
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 22 June 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1177467
This article is part of the Research Topic Molecular Drivers of Immunothrombosis View all 15 articles
 Kateryna Sopova1,2,3,4,5†
Kateryna Sopova1,2,3,4,5† Simon Tual-Chalot6†Matthias Mueller-Hennessen4,7
Simon Tual-Chalot6†Matthias Mueller-Hennessen4,7 Nikolaos I. Vlachogiannis6
Nikolaos I. Vlachogiannis6 Georgios Georgiopoulos8Moritz Biener4,7
Georgios Georgiopoulos8Moritz Biener4,7 Marco Sachse5
Marco Sachse5 Andrey Turchinovich5Maria Polycarpou-Schwarz5Luke Spray3,6
Andrey Turchinovich5Maria Polycarpou-Schwarz5Luke Spray3,6 Eleni Maneta8Karim Bennaceur6
Eleni Maneta8Karim Bennaceur6 Ashfaq Mohammad3,6
Ashfaq Mohammad3,6 Gavin David Richardson6Aikaterini Gatsiou6Harald F. Langer2,4
Gavin David Richardson6Aikaterini Gatsiou6Harald F. Langer2,4 Norbert Frey4,7
Norbert Frey4,7 Kimon Stamatelopoulos6,8
Kimon Stamatelopoulos6,8 Joerg Heineke4,9Daniel Duerschmied2,4Evangelos Giannitsis7
Joerg Heineke4,9Daniel Duerschmied2,4Evangelos Giannitsis7 Ioakim Spyridopoulos1,3*‡
Ioakim Spyridopoulos1,3*‡ Konstantinos Stellos2,4,5,6,7,9*‡
Konstantinos Stellos2,4,5,6,7,9*‡Background and aims: Preclinical data suggest that activation of the adaptive immune system is critical for myocardial repair processes in acute myocardial infarction. The aim of the present study was to determine the clinical value of baseline effector T cell chemokine IP-10 blood levels in the acute phase of ST-segment elevation myocardial infarction (STEMI) for the prediction of the left ventricular function changes and cardiovascular outcomes after STEMI.
Methods: Serum IP-10 levels were retrospectively quantified in two independent cohorts of STEMI patients undergoing primary percutaneous coronary intervention.
Results: We report a biphasic response of the effector T cell trafficking chemokine IP-10 characterized by an initial increase of its serum levels in the acute phase of STEMI followed by a rapid reduction at 90min post reperfusion. Patients at the highest IP-10 tertile presented also with more CD4 effector memory T cells (CD4 TEM cells), but not other T cell subtypes, in blood. In the Newcastle cohort (n=47), patients in the highest IP-10 tertile or CD4 TEM cells at admission exhibited an improved cardiac systolic function 12 weeks after STEMI compared to patients in the lowest IP-10 tertile. In the Heidelberg cohort (n=331), STEMI patients were followed for a median of 540 days for major adverse cardiovascular events (MACE). Patients presenting with higher serum IP-10 levels at admission had a lower risk for MACE after adjustment for traditional risk factors, CRP and high-sensitivity troponin-T levels (highest vs. rest quarters: HR [95% CI]=0.420 [0.218-0.808]).
Conclusion: Increased serum levels of IP-10 in the acute phase of STEMI predict a better recovery in cardiac systolic function and less adverse events in patients after STEMI.
Coronary artery disease (CAD) is the single largest cause of mortality worldwide, directly accountable for 9.4 million of deaths every year (1). Acute ST-segment elevation myocardial infarction (STEMI) is one of the most severe manifestations of CAD which may lead to left ventricular systolic dysfunction and subsequent heart failure. Although the management of STEMI has significantly advanced in recent years, 6-month mortality remains elevated at 13%, while a third of surviving patients develop heart failure over the following 5-8 years after MI (2–4). The persistence of left ventricular (LV) systolic dysfunction a few months after STEMI is a strong predictor of morbidity and mortality among STEMI patients (5, 6). Therefore, there is an urgent need to identify early predictors of LV function recovery and of cardiovascular outcomes to identify the residual risk in patients with STEMI and to correctly risk stratify these patients in order to offer a tailored individualised approach of patient management and disease treatment.
While early primary percutaneous intervention can limit myocardial infarct size in patients with STEMI, reperfusion triggers an intense inflammatory response, characterised by mobilisation of leukocytes in the blood followed by an abundant leukocyte infiltration in the infarcted region (7). Although inflammation is a determinant of heart function after STEMI in experimental acute myocardial infarction (AMI) models, the contribution of adaptive immunity in human cardiac repair remains poorly understood. We have previously reported that circulating levels of total T lymphocytes in the acute phase of STEMI predict long-term survival in these patients (8). However, the underlying mechanisms remain unknown.
Preclinical studies have reported distinct roles of the two main subsets of T lymphocytes, CD4+ and CD8+, in acute ischemia. CD4+ T cells have been shown to exert cardioprotective effects in a preclinical model of AMI by acquiring a T-regulatory (Treg) phenotype and suppressing the local inflammatory response (9, 10). On the other hand, the role of CD8+ T cells post-myocardial infarction remains unclear. From one side, several studies suggested that the presence of CD8+ T-cells in the heart may lead to myocardial necrosis, cardiac dysfunction and poor survival post-MI (11–13). However, another study reported that CD8+ T cells may play both a beneficial and a detrimental role in cardiac remodeling, depending on the stage of left ventricular remodeling. In specific, the latter study reported that CD8+ T cells may initiate the removal of necrotic tissue and decrease the incidence of cardiac rupture in a murine model of MI after permanent ligation of the left anterior descending coronary artery (14). At a later stage, CD8+ T cells induce cytotoxic effects on healthy cardiomyocytes and enhance neutrophil and macrophage-mediated inflammation leading to left ventricle dilation and reduced cardiac function (14). The CD8+ T cell responses may be beneficial in the acute phase of AMI in the regulation of scar formation but may be maladaptive in the long term. Understanding the temporal and spatial regulation of these immune cells is of utmost importance for the identification of residual risk and risk stratification of patients with STEMI and the development of novel immunomodulatory therapeutic strategies against LV remodelling post-MI.
Interferon-γ-inducible protein-10 (IP-10, also known as CXCL10) is a chemokine secreted by innate immune cells (monocytes, neutrophils, natural killer (NK), and NKT-like cells) and sentinel cells of the innate immune system, such as the endothelial cells and fibroblasts, which are dedicated to respond to cytokines and damage-associated molecular pattern molecules emitted upon tissue injury (15, 16). IP-10 elicits its biological effects by binding to its receptor CXCR3 and is integral for recruiting activated (effector) T cells to the sites of tissue inflammation (15). IP-10 levels are known to be elevated in animal models of experimental myocardial infarction and in patients with coronary heart disease (17–19). Preclinical studies suggest that IP-10 is integral for CXCR3-positive immune cell recruitment to the ischemic myocardial tissue and regulation of reparative response following experimental AMI in mice (19, 20). However, its translational value in myocardial repair processes in patients with STEMI remains unknown. Herein we tested the hypothesis that serum IP-10 levels on admission may predict recovery of left ventricular systolic function and future cardiovascular outcomes in patients after STEMI.
The Figure 1 introduces in a flow chart the two patient cohorts and the methodology applied in this study.
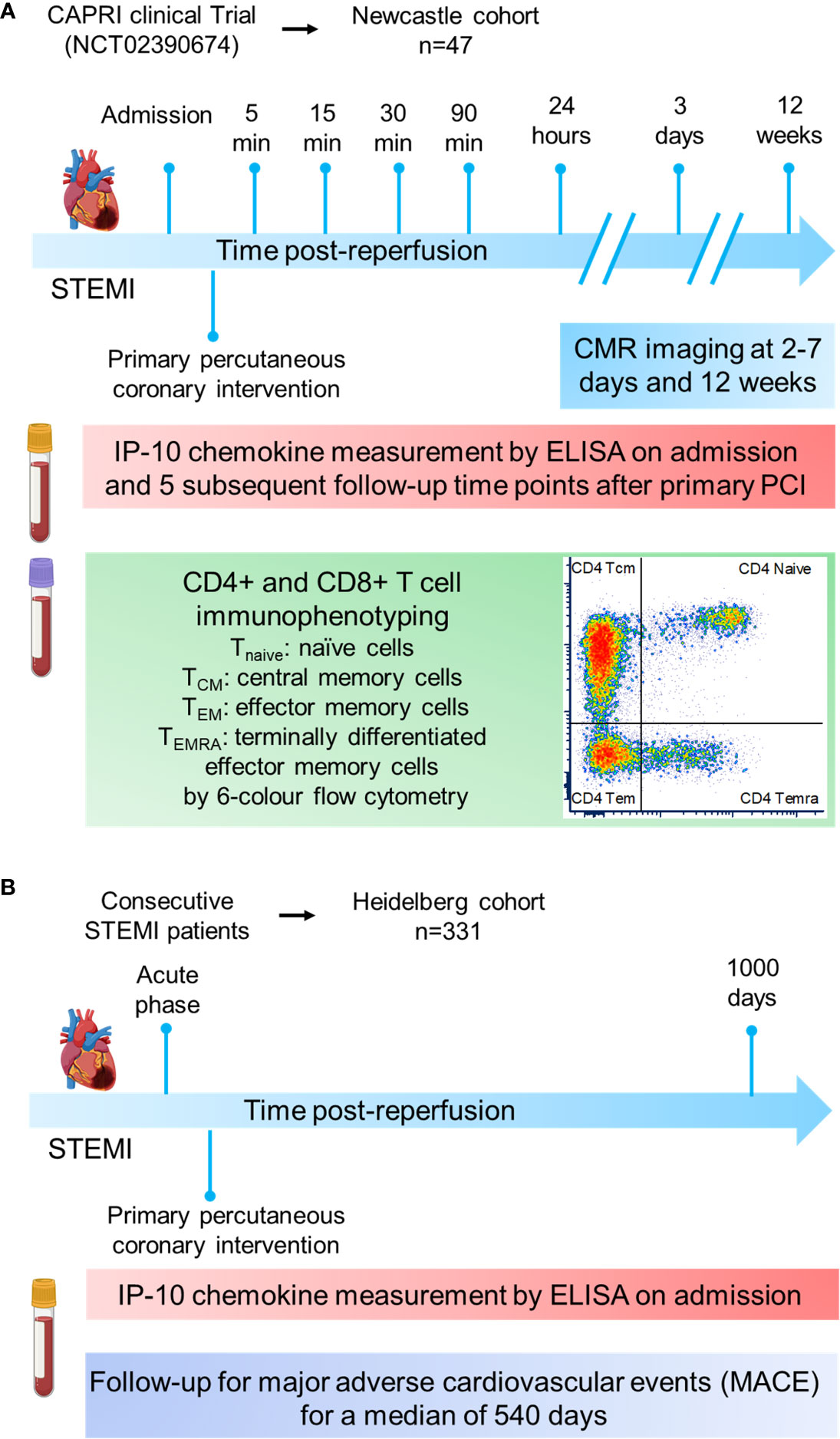
Figure 1 Cohort study design. (A) The Newcastle cohort is derived from the CAPRI clinical trial (NCT02390674). Blood from 47 STEMI patients was collected at admission and at sequential time-points post-reperfusion (5, 15, 30, 90min and 24h) to measure IP-10 by ELISA and proceed to CD4+ or CD8+ immunophenotyping by flow cytometry. Cardiac MRI scans were obtained 3 days as well as 12 weeks post‐myocardial infarction. (B) The Heidelberg cohort constited of 331 STEMI patients. Blood was collected at admission to measure IP-10 by ELISA. Patients were followed up for major adverse cardiovascular events for a median of 540 days.
The first cohort consisted of 47 out of 52 patients with available serum probes from the double-blind, randomised controlled trial (Cyclosporin to Reduce Reperfusion Injury in Primary PCI Clinical Trial; “CAPRI”, NCT02390674) (demographics in Table 1, named as Newcastle cohort) (21, 22). All participants had to present with STEMI and undergo primary percutaneous coronary intervention (primary PCI), be at least 18 years of age, and present within 6 hours of the chest pain onset. The culprit coronary artery had to be a major coronary artery with a diameter of at least 3 mm and had to be proximally occluded at the time of admission identified by coronary angiography. Exclusion criteria for this trial were patients presenting with immunological disorders, cardiogenic shock, unconscious patients, evidence of coronary collaterals to the infarct area, open culprit coronary artery at the time of angiography, previous myocardial infarction or thrombolytic therapy, sensitivity to cyclosporin or egg, peanut or soya-bean proteins, treatment with any compound which may modify blood concentration of cyclosporin, known renal or liver insufficiency, uncontrolled hypertension (>180/110 mmHg), female patients either currently pregnant or of child-bearing age with no current contraceptive use (verbal diagnosis), use of investigational study drug within the previous 30 days, life expectancy of less than one year due to non-cardiac illness, or patients with contraindication to cardiac MRI. Following angiography, participants were randomised in a 1:1 ratio to either cyclosporin (n=26) or control (saline, n=26) using a blocked allocation (permuted random blocks of variable length) system immediately before reperfusion implemented using Newcastle Clinical Trials Unit’s online randomisation service. Cyclosporin (Sandimmun®, Novartis) was given as an intravenous infusion dissolved in saline (maximum concentration 2.5mg per millilitre) of 2.5mg per kilogram of body weight (maximum total amount 250 mg). After successful infusion, the coronary wire was advanced and the culprit lesion crossed, followed by revascularisation. Patients underwent primary PCI according to standard guidelines. All patients received a loading dose of two antiplatelet drugs and heparin before PCI. Use of thrombus aspiration and/or glycoprotein IIb/IIIa inhibition were used according to the operator’s discretion. Zotarolimus- or everolimus-eluting stents were implanted.
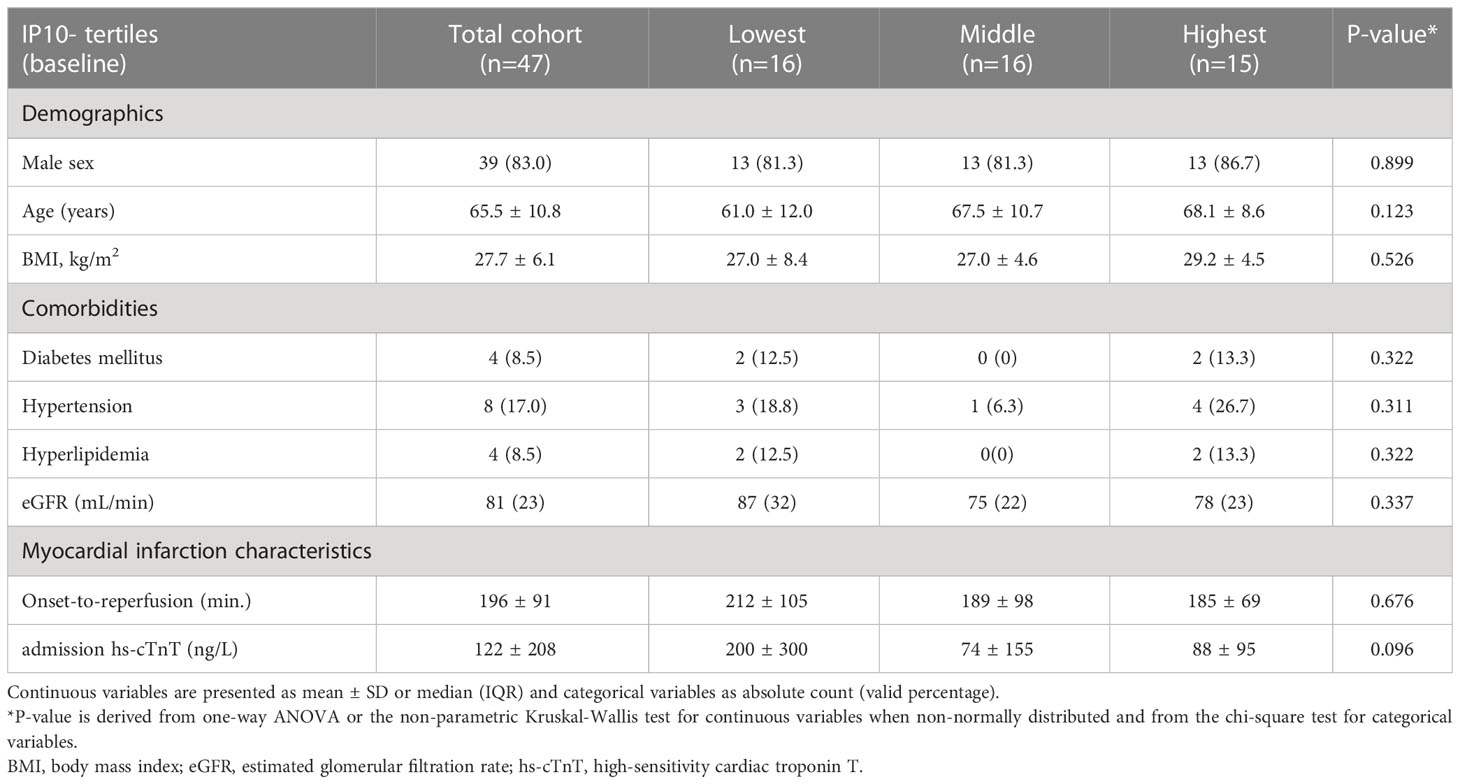
Table 1 Patient characteristics of the cardiac recovery post-STEMI (Newcastle) cohort according to baseline IP-10 tertiles.
Patients presenting to the emergency department of Heidelberg University Hospital, Germany with a working diagnosis of acute coronary syndrome (ACS) were consecutively recruited from the 9th June 2009 to the 10th May 2014 (23). Patients were retrospectively adjudicated, and analysis was restricted to a final diagnosis of STEMI with available follow-up for major adverse cardiovascular events (MACE) and available blood samples at admission, leaving 331 for measurement of serum IP-10 (demographics in Table 2, named as Heidelberg cohort). In brief, MI was diagnosed using high-sensitivity (hs) cardiac troponin T (cTnT) in accordance with the universal MI definition (24). Only patients with complete follow-up information were screened for eligibility. Follow-up was performed by telephone contact and/or questionnaires sent by email or land mail. Information on mortality events was further obtained by the local residents’ registry. Follow-up was executed at different time points since this is a registry with repetitive adjudication of diagnoses and events. All main analyses were performed for a maximum follow-up time of 1000 days (median 540 days) and vital status was censored at this time point. This time point was selected because it provided the optimal balance between the longest possible follow-up and missing information. Specifically, medical records were reviewed and evaluated regarding the incidence of MACE, which were defined as CVD death, AMI and performance of coronary revascularisation procedure. CV death was defined as death resulting from MI, sudden cardiac death, death due to heart failure, stroke and other CVD causes (25). Coronary revascularisation procedures included percutaneous coronary intervention or coronary artery bypass grafting. The research protocol did not interfere with the management of the study patients. Treatment decisions were made by the attending cardiologists.
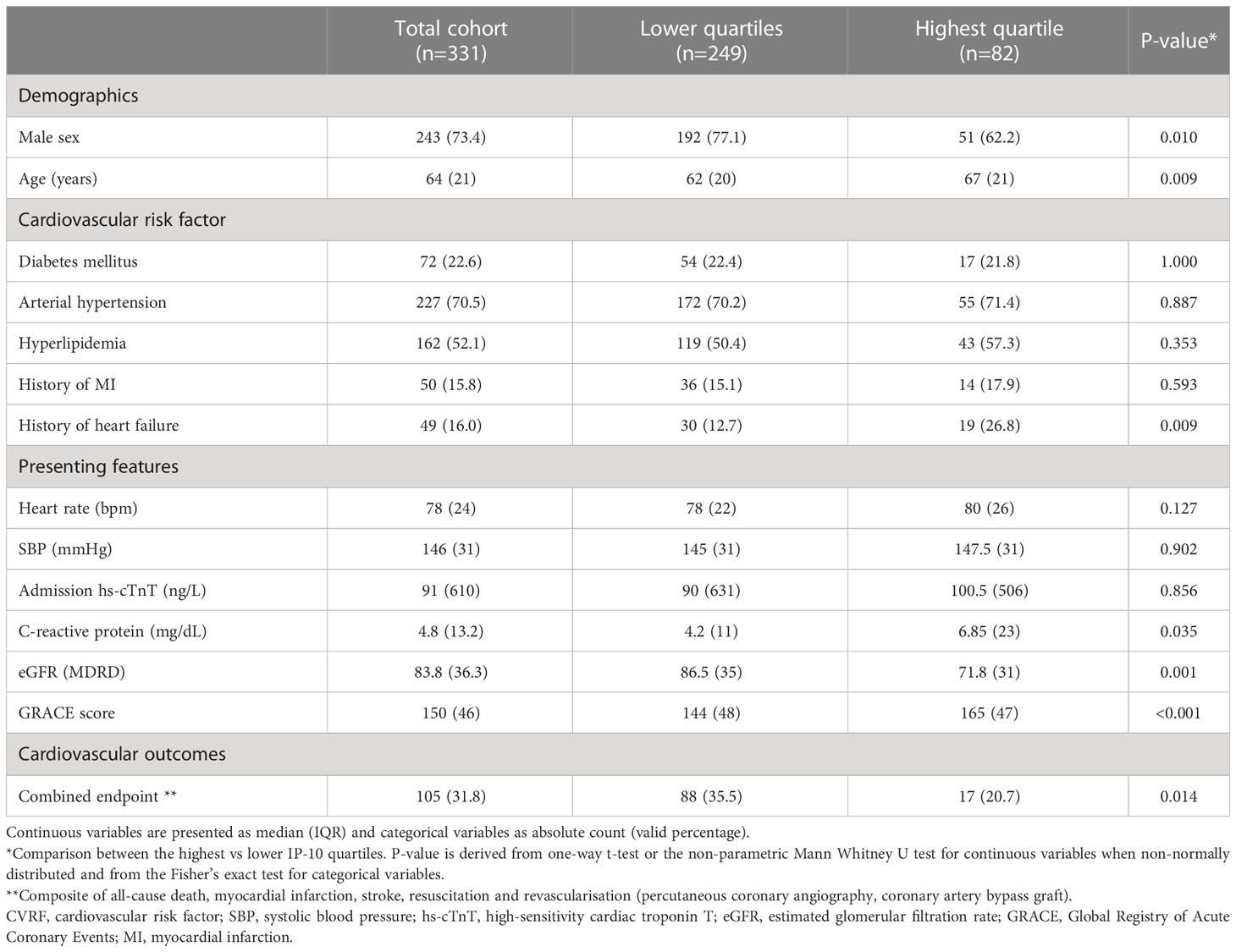
Table 2 Descriptive characteristics of the prospective STEMI cohort for MACE (Heidelberg cohort) by highest vs lower quarters of baseline IP-10 levels.
In the Newcastle cohort, CMR imaging was obtained with a Siemens Avanto 1.5 Telsa MRI scanner, using a phased array body coil combined with a spine coil at 2-7 days post-reperfusion, and repeat CMR at 12 ± 2 weeks was obtained to assess myocardial function improvement by calculating change in LV ejection fraction (ΔLVEF) (21). All images were obtained during breath-holding. Localiser images were acquired as well as axial black blood HASTE images to define the anatomy. Cine images of the heart in 2, 3 and 4 chamber views were obtained using a steady-state free precession pulse (SSFP) sequence (repetition time [TR]: set according to heart rate, image matrix 144x192, echo time (TE): 1.19ms, flip angle: 80°). T2 weighted STIR (short inversion time [TI] inversion recovery) images were then obtained in the same projections, using a black-blood segmented turbo spin-echo technique (TR according to heart rate, TE 47ms, flip angle 180°, TI 140ms, image matrix 208x256). Further sequential end-diastolic STIR images were acquired along the short-axis of the heart, covering the full extent of the left ventricle in parallel slices (each 8mm with 0mm gap). Corresponding short-axis SSFP cine images were then obtained to allow quantification of chamber volumes and function. Intravenous Gadobutrol contrast (Gadovist, Bayer Schering Pharma AG, Berlin, Germany) was administered at a dose of 0.1mmol/kg. After 10 minutes short-axis end-diastolic late gadolinium enhancement images (in corresponding locations to cine and STIR images) were obtained using an inversion recovery (IR) segmented gradient echo sequence (TR: according to heart rate, TE: 3.41ms, flip angle: 25°, image matrix: 196x256). The inversion time (TI) for late gadolinium enhancement imaging was selected in order to null normal myocardium (giving it a dark appearance), and adjusted throughout acquisition (increased approximately every second slice) to maintain nulling. As previously described, all analyses were performed using validated cardiac MRI analysis software (cvi42, Circle Cardiovascular Imaging Inc., Calgary, Canada) (8). LV volumes and mass were determined using the short axis SSFP cine images, following the determination of the longitudinal extent of the chamber by cross-referencing with the 4 and 2 chamber images (8). Epicardial and endocardial borders were traced automatically on each end-systolic and end-diastolic short-axis cine frame with manual correction where necessary, allowing automated calculation of left ventricular mass, dimensions and ejection fraction (LVEF), as previously described and validated (21, 22).
A 6-colour assay was used to measure the expression of CD3, CD4, CD8, CD45RA, CCR7 and CX3CR1 in blood samples as previously described and validated (26). All flow cytometric measurements were carried out on fresh blood within hours of collection. 50μL fresh blood aliquots from every time point were added to their corresponding labelled 5mL Falcon tubes. A cocktail of antibodies (Mouse Anti-Human CD3, 555333, BD Biosciences; Mouse Anti-Human CD4, 560768, BD Biosciences; Mouse Anti-Human CD8, 555366, BD Biosciences; anti-human CD197 (CCR7), 353208, BioLegend; CD45RA (L48) PE- Cy7, 337186, BD Biosciences; anti-human CX3CR1, 341610, BioLegend) were added to the sample. After a 20-minute incubation, 1mL of fresh lysis buffer was added to each mixture. Following another 20-minute incubation, the sample was washed and run through a BD FACSCanto-II Flow Cytometer (339473, BD Biosciences) using the BD FACSDiva acquisition software. Cells were gated on CD3+ T-lymphocytes and 20,000 events were recorded allowing for representative data. We gated T-lymphocytes (CD3+) first into CD4+ and CD8+ T cells, then into four subsets of each by expression of CCR7, the chemokine receptor for CCL19/21, and CD45RA, as previously described (27). Four sub-populations of CD4+ and CD8+ T cells were investigated: naïve (TNaive), central memory (TCM), effector memory (TEM) and terminally differentiated effector memory cells (TEMRA). No significant amount of non-viable cells (<1%) was detected. Percentages and absolute count of T lymphocyte subtypes determined in peripheral blood on admission, at 5, 15, 30 and 90 minutes and 24 hours post-reperfusion are reported.
Levels of IP-10 were measured in serum samples using a commercial enzyme-linked immunosorbent assay (ELISA) (R&D Systems, Minneapolis, MN, USA), which is commonly considered the gold-standard assay for analyte quantification in clinical research (28). In the Newcastle cohort, IP-10 concentration was measured retrospectively on admission and at various sequential time-points post-reperfusion (5, 15, 30, 90min and 24h). In the Heidelberg cohort, IP-10 levels were retrospectively measured at admission. The intra- and inter-assay coefficients of variance of the ELISA measurements were reported to be less than 3.6% and 6.7% respectively, and the minimum detectable concentration of human IP-10 was 4.46 pg/mL (R&D Systems). All measurements were performed by experienced personnel who were blinded to patients’ characteristics.
Favourable ethical opinions were received for the CAPRI study (National Research Ethics Committee North-East – Newcastle and North Tyneside 2; REC reference: 14/NE/1070), and the Heidelberg cohort (21, 23). CAPRI received a clinical trial authorisation from the Medicines and Healthcare products Regulatory Agency (MHRA). All STEMI/PPCI patients analysed retrospectively had provided written informed consent to their clinical data included in a database of PPCI procedures for research. All studies used complied with the principles laid out in the declaration of Helsinki, and written informed consent was obtained from all participants.
Data are presented as mean ± standard deviation or median (interquartile range) for continuous variables and count (percentage) for nominal variables. Normal distribution of all continuous variables was graphically assessed by distributional plots, including histograms, symmetry plots and normal-quantile plots, as well as by Kolmogorov-Smirnov and Shapiro-Wilk tests. Baseline characteristics were compared with analysis of variance or with the non-parametric Kruskal-Wallis test for continuous variables and with the Pearson’s chi-square test for nominal variables. Pair-wise comparisons (highest vs lower IP-10 quarters) of continuous variables were performed by Dunnett’s (parametric) or Dunn’s (non-parametric) multiple comparisons test, while chi square test was used to compare categorical variables between contrast groups. In the Newcastle cohort, the primary endpoint of our study was a change in LVEF at 12 weeks post-STEMI versus LVEF at 2-7 days (median: 3 days) post-STEMI (ΔLVEF). Linear regression was used to examine the association of IP-10 levels with ΔLVEF, as a cardiac repair marker, after adjustment for age, sex, body mass index (BMI), diabetes mellitus (DM), arterial hypertension, hyperlipidemia, estimated glomerular filtration rate (eGFR) and admission high sensitivity cardiac troponin T (hs-cTnT) as biologically plausible variables affecting cardiac repair.
In the Heidelberg cohort, the primary endpoint was a composite of all-cause death, AMI, stroke, resuscitation and revascularisation (percutaneous coronary angiography, coronary artery bypass graft). Nelson–Aalen curves were generated to depict the cumulative incidence of outcomes. Subjects were censored at the time of the first event. Log-rank test was used in the Nelson Aalen curves figure. Cox proportional-hazards models were used to examine the association between IP-10 levels and the primary outcome. Associations are presented as Hazard Ratio (HR) with 95% confidence intervals (CI). Statistical analysis was performed with SPSS v. 26 (IBM) and STATA v. 13 (StataCorp). All tests were two-tailed. We deemed statistical significance at P<0.05.
A priori calculations showed that a sample size of 47 subjects would be adequately powered at 0.85 level to detect a minimum of 3% increase in ΔLVEF post STEMI between patients at highest and lower tertiles of IP-10 by the non-parametric Mann Whitney test. Dispersion measures for ΔLVEF were calculated based on previously published method (29). Type I error was set at 0.05. Power calculation was performed with G Power 3.1.
The descriptive characteristics of the study population are reported in Table 1. IP-10 levels were determined during the acute phase of STEMI at admission before primary PCI-mediated reperfusion as well as at various sequential time points post-reperfusion (5, 15, 30, 90min and 24h). We observed that serum levels of IP-10 peaked at 15min after reperfusion, followed by an approximately 0.5-fold decrease at 90min post-reperfusion compared to admission levels (Figure 2A). IP-10 concentrations reached their lowest point after 24 hours (Figure 2A).
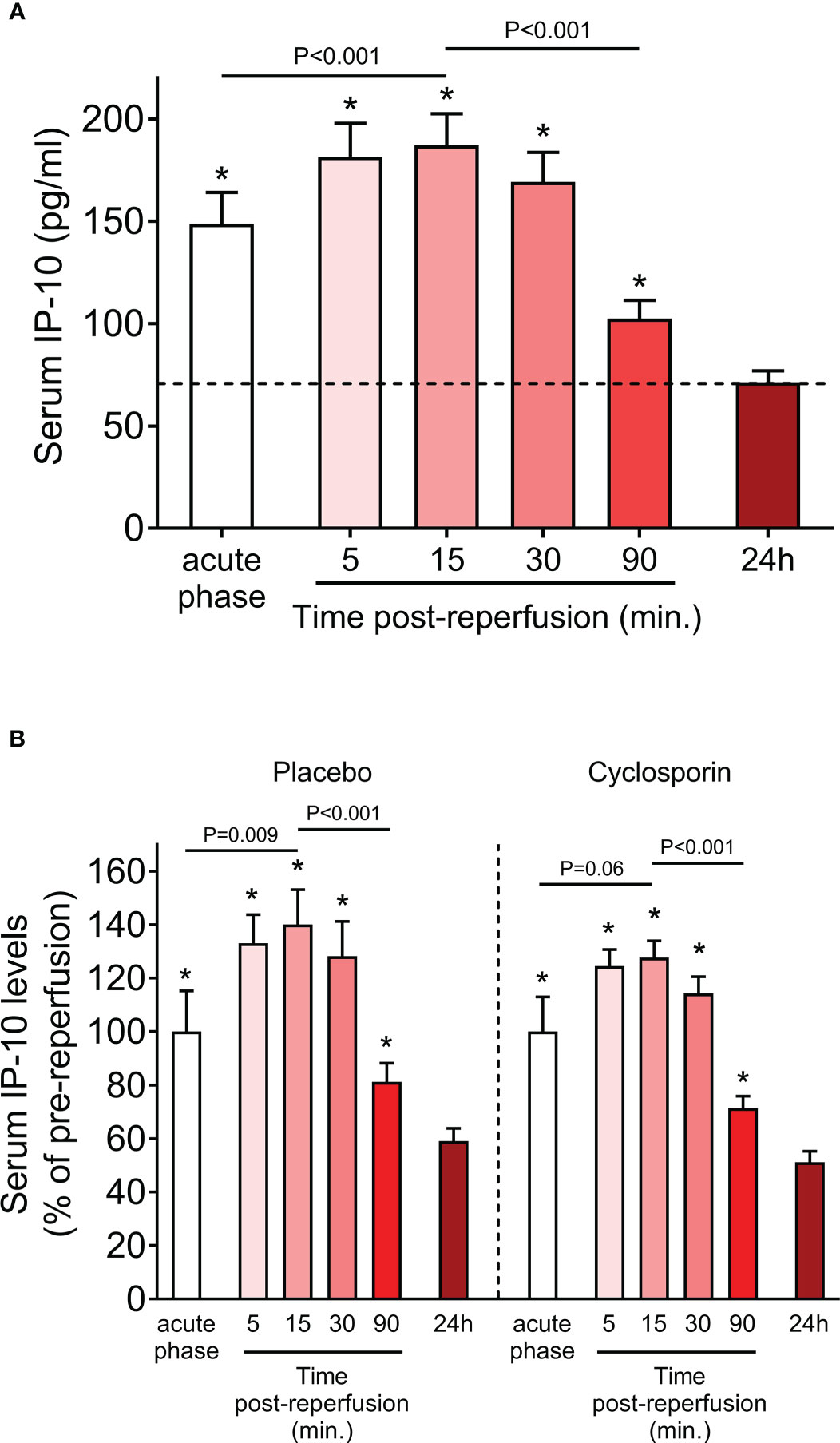
Figure 2 Serum IP-10 levels in STEMI patients. (A) Serum IP-10 kinetics at admission (acute phase) and at sequential time-points post-reperfusion (5, 15, 30, 90min and 24h) in 47 patients with ST-segment elevation myocardial infarction (STEMI). *P<0.001 for each pair-wise analysis compared to the 24h time-point. (Wilcoxon signed-rank test). (B) Serum IP-10 kinetics at admission (acute phase) and at sequential time-points post-reperfusion (5, 15, 30, 90min and 24h) in placebo (n=23) and cyclosporin-treated (n=24) patients with ST-segment elevation myocardial infarction (STEMI). *P<0.05 for each pair-wise analysis compared to the 24h time-point (Wilcoxon signed-rank test). Dunn’s multiple comparisons test between placebo and cyclosporin-treated patients for each time point are all non-significant.
The primary objective of the “CAPRI” trial was to determine whether administration of cyclosporine before primary percutaneous coronary intervention (PPCI) reduces the amount of damage to the heart relative to treatment with placebo. We, therefore, assessed whether cyclosporin treatment affected IP-10 levels. We found that IP-10 kinetics were remarkably similar in the cyclosporin and placebo group (Figure 2B) and pair-wise analysis at each time point showed that cyclosporin did not affect IP-10 levels at any studied time point. Therefore, we used the whole study cohort for subsequent analyses, and we adjusted for randomization treatment where applicable.
IP-10 is a major chemotactic factor for activated T-cells in inflamed tissues and is a homing chemokine for T cells that express CXCR3 (30). We first establish the kinetics of T cell subsets in fresh blood samples from STEMI patients, drawn at different time points. Using multicolour flow cytometry, we quantified the absolute count and the percentage of CD4+ and CD8+ subsets, including TNaive, TCM, TEM and TEMRA within the total CD4+ and CD8+ cells (Figure 3A). We observed a similar trend of the overall kinetics between CD4+ and CD8+ subsets. All cell subset counts were lower at 90 min compared with the admission levels, indicating they dropped during the first 90min post coronary revascularization (primary percutaneous coronary intervention). The 90 min drop observed in CCR7- effector memory cells compared to admission was more drastic than in CCR7+ subsets (CD4+ TEM: 48.8%, CD4+ TEMRA: 64.3%, CD8+ TEM: 59.7%, and CD8+ TEMRA: 62.3% vs CD4+ TNaive: 13.1%, CD4+ TCM: 18.9%, CD8+ TNaive: 22.7%, and CD8+ TCM: 28.4%) (Figure 3B). Similarly, CCR7- effector memory cells percentage of CD4+ or CD8+ T cells decreased at 90 min compared to admission, while the percentage of CCR7+ subsets increased slightly (Figure 3C). Furthermore, all counts of T cell subsets were all significantly higher at 24 hours compared to 90 min (Figure 3B).
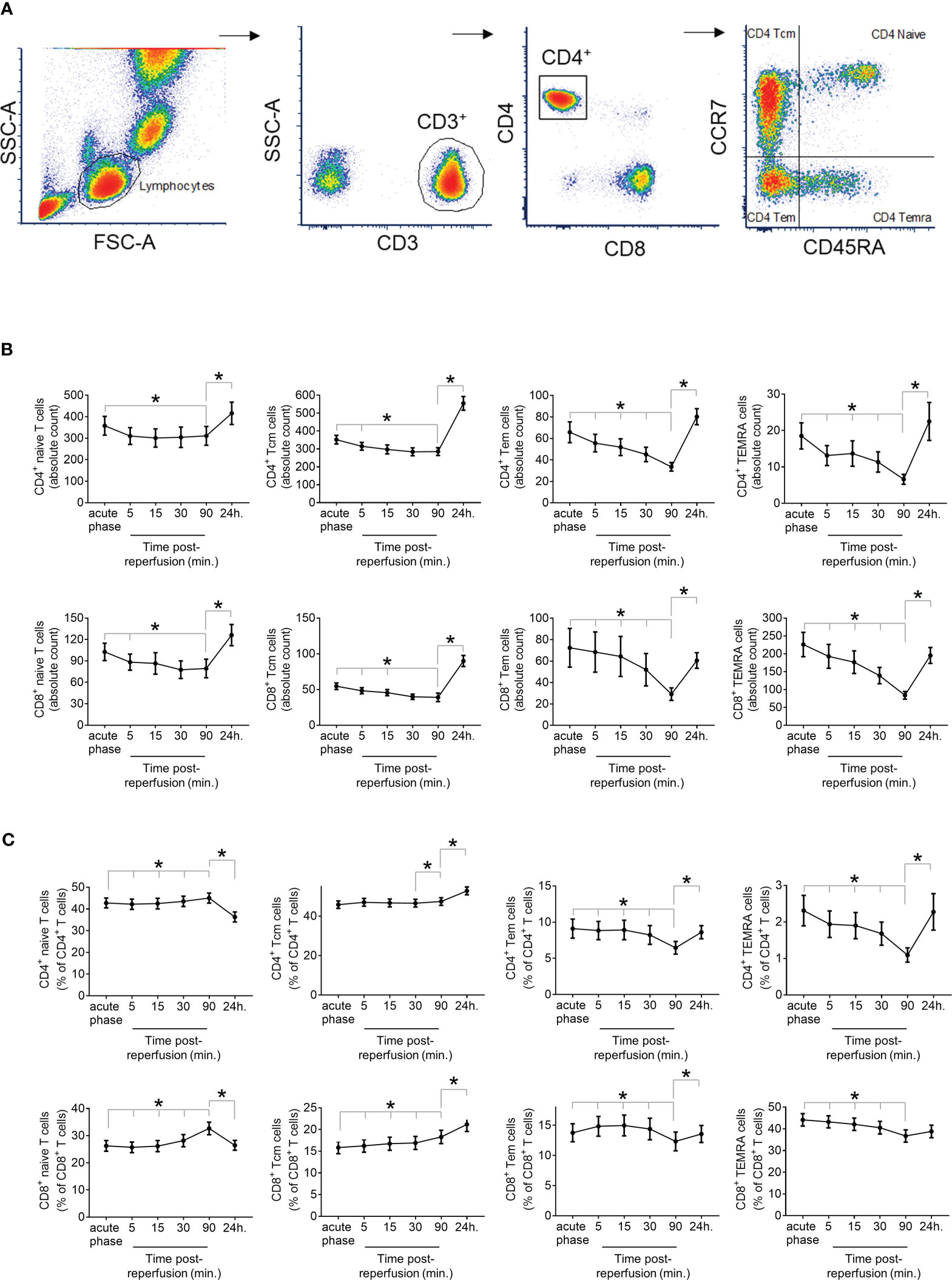
Figure 3 Dynamics of T cell response in STEMI Patients. (A) Immunophenotyping of the T-lymphocyte subtypes CD4+ or CD8+ naïve T cells, central memory T cells (Tcm), effector memory T cells (TEM), or effector memory T cells re-expressing CD45RA (TEMRA) was based on the expression of CD45RA and CCR7 within CD4+ and CD8+ subsets of CD3+ peripheral blood mononuclear cells determined by 6-colour flow cytometry. (B) Absolute count presented at cells/µl of correctly peripheral blood and (C) Percentage of lymphocyte subset at admission (acute phase) and at sequential time-points post-reperfusion (5, 15, 30, 90min and 24h) in 47 STEMI patients. *P<0.05 for time-point comparison by Wilcoxon signed-rank test.
In an attempt to provide a mechanistic explanation for the observed differences in the temporal T cell responses, we examined the potential differences in CD4+ or CD8+ T cell subtypes. Remarkably we observed that patients at the highest IP-10 tertile have a significant increase of CD4+ effector memory T cells (CD4+ TEM) within the CD4+ T cell compartment compared to the lowest tertile (Figure 4A). In contrast, there was no clear association between serum IP-10 levels and the percentage of CD8+ effector memory T cells (CD8+ TEM) within the CD8+ T cell compartment in blood. No association was observed between IP-10 serum levels and the other CD4+ or CD8+ T cell subtypes across all time points (data not shown).
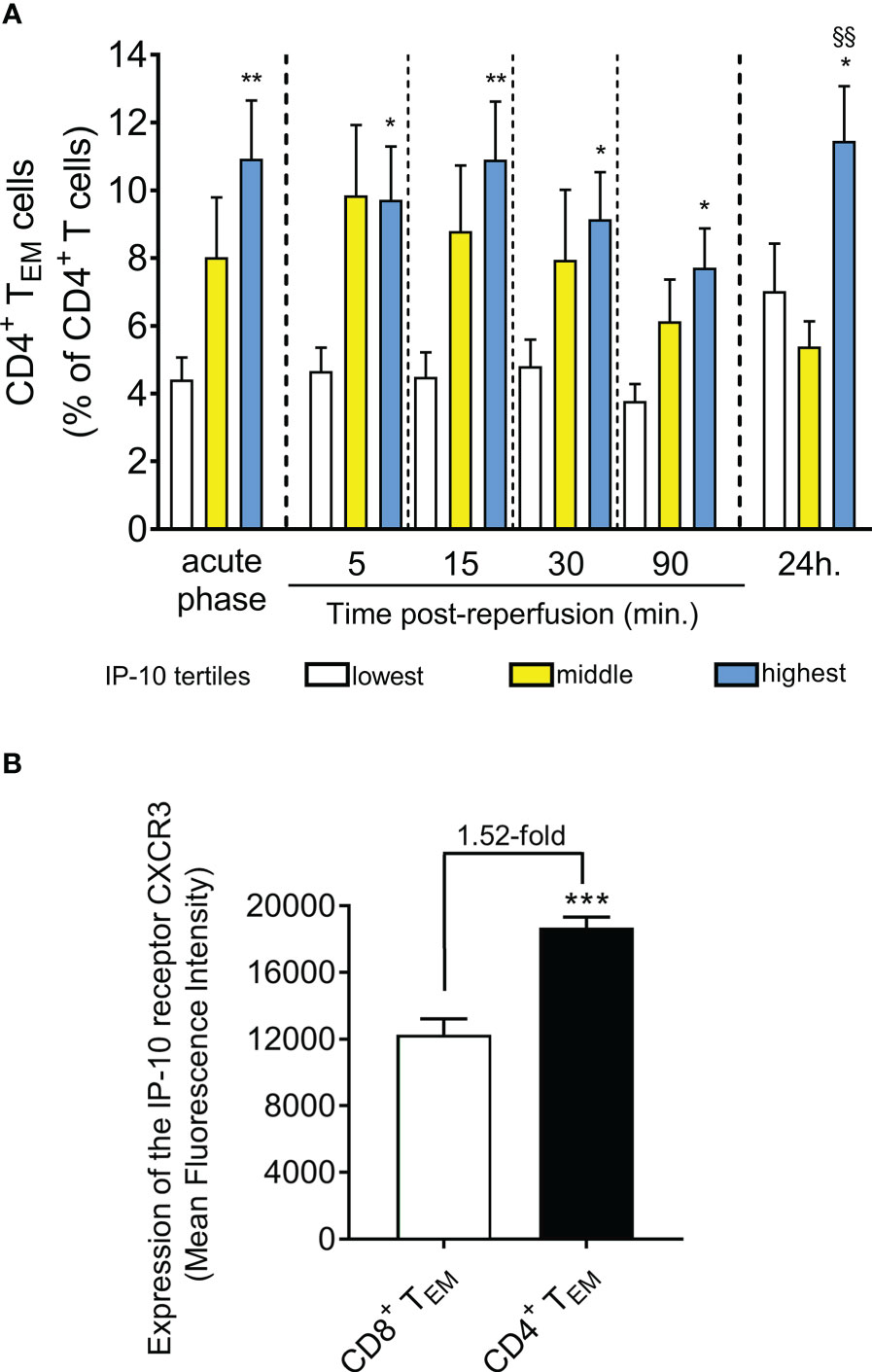
Figure 4 STEMI patients with higher IP-10 serum levels are presented with higher effector CD4+ T cell levels in blood. (A) Levels of CD4+ TEM cells are shown based on IP-10 serum tertiles at admission (acute phase) and five time-points post-reperfusion. n=47, *P<0.05 and **P<0.01 compared to the lowest tertile, §§P<0.01 compared to the middle tertile by Mann Whitney U test. (B) Expression of the IP-10 receptor CXCR3 was determined by flow-cytometry in CD4+ and CD8+ TEM cells derived from 10 STEMI patients. ***P<0.001 by independent samples t-test.
To understand the positive association of IP-10 levels specifically with CD4+ TEM, we quantified the levels of the IP-10 receptor CXCR3 on CD4+ and CD8+ TEM cells. Interestingly, we found that IP-10 homing receptor CXCR3 expression was 1.52-fold higher on the surface of CD4+ TEM cells compared to CD8+ TEM cells (Figure 4B) suggesting that CD4+ TEM cells are more responsive to IP-10 changes in blood.
We then examined the relationship between IP-10 tertiles at admission and left ventricular systolic function improvement by calculating the change (Δ) in LV ejection fraction (LVEF), a predictor of heart failure (31–33), as determined by CMR imaging at 12 weeks minus the LVEF obtained 2-7 days after STEMI (ΔLVEF= LVEF at 12 weeks – LVEF at 2-7 days). Remarkably, high IP-10 levels on admission (acute phase) and within the first 24h after STEMI were associated with left ventricular function recovery 12 weeks after STEMI (Figure 5A). We also estimated the relationship between IP-10 tertiles at admission and end-systolic volume (ESV), another predictor of heart failure (34–36). We estimated the ESV improvement by calculating the change (Δ) in ESV as determined by CMR imaging at 12 weeks minus the ESV obtained 2-7 days after STEMI (ΔESV= ESV at 12 weeks – ESV at 2-7 days). High IP-10 levels on admission and within the first 24h after STEMI were associated with end-systolic volume decrease 12 weeks after STEMI, suggesting that the dynamic biphasic regulation of IP-10 in blood at the acute phase of myocardial infarction possibly confers a cardioprotective role (Figure 5B).
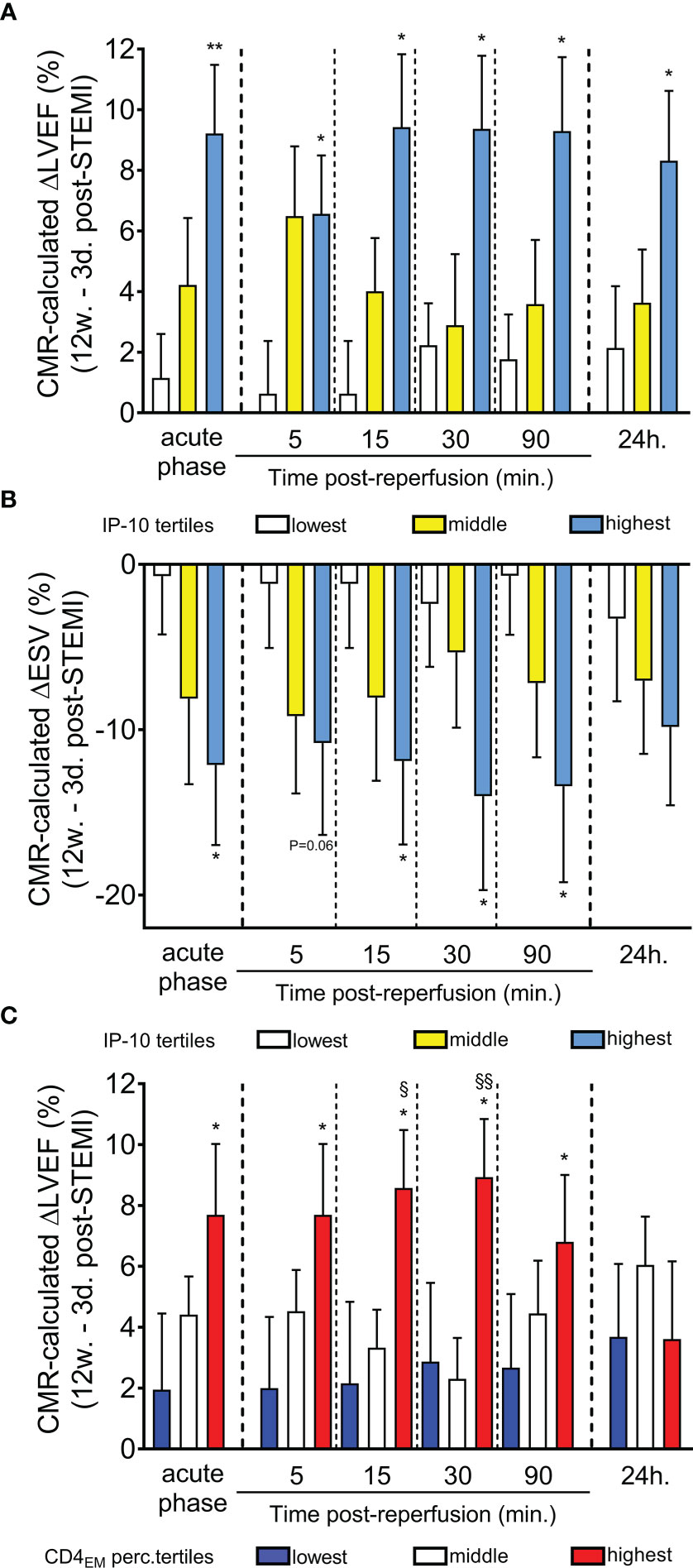
Figure 5 Predictive value of serum IP-10 levels and circulating CD4+ TEM cells for recovery of left ventricular systolic function post-STEMI. (A) Cardiac magnetic resonance imaging-based ΔLVEF (12-week LVEF – 3-day LVEF) and (B) ΔESV (12-week ESV – 3-day ESV) is shown according to IP-10 serum tertiles at admission (acute phase) and at five time-points post primary percutaneous coronary intervention. *P<0.05 and **P<0.01 compared to lowest tertile by Mann Whitney U test. (C) CMR-based ΔLVEF (12-week LVEF – 3-day LVEF) is shown at six time-points based on CD4+ TEM cell tertiles at admission. n=47, *P<0.05 compared to the lowest tertile, §P<0.05 and §§P<0.01 compared to the middle tertile by Mann Whitney U test.
To examine the association of continuous IP-10 levels with myocardial function improvement (ΔLVEF), we used multivariable linear regression analysis adjusting for randomized intervention (cyclosporine vs. placebo), age, sex, body mass index, traditional cardiovascular risk factors including diabetes mellitus, arterial hypertension and hypercholesterolemia, as well as high-sensitivity cardiac troponin T (hs-cTnT) (Table 3). We found that IP-10 levels at admission were independently associated with higher ΔLVEF after adjustment of all these factors.
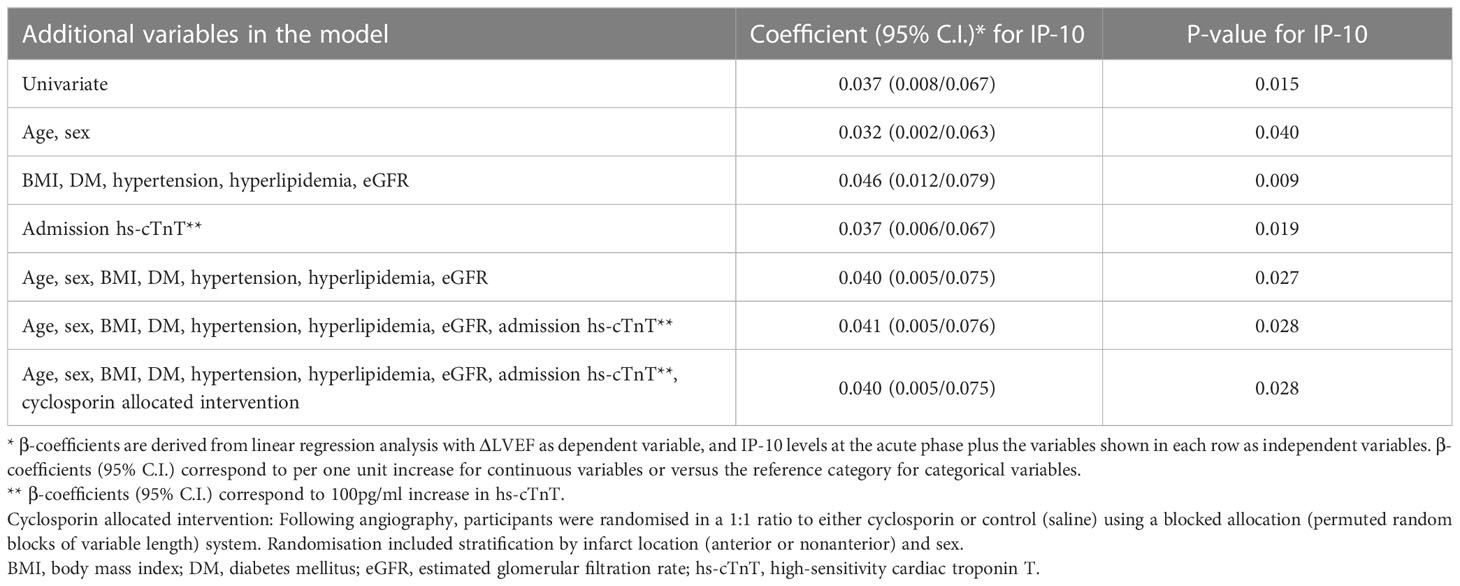
Table 3 Association of continuous IP-10 levels with ΔLVEF 12 weeks after STEMI in the Newcastle cohort by linear regression analysis.
Importantly and in a similar manner to IP-10 tertiles, patients at the highest tertile of CD4+ TEM cells on admission had a higher left ventricular systolic function recovery 12 weeks after STEMI (Figure 5C). No association between CD8+ TEM cells and cardiac recovery was found.
We next evaluated the prognostic value of IP-10 for the prediction of major cardiovascular events (MACE) including death, new myocardial infarction, stroke, revascularisation (CABG or PCI) or resuscitation, in an independent cohort of 331 consecutive STEMI patients (Figure 6), as described in the methods (Table 2). Patients were followed over a median of 540 days for MACE, and the primary endpoint occurred in 105 (31.8%) patients. Patients at the highest IP-10 quarter at admission had reduced risk for the primary endpoint (HR=0.536 for highest vs lower quarters, 95% CI: [0.309-0.93], p=0.027 by Cox regression analysis, Table 4). After adjusting for age, sex, traditional cardiovascular risk factors (diabetes mellitus, arterial hypertension and hypercholesterolemia), eGFR, CRP and high-sensitivity troponin T levels at admission, we observed that higher IP-10 remained an independent predictor of lower incidence of MACE in STEMI patients (adjusted HR=0.420 for highest vs lower quarters, 95% CI: [0.218-0.808], p=0.009; Table 4).
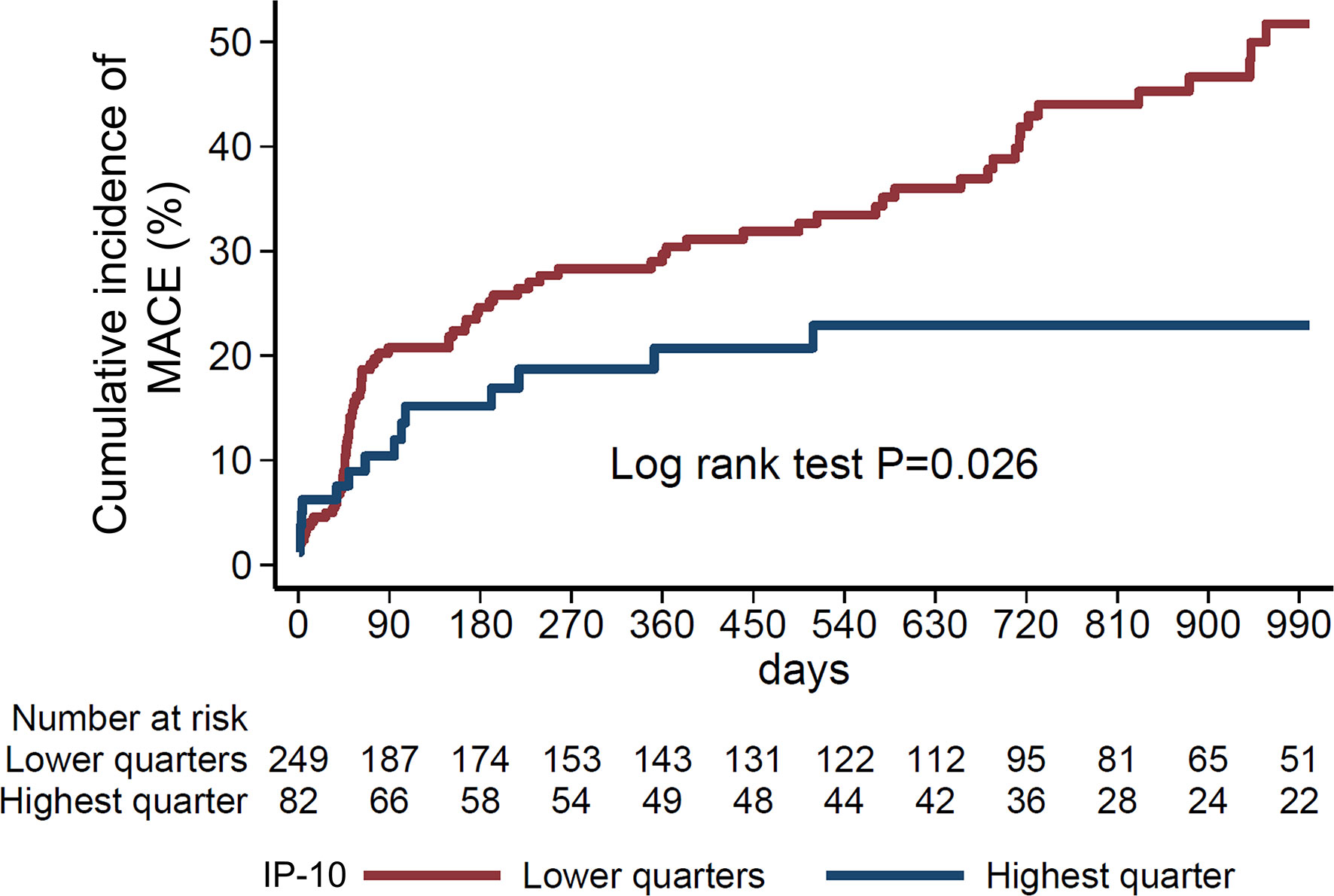
Figure 6 Nelson-Aalen curves of IP-10 quarters with the study endpoint. Cumulative incidence of the study endpoint (composite of all-cause death, myocardial infarction, stroke, resuscitation or revascularisation (percutaneous coronary angiography, coronary artery bypass graft) according to IP-10 quarters (highest vs. lower quarters) in 331 consecutive STEMI patients of the Heidelberg cohort. P=0.026 by Log-rank test.
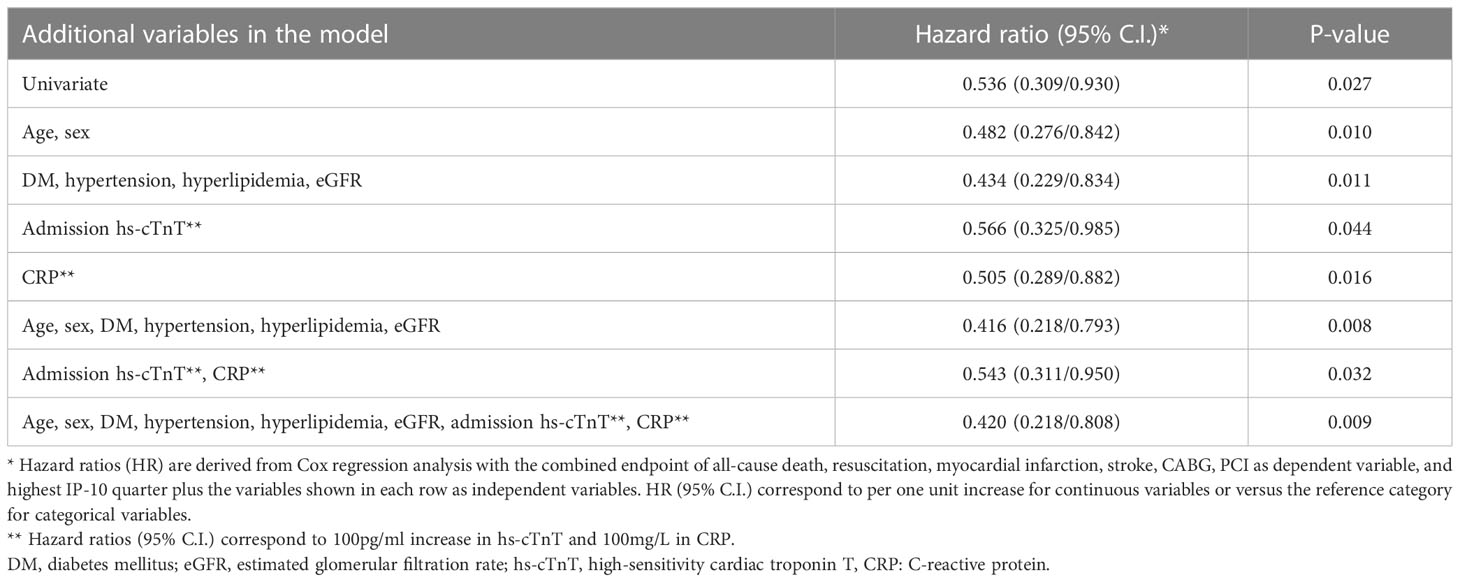
Table 4 Association of continuous IP-10 levels with the study endpoint (MACE) in the Heidelberg cohort by Cox regression analysis.
The major findings of the present study are: 1) increased serum IP-10 levels before and early after reperfusion are associated with improved cardiac systolic function in patients after STEMI; 2) increased serum IP-10 levels are associated with higher blood levels of CD4+ TEM cells, but not other CD4+ or CD8+ cell subtypes; 3) higher CD4+ TEM cells on admission are associated with LVEF increase 12 weeks after STEMI and 4) higher IP-10 serum levels on admission predict a lower long-term risk for MACE in patients with STEMI (Figure 7).
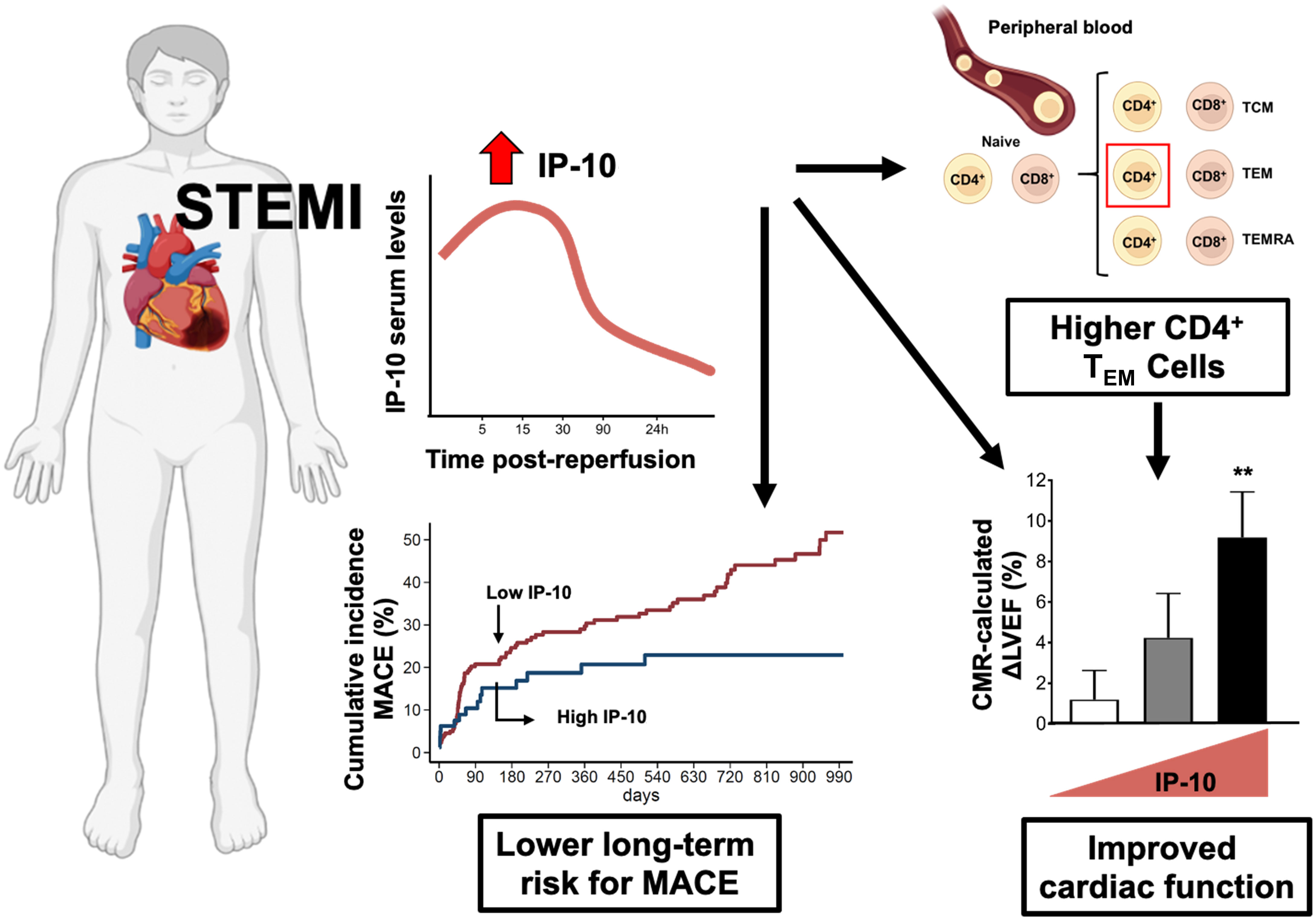
Figure 7 Summary figure of the potential role of IP-10 in STEMI patients. Effector T cell chemokine IP-10 levels predict recovery of left ventricular systolic function and cardiovascular outcomes after STEMI. IP-10 – CXCR3+ effector CD4+ T cells axis is critically involved in myocardial repair and overall disease prognosis after STEMI.
Chemokine release during acute myocardial infarction triggers the mobilisation of leukocytes, trafficking and domiciliation to ischemic tissues controlling wound healing processes, which critically determine cardiac repair post-myocardial infarction (37, 38). In the present study, we report a biphasic response of the effector T cell trafficking chemokine IP-10 to acute STEMI as follows: the serum IP-10 levels were high in the acute phase of STEMI (from patient admission to the hospital up to the first 15min post reperfusion of the occluded coronary artery), followed by a rapid profound reduction already by 90min post reperfusion. This is in accordance with previous preclinical animal data demonstrating that IP-10 peaks within 1–3 h of reperfusion and virtually disappears 24 h after reperfusion (19). This biphasic response may reflect the myocardial tissue ischemia level in STEMI. The initial high myocardial tissue ischemia caused by the infarcted coronary artery is followed by heart tissue reoxygenation after the reperfusion of the coronary circulation of the infarcted coronary artery following PPCI. This IP-10 early rise during acute ischemia indicates that hypoxia may trigger the increased serum levels of IP-10 chemokine. Indeed, the IP-10 gene promoter contains binding sites for both HIF1α and NF-κB, leading to increased IP-10 expression in cardiac microvascular endothelial cells upon hypoxia or ischemia (39).
IP-10 is an essential component of the adaptive immune response regulating the clearance of necrotic tissue in the heart. Absence of IP-10 in mice subjected to experimental myocardial infarction results in a deficient inflammation resolution and adverse cardiac remodelling (20). IP-10 knockout mice exhibit early dilation, followed by a rapid wall thinning during infarct maturation leading to systolic dysfunction and delayed scar contraction (20). In accordance with these preclinical data, we report that high levels of IP-10 during the acute phase of a myocardial infarction predict cardiac repair defined as the increase of LVEF and a decrease of ESV 12 weeks after STEMI, as quantified by cardiac MRI. Importantly, high IP-10 levels on admission predict a lower risk for MACE in an independent cohort of consecutive patients with STEMI. These findings support the notion that high IP-10 in the early acute phase of STEMI is integral for the reparative response in STEMI. Our study suggests that measurement of IP-10 serum levels may serve as a predictor of cardiac outcomes in STEMI patients.
IP-10 is a chemokine involved in the recruitment of activated T cells into sites of tissue inflammation (40). However, T cell temporal and spatial dynamics following myocardial ischemia and their contribution to inflammation resolution remain poorly understood. We have previously reported that T cells are specifically depleted from peripheral blood during myocardial reperfusion and lymphopenia is a critical determinant of survival in patients with STEMI (8, 22, 41). Here we conducted immunophenotyping of T cells at admission and within the first 24 hours post-reperfusion. Our results confirmed a rapid depletion of activated circulating CD4+ and CD8+ T cell subsets from admission to 90 minutes post-reperfusion in STEMI patients. This fall is probably followed by a rapid proliferative response in all T cell subsets after 24 hours, as suggested by previous studies in mice (9, 42). T effector memory cells are known for their migratory ability to inflamed tissue and the release of potent inflammatory mediators (43). In our study, these cells were drastically depleted from peripheral blood during the first 90 minutes of reperfusion, suggesting a potential transmigration of these cells to the infarcted heart during reperfusion. Our findings are in agreement with previous pre-clinical murine disease models showing that during ischemia/reperfusion, CD4+ and CD8+ T cells rapidly transmigrate to the infarcted areas of the heart (14, 42–46). CD4+ T cells become activated and are required for the clearance of necrotic tissue and subsequent remodeling of the myocardium through regulation of the local cytokine/chemokine environment (9, 10, 13, 47). On the other hand, CD8+ T cells may be largely detrimental to cardiac recovery post-MI by amplifying neutrophil and macrophage-mediated inflammation, thus resulting in increased left ventricle dilation and decreased cardiac function (13, 14). The increase in all T cell subsets after 24 hours in peripheral blood could be partly explained by the increased haematopoiesis observed after acute myocardial infarction and/or further mobilisation of lymphocytes from spleen, a secondary lymphoid organ acting as a T cell reservoir, lymph nodes or the bone marrow (48–52).
IP-10 homing receptor CXCR3 is the receptor with the highest expression in CD4+ and CD8+ T cells compared to other chemokine receptors (8). In our study, we report that CXCR3 expression is higher on CD4+ TEM cells compared to CD8+ TEM cells, suggesting that IP-10 promotes CXCR3+CD4+ T cell cardiotropism. Furthermore, the massive drop in IP-10 at 90 min is associated with falling CD4+ TEM cells at 90min post reperfusion suggesting that the IP-10 response may reflect the levels of circulating CD4+ TEM cells. Indeed, CXCR3+ T cells express higher levels of the integrin LFA-1, increasing their potential to adhere more efficiently to ICAM-1 and infiltrate the infarcted heart (53). Interestingly, IP-10 deficiency was associated with reduced recruitment of cell subpopulations that expressed CXCR3 (20). Although CXCR3 is highly expressed in CD8+ cells, it appears that the chemokine fractalkine and its receptor CX3CR1 are the driver of CD8+ TEM cells dynamics in STEMI patients (8). Furthermore, the low expression of CXCR3 and CX3CR1 on naïve cells may explain the unique association in our study between IP-10 and CD4+ TEM cells (8, 54). The data in our present study therefore highlights that several chemokine mechanisms are in play in the lymphocyte kinetics in ischemia-reperfusion.
Taken together, our data suggest that trafficking of CD4+ TEM cells in response to acute myocardial ischemia may be orchestrated in an IP-10 – CXCR3 dependent manner controlling cardiac repair in AMI. This is in accordance with previous preclinical reports in animal models showing that CD4+ effector T cells are rapidly recruited into ischemic myocardium acquiring a cardioprotective phenotype (9, 10, 42–46). It is important to consider that the role of T cells in STEMI is likely time dependent. CD4+ T lymphocytes seem to exhibit biphasic kinetics post-myocardial infarction whereby CD4+ T cells facilitate wound healing in the acute phase of myocardial infarction but their further recruitment in the heart from the 10th day to 8 weeks after STEMI promotes left-ventricular dysfunction (55). IP-10 kinetics several weeks after STEMI and its contribution to this second wave, remains, however, to be determined. Given the observational, retrospective design of our study, our findings cannot prove causality. Nevertheless, our findings encourage further experimental research on the beneficial role of the increased blood levels of IP-10 in the acute phase of STEMI and the relative contribution of CD4+ TEM cells in IP-10-related cardioprotective effects. There is therefore a need to further establish IP-10 as a prognostic marker in a larger STEMI cohort with heart failure as an end-point to relate individual IP-10 levels found in patients at admission to the risk of developing heart failure in the long-term.
In conclusion, the current study adds new translational evidence for the role of IP-10 in left ventricular function recovery and cardiovascular outcomes after STEMI. Understanding the differential effects of T cell subsets and their homing mechanisms to the ischemic heart is an essential first step for developing future targeted immunomodulatory therapies in patients with STEMI. Future studies are warranted to elucidate the underlying mechanisms and investigate the prognostic and therapeutic value of IP-10 modulation in patients with STEMI.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by NCT02390674. The patients/participants provided their written informed consent to participate in this study.
Conception and design: IS, KSte. Analysis and interpretation of the data: KSo, ST-C, MM-H, NV, GG, EG, KB, AM, EG, IS, KSte. Drafting of the article: KSo, ST-C, IS, KSte. Critical revision of the article for important intellectual content: MM-H, NV, GG, MB, GR, EM, GR, AG, JH, HL, NF, KSta, JH, DD, EG. Provision of study materials or patients: MM-H, EG, IS. Statistical expertise: NV, GG, KSta. Obtaining of funding: KSo, ST-C, IS, KSte. Administrative, technical, or logistic support: MM-H, AT, MP-S, LS, KB. Collection and assembly of data: KSo, ST-C, MM-H, MB, MS, KB, AM. All authors contributed to the article and approved the submitted version.
The research was funded/supported by a research scholarship from the Deutsche Herzstiftung (German Heart Foundation) to KSo, by a Wellcome Trust Institutional Strategic Support and a British Heart Foundation grant (PG/23/11093) to ST-C, by the Biotechnology and Biological Sciences Research Council (BBSRC) of the UK Research and Innovation (UKRI) to KSte and AG by grants from the National Institute for Health Research Newcastle Biomedical Research Centre based at Newcastle upon Tyne Hospitals NHS Foundation Trust and Newcastle University, and the British Heart Foundation (PG/18/25/33587) to IS and by grants from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No 759248) and the DFG (CRC1366 C07, project number 394046768) to KSte. DD is supported by the DFG (CRC1366 B08, project number 394046768, and CRC1425 P07, project number 422681845) and the DZHK (MaBo-05). The views expressed are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health.
The authors would like to thank the study participants for enabling the two cohort studies. We acknowledge the Newcastle University Flow Cytometry Core Facility (FCCF) for assistance with the generation of Flow Cytometry data. Figures were created using Servier Medical Art and BioRender.
MM-H. reports consulting fees for Zoll CMS GmbH, research funding from Roche Diagnostics and BRAHMS Thermo Scientific. NF has received lecture fees from AstraZeneca, Boehringer Ingelheim, Bayer Vital and consulting fees for Boehringer Ingelheim, all of which not related to the subject in this manuscript. DD reports consulting and speaker fees from Bayer Healthcare, Boston Scientific, Daiichi Sankyo, AstraZeneca, and research grants from DFG, DZHK. EG has received honoraria for lectures from AstraZeneca, Daiichi Sankyo and Roche Diagnostics, consulting fees for BRAHMS Deutschland, Roche Diagnostics, AstraZeneca, Daiichi and Novo Nordisk, research funding from Daiichi and Roche Diagnostics. IS receives research grants from Kancera AB and Astra Zaneca.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Vaduganathan M, Mensah GA, Turco JV, Fuster V, Roth GA. The global burden of cardiovascular diseases and risk: a compass for future health. J Am Coll Cardiol (2022) 80(25):2361–71. doi: 10.1016/j.jacc.2022.11.005
2. Fox KAA, Steg PG, Eagle KA, Goodman SG, Anderson FA, Granger CB, et al. Decline in rates of death and heart failure in acute coronary syndromes, 1999-2006. JAMA (2007) 297:1892–900. doi: 10.1001/jama.297.17.1892
3. Nallamothu BK, Normand S-LT, Wang Y, Hofer TP, Brush JE, Messenger JC, et al. Relation between door-to-balloon times and mortality after primary percutaneous coronary intervention over time: a retrospective study. Lancet (2015) 385:1114–22. doi: 10.1016/S0140-6736(14)61932-2
4. Hellermann JP, Goraya TY, Jacobsen SJ, Weston SA, Reeder GS, Gersh BJ, et al. Incidence of heart failure after myocardial infarction: is it changing over time? Am J Epidemiol (2003) 157:1101–7. doi: 10.1093/aje/kwg078
5. St John Sutton M, Lee D, Rouleau JL, Goldman S, Plappert T, Braunwald E, et al. Left ventricular remodeling and ventricular arrhythmias after myocardial infarction. Circulation (2003) 107:2577–82. doi: 10.1161/01.CIR.0000070420.51787.A8
6. van der Bijl P, Abou R, Goedemans L, Gersh BJ, Holmes DR, Ajmone Marsan N, et al. Left ventricular post-infarct remodeling: implications for systolic function improvement and outcomes in the modern era. JACC Heart Fail (2020) 8:131–40. doi: 10.1016/j.jchf.2019.08.014
7. Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res (2002) 53:31–47. doi: 10.1016/S0008-6363(01)00434-5
8. Boag SE, Das R, Shmeleva EV, Bagnall A, Egred M, Howard N, et al. T Lymphocytes and fractalkine contribute to myocardial ischemia/reperfusion injury in patients. J Clin Invest (2015) 125:3063–76. doi: 10.1172/JCI80055
9. Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G, et al. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation (2012) 125(13):1652–63. doi: 10.1161/CIRCULATIONAHA.111.044164
10. Weirather J, Hofmann UDW, Beyersdorf N, Ramos GC, Vogel B, Frey A, et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res (2014) 115:55–67. doi: 10.1161/CIRCRESAHA.115.303895
11. Zhang L, Wang Z, Wang D, Zhu J, Wang Y. CD8(+)CD28(+) T cells might mediate injury of cardiomyocytes in acute myocardial infarction. Mol Immunol (2018) 101:74–9. doi: 10.1016/j.molimm.2018.05.015
12. Tae Yu H, Youn JC, Lee J, Park S, Chi HS, Lee J, et al. Characterization of CD8(+)CD57(+) T cells in patients with acute myocardial infarction. Cell Mol Immunol (2015) 12(4):466–73. doi: 10.1038/cmi.2014.74
13. Santos-Zas I, Lemarié J, Zlatanova I, Cachanado M, Seghezzi JC, Benamer H, et al. Cytotoxic CD8(+) T cells promote granzyme b-dependent adverse post-ischemic cardiac remodeling. Nat Commun (2021) 12(1):1483. doi: 10.1038/s41467-021-21737-9
14. Ilatovskaya DV, Pitts C, Clayton J, Domondon M, Troncoso M, Pippin S, et al. CD8+ T-cells negatively regulate inflammation post-myocardial infarction. Am J Physiol Heart Circ Physiol (2019) 317:H581–96. doi: 10.1152/ajpheart.00112.2019
15. Altara R, Mallat Z, Booz GW, Zouein FA. The CXCL10/CXCR3 axis and cardiac inflammation: implications for immunotherapy to treat infectious and noninfectious diseases of the heart. J Immunol Res (2016) 2016:4396368. doi: 10.1155/2016/4396368
16. Liu M, Guo S, Hibbert JM, Jain V, Singh N, Wilson NO, et al. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev (2011) 22(3):121–30. doi: 10.1016/j.cytogfr.2011.06.001
17. Safa A, Rashidinejad HR, Khalili M, Dabiri S, Nemati M, Mohammadi MM, et al. Higher circulating levels of chemokines CXCL10, CCL20 and CCL22 in patients with ischemic heart disease. Cytokine (2016) 83:147–57. doi: 10.1016/j.cyto.2016.04.006
18. Dookun E, Walaszczyk A, Redgrave R, Palmowski P, Tual-Chalot S, Suwana A, et al. Clearance of senescent cells during cardiac ischemia-reperfusion injury improves recovery. Aging Cell (2020) 19(10):e13249. doi: 10.1111/acel.13249
19. Frangogiannis NG, Mendoza LH, Lewallen M, Michael LH, Smith CW, Entman ML. Induction and suppression of interferon-inducible protein 10 in reperfused myocardial infarcts may regulate angiogenesis. FASEB J (2001) 15:1428–30. doi: 10.1096/fj.00-0745fje
20. Bujak M, Dobaczewski M, Gonzalez-Quesada C, Xia Y, Leucker T, Zymek P, et al. Induction of the CXC chemokine interferon-gamma-inducible protein 10 regulates the reparative response following myocardial infarction. Circ Res (2009) 105:973–83. doi: 10.1161/CIRCRESAHA.109.199471
21. Cormack S, Mohammed A, Panahi P, Das R, Steel AJ, Chadwick T, et al. Effect of ciclosporin on safety, lymphocyte kinetics and left ventricular remodelling in acute myocardial infarction. Br J Clin Pharmacol (2020) 86:1387–97. doi: 10.1111/bcp.14252
22. Spray L, Park C, Cormack S, Mohammed A, Panahi P, Boag S, et al. The fractalkine receptor CX3CR1 links lymphocyte kinetics in CMV-seropositive patients and acute myocardial infarction with adverse left ventricular remodeling. Front Immunol (2021) 12:605857. doi: 10.3389/fimmu.2021.605857
23. Stamatelopoulos K, Mueller-Hennessen M, Georgiopoulos G, Sachse M, Boeddinghaus J, Sopova K, et al. Amyloid-β (1-40) and mortality in patients with non-ST-Segment elevation acute coronary syndrome: a cohort study. Ann Intern Med (2018) 168:855–65. doi: 10.7326/M17-1540
24. Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, et al. Executive group on behalf of the joint European society of cardiology (ESC)/American college of cardiology (ACC)/American heart association (AHA)/World heart federation (WHF) task force for the universal definition of myocardial infarction. fourth universal definition of myocardial infarction (2018). J Am Coll Cardiol (2018) 72:2231–64. doi: 10.1016/j.jacc.2018.08.1038
25. Hicks KA, Mahaffey KW, Mehran R, Nissen SE, Wiviott SD, Dunn B, et al. 2017 cardiovascular and stroke endpoint definitions for clinical trials. Circulation (2018) 137:961–72. doi: 10.1161/CIRCULATIONAHA.117.033502
26. Koch S, Larbi A, Derhovanessian E, Ozcelik D, Naumova E, Pawelec G. Multiparameter flow cytometric analysis of CD4 and CD8 T cell subsets in young and old people. Immun Ageing (2008) 5:6. doi: 10.1186/1742-4933-5-6
27. Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401:708–12. doi: 10.1038/44385
28. Bernhard OK, Mathias RA, Barnes TW, Simpson RJ. A fluorescent microsphere-based method for assay of multiple analytes in plasma. Methods Mol Biol (2011) 728:195–206. doi: 10.1007/978-1-61779-068-3_12
29. Kim SH, Cho JH, Lee YH, Lee JH, Kim SS, Kim MY, et al. Improvement in left ventricular function with intracoronary mesenchymal stem cell therapy in a patient with anterior wall ST-segment elevation myocardial infarction. Cardiovasc Drugs Ther (2018) 32:329–38. doi: 10.1007/s10557-018-6804-z
30. Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, et al. Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J Exp Med (1996) 184(3):963–9. doi: 10.1084/jem.184.3.963
31. Lewis EF, Velazquez EJ, Solomon SD, Hellkamp AS, McMurray JJ, Mathias J, et al. Predictors of the first heart failure hospitalization in patients who are stable survivors of myocardial infarction complicated by pulmonary congestion and/or left ventricular dysfunction: a VALIANT study. Eur Heart J (2008) 29:748–56. doi: 10.1093/eurheartj/ehn062
32. Lewis EF, Moye LA, Rouleau JL, Sacks FM, Arnold JM, Warnica JW, et al. Predictors of late development of heart failure in stable survivors of myocardial infarction: the CARE study. J Am Coll Cardiol (2003) 42:1446–53. doi: 10.1016/S0735-1097(03)01057-X
33. Kelly DJ, Gershlick T, Witzenbichler B, Guagliumi G, Fahy M, Dangas G, et al. Incidence and predictors of heart failure following percutaneous coronary intervention in ST-segment elevation myocardial infarction: the HORIZONS-AMI trial. Am Heart J (2011) 162:663–70. doi: 10.1016/j.ahj.2011.08.002
34. White HD, Norris RM, Brown MA, Brandt PW, Whitlock RM, Wild CJ. Left ventricular end-systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation (1987) 76(1):44–51. doi: 10.1161/01.CIR.76.1.44
35. Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. behalf of an international forum on cardiac remodeling. J Am Coll Cardiol (2000) 35(3):569–82.4. doi: 10.1016/S0735-1097(99)00630-0
36. Corral Acero J, Schuster A, Zacur E, Lange T, Stiermaier T, Backhaus SJ, et al. Understanding and improving risk assessment after myocardial infarction using automated left ventricular shape analysis. JACC Cardiovasc Imaging (2022) 15(9):1563–74. doi: 10.1016/j.jcmg.2021.11.027
37. Cavalera M, Frangogiannis NG. Targeting the chemokines in cardiac repair. Curr Pharm Des (2014) 20(12):1971–9. doi: 10.2174/13816128113199990449
38. Chen B, Frangogiannis NG. Chemokines in myocardial infarction. J Cardiovasc Transl Res (2021) 14(1):35–52. doi: 10.1007/s12265-020-10006-7
39. Xia J-B, Liu G-H, Chen Z-Y, Mao C-Z, Zhou D-C, Wu H-Y, et al. Hypoxia/ischemia promotes CXCL10 expression in cardiac microvascular endothelial cells by NFkB activation. Cytokine (2016) 81:63–70. doi: 10.1016/j.cyto.2016.02.007
40. Khan IA, MacLean JA, Lee FS, Casciotti L, DeHaan E, Schwartzman JD, et al. IP-10 is critical for effector T cell trafficking and host survival in toxoplasma gondii infection. Immunity (2000) 12:483–94. doi: 10.1016/S1074-7613(00)80200-9
41. Hoffmann J, Fiser K, Weaver J, Dimmick I, Loeher M, Pircher H, et al. High-throughput 13-parameter immunophenotyping identifies shifts in the circulating T-cell compartment following reperfusion in patients with acute myocardial infarction. PLoS One (2012) 7(10):e47155. doi: 10.1371/journal.pone.0047155
42. Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol (2013) 62:24–35. doi: 10.1016/j.yjmcc.2013.04.023
43. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol (2004) 22:745–63. doi: 10.1146/annurev.immunol.22.012703.104702
44. Curato C, Slavic S, Dong J, Skorska A, Altarche-Xifró W, Miteva K, et al. Identification of noncytotoxic and IL-10-producing CD8+AT2R+ T cell population in response to ischemic heart injury. J Immunol (2010) 185(10):6286–93. doi: 10.4049/jimmunol.0903681
45. Yang Z, Day YJ, Toufektsian MC, Xu Y, Ramos SI, Marshall MA, et al. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation (2006) 114(19):2056–64. doi: 10.1161/CIRCULATIONAHA.106.649244
46. Forteza MJ, Trapero I, Hervas A, de Dios E, Ruiz-Sauri A, Minana G, et al. Apoptosis and mobilization of lymphocytes to cardiac tissue is associated with myocardial infarction in a reperfused porcine model and infarct size in post-PCI patients. Oxid Med Cell Longev (2018) 2018:1975167. doi: 10.1155/2018/1975167
47. Rieckmann M, Delgobo M, Gaal C, Büchner L, Steinau P, Reshef D, et al. Myocardial infarction triggers cardioprotective antigen-specific T helper cell responses. J Clin Invest (2019) 129(11):4922–36. doi: 10.1172/JCI123859
48. Ma Y, Yang X, Villalba N, Chatterjee V, Reynolds A, Spence S, et al. Circulating lymphocyte trafficking to the bone marrow contributes to lymphopenia in myocardial infarction. Am J Physiol Heart Circ Physiol (2022) 322(4):H622–35. doi: 10.1152/ajpheart.00003.2022
49. Zou L, Barnett B, Safah H, Larussa VF, Evdemon-Hogan M, Mottram P, et al. Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res (2004) 64(22):8451–5. doi: 10.1158/0008-5472.CAN-04-1987
50. Mazo IB, Honczarenko M, Leung H, Cavanagh LL, Bonasio R, Weninger W, et al. Bone marrow is a major reservoir and site of recruitment for central memory CD8+ T cells. Immunity (2005) 22(2):259–70. doi: 10.1016/j.immuni.2005.01.008
51. Nahrendorf M. Myeloid cell contributions to cardiovascular health and disease. Nat Med (2018) 24(6):711–20. doi: 10.1038/s41591-018-0064-0
52. Cremer S, Schloss MJ, Vinegoni C, Foy BH, Zhang S, Rohde D, et al. Diminished reactive hematopoiesis and cardiac inflammation in a mouse model of recurrent myocardial infarction. J Am Coll Cardiol (2020) 75(8):901–15. doi: 10.1016/j.jacc.2019.12.056
53. Ngwenyama N, Salvador AM, Velázquez F, Nevers T, Levy A, Aronovitz M, et al. CXCR3 regulates CD4+ T cell cardiotropism in pressure overload-induced cardiac dysfunction. JCI Insight (2019) 4(7):e125527. doi: 10.1172/jci.insight.125527
54. Sallusto F, Lenig D, Mackay CR, Lanzavecchia A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med (1998) 187(6):875–83. doi: 10.1084/jem.187.6.875
Keywords: IP-10, myocardial infarction, T cells, prognostic value, lymphocytes, heart failure, remodeling, cardiac MRI
Citation: Sopova K, Tual-Chalot S, Mueller-Hennessen M, Vlachogiannis NI, Georgiopoulos G, Biener M, Sachse M, Turchinovich A, Polycarpou-Schwarz M, Spray L, Maneta E, Bennaceur K, Mohammad A, Richardson GD, Gatsiou A, Langer HF, Frey N, Stamatelopoulos K, Heineke J, Duerschmied D, Giannitsis E, Spyridopoulos I and Stellos K (2023) Effector T cell chemokine IP-10 predicts cardiac recovery and clinical outcomes post-myocardial infarction. Front. Immunol. 14:1177467. doi: 10.3389/fimmu.2023.1177467
Received: 01 March 2023; Accepted: 23 May 2023;
Published: 22 June 2023.
Edited by:
Madhumita Chatterjee, Experimental Therapy and Toxicology, GermanyReviewed by:
Daniel Harding, Queen Mary University of London, United KingdomCopyright © 2023 Sopova, Tual-Chalot, Mueller-Hennessen, Vlachogiannis, Georgiopoulos, Biener, Sachse, Turchinovich, Polycarpou-Schwarz, Spray, Maneta, Bennaceur, Mohammad, Richardson, Gatsiou, Langer, Frey, Stamatelopoulos, Heineke, Duerschmied, Giannitsis, Spyridopoulos and Stellos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ioakim Spyridopoulos, aW9ha2ltLnNweXJpZG9wb3Vsb3NAbmV3Y2FzdGxlLmFjLnVr; Konstantinos Stellos, a29uc3RhbnRpbm9zLnN0ZWxsb3NAbWVkbWEudW5pLWhlaWRlbGJlcmcuZGU=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.