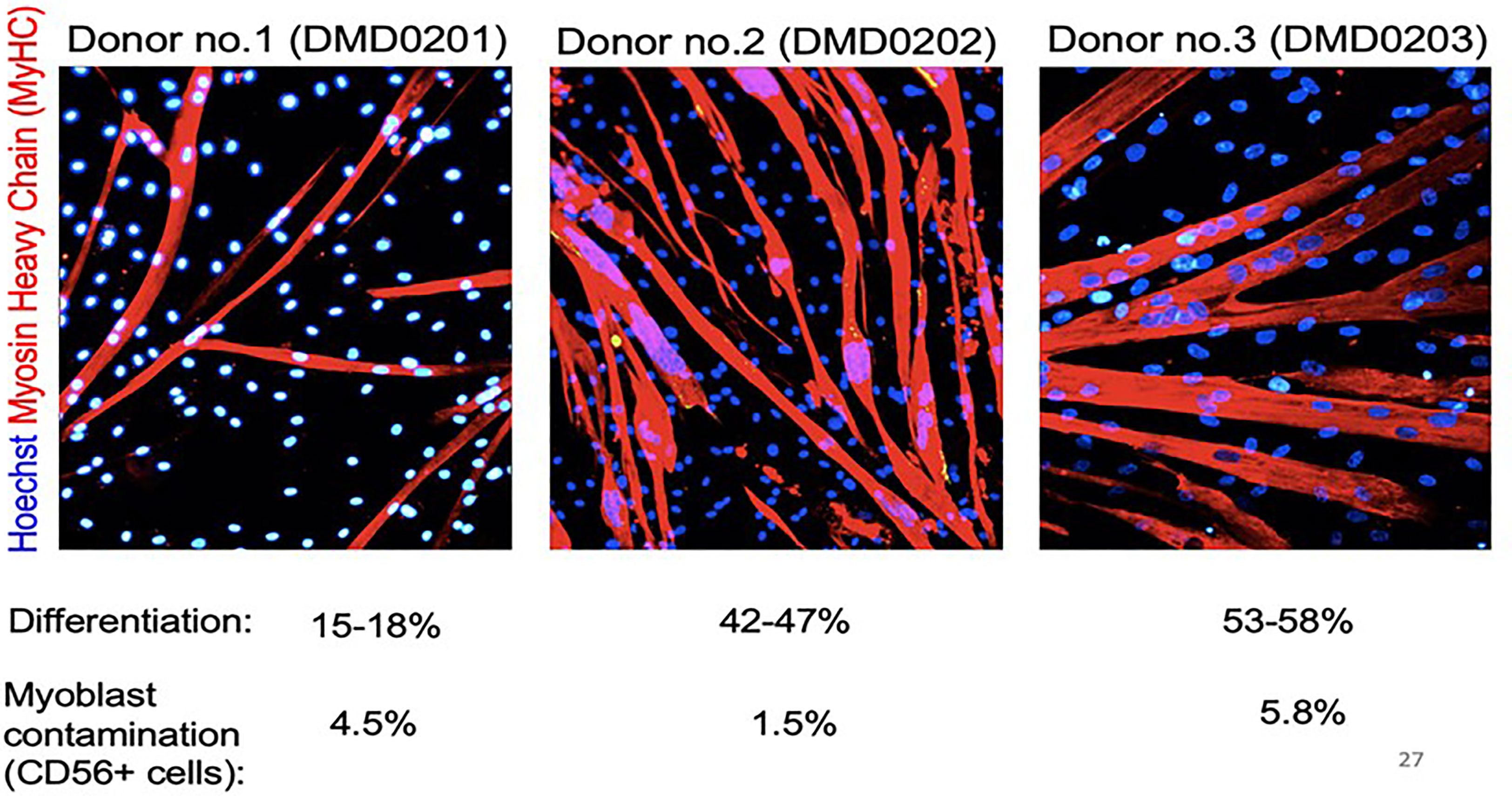
Figure 2 Donor MABs form myotubes in vitro. Spontaneous differentiation of donor MABs obtained from the medicinal product before infusion. 2x105 MABs were plated on low-growth factor matrigel coated 3.5 cm Petri dish, in proliferation medium (Megacell). After an O/N incubation at 37°C, 5% CO2, proliferation medium was replaced by differentiation medium (DMEM supplemented with 2% horse serum), and differentiation extended for 10 days. Cells were stained with anti-myosin heavy chain antibody and Dapi. Reprinted from (204).
Clearly future work will need to define more rapid, quantitative and reliable markers of myogenic stem cell able to predict the differentiation potential of MABs generated in vitro for tissue repair purposes. MABs are only in their first clinical application and future work will also hopefully allow to define whether markers of potency correlate with the capacity of these cells to regenerate muscle tissue in vivo. Finally, a better understanding of the different cell types and soluble players that together orchestrate the regeneration of a complex tissues like muscles will be required.
8 Summary and conclusions
ATMPs are promising drugs in many disease contexts. Potency assays for cell-based ATMPs are complex, often requiring several days or weeks to perform. Due to their complexity, they are not standardized and vary quite widely in different laboratories with little consensus about what specifications should applied for GMP release. Specifications are usually based on the preclinical experience of each laboratory. In some cases, potency assays are qualitative rather than quantitative, for example the capacity of stem cells to differentiate into different cell types. Another variable is whether potency assays are performed on fresh or frozen products or both. Cell numbers or costs may not allow the latter to be performed on each batch and may therefore rather be part of the process validation and of stability studies, as recently reviewed by our group (31).
It is important to establish for each cell type which potency assays should be performed for each use, either for batch release or for information only. Which assays to perform depends on the known predictive potential of the assays and whether the test is performed to demonstrate the fitness of the cell product or to predict in vivo efficacy. Indeed, the evidence that potency assays in vitro correlate with efficacy in vivo is still scarce and depends upon the specific ATMP, disease being treated as well as on the potency assays being performed. Correlations are particularly difficult to demonstrate when disease pathogenesis is complex, when multiple mechanisms of action of the ATMP may apply, and when uncertainty remains about the most important mechanism of action of the drug. Furthermore, patient specific factors (disease stage, burden or localization, tumor microenvironment, patient fitness, etc.), often play an important role in the clinical response to cell therapy, making correlation studies between potency assays in vitro and clinical response particularly arduous. Nonetheless potency assays and analyses of surrogate markers of potency are important to perform systematically during the development phase of an ATMP, using the most appropriate disease models in vitro and in vivo in order to try and define the mechanisms of action of ATMPs in different disease contexts and establish criteria for potency assays that may predict efficacy. Similarly, potency assays are important to include in clinical studies, so that possible correlations with efficacy may be performed. The feasibility of the potency assays obviously depends also on the size of the batches that are being produced and the complexity, cost and number of cells required to perform the assays. Thus, performing relevant in vivo potency assays in small animal models, for large ATMP batches used to treat hundreds of patients, is important, whereas, if possible, standardized simple assays, such as the identification of a biomarker or standardized functional assay in vitro are more suitable for small batches used to treat one or few patients.
Ideally surrogate markers of potency should be established, in order to provide fast, reproducible and more quantitative assays for potency definition. Unfortunately, very few markers of potency have yet been defined. The best example is the high expression of ΔNp63α in epithelial stem cells from different tissues, which is widely used since it correlates with tissue repair capacity. Nonetheless standardization of this marker should be aimed at for future clinical studies, as proposed by some groups (170). On the other end of the spectrum, some cells such as MSCs do not yet have any marker of potency or stemness or biological activity, which may define their capacity to mediate either immunosuppression or tissue repair. Nonetheless the recent technological progress for single cell analyses, such as scRNAseq and other “omics” analyses, should allow to identify in the near future more specific markers predicting the quality and efficacy for each ATMP and each specific use (206). Progress in this sense has already been made for example for CAR-T cell products as discussed above (76).
It is worth noting that, for many autologous or patient-dedicated ATMPs, it is important to speed up the delivery of the drug, either due to the gravity of the patients or to the fitness of the cellular product. For this reason shorter culture times and rapid administration of fresh rather than frozen products are approaches that are currently being introduced in several clinical contexts (209). In these cases, potency assays for release may not be feasible and may be more appropriately carried out only during the development and validation phase of the cell-based drugs, and subsequently for information only, rather than as a formal release assay.
Author contributions
JG, MI have conceived and designed the work. all authors have contributed to the writing and critical revision of the manuscript, each for their expertise on specific ATMPs. MI, AB and GM have contributed to funding. All authors contributed to the article and approved the submitted version.
Funding
This work was fully supported by the Fondazione Regionale per la Ricerca Biomedica (FRRB, Regione Lombardia), Project N°CP2_10/2018 (Plagencell).
Acknowledgments
We also thank the Associazione italiana contro le leucemie-linfomi e mieloma (AIL, sezione Paolo Belli Bergamo) for their continuous support to JG and to the Center of Cellular Therapy “G. Lanzani”, Bergamo, Italy.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ramezankhani R, Torabi S, Minaei N, Madani H, Rezaeiani S, Hassani SN, et al. Two decades of global progress in authorized advanced therapy medicinal products: an emerging revolution in therapeutic strategies. Front Cell Dev Biol (2020) 8:547653. doi: 10.3389/fcell.2020.547653
2. Priesner C, Hildebrandt M. Advanced therapy medicinal products and the changing role of academia. Transfus Med Hemotherapy (2022) 49(3):158–62. doi: 10.1159/000524392
3. Iglesias-López C, Agustí A, Obach M, Vallano A. Regulatory framework for advanced therapy medicinal products in Europe and united states. Front Pharmacol (2019) 10:921. doi: 10.3389/fphar.2019.00921
4. Fernández-Santos ME, Garcia-Arranz M, Andreu EJ, García-Hernández AM, López-Parra M, Villarón E, et al. Optimization of mesenchymal stromal cell (MSC) manufacturing processes for a better therapeutic outcome. Front Immunol (2022) 13:918565. doi: 10.3389/fimmu.2022.918565
5. Wagner DL, Koehl U, Chmielewski M, Scheid C, Stripecke R. Review: sustainable clinical development of CAR-T cells - switching from viral transduction towards CRISPR-cas gene editing. Front Immunol (2022) 13:865424. doi: 10.3389/fimmu.2022.865424
6. Ruppel KE, Fricke S, Köhl U, Schmiedel D. Taking lessons from CAR-T cells and going beyond: tailoring design and signaling for CAR-NK cells in cancer therapy. Front Immunol (2022) 13:822298. doi: 10.3389/fimmu.2022.822298
7. Betts MR, Koup RA. Detection of T-cell degranulation: CD107a and b. Methods Cell Biol (2004) 75:497–512. doi: 10.1016/S0091-679X(04)75020-7
8. Muftuoglu M, Olson A, Marin D, Ahmed S, Mulanovich V, Tummala S, et al. Allogeneic BK virus-specific T cells for progressive multifocal leukoencephalopathy. N Engl J Med (2018) 379(15):1443–51. doi: 10.1056/NEJMoa1801540
9. Papadopoulou A, Gerdemann U, Katari UL, Tzannou I, Liu H, Martinez C, et al. Activity of broad-spectrum T cells as treatment for AdV, EBV, CMV, BKV, and HHV6 infections after HSCT. Sci Transl Med (2014) 6(242):242ra83. doi: 10.1126/scitranslmed.3008825
10. Trivedi D, Williams RY, O’Reilly RJ, Koehne G. Generation of CMV-specific T lymphocytes using protein-spanning pools of pp65-derived overlapping pentadecapeptides for adoptive immunotherapy. Blood. (2005) 105(7):2793–801. doi: 10.1182/blood-2003-05-1433
11. Balduzzi A, Lucchini G, Hirsch HH, Basso S, Cioni M, Rovelli A, et al. Polyomavirus JC-targeted T-cell therapy for progressive multiple leukoencephalopathy in a hematopoietic cell transplantation recipient. Bone Marrow Transplant. (2011) 46(7):987–92. doi: 10.1038/bmt.2010.221
12. Berzero G, Basso S, Stoppini L, Palermo A, Pichiecchio A, Paoletti M, et al. Adoptive transfer of JC virus-specific T lymphocytes for the treatment of progressive multifocal leukoencephalopathy. Ann Neurol (2021) 89(4):769–79. doi: 10.1002/ana.26020
13. Koehne G, Hasan A, Doubrovina E, Prockop S, Tyler E, Wasilewski G, et al. Immunotherapy with donor T cells sensitized with overlapping pentadecapeptides for treatment of persistent cytomegalovirus infection or viremia. Biol Blood Marrow Transplant (2015) 21(9):1663–78. doi: 10.1016/j.bbmt.2015.05.015
14. Ma CKK, Blyth E, Clancy L, Simms R, Burgess J, Brown R, et al. Addition of varicella zoster virus-specific T cells to cytomegalovirus, Epstein-Barr virus and adenovirus tri-specific T cells as adoptive immunotherapy in patients undergoing allogeneic hematopoietic stem cell transplantation. Cytotherapy. (2015) 17(10):1406–20. doi: 10.1016/j.jcyt.2015.07.005
15. Comoli P, Schilham MW, Basso S, van Vreeswijk T, Bernardo ME, Maccario R, et al. T-Cell lines specific for peptides of adenovirus hexon protein and devoid of alloreactivity against recipient cells can be obtained from HLA-haploidentical donors. J Immunother (2008) 31(6):529–36. doi: 10.1097/CJI.0b013e31817b9c6b
16. Feuchtinger T, Opherk K, Bethge WA, Topp MS, Schuster FR, Weissinger EM, et al. Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood. (2010) 116(20):4360–7. doi: 10.1182/blood-2010-01-262089
17. Gerdemann U, Keirnan JM, Katari UL, Yanagisawa R, Christin AS, Huye LE, et al. Rapidly generated multivirus-specific cytotoxic T lymphocytes for the prophylaxis and treatment of viral infections. Mol Ther (2012) 20(8):1622–32. doi: 10.1038/mt.2012.130
18. Tzannou I, Papadopoulou A, Naik S, Leung K, Martinez CA, Ramos CA, et al. Off-the-Shelf virus-specific T cells to treat BK virus, human herpesvirus 6, cytomegalovirus, Epstein-Barr virus, and adenovirus infections after allogeneic hematopoietic stem-cell transplantation. J Clin Oncol (2017) 35(31):3547–57. doi: 10.1200/JCO.2017.73.0655
19. Prockop S, Doubrovina E, Suser S, Heller G, Barker J, Dahi P, et al. Off-the-shelf EBV-specific T cell immunotherapy for rituximab-refractory EBV-associated lymphoma following transplantation. J Clin Invest. (2020) 130(2):733–47. doi: 10.1172/JCI121127
20. Lucas KG, Small TN, Heller G, Dupont B, O’Reilly RJ. The development of cellular immunity to Epstein-Barr virus after allogeneic bone marrow transplantation. Blood. (1996) 87(6):2594–603. doi: 10.1182/blood.V87.6.2594.bloodjournal8762594
21. Bollard CM, Huls MH, Buza E, Weiss H, Torrano V, Gresik MV, et al. Administration of latent membrane protein 2-specific cytotoxic T lymphocytes to patients with relapsed Epstein-Barr virus-positive lymphoma. Clin Lymphoma Myeloma. (2006) 6(4):342–7. doi: 10.3816/CLM.2006.n.011
22. Doubrovina E, Oflaz-Sozmen B, Prockop SE, Kernan NA, Abramson S, Teruya-Feldstein J, et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood. (2012) 119(11):2644–56. doi: 10.1182/blood-2011-08-371971
23. Koehne G, Smith KM, Ferguson TL, Williams RY, Heller G, Pamer EG, et al. Quantitation, selection, and functional characterization of Epstein-Barr virus-specific and alloreactive T cells detected by intracellular interferon-gamma production and growth of cytotoxic precursors. Blood. (2002) 99(5):1730–40. doi: 10.1182/blood.V99.5.1730
24. Basso S, Zecca M, Calafiore L, Rubert L, Fiocchi R, Paulli M, et al. Successful treatment of a classic Hodgkin lymphoma-type post-transplant lymphoproliferative disorder with tailored chemotherapy and Epstein-Barr virus-specific cytotoxic T lymphocytes in a pediatric heart transplant recipient. Pediatr Transplant. (2013) 17(7):E168–173. doi: 10.1111/petr.12146
25. Comoli P, Pedrazzoli P, Maccario R, Basso S, Carminati O, Labirio M, et al. Cell therapy of stage IV nasopharyngeal carcinoma with autologous Epstein-Barr virus-targeted cytotoxic T lymphocytes. J Clin Oncol (2005) 23(35):8942–9. doi: 10.1200/JCO.2005.02.6195
26. Secondino S, Zecca M, Licitra L, Gurrado A, Schiavetto I, Bossi P, et al. T-Cell therapy for EBV-associated nasopharyngeal carcinoma: preparative lymphodepleting chemotherapy does not improve clinical results. Ann Oncol (2012) 23(2):435–41. doi: 10.1093/annonc/mdr134
27. Louis CU, Straathof K, Bollard CM, Ennamuri S, Gerken C, Lopez TT, et al. Adoptive transfer of EBV-specific T cells results in sustained clinical responses in patients with locoregional nasopharyngeal carcinoma. J Immunother (2010) 33(9):983–90. doi: 10.1097/CJI.0b013e3181f3cbf4
28. Comoli P, Labirio M, Basso S, Baldanti F, Grossi P, Furione M, et al. Infusion of autologous Epstein-Barr virus (EBV)-specific cytotoxic T cells for prevention of EBV-related lymphoproliferative disorder in solid organ transplant recipients with evidence of active virus replication. Blood. (2002) 99(7):2592–8. doi: 10.1182/blood.V99.7.2592
29. Comoli P, Basso S, Zecca M, Pagliara D, Baldanti F, Bernardo ME, et al. Preemptive therapy of EBV-related lymphoproliferative disease after pediatric haploidentical stem cell transplantation. Am J Transplant (2007) 7(6):1648–55. doi: 10.1111/j.1600-6143.2007.01823.x
30. Savoldo B, Goss JA, Hammer MM, Zhang L, Lopez T, Gee AP, et al. Treatment of solid organ transplant recipients with autologous Epstein Barr virus-specific cytotoxic T lymphocytes (CTLs). Blood. (2006) 108(9):2942–9. doi: 10.1182/blood-2006-05-021782
31. Capelli C, Frigerio S, Lisini D, Nava S, Gaipa G, Belotti D, et al. A comprehensive report of long-term stability data for a range ATMPs: a need to develop guidelines for safe and harmonized stability studies. Cytotherapy. (2022) 24(5):544–56. doi: 10.1016/j.jcyt.2021.12.004
32. van den Berg JH, Heemskerk B, van Rooij N, Gomez-Eerland R, Michels S, van Zon M, et al. Tumor infiltrating lymphocytes (TIL) therapy in metastatic melanoma: boosting of neoantigen-specific T cell reactivity and long-term follow-up. J Immunother Cancer. (2020) 8(2):e000848. doi: 10.1136/jitc-2020-000848
33. Saberian C, Amaria RN, Najjar AM, Radvanyi LG, Haymaker CL, Forget MA, et al. Randomized phase II trial of lymphodepletion plus adoptive cell transfer of tumor-infiltrating lymphocytes, with or without dendritic cell vaccination, in patients with metastatic melanoma. J Immunother Cancer (2021) 9(5):e002449. doi: 10.1136/jitc-2021-002449
34. Comoli P, Basso S, Riva G, Barozzi P, Guido I, Gurrado A, et al. BCR-ABL-specific T-cell therapy in ph+ ALL patients on tyrosine-kinase inhibitors. Blood. (2017) 129(5):582–6. doi: 10.1182/blood-2016-07-731091
35. Mackensen A, Meidenbauer N, Vogl S, Laumer M, Berger J, Andreesen R. Phase I study of adoptive T-cell therapy using antigen-specific CD8+ T cells for the treatment of patients with metastatic melanoma. J Clin Oncol (2006) 24(31):5060–9. doi: 10.1200/JCO.2006.07.1100
36. Linn YC, Yong HX, Niam M, Lim TJ, Chu S, Choong A, et al. A phase I/II clinical trial of autologous cytokine-induced killer cells as adjuvant immunotherapy for acute and chronic myeloid leukemia in clinical remission. Cytotherapy. (2012) 14(7):851–9. doi: 10.3109/14653249.2012.694419
37. Mareschi K, Adamini A, Castiglia S, Rustichelli D, Castello L, Mandese A, et al. Cytokine-induced killer (CIK) cells, In vitro expanded under good manufacturing process (GMP) conditions, remain stable over time after cryopreservation. Pharm Basel Switz. (2020) 13(5):93. doi: 10.3390/ph13050093
38. Gotti E, Tettamanti S, Zaninelli S, Cuofano C, Cattaneo I, Rotiroti MC, et al. Optimization of therapeutic T cell expansion in G-Rex device and applicability to large-scale production for clinical use. Cytotherapy. (2022) 24(3):334–43. doi: 10.1016/j.jcyt.2021.11.004
39. Rutella S, Iudicone P, Bonanno G, Fioravanti D, Procoli A, Lavorino C, et al. Adoptive immunotherapy with cytokine-induced killer cells generated with a new good manufacturing practice-grade protocol. Cytotherapy. (2012) 14(7):841–50. doi: 10.3109/14653249.2012.681038
40. Narayan R, Benjamin JE, Shah O, Tian L, Tate K, Armstrong R, et al. Donor-derived cytokine-induced killer cell infusion as consolidation after nonmyeloablative allogeneic transplantation for myeloid neoplasms. Biol Blood Marrow Transplant (2019) 25(7):1293–303. doi: 10.1016/j.bbmt.2019.03.027
41. Introna M, Lussana F, Algarotti A, Gotti E, Valgardsdottir R, Micò C, et al. Phase II study of sequential infusion of donor lymphocyte infusion and cytokine-induced killer cells for patients relapsed after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2017) 23(12):2070–8. doi: 10.1016/j.bbmt.2017.07.005
42. Li R, Wang C, Liu L, Du C, Cao S, Yu J, et al. Autologous cytokine-induced killer cell immunotherapy in lung cancer: a phase II clinical study. Cancer Immunol Immunother (2012) 61(11):2125–33. doi: 10.1007/s00262-012-1260-2
43. Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory b-cell leukemias. Blood. (2011) 118(18):4817–28. doi: 10.1182/blood-2011-04-348540
44. Xiao Z, Wang CQ, Feng JH, Zhou MH, Wang YZ, Li NN, et al. Effectiveness and safety of chemotherapy with cytokine-induced killer cells in non-small cell lung cancer: a systematic review and meta-analysis of 32 randomized controlled trials. Cytotherapy. (2019) 21(2):125–47. doi: 10.1016/j.jcyt.2018.10.011
45. Fernández L, Fernández A, Mirones I, Escudero A, Cardoso L, Vela M, et al. GMP-compliant manufacturing of NKG2D CAR memory T cells using CliniMACS prodigy. Front Immunol (2019) 10:2361. doi: 10.3389/fimmu.2019.02361
46. Gee AP. Manufacturing genetically modified T cells for clinical trials. Cancer Gene Ther (2015) 22(2):67–71. doi: 10.1038/cgt.2014.71
47. Castella M, Caballero-Baños M, Ortiz-Maldonado V, González-Navarro EA, Suñé G, Antoñana-Vidósola A, et al. Point-Of-Care CAR T-cell production (ARI-0001) using a closed semi-automatic bioreactor: experience from an academic phase I clinical trial. Front Immunol (2020) 11:482. doi: 10.3389/fimmu.2020.00482
48. Qu X, An G, Sui W, Wang T, Zhang X, Yang J, et al. Phase 1 study of c-CAR088, a novel humanized anti-BCMA CAR T-cell therapy in relapsed/refractory multiple myeloma. J Immunother Cancer. (2022) 10(9):e005145. doi: 10.1136/jitc-2022.005145
49. Mailankody S, Devlin SM, Landa J, Nath K, Diamonte C, Carstens EJ, et al. GPRC5D-targeted CAR T cells for myeloma. N Engl J Med (2022) (13):1196–206. doi: 10.1056/NEJMoa2209900
50. Zhang J, Hu Y, Yang J, Li W, Zhang M, Wang Q, et al. Non-viral, specifically targeted CAR-T cells achieve high safety and efficacy in b-NHL. Nature. (2022) 609(7926):369–74. doi: 10.1038/s41586-022-05140-y
51. Jin X, Zhang M, Sun R, Lyu H, Xiao X, Zhang X, et al. First-in-human phase I study of CLL-1 CAR-T cells in adults with relapsed/refractory acute myeloid leukemia. J Hematol Oncol (2022) 15(1):88. doi: 10.1186/s13045-022-01308-1
52. Yu L, Huang L, Lin D, Lai X, Wu L, Liao X, et al. GD2-specific chimeric antigen receptor-modified T cells for the treatment of refractory and/or recurrent neuroblastoma in pediatric patients. J Cancer Res Clin Oncol (2022) 148(10):2643–52. doi: 10.1007/s00432-021-03839-5
53. Magnani CF, Gaipa G, Lussana F, Belotti D, Gritti G, Napolitano S, et al. Sleeping beauty-engineered CAR T cells achieve antileukemic activity without severe toxicities. J Clin Invest. (2020) 130(11):6021–33. doi: 10.1172/JCI138473
54. Ishihara M, Kitano S, Kageyama S, Miyahara Y, Yamamoto N, Kato H, et al. NY-ESO-1-specific redirected T cells with endogenous TCR knockdown mediate tumor response and cytokine release syndrome. J Immunother Cancer. (2022) 10(6):e003811. doi: 10.1136/jitc-2021-003811
55. Kageyama S, Ikeda H, Miyahara Y, Imai N, Ishihara M, Saito K, et al. Adoptive transfer of MAGE-A4 T-cell receptor gene-transduced lymphocytes in patients with recurrent esophageal cancer. Clin Cancer Res (2015) 21(10):2268–77. doi: 10.1158/1078-0432.CCR-14-1559
56. Van den Hove LE, Van Gool SW, Vandenberghe P, Boogaerts MA, Ceuppens JL. CD57+/CD28- T cells in untreated hemato-oncological patients are expanded and display a Th1-type cytokine secretion profile, ex vivo cytolytic activity and enhanced tendency to apoptosis. Leukemia. (1998) 12(10):1573–82. doi: 10.1038/sj.leu.2401146
57. Guedan S, Luu M, Ammar D, Barbao P, Bonini C, Bousso P, et al. Time 2EVOLVE: predicting efficacy of engineered T-cells - how far is the bench from the bedside? J Immunother Cancer (2022) 10(5):e003487. doi: 10.1136/jitc-2021-003487
58. Hong R, Hu Y, Huang H. Biomarkers for chimeric antigen receptor T cell therapy in acute lymphoblastic leukemia: prospects for personalized management and prognostic prediction. Front Immunol (2021) 12:627764. doi: 10.3389/fimmu.2021.627764
59. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large b-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet (2020) 396(10254):839–52. doi: 10.1016/S0140-6736(20)31366-0
60. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med (2018) 24(5):563–71. doi: 10.1038/s41591-018-0010-1
61. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory Large b-cell lymphoma. N Engl J Med (2017) 377(26):2531–44. doi: 10.1056/NEJMoa1707447
62. Shi N, Luo Y, Xu Y, Liang J, Ma A, Gan Y, et al. DAP10 predicted the outcome of pediatric b-cell acute lymphoblastic leukemia and was associated with the T-cell exhaustion. J Oncol (2021) 2021:4824868. doi: 10.1155/2021/4824868
63. Good Z, Spiegel JY, Sahaf B, Malipatlolla MB, Ehlinger ZJ, Kurra S, et al. Post-infusion CAR TReg cells identify patients resistant to CD19-CAR therapy. Nat Med (2022) 28(9):1860–71. doi: 10.1038/s41591-022-01960-7
64. Tan JY, Low MH, Chen Y, Lim FLWI. CAR T cell therapy in hematological malignancies: implications of the tumor microenvironment and biomarkers on efficacy and toxicity. Int J Mol Sci (2022) 23(13):6931. doi: 10.3390/ijms23136931
65. Caballero AC, Escribà-Garcia L, Alvarez-Fernández C, Briones J. CAR T-cell therapy predictive response markers in diffuse Large b-cell lymphoma and therapeutic options after CART19 failure. Front Immunol (2022) 13:904497. doi: 10.3389/fimmu.2022.904497
66. Lamble AJ, Myers RM, Taraseviciute A, John S, Yates B, Steinberg SM, et al. Preinfusion factors impacting relapse immunophenotype following CD19 CAR T cells. Blood Adv (2023) 7(4):575–85. doi: 10.1182/bloodadvances.2022007423
67. Bai Z, Woodhouse S, Zhao Z, Arya R, Govek K, Kim D, et al. Single-cell antigen-specific landscape of CAR T infusion product identifies determinants of CD19-positive relapse in patients with ALL. Sci Adv (2022) 8(23):eabj2820. doi: 10.1126/sciadv.abj2820
68. Chen GM, Chen C, Das RK, Gao P, Chen CH, Bandyopadhyay S, et al. Integrative bulk and single-cell profiling of premanufacture T-cell populations reveals factors mediating long-term persistence of CAR T-cell therapy. Cancer Discovery (2021) 11(9):2186–99. doi: 10.1158/2159-8290.CD-20-1677
69. Deng Q, Han G, Puebla-Osorio N, Ma MCJ, Strati P, Chasen B, et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large b cell lymphomas. Nat Med (2020) 26(12):1878–87. doi: 10.1038/s41591-020-1061-7
70. Rossi J, Paczkowski P, Shen YW, Morse K, Flynn B, Kaiser A, et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood. (2018) 132(8):804–14. doi: 10.1182/blood-2018-01-828343
71. Romain G, Strati P, Rezvan A, Fathi M, Bandey IN, Adolacion JRT, et al. Multidimensional single-cell analysis identifies a role for CD2-CD58 interactions in clinical antitumor T cell responses. J Clin Invest. (2022) 132(17):e159402. doi: 10.1172/JCI159402
72. Garcia-Prieto CA, Villanueva L, Bueno-Costa A, Davalos V, González-Navarro EA, Juan M, et al. Epigenetic profiling and response to CD19 chimeric antigen receptor T-cell therapy in b-cell malignancies. J Natl Cancer Inst (2022) 114(3):436–45. doi: 10.1093/jnci/djab194
73. Ran GH, Lin YQ, Tian L, Zhang T, Yan DM, Yu JH, et al. Natural killer cell homing and trafficking in tissues and tumors: from biology to application. Signal Transduct Target Ther (2022) 7(1):205. doi: 10.1038/s41392-022-01058-z
74. Zhang DKY, Adu-Berchie K, Iyer S, Liu Y, Cieri N, Brockman JM, et al. Enhancing CAR-T cell functionality in a patient-specific manner. Nat Commun (2023) 14(1):506. doi: 10.1038/s41467-023-36126-7
75. Finney OC, Brakke HM, Rawlings-Rhea S, Hicks R, Doolittle D, Lopez M, et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J Clin Invest. (2019) 129(5):2123–32. doi: 10.1172/JCI125423
76. Yang J, Chen Y, Jing Y, Green MR, Han L. Advancing CAR T cell therapy through the use of multidimensional omics data. Nat Rev Clin Oncol (2023) 20(4):211–228. doi: 10.1038/s41571-023-00729-2
77. Yan C, Yang Q, Zhang S, Millar DG, Alpert EJ, Do D, et al. Single-cell imaging of T cell immunotherapy responses in vivo. J Exp Med (2021) 218(10):e20210314. doi: 10.1084/jem.20210314
78. Dimitri A, Herbst F, Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer. (2022) 21(1):78. doi: 10.1186/s12943-022-01559-z
79. Cichocki F, van der Stegen SJC, Miller JS. Engineered and banked iPSCs for advanced NK- and T-cell immunotherapies. Blood. (2023) 141(8):846–55. doi: 10.1182/blood.2022016205
80. Hang S, Wang N, Sugimura RT. NK, then macrophages: recent advances and challenges in adaptive immunotherapy from human pluripotent stem cells. Differ Res Biol Divers (2023) 130:51–7. doi: 10.1016/j.diff.2023.01.001
81. Boyd-Gibbins N, Karagiannis P, Hwang DW, Kim SI. iPSCs in NK cell manufacturing and NKEV development. Front Immunol (2022) 13:890894. doi: 10.3389/fimmu.2022.890894
82. Aoki T, Motohashi S, Koseki H. Regeneration of invariant natural killer T (iNKT) cells: application of iPSC technology for iNKT cell-targeted tumor immunotherapy. Inflammation Regen. (2023) 43(1):27. doi: 10.1186/s41232-023-00275-5
83. Bachanova V, Ghobadi A, Patel K, Park JH, Flinn IW, Shah P, et al. Safety and efficacy of FT596, a first-in-Class, multi-antigen targeted, off-the-Shelf, iPSC-derived CD19 CAR NK cell therapy in Relapsed/Refractory b-cell lymphoma. Blood. (2021) 138:823. doi: 10.1182/blood-2021-151185
84. Patel K, Bachanova V, Goodman AM, Pagel JM, Griffis K, Anderson M, et al. Phase I study of FT516, an off-the-Shelf iPSC-derived NK cell therapy, in combination with rituximab in patients with Relapsed/Refractory b-cell lymphoma. Blood (2021) 138:3873. doi: 10.1182/blood-2021-151520
85. Miki S, Kawai Y, Nakayama-Hosoya K, Iwabuchi R, Terahara K, Tsunetsugu-Yokota Y, et al. Sustainable antiviral efficacy of rejuvenated HIV-specific cytotoxic T lymphocytes generated from induced pluripotent stem cells. J Virol (2022) 96(6):e0221721. doi: 10.1128/jvi.02217-21
86. Vizcardo R, Masuda K, Yamada D, Ikawa T, Shimizu K, Fujii SI, et al. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell (2013) 12(1):31–6. doi: 10.1016/j.stem.2012.12.006
87. Nishimura T, Kaneko S, Kawana-Tachikawa A, Tajima Y, Goto H, Zhu D, et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell (2013) 12(1):114–26. doi: 10.1016/j.stem.2012.11.002
88. Ito T, Kawai Y, Yasui Y, Iriguchi S, Minagawa A, Ishii T, et al. The therapeutic potential of multiclonal tumoricidal T cells derived from tumor infiltrating lymphocyte-1derived iPS cells. Commun Biol (2021) 4(1):694. doi: 10.1038/s42003-021-02195-x
89. Maeda T, Nagano S, Ichise H, Kataoka K, Yamada D, Ogawa S, et al. Regeneration of CD8αβ T cells from T-cell-Derived iPSC imparts potent tumor antigen-specific cytotoxicity. Cancer Res (2016) 76(23):6839–50. doi: 10.1158/0008-5472.CAN-16-1149
90. Yu S, Yi M, Qin S, Wu K. Next generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Mol Cancer. (2019) 18(1):125. doi: 10.1186/s12943-019-1057-4
91. Zhang P, Raju J, Ullah MA, Au R, Varelias A, Gartlan KH, et al. Phase I trial of inducible caspase 9 T cells in adult stem cell transplant demonstrates massive clonotypic proliferative potential and long-term persistence of transgenic T cells. Clin Cancer Res (2019) 25(6):1749–55. doi: 10.1158/1078-0432.CCR-18-3069
92. Zhou X, Tu S, Wang C, Huang R, Deng L, Song C, et al. Phase I trial of fourth-generation anti-CD19 chimeric antigen receptor T cells against relapsed or refractory b cell non-Hodgkin lymphomas. Front Immunol (2020) 11:564099. doi: 10.3389/fimmu.2020.564099
93. Thurner B, Röder C, Dieckmann D, Heuer M, Kruse M, Glaser A, et al. Generation of large numbers of fully mature and stable dendritic cells from leukapheresis products for clinical application. J Immunol Methods (1999) 223(1):1–15. doi: 10.1016/S0022-1759(98)00208-7
94. Kolostova K, Pospisilova E, Matkowski R, Szelachowska J, Bobek V. Immune activation of the monocyte-derived dendritic cells using patients own circulating tumor cells. Cancer Immunol Immunother (2022) 71(12):2901–11. doi: 10.1007/s00262-022-03189-2
95. Ni M, Hoffmann JM, Schmitt M, Schmitt A. Progress of dendritic cell-based cancer vaccines for patients with hematological malignancies. Expert Opin Biol Ther (2016) 16(9):1113–23. doi: 10.1080/14712598.2016.1196181
96. Liau LM, Ashkan K, Brem S, Campian JL, Trusheim JE, Iwamoto FM, et al. Association of autologous tumor lysate-loaded dendritic cell vaccination with extension of survival among patients with newly diagnosed and recurrent glioblastoma: a phase 3 prospective externally controlled cohort trial. JAMA Oncol (2023) 9(1):112–21. doi: 10.1001/jamaoncol.2022.5370
97. Goyvaerts C, Breckpot K. The journey of in vivo virus engineered dendritic cells from bench to bedside: a bumpy road. Front Immunol (2018) 9:2052. doi: 10.3389/fimmu.2018.02052
98. Figlin RA, Tannir NM, Uzzo RG, Tykodi SS, Chen DYT, Master V, et al. Results of the ADAPT phase 3 study of rocapuldencel-T in combination with sunitinib as first-line therapy in patients with metastatic renal cell carcinoma. Clin Cancer Res (2020) 26(10):2327–36. doi: 10.1158/1078-0432.CCR-19-2427
99. Sundarasetty BS, Chan L, Darling D, Giunti G, Farzaneh F, Schenck F, et al. Lentivirus-induced ‘Smart’ dendritic cells: pharmacodynamics and GMP-compliant production for immunotherapy against TRP2-positive melanoma. Gene Ther (2015) 22(9):707–20. doi: 10.1038/gt.2015.43
100. Sundarasetty BS, Kloess S, Oberschmidt O, Naundorf S, Kuehlcke K, Daenthanasanmak A, et al. Generation of lentivirus-induced dendritic cells under GMP-compliant conditions for adaptive immune reconstitution against cytomegalovirus after stem cell transplantation. J Transl Med (2015) 13:240. doi: 10.1186/s12967-015-0599-5
101. De Keersmaecker B, Claerhout S, Carrasco J, Bar I, Corthals J, Wilgenhof S, et al. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: link between T-cell activation and clinical responses in advanced melanoma. J Immunother Cancer. (2020) 8(1):e000329. doi: 10.1136/jitc-2019-000329
102. Wang D, Huang XF, Hong B, Song XT, Hu L, Jiang M, et al. Efficacy of intracellular immune checkpoint-silenced DC vaccine. JCI Insight (2018) 3(3):e98368. doi: 10.1172/jci.insight.98368
103. Ge C, Li R, Song H, Geng T, Yang J, Tan Q, et al. Phase I clinical trial of a novel autologous modified-DC vaccine in patients with resected NSCLC. BMC Cancer. (2017) 17(1):884. doi: 10.1186/s12885-017-3859-3
104. Nava S, Lisini D, Pogliani S, Dossena M, Bersano A, Pellegatta S, et al. Safe and reproducible preparation of functional dendritic cells for immunotherapy in glioblastoma patients. Stem Cells Transl Med (2015) 4(10):1164–72. doi: 10.5966/sctm.2015-0091
105. Carloni S, Piccinini C, Pancisi E, Soldati V, Stefanelli M, Granato AM, et al. Potency assessment of dendritic cell anticancer vaccine: validation of the Co-flow DC assay. Int J Mol Sci (2021) 22(11):5824. doi: 10.3390/ijms22115824
106. Westdorp H, Creemers JHA, van Oort IM, Schreibelt G, Gorris MAJ, Mehra N, et al. Blood-derived dendritic cell vaccinations induce immune responses that correlate with clinical outcome in patients with chemo-naive castration-resistant prostate cancer. J Immunother Cancer. (2019) 7(1):302. doi: 10.1186/s40425-019-0787-6
107. Lau SP, Klaase L, Vink M, Dumas J, Bezemer K, van Krimpen A, et al. Autologous dendritic cells pulsed with allogeneic tumour cell lysate induce tumour-reactive T-cell responses in patients with pancreatic cancer: a phase I study. Eur J Cancer (2022) 169:20–31. doi: 10.1016/j.ejca.2022.03.015
108. Surenaud M, Montes M, Lindestam Arlehamn CS, Sette A, Banchereau J, Palucka K, et al. Anti-HIV potency of T-cell responses elicited by dendritic cell therapeutic vaccination. PloS Pathog (2019) 15(9):e1008011. doi: 10.1371/journal.ppat.1008011
109. Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res (2011) 17(6):1603–15. doi: 10.1158/1078-0432.CCR-10-2563
110. Lim JH, Park CJ, Kim MJ, Jang S, Chi HS, Lee JH, et al. Generation of lymphocytes potentiated against leukemic lymphoblasts by stimulation using leukemic cell lysate-pulsed dendritic cells in patients with acute lymphoblastic leukemia and measurement of in vitro anti-leukemic cytotoxicity. Hematol Amst Neth. (2012) 17(1):15–22. doi: 10.1179/102453312X13221316477453
111. Castiello L, Sabatino M, Ren J, Terabe M, Khuu H, Wood LV, et al. Expression of CD14, IL10, and tolerogenic signature in dendritic cells inversely correlate with clinical and immunologic response to TARP vaccination in prostate cancer patients. Clin Cancer Res (2017) 23(13):3352–64. doi: 10.1158/1078-0432.CCR-16-2199
112. Capelli C, Pedrini O, Valgardsdottir R, Da Roit F, Golay J, Introna M. Clinical grade expansion of MSCs. Immunol Lett (2015) 168(2):222–7. doi: 10.1016/j.imlet.2015.06.006
113. Introna M, Golay J. Tolerance to bone marrow transplantation: do mesenchymal stromal cells still have a future for acute or chronic GvHD? Front Immunol (2020) 11:609063. doi: 10.3389/fimmu.2020.609063
114. Squillaro T, Peluso G, Galderisi U. Clinical trials with mesenchymal stem cells: an update. Cell Transplant. (2016) 25(5):829–48. doi: 10.3727/096368915X689622
115. Pittenger MF, Discher DE, Péault BM, Phinney DG, Hare JM, Caplan AI. Mesenchymal stem cell perspective: cell biology to clinical progress. NPJ Regener Med (2019) 4:22. doi: 10.1038/s41536-019-0083-6
116. Jayaraman P, Lim R, Ng J, Vemuri MC. Acceleration of translational mesenchymal stromal cell therapy through consistent quality GMP manufacturing. Front Cell Dev Biol (2021) 9:648472. doi: 10.3389/fcell.2021.648472
117. Casiraghi F, Perico N, Podestà MA, Todeschini M, Zambelli M, Colledan M, et al. Third-party bone marrow-derived mesenchymal stromal cell infusion before liver transplantation: a randomized controlled trial. Am J Transplant (2021) 21(8):2795–809. doi: 10.1111/ajt.16468
118. Li M, Chen H, Zhu M. Mesenchymal stem cells for regenerative medicine in central nervous system. Front Neurosci (2022) 16:1068114. doi: 10.3389/fnins.2022.1068114
119. Dunn CM, Kameishi S, Grainger DW, Okano T. Strategies to address mesenchymal stem/stromal cell heterogeneity in immunomodulatory profiles to improve cell-based therapies. Acta Biomater. (2021) 133:114–25. doi: 10.1016/j.actbio.2021.03.069
120. Lim HC, Park YB, Ha CW, Cole BJ, Lee BK, Jeong HJ, et al. Allogeneic umbilical cord blood-derived mesenchymal stem cell implantation versus microfracture for Large, full-thickness cartilage defects in older patients: a multicenter randomized clinical trial and extended 5-year clinical follow-up. Orthop J Sports Med (2021) 9(1):2325967120973052. doi: 10.1177/2325967120973052
121. Wright A, Arthaud-Day ML, Weiss ML. Therapeutic use of mesenchymal stromal cells: the need for inclusive characterization guidelines to accommodate all tissue sources and species. Front Cell Dev Biol (2021) 9:632717. doi: 10.3389/fcell.2021.632717
122. Lechanteur C, Briquet A, Bettonville V, Baudoux E, Beguin Y. MSC manufacturing for academic clinical trials: from a clinical-grade to a full GMP-compliant process. Cells. (2021) 10(6):1320. doi: 10.3390/cells10061320
123. Herzig MC, Delavan CP, Jensen KJ, Cantu C, Montgomery RK, Christy BA, et al. A streamlined proliferation assay using mixed lymphocytes for evaluation of human mesenchymal stem cell immunomodulation activity. J Immunol Methods (2021) 488:112915. doi: 10.1016/j.jim.2020.112915
124. Wang J, Gao S, Zhao Y, Fan T, Zhang M, Chang D. Manufacture and quality control of human umbilical cord-derived mesenchymal stem cell sheets for clinical use. Cells. (2022) 11(17):2732. doi: 10.3390/cells11172732
125. Galipeau J, Krampera M, Barrett J, Dazzi F, Deans RJ, DeBruijn J, et al. International society for cellular therapy perspective on immune functional assays for mesenchymal stromal cells as potency release criterion for advanced phase clinical trials. Cytotherapy. (2016) 18(2):151–9. doi: 10.1016/j.jcyt.2015.11.008
126. Labedz-Maslowska A, Szkaradek A, Mierzwinski T, Madeja Z, Zuba-Surma E. Processing and ex vivo expansion of adipose tissue-derived mesenchymal Stem/Stromal cells for the development of an advanced therapy medicinal product for use in humans. Cells. (2021) 10(8):1908. doi: 10.3390/cells10081908
127. Aghayan HR, Salimian F, Abedini A, Fattah Ghazi S, Yunesian M, Alavi-Moghadam S, et al. Human placenta-derived mesenchymal stem cells transplantation in patients with acute respiratory distress syndrome (ARDS) caused by COVID-19 (phase I clinical trial): safety profile assessment. Stem Cell Res Ther (2022) 13(1):365. doi: 10.1186/s13287-022-02953-6
128. Mebarki M, Iglicki N, Marigny C, Abadie C, Nicolet C, Churlaud G, et al. Development of a human umbilical cord-derived mesenchymal stromal cell-based advanced therapy medicinal product to treat immune and/or inflammatory diseases. Stem Cell Res Ther (2021) 12(1):571. doi: 10.1186/s13287-021-02637-7
129. Chinnadurai R, Rajan D, Qayed M, Arafat D, Garcia M, Liu Y, et al. Potency analysis of mesenchymal stromal cells using a combinatorial assay matrix approach. Cell Rep (2018) 22(9):2504–17. doi: 10.1016/j.celrep.2018.02.013
130. Karanu F, Ott L, Webster DA, Stehno-Bittel L. Improved harmonization of critical characterization assays across cell therapies. Regener Med (2020) 15(5):1661–78. doi: 10.2217/rme-2020-0003
131. Trento C, Bernardo ME, Nagler A, Kuçi S, Bornhäuser M, Köhl U, et al. Manufacturing mesenchymal stromal cells for the treatment of graft-versus-Host disease: a survey among centers affiliated with the European society for blood and marrow transplantation. Biol Blood Marrow Transplant (2018) 24(11):2365–70. doi: 10.1016/j.bbmt.2018.07.015
132. de Wolf C, van de Bovenkamp M, Hoefnagel M. Regulatory perspective on in vitro potency assays for human mesenchymal stromal cells used in immunotherapy. Cytotherapy. (2017) 19(7):784–97. doi: 10.1016/j.jcyt.2017.03.076
133. Cheung TS, Bertolino GM, Giacomini C, Bornhäuser M, Dazzi F, Galleu A. Mesenchymal stromal cells for graft versus host disease: mechanism-based biomarkers. Front Immunol (2020) 11:1338. doi: 10.3389/fimmu.2020.01338
134. Hansen SB, Højgaard LD, Kastrup J, Ekblond A, Follin B, Juhl M. Optimizing an immunomodulatory potency assay for mesenchymal stromal cell. Front Immunol (2022) 13:1085312. doi: 10.3389/fimmu.2022.1085312
135. Capelli C, Zaccara E, Cipriani P, Di Benedetto P, Maglione W, Andracco R, et al. Phenotypical and functional characteristics of In vitro-expanded adipose-derived mesenchymal stromal cells from patients with systematic sclerosis. Cell Transplant. (2017) 26(5):841–54. doi: 10.3727/096368917X694822
136. De Luca M, Aiuti A, Cossu G, Parmar M, Pellegrini G, Robey PG. Advances in stem cell research and therapeutic development. Nat Cell Biol (2019) 21(7):801–11. doi: 10.1038/s41556-019-0344-z
137. Galleu A, Riffo-Vasquez Y, Trento C, Lomas C, Dolcetti L, Cheung TS, et al. Apoptosis in mesenchymal stromal cells induces in vivo recipient-mediated immunomodulation. Sci Transl Med (2017) 9(416):eaam7828. doi: 10.1126/scitranslmed.aam7828
138. Cheung TS, Galleu A, von Bonin M, Bornhäuser M, Dazzi F. Apoptotic mesenchymal stromal cells induce prostaglandin E2 in monocytes: implications for the monitoring of mesenchymal stromal cell activity. Haematologica. (2019) 104(10):e438–41. doi: 10.3324/haematol.2018.214767
139. Krampera M, Le Blanc K. Mesenchymal stromal cells: putative microenvironmental modulators become cell therapy. Cell Stem Cell (2021) 28(10):1708–25. doi: 10.1016/j.stem.2021.09.006
140. Bonab MM, Alimoghaddam K, Talebian F, Ghaffari SH, Ghavamzadeh A, Nikbin B. Aging of mesenchymal stem cell in vitro. BMC Cell Biol (2006) 7:14. doi: 10.1186/1471-2121-7-14
141. Bork S, Pfister S, Witt H, Horn P, Korn B, Ho AD, et al. DNA Methylation pattern changes upon long-term culture and aging of human mesenchymal stromal cells. Aging Cell (2010) 9(1):54–63. doi: 10.1111/j.1474-9726.2009.00535.x
142. Wagner W, Horn P, Castoldi M, Diehlmann A, Bork S, Saffrich R, et al. Replicative senescence of mesenchymal stem cells: a continuous and organized process. PloS One (2008) 3(5):e2213. doi: 10.1371/journal.pone.0002213
143. Horwitz EM, Le Blanc K, Dominici M, Mueller I, Slaper-Cortenbach I, Marini FC, et al. Clarification of the nomenclature for MSC: the international society for cellular therapy position statement. Cytotherapy. (2005) 7(5):393–5. doi: 10.1080/14653240500319234
144. Cancedda R, Bianchi G, Derubeis A, Quarto R. Cell therapy for bone disease: a review of current status. Stem Cells Dayt Ohio. (2003) 21(5):610–9. doi: 10.1634/stemcells.21-5-610
145. Miclau K, Hambright WS, Huard J, Stoddart MJ, Bahney CS. Cellular expansion of MSCs: shifting the regenerative potential. Aging Cell (2023) 22(1):e13759. doi: 10.1111/acel.13759
146. Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. the international society for cellular therapy position statement. Cytotherapy. (2006) 8(4):315–7. doi: 10.1080/146532240600855905
147. Ahn SH, Ryu SW, Choi H, You S, Park J, Choi C. Manufacturing therapeutic exosomes: from bench to industry. Mol Cells (2022) 45(5):284–90. doi: 10.14348/molcells.2022.2033
148. Zhang K, Cheng K. Stem cell-derived exosome versus stem cell therapy. Nat Rev Bioeng (2023), 1–2. doi: 10.1038/s44222-023-00064-2
149. Rezaie J, Feghhi M, Etemadi T. A review on exosomes application in clinical trials: perspective, questions, and challenges. Cell Commun Signal CCS. (2022) 20(1):145. doi: 10.1186/s12964-022-00959-4
150. Muthu S, Bapat A, Jain R, Jeyaraman N, Jeyaraman M. Exosomal therapy-a new frontier in regenerative medicine. Stem Cell Investig (2021) 8:7. doi: 10.21037/sci-2020-037
151. Lotfy A, AboQuella NM, Wang H. Mesenchymal stromal/stem cell (MSC)-derived exosomes in clinical trials. Stem Cell Res Ther (2023) 14(1):66. doi: 10.1186/s13287-023-03287-7
152. Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. (2018) 7(1):1535750. doi: 10.1080/20013078.2018.1535750
153. Grafting of burns with cultured epithelium prepared from autologous epidermal cells. Lancet Lond Engl (1981) 1(8211):75–8.
154. Jurkunas U, Johns L, Armant M. Cultivated autologous limbal epithelial cell transplantation: new frontier in the treatment of limbal stem cell deficiency. Am J Ophthalmol (2022) 239:244–68. doi: 10.1016/j.ajo.2022.03.015
155. Ohki T, Yamamoto M. Esophageal regenerative therapy using cell sheet technology. Regener Ther (2020) 13:8–17. doi: 10.1016/j.reth.2020.04.009
156. De Luca M, Pellegrini G, Green H. Regeneration of squamous epithelia from stem cells of cultured grafts. Regener Med (2006) 1(1):45–57. doi: 10.2217/17460751.1.1.45
157. Mascré G, Dekoninck S, Drogat B, Youssef KK, Broheé S, Sotiropoulou PA, et al. Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature. (2012) 489(7415):257–62. doi: 10.1038/nature11393
158. Pellegrini G, Golisano O, Paterna P, Lambiase A, Bonini S, Rama P, et al. Location and clonal analysis of stem cells and their differentiated progeny in the human ocular surface. J Cell Biol (1999) 145(4):769–82. doi: 10.1083/jcb.145.4.769
159. Barrandon Y, Green H. Three clonal types of keratinocyte with different capacities for multiplication. Proc Natl Acad Sci U S A. (1987) 84(8):2302–6. doi: 10.1073/pnas.84.8.2302
160. Sun X, Joost S, Kasper M. Plasticity of epithelial cells during skin wound healing. Cold Spring Harb Perspect Biol (2023) 15(5):a041232. doi: 10.1101/cshperspect.a041231
161. Tait A, Proctor T, Hamilton NJI, Birchall MA, Lowdell MW. GMP compliant isolation of mucosal epithelial cells and fibroblasts from biopsy samples for clinical tissue engineering. Sci Rep (2021) 11(1):12392. doi: 10.1038/s41598-021-91939-0
162. Zsebik B, Ujlaky-Nagy L, Losonczy G, Vereb G, Takács L. Cultivation of human oral mucosal explants on contact lenses. Curr Eye Res (2017) 42(8):1094–9. doi: 10.1080/02713683.2017.1279635
163. Liu L, Nielsen FM, Emmersen J, Bath C, Østergaard Hjortdal J, Riis S, et al. Pigmentation is associated with stemness hierarchy of progenitor cells within cultured limbal epithelial cells. Stem Cells Dayt Ohio. (2018) 36(9):1411–20. doi: 10.1002/stem.2857
164. Nair DSR, Thomas BB. Stem cell-based treatment strategies for degenerative diseases of the retina. Curr Stem Cell Res Ther (2022) 17(3):214–25. doi: 10.2174/1574888X16666210804112104
165. Reichman S, Slembrouck A, Gagliardi G, Chaffiol A, Terray A, Nanteau C, et al. Generation of storable retinal organoids and retinal pigmented epithelium from adherent human iPS cells in xeno-free and feeder-free conditions. Stem Cells Dayt Ohio. (2017) 35(5):1176–88. doi: 10.1002/stem.2586
166. Mikhailova A, Ilmarinen T, Ratnayake A, Petrovski G, Uusitalo H, Skottman H, et al. Human pluripotent stem cell-derived limbal epithelial stem cells on bioengineered matrices for corneal reconstruction. Exp Eye Res (2016) 146:26–34. doi: 10.1016/j.exer.2015.11.021
167. Wood FM. The role of cell-based therapies in acute burn wound skin repair: a review. J Burn Care Res (2023) 44(Supplement_1):S42–7. doi: 10.1093/jbcr/irac146
168. Holmes JH, Schurr MJ, King BT, Foster K, Faucher LD, Lokuta MA, et al. An open-label, prospective, randomized, controlled, multicenter, phase 1b study of StrataGraft skin tissue versus autografting in patients with deep partial-thickness thermal burns. Burns J Int Soc Burn Inj. (2019) 45(8):1749–58. doi: 10.1016/j.burns.2019.07.021
169. Pellegrini G, Dellambra E, Golisano O, Martinelli E, Fantozzi I, Bondanza S, et al. p63 identifies keratinocyte stem cells. Proc Natl Acad Sci U S A. (2001) 98(6):3156–61. doi: 10.1073/pnas.061032098
170. Bertolin M, Breda C, Ferrari S, Lamon M, Ponzin D, Ferrari B, et al. A new standardized immunofluorescence method for potency quantification (SMPQ) of human conjunctival cell cultures. Cell Tissue Bank. (2021) 22(1):145–59. doi: 10.1007/s10561-020-09874-9
171. Rama P, Matuska S, Paganoni G, Spinelli A, De Luca M, Pellegrini G. Limbal stem-cell therapy and long-term corneal regeneration. N Engl J Med (2010) 363(2):147–55. doi: 10.1056/NEJMoa0905955
172. Pellegrini G, Rama P, Matuska S, Lambiase A, Bonini S, Pocobelli A, et al. Biological parameters determining the clinical outcome of autologous cultures of limbal stem cells. Regener Med (2013) 8(5):553–67. doi: 10.2217/rme.13.43
173. Ramírez BE, Sánchez A, Herreras JM, Fernández I, García-Sancho J, Nieto-Miguel T, et al. Stem cell therapy for corneal epithelium regeneration following good manufacturing and clinical procedures. BioMed Res Int (2015) 2015:408495. doi: 10.1155/2015/408495
174. Campbell JDM, Ahmad S, Agrawal A, Bienek C, Atkinson A, Mcgowan NWA, et al. Allogeneic ex vivo expanded corneal epithelial stem cell transplantation: a randomized controlled clinical trial. Stem Cells Transl Med (2019) 8(4):323–31. doi: 10.1002/sctm.18-0140
175. Kolli S, Ahmad S, Mudhar HS, Meeny A, Lako M, Figueiredo FC. Successful application of ex vivo expanded human autologous oral mucosal epithelium for the treatment of total bilateral limbal stem cell deficiency. Stem Cells Dayt Ohio. (2014) 32(8):2135–46. doi: 10.1002/stem.1694
176. Zakaria N, Possemiers T, Dhubhghaill SN, Leysen I, Rozema J, Koppen C, et al. Results of a phase I/II clinical trial: standardized, non-xenogenic, cultivated limbal stem cell transplantation. J Transl Med (2014) 12:58. doi: 10.1186/1479-5876-12-58
177. Burillon C, Huot L, Justin V, Nataf S, Chapuis F, Decullier E, et al. Cultured autologous oral mucosal epithelial cell sheet (CAOMECS) transplantation for the treatment of corneal limbal epithelial stem cell deficiency. Invest Ophthalmol Vis Sci (2012) 53(3):1325–31. doi: 10.1167/iovs.11-7744
178. Baba K, Sasaki K, Morita M, Tanaka T, Teranishi Y, Ogasawara T, et al. Cell jamming, stratification and p63 expression in cultivated human corneal epithelial cell sheets. Sci Rep (2020) 10(1):9282. doi: 10.1038/s41598-020-64394-6
179. Hirsch T, Rothoeft T, Teig N, Bauer JW, Pellegrini G, De Rosa L, et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature. (2017) 551(7680):327–32. doi: 10.1038/nature24487
180. Guo W, Zhang X, Zhai J, Xue J. The roles and applications of neural stem cells in spinal cord injury repair. Front Bioeng Biotechnol (2022) 10:966866. doi: 10.3389/fbioe.2022.966866
181. Zhao L, Liu JW, Shi HY, Ma YM. Neural stem cell therapy for brain disease. World J Stem Cells (2021) 13(9):1278–92. doi: 10.4252/wjsc.v13.i9.1278
182. Garitaonandia I, Gonzalez R, Christiansen-Weber T, Abramihina T, Poustovoitov M, Noskov A, et al. Neural stem cell tumorigenicity and biodistribution assessment for phase I clinical trial in parkinson’s disease. Sci Rep (2016) 6:34478. doi: 10.1038/srep34478
183. Mazzini L, Gelati M, Profico DC, Sorarù G, Ferrari D, Copetti M, et al. Results from phase I clinical trial with intraspinal injection of neural stem cells in amyotrophic lateral sclerosis: a long-term outcome. Stem Cells Transl Med (2019) 8(9):887–97. doi: 10.1002/sctm.18-0154
184. Curtis E, Martin JR, Gabel B, Sidhu N, Rzesiewicz TK, Mandeville R, et al. A first-in-Human, phase I study of neural stem cell transplantation for chronic spinal cord injury. Cell Stem Cell (2018) 22(6):941–950.e6. doi: 10.1016/j.stem.2018.05.014
185. Genchi A, Brambilla E, Sangalli F, Radaelli M, Bacigaluppi M, Furlan R, et al. Neural stem cell transplantation in patients with progressive multiple sclerosis: an open-label, phase 1 study. Nat Med (2023) 29(1):75–85. doi: 10.1038/s41591-022-02097-3
186. Profico DC, Gelati M, Ferrari D, Sgaravizzi G, Ricciolini C, Projetti Pensi M, et al. Human neural stem cell-based drug product: clinical and nonclinical characterization. Int J Mol Sci (2022) 23(21):13425. doi: 10.3390/ijms232113425
187. Zou Y, Mu D, Ma X, Wang D, Zhong J, Gao J, et al. Review on the roles of specific cell-derived exosomes in alzheimer’s disease. Front Neurosci (2022) 16:936760. doi: 10.3389/fnins.2022.936760
188. Anderson AJ, Piltti KM, Hooshmand MJ, Nishi RA, Cummings BJ. Preclinical efficacy failure of human CNS-derived stem cells for use in the pathway study of cervical spinal cord injury. Stem Cell Rep (2017) 8(2):249–63. doi: 10.1016/j.stemcr.2016.12.018
189. D’Aiuto L, Zhi Y, Kumar Das D, Wilcox MR, Johnson JW, McClain L, et al. Large-Scale generation of human iPSC-derived neural stem cells/early neural progenitor cells and their neuronal differentiation. Organogenesis. (2014) 10(4):365–77. doi: 10.1080/15476278.2015.1011921
190. Kobold S, Guhr A, Mah N, Bultjer N, Seltmann S, Seiler Wulczyn AEM, et al. A manually curated database on clinical studies involving cell products derived from human pluripotent stem cells. Stem Cell Rep (2020) 15(2):546–55. doi: 10.1016/j.stemcr.2020.06.014
191. Schweitzer JS, Song B, Herrington TM, Park TY, Lee N, Ko S, et al. Personalized iPSC-derived dopamine progenitor cells for parkinson’s disease. N Engl J Med (2020) 382(20):1926–32. doi: 10.1056/NEJMoa1915872
192. Lee S, Huh JY, Turner DM, Lee S, Robinson J, Stein JE, et al. Repurposing the cord blood bank for haplobanking of HLA-homozygous iPSCs and their usefulness to multiple populations. Stem Cells Dayt Ohio. (2018) 36(10):1552–66. doi: 10.1002/stem.2865
193. Hanatani T, Takasu N. CiRA iPSC seed stocks (CiRA’s iPSC stock project). Stem Cell Res (2020) 50:102033. doi: 10.1016/j.scr.2020.102033
194. Popp B, Krumbiegel M, Grosch J, Sommer A, Uebe S, Kohl Z, et al. Need for high-resolution genetic analysis in iPSC: results and lessons from the ForIPS consortium. Sci Rep (2018) 8(1):17201. doi: 10.1038/s41598-018-35506-0
195. Li X, Zhang Y, Yan Y, Ciric B, Ma CG, Gran B, et al. Neural stem cells engineered to express three therapeutic factors mediate recovery from chronic stage CNS autoimmunity. Mol Ther (2016) 24(8):1456–69. doi: 10.1038/mt.2016.104
196. Velychko S, Kang K, Kim SM, Kwak TH, Kim KP, Park C, et al. Fusion of reprogramming factors alters the trajectory of somatic lineage conversion. Cell Rep (2019) 27(1):30–39.e4. doi: 10.1016/j.celrep.2019.03.023
197. Zhang M, Lin YH, Sun YJ, Zhu S, Zheng J, Liu K, et al. Pharmacological reprogramming of fibroblasts into neural stem cells by signaling-directed transcriptional activation. Cell Stem Cell (2016) 18(5):653–67. doi: 10.1016/j.stem.2016.03.020
198. Erharter A, Rizzi S, Mertens J, Edenhofer F. Take the shortcut - direct conversion of somatic cells into induced neural stem cells and their biomedical applications. FEBS Lett (2019) 593(23):3353–69. doi: 10.1002/1873-3468.13656
199. Périé S, Trollet C, Mouly V, Vanneaux V, Mamchaoui K, Bouazza B, et al. Autologous myoblast transplantation for oculopharyngeal muscular dystrophy: a phase I/IIa clinical study. Mol Ther (2014) 22(1):219–25. doi: 10.1038/mt.2013.155
200. Péault B, Rudnicki M, Torrente Y, Cossu G, Tremblay JP, Partridge T, et al. Stem and progenitor cells in skeletal muscle development, maintenance, and therapy. Mol Ther (2007) 15(5):867–77. doi: 10.1038/mt.sj.6300145
201. Dellavalle A, Sampaolesi M, Tonlorenzi R, Tagliafico E, Sacchetti B, Perani L, et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat Cell Biol (2007) 9(3):255–67. doi: 10.1038/ncb1542
202. Sampaolesi M, Torrente Y, Innocenzi A, Tonlorenzi R, D’Antona G, Pellegrino MA, et al. Cell therapy of alpha-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science. (2003) 301(5632):487–92. doi: 10.1126/science.1082254
203. Sampaolesi M, Blot S, D’Antona G, Granger N, Tonlorenzi R, Innocenzi A, et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature. (2006) 444(7119):574–9. doi: 10.1038/nature05282
204. Cossu G, Previtali SC, Napolitano S, Cicalese MP, Tedesco FS, Nicastro F, et al. Intra-arterial transplantation of HLA-matched donor mesoangioblasts in duchenne muscular dystrophy. EMBO Mol Med (2015) 7(12):1513–28. doi: 10.15252/emmm.201505636
205. Ausems CRM, van Engelen BGM, van Bokhoven H, Wansink DG. Systemic cell therapy for muscular dystrophies: the ultimate transplantable muscle progenitor cell and current challenges for clinical efficacy. Stem Cell Rev Rep (2021) 17(3):878–99. doi: 10.1007/s12015-020-10100-y
206. Cossu G, Tonlorenzi R, Brunelli S, Sampaolesi M, Messina G, Azzoni E, et al. Mesoangioblasts at 20: from the embryonic aorta to the patient bed. Front Genet (2022) 13:1056114. doi: 10.3389/fgene.2022.1056114
207. Tonlorenzi R, Dellavalle A, Schnapp E, Cossu G, Sampaolesi M. Isolation and characterization of mesoangioblasts from mouse, dog, and human tissues. Curr Protoc Stem Cell Biol (2007). doi: 10.1002/9780470151808.sc02b01s3
208. Lee-Wing M, Szwajcer D, Lockwood A, Flynn A, Anjos K, Tulloch M, et al. Manufacturing autologous myoblast for regenerative medicine applications. Cytotechnology. (2020) 72(5):605–14. doi: 10.1007/s10616-020-00420-9
Keywords: advanced therapy medicinal product (ATMP), potency, CAR (chimeric antigen receptor), T cell therapy, stem cell, tissue regeneration, biomarker
Citation: Capelli C, Cuofano C, Pavoni C, Frigerio S, Lisini D, Nava S, Quaroni M, Colombo V, Galli F, Bezukladova S, Panina-Bordignon P, Gaipa G, Comoli P, Cossu G, Martino G, Biondi A, Introna M and Golay J (2023) Potency assays and biomarkers for cell-based advanced therapy medicinal products. Front. Immunol. 14:1186224. doi: 10.3389/fimmu.2023.1186224
Received: 14 March 2023; Accepted: 24 May 2023;
Published: 09 June 2023.
Edited by:
Alasdair R. Fraser, University of Glasgow, United KingdomReviewed by:
Jyothi S. Prasanna, Manipal Academy of Higher Education, IndiaHong Lei, The Affiliated Children’s Hospital of Xi’an Jiaotong University, China
Copyright © 2023 Capelli, Cuofano, Pavoni, Frigerio, Lisini, Nava, Quaroni, Colombo, Galli, Bezukladova, Panina-Bordignon, Gaipa, Comoli, Cossu, Martino, Biondi, Introna and Golay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martino Introna, bWludHJvbmFAYXNzdC1wZzIzLml0; Josée Golay, amdvbGF5NTlAZ21haWwuY29t
†These authors have contributed equally to this work