
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 12 May 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1175387
This article is part of the Research TopicCellular and Systemic Interplay of Metabolism and Inflammation in the Pathogenesis of Lung DiseasesView all 11 articles
 Yunhong Yu
Yunhong Yu Zhengqiu Lian*
Zhengqiu Lian*Transfusion-related acute lung injury (TRALI) is a severe adverse event and a leading cause of transfusion-associated death. Its poor associated prognosis is due, in large part, to the current dearth of effective therapeutic strategies. Hence, an urgent need exists for effective management strategies for the prevention and treatment of associated lung edema. Recently, various preclinical and clinical studies have advanced the current knowledge regarding TRALI pathogenesis. In fact, the application of this knowledge to patient management has successfully decreased TRALI-associated morbidity. This article reviews the most relevant data and recent progress related to TRALI pathogenesis. Based on the existing two-hit theory, a novel three-step pathogenesis model composed of a priming step, pulmonary reaction, and effector phase is postulated to explain the process of TRALI. TRALI pathogenesis stage-specific management strategies based on clinical studies and preclinical models are summarized with an explication of their models of prevention and experimental drugs. The primary aim of this review is to provide useful insights regarding the underlying pathogenesis of TRALI to inform the development of preventive or therapeutic alternatives.
Transfusion of blood components is a widely employed life-saving treatment in clinical settings; however, it can result in potentially life-threatening adverse reactions (1). For example, transfusion-related acute lung injury (TRALI) is a rare but severe adverse event that occurs during—or within—6 hours of blood transfusion and is characterized by hypoxia and non-cardiogenic pulmonary edema, known as respiratory distress (2). In 2019, a new diagnostic criteria was established using the Delphi approach, effectively subdividing TRALI into Type I and II, which occur in the absence or presence of acute respiratory distress syndrome (ARDS) risk factors, respectively (3). Although TRALI represents a main cause of transfusion-associated mortality in developed countries (4), it is likely underdiagnosed, particularly in intensive care patients, owing to the synergetic action of multiple factors (5, 6). Although estimates of TRALI mortality vary markedly between 5% and 25% (7, 8), they can reach 47% in intensive care and surgical patients (9). Even so, specific therapeutics remain unavailable.
When TRALI was first reported in 1957, it was thought to be related to the passive transfer of high-titer leucoagglutinins (10). It was not until 1983 that TRALI was formally identified as a distinctive clinical syndrome caused by transfused anti-human leukocyte antigen (HLA) antibodies (11). However, in addition to the transfer of anti-HLA antibodies, anti-human neutrophil antigen (HNA) antibodies also contribute to TRALI induction. In fact, antibody-dependent mechanisms are estimated to be responsible for 50–80% of TRALI cases (12); however, because of the inaccuracy of current laboratory testing techniques, their contribution has likely been underestimated (13). Moreover, TRALI cases that meet the diagnostic criteria have been reported in patients who received transfused products without antibodies (14). More specifically, approximately 10–15% of TRALI cases are associated with biological response modifiers (BRMs), such as soluble CD40 ligand (sCD40L) or lyso-phosphatidylcholines (lyso-PCs), which originate from the storage of blood components. Thus, non-antibody-mediated TRALI has also been proposed. Although significant progress has been made in the field, the complex pathogenesis of TRALI has not yet been fully characterized.
In this review, we highlight the recent advances in the field regarding TRALI pathogenesis and management based on clinical studies and preclinical models. The findings of these studies provide a clearer understanding of the underlying pathogenesis, which will assist in developing effective prevention or therapeutic strategies.
The pathogenesis of TRALI has been described as a two-hit theory, in which recipient predisposition, together with the presence of detrimental factors in blood components, play significant roles (15). That is, the first hit focusing on recipient predisposition, in which pulmonary endothelial cells (ECs) are activated and polymorphonuclear neutrophils (PMNs) are primed. The second hit comprises mediators in transfused stored units, which trigger the primed PMNs and other cells including ECs,monocytes, macrophages, and platelets to release pathogenetic factors and induce coagulopathy, ultimately resulting in fluid intrapulmonary infiltration. The second hit can be classified into antibody-dependent and -independent TRALI based on the detection of differential pathogenic mediators in the blood components. While the two-hit hypothesis clearly explains TRALI pathogenic factors, the disease pathogenesis can be further characterized as three overlapping phases (Figure 1), i.e., the priming step, pulmonary reaction, and effector phase. During the priming step, recipient-related risk factors mainly lead to endothelial activation and PMN priming via the EC-PMN interaction. Subsequently, in the pulmonary reaction, antibodies or BRMs bind to target cells, such as ECs, PMNs, or mononuclear cells, thereby inducing a host response. In the final effector phase, the pulmonary vascular endothelial barrier may become damaged by the release of neutrophil extracellular traps (NETs) and reactive oxygen species (ROS); the resulting coagulopathy might aggravate lung injury. The diverse phase characteristics of the three-step pathogenesis model will be described in detail to provide a better understanding of TRALI pathogenesis.
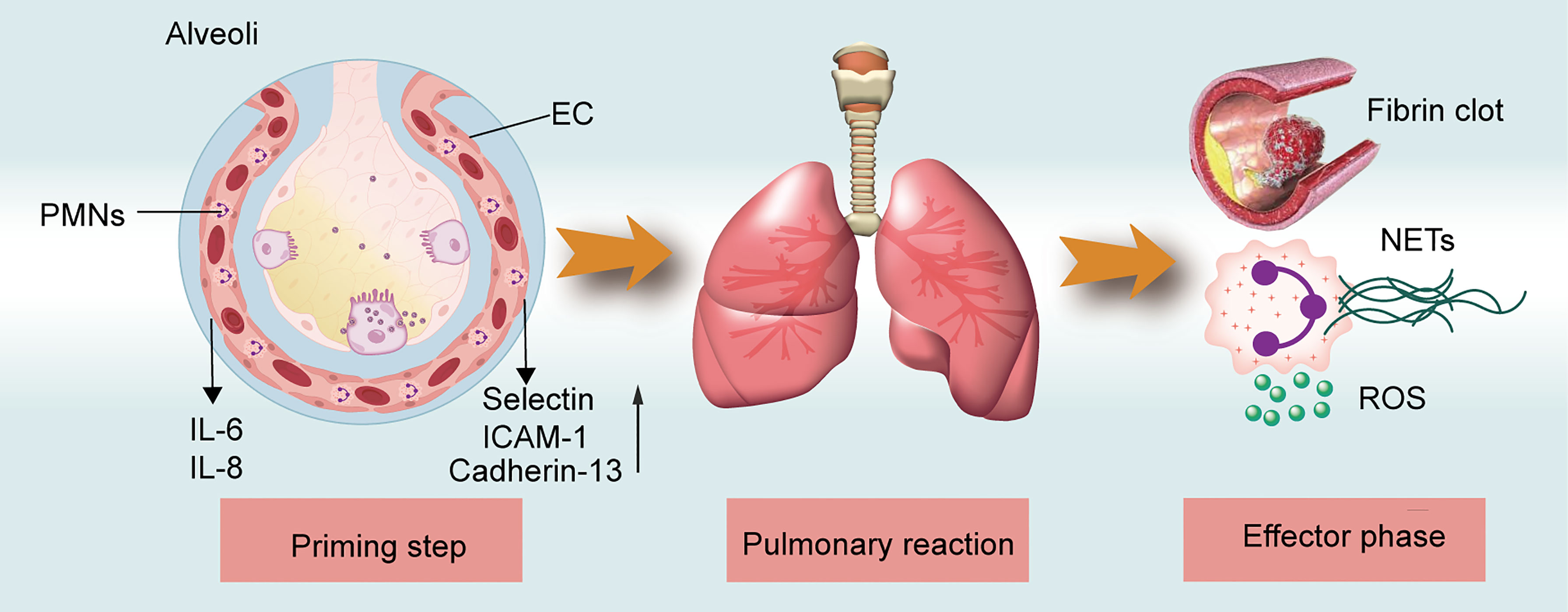
Figure 1 TRALI pathogenesis model. The three-step of TRALI pathogenesis: prime step, pulmonary reaction, and effector step. The pathogenesis of each step is described.
As early as 1997, the results of a retrospective study in combination with an in vivo rat lung model suggested that stronger PMN-priming activity occurred in patients with TRALI and underlying clinical conditions than in those not diagnosed with TRALI (16). A prospective study revealed that patients diagnosed with hematologic malignance or cardiac disease are at increased risk for developing TRALI (15). Subsequent studies have identified myriad complications that function as first hits, including hypertension, end-stage liver disease, chronic alcohol abuse, surgery, mechanical ventilation, trauma, massive transfusion, and systemic inflammation (13, 17). Based on retrospective cohort studies, pulmonary diseases, including pulmonary fibrosis, interstitial lung disease, and tobacco abuse, are identified as potential high-risk factors for TRALI occurrence (18, 19). Moreover, critical patients with these underlying diseases conveyed a serious first hit, requiring only a minor second hit to trigger TRALI (20). Patients with TRALI in an intensive care unit (ICU) exhibited 70% mortality compared with typical 5–25% rates (20, 21). Thus, underlying clinical events play a pivotal pathogenic role in TRALI onset; screening these events in transfusion recipients might improve clinical outcomes.
In the initial TRALI mouse model, transfusion of major histocompatibility complex (MHC) I antibodies 1 to BALB/c mice resulted in TRALI induction (22). In a subsequent study, housing the mice in a specific pathogen-free barrier facility prevented the same lung injury, unless the mice were primed by treatment with lipopolysaccharide (LPS) (23). The difference in results according to housing conditions may be explained by gut flora (24). Therefore, gut flora can be considered a pivotal factor in the priming of TRALI. However, the impact of gut flora on TRALI pathogenesis requires further study.
The role of risk factors of recipients in TRALI pathogenesis has been investigated in preclinical studies, employing low-dose LPS or C-reactive protein (CRP) as the first hit in two-hit TRALI models to represent systemic inflammation in transfusion recipients (25, 26). However, the nature of TRALI first hits, e.g., LPS, is different from CRP. For instance, LPS activates cells by triggering a toll-like receptor four-signal pathway (27). Alternatively, CRP can represent the first hit by benefiting PMN sequestration in the lungs and activating ECs (27). Collectively, these first hits facilitate the activation of pulmonary ECs and macrophage to release interleukin (IL)-6, macrophage inflammatory protein-2 (MIP-2), the murine homolog of human IL-8, and osteopontin (OPN) and increase the expression of adhesion molecules and cadherin-13 due to endothelial activation (14, 26, 28, 29) (Figure 2). Circulating PMNs retained within the lung microvasculature constitute a lung-marginated pool that maintains a dynamic equilibrium with the circulating pool (30). These elevated molecules attract PMNs from the circulating pool to travel to the injured lung and firmly adhere to ECs, creating a predisposition for developing TRALI (Figure 2). These mechanisms might account, in part, for why patients with underlying clinical conditions, such as systemic inflammation, are at a high risk of developing TRALI. However, multiple underlying conditions that are not associated with systemic inflammation, such as chronic alcohol abuse and hypertension, among others, also represent high-risk factors for TRALI onset. In particular, reduced production of nitric oxide, an endothelial anti-adhesive molecule, strengthens endothelial intercellular cell adhesion molecule-1-dependent PMN adhesion, thus providing an alternate mechanism for PMN priming in the absence of systemic inflammation (31).

Figure 2 First hits in the pathogenesis of TRALI. The underlying diseases of patients stimulate the activation of ECs and macrophages, thus attracting PMNs from circulating pool to the injured lung.
Taken together, these data indicate that endothelial activation and pulmonary sequestration are key process in the priming step, where direct activation of pulmonary ECs leading to PMN priming initiates the first phase of TRALI development.
Following pulmonary endothelial activation and PMN priming caused by underlying diseases, donor-derived risk factors, including pathogenic antibodies and BRMs, target different cell populations, such as hematopoietic and non-hematopoietic cells. Thus, these risk factors make specific contributions to the host response and uniquely influence TRALI progression (13, 14), representing the pulmonary reaction phase.
In wild-type BALB/c or C57BL/6 mice, the development of pulmonary edema requires monocytes/macrophages, while PMNs and platelets appear to be dispensable. Conversely, regulatory T cells (Tregs) and dendritic cells (DCs) are associated with lung injury amelioration (32). Meanwhile, given that ECs represent the key regulator of PMN transmigration, they may represent pivotal targets for TRALI induction (33). However, the interplay between pathogenic antibodies and their target cells remains unclear in the context of antibody-dependent TRALI pathogenesis. In this section, we discuss how antibodies or BRMs in transfused blood components respond to target cells and how the induced aberrant host response delineates the different pathogenic scenarios of TRALI (Figure 3).
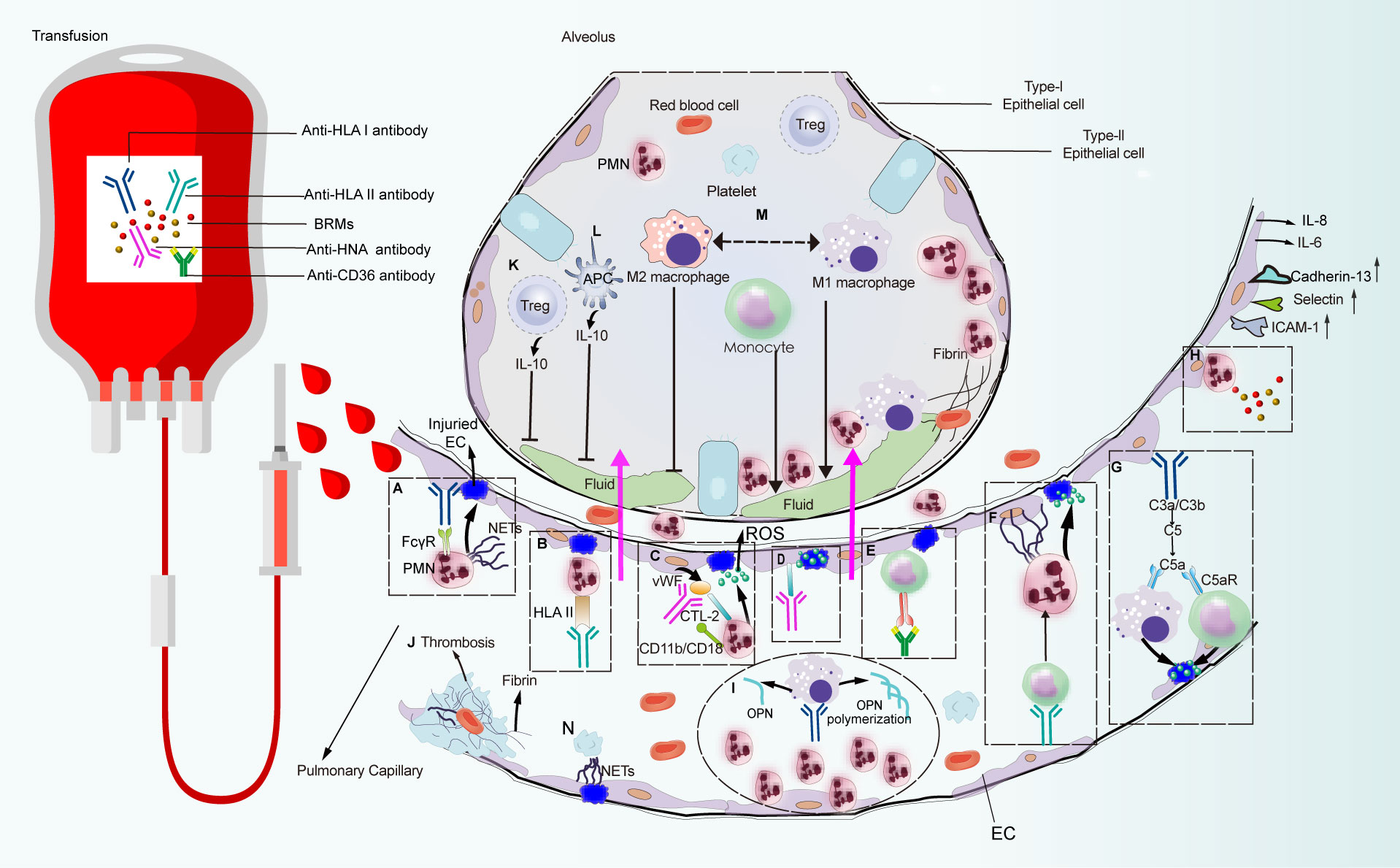
Figure 3 Pulmonary reaction and effector phase of TRALI pathogenesis. Pathways that are constituted with detrimental factors from blood products which connect with immune cells and non-hematopoietic cell lead to pulmonary edema or lung injury alleviation. Pathways (A–N) are systematically discussed in the main text. CTL-2, choline transporter-like protein-2.
Based on patient autopsy reports and preclinical studies on the TRALI reaction, PMNs are regarded as the main pathogenic cells mediating lung injury (13, 26). Among the initially reported TRALI cases, anti-HLA class antibodies were the most frequently identified antibody types in blood products, of which anti-HLA class I antibodies were the most prevalent (21, 34). Indeed murine models that mimic human anti-HLA class I antibody-dependent TRALI—administration of anti-MHC class I mAbs (clone 34–1–2S) against murine H2Kd, or the H-2d antigen—has advanced our understanding of this disease. The essential role of PMNs in TRALI was first identified in preclinical models, in which in vivo ablation of PMNs reportedly protected mice from anti-MHC-I-mediated TRALI development (23, 32). In the first genetically manipulated in vivo murine TRALI model, endothelial-bound 34-1-2S reacted with the pulmonary PMN Fc gamma receptor (FcγR), leading to PMN activation and lung injury (22) (Figure 3A). Although resident PMNs do not regularly express HLA class II, high levels are expressed on the surface of activated PMNs (35). Moreover, evidence from two-hit rat models revealed that transfusion of donor antibodies against HLA class II stimulated PMN activation and TRALI induction (35, 36) (Figure 3B). In a rat anti-HNA-2a-mediated TRALI model, the antibody directly activated PMNs in the presence of cognate antigen on the surface of PMNs in an EC-independent manner (37). Unlike other anti-leukocyte antibodies that participated in TRALI development, anti-HNA-3a antibodies failed to induce direct PMN activation and appeared to primarily interact with ECs (38) (Figure 3D). Following the activation of ECs, von Willebrand factor (vWF) was produced and released into circulation; subsequently, anti-HNA-3a antibodies bound to the trimolecular complex comprising choline transporter-like protein-2, vWF, and CD11b/CD18 on the surface of PMNs, leading to PMN activation and agglutination via CD11b/CD18 signal transduction, which may promote TRALI-associated endothelial leakage (38) (Figure 3C).
In the case of antibody-independent TRALI, lyso-PCs generated during blood storage caused PMN-mediated EC leakage in an in vitro TRALI model, in which human pulmonary microvascular endothelial cells (HPMVECs) were primed with LPS and co-cultured with PMNs followed by the addition of lyso-PCs as the second hit (39) (Figure 3H). CD40—the CD40L receptor—is distributed on the surface of leukocytes, platelets, and ECs (40). The interaction between CD40 and CD40L has been proposed as a proinflammatory feature and a pivotal pathogenic response to inflammation and organ injury (41). Stored platelet microparticles that released sCD40L boosted PMN-dependent HPMVEC damage, which may contribute to TRALI onset (40). Indeed, blocking the CD40-CD40L interaction with an anti-CD40L antibody protected mice against 34-1-2S-mediated TRALI development by suppressing PMN migration into the alveolar space (42, 43).
Although the aberrant activation of PMNs is recognized as the main feature of TRALI, the underlying molecular mechanisms driving PMN activation remain elusive. MicroRNAs (miRNAs)—single-stranded non-coding RNAs constituting approximately 22 nucleotides—contributed to lung injury by interacting with the 3′-untranslated region of mRNA (44). More specifically, miR-144 caused 34-1-2S-mediated TRALI by activating the NF-κB/CXCR1 signaling pathway via KLF2 in a PMN-dependent manner (45) (Figure 4A). Additionally, protein tyrosine phosphatase-1B (PTP1B) activated the PI3Kγ/AKT/mTOR-dependent CXCR axis, which may be involved in PMN activation (46) (Figure 4B). Anti-human leukocyte antigen-A2 (HLA-A2) antibody can function as an initiator of TRALI (47). In vitro coculture of anti-HLA-A2 antibody with PMNs resulted in PMN activation and subsequent endothelial permeability (48). The anti-HLA-A2 antibody may activate PMNs by stimulating NF-κB/NLRP3 inflammasome activation via increased abundance of phosphorylated-Src, thus aggravating TRALI (49) (Figure 4C).
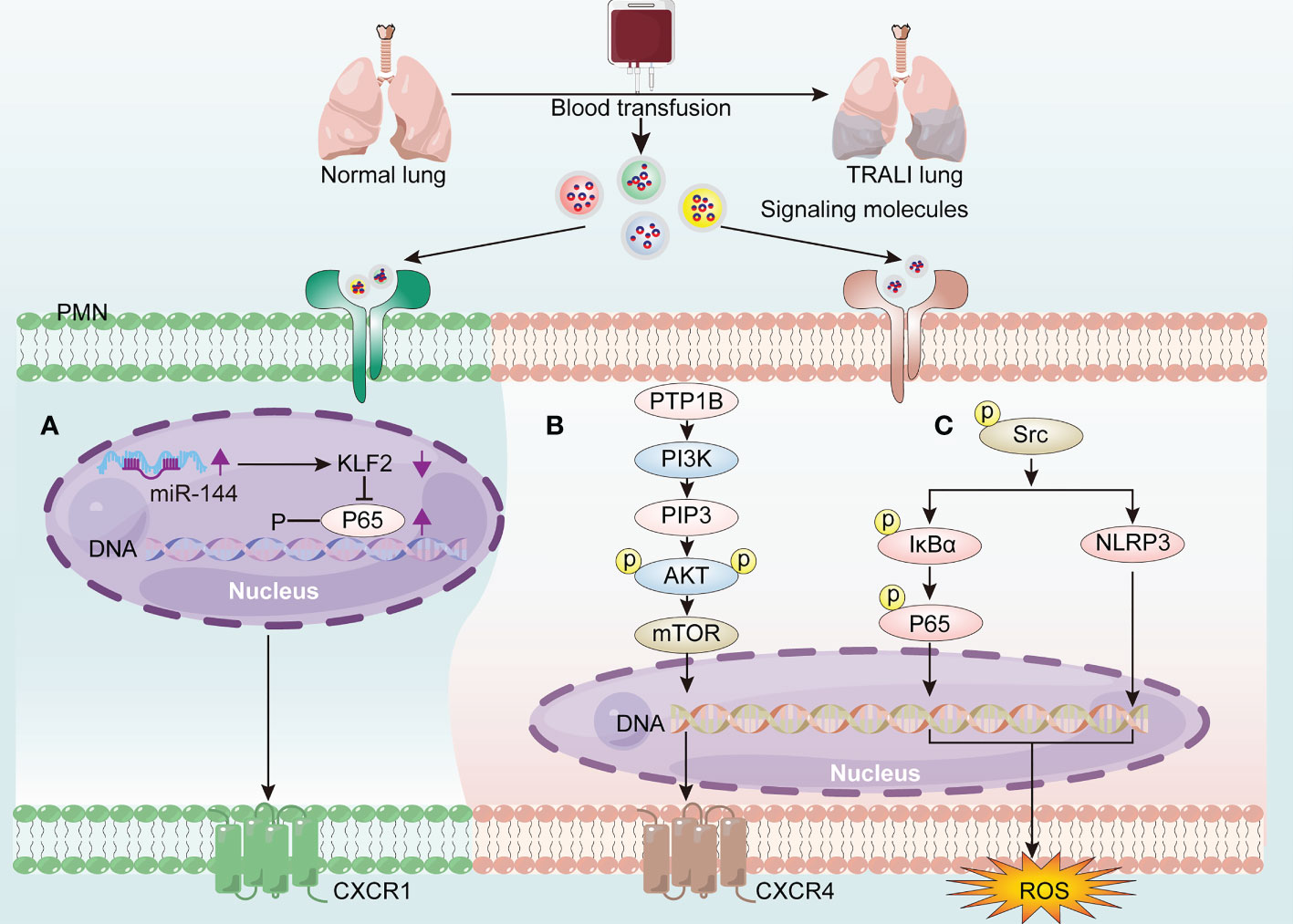
Figure 4 The molecular mechanisms of driving PMN activation in TRALI pathogenesis. Blood transfusion induces PMN activation via the generation of signaling molecules activating the receptors on PMNs, thereby exacerbating TRALI. (A) Up-regulated miR-144 promotes PMN activation through activating the NF-κB/CXCR1 signaling pathway via down-regulation of KLF2. (B) PTP1B activates PMNs via PI3Kγ/AKT/mTOR-dependent CXCR4 signaling. (C) HLA-A2 induces PMN-derived ROS production via activating NF-κB/NLRP3 inflammasome via phoosphorylate-Src elevation.
Anti-leukocyte antibodies are recognized as the main contributor to TRALI pathogenesis; however, the critical binding site of these antibodies remains unclear. HLA expression on nonleukocytic cells suggests ECs as probable targets for antibody binding. As early as 2011, infusion of 34-1-2S to a murine model of human TRALI suggested that the reaction between the antibody and MHC class I molecules on pulmonary ECs represented a probable initiating event for TRALI (50). This may be explained by the high expression of MHC-I on the surfaces of murine pulmonary ECs (51). Similarly, high pulmonary endothelial MHC-I expression has been observed in clinical TRALI (51). Given that the lung capillaries represent the first location encountered by the infused antibodies, the ECs may serve as a key site for antibody binding. Furthermore, targeted deletion of endothelial MHC-I alleviated 34-1-2S-mediated lung injury, while its restoration rendered mice susceptible to lung edema (51). Additionally, the interaction between 34-1-2S and pulmonary ECs may drive the production of complement component 5a (C5a) via activation of the complement cascade, thus contributing to TRALI development (50). Collectively, these findings highlight the importance of 34-1-2S engagement with endothelial MHC-I in the induction of TRALI.
ECs also express other leukocyte antigens, including HLA class II and HNA-3, the interaction of which, with their respective antibodies, reportedly contributed to TRALI development, i.e., alloantibodies against HNA-3a, not HNA-3b, have been linked to severe TRALI (52, 53). Anti-HNA-3a antibodies interacted with cognate antigens on the surface of pulmonary ECs in a PMN-independent manner, thereby precipitating TRALI (54, 55).
Moreover, ECs participate in antibody-independent TRALI via direct endothelial dysfunction. Transfusion of aged platelets elicited TRALI via acid sphingomyelinase (ASM)-forming ceramide-mediated endothelial apoptosis, further accelerating endothelial barrier failure (56). Furthermore, stored platelet-derived injurious ceramide enhanced the formation of extracellular vesicles in an ASM-dependent manner, which transported ceramide to the lung, resulting in endothelial barrier failure and TRALI onset (57, 58). Moreover, red blood cell (RBC) transfusion was a hazard factor for the induction of pulmonary respiratory failure (59, 60). RBC transfusion-mediated lung injury may be related to pulmonary ECs undergoing necrotic cell death, i.e., necroptosis (61, 62).
Collectively, these data support the evidence that ECs may be crucial regulators of TRALI.
The binding of anti-leukocyte antibodies to corresponding cognate antigens distributed on PMNs or pulmonary ECs may lead to PMN activation and induction of lung injury. However, given that PMN depletion did not prevent TRALI development in murine antibody-mediated TRALI (54), other cells have also been implicated. Although HLA class II antigen is not specifically expressed on PMNs or ECs, anti-HLA class II antibodies are consistently involved in TRALI onset. Indeed, plasma containing anti-HLA class II antibodies effectively sensitized peripheral blood mononuclear cells, leading to severe transfusion reactions via the production of inflammatory cytokines, such as IL-1β and IL-6, and chemokines (IL-8) by monocytes (63). Moreover, evidence from an ex vivo model showed that anti-HLA class II antibody-initiated monocyte activation was attributable to exaggerated HPMVEC permeability, which reflected endothelial damage in patients suffering from TRALI (64). Consistent with this result, anti-HLA class II antibody-containing plasma can cause matched monocyte activation, resulting in the initiation of a PMN amplification cascade and lung endothelial damage in an in vitro two-hit model (65) (Figure 3F). This finding has been further verified in an ex vivo lung model, which showed that anti-HLA class II antibody-mediated TRALI was dependent on monocytes, while PMNs were apparently dispensable (65). Following injection of 34-1-2S to severe combined immunodeficient (SCID) mice, the intact antibody reacted with its cognate antigen on peripheral blood monocytes, resulting in monocyte-dependent upregulation of MIP-2 expression, leading to recruitment of PMNs to the lung microvasculature, where the Fc domain of the antibody led to full TRALI initiation (66). Additionally, in place of FcγR-mediated PMNs activation, the C5a receptor on the surface of peripheral blood monocytes/macrophages responded to C5a derived from complement cascade activation, causing disease induction (50) (Figure 3G). Furthermore, individuals with CD36-deficient platelets and monocytes can develop isoantibodies against CD36 because of incompatible immune stimulation, which contributes to TRALI onset (67–69). The preliminary mechanism may be associated with anti-CD36 antibody-Fc receptor (FcR) binding to monocytes, thus inducing the TRALI reaction (70) (Figure 3E).
Macrophages have also been identified as major opsonins involved in TRALI induction. In vivo macrophage-derived OPN polymerization was involved in PMN chemotaxis (71), whereas macrophage elimination protected mice from antibody-mediated TRALI (26) (Figure 3I). Macrophages exert diverse functions, facilitated in part by their ability to undergo polarization (72), which enables them to readily adopt phenotypes based on stimulation within their microenvironment (73). Macrophages are roughly categorized as M1 or M2 according to phenotype. M1 macrophages are regarded as the proinflammatory phenotype with microbicidal potential, while M2 macrophages have a propensity to facilitate tissue repair and cell proliferation (72). Using a murine TRALI model, Wang et al. (74) confirmed that M1-polarized alveolar macrophages (AMs)—the proinflammatory phenotype—played a critical role in lung injury. Lung edema in TRALI mice can be abrogated by inhibiting the polarization of AMs toward the M1 phenotype (74) (Figure 3M).
These data suggest a clear role for monocytes/macrophages in TRALI pathogenesis.
Platelets derived from bone marrow megakaryocytes are commonly associated with thrombosis and hemostasis (75). However, beyond these traditional roles, platelets are key effectors of thromboinflammation and immune responses via crosstalk with various immune cells, such as PMNs (76). Moreover, considering the observed thrombocytopenia in TRALI patients, as well as the results of murine antibody-mediated TRALI models, platelets appear to be drivers of TRALI (13, 77). However, the evidence regarding the role of recipient platelets in aggravating lung injury is conflicting, i.e., the role of recipient platelets as either pathogenic or dispensable, in the context of TRALI development, within murine models injected with 34-1-2S is dependent on the experimental designs for assessing lung injury. On the one hand, treatment with rabbit polyclonal anti-mouse platelet serum to eliminate platelets, as well as 100 mg kg−1 aspirin or tirofiban to inhibit platelet reactivity, effectively prevented alveolar edema (23, 78, 79). Moreover, GPIb—a major platelet receptor—has been implicated in platelet activation and aggregation (76). However, platelet depletion, using an anti-GPIbα antibody, improved TRALI outcomes, without influencing its progression (80). On the other hand, several studies using different animal models have shown that platelet depletion or inhibition was dispensable in TRALI development. For example, neuraminidase-induced thrombocytopenia failed to effectively protect mice from TRALI (50, 81), diphtheria toxin-induced immunologic thrombocytopenia did not prevent PF4-cre/iDTR mice from 34-1-2S-associated lung edema (82), and pretreatment of mice with ML354—a protease receptor 4 pathway inhibitor—failed to prevent TRALI induction following activation of platelets via ex vivo stimulation (80).
Recently, novel insights regarding recipient platelets have been reported. Receptors bound to immunoglobulin G (IgG) antibody are uniquely expressed on human platelets and include IgG receptor I (FcγRI) and FcγRIIA, among others (83). Among these receptors, FcγRIIA/CD32A is the most extensively distributed FcγR on the surface of human platelets (83). Yet, its effects on TRALI have not been reported in mouse models, suggesting that the previous conclusions regarding the role of recipient platelets as main contributors to the pathogenicity of antibody-dependent TRALI are potentially incomplete. To address this gap in knowledge, EI Mdawar et al. (84) established an FcγRIIA/CD32A transgenic mouse model. When the humanized mouse model expressing the FcγRIIA/CD32A receptor was challenged with 34-1-2S, aggravated lung edema was observed (84). The authors concluded that the associated pathogenesis was caused by TRALI induction triggering platelet activation and subsequent 5-hydroxytryptamine 2A serotonin release. Thus, recipient platelets appear to represent key effector cells in exaggerated lung injury; however, their contribution to TRALI onset and pathogenesis requires further investigation.
Conflicting evidence has been reported regarding the role of recipient T cells in other inflammatory lung injury models. For example, T-cell activation may aggravate endotoxin-induced acute lung injury (ALI) (85). Meanwhile, mice treated with LPS developed ARDS, whereas lethal lung injury was significantly ameliorated by decreasing CD3+ T cells in vivo (86). Conversely, murine ARDS induced by reovirus was critically dependent on T cells and their ability to secret interferon-γ (17). In contrast, reconstituted T cells protected SCID mice from lung injury, implicating T cells as protective factors against TRALI development (17). This was further confirmed via removal of Tregs, DCs, and IL-10, resulting in murine TRALI initiation (32), which was likely related to the protective effect elicited by Treg- and DC-secreted IL-10. Indeed, both patients with low levels of IL-10 and mice lacking IL-10 were susceptible to TRALI development (32, 87). Hence, the Treg-DC-IL-10 axis has been proposed as a protective mechanism against TRALI induction (Figures 3K, L). However, to date, the mechanism underlying the protective effect of IL-10 remains unclear and warrants further investigation.
In this phase, activated effector cells, including PMNs, ECs, monocytes/macrophages, and platelets, produced effector molecules, such as NETs (Figures 3A, N) and ROS (Figures 3C, D, F, G), which directly damage pulmonary vascular ECs, resulting in lung edema. Additionally, EC dysfunction may aggravate pulmonary vascular leakage. Moreover, dysregulated coagulation has been implicated in lung injury, including that associated with TRALI and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) mediated ALI (77, 88).
NETs can effectively restrict pathogen spreading and kill microorganisms via the release of high doses of histones and granule contents by active immune cells (89). The role of NETs in TRALI was first reported in 2012 by Thomas, who observed NETs in the blood of patients and mice with TRALI (81). They further observed that PMN-derived NETs were released in vitro following challenge by an anti-leukocyte antibody. More recently, the formation of NETs in vivo, as well as their detrimental contributions, have been further validated in TRALI models (90, 91). The complement system has also been shown to function as a key trigger of NET production by assisting the 34-1-2S in PMN activation (51, 92). However, NETs are not exclusively produced by PMNs. Platelet-derived NETs can directly damage pulmonary ECs, further aggravating TRALI (93). Moreover, within a murine TRALI model, an increase in platelet–neutrophil interactions and NET formation was observed, with fewer NET produced following the administration of platelet-targeting therapeutics (78). Additionally, NET formation has been observed in antibody-independent TRALI. For example, under long-term storage, RBC hemolysis can occur and cause sufficient hemin accumulation in the solution (94). Hemin induced NET release, resulting in TRALI induction (94). Hence, NETs function as pivotal effector molecules of TRALI induction.
Upon exposure to inflammatory factors, the NADPH oxidase family members are activated, resulting in the production and release of a large amount of ROS, leading to lung disease (95). ROS are important effector molecules involved in TRALI pathogenesis. That is, trapped PMNs within the lung capillary bed being activated by anti-leukocyte antibodies and BRMs from transfused blood components, resulting in the production and release of ROS, which further destroy the ECs, causing capillary leakage and pulmonary edema (48, 96). PMNs are also recruited to the lungs, where they produce ROS that damage the ECs (26). Thus, PMN-derived ROS production blockade was an effective strategy for protecting mice from antibody-mediated TRALI (50, 97, 98). Activated monocytes/macrophages and pulmonary ECs also generated ROS, thus inducing deterioration of endothelial permeability and TRALI onset (50, 54). More importantly, gp91phox-KO mice, which lacked the ability to induce ROS, were protected from antibody-mediated TRALI, indicating that ROS were essential for TRALI induction (32). Storage of blood products can cause vesicle shedding and accumulation of hemin and lysoPC, which contributed to aberrant oxidative bursts, ROS production, and lung endothelial damage in vitro (39, 99–101). This may represent a potential pathogenesis for antibody-independent TRALI development.
Microthrombosis occured in the pulmonary vascular walls of animals with TRALI (84). Hence, considering that ECs are the key initiator and regulator of coagulation (102), coagulopathy may contribute to deteriorated lung function following transfusion of pathogenic antibodies or BRMs that would destroy pulmonary ECs. Indeed, coagulopathy has been identified as a pivotal pathogenic characteristic in rat models of aged erythrocyte- or platelet-mediated TRALI (103, 104). A case-control study reported that blood transfusions in patients who underwent cardiovascular surgery and exhibited obvious pulmonary inflammatory reactions were accompanied by coagulation and fibrinolysis dysregulation, leading to worsened prognosis in the ICU (105), thus further demonstrating the potential role of coagulopathy in TRALI development. Moreover, intravital lung imaging revealed platelet adhesion to the pulmonary ECs as well as occasional thrombotic events in a murine 34-1-2S-mediated TRALI model (51). Coagulopathy was involved in the dysregulation of coagulation and fibrinolytic pathways (106), leading to fibrin deposition in the lungs (77). Fibrin effusion in the lung alveoli was recognized as a main contributing factor to ALI and ARDS pathogenesis (107–109). Additionally, in a murine 34-1-2S-mediated TRALI model, increased coagulation, impaired fibrinolysis, and fibrin deposition were observed in the lungs (77) (Figure 3J). In fact, fibrin deposition in the lungs may activate ECs, thereby promoting the release of proinflammatory mediators and increasing vascular permeability and lung injury (77).
The morbidity associated with TRALI has plummeted with the introduction of mitigation strategies that include donor management, blood component processing, and patient blood management (PBM). However, no specific therapies are available and therapeutic options primarily focus on supportive measures. Hence, additional research is needed to develop, standardize, and evaluate novel drugs for the treatment of TRALI. Recently, a full spectrum of potential targets, including anti-inflammatory and immunoregulatory factors, have been investigated as potential TRALI therapeutics. Recent progress in prevention strategies (Table 1) and treatments (Table 2) has been made and is described below.
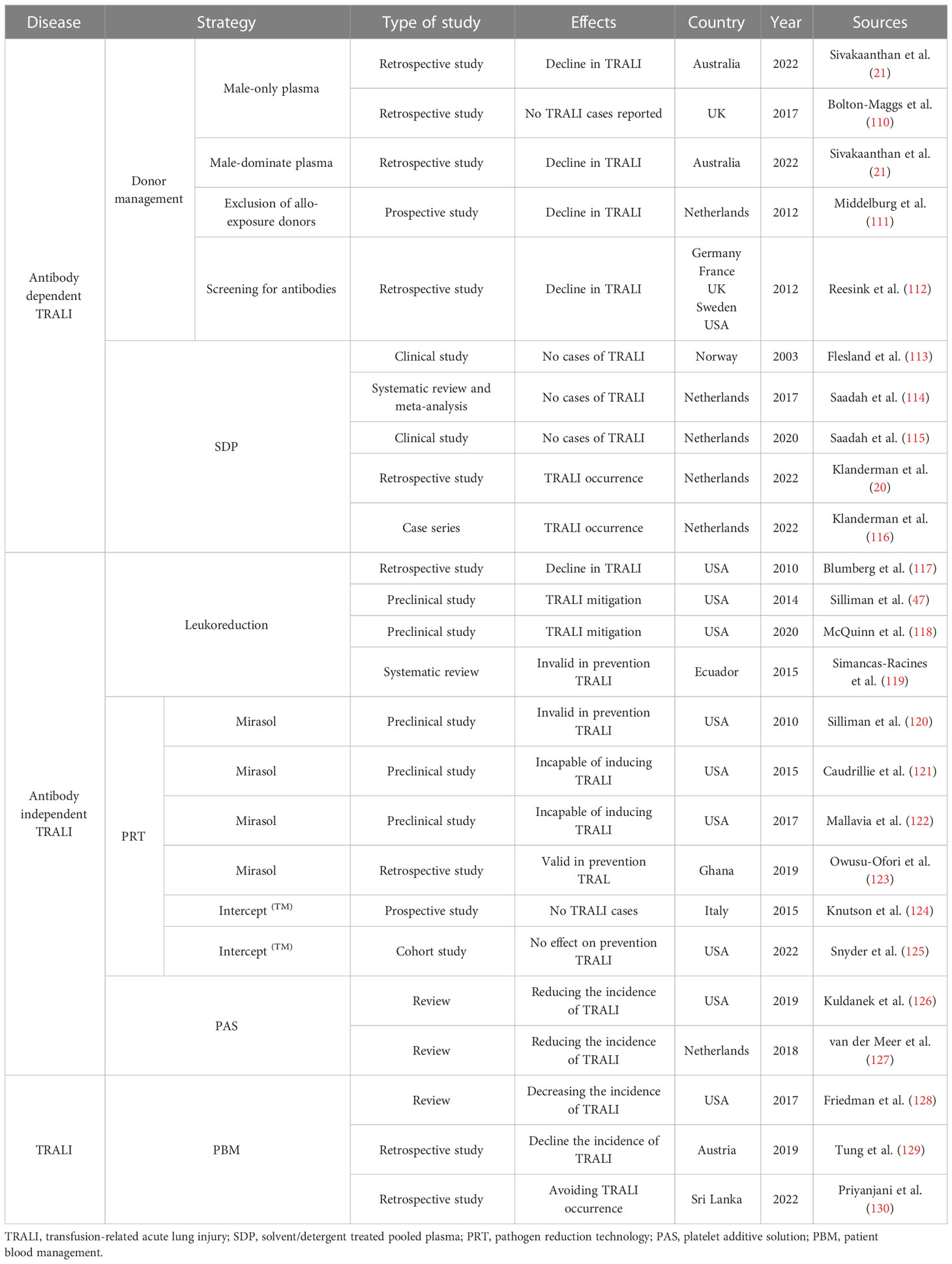
Table 1 Summary of the prevention strategies of TRALI.
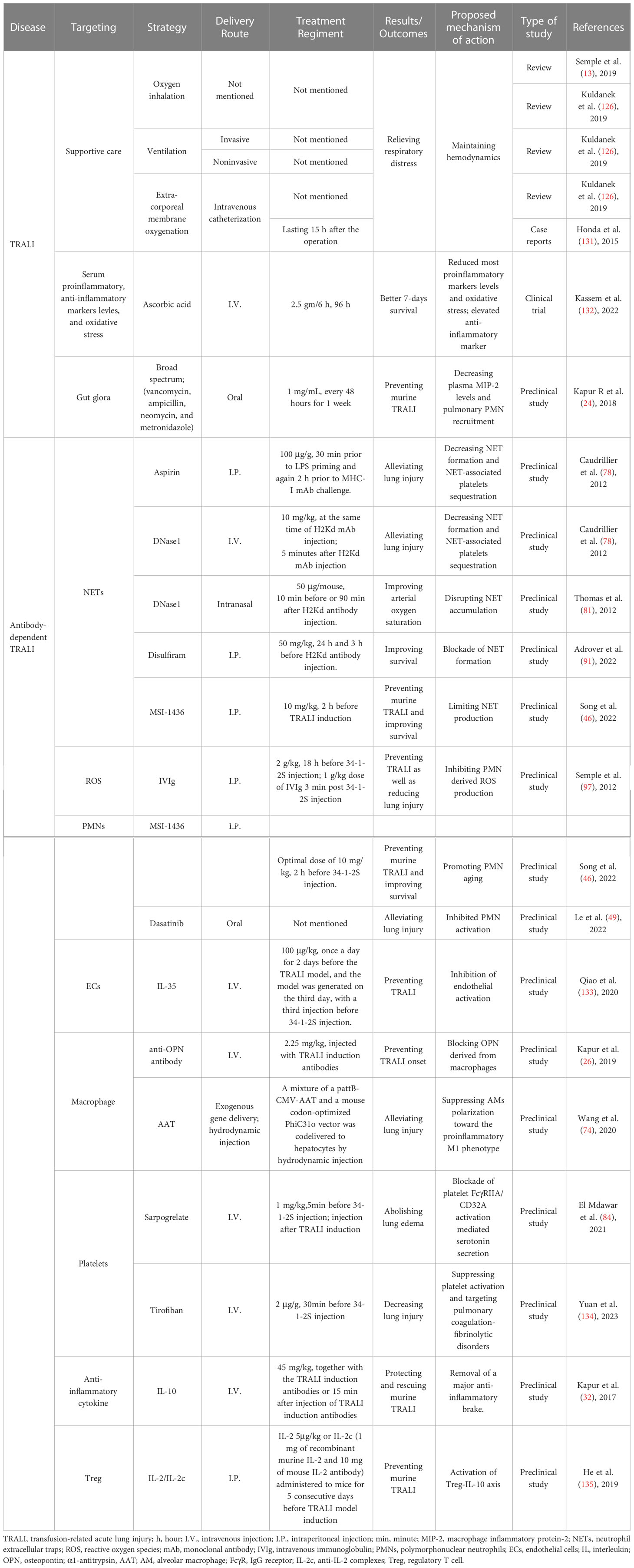
Table 2 Summary of innovative treatments undergoing TRALI.
Considering that anti-HLA or anti-HNA antibodies are detected most frequently in multiparous donors, TRALI risk-reduction strategies, including the introduction of male-only donors, male-dominated plasma, exclusion of all-exposure donors, and antibody screening, have been introduced, resulting in a significant reduction in the morbidity associated with antibody-dependent TRALI in developed countries (4, 21, 110–112). However, anti-leukocyte antibody screening has not yet been routinely implemented because of financial complexities (136). Despite the use of these prevention strategies, additional research is needed, as anti-HLA antibodies were reportedly triggered in coronavirus disease 19 convalescent plasma donors and SARS-CoV-2 vaccination volunteers (137, 138). Therefore, TRALI prevention may also require screening antibodies against HLA antigens in donors following SARS-CoV-2 sensitization.
Solvent/detergent-treated pooled plasma (SDP) is manufactured by treating large pools of plasma with solvent detergent to ensure that the anti-leukocyte antibody levels are below the limit of detection (116). Since the adoption of SDP in Norway in 1993, no TRALI cases were reported over a 10-year period (113). As of 2020, no TRALI cases had been reported in association with SDP transfusion (115). However, in 2022, a definite TRALI case was observed following SDP transfusion (20, 116), resulting in a total of three reported TRALI cases following SDP transfusion to date (116). Although implementation of SDP transfusion has markedly reduced the incidence of TRALI, medical staff associated with SDP transfusions must realize the potential risk regarding severe complications, i.e., TRALI.
Leukoreduction is a procedure that intentionally filters leukocytes in donated samples to decrease the abundance of mediators derived from leukocytes and platelets, thereby diminishing transfusion adverse reactions (117, 119). A retrospective analysis revealed that leukoreduction of blood components reduced the occurrence of TRALI cases by 83% (117). Furthermore, previous in vivo animal studies demonstrated that TRALI could be attenuated by leukoreduction before storage because of the reduction of proinflammatory mediators (47, 118). Conversely, leukoreduction of packed RBC failed to efficiently prevent TRALI, as reported in a systematic review (119). Hence, the effect of blood component leukoreduction on TRALI incidence should continue to be monitored.
The pathogen reduction technology (PRT) of Mirasol and Intercept(TM) has been proven effective in improving blood component safety by targeting pathogen nucleic acid or membrane lipid structures in donor samples (124, 139). However, the efficiency of PRT to reduce the risk of TRALI has been proven inconsistent in preclinical studies. An earlier in vivo study reported that blood products treated with Mirasol PRT failed to prevent rat TRALI (120). However, transfusion of Mirasol PRT-treated platelets has also prevented lung injury in immunodeficient mice (121). Meanwhile, transfusion of aged Mirasol PRT-treated RBC did not deteriorate this disease in a murine TRALI model parallel with standard-delivery RBC (122). These contradictory in vivo results may be because of discrepancies in experimental design.
The role of PRT in TRALI risk-reduction was also debated in clinical studies. An open label, prospective hemovigilance program confirmed that no TRALI cases occurred following transfusion with Intercept(TM)-processed platelets (124). Similarly, data from a hemovigilance system involving 2181 transfusion records in Ghana demonstrated that Mirasol PRT-treated whole blood effectively prevented TRALI (123). However, a recent open label, sequential cohort study found that ARDS-associated morbidity did not differ significantly between patients receiving Intercept (TM) PRT platelets and those receiving conventional platelets (125). Therefore, comprehensive clinical trials are required to validate the effect of PRT blood components on TRALI risk-reduction.
Following the introduction of risk-reduction strategies in 2014, the rate of TRALI did not differ between blood components (140). However, a significantly higher incidence of TRALI was sustained in female-donated platelets compared with that in male-donated platelets (140), which may be because of the platelets suspended in female plasma containing anti-HLA or anti-HNA antibodies. Meanwhile, buffy-coat-derived platelets suspended in platelet additive solution (PAS) can reduce TRALI-associated morbidity, which was similar to that reported in single-donor platelets (126, 127). Thus, additional strategies that may decrease the incidence of TRALI must be implemented, including replacement of traditional residual plasma with novel PAS in buffy coat pooled platelets.
PBM attempts to optimize the patient’s hematologic capabilities, decrease bleeding, and limit unnecessary transfusions (141). Findings from the hemovigilance network in the United States in 2015 highlighted the ability of PBM to decrease the incidence of TRALI (128). Similarly, a decrease in TRALI incidence was observed in Austria following the introduction of PBM (129). A recent study further emphasized that proper PBM represented the standard tool for avoiding TRALI occurrence (130).
To date, there are no established treatments for TRALI beyond supportive care, which comprises oxygen inhalation, ventilation, and extra-corporeal membrane oxygenation to effectively maintain hemodynamics (13, 126, 131). In rare cases of sickle cell disease, patients experiencing TRALI also receive 5% albumin, erythropoietin, and iron as supportive modalities (142). Although pulmonary edema is common, diuretics should be avoided because of the associated hypotension (143).
Ascorbic acid, a water-soluble vitamin, can limit ROS damage to the epithelial barrier. A randomized controlled trial revealed that critically ill patients with TRALI exhibited a better 7-day survival following intravenous administration of high-dose ascorbic acid (132). To fully explore more effective TRALI treatments, future large-scale multi-center randomized controlled trials and in-depth clinical studies are needed.
Activation of ECs, PMNs, monocytes/macrophages and platelets leading to effector molecule production and pulmonary fibrin deposition is the key process of the onset of pulmonary edema, the hallmark of TRALI. Hence, blocking these pathways may effectively prevent TRALI initiation. Additionally, targeting gut flora and Tregs/IL-10 seems to be prospective prevention strategies or treatments.
Since gut flora resulted in mice susceptibility to 34-1-2S-dependent TRALI, depletion of gut flora in mice may be a promising strategy for relieving TRALI. Utilizing broad-spectrum antibiotics composed of vancomycin, ampicillin, neomycin, and metronidazole in drinking water to kill gut flora can prevent TRALI in mice via decreasing plasma MIP-2 levels and PMN recruitment in the lungs (24).
Various compounds have been found to halt the effect of NETs. For instance, DNase I is an effective therapeutic drug for digesting NET present in the lungs (91). Combined aspirin and DNase1 treatment reduced NET formation and NET-associated platelet sequestration, thus alleviating TRALI (78). Moreover, intranasal DNase 1 treatment of mice before 34-1-2S infusion, or injection 90 min after TRALI occurrence, can disrupt NET accumulation in the alveoli and improve blood oxygenation (81). Meanwhile, inhibition of NET formation is also an effective means to prevent TRALI. Disulfiram—an existing FDA-approved drug for alcohol use disorder—effectively improved the survival of murine 34-1-2S-induced TRALI via blockade of NET formation (91).
Intravenous immunoglobulin (IVIg) is biologic components of polyclonal antibodies, extracted from human plasma of large healthy donor cohort blood banks. IVIg has proven beneficial for the clinical treatment of autoimmune diseases, chronic lymphocytic leukemia, and sepsis (144). To date, IVIg therapy remains off-label for patients with TRALI. Nevertheless, one study has assessed the efficacy of IVIg in a TRALI model and found that it prevented murine antibody-mediated TRALI and reduced lung injury at the level of PMN-derived ROS production, thus improving lung injury (97). Conversely, several clinical lines of evidence have reported that IVIg may contribute to TRALI induction (145, 146). Although IVIg may contribute to TRALI induction, this is extremely rare given the amount of IVIg infused annually. Due to these controversial findings, the therapeutic pathway associated with IVIg requires further elucidation.
The pivotal role of PMNs in TRALI provides a basic principle for exploiting PMN-targeted therapies. PMN aging is described as a “programmed disarming” that decreases the capacity of PMNs to inflict damage after infiltrating target tissues (147). Based on this theory, it is proposed that therapeutic intervention to regulate PMN aging may protect against tissue damage. PTP1B regulates signaling events that are of fundamental importance to homeostatic control (46). PTP1B inhibitors, including MSI-1436 and DPM-1003, have been tested in clinical trials for obesity and metastatic breast cancer (46). Inhibition of PTP1B with MSI-1436 effectively promoted PMN aging, thus preventing murine TRALI and improving survival (46). Dasatinib—a tyrosine kinase inhibitor commonly used to improve leukemia survival rates (148)—provided a clinical benefit in PMN-mediated inflammatory diseases by influencing the proinflammatory functions of mature PMNs (148). Moreover, dasatinib inhibited PMN activation in a TRALI mouse model by negatively regulating the NF-κB/NLRP3 inflammasome activation (49).
As endothelial activation is crucial to TRALI onset, it may represent an effective therapeutic target for preventing lung injury caused by blood transfusion. IL-35, which belongs to the IL-12 cytokine family, is regarded as a novel immune-suppressive cytokine (149). IL-35 may alleviate disease states by inhibiting pulmonary endothelial proliferation, apoptosis, and activation (133, 150). IL-35 treatment has been proven to prevent murine TRALI via inhibition of endothelial activation (133), which could provide new insights into therapeutic targets.
The treatments for targeting macrophages, including blocking the production of key effectors by macrophages and regulating macrophage polarization, represent potential TRALI therapies. OPN derived from macrophages is a matricellular protein that served a crucial role in PMN migration (26). Meanwhile, inhibition of OPN with an anti-OPN antibody in the presence of macrophages can prevent TRALI onset (26). Likewise, inducing polarization of macrophages toward the anti-inflammatory M2 phenotype has improved various disease states in preclinical models (151, 152). In particular, α1-antitrypsin (AAT) elicits tissue-protective effects as well as anti-inflammatory and immunomodulatory properties (74). Application of human AAT exogenous gene delivery technology to 34-1-2S-treated mouse livers successfully alleviated lung injury via suppressing M1 polarization (74). Hence, targeting macrophage-associated inflammatory proteins or promoting polarization toward the M2 phenotype may serve as prospective therapeutic strategies.
Sarpogrelate has been widely used in treating cardiovascular disorders via specifically antagonizing 5-HT2A receptor signaling (153). Thus, to investigate the role of 5-HT2A receptor in serotonin-mediated deterioration that caused pulmonary capillary leakage in humanized mice, sarpogrelate was employed (84). Administration of this drug to antibody-mediated TRALI in humanized mice abolished the exacerbation of lung edema (84). The significant effect of sarpogrelate on TRALI suggested that targeting activated platelet-mediated serotonin secretion may represent a potential TRALI therapy. Alternatively, tirofiban—a platelet receptor antagonist—possessed potential for preventing murine 34-1-2S-mediated TRALI via suppressing platelet activation and targeting pulmonary coagulation-fibrinolytic disorders (134).
Tregs are a subpopulation of CD4+ T cells that maintain tissue tolerance via secretion of anti-inflammatory cytokines (e.g., IL-10) (154, 155). Treg-based therapies have been proven promising for autoimmune diseases and allograft rejection (155, 156). In particular, IL-2 is a trophic cytokine required for the expansion of effector cells as well as Tregs (135), and the IL-2/anti-IL-2 antibody complex (IL-2c) induces vigorous T-cell proliferation in vivo (135). In a murine antibody-mediated TRALI model, administration of IL-2/IL-2c rapidly expanded the Treg population, thereby increasing the level of IL-10, which enabled mice to recover from lung injury (157). Given that IL-10 possesses potent anti-inflammatory and tissue regenerative capabilities, IL-10 administration prophylactically protected and therapeutically rescued against TRALI in vivo (13, 32, 158). These findings indicate that induction of Treg IL-10 production, or direct IL-10 treatment represent promising therapeutic strategies for TRALI.
Although TRALI is a leading cause of transfusion-associated death, specific treatments are still lacking. Thus, understanding its pathogenesis is key to establishing effective disease management strategies. Recently, significant progress has been made in defining TRALI pathophysiology. In particular, based on the two-hit theory, we innovatively define a three-step pathogenesis model for TRALI that is composed of the priming step, pulmonary reaction, and effector phase. As depicted in this model, the patient risk factors, in combination with detrimental factors within the blood transfusion components, trigger a dynamic spectrum of clinical manifestations based on complex host responses. These responses involve immune cells, including PMNs, platelets, Tregs, monocytes, macrophages; non-immune cells, such as ECs; and effector molecules, such as NETs, ROS, and coagulation-fibrinolytic disorders. In fact, the three-step pathogenesis model especially in pulmonary reaction and effector phase may be potentially overlapping.As such, application of a TRALI pathogenesis stage-specific management strategies may serve to improve disease progression. Hence, a better understanding of modifiable risk factors may help identify those patients most susceptible to TRALI onset. Following the introduction of strategies to exclude anti-leukocyte antibodies, BRMs, and associated pathogenic molecules from blood products, the incidence of TRALI in clinical studies and preclinical models has decreased rapidly. Additionally, a large number of in vivo drug experiments targeting PMNs, macrophages, platelets, Tregs, ECs, NETs, ROS, and coagulation-fibrinolytic disorders have effectively prevented the occurrence of lung edema. As these interventions have demonstrated efficacy in preventing TRALI occurrence in preclinical studies, their early implementation might benefit disease prognosis. One potential therapeutic agent is ascorbic acid, which has been proven beneficial for TRALI recovery in critical patients within a clinical trial. Further in-depth research is required to investigate effective drugs for targeted therapies to achieve better clinical outcomes.
YY and ZL conceptualized the review. YY drafted the manuscript, created the figures, and constructed the tables. ZL edited the final version of manuscript. ZL obtained the funding and provided the financial resource. All authors contributed to the article and approved the submitted version.
This study was supported by the Technology Innovation Project of Chengdu Science and Technology Bureau (2022-YF05-02122-SN).
We gratefully appreciate the generous assistance from colleagues at the Department of Blood Transfusion, the Third People’s Hospital of Chengdu, Affiliated Hospital of Southwest Jiaotong University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Morsing SKH, Zeeuw van der Laan E, van Stalborch AD, van Buul JD, Kapur R, Vlaar AP. A pulmonary endothelial amplification loop aggravates ex-vivo transfusion-related acute lung injury Via increased toll-like receptor 4 and intra-cellular adhesion molecule-1 expression. Transfusion (2022) 62(10):1961–6. doi: 10.1111/trf.17076
2. Guo K, Ma S. The immune system in transfusion-related acute lung injury prevention and therapy: update and perspective. Front Mol Biosci (2021) 8:639976. doi: 10.3389/fmolb.2021.639976
3. Vlaar APJ, Toy P, Fung M, Looney MR, Juffermans NP, Bux J, et al. A consensus redefinition of transfusion-related acute lung injury. Transfusion (2019) 59(7):2465–76. doi: 10.1111/trf.15311
4. US Food and Drug Administration. Fatalities reported to fda following blood collection and transfusion annual summary for fy 2020 (2021). Available at: https://www.fda.gov/media/160859/download.
5. Zeeuw van der Laan EAN, van der Velden S, Porcelijn L, Semple JW, van der Schoot CE, Kapur R. Update on the pathophysiology of transfusion-related acute lung injury. Curr Opin Hematol (2020) 27(6):386–91. doi: 10.1097/moh.0000000000000607
6. Benson AB, Moss M, Silliman CC. Transfusion-related acute lung injury (Trali): a clinical review with emphasis on the critically ill. Br J haematology (2009) 147(4):431–43. doi: 10.1111/j.1365-2141.2009.07840.x
7. Vlaar AP, Juffermans NP. Transfusion-related acute lung injury: a clinical review. Lancet (London England) (2013) 382(9896):984–94. doi: 10.1016/s0140-6736(12)62197-7
8. Wallis JP. Transfusion-related acute lung injury (Trali)–under-Diagnosed and under-reported. Br J anaesthesia (2003) 90(5):573–6. doi: 10.1093/bja/aeg101
9. Vlaar AP, Binnekade JM, Prins D, van Stein D, Hofstra JJ, Schultz MJ, et al. Risk factors and outcome of transfusion-related acute lung injury in the critically ill: a nested case-control study. Crit Care Med (2010) 38(3):771–8. doi: 10.1097/CCM.0b013e3181cc4d4b
10. Brittingham TE. Immunologic studies on leukocytes. Vox sanguinis (1957) 2(4):242–8. doi: 10.1111/j.1423-0410.1957.tb03699.x
11. Popovsky MA, Abel MD, Moore SB. Transfusion-related acute lung injury associated with passive transfer of antileukocyte antibodies. Am Rev Respir Dis (1983) 128(1):185–9. doi: 10.1164/arrd.1983.128.1.185
12. Bayat B, Nielsen KR, Bein G, Traum A, Burg-Roderfeld M, Sachs UJ. Transfusion of target antigens to preimmunized recipients: a new mechanism in transfusion-related acute lung injury. Blood Adv (2021) 5(20):3975–85. doi: 10.1182/bloodadvances.2020003843
13. Semple JW, Rebetz J, Kapur R. Transfusion-associated circulatory overload and transfusion-related acute lung injury. Blood (2019) 133(17):1840–53. doi: 10.1182/blood-2018-10-860809
14. Peters AL, van Hezel ME, Juffermans NP, Vlaar AP. Pathogenesis of non-antibody mediated transfusion-related acute lung injury from bench to bedside. Blood Rev (2015) 29(1):51–61. doi: 10.1016/j.blre.2014.09.007
15. Silliman CC, Boshkov LK, Mehdizadehkashi Z, Elzi DJ, Dickey WO, Podlosky L, et al. Transfusion-related acute lung injury: epidemiology and a prospective analysis of etiologic factors. Blood (2003) 101(2):454–62. doi: 10.1182/blood-2002-03-0958
16. Silliman CC, Paterson AJ, Dickey WO, Stroneck DF, Popovsky MA, Caldwell SA, et al. The association of biologically active lipids with the development of transfusion-related acute lung injury: a retrospective study. Transfusion (1997) 37(7):719–26. doi: 10.1046/j.1537-2995.1997.37797369448.x
17. Fung YL, Kim M, Tabuchi A, Aslam R, Speck ER, Chow L, et al. Recipient T lymphocytes modulate the severity of antibody-mediated transfusion-related acute lung injury. Blood (2010) 116(16):3073–9. doi: 10.1182/blood-2010-05-284570
18. Menis M, Anderson SA, Forshee RA, McKean S, Johnson C, Warnock R, et al. Transfusion-related acute lung injury and potential risk factors among the inpatient us elderly as recorded in Medicare claims data, during 2007 through 2011. Transfusion (2014) 54(9):2182–93. doi: 10.1111/trf.12626
19. Yokoyama A, Sakamoto Y, Jo T, Urushiyama H, Tamiya H, Tanaka G, et al. Pulmonary disease as a risk factor for transfusion-related acute lung injury. ERJ Open Res (2021) 7(3):00039–2021. doi: 10.1183/23120541.00039-2021
20. Klanderman RB, van Mourik N, Eggermont D, Peters AL, Tuinman PR, Bosman R, et al. Incidence of transfusion-related acute lung injury temporally associated with Solvent/Detergent plasma use in the icu: a retrospective before and after implementation study. Transfusion (2022) 62(9):1752–62. doi: 10.1111/trf.17049
21. Sivakaanthan A, Swain F, Pahn G, Goodison K, Gutta N, Holdsworth R, et al. Transfusion-related acute lung injury (Trali): a retrospective review of reported cases in Queensland, Australia over 20 years. Blood transfusion = Trasfusione del sangue (2022) 20(6):454–64. doi: 10.2450/2022.0020-22
22. Looney MR, Su X, Van Ziffle JA, Lowell CA, Matthay MA. Neutrophils and their fc gamma receptors are essential in a mouse model of transfusion-related acute lung injury. J Clin Invest (2006) 116(6):1615–23. doi: 10.1172/jci27238
23. Looney MR, Nguyen JX, Hu Y, Van Ziffle JA, Lowell CA, Matthay MA. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J Clin Invest (2009) 119(11):3450–61. doi: 10.1172/jci38432
24. Kapur R, Kim M, Rebetz J, Hallström B, Björkman JT, Takabe-French A, et al. Gastrointestinal microbiota contributes to the development of murine transfusion-related acute lung injury. Blood Adv (2018) 2(13):1651–63. doi: 10.1182/bloodadvances.2018018903
25. Kapur R, Kim M, Shanmugabhavananthan S, Liu J, Li Y, Semple JW. C-reactive protein enhances murine antibody-mediated transfusion-related acute lung injury. Blood (2015) 126(25):2747–51. doi: 10.1182/blood-2015-09-672592
26. Kapur R, Kasetty G, Rebetz J, Egesten A, Semple JW. Osteopontin mediates murine transfusion-related acute lung injury Via stimulation of pulmonary neutrophil accumulation. Blood (2019) 134(1):74–84. doi: 10.1182/blood.2019000972
27. Tung JP, Chiaretti S, Dean MM, Sultana AJ, Reade MC, Fung YL. Transfusion-related acute lung injury (Trali): potential pathways of development, strategies for prevention and treatment, and future research directions. Blood Rev (2022) 53:100926. doi: 10.1016/j.blre.2021.100926
28. Peters AL, Van Stein D, Vlaar AP. Antibody-mediated transfusion-related acute lung injury; from discovery to prevention. Br J haematology (2015) 170(5):597–614. doi: 10.1111/bjh.13459
29. Morsing SKH, Zeeuw van der Laan E, van Stalborch AD, van Buul JD, Vlaar APJ, Kapur R. Endothelial cells of pulmonary origin display unique sensitivity to the bacterial endotoxin lipopolysaccharide. Physiol Rep (2022) 10(8):e15271. doi: 10.14814/phy2.15271
30. Giacalone VD, Margaroli C, Mall MA, Tirouvanziam R. Neutrophil adaptations upon recruitment to the lung: new concepts and implications for homeostasis and disease. Int J Mol Sci (2020) 21(3):851. doi: 10.3390/ijms21030851
31. Gao F, Lucke-Wold BP, Li X, Logsdon AF, Xu LC, Xu S, et al. Reduction of endothelial nitric oxide increases the adhesiveness of constitutive endothelial membrane icam-1 through src-mediated phosphorylation. Front Physiol (2017) 8:1124. doi: 10.3389/fphys.2017.01124
32. Kapur R, Kim M, Aslam R, McVey MJ, Tabuchi A, Luo A, et al. T Regulatory cells and dendritic cells protect against transfusion-related acute lung injury Via il-10. Blood (2017) 129(18):2557–69. doi: 10.1182/blood-2016-12-758185
33. Qian Y, Wang Z, Lin H, Lei T, Zhou Z, Huang W, et al. Trim47 is a novel endothelial activation factor that aggravates lipopolysaccharide-induced acute lung injury in mice Via K63-linked ubiquitination of Traf2. Signal transduction targeted Ther (2022) 7(1):148. doi: 10.1038/s41392-022-00953-9
34. Simtong P, Sudwilai Y, Cheunta S, Leelayuwat C, Romphruk AV. Prevalence of leucocyte antibodies in non-transfused Male and female platelet apheresis donors. Transfusion Med (Oxford England) (2021) 31(3):186–92. doi: 10.1111/tme.12781
35. Kelher MR, Banerjee A, Gamboni F, Anderson C, Silliman CC. Antibodies to major histocompatibility complex class ii antigens directly prime neutrophils and cause acute lung injury in a two-event in vivo rat model. Transfusion (2016) 56(12):3004–11. doi: 10.1111/trf.13817
36. Kelher MR, Silliman CC. Antibodies specific for mhc class ii antigens cause trali directly in a two-event in vivo model. Blood (2012) 120:21. doi: 10.1182/blood.V120.21.845.845
37. Sachs UJ, Hattar K, Weissmann N, Bohle RM, Weiss T, Sibelius U, et al. Antibody-induced neutrophil activation as a trigger for transfusion-related acute lung injury in an ex vivo rat lung model. Blood (2006) 107(3):1217–9. doi: 10.1182/blood-2005-04-1744
38. Bayat B, Tjahjono Y, Berghöfer H, Werth S, Deckmyn H, De Meyer SF, et al. Choline transporter-like protein-2: new Von willebrand factor-binding partner involved in antibody-mediated neutrophil activation and transfusion-related acute lung injury. Arteriosclerosis thrombosis Vasc Biol (2015) 35(7):1616–22. doi: 10.1161/atvbaha.115.305259
39. Wyman TH, Bjornsen AJ, Elzi DJ, Smith CW, England KM, Kelher M, et al. A two-insult in vitro model of pmn-mediated pulmonary endothelial damage: requirements for adherence and chemokine release. Am J Physiol Cell Physiol (2002) 283(6):C1592–603. doi: 10.1152/ajpcell.00540.2001
40. Xie RF, Hu P, Wang ZC, Yang J, Yang YM, Gao L, et al. Platelet-derived microparticles induce polymorphonuclear leukocyte-mediated damage of human pulmonary microvascular endothelial cells. Transfusion (2015) 55(5):1051–7. doi: 10.1111/trf.12952
41. Ots HD, Tracz JA, Vinokuroff KE, Musto AE. Cd40-Cd40l in neurological disease. Int J Mol Sci (2022) 23(8):4115. doi: 10.3390/ijms23084115
42. Tariket S, Hamzeh-Cognasse H, Laradi S, Arthaud CA, Eyraud MA, Bourlet T, et al. Evidence of Cd40l/Cd40 pathway involvement in experimental transfusion-related acute lung injury. Sci Rep (2019) 9(1):12536. doi: 10.1038/s41598-019-49040-0
43. Tariket S, Hamzeh-Cognasse H, Arthaud CA, Laradi S, Bourlet T, Berthelot P, et al. Inhibition of the Cd40/Cd40l complex protects mice against Ali-induced pancreas degradation. Transfusion (2019) 59(3):1090–101. doi: 10.1111/trf.15206
44. Lu Q, Yu S, Meng X, Shi M, Huang S, Li J, et al. Micrornas: important regulatory molecules in acute lung Injury/Acute respiratory distress syndrome. Int J Mol Sci (2022) 23(10):5545. doi: 10.3390/ijms23105545
45. Le A, Wu Y, Liu W, Wu C, Hu P, Zou J, et al. Mir-144-Induced Klf2 inhibition and nf-Kappab/Cxcr1 activation promote neutrophil extracellular trap-induced transfusion-related acute lung injury. J Cell Mol Med (2021) 25(14):6511–23. doi: 10.1111/jcmm.16650
46. Song D, Adrover JM, Felice C, Christensen LN, He XY, Merrill JR, et al. Ptp1b inhibitors protect against acute lung injury and regulate Cxcr4 signaling in neutrophils. JCI Insight (2022) 7(14):e158199. doi: 10.1172/jci.insight.158199
47. Silliman CC, Kelher MR, Khan SY, LaSarre M, West FB, Land KJ, et al. Experimental prestorage filtration removes antibodies and decreases lipids in rbc supernatants mitigating trali in vivo. Blood (2014) 123(22):3488–95. doi: 10.1182/blood-2013-10-532424
48. Khoy K, Nguyen MVC, Masson D, Bardy B, Drouet C, Paclet MH. Transfusion-related acute lung injury: critical neutrophil activation by anti-Hla-A2 antibodies for endothelial permeability. Transfusion (2017) 57(7):1699–708. doi: 10.1111/trf.14134
49. Le A, Liu W, Wu C, Hu P, Zou J, Wu Y, et al. Polymorphonuclear neutrophil activation by src phosphorylation contributes to hla-A2 antibody-induced transfusion-related acute lung injury. Mol Immunol (2022) 150:9–19. doi: 10.1016/j.molimm.2022.04.010
50. Strait RT, Hicks W, Barasa N, Mahler A, Khodoun M, Köhl J, et al. Mhc class I-specific antibody binding to nonhematopoietic cells drives complement activation to induce transfusion-related acute lung injury in mice. J Exp Med (2011) 208(12):2525–44. doi: 10.1084/jem.20110159
51. Cleary SJ, Kwaan N, Tian JJ, Calabrese DR, Mallavia B, Magnen M, et al. Complement activation on endothelium initiates antibody-mediated acute lung injury. J Clin Invest (2020) 130(11):5909–23. doi: 10.1172/jci138136
52. Gottschall JL, Triulzi DJ, Curtis B, Kakaiya RM, Busch MP, Norris PJ, et al. The frequency and specificity of human neutrophil antigen antibodies in a blood donor population. Transfusion (2011) 51(4):820–7. doi: 10.1111/j.1537-2995.2010.02913.x
53. Reil A, Wesche J, Greinacher A, Bux J. Geno- and phenotyping and immunogenicity of hna-3. Transfusion (2011) 51(1):18–24. doi: 10.1111/j.1537-2995.2010.02751.x
54. Bayat B, Tjahjono Y, Sydykov A, Werth S, Hippenstiel S, Weissmann N, et al. Anti-human neutrophil antigen-3a induced transfusion-related acute lung injury in mice by direct disturbance of lung endothelial cells. Arteriosclerosis thrombosis Vasc Biol (2013) 33(11):2538–48. doi: 10.1161/atvbaha.113.301206
55. Chiaretti S, Sultana A, Temple F, Burton M, Pahn G, Flower R, et al. Donor anti-Hna-3a antibodies induce monocytemediated hlmvec damage in a 2 insult in vitro model. Internal Med J (2019) 49:34. doi: 10.1111/imj.14678
56. McVey MJ, Kim M, Tabuchi A, Srbely V, Japtok L, Arenz C, et al. Acid sphingomyelinase mediates murine acute lung injury following transfusion of aged platelets. Am J Physiol Lung Cell Mol Physiol (2017) 312(5):L625–l37. doi: 10.1152/ajplung.00317.2016
57. McVey MJ, Weidenfeld S, Maishan M, Spring C, Kim M, Tabuchi A, et al. Platelet extracellular vesicles mediate transfusion-related acute lung injury by imbalancing the sphingolipid rheostat. Blood (2021) 137(5):690–701. doi: 10.1182/blood.2020005985
58. Cleary SJ, Looney MR. Chewing the fat on trali. Blood (2021) 137(5):586–7. doi: 10.1182/blood.2020010034
59. Sampson AC, Lassiter BP, Gregory Rivera M, Hair PS, Jackson KG, Enos AI, et al. Peptide inhibition of acute lung injury in a novel two-hit rat model. PloS One (2021) 16(10):e0259133. doi: 10.1371/journal.pone.0259133
60. Hu A, Chen W, Wu S, Pan B, Zhu A, Yu X, et al. An animal model of transfusion-related acute lung injury and the role of soluble Cd40 ligand. Vox sanguinis (2020) 115(4):303–13. doi: 10.1111/vox.12895
61. Qing DY, Conegliano D, Shashaty MG, Seo J, Reilly JP, Worthen GS, et al. Red blood cells induce necroptosis of lung endothelial cells and increase susceptibility to lung inflammation. Am J Respir Crit Care Med (2014) 190(11):1243–54. doi: 10.1164/rccm.201406-1095OC
62. Faust H, Lam LM, Hotz MJ, Qing D, Mangalmurti NS. Rage interacts with the necroptotic protein Ripk3 and mediates transfusion-induced danger signal release. Vox sanguinis (2020) 115(8):729–34. doi: 10.1111/vox.12946
63. Sakagawa H, Miyazaki T, Fujihara M, Sato S, Yamaguchi M, Fukai K, et al. Generation of inflammatory cytokines and chemokines from peripheral blood mononuclear cells by hla class ii antibody-containing plasma unit that was associated with severe nonhemolytic transfusion reactions. Transfusion (2007) 47(1):154–61. doi: 10.1111/j.1537-2995.2007.01078.x
64. Wakamoto S, Fujihara M, Takahashi D, Niwa K, Sato S, Kato T, et al. Enhancement of endothelial permeability by coculture with peripheral blood mononuclear cells in the presence of hla class ii antibody that was associated with transfusion-related acute lung injury. Transfusion (2011) 51(5):993–1001. doi: 10.1111/j.1537-2995.2010.02910.x
65. Sachs UJ, Wasel W, Bayat B, Bohle RM, Hattar K, Berghöfer H, et al. Mechanism of transfusion-related acute lung injury induced by hla class ii antibodies. Blood (2011) 117(2):669–77. doi: 10.1182/blood-2010-05-286146
66. McKenzie CG, Kim M, Singh TK, Milev Y, Freedman J, Semple JW. Peripheral blood monocyte-derived chemokine blockade prevents murine transfusion-related acute lung injury (Trali). Blood (2014) 123(22):3496–503. doi: 10.1182/blood-2013-11-536755
67. Xu X, Chen D, Ye X, Xia W, Shao Y, Deng J, et al. Improvement of anti-Cd36 antibody detection Via monoclonal antibody immobilization of platelet antigens assay by using selected monoclonal antibodies. Ann Lab Med (2023) 43(1):86–91. doi: 10.3343/alm.2023.43.1.86
68. Ando M. Cd36, an important antigen on platelets and monocytes. Vox sanguinis (2020) 115(SUPPL 1):27. doi: 10.1111/vox.13031
69. Nakajima F, Nishimura M, Hashimoto S, Okazaki H, Tadokoro K. Role of anti-nak(a) antibody, monocytes and platelets in the development of transfusion-related acute lung injury. Vox sanguinis (2008) 95(4):318–23. doi: 10.1111/j.1423-0410.2008.01095.x
70. Wu Y, Santoso S, Fu Y. Anti-Cd36 antibodies induced transfusion-related acute lung injury in mice. Vox sanguinis (2020) 115(SUPPL 1):55–6. doi: 10.1111/vox.13031
71. Nishimichi N, Hayashita-Kinoh H, Chen C, Matsuda H, Sheppard D, Yokosaki Y. Osteopontin undergoes polymerization in vivo and gains chemotactic activity for neutrophils mediated by integrin Alpha9beta1. J Biol Chem (2011) 286(13):11170–8. doi: 10.1074/jbc.M110.189258
72. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: different gene signatures in M1(Lps+) vs. classically and M2(Lps-) vs. alternatively activated macrophages. Front Immunol (2019) 10:1084. doi: 10.3389/fimmu.2019.01084
73. Gharavi AT, Hanjani NA, Movahed E, Doroudian M. The role of macrophage subtypes and exosomes in immunomodulation. Cell Mol Biol Lett (2022) 27(1):83. doi: 10.1186/s11658-022-00384-y
74. Wang L, Wu T, Yan S, Wang Y, An J, Wu C, et al. M1-polarized alveolar macrophages are crucial in a mouse model of transfusion-related acute lung injury. Transfusion (2020) 60(2):303–16. doi: 10.1111/trf.15609
75. Burnouf T, Walker TL. The multifaceted role of platelets in mediating brain function. Blood (2022) 140(8):815–27. doi: 10.1182/blood.2022015970
76. Mandel J, Casari M, Stepanyan M, Martyanov A, Deppermann C. Beyond hemostasis: platelet innate immune interactions and thromboinflammation. Int J Mol Sci (2022) 23(7):3868. doi: 10.3390/ijms23073868
77. Yu Y, Jiang P, Sun P, Su N, Lin F. Pulmonary coagulation and fibrinolysis abnormalities that favor fibrin deposition in the lungs of mouse antibody-mediated transfusion-related acute lung injury. Mol Med Rep (2021) 24(2):601. doi: 10.3892/mmr.2021.12239
78. Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest (2012) 122(7):2661–71. doi: 10.1172/jci61303
79. Ortiz-Muñoz G, Mallavia B, Bins A, Headley M, Krummel MF, Looney MR. Aspirin-triggered 15-Epi-Lipoxin A4 regulates neutrophil-platelet aggregation and attenuates acute lung injury in mice. Blood (2014) 124(17):2625–34. doi: 10.1182/blood-2014-03-562876
80. Cognasse F, Tariket S, Hamzeh-Cognasse H, Arthaud CA, Eyraud MA, Bourlet T, et al. Platelet depletion limits the severity but does not prevent the occurrence of experimental transfusion-related acute lung injury. Transfusion (2020) 60(4):713–23. doi: 10.1111/trf.15738
81. Thomas GM, Carbo C, Curtis BR, Martinod K, Mazo IB, Schatzberg D, et al. Extracellular DNA traps are associated with the pathogenesis of trali in humans and mice. Blood (2012) 119(26):6335–43. doi: 10.1182/blood-2012-01-405183
82. Hechler B, Maître B, Magnenat S, Heim V, El Mdawar MB, Gachet C, et al. Platelets are dispensable for antibody-mediated transfusion-related acute lung injury in the mouse. J Thromb haemostasis JTH (2016) 14(6):1255–67. doi: 10.1111/jth.13335
83. Beutier H, Hechler B, Godon O, Wang Y, Gillis CM, de Chaisemartin L, et al. Platelets expressing igg receptor Fcγriia/Cd32a determine the severity of experimental anaphylaxis. Sci Immunol (2018) 3(22):eaan5997. doi: 10.1126/sciimmunol.aan5997
84. El Mdawar MB, Maître B, Magnenat S, Tupin F, Jönsson F, Gachet C, et al. Platelet fcγriia-induced serotonin release exacerbates the severity of transfusion-related acute lung injury in mice. Blood Adv (2021) 5(23):4817–30. doi: 10.1182/bloodadvances.2021004336
85. Nakajima T, Lin KW, Li J, McGee HS, Kwan JM, Perkins DL, et al. T Cells and lung injury. Impact Rapamycin Am J Respir Cell Mol Biol (2014) 51(2):294–9. doi: 10.1165/rcmb.2013-0171OC
86. Yuan R, Li Y, Han S, Chen X, Chen J, He J, et al. Fe-curcumin nanozyme-mediated reactive oxygen species scavenging and anti-inflammation for acute lung injury. ACS Cent Sci (2022) 8(1):10–21. doi: 10.1021/acscentsci.1c00866
87. Kapur R, Kim M, Rebetz J, Rondina MT, Porcelijn L, Semple JW. Low levels of interleukin-10 in patients with transfusion-related acute lung injury. Ann Trans Med (2017) 5(16):339. doi: 10.21037/atm.2017.04.37
88. Lee C, Choi WJ. Overview of covid-19 inflammatory pathogenesis from the therapeutic perspective. Arch pharmacal Res (2021) 44(1):99–116. doi: 10.1007/s12272-020-01301-7
89. Herre M, Cedervall J, Mackman N, Olsson AK. Neutrophil extracellular traps in the pathology of cancer and other inflammatory diseases. Physiol Rev (2023) 103(1):277–312. doi: 10.1152/physrev.00062.2021
90. Aroca-Crevillén A, Hidalgo A, Adrover JM. In vivo imaging of circadian net formation during lung injury by four-dimensional intravital microscopy. Methods Mol Biol (Clifton NJ) (2022) 2482:285–300. doi: 10.1007/978-1-0716-2249-0_19
91. Adrover JM, Carrau L, Daßler-Plenker J, Bram Y, Chandar V, Houghton S, et al. Disulfiram inhibits neutrophil extracellular trap formation and protects rodents from acute lung injury and sars-Cov-2 infection. JCI Insight (2022) 7(5):e157342. doi: 10.1172/jci.insight.157342
92. Jongerius I, Porcelijn L, van Beek AE, Semple JW, van der Schoot CE, Vlaar APJ, et al. The role of complement in transfusion-related acute lung injury. Transfusion Med Rev (2019) 33(4):236–42. doi: 10.1016/j.tmrv.2019.09.002
93. Chen D, Xu X, Xia W, Ye X, Bayat B, Bein G, et al. Anti-Cd36 antibodies induced endothelial disturbance via activation of pbmc: evidence of crucial Epitope(S). Vox sanguinis (2020) 115(SUPPL 1):58. doi: 10.1111/vox.13031
94. Kono M, Saigo K, Takagi Y, Takahashi T, Kawauchi S, Wada A, et al. Heme-related molecules induce rapid production of neutrophil extracellular traps. Transfusion (2014) 54(11):2811–9. doi: 10.1111/trf.12700
95. Li D, Cong Z, Lyu X, Wu C, Tao Y, Zhu X. [the role of nicotinamide-adenine dinucleotide phosphate oxidase nox family in acute lung injury]. Zhonghua wei zhong bing ji jiu yi xue (2019) 31(2):244–7. doi: 10.3760/cma.j.issn.2095-4352.2019.02.026
96. Segel GB, Halterman MW, Lichtman MA. The paradox of the neutrophil's role in tissue injury. J leukocyte Biol (2011) 89(3):359–72. doi: 10.1189/jlb.0910538
97. Semple JW, Kim M, Hou J, McVey M, Lee YJ, Tabuchi A, et al. Intravenous immunoglobulin prevents murine antibody-mediated acute lung injury at the level of neutrophil reactive oxygen species (Ros) production. PloS One (2012) 7(2):e31357. doi: 10.1371/journal.pone.0031357
98. Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY, Frenette PS. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med (2009) 15(4):384–91. doi: 10.1038/nm.1939
99. Jank H, Salzer U. Vesicles generated during storage of red blood cells enhance the generation of radical oxygen species in activated neutrophils. TheScientificWorldJournal (2011) 11:173–85. doi: 10.1100/tsw.2011.25
100. Kono M, Saigo K, Takagi Y, Kawauchi S, Wada A, Hashimoto M, et al. Morphological and flow-cytometric analysis of haemin-induced human neutrophil activation: implications for transfusion-related acute lung injury. Blood transfusion = Trasfusione del sangue (2013) 11(1):53–60. doi: 10.2450/2012.0141-11
101. Yu C, Xu L, Chen LF, Guan YJ, Kim M, Biffl WL, et al. Prbc-derived plasma induces non-muscle myosin type iia-mediated neutrophil migration and morphologic change. Immunopharmacol immunotoxicology (2013) 35(1):71–9. doi: 10.3109/08923973.2012.677046
102. De Pablo-Moreno JA, Serrano LJ, Revuelta L, Sánchez MJ, Liras A. The vascular endothelium and coagulation: homeostasis, disease, and treatment, with a focus on the Von willebrand factor and factors viii and V. Int J Mol Sci (2022) 23(15):8283. doi: 10.3390/ijms23158283
103. Vlaar AP, Hofstra JJ, Levi M, Kulik W, Nieuwland R, Tool AT, et al. Supernatant of aged erythrocytes causes lung inflammation and coagulopathy in a "Two-hit" in vivo syngeneic transfusion model. Anesthesiology (2010) 113(1):92–103. doi: 10.1097/ALN.0b013e3181de6f25
104. Vlaar AP, Hofstra JJ, Kulik W, van Lenthe H, Nieuwland R, Schultz MJ, et al. Supernatant of stored platelets causes lung inflammation and coagulopathy in a novel in vivo transfusion model. Blood (2010) 116(8):1360–8. doi: 10.1182/blood-2009-10-248732
105. Tuinman PR, Vlaar AP, Cornet AD, Hofstra JJ, Levi M, Meijers JC, et al. Blood transfusion during cardiac surgery is associated with inflammation and coagulation in the lung: a case control study. Crit Care (London England) (2011) 15(1):R59. doi: 10.1186/cc10032
106. Conway EM, Mackman N, Warren RQ, Wolberg AS, Mosnier LO, Campbell RA, et al. Understanding covid-19-Associated coagulopathy. Nat Rev Immunol (2022) 22(10):639–49. doi: 10.1038/s41577-022-00762-9
107. Sebag SC, Bastarache JA, Ware LB. Therapeutic modulation of coagulation and fibrinolysis in acute lung injury and the acute respiratory distress syndrome. Curr Pharm Biotechnol (2011) 12(9):1481–96. doi: 10.2174/138920111798281171
108. Ware LB, Matthay MA. The acute respiratory distress syndrome. New Engl J Med (2000) 342(18):1334–49. doi: 10.1056/nejm200005043421806
109. Bos LDJ, Ware LB. Acute respiratory distress syndrome: causes, pathophysiology, and phenotypes. Lancet (London England) (2022) 400(10358):1145–56. doi: 10.1016/s0140-6736(22)01485-4
110. Bolton-Maggs PHB. Serious hazards of transfusion - conference report: celebration of 20 years of uk haemovigilance. Transfusion Med (Oxford England) (2017) 27(6):393–400. doi: 10.1111/tme.12502
111. Middelburg RA, Beckers EA, Porcelijn L, Lardy N, Wiersum-Osselton JC, Schipperus MR, et al. Allo-exposure status and leucocyte antibody positivity of blood donors show a similar relation with trali. Transfusion Med (Oxford England) (2012) 22(2):128–32. doi: 10.1111/j.1365-3148.2012.01140.x
112. Reesink HW, Lee J, Keller A, Dennington P, Pink J, Holdsworth R, et al. Measures to prevent transfusion-related acute lung injury (Trali). Vox sanguinis (2012) 103(3):231–59. doi: 10.1111/j.1423-0410.2012.01596.x
113. Flesland O, Seghatchian J, Solheim BG. The Norwegian plasma fractionation project–a 12 year clinical and economic success story. Transfusion apheresis Sci Off J World Apheresis Assoc Off J Eur Soc Haemapheresis (2003) 28(1):93–100. doi: 10.1016/s1473-0502(02)00104-0
114. Saadah NH, van Hout FMA, Schipperus MR, le Cessie S, Middelburg RA, Wiersum-Osselton JC, et al. Comparing transfusion reaction rates for various plasma types: a systematic review and meta-Analysis/Regression. Transfusion (2017) 57(9):2104–14. doi: 10.1111/trf.14245
115. Saadah NH, Schipperus MR, Wiersum-Osselton JC, van Kraaij MG, Caram-Deelder C, Beckers EAM, et al. Transition from fresh frozen plasma to Solvent/Detergent plasma in the Netherlands: comparing clinical use and transfusion reaction risks. Haematologica (2020) 105(4):1158–65. doi: 10.3324/haematol.2019.222083
116. Klanderman RB, Bulle EB, Heijnen JWM, Allen J, Purmer IM, Kerkhoffs JH, et al. Reported transfusion-related acute lung injury associated with Solvent/Detergent plasma - a case series. Transfusion (2022) 62(3):594–9. doi: 10.1111/trf.16822
117. Blumberg N, Heal JM, Gettings KF, Phipps RP, Masel D, Refaai MA, et al. An association between decreased cardiopulmonary complications (Transfusion-related acute lung injury and transfusion-associated circulatory overload) and implementation of universal leukoreduction of blood transfusions. Transfusion (2010) 50(12):2738–44. doi: 10.1111/j.1537-2995.2010.02748.x
118. McQuinn ER, Smith SA, Viall AK, Wang C, LeVine DN. Neutrophil extracellular traps in stored canine red blood cell units. J veterinary Internal Med (2020) 34(5):1894–902. doi: 10.1111/jvim.15876
119. Simancas-Racines D, Osorio D, Martí-Carvajal AJ, Arevalo-Rodriguez I. Leukoreduction for the prevention of adverse reactions from allogeneic blood transfusion. Cochrane Database Systematic Rev (2015) 2015(12):CD009745. doi: 10.1002/14651858.CD009745.pub2
120. Silliman CC, Khan SY, Ball JB, Kelher MR, Marschner S. Mirasol pathogen reduction technology treatment does not affect acute lung injury in a two-event in vivo model caused by stored blood components. Vox sanguinis (2010) 98(4):525–30. doi: 10.1111/j.1423-0410.2009.01289.x
121. Caudrillier A, Mallavia B, Rouse L, Marschner S, Looney MR. Transfusion of human platelets treated with mirasol pathogen reduction technology does not induce acute lung injury in mice. PloS One (2015) 10(7):e0133022. doi: 10.1371/journal.pone.0133022
122. Mallavia B, Kwaan N, Marschner S, Yonemura S, Looney MR. Mirasol pathogen reduction technology treatment of human whole blood does not induce acute lung injury in mice. PloS One (2017) 12(6):e0178725. doi: 10.1371/journal.pone.0178725
123. Owusu-Ofori A, Asamoah-Akuoko L, Acquah M, Wilkinson S, Ansa J, Brown B, et al. Hemovigilance on mirasol pathogen-reduced whole blood in Ghana. Vox sanguinis (2019) 114:67. doi: 10.1111/vox.12792
124. Knutson F, Osselaer J, Pierelli L, Lozano M, Cid J, Tardivel R, et al. A prospective, active haemovigilance study with combined cohort analysis of 19,175 transfusions of platelet components prepared with amotosalen-uva photochemical treatment. Vox sanguinis (2015) 109(4):343–52. doi: 10.1111/vox.12287
125. Snyder EL, Wheeler AP, Refaai M, Cohn CS, Poisson J, Fontaine M, et al. Comparative risk of pulmonary adverse events with transfusion of pathogen reduced and conventional platelet components. Transfusion (2022) 62(7):1365–76. doi: 10.1111/trf.16987
126. Kuldanek SA, Kelher M, Silliman CC. Risk factors, management and prevention of transfusion-related acute lung injury: a comprehensive update. Expert Rev Hematol (2019) 12(9):773–85. doi: 10.1080/17474086.2019.1640599
127. van der Meer PF, de Korte D. Platelet additive solutions: a review of the latest developments and their clinical implications. Transfusion Med hemotherapy offizielles Organ der Deutschen Gesellschaft fur Transfusionsmedizin und Immunhamatologie (2018) 45(2):98–102. doi: 10.1159/000487513
128. Friedman T, Javidroozi M, Lobel G, Shander A. Complications of allogeneic blood product administration, with emphasis on transfusion-related acute lung injury and transfusion-associated circulatory overload. Adv Anesth (2017) 35(1):159–73. doi: 10.1016/j.aan.2017.07.008
129. Tung JP. Transfusion-related acute lung injury (Trali): pathogenesis and diagnosis. Pathology (2019) 51:S44. doi: 10.1016/j.pathol.2018.12.103
130. Priyanjani R, Naotunna S, Senanayake D, Kuruppu D. How we investigated a case of transfusion related acute lung injury (Trali) at the histocompatibility reference laboratory, national blood centre, Sri Lanka. Vox sanguinis (2022) 117(SUPPL 1):231–2. doi: 10.1111/vox.13285
131. Honda A, Morita M, Taniguchi A, Tabuchi A, Kubo S. [Successful extracorporeal membrane oxygenation for a patient with nearly fatal hypoxemia induced by transfusion-related acute lung injury]. Masui Japanese J anesthesiology (2015) 64(11):1181–5.
132. Kassem AB, Ahmed I, Omran G, Megahed M, Habib T. Role of ascorbic acid infusion in critically ill patients with transfusion-related acute lung injury. Br J Clin Pharmacol (2022) 88(5):2327–39. doi: 10.1111/bcp.15167
133. Qiao J, He R, Yin Y, Tian L, Li L, Lian Z, et al. Ril-35 prevents murine transfusion-related acute lung injury by inhibiting the activation of endothelial cells. Transfusion (2020) 60(7):1434–42. doi: 10.1111/trf.15805
134. Yuan X, Jiang P, Qiao C, Su N, Sun P, Lin F, et al. Platelet suppression by tirofiban ameliorates pulmonary coagulation and fibrinolysis abnormalities in the lungs of mouse antibody-mediated transfusion-related acute lung injury. Shock (Augusta Ga) (2023) 59(4):603–11. doi: 10.1097/shk.0000000000002080
135. Hong SW O E, Lee JY, Yi J, Cho K, Kim J, et al. Interleukin-2/Antibody complex expanding Foxp3(+) regulatory T cells exacerbates Th2-mediated allergic airway inflammation. BMB Rep (2019) 52(4):283–8. doi: 10.5483/BMBRep.2019.52.4.271
136. Schönbacher M, Aichinger N, Weidner L, Jungbauer C, Grabmer C, Schuha B, et al. Leukocyte-reactive antibodies in female blood donors: the Austrian experience. Transfusion Med hemotherapy offizielles Organ der Deutschen Gesellschaft fur Transfusionsmedizin und Immunhamatologie (2021) 48(2):99–108. doi: 10.1159/000509946
137. Dada A, Elhassan K, Bawayan RM, Albishi G, Hefni L, Bassi S, et al. Sars-Cov-2 triggers the development of class I and class ii hla antibodies in recovered convalescent plasma donors. Intervirology (2022) 65(4):230–5. doi: 10.1159/000524016
138. Nissen-Meyer LSH, Czapp E, Naper C, Jensen T, Boulland LML. Screening for antibodies to hla class I in apheresis donors following covid-19 or sars-Cov-2 vaccination. Transfusion apheresis Sci Off J World Apheresis Assoc Off J Eur Soc Haemapheresis (2022) 61(5):103567. doi: 10.1016/j.transci.2022.103567
139. Devine DV, Schubert P. Pathogen inactivation technologies: the advent of pathogen-reduced blood components to reduce blood safety risk. Hematology/oncology Clinics North America (2016) 30(3):609–17. doi: 10.1016/j.hoc.2016.01.005
140. Vossoughi S, Gorlin J, Kessler DA, Hillyer CD, Van Buren NL, Jimenez A, et al. Ten years of trali mitigation: measuring our progress. Transfusion (2019) 59(8):2567–74. doi: 10.1111/trf.15387
141. Roman M, Fashina O, Tomassini S, Abbasciano RG, Lai F, Richards T, et al. Reporting conflicts of interest in randomised trials of patient blood management interventions in patients requiring major surgery: a systematic review and meta-analysis. BMJ Open (2022) 12(8):e054582. doi: 10.1136/bmjopen-2021-054582
142. Arzoun H, Srinivasan M, Adam M, Thomas SS, Lee B, Yarema A. A systematic review on the management of transfusion-related acute lung injury in transfusion-dependent sickle cell disease. Cureus (2022) 14(2):e22101. doi: 10.7759/cureus.22101
143. Ackfeld T, Schmutz T, Guechi Y, Le Terrier C. Blood transfusion reactions-a comprehensive review of the literature including a Swiss perspective. J Clin Med (2022) 11(10):2859. doi: 10.3390/jcm11102859
144. Tocut M, Kolitz T, Shovman O, Haviv Y, Boaz M, Laviel S, et al. Outcomes of icu patients treated with intravenous immunoglobulin for sepsis or autoimmune diseases. Autoimmun Rev (2022) 21(12):103205. doi: 10.1016/j.autrev.2022.103205
145. Baudel JL, Vigneron C, Pras-Landre V, Joffre J, Marjot F, Ait-Oufella H, et al. Transfusion-related acute lung injury (Trali) after intravenous immunoglobulins: French multicentre study and literature review. Clin Rheumatol (2020) 39(2):541–6. doi: 10.1007/s10067-019-04832-7
146. Guo Y, Tian X, Wang X, Xiao Z. Adverse effects of immunoglobulin therapy. Front Immunol (2018) 9:1299. doi: 10.3389/fimmu.2018.01299
147. Adrover JM, Aroca-Crevillén A, Crainiciuc G, Ostos F, Rojas-Vega Y, Rubio-Ponce A, et al. Programmed 'Disarming' of the neutrophil proteome reduces the magnitude of inflammation. Nat Immunol (2020) 21(2):135–44. doi: 10.1038/s41590-019-0571-2
148. Futosi K, Németh T, Pick R, Vántus T, Walzog B, Mócsai A. Dasatinib inhibits proinflammatory functions of mature human neutrophils. Blood (2012) 119(21):4981–91. doi: 10.1182/blood-2011-07-369041
149. Wan N, Rong W, Zhu W, Jia D, Bai P, Liu G, et al. Tregs-derived interleukin 35 attenuates endothelial proliferation through Stat1 in pulmonary hypertension. Ann Trans Med (2021) 9(11):926. doi: 10.21037/atm-21-1952
150. Li M, Liu Y, Fu Y, Gong R, Xia H, Huang X, et al. Interleukin-35 inhibits lipopolysaccharide-induced endothelial cell activation by downregulating inflammation and apoptosis. Exp Cell Res (2021) 407(2):112784. doi: 10.1016/j.yexcr.2021.112784
151. Liu M, Meng X, Xuan Z, Chen S, Wang J, Chen Z, et al. Effect of er miao San on peritoneal macrophage polarisation through the mirna-33/Nlrp3 signalling pathway in a rat model of adjuvant arthritis. Pharm Biol (2022) 60(1):846–53. doi: 10.1080/13880209.2022.2066700
152. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. Pi3kγ is a molecular switch that controls immune suppression. Nature (2016) 539(7629):437–42. doi: 10.1038/nature19834
153. Han A, Lee T, Lee J, Song SW, Lee SS, Jung IM, et al. A multicenter, randomized, open-labelled, non-inferiority trial of sustained-release sarpogrelate versus clopidogrel after femoropopliteal artery intervention. Sci Rep (2023) 13(1):2502. doi: 10.1038/s41598-023-29006-z
154. Sugiyama D, Hinohara K, Nishikawa H. Significance of regulatory T cells in cancer immunology and immunotherapy. Exp Dermatol (2023) 32(3):256–63. doi: 10.1111/exd.14721
155. Yu Y, Jiang P, Sun P, Su N, Lin F. Analysis of therapeutic potential of preclinical models based on Dr3/Tl1a pathway modulation (Review). Exp Ther Med (2021) 22(1):693. doi: 10.3892/etm.2021.10125
156. Goswami TK, Singh M, Dhawan M, Mitra S, Emran TB, Rabaan AA, et al. Regulatory T cells (Tregs) and their therapeutic potential against autoimmune disorders - advances and challenges. Hum Vaccines immunotherapeutics (2022) 18(1):2035117. doi: 10.1080/21645515.2022.2035117
157. He R, Li L, Kong Y, Tian L, Tian X, Fang P, et al. Preventing murine transfusion-related acute lung injury by expansion of Cd4(+) Cd25(+) Foxp3(+) tregs using il-2/Anti-Il-2 complexes. Transfusion (2019) 59(2):534–44. doi: 10.1111/trf.15064
Keywords: transfusion-related acute lung injury (TRALI), pathogenesis, priming step, pulmonary reaction, effector phase, management
Citation: Yu Y and Lian Z (2023) Update on transfusion-related acute lung injury: an overview of its pathogenesis and management. Front. Immunol. 14:1175387. doi: 10.3389/fimmu.2023.1175387
Received: 27 February 2023; Accepted: 27 April 2023;
Published: 12 May 2023.
Edited by:
Miguel Angel Alejandre Alcazar, University Hospital of Cologne, GermanyReviewed by:
Yue Li, First Affiliated Hospital of Zhengzhou University, ChinaCopyright © 2023 Yu and Lian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhengqiu Lian, MjIzNzkzMjUzMUBxcS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.