 Fadi Sheban
Fadi Sheban
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol. , 09 June 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1183578
This article is part of the Research Topic Emerging Talents in Cancer Immunity and Immunotherapy: 2022 View all 7 articles
Immunotherapy has revolutionized cancer care in the past decade. Treatment with immune checkpoint inhibitors has demonstrated promising clinical activity against tumors. However, only a subset of patients responds to these treatments, limiting their potential benefit. Efforts to understand, predict, and overcome the lack of response in patients, have thus far focused mainly on the tumor immunogenicity and the quantity and characteristics of tumor-infiltrating T cells, since these cells are the main effectors of immunotherapies. However, recent comprehensive analyses of the tumor microenvironment (TME) in the context of immune checkpoint blockade (ICB) therapy have revealed critical functions of other immune cells in the effective anti-tumor response, highlighting the need to account for complex cell-cell interaction and communication underlying clinical outputs. In this perspective, I discuss the current understanding of the crucial roles of tumor-associated macrophages (TAMs) in the success of T cell-directed immune checkpoint blockade therapies, as well as the present, and the future of clinical trials on combinatorial therapies targeting both cell types.
Cancer immunotherapy has emerged in the late 19th century when Dr. William B Coley treated cancer patients with intratumoral injections of live pathogens (1). The mechanism by which Coley’s pathogens led to the eradication of cancer was not clear at the time. However, today we understand that they elicit a local immune response that fights cancerous cells incidentally (2). In 1976, Coley’s work was followed by the first report of successful cancer immunotherapy using Bacillus Calmette-Guerin (BCG) to treat bladder cancer which is still in use until this day (3). A very important milestone in cancer immunotherapy was the discovery of immune checkpoints (4). These checkpoints are negative regulators of the immune system and particularly T cell activation and are evolutionarily conserved to fine-tune the duration and extent of immune responses to ensure self-tolerance (5, 6). Two prominent examples of immune checkpoints are the Programmed cell Death 1 (PD-1) and Cytotoxic T Lymphocyte Antigen 4 (CTLA-4). These receptors are expressed by activated T cells (among other cell types) and act as natural brakes allowing T cells to exert their function in a timely manner that ensures effective protection from cancer cells and pathogens while preventing autoimmunity (7, 8) In the last decade, inhibitory molecules that target these checkpoints, namely ICB therapies, have been developed and successfully used for the treatment of various cancer types. The success of ICB molecules in cancer treatment eventually earned James P. Allison and Tasuku Honjo the 2018 Nobel Prize in Physiology or Medicine (9).
Despite the promising results of cancer ICB therapy, durable clinical responses are limited to a subset of individuals with specific cancer types (10–12). Therefore, tremendous efforts were made to understand and predict the lack of response in patients. One of the most prominent biomarkers used to predict ICB response in some cancer types is tumor immunogenicity, which is primarily determined by tumor mutational burden, genomic instability, and efficiency of antigen presentation (13–16). The stronger the immunogenicity of the tumor, the higher the chances for a patient to benefit from ICB treatment. Additional biomarkers include the tumor immunophenotype of patients, which is mainly characterized by T cell infiltration into the TME. Hot tumors (immune-inflamed) are defined by high infiltration of T cells, whereas cold tumors (immune-desert) lack or have low T cell percentages (17–19). Patients with hot tumors were shown to be more likely to have a beneficial clinical response to ICB. Moreover, high expression levels of molecules that are targeted by ICB therapies such as PD-1 ligand (PD-L1) within the TME are considered as a biomarker for positive treatment responses (20). However, these parameters are limited to certain types of cancers and are not perfectly accurate. For instance, some patients which are considered negative for PD-L1 expression can still benefit from anti-PD-1/L1 treatment (21–24). This emphasizes that we still do not fully understand the mechanism of action (MOA) of such therapies and the need for further research to improve ICB therapy outcomes.
Recently, several comprehensive analyses of the TME have shed the light on the involvement of different immune cells, besides the extensively studied T cells, in the MOA of different immunotherapies. Specifically, TAMs were described as critical in shaping the TME and being directly involved in the treatment outcomes of immunotherapies. In this perspective, I describe the recent advancements in the understanding of the roles of TAMs in shaping anti-tumor responses elicited by cancer ICB therapies. Further, I highlight the potential of combination therapies targeting TAMs together with T cell-directed ICB molecules and the ongoing and future clinical trials.
TAMs are an essential component of the TME and are usually key tumor-promoting players in most solid cancers. By orchestrating immunosuppression, angiogenesis, tissue remodeling, and tumor cell proliferation they support tumor growth and the formation of metastasis (25–28). As a consequence, high TAMs infiltration in solid tumors is usually associated with poor prognosis and resistance to chemotherapy and immunotherapy such as ICB therapies (29–37) Historically, macrophages have been divided into two functional phenotypes, referred to as M1 and M2 (38). This dichotomic model is based on the stimulation used to activate macrophages in vitro. M1 polarized macrophages are associated with inflammatory and anti-tumor activities, whereas M2 polarized macrophages are associated with resolution of inflammation and pro-tumor activities. However, in vivo in general, and specifically in the context of cancer, the phenotypes of macrophages are much more complicated. Single-cell analyses of human and murine TAMs have revealed several TAMs phenotypes that can even co-exist, indicating a more complex scenario beyond the simplistic M1/M2 classification (39, 40).
Manipulation or depletion of TAMs in the TME can potentiate several immunotherapeutic strategies, including ICB, CAR-T cell therapies, and tumor vaccination (25, 41–43). This demonstrates the paramount role of TAMs in shaping treatment responses to immunotherapies. In the past decade, utilizing multi-omics techniques, several research groups have dissected the different functions of TAMs in the context of immunotherapy. In the settings of ICB therapy, TAMs have been shown to play critical expected, and unexpected roles in the effectiveness of various therapeutic molecules. Understanding these roles would help with finding novel biomarkers for response, defining potential therapeutic targets expressed by TAMs, and developing combinatorial therapeutic approaches to enhance the efficacy of the existing ICB therapies.
The foremost goal of ICB therapy is to unleash the cytotoxic activity of T cells which is limited by the immunosuppressive nature of the TME. TAMs are key players in suppressing adaptive anti-tumor immune responses (29, 44). Among the various mechanisms which TAMs apply to attenuate T cell activities, is the high expression of checkpoint ligands such as CD80, CD86, PD-L1, PD-L2, and CD155, etc. (Figure 1A) (45–47). The interaction of some of these ligands with their receptors on T cells was shown to downmodulate the amplitude of T cell activation and proliferation. For instance, PD-1-PD-L1 interaction drives inhibitory intracellular signals in T cells that eventually lead to T cell exhaustion (9). Assessment of PD-L1 expression in tumors is used as a diagnostic marker for anti-PD1 therapy in non-small cell lung cancer (NSCLC) and several other malignancies (17, 48). Both tumor cells and myeloid antigen-presenting cells (APCs) such as TAMs express PD-L1 in the TME. Yet, it was shown in animal models that PD-L1 on tumor cells was largely dispensable for the response to anti-PD1 therapy, whereas PD-L1 in host myeloid cells, was essential for this response (49). Further, it was shown that among myeloid cells, TAMs are the main source of PD-L1 in the TME. Depletion of PD-L1 in TAMs resulted in a reduction in tumor growth compared to control group, yet these effects were much stronger when depleted in dendritic cells (50). Interestingly, besides these extensively studied effects of the immune checkpoint-ligand interactions, it was shown that TAMs can also trap T cells by forming long-lasting interactions and impede them from reaching tumor cells (51). Importantly, the inability of T cells to infiltrate tumors is considered an important mechanism of resistance to ICB (17). Likewise, FasL+ TAMs promote liver metastases by inducing apoptosis of Fas+CD8+ T cells through Fas-FasL interaction, leading to an immune-desert TME (52).
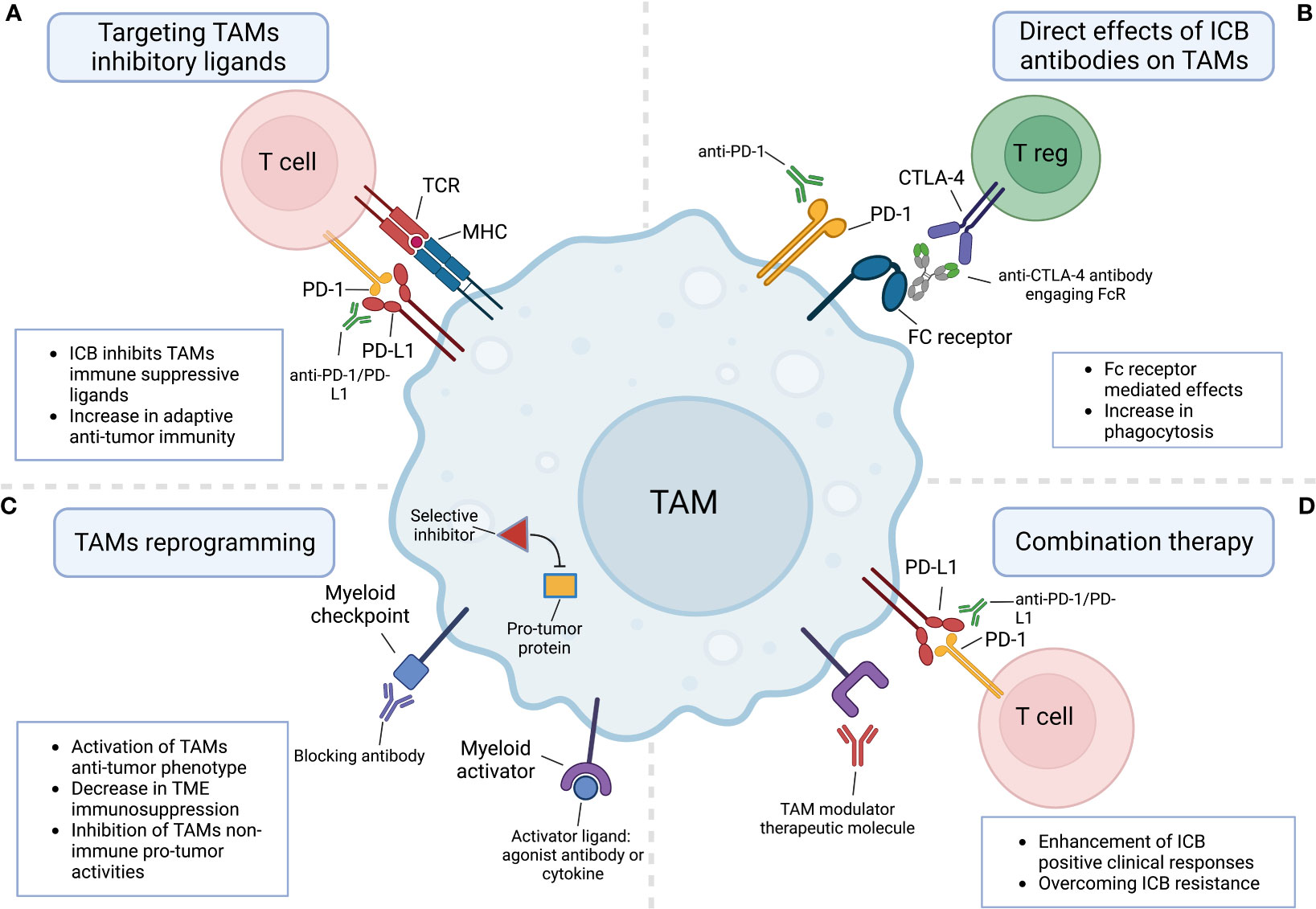
Figure 1 The involvement of tumor-associated macrophages in responses to immune checkpoint blockade therapies. (A) ICB molecules block TAMs cell surface immunosuppressive checkpoint ligands such as PD-L1 from interacting with their counterpart receptors on T cells. (B) TAMs directly respond to ICB molecules either by a direct interaction with the targeted receptor or through interaction with FC receptors expressed by TAMs. (C) TAMs can be reprogrammed by targeting their myeloid checkpoints receptors and pathways or by engaging their activating receptors. (D) Combination therapy combining ICB molecules and TAMs modulator therapeutic molecules boosts ICB clinical positive responses and overcomes resistance. Created with BioRender.com.
More immune checkpoints ligands expressed by macrophages are being discovered and assessed for their ability to suppress T cell responses and thus their potential use as targets for ICB. V-domain Ig suppressor of T cell activation (VISTA) is a novel distinct immunoglobulin inhibitory ligand whose extracellular domain bears homology to the B7 family ligand PD-L1 (53, 54). By interacting with the adhesion and co-inhibitory receptor P-selectin glycoprotein ligand-1 (PSGL-1) on T cells, VISTA suppresses T cells selectively at acidic pH such as that found in TME (54, 55). Hence, VISTA-PSGL-1 axis is considered a new promising target for ICB and is now being assessed in clinical trials. Further research and screening for ligands expressed on TAMs that interfere with anti-tumor T cell responses will open the door for more discoveries and potential targets for ICB therapies.
ICB molecules that target PD-1-PD-L1 interaction have shown remarkable success in treating melanoma and NSCLC. Many monoclonal antibodies (mABs) targeting the PD-1-PD-L1 axis are FDA-approved and widely used to treat cancer patients. Yet, the MOA of these mABs is still not fully understood, limiting the advancement of novel therapeutic approaches and the improvement of the current clinical response rates. Early explanations of how these mABs work were based on the understanding that the two key player cells in the targeted axis are a T cell and another interacting cell expressing PD-1 and PD-L1 respectively. In this interaction, tumor or myeloid cells expressing PD-L1 suppress T cell responses by interacting with PD-1 on these cells.
Previously, it was reported that macrophages express PD-1 in the context of pathogen infection (56). In a more recent study, Gordon et al. aimed to assess whether macrophages might also express PD-1 in the context of cancer. Surprisingly, they showed that both human and mouse TAMs do express PD-1 in the TME. Interestingly, PD-1 expression levels negatively correlated with the phagocytic potency of TAMs against tumor cells, and in vivo blockade of PD-1 on TAMs improved their phagocytic activity, reduced tumor growth, and increased the survival of the mice (57). This study suggested that anti-PD-1 ICB therapy may also function through direct effects on TAMs and opened the door for further investigations. More recent studies confirmed this observation and showed in more detail how the deletion of PD-1 in TAMs induces anti-tumor immunity and suppresses tumor growth (58, 59). Interestingly, a recent study indicated that the functional effects of anti-PD-L1 therapy also are not only mediated by T cells. Instead, it seems that anti-PD-L1 treatment can reprogram TAMs into pro-inflammatory, antigen-presenting cells that help sustain and enhance effector CD8+ T cell activity (60, 61). However, it is not yet clear whether this is a direct or a secondary effect of anti-PD-L1 therapy.
These discoveries twisted the traditional view of the PD-1-PD-L1 axis as a T cell-specific immune checkpoint and brought light to a new role of TAMs as key effector cells in ICB therapy targeting the PD-1-PD-L1 axis.
As for anti-PD-1/PD-L1 therapy, the MOA of anti-CTLA-4 therapy is still under debate. The initial goal behind mABs targeting this molecule was to block inhibitory signals in activated effector T cells upregulating CTLA-4 and thereby unleashing their anti-tumor responses. However, more recently, several studies raised the possibility that these mABs might function by depleting or affecting the suppressive activity of regulatory T cells (Tregs) which constitutively express high levels of CTLA-4 (62). Moreover, it was shown that Fcγ receptor (FcγR)-dependent depletion of Tregs is crucial for the anti-tumor response elicited by anti-CTLA-4 (63). More recently, this (FcγR)-dependent depletion was shown to be accompanied with the remodeling of the myeloid compartment in the TME. Importantly, this immune remodeling was not driven solely by Treg depletion or CTLA-4 blockade, but mainly through FcγR engagement and downstream activation of monocytes and TAMs through type I interferon signaling (64). These findings indicated that FcγR engagement and TAMs remodeling are involved in successful anti-CTLA-4 treatment, emphasizing again the crucial emerging role of TAMs as direct effector cells in ICB therapies (Figure 1B).
TAMs have been shown to regulate the therapeutic resistance mechanisms of different cancer therapies (33, 65–67). Therefore, whether TAMs regulate therapeutic resistance to ICB was an inevitable question. In one of the first studies assessing this, Zhu et al. showed that targeting TAMs by CSF-1R inhibition, enhanced the efficacy of either anti-PD1 or anti-CTLA4 therapies in an immunotherapy-resistant pancreatic cancer model (68). As of today, owing to several additional studies we understand that TAMs play a crucial role in resistance to ICB therapies (37). Thus, combining ICB with therapeutic agents impacting TAMs infiltration and/or activity has attracted particular attention and is being evaluated in clinical trials.
Even though TAMs are usually promoting tumor growth, and their infiltration is correlated with negative outcomes, eliminating them in the TME by blocking their recruitment or depleting them might be not the optimal solution to augment ICB efficacy (25). Instead, reprogramming them into anti-tumor TAMs would exploit their beneficial abilities to fight tumors such as activating T cells (rather than suppressing them), mediating direct cytotoxic tumor killing, and phagocytosis of dying tumor cells (Figure 1C). There are two strategies to functionally reprogram TAMs from pro-tumor into anti-tumor phenotype. The first one is by activating them towards an M1-like phenotype using receptors sensing pathogenic molecules and stimulation with inflammatory cytokines. The second strategy is by modulating their myeloid checkpoints and negative regulators. One prominent activator reported to repolarize TAMs and other myeloid cells is the CD40 receptor which is a member of the TNF receptor family expressed by APCs. When activated by its ligand CD40L, it triggers the production of anti-tumor cytokines and factors such as TNF and reactive oxygen species. Targeting CD40 by agonistic mABs powerfully enhanced responses of both anti-PD1 and anti-CTLA4 therapies in an immunotherapy-resistant pancreatic cancer model (69). Other macrophage activators include engagers of Toll-like receptors (TLRs), interferon receptors (IFNR), stimulator of interferon genes (STING), and FC receptors (25).
The physiological role of myeloid checkpoints and negative regulators of macrophage polarization is the protection of tissues from excessive inflammation and damage. Hijacking of such regulators by tumor cells results in immunosuppressive and tumor-promoting TAMs. One example of a frequently hijacked regulator is the CD47 “don’t eat me” signal (70). This protein is expressed on normal cells and interacts with SIRPa which is found on professional phagocytes to inhibit them from phagocytizing host cells. Tumor cells overexpress CD47 in many cancers and by this, they avoid their removal by TAMs and other phagocytes. Treatment with anti-CD47 or anti-SIRPa antibodies increased phagocytosis of cancer cells by TAMs and resulted in increased priming of CD8+ T to exhibit cytotoxic functions (71, 72). Other phagocytosis inhibitory receptor-ligand pairs include LILRB1–HLA1, SIGLEC10–CD24, and PD1–PDL1 (73, 74).
Scavenger receptors are highly expressed on TAMs and are associated with an immunosuppressive phenotype. Targeting scavenger receptors is a promising approach to reprogramming TAMs. For instance, the engagement of mannose receptor 1 (CD206) by a selective peptide (RP-192) that changes the receptor conformation, reprogrammed TAMs into an M1-like phenotype. Importantly, the combination of RP-192 and anti-PD-L1 therapies allowed to overcome ICB resistance in a pancreatic cancer model not known to respond to single agent of anti-PD-1/PD-L1 therapy (75). Macrophage receptor with collagenous structure (MARCO), macrophage scavenger receptor 1 (MSR1), and CLEVER-1 are other examples of scavenger receptors that mediate TAM reprogramming upon their targeting and are promising candidates for combination therapies with ICB (76–78). Recently, single-cell analysis of suppressive myeloid cells in tumors identified triggering receptor expressed on myeloid cells 2 (TREM2) to be a novel immunosuppressive gene expressed by myeloid subsets and especially TAMs in the TME (79, 80). Importantly, the modulation of the TREM2 pathway using blocking antibody remodeled the tumor myeloid landscape and enhanced anti-PD-1 therapy in a responsive tumor model (80, 81).
TAMs can be reprogrammed not only by agonizing or antagonizing their surface receptors but also by targeting intracellular proteins and pathways that are involved in the regulation of suppressive programs. Macrophage PI3Kγ was shown to control a critical switch between immune stimulation and suppression during inflammation and cancer, making it an ideal candidate for clinical purposes (82). Pharmacological inhibition of PI3Kγ resulted in efficient reprogramming of TAMs which synergized with anti-PD-1 treatments to reduce tumor growth. Another example is the epigenetic reprogramming of TAMs by inhibiting class IIa histone deacetylase (HDAC) (83). The inhibitor of HDAC, TMP195, was shown to alter the TME and reduce tumor growth and pulmonary metastases by modulating the phenotype of TAMs. Furthermore, combining TMP195 with anti-PD1 blockade significantly enhanced tumor size reduction in an otherwise resistant tumor model. Lastly, a recent report showed that the N6-methyladenosine reader YTHDF2 regulates the anti-tumor functions of TAMs (84). Ablation of YTHDF2 in TAMs suppressed tumor growth by reprogramming TAMs toward an antitumoral phenotype and increasing their antigen cross-presentation abilities, which in turn enhanced CD8+ T cell-mediated anti-tumor immune responses. In line with this observation, the ablation of YTHDF2 enhanced the efficacy of anti-PD-L1 therapy.
Standard of care cancer treatment such as chemotherapy was also shown to affect TAMs phenotype (66, 85, 86). Paclitaxel is a common chemotherapeutic drug used to treat various types of solid tumors. Recent studies have revealed that Paclitaxel not only inhibits cancer growth through its traditional cell-cycle arrest mechanism, but it can also reprogram TAMs in a TLR4-dependent manner, resulting in an enhanced immune response against the tumor (87, 88). Thus, such immunostimulatory chemotherapies appear to be a promising combination partners of ICB therapies, although further research is needed to optimize such treatment regimens (89).
Together, the different examples presented here demonstrate the paramount importance of TAMs modulation in gaining a successful clinical response to different T-cell-mediated immunotherapies. Future studies that will discover additional suppressive genes and pathways, will broaden the arsenal of possible clinical TAMs targets, and increase the chances for successful ICB combinatorial treatment in patients.
The sum of the mentioned discoveries encouraged clinical trials combining TAM modulators with different ICB therapeutic agents (Figure 1D). Most of the selected ongoing clinical trials (Table 1) evaluate the combination of a novel TAM modulator agent with an existing ICB therapy. The more data and clinical parameters that will be collected from these clinical trials the deeper our understanding will be of such combination therapy approaches in humans. For instance, single-cell profiling of tumor biopsies before treatment (if possible) would enable us to correlate TAMs infiltration and expression of specific markers by TAMs with clinical outcomes. This would allow the acquisition of more detailed insights on the patients that are more likely to respond to combination therapy and help to design future data-driven clinical trials. Moreover, as discussed earlier, the binding of therapeutic antibodies to macrophage Fc receptors is critical for their clinical output (90). Thus, profiling of Fc receptors on myeloid cells in the TME would boost our knowledge of the correlation between the Fc receptors landscape and clinical responses. Additionally, understanding these basic concepts will critically improve the way we design antibody-based drugs in the future.
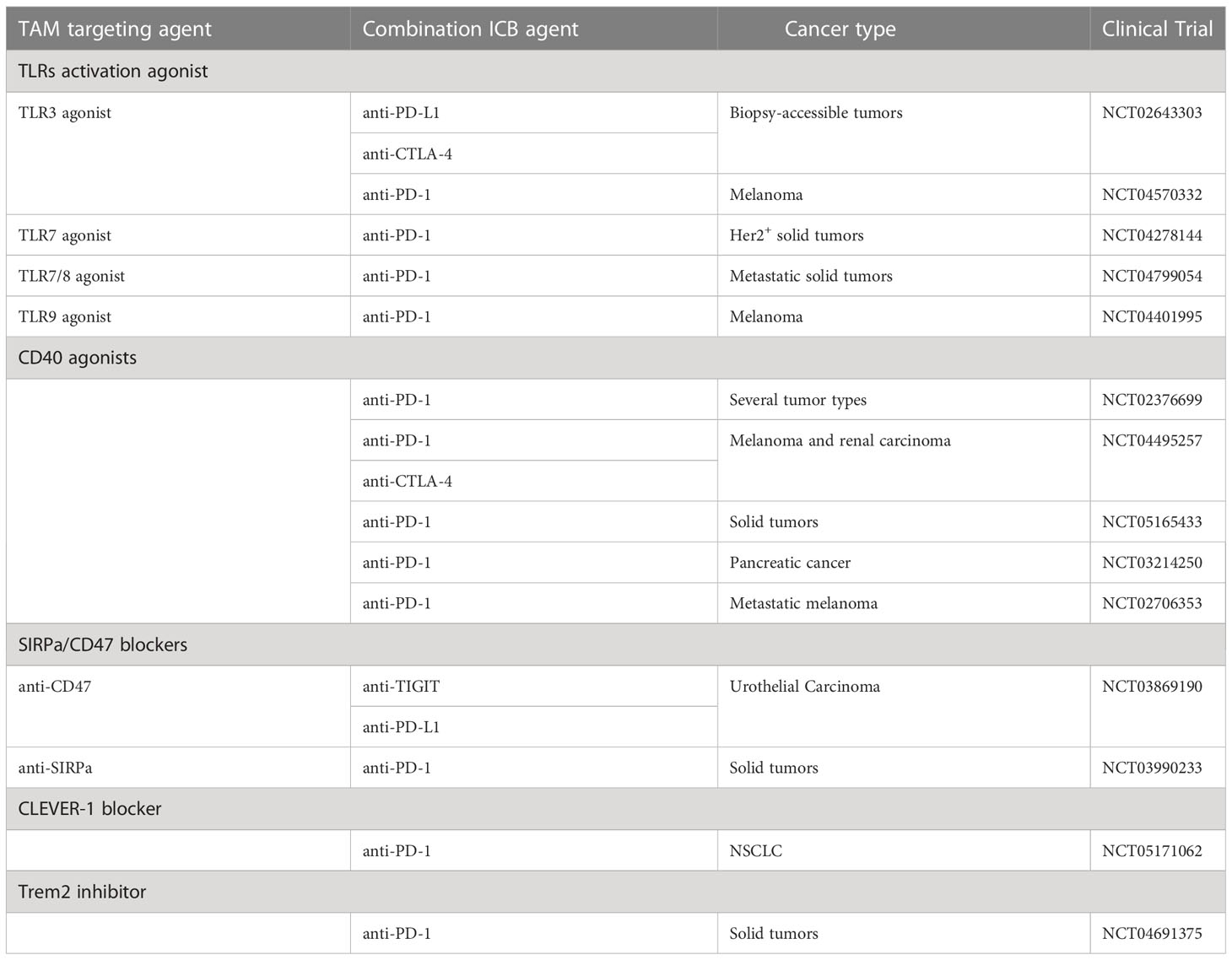
Table 1 Selected clinical trials of combination therapies targeting tumor-associated macrophages together with immune checkpoint blockade agents.
The clinical interest in combination therapy is already high due to the resistance of certain cancer types and individuals to ICB monotherapy. If some of the current combination therapy clinical trials will prove their enhanced efficacy over monotherapies, this will further fuel the enthusiasm for combination therapies, and we will see much more clinical trials in the next years. With the ongoing advances in antibody design, future clinical trials will probably include more sophisticated antibodies. For instance, bi-specific antibodies that target a myeloid checkpoint while activating T-cells or engaging Fcγ receptors could be of high clinical potential. Finally, further basic research that involves large and well-designed perturbation screens will allow us to bring more TAM-specific targets to clinical trials.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
The author confirms being the sole contributor of this work and has approved it for publication.
This perspective was supported by the Israel Council for Higher Education - VATAT scholarship for outstanding doctoral students.
I thank my colleagues, Dr. Pascale Zwicky and Dr. Ido Yofe for reviewing the manuscript and my mentor Prof. Ido Amit for his generous support. Moreover, I am grateful to Prof. Özlem Türeci and the CIMT Summer School 2022 faculty for inviting me to contribute to this article collection. FS is a recipient of the Israeli Council for Higher Education MSc and PhD fellowships. Most of all, I warmly thank my late mother, Nibal Sheban (1963-2020) for her endless support and encouragement throughout my research career.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. with a report of ten original cases. Clin Orthop Relat Res (1893) 262:3–11. doi: 10.1097/00000658-189307000-00009
2. Redelman-Sidi G, Glickman MS, Bochner BH. The mechanism of action of BCG therapy for bladder cancer-a current perspective. Nat Rev Urol (2014) 11:153–62. doi: 10.1038/nrurol.2014.15
3. Morales A, Eidinger D, Bruce AW. Intracavitary bacillus calmette guerin in the treatment of superficial bladder tumors. J Urol (1976) 116:180–3. doi: 10.1016/s0022-5347(17)58737-6
4. Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, et al. A new member of the immunoglobulin superfamily—CTLA-4. Nature (1987) 328:260–70. doi: 10.1038/328267a0
5. Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T ceils to stimulation. J Exp Med (1995) 182:459–65. doi: 10.1084/jem.182.2.459
6. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med (2000) 192:1027–34. doi: 10.1084/jem.192.7.1027
7. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U.S.A. (2002) 99:12293–7. doi: 10.1073/pnas.192461099
8. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Sci (1979) (1996) 271:1734–6. doi: 10.1126/science.271.5256.1734
9. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol (2020) 20:651–68. doi: 10.1038/s41577-020-0306-5
10. Pilard C, Ancion M, Delvenne P, Jerusalem G, Hubert P, Herfs M. Cancer immunotherapy: it’s time to better predict patients’ response. Br J Cancer (2021) 125:927–38. doi: 10.1038/s41416-021-01413-x
11. Hamid O, Robert C, Daud A, Stephen Hodi F, Hwu W-J, Kefford R, et al. Safety and tumor responses with lambrolizumab (Anti-PD-1) in melanoma a bs t r ac t. N Engl J Med (2013) 369:134–78. doi: 10.1056/NEJMoa1305133
12. Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity (2020) 52:17–35. doi: 10.1016/j.immuni.2019.12.011
13. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell (2016) 165:35–44. doi: 10.1016/J.CELL.2016.02.065
14. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Sci (1979) (2015) 348:124–8. doi: 10.1126/SCIENCE.AAA1348
15. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma a BS TR AC T. N Engl J Med (2014) 371:2189–99. doi: 10.1056/NEJMoa1406498
16. Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane J-P, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun (2020) 8:1–12. doi: 10.1038/s41467-017-01062-w
17. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer (2019) 19:133–50. doi: 10.1038/s41568-019-0116-x
18. Herbst RS, Soria J-C, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature (2014) 515:563–7. doi: 10.1038/nature14011
19. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515:568–71. doi: 10.1038/nature13954
20. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. New Engl J Med (2012) 366:2443–54. doi: 10.1056/nejmoa1200690
21. Zhang F, Zhang J, Zhao L, Zhai M, Zhang T, Yu D. A PD-L1 negative advanced gastric cancer patient with a long response to PD-1 blockade after failure of systematic treatment: a case report. Front Immunol (2021) 12:759250. doi: 10.3389/fimmu.2021.759250
22. Li H, van der Merwe PA, Sivakumar S. Biomarkers of response to PD-1 pathway blockade. Br J Cancer (2022) 126:1663–75. doi: 10.1038/s41416-022-01743-4
23. Wendel Naumann R, Hollebecque A, Meyer T, Devlin MJ, Oaknin A, Kerger J, et al. Safety and efficacy of nivolumab monotherapy in recurrent or metastatic cervical, vaginal, or vulvar carcinoma: results from the phase I/II CheckMate 358 trial. J Clin Oncol (2019) 37:2825–34. doi: 10.1200/JCO.19.00739
24. Grossman JE, Vasudevan D, Cailin • JE, Hildago • M. Is PD-L1 a consistent biomarker for anti-PD-1 therapy? the model of balstilimab in a virally-driven tumor. Oncogene (2021) 40:1393–5. doi: 10.1038/s41388-020-01611-6
25. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discovery (2022) 21:799–820. doi: 10.1038/s41573-022-00520-5
26. Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med (2015) 212:1043–59. doi: 10.1084/jem.20141836
27. Bieniasz-Krzywiec P, Martín-Pérez R, Ehling M, García-Caballero M, Pinioti S, Pretto S, et al. Podoplanin-expressing macrophages promote lymphangiogenesis and lymphoinvasion in breast cancer. Cell Metab (2019) 30:917–936.e10. doi: 10.1016/j.cmet.2019.07.015
28. Casanova-Acebes M, Dalla E, Leader AM, LeBerichel J, Nikolic J, Morales BM, et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature (2021) 595:578–84. doi: 10.1038/S41586-021-03651-8
29. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discovery (2018) 17:887–904. doi: 10.1038/nrd.2018.169
30. jie ZW, hua WX, ting GS, Chen C, yun X, sun Q, et al. Tumor-associated macrophages correlate with phenomenon of epithelial-mesenchymal transition and contribute to poor prognosis in triple-negative breast cancer patients. J Surg Res (2018) 222:93–101. doi: 10.1016/J.JSS.2017.09.035
31. Yuan ZY, Luo RZ, Peng RJ, Sen WS, Xue C. High infiltration of tumor-associated macrophages in triple-negative breast cancer is associated with a higher risk of distant metastasis. Onco Targets Ther (2014) 7:1475–80. doi: 10.2147/OTT.S61838
32. Chen JJ, Yao P-L, Yuan A, Hong T-M, Shun C-T, Kuo M-L, et al. Up-regulation of tumor interleukin-8 expression by infiltrating macrophages: its correlation with tumor angiogenesis and patient survival in non-small cell lung cancer 1. Clin Cancer Res (2003) 9:729–37.
33. Weizman N, Krelin Y, Shabtay-Orbach A, Amit M, Binenbaum Y, Wong RJ, et al. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene (2013) 33:3812–9. doi: 10.1038/onc.2013.357
34. Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol (2019) 21:1113–26. doi: 10.1038/s41556-019-0373-7
35. Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer (2016) 16:447–62. doi: 10.1038/nrc.2016.54
36. Neophytou CM, Pierides C, Christodoulou MI, Costeas P, Kyriakou TC, Papageorgis P. The role of tumor-associated myeloid cells in modulating cancer therapy. Front Oncol (2020) 10:899. doi: 10.3389/fonc.2020.00899
37. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol (2019) 19:369–82. doi: 10.1038/s41577-019-0127-6
38. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23:549–55. doi: 10.1016/S1471-4906(02)02302-5
39. Ma RY, Black A, Qian BZ. Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol (2022) 43:546–63. doi: 10.1016/j.it.2022.04.008
40. Mulder K, Patel AA, Kong WT, Piot C, Halitzki E, Dunsmore G, et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity (2021) 54:1883–900. doi: 10.1016/j.immuni.2021.07.007
41. Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, et al. Erratum: high-dimensional analysis delineates myeloid and lymphoid compartment remodeling during successful immune-checkpoint cancer therapy (Cell (2018) 175(4) (1014–1030.e19), (S009286741831242X) (10.1016/j.cell.2018.09.030)). Cell (2018) 175:1443. doi: 10.1016/j.cell.2018.11.003
42. Luo W, Napoleon JV, Zhang F, Lee YG, Wang B, Putt KS, et al. Repolarization of tumor-infiltrating myeloid cells for augmentation of CAR T cell therapies. Front Immunol (2022) 13:816761. doi: 10.3389/fimmu.2022.816761
43. Baharom F, Ramirez-Valdez RA, Khalilnezhad A, Khalilnezhad S, Dillon M, Hermans D, et al. Systemic vaccination induces CD8+ T cells and remodels the tumor microenvironment. Cell (2022) 185:4317–4332.e15. doi: 10.1016/j.cell.2022.10.006
44. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
45. Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med (2009) 206:1327–37. doi: 10.1084/jem.20082173
46. Kryczek I, Zou L, Rodriguez P, Zhu G, Wei S, Mottram P, et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med (2006) 203:871–81. doi: 10.1084/jem.20050930
47. Huber S, Hoffmann R, Muskens F, Voehringer D. Alternatively activated macrophages inhibit T-cell proliferation by Stat6-dependent expression of PD-L2. Blood (2010) 116:3311–20. doi: 10.1182/blood-2010-02-271981
48. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer (2016) 16:275–87. doi: 10.1038/nrc.2016.36
49. Tang H, Liang Y, Anders RA, Taube JM, Qiu X, Mulgaonkar A, et al. PD-L1 on host cells is essential for PD-L1 blockade–mediated tumor regression. J Clin Invest (2018) 128:580–8. doi: 10.1172/JCI96061
50. Oh SA, Wu DC, Cheung J, Navarro A, Xiong H, Cubas R, et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat Cancer (2020) 1:681–91. doi: 10.1038/s43018-020-0075-x
51. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc Natl Acad Sci U.S.A. (2018) 115:E4041–50. doi: 10.1073/pnas.1720948115
52. Yu J, Green MD, Li S, Sun Y, Journey SN, Choi JE, et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat Med (2021) 27:152–64. doi: 10.1038/s41591-020-1131-x
53. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse ig superfamily ligand that negatively regulates T cell responses. J Exp Med (2011) 208:577–92. doi: 10.1084/jem.20100619
54. Yuan L, Tatineni J, Mahoney KM, Freeman GJ. VISTA: a mediator of quiescence and a promising target in cancer immunotherapy. Trends Immunol (2021) 42:209–27. doi: 10.1016/j.it.2020.12.008
55. Johnston RJ, Su LJ, Pinckney J, Critton D, Boyer E, Krishnakumar A, et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature (2019) 574:565–70. doi: 10.1038/s41586-019-1674-5
56. Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci U.S.A. (2009) 106:6303–8. doi: 10.1073/pnas.0809422106
57. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature (2017) 545:495–9. doi: 10.1038/nature22396
58. Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol (2020) 5:1–15. doi: 10.1126/sciimmunol.aay1863
59. Christofides A, Katopodi XL, Cao C, Karagkouni D, Aliazis K, Yenyuwadee S, et al. SHP-2 and PD-1-SHP-2 signaling regulate myeloid cell differentiation and antitumor responses. Nat Immunol (2023) 24:55–68. doi: 10.1038/s41590-022-01385-x
60. Xiong H, Mittman S, Rodriguez R, Moskalenko M, Pacheco-Sanchez P, Yang Y, et al. Anti-PD-L1 treatment results in functional remodeling of the macrophage compartment. Cancer Res (2019) 79:1493–506. doi: 10.1158/0008-5472.CAN-18-3208
61. Tichet M, Wullschleger S, Chryplewicz A, Fournier N, Marcone R, Kauzlaric A, et al. Bispecific PD1-IL2v and anti-PD-L1 break tumor immunity resistance by enhancing stem-like tumor-reactive CD8+ T cells and reprogramming macrophages. Immunity (2023) 56:162–179.e6. doi: 10.1016/j.immuni.2022.12.006
62. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+Regulatory T cell function. Sci (1979) (2008) 322:271–5. doi: 10.1126/science.1160062
63. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory t cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med (2013) 210:1695–710. doi: 10.1084/jem.20130579
64. Yofe I, Landsberger T, Yalin A, Solomon I, Costoya C, Demane DF, et al. Anti-CTLA-4 antibodies drive myeloid activation and reprogram the tumor microenvironment through FcγR engagement and type I interferon signaling. Nat Cancer (2022) 11:1336–50. doi: 10.1038/s43018-022-00447-1
65. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell (2015) 27:462–72. doi: 10.1016/j.ccell.2015.02.015
66. Larionova I, Cherdyntseva N, Liu T, Patysheva M, Rakina M, Kzhyshkowska J. Interaction of tumor-associated macrophages and cancer chemotherapy. Oncoimmunology (2019) 8:1–15. doi: 10.1080/2162402X.2019.1596004
67. De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell (2013) 23:277–86. doi: 10.1016/j.ccr.2013.02.013
68. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res (2014) 74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723
69. Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer ARL, Bajor DL, et al. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res (2015) 3:399–411. doi: 10.1158/2326-6066.CIR-14-0215
70. Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell (2009) 138:286–99. doi: 10.1016/j.cell.2009.05.045
71. Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U.S.A. (2013) 110:11103–8. doi: 10.1073/pnas.1305569110
72. Ring NG, Herndler-Brandstetter D, Weiskopf K, Shan L, Volkmer JP, George BM, et al. Anti-SIRPα antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc Natl Acad Sci U.S.A. (2017) 114:E10578–85. doi: 10.1073/pnas.1710877114
73. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage siglec-10 is a target for cancer immunotherapy. Nature (2019) 572:392–6. doi: 10.1038/s41586-019-1456-0
74. Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY, et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol (2017) 19:76–84. doi: 10.1038/s41590-017-0004-z
75. Jaynes JM, Sable R, Ronzetti M, Bautista W, Knotts Z, Abisoye-Ogunniyan A, et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci Transl Med (2020) 12:1–20. doi: 10.1126/scitranslmed.aax6337
76. Etzerodt A, Tsalkitzi K, Maniecki M, Damsky W, Delfini M, Baudoin E, et al. Specific targeting of CD163+ TAMs mobilizes inflammatory monocytes and promotes T cell-mediated tumor regression. J Exp Med (2019) 216:2394–411. doi: 10.1084/jem.20182124
77. Palani S, Maksimow M, Miiluniemi M, Auvinen K, Jalkanen S, Salmi M. Stabilin-1/CLEVER-1, a type 2 macrophage marker, is an adhesion and scavenging molecule on human placental macrophages. Eur J Immunol (2011) 41:2052–63. doi: 10.1002/eji.201041376
78. Hollmén M, Figueiredo CR, Jalkanen S. New tools to prevent cancer growth and spread: a ‘Clever’ approach. Br J Cancer (2020) 123:501–9. doi: 10.1038/s41416-020-0953-0
79. Katzenelenbogen Y, Sheban F, Yalin A, Yofe I, Svetlichnyy D, Jaitin DA, et al. Coupled scRNA-seq and intracellular protein activity reveal an immunosuppressive role of TREM2 in cancer. Cell (2020) 182:872–885.e19. doi: 10.1016/j.cell.2020.06.032
80. Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J, et al. TREM2 modulation remodels the tumor myeloid landscape enhancing anti-PD-1 immunotherapy. Cell (2020) 182:886–900.e17. doi: 10.1016/j.cell.2020.07.013
81. Xiong D, Wang Y, You M. A gene expression signature of TREM2hi macrophages and γδ T cells predicts immunotherapy response. Nat Commun (2020) 11:1–12. doi: 10.1038/s41467-020-18546-x
82. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kγ 3 is a molecular switch that controls immune suppression. Nature (2016) 539:437–42. doi: 10.1038/nature19834
83. Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature (2017) 543:428–32. doi: 10.1038/nature21409
84. Ma S, Sun B, Duan S, Han J, He C, Chen J, et al. YTHDF2 orchestrates tumor-associated macrophage reprogramming and controls antitumor immunity through CD8 + T cells. Nat Immunol (2022) 24:255–66. doi: 10.1038/s41590-022-01398-6
85. Buhtoiarov IN, Sondel PM, Wigginton JM, Buhtoiarova TN, Yanke EM, Mahvi DA, et al. Anti-tumour synergy of cytotoxic chemotherapy and anti-CD40 plus CpG-ODN immunotherapy through repolarization of tumour-associated macrophages. Immunology (2011) 132:226–39. doi: 10.1111/j.1365-2567.2010.03357.x
86. Kodumudi KN, Woan K, Gilvary DL, Sahakian E, Wei S, Djeu JY. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res (2010) 16:4583–94. doi: 10.1158/1078-0432.CCR-10-0733
87. Wanderley CW, Colón DF, Luiz JPM, Oliveira FF, Viacava PR, Leite CA, et al. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1 profile in a TLR4-dependent manner. Cancer Res (2018) 78:5891–900. doi: 10.1158/0008-5472.CAN-17-3480
88. Heath O, Berlato C, Maniati E, Lakhani A, Pegrum C, Kotantaki P, et al. Chemotherapy induces tumor-associated macrophages that aid adaptive immune responses in ovarian cancer. Cancer Immunol Res (2021) 9:665–81. doi: 10.1158/2326-6066.CIR-20-0968
89. Galluzzi L, Humeau J, Buqué A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol (2020) 17:725–41. doi: 10.1038/s41571-020-0413-z
Keywords: tumor-associated macrophages, immunotherapy, cancer, combination therapy, tumor microenvironment
Citation: Sheban F (2023) It takes two to tango: the role of tumor-associated macrophages in T cell-directed immune checkpoint blockade therapy. Front. Immunol. 14:1183578. doi: 10.3389/fimmu.2023.1183578
Received: 10 March 2023; Accepted: 26 May 2023;
Published: 09 June 2023.
Edited by:
Cara Haymaker, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Lisa Sevenich, Georg Speyer Haus, GermanyCopyright © 2023 Sheban. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fadi Sheban, ZmFkaS5zaGViYW5Ad2Vpem1hbm4uYWMuaWw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.