 Rongbo Wen†
Rongbo Wen† Leqi Zhou†
Leqi Zhou† Zhiying Peng†Hao FanTianshuai ZhangHang Jia
Zhiying Peng†Hao FanTianshuai ZhangHang Jia Xianhua Gao
Xianhua Gao Liqiang HaoZheng LouFuao Cao*
Liqiang HaoZheng LouFuao Cao* Guanyu Yu*
Guanyu Yu* Wei Zhang*
Wei Zhang*- Department of Colorectal Surgery, Changhai Hospital, Naval Medical University, Shanghai, China
Colorectal Cancer (CRC) is one of the most common gastrointestinal tumors, and its high tumor heterogeneity makes traditional sequencing methods incapable of obtaining information about the heterogeneity of individual cancer cells in CRC. Therefore, single-cell sequencing technology can be applied to better analyze the differences in genetic and protein information between cells, to obtain genomic sequence information of single cells, and to more thoroughly analyze the cellular characteristics and interactions in the CRC microenvironment. This will provide a more comprehensive understanding of colorectal cancer development and metastasis and indicate the treatment plan and prognosis. In this study, we review the application of single-cell sequencing to analyze the tumor microenvironment of CRC, explore the mechanisms involved in CRC metastasis and progression, and provide a reference for potential treatment options.
1 Introduction
Colorectal cancer (CRC) is one of the most common gastrointestinal tumors and is reported to have the third-highest morbidity and second-highest mortality worldwide (1). Additionally, approximately 50% of patients are diagnosed with metastatic CRC (mCRC) at the first visit, and the 5-year survival rate is no more than 50% (2). Tumor heterogeneity is a result of differences in genetic and molecular characteristics between individual cancer cells due to different degrees of cell differentiation in the same tumor tissue (3). The tumor microenvironment (TME) interacts with tumor heterogeneity to promote differentiation into different cell subtypes and metastatic spread, thus participating in the cancer development process (4, 5). CRC is highly heterogeneous; however, traditional sequencing methods target the entire tumor tissue, which can only reflect the total characteristics of the cell population and cannot obtain information on cell heterogeneity (6). Single-cell sequencing (SCS) sequences single cells at the genome or transcriptome level to obtain genomic, transcriptomic, or other information about individual cells, thereby revealing cell population differences and evolutionary relationships (7, 8). Therefore, studies conducted on individual cells can be more precisely localized to cell-to-cell interactions and are more conducive to detecting heterogeneity among individual cancer cells, thus exploring the complex heterogeneous mechanisms involved in the development of CRC, further clarifying diagnosis, improving prognostic analysis, and monitoring drug efficacy. Recently, SCS technology has been applicated in more and more studies and reported to help achieve considerable breakthrough in many other tumors including lung cancer, breast cancer and prostate cancer (9–11). Here, we review the studies of SCS in colorectal cancer and provide researchers with a reference for better understanding the tumor characteristics from multiple dimensions (Table 1) exploring its clinical application potential, and directing precise treatment.
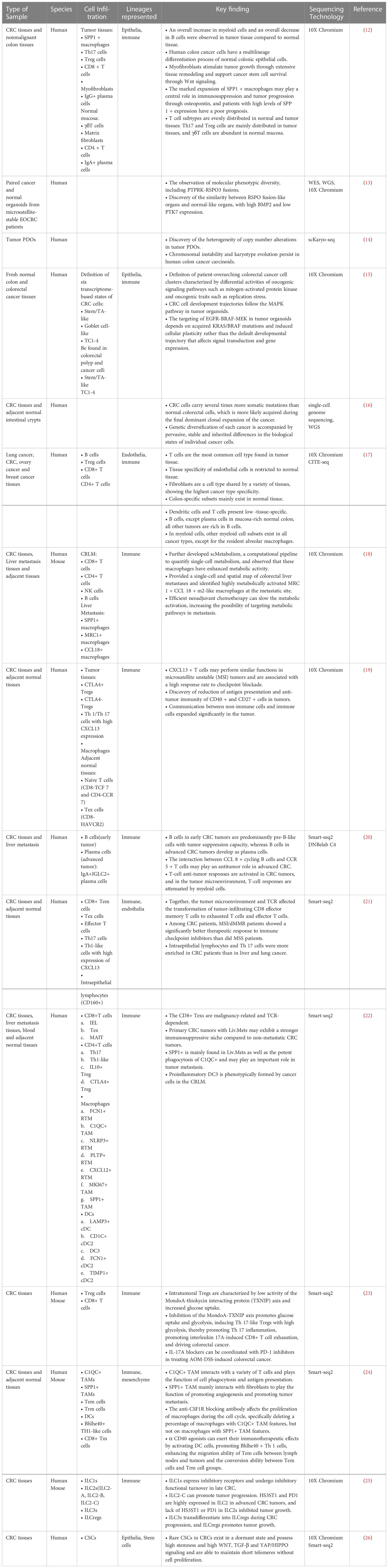
Table 1 Overview of key findings in CRC SCS technology.
2 Application of SCS for in-depth analysis of CRC TME
The TME is a complex integrated system composed of tumor cells, various cytokines, chemokines, and multiple stromal cells, including immune inflammatory cells, cancer-associated fibroblasts, and endothelial cells. These can be divided into immune microenvironments, which are dominated by immune cells, and non-immune microenvironments, which are dominated by fibroblasts (27). Tumor cells are closely related to TME. Tumor cells can influence its microenvironment by releasing cell signaling molecules to promote tumor angiogenesis and induce immune tolerance, while immune cells in the microenvironment can influence tumor cells growth and development. Several studies have shown that changes of TME are closely related to cancer development, invasion, and metastasis, and immunotherapy efficacy can be predicted by distinguishing different TME subtypes (28–30). Therefore, the application of SCS technology for in-depth analysis of the CRC TME is a hot research area, and many research results have laid the foundation for future molecular typing and treatment of CRC.
2.1 Tumor cells
CRC is characterized by genomic instability, epigenetic abnormalities, and abnormal gene expression (31). This has led to high tumor heterogeneity in patients with CRC. Tumor heterogeneity in CRC can be divided into inter- and intra-tumor heterogeneity. CRC inter-tumor heterogeneity may originate from human embryonic development, where different embryonic layers develop as proximal and distal colon, and hypermutated tumors with microsatellite instability (MSI) are often located in the proximal colon, while tumors in the distal colon or rectum usually exhibit microsatellite stability (MSS) and chromosomal instability (CIN) (32). CRC intra-tumor heterogeneity (ITH) may also occur due to differences in cancer cells and TME within the tumor, further complicating the development of new therapeutic strategies and biomarker identification (33). ITH is detectable within a single tumor in which cancer cell subpopulations with different genome features coexist in a patient in different tumor areas or may differ over time (34–36). Exploring the ITH within single tumor is the advantage of SCS technology.
CRC cells are similar to normal cells to some extent but exhibit individualized phenotypic diversity at the same time. Through analyzing the single-cell RNA sequencing (scRNA-seq) data of metastatic CRC patients, Lee HO et al. found that the differentiation trajectory of CRC cells was similar to that of normal epithelial cells, with aggregation of tumor cells with normal stem cell-like/transporter-amplified cell populations. This suggests that tumor epithelial cells have regenerative and proliferative potential, while clustering analysis showed that tumor epithelial cells have highly variable transcriptional status and individual variability (12). Another study also confirmed the similarity between CRC-like organs and normal-like organs at the patient-derived tumor organoids (tumor PDOs) level, but with molecular phenotypic diversity. scRNA-seq analysis of paired cancer and normal tissue organoid models from patients with MSS sporadic early-onset CRC (EOCRC) resulted in the observation of significant molecular phenotypic diversity, including PTPRK-RSPO3 fusions, and RSPO fusion organoids were similar to normal colon organoids with high BMP2 and low PTK7 expression, which confirms the similarity between RSPO fusion organs and normal organs (13). In addition, a study confirmed the heterogeneity of copy number alterations in tumor PDO by single-cell karyotype sequencing and showed that monoclonal lines evolved new karyotypes over time in vitro, suggesting that chromosomal instability and karyotype evolution persist in human colon cancer carcinoids (14). In another study using tumor PDOs and biopsy tumors, it was demonstrated that karyotypic alterations of varying complexities are prevalent and can occur within several cell generations (37). Both studies corroborate the existence of complex karyotypic alterations and evolution in CRC, resulting in high intratumoral heterogeneity.
Meanwhile, RNA metabolic markers and RNA velocity assessments revealed CRC cell development trajectories along the mitogen-activated protein kinase (MAPK) pathway in tumor PDOs, whereas normal colon organoid cells develop along a hierarchy of WNT activity, highlighting that cancer cell development is a driver of non-genetic cancer cell heterogeneity and that changes in trajectories affect the effectiveness of targeted therapies (15). Roerink et al. concluded that CRC cells exhibit extensive mutational diversity and carry several times more somatic mutations than normal colorectal cells. Most mutations are acquired during the final dominant clonal expansion of cancer cells and are caused by mutations that are absent in normal colorectal cells. Specific somatic mutations can result in different responses to chemotherapy and targeted therapy (16). This suggests that CRC cell development, clonal expansion, and mutations continue to drive intratumor heterogeneity in CRC, making chemotherapy and targeted therapy difficult and promoting drug resistance in patients.
To deeply explore CRC intra-tumor heterogeneity, a study performed optimized single-cell multi-omics sequencing, including DNA, DNA methylation, and transcriptome sequencing, on CRC patients with CRC. This showed that DNA methylation levels in CRC cells were lower than those in normal epithelial cells adjacent to the cancer cells, with different methylation levels varying from different spectra in the same tumor tissue, suggesting that methylation heterogeneity mainly results from differences in DNA methylation between different subclones within the same patient’s tumor. This study also elucidated the demethylation characteristics of CRC, where the degree of demethylation was consistent within each subtype but varied across subtypes. Interestingly, long interspersed nuclear element 1 (LINE-1, L1) shows stronger demethylation than L2 in cancer cells, in contrast to embryonic development, suggesting that abnormal demethylation processes may arise in the L1 and heterochromatin regions during tumorigenesis and progression, breaking the normal developmental pattern (38).
2.2 Immune cells
2.2.1 T cell
Various immune cells in the TME interact with tumor cells and mediate immune tolerance to tumors, affecting tumor progression and metastasis, and thus immunotherapy efficacy. T cells, one of the major cellular components involved in the body’s immune response and the most common cell type in tumor tissue (17), can kill tumor cells. In order to escape the pursuit of T cells, tumor cells could produce some inhibitory signals on their own surface, and inhibit the immune function of T cells through immune checkpoint (39). Ever since Allison et al. discovered the immunosuppressive effects of CTLA4 on T cells in 1996, immunotherapy drugs, notably immune checkpoint inhibitors (ICIs), have taken off and become a lifesaving drug for tumor patients (40). Various ICIs have been applicated in cancer immunotherapy, including PD1/PDL1 and CTLA4 (41, 42).
However, the efficacy of immunotherapy in patients with CRC is not as good today. Thus, an increasing number of studies have been conducted to analyze the TME in depth using SCS technology.
Several studies have mapped the global cellular landscape in CRC, with an overall increase in myeloid cells and an overall decrease in B-cell numbers observed in tumor tissues compared to normal tissues, suggesting a redirected immune response. This indicates that the immune response undergoes dynamic changes during cancer development and that the transcriptional profile of cancer cells is similar to that of normal human differentiation, with genetic alterations creating an immunosuppressive microenvironment directed by regulatory T cells (Tregs), myofibroblasts, and myeloid cells (12, 18–20). Meanwhile, T cell subtypes are unevenly distributed in normal and tumor tissues, Th17 and Treg cells are mainly distributed in tumor tissues, while γδT cells are more abundant in normal mucosa. The TME and T-cell receptors (TCR) affect the transformation of tumor-infiltrating CD8+ effector memory T cells (Tems) into exhausted T cells (Texs) and effective T cells (Teffs), indicating the transformation of the organism from mucosal immunity to inhibiting cellular immunity (12, 21).
A study analyzing CD45+ cells from multiple matched tissues of patients with untreated primary hepatocellular carcinoma (HCC), CRC, and CRC liver metastases (CLM) found that Texs and activated Tregs originate from primary CRC tumors with a malignancy-related phenotype and are TCR-dependent. There is a high degree of TCR sharing between Tems and Texs. Natural killer (NK) cells and mucosa-associated T cells are mainly derived from the liver tissue at metastatic foci, and their phenotypes are associated with the liver TME (22). Another study similarly found decreased B-cell antigen presentation as well as tumor-specific Tregs and their two subtypes, proliferative exhausted T cells and a predominance of naive T cells (CD8-TCF7 and CD4-CCR7) in adjacent tissues and exhausted T cells in tumors (CD8-HAVCR2). Th1/Th17 cells with high CXCL13 expression are preferentially enriched in patients with a high tumor mutational burden (TMB) and respond well to immunotherapy (19), suggesting that CXCL13+ T cells may perform functions similar to those of MSI tumors and are associated with a high response rate to checkpoint blockade. The results of these two studies are consistent, suggesting that the exhausted T cell phenotype is associated with malignancy and that CXCL13+ T cells may be associated with immunotherapy. Moreover, CXCL13 expression accurately identifies both tumor-specific T cell clones that are terminally differentiated and highly exhausted and tumor-specific T cell precursor cell clones that are abundantly present in responding tumors after immune checkpoint blockade (ICB) treatment, demonstrating that tumor-specific CXCL13+CD8+ T cells play a key role in the treatment process and that the degree of infiltration before treatment can predict ICB efficacy (43).
It was also found that in tissue samples from CRC patients, impaired T cell proliferation and activation were associated with ZFP91, which disrupts the metabolic pathways and antitumor activity of tumor-infiltrating T cells, suggesting that targeting ZFP91 may improve the effectiveness of tumor immunotherapy (44). The transcription factor TCF-1 is critical for Treg development and function and primarily inhibits the transcription of genes that co-bind with FOXP3. Deficiency of TCF-1 could activate Treg cells and make Th17 cells acquire the intestinal homing characteristics, leading to more dangerous and dramatic CRC, and the specific TCF-1 expression of Tregs regulates inflammation and CD8+ T cell toxicity and may determine the prognosis of CRC (45). Similarly, the glucose-responsive transcription factor MondoA is highly expressed in Tregs, and inhibition of the MondoA-TXNIP axis promotes glucose uptake and glycolysis, inducing highly glycolytic Th17-like Tregs, which promotes Th17 inflammation and CRC development (23).
2.2.2 Cancer-associated fibroblasts
In addition to T cells, the most common cells in the TME, cancer-associated fibroblasts (CAF), and tumor-associated macrophages (TAMs), are also being explored and analyzed with the development of SCS technology.
Somatic copy number alterations (SCNAs) are prevalent in the TME and immune cells, fibroblasts, and endothelial cells in the normal tissues of each individual, and the proportion of fibroblasts with genomic copy number variants is much higher in tumor tissues than in adjacent issues, so that it can predict the prognosis of CRC by screening the differentially expressed genes of CAFs in tumor tissues. Five genes (BGN, RCN3, TAGLN, MYL9, and TPM2) have been identified as specific CAFs biomarkers of poor prognosis in CRC (46). Fibroblasts are a cell type common to multiple tissues and exhibit the highest cancer-type specificity (17). In addition, myofibroblasts have been shown to stimulate tumor growth through extensive tissue remodeling and support cancer stem cell survival through WNT signaling (12), indicating that fibroblasts also play a role in promoting tumor growth, and that genetic alterations in fibroblasts could incur CRC via paracrine signaling in epithelial cells (47).
2.2.3 Tumor-associated macrophages
TAMs are infiltrating macrophages in tumor tissue, mainly derived from monocyte differentiation. TAMs can interact with tumor cells through exosomes or secrete multiple cytokines to promote tumor cell proliferation, invasion, migration, and angiogenesis.
TAMs recruit Tregs through chemokine CCL2 secretion, which inhibits the antitumor immune response of T cells and interferes with immune cell interactions, thus leading to an immunosuppressive microenvironment in CRC (48). TAMs in CRC can be divided into two cell groups, SPP1+ TAM and C1QC+ TAM. C1QC+ TAM interacts with various T cells and performs cytophagic and antigen-presenting functions, whereas SPP1+ TAM interacts mainly with fibroblasts and performs pro-angiogenic and tumor-promoting functions (24). Single-cell analysis of CLM samples showed that a subpopulation of dendritic cells (DC3s) and SPP1+ macrophages is associated with malignancy and plays a critical role in liver metastasis (22). Additionally, a study detected numerous immunosuppressive cells in CRC liver metastatic tumors, with a dramatic increase in SPP1+ macrophages and MRC1+ CCL18+ macrophages and an enrichment of neutrophils as potential participants in liver metastases (18). A previous study also showed that significant expansion of SPP1+ macrophages may play a central role in immunosuppression and tumor progression through bone bridge proteins and that CRC patients with high SPP1+ expression levels have a poorer prognosis (12). These studies confirm the role of SPP1+ macrophages in promoting CRC progression and the potential suppressive TME in liver metastasis. In addition, one study showed that the density of TAMs was not associated with survival in patients with CLM, but the area and circumference of TAMs were significantly higher in CLMs, while larger morphologies of TAMs were mostly observed in patients with a poorer prognosis (49), which demonstrated the strong prognostic significance of the morphological representation of TAM.
2.2.4 Intrinsic lymph-like cell
Intrinsic lymphocytes (ILCs) are located on the mucosal surface and include NK cells; helper classes ILC1s, ILC2s, and ILC3s; and lymphoid tissue-inducing (LTi) cells that enhance the immune response, maintain mucosal integrity, and sustain tissue homeostasis (25, 50).
One study analyzed tumor-infiltrating ILCs during CRC progression using scRNA-seq and classified them into six clusters. ILC1 expressed inhibitory receptors and underwent an inhibitory functional transformation in advanced CRC, and ILC2 was divided into three subgroups (ILC2-A, -B, and -C), of which the ILC2-C subgroup promoted tumor progression. HS3ST1 and PD1 are highly expressed in ILC2 of advanced CRC In addition, ILC3 transdifferentiates into ILCregs during CRC progression and promotes tumor growth. Notably, the TGF-β signaling pathway initiated the conversion of ILC3 to ILCregs. Therefore, this study suggests that interfering with ILC conversion may be a potential strategy for CRC immunotherapy (25). Moreover, another study reported single-cell characteristics of blood and intestinal helper ILC subtypes in healthy conditions and CRC, where the healthy intestine contained ILC1s, ILC3s, and ILC3/NKs, but not ILC2s, while additional tumor-specific ILC1 and ILC2 subtypes were identified in CRC patients. SLAMF1 (signaling lymphocyte-activating molecule family member 1, CD150) was selectively expressed on tumor-specific ILCs, and higher levels of SLAMF1+ ILCs were observed in the blood of patients with CRC. The survival rate of patients with CRC in the high SLAMF1 group was significantly higher than that in the low SLAMF1 group, indicating that SLAMF1 is an antitumor biomarker of CRC (50).
3 Application of SCS to explore mechanisms affecting CRC metastasis & progression
3.1 CRC liver metastasis
CLM is the leading cause of death from CRC and a major factor in reducing the survival time of CRC patients (51). Besides the rapid metastatic spread of cancer cells, TME with liver metastasis exhibits a highly immunosuppressive phenotype (52). A previous study observed a dramatic increase in SPP1+ and MRC1+ CCL18+ macrophages in metastatic tumors, which corroborated the potentially suppressive TME in liver metastases. Meanwhile, TAM may be suppressed in metastatic tumors, liver metastatic cells may preferentially reprogram macrophages and induce their specific functional states, and metastatic tumor cells in liver metastases preferentially express the ligand CD47 and thus may recruit or activate MRC1+ CCL18+ macrophages through the corresponding receptor SIRPA, suggesting that specific macrophage subpopulations may play a fundamental role in the formation of premetastatic niche in CLM (18).
In addition, studies have revealed rare mutations in metastatic tumors and defined two separate cell populations by analyzing single-cell sequencing data from primary and metastatic foci in patients with CRC and liver metastases, suggesting different evolutionary trajectories between primary and metastatic tumor cells. Meanwhile, extensive WES data reflect different mutant allele frequencies between primary and metastatic foci. TP53, APC, and SNVs in SMAD4 show an increase in variant allele frequency (VAF) in metastatic samples (53).
3.2 Cancer stem cells
Cancer stem cells (CSCs) are thought to proliferate extensively and drive tumor growth, indicating that malignant cell populations in tumors are generated by CSCs (54). A previous study found that each tumor gland was derived from a stem cell by genomic analysis of 349 individual tumor glands, and they found that after initial transformation, CRC tumors grew primarily as a single expansion comprising many intermixed subclones, and it was then proposed that most of the mutations driving tumor growth occurred during early tumor expansion and led to clonal diversity and intra-tumor heterogeneity (55).
Currently, it has been suggested that CSCs may contribute to tumor progression and drug resistance. By single-cell sequencing of telomerase and transcriptome in 8 primary foci of untreated CRC, it was shown that CSCs can be remodeled into cancer epithelial cells and both of them retain the important signaling pathway such as WNT, TGF-β, and HIPPO/YAP. In addition, proliferating tumor epithelial cells were found to be derived from resting CSCs, which are related to the recurrence and metastasis of tumors, and resting CSCs may develop drug resistance through mutations (26).
3.3 CRC genomic/chromosomal mutations
Continued high-frequency chromosomal instability (CIN) has a dramatic impact on tumor evolution and treatment response. A previous study showed that CIN is prevalent in CRC, and single-cell karyotyping sequencing confirmed the heterogeneity of copy number alterations in tumor PDOs and showed that monoclonal lines evolved new karyotypes over time in vitro (14). Abnormal DNA methylation at the chromosomal level has also been found in CRC cells, where six chromosomes (chromosomes 4, 5, 8, 13, 18, and X) tend to undergo intense DNA demethylation, with three hypomethylated chromosomes (chromosomes 8, 13, and 18) (46), which confirms that CRC is characterized by genomic instability.
In addition, it has been found that CRC cells usually develop along the mitogen-activated protein kinase (MAPK) pathway, while MAPK activity will drive the cellular trajectory of cancer cells, and the targeting of EGFR-BRAF-MEK in tumor-like organs depends on acquired KRAS/BRAF mutations and induced cellular plasticity, which affects signal transduction and gene expression (15). Furthermore, a study showed a significantly increased somatic mutation rate in CRC cells compared to normal colorectal cells; the presence of driver mutations such as BRAF (V600E), PIK3CA (E81K), and ACVR2A (protein truncated small indel); as well as MLH1 methylation and genetic diversification in each cancer, accompanied by generalized, stable and genetic differences (16).
4 Application of SCS to explore and improve CRC treatment
4.1 Immunotherapy targets
Current immunotherapies for metastatic CRC are effective only in tumors with high microsatellite instability or mismatch repair defects. As tumor cells can determine their immune microenvironment and often form an immunosuppressive microenvironment (12), current immune checkpoint inhibitors (ICIs) are not effective against tumors with proficient mismatch repair (pMMR), MSS, or low-frequency microsatellite instability (MSI-L) (called pMMR-MSI-L tumors) (56). Therefore, finding new immunotherapy targets or improving current immunotherapy to expand the range of CRC immunotherapies has become an area of research interest.
A study found that T-cell antigen receptor-dependent cytoplasmic translocation of ZFP91 promotes the assembly of the PP2A complex, thereby limiting mTORC1-mediated metabolic reprogramming, suggesting that ZFP91 interferes with the metabolic and functional state of T cells in the TME, suggesting that targeting ZFP91 may improve the efficacy of CRC immunotherapy (44).
In addition, a study indicated that anti-CSF1R treatment preferentially depletes macrophages with inflammatory features but avoids macrophage populations expressing pro-angiogenic/tumorigenic genes in mice and humans. Treatment with CD40 agonist antibodies preferentially activates conventional dendritic cell (cDC) populations with increased Bhlhe40+ Th1-like cells and CD8+ memory T cells and identified key cellular interactions that regulate tumor immunity and mechanisms for myeloid-targeted immunotherapy currently in clinical trials (24).
4.2 Causes of chemotherapy sensitivity
Currently, the primary treatment for CRC is surgical resection combined with radiotherapy, chemotherapy, and targeted therapy. However, some patients undergoing chemotherapy may develop drug resistance, resulting in reduced efficacy.
One study developed a scMetabolism system to provide a single-cell and spatial atlas of colorectal liver metastases and identified highly metabolically activated MRC1+ CCL18+ M2-like macrophages at metastatic sites. Efficient neoadjuvant chemotherapy can slow metabolic activation and increase the possibility of targeting metabolic pathways in metastases (18).
In addition, another study confirmed the strong response of the CMS2 epithelial/typical group to EGFR and HER2 inhibitors by translating consensus molecular typing (CMS) preclinical models of developing cancer cells adapted to CMS classifiers combined with high-throughput drug sensitivity screening and revealed that cells with CMS1 microsatellite instability/immunity and CMS4 mesenchymal phenotypes were strongly sensitive to HSP90 inhibitors. A combination of 5-fluorouracil and HSP90 inhibitor has the potential to relieve drug resistance and improve treatment efficacy in a CMS4 patient-derived xenograft (PDX) model (57).
5 Summary
Tumor heterogeneity is widespread in CRC patients, and cluster analysis shows that tumor epithelial cells are individualized for each patient and are highly mutated Various immune cells and inflammatory chemokines in the TME interact and influence each other to promote tumor progression, thus affecting tumor recurrence and treatment response and adversely affecting the prognosis of CRC patients. While the impact of the TME on CRC can be fully investigated by obtaining information on cancer cell characteristics through SCS, it is also possible to identify relevant predictive markers for CRC prognosis and potential immunotherapeutic targets through SCS, thus improving patient prognosis and therapeutic effects. In addition, the use of SCS technology for drug development and for addressing the problem of chemotherapy resistance in some patients is now emerging (31). It is expected to be applied in precision medicine to develop and personalize cancer medical treatments, providing a more accurate diagnosis and the best-individualized treatment plan for cancer patients.
However, SCS technology still has some limitations in current platform. First, SCS technology requires high levels of sample preparation and sample quality, including cell quantity and activity, which increased the combined cost. Second, although the number of cells that can be detected from SCS has increased from 10~100 to tens of thousands with the development of technology, the tedious process still results in the loss of some cell populations, which can bias the results. In addition, SCS technology lacks of spatial information. Thus, a combination of multiple sequencing methods including bulk sequencing, spatial transcriptomics and SCS technology could be a solution and future direction of development.
Author contributions
WZ, GY and FC designed the research. RW, LZ and ZP made substantial contributions to acquisition, analysis and interpretation of data, and wrote the manuscript. HF, TZ, HJ, XG, ZL and LH coordinated and were involved in acquisition, interpretation of the data. WZ, GY and FC revised it critically for important intellectual content and gave final approval of the version to be published.
Funding
National Natural Science Foundation of China(82072750, 82203137), Natural Science Fund of Shanghai(20ZR1457200), Shanghai Sailing Program(21YF1459300), 71st Batch of China Postdoctoral Science Foundation (48804), and Excellent doctoral dissertation training program of Naval Medical University.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Engstrand J, Nilsson H, Stromberg C, Jonas E, Freedman J. Colorectal cancer liver metastases - a population-based study on incidence, management and survival. BMC Cancer (2018) 18(1):78. doi: 10.1186/s12885-017-3925-x
3. Zhang Y, Song J, Zhao Z, Yang M, Chen M, Liu C, et al. Single-cell transcriptome analysis reveals tumor immune microenvironment heterogenicity and granulocytes enrichment in colorectal cancer liver metastases. Cancer Lett (2020) 470:84–94. doi: 10.1016/j.canlet.2019.10.016
4. Hui L, Chen Y. Tumor microenvironment: sanctuary of the devil. Cancer Lett (2015) 368(1):7–13. doi: 10.1016/j.canlet.2015.07.039
5. Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett (2017) 387:61–8. doi: 10.1016/j.canlet.2016.01.043
6. Kulkarni A, Anderson AG, Merullo DP, Konopka G. Beyond bulk: a review of single cell transcriptomics methodologies and applications. Curr Opin Biotechnol (2019) 58:129–36. doi: 10.1016/j.copbio.2019.03.001
7. Gawad C, Koh W, Quake SR. Single-cell genome sequencing: current state of the science. Nat Rev Genet (2016) 17(3):175–88. doi: 10.1038/nrg.2015.16
8. Lawson DA, Kessenbrock K, Davis RT, Pervolarakis N, Werb Z. Tumour heterogeneity and metastasis at single-cell resolution. Nat Cell Biol (2018) 20(12):1349–60. doi: 10.1038/s41556-018-0236-7
9. Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, et al. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell (2018) 173(4):879–93.e13. doi: 10.1016/j.cell.2018.03.041
10. Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med (2018) 24(7):978–85. doi: 10.1038/s41591-018-0045-3
11. Karthaus WR, Hofree M, Choi D, Linton EL, Turkekul M, Bejnood A, et al. Regenerative potential of prostate luminal cells revealed by single-cell analysis. Science (2020) 368(6490):497–505. doi: 10.1126/science.aay0267
12. Lee HO, Hong Y, Etlioglu HE, Cho YB, Pomella V, Van den Bosch B, et al. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat Genet (2020) 52(6):594–603. doi: 10.1038/s41588-020-0636-z
13. Yan HHN, Siu HC, Ho SL, Yue SSK, Gao Y, Tsui WY, et al. Organoid cultures of early-onset colorectal cancers reveal distinct and rare genetic profiles. Gut (2020) 69(12):2165–79. doi: 10.1136/gutjnl-2019-320019
14. Bolhaqueiro ACF, Ponsioen B, Bakker B, Klaasen SJ, Kucukkose E, van Jaarsveld RH, et al. Ongoing chromosomal instability and karyotype evolution in human colorectal cancer organoids. Nat Genet (2019) 51(5):824–34. doi: 10.1038/s41588-019-0399-6
15. Uhlitz F, Bischoff P, Peidli S, Sieber A, Trinks A, Luthen M, et al. Mitogen-activated protein kinase activity drives cell trajectories in colorectal cancer. EMBO Mol Med (2021) 13(10):e14123. doi: 10.15252/emmm.202114123
16. Roerink SF, Sasaki N, Lee-Six H, Young MD, Alexandrov LB, Behjati S, et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature (2018) 556(7702):457–62. doi: 10.1038/s41586-018-0024-3
17. Qian J, Olbrecht S, Boeckx B, Vos H, Laoui D, Etlioglu E, et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res (2020) 30(9):745–62. doi: 10.1038/s41422-020-0355-0
18. Wu Y, Yang S, Ma J, Chen Z, Song G, Rao D, et al. Spatiotemporal immune landscape of colorectal cancer liver metastasis at single-cell level. Cancer Discov (2022) 12(1):134–53. doi: 10.1158/2159-8290.CD-21-0316
19. Mei Y, Xiao W, Hu H, Lu G, Chen L, Sun Z, et al. Single-cell analyses reveal suppressive tumor microenvironment of human colorectal cancer. Clin Transl Med (2021) 11(6):e422. doi: 10.1002/ctm2.422
20. Wang W, Zhong Y, Zhuang Z, Xie J, Lu Y, Huang C, et al. Multiregion single-cell sequencing reveals the transcriptional landscape of the immune microenvironment of colorectal cancer. Clin Transl Med (2021) 11(1):e253. doi: 10.1002/ctm2.253
21. Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature (2018) 564(7735):268–72. doi: 10.1038/s41586-018-0694-x
22. Liu Y, Zhang Q, Xing B, Luo N, Gao R, Yu K, et al. Immune phenotypic linkage between colorectal cancer and liver metastasis. Cancer Cell (2022) 40(4):424–37.e5. doi: 10.1016/j.ccell.2022.02.013
23. Lu Y, Li Y, Liu Q, Tian N, Du P, Zhu F, et al. MondoA-Thioredoxin-Interacting protein axis maintains regulatory T-cell identity and function in colorectal cancer microenvironment. Gastroenterology (2021) 161(2):575–91.e16. doi: 10.1053/j.gastro.2021.04.041
24. Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O'Brien SA, et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell (2020) 181(2):442–59.e29. doi: 10.1016/j.cell.2020.03.048
25. Wang S, Qu Y, Xia P, Chen Y, Zhu X, Zhang J, et al. Transdifferentiation of tumor infiltrating innate lymphoid cells during progression of colorectal cancer. Cell Res (2020) 30(7):610–22. doi: 10.1038/s41422-020-0312-y
26. Wang H, Gong P, Chen T, Gao S, Wu Z, Wang X, et al. Colorectal cancer stem cell states uncovered by simultaneous single-cell analysis of transcriptome and telomeres. Advanced Sci (Weinheim Baden-Wurttemberg Germany) (2021) 8(8):2004320. doi: 10.1002/advs.202004320
27. Huang HW, Chang CC, Wang CS, Lin KH. Association between inflammation and function of cell adhesion molecules influence on gastrointestinal cancer development. Cells (2021) 10(1):67. doi: 10.3390/cells10010067
28. Bader JE, Voss K, Rathmell JC. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol Cell (2020) 78(6):1019–33. doi: 10.1016/j.molcel.2020.05.034
29. Bagaev A, Kotlov N, Nomie K, Svekolkin V, Gafurov A, Isaeva O, et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell (2021) 39(6):845–65 e7. doi: 10.1016/j.ccell.2021.04.014
30. Schulz M, Salamero-Boix A, Niesel K, Alekseeva T, Sevenich L. Microenvironmental regulation of tumor progression and therapeutic response in brain metastasis. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.01713
31. Angius A, Scanu AM, Arru C, Muroni MR, Carru C, Porcu A, et al. A portrait of intratumoral genomic and transcriptomic heterogeneity at single-cell level in colorectal cancer. Medicina (Kaunas Lithuania) (2021) 57(11):1257. doi: 10.3390/medicina57111257
32. Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature (2012) 487(7407):330–7. doi: 10.1038/nature11252
33. Molinari C, Marisi G, Passardi A, Matteucci L, De Maio G, Ulivi P. Heterogeneity in colorectal cancer: a challenge for personalized medicine? Int J Mol Sci (2018) 19(12):3733. doi: 10.3390/ijms19123733
34. Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour evolution inferred by single-cell sequencing. Nature (2011) 472(7341):90–4. doi: 10.1038/nature09807
35. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med (2012) 366(10):883–92. doi: 10.1056/NEJMoa1113205
36. Driessens G, Beck B, Caauwe A, Simons BD, Blanpain C. Defining the mode of tumour growth by clonal analysis. Nature (2012) 488(7412):527–30. doi: 10.1038/nature11344
37. Bollen Y, Stelloo E, Van Leenen P, van den Bos M, Ponsioen B, Lu B, et al. Reconstructing single-cell karyotype alterations in colorectal cancer identifies punctuated and gradual diversification patterns. Nat Genet (2021) 53(8):1187–95. doi: 10.1038/s41588-021-00891-2
38. Bian S, Hou Y, Zhou X, Li X, Yong J, Wang Y, et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science (2018) 362(6418):1060–3. doi: 10.1126/science.aao3791
39. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
40. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (1996) 271(5256):1734–6. doi: 10.1126/science.271.5256.1734
41. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature (2011) 480(7378):480–9. doi: 10.1038/nature10673
42. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
43. Liu B, Zhang Y, Wang D, Hu X, Zhang Z. Single-cell meta-analyses reveal responses of tumor-reactive CXCL13(+) T cells to immune-checkpoint blockade. Nat Cancer (2022) 3(9):1123–36. doi: 10.1038/s43018-022-00433-7
44. Wang F, Zhang Y, Yu X, Teng XL, Ding R, Hu Z, et al. ZFP91 disturbs metabolic fitness and antitumor activity of tumor-infiltrating T cells. J Clin Invest (2021) 131(19):e144318. doi: 10.1172/JCI144318
45. Osman A, Yan B, Li Y, Pavelko KD, Quandt J, Saadalla A, et al. TCF-1 controls t(reg) cell functions that regulate inflammation, CD8(+) T cell cytotoxicity and severity of colon cancer. Nat Immunol (2021) 22(9):1152–62. doi: 10.1038/s41590-021-00987-1
46. Zhou Y, Bian S, Zhou X, Cui Y, Wang W, Wen L, et al. Single-cell multiomics sequencing reveals prevalent genomic alterations in tumor stromal cells of human colorectal cancer. Cancer Cell (2020) 38(6):818–28.e5. doi: 10.1016/j.ccell.2020.09.015
47. Roulis M, Kaklamanos A, Schernthanner M, Bielecki P, Zhao J, Kaffe E, et al. Paracrine orchestration of intestinal tumorigenesis by a mesenchymal niche. Nature (2020) 580(7804):524–9. doi: 10.1038/s41586-020-2166-3
48. Wang H, Tian T, Zhang J. Tumor-associated macrophages (TAMs) in colorectal cancer (CRC): from mechanism to therapy and prognosis. Int J Mol Sci (2021) 22(16):8470. doi: 10.3390/ijms22168470
49. Donadon M, Torzilli G, Cortese N, Soldani C, Di Tommaso L, Franceschini B, et al. Macrophage morphology correlates with single-cell diversity and prognosis in colorectal liver metastasis. J Exp Med (2020) 217(11):e20191847. doi: 10.1084/jem.20191847
50. Qi J, Crinier A, Escaliere B, Ye Y, Wang Z, Zhang T, et al. Single-cell transcriptomic landscape reveals tumor specific innate lymphoid cells associated with colorectal cancer progression. Cell Rep Med (2021) 2(8):100353. doi: 10.1016/j.xcrm.2021.100353
51. Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, Wallace MB. Colorectal cancer. Lancet (2019) 394(10207):1467–80. doi: 10.1016/S0140-6736(19)32319-0
52. Zhou SN, Pan WT, Pan MX, Luo QY, Zhang L, Lin JZ, et al. Comparison of immune microenvironment between colon and liver metastatic tissue in colon cancer patients with liver metastasis. Digestive Dis Sci (2021) 66(2):474–82. doi: 10.1007/s10620-020-06203-8
53. Tang J, Tu K, Lu K, Zhang J, Luo K, Jin H, et al. Single-cell exome sequencing reveals multiple subclones in metastatic colorectal carcinoma. Genome Med (2021) 13(1):148. doi: 10.1186/s13073-021-00962-3
54. Frank MH, Wilson BJ, Gold JS, Frank NY. Clinical implications of colorectal cancer stem cells in the age of single-cell omics and targeted therapies. Gastroenterology (2021) 160(6):1947–60. doi: 10.1053/j.gastro.2020.12.080
55. Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J, et al. A big bang model of human colorectal tumor growth. Nat Genet (2015) 47(3):209–16. doi: 10.1038/ng.3214
56. Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol (2019) 16(6):361–75. doi: 10.1038/s41575-019-0126-x
Keywords: colorectal cancer, single-cell sequencing, tumor heterogeneity, tumor microenvironment, precise treatment
Citation: Wen R, Zhou L, Peng Z, Fan H, Zhang T, Jia H, Gao X, Hao L, Lou Z, Cao F, Yu G and Zhang W (2023) Single-cell sequencing technology in colorectal cancer: a new technology to disclose the tumor heterogeneity and target precise treatment. Front. Immunol. 14:1175343. doi: 10.3389/fimmu.2023.1175343
Received: 27 February 2023; Accepted: 24 April 2023;
Published: 15 May 2023.
Edited by:
Xi Cheng, Shanghai Jiao Tong University, ChinaReviewed by:
Bo Feng, Shanghai Jiao Tong University, ChinaCheng-Cheng Zhang, Army Medical University, China
Copyright © 2023 Wen, Zhou, Peng, Fan, Zhang, Jia, Gao, Hao, Lou, Cao, Yu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Zhang, d2VpemhhbmcyMDAwY25AMTYzLmNvbQ==; Guanyu Yu, eXVndWFueXUwNDUxQDE2My5jb20=; Fuao Cao, Y2FvZnVhb0BnbWFpbC5jb20=
†These authors have contributed equally to this work