 Soon Sung Kwon1
Soon Sung Kwon1 Youn Keong Cho1
Youn Keong Cho1 Seungmin Hahn2
Seungmin Hahn2 Jiyoung Oh3
Jiyoung Oh3 Dongju Won1
Dongju Won1 Saeam Shin1*
Saeam Shin1* Ji-Man Kang4,5*
Ji-Man Kang4,5* Jong Gyun Ahn4,5
Jong Gyun Ahn4,5 Seung-Tae Lee1,6
Seung-Tae Lee1,6 Jong Rak Choi1,6
Jong Rak Choi1,6- 1Department of Laboratory Medicine, Yonsei University College of Medicine, Seoul, Republic of Korea
- 2Department of Pediatric Hemato-oncology, Yonsei Cancer Center, Severance Hospital, Yonsei University College of Medicine, Seoul, Republic of Korea
- 3Division of Clinical Genetics, Department of Pediatrics, Severance Children’s Hospital, Yonsei University College of Medicine, Seoul, Republic of Korea
- 4Department of Pediatrics, Severance Children’s Hospital, Yonsei University College of Medicine, Seoul, Republic of Korea
- 5Institute for Immunology and Immunological Diseases, Yonsei University College of Medicine, Seoul, Republic of Korea
- 6Dxome, Seoul, Republic of Korea
Inborn errors of immunity (IEI) include a variety of heterogeneous genetic disorders in which defects in the immune system lead to an increased susceptibility to infections and other complications. Accurate, prompt diagnosis of IEI is crucial for treatment plan and prognostication. In this study, clinical utility of clinical exome sequencing (CES) for diagnosis of IEI was evaluated. For 37 Korean patients with suspected symptoms, signs, or laboratory abnormalities associated with IEI, CES that covers 4,894 genes including genes related to IEI was performed. Their clinical diagnosis, clinical characteristics, family history of infection, and laboratory results, as well as detected variants, were reviewed. With CES, genetic diagnosis of IEI was made in 15 out of 37 patients (40.5%). Seventeen pathogenic variants were detected from IEI-related genes, BTK, UNC13D, STAT3, IL2RG, IL10RA, NRAS, SH2D1A, GATA2, TET2, PRF1, and UBA1, of which four variants were previously unreported. Among them, somatic causative variants were identified from GATA2, TET2, and UBA1. In addition, we identified two patients incidentally diagnosed IEI by CES, which was performed to diagnose other diseases of patients with unrecognized IEI. Taken together, these results demonstrate the utility of CES for the diagnosis of IEI, which contributes to accurate diagnosis and proper treatments.
Introduction
Inborn errors of immunity (IEI) include a variety of heterogeneous genetic disorders in which defects in the immune system lead to an increased susceptibility to infections and other complications. The prevalence of IEI has been known to be rare. However, recent estimated prevalence is 1/1,100 to 1/1,500 and has been increasing over the years with increased recognition of IEI including universal neonatal screening programs in several countries (1, 2).
Defect or dysregulation of the immune system is usually suspected in patients with recurrent or persistent infections, unusual infection, and severe infection. In addition, it is also suspected when recurrent fever and inflammation present. Often, laboratory abnormalities such as low immunoglobulin and abnormal lymphocyte subsets and leukopenia including neutropenia and/or lymphopenia can lead to presumed diagnosis of IEI. Above all, the family history of IEI provides very important clues in suspecting the patient (3).
In the 2022 classification of the International Union of Immunological Societies (IUIS), 485 diseases that affect various components of immune systems are presented. These include immunodeficiencies, antibody deficiencies, complement deficiencies, immune dysregulations, phagocyte defects, and autoinflammatory disorders (4). Most IEI are associated with specific gene defects. When IEI is suspected, it is important to find underlying genetic etiology. Detection of causative pathogenic variants from genes related to IEI is crucial for developing an appropriate treatment strategy and determining the patient’s prognosis.
Because of the wide application of next-generation sequencing (NGS) for detecting genetic variants in IEI, genetic causes of IEI are now more correctly identified and more patients have been diagnosed (5–8). Here, we present the genetic analysis of patients with IEI at a tertiary care hospital in Korea. Genetic causes were identified in 40.5% of the patients (15/37), and four novel pathogenic variants of genes related to IEI were detected. Also, somatic variants were identified in three patients including a patient with VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome, which emphasize the utility of genetic test using in-depth NGS, especially clinical exome sequencing (CES) for the diagnosis of IEI.
Materials and methods
Subjects
This retrospective study was conducted on patients who received NGS testing for suspected IEI from December 2018 to October 2021 at Severance Hospital, Seoul, South Korea. Their clinical data, clinical manifestations, laboratory test results and genetic analysis results were collected and analyzed.
This study was reviewed and approved by the Institutional Review Board of Yonsei University Health System (4-2022-1558).
Genetic analysis and variant interpretation
For the genetic diagnosis of IEI, we used a custom-designed clinical exome panel (Dxome, Seoul, Republic of Korea) including 4,894 genes related to human genetic diseases, including IEI (Supplementary Table S1). In our panel, we incorporated the majority of the key genes associated with IEI as suggested by IUIS (4); however, 69 genes were not included (see Supplementary Table S2 for the list of these genes). With our panel, an average depth of 242.6× was achieved (range, 115.7× to 481.5×). Genomic DNA (gDNA) was extracted from peripheral blood of each patient using the QIAamp Blood Mini Kit (Qiagen, Hilden, Germany) according to manufacturer’s guidelines. gDNA was quantified using the Qubit BR dsDNA kit (Invitrogen, Carlsbad, CA, United States). Then, gDNA was fragmented, end-repaired, and ligated to adapters followed by hybridization with probes. Enriched, prepared libraries were sequenced using NextSeq 550Dx instrument (Illumina, San Diego, CA, United States). Alignment, variant calling, and annotation of sequenced data were done by our custom pipeline as described previously (9, 10). Briefly, our study utilized the Burrows-Wheeler Aligner algorithm to map raw sequence data to the GRCh37 (hg19) reference genome. We then processed the data, removing duplicate reads, realigning insertions and deletions, recalibrating base quality, and calling variants using the Genome Analysis Toolkit. Any variants of potential clinical significance were confirmed by visual inspection using Integrated Genomics Viewer (Broad Institute, Cambridge, MA, USA). Large insertions and deletions were detected using split-read approaches with Pindel and Manta algorithms, while structural rearrangements were identified through read-depth analysis using ExomeDepth and combined custom tool (11). Chromosomal copy number variations were cross-checked using a custom pipeline, which included normalizing base-level depth of coverage against other samples in the same batch.
All detected variants were classified based on the recommendation of the American College of Medical Genetics and Genomics (12). For the assessment of pathogenicity, evidence was collected by using population frequency data from multiple databases, 1000 Genomes, the Genome Aggregation Database (gnomAD), the Exome Sequencing Project (ESP), and the Korean Reference Genome Database (KRGDB). In silico analysis were conducted by SIFT, MutationTaster, FATHMM, and MetaSVM. Literature and database search for the collection of evidence were conducted using ClinVar, Online Mendelian Inheritance in Man (OMIM), the Human Gene Mutation Database (HGMD), and relevant scientific publications found through online academic databases.
If needed, peripheral blood samples of parents were obtained and tested by Sanger sequencing or CES to determine the de novo occurrence of the pathogenic variants (autosomal dominant disorders) or maternal inheritance (X-linked recessive disorders) or whether the suspected variants occurred in cis or in trans (autosomal recessive disorders), for the confirmation of pathogenicity of detected variants.
Statistical analysis
For statistical analysis, SPSS version 26 was used (IBM corp., Armonk, NY, USA). Patients with causal variants and without causal variant were compared by Fisher’s exact test, where p-values less than 0.05 were considered as significant.
Results
Patient characteristics
The demographics of patients included in this study are summarized in Table 1. Of the 37 patients who underwent the test, 22 were male (59.5%) and 15 were female (40.5%), aged from 1 month to 64 years old (mean 7.1 and median 2.5 years old). The most common diagnostic category was immune dysregulation (27.0%, 10/37) and periodic fever syndrome (27.0%, 10/37), followed by antibody deficiency (13.5%, 5/37) and combined immunodeficiency (8.1%, 3/37). Twenty-five (67.6%) patients had a history of recurrent or persistent infection. Genetic diagnosis was made in 15 patients (40.5%, 15/37).
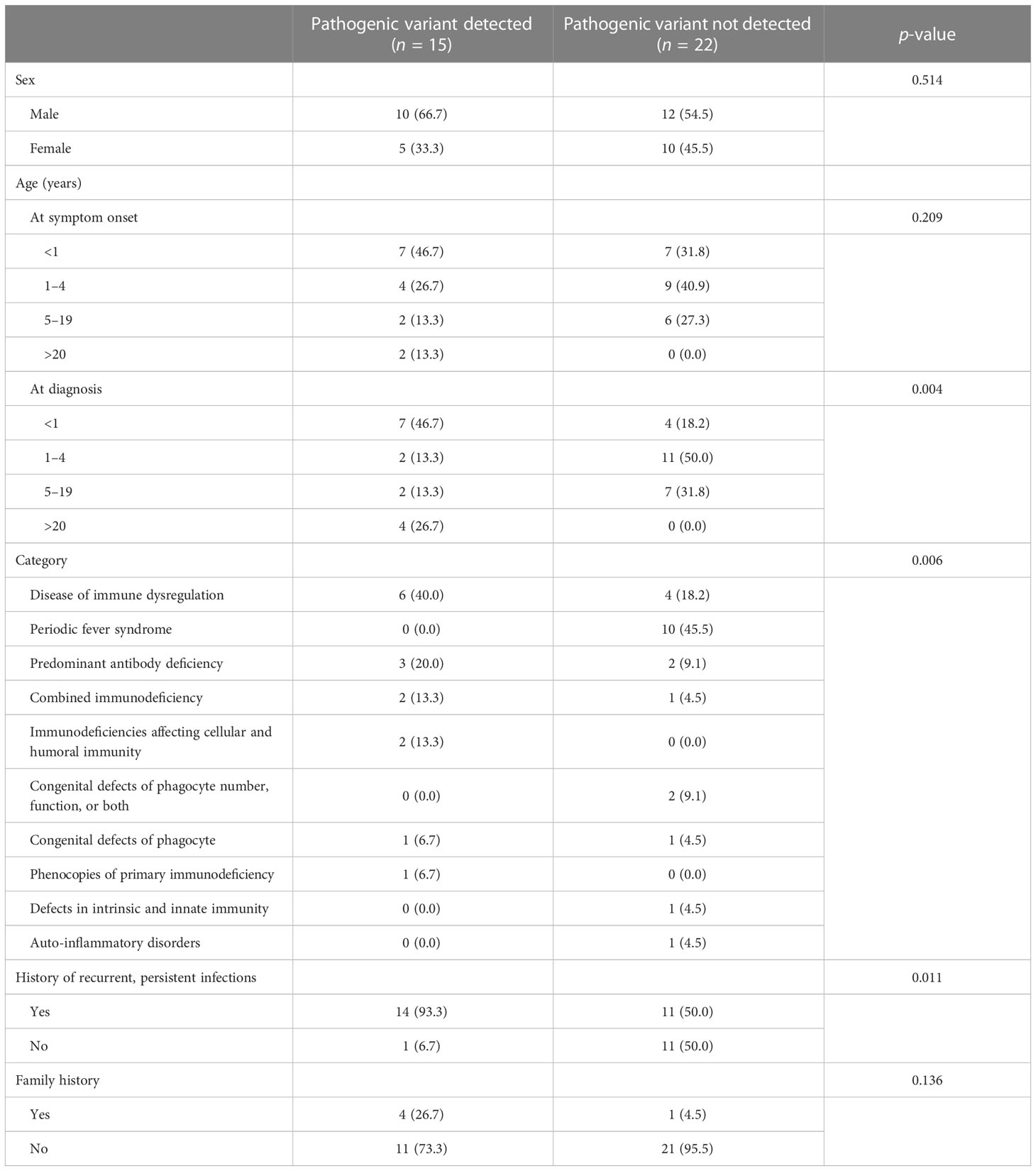
Table 1 Demographics of patients included in this study (n = 37).
Genetic diagnosis of IEI
Detailed clinical characteristics and laboratory test results of patients with genetic diagnosis are shown in Table 2. Genetic diagnosis was made promptly after symptom onset in eight cases (60.0%, 9/15). Mean turnaround time of CES for IEI was 33.1 days (range, 13–66 days). In four patients, clinical diagnosis was made several years ago and later confirmed by genetic test (Cases 3, 4, 6, and 14). In two patients, precise diagnosis could not be made before CES, which solved the cases by detecting causative pathogenic variants (Cases 9 and 15). Except for a patient with hypogammaglobulinemia only (Case 1), patients had a history of recurrent or persistent infections. Ten patients had hypogammaglobulinemia.
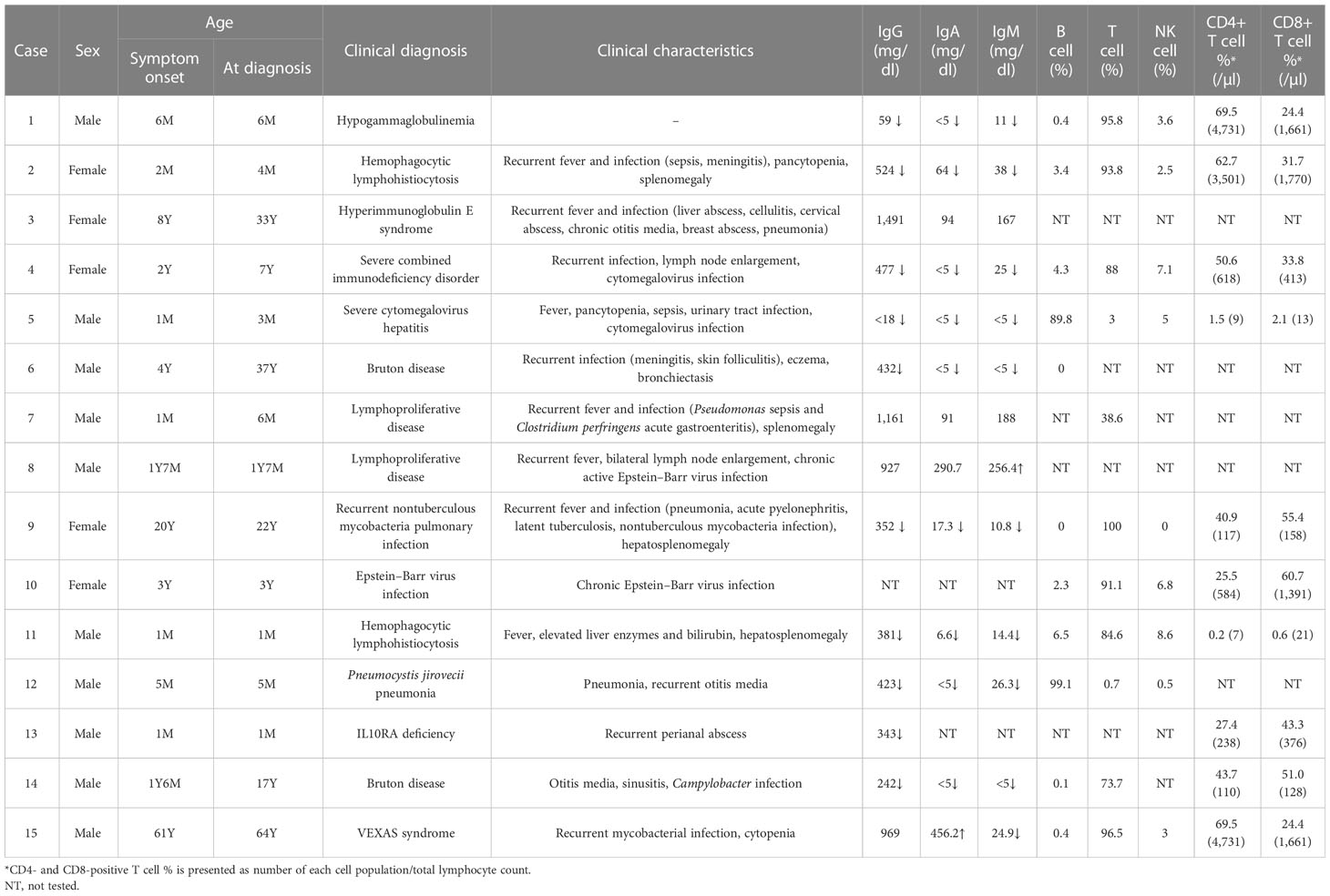
Table 2 Clinical and laboratory data of patients with genetic diagnosis.
In 10 different genes, a total of 17 clinically significant pathogenic variants were found, of which 5 were novel variants (Table 3). The most common gene found to have clinically significant pathogenic variants was the BTK gene (3/17, 17.6%) followed by the UNC13D, STAT3, IL2RG, and IL10RA genes (2/17, 11.8% for each). The rest included NRAS, SH2D1A, GATA2, TET2, PRF1, and UBA1 (1/17, 5.9% for each). None of the variants were found in common among the patients. Of 17 pathogenic variants, missense variants were most common (6/17, 35.3%), followed by frameshift variants (4/17, 23.5%) and nonsense variants (2/17, 11.8%). A canonical splice site variant, a translation initiation codon variant, a synonymous variant, an intronic variant, and an exon deletion were detected once each.
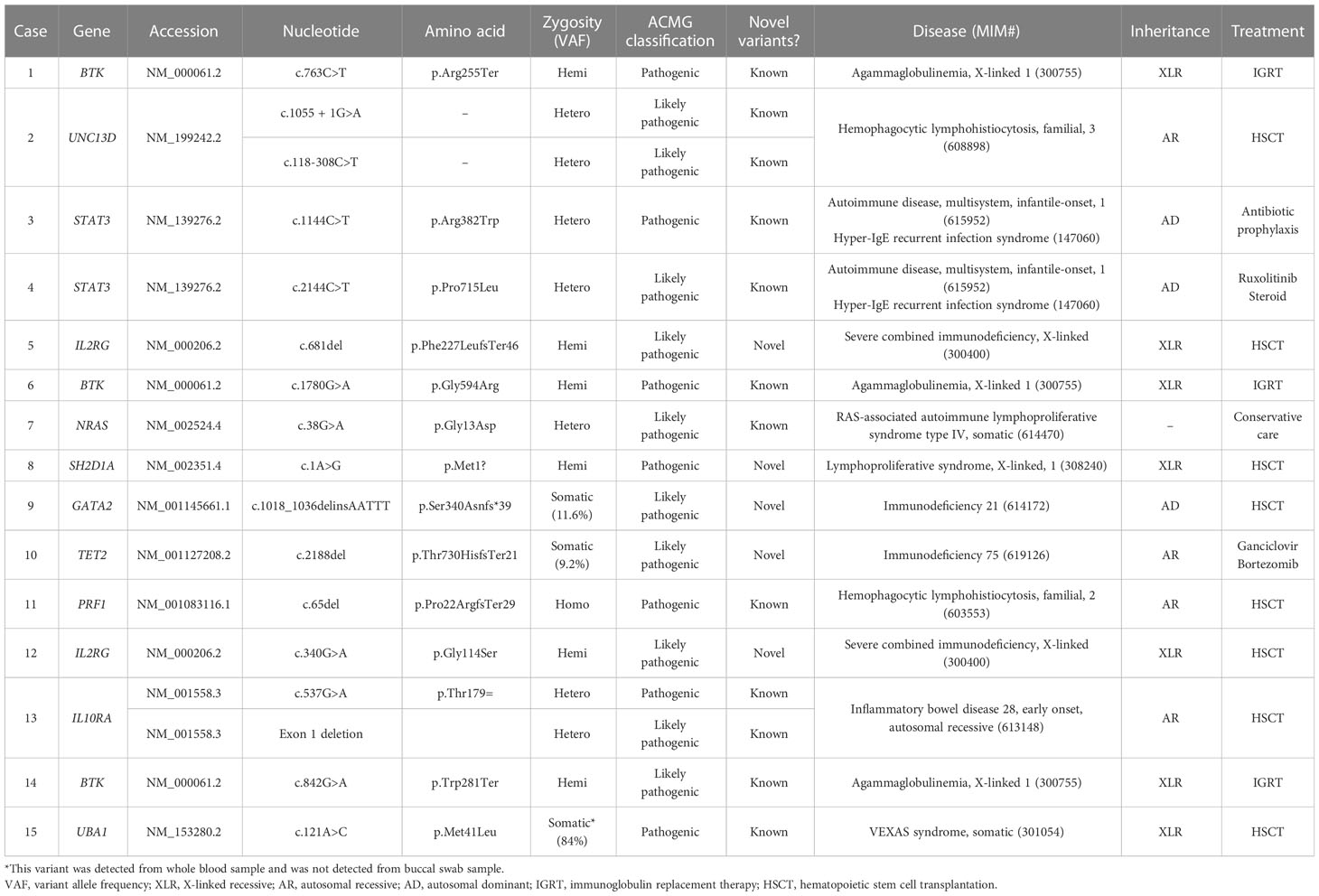
Table 3 Identified pathogenic variants from patients with genetic diagnosis and treatments.
Genetic diagnosis led to appropriate treatment for IEI diagnosed in each patient. Immunoglobulin replacement was done in three patients diagnosed with Bruton disease. Eight patients were treated with allogenic hematopoietic stem cell transplantation.
Patients with germline pathogenic variants
Pathogenic variants of BTK are causes of X-linked agammaglobulinemia, also known as Bruton disease. Known pathogenic variants of BTK (Arg255Ter, Gly594Arg, and Trp281Ter) were detected from three patients (Cases 1, 6, and 14) with immunodeficiency. All patients had hypogammaglobulinemia with significantly reduced B-cell number. Of three patients, two patients (Cases 6 and 14) were previously diagnosed with X-linked agammaglobulinemia based on laboratory results and clinical history, while one patient (Case 1) was newly diagnosed by CES performed to identify the cause of hypogammaglobulinemia. The patient did not have clinical symptoms or signs (recurrent infections) suggestive of immunodeficiency.
Two patients were diagnosed with familial hemophagocytic lymphohistiocytosis (fHLH) by CES. In both patients, the laboratory findings, clinical symptoms, and signs suggested HLH. Two pathogenic variants of UNC13D were detected from a patient (Case 2) with fHLH. Although HLH was suspected in this patient, HLH was not confirmed because diagnostic criteria (13) were not fulfilled before the molecular diagnosis. In the other patient, Case 11, detection of a homozygous pathogenic variant of PRF1 confirmed the diagnosis of fHLH, which was made before CES by fulfilling other diagnostic criteria.
A pathogenic STAT3 variant was detected from an adult female patient (Case 3) with a history of recurrent fever, pneumonia, and breast abscess who was diagnosed with hyperimmunoglobulin E syndrome in childhood. The diagnosis was made based on clinical symptoms and confirmed by the detection of the variant, which was previously reported from patients with hyperimmunoglobulin E syndrome (14, 15). Also, the dominant-negative effect of the variant has been previously observed (14).
A pathogenic STAT3 variant with several previous reports of autoimmunity and immunodeficiency was detected from a patient (Case 4) with recurrent infection history, lymph node enlargement, pancytopenia, and splenomegaly. A previous study has shown that the variant is a gain-of-function variant (16).
Two novel pathogenic variants of IL2RG were identified by CES from patients with severe combined immunodeficiency (SCID). One of the patients (Case 5) had recurrent fever from 2 months old and also had leukopenia, thrombocytopenia, and persistent CMV infection (17). Laboratory findings showed significantly decreased T-cell population and a bone marrow study revealed slightly increased number of histiocytes. Precise diagnosis of the patient was made by genetic test, which revealed a frameshift variant of IL2RG. Another novel pathogenic variant of IL2RG was detected from a patient with SCID (Case 12). There was a family history of two male brothers of the mother who died of unknown causes when they were young.
A novel, pathogenic SH2D1A was detected from a patient (Case 8) with chronic active EBV infection. From 1 year old, the patient had suffered from recurrent fever and bilateral lymph node enlargement. Later, EBV infection was confirmed and persisted despite the treatment.
From an infant (Case 13) with recurrent perianal abscess, genetic diagnosis was made by the detection of two heterozygous pathogenic variants of IL10RA. A pathogenic variant (c.537G>A, p.Thr179Thr) was inherited from the mother and IL10RA exon 1 deletion was confirmed to occur de novo. The silent variant of IL10RA has been reported in patients with IL10RA-related inflammatory bowel disease (18, 19). One of these studies has shown the aberrant splicing caused by the variant (18).
In a patient (Case 7) with recurrent fever and infection, genetic testing was requested to find the cause of immune deficiency. A known pathogenic variant of NRAS (c.38G>A, p.Gly13Asp) was detected, which caused Ras-associated lymphoproliferative disease.
Patients with somatic pathogenic variants
Most of the pathogenic variants were germline variants; however, three somatic pathogenic variants were also identified from some patients (3/15, 20.0%).
A somatic GATA2 pathogenic, frameshift variant (p.Ser340AsnfsTer39, variant allele frequency 11.6%) was detected from a young female adult patient (Case 9) with a history of recurrent infection (tuberculosis, pneumonia, EBV, CMV, and BKV). At initial assessment, the patient had pancytopenia with lymphopenia and monocytopenia. The patient also had B and NK cell deficiency, which was consistent with GATA2-associated immunodeficiency.
A somatic TET2 pathogenic variant (c.2188del, p.Thr730HisfsTer21, variant allele frequency 9.2%) was detected from a patient (Case 10) with chronic EBV infection. This patient was treated for infectious mononucleosis, and EBV infection persisted despite the treatments. CES, which was requested to evaluate the etiology of chronic EBV infection, detected the somatic variant of TET2.
A pathogenic variant of UBA1, which is the cause of VEXAS syndrome, was identified by CES from a 64-year-old male patient (Case 15). At initial assessment, the patient suffered from multiple arthralgia, leukopenia, and anemia. During follow-up, chronic granulomatous inflammation with necrosis of bone marrow and tuberculosis lymphadenitis were detected. Various symptoms persisted, however, since the cause was not clearly identified, and CES was performed to diagnose the cause of immunodeficiency and autoinflammatory conditions. A known pathogenic UBA1 variant (p.Met41Leu, variant allele frequency 84.0%) was detected, which led to an accurate diagnosis of the patient.
Patients with incidentally identified IEI
There were two patients with incidentally identified pathogenic variants causing IEI, which were identified by CES during genetic workup for other diseases (Tables 4, 5).
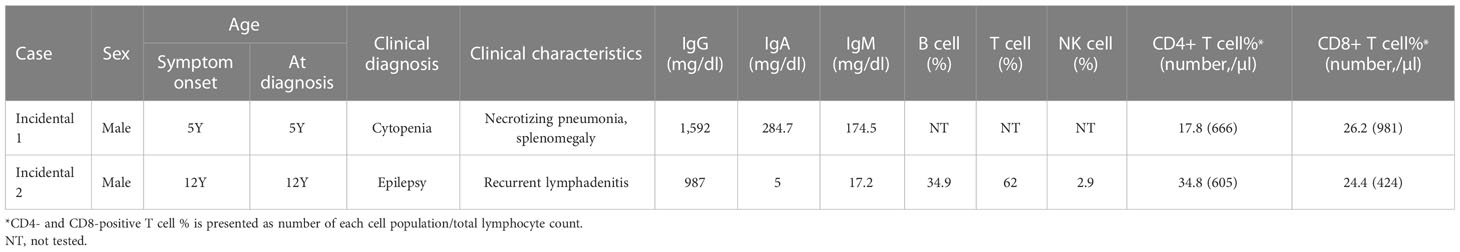
Table 4 Clinical and laboratory data of patients with incidentally detected inborn errors of immunity.

Table 5 Incidental genetic findings and treatment of patients with inborn errors of immunity.
CES identified a somatic, pathogenic variant of KRAS (c.37G>T, p.Gly13Cys, variant allele frequency 19.6%) from a patient with unknown cause of anemia and thrombocytopenia (Incidental case 1), which made the genetic diagnosis of RAS-associated lymphoproliferative disorder.
To identify a cause of neurodevelopmental disorder, CES was performed for a patient with epilepsy, delayed development, dystonia, spasticity, and brain atrophic changes (Incidental case 2). CES revealed uniparental disomy of chromosome 1 and homozygous ISG15 pathogenic variants. The patient had a history of CMV infection and recurrent lymphadenitis.
Discussion
Genetic causes of IEI are increasingly being diagnosed. IUIS have regularly updated the list of IEI, which are caused by single gene defects, and a growing number of genetic defects have been reported (4). Although certain IEI can be suspected through the patient’s history, clinical symptoms, and laboratory tests, it is difficult to make precise diagnosis since there are various genes that cause similar disorders. Thus, NGS, which can test various causative genes at the same time, is very useful for diagnosing IEI.
Diagnostic yield varies among reports, which is due to the difference in the characteristics of study populations and the methods used (Supplementary Table S3). Previous studies used targeted panel sequencing (20, 21), CES (22), whole exome sequencing (WES) (23, 24), or whole genome sequencing (25), and each method has its pros and cons. With targeted gene panel sequencing, in-depth sequencing of selected genes related to IEI is possible, which enables detection of low-level mosaic variants, whereas with methods that can test a large number of genes, diagnostic yield would be increased. In addition, as well as newly identified genes related to IEI, previous unknown related genes can be assessed. In this study, CES was used for genetic diagnosis, which showed a 40.5% positive rate. Pathogenic variants of a gene not included in targeted panels used in other studies (20, 21), UBA1, were detected by CES. In addition, two mosaic variants with low variant allele frequency were successfully detected. Taken together, these showed the utility of CES for diagnosis of IEI.
For patients with IEI, early diagnosis is crucial for proper management and avoiding severe, recurrent infections. In our cases, genetic diagnosis was made in a few months after symptom onset in patients younger than 2 years old, which emphasizes the clinical utility of CES for the diagnosis of IEI. CES is also useful for the diagnosis of unsolved cases. Two patients with a long history of undiagnosed recurrent infection and inflammations were diagnosed by CES after 2 years and 3 years after the onset of symptoms, respectively, which led to appropriate curative therapy for the patients. In particular, in the case of patients with GATA2 and IL10RA variants, prior to genetic diagnosis, the current ongoing infection treatment was given priority, but after genetic diagnosis, allogeneic hematopoietic stem cell transplantation, a curative treatment option, could be attempted with more confidence.
Although disorders of the immune system can be suspected in patients with various conditions, diagnostic yield seemed to be different among conditions. Based on the clinical diagnoses and clinical features of patients without a genetic diagnosis, it appears that they tend to have a milder disease or exhibit a lower frequency of infections (Supplementary Table S4). Recurrent fever can be evidence of immune disorder; however, in our study population, genetic analysis did not yield any positive result from the patients with recurrent fever without apparent infections. Several diseases were associated with periodic fever syndrome, such as cryopyrin-associated periodic syndromes (caused by NLRP3), familial Mediterranean fever (caused by MEFV), tumor necrosis factor receptor-associated periodic syndrome (caused by TNFRSF1A), and mevalonate kinase deficiency (caused by MVK) (26). Our patients were diagnosed with PFAPA syndrome, by combining clinical features and negative genetic test results. Although there might be unknown genetic causes, genetic testing for patients with periodic fever without identifiable infection is more likely to yield negative results.
Previous studies have reported the presence of mosaic variants in patients with various IEI (27, 28). For example, a somatic, heterozygous pathogenic variant of FAS is a known cause of autoimmune lymphoproliferative syndrome (29). There was a recent report of mosaic TLR8 gain-of-function variants that cause immunodeficiency (30). In this study, we also revealed three patients with somatic pathogenic variants of GATA2, TET2, and UBA1. Pathogenic variants of GATA2 were known to cause immunodeficiency with variable onset and susceptibility to mycobacteria and fungal infections (31). The patient with the GATA2 variant in this study also showed similar features, with a history of recurrent infection with mycobacterial infection, hypogammaglobulinemia, and B and NK cell deficiency. Somatic pathogenic variants of TET2 have been reported from patients with various hematologic malignancies (32). Although the association between TET2 variant and IEI has not been elucidated yet, a recent study reported that somatic TET2 variants were detected in CD4+ T lymphocytes of a patient with combined variable immunodeficiency (33). The UBA1 somatic variant was detected from a patient with a long history of recurrent infections and systemic inflammation of unknown etiology, whose diagnosis of VEXAS syndrome was made by CES. VEXAS syndrome is an adult-onset autoinflammatory disease defined recently, caused by myeloid lineage restricted somatic variants of UBA1 (34). Since various studies have revealed the presence of low-level mosaic variants and variants detected only in specific population of peripheral mononuclear cells, simple, routine genetic analysis without cell sorting or in-depth sequencing can lead to missed detection of some variants related to IEI.
Among the known indicators that suggest the presence of IEI, a recurrent and/or persistent history of infection and a family history of IEI have been previously reported (35–37). The results of this study are consistent with these earlier findings, demonstrating a correlation between infection history and IEI (35, 36). However, the relationship between family history and IEI remains a subject of debate, with some studies reporting a connection and others finding no association (35–37). In this research, no statistically significant correlation was found between family history and molecular diagnosis of IEI. Nevertheless, a higher proportion of cases with a family history of IEI were observed among those who had a genetic diagnosis. This observation suggests that family history may be a useful factor to consider when deciding whether to perform genetic testing for IEI and for supporting the diagnostic process, potentially providing valuable information for a more comprehensive understanding of the patient’s condition.
In this study, we also described patients with IEI, who were diagnosed incidentally. Owing to CES, which was used for genetic diagnosis for various diseases in our institute, IEI could be diagnosed in previously unrecognized patients. This shows the benefits of using CES for various disease groups as well as IEI.
In summary, this study showed the clinical utility of CES for the diagnosis of IEI. In addition to pediatric patients suspected of primary immunodeficiency, early CES should be considered in adult patients with suspected symptoms that may be caused by immunodeficiency or immune dysregulation. Early, accurate diagnosis made by CES results in the reduction of labor and cost for other diagnostic methods and can improve patient’s prognosis and quality of life by reducing time before diagnosis is made.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: SCV003837562 - SCV003837580 (ClinVar).
Ethics statement
The studies involving human participants were reviewed and approved by Institutional Review Board of Yonsei University Health System. Written informed consent from the participants’ legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
Author contributions
Conceptualization: SK, SS, and J-MK. Data curation: SK, YC, JO, DW, SS, and J-MK. Funding acquisition: SS and J-MK. Investigation: SK, YC, SH, JO, SS, J-MK, JA, S-TL, and JC. Methodology: SK, JO, DW, SS, and J-MK. Project administration: SS, J-MK, JA, S-TL, and JC. Supervision: SS and J-MK. Writing—original draft: SK. Writing—review and editing: SK, YC, SH, JO, DW, SS, J-MK, JA, S-TL, and JC. All authors contributed to the final article and approved the submitted version.
Funding
This research was supported by a grant from the National Research Foundation of Korea (2021R1I1A1A01045980) and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2019R1A6A1A03032869).
Conflict of interest
Authors S-TL and JC were employed by company Dxome.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1178582/full#supplementary-material
References
1. Kobrynski L, Powell RW, Bowen S. Prevalence and morbidity of primary immunodeficiency diseases, united states 2001-2007. J Clin Immunol (2014) 34(8):954–61. doi: 10.1007/s10875-014-0102-8
2. Currier R, Puck JM. SCID newborn screening: what we've learned. J Allergy Clin Immunol (2021) 147(2):417–26. doi: 10.1016/j.jaci.2020.10.020
3. Subbarayan A, Colarusso G, Hughes SM, Gennery AR, Slatter M, Cant AJ, et al. Clinical features that identify children with primary immunodeficiency diseases. Pediatrics (2011) 127(5):810–6. doi: 10.1542/peds.2010-3680
4. Bousfiha A, Moundir A, Tangye SG, Picard C, Jeddane L, Al-Herz W, et al. The 2022 update of IUIS phenotypical classification for human inborn errors of immunity. J Clin Immunol (2022) 42(7):1508–20. doi: 10.1007/s10875-022-01352-z
5. Rojas-Restrepo J, Caballero-Oteyza A, Huebscher K, Haberstroh H, Fliegauf M, Keller B, et al. Establishing the molecular diagnoses in a cohort of 291 patients with predominantly antibody deficiency by targeted next-generation sequencing: experience from a monocentric study. Front Immunol (2021) 12:786516. doi: 10.3389/fimmu.2021.786516
6. Al-Tamemi S, Al-Zadjali S, Bruwer Z, Naseem SU, Al-Siyabi N, ALRawahi M, et al. Genetic causes, clinical features, and survival of underlying inborn errors of immunity in omani patients: a single-center study. J Clin Immunol (2023) 43(2):452–65. doi: 10.1007/s10875-022-01394-3
7. Moundir A, Ouair H, Benhsaien I, Jeddane L, Rada N, Amenzoui N, et al. Genetic diagnosis of inborn errors of immunity in an emerging country: a retrospective study of 216 Moroccan patients. J Clin Immunol (2023) 43(2):485–94. doi: 10.1007/s10875-022-01398-z
8. Engelbrecht C, Urban M, Schoeman M, Paarwater B, van Coller A, Abraham DR, et al. Clinical utility of whole exome sequencing and targeted panels for the identification of inborn errors of immunity in a resource-constrained setting. Front Immunol (2021) 12:665621. doi: 10.3389/fimmu.2021.665621
9. Rim JH, Kim SH, Hwang IS, Kwon SS, Kim J, Kim HW, et al. Efficient strategy for the molecular diagnosis of intractable early-onset epilepsy using targeted gene sequencing. BMC Med Genomics (2018) 11(1):6. doi: 10.1186/s12920-018-0320-7
10. Kim SH, Kim B, Lee JS, Kim HD, Choi JR, Lee ST, et al. Proband-only clinical exome sequencing for neurodevelopmental disabilities. Pediatr Neurol (2019) 99:47–54. doi: 10.1016/j.pediatrneurol.2019.02.017
11. Kim H, Shim Y, Lee TG, Won D, Choi JR, Shin S, et al. Copy-number analysis by base-level normalization: an intuitive visualization tool for evaluating copy number variations. Clin Genet (2023) 103(1):35–44. doi: 10.1111/cge.14236
12. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
13. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer (2007) 48(2):124–31. doi: 10.1002/pbc.21039
14. Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature (2007) 448(7157):1058–62. doi: 10.1038/nature06096
15. Wolach O, Kuijpers T, Ben-Ari J, Gavrieli R, Feinstein-Goren N, Alders M, et al. Variable clinical expressivity of STAT3 mutation in hyperimmunoglobulin e syndrome: genetic and clinical studies of six patients. J Clin Immunol (2014) 34(2):163–70. doi: 10.1007/s10875-014-9988-4
16. Jägle S, Heeg M, Grün S, Rensing-Ehl A, Maccari ME, Klemann C, et al. Distinct molecular response patterns of activating STAT3 mutations associate with penetrance of lymphoproliferation and autoimmunity. Clin Immunol (2020) 210:108316. doi: 10.1016/j.clim.2019.108316
17. Koh JY, Lee SB, Kim B, Park Y, Choi JR, Son S, et al. Impact of maternal engrafted cytomegalovirus-specific CD8+ T cells in a patient with severe combined immunodeficiency. Clin Transl Immunol (2021) 10(4):e1272. doi: 10.1002/cti2.1272
18. Yanagi T, Mizuochi T, Takaki Y, Eda K, Mitsuyama K, Ishimura M, et al. Novel exonic mutation inducing aberrant splicing in the IL10RA gene and resulting in infantile-onset inflammatory bowel disease: a case report. BMC Gastroenterol (2016) 16:10. doi: 10.1186/s12876-016-0424-5
19. Ye Z, Zhou Y, Huang Y, Wang Y, Lu J, Tang Z, et al. Phenotype and management of infantile-onset inflammatory bowel disease: experience from a tertiary care center in China. Inflamm Bowel Dis (2017) 23(12):2154–64. doi: 10.1097/MIB.0000000000001269
20. Rawat A, Sharma M, Vignesh P, Jindal AK, Suri D, Das J, et al. Utility of targeted next generation sequencing for inborn errors of immunity at a tertiary care centre in north India. Sci Rep (2022) 12(1):10416. doi: 10.1038/s41598-022-14522-1
21. Rae W, Ward D, Mattocks C, Pengelly RJ, Eren E, Patel SV, et al. Clinical efficacy of a next-generation sequencing gene panel for primary immunodeficiency diagnostics. Clin Genet (2018) 93(3):647–55. doi: 10.1111/cge.13163
22. Rudilla F, Franco-Jarava C, Martínez-Gallo M, Garcia-Prat M, Martín-Nalda A, Rivière J, et al. Expanding the clinical and genetic spectra of primary immunodeficiency-related disorders with clinical exome sequencing: expected and unexpected findings. Front Immunol (2019) 10:2325. doi: 10.3389/fimmu.2019.02325
23. Zhu T, Gong X, Bei F, Ma L, Sun J, Wang J, et al. Primary immunodeficiency-related genes in neonatal intensive care unit patients with various genetic immune abnormalities: a multicentre study in China. Clin Transl Immunol (2021) 10(3):e1266. doi: 10.1002/cti2.1266
24. Erman B, Çipe F. Genetic screening of the patients with primary immunodeficiency by whole-exome sequencing. Pediatr Allergy Immunol Pulmonol (2020) 33(1):19–24. doi: 10.1089/ped.2019.1097
25. Thaventhiran JED, Lango Allen H, Burren OS, Rae W, Greene D, Staples E, et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature (2020) 583(7814):90–5. doi: 10.1038/s41586-020-2265-1
26. Lachmann HJ. Periodic fever syndromes. Best Pract Res Clin Rheumatol (2017) 31(4):596–609. doi: 10.1016/j.berh.2017.12.001
27. Wada T, Candotti F. Somatic mosaicism in primary immune deficiencies. Curr Opin Allergy Clin Immunol (2008) 8(6):510–4. doi: 10.1097/ACI.0b013e328314b651
28. Mensa-Vilaró A, Bravo García-Morato M, de la Calle-Martin O, Franco-Jarava C, Martínez-Saavedra MT, González-Granado LI, et al. Unexpected relevant role of gene mosaicism in patients with primary immunodeficiency diseases. J Allergy Clin Immunol (2019) 143(1):359–68. doi: 10.1016/j.jaci.2018.09.009
29. Holzelova E, Vonarbourg C, Stolzenberg MC, Arkwright PD, Selz F, Prieur AM, et al. Autoimmune lymphoproliferative syndrome with somatic fas mutations. N Engl J Med (2004) 351(14):1409–18. doi: 10.1056/NEJMoa040036
30. Aluri J, Bach A, Kaviany S, Chiquetto Paracatu L, Kitcharoensakkul M, Walkiewicz MA, et al. Immunodeficiency and bone marrow failure with mosaic and germline TLR8 gain of function. Blood (2021) 137(18):2450–62. doi: 10.1182/blood.2020009620
31. Donadieu J, Lamant M, Fieschi C, de Fontbrune FS, Caye A, Ouachee M, et al. Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica (2018) 103(8):1278–87. doi: 10.3324/haematol.2017.181909
32. Nakajima H, Kunimoto H. TET2 as an epigenetic master regulator for normal and malignant hematopoiesis. Cancer Sci (2014) 105(9):1093–9. doi: 10.1111/cas.12484
33. Savola P, Martelius T, Kankainen M, Huuhtanen J, Lundgren S, Koski Y, et al. Somatic mutations and T-cell clonality in patients with immunodeficiency. Haematologica (2020) 105(12):2757–68. doi: 10.3324/haematol.2019.220889
34. Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med (2020) 383(27):2628–38. doi: 10.1056/NEJMoa2026834
35. Eldeniz FC, Gul Y, Yorulmaz A, Guner SN, Keles S, Reisli I. Evaluation of the 10 warning signs in primary and secondary immunodeficient patients. Front Immunol (2022) 13:900055. doi: 10.3389/fimmu.2022.900055
36. Reda SM, El-Ghoneimy DH, Afifi HM. Clinical predictors of primary immunodeficiency diseases in children. Allergy Asthma Immunol Res (2013) 5(2):88–95. doi: 10.4168/aair.2013.5.2.88
Keywords: inborn errors of immunity, next generation sequencing, clinical exome sequencing, genetic diagnosis, somatic variant, incidental finding
Citation: Kwon SS, Cho YK, Hahn S, Oh J, Won D, Shin S, Kang J-M, Ahn JG, Lee S-T and Choi JR (2023) Genetic diagnosis of inborn errors of immunity using clinical exome sequencing. Front. Immunol. 14:1178582. doi: 10.3389/fimmu.2023.1178582
Received: 03 March 2023; Accepted: 15 May 2023;
Published: 31 May 2023.
Edited by:
Attila Kumanovics, Mayo Clinic, United StatesReviewed by:
Saul Oswaldo Lugo Reyes, National Institute of Pediatrics, MexicoAnn M Moyer, Mayo Clinic, United States
Copyright © 2023 Kwon, Cho, Hahn, Oh, Won, Shin, Kang, Ahn, Lee and Choi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saeam Shin, c2FlYW0wMzA0QHl1aHMuYWM=; Ji-Man Kang, dW1pODdjQHl1aHMuYWM=