 Tsukasa Aoki1,2
Tsukasa Aoki1,2 Fumitaka Hiura1Aonan Li1
Fumitaka Hiura1Aonan Li1 Nan Yang1Nana Takakura-Hino1Satoru Mukai3
Nan Yang1Nana Takakura-Hino1Satoru Mukai3 Miho Matsuda1
Miho Matsuda1 Fusanori Nishimura2
Fusanori Nishimura2 Eijiro Jimi1,4*
Eijiro Jimi1,4*- 1Laboratory of Molecular and Cellular Biochemistry, Division of Oral Biological Sciences, Faculty of Dental Science, Kyushu University, Fukuoka, Japan
- 2Department of Periodontology, Division of Oral Rehabilitation, Faculty of Dental Science, Kyushu University, Fukuoka, Japan
- 3Department of Health and Nutrition Care, Faculty of Allied Health Sciences, University of East Asia, Shimonoseki, Japan
- 4Oral Health/Brain Health/Total Health Research Center, Faculty of Dental Science, Kyushu University, Fukuoka, Japan
Periodontal disease is an infectious disease that affects many people worldwide. Disease progression destroys the alveolar bone and causes tooth loss. We have previously shown that alymphoplasia (aly/aly) mice harboring a loss-of-function mutation in the map3k14 gene, which is involved in p100 to p52 processing of the alternative NF-κB pathway, exhibited mild osteopetrosis due to decreased number of osteoclasts, suggesting the alternative NF-κB pathway as a potential drug target for the amelioration of bone disease. In the present study, wild-type (WT) and aly/aly mice were subjected to silk ligation to establish a periodontitis model. Alveolar bone resorption was suppressed in aly/aly mice by decreased numbers of osteoclasts in the alveolar bone in comparison to WT mice. Furthermore, the expression of receptor activator of NF-κB ligand (RANKL) and TNFα (cytokines involved in osteoclast induction in periligative gingival tissue) was decreased. When primary osteoblasts (POBs) and bone marrow cells (BMCs) derived from WT and aly/aly mice were prepared and co-cultured, osteoclasts were induced from WT-derived BMCs, regardless of the origin of the POBs, but hardly formed from aly/aly mouse-derived BMCs. Furthermore, the local administration of an NIK inhibitor, Cpd33, inhibited osteoclast formation and thereby inhibited alveolar bone resorption in the periodontitis model. Therefore, the NIK-mediated NF-κB alternative pathway can be a therapeutic target for periodontal disease.
Introduction
Periodontal disease is an inflammatory disease caused by periodontal pathogens (1–3). As inflammation progresses, the alveolar bone is destroyed and the disease becomes chronic. Periodontal tissue possesses anatomical features and immune mechanisms that protect against invasion by periodontal pathogens (1–3). When bacterial plaque adheres to the gingival sulcus for a long time, lymphocytes infiltrate and inflammation occurs in the gingiva. The invasion of periodontal tissue by periodontal pathogens leads to infiltration by lymphocytes, neutrophils, macrophages, and plasma cells, the disappearance of collagen fibers, and the subsequent expansion of inflammation. Cellular components of periodontal pathogens, proteases, bacterial toxins, and proteases, cytokines, and inflammatory chemical mediators released by host cells, expand inflammatory infiltration in the apical and lateral directions, leading to the formation of periodontal pockets and the destruction of alveolar bone (1–3). Excessive alveolar bone resorption leads to tooth loss and significantly impacts quality of life (QOL) (1–3).
Osteoclasts are essential for alveolar bone resorption in periodontitis (4). Osteoclast precursor cells derived from hematopoietic stem cells differentiate into osteoclasts (4, 5). In periodontitis, lipopolysaccharide (LPS) produced by periodontal pathogens, and inflammatory cytokines produced by host immune cells act on the periodontal ligament cells and osteoblasts to induce the receptor activator of nuclear factor κB (NF-κB) ligand (RANKL) to differentiate into osteoclasts (4, 6). When RANKL binds to RANK expressed on the plasma membrane of osteoclast progenitors, NF-κB and mitogen-activated protein kinase (MAPK) are activated within the cells, and NFATc1, a master regulator of osteoclastogenesis, resulting in osteoclast differentiation (7). In addition, since NF-κB regulates the induction of inflammatory cytokine genes, NF-κB are therapeutic targets for diseases associated with inflammatory bone destruction, including rheumatoid arthritis (7).
There are two pathways for NF-κB activation. One is a so-called “classical pathway” that is activated shortly after stimulation with inflammatory cytokines (e.g., tumor necrosis factor-α [TNF-α] and interleukin-1β [IL-1β]) accompanied by IκBα degradation (7, 8). The other is a so-called “alternative pathway” that is activated several hours after stimulation with cytokines involved in the development of lymph nodes (e.g., CD40L and lymphotoxin-β [LTβ]) mediated by the NF-κB inducing kinase (NIK)-IκB kinase α (IKKα) pathway (7–9). RANKL activates these two pathways, and mice with a double knockout of the two important subunits of each pathway (NF-κB1 and NF-κB2) exhibit osteopetrosis a total lack of osteoclasts (7, 10, 11). These indicate that both NF-κB activation pathways play an important role in RNAKL-induced osteoclast bone resorption.
We previously showed that lymphoid hypoplasia (aly/aly) mice with a natural loss-of-function mutation in the map3k14 gene, which encodes a kinase essential for p100 to p52 processing in the NF-κB alternative pathway, exhibit osteopetrosis in association with decreased osteoclast numbers (12, 13). Furthermore, stimulation of osteoclast precursors from aly/aly mice with RANKL suppressed the expression of NFATc1 due to the inhibition of p100 to p52 processing. Compound 33 (Cpd33), which was developed as a selective inhibitor of NIK, inhibited RANKL-induced osteoclastogenesis in a dose-dependent manner without affecting cell viability (14). Moreover, Cdp33 treatment inhibited osteoclast formation, thereby suppressing bone loss in ovariectomized mice (14). These results suggest that the alternative NF-κB pathway is a suitable therapeutic target for diseases with excessive bone loss. In this study, we used aly/aly mice and Cpd33 to investigate the possibility that the alternative pathway of NF-κB could be a therapeutic target for periodontal disease.
Materials and methods
Reagents
Glutathione S-transferase (GST)-RANKL and recombinant human macrophage-stimulating factor (M-CSF) were purchased from Oriental Yeast Company, Ltd. (Shiga, Japan) and FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan), respectively. LPS (Escherichia coli 055: B5) was purchased from Sigma-Aldrich (St Louis, MO, USA). Cpd33 was kindly provided by Genentech Inc. (South San Francisco, CA, USA).
Mice
Mouse handling and all procedures were approved by the Animal Care Committee of Kyushu University, according to the guidelines of the Japanese Council on Animal Care (Approval Number: A21-284-3, A22-233-2). All experiments were performed in accordance with ARRIVE guidelines. Eight-week-old male C57BL/6J and aly/aly mice were purchased from CLEA Japan (Tokyo, Japan).
Mouse periodontal disease model
To induce periodontal bone loss in 8-week-old C57BL6/J (wild-type:WT) and aly/aly mice, 6-0 silk ligatures (Akiyama Medical Mfg. Co., Ltd., Tokyo, Japan) were tied around the right maxillary second molar (15). Meanwhile, the contralateral molar of each mouse was left non-ligated (baseline control for bone loss measurements). In some experiments, C57BL6/J mice were divided into two groups, a control group (treated with phosphate-buffered saline [PBS]) and a Cpd33-treated group (0.5 mg/kg dissolved in DMSO, diluted with PBS). Ligation followed by a Hamilton syringe with a D33 gauge needle (Hamilton Company, Reno, NV, USA) was used to inject 10 μl of DMSO with PBS or Cpd33 into the palatal gingiva of the ligated second maxillary molar 3 times for 7 days. The ligation remained intact in all mice for the duration of the experiment. The mice were euthanized after 7 days and the maxilla was removed for a further analysis.
Radiological assesments
Excised maxillae were fixed in 4% paraformaldehyde in PBS (pH 7.4) at 4°C for 24 h, and then washed several times with ice-cold PBS. Bone mineral density (BMD) and three-dimensional (3D) reconstruction images of the maxilla were acquired using microfocal computed tomography (μCT; ScanXmate-L090T, Comscan Co, Kanagawa, Japan). Alveolar bone resorption was calculated by measuring the distance from the cement/enamel junction (CEJ) to the alveolar bone surface at three locations, mesial, central, and distal, of the maxillary second molars (Figure 1A), and expressed as the mean value.
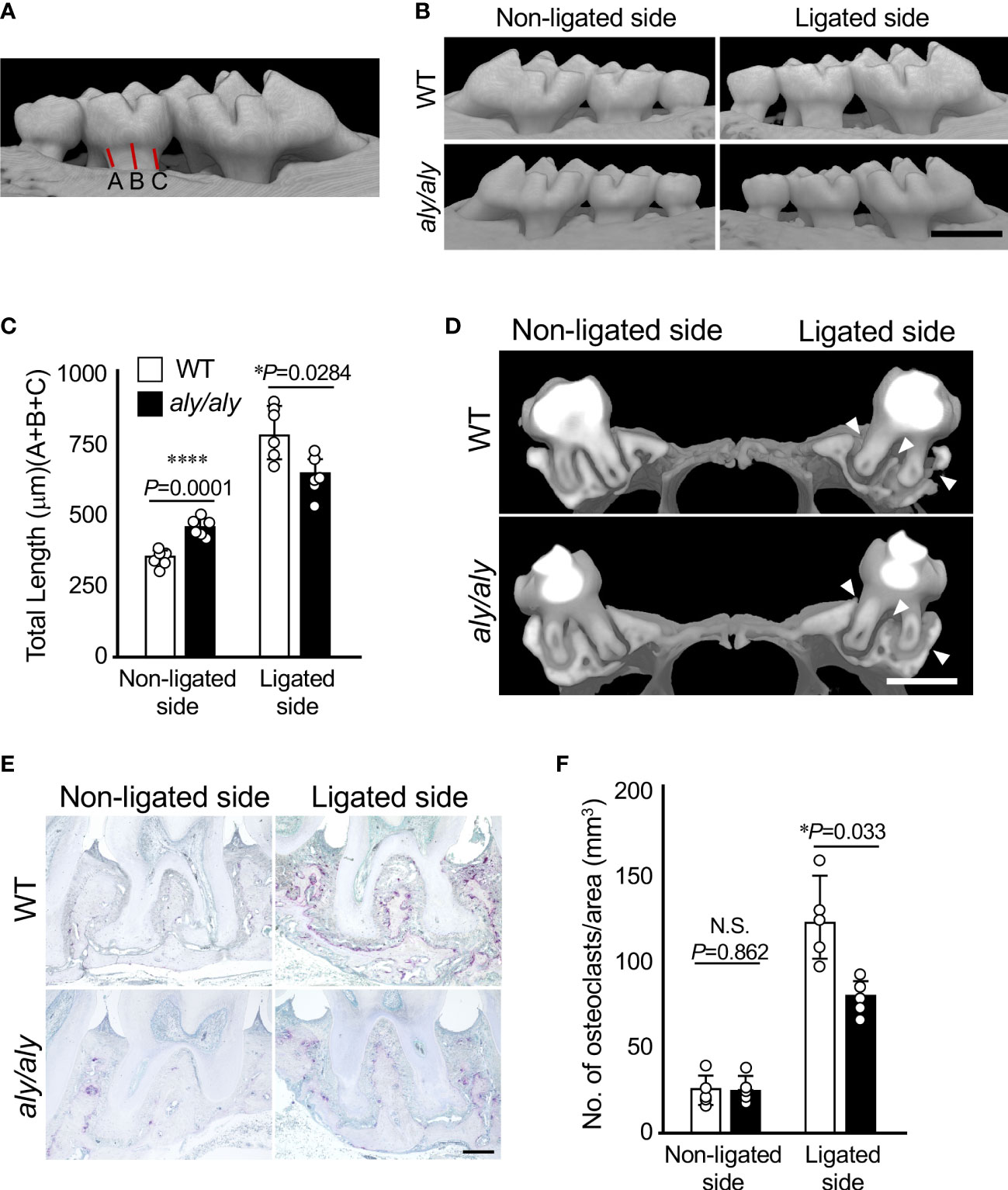
Figure 1 Alveolar bone resorption was inhibited in a ligation-induced periodontitis model in aly/aly mice. (A) Measured distance from CEJ to alveolar bone was shown. (B) 6-0 silk ligature was ligated around the right maxillary second molar of C57BL/6 (WT) and aly/aly mice for 7 days. (B) Representative three-dimensional μCT images of the maxilla of WT or aly/aly mice 7 days after ligation. Scale bar = 1 mm. (C) The distance from the cement-enamel junction (CEJ) to the apex of alveolar bone (AB) on µCT images was measured using an image analysis software program. Data are expressed as the mean ± SD (n = 6). (D) Representative three-dimensional μCT images of the mid-to-sagittal section of a maxillary second molar of WT or aly/aly mice. Scale bar = 1 mm. (E) Bone resorption was confirmed by the detection of osteoclasts by TRAP staining. Scale bar = 200 μm. (F) The number of TRAP-positive osteoclasts per area (mm2) was counted. TRAP-positive MNCs were counted from her two random coronal sections of ligated molars of five male mice in each group. Data are expressed as the mean ± SD (n = 5). *p < 0.05, ****p < 0.0001. N.S., not significant.
Histrogical analysis
After the completion of μCT imaging, maxillae were decalcified with Osteosoft (Merck, Darmstadt, Germany), sagittal paraffin sections (thickness: 5 μm) were prepared, and then tartrate-resistant acid phosphatase (TRAP) staining was performed to detect osteoclasts. Nuclei were stained with methyl green. Each tissue section was observed using a BZ-9000 (Keyence), and TRAP+ multinucleated cells (MNCs) containing ≥ 3 nuclei on the bone surface were counted as osteoclasts.
Quantitative real-time polymerase -chain reaction
The palatal gingiva of the mouse maxillary second molar on the ligated side and the non-ligated side was cut into 1-mm squares, and total RNA was extracted using ISOGEN II (Nippon Gene, Tokyo) according to the protocol. Total RNA from cultured osteoblasts was extracted using TRIZOL (Invitrogen, Waltham, MA, USA). Total RNA (1 µg) was transcribed into cDNA using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Real-time PCR was performed using the KOD SYBR qPCR Mix (Toyobo, Osaka, Japan) using a Thermal Cycler Dice Real Time System II (TaKaRa, Shiga, Japan). The GAPDH was used as an internal control. Primer sequences were as follows: tnfs11, 5′- CAGCATCGCTCTGTTCCTGTA -3′ (forward) and 5′- CTGCGTTTTCATGGAGTCTCA -3′ (reverse), Il-1b, 5′- TCGCTCAGGGTCACAAGAAA -3′ (forward) and 5′- CATCAGAGGCAAGGAGGAAAAC -3′ (reverse); Il-6, 5′- GCTACCAAACTGGATATAATCAGGA -3′ (forward) and 5′- CCAGGTAGCTATGGTACTCCAGAA -3′ (reverse); tnfa5′- TCTTCTCATTCCTGCTTGTGG -3′ (forward) and 5′- GGTCTGGGCCATAGAACTGA -3′ (reverse), and gapdh, 5′-AACTTTGGCATTGTGGAAGG-3′ (forward) and 5′-ACACATTGGGGGTAGGAACA-3′ (reverse).
Osteoclastogenesis
Primary osteoblasts (POB) were obtained by the calvaria of 1-day-old neonatal WT or aly/aly mice by digesting with 0.1% collagenase (Fujifilm Wako Pure Chemical Industries, Ltd.) and 0.2% dispase (Godo Shusei, Tokyo, Japan). Bone marrow cells (BMCs) obtained from 8-week-old WT or aly/aly mice were co-cultured in α-MEM containing 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin, together with or without LPS (100 ng/ml) for 7 days. In some experiments, bone marrow cells obtained from 8-week-old WT or aly/aly mice were cultured with M-CSF (50 ng/ml) and RANKL (50 ng/ml) for 5 days. The medium was changed every 2 days. After culturing, the cells were fixed with 3.7% formaldehyde in PBS for 10 min. After permeabilization with ethanol/acetone, cells were stained with TRAP, and TRAP+ MNCs were observed under a microscope and counted as osteoclasts.
Osteoblast differentiation
After the POBs became confluent, they were pretreated with or without Cpd33 (1 μM) and then cultured in osteoblast differentiation medium (α-MEM containing 5% FBS, 10 mM β-glycerophosphate (β-GP), and 50 μg/ml ascorbic acid) in the presence or absence of LPS (100 ng/ml). After fixing the cells with ethanol/acetone on day 7 of culture, they were incubated with alkaline phosphatase (ALP) substrate buffer (10 mg/ml p-nitrophenyl phosphate, 0.1 M diethanol amine, and 1 mM MgCl2, pH 8.0). After 15 minutes, the reaction was stopped by adding 5M NaOH and then the absorbance at a wavelength of 405 nm was measured with a plate reader (Bio-Rad Laboratories, Inc, Hercules, CA).
Statistical analysis
Data are presented as the mean ± standard deviation (SD). Means were compared between two groups using the equal variance t-test (versus control; * P < 0.05, ** P < 0.01). Statistical comparisons were performed by an analysis of variance (ANOVA) with Tukey’s test for multiple comparisons, when evaluating more than two groups. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Statistical analysis was performed using the SPSS software package version 27.0 (IBM, Armonk, NY, USA).
Results
Alveolar bone resorption in a ligation-induced periodontitis model was inhibited in aly/aly mice
First, we established a periodontitis model by ligating the right maxillary second molars of either WT or aly/aly mice with silk thread, and examined the degree of alveolar bone resorption at 7 days after ligation using μCT. The distance from the CEJ of the second molar to the alveolar bone surface was shorter in aly/aly mice than in WT mice, and the buccal alveolar bone thinning observed in WT mice was suppressed in aly/aly mice (Figures 1B, C). Alveolar bone resorption on the non-ligated side was significantly longer in aly/aly mice than in WT mice (Figure 1C). The sagittal analysis of the maxillary second molar region revealed significant thinning of the buccal alveolar bone and significant resorption of the furcation and palatal alveolar bone in the WT mice, whereas these changes were not observed in aly/aly mice (Figure 1D, arrowheads). Consistent with these results, the number of osteoclasts in the alveolar bone of aly/aly mice was lower than that of WT mice in a histological analysis (Figures 1E, F). These results indicate that the inhibition of NIK suppresses ligation-induced periodontitis.
Expression of ligation-induced inflammatory cytokines was suppressed in the gingival tissue of aly/aly mice
Next, we compared the expression changes of NF-κB-dependent inflammatory cytokines in the palatal gingiva between WT and aly/aly mice. In the gingival tissue of aly/aly mice, the expression of RANKL, which is essential for osteoclast differentiation, TNFα, and IL-6, which induces osteoclastogenesis (16, 17), were suppressed (Figure 2). The expression of IL-1β did not change between the non-ligated side and the ligated side of WT and aly/aly mice (Figure 2).
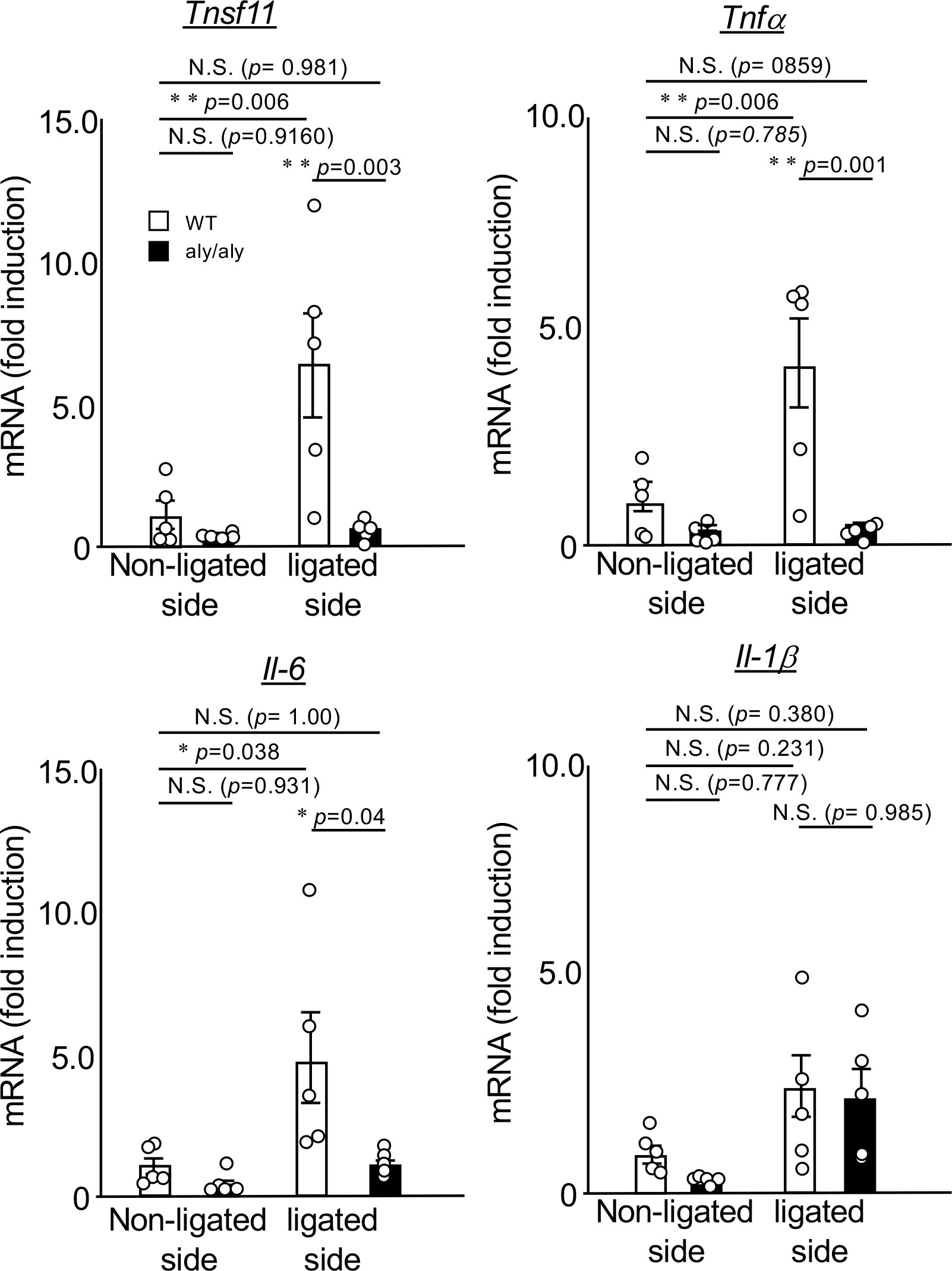
Figure 2 The expression of ligation-induced inflammatory cytokines was suppressed in the gingival tissue of aly/aly mice. On the 7th day after ligating the upper right maxillary second molar of WT and aly/aly mice, the palatal gingiva of the ligated tooth from each mouse was collected and total RNA was prepared. The mRNA expression of tnfsf11, tnfα, IL-6, IL-1β and gapdh was analyzed by real-time polymerase chain reaction. Data are expressed as the mean ± SD (WT:n = 5, aly/aly:n = 5). Similar results were obtained from three independent experiments. *P<0.05, **P<0.01. N.S., not significant.
Suppression of osteoclastogenesis in the aly/aly mouse periodontitis model was primarily attributed to osteoclast precursors
To clarify the cellular mechanism by which the number of osteoclasts in aly/aly mice was reduced in comparison to WT mice in a periodontitis model, primary osteoblasts (POBs) and bone marrow cells (BMCs) from WT and aly/aly mice were prepared and cocultured in the presence or absence of LPS. When POBs and BMCs from WT mice and aly/aly mice were examined, aly/aly mice were found to produce smaller osteoclasts. On the other hand, osteoclasts were hardly detected in the co-culture of BMCs from aly/aly mice with POBs from either WT or aly/aly mice (Figures 3A, B). As we previously reported, the stimulation of BMCs from either WT or aly/aly mice with M-CSF and RANKL resulted in the formation of numerous large osteoclasts, whereas a few small osteoclasts were formed in BMCs from aly/aly mice (Figures 3C, D). These results suggest that the low osteoclast differentiation potential of osteoclast precursors in aly/aly mice is mainly responsible for the decrease in the number of osteoclasts in the periodontitis model in comparison to WT mice.
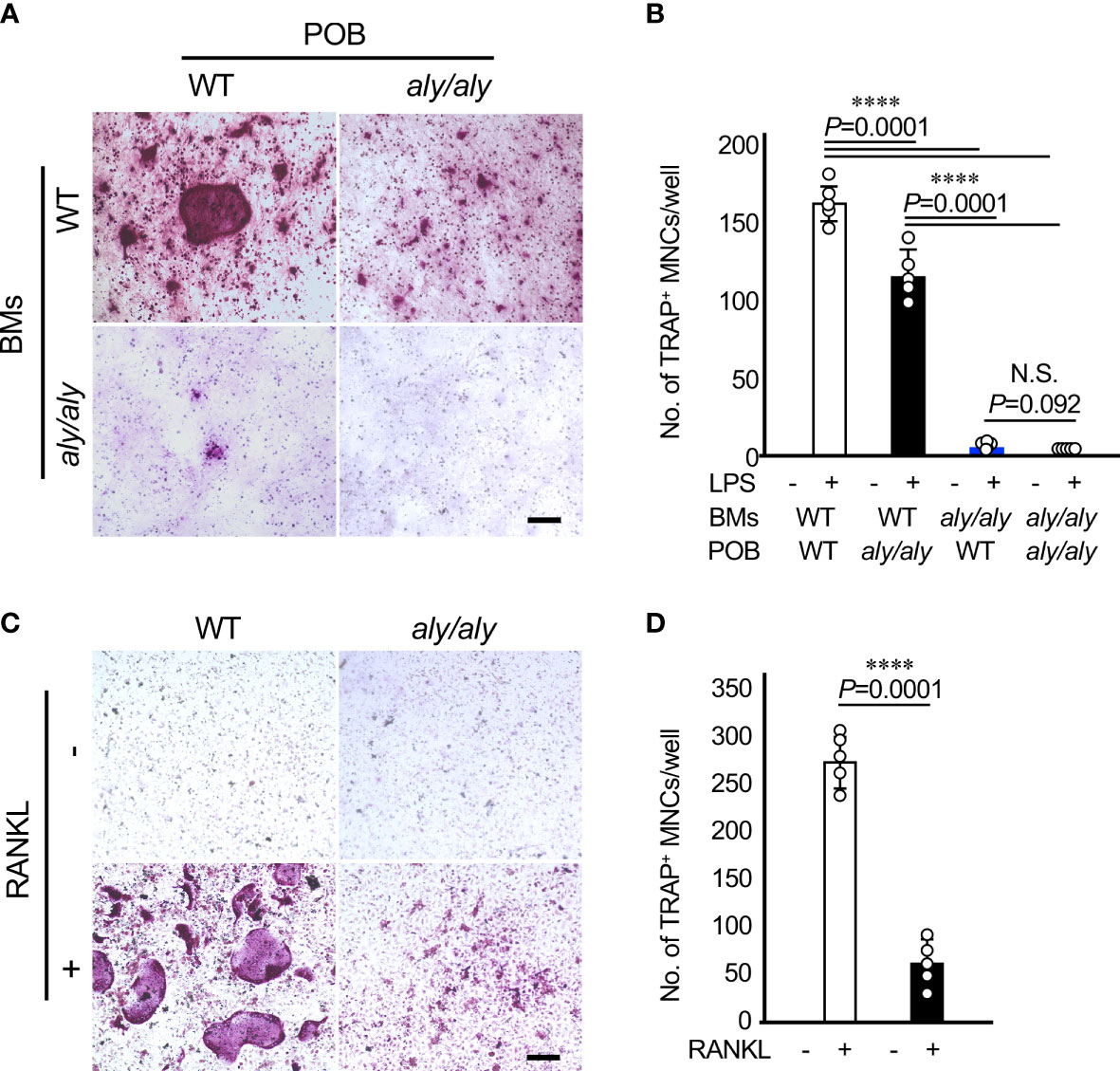
Figure 3 The suppression of osteoclastogenesis in aly/aly mice in the periodontitis model was primarily attributed to osteoclast precursors. (A) Bone marrow cells (BMCs) and primary osteoblasts (POB) from either WT or aly/aly mice were cocultured to differentiate into osteoclasts in the presence or absence of LPS (100 ng/ml) for 7 days. Cells were stained by TRAP. Scale bar = 200 μm. (B) The number of TRAP+MNCs was counted. Data are expressed as the mean ± SD (n = 5). (C) BMCs from either WT or aly/aly mice were cultured in the presence or absence of RANKL (50 ng/ml) together with M-CSF (50 ng/ml) for 5 days. Cells were stained by TRAP. Scale bar = 200 μm. (D) The number of TRAP+MNCs was counted. Data are expressed as the mean ± SD (n = 5). ****P<0.0001. Similar results were obtained from three independent experiments. N.S., not significant.
Cpd33 inhibited the LPS-induced expression of RANKL and inflammatory cytokines in osteoblasts, and restored the LPS-induced suppression of osteoblastic differentiation
We next investigated the possibility of Cpd33, a selective inhibitor of NIK, as a therapeutic agent for periodontitis. Since we have already reported that Cpd33 suppresses RANKL-induced osteoclastogenesis in a dose-dependent manner (14), we examined the effects of Cpd33 on the induction of RANKL and inflammatory cytokines induced by LPS stimulation in osteoblasts. Cpd33 at a concentration of 1 μM (which completely suppresses RANKL-induced osteoclastogenesis) (14) suppressed the expression of RANKL and TNFα induced by LPS in osteoblasts (Figure 4A). Cpd33 tended to suppress LPS-stimulated IL-6 expression, but the difference was not significant. IL-1β levels were almost unchanged after 48 hours of LPS stimulation with or without CPd33 pretreatment. We next investigated the possibility that Cpd33 could restore the suppression of LPS-stimulated osteoblastic differentiation. When POBs were cultured in a medium containing ascorbic acid and β-glycerophosphate and pretreated with Cpd33 for 1 hour before LPS stimulation, the inhibition of ALP activity was restored in comparison to that without pretreatment with Cpd33 (Figure 4B). Cpd33 alone slightly increased ALP activity, but the difference was not statistically significant.
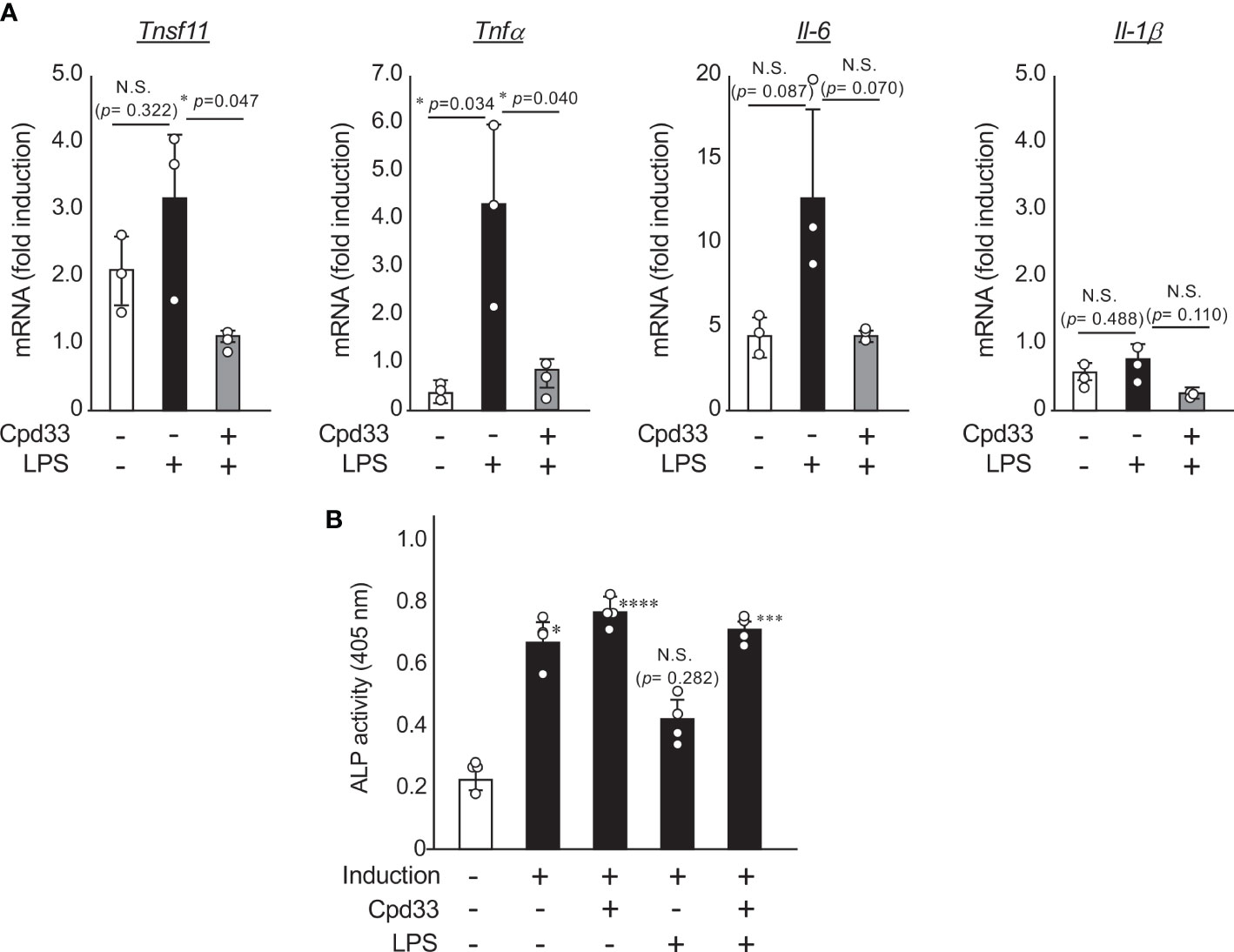
Figure 4 Cpd33 inhibited the LPS-induced expression of RANKL and inflammatory cytokines in osteoblasts, and restored the LPS-induced suppression of osteoblastic differentiation. (A) POBs were pretreated with or without Cpd33 (1 μM) for 1h and then treated with LPS (100 ng/ml) for 48 (h) Total RNA was prepared and then subjected to real-time polymerase chain reaction to examine the mRNA expression of tnfsf11, tnfα, IL-6, IL-1β and gapdh. Fold-induction was displayed with the expression level of each cytokine before stimulation (time=0) set to 1. Data are expressed as the mean ± SD (n = 3). *P<0.05. (B) POBs were cultured with osteoblast inducetion media (αMEM containing 5% FBS, 50 μg/ml ascorbic acid and 10 mM β-grycelophosphate) for 7 days. Pretreatment with or without Cpd33(1 μM) was followed by stimulation with or without LPS (100 ng/ml). After 7 days, alkaline phosphatase (ALP) activity was measured to assess osteoblast differentiation. Absorbance was measured at a wavelength of 405 nm. Data are expressed as the mean ± SD (n = 4). Similar results were obtained from three independent experiments. *P<0.05, ***P<0.001, ****P<0.0001 (vs vehicle). N.S., not significant.
Cpd33 prevented alveolar bone loss in the periodontitis model
We finally examined the possibility of suppressing alveolar bone resorption in a periodontitis model. Cpd33 (0.5 mg/kg) was injected through the palatal gingiva three times a week, and the degree of alveolar bone resorption was evaluated 7 days later (Figure 5A). Cpd33 significantly inhibited alveolar bone resorption (Figures 5B, C). There was no difference in the amount of alveolar bone resorption on the non-ligated side between the vehicle group and Cpd33-treated group. The sagittal analysis of the maxillary second molar region revealed significant thinning of the buccal alveolar bone and significant resorption of the furcation and palatal alveolar bone in the vehicle group. On the other hand, even in the Cpd33-treated group, thinning of the buccal alveolar bone was observed, while sufficient alveolar bone remained on the furcation and palatal side. (Figure 5D). In the histological analysis, the number of osteoclasts in the alveolar bone of Cpd33-treated group was lower than that of the vehicle group (Figures 5E, F). These results further supported that the inhibition of NIK suppresses alveolar bone resorption in ligation-induced periodontitis model.
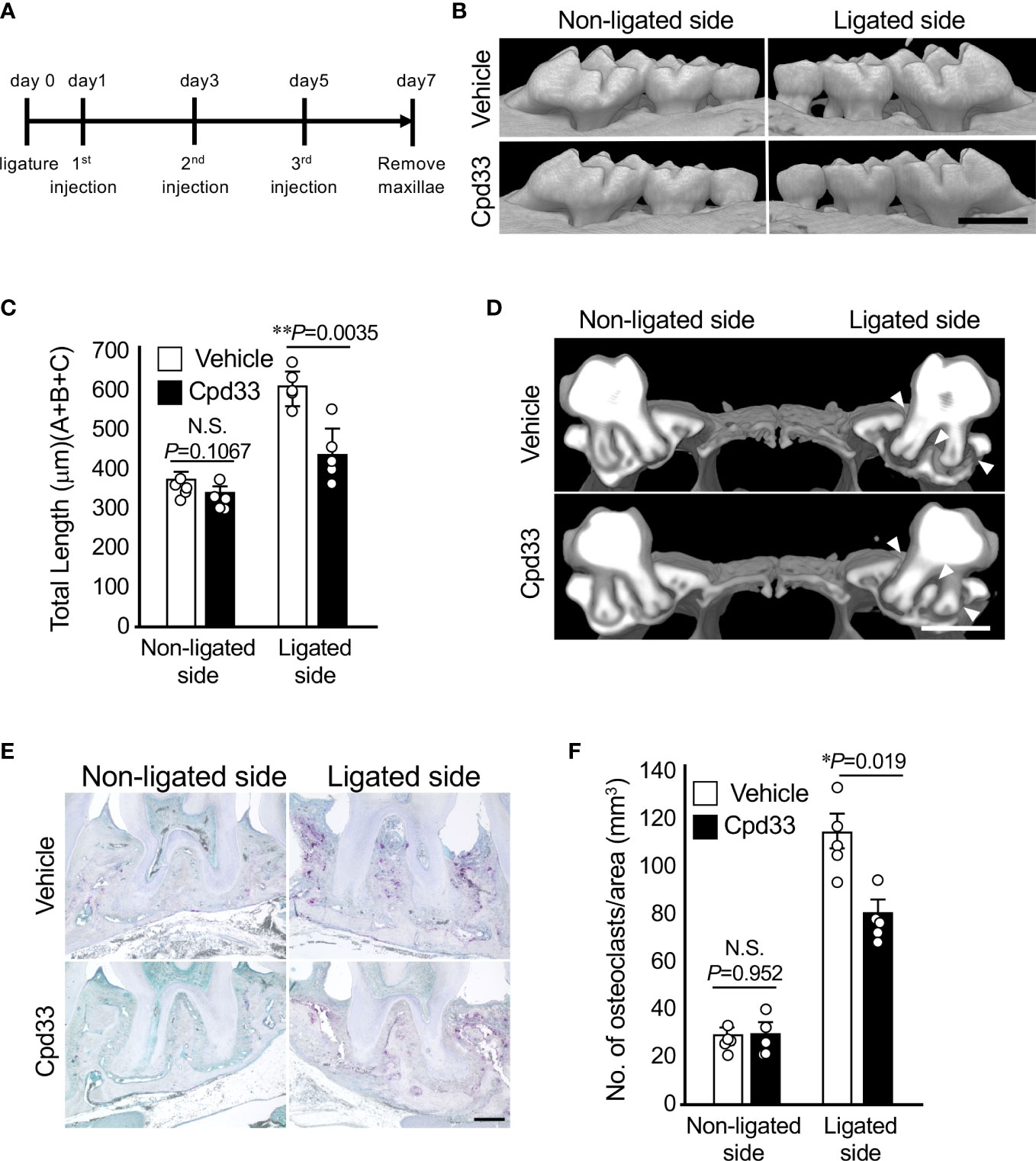
Figure 5 Cpd33 prevented alveolar bone loss in the periodontitis model. (A) Experimental schedule was shown. (B) 6-0 silk ligature was ligated around the right maxillary second molar of C57BL/6 mice, and from the same day, control group (DMSO) and Cpd33 (0.5 mg/kg) were administered once every two days for 7 days. (B) Representative three-dimensional μCT images of the maxilla in each treatment group at 7 days after ligation. Scale bar = 1 mm. (C) The distance from the cement-enamel junction (CEJ) to the apex of alveolar bone (AB) on µCT images was measured using an image analysis software program. Data are expressed as the mean ± SD (n = 5). (D) Representative three-dimensional μCT images of the mid-to-sagittal section of a maxillary second molar in each treatment group. Scale bar = 1 mm. (E) Bone resorption was confirmed by the detection of osteoclasts by TRAP staining. Scale bar = 200 μm. (F) The number of TRAP-positive osteoclasts per area (mm2) was counted. TRAP-positive MNCs were counted from two random coronal sections of ligated molars of five male mice in each group. Data are expressed as the mean ± SD (n = 10). *p < 0.05, **p < 0.01. N.S., not significant.
Discussion
Based on our previous findings that NIK inhibition is effective for suppressing osteoclast differentiation using aly/aly mice (12, 13) and the NIK inhibitor Cpd33 (14), the present study demonstrated that NIK inhibition is also effective in alveolar bone resorption, even in an experimental periodontitis model, by inhibiting osteoclastogenesis at histological and cellular levels. When the right upper maxillary second molar of WT mice were ligated using silk thread, osteoclasts appeared and alveolar bone resorption was observed, but the number of osteoclasts was low, and the expression of RANKL and inflammatory cytokines in the gingiva around the ligation was decreased in aly/aly mice. The decreases in the number of osteoclasts in the aly/aly mouse periodontitis model, were mainly due to cell autonomous defects of osteoclast precursors. Furthermore, the local administration of Cpd33 inhibited alveolar bone resorption by reducing the number of osteoclasts and the expression of inflammatory cytokines, including RANKL, in the surrounding gingiva. in the surrounding gingiva, as seen in aly/aly mice. Taken together, NIK can be a suitable target molecule for the treatment of periodontal disease.
As described above, the expression of RANKL in the periligative gingival tissue was suppressed in the aly/aly mouse periodontitis model. Thus, we further investigated the possibility that the decrease in the number of osteoclasts observed in the aly/aly mouse periodontitis model was caused by the decreased expression of RANKL. POBs and BMCs derived from WT and aly/aly mice were prepared, co-cultured in four combinations, and osteoclasts were induced by LPS stimulation. WT mouse-derived BMCs induced osteoclasts in both mouse-derived POBs. However, POBs from aly/aly mice induced smaller osteoclasts than POBs from WT mice. As RANKL expression was decreased in the periligative gingival tissues of aly/aly mice, LPS-induced RANKL expression in POB from aly/aly mice may suppress compared with POB from WT mice. On the other hand, when BMCs derived from aly/aly mice were used, few osteoclasts were formed regardless of whether the POBs were derived from WT or aly/aly mice. Furthermore, even when osteoclasts were induced with M-CSF and RANKL using BMCs derived from WT or aly/aly mice, only a few osteoclasts were formed from BMCs derived from aly/aly mice. These results suggested that RANKL signaling is inhibited in osteoclast progenitor cells derived from aly/aly mice. We have already shown that NIK inhibition by aly/aly mice and Cpd33 suppresses the RANKL-induced expression of NFATc1 (12–14).
NIK was originally identified as a protein that binds to TRAF2 using the yeast two hybrid system with TRAF2 as a bait (18). Furthermore, the overexpression of NIK activated the canonical pathway of NF-κB (18). On the other hand, Xiao et al. reported that the stimulation of cytokines involved in lymphadenopathy, such as CD40L and BAFF, plays an important role for NIK in the activation of alternative pathways through p100 to p52 processing (19). Despite there seems to be a seemingly clear line between the canonical and alternative NF-κB pathways, NIK may be involved in both. LPS activates canonical NF-κB signaling through TLR4 (20, 21). Among several inflammatory cytokines induced by LPS stimulation, RANKL, TNFαN and IL-6 were decreased in aly/aly mouse-derived or Cpd33-pretreated osteoblasts, whereas, expression of IL-1b was not changed. Since, changes in the expression of inflammatory cytokines, including RANKL, were examined 7 days after ligation and 48 hours after LPS stimulation, detailed time-course changes may yield different results. RANKL is a rare cytokine that activates both the classical and alternative pathways (7), and stimulation of BMCs from aly/aly mice or Cpd33-pretreated BMCs with RANKL did not affect the degradation of IκBα, only p52 processing was inhibited (12–14). Moreover, overexpression of p52 or RelB in BMCs from aly/aly mice induced expression of Cot (cancer ocular thyroid) and NFATc1, thereby rescuing osteoclastogenesis (22). Analysis of the Cot promoter showed that p65 and RelB bind to distal NF-κB binding sites, and that RelB, but not p65, binds to her NF-κB binding sites proximal to her Cot promoter. The alternative pathway is thought to induce osteoclast differentiation through long-term maintenance of Cot expression (22). Thus, the inhibition of alveolar bone resorption in the periodontal disease model by the inhibition of NIK is considered to be mainly due to the inhibition of osteoclast differentiation rather than suppression of inflammation.
From the results of the bone morphometric analysis of the long bones of aly/aly mice, the increase in bone mass in aly/aly mice was due to not only to the suppression of bone resorption by a decreasing number of osteoclasts, but also to the enhancement of bone formation (12, 13). Furthermore, POBs prepared from aly/aly mouse calvaria showed enhanced osteoblastic differentiation in comparison to POBs from WT mice (23). However, the injection of Cpp33 into ovariectomized mice, mainly suppressed bone resorption and had almost no effect on bone formation (14). The reason for this may be that ovariectomized mice are in a state of high turnover where bone resorption and formation are accelerated; thus, the effect of enhancing bone formation may not have been so obvious. In this study, adding Cpd33 to POB from WT mice tended to increase the ALP activity in comparison to the control group, but the difference was not significant. On the other hand, the stimulation of POB with LPS suppressed osteoblastic differentiation, whereas pretreatment with Cpd33 partially restored the suppression of osteoblastic differentiation. These results suggest that the suppression of alveolar bone resorption by aly/aly mice and Cpp33 treatment may also involve the effect of preventing the suppression of bone formation under inflammation.
Since the crystal structure analysis of NIK was clarified (24, 25), selective inhibitors of NIK have been developed, and in addition to regulating cell proliferation and differentiation, they are effective against liver inflammation/steatosis, rheumatoid arthritis, lupus, osteoporosis, metabolic disorder, and cancer in animal models (26, 27). NIK-SM1 and NIK inhibitor, 4-(3-((7H-pyrrolo [2,3-d] pyrimidin-4-yl) amino)-4-morpholinophenyl)-2-(thiazol-2-yl) -but-3-yn-2-ol, suppressed alveolar bone resorption in the mouse periodontitis model by inhibiting osteoclast formation and the expression of inflammatory cytokines in the gingiva (28, 29). Injection of LPS into the calvaria of aly/aly mice also inhibited osteoclast induction, thereby suppressing bone destruction compared to WT mice (30). In this study, we not only examined the inhibition of alveolar bone resorption by NIK inhibition at the genetic level, but also clarified the structure, cell and histological analysis of the internal alveolar bone. The local administration of Cpd33 to a periodontitis model also inhibited alveolar bone resorption by suppressing osteoclast formation, as in the case of applying a periodontal disease model to aly/aly mice. Since we administered Cpd33 locally from the palatal gingiva, Cpd33 could not sufficiently suppress buccal alveolar bone resorption, but Cpd33 was able to suppress palatal and furcation alveolar bone resorption. Since the defect of alveolar bone at the furcation (class II defect) is particularly difficult to regenerate (31), the suppression of alveolar bone resorption at the furcation is considered clinically useful.
As aly/aly mice (32) and NIK-/- mice (33) exhibit abnormal spleen and thymus architecture, complete lack of lymph nodes, and poor antibody production, the long-term administration of inhibitors of NIK might be risk of weakened immune responses. We previously reported that intraperitoneal administration of Cpd33 for 4 weeks in OVX mice inhibited osteoclast formation, thereby suppressing bone loss (14). No significant body weight loss was observed during treatment, and neither structural abnormalities in the spleen and thymus, nor damage to the liver and kidneys were bserved in histological analysis (14). In this study, the local administration of Cpd33, no abnormalities such as gingival redness or necrosis were observed. Furthermore, a small volume of liquid was required for gingival injection, and at concentrations above 0.5 mg/kg, Cpd33 was not sufficiently soluble. In the future, in order to consider the clinical application of Cpd33, it is necessary to investigate in detail the solvent that dissolves Cpd33, the administration method, and the administration period. Taken together, the inhibition of NIK is relatively effective in suppressing alveolar bone resorption due to periodontal disease.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by The Animal Care Committee of Kyushu University.
Author contributions
The authors TA, and FH performed the experiments, µCT, and histological analysis and prepared the initial version of the paper. AL, NY, and NT-H performed the experiments. EJ designed the study, performed the experiments and the literature review, prepared the initial and the final versions of the paper, and submitted the document. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI grants (20K09890 to SM, and 21H03108 to EJ).
Acknowledgments
The authors thank Mr. Hiroshi Otowa for his technical assistance in data collection of histology. We would like to thank Japan Medical Communication (https://www.japan-mc.co.jp) for English language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hajishengallis G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol (2015) 15(1):30–44. doi: 10.1038/nri3785
2. Kajiya M, Kurihara H. Molecular mechanisms of periodontal disease. Int J Mol Sci (2021) 22(2):930. doi: 10.3390/ijms22020930
3. Li Y, Ling J, Jiang Q. Inflammasomes in alveolar bone loss. Front Immunol (2021) 12):691013. doi: 10.3389/fimmu.2021.691013
4. Tsukasaki M. RANKL and osteoimmunology in periodontitisJ Bone Miner Metab (2021) 39(1):82–90 doi: 10.1007/s00774-020-01165-3
5. Udagawa N, Koide M, Nakamura M, Nakamichi Y, Yamashita T, Uehara S, et al. Osteoclast differentiation by RANKL and OPG signaling pathways. J Bone Miner Metab (2021) 39(1):19–26. doi: 10.1007/s00774-020-01162-6
6. Suda K, Udagawa N, Sato N, Takami M, Itoh K, Woo JT, et al. Suppression of osteoprotegerin expression by prostaglandin E2 is crucially involved in lipopolysaccharide-induced osteoclast formation. J Immunol (2004) 172(4):2504–10. doi: 10.4049/jimmunol.172.4.2504
7. Jimi E, Takakura N, Hiura F, Nakamura I, Hirata-Tsuchiya S. The role of NF-κB in physiological bone development and inflammatory bone diseases: is NF-κB inhibition “Killing two birds with one stone”? Cells (2019) 8(12):1636. doi: 10.3390/cells8121636
8. Hayden MS, Ghosh S. Regulation of NF-κB by TNF family cytokines. Semin Immunol (2014) 26(3):253–66. doi: 10.1016/j.smim.2014.05.004
9. Sun SC. The non-canonical NF-κB pathway in immunity and inflammation. Nat Rev Immunol (2017) 17(9):545–58. doi: 10.1038/nri.2017.52
10. Iotsova V, Caamaño J, Loy J, Yang Y, Lewin A, Bravo R. Osteopetrosis in mice lacking NF-κB1 and NF-κB2. Nat Med (1997) 3(11):1285–9. doi: 10.1038/nm1197-1285
11. Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, et al. Requirement for NF-κB in osteoclast and b-cell development. Genes Dev (1997) 11(24):3482–96. doi: 10.1101/gad.11.24.3482
12. Soysa NS, Alles N, Weih D, Lovas A, Mian AH, Shimokawa H, et al. The pivotal role of the alternative NF-κB pathway in maintenance of basal bone homeostasis and osteoclastogenesis. J Bone Miner Res (2010) 25(4):809–18. doi: 10.1359/jbmr.091030
13. Maruyama T, Fukushima H, Nakao K, Shin M, Yasuda H, Weih F, et al. Processing of the NF-κB2 precursor p100 to p52 is critical for RANKL-induced osteoclast differentiation. J Bone Miner Res (2010) 25(5):1058–67. doi: 10.1359/jbmr.091032
14. Takakura N, Matsuda M, Khan M, Hiura F, Aoki K, Hirohashi Y, et al. A novel inhibitor of NF-κB-Inducing kinase prevents bone loss by inhibiting osteoclastic bone resorption in ovariectomized mice. Bone (2020) 135:115316. doi: 10.1016/j.bone.2020.115316
15. Abe T, Hajishengallis G. Optimization of the ligature-induced periodontitis model in mice. J Immunol Methods (2013) 394(1-2):49–54. doi: 10.1016/j.jim.2013.05.002
16. Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, et al. Tumor necrosis factor α stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med (2000) 191(2):275–86. doi: 10.1084/jem.191.2.275
17. Udagawa N, Takahashi N, Katagiri T, Tamura T, Wada S, Findlay DM, et al. Interleukin (IL)-6 induction of osteoclast differentiation depends on IL-6 receptors expressed on osteoblastic cells but not on osteoclast progenitors. J Exp Med (1995) 182(5):1461–8. doi: 10.1084/jem.182.5.1461
18. Malinin NL, Boldin MP, Kovalenko AV, Wallach D. MAP3K-related kinase involved in NF-κB induction by TNF, CD95 and IL-1. Nature (1997) 385(6616):540–4. doi: 10.1038/385540a0
19. Xiao G, Harhaj EW, Sun SC. NF-κB-Inducing kinase regulates the processing of NF-κB2 p100. Mol Cell (2001) 7(2):401–9. doi: 10.1016/s1097-2765(01)00187-3
20. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4gene. Science (1998) 282(5396):2085–8. doi: 10.1126/science.282.5396.2085
21. Qureshi ST, Larivière L, Leveque G, Clermont S, Moore KJ, Gros P, et al. Endotoxin-tolerant mice have mutations in toll-like receptor 4 (Tlr4). J Exp Med (1999) 189(4):615–25. doi: 10.1084/jem.189.4.615
22. Taniguchi R, Fukushima H, Osawa K, Maruyama T, Yasuda H, Weih F, et al. RelB-induced expression of cot, an MAP3K family member, rescues RANKL-induced osteoclastogenesis in alymphoplasia mice by promoting NF-κB2 processing by IKKα. J Biol Chem (2014) 289(11):7349–61. doi: 10.1074/jbc.M113.538314
23. Seo Y, Fukushima H, Maruyama T, Kuroishi KN, Osawa K, Nagano K, et al. Accumulation of p100, a precursor of NF-κB2, rnhances osteoblastic differentiation in vitro and bone formation in vivo in aly/aly mice. Mol Endocrinol (2012) 26(3):414–22. doi: 10.1210/me.2011-1241
24. de Leon-Boenig G, Bowman KK, Feng JA, Crawford T, Everett C, Franke Y, et al. The crystal structure of the catalytic domain of the NF-κB inducing kinase reveals a narrow but flexible active site. Structure (2012) 20(10):1704–14. doi: 10.1016/j.str.2012.07.013
25. Liu J, Sudom A, Min X, Cao Z, Gao X, Ayres M, et al. Structure of the nuclear factor κB-inducing kinase (NIK) kinase domain reveals a constitutively active conformation. J Biol Chem (2012) 287(33):27326–34. doi: 10.1074/jbc.M112.366658
26. Pflug KM, Sitcheran R. Targeting NF-κB-Inducing kinase (NIK) in immunity, inflammation, and cancer. Int J Mol Sci (2020) 21(22):8470. doi: 10.3390/ijms21228470
27. Haselager MV, Eldering E. The therapeutic potential of targeting NIK in b cell malignancies. Front Immunol (2022) 13:930986. doi: 10.3389/fimmu.2022.930986
28. Wang J, Wang B, Lv X, Wang L. NIK inhibitor impairs chronic periodontitis via suppressing non-canonical NF-κB and osteoclastogenesis. Pathog Dis (2020) 78(7):ftaa045. doi: 10.1093/femspd/ftaa045
29. Wang J, Wu S, Li Z, Liu L, Pang Y, Wei J. Inhibition of nuclear factor κB inducing kinase suppresses inflammatory responses and the symptoms of chronic periodontitis in a mouse model. Int J Biochem Cell Biol (2021) 139:106052. doi: 10.1016/j.biocel.2021.106052
30. Soysa NS, Alles N, Takahashi M, Aoki K, Ohya K. Defective nuclear factor-κB-Inducing kinase in aly/aly mice prevents bone resorption induced by local injection of lipopolysaccharide. J Periodontal Res (2011) 46(2):280–4. doi: 10.1111/j.1600-0765.2010.01333.x
31. Graziani F, Gennai S, Karapetsa D, Rosini S, Filice N, Gabriele M, et al. Clinical performance of access flap in the treatment of class II furcation defects. a systematic review and meta-analysis of randomized clinical trials. J Clin Periodontol (2015) 42(2):169–81. doi: 10.1111/jcpe.12327
32. Miyawaki S, Nakamura Y, Suzuka H, Koba M, Yasumizu R, Ikehara S, et al. A new mutation, aly, that induces a generalized lack of lymph nodes accompanied by immunodeficiency in mice. Eur J Immunol (1994) 24(2):429–34. doi: 10.1002/eji.1830240224
Keywords: nuclear factor-k B, periodonititis, osteoclasts (OCs), inflammation, bone, NIK
Citation: Aoki T, Hiura F, Li A, Yang N, Takakura-Hino N, Mukai S, Matsuda M, Nishimura F and Jimi E (2023) Inhibition of non-canonical NF-κB signaling suppresses periodontal inflammation and bone loss. Front. Immunol. 14:1179007. doi: 10.3389/fimmu.2023.1179007
Received: 03 March 2023; Accepted: 04 April 2023;
Published: 18 April 2023.
Edited by:
Sander Tas, Academic Medical Center, NetherlandsReviewed by:
Robert Weil, INSERM U1135 Centre d’Immunologie et de Maladies Infectieuses, FranceYanchuan Li, University of Texas MD Anderson Cancer Center, United States
Copyright © 2023 Aoki, Hiura, Li, Yang, Takakura-Hino, Mukai, Matsuda, Nishimura and Jimi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eijiro Jimi, ZWppbWlAZGVudC5reXVzaHUtdS5hYy5qcA==