 Man Wang
Man Wang Fei Yu
Fei Yu Wenguang Chang
Wenguang Chang Yuan Zhang
Yuan Zhang Lei Zhang
Lei Zhang Peifeng Li
Peifeng Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 12 May 2023
Sec. Viral Immunology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1185233
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a contagious respiratory virus that is the cause of the coronavirus disease 2019 (COVID-19) pandemic which has posed a serious threat to public health. COVID-19 is characterized by a wide spectrum of clinical manifestations, ranging from asymptomatic infection to mild cold-like symptoms, severe pneumonia or even death. Inflammasomes are supramolecular signaling platforms that assemble in response to danger or microbial signals. Upon activation, inflammasomes mediate innate immune defense by favoring the release of proinflammatory cytokines and triggering pyroptotic cell death. Nevertheless, abnormalities in inflammasome functioning can result in a variety of human diseases such as autoimmune disorders and cancer. A growing body of evidence has showed that SARS-CoV-2 infection can induce inflammasome assembly. Dysregulated inflammasome activation and consequent cytokine burst have been associated with COVID-19 severity, alluding to the implication of inflammasomes in COVID-19 pathophysiology. Accordingly, an improved understanding of inflammasome-mediated inflammatory cascades in COVID-19 is essential to uncover the immunological mechanisms of COVID-19 pathology and identify effective therapeutic approaches for this devastating disease. In this review, we summarize the most recent findings on the interplay between SARS-CoV-2 and inflammasomes and the contribution of activated inflammasomes to COVID-19 progression. We dissect the mechanisms involving the inflammasome machinery in COVID-19 immunopathogenesis. In addition, we provide an overview of inflammasome-targeted therapies or antagonists that have potential clinical utility in COVID-19 treatment.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent responsible for the ongoing coronavirus disease 2019 (COVID-19) pandemic (1). SARS-CoV-2 is an enveloped single-stranded positive-sense RNA (+ssRNA) virus with a genome of approximately 30 kb (2) (Figure 1). Like other coronaviruses, SARS-CoV-2 virions are comprised of four major structural proteins, spike (S), membrane (M), nucleocapsid (N) and envelope (E) proteins (3). The viral genome contains various open reading frames (ORFs) encoding for nonstructural proteins (NSPs, e.g., 3-chymotrypsin-like cysteine (3CL) protease and RNA-dependent RNA polymerase) and the four essential structural proteins (4). SARS-CoV-2 is principally transmitted via respiratory droplets, aerosols, and to a lesser extent, direct contact with fomites from infected people (5). Albeit the respiratory system is the primary target of SARS-CoV-2, this virus can also damage other vital organs including brain, gut, heart and kidney (6, 7). Typical manifestations of COVID-19 encompass cough, fatigue, fever, headache and shortness of breath (8). Most SARS-CoV-2-infected individuals have mild symptoms such as cough and fever, while some experience severe pneumonia and acute respiratory distress syndrome (ARDS) with systemic inflammation (9). The systemic inflammatory syndrome is triggered by over-activation of innate immune system and intense proinflammatory cytokine responses (10). Generally, mild cases of COVID-19 can be cured using over-the-counter medications (e.g., paracetamol), but patients with severe COVID-19 have a high risk of life-threatening complications and death due to a lack of specific therapeutic options (11). The nature of host immune response to SARS-CoV-2 could have a tremendous impact on COVID-19 progression. Elucidating the immunological mechanisms responsible for disease severity should be a focus of SARS-CoV-2 research with the aim of improving clinical outcomes in COVID-19 patients.
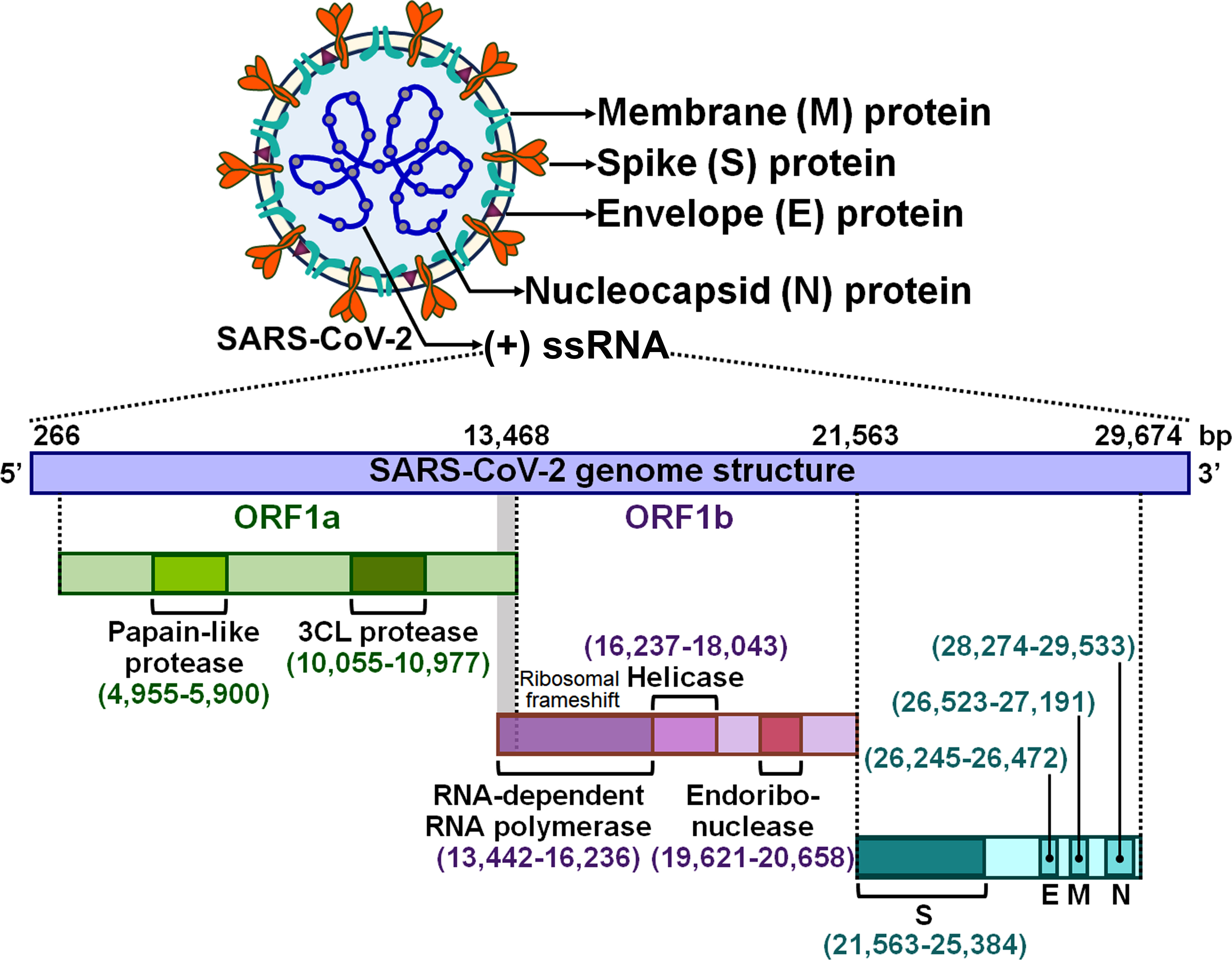
Figure 1 Schematic representation of the SARS-CoV-2 virion and genomic structure. SARS-CoV-2 is an enveloped +ssRNA virus with a genome of 29,674 bp. The SARS-CoV-2 virion has four main structural proteins, which are spike (S), envelope (E), membrane (M) and nucleocapsid (N) glycoproteins. The viral genome contains several ORFs that encode nonstructural proteins and structural proteins. Two large overlapping ORFs (ORF1a and ORF1b) occupy two-thirds of the viral genome at the 5’-end. ORF1a encodes papain-like protease and 3CL protease, while ORF1b encodes RNA-dependent RNA polymerase, helicase and endoribonuclease. A third of the genome at the 3’-end encodes the four essential structural proteins (S, E, M and N). +ssRNA, single-stranded positive-sense RNA; ORF, open reading frame; 3CL protease, 3-chymotrypsin-like protease; S, spike; E, envelope; M, membrane; N, nucleocapsid.
Established studies have proven that SARS-CoV-2 represents a pathogen-associated molecular pattern (PAMP) able to trigger inflammasome activation (12, 13). Inflammasomes are macromolecular immune complexes formed in the cytosol in response to an array of stimuli (14). The assembly of inflammasomes is initiated by pattern-recognition receptors (PRRs) following the detection of endogenous or pathogenic danger signals (15). Activated inflammasomes fuel the inflammatory caspase to induce pyroptosis and cytokine production, thereby removing virus-infected cells from the host (16–18). However, aberrant inflammasome activation can drive metainflammation and bring about deleterious consequences. Currently, considerable attention has been paid to the involvement of inflammasomes in the pathogenesis of various diseases such as cancer, cardiovascular disorders and diabetes (19–21). Aberrant inflammasome activation and concomitant cytokine burst also occur in COVID-19 patients (22, 23). More importantly, COVID-19 progression and severity have been closely correlated with the active status of inflammasomes and the levels of proinflammatory cytokines including interleukin (IL)-1β and IL-18 (24, 25), stressing the potential role of inflammasomes in the pathophysiology of COVID-19-associated systemic complications. Recent studies have contributed towards an advanced understanding of the interaction between SARS-CoV-2 and inflammasomes throughout the course of infection. Here, we review the literature concerning the mechanisms behind SARS-CoV-2-mediated regulation of inflammasome activation, the role of inflammasomes in COVID-19 immunopathogenesis and the potential mechanisms that coordinate inflammasome-directed pathologies in COVID-19. In addition, we discuss the therapeutic potential of inflammasome-centered therapies. Future research into inflammasome activity will provide new perspectives for illuminating the pathological processes underlying COVID-19 progression and facilitate the development of more targeted therapeutics for this infectious disease.
Inflammasomes are intracellular multimeric innate immune rheostats that trigger inflammation in response to multiple physiological or pathogenic factors (26). Inflammasomes mediate the proteolytic maturation of proinflammatory cytokines IL-1β and IL-18, culminating in an expeditious inflammatory form of programmed cell death termed pyroptosis (27). The inflammasomes can be categorized into canonical and noncanonical inflammasomes (28) (Figure 2). Canonical inflammasomes encompassing absent in melanoma 2 (AIM2), nucleotide-binding oligomerization domain (NOD)-like receptor family pyrin domain (PYD)-containing protein 1 (NLRP1), NLRP3, NOD-like receptor (NLR) family caspase recruitment domain (CARD)-containing protein 4 (NLRC4) and pyrin can motivate caspase-1 (29). Noncanonical inflammasomes serve as platforms for the activation of caspase-4/5 (human) or caspase-11 (mouse) (30). Canonical inflammasomes typically consist of three major components: an upstream cytosolic sensor (e.g., PRR), an adaptor protein (e.g., apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC)) and a downstream effector protein (e.g., procaspase-1) (31). The structures of canonical inflammasomes are similar, and the main discrepancy lies in the diversity of cytosolic sensors (32). Cytosolic PRRs expressed by innate immune cells include C-type lectin receptors (CLRs), NLRs and Toll-like receptors (TLRs) that can sense damage-associated molecular patterns (DAMPs) or PAMPs to prime inflammasome assembly (33). The sensor proteins NLRP1, NLRP3 and NLRC4, which form canonical inflammasomes, are members of the NLR family, while AIM2 belongs to the ALR family (34). The sensor proteins react to a multitude of signals. NLRP1 is able to detect anthrax lethal toxin and muramyl dipeptide (35, 36). NLRP3, one of the most studied sensor proteins, responds to a number of agonists including adenosine triphosphate (ATP), nucleic acids, bacteria, fungi and viruses (37). NLRC4 senses bacterial flagellin and type III secretion apparatus in the cytosol (38, 39). AIM2 is activated by cytosolic double-stranded DNA (dsDNA) (40), while pyrin recognizes inactivating modifications of host Rho GTPases by diverse bacterial effectors or toxins, including adenylylation, deamidation and glucosylation (41). CARD-carrying PRRs (e.g., NLRP1 and NLRC4) directly recruit procaspase-1 to initiate inflammasome assembly. In ASC-independent inflammasomes, caspase-1 is activated via dimerization-induced autoproteolysis (42). ASC, which contains an N-terminal PYD and a C-terminal CARD, serves as a scaffold protein bridging PYD-containing PRRs (e.g., AIM2, NLRP3 and pyrin) and procaspase-1 (43). Upon activation, PYD-carrying PRRs couple to ASC via homotypic interactions and drive the supramolecular assembly of ASC, leading to the formation of ASC specks (44). ASC specks subsequently recruit inactive zymogen procaspase-1 via CARD-CARD interactions, which brings monomers of procaspase-1 in close proximity and facilitates its autoproteolysis (45). Active caspase-1 not only proteolytically processes the biologically inactive procytokines (pro-IL-1β and pro-IL-18) into the active mature forms, but also cleaves gasdermin D (GSDMD) to unmask its N-terminal pore-forming domain (GSDMD-NT) (46, 47). GSDMD-NT perforates the cellular membrane and induces pyroptosis, leading to the secretion of proinflammatory cytokines into the extracellular milieu (48). The canonical inflammasome-dependent pyroptosis constitutes a significant innate defense mechanism against pathogen invasion. In the noncanonical inflammasome pathway, procaspase-4/5/11 can directly bind to bacterial lipopolysaccharide (LPS) to activate their proteolytic activity (49). Caspase-4/5/11 cleave GSDMD to induce pyroptosis (46, 50). Caspase-11 is capable of stimulating the NLRP3 inflammasome by fostering potassium (K+) efflux (51). After that, active caspase-1 promotes the maturation of proinflammatory cytokines, which can be released to the outside of the cell through GSDMD-NT pores.
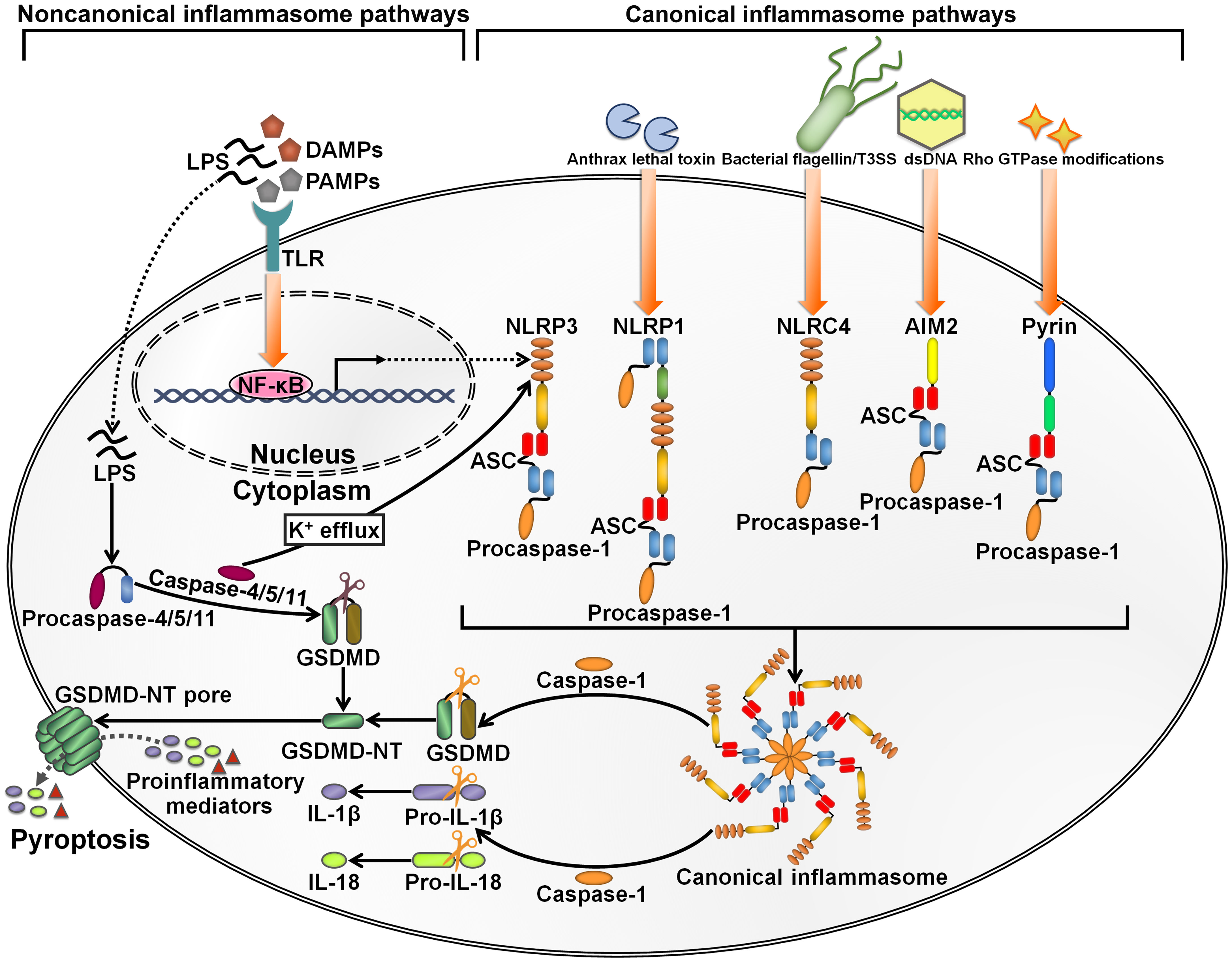
Figure 2 Schematic illustration of key inflammasome pathways. Canonical inflammasomes are multimeric protein complexes in the cytosol that can be activated by diverse signals. They are generally composed of a cytosolic sensor (e.g., PRR), an adaptor protein (ASC) and an effector protein (e.g., procaspase-1). NLRP3 inflammasome activation is caused by a number of DAMPs and PAMPs, such as ATP, nucleic acids, bacteria, fungi and viruses. NLRP1 is mainly triggered by anthrax lethal toxin and muramyl dipeptide. NLRC4 is activated by bacterial flagellin and type III secretion apparatus. AIM2 typically responds to cytosolic dsDNA. Pyrin detects inactivating modifications of host Rho GTPases by diverse bacterial effectors or toxins. The assembly of canonical inflammasomes leads to caspase-1 activation. Caspase-1 then cleaves GSDMD to unleash its N-terminal pore-forming domain (GSDMD-NT). GSDMD-NT punches holes in the cellular membrane, which induces water influx, cell swelling and cytomembrane rupture, ultimately leading to the occurrence of an inflammatory cell death termed pyroptosis. Caspase-1 also mediates the conversion of pro-IL-1β and pro-IL-18 into their active forms. The bioactive IL-1β and IL-18 are released into the extracellular space via the GDDMD-NT pores. Cell lysis further enhances the inflammatory signaling through extravasation of DAMPs. The noncanonical inflammasome pathway is initiated by LPS from extracellular Gram-negative bacteria. LPS directly binds to procaspase-4/5 (human) or procaspase-11 (mouse) to induce their activation. Active caspase-4/5/11 cleave GSDMD into GSDMD-NT, hence actuating the pyroptosis program. Furthermore, caspase-4/5/11 can motivate the NLRP3 inflammasome pathway for inflammatory cytokine processing by promoting K+ efflux. LPS, lipopolysaccharide; DAMPs, damage-associated molecular patterns; PAMPs, pathogen-associated molecular patterns; TLR, Toll-like receptor; NF-κB, nuclear factor-κB; GSDMD, gasdermin D; GSDMD-NT, the N-terminal fragment of gasdermin D; IL-1β, interleukin-1β; IL-18, interleukin-18; NLRP3, nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing protein 3; ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain; NLRP1, nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing protein 1; NLRC4, nucleotide-binding oligomerization domain-like receptor family caspase recruitment domain-containing protein 4; dsDNA, double-stranded DNA; AIM2, absent in melanoma 2.
Inflammasomes play a double-edged role in viral infection. Appropriate activation of inflammasomes instigates innate antiviral responses, but the dysregulation of the inflammasome signaling due to defective host immunity or comorbidities can lead to the cytokine storm and hyperinflammation. NLRP3 has been detected in various cell types, including innate immune cells, lung epithelial cells and cardiac cells (52). It is thus inferred that SARS-CoV-2 infection could cause NLRP3 inflammasome activation in these cells.
Reportedly, NLRP3 inflammasome was activated in peripheral blood mononuclear cells (PBMCs) from COVID-19 patients (12). The levels of active caspase-1 and IL-18 were higher in the sera of COVID-19 patients than the controls (Table 1). The serum concentration of active caspase-1 showed a positive association with the levels of C-reactive protein (CRP), IL-6, IL-18 and lactate dehydrogenase (LDH). Moreover, NLRP3 inflammasome activation had an impact on the clinical outcome of COVID-19. Particularly, the level of active caspase-1 was elevated as illness severity increased. The level of IL-18 was higher in patients who required mechanical ventilation than patients who did not. Likewise, lethal cases had an increased level of IL-18 relative to survivors. Since caspase-1-independent pathway also gives rise to enhanced IL-18 secretion (61), the relationship between NLRP3 inflammasome activation and COVID-19 development is an important subject to be further addressed. The robust activation of NLRP3 inflammasome may aggrandize inflammation in patients with severe COVID-19, culminating in worse clinical results. The NLRP3 inflammasome could be used as a prospective predictor of COVID-19 severity and outcomes. Blockade of inflammasome pathways represents a promising approach to treat patients with severe COVID-19. An improved understanding of activated inflammasome complexes during the course of COVID-19 will offer new opportunities for therapeutic intervention.
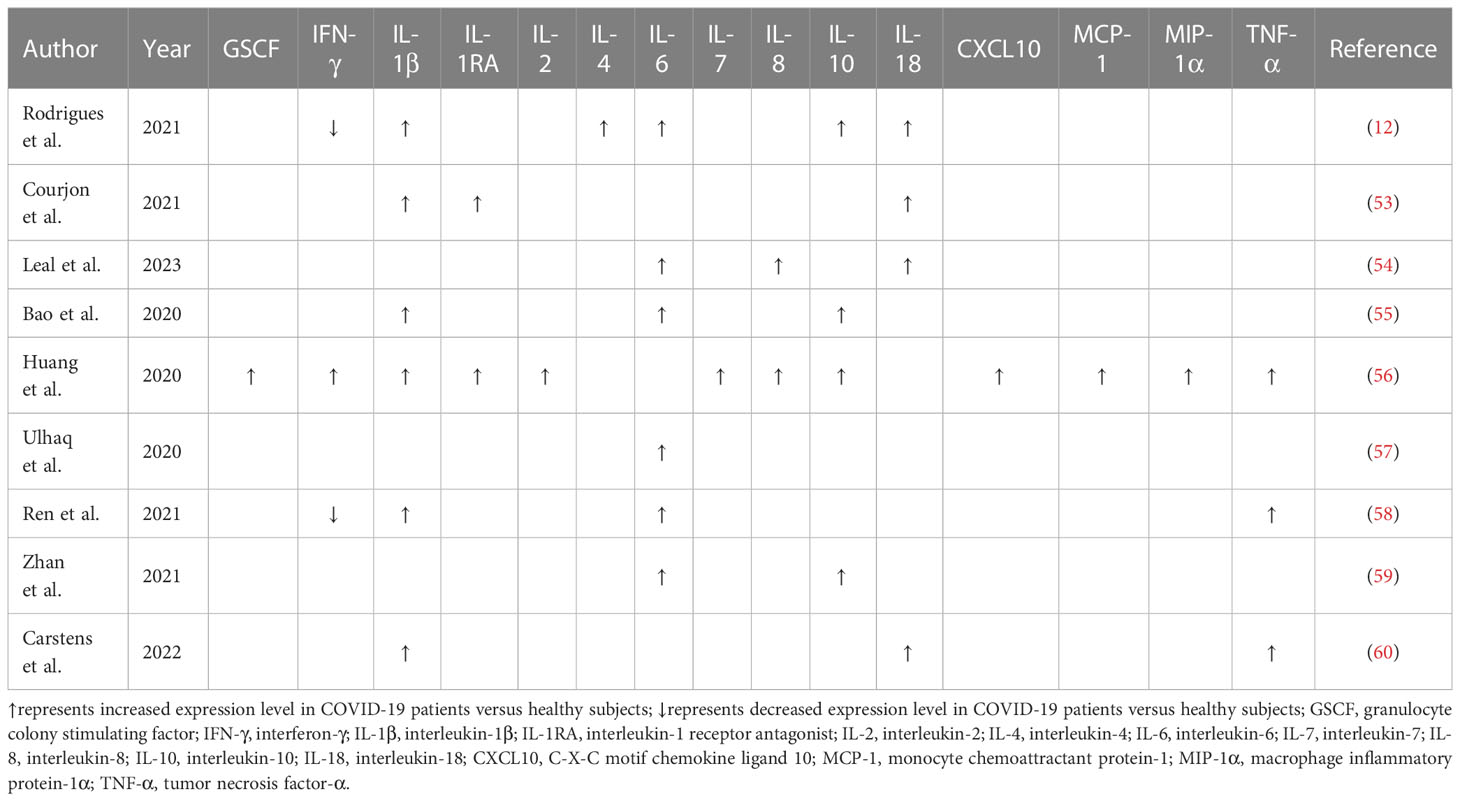
Table 1 Overview of altered cytokine profiles in COVID-19 patients.
Myeloid cells are a vital cellular compartment of innate immunity and consist of monocytes, macrophages and neutrophils (62). Myeloid cells isolated from COVID-19 patients showed heterogeneous NLRP3 inflammasome activation potentials (53). Opposed potentials of caspase-1 activation were observed between nonclassical monocytes and immature neutrophils from severe/critical COVID-19 patients upon NLRP3 inflammasome stimulation. NLRP3 inflammasome activation of nonclassical monocytes was elevated with increasing COVID-19 severity, while CD66b+CD16dim granulocytes exhibited lower NLRP3 activation potential in severe forms of COVID-19 than mild cases. The number of nonclassical monocytes was remarkably decreased in severe to critical patients compared with healthy controls, while immature neutrophils exhibited the opposite trend. In recovered patients, caspase-1 activation in CD66b+CD16dim granulocytes was restored and the percentage of immature neutrophils was comparable to the controls. Altogether, the NLRP3 inflammasome signatures in myeloid cells may represent a predictive biomarker for COVID-19 development and patient outcome and provide important guidance for therapeutic decisions involving immunoregulatory agents.
Monocytes and macrophages act as sentinel cells that detect SARS-CoV-2 infection to assemble inflammasomes (e.g., AIM2 and NLRP3), resulting in pyroptosis and the extravasation of proinflammatory mediators (63). CD14highCD16- monocytes derived from severe COVID-19 patients showed increased NLRP3 inflammasome activation potential, as shown by the formation of ASC speck/caspase-1 complexes and increased IL-1β secretion (64). Monocytes from critically ill patients with COVID-19 exhibited higher caspase-1 activation than those from healthy controls (13). Accordingly, intense monocyte death was documented in critically ill patients. The plasma concentration of IL-1β was higher in critically ill patients than healthy controls. Inflammasome activation in monocytes caused massive production of proinflammatory cytokines, leading to hyperinflammation in severe COVID-19. These findings implied that COVID-19 deterioration was associated with inflammasome activation in monocytes. Monocytes may be pivotal cells in the detrimental proinflammatory cascades that reflect COVID-19 progression and severity. SARS-CoV-2 infection instigated the NLRP3 inflammasome pathway in human lung macrophages (24). MCC950, a selective inhibitor of NLRP3, could dampen the release of proinflammatory cytokines and chemokines; in parallel, it reduced inflammation and reversed chronic lung pathology in SARS-CoV-2-infected mice. In addition, SARS-CoV-2 open reading frame 3a (ORF3a) protein was able to activate the NLRP3 inflammasome in LPS-primed macrophages, and it could promote the expression of pro-IL-1β via nuclear factor-κB (NF-κB) (65). A recent study indicated that NLRP3 inflammasome was hyperactivated in neutrophils from severe COVID-19 patients (54). Severe COVID-19 neutrophils exhibited impaired responsiveness to NLRP3 insults (e.g., ATP and bacterial LPS). These NLRP stimuli failed to induce the expression of NLRP3 inflammasome components, which might involve an epigenetic mechanism. The presence of numerous immature neutrophils may be an explanation for COVID-19 neutrophil dysfunction. The reduced neutrophilic responsiveness towards common NLRP3 stimuli could predispose severe COVID-19 patients to secondary pathogen infections. The mechanism responsible for alterations in neutrophilic response to NLRP3 stimulation necessitates considerable attention. It will be of paramount importance to clarify the causal link between neutrophil impairment and COVID-19 immunopathogenesis.
Notably, circulatory exosomes from severe COVID-19 patients were able to increase the expression of NLRP3, caspase-1 and IL-1β in endothelial cells (66). COVID-19 exosomes markedly promoted IL-1β secretion. These results demonstrated that exosomes from COVID-19 plasma stimulated NLRP3 inflammasome in distant endothelial cells, leading to amplified inflammatory reactions. Endothelial cells function as a selectively permeable barrier to blood and dominate the transfer of proteins from the circulation to tissues (67). Endothelial cells act to oppose inflammation. Dysfunction of endothelial cells can give rise to multiorgan damage that is a general characteristic of viral infections (68). Exosomes act as a crucial mediator for endothelial cell dysfunction and multiorgan inflammation during SARS-CoV-2 infection. As exosomes carry a number of bioactive materials, it is intriguing which components have the ability to ignite NLRP3 inflammasome. The mechanism responsible for COVID-19 exosome-induced NLRP3 inflammasome activation is worthy of more detailed investigation.
Accumulating evidence has indicated the complex interaction between SARS-CoV-2 and inflammasomes. At the initial stage of infection, SARS-CoV-2 and its derived products stimulate inflammasome formation, inducing inflammatory responses to fight against viral infection. SARS-CoV-2 employs diverse strategies to hinder inflammasome activation, ensuring a beneficial environment for its replication. After that, SARS-CoV-2 inflames the inflammatory reaction to propel viral pathogenicity. Highly sophisticated mechanisms involving various viral components contribute to regulation of inflammasome functionality during SARS-CoV-2 infection (Figure 3).
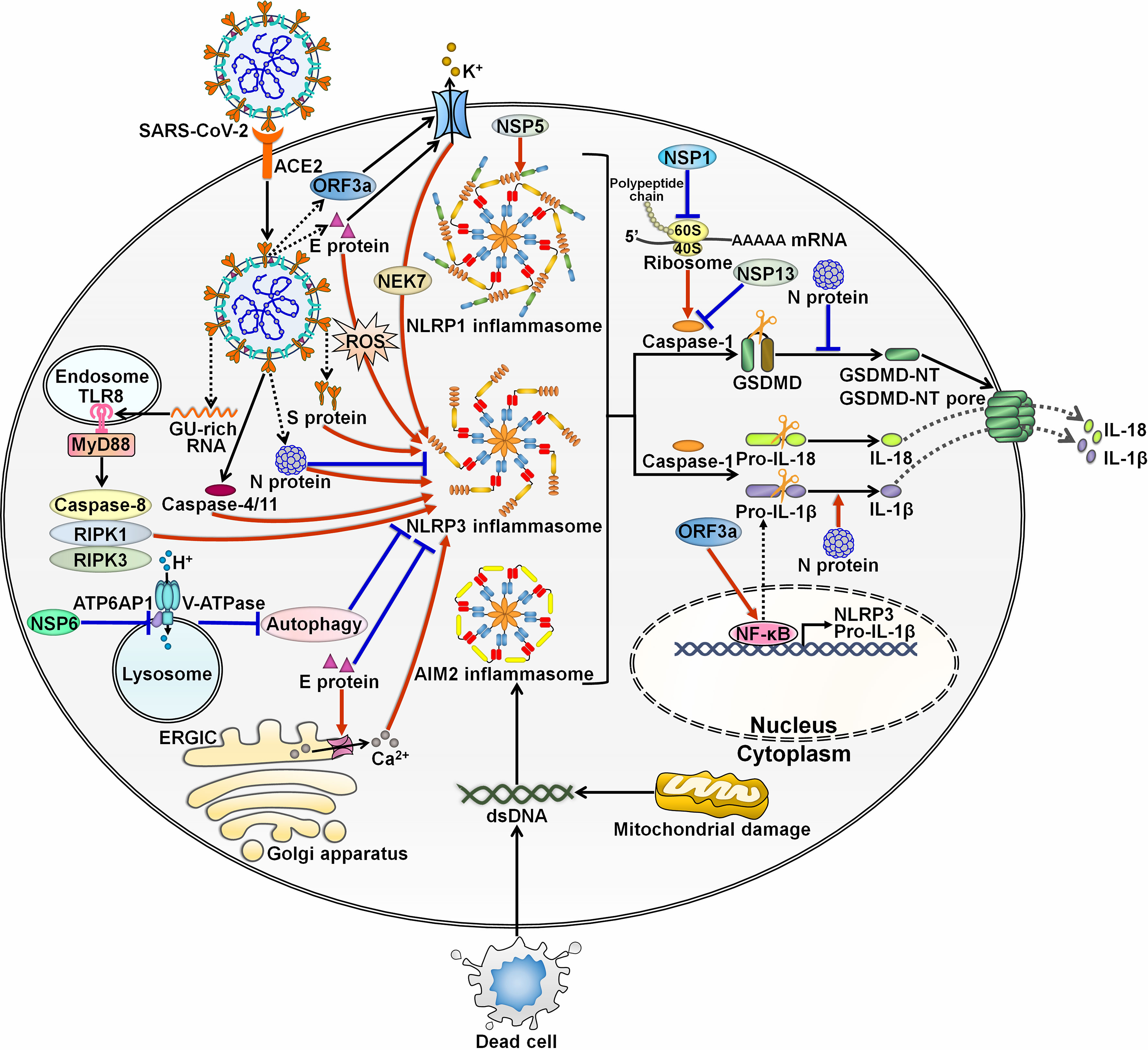
Figure 3 Regulation of inflammasome pathways by SARS-CoV-2. SARS-CoV-2 infection triggers the assembly of inflammasomes. These molecular platforms impel caspase-1 activation, hence promoting the extracellular secretion of IL-1β and IL-18. SARS-CoV-2 has evolved a multitude of mechanisms to interfere with inflammasome activity. Viral ORF3a and E proteins can activate NLRP3 inflammasome by inducing K+ efflux. ORF3a increases the expression of pro-IL-1β by motivating the NF-κB signaling cascade. E protein triggers the formation of Ca2+ ion channels in ERGIC/Golgi membranes and initiates ROS-dependent activation of NLRP3 inflammasome. Paradoxically, E protein is found to exert inhibitory effects on NLRP3 inflammasome priming by blunting ER stress. S protein acts as an agonist of NLRP3 inflammasome. The GU-rich RNA derived from S protein actuates NLRP3 inflammasome through the caspase-8/RIPK1/RIPK3 signaling pathway. SARS-CoV-2 infection can induce NLRP3 inflammasome formation by upregulating caspase-4/11. N protein plays dual roles in regulating NLRP3 inflammation. N protein favors the assembly of NLRP3 inflammasome and drives the maturation of the proinflammatory cytokine IL-1β. N protein blocks the NLRP3 inflammasome signaling by preventing caspase-1-mediated GSDMD activation. NSP6 suppresses lysosome acidification through direct interaction with ATP6AP1, causing autophagic flux stagnation to activate NLRP3 inflammasome. NSP1 and NSP13 repress NLRP3 inflammasome activation and IL-1β maturation via attenuation of caspase-1 activity. Specifically, NSP1 impedes host translation by binding to the 40S ribosome subunit, which is required for caspase-1 inhibition. Notably, viral NSP5 can induce NLRP1 inflammasome activation. SARS-CoV-2 infection also contributes to the assembly of AIM2 inflammasome. SARS-CoV-2-induced excessive cell death results in the liberation of endogenous DNA. Moreover, SARS-CoV-2 may favor the release of mtDNA into the cytoplasm by impairing mitochondrial membranes. Either free DNA or mtDNA can activate AIM2 inflammasome. SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; ACE2, angiotensin-converting enzyme 2; ORF3a, open reading frame 3a; E protein, envelope protein; TLR8, Toll-like receptor 8; MyD88, myeloid differentiation factor 88; RIPK1, receptor-interacting protein kinase 1; RIPK3, receptor-interacting protein kinase 3; NSP6, nonstructural protein 6; ATP6AP1, ATPase H+ transporting accessory protein 1; ERGIC, endoplasmic reticulum Golgi apparatus intermediate compartment; ROS, reactive oxygen species; S protein, spike protein; N protein, nucleocapsid protein; NSP5, nonstructural protein 5; NEK7, NIMA-related kinase 7; NLRP1, nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing protein 1; NLRP3, nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing protein 3; AIM2, absent in melanoma 2; dsDNA, double-stranded DNA; NSP1, nonstructural protein 1; NSP13, nonstructural protein 13; GSDMD, gasdermin D; GSDMD-NT, the N-terminal fragment of gasdermin D; IL-1β, interleukin-1β; IL-18, interleukin-18; NF-κB, nuclear factor-κB.
The inflammasome signaling performs important functions in pathogen defense. It is conceived that SARS-CoV-2 has evolved multiple mechanisms to counteract inflammasome-dependent cascades, thereby allowing for viral survival and pathogenesis. SARS-CoV-2 N protein was released into the cytosol immediately after viral entry into the host cell (69). N protein inhibited pyroptotic cell death in infected human monocytes by binding to GSDMD linker region and impeding caspase-1-mediated GSDMD activation. This event prevented the release of immune signaling factors into the extracellular milieu. Because of GSDMD inhibition, excessive proinflammatory cytokines might accumulate in the host cytosol. The release of massive proinflammatory cytokines could result in severe disease in COVID-19 patients due to the lytic infection of SARS-CoV-2. The interfaces between viral N protein and GSDMD were diverse, and both the N-terminal and C-terminal domains of GSDMD were required for the suppression of GSDMD activation, suggesting a higher tertiary structure employed by N protein to seize GSDMD. The impact of SARS-CoV-2 N protein on the inflammatory response and COVID-19 progression should be further explored. SARS-CoV-2 E protein had the ability to inhibit endoplasmic reticulum (ER) stress and NLRP3 inflammasome priming in macrophages (70). In mice treated with poly(I:C) to simulate the effects of viral infection, E protein decreased the expression levels of IL-1β and IL-18 in bronchoalveolar lavage fluid (BALF) and suppressed macrophage infiltration in the lung tissues. Thus, E protein prevented NLRP3 inflammasome activation and pulmonary inflammation. Two early products of SARS-CoV-2 infection, NSP1 and NSP13, were revealed to suppress NLRP3 inflammasome activation and IL-1β maturation via reduction of caspase-1 activity (71). Mechanistic investigation indicated that NSP1 induced RNA cleavage and suppressed host translation by binding to the 40S ribosome subunit, which was required for caspase-1 inhibition. NSP1 potentially prevented NLRP3 inflammasome assembly by downregulating its constituents, including NLRP3, ASC and caspase-1. As for NSP13, its N-terminal zinc-binding domain (ZBD) and C-terminal helicase domain were critical for caspase-1 inhibition. The mechanisms through which SARS-CoV-2 NSPs inhibit NLRP3 inflammasome merit further study. Altogether, early inhibition of inflammasome activation renders SARS-CoV-2 to evade host immune surveillance and may be beneficial for continued viral infection leading to enhanced pathogenicity and potential detrimental effects to the host. These results were in line with the previous report indicating reduced inflammatory responses as a predominant effect of early SARS-CoV-2 infection (72). Further investigation of the intracellular activities of viral early products would shed new light on the detailed mechanisms governing SARS-CoV-2 pathogenesis.
Intracellular ionic imbalance is a critical inducer of NLRP3 inflammasome activation (37). Viral proteins, which were translated during the late stage of SARS-CoV-2 infection, were able to induce NLRP3 inflammasome complex by fostering intracellular ionic disturbances (71). Intriguingly, SARS-CoV-2 E protein and ORF3a possess ion channel activity and contribute to ionic imbalances within infected cells. Calcium (Ca2+) flux is an important signaling event for NLRP3 inflammasome activation (73). E protein formed Ca2+ ion channels in ER Golgi apparatus intermediate compartment (ERGIC)/Golgi membranes and impelled reactive oxygen species (ROS)-dependent activation of NLRP3 inflammasome at the later stages of viral infection, which might contribute to advanced or complicated diseases (17, 74). It can be concluded that E protein plays a dichotomous role in NLRP3 inflammasome activation during viral infection. SARS-CoV-2 viroporin ORF3a primed NLRP3 inflammasome, inducing GSDMD activation and NF-κB-driven IL-1β expression (75). Depletion of NLRP3 abrogated ORF3a-driven caspase-1 cleavage, hinting the activation of NLRP3 inflammasome by ORF3a. NIMA-related kinase 7 (NEK7) was a critical mediator of NLRP3 assembly downstream of K+ efflux (76). Knockdown of NEK7 curtailed ORF3a protein’s ability to motivate NLRP3 inflammasome (75). Consistently, inhibiting K+ efflux obliterated ORF3a-mediated caspase-1 activation. Further study indicated that ORF3a facilitated the generation of inward-rectifier K+ channels in cellular membrane to drive K+ efflux, thereby inducing the oligomerization between NEK7 and NLRP3. This event contributed to the ignition of NLRP3 inflammasome. Similar to E protein, viral ORF3a also induced mitochondrial injury and drove mitochondrial ROS production, which potentially actuated NLRP3 inflammasome assembly and proinflammatory response (75, 77).
Human caspase-4 was shown to activate the NLRP3 inflammasome by promoting K+ efflux (78). A recent study indicated that SARS-CoV-2 infection induced NLRP3 activation by upregulating caspase-4 (79). The expression of caspase-4 in the lungs of COVID-19 patients showed an association with the levels of inflammasome activation markers such as caspase-1, IL-1β, IL-6 and IL-18. Deletion of caspase-11, the murine counterpart of human caspase-4, impeded disease development and reduced mortality in mice. Collectively, caspase-4 acted as a master factor mediating NLRP3 inflammasome activation and COVID-19 pathology. It is proposed that caspase-4 may be involved in viral protein-induced NLRP3 inflammasome activation. This hypothesis merits further validation. Increasing evidence indicated that SARS-CoV-2 S protein served as an inflammasome agonist (80–82). S protein selectively promoted inflammasome formation and IL-1β secretion in macrophages isolated from COVID-19 patients but not in non-COVID-19 subject-derived macrophages (83). Intriguingly, inflammasome activation was primed and the formation of IL-1β was robustly increased in macrophages from convalescent COVID-19 patients upon S protein stimulation, suggesting SARS-CoV-2-specific innate immune memory following recovery, which declined over time. Notably, S protein caused the reprogramming of human macrophages from convalescent subjects, as evidenced by the induction of inflammation- and cytokine-related pathways and inflammation/myeloid cell activation-associated epigenetic histone modifications. Such signatures and corresponding intercellular environment could permit long-lived and selective priming of the inflammasome pathway in SARS-CoV-2-infected individuals. Collectively, SARS-CoV-2 S protein acts as an ideal target against hyperinflammation in COVID-19 patients. The GU-rich single-stranded RNA (GU-rich RNA) derived from SARS-CoV-2 S protein actuated NLRP3 inflammasome and promoted the secretion of IL-1β, IL-6 and tumor necrosis factor (TNF) from human macrophages in the absence of pyroptotic cell death (84). Mechanistically, K+ efflux was required for GU-rich RNA-mediated NLRP3 inflammasome activation. The effect of GU-rich RNA on NLRP3 inflammasome activation also engaged the caspase-8/receptor-interacting protein kinase 1 (RIPK1)/RIPK3 signaling pathway. The TLR8/myeloid differentiation factor 88 (MyD88) pathway was necessary for SARS-CoV-2 GU-rich RNA-stimulated IL-1β maturation. Consistently, endosomal acidification blocker chloroquine prevented GU-rich RNA-induced cytokine secretion by inhibiting endosomal TLR signaling (Table 2). Alternative inflammasomes may contribute to cytokine burst in macrophages, which warrants additional study. It has been established that inflammasome activation and IL-1β release are not necessarily coupled to the occurrence of pyroptosis (99). Reportedly, GU-rich RNA-treated human macrophages had high expression levels of triggering receptor expressed on myeloid cells 1 (TREM1) that was essential for macrophage survival under certain circumstances (100). Human macrophages could enhance TREM1 expression to antagonize GU-rich RNA-induced pyroptosis. This may be an explanation for the enrichment of macrophages in the BALF from severe COVID-19 patients (101). IL-1 receptor-associated kinases (IRAKs) are integral to TLR-dependent activation of NLRP3 inflammasome and subsequent cytokine production (102). Pacritinib exerted inhibitory effects on TLR8-dependent inflammatory responses elicited by GU-rich RNA, contributing to the suppression of proinflammatory cytokine release from human macrophages (85). Mechanistically, pacritinib blocked IRAK1 phosphorylation and ubiquitination to suppress its association with the transforming growth factor β-activated kinase 1 (TAK1) complex, hence inhibiting TLR8-dependent NLRP3 inflammasome assembly and proinflammatory cytokine response. The anti-inflammatory activity of pacritinib in the clinical setting remains to be further studied. SARS-CoV-2 NSP6 induced NLRP3 inflammasome-mediated caspase-1 activation, IL-1β and IL-18 production, and pyroptotic cell death in lung epithelial cells (103). Mechanistically, NSP6 inhibited lysosome acidification through direct interaction with ATPase H+ transporting accessory protein 1 (ATP6AP1), thus resulting in autophagic flux stagnation to activate NLRP3 inflammasome. Pharmacological restoration of autophagic flux dampened NSP6-mediated NLRP3 inflammasome activation. The dysfunction of autophagic flux has been associated with the priming of AIM2 inflammasome (104). It would be rewarding to investigate the potential engagement of alternative inflammasome pathways in the pathogenicity of NSP6. The role of other viral NSPs in stimulating inflammasome activation also deserves special attention.
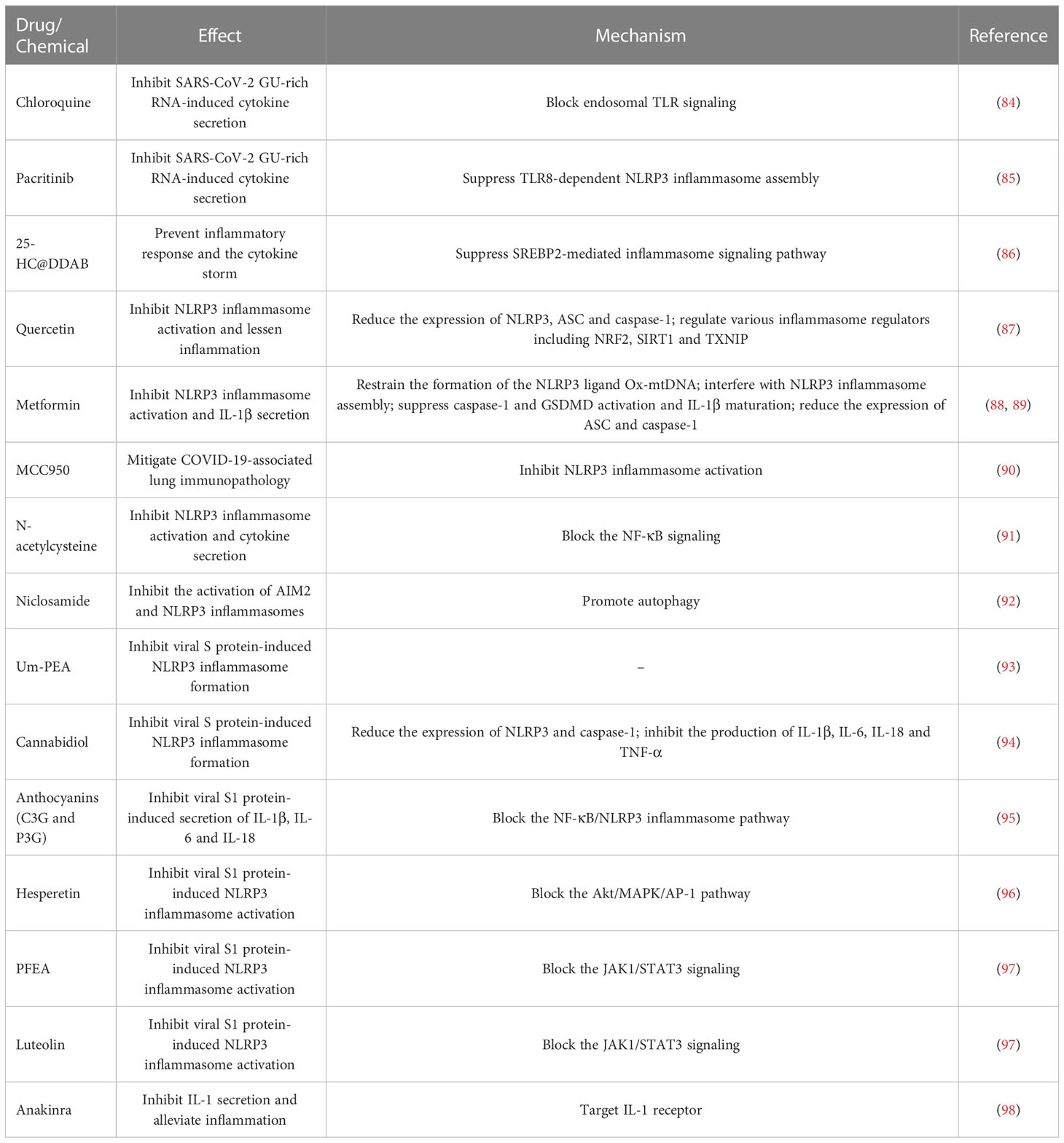
Table 2 Overview of inflammasome inhibitors and their mechanisms of action.
SARS-CoV-2 N protein served as an activator of NLRP3 inflammasome and could trigger excessive inflammatory responses (105). In terms of mechanism, N protein directly bound to NLRP3, fostered the interaction between NLRP3 and ASC, and primed NLRP3 inflammasome assembly. The C-terminal domain of N protein promoted its interaction between NLRP3. In addition, N protein favored the conversion of pro-IL-1β and pro-IL-6 into their active forms. As a result, N protein exacerbated lung damage and facilitated death in acute inflammation mouse models through induction of NLRP3 inflammasome activation. SARS-CoV-2 infection triggered NLRP1 inflammasome response in human lung epithelial cells (106). NLRP1 inflammasome acted as an innate immune sensor of the SARS-CoV-2 3CL protease NSP5. Specifically, NSP5 could cleave NLRP1 at the Q333 site. The repressive N-terminal fragment of NLRP1 then underwent proteasomal degradation, leading to the release of its inflammatory C-terminal fragment. This bioactive domain self-assembled and acted as a platform for caspase-1 recruitment and activation. SARS-CoV-2 NSP5-driven NLRP1 cleavage contributed to proteasome-dependent inflammasome activation. NLRP1-induced inflammation might exert a double-edged sword effect on SARS-CoV-2 immunopathogenesis. On the one hand, NLRP1 inflammasome-dependent pyroptosis restricted viral replication and potentiated alarmin/DAMP responses. On the other hand, NLRP1 inflammasome might be involved in the development of a cytokine storm. The linkage between NLRP1 inflammasome and COVID-19 immunopathology warrants further research.
Collectively, these findings suggest the existence of stage-specific mechanisms controlling inflammasome activity during viral infection. It is necessary to elucidate how SARS-CoV-2 subtly dominates NLRP3 inflammasome activation to facilitate its survival and pathogenicity. The switch molecules regulating NLRP3 inflammasome activation remain to be determined. The activation status and mechanism of other inflammasomes at different stages of SARS-CoV-2 infection are worthy of great attention. Further elucidation of the mechanisms underlying the immunoregulatory effects of viral proteins would broaden the understanding of SARS-CoV-2 immunopathogenesis.
COVID-19 has suggested as a complex host-driven disorder that can be primarily attributable to dysregulation of host immune responses. Over-reaction of the inflammasome signaling has been associated with the pathogenesis of COVID-19-related complications, including the cytokine storm, pneumonia, ARDS, coagulopathy and neuroinflammation (Figure 4).
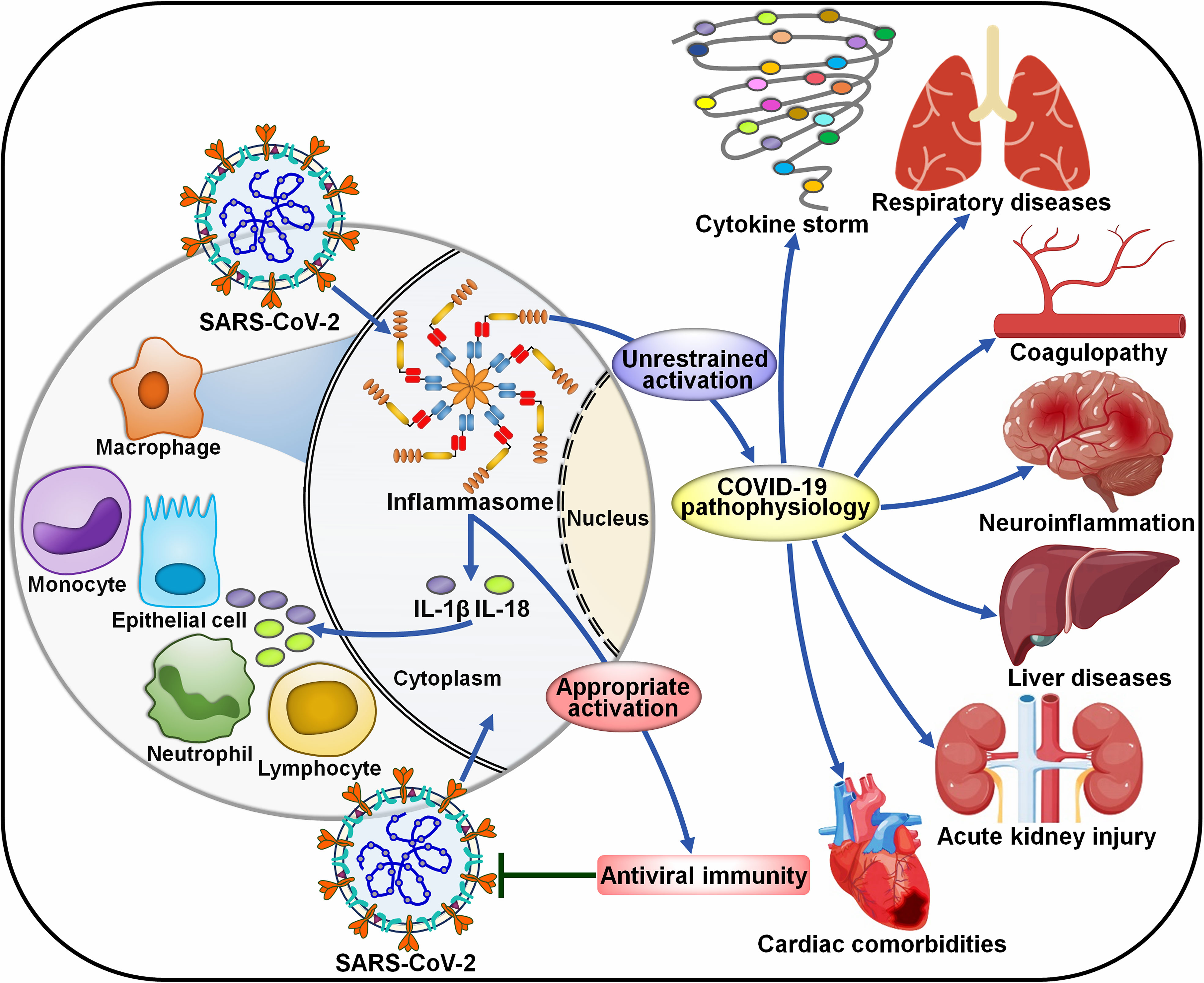
Figure 4 Roles of inflammasome pathways in the pathogenesis of COVID-19-associated complications. SARS-CoV-2 infection can activate inflammasomes in infected cells, especially macrophages. The inflammasome pathway constitutes a fundamental intracellular defense mechanism against pathogen invasion. However, uncontrolled activation of inflammasomes has emerged as a major driver of COVID-19-associated complications. The massive release of proinflammaotry cytokines by infected macrophages results in extensive cell damage and death, immune cell recruitment and widespread inflammasome activation, forming a proinflammatory positive feedback cascade. The over-reacted inflammasome pathway eventually leads to a cytokine storm, respiratory diseases, coagulopathy, neuroinflammation, liver diseases, acute kidney injury and cardiac comorbidities. SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; IL-1β, interleukin-1β; IL-18, interleukin-18.
Cytokine secretion plays an important role in the body’s defense against viral infections, while abnormal and uncontrolled activation of host immune system can bring about serious consequences. The cytokine storm is an exuberant systemic immune response characterized by an unrestrained overproduction of proinflammatory factors such as C-X-C motif chemokine ligand 8 (CXCL8), CXCL10, interferon-γ (IFN-γ), IL-1β, IL-6, IL-18 and TNF-α (107). Available evidence has verified that COVID-19 patients had increased serum levels of proinflammatory cytokines, such as IL-1β, IL-6 and IL-10, alluding to the implication of a cytokine storm (55, 56) (Table 3). Patients that undergo an exacerbated cytokine response commonly present with fever, hypotension and hypoxemia (128). The syndrome can either be mitigated or progress into persistent high-grade fever and severe complications (129). As shown by previous reports, elevated levels of proinflammatory cytokines and chemokines have emerged as a major factor driving pulmonary inflammation (130). It was reported that COVID-19 patients exhibited high plasma levels of C-C motif chemokine ligand 2 (CCL2), CXCL10, IFN-γ, IL-1β, IL-7, IL-8, IL-10 and TNF-α (56). Elevated levels of CCL2, CXCL10, IFN-γ and IL-1β subsequently mounted T helper type 1 (Th1) cell responses. Remarkably, COVID-19 patients requiring intensive care unit (ICU) admission presented higher levels of CCL2, CXCL10, IL-2, IL-7, IL-10 and TNF-α than non-ICU patients, hinting the linkage between the cytokine storm and COVID-19 deterioration.
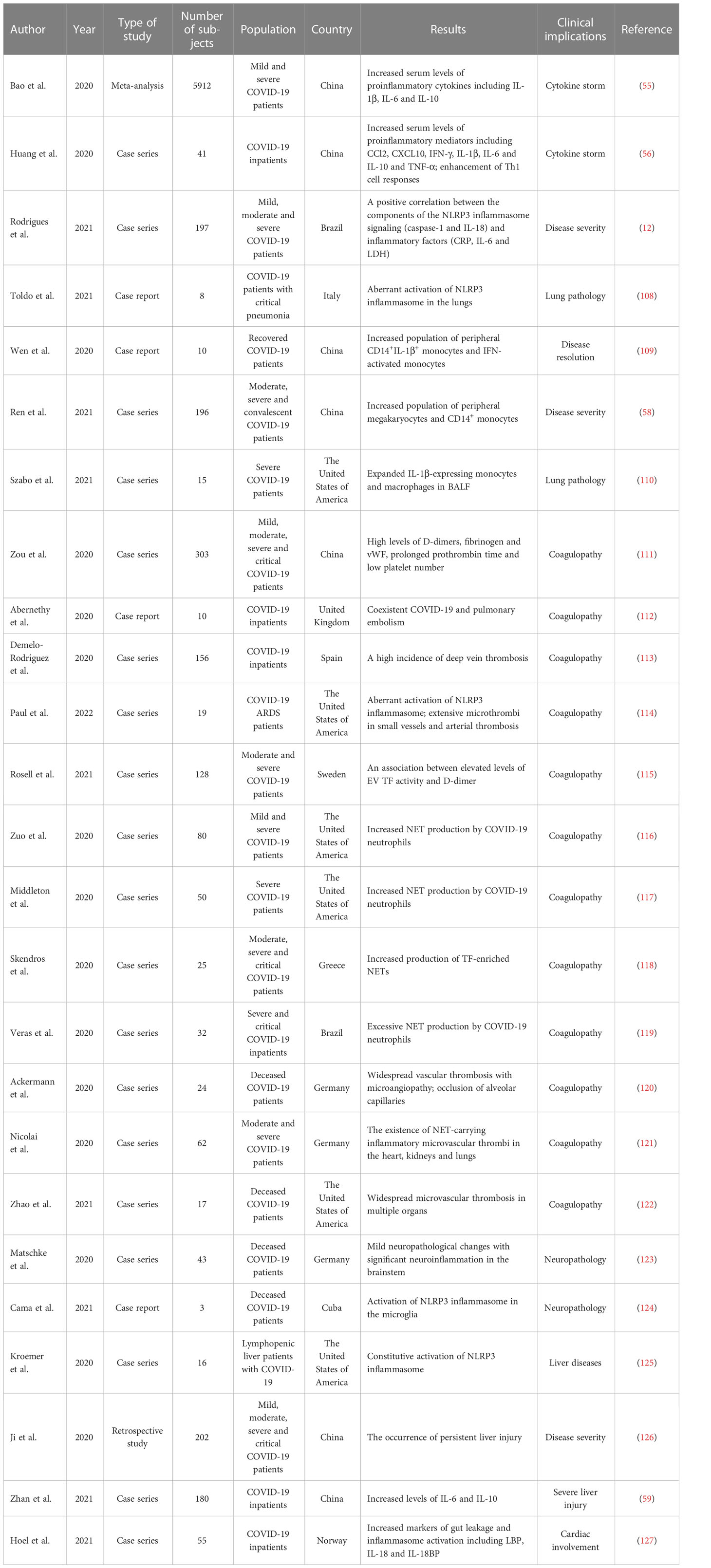
Table 3 Altered laboratory/clinical parameters with their clinical implications.
The inflammasome machineries are needed for the synthesis of inflammatory cytokines (73). Particularly, NLRP3 inflammasome can induce host immune responses via caspase-1-dependent pyroptosis, which results in extravasation of proinflammatory cytokines (e.g., IL-1β and IL-18) and biologically active DAMPs (131). IL-1β and IL-18 further enhance the secretion of the key cytokine IL-6 (132). The IL-6 level, a crucial mediator of acute inflammation, is deemed as a pathophysiological feature of the cytokine storm (133). It has been established that high serum IL-6 levels are positively associated with more severe clinical courses in COVID-19 patients (134, 135). IL-6 can be a predictor for COVID-19 progression (57). As expected, the components of the NLRP3 inflammasome signaling, caspase-1 and IL-18, had a positive relationship with the inflammatory factors CRP, IL-6 and LDH (12). These inflammasome-related products were also related to disease progression and patient outcome. Excessive release of proinflammatory mediators is responsible for aberrant systemic inflammatory response, which eventually results in ARDS, multiorgan failure and even death (136). Accordingly, ARDS has been considered as a deleterious outcome of the cytokine storm that can be flamed by inflammasome activation.
It was reported that the NLRP3 inflammasome was over-activated in elderly patients with COVID-19, which could be attributable to defects in mitochondrial functioning, enhanced production of mitochondrial ROS (mtROS) and massive release of mitochondrial DNA (mtDNA) (137). This caused the increased secretion of IL-1β and IL-18 in response to SARS-CoV-2 infection. Consistently, elderly patients were prone to over-react to SARS-CoV-2 infection and developed hyperinflammatory syndromes such as ARDS. Moreover, IL-1β and IL-18 have been considered as rapid inducers of lung fibrosis. Thus, over-activation of NLRP3 could be a predisposing factor contributing to high mortality in elderly patients. Compared with healthy controls, patients with fatal COVID-19 pneumonia exhibited intense expression of NLRP3 inflammasome, as manifested by increased numbers of ASC specks and high levels of NLRP3 and caspase-1 in the area of lung injury (108). These findings provided evidence linking inflammasome activation to lung pathology in COVID-19. NLRP3 is related to many inflammatory lung pathologies and abnormal activation of NLRP3 inflammasome leads to ARDS-induced pulmonary inflammation and damage (138–140). Antagonists or inhibitors of NLRP3 inflammasome could be effective in ameliorating COVID-19-related inflammation. However, the time course of NLRP3 inflammasome formation and the type of cells producing the inflammasome configuration remain to be investigated. Successive research efforts should be directed to elucidating the mechanisms of action of NLRP3 inflammasome in COVID-19 pneumonia.
The secretion of massive IL-1β into the peripheral blood enhanced the expression of other proinflammatory cytokines (e.g., IL-6), thus exaggerating SARS-CoV-2-induced cytokine storm (65). IL-1β coupling to IL-1 receptor (IL-1R) instigates the NF-κB response to enhance pro-IL-1β transcription in myeloid cells recruited to the lung tissues (141). Severe COVID-19 patients had expanded IL-1β-expressing monocytes and macrophages in BALF and increased population of peripheral CD14+IL-1β+ monocytes and macrophages (58, 109, 110). Inflammasome activation and subsequent cytokine response can be amplified through positive feedback loops, leading to the creation of a hyperinflammatory environment comprising multitudinous cytokine-producing immune cells associated with severe COVID-19 (142). Intense immune cell infiltration and cytokine secretion in lung tissues are representative features of ARDS and may ultimately lead to severe lung injury in COVID-19 patients. The cytokine storm may also form a positive feedforward loop to escalate lung damage. IL-1β-induced activation of endothelial cells inhibited the transcription of vascular endothelial cadherin (VE-cadherin), contributing to the disruption of vascular integrity (143). IL-1β-induced IL-6 release enhanced the formation of vascular endothelial growth factor (VEGF) that damaged the pulmonary endothelium via VE-cadherin internalization (144). This event facilitated accumulation of interstitial and alveolar fluid that compromised pulmonary gas exchange (145, 146). Because of the reduction of resorptive activity from infected alveolar epithelium, alveolar fluid accumulation could result in a series of unfavorable consequences such as disturbance of pulmonary surfactant, and alveolar instability and breakdown (147, 148). Immune cell recruitment and tissue failure could cooperatively exaggerate lung damage in response to aberrant release of IL-1-directed proinflammatory cytokines.
SARS-CoV-2 infection causes inflammation-directed coagulopathy, which may account for venous/arterial thrombosis and unfavorable outcomes in COVID-19 patients (Figure 4). A previous study demonstrated that a majority of COVID-19 patients had coagulation aberrations that manifested as high levels of D-dimers, fibrinogen and von Willebrand factor (vWF), prolonged prothrombin time and low platelet number (111). Thrombi have been described in the brain, heart, lungs, liver and kidneys of COVID-19 patients (149). The degree of thrombotic complications was heightened with increasing illness severity (150, 151). Severe cases of COVID-19 presented with deep vein thrombosis, pulmonary embolism and disseminated intravascular coagulation (112, 113). Inflammasomes have emerged as part of the mechanism responsible for SARS-CoV-2-induced thrombosis due to their regulatory effects on clot retraction and arterial thrombosis (152). Compared with non-COVID-19 subjects, the expression of NLRP3 and caspase-1 was increased along the vascular wall in COVID-19 ARDS cases, suggesting the activation of NLRP3 inflammasome in vessel wall (114). Vascular alternations including arterial thrombosis, microthrombi in small vessels and organization were widespread in lungs from COVID-19 patients compared with healthy controls. After viral entry, SARS-CoV-2 attaches to the alveolar epithelium. The infected lung cells then release cytokines and chemokines, which recruit neutrophils to the alveolar space. These events may contribute to the collapse of the alveolar wall. Moreover, the NLRP3 inflammasome was also activated in endothelial cells in response to various stimuli (e.g., SARS-CoV-2 infection, cytokines or chemokines), leading to endothelial pyroptosis. As a result, the endothelial-alveolar barrier was destroyed accompanied by the flooding of interstitial and alveolar spaces. Endothelial cell death and debris act as an inducer of coagulation cascades that foster thrombi formation.
Inflammasome activation in monocytes and neutrophils propels coagulation via distinct mechanisms. Inflammasome-mediated pyroptosis causes extensive coagulopathy independent of cytokine release. Previous studies suggested that induction of the inflammasome pathway contributed to systemic blood clotting and extensive thrombosis in tissues, and GSDMD but not proinflammatory cytokine was required for these coagulation cascades. Following inflammasome formation, GSDMD-executed pyroptosis of macrophages promoted the release of extracellular vesicles carrying high levels of membrane-bound tissue factor (TF) that subsequently entered the circulation and drove coagulation through activation of prothrombin (153). As expected, COVID-19 patients had elevated levels of circulating TF-positive extracellular vesicles, which correlated with disease severity and outcome (115). In addition, GSDMD pores that were formed downstream of inflammasome activation acted as the conduit for Ca2+ influx. This activated the phospholipid scramblase transmembrane protein 16F (TMEM16F), which increased TF activity through phosphatidylserine externalization (154).
Extracellular traps (NETs), extracellular DNA fibers decorated with antimicrobial proteins, are extruded from activated neutrophils (155). NETs are responsible for ensnaring and immobilizing extracellular pathogens and form an effector mechanism of host innate immune defense (156). Dysregulated NET formation during the course of COVID-19 can be a trigger of inflammation, blood coagulation and immunothrombosis (116). As NLRP3 inflammasome induced the rupture of nuclear envelope and cellular membrane, it was needed for NET protruding from neutrophil membrane (157, 158). It is conceivable that inflammasomes play an important role in COVID-19 coagulopathy. Reportedly, the circulating neutrophils of COVID-19 patients displayed elevated activation of NETs (117–119). Extensive neutrophil infiltration and abundant NETs in the lungs have been described in COVID-19 patients, which may trigger the occlusion of the pulmonary vasculature (120, 159, 160). Importantly, increased levels of NET-producing neutrophils positively correlated with the risk of morbid thrombotic complications in COVID-19 patients (116). NET-induced endothelial microvascular thrombosis was likely an explanation for thrombosis in COVID-19 (161). NET-released histones and cytotoxic enzymes caused endothelial cell death and endothelial dysfunction, culminating in organ damage in COVID-19 patients (162, 163). NETs induced acute lung injury in COVID-19 patients by accelerating lung epithelial cell death and intravascular thrombus synthesis (164, 165). In addition, NET-carrying inflammatory microvascular thrombi were detected in the heart, kidneys and lungs of COVID-19 patients (121). The pro-thrombiotic effect of NETs might be a cause of organ injury in COVID-19 patients (122). The inflammasome pathway could contribute to COVID-19 thrombus by regulating NET formation (116, 166). There is a great need for more research into the pathogenic mechanisms underpinning inflammasome-mediated immunothrombosis in COVID-19. Therapeutic interventions that aim to suppress NET formation by neutrophils will be necessary to prevent immunothrombosis and organ failure in COVID-19 patients.
SARS-CoV-2 RNA or proteins could be detected in many typical cells of central nervous system (CNS) (123, 124). Importantly, NLRP3 inflammasome was activated in monocytes or macrophages from lungs and brain, suggesting the important role of NLRP3 inflammasome in SARS-CoV-2-induced injuries of both the respiratory and nervous systems. Neurotropism of SARS-CoV-2 might foster blood-brain barrier leakage, leading to the impairment of the CNS vascular endothelium. The receptors angiotensin-converting enzyme 2 (ACE2) and cluster of differentiation 147 (CD147) mediated SARS-CoV-2 entry into cells of neuronal origin (e.g., astrocytes, microglia and neurons) (167, 168). Astrocytes and microglia are two main sources of proinflammatory cytokines and are critical for SARS-CoV-2-induced neuroinflammation (169). SARS-CoV-2 infection initiated robust NLRP3 inflammasome activation in microglia (170). Moreover, SARS-CoV-2 S protein primed inflammasome assembly in response to diverse stimuli (e.g., ATP and nigericin) via the NF-κB signaling pathway. Depletion of NLRP3 dampened viral S protein-induced inflammasome activation. SARS-CoV-2 S protein subunit 1 (S1) could elevate the expression level of NLRP3 inflammasome and increased caspase-1 activity in microglia (171). Moreover, S1 increased the production of IL-1β, IL-6 and TNF-α that were hallmarks of neuroinflammation, suggesting the role of S protein in the pathophysiology of neurological diseases. Knowledge of inflammasome-mediated neuroinflammation during SARS-CoV-2 infection is warranted to dissect the pathogenic mechanisms of neurological disorders in COVID-19 patients. Furthermore, S1 was able to stimulate the NF-κB and p38 mitogen-activated protein kinase (MAPK) signaling pathways through its regulatory action on TLR4 (171). Inhibition of p38 could block S1-mediated secretion of IL-6 and TNF-α, implying that the p38 MAPK pathway was implicated in S1-induced hyperinflammation. Reportedly, NF-κB triggered NLRP3 through caspase-1 (172). It is equivocal whether S1-mediated NLRP3 inflammasome activation is ascribed to its direct influence on NLRP3 activity or an indirect effect as a consequence of NF-κB activation. The contribution of NLRP3 inflammasome to S1-mediated neuroinflammation needs thorough investigation. Similarly, SARS-CoV-2 infection caused the priming of NLRP3 inflammasome and induced caspase-1-mediated secretion of IL-1β and IL-18 from human astrocytes (173). The inflammasome-derived product caspase-1 could facilitate neuroinflammation by interrupting the blood-brain barrier (174). IL-1β and IL-18 facilitated the production of other proinflammatory cytokines by astrocytes, microglia and neurons (175). Proinflammatory cytokines increased the permeability of the blood-brain barrier, activated astrocytes and microglia, and eventually promoted the infiltration of immune cells into the CNS (176). In addition, activated astrocytes and microglia triggered the release of cytokines and chemokines, including CCL2, CXCL10, IL-6 and TNF-α (177). IL-18 enhanced caspase-1 activity and potentiated proinflammatory cytokine responses in the CNS (178). Such pathological processes further exaggerated neuroinflammation-induced neuronal damage and led to neurological presentations in COVID-19 patients. Altogether, these studies provide scientific-based evidence to support hidden mechanisms underlying innate immune activation following SARS-CoV-2 infection in the CNS and open up new opportunities for developing new therapeutic avenues to relieve SARS-CoV-2-induced neurological symptoms.
The NLRP3 inflammasome was constitutively activated in COVID-19 patients with underlying liver diseases such as cirrhosis, acute live failure and non-alcoholic fatty liver disease (NAFLD) (125). SARS-CoV-2 infection further motivated NLRP3 inflammasome, as evidenced by increased caspase-1 activity in T cells and high levels of IL-18 and LDH. The inflammasome-dependent pyroptosis of T cells might not only cause defective adaptive immunity but also induce lethal inflammatory responses through extravasation of proinflammatory factors. It is likely that extravagant inflammasome activity exacerbates inflammation and immune dysfunction, ultimately leading to severe illnesses in patients with liver diseases. Collectively, inflammasome activation is a significant contributing factor to poor COVID-19 outcomes in comorbid patients. Liver injury was observed in COVID-19 patients (126). Increased levels of IL-6 and IL-10 were associated with a high risk of severe liver damage in COVID-19 patients (59). The over-activation of inflammasomes induces excessive cytokine release by immune cells, which can trigger a systematic inflammatory cytokine storm, leading to liver injury in infected individuals (179). Further research is still required to disclose the exact mechanisms of COVID-19-associated liver injury. Acute kidney injury (AKI) is a critical issue in COVID-19 patients (180). Severe COVID-19 patients that developed AKI had increased levels of proinflammatory cytokines (e.g., IFN-γ, IL-1β, IL-8 and TNF-α) compared with non-severe patients (181). Inflammasomes are likely an underlying contributor to hyperactivated inflammatory response and vascular thrombosis that consequently lead to AKI in COVID-19 patients (182). Moreover, the cytokine storm may cause renal impairment by triggering intrarenal inflammation (183). However, whether the inflammasome pathway is an important cause of SARS-CoV-2-associated AKI has yet to be examined.
SARS-CoV-2 infection has been connected with various cardiovascular diseases such as acute coronary syndrome, myocardial injury, arrhythmia and myocarditis (184). Inflammasome activation has been documented in COVID-19 patients with cardiac involvement, as shown by increased plasma levels of inflammasome markers including IL-1β, IL-1R antagonist (IL-1RA), IL-18 and IL-18-binding protein (IL-18BP) (127). NLRP3 inflammasome triggered hyperinflammatory responses by intensifying COVID-19-associated inflammation or coordinating the ACE2/angiotensin signaling cascade (185). Over-inflammation likely facilitates viral infiltration of other organs including the cardiovascular system. Cardiac cells with high expression of ACE2 can be a direct target of SARS-CoV-2 infection. Viral S protein-mediated ACE2 dysregulation in cardiac cells triggered NLRP3 inflammasome activation and proinflammatory cytokine response, hence aggravating cardiovascular disease comorbidity in COVID-19 (186). Meanwhile, ACE2-relevant signaling pathway may contribute to myocardial injury. Excessive cytokine release downstream of inflammasome priming gave rise to an extremely high demand for metabolites and energy, thus exacerbating myocardial damage (186). IL-1RA, IL-18 and IL-18BP were intertwined with increased levels of troponin, suggesting that inflammasome activation had a relationship with myocardial injury (127). The levels of IL-1, IL-6 and TNF-α were positively associated with increased risk of arrhythmia in COVID-19 patients (187). The mechanism underlying inflammasome activation in COVID-19 patients with cardiac involvement needs to be revealed. Unfavorable cardiovascular disorders following viral infection can be mainly attributable to inflammasome action, but it is still essential to define the mechanistic linkage between inflammasome activation and the development of cardiac comorbidities in COVID-19 patients.
It is increasingly acknowledged that host genetic variation is a crucial factor affecting COVID-19 severity (188–190). The genome-wide association meta-analyses that consisted of 49,562 COVID-19 patients from 46 studies across 19 countries indicated that specific mutations of the tyrosine kinase 2 (TYK2) gene could enhance the risk of becoming critically ill with COVID-19, while individuals who carried a variant in the dipeptidyl peptidase 9 (DPP9) gene were at elevated risk of developing critical disease (191, 192). Loss-of-function mutations in type I IFN (IFN-I) immunity genes (TLR3 and TLR7) might be connected with severe forms of COVID-19 in European populations (193–195). NLRP3 inflammasome gene variants were correlated with critical COVID-19 in the Brazilian population (196). The genome-wide association study among the Chinese population showed that the most pronounced gene variant related to COVID-19 severity was located in TMEM189-UBE2V1 that was implicated in the IL-1 signaling cascade (197). The variability in the frequency of inflammasome-relevant gene variants among individuals from other ancestry groups remains to be comprehensively evaluated. The identified host genetic factors are mainly intertwined with essential pathophysiological processes, including immune responses and inflammation (198). It is worth noting that these associations must be interpreted with caution and need to be further verified. Future studies including genetically diverse study groups will be required to identify the genomic loci associated with COVID-19 outcomes. A thorough investigation of COVID-19 host genetics will provide new insight into pathogenesis, risk stratification, therapy response as well as precision medicine.
Dysregulation of the inflammasome signalings could result in tissue and organ impairment in various systems of the human body (199). Harnessing the inflammasome to restrict the proinflammatory milieu may provide novel therapeutic modalities for COVID-19. So far, a multitude of inhibitors have been introduced for the key components of the inflammasome pathways (Figure 5). Kim et al. (86) developed 25-hydroxycholesterol (25-HC) and didodecyldimethylammonium bromide (DDAB) nanovesicles, namely 25-HC@DDAB, as an anti-COVID-19 drug candidate (Table 2). The in vivo study indicated that 25-HC@DDAB showed preferential accumulation to the lung tissues. Particularly, 25-HC@DDAB suppressed sterol regulatory element-binding protein 2 (SREBP2)-mediated inflammasome signaling pathway in PBMCs isolated from COVID-19 patients, hence ameliorating excessive inflammatory response and the cytokine storm. Accordingly, the nanotherapeutics may be an effective therapeutic approach for COVID-19 treatment. However, further clinical studies are necessary to comprehensively evaluate the safety and therapeutic efficiency of 25-HC@DDAB. Quercetin was able to restrict the production of NLRP3 inflammasome components such as NLRP3, ASC and caspase-1 and coordinate various inflammasome regulators (e.g., NRF2, SIRT1 and TXNIP) (87). Thus, quercetin acted as an NLRP3 inflammasome inhibitor and could be exploited to dampen severe inflammation in COVID-19 patients. Metformin was reported to repress NLRP3 inflammasome activation and IL-1β generation in macrophages (88). Mechanistically, metformin could restrain new mtDNA synthesis, which was essential for the formation of the NLRP3 ligand oxidized (Ox)-mtDNA, leading to the inhibition of NLRP3 inflammasome activation. Moreover, metformin exerted direct effects on NLRP3 inflammasome. Metformin affected the assembly of NLRP3 inflammasome instead of the expression of its components. Furthermore, metformin negatively modulated caspase-1 and GSDMD activation and IL-1β maturation. Administration of metformin reduced SARS-CoV-2-induced pulmonary inflammation and ARDS in SARS-CoV-2 infectable hACE2 transgenic mice. Another study demonstrated that melatonin treatment was able to restrain NLRP3 inflammasome priming by downregulating ASC and caspase-1 (89). It also inhibited the production of IL-1β and TNF-α in COVID-19 patients. Melatonin may represent an attractive supplemental treatment to alleviate immune over-activation and the cytokine storm in COVID-19 patients. Continued research efforts are needed to assess the therapeutic efficacy of metformin in COVID-19 patients.
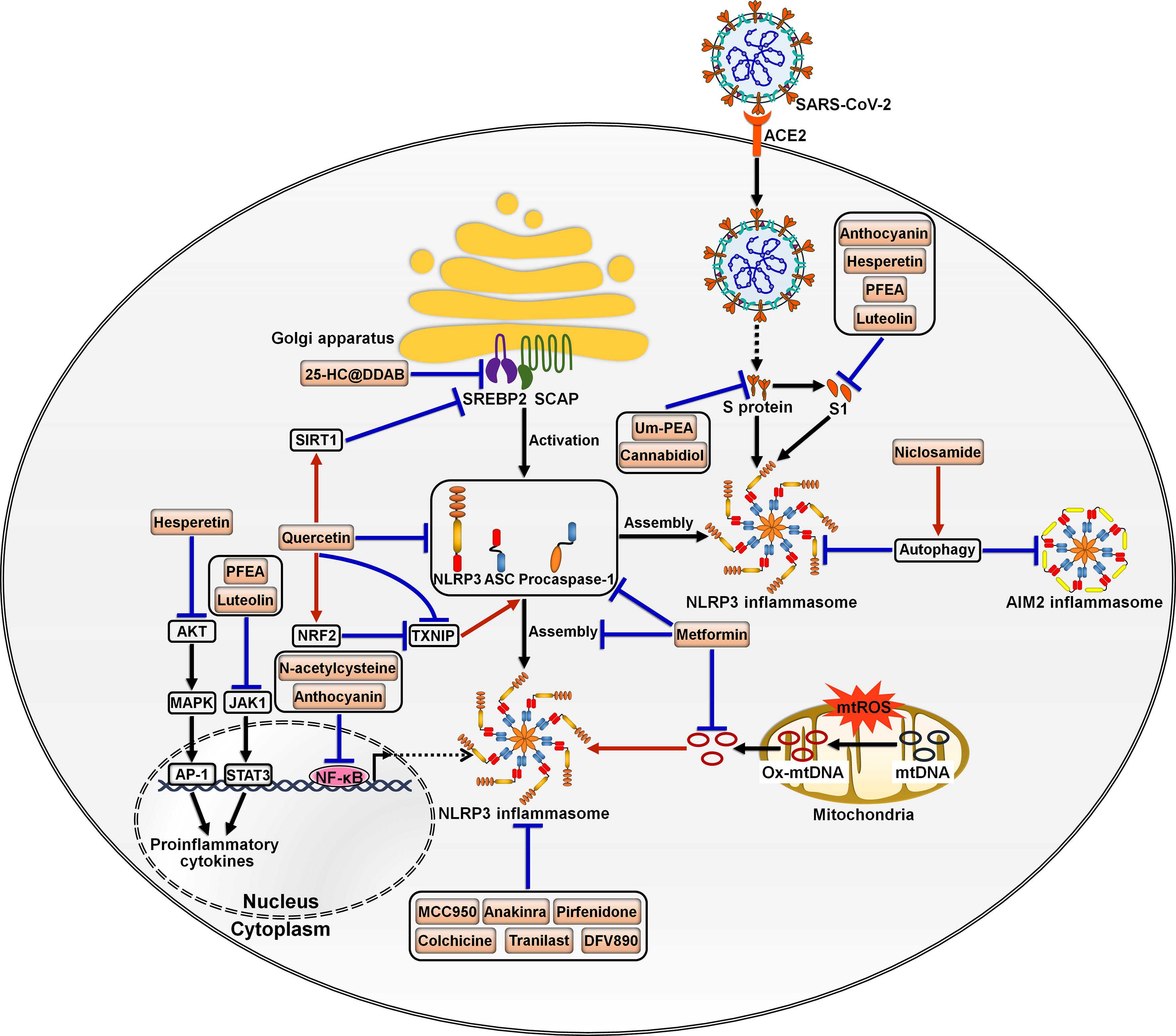
Figure 5 Potential inflammasome-targeted therapies for COVID-19. Various chemicals have an inhibitory effect on inflammasome activation and hold great promise in the treatment of COVID-19-associated complications. 25-HC@DDAB suppresses SREBP2-mediated inflammasome signaling pathway. Um-PEA and cannabidiol can abrogate SARS-CoV-2 S protein-stimulated NLRP3 inflammasome formation. Anthocyanin inhibits viral S1-induced inflammatory responses by targeting the NF-κB/NLRP3 inflammasome pathway. Likewise, N-acetylcysteine impedes SARS-CoV-2-induced inflammasome activation and cytokine secretion by targeting NF-κB. Hesperetin represses S1-induced NLRP3 inflammasome activation by blocking the Akt/MAPK/AP-1 signaling pathway. PFEA and luteolin restrain S1-driven proinflammatory response by downregulating the JAK1/STAT3 signaling cascade. Niclosamide restricts the activation of AIM2 and NLRP3 inflammasomes, which can be partially ascribed to its pro-autophagic property. Quercetin reduces the production of NLRP3 inflammasome components such as NLRP3, ASC and caspase-1 and coordinates inflammasome regulators SIRT1, NRF2 and TXNIP. Metformin prevents the formation of the NLRP3 ligand Ox-mtDNA, leading to the inhibition of NLRP3 inflammasome activation. Moreover, metformin decreases the expression of NLRP3 inflammasome components ASC and caspase-1 and inhibits the assembly of NLRP3 inflammasome. MCC950 and anakinra also act as NLRP3 inflammasome inhibitors and may be of potential translational value in severe COVID-19 patients. Some inflammasome antagonists including pirfenidone, colchicine, Tranilast and DFV890 are under development and being explored for their therapeutic efficacy in COVID-19 patients. SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; ACE2, angiotensin-converting enzyme 2; SREBP2, sterol regulatory element-binding protein 2; SCAP, SREBP cleavage-activating protein; S protein, spike protein; S1, S protein subunit 1; SIRT1, sirtuin 1; NLRP3, nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing protein 3; ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain; AIM2, absent in melanoma 2; AKT, protein kinase B; NRF2, nuclear factor erythroid 2-related factor 2; TXNIP, thioredoxin-interacting protein; MAPK, mitogen-activated protein kinase; JAK1, Janus kinase 1; mtROS, mitochondrial reactive oxygen species; mtDNA, mitochondrial DNA; Ox-mtDNA, oxidized-mitochondrial DNA; AP-1, activator protein-1; STAT3, signal transducer and activator of transcription 3; NF-κB, nuclear factor-κB.
SARS-CoV-2 infection triggered NLRP3 inflammasome activation and increased the levels of caspase-1, IL-1β and IL-18 in the lung tissues, which precipitated systemic inflammation and exacerbated lung immunopathology in vivo (60, 90). Blockade of NLRP3 inflammasome via genetic knockout and specific inhibitor MCC950 apparently restrained NLRP3 inflammasome activation induced by SARS-CoV-2 infection (90). Furthermore, MCC950 abated COVID-19-associated lung immunopathology in SARS-CoV-2-infected mice, as evidenced by fewer signs of interstitial pneumonia, reduced accumulation of immune cells (e.g., macrophages and neutrophils) and decreased levels of proinflammatory mediators (e.g., IL-1β, IL-6, IL-18, TNF-α, monocyte chemoattractant protein-1 (MCP-1), CCL2 and CXCL9) in lung tissues. It is proposed that NLRP3 is directly involved in production of SARS-CoV-2 replication complex. Activation of NLRP3 inflammasome may also contribute to the formation of a favorable environment for viral replication. The implication of NLRP3 inflammasome in SARS-CoV-2 replication awaits follow-up research. NLRP3 inflammasome favors SARS-CoV-2 propagation, leading to high viral loads and increasing infectiousness, which results in direct pulmonary injury. Moreover, SARS-CoV-2 can trigger a systematic cytokine storm that impels the progression of more advanced diseases in COVID-19 patients (200). Over-activation of NLRP3 inflammasome has been suggested as a pathological event responsible for the formation of SARS-CoV-2-induced cytokine storm (201). Targeting NLRP3 inflammasome by MCC950 could be a promising immunotherapeutic approach for treating COVID-19. NLRP3 inflammasome was over-activated in airway tissues from severe COVID-19 patients evidenced by increased levels of the inflammasome components including NLRP3, ASC and caspase-1 and massive production of IL-1β and IL-18 (91). The activation of NLRP3 inflammasome was connected with systemic inflammation and mortality. Mechanistic investigation showed that SARS-CoV-2 proteins primed NLRP3 inflammasome in human macrophages and bronchial epithelial cells by activating the TLR2/MyD88/NF-κB axis. N-acetylcysteine inhibited SARS-CoV-2-induced inflammasome activation and cytokine secretion by targeting NF-κB. N-acetylcysteine may have the potential to curb hyperinflammation and the cytokine storm in COVID-19 patients, mandating further study of the therapeutic efficacy and mechanism of N-acetylcysteine. Niclosamide effectively repressed the activation of AIM2 and NLRP3 inflammasomes, which was partially ascribed to its pro-autophagic property (92). It was worth noting that diverse mechanisms might operate in niclosamide-mediated suppression of inflammasome activation. The pathways that contribute to its anti-inflammatory action need to be further studied. Whether niclosamide is effective in preventing inflammation in COVID-19 patients is a vital future question to pursue.
SARS-CoV-2 S protein caused NLRP3 inflammasome activation and the extravasation of proinflammatory cytokines (IL-1β and IL-6) by upregulating NLRP3 and caspase-1 (93). Ultramicronized palmitoylethanolamide (um-PEA) abrogated S protein-stimulated NLRP3 inflammasome formation. Therefore, um-PEA held promise in the fight against COVID-19. Similarly, cannabidiol (CBD) had the ability to resist excess inflammatory response induced by S protein (94). Mechanistically, CBD was capable of downregulating the expression of NLRP3 and caspase-1 and preventing the production of IL-1β, IL-6, IL-18 and TNF-α. Thus, CBD functioned as an inhibitor of SARS-CoV-2 S protein. It is worth noting that the aforementioned conclusions were drawn from in vitro experimental results. Further in vivo and clinical studies are critically needed to substantiate the protective potential of um-PEA and CBD in COVID-19 patients. Active anthocyanins (C3G and P3G) derived from black rice germ and bran (BR extract) could reduce SARS-CoV-2 S1-induced secretion of IL-1β, IL-6 and IL-18 in macrophages by targeting the NF-κB/NLRP3 inflammasome pathway (95). C3G and P3G exerted anti-inflammatory effects and could be developed into preventive agents for SARS-CoV-2-induced inflammation. Continual efforts should be made to identify the upstream molecules (e.g., TLR and MyD88) involved in the anti-inflammatory actions of C3G and P3G. The health benefits of these anthocyanins should be confirmed in COVID-19 patients. Hesperetin from root extract of Clerodendrum petasites S. Moore decreased SARS-CoV-2 S1-induced expression of NLRP3, ASC and caspase-1 through counteraction with the protein kinase B (Akt)/MAPK/activator protein-1 (AP-1) pathway (96). Hesperetin markedly attenuated the production of proinflammatory cytokines including IL-1β and IL-8. These findings provided evidence for the potential application of hesperetin in the prevention of SARS-CoV-2-induced chronic inflammation. The ethyl acetate fraction of Perilla frutescens (PFEA) and luteolin were found to quench SARS-CoV-2 S1-induced NLRP3 inflammasome activation in lung cells via downregulation of the Janus kinase 1 (JAK1)/signal transducer and activator of transcription 3 (STAT3) signaling, as shown by decreased expression of NLRP3, ASC and caspase-1 (97). PFEA and luteolin could be introduced as preventive agents against inflammation-associated post-acute sequelae in COVID-19 patients. Consistent with previous studies, activated NLRP3 inflammasome, accompanied by IL-1β release, was observed in circulating monocytes from severe COVID-19 patients (98). The inflammasome activation was retarded by administration of the IL-1 receptor antagonist anakinra, which might be of potential translational value in severe COVID-19 patients.
Some inflammasome inhibitors are under development and being explored for their therapeutic benefits in COVID-19 intervention (Table 4). Pirfenidone, an approved drug for the treatment of idiopathic pulmonary fibrosis, could inhibit the assembly of NLRP3 inflammasome and alleviate lung inflammation and the duration of hospital stay in COVID-19 patients (202). Inhibition of NLRP3 inflammasome by pirfenidone may be a prospective therapeutic option for COVID-19. Colchicine, an alkaloid derived from Colchicum autumnale, can inhibit NLRP3 inflammasome activation (209, 210). Colchicine may be used as a mediation that acts on NLRP3 infammasome-mediated cytokine storm in COVID-19. The therapeutic potential of colchicine was previously investigated in nine COVID-19 patients (204). The results indicated that colchicine was well tolerated and effective in lessening fever. A randomized controlled clinical study including 105 COVID-19 patients showed that colchicine decreased the clinical deterioration rate. In addition, colchicine could reduce the duration of hospital stay in COVID-19 patients (205). Given the great potential of colchicine to reduce COVID-19-associated inflammation, the efficacy of colchicine plus the IL-6 receptor antagonist (IL-6Ra) tocilizumab is being evaluated in an open-label randomized controlled trial (206). Larger randomized controlled clinical trials will be necessary to corroborate the beneficial effects of colchicine in COVID-19 treatment. Of note, a minority of patients treated with colchicine developed gastrointestinal symptoms such as diarrhoea, nausea or vomit. The adverse effects relevant to the intake of colchicine are correlated with prolonged use, pre-existing diseases and potential interaction with other agents. Thus, the safety and side effects of colchicine are worthy of detailed investigation. In a randomized controlled trial, Tranilast, a potential NLRP3 inflammasome inhibitor, was used as a complementary treatment in combination with standard therapeutic measures (e.g., antivirals and supportive care) in COVID-19 patients (207). Tranilast treatment could ameliorate clinical symptoms including weakness and fatigue and reduce hospital stay. COVID-19 patients treated with Tranilast had apparently lower levels of proinflammatory cytokines (e.g., IL-1β, IL-6 and TNF-α) than the controls. Furthermore, the results of the quantitative outcome of blood oxygen saturation showed that Tranilast treatment produced significant effectiveness. Collectively, Tranilast could improve the clinical outcome of COVID-19 patients. Nevertheless, more clinical studies are warranted to assess the protective potency of Tranilast. It would be equally important to illuminate the mechanisms contributing to the therapeutic effect of Tranilast. An early phase 2a randomized clinical trial including 143 patients with COVID-19 pneumonia and impaired respiratory function was previously conducted to assess the clinical efficacy of the NLRP3 inhibitor DFV890 (208). As a result, DFV890 was well-tolerated and had an acceptable safety profile. Patients treated with DFV890 showed early viral clearance, improved clinical status and in-hospital outcomes and decreased mortality rate compared with the standard-of-care group. These results provided evidence to support the utilization of DFV890 as a complementary therapy for COVID-19-related ARDS. Collectively, these studies suggest that inflammasome-based therapy holds great promise in the treatment of COVID-19-associated complications. However, some limitations such as the small sample size and the short duration of follow-up may increase the probability of bias. Well-designed multi-center trials with a large sample size will be able to verify existing clinical results in the future.
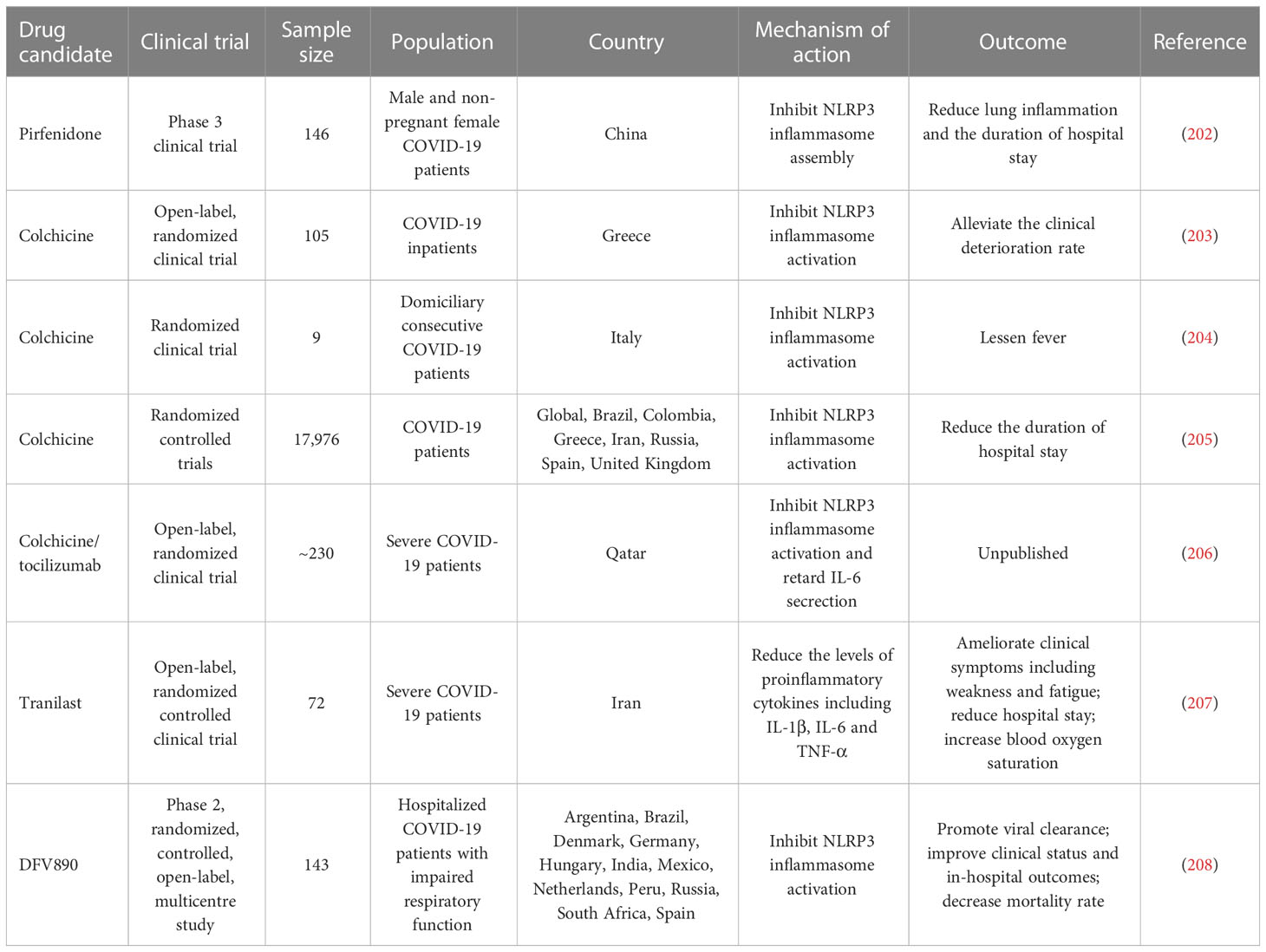
Table 4 Clinical trials of inflammasome-targeted therapies in COVID-19.
The formation and activation of inflammasomes are crucial for counteracting pathogen invasion. However, unrestrained activation of inflammasomes may cause a cytokine storm, systemic hypercoagulation, ARDS and even death. Over-exuberant inflammasome response has emerged as an important predisposing factor for disease severity and poor clinical outcome in COVID-19 patients. So far, NLRP3 is the most extensively studied inflammasome in COVID-19 owing to its critical role in mediating infection-induced inflammation. SARS-CoV-2 plays opposite roles in controlling NLRP3 inflammasome function. SARS-CoV-2 infection induces the activation of NLRP3 inflammasome that triggers antiviral immune response. In vivo studies are expected to be carried out to determine the protective role of the inflammasome signaling during the early stage of SARS-CoV-2 infection. SARS-CoV-2 have evolved multiple mechanisms to antagonize the priming of NLRP3 inflammasome. On the contrary, the virus can fuel unbridled inflammatory reaction by triggering NLRP3 inflammasome. As there is relatively little literature available on this topic, our understanding of the intricate interconnection between SARS-CoV-2 and NLRP3 inflammasome is still incomplete. It is of value to figure out how SARS-CoV-2 takes advantage of NLRP3 inflammasome to ensure its own survival and pathogenesis. NLRP1, a sensor for RNA virus infection, was found to be activated in SARS-CoV-2-infected subjects (106, 211). It is proposed that AIM2 inflammasome plays a fundamental role in antiviral response against SARS-CoV-2 infection (63). SARS-CoV-2 infection could facilitate nutrient deficiency and induce excessive cell death, contributing to the liberation of endogenous DNA. Moreover, SARS-CoV-2 invasion may cause the release of mtDNA into the cytoplasm by impairing mitochondrial membranes. Both free DNA and mtDNA could act as activators of AIM2 inflammasome. However, proinflammatory products resulting from NLRP1 and AIM2 inflammasomes may overlap with those of NLRP3 inflammasome activation. Thus, it is not surprising that previous studies do not center on the interrelation between these inflammasomes and COVID-19. Considerably more work will be needed to investigate the potential engagement of other inflammasomes in COVID-19 and unravel the mechanisms associated with their activation and synergistic functions. It appears that the lung tissues secrete inflammasome component-carrying extracellular vesicles into the circulation following SARS-CoV-2 infection, leading to non-respiratory manifestations. More evidence is required to determine whether extracellular vesicles act as a key mediator of SARS-CoV-2-induced pathologies. It is worth noting that the prolonged activation of inflammasomes does not always cause hyperinflammation in some COVID-19 patients (212). The discrepancy may be ascribed to the duration or magnitude of inflammasome-activating insults, the location of inflammation or the type of cells that are inflamed. There may exist negative feedback mechanisms that hinder the inflammasome signaling. Continual efforts must be made to address the variability in inflammasome-induced immunopathology in COVID-19 patients. It is believed that new discoveries in the specific mechanisms linking inflammasomes with COVID-19 development could provide a more thorough understanding of SARS-CoV-2 immunopathogenesis.
Manipulation of the inflammasome signaling exerts a protective effect against COVID-19 by restricting inflammasome activation, cytokine release or SARS-CoV-2-induced pyroptotic cell death. Inflammasomes have become an attractive drug target to conquer COVID-19. Although some inflammasome-targeted molecules have potential clinical utility in treating COVID-19, their mechanisms of action, toxicity and adverse effects remain to be adequately explored. Currently, a multitude of drug candidates are still in preclinical trials, and well-controlled clinical studies are urgently warranted to evaluate their genuine therapeutic effect. Given the crucial role of the inflammasome pathway in resisting viral infection, inflammasome-directed interventions should be used with caution. The timing of intervention with inflammasome antagonists must be carefully determined. Furthermore, the upstream signaling cascades that coordinate the inflammasome machinery may also represent a therapeutic target in COVID-19 treatment. It is reported that the inflammasome priming is dictated by diverse signaling pathways, such as the autophagic flux, IL-1R, MAPK, NF-κB, TLR and TNF receptor (TNFR) (213). Specific signaling pathways (e.g., MAPK and TLR) can activate inflammasomes in response to viral infections, while negative regulatory cascades (e.g., autophagy and AMP-activated protein kinase (AMPK)) work to sustain an appropriate immune defense and protect against inflammation-inflicted organ damage by suppressing over-activation of inflammasomes. These signal transduction cascades may interact with each other to control inflammasome assembly. It is necessary to characterize the complex regulatory networks underlying inflammasome activation. The impact of SARS-CoV-2 on the activity of inflammasome-regulating pathways is an important area for further research. Importantly, autophagy and the inflammatory response have a mutually antagonistic relationship. Inducing autophagy can inhibit SARS-CoV-2-induced inflammasome formation, while blocking autophagy prevents viral immune evasion (214). The interconnection between autophagy and inflammation during COVID-19 pathogenesis deserves considerable attention. It is supposed that the combinational therapies of NLRP3 inhibitors with autophagy regulators hold great promise for strengthening antiviral responses to achieve a better therapeutic effect, which awaits further verification in the clinic. In conclusion, an in-depth investigation on the host-SARS-CoV-2 interaction will provide a theoretical basis for unearthing new treatment options in the future that could be exploited to relieve COVID-19-related complications.
MW and PL conceived and supervised the project. MW wrote the manuscript and drew the figures. FY and WC searched and analyzed the literatures. YZ and LZ revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Natural Science Foundation of Shandong Province, China (Grant No. ZR2021MH018).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hu B, Guo H, Zhou P, Shi ZL. Characteristics of sars-Cov-2 and covid-19. Nat Rev Microbiol (2021) 19(3):141–54. doi: 10.1038/s41579-020-00459-7
2. Zheng Y, Zhuang MW, Han L, Zhang J, Nan ML, Zhan P, et al. Severe acute respiratory syndrome coronavirus 2 (Sars-Cov-2) membrane (M) protein inhibits type I and iii interferon production by targeting rig-I/Mda-5 signaling. Signal transduction targeted Ther (2020) 5(1):299. doi: 10.1038/s41392-020-00438-7
3. Boson B, Legros V, Zhou B, Siret E, Mathieu C, Cosset FL, et al. The sars-Cov-2 envelope and membrane proteins modulate maturation and retention of the spike protein, allowing assembly of virus-like particles. J Biol Chem (2021) 296:100111. doi: 10.1074/jbc.RA120.016175
4. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. New Engl J Med (2020) 382(8):727–33. doi: 10.1056/NEJMoa2001017
5. Moschovis PP, Yonker LM, Shah J, Singh D, Demokritou P, Kinane TB. Aerosol transmission of sars-Cov-2 by children and adults during the covid-19 pandemic. Pediatr pulmonol (2021) 56(6):1389–94. doi: 10.1002/ppul.25330
6. Tay MZ, Poh CM, Renia L, MacAry PA, Ng LFP. The trinity of covid-19: immunity, inflammation and intervention. Nat Rev Immunol (2020) 20(6):363–74. doi: 10.1038/s41577-020-0311-8
7. Singh S, Meher N, Mohammed A, Razab M, Bhaskar L, Nawi NM. Neurological infection and complications of sars-Cov-2: a review. Medicine (2023) 102(5):e30284. doi: 10.1097/MD.0000000000030284
8. Adil MT, Rahman R, Whitelaw D, Jain V, Al-Taan O, Rashid F, et al. Sars-Cov-2 and the pandemic of covid-19. Postgraduate Med J (2021) 97(1144):110–6. doi: 10.1136/postgradmedj-2020-138386
9. Yap JKY, Moriyama M, Iwasaki A. Inflammasomes and pyroptosis as therapeutic targets for covid-19. J Immunol (2020) 205(2):307–12. doi: 10.4049/jimmunol.2000513
10. Cheng T, Feng Y, Chen X, Zhou J, Song Y. Lung-resident mesenchymal stem cells regulated the inflammatory responses in innate and adaptive immune cells through hvem-btla pathway during Ards. Exp Cell Res (2020) 395(1):112155. doi: 10.1016/j.yexcr.2020.112155
11. Hooftman A, O'Neill LAJ. Can Nlrp3 inhibitors improve on dexamethasone for the treatment of covid-19? Curr Res Pharmacol Drug Discovery (2021) 2:100048. doi: 10.1016/j.crphar.2021.100048
12. Rodrigues TS, de Sa KSG, Ishimoto AY, Becerra A, Oliveira S, Almeida L, et al. Inflammasomes are activated in response to sars-Cov-2 infection and are associated with covid-19 severity in patients. J Exp Med (2021) 218(3):e20201707. doi: 10.1084/jem.20201707
13. Ferreira AC, Soares VC, de Azevedo-Quintanilha IG, Dias S, Fintelman-Rodrigues N, Sacramento CQ, et al. Sars-Cov-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discovery (2021) 7(1):43. doi: 10.1038/s41420-021-00428-w
14. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16(7):407–20. doi: 10.1038/nri.2016.58
15. Alnemri ES. Sensing cytoplasmic danger signals by the inflammasome. J Clin Immunol (2010) 30(4):512–9. doi: 10.1007/s10875-010-9419-0
16. Chen IY, Ichinohe T. Response of host inflammasomes to viral infection. Trends Microbiol (2015) 23(1):55–63. doi: 10.1016/j.tim.2014.09.007
17. Nieto-Torres JL, Verdia-Baguena C, Jimenez-Guardeno JM, Regla-Nava JA, Castano-Rodriguez C, Fernandez-Delgado R, et al. Severe acute respiratory syndrome coronavirus e protein transports calcium ions and activates the Nlrp3 inflammasome. Virology (2015) 485:330–9. doi: 10.1016/j.virol.2015.08.010
18. Rawat P, Teodorof-Diedrich C, Spector SA. Human immunodeficiency virus type-1 single-stranded rna activates the Nlrp3 inflammasome and impairs autophagic clearance of damaged mitochondria in human microglia. Glia (2019) 67(5):802–24. doi: 10.1002/glia.23568
19. Lu F, Zhao Y, Pang Y, Ji M, Sun Y, Wang H, et al. Nlrp3 inflammasome upregulates pd-L1 expression and contributes to immune suppression in lymphoma. Cancer Lett (2021) 497:178–89. doi: 10.1016/j.canlet.2020.10.024
20. He J, Yang Y, Peng DQ. Monosodium urate (Msu) crystals increase gout associated coronary heart disease (Chd) risk through the activation of Nlrp3 inflammasome. Int J Cardiol (2012) 160(1):72–3. doi: 10.1016/j.ijcard.2012.05.083
21. Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the Nlrp3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced il-1beta in type 2 diabetes. Nat Immunol (2010) 11(10):897–904. doi: 10.1038/ni.1935
22. Aymonnier K, Ng J, Fredenburgh LE, Zambrano-Vera K, Munzer P, Gutch S, et al. Inflammasome activation in neutrophils of patients with severe covid-19. Blood Adv (2022) 6(7):2001–13. doi: 10.1182/bloodadvances.2021005949
23. Han H, Ma Q, Li C, Liu R, Zhao L, Wang W, et al. Profiling serum cytokines in covid-19 patients reveals il-6 and il-10 are disease severity predictors. Emerging Microbes infections (2020) 9(1):1123–30. doi: 10.1080/22221751.2020.1770129
24. Sefik E, Qu R, Junqueira C, Kaffe E, Mirza H, Zhao J, et al. Inflammasome activation in infected macrophages drives covid-19 pathology. Nature (2022) 606(7914):585–93. doi: 10.1038/s41586-022-04802-1
25. Ling L, Chen Z, Lui G, Wong CK, Wong WT, Ng RWY, et al. Longitudinal cytokine profile in patients with mild to critical covid-19. Front Immunol (2021) 12:763292. doi: 10.3389/fimmu.2021.763292
26. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21(7):677–87. doi: 10.1038/nm.3893
27. Rathinam VA, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nat Immunol (2012) 13(4):333–42. doi: 10.1038/ni.2237
28. Jiang Q, Wang X, Huang E, Wang Q, Wen C, Yang G, et al. Inflammasome and its therapeutic targeting in rheumatoid arthritis. Front Immunol (2021) 12:816839. doi: 10.3389/fimmu.2021.816839
29. Xue Y, Enosi Tuipulotu D, Tan WH, Kay C, Man SM. Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol (2019) 40(11):1035–52. doi: 10.1016/j.it.2019.09.005
30. Matikainen S, Nyman TA, Cypryk W. Function and regulation of noncanonical caspase-4/5/11 inflammasome. J Immunol (2020) 204(12):3063–9. doi: 10.4049/jimmunol.2000373
31. von Moltke J, Ayres JS, Kofoed EM, Chavarria-Smith J, Vance RE. Recognition of bacteria by inflammasomes. Annu Rev Immunol (2013) 31:73–106. doi: 10.1146/annurev-immunol-032712-095944
32. Chen L, Cao SQ, Lin ZM, He SJ, Zuo JP. Nod-like receptors in autoimmune diseases. Acta pharmacologica Sin (2021) 42(11):1742–56. doi: 10.1038/s41401-020-00603-2
33. Li D, Wu M. Pattern recognition receptors in health and diseases. Signal transduction targeted Ther (2021) 6(1):291. doi: 10.1038/s41392-021-00687-0
34. Sharma D, Kanneganti TD. The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J Cell Biol (2016) 213(6):617–29. doi: 10.1083/jcb.201602089
35. Fink SL, Bergsbaken T, Cookson BT. Anthrax lethal toxin and salmonella elicit the common cell death pathway of caspase-1-Dependent pyroptosis Via distinct mechanisms. Proc Natl Acad Sci United States America (2008) 105(11):4312–7. doi: 10.1073/pnas.0707370105
36. Hsu LC, Ali SR, McGillivray S, Tseng PH, Mariathasan S, Humke EW, et al. A Nod2-Nalp1 complex mediates caspase-1-Dependent il-1beta secretion in response to bacillus anthracis infection and muramyl dipeptide. Proc Natl Acad Sci United States America (2008) 105(22):7803–8. doi: 10.1073/pnas.0802726105
37. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol (2013) 13(6):397–411. doi: 10.1038/nri3452
38. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The Nlrc4 inflammasome receptors for bacterial flagellin and type iii secretion apparatus. Nature (2011) 477(7366):596–600. doi: 10.1038/nature10510
39. Yang J, Zhao Y, Shi J, Shao F. Human naip and mouse Naip1 recognize bacterial type iii secretion needle protein for inflammasome activation. Proc Natl Acad Sci United States America (2013) 110(35):14408–13. doi: 10.1073/pnas.1306376110
40. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. Aim2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature (2009) 458(7237):509–13. doi: 10.1038/nature07710
41. Xu H, Yang J, Gao W, Li L, Li P, Zhang L, et al. Innate immune sensing of bacterial modifications of rho gtpases by the pyrin inflammasome. Nature (2014) 513(7517):237–41. doi: 10.1038/nature13449
42. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors asc and ipaf. Nature (2004) 430(6996):213–8. doi: 10.1038/nature02664
43. Stehlik C, Lee SH, Dorfleutner A, Stassinopoulos A, Sagara J, Reed JC. Apoptosis-associated speck-like protein containing a caspase recruitment domain is a regulator of procaspase-1 activation. J Immunol (2003) 171(11):6154–63. doi: 10.4049/jimmunol.171.11.6154
44. Srinivasula SM, Poyet JL, Razmara M, Datta P, Zhang Z, Alnemri ES. The pyrin-card protein asc is an activating adaptor for caspase-1. J Biol Chem (2002) 277(24):21119–22. doi: 10.1074/jbc.C200179200
45. Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med (2018) 215(3):827–40. doi: 10.1084/jem.20172222
46. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of gsdmd by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
47. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin d is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res (2015) 25(12):1285–98. doi: 10.1038/cr.2015.139
48. Evavold CL, Kagan JC. Inflammasomes: threat-assessment organelles of the innate immune system. Immunity (2019) 51(4):609–24. doi: 10.1016/j.immuni.2019.08.005
49. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular lps. Nature (2014) 514(7521):187–92. doi: 10.1038/nature13683
50. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin d for non-canonical inflammasome signalling. Nature (2015) 526(7575):666–71. doi: 10.1038/nature15541
51. Ruhl S, Broz P. Caspase-11 activates a canonical Nlrp3 inflammasome by promoting k(+) efflux. Eur J Immunol (2015) 45(10):2927–36. doi: 10.1002/eji.201545772
52. Place DE, Kanneganti TD. Recent advances in inflammasome biology. Curr Opin Immunol (2018) 50:32–8. doi: 10.1016/j.coi.2017.10.011
53. Courjon J, Dufies O, Robert A, Bailly L, Torre C, Chirio D, et al. Heterogeneous Nlrp3 inflammasome signature in circulating myeloid cells as a biomarker of covid-19 severity. Blood Adv (2021) 5(5):1523–34. doi: 10.1182/bloodadvances.2020003918
54. Leal VNC, Andrade MMS, Teixeira FME, Cambui RAG, Roa M, Marra LG, et al. Severe covid-19 patients show a dysregulation of the Nlrp3 inflammasome in circulating neutrophils. Scandinavian J Immunol (2023) 97(3):e13247. doi: 10.1111/sji.13247
55. Bao J, Li C, Zhang K, Kang H, Chen W, Gu B. Comparative analysis of laboratory indexes of severe and non-severe patients infected with covid-19. Clinica chimica acta; Int J Clin Chem (2020) 509:180–94. doi: 10.1016/j.cca.2020.06.009
56. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
57. Ulhaq ZS, Soraya GV. Interleukin-6 as a potential biomarker of covid-19 progression. Medecine maladies infectieuses (2020) 50(4):382–3. doi: 10.1016/j.medmal.2020.04.002
58. Ren X, Wen W, Fan X, Hou W, Su B, Cai P, et al. Covid-19 immune features revealed by a Large-scale single-cell transcriptome atlas. Cell (2021) 184(7):1895–913 e19. doi: 10.1016/j.cell.2021.01.053
59. Zhan K, Liao S, Li J, Bai Y, Lv L, Yu K, et al. Risk factors in patients with covid-19 developing severe liver injury during hospitalisation. Gut (2021) 70(3):628–9. doi: 10.1136/gutjnl-2020-321913
60. Baena Carstens L, Campos D'amico R, Fernandes de Moura K, Morais de Castro E, Centenaro F, Silva Barbosa G, et al. Lung inflammasome activation in sars-Cov-2 post-mortem biopsies. Int J Mol Sci (2022) 23(21):13033. doi: 10.3390/ijms232113033
61. Tsuchiya K, Hara H, Fang R, Hernandez-Cuellar E, Sakai S, Daim S, et al. The adaptor asc exacerbates lethal listeria monocytogenes infection by mediating il-18 production in an inflammasome-dependent and -independent manner. Eur J Immunol (2014) 44(12):3696–707. doi: 10.1002/eji.201444673
62. Bassler K, Schulte-Schrepping J, Warnat-Herresthal S, Aschenbrenner AC, Schultze JL. The myeloid cell compartment-cell by cell. Annu Rev Immunol (2019) 37:269–93. doi: 10.1146/annurev-immunol-042718-041728
63. Junqueira C, Crespo A, Ranjbar S, de Lacerda LB, Lewandrowski M, Ingber J, et al. Fcgammar-mediated sars-Cov-2 infection of monocytes activates inflammation. Nature (2022) 606(7914):576–84. doi: 10.1038/s41586-022-04702-4
64. Lage SL, Amaral EP, Hilligan KL, Laidlaw E, Rupert A, Namasivayan S, et al. Persistent oxidative stress and inflammasome activation in Cd14(High)Cd16(-) monocytes from covid-19 patients. Front Immunol (2021) 12:799558. doi: 10.3389/fimmu.2021.799558
65. Lucchesi A, Silimbani P, Musuraca G, Cerchione C, Martinelli G, Di Carlo P, et al. Clinical and biological data on the use of hydroxychloroquine against sars-Cov-2 could support the role of the Nlrp3 inflammasome in the pathogenesis of respiratory disease. J Med Virol (2021) 93(1):124–6. doi: 10.1002/jmv.26217
66. Sur S, Steele R, Isbell TS, Ray R, Ray RB. Circulatory exosomes from covid-19 patients trigger Nlrp3 inflammasome in endothelial cells. mBio (2022) 13(3):e0095122. doi: 10.1128/mbio.00951-22
67. Schwartz BG, Economides C, Mayeda GS, Burstein S, Kloner RA. The endothelial cell in health and disease: its function, dysfunction, measurement and therapy. Int J impotence Res (2010) 22(2):77–90. doi: 10.1038/ijir.2009.59
68. Shrivastava G, Valenzuela Leon PC, Calvo E. Inflammasome fuels dengue severity. Front Cell infection Microbiol (2020) 10:489. doi: 10.3389/fcimb.2020.00489
69. Ma J, Zhu F, Zhao M, Shao F, Yu D, Ma J, et al. Sars-Cov-2 nucleocapsid suppresses host pyroptosis by blocking gasdermin d cleavage. EMBO J (2021) 40(18):e108249. doi: 10.15252/embj.2021108249
70. Yalcinkaya M, Liu W, Islam MN, Kotini AG, Gusarova GA, Fidler TP, et al. Modulation of the Nlrp3 inflammasome by sars-Cov-2 envelope protein. Sci Rep (2021) 11(1):24432. doi: 10.1038/s41598-021-04133-7
71. Kim NE, Kim DK, Song YJ. Sars-Cov-2 nonstructural proteins 1 and 13 suppress caspase-1 and the Nlrp3 inflammasome activation. Microorganisms (2021) 9(3):494. doi: 10.3390/microorganisms9030494
72. Mick E, Kamm J, Pisco AO, Ratnasiri K, Babik JM, Castaneda G, et al. Upper airway gene expression reveals suppressed immune responses to sars-Cov-2 compared with other respiratory viruses. Nat Commun (2020) 11(1):5854. doi: 10.1038/s41467-020-19587-y
73. Zhao N, Di B, Xu LL. The Nlrp3 inflammasome and covid-19: activation, pathogenesis and therapeutic strategies. Cytokine Growth factor Rev (2021) 61:2–15. doi: 10.1016/j.cytogfr.2021.06.002
74. Cabrera-Garcia D, Bekdash R, Abbott GW, Yazawa M, Harrison NL. The envelope protein of sars-Cov-2 increases intra-golgi ph and forms a cation channel that is regulated by ph. J Physiol (2021) 599(11):2851–68. doi: 10.1113/JP281037
75. Xu H, Akinyemi IA, Chitre SA, Loeb JC, Lednicky JA, McIntosh MT, et al. Sars-Cov-2 viroporin encoded by Orf3a triggers the Nlrp3 inflammatory pathway. Virology (2022) 568:13–22. doi: 10.1016/j.virol.2022.01.003
76. He Y, Zeng MY, Yang D, Motro B, Nunez G. Nek7 is an essential mediator of Nlrp3 activation downstream of potassium efflux. Nature (2016) 530(7590):354–7. doi: 10.1038/nature16959
77. Tian M, Liu W, Li X, Zhao P, Shereen MA, Zhu C, et al. Hif-1alpha promotes sars-Cov-2 infection and aggravates inflammatory responses to covid-19. Signal transduction targeted Ther (2021) 6(1):308. doi: 10.1038/s41392-021-00726-w
78. Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V. Caspase-4 mediates non-canonical activation of the Nlrp3 inflammasome in human myeloid cells. Eur J Immunol (2015) 45(10):2911–7. doi: 10.1002/eji.201545523
79. Rodrigues TS, Caetano CCS, de Sa KSG, Almeida L, Becerra A, Goncalves AV, et al. Casp4/11 contributes to Nlrp3 activation and covid-19 exacerbation. J Infect Dis (2023). doi: 10.1093/infdis/jiad037
80. Eisfeld HS, Simonis A, Winter S, Chhen J, Stroh LJ, Krey T, et al. Viral glycoproteins induce Nlrp3 inflammasome activation and pyroptosis in macrophages. Viruses (2021) 13(10):2076. doi: 10.3390/v13102076
81. Olajide OA, Iwuanyanwu VU, Lepiarz-Raba I, Al-Hindawi AA. Induction of exaggerated cytokine production in human peripheral blood mononuclear cells by a recombinant sars-Cov-2 spike glycoprotein S1 and its inhibition by dexamethasone. Inflammation (2021) 44(5):1865–77. doi: 10.1007/s10753-021-01464-5
82. Kucia M, Ratajczak J, Bujko K, Adamiak M, Ciechanowicz A, Chumak V, et al. An evidence that sars-Cov-2/Covid-19 spike protein (Sp) damages hematopoietic Stem/Progenitor cells in the mechanism of pyroptosis in Nlrp3 inflammasome-dependent manner. Leukemia (2021) 35(10):3026–9. doi: 10.1038/s41375-021-01332-z
83. Theobald SJ, Simonis A, Georgomanolis T, Kreer C, Zehner M, Eisfeld HS, et al. Long-lived macrophage reprogramming drives spike protein-mediated inflammasome activation in covid-19. EMBO Mol Med (2021) 13(8):e14150. doi: 10.15252/emmm.202114150
84. Campbell GR, To RK, Hanna J, Spector SA. Sars-Cov-2, sars-Cov-1, and hiv-1 derived ssrna sequences activate the Nlrp3 inflammasome in human macrophages through a non-classical pathway. iScience (2021) 24(4):102295. doi: 10.1016/j.isci.2021.102295
85. Campbell GR, Rawat P, Spector SA. Pacritinib inhibition of Irak1 blocks aberrant Tlr8 signalling by sars-Cov-2 and hiv-1-Derived rna. J innate Immun (2022) 15:96–106. doi: 10.1159/000525292
86. Kim H, Lee HS, Ahn JH, Hong KS, Jang JG, An J, et al. Lung-selective 25-hydroxycholesterol nanotherapeutics as a suppressor of covid-19-Associated cytokine storm. Nano Today (2021) 38:101149. doi: 10.1016/j.nantod.2021.101149
87. Saeedi-Boroujeni A, Mahmoudian-Sani MR. Anti-inflammatory potential of quercetin in covid-19 treatment. J Inflammation (2021) 18(1):3. doi: 10.1186/s12950-021-00268-6
88. Xian H, Liu Y, Rundberg Nilsson A, Gatchalian R, Crother TR, Tourtellotte WG, et al. Metformin inhibition of mitochondrial atp and DNA synthesis abrogates Nlrp3 inflammasome activation and pulmonary inflammation. Immunity (2021) 54(7):1463–77 e11. doi: 10.1016/j.immuni.2021.05.004
89. Ghaleh HEG, Hosseini A, Aghamollaei H, Fasihi-Ramandi M, Alishiri G, Saeedi-Boroujeni A, et al. Nlrp3 inflammasome activation and oxidative stress status in the mild and moderate sars-Cov-2 infected patients: impact of melatonin as a medicinal supplement. Z fur Naturforschung C J Biosci (2022) 77(1-2):37–42. doi: 10.1515/znc-2021-0101
90. Zeng J, Xie X, Feng XL, Xu L, Han JB, Yu D, et al. Specific inhibition of the Nlrp3 inflammasome suppresses immune overactivation and alleviates covid-19 like pathology in mice. EBioMedicine (2022) 75:103803. doi: 10.1016/j.ebiom.2021.103803
91. Milara J, Martinez-Exposito F, Montero P, Roger I, Bayarri MA, Ribera P, et al. N-acetylcysteine reduces inflammasome activation induced by sars-Cov-2 proteins in vitro. Int J Mol Sci (2022) 23(23):14518. doi: 10.3390/ijms232314518
92. de Almeida L, da Silva ALN, Rodrigues TS, Oliveira S, Ishimoto AY, Seribelli AA, et al. Identification of immunomodulatory drugs that inhibit multiple inflammasomes and impair sars-Cov-2 infection. Sci Adv (2022) 8(37):eabo5400. doi: 10.1126/sciadv.abo5400
93. Del Re A, Corpetti C, Pesce M, Seguella L, Steardo L, Palenca I, et al. Ultramicronized palmitoylethanolamide inhibits Nlrp3 inflammasome expression and pro-inflammatory response activated by sars-Cov-2 spike protein in cultured murine alveolar macrophages. Metabolites (2021) 11(9):592. doi: 10.3390/metabo11090592
94. Corpetti C, Del Re A, Seguella L, Palenca I, Rurgo S, De Conno B, et al. Cannabidiol inhibits sars-Cov-2 spike (S) protein-induced cytotoxicity and inflammation through a ppargamma-dependent Tlr4/Nlrp3/Caspase-1 signaling suppression in caco-2 cell line. Phytother Res (2021) 35(12):6893–903. doi: 10.1002/ptr.7302
95. Semmarath W, Mapoung S, Umsumarng S, Arjsri P, Srisawad K, Thippraphan P, et al. Cyanidin-3-O-Glucoside and peonidin-3-O-Glucoside-Rich fraction of black rice germ and bran suppresses inflammatory responses from sars-Cov-2 spike glycoprotein S1-induction in vitro in A549 lung cells and thp-1 macrophages Via inhibition of the Nlrp3 inflammasome pathway. Nutrients (2022) 14(13):2738. doi: 10.3390/nu14132738
96. Arjsri P, Srisawad K, Mapoung S, Semmarath W, Thippraphan P, Umsumarng S, et al. Hesperetin from root extract of clerodendrum petasites s. Moore inhibits sars-Cov-2 spike protein S1 subunit-induced Nlrp3 inflammasome in A549 lung cells Via modulation of the Akt/Mapk/Ap-1 pathway. Int J Mol Sci (2022) 23(18):10346. doi: 10.3390/ijms231810346
97. Dissook S, Umsumarng S, Mapoung S, Semmarath W, Arjsri P, Srisawad K, et al. Luteolin-rich fraction from perilla frutescens seed meal inhibits spike glycoprotein S1 of sars-Cov-2-Induced Nlrp3 inflammasome lung cell inflammation Via regulation of Jak1/Stat3 pathway: a potential anti-inflammatory compound against inflammation-induced long-covid. Front Med (2022) 9:1072056. doi: 10.3389/fmed.2022.1072056
98. Bertoni A, Penco F, Mollica H, Bocca P, Prigione I, Corcione A, et al. Spontaneous Nlrp3 inflammasome-driven il-1β secretion is induced in severe covid-19 patients and responds to anakinra treatment. J Allergy Clin Immunol (2022) 150(4):796–805. doi: 10.1016/j.jaci.2022.05.029
99. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin d regulates interleukin-1 secretion from living macrophages. Immunity (2018) 48(1):35–44 e6. doi: 10.1016/j.immuni.2017.11.013
100. Campbell GR, To RK, Spector SA. Trem-1 protects hiv-1-Infected macrophages from apoptosis through maintenance of mitochondrial function. mBio (2019) 10(6):e02638–19. doi: 10.1128/mBio.02638-19
101. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with covid-19. Nat Med (2020) 26(6):842–4. doi: 10.1038/s41591-020-0901-9
102. Lin KM, Hu W, Troutman TD, Jennings M, Brewer T, Li X, et al. Irak-1 bypasses priming and directly links tlrs to rapid Nlrp3 inflammasome activation. Proc Natl Acad Sci United States America (2014) 111(2):775–80. doi: 10.1073/pnas.1320294111
103. Sun X, Liu Y, Huang Z, Xu W, Hu W, Yi L, et al. Sars-Cov-2 non-structural protein 6 triggers Nlrp3-dependent pyroptosis by targeting Atp6ap1. Cell Death differentiation (2022) 29(6):1240–54. doi: 10.1038/s41418-021-00916-7
104. Sun Q, Fan J, Billiar TR, Scott MJ. Inflammasome and autophagy regulation - a two-way Street. Mol Med (2017) 23:188–95. doi: 10.2119/molmed.2017.00077
105. Pan P, Shen M, Yu Z, Ge W, Chen K, Tian M, et al. Sars-Cov-2 n protein promotes Nlrp3 inflammasome activation to induce hyperinflammation. Nat Commun (2021) 12(1):4664. doi: 10.1038/s41467-021-25015-6
106. Planes R, Pinilla M, Santoni K, Hessel A, Passemar C, Lay K, et al. Human Nlrp1 is a sensor of pathogenic coronavirus 3cl proteases in lung epithelial cells. Mol Cell (2022) 82(13):2385–400 e9. doi: 10.1016/j.molcel.2022.04.033
107. Karki R, Kanneganti TD. The 'Cytokine storm': molecular mechanisms and therapeutic prospects. Trends Immunol (2021) 42(8):681–705. doi: 10.1016/j.it.2021.06.001
108. Toldo S, Bussani R, Nuzzi V, Bonaventura A, Mauro AG, Cannata A, et al. Inflammasome formation in the lungs of patients with fatal covid-19. Inflammation Res (2021) 70(1):7–10. doi: 10.1007/s00011-020-01413-2
109. Wen W, Su W, Tang H, Le W, Zhang X, Zheng Y, et al. Immune cell profiling of covid-19 patients in the recovery stage by single-cell sequencing. Cell Discovery (2020) 6:31. doi: 10.1038/s41421-020-0168-9
110. Szabo PA, Dogra P, Gray JI, Wells SB, Connors TJ, Weisberg SP, et al. Longitudinal profiling of respiratory and systemic immune responses reveals myeloid cell-driven lung inflammation in severe covid-19. Immunity (2021) 54(4):797–814 e6. doi: 10.1016/j.immuni.2021.03.005
111. Zou Y, Guo H, Zhang Y, Zhang Z, Liu Y, Wang J, et al. Analysis of coagulation parameters in patients with covid-19 in shanghai, China. Bioscience Trends (2020) 14(4):285–9. doi: 10.5582/bst.2020.03086
112. Abernethy K, Sivakumar P, Patrick T, Robbie H, Periselneris J. Coexistent covid-19 pneumonia and pulmonary embolism: challenges in identifying dual pathology. Thorax (2020) 75(9):812–4. doi: 10.1136/thoraxjnl-2020-215011
113. Demelo-Rodriguez P, Cervilla-Munoz E, Ordieres-Ortega L, Parra-Virto A, Toledano-Macias M, Toledo-Samaniego N, et al. Incidence of asymptomatic deep vein thrombosis in patients with covid-19 pneumonia and elevated d-dimer levels. Thromb Res (2020) 192:23–6. doi: 10.1016/j.thromres.2020.05.018
114. Paul O, Tao JQ, West E, Litzky L, Feldman M, Montone K, et al. Pulmonary vascular inflammation with fatal coronavirus disease 2019 (Covid-19): possible role for the Nlrp3 inflammasome. Respir Res (2022) 23(1):25. doi: 10.1186/s12931-022-01944-8
115. Rosell A, Havervall S, von Meijenfeldt F, Hisada Y, Aguilera K, Grover SP, et al. Patients with covid-19 have elevated levels of circulating extracellular vesicle tissue factor activity that is associated with severity and mortality-brief report. Arteriosclerosis thrombosis Vasc Biol (2021) 41(2):878–82. doi: 10.1161/ATVBAHA.120.315547
116. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil extracellular traps in covid-19. JCI Insight (2020) 5(11):e138999. doi: 10.1172/jci.insight.138999
117. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in covid-19 acute respiratory distress syndrome. Blood (2020) 136(10):1169–79. doi: 10.1182/blood.2020007008
118. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in covid-19 immunothrombosis. J Clin Invest (2020) 130(11):6151–7. doi: 10.1172/JCI141374
119. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. Sars-Cov-2-Triggered neutrophil extracellular traps mediate covid-19 pathology. J Exp Med (2020) 217(12):e20201129. doi: 10.1084/jem.20201129
120. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in covid-19. New Engl J Med (2020) 383(2):120–8. doi: 10.1056/NEJMoa2015432
121. Nicolai L, Leunig A, Brambs S, Kaiser R, Weinberger T, Weigand M, et al. Immunothrombotic dysregulation in covid-19 pneumonia is associated with respiratory failure and coagulopathy. Circulation (2020) 142(12):1176–89. doi: 10.1161/CIRCULATIONAHA.120.048488
122. Zhao CL, Rapkiewicz A, Maghsoodi-Deerwester M, Gupta M, Cao W, Palaia T, et al. Pathological findings in the postmortem liver of patients with coronavirus disease 2019 (Covid-19). Hum Pathol (2021) 109:59–68. doi: 10.1016/j.humpath.2020.11.015
123. Matschke J, Lutgehetmann M, Hagel C, Sperhake JP, Schroder AS, Edler C, et al. Neuropathology of patients with covid-19 in Germany: a post-mortem case series. Lancet Neurol (2020) 19(11):919–29. doi: 10.1016/S1474-4422(20)30308-2
124. Cama VF, Marin-Prida J, Acosta-Rivero N, Acosta EF, Diaz LO, Casadesus AV, et al. The microglial Nlrp3 inflammasome is involved in human sars-Cov-2 cerebral pathogenicity: a report of three post-mortem cases. J neuroimmunol (2021) 361:577728. doi: 10.1016/j.jneuroim.2021.577728
125. Kroemer A, Khan K, Plassmeyer M, Alpan O, Haseeb MA, Gupta R, et al. Inflammasome activation and pyroptosis in lymphopenic liver patients with covid-19. J Hepatol (2020) 73(5):1258–62. doi: 10.1016/j.jhep.2020.06.034
126. Ji D, Qin E, Xu J, Zhang D, Cheng G, Wang Y, et al. Non-alcoholic fatty liver diseases in patients with covid-19: a retrospective study. J Hepatol (2020) 73(2):451–3. doi: 10.1016/j.jhep.2020.03.044
127. Hoel H, Heggelund L, Reikvam DH, Stiksrud B, Ueland T, Michelsen AE, et al. Elevated markers of gut leakage and inflammasome activation in covid-19 patients with cardiac involvement. J Internal Med (2021) 289(4):523–31. doi: 10.1111/joim.13178
128. Shimabukuro-Vornhagen A, Godel P, Subklewe M, Stemmler HJ, Schlosser HA, Schlaak M, et al. Cytokine release syndrome. J immunotherapy Cancer (2018) 6(1):56. doi: 10.1186/s40425-018-0343-9
129. Freeman TL, Swartz TH. Targeting the Nlrp3 inflammasome in severe covid-19. Front Immunol (2020) 11:1518. doi: 10.3389/fimmu.2020.01518
130. Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin immunopathol (2017) 39(5):529–39. doi: 10.1007/s00281-017-0629-x
131. Ratajczak MZ, Kucia M. Sars-Cov-2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine "Storm" and risk factor for damage of hematopoietic stem cells. Leukemia (2020) 34(7):1726–9. doi: 10.1038/s41375-020-0887-9
132. Barker BR, Taxman DJ, Ting JP. Cross-regulation between the il-1beta/Il-18 processing inflammasome and other inflammatory cytokines. Curr Opin Immunol (2011) 23(5):591–7. doi: 10.1016/j.coi.2011.07.005
133. Uciechowski P, Dempke WCM. Interleukin-6: a masterplayer in the cytokine network. Oncology (2020) 98(3):131–7. doi: 10.1159/000505099
134. Zhang C, Wu Z, Li JW, Zhao H, Wang GQ. Cytokine release syndrome in severe covid-19: interleukin-6 receptor antagonist tocilizumab may be the key to reduce mortality. Int J antimicrobial Agents (2020) 55(5):105954. doi: 10.1016/j.ijantimicag.2020.105954
135. McGonagle D, Sharif K, O'Regan A, Bridgewood C. The role of cytokines including interleukin-6 in covid-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev (2020) 19(6):102537. doi: 10.1016/j.autrev.2020.102537
136. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of covid-19 associated with acute respiratory distress syndrome. Lancet Respir Med (2020) 8(4):420–2. doi: 10.1016/S2213-2600(20)30076-X
137. Lara PC, Macias-Verde D, Burgos-Burgos J. Age-induced Nlrp3 inflammasome over-activation increases lethality of sars-Cov-2 pneumonia in elderly patients. Aging Dis (2020) 11(4):756–62. doi: 10.14336/AD.2020.0601
138. Jones HD, Crother TR, Gonzalez-Villalobos RA, Jupelli M, Chen S, Dagvadorj J, et al. The Nlrp3 inflammasome is required for the development of hypoxemia in Lps/Mechanical ventilation acute lung injury. Am J Respir Cell Mol Biol (2014) 50(2):270–80. doi: 10.1165/rcmb.2013-0087OC
139. Dolinay T, Kim YS, Howrylak J, Hunninghake GM, An CH, Fredenburgh L, et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med (2012) 185(11):1225–34. doi: 10.1164/rccm.201201-0003OC
140. Grailer JJ, Canning BA, Kalbitz M, Haggadone MD, Dhond RM, Andjelkovic AV, et al. Critical role for the Nlrp3 inflammasome during acute lung injury. J Immunol (2014) 192(12):5974–83. doi: 10.4049/jimmunol.1400368
141. van de Veerdonk FL, Netea MG. Blocking il-1 to prevent respiratory failure in covid-19. Crit Care (2020) 24(1):445. doi: 10.1186/s13054-020-03166-0
142. Vora SM, Lieberman J, Wu H. Inflammasome activation at the crux of severe covid-19. Nat Rev Immunol (2021) 21(11):694–703. doi: 10.1038/s41577-021-00588-x
143. Xiong S, Hong Z, Huang LS, Tsukasaki Y, Nepal S, Di A, et al. Il-1beta suppression of ve-cadherin transcription underlies sepsis-induced inflammatory lung injury. J Clin Invest (2020) 130(7):3684–98. doi: 10.1172/JCI136908
144. Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial vegf receptor signalling. Nat Rev Mol Cell Biol (2016) 17(10):611–25. doi: 10.1038/nrm.2016.87
145. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers (2019) 5(1):18. doi: 10.1038/s41572-019-0069-0
146. Costela-Ruiz VJ, Illescas-Montes R, Puerta-Puerta JM, Ruiz C, Melguizo-Rodriguez L. Sars-Cov-2 infection: the role of cytokines in covid-19 disease. Cytokine Growth factor Rev (2020) 54:62–75. doi: 10.1016/j.cytogfr.2020.06.001
147. Mason RJ. Thoughts on the alveolar phase of covid-19. Am J Physiol Lung Cell Mol Physiol (2020) 319(1):L115–L20. doi: 10.1152/ajplung.00126.2020
148. Gunther A, Ruppert C, Schmidt R, Markart P, Grimminger F, Walmrath D, et al. Surfactant alteration and replacement in acute respiratory distress syndrome. Respir Res (2001) 2(6):353–64. doi: 10.1186/rr86
149. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, et al. Covid-19: consider cytokine storm syndromes and immunosuppression. Lancet (2020) 395(10229):1033–4. doi: 10.1016/S0140-6736(20)30628-0
150. Joly BS, Siguret V, Veyradier A. Understanding pathophysiology of hemostasis disorders in critically ill patients with covid-19. Intensive Care Med (2020) 46(8):1603–6. doi: 10.1007/s00134-020-06088-1
151. Stefan N, Birkenfeld AL, Schulze MB, Ludwig DS. Obesity and impaired metabolic health in patients with covid-19. Nat Rev Endocrinol (2020) 16(7):341–2. doi: 10.1038/s41574-020-0364-6
152. Qiao J, Wu X, Luo Q, Wei G, Xu M, Wu Y, et al. Nlrp3 regulates platelet integrin Alphaiibbeta3 outside-in signaling, hemostasis and arterial thrombosis. Haematologica (2018) 103(9):1568–76. doi: 10.3324/haematol.2018.191700
153. Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, et al. Inflammasome activation triggers blood clotting and host death through pyroptosis. Immunity (2019) 50(6):1401–11 e4. doi: 10.1016/j.immuni.2019.04.003
154. Yang X, Cheng X, Tang Y, Qiu X, Wang Y, Kang H, et al. Bacterial endotoxin activates the coagulation cascade through gasdermin d-dependent phosphatidylserine exposure. Immunity (2019) 51(6):983–96 e6. doi: 10.1016/j.immuni.2019.11.005
155. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr., et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci United States America (2010) 107(36):15880–5. doi: 10.1073/pnas.1005743107
156. Sorvillo N, Cherpokova D, Martinod K, Wagner DD. Extracellular DNA net-works with dire consequences for health. Circ Res (2019) 125(4):470–88. doi: 10.1161/CIRCRESAHA.119.314581
157. Munzer P, Negro R, Fukui S, di Meglio L, Aymonnier K, Chu L, et al. Nlrp3 inflammasome assembly in neutrophils is supported by Pad4 and promotes netosis under sterile conditions. Front Immunol (2021) 12:683803. doi: 10.3389/fimmu.2021.683803
158. Huang W, Jiao J, Liu J, Huang M, Hu Y, Ran W, et al. Mfg-E8 accelerates wound healing in diabetes by regulating "Nlrp3 inflammasome-neutrophil extracellular traps" axis. Cell Death Discovery (2020) 6:84. doi: 10.1038/s41420-020-00318-7
159. de Bont CM, Boelens WC, Pruijn GJM. Netosis, complement, and coagulation: a triangular relationship. Cell Mol Immunol (2019) 16(1):19–27. doi: 10.1038/s41423-018-0024-0
160. Laforge M, Elbim C, Frere C, Hemadi M, Massaad C, Nuss P, et al. Tissue damage from neutrophil-induced oxidative stress in covid-19. Nat Rev Immunol (2020) 20(9):515–6. doi: 10.1038/s41577-020-0407-1
161. Poor HD. Pulmonary thrombosis and thromboembolism in covid-19. Chest (2021) 160(4):1471–80. doi: 10.1016/j.chest.2021.06.016
162. Pieterse E, Rother N, Garsen M, Hofstra JM, Satchell SC, Hoffmann M, et al. Neutrophil extracellular traps drive endothelial-to-Mesenchymal transition. Arteriosclerosis thrombosis Vasc Biol (2017) 37(7):1371–9. doi: 10.1161/ATVBAHA.117.309002
163. Becker K, Beythien G, de Buhr N, Stanelle-Bertram S, Tuku B, Kouassi NM, et al. Vasculitis and neutrophil extracellular traps in lungs of golden Syrian hamsters with sars-Cov-2. Front Immunol (2021) 12:640842. doi: 10.3389/fimmu.2021.640842
164. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting potential drivers of covid-19: neutrophil extracellular traps. J Exp Med (2020) 217(6):e20200652. doi: 10.1084/jem.20200652
165. Szturmowicz M, Demkow U. Neutrophil extracellular traps (Nets) in severe sars-Cov-2 lung disease. Int J Mol Sci (2021) 22(16):8854. doi: 10.3390/ijms22168854
166. Ackermann M, Anders HJ, Bilyy R, Bowlin GL, Daniel C, De Lorenzo R, et al. Patients with covid-19: in the dark-nets of neutrophils. Cell Death differentiation (2021) 28(11):3125–39. doi: 10.1038/s41418-021-00805-z
167. Chen R, Wang K, Yu J, Howard D, French L, Chen Z, et al. The spatial and cell-type distribution of sars-Cov-2 receptor Ace2 in the human and mouse brains. Front Neurol (2020) 11:573095. doi: 10.3389/fneur.2020.573095
168. Ribeiro DE, Oliveira-Giacomelli A, Glaser T, Arnaud-Sampaio VF, Andrejew R, Dieckmann L, et al. Hyperactivation of P2x7 receptors as a culprit of covid-19 neuropathology. Mol Psychiatry (2021) 26(4):1044–59. doi: 10.1038/s41380-020-00965-3
169. Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Trans neurodegeneration (2020) 9(1):42. doi: 10.1186/s40035-020-00221-2
170. Albornoz EA, Amarilla AA, Modhiran N, Parker S, Li XX, Wijesundara DK, et al. Sars-Cov-2 drives Nlrp3 inflammasome activation in human microglia through spike protein. Mol Psychiatry (2022). doi: 10.1038/s41380-022-01831-0
171. Olajide OA, Iwuanyanwu VU, Adegbola OD, Al-Hindawi AA. Sars-Cov-2 spike glycoprotein S1 induces neuroinflammation in bv-2 microglia. Mol Neurobiol (2022) 59(1):445–58. doi: 10.1007/s12035-021-02593-6
172. Kersse K, Bertrand MJ, Lamkanfi M, Vandenabeele P. Nod-like receptors and the innate immune system: coping with danger, damage and death. Cytokine Growth factor Rev (2011) 22(5-6):257–76. doi: 10.1016/j.cytogfr.2011.09.003
173. Sepehrinezhad A, Gorji A, Sahab Negah S. Sars-Cov-2 may trigger inflammasome and pyroptosis in the central nervous system: a mechanistic view of neurotropism. Inflammopharmacology (2021) 29(4):1049–59. doi: 10.1007/s10787-021-00845-4
174. Israelov H, Ravid O, Atrakchi D, Rand D, Elhaik S, Bresler Y, et al. Caspase-1 has a critical role in blood-brain barrier injury and its inhibition contributes to multifaceted repair. J Neuroinflamm (2020) 17(1):267. doi: 10.1186/s12974-020-01927-w
175. Hauptmann J, Johann L, Marini F, Kitic M, Colombo E, Mufazalov IA, et al. Interleukin-1 promotes autoimmune neuroinflammation by suppressing endothelial heme oxygenase-1 at the blood-brain barrier. Acta neuropathologica (2020) 140(4):549–67. doi: 10.1007/s00401-020-02187-x
176. Wang Y, Jin S, Sonobe Y, Cheng Y, Horiuchi H, Parajuli B, et al. Interleukin-1beta induces blood-brain barrier disruption by downregulating sonic hedgehog in astrocytes. PloS One (2014) 9(10):e110024. doi: 10.1371/journal.pone.0110024
177. Thelin EP, Hall CE, Gupta K, Carpenter KLH, Chandran S, Hutchinson PJ, et al. Elucidating pro-inflammatory cytokine responses after traumatic brain injury in a human stem cell model. J neurotrauma (2018) 35(2):341–52. doi: 10.1089/neu.2017.5155
178. Gong Q, Lin Y, Lu Z, Xiao Z. Microglia-astrocyte cross talk through il-18/Il-18r signaling modulates migraine-like behavior in experimental models of migraine. Neuroscience (2020) 451:207–15. doi: 10.1016/j.neuroscience.2020.10.019
179. Zhong P, Xu J, Yang D, Shen Y, Wang L, Feng Y, et al. Covid-19-Associated gastrointestinal and liver injury: clinical features and potential mechanisms. Signal transduction targeted Ther (2020) 5(1):256. doi: 10.1038/s41392-020-00373-7
180. Gabarre P, Dumas G, Dupont T, Darmon M, Azoulay E, Zafrani L. Acute kidney injury in critically ill patients with covid-19. Intensive Care Med (2020) 46(7):1339–48. doi: 10.1007/s00134-020-06153-9
181. Martinez-Rojas MA, Vega-Vega O, Bobadilla NA. Is the kidney a target of sars-Cov-2? Am J Physiol Renal Physiol (2020) 318(6):F1454–F62. doi: 10.1152/ajprenal.00160.2020
182. Cao Y, Fei D, Chen M, Sun M, Xu J, Kang K, et al. Role of the nucleotide-binding domain-like receptor protein 3 inflammasome in acute kidney injury. FEBS J (2015) 282(19):3799–807. doi: 10.1111/febs.13379
183. da Mata GF, Fernandes DE, Luciano EP, Sales GTM, Riguetti MTP, Kirsztajn GM. Inflammation and kidney involvement in human viral diseases caused by sars-Cov-2, hiv, hcv and hbv. J venomous Anim toxins including Trop Dis (2021) 27:e20200154. doi: 10.1590/1678-9199-JVATITD-2020-0154
184. Gedefaw L, Ullah S, Leung PHM, Cai Y, Yip SP, Huang CL. Inflammasome activation-induced hypercoagulopathy: impact on cardiovascular dysfunction triggered in covid-19 patients. Cells (2021) 10(4):916. doi: 10.3390/cells10040916
185. Kadosh BS, Garshick MS, Gaztanaga J, Moore KJ, Newman JD, Pillinger M, et al. Covid-19 and the heart and vasculature: novel approaches to reduce virus-induced inflammation in patients with cardiovascular disease. Arteriosclerosis thrombosis Vasc Biol (2020) 40(9):2045–53. doi: 10.1161/ATVBAHA.120.314513
186. Thankam FG, Agrawal DK. Molecular chronicles of cytokine burst in patients with coronavirus disease 2019 (Covid-19) with cardiovascular diseases. J Thorac Cardiovasc Surg (2021) 161(2):e217–e26. doi: 10.1016/j.jtcvs.2020.05.083
187. Lazzerini PE, Boutjdir M, Capecchi PL. Covid-19, arrhythmic risk, and inflammation: mind the gap! Circulation (2020) 142(1):7–9. doi: 10.1161/CIRCULATIONAHA.120.047293
188. Casanova JL, Su HC, Effort CHG. A global effort to define the human genetics of protective immunity to sars-Cov-2 infection. Cell (2020) 181(6):1194–9. doi: 10.1016/j.cell.2020.05.016
189. Pairo-Castineira E, Clohisey S, Klaric L, Bretherick AD, Rawlik K, Pasko D, et al. Genetic mechanisms of critical illness in covid-19. Nature (2021) 591(7848):92–8. doi: 10.1038/s41586-020-03065-y
190. Severe Covid GG, Ellinghaus D, Degenhardt F, Bujanda L, Buti M, Albillos A, et al. Genomewide association study of severe covid-19 with respiratory failure. New Engl J Med (2020) 383(16):1522–34. doi: 10.1056/NEJMoa2020283
191. Initiative C-HG. Mapping the human genetic architecture of covid-19. Nature (2021) 600(7889):472–7. doi: 10.1038/s41586-021-03767-x
192. Asgari S, Pousaz LA. Human genetic variants identified that affect covid susceptibility and severity. Nature (2021) 600(7889):390–1. doi: 10.1038/d41586-021-01773-7
193. Povysil G, Butler-Laporte G, Shang N, Wang C, Khan A, Alaamery M, et al. Rare loss-of-Function variants in type I ifn immunity genes are not associated with severe covid-19. J Clin Invest (2021) 131(14):e147834. doi: 10.1172/JCI147834
194. Fallerini C, Daga S, Mantovani S, Benetti E, Picchiotti N, Francisci D, et al. Association of toll-like receptor 7 variants with life-threatening covid-19 disease in males: findings from a nested case-control study. eLife (2021) 10:e67569. doi: 10.7554/eLife.67569
195. Matuozzo D, Talouarn E, Marchal A, Zhang P, Manry J, Seeleuthner Y, et al. Rare predicted loss-of-Function variants of type I ifn immunity genes are associated with life-threatening covid-19. Genome Med (2023) 15(1):22. doi: 10.1186/s13073-023-01173-8
196. Maes M, Tedesco Junior WLD, Lozovoy MAB, Mori MTE, Danelli T, Almeida ERD, et al. In covid-19, Nlrp3 inflammasome genetic variants are associated with critical disease and these effects are partly mediated by the sickness symptom complex: a nomothetic network approach. Mol Psychiatry (2022) 27(4):1945–55. doi: 10.1038/s41380-021-01431-4
197. Wang F, Huang S, Gao R, Zhou Y, Lai C, Li Z, et al. Initial whole-genome sequencing and analysis of the host genetic contribution to covid-19 severity and susceptibility. Cell Discovery (2020) 6(1):83. doi: 10.1038/s41421-020-00231-4
198. Velavan TP, Pallerla SR, Ruter J, Augustin Y, Kremsner PG, Krishna S, et al. Host genetic factors determining covid-19 susceptibility and severity. EBioMedicine (2021) 72:103629. doi: 10.1016/j.ebiom.2021.103629
199. Bazrafkan M, Hosseini E, Nazari M, Amorim CA, Sadeghi MR. Nlrp3 inflammasome: a joint, potential therapeutic target in management of covid-19 and fertility problems. J Reprod Immunol (2021) 148:103427. doi: 10.1016/j.jri.2021.103427
200. Mangalmurti N, Hunter CA. Cytokine storms: understanding covid-19. Immunity (2020) 53(1):19–25. doi: 10.1016/j.immuni.2020.06.017
201. van den Berg DF, Te Velde AA. Severe covid-19: Nlrp3 inflammasome dysregulated. Front Immunol (2020) 11:1580. doi: 10.3389/fimmu.2020.01580
202. Zhang F, Wei Y, He L, Zhang H, Hu Q, Yue H, et al. A trial of pirfenidone in hospitalized adult patients with severe coronavirus disease 2019. Chin Med J (2021) 135(3):368–70. doi: 10.1097/CM9.0000000000001614
203. Deftereos SG, Giannopoulos G, Vrachatis DA, Siasos GD, Giotaki SG, Gargalianos P, et al. Effect of colchicine vs standard care on cardiac and inflammatory biomarkers and clinical outcomes in patients hospitalized with coronavirus disease 2019: the grecco-19 randomized clinical trial. JAMA network Open (2020) 3(6):e2013136. doi: 10.1001/jamanetworkopen.2020.13136
204. Della-Torre E, Della-Torre F, Kusanovic M, Scotti R, Ramirez GA, Dagna L, et al. Treating covid-19 with colchicine in community healthcare setting. Clin Immunol (2020) 217:108490. doi: 10.1016/j.clim.2020.108490
205. Kow CS, Lee LH, Ramachandram DS, Hasan SS, Ming LC, Goh HP. The effect of colchicine on mortality outcome and duration of hospital stay in patients with covid-19: a meta-analysis of randomized trials. Immunity Inflammation Dis (2022) 10(2):255–64. doi: 10.1002/iid3.562
206. Rahhal A, Najim M, Aljundi AH, Mahfouz A, Alyafei SM, Awaisu A, et al. Adding colchicine to tocilizumab in hospitalized patients with severe covid-19 pneumonia: an open-label randomized controlled trial. Medicine (2022) 101(39):e30618. doi: 10.1097/MD.0000000000030843
207. Saeedi-Boroujeni A, Nashibi R, Ghadiri AA, Nakajima M, Salmanzadeh S, Mahmoudian-Sani MR, et al. Tranilast as an adjunctive therapy in hospitalized patients with severe covid- 19: a randomized controlled trial. Arch Med Res (2022) 53(4):368–77. doi: 10.1016/j.arcmed.2022.03.002
208. Madurka I, Vishnevsky A, Soriano JB, Gans SJ, Ore DJS, Rendon A, et al. Dfv890: a new oral Nlrp3 inhibitor-tested in an early phase 2a randomised clinical trial in patients with covid-19 pneumonia and impaired respiratory function. Infection (2022). doi: 10.1007/s15010-022-01904-w
209. Ribeiro SA, Lopes C, Amaral R, Amaral A. The therapeutic potential of colchicine in the complications of Covid19. could the immunometabolic properties of an old and cheap drug help? Metab Open (2020) 7:100045. doi: 10.1016/j.metop.2020.100045
210. Huang Q, Wu X, Zheng X, Luo S, Xu S, Weng J. Targeting inflammation and cytokine storm in covid-19. Pharmacol Res (2020) 159:105051. doi: 10.1016/j.phrs.2020.105051
211. Bauernfried S, Scherr MJ, Pichlmair A, Duderstadt KE, Hornung V. Human Nlrp1 is a sensor for double-stranded rna. Science (2021) 371(6528):eabd0811. doi: 10.1126/science.abd0811
212. Swanson KV, Deng M, Ting JP. The Nlrp3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
213. Kang H, Seo E, Oh YS, Jun HS. Tgf-beta activates Nlrp3 inflammasome by an autocrine production of tgf-beta in lx-2 human hepatic stellate cells. Mol Cell Biochem (2022) 477(5):1329–38. doi: 10.1007/s11010-022-04369-5
Keywords: SARS-CoV-2, COVID-19, inflammasomes, proinflammatory cytokines, immunopathogenesis, inflammasome-targeted therapies
Citation: Wang M, Yu F, Chang W, Zhang Y, Zhang L and Li P (2023) Inflammasomes: a rising star on the horizon of COVID-19 pathophysiology. Front. Immunol. 14:1185233. doi: 10.3389/fimmu.2023.1185233
Received: 13 March 2023; Accepted: 02 May 2023;
Published: 12 May 2023.
Edited by:
Shailendra Saxena, King George’s Medical University, IndiaReviewed by:
Rui Song, Capital Medical University, ChinaCopyright © 2023 Wang, Yu, Chang, Zhang, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Man Wang, d2FuZ21hbkBxZHUuZWR1LmNu; Peifeng Li, cGVpZmxpQHFkdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.