 Shankun Zhao
Shankun Zhao Jian Liao2†
Jian Liao2†
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 05 May 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1180866
This article is part of the Research TopicInsights in Inflammation: 2022View all 13 articles
Sepsis-induced acute kidney injury (SI-AKI), a common critically ill, represents one of the leading causes of global death. Emerging evidence reveals autophagy as a pivotal modulator of SI-AKI. Autophagy affects the cellular processes of renal lesions, including cell death, inflammation, and immune responses. Herein, we conducted a systematic and comprehensive review on the topic of the proposed roles of autophagy in SI-AKI. Forty-one relevant studies were finally included and further summarized and analyzed. This review revealed that a majority of included studies (24/41, 58.5%) showed an elevation of the autophagy level during SI-AKI, while 22% and 19.5% of the included studies reported an inhibition and an elevation at the early stage but a declination of renal autophagy in SI-AKI, respectively. Multiple intracellular signaling molecules and pathways targeting autophagy (e.g. mTOR, non-coding RNA, Sirtuins family, mitophagy, AMPK, ROS, NF-Kb, and Parkin) involved in the process of SI-AKI, exerting multiple biological effects on the kidney. Multiple treatment modalities (e.g. small molecule inhibitors, temsirolimus, rapamycin, polydatin, ascorbate, recombinant human erythropoietin, stem cells, Procyanidin B2, and dexmedetomidine) have been found to improve renal function, which may be attributed to the elevation of the autophagy level in SI-AKI. Though the exact roles of autophagy in SI-AKI have not been well elucidated, it may be implicated in preventing SI-AKI through various molecular pathways. Targeting the autophagy-associated proteins and pathways may hint towards a new prospective in the treatment of critically ill patients with SI-AKI, but more preclinical studies are still warranted to validate this hypothesis.
Sepsis, one of the common diseases in the intensive care units, seriously threatens the lives of sufferers, contributing to 30-50% of deaths in hospitals (1). As a result of the dysregulated host response to infection, severe systemic inflammation may induce septic shock, disseminated intravascular coagulation (DIC), and progressive multi-organ dysfunction syndrome (MODS) (2). The commonly affected organs include the heart, lungs, liver, brain, intestine, and kidneys. During sepsis, activation of the sympathetic nervous system, the release of vasoactive substances, and endothelial injury, together contribute to the redistribution of blood flow and microcirculation disturbances (3). These factors remarkably damage the kidney tissue (e.g. renal tubular) and therefore induce acute kidney injury (AKI) and even acute renal failure (ARF). ARF was found to be the most common complication of sepsis, accounting for nearly 50% of the incidence rate (4). On the other hand, it was reported that approximately 76% of in-hospital deaths are caused by sepsis-induced AKI (SI-AKI) (5). Since multidrug-resistant bacteria and adverse events are common in sepsis, septic ARF has a significantly higher mortality rate than that of non-sepsis-related ARF (caused by other pathogenic factors or some diseases) (6). To effectively prevent and treat septic AKI or ARF, intensive efforts have been made to develop innovative therapeutic measures and explore the detailed molecular pathological mechanisms underlying SI-AKI. However, due to the complex character of SI-AKI, the exact pathogenesis mechanisms for septic AKI are not completely addressed, prohibiting or arresting effective treatments for septic AKI.
At present, there is growing evidence that autophagy plays a role in the pathogenesis process of SI-AKI (7). Autophagy is an adaptive catabolic process and is commonly correlated to cellular death, protection, or survival. It conserves the degradation of eukaryotic cells and the recycling process, maintaining cellular homeostasis by engulfing cellular targets (i.e., pathogens, unfolded proteins, carbohydrates, lipids, nucleic acids, and damaged organelles) (8). Autophagy can be regulated by a complex signaling network comprised of autophagy-related genes (ATC). Mitophagy and lysophagy are the common types of selective autophagy. Autophagy is considered to be a cellular stress triggered by a multiplicity of adverse environmental cues, i.e., hypoxia, oxidative stress, and nutrient depletion (9). Experimental and clinical findings reveal that autophagy may serve as a pathogenic mediator of human diseases by regulating inflammation, innate immunity, and host defense. Dysregulated or maladaptive autophagy with propathogenic responses was found to be associated with the pathogenesis of disorders (10).
For the kidneys to function normally, autophagy is necessary (11). Besides, the presence of kidney pathologies in mice bearing genetic deletions of key autophagy regulator proteins. For example, in an animal model with targeted deletions of Atg5 or Atg7, chronic kidney disease could be induced (12). Therefore, autophagy may act as a key safeguard against the declination of kidney function. Also, it was reported that autophagy generally protects the kidney from various injuries, e.g. sepsis, renal ischemia-reperfusion (I/R), or exposure to nephrotoxins (13). However, the exact molecular mechanisms underlying the action of autophagy in AKI are still exploring.
In this review, we focused specifically on the roles of autophagy in sepsis-induced AKI according to the current evidence. Elucidating the biological effects of autophagy in septic AKI is of pivotal importance, which may provide strategies and targets for therapeutic interventions in clinical practice.
To identify the studies that reported the association between autophagy and sepsis-induced AKI, we conducted a comprehensive literature review in the common-used databases, i.e., MEDLINE, Web of Science, Google Scholar, EMBASE, and Cochrane Library. The reference list in the review or original studies was also retrieved to identify additional relevant studies. Data from relevant studies were extracted using a data collection table. The following information was noted, including the first author of the included studies, year of publication, experimental model or participant, methods for establishing sepsis-induced AKI, the status of autophagy, associated genes or pathways in the action of autophagy, and the main findings of the relevant studies. Finally, there were 41 experimental and clinical studies (7, 14–33) (34–53) included in the review. The selection process for screening the relevant studies shown in Supplementary Figure 1. For the methods for establishing the SI-AKI, cecal ligation and puncture (CLP) were used in in-vivo studies, while lipopolysaccharide (LPS) was applied in in vitro studies.
Among the 41 relevant studies, 24 of them (24/41, 58.5%) reported that the autophagy status was activated in septic AKI, while eight eligible studies showed that autophagy inhibition occurred in the kidney under sepsis and the remaining nine studies reported that autophagy elevated in early stage but declined in the later phase during sepsis-induced AKI.
Autophagy, a form of programmed cell death different from apoptosis, occurs in all eukaryotic cells and is associated with cellular turnover and energy balance. Death of cells occurs when apoptosis appears, whereas autophagy is a “double-edged sword” for both survival and death (54, 55). Apoptosis and autophagy are interconnected in some way. To protect cells from apoptosis and necrosis under stress, autophagy is commonly activated. Nevertheless, excessive autophagy can increase apoptosis due to mitochondrial damage. Autophagy plays a protective role in proximal tubular cells of the kidney against AKI (11). Of note, autophagy is considered to play a double role during sepsis. Basal autophagy functions the protection of the injuried kidney by eliminating toxic oxidative proteins. However, severe stress (i.e., ROS eruption) may induce excessive autophagy, leading to autophagic cell death (56). It is reported that autophagy is initiated early after sepsis, protecting against endotoxic kidney damage (57). Afterward, autophagic cell death may cause a phase of dysfunction, aggravating the sepsis-induced oxidative injury.
At present, specific molecular mechanisms underlying sepsis-associated AKI are not fully understood. Several potential mechanisms could be involved (58). First, autophagy can protect proximal tubular cells from mitochondrial oxidative stress and ischemic injury. ‘Mitophagy’ is a specific type of autophagy, which is characterized by the clearance of mitochondria by autophagy. Second, autophagy also plays role in protecting proximal tubular cells from DNA damage. Third, autophagy can reduce the abnormal protein accumulation of proximal tubular cells.
Since the level of autophagy flux was different among the 41 included studies, exploring the underlying mechanisms of autophagy is of great interest and has important implications for septic AKI research. Here, we summarize recent laboratory and clinical studies, focusing on critical factors in the pathophysiology of sepsis-associated AKI: microcirculatory dysfunction, inflammation, NOD-like receptor protein 3 inflammasome, microRNAs, extracellular vesicles, autophagy and efferocytosis, inflammatory reflex pathway, vitamin D, and metabolic reprogramming. Lastly, identifying these molecular targets and defining clinical subphenotypes will permit precision approaches in the prevention and treatment of SI-AKI.
It is possible that autophagy plays a key role in the recovery of AKI by promoting cell survival. Autophagy can be triggered in response to various types of stress, including sepsis. A critical cytoprotective role for autophagy in sepsis-mediated AKI has been found in recent years (11). The protective effects exerted by autophagy may be associated with the removal of damaged mitochondria or mitophagy, which predominantly affects the mitochondria-rich proximal tubule cells (58). Currently, evidence that harnessing the autophagic machinery on SI-AKI is still controversial. The levels of autophagy flux during SI-AKI are different among studies.
Twenty-four publications reported the autophagy level was activated during sepsis-induced AKI. Alexander et al. (27) performed the autopsy on 17 patients who died from coronavirus disease 2019 (COVID-19) and molecular characteristics were compared with archived cases of S-AKI and non-sepsis causes of AKI. They found that the autophagy level was significantly higher in sepsis-induced AKI than in non-sepsis-related AKI (P=0.023). However, the autophagy status was comparable between sepsis AKI and COVID AKI (P=0.621). This clinical trial indicated that mitochondrial autophagy dysfunction might play a pivotal role in SI-AKI, which might provide novel diagnostic and therapeutic targets for treating SI-AKI. In another clinical study developed by Feng et al. (23), the investigators also revealed the autophagy status was elevated in SI-AKI patients. As a result, exploring the molecular mechanisms underlying the actions of autophagy dysfunction in septic AKI may be clinically instructive. Currently, 24 experimental studies had investigated the association between autophagy and SI-AKI. Nevertheless, the detailed pathomechanisms of autophagy contributed to the pathogenesis of septic AKI.
The characteristics and the main findings of the 24 included studies were summarized in Table 1. Figure 1 (left column) displayed the molecular mechanisms of activated autophagy during SI-AKI.
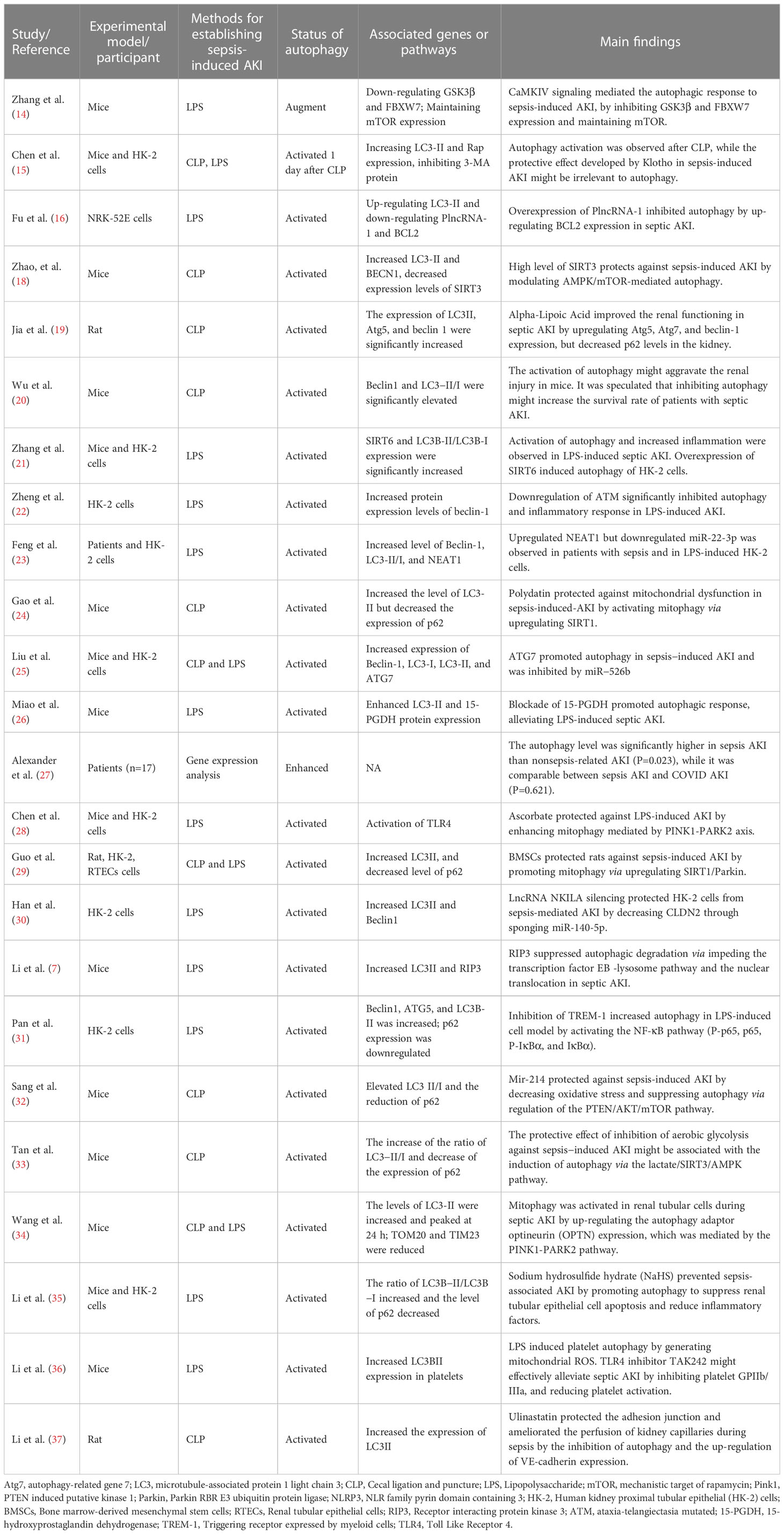
Table 1 The characteristics and the main findings of the 24 relevant studies reporting activation of autophagy in SI-AKI.
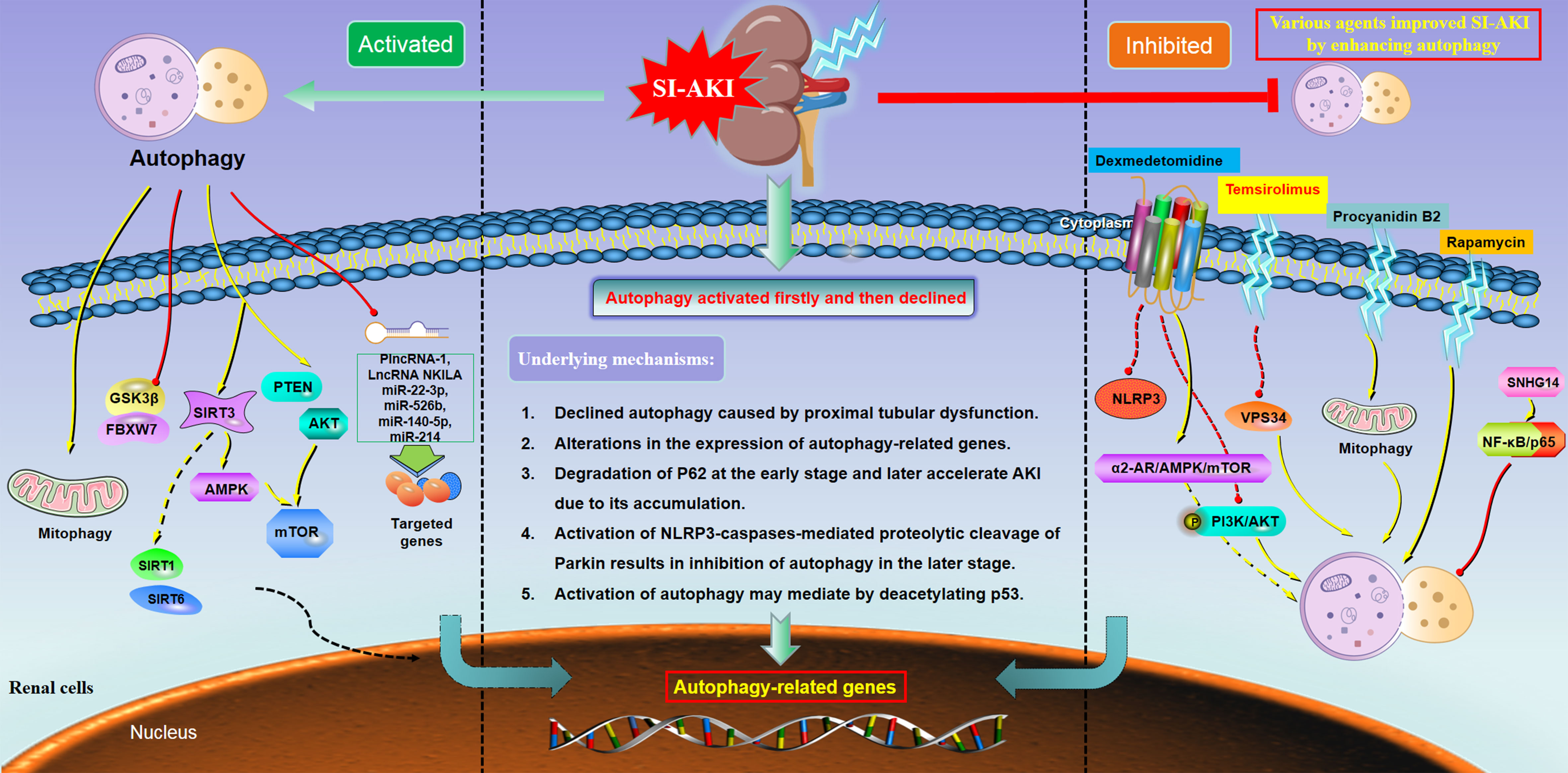
Figure 1 Main molecular mechanisms underlying the dual role of autophagy in sepsis-induced acute kidney injury. Under the condition of SI-AKI, the autophagy level presented with three tendencies, including activation (left column), activated first and then declined (middle column), and inhibition (right column). These distinct patterns of autophagy in SI-AKI might be regulated by multiple associated genes and a series of downstream signaling. Abbreviation: mTOR= mechanistic target of rapamycin; NLRP3, NLR family pyrin domain containing 3; Sirt1, Sirtuin 1; Sirt3, Sirtuin 3; Sirt6, Sirtuin 6; GSK3β, Glycogen Synthase Kinase 3β; AMPK, Adenosine 5’-monophosphate-activated protein kinase; PI3K, Phosphatidylinositol 3-kinase; VPS34, Vacuolar protein sorting 34; PTEN, Phosphatase and tensin homolog.
The mammalian target of the rapamycin (mTOR) pathway is one of the pivotal signaling pathways with critical biological function in multiple diseases (59), including SI-AKI (60). mTOR has been reported to play a central role in the regulation of autophagy, characterized by inhibiting autophagy in the biological process of growth factors and abundant nutrients (61). The calcium/calmodulin-dependent protein kinases (CaMK) were found to regulate septic inflammation. Zhang et al. (14) demonstrated that CaMKIV signaling mediated the autophagic response to LPS-associated septic AKI by inhibiting GSK3β and FBXW7 expression and maintaining mTOR levels. mTOR and AMP-activated protein kinase (AMPK) have been reported to correlate with in the development of autophagy in AKI (62). BECN1, Bcl-2, and LC3-II are pivotal autophagy-related proteins. Increased LC3-II and BECN1 expression have been found in SI-AKI, indicating the autophagy status enhanced during sepsis. Zhao et al. (18) showed that elevated autophagy occurred in SI-AKI, while a high level of SIRT3 could protect against AKI by modulating AMPK/mTOR-mediated autophagy. In a mouse model of SI-AKI conducted by CLP, Sang et al. (32) also confirmed that the kidney autophagy level was elevated. They further found that PTEN/AKT/mTOR signaling pathway was involved in this action. The above studies indicated that mTOR was one of the key targets for the activation of autophagy during SI-AKI.
Both long non-coding RNAs (lncRNAs) and microRNA (miRNA) have been suggested to play essential roles in various diseases, including SI-AKI (63, 64). It is known that lncRNA regulates the activities of miRNA through the lncRNA-miRNA interactions. On the topic of the present study, we also found that both lncRNA and miRNA participated in the development of autophagy-mediated SI-AKI. PlncRNA-1 was reported to initiate malignancy in multiple cancers and play roles in inflammatory diseases (65). Fu et al. (16) showed that renal autophagy was activated in SI-AKI by up-regulating LC3-II and down-regulating PlncRNA-1 and BCL2 levels, while overexpression of PlncRNA-1 inhibited autophagy by up-regulating BCL2 expression. Enhanced autophagy was also observed in Feng et al.’ study (23). Upregulated NEAT1 but downregulated miR-22-3p was found in patients with SI-AKI and in LPS-induced HK-2 cells. Liu et al. (23) suggested that ATG7 promoted autophagy (characterized by increasing levels of Beclin-1, LC3-I, and LC3-II) in SI-AKI, which could be inhibited by miR−526b. In a recent study conducted by Han et al. (30), the authors demonstrated that kidney autophagy was activated, while LncRNA NKILA silencing could protect HK-2 cells from SI-AKI by decreasing CLDN2 by sponging miR-140-5p. Sang et al. (32) reported that an increased level of LC3-II was found in CLP-induced SI-AKI, indicating the autophagic level was active. They also observed that miR-214 protected against sepsis-induced AKI by decreasing oxidative stress and suppressing autophagy by regulating the PTEN/AKT/mTOR pathway. These data collectively implied that the enhanced kidney autophagy occurred in SI-AKI, which was partially mediated by several specific non-coding RNAs, e.g. PlncRNA-1, miR-22-3p, miR−526b, LncRNA NKILA, miR-140-5p, and miR-214.
Mitophagy, an evolutionarily conserved biological process, is one of the pivotal cytoprotective mechanisms. It functions to remove the damaged mitochondria and maintain a healthy mitochondrial population (66). Gao et al. (24) reported that polydatin could protect against mitochondrial dysfunction in SI-AKI by activating Parkin-dependent mitophagy via upregulating SIRT1 and inhibiting NLRP3 activation. Ascorbate is a precursor for carnitine and catecholamine synthesis. It protects against oxidative stress in various diseases (67). Recently, Chen et al. (28) showed that ascorbate protected against LPS-induced AKI by enhancing mitophagy mediated by the PINK1-PARK2 axis. Bone marrow-derived mesenchymal stem cells (BMSCs) play roles in self-renewal and multi-differentiation, functioning in tissue repair and regeneration (68). It was reported that BMSCs protected rats against SI-AKI by promoting mitophagy via upregulating SIRT1/Parkin (29). In line with the above studies, Wang et al. (34) also confirmed that autophagy was increased (characterized by a high level of LC3-II) during SI-AKI. They found that mitophagy was activated in renal tubular cells in SI-AKI by up-regulating the autophagy adaptor optineurin (OPTN) expression, which was affected by the PINK1-PARK2 pathway. The above studies demonstrated that several substances (i.e., polydatin, ascorbate, and BMSCs) significantly protected against SI-AKI, which might partially mediate by the activation of mitophagy.
Sirtuins belong to the family of NAD+-dependent histone deacetylases, which play roles in energy metabolism, inflammation, and tumorigenesis (69). According to some included studies, Sirtuins (e.g. SIRT3, SIRT6, and SIRT1) have been implicated in involving in the activation of autophagy during SI-AKI. Upregulation of SIRT3 has been reported to protect against sepsis-induced AKI (18). Similarly, the protective effects of inhibition of aerobic glycolysis against SI-AKI were found to be associated with the induction of autophagy, which might be partly due to the upregulation of the SIRT3/AMPK pathway (33). In Zhang et al.’s study (21), activation of autophagy (increased LC3B-II/LC3B-I expression) and increased inflammation were observed in LPS-induced septic AKI. The authors further indicated that overexpression of SIRT6 might induce autophagy of HK-2 cells (21). Gao et al. (24) showed that polydatin could protect against mitochondrial dysfunction in SI-AKI by upregulating the expression of SIRT1. These studies suggest that Sirtuins are important in autophagic changes during SI-AKI and are the potential therapeutic targets of SI-AKI.
In addition to the above molecular molecules and pathways, the roles of autophagic activation in SI-AKI might also cause or mediate by some other biological agents. Chen et al. (15) showed that SI-AKI was accompanied by increasing LC3-II and Rap expression but inhibiting 3-MA protein. They observed that autophagy activation presented after CLP, while the protective effect developed by Klotho in SI-AKI might be irrelevant to autophagy. Jia et al. (19) reported that Alpha-Lipoic Acid could improve renal functioning in SI-AKI by upregulating the expression of autophagy-associated genes, such as Atg5, Atg7, and beclin-1. It was suggested that Beclin1 and LC3−II/I were significantly increased in SI-AKI, while the activation of autophagy might aggravate the renal injury (20). Zheng et al. (22) also confirmed that autophagy was enhanced in SI-AKI. They next found that downregulation of ataxia-telangiectasia mutated (ATM) significantly suppressed autophagy and inflammatory response in LPS-induced AKI. 15-hydroxyprostaglandin dehydrogenase (15-PGDH) is an important enzyme in the degradation of prostaglandins. Miao et al. (26) demonstrated that the blockade of 15-PGDH promoted autophagic response, alleviating LPS-induced SI-AKI. Receptor interacting-protein kinase 3 (RIP3) has been found to function as protect against renal tubular injury and renal dysfunction during septic AKI. Li et al. (7) revealed that RIP3 suppressed autophagic degradation by impeding the transcription factor EB-lysosome pathway and the nuclear translocation in SI-AKI.
Triggering receptor expressed by myeloid cells (TREM-1) is an amplifier of inflammatory responses induced by infections. Pan et al. demonstrated that inhibition of TREM-1 elevated autophagy in SI-AKI by activating the NF-κB pathway (P-p65, p65, P-IκBα, and IκBα). Sodium hydrosulfide hydrate (NaHS) has been implicated in preventing SI-AKI by promoting autophagy to suppress renal tubular epithelial cell apoptosis and reduce inflammatory factors (35). LPS can increase the production of intracellular ROS via Toll-like Receptor 4 (TLR4), which can lead to mitochondrial damage and activate platelets. LPS-induced platelet autophagy by generating mitochondrial ROS. Li et al. (36) showed that TLR4 inhibitor TAK242 might effectively alleviate SI-AKI by inhibiting platelet GPIIb/IIIa and platelet activation. Ulinastatin, a urinary trypsin inhibitor, functions to control a series of proinflammatory mediators and cytokines. A recent study showed that Ulinastatin protected the adhesion junction and ameliorated the perfusion of kidney capillaries during SI-AKI by suppressing autophagy and elevating VE-cadherin expression (37). Taken together, all the above-mentioned genes and substances might be involved in the biogenesis and biological functions of autophagy activation in SI-AKI. Targeting these affected proteins may be one of the effective therapeutic regimens that can protect against SI-AKI.
In summary, the above 24 included studies demonstrated that autophagy was increased during SI-AKI, while some drugs, substances, or molecules exert protective effects against SI-AKI also due to their properties on the promotion and enhancement of autophagy. One possible mechanism is that increased autophagy may be one of the phenotypes of the early stage of SI-AKI, which can be considered as a physiological compensatory response. Nevertheless, some specific external interventions applied for enhancing the autophagy flux may contribute to the renoprotective effects on SI-AKI, which may be partially related to a high level of autophagy in the later stage of SI-AKI may promote renal cell survival.
Among the 41 included studies, a majority of them indicated that the autophagy flux was increased in SI-AKI, while there were eight studies (8/41, 19.5%) suggested that autophagy was inhibited during SI-AKI (Table 2). These experimental animal and cellular models indicated that autophagy was diminished in the kidney of SI-AKI and that proximal tubule cells fail to promote autophagy. Since the inhibition of autophagy was observed in SI-AKI, enhanced autophagy flux may effectively improve renal recovery.
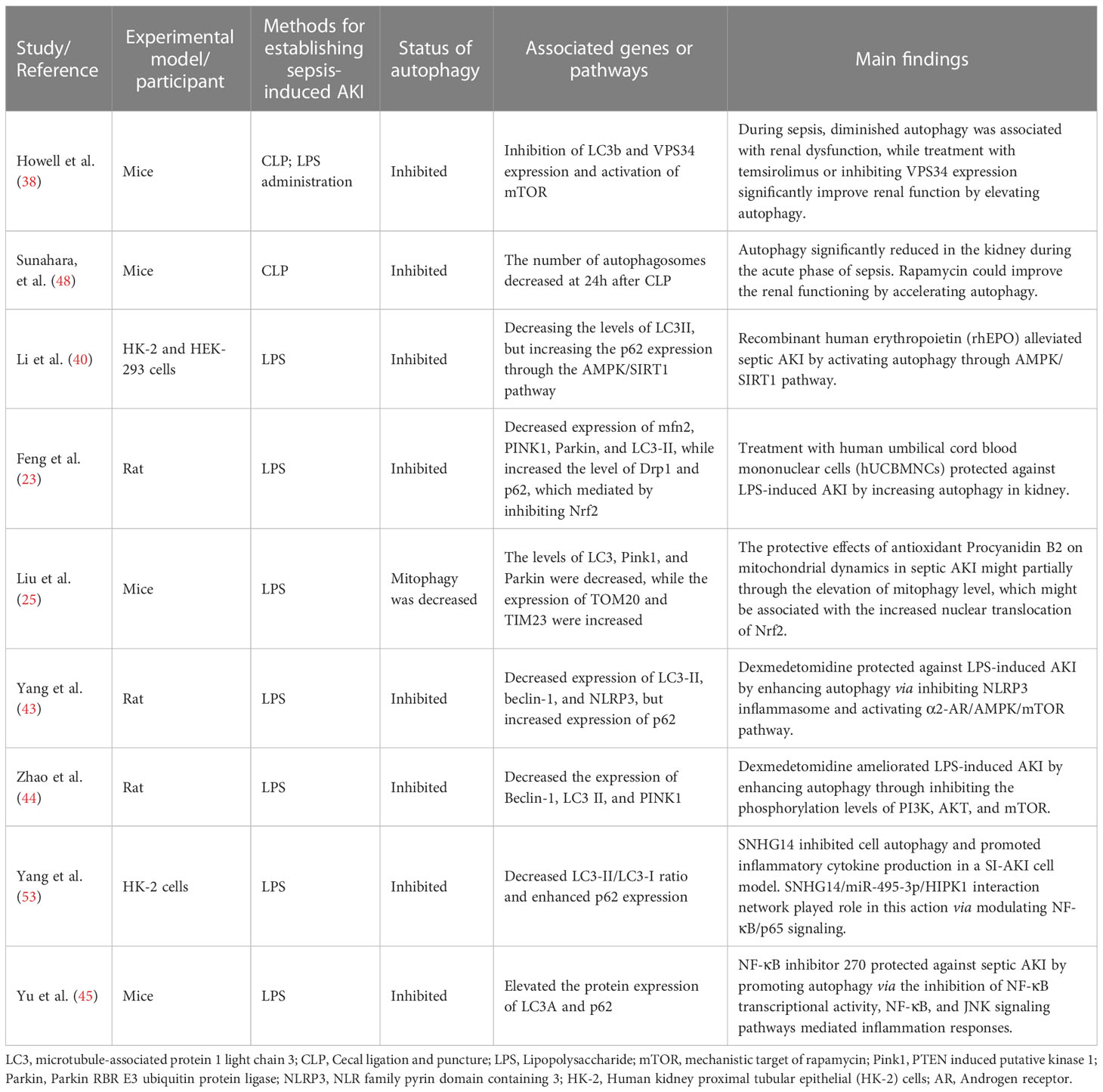
Table 2 The characteristics and the main findings of the 9 relevant studies reporting inhibition of autophagy in SI-AKI.
Howell et al. (38) found that diminished autophagy was associated with renal dysfunction during SI-AKI. Meanwhile, the authors also observed that VPS34 expression was inhibited and the mTOR was activated. Further, they discovered that SI-AKI animals treated with temsirolimus (an mTOR inhibitor) or inhibiting VPS34 expression significantly improve renal function by elevating autophagy. Yang et al. (53) reported that the autophagy level was inhibited during SI-AKI, which was characterized by decreased LC3-II/LC3-I ratio and enhanced p62 expression. In this study, SNHG14 inhibited cell autophagy and promoted inflammatory cytokine production in SI-AKI. SNHG14/miR-495-3p/HIPK1 interaction network plays a key role in the septic process, which might be modulated via the NF-κB/p65 signaling. Rapamycin, an inducer of autophagy, has been found to reduce the extent of SI-AKI. Sunahara et al. (48) reported that the number of autophagosomes decreased at 24h after CLP, indicating that autophagy was restrained during SI-AKI. They next found that rapamycin could improve renal functioning by accelerating autophagy. Li et al. (40) showed that the levels of LC3II were reduced in SI-AKI compared to the controls, but the expression of p62 was increased. The authors revealed that recombinant human erythropoietin (rhEPO) could alleviate SI-AKI by activating autophagy through AMPK/SIRT1 pathway. Feng et al. (50) reported that treatment with human umbilical cord blood mononuclear cells (hUCBMNCs) protected against LPS-induced AKI by increasing autophagy in the kidney. The underlying mechanisms might be associated with the decreased expression of several proteins (e.g. mfn2, PINK1, Parkin, and LC3-II) and the elevated expression of Drp1 and p62, which might be partly mediated by inhibiting Nrf2. A more recent study developed by Yu et al. (45) showed that NF-κB inhibitor 270 could protect against SI-AKI by promoting autophagy by reducing inflammation responses, which might be associated with the inhibition of NF-κB transcriptional activity, NF-κB, and JNK signaling pathways.
Mitophagy also plays a key role in the inhibition of autophagy in SI-AKI. Procyanidin B2 (PB2), one of the common antioxidants, exerts excellent anti-oxidative and anti-inflammatory effects on multiple diseases (70). Decreased LC3, Pink1, and Parkin, while increased TOM20 and TIM23 were identified in Liu et al.’s study (42). The authors further suggested that the protective effects of antioxidant Procyanidin B2 on mitochondrial dynamics in SI-AKI might be partially through the elevation of mitophagy level, which might be associated with the increased nuclear translocation of Nrf2.
Dexmedetomidine (DEX), a selective α2-adrenoreceptor agonist, functions with the effects of the sedative, analgesic, and anti-anxiety (71). Besides, mounting experimental studies demonstrated that DEX has outstanding antioxidant, anti-apoptosis, and anti-inflammatory effects (72). Yang et al. (43) reported that the autophagy level was decreased in SI-AKI (characterized by decreased expression of LC3-II and Beclin-1). The investigators subsequently found that DEX protected against LPS-induced AKI by enhancing autophagy, which might be correlated to the inhibition of NLRP3 inflammasome and the activation of the α2-AR/AMPK/mTOR pathway. Consistent with Yang et al.’ study, Zhao also observed a reduced autophagy flux during SI-AKI (characterized by decreased Beclin-1 and LC3 II expression). They showed that DEX ameliorated LPS-induced AKI by promoting autophagy by inhibiting the phosphorylation levels of PI3K, AKT, and mTOR.
Taken together, the above eight included studies demonstrated that the autophagy level was decreased during SI-AKI, while the promotion of autophagy flux might significantly improve the renal function, which suggested that autophagy played a protective role against SI-AKI. The mechanisms of autophagy inhibition in SI-AKI and the potential molecular mechanisms were illustrated in Figure 1 (right column).
Within the topic of this study, nine included studies demonstrated that autophagy rose firstly and decreased later in SI-AKI (Table 3). Besides, some eligible studies also showed that the autophagy level increased early, then declined, and increased again later. Autophagy is commonly upregulated by environmental stress, such as inflammatory mediators, mitochondrial dysfunction, and ATP depletion, to maintain homeostasis (73). In addition, the autophagy process is a tightly regulated machinery, which can remove damaged proteins and organelles (74). According to the available data from the included studies in this study, the early stage of SI-AKI could be defined as less than 8h after CLP or LPS treatment, while sepsis >8h could be thought as the late stage of SI-AKI. Hsiao et al. (46) observed that the expression of LC3-II increased at 3h and 6 h after CLP but sequentially decreased to the basal level at 9h and 18 h after CLP. In response to septic insult, the level of autophagy transiently elevated in kidney tissue at CLP3h. Due to renal dysfunction and morphological injury, renal autophagy declined at late sepsis, which contributed to proximal tubular dysfunction in an animal model of SI-AKI. In vitro study, siRNA knockdown of Atg7 on NRK-52E cells significantly declined the level of LC3-II. This is the first in vivo study to detect the decline of autophagy that may be conducive to the pathogenesis of polymicrobial sepsis-mediated AKI. Since then, several following preclinical studies also demonstrated a trend of rising first and then falling of autophagy during SI-AKI.
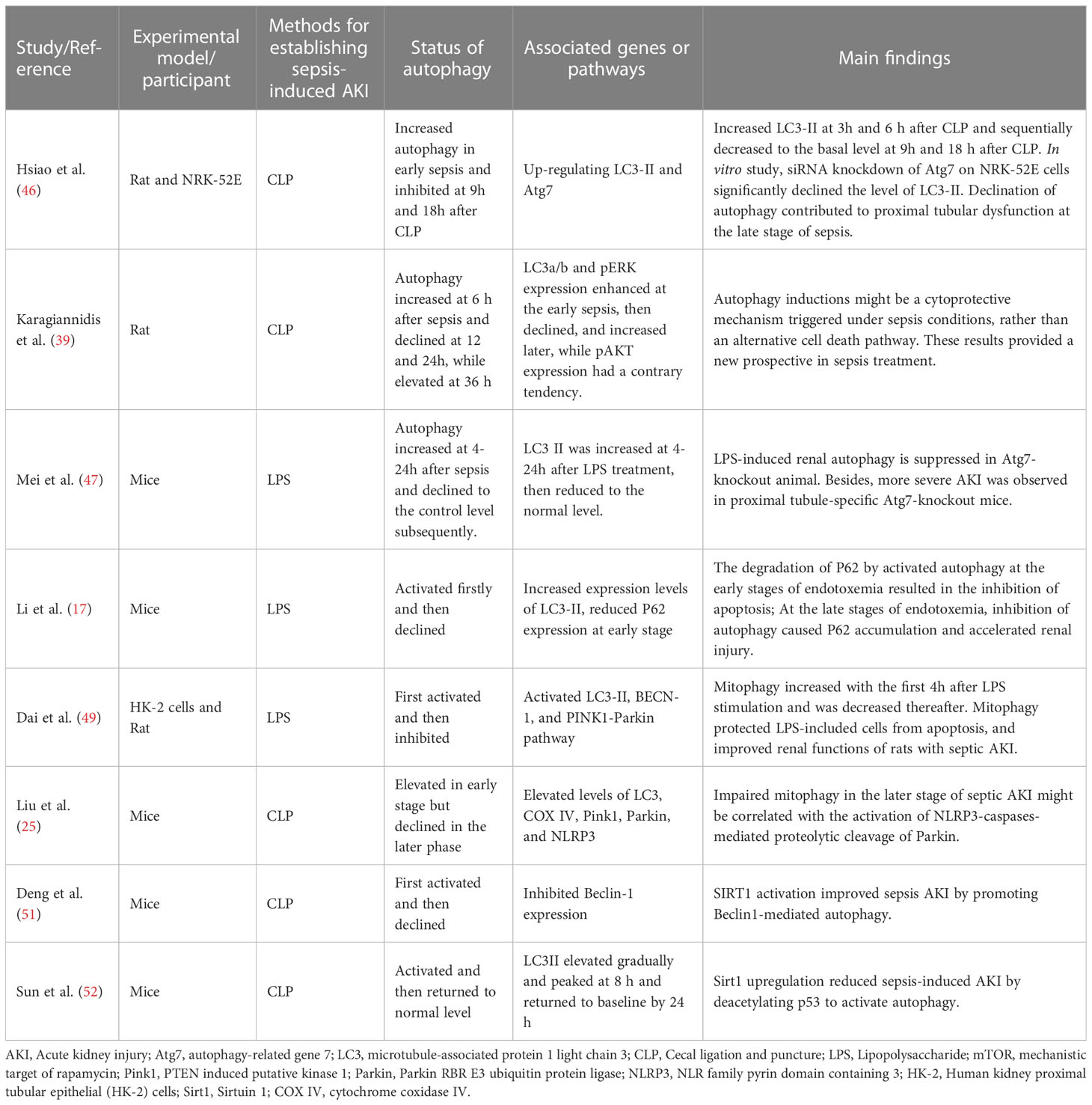
Table 3 The characteristics and the main findings of the relevant 8 studies reporting autophagy is activated firstly and then declined in SI-AKI.
Mei et al. (47) showed that autophagy increased at 4-24h after sepsis and declined to the control level subsequently. LPS-induced renal autophagy was suppressed in Atg7-knockout animals. Additionally, more severe AKI was observed in proximal tubule-specific Atg7-knockout mice. Since the aberrant expression of autophagy-related genes significantly affects the autophagy status under sepsis, this fact may imply that autophagy plays an essential role in SI-AKI. Increased expression levels of LC3-II and reduced P62 expression at an early stage were observed in Li et al.’s study (17). The authors further pointed out that the degradation of P62 by activated autophagy at the early stages of endotoxemia might induce the inhibition of apoptosis. At the late stages of endotoxemia, inhibition of autophagy caused P62 accumulation and accelerated renal injury. Similarly, Dai et al. (49) also found that the autophagy level was first activated and then inhibited during SI-AKI. Mitophagy was increased within the first 4h after LPS stimulation and was decreased thereafter. Mitophagy protected LPS-included cells from apoptosis and improved renal functions of SI-AKI.
Under a similar trend of the autophagy change (increased first and then decreased) in SI-AKI, elevated levels of LC3, COX IV, Pink1, Parkin, and NLRP3 were identified in septic AKI (41). Impaired mitophagy in the later stage of septic AKI might be correlated with the activation of NLRP3-caspases-mediated proteolytic cleavage of Parkin. Sirtuin 1 (Sirt1), an NAD+-dependent protein deacetylase, functions to modify deacetylate histone and nonhistone proteins. Consistently, Deng et al. (51) also demonstrated that kidney autophagy was elevated in the early stage but declined in the later phase. The researchers also found that SIRT1 activation improved SI-AKI by promoting Beclin1-mediated autophagy. A recent study conducted by Sun et al. (52) indicated that the expression of LC3II elevated gradually and peaked at 8 h and returned to baseline by 24 h after SI-AKI, indicating the autophagy level increased first and then declined to normal level subsequently. The authors then showed that Sirt1 upregulation reduced SI-AKI by deacetylating p53 to activate autophagy. Differ from that of the above included studies reporting the autophagy level increased at the early stage and decreased later during SI-AKI, Karagiannidis et al. implied that the autophagy flux increased at 6 h after sepsis and declined at 12 and 24h, while elevated at 36 h. They concluded that autophagy inductions might be a cytoprotective mechanism triggered under sepsis conditions, rather than an alternative cell death pathway.
Based on the above evidence derived from in vitro and in vivo models of SI-AKI, it showed the trend of rising firstly and then falling for the autophagy flux. This may be a molecular mechanism of the renoprotective effect during SI-AKI (Figure 1, middle column). The autophagy feedback may play a protective role in endotoxic AKI, serving as a potential therapeutic target for protecting against the damage of renal tubular epithelial cells.
Based on the different level of autophagy among the 41 included studies and outcomes of the changed autophagic flux, we propose the following hypotheses. First, the autophagic flux during the physiological processes of SI-AKI might elevate firstly (phrase I), next inhibit or decline (phrase II), then elevate (phrase III), and return to the normal level finally (phrase IV). In the 24 studies reporting the elevation of autophagy, it could be explained by the check point time of autophagy in these studies was phrase I or phrase III. The eight studies reporting the elevation first then inhibition could be explained by check point time of autophagy was phrase I and phrase II of SI-AKI. Of note, in the nine included studies reported elevated first and then declined, all of them concluded that the autophagy status was inhibited during SI-AKI, while the renal protective effects exerting by specific interventions (reported in eight studies) might be contributed to the elevation of autophagy. Therefore, the check point time of autophagy in these included studies might be the phrase II. The above interventions exerted the renoprotective effects on SI-AKI might be associated with the acceleration of autophagy from phrase II to phrase III.
As shown in the Table 4, the majority of the included studies (14/15, 93%) showed that the improvement of SI-AKI exhibited by specific interventions might be attributed to the elevation of autophagy, regardless of whether the autophagy activated or inhibited during SI-AKI. The most likely explanation is the elevation of autophagy may be one of the protective mechanisms for SI-AKI. Another possible explanation is that the activation and inhibition of autophagy among different studies might be associated with the different time points of examinations after sepsis in each independent study. As illustrated in Table 3, eight included studies indicated the autophagy was activated firstly (early stage of SI-AKI) and then declined (late stage of SI-AKI) during SI-AKI. Therefore, those studies listed in Table 1 suggested that autophagy was activated, which might be due to the time point for checking the autophagy level being the early stage. On the other hand, the inhibition of autophagy in the studies listed in Table 2 might be correlated to the checking time point was the late stage of SI-AKI.
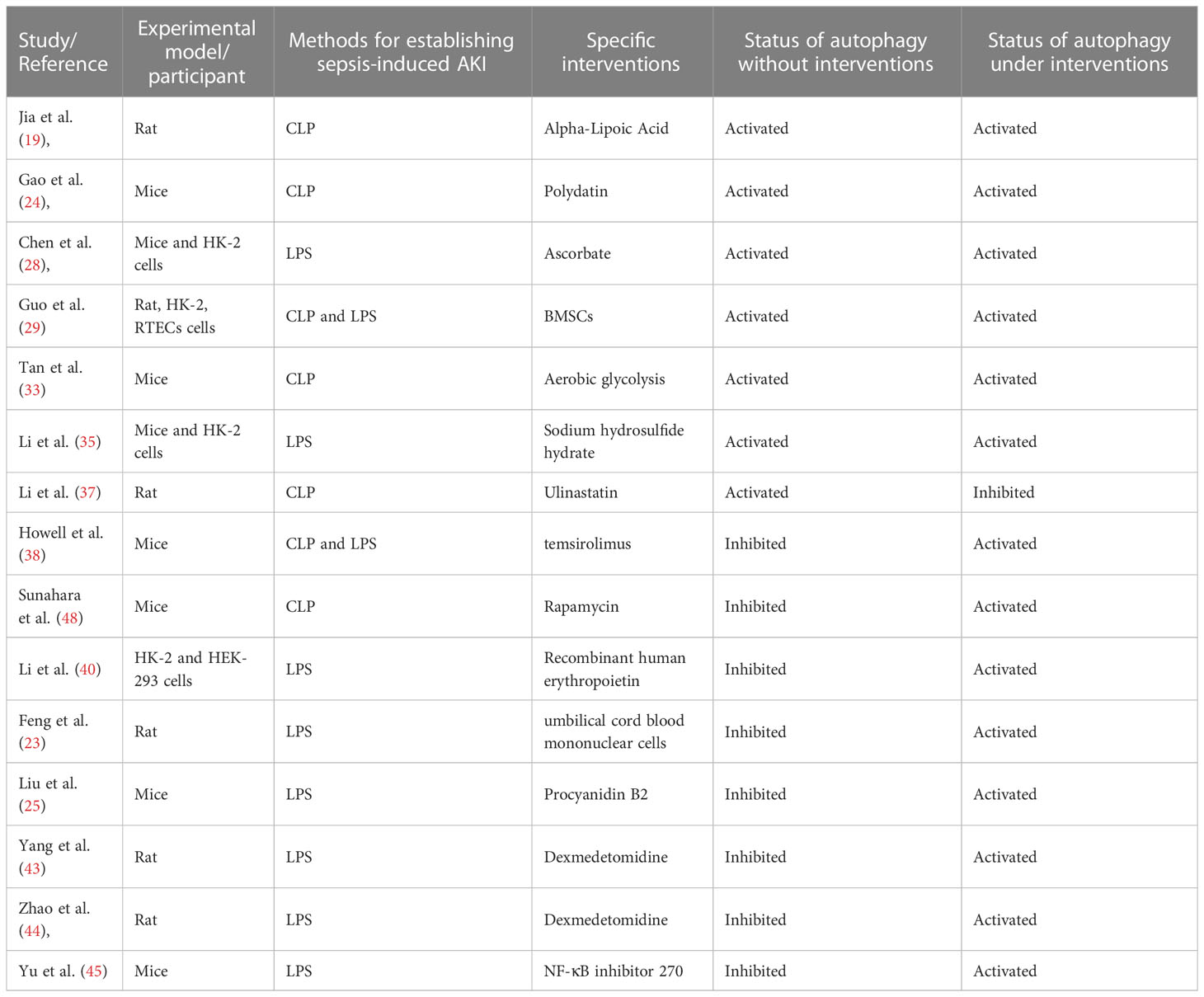
Table 4 The status of autophagy under different interventions in the experimental models of SI-AKI.
In Table 1, there were seven studies reported some interventions for treating SI-AKI. All of the seven included studies indicated CLP or LPS treatment (methods for SI-AKI model establishment) might induce the elevation of autophagy level. Six of them reported that the specific interventions (i.e., alpha-lipoic acid, Polydatin, Ascorbate, BMSCs, aerobic glycolysis, and sodium hydrosulfide hydrate) exhibited the renal protection by increasing autophagy. In Table 2, eight studies reported some interventions for treating SI-AKI. Inconsistent with the above findings, all the eight studies concluded that the autophagy status was inhibited during SI-AKI, while the renal protective effects exerting by specific interventions (i.e., temsirolimus, rapamycin, recombinant human erythropoietin, umbilical cord blood mononuclear cells, Procyanidin B2, dexmedetomidine, and NF-κB inhibitor 270) might be contributed by the elevation of autophagy.
Based on the above evidence, in the aspect of the clinical translational perspective, the strategies for therapeutic intervention should focus on how to elevate the autophagic flux during SI-AKI.
To the best of our knowledge, this is the first systematic and comprehensive review to summarize all the current evidence of the crucial roles of autophagy in SI-AKI. We can notice that a majority of the included studies (about 60%) showed an elevation of the autophagy level during SI-AKI, while 22% and 19.5% of the included studies reported an inhibition and an elevation at the early stage but a declination of renal autophagy in SI-AKI, respectively. As can be seen, the level of autophagy flux in the process of septic AKI is still controversial among different studies. One of the explanations for this inconsistency of the autophagy level in SI-AKI may be caused by the various time points monitored in each study. In addition, different intracellular signaling molecules and pathways involved in the process of SI-AKI may also affect the expression of the autophagy-related genes, resulting in an increase or decrease of autophagy flux. Autophagy is considered to be a “double-edged sword” for both cell survival and cell death in multiple diseases, including SI-AKI. However, this study highlights that one of the main probable mechanisms underlying the multiple treatment modalities (e.g. small molecule inhibitors, temsirolimus, rapamycin, ascorbate, rhEPO, stem cells, Procyanidin B2, and DEX) for improving the renal function may be attributed to the elevation of the autophagy level in SI-AKI. The exact roles of autophagy in SI-AKI have not been well understood, which deserves further investigation. Targeting the autophagy-associated proteins and pathways may provide a new prospective in the treatment of critically ill patients with SI-AKI, but more preclinical studies are still warranted to validate this hypothesis.
SZ, JL, and MW contributed to conceiving and designing the study. XL performed the systematic searching. MLS extracted the data. SZ and JL wrote the manuscript. XL and MW supervised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the grants from the Zhejiang Medical and Health Science and Technology Program (No. 2022RC297); the Natural Science Foundation of Zhejiang Province (No. LQ22H040009).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1180866/full#supplementary-material
1. Liu V, Escobar GJ, Greene JD, Soule J, Whippy A, Angus DC, et al. Hospital deaths in patients with sepsis from 2 independent cohorts. JAMA (2014) 312:90–2. doi: 10.1001/jama.2014.5804
2. Gando S, Shiraishi A, Yamakawa K, Ogura H, Saitoh D, Fujishima S, et al. Role of disseminated intravascular coagulation in severe sepsis. Thromb Res (2019) 178:182–8. doi: 10.1016/j.thromres.2019.04.025
3. He FF, Wang YM, Chen YY, Huang W, Li ZQ, Zhang C. Sepsis-induced AKI: from pathogenesis to therapeutic approaches. Front Pharmacol (2022) 13:981578. doi: 10.3389/fphar.2022.981578
4. Peerapornratana S, Manrique-Caballero CL, Gomez H, Kellum JA. Acute kidney injury from sepsis: current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int (2019) 96:1083–99. doi: 10.1016/j.kint.2019.05.026
5. Huang G, Bao J, Shao X, Zhou W, Wu B, Ni Z, et al. Inhibiting pannexin-1 alleviates sepsis-induced acute kidney injury via decreasing NLRP3 inflammasome activation and cell apoptosis. Life Sci (2020) 254:117791. doi: 10.1016/j.lfs.2020.117791
6. Lorencio CC, Yebenes JC, Vela E, Cleries M, Sirvent JM, Fuster-Bertolin C, et al. Trends in mortality in septic patients according to the different organ failure during 15 years. Crit Care (2022) 26:302. doi: 10.1186/s13054-022-04176-w
7. Li R, Zhao X, Zhang S, Dong W, Zhang L, Chen Y, et al. RIP3 impedes transcription factor EB to suppress autophagic degradation in septic acute kidney injury. Cell Death Dis (2021) 12:593. doi: 10.1038/s41419-021-03865-8
8. Adriaenssens E, Ferrari L, Martens S. Orchestration of selective autophagy by cargo receptors. Curr Biol (2022) 32:R1357–71. doi: 10.1016/j.cub.2022.11.002
9. Vargas J, Hamasaki M, Kawabata T, Youle RJ, Yoshimori T. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol (2023) 24:167–185. doi: 10.1038/s41580-022-00542-2
10. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. CELL (2019) 176:11–42. doi: 10.1016/j.cell.2018.09.048
11. Bhatia D, Choi ME. Autophagy in kidney disease: advances and therapeutic potential. Prog Mol Biol Transl Sci (2020) 172:107–33. doi: 10.1016/bs.pmbts.2020.01.008
12. Kawakami T, Gomez IG, Ren S, Hudkins K, Roach A, Alpers CE, et al. Deficient autophagy results in mitochondrial dysfunction and FSGS. J Am Soc NEPHROL. (2015) 26:1040–52. doi: 10.1681/ASN.2013111202
13. Choi ME. Autophagy in kidney disease. Annu Rev Physiol (2020) 82:297–322. doi: 10.1146/annurev-physiol-021119-034658
14. Zhang X, Howell GM, Guo L, Collage RD, Loughran PA, Zuckerbraun BS, et al. CaMKIV-dependent preservation of mTOR expression is required for autophagy during lipopolysaccharide-induced inflammation and acute kidney injury. J Immunol (2014) 193:2405–15. doi: 10.4049/jimmunol.1302798
15. Chen X, Tong H, Chen Y, Chen C, Ye J, Mo Q, et al. Klotho ameliorates sepsis-induced acute kidney injury but is irrelevant to autophagy. Onco Targets Ther (2018) 11:867–81. doi: 10.2147/OTT.S156891
16. Fu D, Zhou K, Liu J, Zheng P, Li P, Cheng W, et al. Long non-coding RNA PlncRNA-1 regulates cell proliferation, apoptosis, and autophagy in septic acute kidney injury by regulating BCL2. Int J Clin Exp Pathol (2018) 11:314–23.
17. Li T, Zhao J, Miao S, Xu Y, Xiao X, Liu Y. Dynamic expression and roles of sequestome−1/p62 in LPS−induced acute kidney injury in mice. Mol Med Rep (2018) 17:7618–26. doi: 10.3892/mmr.2018.8809
18. Zhao W, Zhang L, Chen R, Lu H, Sui M, Zhu Y, et al. SIRT3 protects against acute kidney injury via AMPK/mTOR-regulated autophagy. Front Physiol (2018) 9:1526. doi: 10.3389/fphys.2018.01526
19. Jia J, Gong X, Zhao Y, Yang Z, Ji K, Luan T, et al. Autophagy enhancing contributes to the organ protective effect of alpha-lipoic acid in septic rats. Front Immunol (2019) 10:1491. doi: 10.3389/fimmu.2019.01491
20. Wu Y, Wang L, Meng L, Cao GK, Zhao YL, Zhang Y. Biological effects of autophagy in mice with sepsis-induced acute kidney injury. Exp Ther Med (2019) 17:316–22. doi: 10.3892/etm.2018.6899
21. Zhang Y, Wang L, Meng L, Cao G, Wu Y. Sirtuin 6 overexpression relieves sepsis-induced acute kidney injury by promoting autophagy. Cell Cycle (2019) 18:425–36. doi: 10.1080/15384101.2019.1568746
22. Zheng C, Zhou Y, Huang Y, Chen B, Wu M, Xie Y, et al. Effect of ATM on inflammatory response and autophagy in renal tubular epithelial cells in LPS-induced septic AKI. Exp Ther Med (2019) 18:4707–17. doi: 10.3892/etm.2019.8115
23. Feng Y, Liu J, Wu R, Yang P, Ye Z, Song F. NEAT1 aggravates sepsis-induced acute kidney injury by sponging miR-22-3p. Open Med (Wars). (2020) 15:333–42. doi: 10.1515/med-2020-0401
24. Gao Y, Dai X, Li Y, Li G, Lin X, Ai C, et al. Role of parkin-mediated mitophagy in the protective effect of polydatin in sepsis-induced acute kidney injury. J Transl Med (2020) 18:114. doi: 10.1186/s12967-020-02283-2
25. Liu Y, Xiao J, Sun J, Chen W, Wang S, Fu R, et al. ATG7 promotes autophagy in sepsis−induced acute kidney injury and is inhibited by miR−526b. Mol Med Rep (2020) 21:2193–201. doi: 10.3892/mmr.2020.11001
26. Miao S, Lv C, Liu Y, Zhao J, Li T, Wang C, et al. Pharmacologic blockade of 15-PGDH protects against acute renal injury induced by LPS in mice. Front Physiol (2020) 11:138. doi: 10.3389/fphys.2020.00138
27. Alexander MP, Mangalaparthi KK, Madugundu AK, Moyer AM, Adam BA, Mengel M, et al. Acute kidney injury in severe COVID-19 has similarities to sepsis-associated kidney injury: a multi-omics study. MAYO Clin Proc (2021) 96:2561–75. doi: 10.1016/j.mayocp.2021.07.001
28. Chen ZD, Hu BC, Shao XP, Hong J, Zheng Y, Zhang R, et al. Ascorbate uptake enables tubular mitophagy to prevent septic AKI by PINK1-PARK2 axis. Biochem Biophys Res Commun (2021) 554:158–65. doi: 10.1016/j.bbrc.2021.03.103
29. Guo J, Wang R, Liu D. Bone marrow-derived mesenchymal stem cells ameliorate sepsis-induced acute kidney injury by promoting mitophagy of renal tubular epithelial cells via the SIRT1/Parkin axis. Front Endocrinol (Lausanne). (2021) 12:639165. doi: 10.3389/fendo.2021.639165
30. Han D, Fang R, Shi R, Jin Y, Wang Q. LncRNA NKILA knockdown promotes cell viability and represses cell apoptosis, autophagy and inflammation in lipopolysaccharide-induced sepsis model by regulating miR-140-5p/CLDN2 axis. Biochem Biophys Res Commun (2021) 559:8–14. doi: 10.1016/j.bbrc.2021.04.074
31. Pan P, Liu X, Wu L, Li X, Wang K, Wang X, et al. TREM-1 promoted apoptosis and inhibited autophagy in LPS-treated HK-2 cells through the NF-kappaB pathway. Int J Med Sci (2021) 18:8–17. doi: 10.7150/ijms.50893
32. Sang Z, Dong S, Zhang P, Wei Y. miR−214 ameliorates sepsis−induced acute kidney injury via PTEN/AKT/mTOR−regulated autophagy. Mol Med Rep (2021) 24:683. doi: 10.3892/mmr.2021.12322
33. Tan C, Gu J, Li T, Chen H, Liu K, Liu M, et al. Inhibition of aerobic glycolysis alleviates sepsis−induced acute kidney injury by promoting lactate/Sirtuin 3/AMPK−regulated autophagy. Int J Mol Med (2021) 47:19. doi: 10.3892/ijmm.2021.4852
34. Wang Y, Zhu J, Liu Z, Shu S, Fu Y, Liu Y, et al. The PINK1/PARK2/optineurin pathway of mitophagy is activated for protection in septic acute kidney injury. Redox Biol (2021) 38:101767. doi: 10.1016/j.redox.2020.101767
35. Li T, Zhao J, Miao S, Chen Y, Xu Y, Liu Y. Protective effect of H(2)S on LPS−induced AKI by promoting autophagy. Mol Med Rep (2022) 25:96. doi: 10.3892/mmr.2022.12612
36. Li Y, Feng G. TLR4 inhibitor alleviates sepsis-induced organ failure by inhibiting platelet mtROS production, autophagy, and GPIIb/IIIa expression. J BIOENERG BIOMEMBR. (2022) 54:155–62. doi: 10.1007/s10863-022-09940-9
37. Li T, Ji X, Liu J, Guo X, Pang R, Zhuang H, et al. Ulinastatin improves renal microcirculation by protecting endothelial cells and inhibiting autophagy in a septic rat model. Kidney Blood Press Res (2022) 47:256–69. doi: 10.1159/000521648
38. Howell GM, Gomez H, Collage RD, Loughran P, Zhang X, Escobar DA, et al. Augmenting autophagy to treat acute kidney injury during endotoxemia in mice. PloS One (2013) 8:e69520. doi: 10.1371/journal.pone.0069520
39. Karagiannidis I, Kataki A, Glustianou G, Memos N, Papalois A, Alexakis N, et al. EXTENDED CYTOPROTECTIVE EFFECT OF AUTOPHAGY IN THE LATE STAGES OF SEPSIS AND FLUCTUATIONS IN SIGNAL TRANSDUCTION PATHWAYS IN a RAT EXPERIMENTAL MODEL OF KIDNEY INJURY. SHOCK (2016) 45:139–47. doi: 10.1097/SHK.0000000000000505
40. Li K, Liu TX, Li JF, Ma YR, Liu ML, Wang YQ, et al. rhEPO inhibited cell apoptosis to alleviate acute kidney injury in sepsis by AMPK/SIRT1 activated autophagy. Biochem Biophys Res Commun (2019) 517:557–65. doi: 10.1016/j.bbrc.2019.07.027
41. Liu JX, Yang C, Zhang WH, Su HY, Liu ZJ, Pan Q, et al. Disturbance of mitochondrial dynamics and mitophagy in sepsis-induced acute kidney injury. Life Sci (2019) 235:116828. doi: 10.1016/j.lfs.2019.116828
42. Liu JX, Yang C, Liu ZJ, Su HY, Zhang WH, Pan Q, et al. Protection of procyanidin B2 on mitochondrial dynamics in sepsis associated acute kidney injury via promoting Nrf2 nuclear translocation. Aging (Albany NY). (2020) 12:15638–55. doi: 10.18632/aging.103726
43. Yang T, Feng X, Zhao Y, Zhang H, Cui H, Wei M, et al. Dexmedetomidine enhances autophagy via alpha2-AR/AMPK/mTOR pathway to inhibit the activation of NLRP3 inflammasome and subsequently alleviates lipopolysaccharide-induced acute kidney injury. Front Pharmacol (2020) 11:790. doi: 10.3389/fphar.2020.00790
44. Zhao Y, Feng X, Li B, Sha J, Wang C, Yang T, et al. Dexmedetomidine protects against lipopolysaccharide-induced acute kidney injury by enhancing autophagy through inhibition of the PI3K/AKT/mTOR pathway. Front Pharmacol (2020) 11:128. doi: 10.3389/fphar.2020.00128
45. Yu YY, Li XQ, Hu WP, Cu SC, Dai JJ, Gao YN, et al. Self-developed NF-kappaB inhibitor 270 protects against LPS-induced acute kidney injury and lung injury through improving inflammation. BioMed PHARMACOTHER. (2022) 147:112615. doi: 10.1016/j.biopha.2022.112615
46. Hsiao HW, Tsai KL, Wang LF, Chen YH, Chiang PC, Chuang SM, et al. The decline of autophagy contributes to proximal tubular dysfunction during sepsis. SHOCK (2012) 37:289–96. doi: 10.1097/SHK.0b013e318240b52a
47. Mei S, Livingston M, Hao J, Li L, Mei C, Dong Z. Autophagy is activated to protect against endotoxic acute kidney injury. Sci Rep (2016) 6:22171. doi: 10.1038/srep22171
48. Sunahara S, Watanabe E, Hatano M, Swanson PE, Oami T, Fujimura L, et al. Influence of autophagy on acute kidney injury in a murine cecal ligation and puncture sepsis model. Sci Rep (2018) 8:1050. doi: 10.1038/s41598-018-19350-w
49. Dai XG, Xu W, Li T, Lu JY, Yang Y, Li Q, et al. Involvement of phosphatase and tensin homolog-induced putative kinase 1-parkin-mediated mitophagy in septic acute kidney injury. Chin Med J (Engl). (2019) 132:2340–7. doi: 10.1097/CM9.0000000000000448
50. Feng LX, Zhao F, Liu Q, Peng JC, Duan XJ, Yan P, et al. Role of Nrf2 in lipopolysaccharide-induced acute kidney injury: protection by human umbilical cord blood mononuclear cells. Oxid Med Cell Longev (2020) 2020:6123459. doi: 10.1155/2020/6123459
51. Deng Z, Sun M, Wu J, Fang H, Cai S, An S, et al. SIRT1 attenuates sepsis-induced acute kidney injury via Beclin1 deacetylation-mediated autophagy activation. Cell Death Dis (2021) 12:217. doi: 10.1038/s41419-021-03508-y
52. Sun M, Li J, Mao L, Wu J, Deng Z, He M, et al. p53 deacetylation alleviates sepsis-induced acute kidney injury by promoting autophagy. Front Immunol (2021) 12:685523. doi: 10.3389/fimmu.2021.685523
53. Yang N, Wang H, Zhang L, Lv J, Niu Z, Liu J, et al. Long non-coding RNA SNHG14 aggravates LPS-induced acute kidney injury through regulating miR-495-3p/HIPK1. Acta Biochim Biophys Sin (Shanghai). (2021) 53:719–28. doi: 10.1093/abbs/gmab034
54. Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. SCIENCE (2004) 306:990–5. doi: 10.1126/science.1099993
55. Vellai T, Toth ML, Kovacs AL. Janus-faced autophagy: a dual role of cellular self-eating in neurodegeneration? AUTOPHAGY (2007) 3:461–3. doi: 10.4161/auto.4282
56. Chang KC, Liu PF, Chang CH, Lin YC, Chen YJ, Shu CW. The interplay of autophagy and oxidative stress in the pathogenesis and therapy of retinal degenerative diseases. Cell Biosci (2022) 12:1. doi: 10.1186/s13578-021-00736-9
57. Kaushal GP, Shah SV. Autophagy in acute kidney injury. Kidney Int (2016) 89:779–91. doi: 10.1016/j.kint.2015.11.021
58. Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc NEPHROL. (2011) 22:902–13. doi: 10.1681/ASN.2010070705
59. Wang Z, Huang Y, Zhang J. Molecularly targeting the PI3K-Akt-mTOR pathway can sensitize cancer cells to radiotherapy and chemotherapy. Cell Mol Biol Lett (2014) 19:233–42. doi: 10.2478/s11658-014-0191-7
60. Xu G, Mo L, Wu C, Shen X, Dong H, Yu L, et al. The miR-15a-5p-XIST-CUL3 regulatory axis is important for sepsis-induced acute kidney injury. Ren Fail (2019) 41:955–66. doi: 10.1080/0886022X.2019.1669460
61. Li X, Li J, Zhang Y, Zhou Y. Di-n-butyl phthalate induced hypospadias relates to autophagy in genital tubercle via the PI3K/Akt/mTOR pathway. J Occup Health (2017) 59:8–16. doi: 10.1539/joh.16-0089-OA
62. Dunlop EA, Tee AR. mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol (2014) 36:121–9. doi: 10.1016/j.semcdb.2014.08.006
63. Cui H, Ren G, Hu X, Xu B, Li Y, Niu Z, et al. Suppression of lncRNA GAS6-AS2 alleviates sepsis-related acute kidney injury through regulating the miR-136-5p/OXSR1 axis in vitro and in vivo. Ren Fail (2022) 44:1070–82. doi: 10.1080/0886022X.2022.2092001
64. Wang H, Mou H, Xu X, Liu C, Zhou G, Gao B. LncRNA KCNQ1OT1 (potassium voltage-gated channel subfamily q member 1 opposite strand/antisense transcript 1) aggravates acute kidney injury by activating p38/NF-kappaB pathway via miR-212-3p/MAPK1 (mitogen-activated protein kinase 1) axis in sepsis. BIOENGINEERED (2021) 12:11353–68. doi: 10.1080/21655979.2021.2005987
65. Chen T, Xue H, Lin R, Huang Z. MiR-34c and PlncRNA1 mediated the function of intestinal epithelial barrier by regulating tight junction proteins in inflammatory bowel disease. Biochem Biophys Res Commun (2017) 486:6–13. doi: 10.1016/j.bbrc.2017.01.115
66. Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol (2018) 20:1013–22. doi: 10.1038/s41556-018-0176-2
67. Spoelstra-de MA, Elbers P, Oudemans-van SH. Making sense of early high-dose intravenous vitamin c in ischemia/reperfusion injury. Crit Care (2018) 22:70. doi: 10.1186/s13054-018-1996-y
68. Ma Y, Qi M, An Y, Zhang L, Yang R, Doro DH, et al. Autophagy controls mesenchymal stem cell properties and senescence during bone aging. Aging Cell (2018) 17:e12709. doi: 10.1111/acel.12709
69. Ng F, Tang BL. Sirtuins' modulation of autophagy. J Cell Physiol (2013) 228:2262–70. doi: 10.1002/jcp.24399
70. Martinez-Micaelo N, Gonzalez-Abuin N, Pinent M, Ardevol A, Blay M. Procyanidin B2 inhibits inflammasome-mediated IL-1beta production in lipopolysaccharide-stimulated macrophages. Mol Nutr Food Res (2015) 59:262–9. doi: 10.1002/mnfr.201400370
71. Poon WH, Ling RR, Yang IX, Luo H, Kofidis T, MacLaren G, et al. Dexmedetomidine for adult cardiac surgery: a systematic review, meta-analysis and trial sequential analysis. ANAESTHESIA (2023) 78:371–380. doi: 10.1111/anae.15947
72. Shi J, Yu T, Song K, Du S, He S, Hu X, et al. Dexmedetomidine ameliorates endotoxin-induced acute lung injury in vivo and in vitro by preserving mitochondrial dynamic equilibrium through the HIF-1a/HO-1 signaling pathway. Redox Biol (2021) 41:101954. doi: 10.1016/j.redox.2021.101954
73. Tang J, Bassham DC. Autophagy during drought: function, regulation, and potential application. Plant J (2022) 109:390–401. doi: 10.1111/tpj.15481
Keywords: sepsis, acute kidney injury, autophagy, protection, mechanism
Citation: Zhao S, Liao J, Shen M, Li X and Wu M (2023) Epigenetic dysregulation of autophagy in sepsis-induced acute kidney injury: the underlying mechanisms for renoprotection. Front. Immunol. 14:1180866. doi: 10.3389/fimmu.2023.1180866
Received: 06 March 2023; Accepted: 19 April 2023;
Published: 05 May 2023.
Edited by:
Pietro Ghezzi, University of Urbino Carlo Bo, ItalyReviewed by:
Qiaobing Huang, Southern Medical University, ChinaCopyright © 2023 Zhao, Liao, Shen, Li and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mei Wu, bWVpd3VkcjIwMTlAMTYzLmNvbQ==
†These authors have contributed equally to this work
‡ORCID: Mei Wu, orcid.org/0000-0002-6881-0670
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.