
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 01 September 2023
Sec. Viral Immunology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1173974
 Meng-Mei Zhong1†
Meng-Mei Zhong1† Jia-Hao Xie2†
Jia-Hao Xie2† Yao Feng1
Yao Feng1 Shao-Hui Zhang3
Shao-Hui Zhang3 Jiang-Nan Xia4
Jiang-Nan Xia4 Li Tan1
Li Tan1 Ning-Xin Chen1
Ning-Xin Chen1 Xiao-Lin Su1
Xiao-Lin Su1 Qian Zhang1
Qian Zhang1 Yun-Zhi Feng1*
Yun-Zhi Feng1* Yue Guo1*
Yue Guo1*Background: The coronavirus disease 2019 (COVID-19) caused a global pandemic, with potential severity. We aimed to investigate whether genetically predicted gut microbiome is associated with susceptibility and severity of COVID-19 risk.
Methods: Mendelian randomization (MR) analysis of two sets with different significance thresholds was carried out to infer the causal relationship between the gut microbiome and COVID-19. SNPs associated with the composition of the gut microbiome (n = 5,717,754) and with COVID-19 susceptibility (n = 14,328,058), COVID-19 severity (n = 11,707,239), and COVID-19 hospitalization (n = 12,018,444) from publicly available genome-wide association studies (GWAS). The random-effect inverse variance weighted (IVW) method was used to determine causality. Three more MR techniques—MR Egger, weighted median, and weighted mode—and a thorough sensitivity analysis were also used to confirm the findings.
Results: IVW showed that 18 known microbial taxa were causally associated with COVID-19. Among them, six microbial taxa were causally associated with COVID-19 susceptibility; seven microbial taxa were causally associated with COVID-19 severity ; five microbial taxa were causally associated with COVID-19 hospitalization. Sensitivity analyses showed no evidence of pleiotropy or heterogeneity. Then, the predicted 37 species of the gut microbiome deserve further study.
Conclusion: This study found that some microbial taxa were protective factors or risky factors for COVID-19, which may provide helpful biomarkers for asymptomatic diagnosis and potential therapeutic targets for COVID-19.
The coronavirus disease 2019 (COVID-19) is an infectious disease of potential severity, with more than 200 symptoms identified and effects on multiple-organ systems (1). According to the World Health Organization (WHO), as of 23 July 2023, over 768 million confirmed cases and over 6.9 million deaths have been reported globally (2). This is due to the highly contagious nature of the respiratory-transmitted virus and the fact that SARS-CoV-2 has evolved variants (e.g., α, β, γ, δ, and Omicron) that may escape from neutralizing antibodies and/or cell-mediated immunity, resulting in general susceptibility to the population (3, 4). The virus mainly affects the respiratory system, but studies have found that several patients (27.3%) had gastrointestinal symptoms but no COVID-19 lung imaging findings, indicating that gastrointestinal tissues are vulnerable to SARS-CoV-2 (5). Moreover, the gastrointestinal tract is the largest immune organ in humans and plays a key role in the host’s fight against pathogenic infection (6). Trillions of microbiota colonize the human gut and are involved in host immunomodulation (7). Therefore, it is important to understand the relationship between the gut microbiome and the susceptibility and severity of SARS CoV-2 to the host, the changes in the gut microbiome after host infection with SARS CoV-2, and the long-term impact on human health (including long-term COVID-19 syndrome, “LongCOVID”).
Recently, research on the correlation between the gut microbiome and COVID-19 has attracted widespread attention. There is evidence that the gut microbiome influences the susceptibility and severity of COVID-19. On the one hand, the recruitment of immune cells to the lung may be adversely affected by gut microbiome disruption, which may therefore raise the susceptibility to respiratory infections (8, 9). On the other hand, COVID-19 has been linked to changes in the gut microbiome and impaired gut barrier function, which may enhance the translocation of bacterial products and toxins into the circulatory system and aggravate the systemic inflammatory response (10, 11). Additionally, some studies have shown that the fecal microbiome of COVID-19 patients shows reduced richness and diversity of the gut microbiome (12, 13). Although most current clinical studies suggest that the gut microbiome may be associated with the pathogenesis and disease outcome of COVID-19, most studies are observational epidemiology and have limitations such as confounding factors and causal inversion (14). Thus, the causal associations between gut microbiome and COVID-19 outcomes remain unconfirmed.
Genetic tools, often single-nucleotide polymorphisms (SNPs), are used in Mendelian randomization (MR) research to identify the causal relationships between exposures and outcomes. As either allele has an equal chance of being randomly inherited by a person, MR investigations are comparable with randomized controlled trials (15). MR investigations are more capable of drawing conclusions about causality and are less prone to confounding problems than observational studies like case–control studies (16). This study used two-sample MR to determine whether there is a causal relationship between the composition of the gut microbiome and the risk of COVID-19, as well as to look for pathogenic bacteria and probiotics for COVID-19.
Summary-level statistics of gut microbiome and COVID-19 (including susceptibility, severity, and hospitalization of COVID-19) for genome-wide association study (GWAS) were obtained from the public database. All studies were approved by their respective institutional review boards (IRBs). No new IRB approval was required. A flowchart briefly presents the whole procedure in Figure 1.
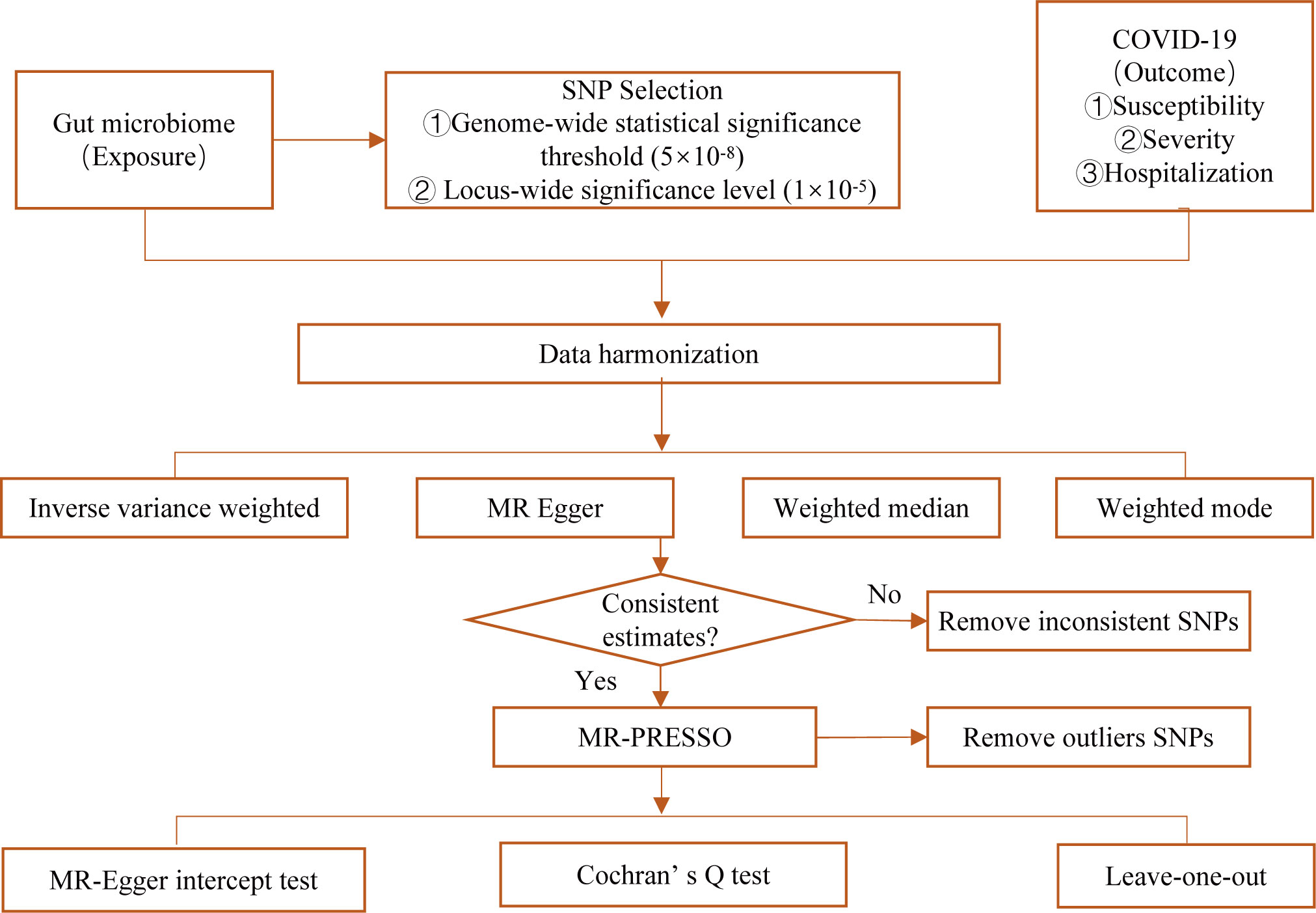
Figure 1 Diagrammatic explanation of the entire MR analysis procedure.
Genetic instruments of the gut microbiome were obtained from 18,340 participants in 24 cohorts, which is the largest, multiethnic, genome-wide meta-analysis of the gut microbiome to date (17) (Table 1). For each cohort, quantitative microbiome characteristic locus (mbQTL) profiling included only taxa in >10% of the sample, containing 211 taxa (9 phyla, 16 classes, 20 orders, 35 families, and 131 genera). Aggregate statistics on the Association’s research are publicly available at https://mibiogen.gcc.rug.nl/.
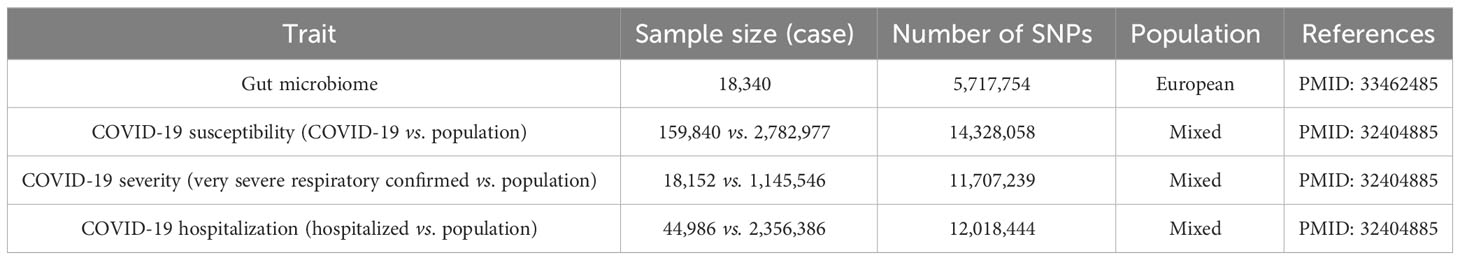
Table 1 Description of the GWAS summary statistics.
Summary-level statistics of COVID-19 susceptibility, severity, and hospitalization for the selected instrument variables were obtained from the COVID-19 Host Genetics Initiative (18) (Table 1). GWAS on COVID-19 susceptibility involved 159,840 patients with COVID-19 and 2,782,977 population controls, whereas GWAS on COVID-19 severity involved 18,152 very severe respiratory confirmed patients with COVID-19 and 1,145,546 population controls. Meanwhile, we also analyzed GWAS on COVID-19 hospitalization which involved 44,986 hospitalized patients with COVID-19 and 2,356,386 population controls. Aggregate statistics on the Association’s research are publicly available at https://www.covid19hg.org/, Release 7.
The following quality control procedures were utilized to choose the best instrument factors to assure the validity and correctness of the results about the causal relationship between gut microbiome and COVID-19 risk. SNPs with a strong correlation to the gut microbiome were first chosen as the instrumental factors. To choose the instrumental variable, two thresholds were applied. The instrumental variables were a group of SNPs that did not meet the genome-wide statistical significance cutoff (). The other group, where SNPs are less than the locus-wide significance threshold (), was chosen as an instrumental factor to acquire more thorough results. The linkage disequilibrium (LD) threshold was set to be , with a clumping window of 10,000 kb. To guarantee a high connection between IVs and exposure variables, the F-statistic of each SNP was utilized to assess the correlation strength and prevent bias brought on by weak IVs. Weak IVs were thought to be bias-free when the F value was more than 10. An important step in MR is ensuring that SNPs’ effect on exposure corresponds to the same allele as the effect on the outcome. According to this principle, palindromic SNPs are removed.
We hypothesized that the gut microbiome influences the risk of COVID-19 and satisfied the assumption that SNP is an instrumental variable correlated with gut microbiome levels; SNP is not associated with any confounding factors; SNP is associated with COVID-19 via gut microbiome (i.e., horizontal pleiotropy should not be present).
We used the two-sample MR analysis utilizing the random-effect inverse variance weighted (IVW) approach, which is the most popular in MR research and could offer reliable causal estimates in the absence of directional pleiotropy, to assess the impact of the gut microbiome on COVID-19 (19). MR Egger, weighted median, and weighted mode were also used to further validate the causal results (20, 21). Also, Mendelian Randomization Pleiotropy RESidual Sum and Outlier (MR-PRESSO) analysis was performed to find outlier instruments and then removed inconsistent SNPs (22). Finally, extensive sensitivity studies were needed to assess any model assumption breaches in the MR study. The Cochran’s Q test was performed to determine if there is heterogeneity among the various causal effects, and MR–Egger intercept analysis was used to assess the horizontal pleiotropy (23). Moreover, leave-one-out analysis was performed to assess the effect of individual variation on the observed association, which refers to eliminating each SNP step by step, calculating the meta-effect of the remaining SNPs, and observing whether the result changes after removing each SNP.
In this MR study, the gut microbiome database’s lowest taxonomic level was genus. However, we need to identify the species of the gut microbiome if we want to explore the mechanism between the gut microbiome and disease development. Therefore, according to the results of the genus, we searched the species included in the genus using the UniProt Knowledgebase (UniProtKB) (24) and determined whether the species belongs to the human gut microbiome through GMrepo (data repository for Gut Microbiota) (25).
All analyses were performed using R software (version 4.2.1) under the Windows environment. The R packages used for all MR-related analysis and image plotting included “TwoSampleMR” and “MR-PRESSO”. Based on the IVW method, a two-sided was considered statistically significant. Additionally, we used the 0.05 threshold for the FDR correction (p-FDR). When and , the causal connection was considered significant. In addition, when but , the causal connection was considered potentially significant.
With the genome-wide statistical significance threshold of , MR analysis was performed with the gut microbiome as a whole, and a total of 36 SNPs were included in the study. For each SNP, the F-statistics varied from 29.81 to 85.38, indicating a low likelihood of weak instruments. In the IVW analysis, we observed evidence of causal associations of the gut microbiome without risks of COVID-19 susceptibility (odds ratio (OR) = 1.042; 95% confidence interval (CI) = 0.970–1.118; p = 0.259), and COVID-19 severity (OR = 1.071; 95% CI = 0.889–1.291; p = 0.469), and COVID-19 hospitalization (OR = 1.067; 95% CI = 0.943–1.207; p = 0.304). MR–Egger regression showed no horizontal pleiotropy between instrumental variables and outcome. IVW heterogeneity testing indicated heterogeneity () but did not affect IVW results. The detailed description is summarized in Table 2.
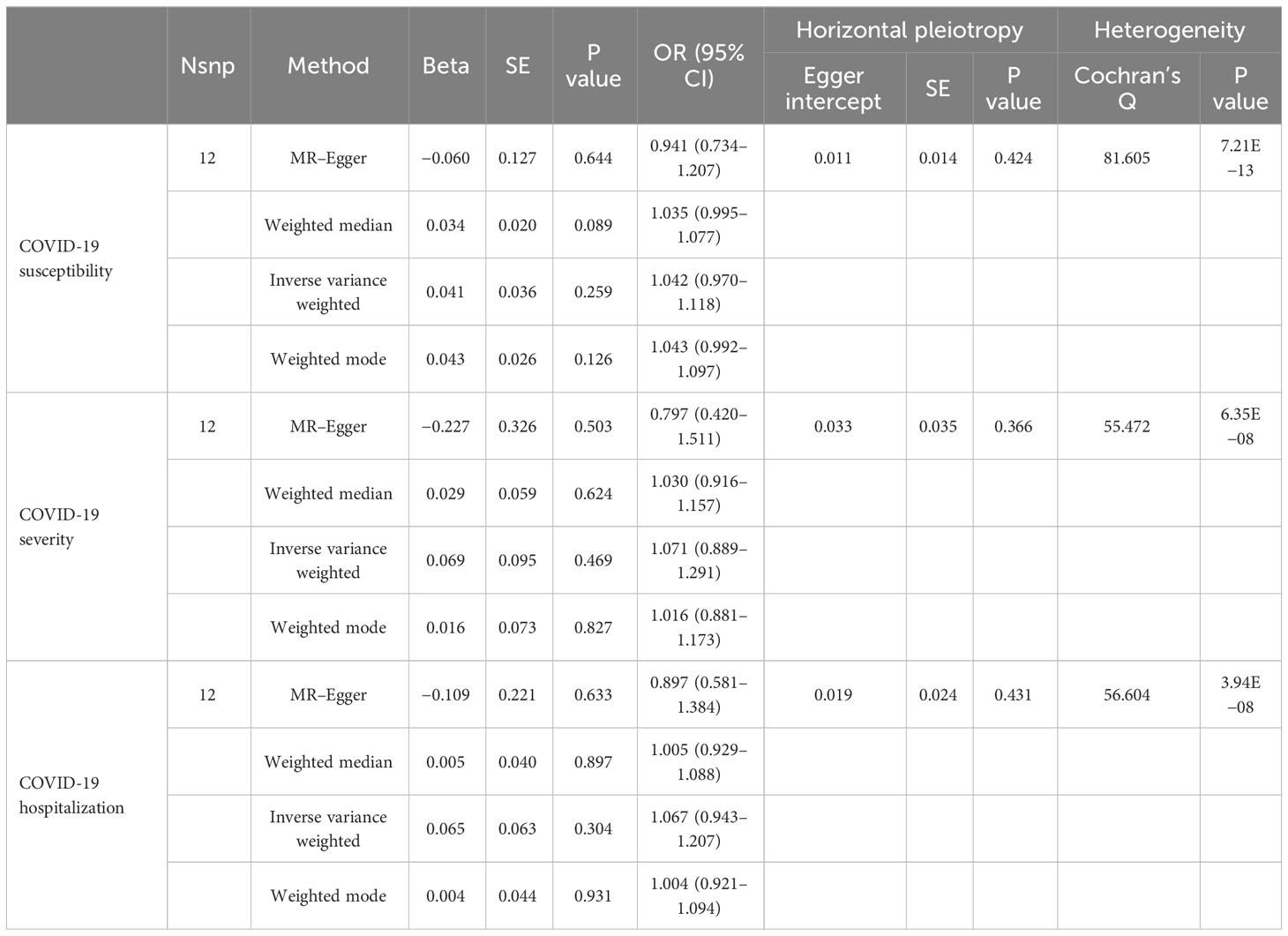
Table 2 MR results of causal links between the gut microbiome and COVID-19 risk p<5×10−8.
A total of 235 SNPs were identified as instrumental variables for predicted gut microbiome mass. For each SNP, the F-statistics varied from 17.68 to 35.42, indicating a low likelihood of weak instruments. Furthermore, we observed some significant evidence for the causal association between gut microbiome and COVID-19 risk, based on IVW methods. Finally, 18 known named microbial taxa were identified at a significance of 0.05, including one phylum, two classes, three orders, three families, and nine genera (Figure 2). Thereinto, two microbial taxa were protective factors and four microbial taxa were risky factors of COVID-19 susceptibility; seven microbial taxa were protective factors, and five microbial taxa were risky factors of COVID-19 severity and hospitalization. The detailed description is summarized in Supplementary Tables 1–3. However, after FDR adjustment, the aforementioned correlations were no longer statistically significant (). This suggests that the above microbial taxa have potential causal suggestions for COVID-19.
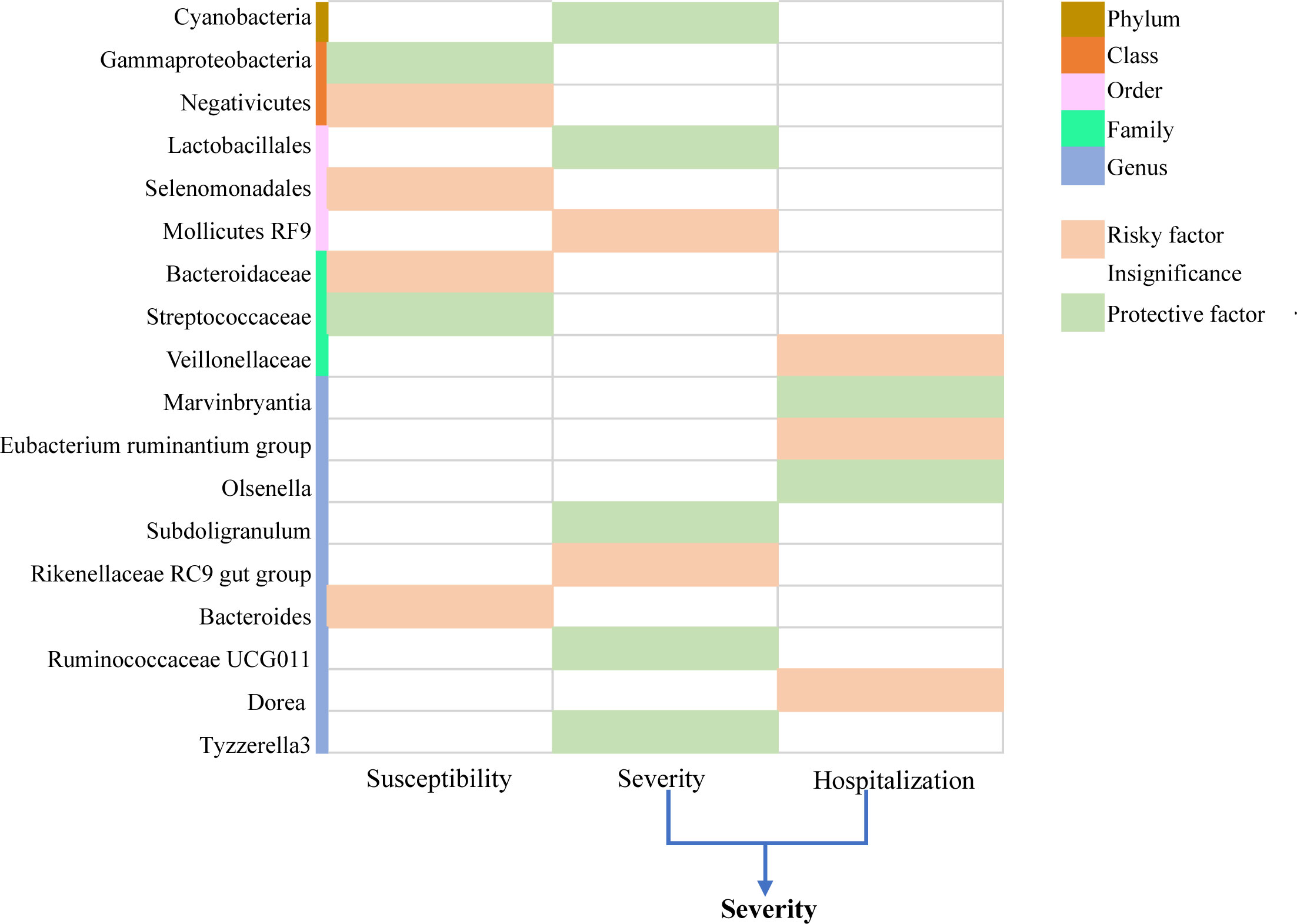
Figure 2 Causal relationship between gut microbiome and COVID-19 susceptibility, COVID-19 severity, and COVID-19 hospitalization at the locus-wide significance level (). .
The results of IVW analysis showed that class Gammaproteobacteria (OR = 0.933; 95% CI = 0.879–0.991; p = 0.023) and family Streptococcaceae (OR = 0.955; 95% CI = 0.916–0.995; p = 0.029) were negatively correlated with the risk of COVID−19 susceptibility; class Negativicutes (OR = 1.054; 95%CI = 1.005–1.105; p = 0.030), order Selenomonadales (OR = 1.054; 95% CI = 1.005–1.105; p = 0.030), family Bacteroidaceae (OR = 1.064; 95% CI = 1.007–1.125; p = 0.028), and genus Bacteroides (OR = 1.064; 95% CI = 1.007–1.125; p = 0.028) were positively correlated with the risk of COVID−19 susceptibility. MR Egger, weighted median, and weighted mode also showed the same direction of influence as IVW, although the P value was not statistically significant (Figure 3).
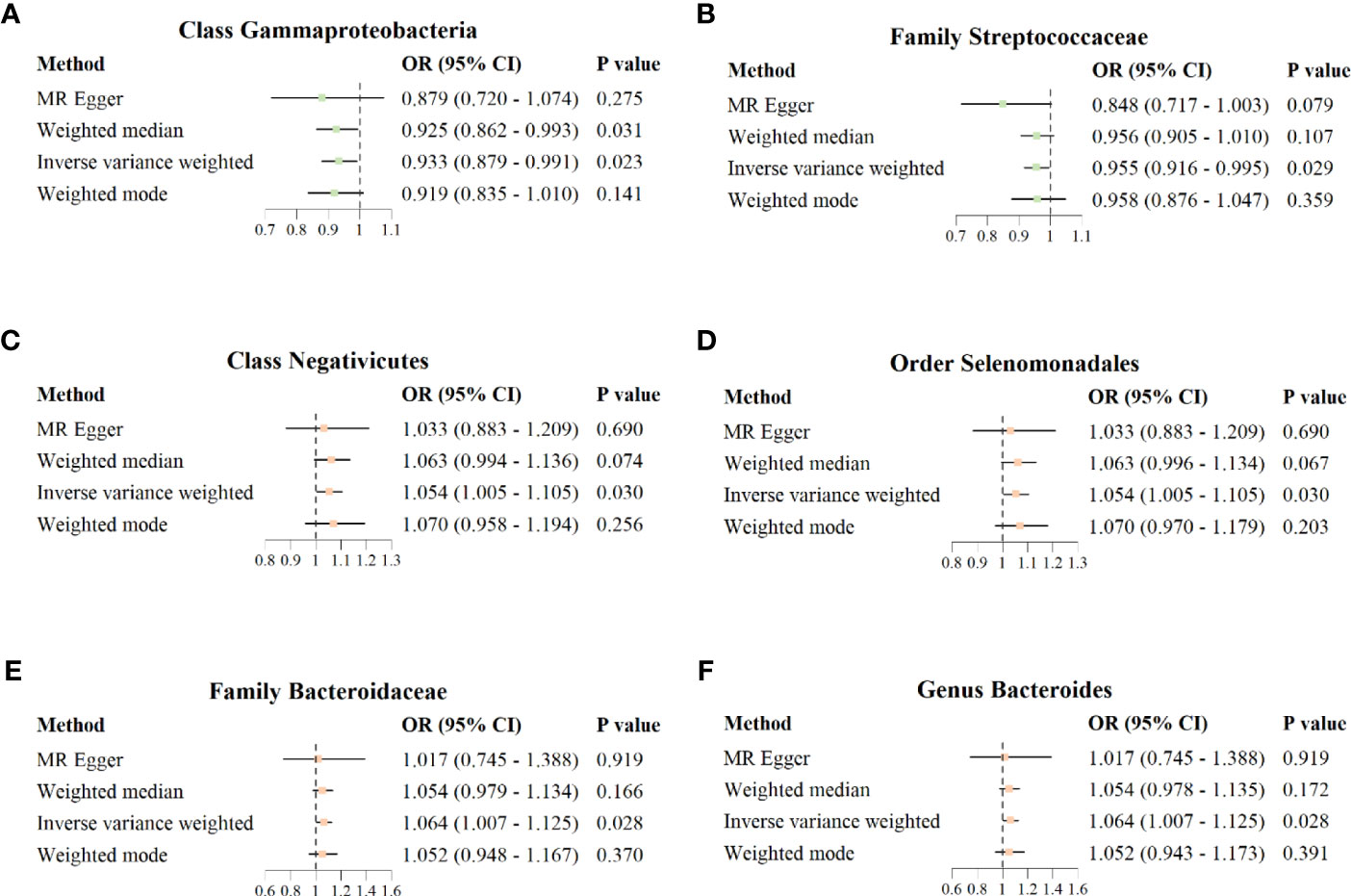
Figure 3 Forest plot for causal association between six unique protective/risky microbial taxa with COVID-19 susceptibility. (A, B) Two protective microbial taxa for COVID-19 susceptibility. (C–F) Four risky microbial taxa for COVID-19 susceptibility.
The results of IVW analysis showed that phylum Cyanobacteria (OR = 0.852; 95% CI = 0.760–0.955; p = 0.006), order Lactobacillales (OR = 0.867; 95% CI = 0.764–0.983; p = 0.026), genus Ruminococcaceae UCG011 (OR = 0.907; 95% CI = 0.832–0.988; p = 0.025), genus Subdoligranulum (OR = 0.807; 95% CI = 0.699–0.932; p = 0.004), and genus Tyzzerella3 (OR = 0.885; 95% CI = 0.810–0.967, p = 0.007) were negatively correlated with the risk of COVID−19 severity; order Mollicutes RF9 (OR = 1.141; 95% CI = 1.009–1.291; p = 0.035) and genus Rikenellaceae RC9 gut group (OR = 1.085; 95% CI = 1.009–1.167; p = 0.028) were positively correlated with the risk of COVID−19 severity (Figure 4). In addition, the results of IVW analysis showed that genus Marvinbryantia (OR = 0.886; 95% CI = 0.812–0.967; p = 0.007) and genus Olsenella (OR = 0.942; 95% CI = 0.897–0.990; p = 0.018) were negatively correlated with the risk of COVID-19 hospitalization; family Veillonellaceae (OR = 1.069; 95% CI = 1.002–1.140; p = 0.044), genus Eubacterium ruminantium group (OR = 1.065; 95% CI = 1.010–1.123; p = 0.021), and genus Dorea (OR = 1.162; 95% CI = 1.055–1.279; p = 0.002) were positively correlated with the risk of COVID-19 hospitalization. MR–Egger, weighted median, and weighted mode also showed the same direction of influence as IVW, although the P value was not statistically significant (Supplementary Figure 1).
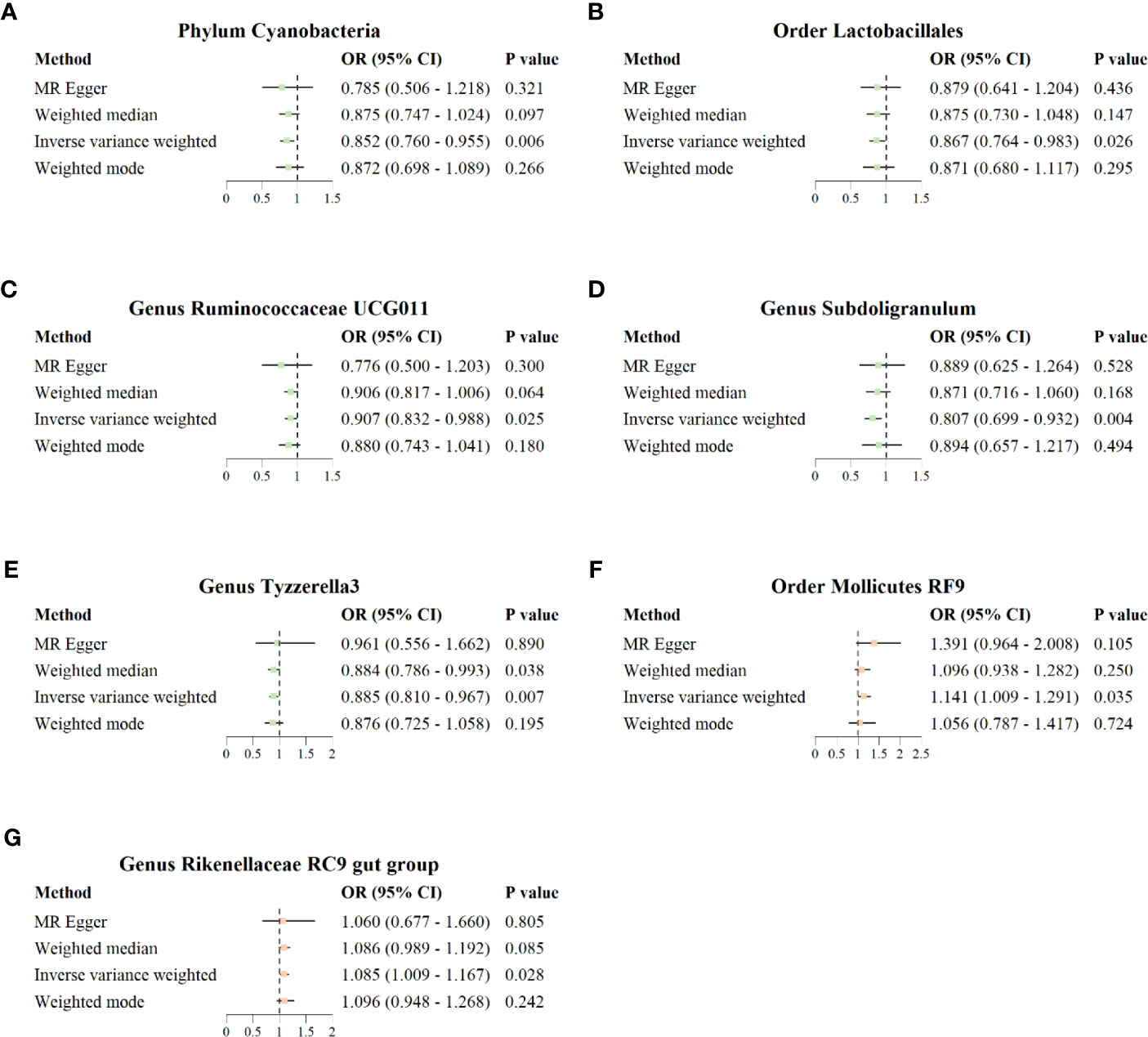
Figure 4 Forest plot for causal association between seven unique protective/risky microbial taxa with COVID-19 severity. (A–E) Five protective microbial taxa for COVID-19 severity. (F, G) Two risky microbial taxa for COVID-19 severity.
A comprehensive sensitivity analysis was then performed to assess the robustness of the causal relationship between the gut microbiome and COVID-19 above. Firstly, the results of the MR-PRESSO trial showed no horizontal pleiotropic outliers. Secondly, no evidence of a horizontal pleiotropic effect was observed using the MR–Egger intercept analysis. In addition, heterogeneity testing also indicated that there is no heterogeneity. The detailed description is summarized in Table 3. Furthermore, the leave-one-out analysis revealed that none of the included SNPs was outliers. The plots of the leave-one-out analysis are shown in Supplementary Figure 2.
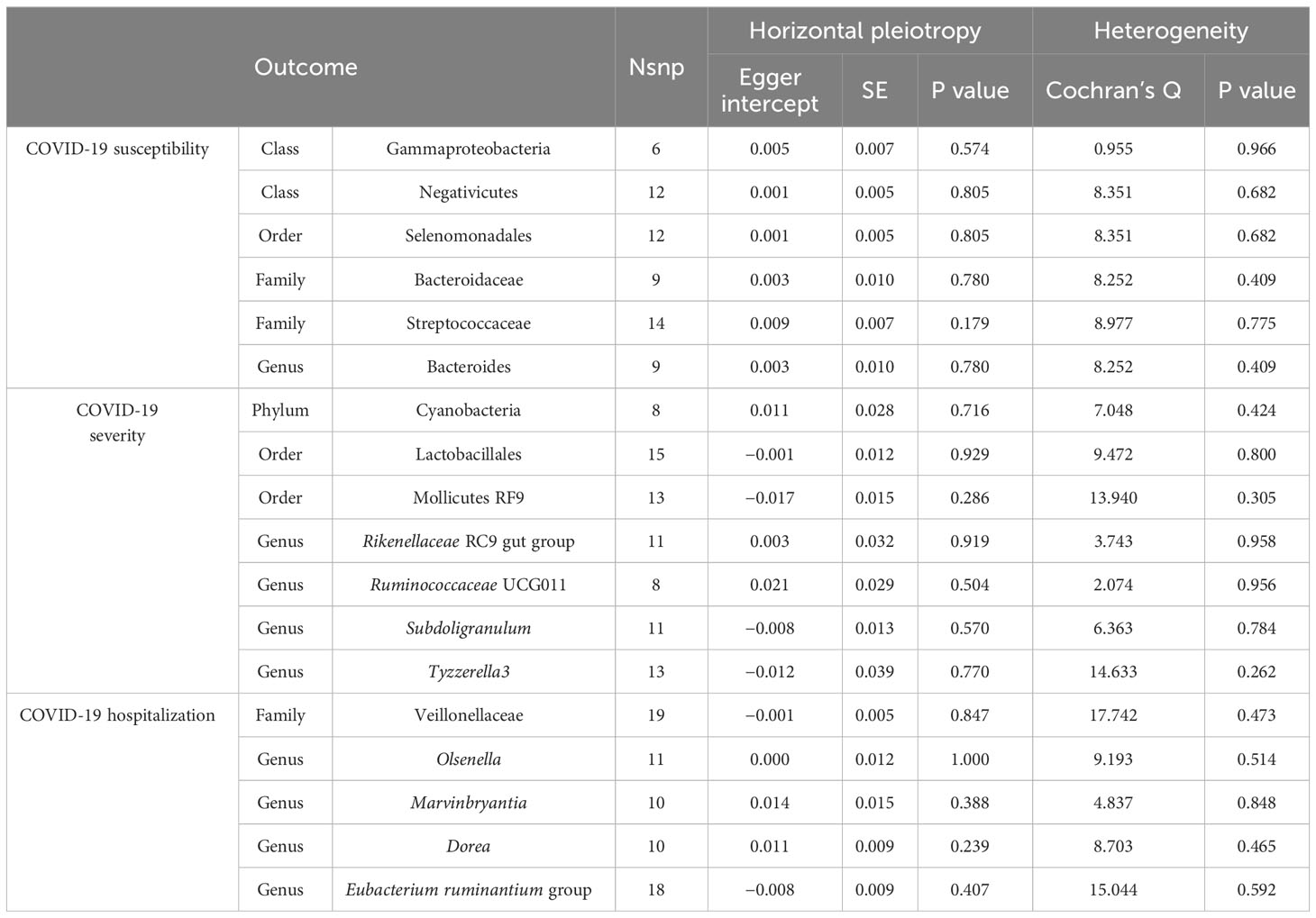
Table 3 Heterogeneity and horizontal pleiotropy analyses results p <1×10-5.
A search on the UniProtKB and GMrepo websites yielded 37 species of the gut microbiome that may have a causal relationship with COVID-19. Among them, 31 species of the gut microbiome affect COVID-19 susceptibility; 1 species of the gut microbiome affects COVID-19 severity; and 5 species of the gut microbiome affect COVID-19 hospitalization. The likely species of the gut microbiome affecting COVID-19 were predicted by the above steps, and the results are visualized in Figure 5.

Figure 5 A visualization of the results for predicting species of gut microbiome that may affect COVID-19 based on the gut microbiome genus level.
To our knowledge, this is the first MR study to assess a causal relationship between the gut microbiome and COVID-19 risk. At the locus-wide significance level (), the gut microbiome was found to be strongly associated with COVID-19 disease. Among them, six microbial taxa were causally associated with COVID-19 susceptibility; seven microbial taxa were causally associated with COVID-19 severity risk; five microbial taxa were causally associated with COVID-19 hospitalization. This provides new ideas for the clinical treatment of COVID-19.
Recently, some clinical evidence has shown that respiratory diseases are often accompanied by dysbiosis or changes in the gut microbiome. In studies of the gut microbiome of asthma patients, a link was found between low gut microbial diversity and childhood asthma (26). Asthma has also been linked closely to certain gut microbiome in addition to microbial diversity, such as asthma sufferers’ guts contained more Clostridium and Eggerthella lenta than healthy controls did (27). A systematic review of 11 studies on respiratory infections and changes in gut microbiome showed that patients with respiratory infections had a 1.45-unit reduction in gut microbiome diversity (Shannon) and a decrease in taxa abundance compared with controls (28). In addition, the antiviral immune response caused by respiratory viral infections such as influenza is associated with changes in the composition and function of gut microbiome (29). Noting that there are alterations in the gut microbiome in respiratory diseases, researchers have sought to prevent and treat respiratory diseases by improving the gut microbiome to modulate the lung immune response (30). A 3-month randomized, double-blind, placebo-controlled human trial in 55 people with asthma demonstrated the adjunct efficacy of Bifidobacterium lactis Probio in the treatment of asthma (31). Fecal microbiota transplantation (FMT) experiments in mice with Streptococcus pneumoniae infection depleted of gut microbiome detected normalization of lung bacterial counts and tumor necrosis factor-α and interleukin-10 levels, indicating that the gut microbiome plays a role in protecting the host during Streptococcus pneumoniae infection (32). A randomized controlled trial of 300 participants found that supplementation with a probiotic formula consisting of Lactobacillus plantarum and Pediococcus acidilactici improved symptoms in COVID-19 patients (33). Furthermore, a large number of observational studies have found changes in the composition of the gut microbiome in COVID-19 patients compared with healthy controls (34) and that COVID-19 infection increases the number of opportunistic pathogens in the gut and reduces the number of beneficial gut microbiome (35). Through studies of different populations, it was found that the fecal microbiota of COVID-19 patients showed a decrease in bacterial diversity (36) and the richness of the gut microbiome did not return to normal even 6 months after the onset of illness (37). In our MR study, alterations in the gut microbiome of patients with respiratory disease (COVID-19) were also found, suggesting that the gut microbiome we obtained also holds promise for clinical treatment.
Furthermore, understanding the mechanism by which the gut microbiome affects COVID-19 through the gut–lung axis can better provide a target site for the prevention and treatment of COVID-19. The human gut microbiome is made up of more than a trillion bacteria in a dynamic ecosystem that controls our whole physiology and immune system (38). Dysbiosis of the gut microbiome affects not only the immune responses of the gastrointestinal tract but also the immunity of distal organs, such as the lung, which further affects lung health and respiratory conditions (30). Based on previous studies, the gut microbiome can influence the severity of SARS-CoV-2 infection and host immune response through several hypothetical mechanisms: (1) Pro-inflammatory opportunistic pathogens become the dominant strain, which is further recognized by innate lymphocytes and causes a decrease in beneficial metabolites or an increase in pro-inflammatory metabolites (39). (2) Intestinal barrier damage and bacterial displacement can induce an increase in intestinal epithelial reactive oxygen species production as well as increased intestinal permeability, thereby enhancing the intestinal pro-inflammatory response (40). (3) Inflammatory factors reach the lungs through blood circulation, causing the aggregation of inflammatory cells represented by neutrophils and macrophages, and then initiate the inflammatory cascade of the lungs (41, 42). (4) In addition to the peripheral circulation, mesenteric lymph can induce lung damage by transporting enterogenic cytotoxic/inflammatory factors to the pulmonary circulation (41). (5) Opportunistic pathogens and toxins may transfer to the circulatory system, leading to bacteremia and exacerbating systemic inflammation and disease severity (10). In addition, gut microbiome could lead to a decreased expression of angiotensin-converting enzyme 2 (ACE2), an entry receptor for SARS-CoV-2, thereby influencing viral invasion and replication (43, 44). The impact of intestinal ACE2 expression on gut microbiome has been investigated in a number of animal studies. For instance, studies using mouse models revealed that the bacteria Bacteroides dorei and Bifidobacterium longum decrease colonic ACE2 expression (11, 45). Hirayama et al. identified GATA4, regulated by the gut microbiome, as a transcription factor that regulates ACE2 expression in the gut (46, 47). Therefore, further research into whether the microbial taxa we found also has a similar pathway can further verify the significant role of gut microbiome in COVID-19 and help in the study of targeted drugs.
In this MR study, we obtained that 18 microbial taxa are causally associated with COVID-19. Since the gut microbiome database’s lowest taxonomic level was genus, we predicted 37 species of gut microbiome that affect COVID-19, through the online website. Among them, some gut microbiome and their species can find evidence of association with COVID-19 in previous studies. The proinflammatory effect of Bacteroides was validated by GM ecology and correlation analysis against blood inflammatory markers, associated with COVID-19 disease severity (48). In addition, it was discovered that there was a positive correlation between Bacteroides cellulosilyticus and the enzymes aspartate transaminase, creatine kinase isoenzymes, and lactate dehydrogenase. This finding suggested that the presence of more gut microorganisms may contribute to severe or critical multiorgan dysfunction (49). Furthermore, there is also evidence of a correlation between Cyanobacteria, Subdolagularum, and COVID-19 (50, 51). The above results suggest that some of the protective/risky gut microbiome in the results of this MR analysis can be found in previous studies and are likely to become important microbiota for future clinical references. In addition, our newly discovered gut microbiome and predicted species need further improvement in clinical, and in vivo experiments to enrich the “gut–lung axis” theories.
The main advantage of this study is that the implementation of the MR method reduces the interference of confounding factors and the reverse causality of results, which may be more persuasive than observational research. However, there are still some limitations: (1) This study only explains the one-way causal relationship between gut microbiome as an exposure factor and COVID-19 as an outcome factor and does not verify the impact of COVID-19 on gut microbiome. (2) The summary-level data of two sample MR was used. Although this method has increased statistical power and obtained interesting results, we cannot adjust important covariates such as diet or drug use. (3) Since most of the participants in GWAS are of European origin, extrapolating the research results to other ethnic groups may be limited. (4) Two GWAS have collected multiple queues, and there may be deviation of overlapping samples. (5) After multiple-testing adjustment, the above association was no longer statistically significant. This MR analysis involved 211 exposures and 3 outcomes, and it is difficult to get meaningful results after multiple validations. Therefore, further experiments are needed to verify the significance of these gut microbiome on COVID-19.
In conclusion, our MR findings suggest a causal effect of the gut microbiome on COVID-19. In addition, genus Marvinbryantia, genus Olsenella, genus Ruminococcaceae UCG011, genus Subdoligranulum, and genus Tyzzerella3 would be potentially beneficial genera for the treatment of COVID-19. This provides new ideas for the development of gut microbiome regulators and new avenues for the treatment of COVID-19 patients.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Conceptualization, M-MZ, J-HX, YF, and YG. Data curation, J-HX, M-MZ, and YG. Formal analysis, S-HZ and J-NX. Funding acquisition, Y-ZF and YG. Investigation, QZ and X-LS. Methodology, M-MZ and J-HX. Project administration, Y-ZF and YG. Resources, J-HX and S-HZ. Supervision, Y-ZF and YG. Validation, LT and N-XC. Visualization, J-NX and J-HX. Writing of original draft, M-MZ and YF. Writing review and editing, M-MZ, YF, and J-HX. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation of China (81800788 and 81773339), Science and Technology Department of Hunan Province, China (2017WK2041, 2018SK52511, and 2022ZK4084), Scientific Research Project of Hunan Provincial Health Commission (202208043514 and B202308056340), Hunan Provincial Natural Science Foundation of China (2022JJ30062), Natural Science Foundation of Changsha City (kq2202403 and kq2202412), Fund for the Xiangya Clinical Medicine Database of Central South University (2014-ZDYZ-1-16), Education and Teaching Reform Research Project of Central South University (2020jy165-3), Research Project on Postgraduate Education and Teaching Reform of Central South University(2021JGB072), Hunan Provincial Innovation Foundation For Postgraduate (CX20220370), and the Fundamental Research Funds for the Central Universities of Central South University (2022ZZTS0913 and 2022ZZTS0912).
We are very grateful to COVID‐19 Host Genetics Initiative and MiBioGen Consortium for their selfless public sharing of GWAS summary data, and thanks are given to UniProtKB and GMrepo websites for providing information about the gut microbiome, which provides us with great convenience to carry out this research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1173974/full#supplementary-material
Supplementary Figure 1 | Forest plot for causal association between 5 unique protective/risky microbial taxa with COVID-19 hospitalization. (A, B) 2 protective microbial taxa for COVID-19 hospitalization; (C–E) 3 risky microbial taxa for COVID-19 hospitalization.
Supplementary Figure 2 | The plots of the leave-one-out analysis ().
1. Davis HE, McCorkell L, Vogel JM, Topol EJ. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol (2023) 21(3):133–46. doi: 10.1038/s41579-022-00846-2
2. Organization WH. Coronavirus disease (COVID-19) (2023). Available at: https://covid19.who.int/.
3. Gang J, Wang H, Xue X, Zhang S. Microbiota and COVID-19: Long-term and complex influencing factors. Front Microbiol (2022) 13:963488. doi: 10.3389/fmicb.2022.963488
4. Hadj Hassine I. Covid-19 vaccines and variants of concern: A review. Rev Med Virol (2022) 32(4):e2313. doi: 10.1002/rmv.2313
5. Lin L, Jiang X, Zhang Z, Huang S, Zhang Z, Fang Z, et al. Gastrointestinal symptoms of 95 cases with SARS-CoV-2 infection. Gut (2020) 69(6):997–1001. doi: 10.1136/gutjnl-2020-321013
6. Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, Knight R. Current understanding of the human microbiome. Nat Med (2018) 24(4):392–400. doi: 10.1038/nm.4517
7. Donaldson GP, Lee SM, Mazmanian SK. Gut biogeography of the bacterial microbiota. Nat Rev Microbiol (2016) 14(1):20–32. doi: 10.1038/nrmicro3552
8. Parrot T, Gorin JB, Ponzetta A, Maleki KT, Kammann T, Emgård J, et al. MAIT cell activation and dynamics associated with COVID-19 disease severity. Sci Immunol (2020) 5(51):eabe1670. doi: 10.1101/2020.08.27.20182550
9. Legoux F, Salou M, Lantz O. MAIT cell development and functions: the microbial connection. Immunity (2020) 53(4):710–23. doi: 10.1016/j.immuni.2020.09.009
10. Sun Z, Song ZG, Liu C, Tan S, Lin S, Zhu J, et al. Gut microbiome alterations and gut barrier dysfunction are associated with host immune homeostasis in COVID-19 patients. BMC Med (2022) 20(1):24. doi: 10.1186/s12916-021-02212-0
11. Vatanen T, Kostic AD, d'Hennezel E, Siljander H, Franzosa EA, Yassour M, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell (2016) 165(4):842–53. doi: 10.1016/j.cell.2016.04.007
12. Gaibani P, D'Amico F, Bartoletti M, Lombardo D, Rampelli S, Fornaro G, et al. The gut microbiota of critically ill patients with COVID-19. Front Cell Infect Microbiol (2021) 11:670424. doi: 10.3389/fcimb.2021.670424
13. Ren Z, Wang H, Cui G, Lu H, Wang L, Luo H, et al. Alterations in the human oral and gut microbiomes and lipidomics in COVID-19. Gut (2021) 70(7):1253–65. doi: 10.1136/gutjnl-2020-323826
14. Arsenault BJ. From the garden to the clinic: how Mendelian randomization is shaping up atherosclerotic cardiovascular disease prevention strategies. Eur Heart J (2022) 43(42):4447–9. doi: 10.1093/eurheartj/ehac394
15. Emdin CA, Khera AV, Kathiresan S. Mendelian randomization. Jama (2017) 318(19):1925–6. doi: 10.1001/jama.2017.17219
16. Bowden J, Holmes MV. Meta-analysis and Mendelian randomization: A review. Res Synth Methods (2019) 10(4):486–96. doi: 10.1002/jrsm.1346
17. Kurilshikov A, Medina-Gomez C, Bacigalupe R, Radjabzadeh D, Wang J, Demirkan A, et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet (2021) 53(2):156–65. doi: 10.1038/s41588-020-00763-1
18. Initiative C-HG. The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur J Hum Genet (2020) 28(6):715–8. doi: 10.1002/jrsm.1346
19. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol (2013) 37(7):658–65. doi: 10.1002/gepi.21758
20. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol (2015) 44(2):512–25. doi: 10.1093/ije/dyv080
21. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol (2016) 40(4):304–14. doi: 10.1002/gepi.21965
22. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet (2018) 50(5):693–8. doi: 10.1038/s41588-018-0099-7
23. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol (2017) 32(5):377–89. doi: 10.1007/s10654-017-0255-x
24. UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res (2023) 51(D1):D523–31. doi: 10.1093/nar/gkac1052
25. Dai D, Zhu J, Sun C, Li M, Liu J, Wu S, et al. GMrepo v2: a curated human gut microbiome database with special focus on disease markers and cross-dataset comparison. Nucleic Acids Res (2022) 50(D1):D777–d84. doi: 10.1093/nar/gkab1019
26. Abrahamsson TR, Jakobsson HE, Andersson AF, Björkstén B, Engstrand L, Jenmalm MC. Low gut microbiota diversity in early infancy precedes asthma at school age. Clin Exp Allergy (2014) 44(6):842–50. doi: 10.1111/cea.12253
27. Wang Q, Li F, Liang B, Liang Y, Chen S, Mo X, et al. A metagenome-wide association study of gut microbiota in asthma in UK adults. BMC Microbiol (2018) 18(1):114. doi: 10.1186/s12866-018-1257-x
28. Woodall CA, McGeoch LJ, Hay AD, Hammond A. Respiratory tract infections and gut microbiome modifications: A systematic review. PloS One (2022) 17(1):e0262057. doi: 10.1371/journal.pone.0262057
29. Hanada S, Pirzadeh M, Carver KY, Deng JC. Respiratory viral infection-induced microbiome alterations and secondary bacterial pneumonia. Front Immunol (2018) 9:2640. doi: 10.3389/fimmu.2018.02640
30. Chunxi L, Haiyue L, Yanxia L, Jianbing P, Jin S. The gut microbiota and respiratory diseases: new evidence. J Immunol Res (2020) 2020:2340670. doi: 10.1155/2020/2340670
31. Liu A, Ma T, Xu N, Jin H, Zhao F, Kwok LY, et al. Adjunctive probiotics alleviates asthmatic symptoms via modulating the gut microbiome and serum metabolome. Microbiol Spectr (2021) 9(2):e0085921. doi: 10.1128/Spectrum.00859-21
32. Schuijt TJ, Lankelma JM, Scicluna BP, de Sousa e Melo F, Roelofs JJ, de Boer JD, et al. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut (2016) 65(4):575–83. doi: 10.1136/gutjnl-2015-309728
33. Gutiérrez-Castrellón P, Gandara-Martí T, Abreu YAAT, Nieto-Rufino CD, López-Orduña E, Jiménez-Escobar I, et al. Probiotic improves symptomatic and viral clearance in Covid19 outpatients: a randomized, quadruple-blinded, placebo-controlled trial. Gut Microbes (2022) 14(1):2018899. doi: 10.1080/19490976.2021.2018899
34. Zhang F, Lau RI, Liu Q, Su Q, Chan FKL, Ng SC. Gut microbiota in COVID-19: key microbial changes, potential mechanisms and clinical applications. Nat Rev Gastroenterol Hepatol (2022) 20(5):323–37. doi: 10.1038/s41575-022-00698-4
35. Zuo T, Zhan H, Zhang F, Liu Q, Tso EYK, Lui GCY, et al. Alterations in fecal fungal microbiome of patients with COVID-19 during time of hospitalization until discharge. Gastroenterology (2020) 159(4):1302–10.e5. doi: 10.1053/j.gastro.2020.06.048
36. Xu R, Lu R, Zhang T, Wu Q, Cai W, Han X, et al. Temporal association between human upper respiratory and gut bacterial microbiomes during the course of COVID-19 in adults. Commun Biol (2021) 4(1):240. doi: 10.1038/s42003-021-01796-w
37. Chen Y, Gu S, Chen Y, Lu H, Shi D, Guo J, et al. Six-month follow-up of gut microbiota richness in patients with COVID-19. Gut (2022) 71(1):222–5. doi: 10.1136/gutjnl-2021-324090
38. de Oliveira GLV, Oliveira CNS, Pinzan CF, de Salis LVV, Cardoso CRB. Microbiota modulation of the gut-lung axis in COVID-19. Front Immunol (2021) 12:635471. doi: 10.3389/fimmu.2021.635471
39. Constantinides MG. Interactions between the microbiota and innate and innate-like lymphocytes. J Leukoc Biol (2018) 103(3):409–19. doi: 10.1002/JLB.3RI0917-378R
40. Uzzan M, Corcos O, Martin JC, Treton X, Bouhnik Y. Why is SARS-CoV-2 infection more severe in obese men? The gut lymphatics - Lung axis hypothesis. Med Hypotheses (2020) 144:110023. doi: 10.1016/j.mehy.2020.110023
41. Ma Y, Yang X, Chatterjee V, Wu MH, Yuan SY. The gut-lung axis in systemic inflammation. Role of mesenteric lymph as a conduit. Am J Respir Cell Mol Biol (2021) 64(1):19–28. doi: 10.1165/rcmb.2020-0196TR
42. Young RP, Hopkins RJ, Marsland B. The gut-liver-lung axis. Modulation of the innate immune response and its possible role in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol (2016) 54(2):161–9. doi: 10.1165/rcmb.2015-0250PS
43. Zhang H, Li HB, Lyu JR, Lei XM, Li W, Wu G, et al. Specific ACE2 expression in small intestinal enterocytes may cause gastrointestinal symptoms and injury after 2019-nCoV infection. Int J Infect Dis (2020) 96:19–24. doi: 10.1016/j.ijid.2020.04.027
44. Viana SD, Nunes S, Reis F. ACE2 imbalance as a key player for the poor outcomes in COVID-19 patients with age-related comorbidities - Role of gut microbiota dysbiosis. Ageing Res Rev (2020) 62:101123. doi: 10.1016/j.arr.2020.101123
45. Geva-Zatorsky N, Sefik E, Kua L, Pasman L, Tan TG, Ortiz-Lopez A, et al. Mining the human gut microbiota for immunomodulatory organisms. Cell (2017) 168(5):928–43.e11. doi: 10.1016/j.cell.2017.01.022
46. Hirayama M, Nishiwaki H, Hamaguchi T, Ito M, Ueyama J, Maeda T, et al. Intestinal Collinsella may mitigate infection and exacerbation of COVID-19 by producing ursodeoxycholate. PloS One (2021) 16(11):e0260451. doi: 10.1371/journal.pone.0260451
47. Liu Y, Zhang H, Tang X, Jiang X, Yan X, Liu X, et al. Distinct metagenomic signatures in the SARS-coV-2 infection. Front Cell Infect Microbiol (2021) 11:706970. doi: 10.3389/fcimb.2021.706970
48. ROmani L, Del Chierico F, Macari G, Pane S, Ristori MV, Guarrasi V, et al. The relationship between pediatric gut microbiota and SARS-coV-2 infection. Front Cell Infect Microbiol (2022) 12:908492. doi: 10.3389/fcimb.2022.908492
49. Xu X, Zhang W, Guo M, Xiao C, Fu Z, Yu S, et al. Integrated analysis of gut microbiome and host immune responses in COVID-19. Front Med (2022) 16(2):263–75. doi: 10.1007/s11684-022-0921-6
50. Barre A, Van Damme EJM, Simplicien M, Le Poder S, Klonjkowski B, Benoist H, et al. Man-specific lectins from plants, fungi, algae and cyanobacteria, as potential blockers for SARS-CoV, MERS-CoV and SARS-CoV-2 (COVID-19) coronaviruses: biomedical perspectives. Cells (2021) 10(7):1619. doi: 10.3390/cells10071619
Keywords: gut microbiome, Mendelian randomization, COVID-19, susceptibility, severity
Citation: Zhong M-M, Xie J-H, Feng Y, Zhang S-H, Xia J-N, Tan L, Chen N-X, Su X-L, Zhang Q, Feng Y-Z and Guo Y (2023) Causal effects of the gut microbiome on COVID-19 susceptibility and severity: a two-sample Mendelian randomization study. Front. Immunol. 14:1173974. doi: 10.3389/fimmu.2023.1173974
Received: 27 February 2023; Accepted: 10 August 2023;
Published: 01 September 2023.
Edited by:
Tao Lin, Baylor College of Medicine, United StatesReviewed by:
Luminița-Smaranda Iancu, Grigore T. Popa University of Medicine and Pharmacy, RomaniaCopyright © 2023 Zhong, Xie, Feng, Zhang, Xia, Tan, Chen, Su, Zhang, Feng and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yue Guo, Z3VveXVlQGNzdS5lZHUuY24=; Yun-Zhi Feng, ZmVuZ3l1bnpoaTAwMUBjc3UuZWR1LmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.