 Kacy S. Yount1†
Kacy S. Yount1† Jesse M. Hall1†
Jesse M. Hall1† Kyle Caution1Mohamed M. Shamseldin1Myra Guo1
Kyle Caution1Mohamed M. Shamseldin1Myra Guo1 Keirsten Marion1Audra R. Fullen1Yimin Huang2
Keirsten Marion1Audra R. Fullen1Yimin Huang2 Jennifer A. Maynard2Sally A. Quataert3
Jennifer A. Maynard2Sally A. Quataert3 Rajendar Deora1,4
Rajendar Deora1,4 Purnima Dubey1*
Purnima Dubey1*- 1Department of Microbial Infection and Immunity, The Ohio State University, Columbus, OH, United States
- 2Department of Chemical Engineering, University of Texas-Austin, Austin, TX, United States
- 3Respiratory Pathogens Research Center, University of Rochester Medical Center, Rochester, NY, United States
- 4Department of Microbiology, The Ohio State University, Columbus, OH, United States
Introduction: Resurgence of pertussis, caused by Bordetella pertussis, necessitates novel vaccines and vaccination strategies to combat this disease. Alum-adjuvanted acellular pertussis vaccines (aPV) delivered intramuscularly reduce bacterial numbers in the lungs of immunized animals and humans, but do not reduce nasal colonization. Thus, aPV-immunized individuals are sources of community transmission. We showed previously that modification of a commercial aPV (Boostrix) by addition of the Th1/17 polarizing adjuvant Bordetella Colonization Factor A (BcfA) attenuated Th2 responses elicited by alum and accelerated clearance of B. pertussis from mouse lungs. Here we tested whether a heterologous immunization strategy with systemic priming and mucosal booster (prime-pull) would reduce nasal colonization.
Methods: Adult male and female mice were immunized intramuscularly (i.m.) with aPV or aPV/BcfA and boosted either i.m. or intranasally (i.n.) with the same formulation. Tissue-resident memory (TRM) responses in the respiratory tract were quantified by flow cytometry, and mucosal and systemic antibodies were quantified by ELISA. Immunized and naïve mice were challenged i.n. with Bordetella pertussis and bacterial load in the nose and lungs enumerated at days 1-14 post-challenge.
Results: We show that prime-pull immunization with Boostrix plus BcfA (aPV/BcfA) generated IFNγ+ and IL-17+ CD4+ lung resident memory T cells (TRM), and CD4+IL-17+ TRM in the nose. In contrast, aPV alone delivered by the same route generated IL-5+ CD4+ resident memory T cells in the lungs and nose. Importantly, nasal colonization was only reduced in mice immunized with aPV/BcfA by the prime-pull regimen.
Conclusions: These results suggest that TH17 polarized TRM generated by aPV/BcfA may reduce nasal colonization thereby preventing pertussis transmission and subsequent resurgence.
Introduction
The Gram-negative obligate human pathogen Bordetella pertussis is the primary cause of whooping cough, or pertussis. Whole cell pertussis vaccines (wPV), used in the United States between the 1940s-1990s, provided long-lived immunity against infection in humans and effectively cleared the bacteria from the entire respiratory tract of mice and non-human primates (1–3). However, reactogenicity and public concerns regarding vaccine side effects led to the replacement of wPV in the 1990s with 3-5 component subunit vaccines adjuvanted with alum (4, 5). Although these acellular pertussis vaccines (aPV) prevent severe disease, they do not prevent establishment of a nasopharyngeal reservoir and subsequent transmission to vulnerable populations (6). Therefore, despite high global vaccine coverage of 85-100%, this disease remains endemic worldwide (7) and multiple recent outbreaks have resulted in significant morbidity and mortality in infants (8–12). While the disease is most severe and sometimes fatal in infants, in recent years there has been a shift in disease and infection burden to adolescents and young adults (13). Globally, estimates suggest that 15% of prolonged cough illnesses are due to B. pertussis infections with a yearly infection rate of 6% (14). However, symptomatic disease is not the only means of bacterial transmission (15). Experimental and computational studies indicate that children and adults vaccinated with aPV are asymptomatic transmitters of disease (16–20). A single primary case of pertussis can cause 12–17 new cases in susceptible individuals. This level of contagion is estimated to be similar to or higher than that of measles, influenza, mumps, rubella, polio, and smallpox (21). In 2015, pertussis and B. pertussis were included in the NIAID emerging infectious diseases and pathogens lists (22), respectively, emphasizing the need for more effective pertussis vaccines to improve global health.
Alum-adjuvanted aPV delivered intramuscularly (i.m.) elicits Th1/Th2 polarized systemic responses and fails to generate mucosal immunity (1). Furthermore, recent studies in mice show that i.m. aPV immunization exacerbates nasal colonization (2, 23). In contrast, natural infection and wPV immunization elicit Th1/17 polarized systemic responses (24) and elicit CD4+ TRM and mucosal antibodies upon challenge with B. pertussis (1). These differences are two potential explanations for the suboptimal protection provided by aPV. Recent studies show that experimental subunit and whole-cell vaccines delivered intranasally (i.n.) elicit IL-17-producing CD4+ resident memory T cells (TRM) and mucosal antibodies that clear the infection from the entire murine respiratory tract (25–27), suggesting that inclusion of a Th1/17 polarizing adjuvant and mucosal vaccine delivery will improve aPV-mediated protection.
Bordetella Colonization Factor A (BcfA) is an outer membrane protein from the animal pathogen Bordetella bronchiseptica (28, 29). It is a pseudogene in the human pathogen B. pertussis (29). We showed that BcfA is an adjuvant that elicits Th1/17 polarized T cell and antibody responses to a variety of protein antigens (30, 31). Additionally, when combined with alum, BcfA attenuated alum primed Th2 responses. We further showed that i.m. priming and booster immunization of mice with a commercial aPV, Boostrix, with the addition of BcfA (aPV/BcfA) accelerated clearance of B. pertussis from mouse lungs.
Here we tested the hypothesis that a prime-pull immunization regimen with i.m. priming and i.n. booster with aPV/BcfA would generate mucosal immune responses and prevent the colonization of B. pertussis in both the upper and lower respiratory tracts of mice. We used the booster aPV, Boostrix, as the test vaccine because adolescents and adults who receive this booster are at risk for nasal colonization and transmission. Thus, modification of this formulation has potential clinical utility. While i.m. prime and boost with aPV/BcfA only cleared the lungs of mice, i.m. priming followed by i.n. booster of the same formula generated CD4+IL-17+ TRM and significantly reduced B. pertussis colonization of the nose. Together, our data show that aPV/BcfA delivered by a prime-pull immunization regimen generates immunity at the site of infection and controls B. pertussis bacterial burden in the mouse upper and lower respiratory tract.
Materials and methods
Bacterial strains, media, and growth conditions
B. pertussis strain Bp536 (32) was maintained on BG agar (Difco) containing 10% defibrinated sheep’s blood (Hemostat, Dixon, CA) supplemented with 100 μg/ml streptomycin. For animal inoculations, liquid cultures were grown at 37°C on a roller drum to OD600 ≈ 1.0 in Stainer-Scholte medium supplemented with 1 mg/mL heptakis (Sigma) and 100 μg/ml streptomycin.
Animals
All experiments were approved by the Ohio State University Institutional Animal Care and Use Committee under protocol number 2017A00000090-R1 and adhered to the NIH Guide for the Care and Use of Laboratory Animals. C57BL/6J mice were purchased from Jackson Labs and bred in-house. Male and female mice (6-12 weeks old) were used for all studies.
Reagents
Boostrix (GSK) was purchased from the Ohio State University Medical Center Pharmacy. Purified filamentous hemagglutinin (FHA) from B. pertussis was purchased from List Biologicals (Campbell, CA) or over-produced in E. coli. Detoxified pertussis toxin (PT) purified from B. pertussis was purchased from List Biologicals. BcfA was over-produced in E. coli and purified as described previously (29) with endotoxin levels as reported in (30). Pertactin (PRN) was purchased from List Biologicals. Endotoxin in FHA and PT was <20 EU/mg. Endotoxin levels in all proteins were below that of Boostrix (33). GentleMACS lung dissociation kit was purchased from Miltenyi Biotec. RPMI, Ca2+/Mg2+-free PBS, ACK red blood cell lysis buffer, protein transport inhibitor, and fixation/permeabilization buffers were purchased from Life Technologies. Fetal bovine serum was purchased from Sigma-Aldrich (St. Louis, MO). U-Plex biomarker assay was purchased from Meso Scale Diagnostics (Rockville, MD). Antibodies and live/dead stains for flow cytometry were purchased from Life Technologies, BioLegend, or BD Biosciences.
Immunizations
Mice were lightly anesthetized with 2.5% isoflurane/O2 for i.m. or i.n. immunization on day 0 and boost on day 28-35 (34) with the following vaccines: a) 1/5th human dose of Boostrix (100 µL) delivered i.m. in the right deltoid and b) 1/5th human dose of Boostrix with the addition of 10 μg BcfA (aPV/BcfA) incubated at room temperature for 30 minutes with rotation to adsorb the BcfA to alum, then resuspended in 100 µL of sterile PBS for i.m. injection or 50 uL sterile PBS for i.n. immunization, divided between both nares.
Intravascular staining for discrimination between circulating and resident cells
Anti-CD45-PE (Clone 30-F11, BD Biosciences) (3 µg in 100 µL sterile PBS) was injected i.v. 10 minutes prior to sacrifice to label circulating lymphocytes, while resident lymphocytes were protected from labeling (35, 36). Peripheral blood was collected at time of sacrifice and checked by flow cytometry to confirm that >90% of circulating lymphocytes were CD45-PE+.
Tissue dissociation and flow cytometry
Lung and nose tissues were enzymatically digested with GentleMACS lung dissociation kit (Miltenyi) according to the manufacturer’s instructions. Cell suspensions were filtered (40-μm) and red blood cells lysed with ACK lysis buffer. To detect cytokines, cells were stimulated with PMA (50 ng/ml) and ionomycin (500 ng/ml) in the presence of a protein transport inhibitor cocktail (eBioscience) for 4 h at 37°C. Cells were incubated with LIVE/DEAD Aqua or Near Infrared viability stains, followed by incubation with α-CD16/CD32 FcγRIII to block IgG Fc receptors. The following antibodies specific for cell surface markers were used: anti-CD3-V450 (clone 17A2), anti-CD4-BV750 (clone H129.19), anti-CD4 PE-CF594, anti-CD44-PerCP-Cy5.5 (clone IM-7), anti-CD62L-BV605 (clone MEL-14), anti-CD69-BV711 (clone HI.2F3), and anti-CD103-PE-CF594 (clone M290). For detection of cytokines, cells were fixed, permeabilized, and stained with antibodies against IFNγ (FITC conjugated clone XMG1.2), IL-17 (PE-Cy7 conjugated clone eBio17B7), and IL-5 (APC conjugated clone TRFK5). Fluorescence minus one or isotype control antibodies were used as negative controls. Samples were acquired on BD Fortessa (Figure 1) or Cytek Aurora (Figure 2) cytometers and data were analyzed with FlowJo software. The number of cells within each population were calculated by multiplying the frequency of live singlets in the population of interest by the total number of cells in each sample.
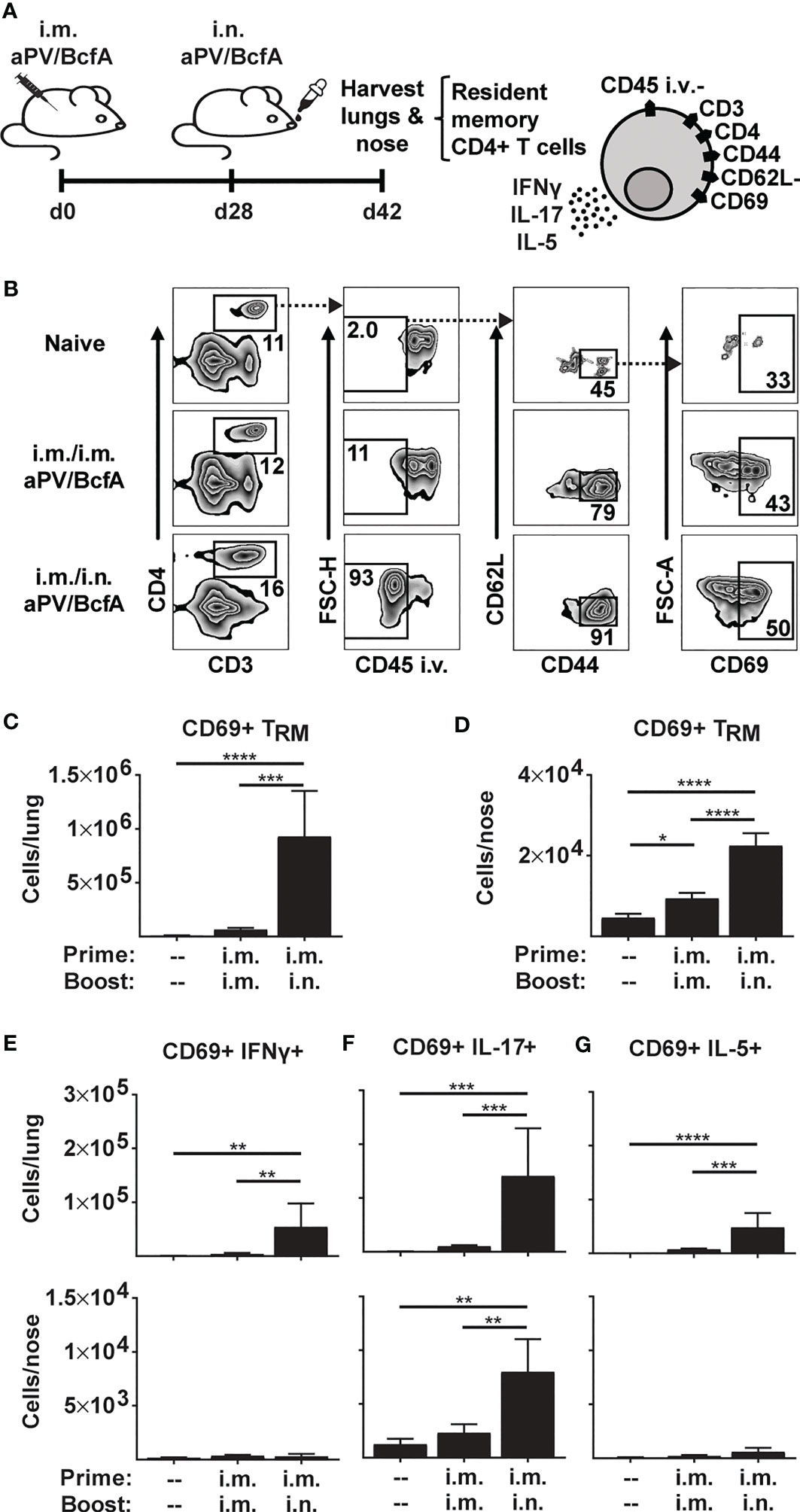
Figure 1 Prime-pull immunization with aPV/BcfA elicits IL-17-producing CD4+ TRM in lungs and nose. (A) C57BL/6 mice (5/group) were immunized on d0 and d28 with 1/5 human dose aPV + 10μg BcfA (aPV/BcfA) delivered i.m./i.m. or i.m./i.n. TRM in lungs and nose were quantified via flow cytometry on a BD Fortessa flow cytometer as live, CD3+, CD4+, CD45 i.v.-, CD44+, CD62L-, CD69+ cells. (B) Representative gating to identify TRM in lungs. Number of CD69+ TRM in (C) lungs and (D) nose. Number of CD69+ TRM that were IFNγ+ (E), IL-17+ (F), or IL-5+ (G) following PMA/I stimulation. Data representative of two independent experiments and presented as mean +/- SEM. Data were analyzed by one-way ANOVA with Holm-Sidak correction for multiple comparisons. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
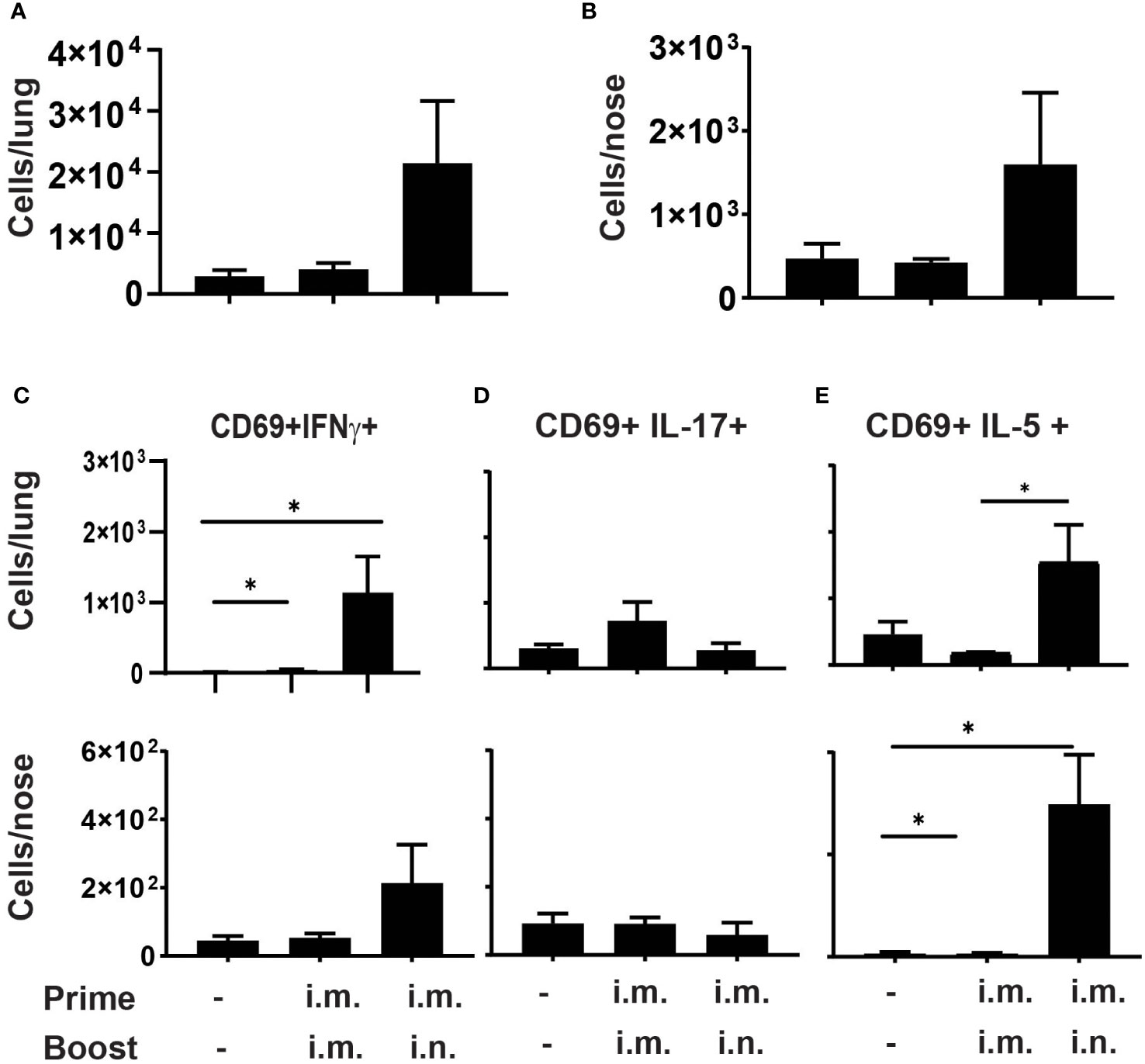
Figure 2 Prime-pull immunization with aPV elicits Th2 polarized CD4+ TRM C57BL/6 mice (5/group) were immunized on d0 and d28 with 1/5 human dose aPV delivered i.m./i.m. or i.m./i.n. TRM in lungs and nose were quantified via flow cytometry on a Cytek Aurora spectral flow cytometer as live, CD3+, CD4+, CD45 i.v.-, CD44+, CD62L-, CD69+ cells. Number of CD69+ TRM in (A) lungs and (B) nose. Number of CD69+ TRM in the lungs and nose that were IFNγ+ (C), IL-17+ (D), or IL-5+ (E) following PMA/I stimulation. Data representative of two independent experiments and presented as mean +/- SEM. Data were analyzed by one-way ANOVA with Holm-Sidak correction for multiple comparisons. *P<0.05.
Bacterial challenge
Bacterial strain Bp536 grown overnight to OD600 ≈ 1.0 was diluted in PBS to 5x105 bacteria per 50 μl. On day 14 post-boost, mice were lightly anesthetized with 2.5% isoflurane/O2 and the 50 μl inoculum was divided between both nares as reported previously (34).
Bacterial enumeration
Mice were sacrificed at 1-, 4-, 7-, and 14-days post challenge. Lungs and nasal tissue (including septum, turbinates, sinuses, and nasal associated lymphoid tissue (NALT)), were harvested, mechanically disrupted in PBS + 1% casein, and plated in serial dilutions on BG agar plates containing 10% sheep’s blood and supplemented with 100 μg/ml streptomycin. Colony forming units (CFUs) were counted after 4 days of incubation at 37° C. Data were transformed to log10.
Antibody analysis
Serum from blood and supernatants from lung digestion were analyzed for the presence of antigen-specific antibodies. Purified antigens FHA, PT, and PRN were coupled through an amine linkage to MagPlex C magnetic microspheres (Luminex Corporation), each with a unique fluorescent bead region address, and combined to form a 5-plex microarray. Mouse serum or lung homogenates were diluted in assay buffer, PBS–0.1% Brij-35–1% bovine serum albumin (BSA), pH 7.2, and incubated with the beads for 2 h at room temperature (r.t.) in the dark while shaking at 800 rpm. After washing, appropriate biotinylated detection antibody was added, i.e., goat anti-mouse total IgG, rat anti-IgG1, rat anti-IgG2b, or goat anti-IgG2c, and rat anti-IgA at a 1:250 dilution in assay buffer for 1 h at r.t. After washing, streptavidin-phycoerythrin (SA-PE) at 1:250 in assay buffer was added for 1 h with shaking. Unbound SA-PE was removed by washing, and the beads were resuspended in 100 μl PBS prior to reading on a Luminex 200 flow cytometer. Antibody isotype and subclass values are reported in arbitrary luminescent intensity units (RLU).
Results
I.m. priming followed by i.n. booster with acellular vaccines elicits CD4+ TRM in the nose and lungs of mice
Mucosal cellular and humoral adaptive immune responses against respiratory pathogens are critical to clear infections from the respiratory tract (37, 38) and studies in murine models showed that CD4+ T cells are necessary for clearing natural B. pertussis infections (39). Experimental pertussis vaccines, when delivered i.n., generate CD4+ TRM cells in mice which prevent nasal colonization (25, 27). However, alum-adjuvanted aPV delivered i.m. alone do not generate CD4+ TRM in the nose or lungs (1).
Here, we tested whether i.m. priming and i.n. booster with aPV/BcfA would elicit CD4+ TRM. We immunized C57BL/6 mice with aPV/BcfA via two different routes: i.m. prime and boost (i.m./i.m.), and i.m. prime followed by i.n. boost (i.m./i.n.). Mice were immunized on d0, boosted on d28, and the number and phenotype of CD4+ T cells in the nose and lungs were evaluated on d42 (2 weeks post-boost) (Figure 1A). Anti-CD45-PE antibody was injected i.v. 10 minutes before sacrifice and tissue harvest to distinguish circulating from tissue-resident memory cells (36). Single cell suspensions of lungs and noses were stimulated with PMA/Ionomycin and stained to identify the number of CD4+ TRM. The flow cytometry gating strategy is shown in Figure 1B. The number of live, CD3+, CD4+, and CD45 i.v.- cells that were CD44+CD62L-CD69+ (CD4+ TRM) were quantified. CD4+ TRM cells were detected in the lungs (Figure 1C) and noses (Figure 1D) of i.m./i.n. immunized mice. However, little induction of TRM in the tissues of i.m./i.m. immunized animals was observed. Lung TRM of i.m./i.n.-vaccinated mice produced a small number of IFNγ+ cells (Figure 1E) that primarily produced IL-17 (Figure 1F) and few IL-5+ TRM (Figure 1G)., while nose TRM had negligible IFNγ- (Figure 1E) or IL-5- (Figure 1G) producing cells and exclusively produced IL-17 (Figure 1F). Thus, the addition of BcfA to aPV generated Th1 and Th17 polarized CD4+ TRM that are correlated with protection against B. pertussis infection (40).
We then tested whether im./i.n. immunization with aPV alone would generate CD4+ TRM. C57BL/6 mice were immunized with aPV alone i.m./i.m. or i.m./i.n. on d0 and d28 and harvested on d42.Lungs and nose cells were stimulated with PMA/Ionomycin, followed by staining for CD4+ TRM. The gating strategy is shown in Figure S1. As observed with aPV/BcfA immunization, i.m./i.m. immunization with aPV alone did not elicit CD4+ TRM in the lungs (Figure 2A) or nose (Figure 2B). I.m./i.n. immunization with aPV elicited CD4+ TRM in the lungs (Figure 2A) and nose (Figure 2B). Intracellular cytokine staining showed that CD4+ TRM from lungs and nose produced IFNγ (Figure 2C), no IL-17 (Figure 2D), and considerable IL-5 (Figure 2E), demonstrating the Th2 polarization of the mucosal T cell response in the nose and lungs in aPV-immunized mice. Thus, the same immunization regimen employed with aPV and aPV/BcfA elicits different CD4+ TRM phenotypes.
Prime-pull immunization generates mucosal and systemic antibodies
Natural infection or i.n. immunization with experimental pertussis vaccines generates both IgG and IgA responses in the lungs (27, 41, 42), which are important for immunity against several respiratory pathogens (37). We quantified antigen-specific IgA and IgG in the lungs of mice immunized with aPV or aPV/BcfA delivered i.m./i.m. or i.m./i.n. aPV delivered by i.m./i.m. or i.m./i.n. immunization did not elicit IgA responses in the lungs against the background of unimmunized mice (Figure 3A). Mice immunized with aPV/BcfA through the i.m./i.n. route elicited higher FHA and PRN specific IgA antibodies in the lungs compared with naïve mice and mice immunized i.m./i.m. with aPV/BcfA (Figure 3A). However, there was no significant difference in IgA levels between aPV- and aPV/BcfA- immunized mice. Serum IgA was not increased compared to naïve mice with either the vaccine or immunization route (data not shown).
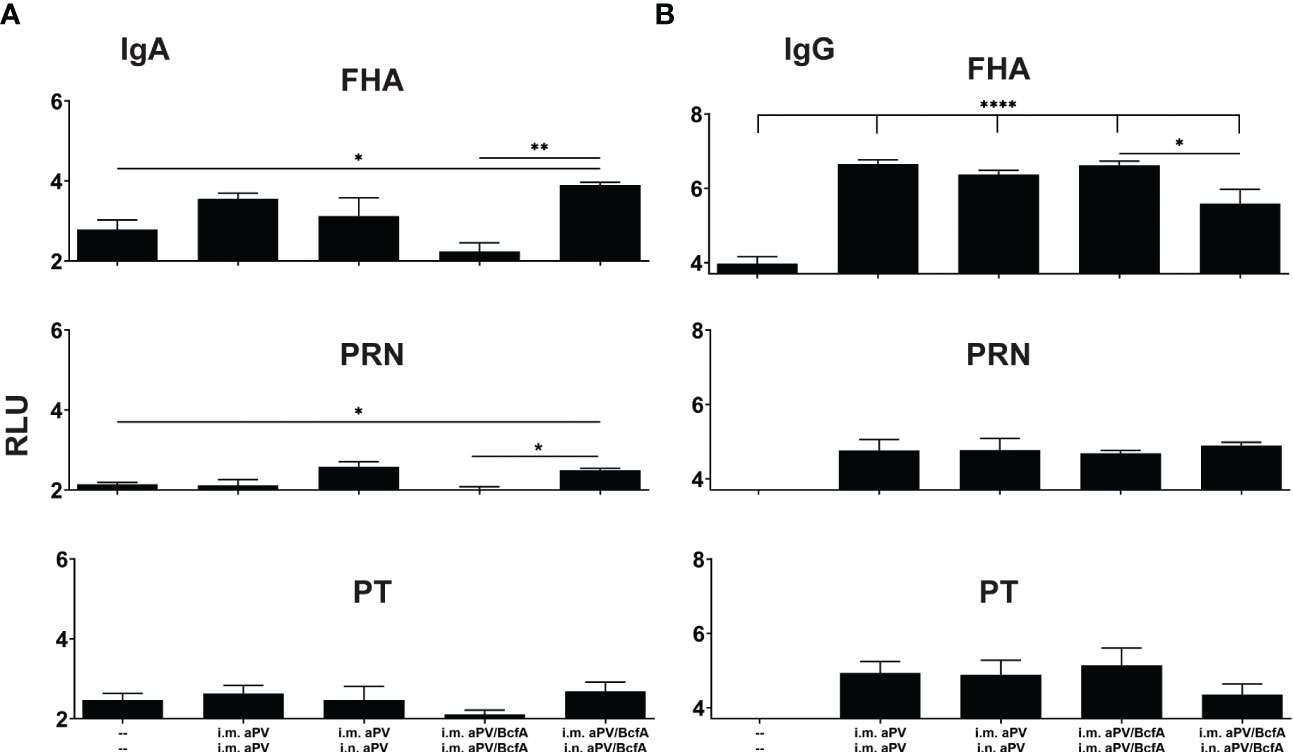
Figure 3 I.m/i.n immunization with aPV and aPV/BcfA generates mucosal and systemic IgA and IgG antibodies. Antibodies in lung homogenates and serum collected at 2 weeks post-boost (d42) were evaluated by an antigen-specific multiplex assay. FHA-, PRN-, and PT-specific IgA (A) and IgG (B) in the lungs. Relative luminescence units were log10 transformed. Data are presented as mean +/- SEM with samples from 5-6 mice/group. One representative experiment of two independent experiments is shown. Data were analyzed by one-way ANOVA with Holm-Sidak correction for multiple comparisons. *P<0.05, **P<0.01, ****P<0.0001.
FHA-specific IgG levels in the lungs were increased with both aPV and aPV/BcfA immunization compared to naïve mice (Figure 3B). There was a slight decrease in FHA-specific IgG in the lungs of mice immunized with i.m./i.n. aPV/BcfA compared to i.m./i.m. delivery of the same vaccine. PRN and PT specific IgG were not increased above naïve mice. FHA, PRN, and PT specific serum IgG levels in immunized mice were higher than in naïve mice. Anti-PT serum IgG levels were slightly lower in mice immunized i.m./i.n. with aPV/BcfA, compared to mice immunized i.m./i.m. with the same formulation (data not shown).
We then quantified the IgG isotypes in the lungs (Figure 4). FHA and PRN-specific IgG1 was higher in vaccinated groups compared to naïve animals but did not differ between vaccines. Anti-PT antibodies were increased in the lungs following i.m./i.m. immunization with aPV/BcfA. Interestingly, compared to naïve mice, anti-PT responses did not increase in the lungs of mice immunized i.m/i/n with aPV/BcfA. Similarly, anti-FHA and anti-PT IgG2c was elevated by immunization in all groups except in mice immunized i.m./i.n.with aPV/BcfA (Figure 4B). In this group, anti-FHA and anti-PT IgG2c was lower in mice immunized i.m./i.n. with aPV/BcfA compared to mice immunized i.m./i.m. with aPV/BcfA. IgG2b did not increase following immunization (data not shown). Overall, these data suggest that i.m./i.n. immunization with aPV/BcfA does not increase antigen-specific IgG responses in the lungs compared to i.m./i.m. immunization with aPV/BcfA or compared to aPV alone delivered i.m./i.m. or i.m./i.n. While IgG isotypes in the serum in all immunization groups were also increased compared to naïve mice (Figure S2), there was little difference in the isotypes between immunization groups. Overall, prime-pull immunization with either aPV or aPV/BcfA did not increase total IgG and IgG isotypes in the serum and lungs compared to systemic prime and boost alone.
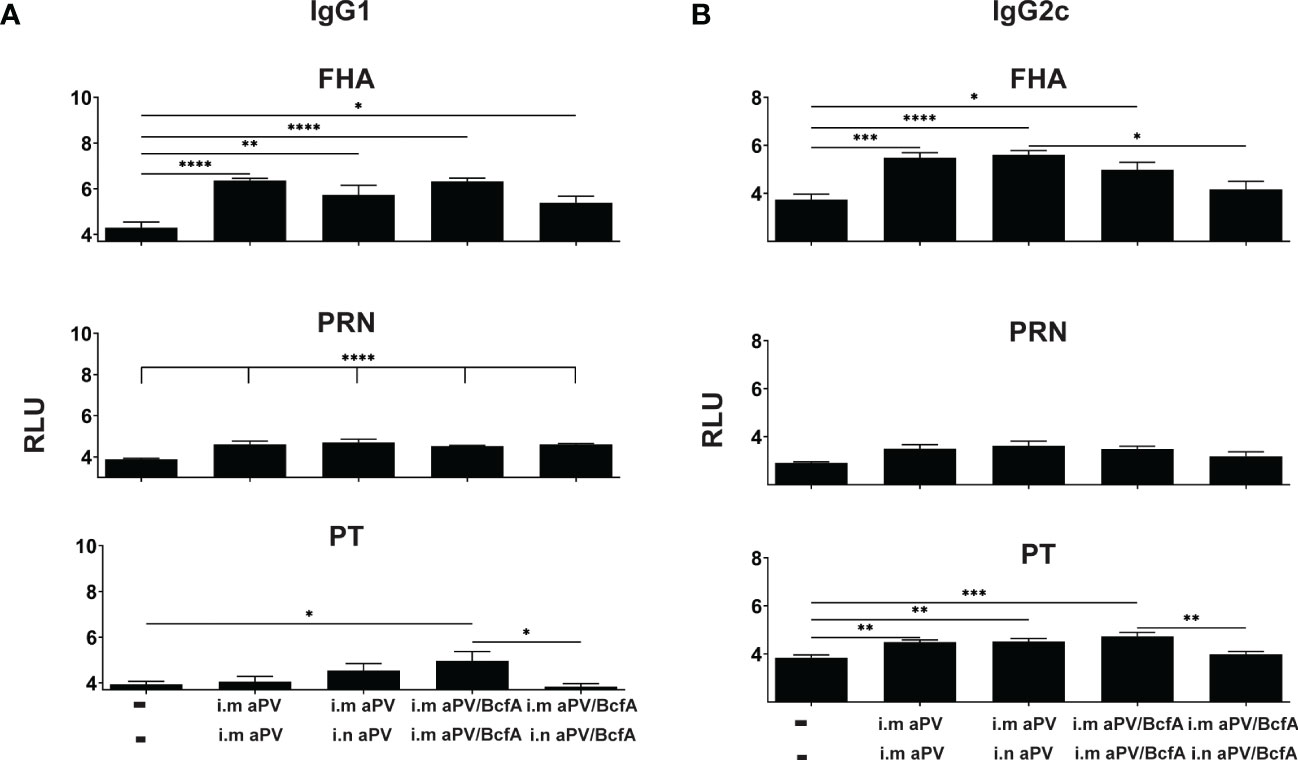
Figure 4 Lung IgG isotypes are similar in aPV and aPV/BcfA immunized mice. IgG1 (A) IgG2c (B) antibody isotypes in lung homogenate were quantified by multiplex assay in samples shown in Figure 3. Data were analyzed by one-way ANOVA with Holm-Sidak correction for multiple comparisons. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Prime-pull immunization with aPV/BcfA reduces B. pertussis nasal colonization
We then measured the effect of systemic priming and mucosal booster on B. pertussis respiratory tract colonization. Mice primed i.m. with aPV alone or aPV/BcfA were boosted i.m. or i.n. with the same formulation. Two weeks post-boost mice were i.n. challenged with 5x105 CFUs of B. pertussis strain Bp536. Lungs and nasal tissue were harvested on days 1-14 post-challenge for enumeration of CFUs. Bacterial burden in the lungs was reduced by more than 2 logs in all immunized mice compared to naïve mice by 1dpi. All lungs of immunized mice had bacterial burden below the limit of detection by 14 dpi while naïve challenged mice remained colonized (Figure 5A). Bacterial burden in the nose was reduced on d4 and d7 post-challenge in mice immunized i.m/i.n. with aPV/BcfA (Figure 5B), compared to mice immunized i.m/i.n. with aPV alone or in unimmunized challenged mice. Interestingly, there was a slight but significant reduction in nasal colonization of mice immunized i.m/i.m with aPV/BcfA compared to mice immunized i.m/i.m with aPV alone (Figure 5B and Figure S3) or naïve mice. (Comparisons between vaccine formulations and immunization routes are separated for clarity in Figure S3). Thus, the immune response elicited by the addition of BcfA to aPV was more effective than aPV alone in reducing B. pertussis colonization of the nose.
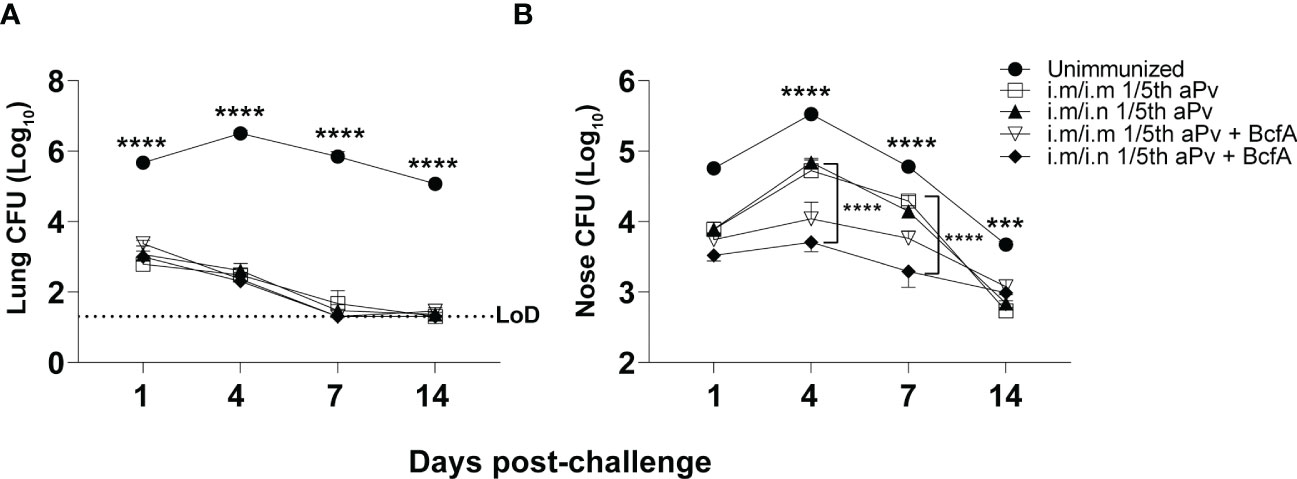
Figure 5 Prime-pull immunization with aPV/BcfA reduces B. pertussis burden in the nose upon challenge. C57BL/6 mice were primed on d0 and boosted on d28 with the indicated vaccines and immunization routes. Unimmunized and immunized mice were challenged i.n. on d42 with 5x105 CFU Bp536. (A) Lungs and (B) nose were harvested at the indicated time points and CFUs enumerated on BG agar plates. 5 mice/group. Dotted line represents limit of detection (LOD) for lung at 20 CFU. LOD for nose is 30 CFU (below the scale). Data were analyzed by two-way ANOVA with Holm-Sidak correction for multiple comparisons. Black asterisks indicate significant differences between unimmunized mice and the immunization groups at all time points post-challenge. The brackets indicate statistical differences between i.m./i.m. aPV and i.m./i.n. aPV/BcfA at day 4 and day 7 post-challenge. ***P<0.001, ****P<0.0001 by ANOVA.
Discussion
Current aPV fail to prevent nasal colonization of B. pertussis and may, in fact, exacerbate nasal colonization (2, 23). Thus, the large cohort of people who have been immunized i.m. with aPV may carry B. pertussis asymptomatically and transmit the infection to infants and other vulnerable populations. This may be due to the immunization route or the Th2 polarized immune responses elicited by alum. Here, we tested whether combining systemic priming with an i.n. booster for a “prime-pull” strategy would elicit CD4+ TRM in the upper and lower respiratory tracts, and whether the addition of BcfA to aPV would change the T-helper phenotype of the resulting TRM cells. Prime-pull immunization with aPV or aPV/BcfA elicited CD4+ TRM, showing that mucosal booster immunization is capable of recruiting T cells to the lungs and nose of mice, regardless of formulation. We previously showed that i.m. immunization of mice with aPV/BcfA elicited Th1/17 systemic T cell responses compared to Th1/2 systemic responses elicited by aPV alone (30). We found a similar trend when comparing the T-helper phenotype of mucosal TRM between vaccine compositions. I.m./i.n. administration of aPV generated IFNγ+ and IL-5+ TRM in the lungs, and primarily generated IL-5+ TRM in the nose, demonstrating a Th2-polarized response. In contrast, i.m./i.n. administration of aPV/BcfA generated IFNγ+, IL-5+, and IL-17+ TRM in the lungs, but exclusively IL-17+ TRM in the nose, demonstrating a Th1/17-polarized response. Notably, IL-5+ TRM were not detected in the nose of i.m./i.n. aPV/BcfA-immunized mice. Thus, adding BcfA to aPV changed the mucosal T cell responses from a Th2 polarized to a Th17 polarized phenotype. This result suggests that the phenotype of the TRM is shaped by the properties of the adjuvant combination.
Systemic immunization with aPV alone does not elicit mucosal antibodies (1, 23, 27). We show that PT-specific IgA was elicited in the lungs of mice that received prime-pull immunization with either aPV or aPV/BcfA, but not i.m. delivery alone. FHA-specific IgA was elicited in the lungs by i.m./i.n. immunization with aPV/BcfA, but not aPV alone, suggesting that adding BcfA to aPV elicits IgA in the mucosa. IgG was elicited in the lungs and serum of mice immunized with both aPV and aPV/BcfA, with a slight reduction in total IgG and some IgG isotypes when aPV/BcfA was delivered i.m./i.n. These data suggest that the composition of mucosal antibodies is determined by the immunization route and the properties of the adjuvant.
We showed previously that i.m. immunization of mice with aPV/BcfA accelerates bacterial clearance of B. pertussis from murine lungs compared to i.m. aPV (30). Here we show that lung colonization is reduced compared to unimmunized mice with both vaccine compositions and immunization routes. In the previous study, aPV/BcfA included a higher dose of BcfA than was used in the current work, which allowed us to discern differences following immunization with 1/5th human dose aPV. Notably, in this study, CFUs in the nose were significantly reduced on day 4 and day 7 in mice immunized i.m./i.n. with aPV/BcfA, compared to unimmunized mice and mice immunized with aPV alone, suggesting that immune responses elicited by aPV/BcfA are more effective in the upper respiratory tract.
These data have several implications. The adjuvant composition of the vaccine determines the T-helper phenotype of the immune response, regardless of immunization route. The observation that i.m./i.n. immunization with aPV/BcfA but not aPV alone reduced B. pertussis CFUs from the nose implies that the Th17 polarized TRM elicited by aPV/BcfA are important for protection of the upper respiratory tract. The systemic and mucosal antibody response generated by aPV and aPV/BcfA was similar, suggesting that the T cells have a greater contribution than antibodies to bacterial clearance from the upper respiratory tract. This conclusion is supported by previous studies in mice which established that CD4+IL-17+ TRM are critical for clearance of B. pertussis from the nose (1). Splenic T cell responses generated by prime-pull immunization with aPV alone were also Th2 polarized, while the splenic T cell responses generated by aPV/BcfA were Th1/Th17 polarized (data not shown). These data also show that the mucosal immune phenotype elicited by alum-adjuvanted aPV can be modified by the addition of BcfA, and imply that modification of the Th2-polarized aPV-primed response to a more Th17-polarized response may improve protection of the upper respiratory tract. In this study we used 1/5th human dose of aPV for mouse immunizations, while other groups deliver as little as 1/100th human dose of aPV (1, 43). Despite this high vaccine dose, the addition of BcfA modified the aPV immune response and reduced nasal colonization. We are testing lower aPV vaccine doses which may provide a larger window of time to observe differences elicited by the addition of BcfA to the vaccine. Overall, this work suggests that immunization of unimmunized individuals using an i.m. priming and i.n. booster regimen with a BcfA-adjuvanted vaccine may be effective in preventing establishment of a B. Pertussis nasopharyngeal reservoir and thereby reduce B. pertussis transmission in humans.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by Ohio State University Institutional Animal Care and Use Committee.
Author contributions
KY, JH, KC, MS, MG, KM, and AF conducted mouse experiments, SQ conducted multiplex antibody analysis, YH and JM produced FHA protein, KY, JH, PD, and RD designed experiments, KY, JH, MS, and PD analyzed data, and KY, JH, and PD wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by 1R01AI125560 and 1R01AI153829 (to PD and RD). KSY was supported by 2 T32 AI112542 through the Infectious Disease Institute at The Ohio State University. JH is supported by NIAID 1T32AI165391-01 from the Infectious Disease Institute at The Ohio State University.
Acknowledgments
We thank Prosper Boyaka and Amy Lovett-Racke for critical review of the manuscript and Jennifer Bruno for technical assistance. We acknowledge use of the Rochester Human Immunology Center Core laboratory.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1181876/full#supplementary-material
Supplementary Figure 1 | Gating strategy to identify CD4+ TRM in the nose and lungs of aPV immunized mice. Lung and nose tissues were enzymatically digested and stained with antibodies and collected on a Cytek Aurora spectral flow cytometry. TRM were identified as live, CD3+, CD4+, CD45 i.v.-, CD44+, CD62L-, CD69+ cells.
Supplementary Figure 2 | Similar systemic IgG responses are elicited by aPV and aPV/BcfA. IgG1 and IgG2c antibody isotypes in serum were quantified by multiplex assay in samples shown in . Data were analyzed by one-way ANOVA with Holm-Sidak correction for multiple comparisons. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Supplementary Figure 3 | aPV/BcfA immunization reduces B. pertussis nasal colonization. Data in were separated according to immunization regimen and vaccine type. (A) Mice immunized i.m./i.m. with aPV alone or aPV/BcfA; (B) Mice immunized i.m./i.n. with aPV or aPV/BcfA. (C) Mice immunized i.m./i.m. or i.m./i.n. with aPV alone (D) Mice immunized i.m./i.m. or i.m./i.n. with aPV/BcfA. *P<0.05, **P<0.01, ****P<0.0001. Black asterisks indicate significant differences between unimmunized mice and the immunization groups at each time point. The bracket indicates the differences between the indicated immunization groups at day 4 and day 7 post-challenge.
References
1. Wilk MM, Borkner L, Misiak A, Curham L, Allen AC, Mills KHG. Immunization with whole cell but not acellular pertussis vaccines primes CD4 TRM cells that sustain protective immunity against nasal colonization with bordetella pertussis. Emerg Microbes Infect (2019) 8:169–85. doi: 10.1080/22221751.2018.1564630
2. Warfel JM, Zimmerman LI, Merkel TJ. Acellular pertussis vaccines protect against disease but fail to prevent infection and transmission in a nonhuman primate model. Proc Natl Acad Sci United States America (2014) 111:787–92. doi: 10.1073/pnas.1314688110
3. Mills KH, Ross PJ, Allen AC, Wilk MM. Do we need a new vaccine to control the re-emergence of pertussis? Trends Microbiol (2014) 22:49–52. doi: 10.1016/j.tim.2013.11.007
4. Cody CL, Baraff LJ, Cherry JD, Marcy SM, Manclark CR. Nature and rates of adverse reactions associated with DTP and DT immunizations in infants and children. Pediatrics (1981) 68:650–60. doi: 10.1542/peds.68.5.650
5. Sato Y, Sato H. Development of acellular pertussis vaccines. Biologicals (1999) 27:61–9. doi: 10.1006/biol.1999.0181
6. Warfel JM, Papin JF, Wolf RF, Zimmerman LI, Merkel TJ. Maternal and neonatal vaccination protects newborn baboons from pertussis infection. J Infect Dis (2014) 210:604–10. doi: 10.1093/infdis/jiu090
7. Feldstein LR, Mariat S, Gacic-Dobo M, Diallo MS, Conklin LM, Wallace AS. Global routine vaccination coverage, 2016. MMWR. Morbidity mortality weekly Rep (2017) 66:1252–5. doi: 10.15585/mmwr.mm6645a3
8. Amirthalingam G, Gupta S, Campbell H. Pertussis immunisation and control in England and Wales, 1957 to 2012: a historical review. Euro surveillance Bull Europeen sur les maladies transmissibles = Eur communicable Dis Bull (2013) 18. doi: 10.2807/1560-7917.ES2013.18.38.20587
9. Barret AS, Ryan A, Breslin A, Cullen L, Murray A, Grogan J, et al. Pertussis outbreak in northwest Ireland, January - June 2010. Euro surveillance Bull Europeen sur les maladies transmissibles = Eur communicable Dis Bull (2010) 15. doi: 10.2807/ese.15.35.19654-en
10. Cherry JD. Epidemic pertussis in 2012–the resurgence of a vaccine-preventable disease. New Engl J Med (2012) 367:785–7. doi: 10.1056/NEJMp1209051
11. de Melker HE, Conyn-van Spaendonck MA, Rumke HC, van Wijngaarden JK, Mooi FR, Schellekens JF. Pertussis in the Netherlands: an outbreak despite high levels of immunization with whole-cell vaccine. Emerg Infect Dis (1997) 3:175–8. doi: 10.3201/eid0302.970211
12. Pillsbury A, Quinn HE, McIntyre PB. Australian Vaccine preventable disease epidemiological review series: pertussis, 2006-2012. Communicable Dis Intell Q Rep (2014) 38:E179–94.
13. Skoff TH, Martin SW. Impact of tetanus toxoid, reduced diphtheria toxoid, and acellular pertussis vaccinations on reported pertussis cases among those 11 to 18 years of age in an era of waning pertussis immunity: a follow-up analysis. JAMA Pediatr (2016) 170:453–8. doi: 10.1001/jamapediatrics.2015.4875
14. Cherry JD. Adult pertussis in the pre- and post-vaccine eras: lifelong vaccine-induced immunity? Expert Rev Vaccines (2014) 13:1073–80. doi: 10.1586/14760584.2014.935765
15. Trainor EA, Nicholson TL, Merkel TJ. Bordetella pertussis transmission. Pathog Dis (2015) 73:ftv068. doi: 10.1093/femspd/ftv068
16. Althouse BM, Scarpino SV. Asymptomatic transmission and the resurgence of bordetella pertussis. BMC Med (2015) 13:146. doi: 10.1186/s12916-015-0382-8
17. Burdin N, Handy LK, Plotkin SA. What is wrong with pertussis vaccine immunity? the problem of waning effectiveness of pertussis vaccines. Cold Spring Harb Perspect Biol (2017) 9. doi: 10.1101/cshperspect.a029454
18. de Melker HE, Versteegh FG, Schellekens JF, Teunis PF, Kretzschmar M. The incidence of bordetella pertussis infections estimated in the population from a combination of serological surveys. J Infect (2006) 53:106–13. doi: 10.1016/j.jinf.2005.10.020
19. Gill C, Rohani P, Thea DM. The relationship between mucosal immunity, nasopharyngeal carriage, asymptomatic transmission and the resurgence of bordetella pertussis. F1000Res (2017) 6:1568. doi: 10.12688/f1000research.11654.1
20. Zhang Q, Yin Z, Li Y, Luo H, Shao Z, Gao Y, et al. Prevalence of asymptomatic bordetella pertussis and bordetella parapertussis infections among school children in China as determined by pooled real-time PCR: a cross-sectional study. Scand J Infect Dis (2014) 46:280–7. doi: 10.3109/00365548.2013.878034
21. Fine PE. Herd immunity: history, theory, practice. Epidemiol Rev (1993) 15:265–302. doi: 10.1093/oxfordjournals.epirev.a036121
23. Holubova J, Stanek O, Brazdilova L, Masin J, Bumba L, Gorringe AR, et al. Acellular pertussis vaccine inhibits bordetella pertussis clearance from the nasal mucosa of mice. Vaccines (2020) 8. doi: 10.3390/vaccines8040695
24. Fedele G, Cassone A, Ausiello CM. T-Cell immune responses to bordetella pertussis infection and vaccination. Pathog Dis (2015) 73. doi: 10.1093/femspd/ftv051
25. Allen AC, Wilk MM, Misiak A, Borkner L, Murphy D, Mills KHG. Sustained protective immunity against bordetella pertussis nasal colonization by intranasal immunization with a vaccine-adjuvant combination that induces IL-17-secreting TRM cells. Mucosal Immunol (2018) 11:1763–76. doi: 10.1038/s41385-018-0080-x
26. Zurita ME, Wilk MM, Carriquiriborde F, Bartel E, Moreno G, Misiak A, et al. A pertussis outer membrane vesicle-based vaccine induces lung-resident memory CD4 T cells and protection against bordetella pertussis, including pertactin deficient strains. Front Cell Infect Microbiol (2019) 9:125. doi: 10.3389/fcimb.2019.00125
27. Solans L, Debrie AS, Borkner L, Aguilo N, Thiriard A, Coutte L, et al. IL-17-dependent SIgA-mediated protection against nasal bordetella pertussis infection by live attenuated BPZE1 vaccine. Mucosal Immunol (2018). 11(6):1753–176. doi: 10.1038/s41385-018-0073-9
28. Sukumar N, Love CF, Conover MS, Kock ND, Dubey P, Deora R. Active and passive immunizations with bordetella colonization factor a protect mice against respiratory challenge with bordetella bronchiseptica. Infect Immun (2009) 77:885–95. doi: 10.1128/IAI.01076-08
29. Sukumar N, Mishra M, Sloan GP, Ogi T, Deora R. Differential bvg phase-dependent regulation and combinatorial role in pathogenesis of two bordetella paralogs, BipA and BcfA. J Bacteriol (2007) 189:3695–704. doi: 10.1128/JB.00009-07
30. Jennings-Gee J, Quataert S, Ganguly T, D'Agostino R Jr., Deora R, Dubey P. The adjuvant bordetella colonization factor a attenuates alum-induced Th2 responses and enhances bordetella pertussis clearance from mouse lungs. Infect Immun (2018) 86. doi: 10.1128/IAI.00935-17
31. Yount KS, Jennings-Gee J, Caution K, Fullen AR, Corps KN, Quataert S, et al. Bordetella colonization factor a (BcfA) elicits protective immunity against bordetella bronchiseptica in the absence of an additional adjuvant. Infect Immun (2019). 87(10):e00506–19. doi: 10.1101/692830
32. Relman DA, Domenighini M, Tuomanen E, Rappuoli R, Falkow S. Filamentous hemagglutinin of bordetella pertussis: nucleotide sequence and crucial role in adherence. Proc Natl Acad Sci United States America (1989) 86:2637–41. doi: 10.1073/pnas.86.8.2637
33. Brito LA, Singh M. Acceptable levels of endotoxin in vaccine formulations during preclinical research. J Pharm Sci (2011) 100:34–7. doi: 10.1002/jps.22267
34. Caution K, Yount K, Deora R, Dubey P. Evaluation of host-pathogen responses and vaccine efficacy in mice. JoVE (2019): (144) e58930. doi: 10.3791/58930
35. Wilk MM, Misiak A, McManus RM, Allen AC, Lynch MA, Mills KHG. Lung CD4 tissue-resident memory T cells mediate adaptive immunity induced by previous infection of mice with <em<Bordetella pertussis</em>. J Immunol (2017) 199:233. doi: 10.4049/jimmunol.1602051
36. Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc (2014) 9:209–22. doi: 10.1038/nprot.2014.005
37. Strugnell RA, Wijburg OL. The role of secretory antibodies in infection immunity. Nat Rev Microbiol (2010) 8:656–67. doi: 10.1038/nrmicro2384
38. Mueller SN, Mackay LK. Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol (2016) 16:79–89. doi: 10.1038/nri.2015.3
39. Mills KH, Barnard A, Watkins J, Redhead K. Cell-mediated immunity to bordetella pertussis: role of Th1 cells in bacterial clearance in a murine respiratory infection model. Infect Immun (1993) 61:399–410. doi: 10.1128/iai.61.2.399-410.1993
40. Ross PJ, Sutton CE, Higgins S, Allen AC, Walsh K, Misiak A, et al. Relative contribution of Th1 and Th17 cells in adaptive immunity to bordetella pertussis: towards the rational design of an improved acellular pertussis vaccine. PloS Pathog (2013) 9:e1003264. doi: 10.1371/journal.ppat.1003264
41. Goodman YE, Wort AJ, Jackson FL. Enzyme-linked immunosorbent assay for detection of pertussis immunoglobulin a in nasopharyngeal secretions as an indicator of recent infection. J Clin Microbiol (1981) 13:286–92. doi: 10.1128/jcm.13.2.286-292.1981
42. Raeven RHM, Rockx-Brouwer D, Kanojia G, van der Maas L, Bindels THE, Ten Have R, et al. Intranasal immunization with outer membrane vesicle pertussis vaccine confers broad protection through mucosal IgA and Th17 responses. Sci Rep (2020) 10:7396. doi: 10.1038/s41598-020-63998-2
Keywords: B. pertussis, mucosal immunity, tissue-resident memory T cells, BcfA, pertussis vaccines, adjuvants
Citation: Yount KS, Hall JM, Caution K, Shamseldin MM, Guo M, Marion K, Fullen AR, Huang Y, Maynard JA, Quataert SA, Deora R and Dubey P (2023) Systemic priming and intranasal booster with a BcfA-adjuvanted acellular pertussis vaccine generates CD4+ IL-17+ nasal tissue resident T cells and reduces B. pertussis nasal colonization. Front. Immunol. 14:1181876. doi: 10.3389/fimmu.2023.1181876
Received: 08 March 2023; Accepted: 04 May 2023;
Published: 18 May 2023.
Edited by:
Luciana C C Leite, Butantan Institute, BrazilReviewed by:
Larry Ellingsworth, Novavax, Inc., United StatesDunia Rodriguez, Butantan Institute, Brazil
Copyright © 2023 Yount, Hall, Caution, Shamseldin, Guo, Marion, Fullen, Huang, Maynard, Quataert, Deora and Dubey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Purnima Dubey, cHVybmltYS5kdWJleUBvc3VtYy5lZHU=
†These authors have contributed equally to this work