
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 26 April 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1172931
This article is part of the Research Topic Targeting Metabolism to active T cells and Enhance the Efficacy of Checkpoint Blockade Immunotherapy in Solid Tumors View all 9 articles
 Shonik Ganjoo1
Shonik Ganjoo1 Priti Gupta1
Priti Gupta1 Halil Ibrahim Corbali1,2Selene Nanez1Thomas S. Riad1
Halil Ibrahim Corbali1,2Selene Nanez1Thomas S. Riad1 Lisa K. Duong1Hampartsoum B. Barsoumian1Fatemeh Masrorpour1
Lisa K. Duong1Hampartsoum B. Barsoumian1Fatemeh Masrorpour1 Hong Jiang1
Hong Jiang1 James W. Welsh1
James W. Welsh1 Maria Angelica Cortez1*
Maria Angelica Cortez1*Immunotherapy has revolutionized cancer treatment and revitalized efforts to harness the power of the immune system to combat a variety of cancer types more effectively. However, low clinical response rates and differences in outcomes due to variations in the immune landscape among patients with cancer continue to be major limitations to immunotherapy. Recent efforts to improve responses to immunotherapy have focused on targeting cellular metabolism, as the metabolic characteristics of cancer cells can directly influence the activity and metabolism of immune cells, particularly T cells. Although the metabolic pathways of various cancer cells and T cells have been extensively reviewed, the intersections among these pathways, and their potential use as targets for improving responses to immune-checkpoint blockade therapies, are not completely understood. This review focuses on the interplay between tumor metabolites and T-cell dysfunction as well as the relationship between several T-cell metabolic patterns and T-cell activity/function in tumor immunology. Understanding these relationships could offer new avenues for improving responses to immunotherapy on a metabolic basis.
Metabolic reprogramming has historically been recognized as a key hallmark of cancer based on its significance for supporting and maintaining a malignant phenotype (1–3). Because cancer cells must survive in the harsh and often nutrient-depleted conditions of solid tumors, their reprogrammed metabolism and overall metabolic plasticity are necessary to meet the bioenergetic demands needed to sustain growth and proliferation (4, 5). Despite enormous genomic, phenotypic, and molecular heterogeneity across cancer types, altered metabolism continues to be a unifying biological feature of cancer that has consistently been linked with tumor growth (6–8). In the 1920s, Otto Warburg’s observation that tumors could rapidly metabolize glucose and produce lactose, even in the presence of oxygen and functioning mitochondria, marked a seminal discovery in cancer metabolism (9). However, this phenomenon, termed the Warburg effect, has historically been the subject of intense debate. Although several proposals have been raised to explain the Warburg effect, its function remains controversial. Nonetheless, the current understanding of cancer metabolism positions the Warburg effect within a larger set of interrelated processes that provide tumors with a metabolic advantage. These processes include the activation of growth-promoting oncogenes, expression of glucose transporters, and loss of function of tumor suppressors. Cumulatively, these molecular and functional processes accelerate glycolytic flux and production of adenosine triphosphate (ATP), but they also support the biosynthesis of organic molecules, maintain redox homeostasis, and drive immunosuppression (10). Crucially, the evolving understanding of cancer metabolism is also important for highlighting the role that metabolism plays in the broader context of tumor immunology.
The complexity of the tumor immune microenvironment is an especially important obstacle to progress in the field of cancer immunotherapy. Despite revolutionary advancements in immune checkpoint inhibitors (ICIs) to improve antitumor T-cell activity (e.g., anti-PD1 and anti-CTLA4), immunologically “cold” tumors remain challenging to treat. Mounting evidence suggests that altered tumor metabolism has key roles in suppressing T-cell activity and reducing the therapeutic efficacy of ICIs (11–13). Also, the tumor microenvironment (TME) and the broader metabolic demands of tumors can both directly interfere with the normal metabolic profiles of activated T cells, dampening antitumor effector functions (14–16). Several ongoing investigations have targeted specific immunosuppressive metabolites or metabolism-regulating molecules as a step towards generating more potent T-cell effectors as well as lasting memory cells (14). Metabolic interventions can also be beneficial for remodeling the tumor immune microenvironment by exploiting differences in the metabolic demands of T-cell subsets (e.g., regulatory T cells [Tregs] vs cytotoxic T cells) (17). Hence, understanding the mechanistic intersections of tumor metabolism and T-cell metabolism is especially important for developing more effective immunotherapeutic strategies.
In this Review, we explore the role of metabolism in mediating the dynamic relationship between T cells and tumors. We also discuss the relevance of specific T-cell metabolic pathways for supporting antitumor T-cell function and examine how the TME can fundamentally interfere with these pathways to modulate T-cell activity, including on an epigenetic basis. We further highlight some of the key metabolites present in the TME that promote T-cell dysfunction, which also have potential as therapeutic targets to overcome immunosuppression. Finally, we present some pertinent molecular strategies for targeting metabolism to bolster antitumor activity and consider the clinical significance of adopting strategies that integrate metabolism with immunotherapy to enable long-term antitumor immune activity.
The independent metabolic profiles of T cells and cancer cells have been extensively characterized (2, 18–20). Indeed, the metabolic patterns of each of these cell types provide important insights into their separate functions and activity. However, the similarities in metabolic demands, pathways, and activities between T cells and cancer cells have drawn significant attention to the dynamic interrelationship between these cells based on metabolism (21). Here, we contextualize this interrelationship by examining the reciprocal interactions between T cells and tumors through three salient metabolic pathways: glycolysis, fatty acid oxidation, and amino acid catabolism (Figure 1).
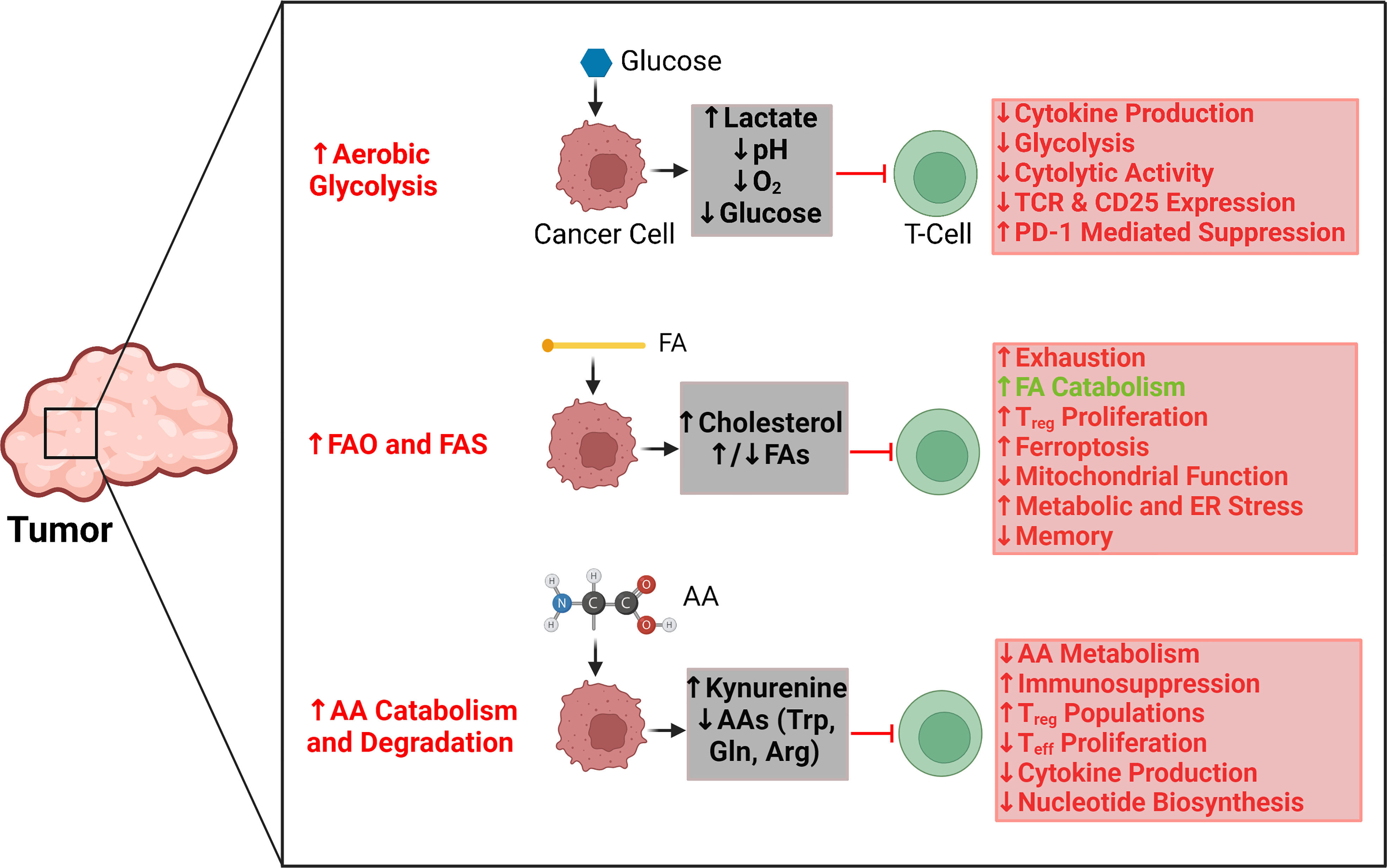
Figure 1 Key regulatory effects and interactions between tumor metabolic pathways and T-cell activity. Aerobic glycolysis in tumors is primarily responsible for creating a hypoxic, acidic, and nutrient-depleted microenvironment that suppresses effector T-cell activity and blunts antitumor immune responses. Similarly, the rapid uptake and catabolism of amino acids by tumors reduces their availability for T cells, which further promotes immunosuppression and supports the metabolic activities of regulatory T cells as opposed to effector T cells. Tumors can upregulate both fatty acid oxidation (FAO) and fatty acid synthesis (FAS), which affect T-cell activity differently. The accumulation of select fatty acids can support FAO in T cells; however, those same fatty acids can also promote exhaustion and inhibit the proliferation of memory T cells. Cholesterol is particularly significant for causing mitochondrial and endoplasmic reticulum (ER) stress in T cells. Red arrows and text indicate tumor metabolism–mediated effects that can suppress antitumor T-cell activity. Green arrows and text indicate tumor metabolism–mediated effects that can support antitumor T-cell activity. AA, amino acid; Arg, arginine; ER, endoplasmic reticulum; FA, fatty acid; FAO, fatty acid oxidation; FAS, fatty acid synthesis; Gln, glutamine; PD1, programmed cell death protein 1; Treg, regulatory T cell; Teff, effector T cell; TCR, T-cell receptor; Trp, tryptophan.
Glycolysis is a fundamental metabolic pathway used by both T cells and cancer cells as a source of energy to support their various activities (22–24). Notably, T cells exhibit metabolic “switching” depending on their activation status (25). Quiescent T cells primarily rely on oxidative phosphorylation to generate ATP, whereas activated T cells upregulate glycolysis to support proliferation and effector functions (26, 27). Glycolysis can also create byproducts that can be used in other macromolecular biosynthetic pathways, such as glycogenesis or the pentose phosphate pathway (22). Glucose is a critical substrate for promoting the normal activity and functioning of effector T cells, and T-cell cytokine production is fundamentally tied to glycolytic flux as well (27, 28). Tumors show similar metabolic reprogramming, although the shift in tumor metabolism does not require an activating or co-stimulatory signal, as is the case for T cells (29). Tumors uniquely rely on “aerobic glycolysis,” which is the use of glycolysis for energy even under aerobic conditions (30, 31). In addition to upregulating various isoforms of glucose transporters (GLUTs) that facilitate glucose uptake from the environment, cancer cells can also chronically obtain glucose through glycogenolysis and gluconeogenic mechanisms (32–35). Due to the abnormally high glycolytic flux in cancer cells, it is estimated that the glucose concentration is approximately 10-fold lower in the tumor interstitium relative to the plasma (36). Furthermore, the rapid consumption of glucose and its conversion into lactic acid in the TME fulfills the anabolic demands of various cancer cells by quickly generating ATP to support proliferation, regardless of oxygen availability (37, 38). Crucially, the metabolic reprogramming of tumors to rapidly generate ATP from aerobic glycolysis is also directly responsible for creating the acidic, hypoxic, and nutrient-depleted conditions of the TME, which have profound consequences for T-cell activity (39, 40).
One of the most significant consequences of aerobic glycolysis is the acidification of the TME (9, 41). The accumulation of lactic acid and corresponding decrease in pH of the TME has numerous inhibitory effects on the activity and function of tumor-infiltrating lymphocytes (TILs), especially cytotoxic T lymphocytes (CTLs) (15, 40). One study showed that lactic acid inhibited the migratory capacities of CD4+ and CD8+ T cells while also inhibiting the cytolytic activity of CD8+ T cells, which collectively blunted their pro-inflammatory activity (42). In mouse melanoma models, lactic acid was also shown to diminish interferon-gamma (IFN-γ) production in T cells, and increased lactate dehydrogenase expression was negatively correlated with T-cell activation (43). The secretion of lactate by tumor cells also affects the gradient across lactate transporters, which can blunt T-cell activation by diminishing their ability to recycle glycolytic byproducts (44, 45). Although lactate demonstrably inhibits the antitumor activity of effector T cells, it also promotes the development of immunosuppressive Treg populations (46–49). Therefore, lactate accumulation in the tumor has a two-pronged immunosuppressive effect: it inhibits the activity of cytotoxic T cells and promotes the activity of Treg populations.
The buildup of lactic acid in the TME is accompanied by a decrease in intratumoral pH, which also inhibits antitumor T-cell activity. As noted above, the acidity of the TME has been shown to decrease cytokine production by T cells and to increase Treg subpopulations (25). In one study, the acidity of the TME was shown to inhibit CTL activity by completely blocking cytokine production and partially blocking lytic granule exocytosis; however, the functional activity of CTLs was recovered when the extracellular pH was neutralized (50). Separately, CD8+ T cells cultured at pH values that correspond to intratumoral pH (pH 6-6.5) exhibited an anergic state with reduced cytokine production and lower expression of CD25 and T-cell receptors (TCRs) (51, 52). Because the acidity of the TME is a major contributor to immune escape, several novel therapeutic strategies have focused on modulating TME pH. For instance, administering sodium bicarbonate has led to improved T-cell infiltration and increased responsiveness to immunotherapy in several murine models (53). Also, T cells genetically modified to overexpress Hvcn1, encoding for a voltage-gated H+ channel that selectively extrudes protons, demonstrated markedly increased antitumor functions (52). Collectively, lactic acid and the increased acidity of the TME are increasingly attractive targets for enhancing antitumor T-cell activity and bolstering immunotherapeutic strategies.
The metabolic competition for glucose, among several other key metabolites, between tumors and T cells is a critical mediator of their relationship (27, 39). Tumor cells have been shown to outcompete T cells for glucose, which has detrimental effects on the cytolytic and effector functions of TILs (29). Mechanistically, restricted glucose consumption by T cells in a mouse sarcoma model led to reduced activity of the mammalian target of rapamycin (mTOR), which in turn led to decreased IFN-γ production and glycolytic capacity (39). Moreover, depletion of glucose in the TME not only reduces glycolytic flux in TILs but also decreases the generation of phosphoenolpyruvate, which has a critical role in sustaining TCR-mediated Ca2+-NFAT [nuclear factor of activated T cells] signaling; consequently, reduced production of phosphoenolpyruvate can markedly impair antitumor T-cell activity (54). Notably, depleted glucose levels in the TME can also provide a metabolic advantage to Tregs over CTLs, as Tregs are more reliant on fatty acid and lactate metabolism (55).
Apart from tumor-specific glucose depletion, limited glucose conditions have been shown to impair T-cell activity in several studies (56–58). Fundamentally, CTLs depend on aerobic glycolysis to supply energy for a vast array of activities and functions; therefore, it is unsurprising that glucose depletion in the TME hinders their cytolytic and effector functions. Notably, the adenosine monophosphate–activated protein kinase (AMPK) pathway has a central role in regulating glycolytic flux in both T cells and tumors. In T cells, activation of AMPK signaling promotes the proliferation of various cellular subsets (59). However, the AMPK pathway also has pleiotropic effects on cancer cells (13). Some studies report that its activation can help cancer cells survive metabolic stress, whereas others indicate that its activation can negatively regulate aerobic glycolysis, suppressing tumor growth (60, 61). As a nexus for T-cell and tumor glucose metabolism, the AMPK pathway is a key energy-sensing system that has been targeted with drugs such as metformin to enhance memory T cells (62). However, because metformin activates the AMPK pathway more globally, it may also promote Treg expansion, consequently driving immunosuppression (13, 63). Nonetheless, it is important to recognize that the glucose-depleted conditions of the TME could be leveraged to develop T-cell priming strategies that improve their antitumor activity. One study reported that depriving T cells of glucose in vitro and inhibiting glycolytic flux improved CD8+ T-cell memory and antitumor activity (62). Transient glucose restriction was also shown to enhance CD8+ effector T-cell function and antitumor activity in mouse tumor models (64). Further research into the mechanisms underlying T-cell metabolic conditioning would provide greater insight into the bioenergetic plasticity of T cells.
The increased glycolytic flux in tumors, coupled with a disorganized vasculature, creates a hypoxic microenvironment that is not conducive to proper T-cell functioning (3). One of the major consequences of the hypoxic TME is T-cell exhaustion; the limited oxygen availability in the TME interferes with mitochondrial dynamics that coordinate TCR functioning and PD1 signaling (3, 65). Indeed, microenvironmental changes can alter pathways that sense oxygen tension, which is a key driver of T-cell exhaustion on a molecular basis (66). Hypoxia can also mediate immunosuppression by driving the recruitment of Tregs via CC-chemokine ligand 28 (CCL28) signaling, which promotes tolerance and angiogenesis (67). Upregulation of the transcription factor hypoxia-inducible factor 1-alpha (HIF-1α) in response to low oxygen conditions is also understood to increase glycolytic flux for cancer cells (68). Although the transcriptional activity of HIF-1α is critical for supporting tumor growth, its contribution to the hypoxic TME is also detrimental to effector T-cell function. For example, T cells have been shown to detect hypoxic conditions through oxygen-sensing prolyl-hydroxylase proteins, which subsequently limited TH1 responses, promoted induction of CD4+ T cells into Tregs, and curtailed CD8+ T-cell effector functions in lung tumor models (69). Separately, HIF-1α was reported to upregulate PDL1 expression in cancer cells, facilitating escape from CTL-mediated lysis and promoting CTL apoptosis (70). Finally, tumor hypoxia was shown to promote the accumulation of extracellular adenosine (which inhibits effector T-cell activity) via HIF-1α-induced expression of the ATP-hydrolyzing ectonucleotidases CD39 and CD73 (71–73).
The role of fatty acid oxidation (FAO) in modulating the activity of CD8+ TILs is controversial, with some studies suggesting that upregulated FAO supplies energy to sustain tumor-killing activity, and others that lipid accumulation in the TME drives T-cell exhaustion (74–76). As is true for glucose metabolism, T cells use FAO differently depending on their phenotype and activation status. For example, memory T cells upregulate fatty acid catabolism to maintain persistence and quiescence, whereas effector T cells rely on fatty acid anabolism to meet their demands for biomolecular mass (77, 78). In the TME, cancer cells have been found to exploit multiple sources of fatty acids. One of the most common mechanisms used to obtain fatty acids is de novo lipogenesis, facilitated by the upregulation of fatty acid synthase (79–81). However, cancer cells may also enhance uptake of lipids from the extracellular environment via receptor-mediated endocytosis of low-density lipoproteins, fatty acid-binding proteins, and/or fatty acid translocases (82, 83). Although altered fatty acid metabolism is an established feature of both T cells and cancer cells, it is important to contextualize how this metabolic change specifically informs the activity of T cells in the TME.
In the glucose-depleted conditions of the TME, CD8+ TILs often rely on fatty acids as an alternative energy source to fuel their antitumor activity; in mouse melanoma models, they were observed to increase fatty acid catabolism via peroxisome proliferator-activated receptor (PPAR)-α signaling (75). Further, because the early glycolytic activity of T cells is inhibited by PD1 signaling in the TME, T cells can switch to FAO to promote their longevity via increased expression of carnitine palmitoyltransferase 1A (CPT1A) (84). However, CD36-mediated uptake of fatty acids has also been shown to impair effector functions of CD8+ TILs by increasing ferroptosis and reducing the expression of pro-inflammatory cytokines (such as IFNγ and tumor necrosis factor [TNF]) (74, 85, 86). Moreover, the accumulation of long-chain fatty acids in the TME has been reported to blunt T-cell activity by reducing FAO (via downregulation of very-long-chain acyl-CoA dehydrogenase) and diminishing mitochondrial functions (76).
Apart from effector T cells, fatty acid metabolism also has a central role in supporting the activity of memory T cells. For instance, mice with T-cell–specific deletion of TNF receptor-associated factor 6 (TRAF6), a protein that modulates FAO, displayed robust CD8+ effector T-cell function, but they had profound defects in the ability to generate memory T cells (87). In the context of cancer, forced expression of proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), a protein that induces transcription factors that promote FAO and mitochondrial biogenesis, was found to improve CD8 T-cell central memory (88). PDL1 blockade in both in vivo and in vitro gastric adenocarcinoma models also increased fatty acid binding protein 4/5 (Fabp4/5) expression in resident memory T cells, improving their survival (89). Notably, the metabolism of Tregs is also characterized by an increased reliance on fatty acids for energy, which is advantageous for their survival in the glucose-depleted TME (90). FAO has also been linked with supporting the differentiation of CD4+ T cells into Tregs, which contributes to the inhibition of antitumor immunity by increasing Tregs in the TME (91).
Although fatty acid metabolism is central to the interplay between T cells and tumors, its different roles in supporting or inhibiting the activities of various T-cell subsets (e.g., effector CD8+ T cells, memory T cells, exhausted T cells, and Tregs) complicate its potential use as a therapeutic target. Nevertheless, several therapeutic strategies tested in preclinical tumor models have shown promising early findings (74). For instance, bezafibrate (an agonist of PGC-1α/PPAR complexes) increased FAO and the mitochondrial respiratory capacity of CTLs, supporting their antitumor immune function when used in conjunction with anti-PD1 therapy in mouse tumor models (92). In addition, genetic ablation of CD36 in Tregs not only decreased intratumoral Treg populations but was also found to enhance the antitumor activities of TILs, especially when coupled with anti-PD1 therapy (90). Finally, inhibition of MEK1/2 was shown to enhance mitochondrial biogenesis and FAO in stem cell-like memory CD8+ T cells, improving their persistence and supporting antitumor activity in tumor-bearing mice (93). Although the overall dynamics of FAO in the context of TIL activity remain controversial, future research that aims to modulate FAO in combination with immunotherapies could reveal new avenues for selectively improving the activity of CD8+ TILs to create targeted antitumor treatment strategies.
Like glucose and fatty acids, amino acids are also subject to metabolic competition between cancer cells and T cells in the TME. Notably, cancer cells can rely on various endogenous and exogenous sources of amino acids to fulfill their bioenergetic demands. As one example, cancer cells have been shown to utilize de novo and/or salvage pathways of amino acid synthesis to produce serine (via phosphoglycerate dehydrogenase) (94). Tumors also depend on exogenous sources of both essential and non-essential amino acids to support their growth, and several amino acid transporters (e.g., SLC7A5 and SLC6A14) have been implicated in facilitating amino acid uptake for cancer cells (95, 96). Furthermore, cancer cells can synthesize amino acids from irregular sources by using transaminases that interconvert amino acids. For example, the aspartate transaminase and glutamic oxaloacetic transaminase were both found to critically support redox balance and growth in human pancreatic ductal adenocarcinoma cells (97, 98). Altogether, the multiple sources of amino acids that cancer cells can exploit to increase biosynthesis elucidate the broader pattern of tumors evolving to optimize overall nutrient consumption. For T cells, amino acids have crucial roles in supporting their activation and proliferation, as demonstrated by the upregulation of various amino acid transporters after activation (99). Glutamine is a particularly important catabolic substrate for activated T cells, as it provides a host of intermediate molecules that enter biosynthetic and mitochondrial metabolic pathways (100). Similarly, tryptophan and arginine have been shown to support T-cell proliferation, cytokine production, and activation marker expression (12, 101–103). Cancer cells not only compete with T cells for these crucial amino acids, but they also evolved mechanisms to degrade or remove them to facilitate immune escape. Several studies have indicated that tumors express tryptophan-degrading enzymes that hinder antitumor immunity by reducing the intratumoral accumulation of T cells (104, 105). These enzymes (particularly indoleamine 2,3-dioxygenase [IDO]) have emerged as novel targets that can be inhibited to bolster T-cell infiltration and activity in tumors. Mechanistically, depleted tryptophan in T cells via IDO activates the stress enzyme GCN2 kinase, which is stimulated when tRNAs are not charged with amino acids. Elevated GCN2 kinase prevents T-cell proliferation and causes anergy (106). In orthotopic and metastatic models of liver cancer, delivery of shRNA for IDO resulted in the upregulation of TH1 cytokines (interleukin [IL] -12 and IFNγ) as a part of improved CD8+ T-cell cytotoxic activity (107). Administration of the receptor tyrosine kinase inhibitor imatinib was shown to downregulate the expression of IDO, resulting in activation of CD8+ T cells and induction of apoptosis of Tregs in mouse models of gastrointestinal stromal tumors (108). Finally, selective inhibition of IDO with the small molecule INCB024360 was found to improve T-cell proliferation and promote IFNγ production, thereby inhibiting tumor growth in a lymphocyte-dependent manner (109). In summary, several types of tumors demonstrably rely on IDO to outcompete T cells for tryptophan, but the emergence of several novel therapeutics that inhibit IDO lend significance to further efforts to combine IDO inhibitors with immunotherapy to strengthen antitumor T-cell activity.
The depletion of glutamine in the TME has similarly profound inhibitory consequences for TILs. However, the overall role of glutamine in mediating T-cell activity, particularly within the TME, is highly complex. T cells utilize glutaminase to convert glutamine into glutamate, supporting protein synthesis, redox balance, and the TCA cycle. However, glutaminase differentially affects the activity and differentiation of helper T-cell subsets via altered chromatin accessibility. Glutaminase deficiency was shown to initially reduce T-cell activation/proliferation and impair TH17 differentiation, but later promoted the effector function of CD4+ TH1 cells and effector CD8+ T cells (110). In the context of the TME, glutaminase inhibition contrarily impaired CD8+ T-cell activation in STK11-/Lkb1-deficient models of lung adenocarcinoma. Lkb1-deficient tumors were shown to have significantly increased glutamine production, suggesting that immunotherapy could be combined with glutamine inhibition to support glutamine availability for CD8+ T cells. However, the authors of this study demonstrated that inhibiting the PD1/PDL1 axis (using immunotherapy) as well as glutaminase activity in T cells in the TME reduced effector differentiation and cytotoxicity (111). A separate study comprehensively inhibited glutamine metabolism, which intriguingly resulted in potent anti-tumor effects but preserved effector T-cell activity. Tumor-bearing mice treated with comprehensive glutamine blockade demonstrated reduced hypoxia, acidosis, and nutrient depletion. On the other hand, glutamine metabolism blockade enhanced the function of anti-tumor immune cells by conditioning TILs toward a long-lived, memory-like phenotype that displayed high proliferation, activity, and effector functioning (112). Apart from T cells, suppressive myeloid cells in the TME can also be targeted via glutamine metabolism blockade. One investigation demonstrated that glutamine metabolism blockade with 6-diazo-5-oxo-L-norleucine (DON) inhibited the generation and recruitment of myeloid-derived suppressor cells (MDSCs). The study also reported that inhibiting glutamine metabolism could reduce immunosuppressive effects in the TME by inhibiting IDO expression and decreasing kynurenine levels, which could indirectly support T cell activation (113).
Glutamine starvation was found to blunt T-cell proliferation and cytokine production, which is consistent with previous findings affirming the importance of glutamine import in supporting T-cell activation (114). Glutamine restriction also impaired the effector activities of CD8+ T cells, but ex vivo culture under glutamine-restricted conditions followed by reinfusion led to extended survival in tumor-inoculated mice (115). Glutamine restriction has also been shown to reduce nucleotide biosynthesis in T cells, promoting the development of T cells with high FOXP3 expression and regulatory properties (116). Several therapeutic strategies centering on glutamine metabolism have been tested such as glutamine transport inhibitors, which prompted reduced tumor growth in lung and prostate cancers (13, 117, 118). Nonetheless, further research is needed to elucidate the dynamics between glutamine uptake in the TME and its consequences for antitumor T-cell activity. A stronger understanding of these dynamics could inform the development of combination therapies that more selectively kill cancer cells while sustaining T-cell function.
Arginine is also critical in modulating T-cell metabolism by enhancing their survival and antitumor functions (119). The expression of arginase by MDSCs in the TME can deplete intratumor L-arginine, resulting in T-cell anergy and cell-cycle arrest of T cells in the G0-G1 phase (120). Moreover, increased expression of arginase by MDSCs in patients with renal cell carcinoma was correlated with decreased cytokine production and lower expression levels of the TCR CD3-ζ chain (121). In organ cultures of human prostate carcinomas, inhibiting the activity of arginase was found to restore TIL responsiveness to tumors by polarizing their cytotoxic granules (122). Aside from arginase, nitric oxide synthase is also responsible for metabolizing arginine. The administration of aspirin, which releases nitric oxide and inhibits nitric oxide synthase in MDSCs, was shown to increase the number and function of tumor-antigen–specific T lymphocytes against primary mammary carcinoma cell lines (123). L-arginine is another crucial metabolic target being explored in several clinical studies with the goal of developing more effective immunotherapies (124).
Branched chain amino acids (BCAAs) also play critical roles in supporting the activity of both cancer cells and T cells. BCAAs, especially leucine, have been shown to promote mTORC1 activity in T-cells, which regulates T-cell differentiation and function (125, 126). Furthermore, the import of BCAAs requires the L-type amino acid transporter (LAT). One member of the LAT family, LAT1, plays an especially important role in facilitating the increased uptake of BCAAs during T-cell activation (127). Two notable branched chain amino transferases, BCATc and BCATm, are responsible for catalyzing the first step in the degradation of the BCAAs isoleucine, leucine, and valine (128). Although several studies have shown that BCAT isoenzymes are highly expressed across different cancer types, the importance of these enzymes in mediating immunosuppressive effects is only recently garnering attention (128–130). One investigation reported that BCAT1 expression was positively correlated with immune checkpoint genes in several cancers (131). Another study demonstrated that BCAT1 was correlated with immunosuppressive status in IDH1 wild-type gliomas (132). The underlying mechanism that explains how BCATs in the TME specifically modulate T-cell activity has not yet been clarified, but early findings have already identified these amino transferases as potential pharmaceutical targets.
The TME is a dynamic and complex environment consisting of resident cells, infiltrating cells, secreted signaling factors, and an extracellular matrix. Tumors depend on the TME for survival and growth; they can remodel the microenvironment by promoting angiogenesis and immune evasion via extracellular factors collectively classified as hallmarks of cancer (1). However, in addition to inducing angiogenesis, increased metabolic activity and the highly proliferative nature of cancer cells cause the depletion of key nutrients in the microenvironment. The hypoxic, acidic, and nutrient-depleted conditions in the TME are not hospitable for immune cells (133–135). Moreover, the reprogramming of cancer cell metabolism allows those cells to adapt to harsh conditions and dampens antitumor immunity through metabolic immune checkpoints (5, 135, 136). The various metabolic pathways exploited by tumors result in either consumption or production of key metabolites that have a variety of salient regulatory effects on T-cell activity (Table 1).
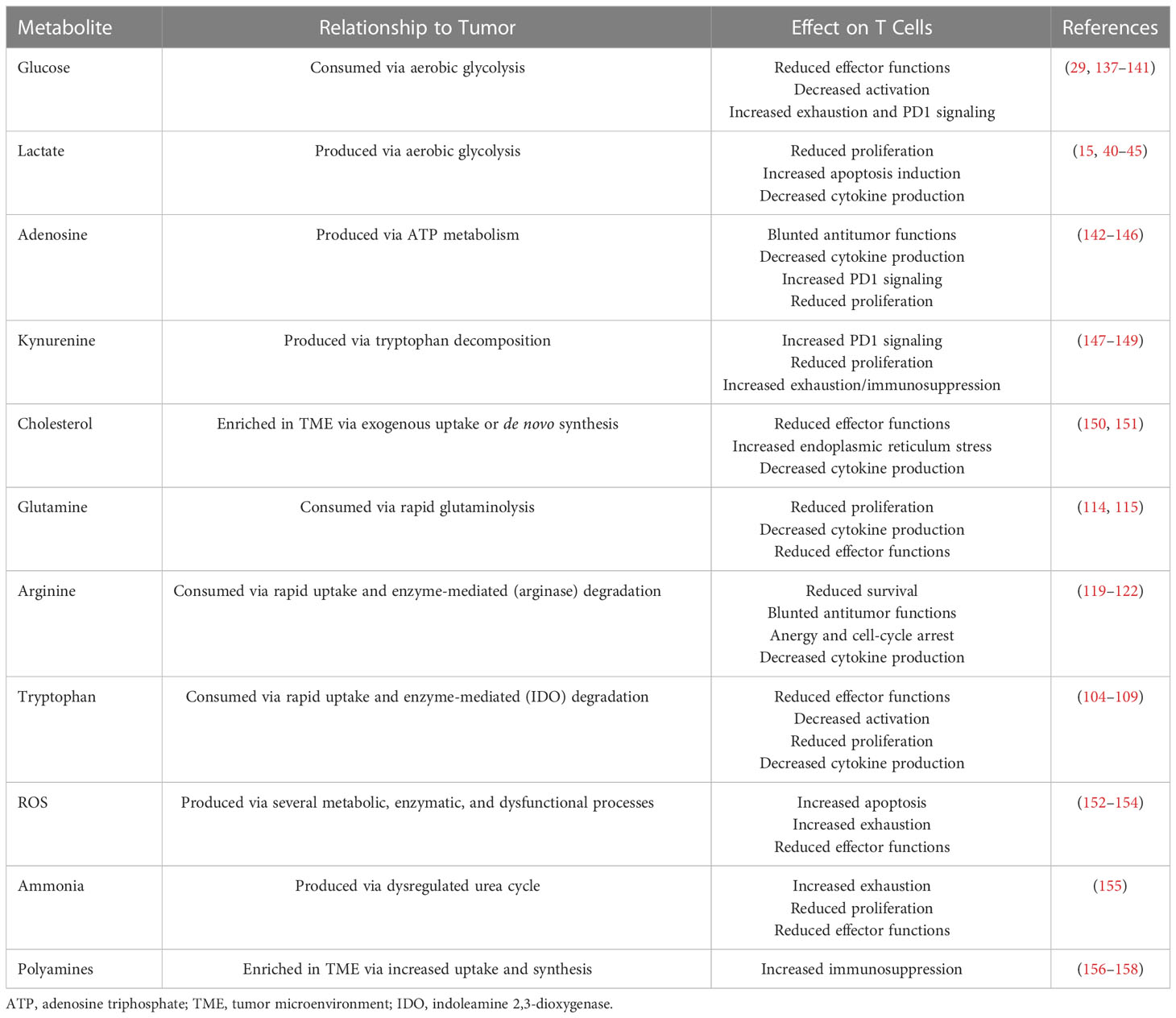
Table 1 Effects of select tumor microenvironment metabolites on T-cell activity.
Cancer cells are well known to increase their glucose uptake and switch from oxidative phosphorylation to aerobic glycolysis. On a clinical level, overexpression of the glucose transporter GLUT1 in cancer cells has been linked with poor prognosis in several types of solid tumors (159). Increased glucose uptake and glycolytic flux by cancer cells limit glucose utilization by T cells and resulted in low T-cell infiltration and impaired intratumoral activity (137, 138). Another study also provided evidence of restricted glucose consumption among T cells in models of ovarian cancer. In that study, the reduction in glucose consumption constrained the expression of the methyltransferase EZH2 in T cells via specific microRNAs. EZH2 is responsible for epigenetic induction of Notch signaling by repressing Notch inhibitors; activated Notch signaling regulates the polyfunctionality and survival of T cells (139). Restricting glucose utilization by T cells also contributes to immune exhaustion, especially because glucose consumption is a limiting factor for T-cell activation. The costimulatory molecule CD28 is required for activation after TCR stimulation, and CD28 increases glycolytic flux and glucose consumption via GLUT1 expression and cell surface trafficking by Akt-dependent and -independent pathways in response to activation (27, 140). PD1 expression similarly affects T-cell glucose metabolism, and reduced glucose levels enhance PD1 expression in T cells (39, 84). Further, restricting glucose increases the susceptibility of T cells to apoptosis via Bcl2 proteins (141). Collectively, these results indicate that glucose is a critical metabolite for sustaining T-cell activity and antitumor T-cell activity, but its rapid consumption by cancer cells can dampen T-cell activity through a variety of immunosuppressive mechanisms.
As previously discussed, increased aerobic glycolysis also results in lactic acid accumulation in and acidification of the microenvironment. Increased amounts of the enzyme lactic acid dehydrogenase have been linked with poor outcomes for patients with various types of cancer (150, 160–163). Notably, excessive lactic acid in the TME affects cancer cells and immune cells differently. Lactic acid accumulation inhibits the proliferation, cytokine production, and chemotaxis of natural killer (NK) cells and T cells and promotes apoptosis of these cells (15, 43). Importantly, the abundance of lactic acid in the TME has profound implications for the activity of tumor associated macrophages (TAMs) in addition to T-cells. Macrophages exhibit phenotypic plasticity and can differentiate into different polarized states depending on specific microenvironmental cues (164). M1 macrophages are largely responsible for mediating pro-inflammatory and anti-tumor responses, whereas M2 macrophages predominantly mediate anti-inflammatory and pro-tumor immune responses (165). TAMs can be polarized into either of these subtypes in response to the various metabolites and signaling molecules present within the TME. For instance, increased lactic acid levels in the TME enhance the polarization of pro-tumorigenic M2 macrophages via the ERK/STAT3 signaling pathway in breast cancer models (166). The acidic environment created by tumor cells, independent of lactate, also contributes to M2 macrophage polarization in the TME in prostate cancer models (167). Lactic acid-induced acidosis also suppresses the differentiation and maturation of dendritic cells; the resulting reduction in antigen presentation to T cells negatively affects adaptive immunity (168, 169). In addition, lactic acid was shown to enhance the activity of the immunosuppressive MDSC population via the HIF-1α pathway as well as disrupting neutrophil antitumor function (170, 171). Separately, lactic acid promotes angiogenesis via the HIF-1α/VEGF axis in the tumor microenvironment, contributing to tumor growth and the development of distant metastases (172). Despite growing evidence implicating lactate as an immunosuppressive metabolite, it is important to recognize that the broader role of lactic acid in T-cell biology is complex (173). On one hand, lactate can support tricarboxylic acid (TCA) cycle anaplerosis in effector T cells by serving as a physiologic carbon source (48). One study reported that lactate is a mitochondrial fuel for CD8+ effector T cells that contributes to TCA metabolism, and when it is readily available, effector T cells will preferentially oxidize lactate to support overall viability (174). On the other hand, the immunosuppressive effects of lactic acid accumulation may be limited to the hypoxic conditions of the TME, making it a promising target for cancer treatment strategies (173). Preclinical findings indicate that blocking lactic acid dehydrogenase limits tumor growth and improves immune checkpoint therapy (175, 176). Another approach involves blocking the MCT4 or MCT1 receptors, which are responsible for transporting lactic acid from inside the cell to the microenvironment. Targeting these receptors can improve T-cell function and immunogenicity by blocking lactic acid accumulation and acidification (177, 178).
Increased inflammation in the TME enhances the metabolism of extracellular ATP or adenosine diphosphate (ADP) to adenosine monophosphate (AMP). AMP is subsequently converted into adenosine by the ectonucleotidases CD39 and CD73, which are highly expressed on stromal cells, tumor cells, immune cells, and endothelial cells. Although extracellular ATP is immunogenic, accumulation of the adenosine metabolite in the TME has inhibitory effects on the immune system. For example, adenosine signaling via A2a receptors, which are G-coupled receptors on immune cells, was shown to dampen the antitumor response (142, 143). Increased protein kinase A activity, because of downstream of A2a receptor signaling, also has suppressive effects on effector T-cell function by reducing cytokine production and cell proliferation. Moreover, A2a receptor signaling increases Treg differentiation and enhances the immunosuppressive activity of Tregs (144). Notably, adenosine also enhances the expression of other immune checkpoints, including CTLA4, PD1, and LAG3 (145, 146). The benefits of targeting the adenosine axis and preventing adenosine accumulation in the TME have been extensively studied in preclinical models. For example, in murine models, blocking the adenosine axis was found to reduce tumor growth and prevent the formation of distant metastases (144, 179, 180). Blocking the adenosine axis also increased CD8+ T-cell infiltration into tumors, enhanced inflammatory cytokine production, and synergized effectively with other immune checkpoint therapies (181–184).
Evidence is growing on the importance of cholesterol in supporting tumor progression, and altered cholesterol biosynthesis programs are increasingly recognized as important features of various cancers, including melanomas and sarcomas (185). Indeed, upregulated de novo biosynthesis of cholesterol, coupled with increased exogenous uptake, are consistently important in facilitating tumor growth and survival (186). On an immunologic basis, accumulation of cholesterol in the TME can induce metabolic and endoplasmic reticulum stress in T cells, resulting in exhaustion and reduced effector functions, including cytokine production (150). Further, a positive correlation was found between tumors enriched in cholesterol and CD8+ T cells with upregulated expression of immunosuppressive molecules such as PD1, TIM3, and LAG3 (150). Separately, inhibition of cholesterol esterification via genetic ablation or pharmacologic inhibition of ACAT1 (a cholesterol esterification enzyme) was found to potentiate an antitumor immune response for CD8+ T cells. These investigators showed that ACAT1-deficient CD8+ T cells were better than wild-type CD8+ T cells at suppressing tumor growth in mouse melanoma models (151).
Increased inflammation within the TME also results in the accumulation of reactive oxygen species (ROS), which can form from a variety of processes including mitochondrial dysfunction, oncogene activity, and increased oxidase activity (187). Excessive ROS and oxidative stress cause field cancerization and metastasis by promoting oncogenic pathways that prevent cancer cell apoptosis (188). Further, ROS affect M2 macrophage polarization in ways that induce T-cell apoptosis (152). Tumor-induced MDSCs promote cancer progression and suppress antitumor immunity via ROS (153). In addition, under hypoxic conditions, T-cell activity is disrupted via abnormal mitochondrial function that creates mitochondrial ROS that increase exhaustion through reduced PGC-1α gene expression, which is responsible for mitochondrial biogenesis (154). Antioxidant inflammation modulators are used to target ROS that bind Keap1 and prevent degradation of the nuclear factor erythroid 2–related factor Nrf2. Increased Nrf2 diminishes ROS levels and inflammation in the TME, consequently controlling tumor growth and metastasis (189).
Accumulation of high levels of ammonia in the microenvironment resulting from disruption in the urea cycle also blunts antitumor immunity by exhausting T cells and reducing their proliferation. In patients with colorectal cancer, increased serum ammonia and ammonia-related genes are associated with poor outcomes and immune checkpoint resistance. In mouse models, enhancing the clearance of tumor-associated ammonia rescued T-cell function and improved the efficacy of anti-PD1 immunotherapy (155).
Kynurenine is another immunosuppressive metabolite produced via tryptophan catabolism, and that IDO enzyme-catalyzed reaction is a rate-limiting step in the kynurenine pathway. Kynurenine is related to aging, inflammation, and modulation of ROS levels via aryl hydrocarbon receptors (190). Kynurenine also increases IDO expression in tumor cells through an autocrine aryl hydrocarbon receptors–IL-6–STAT-3 loop (191). Further, elevated kynurenine levels cause exhaustion and increased expression of immune checkpoint markers by tumor-infiltrating CD8+ T cells. Targeting kynurenine via IDO blockade has synergized with immune checkpoint therapy and improved antitumor immunity in preclinical and clinical studies (147–149).
Polyamines (e.g., putrescine, spermine, spermidine) are aliphatic cationic metabolites that are synthesized from arginine and glutamine and have essential functions in the proliferation of normal and neoplastic cells (192). At physiological concentrations, they also contribute to T-cell activation and differentiation (193). On the other hand, cancer cells show increased polyamine synthesis and uptake. The accumulation of polyamines in the TME produces an intricate immunosuppressive effect (156, 157). Targeting polyamines has been explored in the context of cancer immunotherapy; dietary polyamine deprivation has been shown to deter tumor-induced immunosuppression by increasing IL-2 levels (158). In another study, polyamine-blocking therapy, combined with the inhibition of ornithine decarboxylase (the rate-limiting enzyme of polyamine synthesis), reversed immunosuppression in the TME by reducing MDSCs and increasing infiltrating T-cell populations (157).
Tumors with loss-of-function mutations in succinate dehydrogenase have high levels of succinate present in the TME (194). While the effects of succinate accumulation on innate immune cell activity have been investigated, few studies have explored the role of succinate in modulating T-cell activity within the TME. A key study interrogating this relationship showed that high succinate levels in the TME can inhibit CD4+ and CD8+ T-cell effector function, particularly by blunting cytokine secretion (IFN-γ) and degranulation (195). Mechanistically, increased succinate uptake was found to inhibit T-cell succinyl-CoA synthetase activity, glycolytic flux, and the TCA cycle. These effects cumulatively suppressed T-cell activity, but restoring TCA cycle flux was shown to reverse the succinate-mediated suppression (195).
The metabolic reprogramming of T cells within tumors is inextricably linked to epigenetic modifications, and a growing body of evidence suggests that the metabolism-epigenetics relationship in T cells could also be a therapeutic target for cancer (196). Generally, the epigenetic regulation of certain genes in T cells is understood to be critical for supporting their proliferation, activity, and native functioning. For instance, histone methylation and the corresponding activity of demethylases (e.g., KDM6B) has been shown to support the virus-specific CD8+ T-cell response as well as CTL effector programs (85). Similarly, histone acetylation—namely, acetylation of histone H3 lysine 9 (H3K9)—has also been reported to facilitate a robust CD8+ memory T-cell response by upregulating the expression of various effector molecules, including perforins and granzyme B (197). Although histone acetylation and methylation are among the most widely studied epigenetic modifications that T cells undergo during activation, evidence is accumulating to suggest that certain metabolites can also epigenetically regulate T-cell activity. As one example, β-hydroxybutyrate, a metabolite in the ketogenic pathway, can epigenetically modify H3K9, supporting CD8+ memory T-cell development (198). Separately, there is a growing body of evidence broadly supporting the role of epigenetics in regulating T-cell exhaustion. In fact, exhausted T cells have been characterized by a distinct T-cell chromatin state and shared differentiation program regulated by specific transcription factors (199). One study even reported that transcriptional and epigenetic mechanisms are responsible for a four-stage hierarchical developmental pathway that results in CD8+ T-cell exhaustion in subsets defined by Ly108 and CD69 (200). As the role of epigenetics in regulating T-cell phenotype and activity is further clarified, the dynamic intersection between epigenetics and metabolism is increasingly relevant for guiding therapeutic interventions. Here, we briefly summarize the roles of three epigenetic regulatory mechanisms—acetylation, methylation, and metabolite modulation—in mediating T-cell metabolism, specifically in the context of cancer.
In the glucose-depleted conditions of the TME, T cells can utilize acetate as an alternative energy source to support growth and effector functions (201). As with the aforementioned metabolites, acetate can also be obtained by cancer cells through exogenous or endogenous sources. Acetate in blood plasma, which may be derived from the diet or even the gut microbiome, can be taken up by cancer cells through various transport proteins, including monocarboxylate transporter-1 (202). The nucleocytosolic acetyl-CoA synthetase enzyme, ACSS2, has also been reported to play a key role in facilitating acetate uptake across various human tumors (203). Notably, the findings from this study also suggest that ACSS2 may be responsible for converting acetate released from deacetylated proteins into an acetyl-CoA pool that can be used for epigenetic regulation (203, 204).
Besides serving as a substrate for energy production, acetate can also promote histone acetylation and chromatin accessibility in T cells, enhancing IFN-γ gene transcription and cytokine production (205). Separately, the inhibition of mitochondrial pyruvate carrier (MPC) has been shown to promote acetyl-CoA production, which enhanced histone acetylation and the transcription of pro-memory genes (such as Sell, Tcf7, and Ccr7) that are critical for CD8+ memory T-cell function (206). However, that same study found that ablation of MPC blunted the antitumor function of CD8+ T cells, because MPC sustains lactate oxidation for T cells in the TME. Nevertheless, this antitumor activity could be circumvented by using a small-molecule MPC inhibitor to imprint a memory phenotype during chimeric antigen receptor-T cell (CAR-T) expansion. CAR-T cells are engineered to express synthetic receptors that can recognize specific antigens expressed by cancer cells and potentiate a powerful anti-tumor response (207). However, certain limitations (such as exhaustion and senescence) of CAR-T therapy have brought attention to conditioning strategies that could improve the potency and persistence of CAR-T cells (208). Indeed, the conditioning strategy involving an MPC inhibitor resulted in superior CAR-T antitumor activity upon adoptive cell transfer immunotherapy. These early findings indicate that histone acetylation, particularly acetylation mediated by acetate or acetyl-CoA, could be strategically targeted alongside various metabolic pathways to bolster antitumor T-cell activity (196).
In the hypoxic TME, HIF-1α and von Hippel-Lindau (VHL) proteins are essential for regulating CD8+ T-cell metabolism and function. The VHL-HIF-1α axis (in response to hypoxic conditions) and TCR activation have been shown to upregulate the production of 2-hydroxyglutarate, a molecule that can epigenetically modulate T-cell phenotype and activity, but in contradictory ways (196, 209). One group found that 2-hydroxyglutarate, an oncometabolite, induced global changes in histone H3 methylation by inhibiting H3K27me2/3 demethylase, which promoted CD8+ T-cell lymphocyte proliferation, persistence, and antitumor capacity (209). However, another study reported that the overproduction of 2-hydroxyglutarate in the TME (due to gain-of-function mutations in isocitrate dehydrogenase) decreased CD8+ T-cell cytotoxicity and impaired IFN-γ production, albeit not through epigenetic reprogramming of T-cell metabolism (210). Instead, that study found that 2-hydroxyglutarate inhibited lactate dehydrogenase activity, which decreased the regeneration of cytosolic NAD+ that is ordinarily needed to support T-cell glycolysis.
Tumor cells can also upregulate the methionine transporter SLC43A2 to rapidly take up methionine and prevent T cells from metabolizing it. Reducing intracellular methionine in CD8+ T cells resulted in the loss of demethylation at H3K79me2, causing T-cell apoptosis and dysfunction. Inhibiting SLC43A2 recovered T-cell methionine metabolism and also improved checkpoint blockade antitumor immunity in preclinical models (211). A similar strategy involving metabolic reprogramming through altered methylation patterns was used to decrease the infiltration of Tregs in solid tumor models. Supplying α-ketoglutarate altered the methylation pattern of naïve CD4+ T cells activated under Treg polarizing conditions, which led to reduced Treg differentiation and decreased FOXP3 expression among adoptively transferred cells. These investigators suggested that α-ketoglutarate reprograms T cells to increase mitochondrial metabolism, which may affect the fate of CD4+ T-cell (specifically, Treg) differentiation (212). Further research into the intersection of DNA methylation patterns and T-cell metabolism is warranted to inform a stronger mechanistic understanding of how this intersection can be therapeutically harnessed..
T-cell gene expression can also be regulated epigenetically by select metabolites; however, research on this regulation in the context of cancer is still in early stages. The epigenetic effects of one such metabolite, lactate, are being investigated to clarify its role in modulating anti-cancer T-cell immunity. One recent study found that delivering sodium lactate, but not glucose, to tumor-bearing mice increased the stemness of CD8+ T cells, resulting in tumor growth inhibition. Mechanistically, the lactate inhibited histone deacetylase activity, increasing acetylation at H3K27 and expression of Tcf7, thereby supporting antitumor immunity (213). Arginine is another critical metabolite that can epigenetically influence T-cell metabolism to promote antitumor activity. Increased L-arginine levels have been shown to induce a metabolic shift from glycolysis to oxidative phosphorylation in T cells, promoting a central memory-like phenotype with increased survival capacity. These metabolically reprogrammed T cells had greater antitumor activity in mice bearing B16 melanoma tumors. Strikingly, these investigators also reported that L-arginine induced structural changes in three transcriptional regulators (BAZ1B, PSIP1, and TSN) that ultimately were responsible for mediating pro-survival effects in T cells (119). In short, the growing interest in metabolites and metabolic enzymes as epigenetic regulators of T-cell activity is opening new avenues for the development of more effective cancer immunotherapies. Nevertheless, further research into how specific metabolites regulate T-cell gene expression would be beneficial for informing precise strategies that can bolster antitumor immunity while circumventing T-cell exhaustion and/or dysfunction (196).
The framework of immunometabolism has revealed a new set of therapeutic targets that can be exploited to enhance immune responses. As previously stated, transcriptional regulation of differentiation, effector functions, and longevity of T cells are all fundamentally tied to cellular metabolism. The goal in immunometabolism is to explore and investigate ways to modulate the metabolic programs that govern these effects to manipulate and enhance immune responses against cancer. In this section, we review some of the metabolic pathways that have been explored in strategies to improve antitumor T-cell function.
One of the most salient metabolic targets is the PI3K/AKT/mTOR signaling pathway, which induces glycolysis downstream of T-cell activation. Several studies have shown that sustained activation of this pathway is correlated with terminal differentiation and loss of T-cell memory. An investigation of the AKT inhibitor A-443654 to increase the number of memory CD8+ T cells showed that AKT inhibition led to the transition of a fraction of short-lived effector CD8+ T cells to memory cells (214). Further research into AKT inhibition revealed that TILs that had been expanded with AKT inhibitors had significantly increased expression of the central memory phenotype marker CD62L. The evolutionarily conserved serine/threonine kinase mTOR has a central role in T-cell differentiation and function, as it mediates the induction of glycolysis upon T-cell activation, which is necessary for naïve CD8+ T cells to differentiate into effector CD8+ T cells (215). Administration of the mTOR inhibitor rapamycin, which induces FAO, led to a significant increase in memory CD8+ T-cell development and corresponding recall response (87).
Meanwhile, research on the effect of other mTOR inhibitors on effector T cells has shown mixed results. In one study, the addition of temsirolimus, a rapamycin analogue, enhanced the activation and function of effector T cells stimulated with a heat shock protein–based antitumor vaccine. Temsirolimus also enhanced memory CD8+ T-cell function, corroborating the findings obtained with rapamycin (216). In other studies, however, rapamycin was shown to have immunosuppressive effects that impeded lymphocyte recruitment to tumor-draining lymph nodes as well as recruitment of CD8+ T cells to the tumor (216). FAO is well known for its importance in the formation of memory CD8+ T cells, as evidenced by the deleterious effects on T-cell activity that arise from deficiencies of selected FAO enzymes. Mice with T-cell–specific deletion of TRAF6 exhibit changes in the expression of genes that regulate FAO and show profound defects in the ability to generate memory T cells (87). Conversely, enforced expression of FAO enzymes can yield favorable results.
Another metabolic pathway target is CPT1A, an enzyme essential for FAO. This target can be exploited through adoptive T-cell therapy, a technique in which a patient’s own T cells are isolated, cultured in vitro, and then transferred back to the patient. Adoptive transfer of OT-I T cells transduced ex vivo with CTP1a resulted in increased numbers of memory T cells compared with control transduced OT-I T cells in a mouse model (217). This same mechanism of adoptive cell transfer was used to examine the effect of pathway modulation in targeting glycolysis. Expression of the glycolytic enzyme Pgam1 induced CD8+ T cells to adopt a predominantly glycolytic phenotype, and adoptive transfer of Pgam1-transduced T cells showed decreased long-term survival in both lymphoid and peripheral tissue (62). That group also investigated whether inhibiting glycolysis with the H2K inhibitor 2DG would conversely enhance T-cell survival and the capacity for memory T-cell generation. The phosphorylated form of the glucose analog 2DG-6-P was found to accumulate within cells and to inhibit hexokinase activity. Adoptively transferred 2DG-treated pmel-1 CD8+ T cells were shown to expand robustly in wild-type mice infected with gp100-VV. Moreover, a higher proportion of 2DG-treated cells retained a memory phenotype, whereas most control CD8+ T cells underwent terminal differentiation (62).
The 2DG molecule can also be used in manufacturing CAR-T cells, which include genetically engineered self-CD8+ TILs that are expanded before being re-introduced into the patient. During expansion, the cells can be cultured with nutrients that support any metabolic challenges encountered when competing in the TME (218). Glycolysis inhibition, for example, is crucial for the manufacturing process of CAR-T cells to maintain an undifferentiated state that also persists for prolonged periods in vivo. This can take many forms, including optimizing the medium to enhance mitochondrial function and indirectly inhibiting glycolysis. As one example, the addition of arginine to the medium can promote oxidative phosphorylation and inhibit glycolysis (219). Many cytokines (e.g., IL-7, IL-15, IL-21) can also be added to the medium to shift metabolic programs away from glycolysis (9). Another way of enhancing mitochondrial metabolism in CAR-T cells is by using PGC-1α activators, as discussed earlier (88). Enhancing mitochondrial metabolism can also support memory T cells. CAR-T cells manipulated with the co-stimulatory activator 4-1BB displayed increased mitochondrial synthesis and FAO, supporting their antitumor activity (220). Finally, a CRISPR activation screen identified proline metabolism a potential target for enhancing CAR-T therapy (221). Specifically, the authors of the study identified PRODH2, encoding for proline dehydrogenase 2, as an ideal gain-of-function target that could be leveraged to enhance CAR-T therapy. Indeed, the targeted genomic knock-in or lentiviral overexpression of PRODH2 boosted CAR-T killing of cognate cancer cells.
ICIs can also be used to manipulate T-cell metabolism. PD1 signaling inhibits T-cell activation by altering glucose uptake and glycolysis (222). CTLA4 signaling also impairs glycolysis during effector T-cell activation (84). PDL1 and B7H3 support malignant cells in the TME by enhancing aerobic glycolysis via activation of the HIF-1α and PI3K/AKT/mTOR pathways (223). Blocking these immune checkpoint inhibitors is therefore beneficial for T-cell activation, proliferation, and survival. CTLA4 blockade promotes cytotoxic CD8+ T-cell priming while inhibiting tumor-promoting Tregs (224). PD1 blockade can also aid in the differentiation of progenitor CD8+ T cells (222). Moreover, blocking PD1 inhibits the mTORC1 pathway, which leads to impaired glycolysis in the TME (39). Overall, the progression in our understanding of the metabolic mechanisms underlying T-cell activity will open new avenues for leveraging strategies such as adoptive cell transfer, CAR-T therapy, and ICIs to design more effective therapies.
Finally, apart from directly targeting cancer cells that metabolically modulate T-cell activity, stromal players in the TME can also be targeted due to their adverse effects on T-cell activity. For instance, the elimination of cancer-associated fibroblasts (CAFs) can facilitate the improved metabolic activity and cytotoxic effects of CD8+ T cells (225). One marker of CAFs, fibroblast activation protein–α (FAP), was targeted by combining a traditional cancer vaccine with a vaccine that selectively targets FAP+ fibroblasts. This strategy reduced tumor progression in mouse melanoma models by alleviating metabolic stress on CD8+ T-cells, potentially by increasing glucose availability (226). Furthermore, in prostate cancer models, the release of lactate by CAFs was found to reduce the proportion of anti-tumor TH1 susbsets and increase Treg cells. Targeting the TLR8/miR21 axis, which is activated by this CAF-immunomodulated environment, could therefore be a viable therapeutic strategy (227). Stroma-associated pancreatic stellate cells can also modulate metabolic activity in pancreatic ductal adenocarcinoma by secreting alanine as a carbon source to fuel the TCA cycle in cancer cells. These pancreatic stellate cells can therefore provide a metabolic advantage to cancer cells over effector T-cells in pancreatic ductal adenocarcinoma (228). Altogether, the stromal cells of the TME can serve as important targets for therapeutic intervention (e.g., by vaccination or molecular inhibition) to bolster an anti-tumor immune response.
The advent of ICIs has revolutionized cancer immunotherapy. In less than a decade, significant improvements have also been observed in response rates, as the percentage of patients estimated to respond to checkpoint inhibitor drugs increased to from 0.14% in 2011 to 12.46% in 2018 (229). However, this response rate is still far from optimal, underscoring the need to either develop other treatment options or modify current approaches. As discussed throughout this review, cancer cells can suppress an antitumor response by depleting essential nutrients or reducing the metabolic fitness of tumor-infiltrating cells (230, 231). Therefore, tailoring immune responses by manipulating cellular metabolic pathways may improve clinical outcomes. Indeed, several clinical studies have either been conducted or are currently underway to examine combinations of metabolic targets and immune checkpoint inhibition to bolster antitumor immune responses.
As noted previously, T cells undergo metabolic reprogramming to fuel their bioenergetic needs based on their function. Naïve T cells depend largely on oxidative phosphorylation to survive (19). However, upon exposure to antigens, naïve T cells differentiate into effector cells that utilize amino acids (e.g., glutamine) and glucose for proliferation and cytolytic activity (232). If the antigen is cleared, the T cells differentiate into long-lived memory cells; however, if the T cells fail to clear the antigen, as is often the case in cancer, then the T cells become exhausted. Exhausted T cells generally exhibit dysfunctional mitochondria and decreased mitochondrial mass. Notably, they also exhibit gene signatures that have been correlated with poor prognosis in hepatocellular carcinoma patients, but also represent therapeutic targets (233). Moreover, inhibitory receptors like PD1 and CTLA4 also impair T-cell metabolism. For instance, T cells exposed to PD1 signaling showed decreased rates of glycolysis and glutaminolysis but increased rates of FAO (84, 234).
Metabolic reprogramming has emerged in recent years as a novel means of reversing T-cell exhaustion in the TME, with promising results obtained in both animal models and in vitro studies. However, whether metabolic reprogramming with ICIs has beneficial effects in humans remains unclear. To our knowledge, at least 15 clinical studies either have been completed or are currently underway to evaluate the effects of targeted T-cell metabolic inhibitors in conjunction with ICIs. Three of these studies have been withdrawn (NCT04231864) or terminated (NCT03894540, NCT04265534) for feasibility issues, lack of benefit, or other reasons. However, other studies have reported some important beneficial effects (Table 2). Some of those trials are described briefly in the following paragraphs.
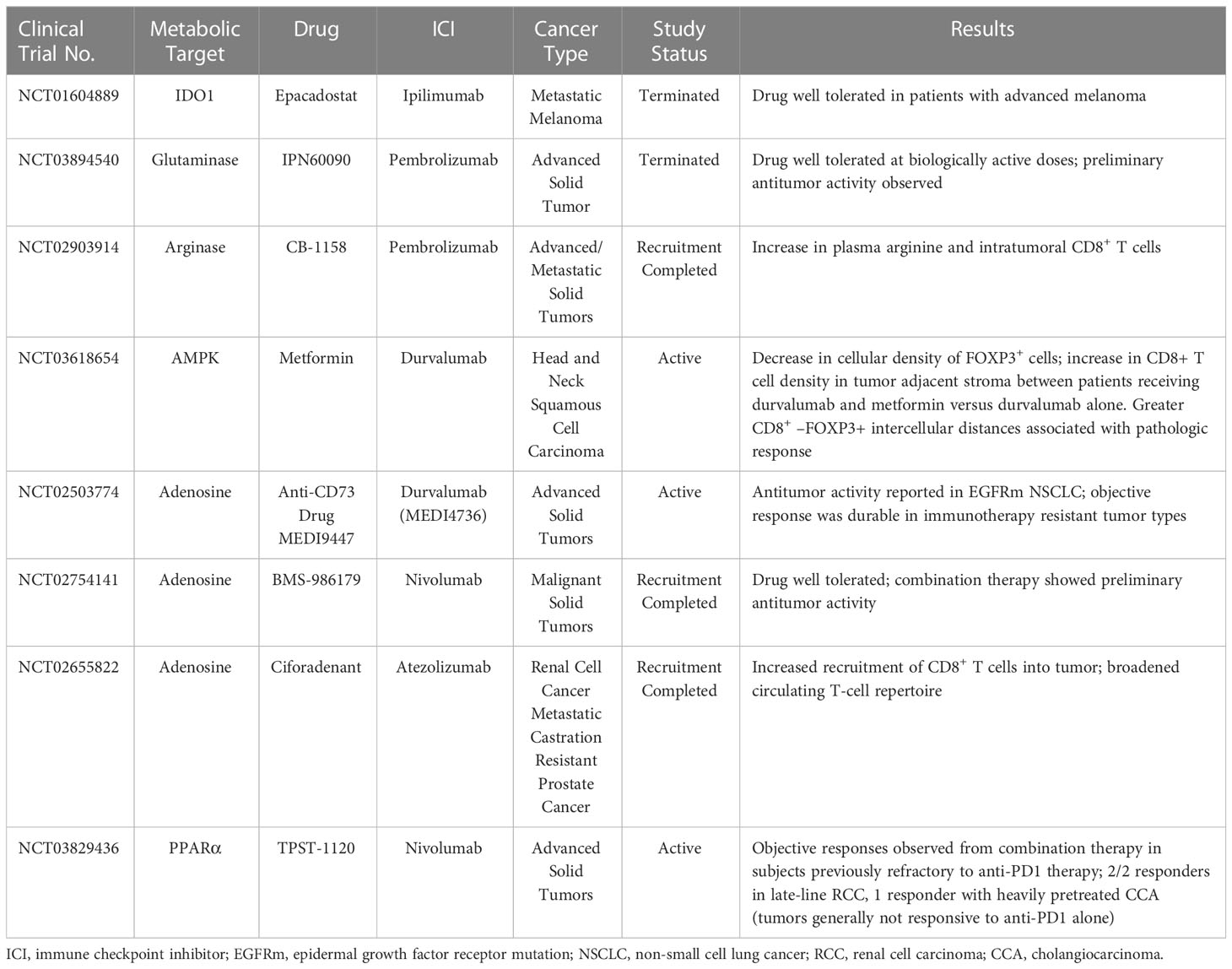
Table 2 Clinical trials of ICIs plus metabolic targets showing promising results.
Arginine is an amino acid required for T-cell activation and proliferation. In the TME, MDSCs consistently secrete arginase, which depletes arginine levels within the tumor, leading to impaired T-cell proliferation and cytokine production (235). CD1158, a molecular inhibitor of arginase, was reported to increase plasma arginine when used in combination with pembrolizumab in an ongoing clinical trial for patients with colorectal carcinoma (NCT02903914). This trial reported an increase in intratumoral CD8+ T cells in patients with microsatellite-stable colorectal cancer after treatment with CB1158 and pembrolizumab.
Metformin is commonly prescribed for the treatment of type II diabetes. Metformin has also been shown to downregulate cytosolic oxidative phosphorylation by inhibiting complex I of the electron transport chain, which ordinarily has an important role in tumor growth (218, 236, 237). Numerous preclinical, epidemiological, and clinical studies have suggested that metformin can inhibit cancer cell growth and proliferation (238). An ongoing phase I trial (NCT03618654) investigating the effect of metformin in combination with durvalumab for head and neck squamous cell carcinoma reported a significant decrease in FOXP3 Tregs and an increase in CD8+ cellular density in the stroma adjacent to the tumor. Greater increases in CD8+ cell density and decreases in FOXP3 cell density were observed in patients receiving metformin with durvalumab relative to durvalumab alone (239). These findings were supported by a retrospective review of patients given FDA-approved nivolumab, pembrolizumab, or atezolizumab, with or without metformin, for newly diagnosed stage IV non-small cell lung cancer (NSCLC). This study reported improved clinical outcomes (overall response rate, disease control rate, overall survival) among patients who received an ICI with metformin; however, this observed improvement was not statistically significant (240). Several clinical trials are currently underway to test metformin with anti-PD1 ICIs. In one such trial sponsored by Northwestern University (NCT03048500), the safety, tolerability, and antitumor efficacy of a metformin-nivolumab combination are being evaluated in patients with NSCLC with or without prior treatment with PD1/PDL1 inhibitors. Another phase IB clinical trial, conducted at Okayama University in Japan (UMIN000028405), is testing the safety, efficacy, and pharmacokinetics of a nivolumab–metformin combination for refractory/recurrent tumors. Another phase II trial (NCT03800602) is underway to evaluate the effects of combining metformin with nivolumab on overall response rate among patients with microsatellite-stable stage IV colorectal cancer that has not responded to previous treatment. Finally, an investigator-initiated phase I clinical trial (NCT03311308) is currently evaluating the effectiveness and safety of pembrolizumab with metformin for advanced-stage melanoma.
Adenosine is produced mainly from ATP catabolism mediated by CD39 and CD73; adenosine binds to adenosine receptors to trigger downstream signaling (142). Adenosine concentrations are significantly elevated in some solid tumors. An elevated level of adenosine in the TME can potentially impair T-cell function by inducing the accumulation of intracellular cAMP (241). The anti-CD73 agent MEDI9447 combined with the anti-PDL1 agent durvalumab is currently being investigated for patients with advanced colorectal or pancreatic cancer (NCT02503774). Also, combinations of the anti-CD73 antibody BMS-986179 and nivolumab are being investigated for the treatment of various advanced solid tumors (NCT02754141). Early findings from the latter study reported that the combination therapy had a more pronounced antitumor effect compared with ICI alone.
Ciforadenant, a molecule targeting the adenosine-A2A receptor on T lymphocytes and other immune cells, may also enhance the antitumor activity of T cells. A clinical trial investigating ciforadenant (NCT02655822) reported that it was well tolerated, alone and in combination with atezolizumab, among patients with advanced metastatic castration-resistant prostate cancer, with 5 of 33 such patients showing had tumor regression (241). This combination has also shown antitumor activity against renal cell carcinoma and NSCLC.
Another adenosine A2a receptor antagonist (AZD4635), given as monotherapy or in combination with durvalumab, was also found to be well tolerated and to have clinical benefit among patients with refractory metastatic castrate-resistant prostate cancer (NCT02740985). Tumor responses were noted in 2 of 39 patients given AZD4635 as monotherapy, and 6 of 37 given combination therapy; prostate-specific antigen responses were found in 3 of 60 patients (monotherapy) and 10 of 45 patients (combination therapy) (242). Several other clinical trials of adenosine antagonists are currently underway, among them a phase II study of the safety and efficacy of AZD4635 with durvalumab or oleclumab for patients with prostate cancer (NCT04089553); and a nonrandomized phase II study of the efficacy and safety of AZD4635 plus durvalumab ± cabazitaxel for patients with progressing advanced prostate cancer (NCT04495179). Finally, the efficacy of a dual adenosine receptor antagonist (AB928) and a PD1 checkpoint inhibitor (AB122) is being tested in combination with short-course radiotherapy and consolidation chemotherapy for patients with rectal cancer (NCT05024097).
Another clinical trial approach currently being evaluated targets the regulation of lipid metabolism and FAO. As noted previously, tumor cells rapidly proliferate, metastasize, quickly utilize existing glucose stores, and undergo metabolic reprogramming shifts toward FAO. Fatty acids in turn support the metabolism of suppressive immune cells in the TME in addition to tumor growth. PPARα is a ligand-activated nuclear transcription factor that regulates lipid metabolism and FAO (243). TPST-1120, a selective PPARα antagonist, blocks the transcription of PPARα target genes, leading to an intracellular metabolism shift from FAO to glycolysis (244). Reduction of fatty acids in the TME leads to the direct killing of tumor cells that depend on FAO and skews macrophages from the immunosuppressive M2 phenotype to an effector M1 phenotype, thereby improving the cytotoxicity of immune effector cells (245). Early findings from an ongoing trial of TPST-1120 in combination with nivolumab have shown that TPST-1120 is well-tolerated, both as a single agent and in combination with nivolumab, and the combination has shown promising objective responses among subjects with disease previously refractory to anti-PD1 therapy, including 2 of 2 responders with pretreated renal cell carcinoma, and a subject with heavily pretreated cholangiocarcinoma, a tumor that generally does not respond to anti-PD1 alone.
Other clinical trials currently underway are targeting different metabolic pathways. One example is a trial of DON, a glutamine antagonist that suppresses cancer cell metabolism but concurrently enhances the metabolic fitness of tumor CD8+ T cells. DRP-104, a peptide prodrug of DON, is as effective as DON in inhibiting tumor growth but has markedly less toxicity. An ongoing clinical trial (NCT04471415) is currently assessing the safety, tolerability, pharmacokinetics, pharmacodynamics, and preliminary antitumor activity of DRP-104, given as single-agent therapy.
These clinical trial results support the contention that metabolic pathways can be exploited for therapeutic purposes. That said, we urge caution in interpreting these findings, as many of these clinical trials are still ongoing and the findings described here are based on journal and conference proceedings with limited sample sizes. More studies are needed to obtain reliable information on the agents’ safety, tolerability, exposure duration, and pharmacokinetics, especially when they are used in combination with chemotherapy or radiation in attempts to improve treatment efficacy. Nevertheless, increasing interest in developing various metabolic targets to enhance antitumor T-cell activity or interfere with aberrant tumor metabolic pathways is a promising step towards designing more effective immunotherapy strategies.
Concept developed by MC and SG. SG: writing–review and editing. PG: writing–review. HC: writing–review. SN: writing–review. TR: writing–review. LD: writing–review. HB: writing–review. FM: writing–review. HJ: writing–review. JW: writing–review. MC: writing–review and editing. All authors have read approved the final version of this manuscript. All authors contributed to the article.
This work was supported by U.S. Department of Defense (DOD) Lung Cancer Research Program (LRP; W81XWH-21-1-0336) to MC and by Cancer Center Support (Core) Grant P30 CA016672 from the National Cancer Institute, National Institutes of Health to The University of Texas MD Anderson Cancer Center (PI: PW Pisters).
The authors thank Christine F. Wogan, MS, ELS, of MD Anderson’s Division of Radiation Oncology, for providing editing support during the development of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
2. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv (2016) 2(5):e1600200. doi: 10.1126/sciadv.1600200
3. Leone RD, Powell JD. Fueling the revolution: targeting metabolism to enhance immunotherapy. Cancer Immunol Res (2021) 9(3):255–60. doi: 10.1158/2326-6066.CIR-20-0791
4. Palm W. Metabolic plasticity allows cancer cells to thrive under nutrient starvation. Proc Natl Acad Sci USA (2021) 118(14):1–3. doi: 10.1073/pnas.2102057118
5. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab (2016) 23(1):27–47. doi: 10.1016/j.cmet.2015.12.006
6. Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discovery (2012) 2(10):881–98. doi: 10.1158/2159-8290.CD-12-0345
7. Kim J, DeBerardinis RJ. Mechanisms and implications of metabolic heterogeneity in cancer. Cell Metab (2019) 30(3):434–46. doi: 10.1016/j.cmet.2019.08.013
8. Dang CV. Links between metabolism and cancer. Genes Dev (2012) 26(9):877–90. doi: 10.1101/gad.189365.112
9. Liberti MV, Locasale JW. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci (2016) 41(3):211–8. doi: 10.1016/j.tibs.2015.12.001
10. Vaupel P, Multhoff G. Revisiting the warburg effect: historical dogma versus current understanding. J Physiol (2021) 599(6):1745–57. doi: 10.1113/JP278810
11. Esfahani K, Roudaia L, Buhlaiga N, Del Rincon SV, Papneja N, Miller WH Jr. A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol (2020) 27(Suppl 2):S87–97. doi: 10.3747/co.27.5223
12. Kouidhi S, Elgaaied AB, Chouaib S. Impact of metabolism on T-cell differentiation and function and cross talk with tumor microenvironment. Front Immunol (2017) 8:270. doi: 10.3389/fimmu.2017.00270
13. Kouidhi S, Ben Ayed F, Benammar Elgaaied A. Targeting tumor metabolism: a new challenge to improve immunotherapy. Front Immunol (2018) 9:353. doi: 10.3389/fimmu.2018.00353
14. Le Bourgeois T, Strauss L, Aksoylar HI, Daneshmandi S, Seth P, Patsoukis N, et al. Targeting T cell metabolism for improvement of cancer immunotherapy. Front Oncol (2018) 8:237. doi: 10.3389/fonc.2018.00237
15. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007) 109(9):3812–9. doi: 10.1182/blood-2006-07-035972
16. Cham CM, Driessens G, O'Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in Cd8+ T cells. Eur J Immunol (2008) 38(9):2438–50. doi: 10.1002/eji.200838289
17. Mockler MB, Conroy MJ, Lysaght J. Targeting T cell immunometabolism for cancer immunotherapy; understanding the impact of the tumor microenvironment. Front Oncol (2014) 4:107. doi: 10.3389/fonc.2014.00107
18. Buck MD, O'Sullivan D, Pearce EL. T Cell metabolism drives immunity. J Exp Med (2015) 212(9):1345–60. doi: 10.1084/jem.20151159
19. Geltink RIK, Kyle RL, Pearce EL. Unraveling the complex interplay between T cell metabolism and function. Annu Rev Immunol (2018) 36:461–88. doi: 10.1146/annurev-immunol-042617-053019
20. Martinez-Reyes I, Chandel NS. Cancer metabolism: looking forward. Nat Rev Cancer (2021) 21(10):669–80. doi: 10.1038/s41568-021-00378-6
21. Molon B, Cali B, Viola A. T Cells and cancer: how metabolism shapes immunity. Front Immunol (2016) 7:20. doi: 10.3389/fimmu.2016.00020
22. van der Windt GJ, Pearce EL. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol Rev (2012) 249(1):27–42. doi: 10.1111/j.1600-065X.2012.01150.x
23. Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer (2013) 12:152. doi: 10.1186/1476-4598-12-152
24. Zhang Y, Yang JM. Altered energy metabolism in cancer: a unique opportunity for therapeutic intervention. Cancer Biol Ther (2013) 14(2):81–9. doi: 10.4161/cbt.22958
25. Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol (2012) 33(4):168–73. doi: 10.1016/j.it.2012.01.010
26. Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol (2005) 5(11):844–52. doi: 10.1038/nri1710
27. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires Cd28-mediated akt-dependent and independent pathways. J Immunol (2008) 180(7):4476–86. doi: 10.4049/jimmunol.180.7.4476
28. Greiner EF, Guppy M, Brand K. Glucose is essential for proliferation and the glycolytic enzyme induction that provokes a transition to glycolytic energy production. J Biol Chem (1994) 269(50):31484–90. doi: 10.1016/S0021-9258(18)31720-4
29. Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153(6):1239–51. doi: 10.1016/j.cell.2013.05.016
30. Amoedo ND, Valencia JP, Rodrigues MF, Galina A, Rumjanek FD. How does the metabolism of tumour cells differ from that of normal cells. Biosci Rep (2013) 33(6):865–73. doi: 10.1042/BSR20130066
31. Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer (2011) 11(5):325–37. doi: 10.1038/nrc3038
32. Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor P53 down-regulates glucose transporters Glut1 and Glut4 gene expression. Cancer Res (2004) 64(7):2627–33. doi: 10.1158/0008-5472.can-03-0846
33. Afonso J, Santos LL, Longatto-Filho A, Baltazar F. Competitive glucose metabolism as a target to boost bladder cancer immunotherapy. Nat Rev Urol (2020) 17(2):77–106. doi: 10.1038/s41585-019-0263-6
34. Balsa-Martinez E, Puigserver P. Cancer cells hijack gluconeogenic enzymes to fuel cell growth. Mol Cell (2015) 60(4):509–11. doi: 10.1016/j.molcel.2015.11.005
35. Semeniuk-Wojtas A, Poddebniak-Strama K, Modzelewska M, Baryla M, Dziag-Dudek E, Syrylo T, et al. Tumour microenvironment as a predictive factor for immunotherapy in non-Muscle-Invasive bladder cancer. Cancer Immunol Immunother (2023) 1–19. doi: 10.1007/s00262-023-03376-9
36. Olenchock BA, Rathmell JC, Vander Heiden MG. Biochemical underpinnings of immune cell metabolic phenotypes. Immunity (2017) 46(5):703–13. doi: 10.1016/j.immuni.2017.04.013
37. Li C, Zhang G, Zhao L, Ma Z, Chen H. Metabolic reprogramming in cancer cells: glycolysis, glutaminolysis, and bcl-2 proteins as novel therapeutic targets for cancer. World J Surg Oncol (2016) 14(1):15. doi: 10.1186/s12957-016-0769-9
38. Zheng J. Energy metabolism of cancer: glycolysis versus oxidative phosphorylation (Review). Oncol Lett (2012) 4(6):1151–7. doi: 10.3892/ol.2012.928
39. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
40. Beckermann KE, Dudzinski SO, Rathmell JC. Dysfunctional T cell metabolism in the tumor microenvironment. Cytokine Growth Factor Rev (2017) 35:7–14. doi: 10.1016/j.cytogfr.2017.04.003
41. Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res (2013) 73(5):1524–35. doi: 10.1158/0008-5472.CAN-12-2796
42. Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D'Acquisto F, et al. Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions. PloS Biol (2015) 13(7):e1002202. doi: 10.1371/journal.pbio.1002202
43. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. Ldha-associated lactic acid production blunts tumor immunosurveillance by T and nk cells. Cell Metab (2016) 24(5):657–71. doi: 10.1016/j.cmet.2016.08.011
44. Broer S. Lactate transportation is required for lymphocyte activation. Nat Chem Biol (2005) 1(7):356–7. doi: 10.1038/nchembio1205-356
45. Rangel Rivera GO, Knochelmann HM, Dwyer CJ, Smith AS, Wyatt MM, Rivera-Reyes AM, et al. Fundamentals of T cell metabolism and strategies to enhance cancer immunotherapy. Front Immunol (2021) 12:645242. doi: 10.3389/fimmu.2021.645242
46. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory Cd4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
47. Scott KE, Cleveland JL. Lactate wreaks havoc on tumor-infiltrating T and nk cells. Cell Metab (2016) 24(5):649–50. doi: 10.1016/j.cmet.2016.10.015
48. Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab (2017) 25(6):1282–93 e7. doi: 10.1016/j.cmet.2016.12.018
49. Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res (2011) 71(22):6921–5. doi: 10.1158/0008-5472.CAN-11-1457
50. Mendler AN, Hu B, Prinz PU, Kreutz M, Gottfried E, Noessner E. Tumor lactic acidosis suppresses ctl function by inhibition of P38 and Jnk/C-jun activation. Int J Cancer (2012) 131(3):633–40. doi: 10.1002/ijc.26410
51. Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res (2012) 72(11):2746–56. doi: 10.1158/0008-5472.CAN-11-1272
52. Navarro F, Casares N, Martin-Otal C, Lasarte-Cia A, Gorraiz M, Sarrion P, et al. Overcoming T cell dysfunction in acidic ph to enhance adoptive T cell transfer immunotherapy. Oncoimmunology (2022) 11(1):2070337. doi: 10.1080/2162402X.2022.2070337
53. Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res (2016) 76(6):1381–90. doi: 10.1158/0008-5472.CAN-15-1743
54. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell (2015) 162(6):1217–28. doi: 10.1016/j.cell.2015.08.012
55. Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature (2021) 591(7851):645–51. doi: 10.1038/s41586-020-03045-2
56. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for Cd4 T cell activation and effector function. Cell Metab (2014) 20(1):61–72. doi: 10.1016/j.cmet.2014.05.004
57. Maciver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leukoc Biol (2008) 84(4):949–57. doi: 10.1189/jlb.0108024
58. Cham CM, Gajewski TF. Glucose availability regulates ifn-gamma production and P70s6 kinase activation in Cd8+ effector T cells. J Immunol (2005) 174(8):4670–7. doi: 10.4049/jimmunol.174.8.4670
59. Ma EH, Poffenberger MC, Wong AH, Jones RG. The role of ampk in T cell metabolism and function. Curr Opin Immunol (2017) 46:45–52. doi: 10.1016/j.coi.2017.04.004
60. Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, et al. Ampk is a negative regulator of the warburg effect and suppresses tumor growth in vivo. Cell Metab (2013) 17(1):113–24. doi: 10.1016/j.cmet.2012.12.001
61. Zhao Y, Hu X, Liu Y, Dong S, Wen Z, He W, et al. Ros signaling under metabolic stress: cross-talk between ampk and akt pathway. Mol Cancer (2017) 16(1):79. doi: 10.1186/s12943-017-0648-1
62. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances Cd8+ T cell memory and antitumor function. J Clin Invest (2013) 123(10):4479–88. doi: 10.1172/JCI69589
63. Lee SY, Lee SH, Yang EJ, Kim EK, Kim JK, Shin DY, et al. Metformin ameliorates inflammatory bowel disease by suppression of the Stat3 signaling pathway and regulation of the between Th17/Treg balance. PloS One (2015) 10(9):e0135858. doi: 10.1371/journal.pone.0135858
64. Klein Geltink RI, Edwards-Hicks J, Apostolova P, O'Sullivan D, Sanin DE, Patterson AE, et al. Metabolic conditioning of Cd8(+) effector T cells for adoptive cell therapy. Nat Metab (2020) 2(8):703–16. doi: 10.1038/s42255-020-0256-z
65. Yu YR, Imrichova H, Wang H, Chao T, Xiao Z, Gao M, et al. Disturbed mitochondrial dynamics in Cd8(+) tils reinforce T cell exhaustion. Nat Immunol (2020) 21(12):1540–51. doi: 10.1038/s41590-020-0793-3
66. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862
67. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis Via Ccl28 and T(Reg) cells. Nature (2011) 475(7355):226–30. doi: 10.1038/nature10169
68. Courtnay R, Ngo DC, Malik N, Ververis K, Tortorella SM, Karagiannis TC. Cancer metabolism and the warburg effect: the role of hif-1 and Pi3k. Mol Biol Rep (2015) 42(4):841–51. doi: 10.1007/s11033-015-3858-x
69. Clever D, Roychoudhuri R, Constantinides MG, Askenase MH, Sukumar M, Klebanoff CA, et al. Oxygen sensing by T cells establishes an immunologically tolerant metastatic niche. Cell (2016) 166(5):1117–31 e14. doi: 10.1016/j.cell.2016.07.032
70. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res (2014) 74(3):665–74. doi: 10.1158/0008-5472.CAN-13-0992
71. Chouaib S, Noman MZ, Kosmatopoulos K, Curran MA. Hypoxic stress: obstacles and opportunities for innovative immunotherapy of cancer. Oncogene (2017) 36(4):439–45. doi: 10.1038/onc.2016.225
72. Lukashev D, Ohta A, Sitkovsky M. Hypoxia-dependent anti-inflammatory pathways in protection of cancerous tissues. Cancer Metastasis Rev (2007) 26(2):273–9. doi: 10.1007/s10555-007-9054-2
73. Hoskin DW, Mader JS, Furlong SJ, Conrad DM, Blay J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review). Int J Oncol (2008) 32(3):527–35. doi: 10.3892/ijo.32.3.527
74. Wu D, Chen Y. Lipids for Cd8(+) tils: beneficial or harmful? Front Immunol (2022) 13:1020422. doi: 10.3389/fimmu.2022.1020422
75. Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing Cd8(+) T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell (2017) 32(3):377–91 e9. doi: 10.1016/j.ccell.2017.08.004
76. Manzo T, Prentice BM, Anderson KG, Raman A, Schalck A, Codreanu GS, et al. Accumulation of long-chain fatty acids in the tumor microenvironment drives dysfunction in intrapancreatic Cd8+ T cells. J Exp Med (2020) 217(8):1–22. doi: 10.1084/jem.20191920
77. Lim SA, Su W, Chapman NM, Chi H. Lipid metabolism in T cell signaling and function. Nat Chem Biol (2022) 18(5):470–81. doi: 10.1038/s41589-022-01017-3
78. Raud B, McGuire PJ, Jones RG, Sparwasser T, Berod L. Fatty acid metabolism in Cd8(+) T cell memory: challenging current concepts. Immunol Rev (2018) 283(1):213–31. doi: 10.1111/imr.12655
79. Kuhajda FP, Jenner K, Wood FD, Hennigar RA, Jacobs LB, Dick JD, et al. Fatty acid synthesis: a potential selective target for antineoplastic therapy. Proc Natl Acad Sci U.S.A. (1994) 91(14):6379–83. doi: 10.1073/pnas.91.14.6379
80. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer (2007) 7(10):763–77. doi: 10.1038/nrc2222
81. Daniels VW, Smans K, Royaux I, Chypre M, Swinnen JV, Zaidi N. Cancer cells differentially activate and thrive on De Novo lipid synthesis pathways in a low-lipid environment. PloS One (2014) 9(9):e106913. doi: 10.1371/journal.pone.0106913
82. Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science (1986) 232(4746):34–47. doi: 10.1126/science.3513311
83. Gyamfi J, Yeo JH, Kwon D, Min BS, Cha YJ, Koo JS, et al. Interaction between Cd36 and Fabp4 modulates adipocyte-induced fatty acid import and metabolism in breast cancer. NPJ Breast Cancer (2021) 7(1):129. doi: 10.1038/s41523-021-00324-7
84. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. Pd-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6:6692. doi: 10.1038/ncomms7692
85. Xu T, Schutte A, Jimenez L, Goncalves ANA, Keller A, Pipkin ME, et al. Kdm6b regulates the generation of effector Cd8(+) T cells by inducing chromatin accessibility in effector-associated genes. J Immunol (2021) 206(9):2170–83. doi: 10.4049/jimmunol.2001459
86. Xu S, Chaudhary O, Rodriguez-Morales P, Sun X, Chen D, Zappasodi R, et al. Uptake of oxidized lipids by the scavenger receptor Cd36 promotes lipid peroxidation and dysfunction in Cd8(+) T cells in tumors. Immunity (2021) 54(7):1561–77 e7. doi: 10.1016/j.immuni.2021.05.003
87. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing Cd8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460(7251):103–7. doi: 10.1038/nature08097
88. Dumauthioz N, Tschumi B, Wenes M, Marti B, Wang H, Franco F, et al. Enforced pgc-1alpha expression promotes Cd8 T cell fitness, memory formation and antitumor immunity. Cell Mol Immunol (2021) 18(7):1761–71. doi: 10.1038/s41423-020-0365-3
89. Lin R, Zhang H, Yuan Y, He Q, Zhou J, Li S, et al. Fatty acid oxidation controls Cd8(+) tissue-resident memory T-cell survival in gastric adenocarcinoma. Cancer Immunol Res (2020) 8(4):479–92. doi: 10.1158/2326-6066.CIR-19-0702
90. Wang H, Franco F, Tsui YC, Xie X, Trefny MP, Zappasodi R, et al. Cd36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat Immunol (2020) 21(3):298–308. doi: 10.1038/s41590-019-0589-5
91. Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: can treg cells be a new therapeutic target? Cancer Sci (2019) 110(7):2080–9. doi: 10.1111/cas.14069
92. Chowdhury PS, Chamoto K, Kumar A, Honjo T. Ppar-induced fatty acid oxidation in T cells increases the number of tumor-reactive Cd8(+) T cells and facilitates anti-Pd-1 therapy. Cancer Immunol Res (2018) 6(11):1375–87. doi: 10.1158/2326-6066.CIR-18-0095
93. Verma V, Jafarzadeh N, Boi S, Kundu S, Jiang Z, Fan Y, et al. Mek inhibition reprograms Cd8(+) T lymphocytes into memory stem cells with potent antitumor effects. Nat Immunol (2021) 22(1):53–66. doi: 10.1038/s41590-020-00818-9
94. Mullarky E, Lucki NC, Beheshti Zavareh R, Anglin JL, Gomes AP, Nicolay BN, et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc Natl Acad Sci U.S.A. (2016) 113(7):1778–83. doi: 10.1073/pnas.1521548113
95. Najumudeen AK, Ceteci F, Fey SK, Hamm G, Steven RT, Hall H, et al. The amino acid transporter Slc7a5 is required for efficient growth of kras-mutant colorectal cancer. Nat Genet (2021) 53(1):16–26. doi: 10.1038/s41588-020-00753-3
96. Gupta N, Miyauchi S, Martindale RG, Herdman AV, Podolsky R, Miyake K, et al. Upregulation of the amino acid transporter Atb0,+ (Slc6a14) in colorectal cancer and metastasis in humans. Biochim Biophys Acta (2005) 1741(1-2):215–23. doi: 10.1016/j.bbadis.2005.04.002
97. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a kras-regulated metabolic pathway. Nature (2013) 496(7443):101–5. doi: 10.1038/nature12040
98. Lieu EL, Nguyen T, Rhyne S, Kim J. Amino acids in cancer. Exp Mol Med (2020) 52(1):15–30. doi: 10.1038/s12276-020-0375-3
99. Michalek RD, Rathmell JC. The metabolic life and times of a T-cell. Immunol Rev (2010) 236:190–202. doi: 10.1111/j.1600-065X.2010.00911.x
100. Newsholme EA, Crabtree B, Ardawi MS. Glutamine metabolism in lymphocytes: its biochemical, physiological and clinical importance. Q J Exp Physiol (1985) 70(4):473–89. doi: 10.1113/expphysiol.1985.sp002935
101. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med (1999) 189(9):1363–72. doi: 10.1084/jem.189.9.1363
102. Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, et al. L-arginine consumption by macrophages modulates the expression of Cd3 zeta chain in T lymphocytes. J Immunol (2003) 171(3):1232–9. doi: 10.4049/jimmunol.171.3.1232
103. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64(16):5839–49. doi: 10.1158/0008-5472.CAN-04-0465
104. van Baren N, Van den Eynde BJ. Tryptophan-degrading enzymes in tumoral immune resistance. Front Immunol (2015) 6:34. doi: 10.3389/fimmu.2015.00034
105. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med (2003) 9(10):1269–74. doi: 10.1038/nm934
106. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. Gcn2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity (2005) 22(5):633–42. doi: 10.1016/j.immuni.2005.03.013
107. Huang TT, Yen MC, Lin CC, Weng TY, Chen YL, Lin CM, et al. Skin delivery of short hairpin rna of indoleamine 2,3 dioxygenase induces antitumor immunity against orthotopic and metastatic liver cancer. Cancer Sci (2011) 102(12):2214–20. doi: 10.1111/j.1349-7006.2011.02094.x
108. Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of ido. Nat Med (2011) 17(9):1094–100. doi: 10.1038/nm.2438
109. Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, et al. Selective inhibition of Ido1 effectively regulates mediators of antitumor immunity. Blood (2010) 115(17):3520–30. doi: 10.1182/blood-2009-09-246124
110. Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell (2018) 175(7):1780–95 e19. doi: 10.1016/j.cell.2018.10.001
111. Best SA, Gubser PM, Sethumadhavan S, Kersbergen A, Negron Abril YL, Goldford J, et al. Glutaminase inhibition impairs Cd8 T cell activation in Stk11-/Lkb1-Deficient lung cancer. Cell Metab (2022) 34(6):874–87 e6. doi: 10.1016/j.cmet.2022.04.003
112. Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science (2019) 366(6468):1013–21. doi: 10.1126/science.aav2588
113. Oh MH, Sun IH, Zhao L, Leone RD, Sun IM, Xu W, et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J Clin Invest (2020) 130(7):3865–84. doi: 10.1172/JCI131859
114. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by Erk/Mapk during T lymphocyte activation. J Immunol (2010) 185(2):1037–44. doi: 10.4049/jimmunol.0903586
115. Nabe S, Yamada T, Suzuki J, Toriyama K, Yasuoka T, Kuwahara M, et al. Reinforce the antitumor activity of Cd8(+) T cells Via glutamine restriction. Cancer Sci (2018) 109(12):3737–50. doi: 10.1111/cas.13827
116. Metzler B, Gfeller P, Guinet E. Restricting glutamine or glutamine-dependent purine and pyrimidine syntheses promotes human T cells with high Foxp3 expression and regulatory properties. J Immunol (2016) 196(9):3618–30. doi: 10.4049/jimmunol.1501756
117. van der Mijn JC, Panka DJ, Geissler AK, Verheul HM, Mier JW. Novel drugs that target the metabolic reprogramming in renal cell cancer. Cancer Metab (2016) 4:14. doi: 10.1186/s40170-016-0154-8
118. Hassanein M, Qian J, Hoeksema MD, Wang J, Jacobovitz M, Ji X, et al. Targeting Slc1a5-mediated glutamine dependence in non-small cell lung cancer. Int J Cancer (2015) 137(7):1587–97. doi: 10.1002/ijc.29535
119. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell (2016) 167(3):829–42 e13. doi: 10.1016/j.cell.2016.09.031
120. Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood (2007) 109(4):1568–73. doi: 10.1182/blood-2006-06-031856
121. Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res (2005) 65(8):3044–8. doi: 10.1158/0008-5472.CAN-04-4505
122. Bronte V, Kasic T, Gri G, Gallana K, Borsellino G, Marigo I, et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J Exp Med (2005) 201(8):1257–68. doi: 10.1084/jem.20042028
123. De Santo C, Serafini P, Marigo I, Dolcetti L, Bolla M, Del Soldato P, et al. Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc Natl Acad Sci U.S.A. (2005) 102(11):4185–90. doi: 10.1073/pnas.0409783102
124. Bronte V, Zanovello P. Regulation of immune responses by l-arginine metabolism. Nat Rev Immunol (2005) 5(8):641–54. doi: 10.1038/nri1668
125. Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol (2013) 14(5):500–8. doi: 10.1038/ni.2556
126. Ananieva EA, Powell JD, Hutson SM. Leucine metabolism in T cell activation: mtor signaling and beyond. Adv Nutr (2016) 7(4):798S–805S. doi: 10.3945/an.115.011221
127. Hayashi K, Jutabha P, Endou H, Sagara H, Anzai N. Lat1 is a critical transporter of essential amino acids for immune reactions in activated human T cells. J Immunol (2013) 191(8):4080–5. doi: 10.4049/jimmunol.1300923
128. Wetzel TJ, Erfan SC, Ananieva EA. The emerging role of the branched chain aminotransferases, bcatc and bcatm, for anti-tumor T-cell immunity. Immunometabolism (Cobham) (2023) 5(1):e00014. doi: 10.1097/IN9.0000000000000014
129. Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant kras-driven cancers. Science (2016) 353(6304):1161–5. doi: 10.1126/science.aaf5171
130. Tonjes M, Barbus S, Park YJ, Wang W, Schlotter M, Lindroth AM, et al. Bcat1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type Idh1. Nat Med (2013) 19(7):901–8. doi: 10.1038/nm.3217
131. Li GS, Huang HQ, Liang Y, Pang QY, Sun HJ, Huang ZG, et al. Bcat1: a risk factor in multiple cancers based on a pan-cancer analysis. Cancer Med (2022) 11(5):1396–412. doi: 10.1002/cam4.4525
132. Yi L, Fan X, Li J, Yuan F, Zhao J, Nister M, et al. Enrichment of branched chain amino acid transaminase 1 correlates with multiple biological processes and contributes to poor survival of Idh1 wild-type gliomas. Aging (Albany NY) (2021) 13(3):3645–60. doi: 10.18632/aging.202328
133. Hu J, Li X, Yang L, Li H. Hypoxia, a key factor in the immune microenvironment. BioMed Pharmacother (2022) 151:113068. doi: 10.1016/j.biopha.2022.113068
134. Wang JX, Choi SYC, Niu X, Kang N, Xue H, Killam J, et al. Lactic acid and an acidic tumor microenvironment suppress anticancer immunity. Int J Mol Sci (2020) 21(21):1–14. doi: 10.3390/ijms21218363
135. Leone RD, Powell JD. Metabolism of immune cells in cancer. Nat Rev Cancer (2020) 20(9):516–31. doi: 10.1038/s41568-020-0273-y
136. Pavlova NN, Zhu J, Thompson CB. The hallmarks of cancer metabolism: still emerging. Cell Metab (2022) 34(3):355–77. doi: 10.1016/j.cmet.2022.01.007
137. Singer K, Kastenberger M, Gottfried E, Hammerschmied CG, Buttner M, Aigner M, et al. Warburg phenotype in renal cell carcinoma: high expression of glucose-transporter 1 (Glut-1) correlates with low Cd8(+) T-cell infiltration in the tumor. Int J Cancer (2011) 128(9):2085–95. doi: 10.1002/ijc.25543
138. Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, et al. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab (2018) 27(5):977–87 e4. doi: 10.1016/j.cmet.2018.02.024
139. Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, et al. Cancer mediates effector T cell dysfunction by targeting micrornas and Ezh2 Via glycolysis restriction. Nat Immunol (2016) 17(1):95–103. doi: 10.1038/ni.3313
140. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The Cd28 signaling pathway regulates glucose metabolism. Immunity (2002) 16(6):769–77. doi: 10.1016/s1074-7613(02)00323-0
141. Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, et al. Noxa, a Bh3-only member of the bcl-2 family and candidate mediator of P53-induced apoptosis. Science (2000) 288(5468):1053–8. doi: 10.1126/science.288.5468.1053
142. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases Cd39 and Cd73: novel checkpoint inhibitor targets. Immunol Rev (2017) 276(1):121–44. doi: 10.1111/imr.12528
143. Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J Immunother Cancer (2018) 6(1):57. doi: 10.1186/s40425-018-0360-8
144. Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-Cd73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci U.S.A. (2010) 107(4):1547–52. doi: 10.1073/pnas.0908801107
145. Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol (2016) 16(3):177–92. doi: 10.1038/nri.2016.4
146. Allard D, Turcotte M, Stagg J. Targeting A2 adenosine receptors in cancer. Immunol Cell Biol (2017) 95(4):333–9. doi: 10.1038/icb.2017.8
147. Wu D, Zhu Y. Role of kynurenine in promoting the generation of exhausted Cd8(+) T cells in colorectal cancer. Am J Transl Res (2021) 13(3):1535–47.
148. Amobi-McCloud A, Muthuswamy R, Battaglia S, Yu H, Liu T, Wang J, et al. Ido1 expression in ovarian cancer induces pd-1 in T cells Via aryl hydrocarbon receptor activation. Front Immunol (2021) 12:678999. doi: 10.3389/fimmu.2021.678999
149. Le Naour J, Galluzzi L, Zitvogel L, Kroemer G, Vacchelli E. Trial watch: ido inhibitors in cancer therapy. Oncoimmunology (2020) 9(1):1777625. doi: 10.1080/2162402X.2020.1777625
150. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces Cd8(+) T cell exhaustion in the tumor microenvironment. Cell Metab (2019) 30(1):143–56 e5. doi: 10.1016/j.cmet.2019.04.002
151. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of Cd8(+) T cells by modulating cholesterol metabolism. Nature (2016) 531(7596):651–5. doi: 10.1038/nature17412
152. Ghosh S, Mukherjee S, Choudhury S, Gupta P, Adhikary A, Baral R, et al. Reactive oxygen species in the tumor niche triggers altered activation of macrophages and immunosuppression: role of fluoxetine. Cell Signal (2015) 27(7):1398–412. doi: 10.1016/j.cellsig.2015.03.013
153. OuYang LY, Wu XJ, Ye SB, Zhang RX, Li ZL, Liao W, et al. Tumor-induced myeloid-derived suppressor cells promote tumor progression through oxidative metabolism in human colorectal cancer. J Transl Med (2015) 13:47. doi: 10.1186/s12967-015-0410-7
154. Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL, et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol (2021) 22(2):205–15. doi: 10.1038/s41590-020-00834-9
155. Bell HN, Huber AK, Singhal R, Korimerla N, Rebernick RJ, Kumar R, et al. Microenvironmental ammonia enhances T cell exhaustion in colorectal cancer. Cell Metab (2023) 35(1):134–49 e6. doi: 10.1016/j.cmet.2022.11.013
156. O'Brien TG. The induction of ornithine decarboxylase as an early, possibly obligatory, event in mouse skin carcinogenesis. Cancer Res (1976) 36(7 PT 2):2644–53. Available at: https://aacrjournals.org/cancerres/article/36/7_Part_2/2644/481573/The-Induction-of-Ornithine-Decarboxylase-as-an.
157. Hayes CS, Shicora AC, Keough MP, Snook AE, Burns MR, Gilmour SK. Polyamine-blocking therapy reverses immunosuppression in the tumor microenvironment. Cancer Immunol Res (2014) 2(3):274–85. doi: 10.1158/2326-6066.CIR-13-0120-T
158. Chamaillard L, Catros-Quemener V, Delcros JG, Bansard JY, Havouis R, Desury D, et al. Polyamine deprivation prevents the development of tumour-induced immune suppression. Br J Cancer (1997) 76(3):365–70. doi: 10.1038/bjc.1997.391
159. Wang J, Ye C, Chen C, Xiong H, Xie B, Zhou J, et al. Glucose transporter Glut1 expression and clinical outcome in solid tumors: a systematic review and meta-analysis. Oncotarget (2017) 8(10):16875–86. doi: 10.18632/oncotarget.15171
160. Fu Y, Lan T, Cai H, Lu A, Yu W. Meta-analysis of serum lactate dehydrogenase and prognosis for osteosarcoma. Med (Baltimore) (2018) 97(19):e0741. doi: 10.1097/MD.0000000000010741
161. Gan J, Wang W, Yang Z, Pan J, Zheng L, Yin L. Prognostic value of pretreatment serum lactate dehydrogenase level in pancreatic cancer patients: a meta-analysis of 18 observational studies. Med (Baltimore) (2018) 97(46):e13151. doi: 10.1097/MD.0000000000013151
162. Girgis H, Masui O, White NM, Scorilas A, Rotondo F, Seivwright A, et al. Lactate dehydrogenase a is a potential prognostic marker in clear cell renal cell carcinoma. Mol Cancer (2014) 13:101. doi: 10.1186/1476-4598-13-101
163. Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfor K, Rofstad EK, et al. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res (2000) 60(4):916–21. Available at: https://aacrjournals.org/cancerres/article/60/4/916/507131/High-Lactate-Levels-Predict-Likelihood-of.
164. Murray PJ. Macrophage polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
165. Ley K. M1 means kill; M2 means heal. J Immunol (2017) 199(7):2191–3. doi: 10.4049/jimmunol.1701135
166. Mu X, Shi W, Xu Y, Xu C, Zhao T, Geng B, et al. Tumor-derived lactate induces M2 macrophage polarization Via the activation of the Erk/Stat3 signaling pathway in breast cancer. Cell Cycle (2018) 17(4):428–38. doi: 10.1080/15384101.2018.1444305
167. El-Kenawi A, Gatenbee C, Robertson-Tessi M, Bravo R, Dhillon J, Balagurunathan Y, et al. Acidity promotes tumour progression by altering macrophage phenotype in prostate cancer. Br J Cancer (2019) 121(7):556–66. doi: 10.1038/s41416-019-0542-2
168. Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood (2006) 107(5):2013–21. doi: 10.1182/blood-2005-05-1795
169. Monti M, Vescovi R, Consoli F, Farina D, Moratto D, Berruti A, et al. Plasmacytoid dendritic cell impairment in metastatic melanoma by lactic acidosis. Cancers (Basel) (2020) 12(8):1–24. doi: 10.3390/cancers12082085
170. Cao S, Liu P, Zhu H, Gong H, Yao J, Sun Y, et al. Extracellular acidification acts as a key modulator of neutrophil apoptosis and functions. PloS One (2015) 10(9):e0137221. doi: 10.1371/journal.pone.0137221
171. Yang X, Lu Y, Hang J, Zhang J, Zhang T, Huo Y, et al. Lactate-modulated immunosuppression of myeloid-derived suppressor cells contributes to the radioresistance of pancreatic cancer. Cancer Immunol Res (2020) 8(11):1440–51. doi: 10.1158/2326-6066.CIR-20-0111
172. Dhup S, Dadhich RK, Porporato PE, Sonveaux P. Multiple biological activities of lactic acid in cancer: influences on tumor growth, angiogenesis and metastasis. Curr Pharm Des (2012) 18(10):1319–30. doi: 10.2174/138161212799504902
173. Tu VY, Ayari A, O'Connor RS. Beyond the lactate paradox: how lactate and acidity impact T cell therapies against cancer. Antibodies (Basel) (2021) 10(3):1–12. doi: 10.3390/antib10030025
174. Kaymak I, Luda KM, Duimstra LR, Ma EH, Longo J, Dahabieh MS, et al. Carbon source availability drives nutrient utilization in Cd8(+) T cells. Cell Metab (2022) 34(9):1298–311 e6. doi: 10.1016/j.cmet.2022.07.012
175. Liu X, Yang Z, Chen Z, Chen R, Zhao D, Zhou Y, et al. Effects of the suppression of lactate dehydrogenase a on the growth and invasion of human gastric cancer cells. Oncol Rep (2015) 33(1):157–62. doi: 10.3892/or.2014.3600
176. Daneshmandi S, Wegiel B, Seth P. Blockade of lactate dehydrogenase-a (Ldh-a) improves efficacy of anti-programmed cell death-1 (Pd-1) therapy in melanoma. Cancers (Basel) (2019) 11(4):1–11. doi: 10.3390/cancers11040450
177. Fang Y, Liu W, Tang Z, Ji X, Zhou Y, Song S, et al. Monocarboxylate transporter 4 inhibition potentiates hepatocellular carcinoma immunotherapy through enhancing T cell infiltration and immune attack. Hepatology (2023) 77(1):109–23. doi: 10.1002/hep.32348
178. Polanski R, Hodgkinson CL, Fusi A, Nonaka D, Priest L, Kelly P, et al. Activity of the monocarboxylate transporter 1 inhibitor Azd3965 in small cell lung cancer. Clin Cancer Res (2014) 20(4):926–37. doi: 10.1158/1078-0432.CCR-13-2270
179. Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, et al. Co-Inhibition of Cd73 and A2ar adenosine signaling improves anti-tumor immune responses. Cancer Cell (2016) 30(3):391–403. doi: 10.1016/j.ccell.2016.06.025
180. Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S, et al. Adenosine receptor 2a blockade increases the efficacy of anti-Pd-1 through enhanced antitumor T-cell responses. Cancer Immunol Res (2015) 3(5):506–17. doi: 10.1158/2326-6066.CIR-14-0211
181. Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, et al. Targeting Cd73 in the tumor microenvironment with Medi9447. Oncoimmunology (2016) 5(8):e1208875. doi: 10.1080/2162402X.2016.1208875
182. Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Adenosine limits the therapeutic effectiveness of anti-Ctla4 mab in a mouse melanoma model. Am J Cancer Res (2014) 4(2):172–81. Available at: https://e-century.us/files/ajcr/4/2/ajcr0000261.pdf.
183. Forte G, Sorrentino R, Montinaro A, Luciano A, Adcock IM, Maiolino P, et al. Inhibition of Cd73 improves b cell-mediated anti-tumor immunity in a mouse model of melanoma. J Immunol (2012) 189(5):2226–33. doi: 10.4049/jimmunol.1200744
184. Ma SR, Deng WW, Liu JF, Mao L, Yu GT, Bu LL, et al. Blockade of adenosine A2a receptor enhances Cd8(+) T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol Cancer (2017) 16(1):99. doi: 10.1186/s12943-017-0665-0
185. Kuzu OF, Noory MA, Robertson GP. The role of cholesterol in cancer. Cancer Res (2016) 76(8):2063–70. doi: 10.1158/0008-5472.CAN-15-2613
186. Wang W, Bai L, Li W, Cui J. The lipid metabolic landscape of cancers and new therapeutic perspectives. Front Oncol (2020) 10:605154. doi: 10.3389/fonc.2020.605154
187. Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res (2010) 44(5):479–96. doi: 10.3109/10715761003667554
188. Liao Z, Chua D, Tan NS. Reactive oxygen species: a volatile driver of field cancerization and metastasis. Mol Cancer (2019) 18(1):65. doi: 10.1186/s12943-019-0961-y
189. Probst BL, Trevino I, McCauley L, Bumeister R, Dulubova I, Wigley WC, et al. Rta 408, a novel synthetic triterpenoid with broad anticancer and anti-inflammatory activity. PloS One (2015) 10(4):e0122942. doi: 10.1371/journal.pone.0122942
190. Kaiser H, Parker E, Hamrick MW. Kynurenine signaling through the aryl hydrocarbon receptor: implications for aging and healthspan. Exp Gerontol (2020) 130:110797. doi: 10.1016/j.exger.2019.110797
191. Litzenburger UM, Opitz CA, Sahm F, Rauschenbach KJ, Trump S, Winter M, et al. Constitutive ido expression in human cancer is sustained by an autocrine signaling loop involving il-6, Stat3 and the ahr. Oncotarget (2014) 5(4):1038–51. doi: 10.18632/oncotarget.1637
192. Pegg AE. Polyamine metabolism and its importance in neoplastic growth and a target for chemotherapy. Cancer Res (1988) 48(4):759–74. Available at: https://aacrjournals.org/cancerres/article/60/4/916/507131/High-Lactate-Levels-Predict-Likelihood-of.
193. Wang Z. Polyamines instruct T-cell differentiation. Nat Cell Biol (2021) 23(8):811. doi: 10.1038/s41556-021-00739-1
194. Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, et al. Sdh mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell (2013) 23(6):739–52. doi: 10.1016/j.ccr.2013.04.018
195. Gudgeon N, Munford H, Bishop EL, Hill J, Fulton-Ward T, Bending D, et al. Succinate uptake by T cells suppresses their effector function Via inhibition of mitochondrial glucose oxidation. Cell Rep (2022) 40(7):111193. doi: 10.1016/j.celrep.2022.111193
196. Soriano-Baguet L, Brenner D. Metabolism and epigenetics at the heart of T cell function. Trends Immunol (2023) 44(3):231–44. doi: 10.1016/j.it.2023.01.002
197. Araki Y, Fann M, Wersto R, Weng NP. Histone acetylation facilitates rapid and robust memory Cd8 T cell response through differential expression of effector molecules (Eomesodermin and its targets: perforin and granzyme b). J Immunol (2008) 180(12):8102–8. doi: 10.4049/jimmunol.180.12.8102
198. Zhang H, Tang K, Ma J, Zhou L, Liu J, Zeng L, et al. Ketogenesis-generated beta-hydroxybutyrate is an epigenetic regulator of Cd8(+) T-cell memory development. Nat Cell Biol (2020) 22(1):18–25. doi: 10.1038/s41556-019-0440-0
199. Belk JA, Daniel B, Satpathy AT. Epigenetic regulation of T cell exhaustion. Nat Immunol (2022) 23(6):848–60. doi: 10.1038/s41590-022-01224-z
200. Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, et al. Developmental relationships of four exhausted Cd8(+) T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity (2020) 52(5):825–41 e8. doi: 10.1016/j.immuni.2020.04.014
201. Balmer ML, Ma EH, Bantug GR, Grahlert J, Pfister S, Glatter T, et al. Memory Cd8(+) T cells require increased concentrations of acetate induced by stress for optimal function. Immunity (2016) 44(6):1312–24. doi: 10.1016/j.immuni.2016.03.016
202. Ferro S, Azevedo-Silva J, Casal M, Corte-Real M, Baltazar F, Preto A. Characterization of acetate transport in colorectal cancer cells and potential therapeutic implications. Oncotarget (2016) 7(43):70639–53. doi: 10.18632/oncotarget.12156
203. Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, et al. Acetate dependence of tumors. Cell (2014) 159(7):1591–602. doi: 10.1016/j.cell.2014.11.020
204. Lyssiotis CA, Cantley LC. Acetate fuels the cancer engine. Cell (2014) 159(7):1492–4. doi: 10.1016/j.cell.2014.12.009
205. Qiu J, Villa M, Sanin DE, Buck MD, O'Sullivan D, Ching R, et al. Acetate promotes T cell effector function during glucose restriction. Cell Rep (2019) 27(7):2063–74 e5. doi: 10.1016/j.celrep.2019.04.022
206. Wenes M, Jaccard A, Wyss T, Maldonado-Perez N, Teoh ST, Lepez A, et al. The mitochondrial pyruvate carrier regulates memory T cell differentiation and antitumor function. Cell Metab (2022) 34(5):731–46 e9. doi: 10.1016/j.cmet.2022.03.013
207. Sterner RC, Sterner RM. Car-T cell therapy: current limitations and potential strategies. Blood Cancer J (2021) 11(4):69. doi: 10.1038/s41408-021-00459-7
208. Poorebrahim M, Melief J, Pico de Coana Y, LW S, Cid-Arregui A, Kiessling R. Counteracting car T cell dysfunction. Oncogene (2021) 40(2):421–35. doi: 10.1038/s41388-020-01501-x
209. Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Velica P, et al. S-2-Hydroxyglutarate regulates Cd8(+) T-lymphocyte fate. Nature (2016) 540(7632):236–41. doi: 10.1038/nature20165
210. Notarangelo G, Spinelli JB, Perez EM, Baker GJ, Kurmi K, Elia I, et al. Oncometabolite d-2hg alters T cell metabolism to impair Cd8(+) T cell function. Science (2022) 377(6614):1519–29. doi: 10.1126/science.abj5104
211. Bian Y, Li W, Kremer DM, Sajjakulnukit P, Li S, Crespo J, et al. Cancer Slc43a2 alters T cell methionine metabolism and histone methylation. Nature (2020) 585(7824):277–82. doi: 10.1038/s41586-020-2682-1
212. Matias MI, Yong CS, Foroushani A, Goldsmith C, Mongellaz C, Sezgin E, et al. Regulatory T cell differentiation is controlled by alphakg-induced alterations in mitochondrial metabolism and lipid homeostasis. Cell Rep (2021) 37(5):109911. doi: 10.1016/j.celrep.2021.109911
213. Feng Q, Liu Z, Yu X, Huang T, Chen J, Wang J, et al. Lactate increases stemness of Cd8 + T cells to augment anti-tumor immunity. Nat Commun (2022) 13(1):4981. doi: 10.1038/s41467-022-32521-8
214. Kim EH, Sullivan JA, Plisch EH, Tejera MM, Jatzek A, Choi KY, et al. Signal integration by akt regulates Cd8 T cell effector and memory differentiation. J Immunol (2012) 188(9):4305–14. doi: 10.4049/jimmunol.1103568
215. Waickman AT, Powell JD. Mtor, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev (2012) 249(1):43–58. doi: 10.1111/j.1600-065X.2012.01152.x
216. Wang Y, Wang XY, Subjeck JR, Shrikant PA, Kim HL. Temsirolimus, an mtor inhibitor, enhances anti-tumour effects of heat shock protein cancer vaccines. Br J Cancer (2011) 104(4):643–52. doi: 10.1038/bjc.2011.15
217. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of Cd8+ T cell memory development. Immunity (2012) 36(1):68–78. doi: 10.1016/j.immuni.2011.12.007
218. Chen C, Wang Z, Ding Y, Qin Y. Manipulating T-cell metabolism to enhance immunotherapy in solid tumor. Front Immunol (2022) 13:1090429. doi: 10.3389/fimmu.2022.1090429
219. Ghassemi S, Martinez-Becerra FJ, Master AM, Richman SA, Heo D, Leferovich J, et al. Enhancing chimeric antigen receptor T cell anti-tumor function through advanced media design. Mol Ther Methods Clin Dev (2020) 18:595–606. doi: 10.1016/j.omtm.2020.07.008
220. Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in car T cells. Immunity (2016) 44(2):380–90. doi: 10.1016/j.immuni.2016.01.021
221. Ye L, Park JJ, Peng L, Yang Q, Chow RD, Dong MB, et al. A genome-scale gain-of-Function crispr screen in Cd8 T cells identifies proline metabolism as a means to enhance car-T therapy. Cell Metab (2022) 34(4):595–614 e14. doi: 10.1016/j.cmet.2022.02.009
222. Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (Glut1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem (2014) 289(11):7884–96. doi: 10.1074/jbc.M113.522037
223. Lim S, Liu H, Madeira da Silva L, Arora R, Liu Z, Phillips JB, et al. Immunoregulatory protein B7-H3 reprograms glucose metabolism in cancer cells by ros-mediated stabilization of Hif1alpha. Cancer Res (2016) 76(8):2231–42. doi: 10.1158/0008-5472.CAN-15-1538
224. Brunner-Weinzierl MC, Rudd CE. Ctla-4 and pd-1 control of T-cell motility and migration: implications for tumor immunotherapy. Front Immunol (2018) 9:2737. doi: 10.3389/fimmu.2018.02737
225. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer (2021) 20(1):131. doi: 10.1186/s12943-021-01428-1
226. Zhang Y, Ertl HC. Depletion of fap+ cells reduces immunosuppressive cells and improves metabolism and functions Cd8+T cells within tumors. Oncotarget (2016) 7(17):23282–99. doi: 10.18632/oncotarget.7818
227. Comito G, Iscaro A, Bacci M, Morandi A, Ippolito L, Parri M, et al. Lactate modulates Cd4(+) T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression Via Tlr8/Mir21 axis. Oncogene (2019) 38(19):3681–95. doi: 10.1038/s41388-019-0688-7
228. Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature (2016) 536(7617):479–83. doi: 10.1038/nature19084
229. Haslam A, Prasad V. Estimation of the percentage of us patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw Open (2019) 2(5):e192535. doi: 10.1001/jamanetworkopen.2019.2535
230. Hurley HJ, Dewald H, Rothkopf ZS, Singh S, Jenkins F, Deb P, et al. Frontline science: ampk regulates metabolic reprogramming necessary for interferon production in human plasmacytoid dendritic cells. J Leukoc Biol (2021) 109(2):299–308. doi: 10.1002/JLB.3HI0220-130
231. Claiborne MD. Manipulation of metabolic pathways to promote stem-like and memory T cell phenotypes for immunotherapy. Front Immunol (2022) 13:1061411. doi: 10.3389/fimmu.2022.1061411
232. Bental M, Deutsch C. Metabolic changes in activated T cells: an nmr study of human peripheral blood lymphocytes. Magn Reson Med (1993) 29(3):317–26. doi: 10.1002/mrm.1910290307
233. Chi H, Zhao S, Yang J, Gao X, Peng G, Zhang J, et al. T-Cell exhaustion signatures characterize the immune landscape and predict hcc prognosis Via integrating single-cell rna-seq and bulk rna-sequencing. Front Immunol (2023) 14:1137025. doi: 10.3389/fimmu.2023.1137025
234. Madden MZ, Rathmell JC. The complex integration of T-cell metabolism and immunotherapy. Cancer Discovery (2021) 11(7):1636–43. doi: 10.1158/2159-8290.CD-20-0569
235. Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev (2008) 222:180–91. doi: 10.1111/j.1600-065X.2008.00608.x
236. Sanchez-Alvarez R, Martinez-Outschoorn UE, Lamb R, Hulit J, Howell A, Gandara R, et al. Mitochondrial dysfunction in breast cancer cells prevents tumor growth: understanding chemoprevention with metformin. Cell Cycle (2013) 12(1):172–82. doi: 10.4161/cc.23058
237. Zacksenhaus E, Shrestha M, Liu JC, Vorobieva I, Chung PED, Ju Y, et al. Mitochondrial oxphos induced by Rb1 deficiency in breast cancer: implications for anabolic metabolism, stemness, and metastasis. Trends Cancer (2017) 3(11):768–79. doi: 10.1016/j.trecan.2017.09.002
238. Saraei P, Asadi I, Kakar MA, Moradi-Kor N. The beneficial effects of metformin on cancer prevention and therapy: a comprehensive review of recent advances. Cancer Manag Res (2019) 11:3295–313. doi: 10.2147/CMAR.S200059
239. Curry J, Alnemri A, Philips R, Fiorella M, Sussman S, Stapp R, et al. Cd8+ and Foxp3+ T-cell cellular density and spatial distribution after programmed death-ligand 1 check point inhibition. Laryngoscope (2022) 1–10. doi: 10.1002/lary.30389
240. Afzal MZ, Dragnev K, Sarwar T, Shirai K. Clinical outcomes in non-Small-Cell lung cancer patients receiving concurrent metformin and immune checkpoint inhibitors. Lung Cancer Manag (2019) 8(2):LMT11. doi: 10.2217/lmt-2018-0016
241. Vigano S, Alatzoglou D, Irving M, Menetrier-Caux C, Caux C, Romero P, et al. Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front Immunol (2019) 10:925. doi: 10.3389/fimmu.2019.00925
242. Lim EA, Bendell JC, Falchook GS, Bauer TM, Drake CG, Choe JH, et al. Phase Ia/B, open-label, multicenter study of Azd4635 (an adenosine A2a receptor antagonist) as monotherapy or combined with durvalumab, in patients with solid tumors. Clin Cancer Res (2022) 28(22):4871–84. doi: 10.1158/1078-0432.CCR-22-0612
243. Pawlak M, Lefebvre P, Staels B. Molecular mechanism of pparalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol (2015) 62(3):720–33. doi: 10.1016/j.jhep.2014.10.039
244. Aloia A, Mullhaupt D, Chabbert CD, Eberhart T, Fluckiger-Mangual S, Vukolic A, et al. A fatty acid oxidation-dependent metabolic shift regulates the adaptation of braf-mutated melanoma to mapk inhibitors. Clin Cancer Res (2019) 25(22):6852–67. doi: 10.1158/1078-0432.CCR-19-0253
Keywords: metabolism, T cell, cancer, immunotherapy, tumor immune microenvironment
Citation: Ganjoo S, Gupta P, Corbali HI, Nanez S, Riad TS, Duong LK, Barsoumian HB, Masrorpour F, Jiang H, Welsh JW and Cortez MA (2023) The role of tumor metabolism in modulating T-Cell activity and in optimizing immunotherapy. Front. Immunol. 14:1172931. doi: 10.3389/fimmu.2023.1172931
Received: 24 February 2023; Accepted: 13 April 2023;
Published: 26 April 2023.
Edited by:
Xiaosong Li, Chongqing Medical University, ChinaReviewed by:
Leonard Maggi, Washington University in St. Louis, United StatesCopyright © 2023 Ganjoo, Gupta, Corbali, Nanez, Riad, Duong, Barsoumian, Masrorpour, Jiang, Welsh and Cortez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Angelica Cortez, bWFjb3J0ZXpAbWRhbmRlcnNvbi5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.