 Jens Wittner
Jens Wittner Wolfgang Schuh
Wolfgang Schuh- Division of Molecular Immunology, Department of Internal Medicine 3, Nikolaus-Fiebiger-Center, University Hospital Erlangen, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany
The development of B cells, their activation and terminal differentiation into antibody-producing plasma cells are characterized by alternating phases of proliferation and quiescence that are controlled by complex transcriptional networks. The spatial and anatomical organization of B cells and plasma cells inside lymphoid organs as well as their migration within lymphoid structures and between organs are prerequisites for the generation and the maintenance of humoral immune responses. Transcription factors of the Krüppel-like family are critical regulators of immune cell differentiation, activation, and migration. Here, we discuss the functional relevance of Krüppel-like factor 2 (KLF2) for B cell development, B cell activation, plasma cell formation and maintenance. We elaborate on KLF2-mediated regulation of B cell and plasmablast migration in the context of immune responses. Moreover, we describe the importance of KLF2 for the onset and the progression of B cell-related diseases and malignancies.
Introduction
Krüppel-like factor 2 (KLF2) is a transcription factor of the Krüppel-like factor (KLF) family whose members are characterized by a C-terminal zinc finger DNA-binding domain. The family name originated from the phenotype of a Drosophila loss-of-function mutant with abnormal segmentation of the abdominal region of the Drosophila larva (“Krüppel” mutant, Krüppel: German word for cripple). In Drosophila, the krüppel gene is one of the so-called gap genes, a group of genes responsible for the development of the Drosophila larvae and their segmentation (1, 2). The KLF family consists of 17 members in vertebrates, all of which are involved in the control of differentiation, proliferation, cell adhesion, and migration processes in a variety of cell types (3, 4). KLF2 was first described by Anderson and colleagues in 1995 and originally named lung Krüppel-like factor (LKLF) due to its high expression in the lung (5). The importance of KLF2 during embryonic development was revealed in 1997 by Kuo and colleagues using a genomic knockout mouse model for the Klf2 gene. Their study demonstrated that KLF2-deficient embryos died between days E12.5 and E14.5 due to hemorrhage, defective blood vessels, and an abnormal tunica media in utero (6). Thus, KLF2 has an essential function in embryonic development and in endothelial cell biology. From the time point of its discovery in the late 1990s, numerous studies have revealed a crucial role for KLF2 during proliferation, differentiation, activation, and positioning of B and T cells, and other immune cells (4, 7). The loss of function of KLF2 is associated with diseases, such as arteriosclerosis, adipogenesis, thrombosis, and lymphoma (3, 4, 7–12). The role of KLF2 has been intensively studied in T-lymphoid cells and it becomes increasingly evident that KLF2 also acts as an important regulator of different aspects of B cell biology. Therefore, in this review article, we discuss the relevance of KLF2 during B cell differentiation and activation as well as its function of KLF2 as a regulator of B cell and plasma cell homing. Finally, we elaborate on how KLF2 contributes to B cell-related diseases and malignancies.
Expression of KLF2 in B-lymphoid cells
Expression of KLF2 in early B cell progenitors in the bone marrow (BM) was discovered in a mouse model with tetracycline-controllable expression of the pre-B cell receptor (pre-BCR) (13). The pre-BCR is part of a critical checkpoint in early B cell development, which tests the ability of newly formed immunoglobulin (Ig) µ-heavy chains (µHC) to functionally pair with the surrogate light chain components VpreB and λ5. Pre-BCR-mediated signals result in clonal expansion of pre-B cells, suppression of apoptosis, targeting of the VDJ-recombination machinery to the Ig light chain (IgL) loci, and allelic exclusion (14, 15). Analyses of changes in the transcriptome upon tetracycline-controlled pre-BCR induction, uncovered KLF2 as a pre-BCR-induced gene (13). KLF2 expression in pre-B cells was confirmed in KLF2:GFP reporter mice (16). Pre-BCR signals result in Erk5 phosphorylation, which in turn activates the transcription factors Mef2c and Mef2d by phosphorylation. Phosphorylated Mef2c and Mef2d, in turn, activate transcription of the Klf2 gene and, in parallel, of immediate-early genes, encoding for the transcription factors Jun and Fos, as well as the early growth response proteins Egr1 and 2 that induce pre-B cell expansion (17). In addition, Mef2c/d transcription factors induce IRF-4, a transcription factor important for the termination of pre-B cell expansion and the initiation of immature B cell differentiation (18). Over time, KLF2 accumulates in proliferating pre-B cells and inhibits the Mef2c/d-mediated transcription of the immediate-early genes Jun and Fos and Egr1/2, thus, contributing to the termination of pre-B cell expansion (17). Along this line, ectopic expression of KLF2 resulted in a block of pre-B cell proliferation concurrent with decreased c-myc and increased p21 and p27 mRNA abundances (19) (Figure 1). However, KLF2-deficient mice displayed normal pre-B and immature B cell compartments (16, 20), suggesting that in the absence of KLF2, termination of pre-B cell expansion still occurs and is presumably mediated through Irf-4 upregulation. As aforementioned, activation of Mef2c/2d by pre-BCR signals results in the upregulation of Irf-4 expression. Subsequently, IRF-4/IRF-8-mediated upregulation of the transcription factors Aiolos and Ikaros was shown to downregulate pre-BCR expression and to impair cell cycle progression and thereby pre-B cell expansion (21).
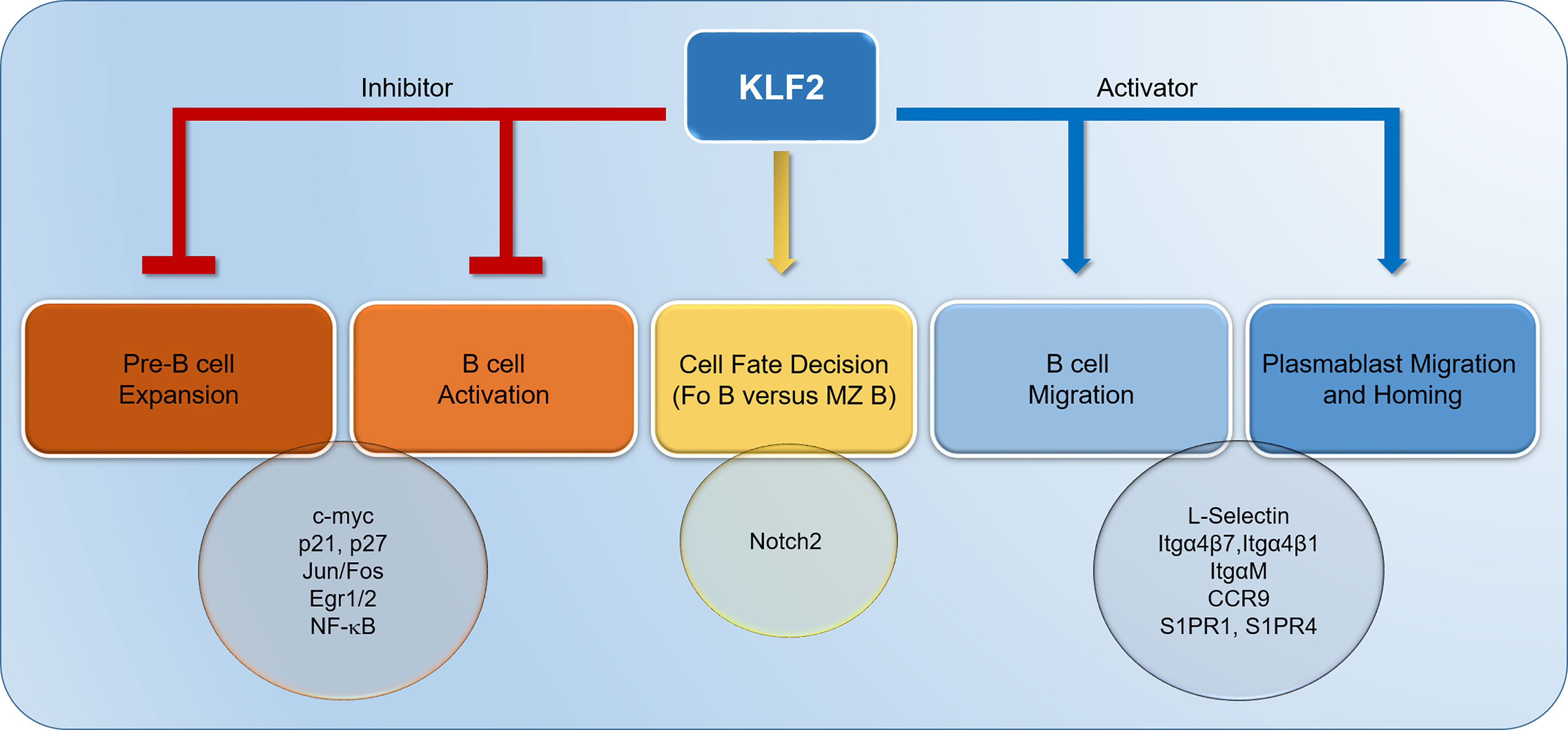
Figure 1 Krüppel-like factor-2 (KLF2) contributes to the termination of pre-B cell expansion through inhibition of Jun/Fos and Egr1/2. Moreover, KLF2 inhibits the proliferation of pre-B cells and the activation of naïve, mature B cells by downregulating c-myc and upregulating p21 and p27. In B cells, KLF2 suppresses NF-κB activation. Furthermore, KLF2 represses Notch2 signaling in naïve B cells, thereby driving B cell differentiation to follicular B cells. KLF2 controls the migration of B cells and plasmablasts by positively regulating L-Selectin, Itgα4β7, Itgα4β1, ItgαM, Chemokine receptor 9 (CCR9), Sphingosine-1-Phosphate-Receptor (S1PR) 1 and S1PR4. IgA plasmablast homing to gut-associated lymphoid tissues (GALT) is mediated by KLF2-regulated factors Itgα4β7 and CCR9. Itg, integrin.
As aforementioned, KLF2 expression is induced by the pre-BCR in early B cell development and is maintained in immature B and follicular (Fo) B cells (13, 16, 20, 22). Marginal zone (MZ) B cells show low abundances of KLF2 mRNA and protein whereas B1 cells in the peritoneum display the highest abundance of KLF2 (16, 20, 22). Activation of splenic B cells in vitro with LPS, anti-CD40/IL-4 or anti-IgM (anti-BCR) led to decreased KLF2 mRNA and protein abundances (16, 20, 22, 23). In this context, ectopic expression of KLF2 in LPS-activated, proliferating B cells led to an inhibition of B cell activation, expansion, and plasmablast differentiation (19). Therefore, KLF2 acts as a quiescence factor that keeps mature B cells in a resting state. The function of KLF2 as an important quiescence regulator was already postulated in 2000, when KLF2 was found in comparative transcriptome analyses to be highly abundant in resting, naïve B, and anergic B cells, but downregulated in activated B cells (24). In mature B cells, Klf2 gene expression might be driven by the transcription factor Foxo1 (similar to the Foxo-1-mediated regulation of the Klf4 gene) as Foxo1-binding sites were found in the Klf2 promoter and Foxo1-binding to the Klf2 promoter was described (25). In support, Klf2 mRNA was reduced in Foxo1-deficient B cells (26). B cell activation results in PI3K-Akt-mediated phosphorylation of Foxo1. Phosphorylated Foxo1 is transported out of the nucleus and becomes transcriptionally inactive (27). Consequently, Klf2 expression is terminated, which in turn, might enable B cell proliferation and differentiation.
B cells in secondary lymphoid organs can be activated by antigen in either a T cell-dependent (TD) or a T cell-independent (TI) manner. TD activation leads to the formation of a germinal center (GC) reaction in which the BCR of the activated B cell undergoes affinity maturation and Ig class switch recombination occurs. As a result of the GC reaction, B cells with a high affinity BCR either differentiate to memory B cells (Bmem) or to plasma cells (28–31). One study unraveled increased Klf2 RNA abundances in CD80+/PD-L2+ Bmem that were shown to quickly differentiate into antibody-secreting cells but did not form new germinal centers (32). Furthermore, single-cell RNAseq of isotype-switched Bmem uncovered a cluster of Klf2-expressing Bmem. The cells in this cluster were characterized by low abundances of Cr2 (CD21), intermediate abundances of Fcer2a (CD23), and expressed Klf2, Vimentin-1 and Prostate androgen-regulated mucin-like protein 1 (Parm-1). Based on these characteristics, the authors of this study defined cluster I cells as transitional Bmem (33). Although KLF2 has been detected in Bmem subsets, its functional relevance for Bmem is so far mostly unknown. We speculate that KLF2 in Bmem might functionally contribute to their tissue distribution and retention. In this context, KLF2 expression in Bmem correlated with expression of factors critical for homing and migration, such as Integrin (Itg)β7, Sphingosine-1-phosphate-receptor 1 (S1PR1) and C-C chemokine receptor CCR6 expression (34). Additionally, it is tempting to speculate that KLF2 might keep Bmem in the resting state until they encounter their specific antigen.
GC B cells that differentiate to plasma cells undergo a dramatic morphological change characterized by an increase in cell size and an enlargement of the endoplasmatic reticulum (ER) (31). This process is controlled by a complex regulatory network of transcription factors. Blimp-1 (encoded by the Prdm-1 gene) is the key transcription factor that drives plasma cell differentiation by promoting Ig production and secretion, and by repressing B ell activation-signature transcription factors Pax5, Bcl-6, Bach2 and the enzyme Activation-induced cytidine deaminase (AID, encoded by the Aicda gene) (31). Activated B cells first differentiate into proliferating plasmablasts that are migratory and then into mature, resting plasma cells (35). In plasmablasts in the blood, expression of KLF2 and its target gene S1pr1 was detected (36). Migration along the sphingosine-1-phosphate (S1P) gradient guides plasmablasts from lymph nodes and spleen to lymph and blood (36). Analysis of KLF2:GFP reporter mice revealed KLF2 expression in IgM and IgA plasmablasts in the blood. In lymphoid organs, the highest frequency of KLF2-positive cells was found within the IgA plasmablast population in mesenteric lymph nodes (mLN), suggesting a pivotal role of KLF2 for IgA plasmablasts and IgA plasma cells (37).
Functional role of KLF2 in peripheral B cell subsets
The regulatory role of KLF2 in B cell proliferation and activation was primarily analyzed in vitro by overexpression approaches and by studying loss-of-function mutants of KLF2 and their ability to activate NF-κB signaling. Regarding the regulation of quiescence, ectopic expression of KLF2 in pre-B cell cultures and in LPS-activated B cells led to the downregulation of c-myc and upregulation of the cell cycle inhibitors p27 and p21 (19). Moreover, as shown in monocytes, KLF2 interferes with NF-κB activation (4, 38), a mechanism that might also apply for B cells and B lymphoma cells. Accordingly, KLF2 loss-of-function mutations as found in human lymphoma cells impaired KLF2-mediated NF-κB suppression in a B lymphoma cell line (11), a topic that will be discussed later in the review article.
To study the functional relevance of KLF2 during B cell development and activation in vivo, mouse models with a conditional B cell-specific deletion of a floxed KLF2 gene were generated. To achieve B cell-specific deletion, either mb1cre or CD19cre deleter mouse strains were used (16, 20, 22). The B cell-specific deletion of KLF2 resulted in enlarged spleens with an expansion of Fo B cells and MZ B cells (16, 20, 22). KLF2-deficient Fo B cells showed enhanced CD21 surface expression and altered BCR-mediated calcium signals, and thus, as concluded from these parameters and changes in the global gene expression profile partially resembled MZ B cells (16, 20, 22). Fo B and MZ B cells are functionally distinct B cell subsets. Fo B cells migrate between lymphoid organs and give rise to GC upon activation. MZ B cells are a specialized B cell subset located in the splenic marginal zone and their mobility, in contrast to Fo B cells, is limited to shuttling between the marginal zone and the B cell follicle to facilitate antigen transport (39). MZ B cells can develop either from transitional B cells or from follicular B cells (40). Their differentiation is driven by Notch2 signaling. Deletion of Notch2 or its ligand Dll-1 resulted in a loss of MZ B cells (41, 42). In an elegant study, induction of Notch2IC (intracellular domain of Notch2 that interacts with DNA-binding protein RBPJ and regulates transcription) resulted in the conversion of Fo B cells to MZ B cells. Upon induction of Notch2IC signaling, Klf2 (besides Irf-8 and Foxo1) was downregulated (43). These findings are supported by the expansion of MZ B cells observed in KLF2-deficient mice and suggest a role of KLF2 in the cell fate decision and the imprinting of the cellular identity of Fo B versus MZ B cells (Figure 1). As described later, loss-of-function mutations of human KLF2 are frequently found in splenic marginal cell lymphoma (SMZL) and play a role in disease onset and/or progression. Immunization experiments showed an increased immune response to TI antigen type 2 (TNP-Ficoll) antigens in B cell-specific KLF2-deficient animals compared to controls, which might be due to the observed expansion of MZ B cells and the altered phenotype of KLF2-deficient Fo B cells (22). Immunization with the TD antigen TNP-KLH, however, resulted in reduced antigen-specific IgG titers upon boost immunization. Antigen-specific IgG plasma cells as determined by ELISpot analyses were unaffected in the spleen but were virtually absent in the BM, indicating that loss of KLF2 affects plasmablast homing and/or plasma cell survival in the BM (20).
Importantly, KLF2 deletion profoundly affected mucosal immune responses. KLF2-deficiency resulted in reduction and phenotypic alterations of peritoneal B1 cells (16, 20, 44). Mice with a B cell-specific KLF2 deletion develop fewer and smaller Peyer’s patches (PP) and natural IgA in the serum was reduced (16, 20, 22). Furthermore, B cell-specific deletion of KLF2 resulted in drastically reduced secretory IgA (SIgA) in the gut lumen concomitant with reduced IgA plasma cells in the intestinal lamina propria (LP). IgA plasmablasts and plasma cells, however, accumulated in the mLN and PP, although PP were smaller in size and numbers. Immune responses to immunization with soluble recombinant Flagellin, an immunodominant protein of Salmonella typhimurium, were blunted. In summary, B cell-specific deletion of KLF2 in B cells in mice led to a phenotype similar to that observed in human IgA deficiencies (37).
KLF2-regulated genes in B cells and plasma cells
KLF2 acts a major regulator of thymic exit and T cell migration by regulating S1PR1 (45–47). In peripheral murine B cell subsets, one study also described direct binding of KLF2 to the edg1 promoter (the edg1 gene encodes for S1PR1) in murine MZ B cells by chromatin immunoprecipitation (ChIP) (22). In this study, S1pr1 mRNA was shown to be downregulated in KLF2-deficient MZ B cells and upregulated in Fo-deficient B cells despite the lack of KLF2 binding to the edg1 promoter in Fo B cells (22). Two other independent studies demonstrated that S1PR1 mRNA and protein were not significantly altered in KLF2-deficient Fo B cells (16, 20). Therefore, the involvement of KLF2 in the regulation of S1pr1 expression in MZ B cells and Fo B cells remains unresolved. In IgA plasmablasts, however, RNASeq data confirmed the KLF2-dependent regulation of S1pr1 and S1pr4 mRNAs, which were both significantly reduced in KLF2-deficient IgA plasmablasts in the mLN (37). Therefore, KLF2-mediated regulation of S1PRs might contribute to plasmablast migration and homing to the bone marrow as well as mucosal effector sides (Figures 1, 2).
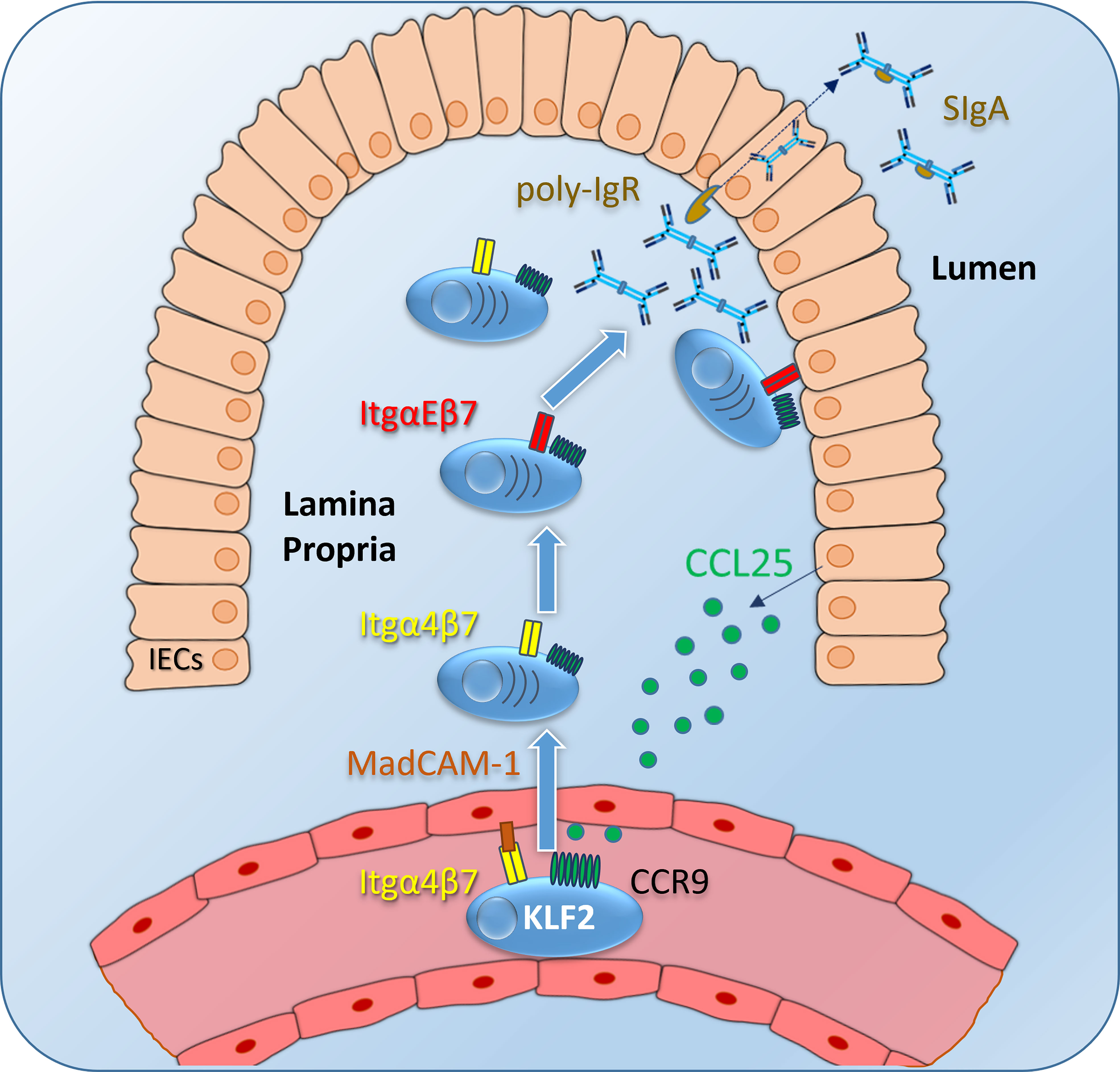
Figure 2 IgA plasmablast homing to the intestinal lamina propria (LP): Intestinal epithelial cells (IECs) express CCL25, which is presented on glucosamine-glycans on endothelial cells of venules as a ligand for the CCR9 receptor on IgA-expressing plasmablasts (and other immune cells). Integrin α4β7 is activated upon CCR9 signaling and binds to its ligand MadCAM-1, followed by plasmablast migration to the intestinal LP. Itgα4β7 and CCR9 expression is induced by KLF2 in IgA plasmablasts. Inside the LP, IgA plasmablasts differentiate into IgA-secreting plasma cells, a subset of those express ItgαEβ7 to localize close to the IECs. This mechanism might facilitate the binding of dimeric IgA to the poly-Ig receptor and might subsequently promote the transcytosis of dimeric IgA through the epithelial layer to the gut lumen. Itg, integrin; SIgA, secretory IgA.
The chemokine receptor CXCR5 recognizes the chemokine CXCL13 and is important for the positioning of B and T cells inside the B cell follicles in lymph nodes and the spleen (48) and for the shuttling of MZ B cells between the follicle and the marginal zone of the spleen (39). In T follicular helper (TFH) cells, KLF2 binds directly to the Cxcr5 promoter (as shown by ChIP) and represses Cxcr5 expression. Downregulation of KLF2 caused by ICOS signals via Foxo1 resulted in Cxcr5 upregulation that is critical for TFH-positioning in the B cell follicle (49). In contrast to the well described regulation in TFH cells, KLF2-mediated regulation of Cxcr5 in B cells remains controversial: one study described downregulation of CXCR5 mRNA and protein in KLF2-deficient MZ B cells and an upregulation in KLF2-deficient Fo B cells (22). However, two other studies were not able to confirm this regulation (16, 20). Therefore, it remains unclear whether KLF2 might be involved in the regulation of MZ B cell-shuttling between the marginal zone and the follicle, or in Fo B cell-positioning within the follicle as shown for TFH cells. Hence, resolving the role of KLF2 in MZ B-shuttling and Fo B cell migration within the follicle will require more sophisticated spatial and temporal analyses.
Genome-wide microarray RNA expression analyses in Fo B cells in two different mouse strains with a B cell-specific Klf2 deletion (either CD19Cre- or mb1Cre-mediated) identified the surface receptors L-Selectin (CD62L) and Integrin (Itg) β7, which are important for migration and homing, as KLF2-regulated factors (16, 20) (Figure 1). While L-Selectin as a major factor of leucocyte extravasation, plays an important role in B cell migration from blood to lymph nodes, Itgβ7 is known for its specific role in mucosal lymphocyte migration. It was demonstrated by chromatin immunoprecipitation (ChIP) that KLF2 directly binds to the Itgβ7 promoter in B cell lines (50). On protein level, loss of surface L-Selectin and surface Itgα4β7 was demonstrated in KLF2-deficient splenic Fo B cells and B cells in the blood (16, 20). Moreover, in KLF2-deficient TACI+/CD138+ IgA plasmablasts, Itgα4β7 was downregulated (37). As the Itgα4 chain was virtually absent on the surface of KLF2-deficient IgA plasmablasts, not only Itgα4β7 but also surface expression of Itgα4β1, which is critical for BM homing, is impaired (37). Besides downregulation of L-Selectin and Itgβ7, a significant reduction of S1pr4 and an increase of S1pr3 transcripts in KLF2-deficient Fo B cells was detected (16). While S1PR3 plays a role for MZ B cell positioning but is dispensable for lymph node motility, the function of S1PR4 in B cells is unclear (39, 51).
KLF2 directly induces Blimp1 during Th1 cell differentiation by binding to the Prdm1 promoter (52) but it remains unclear whether Blimp1 is also controlled by KLF2 during plasma cell differentiation. Based on the findings that KLF2-deficent mice had reduced numbers of antigen-specific IgG-secreting plasma cells in the BM and that natural IgA was reduced in their serum, the effect of KLF2 deletion on plasmablast and plasma cells subsets was thoroughly assessed by our group (20, 37). Plasmablasts were defined as CD19+/B220+/TACI+/CD138+ cells with a high frequency of proliferating Ki67+ cells, whereas plasma cells were identified as CD19lo/neg/B220-/TACI+/CD138+ which are non-proliferating (35, 37). Analysis of plasma cell compartments in B cell-specific KLF2-deficient mice revealed a severe dysregulation of the compartmentalization of IgA plasmablasts and IgA plasma cells. In these mice, IgA plasmablasts and IgA plasma cells were virtually absent in the BM, reduced in the blood, the spleen and importantly, the intestinal LP. However, IgA plasmablasts and IgA plasma cells accumulated in mLN of KLF2-deficient mice (37). RNAseq as well as flow cytometric analyses of KLF2-deficient IgA plasmablasts compared to controls identified L-Selectin, Itgβ7, ItgαM, and chemokine receptor CCR9 as KLF2-regulated factors (37). Surface CCR9 on IgA plasmablasts was significantly reduced concomitant with an impaired migration towards a CCL25 gradient in vitro (37). Together, reductions of Itgβ7 and CCR9 expression in KLF2-deficient IgA plasmablasts led to compromised IgA responses caused by impaired migration from mLN to the LP of the small intestine and colon (37). Hence, KLF2 regulates the expression of the important gut-associated lymphoid tissue (GALT)-homing factors Itgβ7 and CCR9 (Figures 1, 2). Upon KLF2-regulated expression of CCR9 and Itgβ7, IgA plasmablasts are attracted to the LP by gradients of CCL25, the ligand of CCR9. CCL25 is secreted by e.g., intestinal epithelial cells (IEC) (53). CCL25-binding to CCR9 activates Itgα4β7 that binds to MadCAM-1 on endothelial cells and leads to the extravasation of plasmablasts to the mucosal LP (54, 55). Inside the intestinal LP, IgA plasmablasts differentiate to mature IgA plasma cells. A subset of those express ItgαEβ7 which enables them to bind to E-Cadherin on IECs, a mechanism that might promote dimeric IgA binding to the poly-IgR and facilitate transcytosis of dimeric IgA to the gut lumen [(56), Figure 2].
In addition to the regulation of Itgα4β7, the expression of the ItgαM chain was also affected in KLF2-deficient IgA plasmablasts. ItgαM is a binding partner of Itgβ2, which is important for lymph node egress of B cells (57). Moreover, ItgαM was absent on KLF2-deficient IgA plasmablasts compared to their wildtype counterparts (37). The dysregulation of ItgαM together with the aforementioned reduction of S1PR1 might be the cause for the observed accumulation of IgA plasmablasts/plasma cells in the mLN and in the remaining PP of KLF2-deficient mice (37). Hence, KLF2 might be involved in the process of lymph node exit of IgA plasmablasts presumably by regulating ItgαMβ2 and S1PR1.
In summary, KLF2 contributes to the control of the quiescent, resting state of mature B cells and pre-B cells by controlling cell cycle regulators (c-myc, p21, and p27) and immediate-early transcription factors (such as Jun, Fos, and Egr1/2), respectively. Moreover, KLF2-regulated genes are crucial for migration and homing of naïve B cells, activated B cells, and plasmablasts. KLF2-regulated gene products include integrins (Itgα4β7, Itgα4β1, and ItgαM), selectins (L-Selectin), and chemokine receptors (CCR9) as well as Sphingosin-1-phosphat-receptors (S1PR1, S1PR3, and S1PR4) in IgA plasmablasts. By regulating the expression of these factors, KLF2 controls the exit of IgA plasmablasts from the lymph node as well as their homing to the intestinal LP.
KLF2 in B cell-related diseases and malignancies
Splenic marginal zone lymphoma
In humans, splenic marginal zone lymphoma (SMZL) is a low-grade B cell lymphoma, with variable clinical course. Clinical diagnosis is rather difficult as specific phenotypic and genetic markers are lacking. In approximately one third of SMZL cases, the IgHV1-2 heavy chain that harbors few somatic mutations and a long CDR3 region is expressed (58, 59) and approximately one third of SMZL cases harbor a hemizygous deletion of chromosome 7q with a so far unsolved role in the pathogenesis of SMZL (60–62). Transcriptome and mutational analyses have revealed candidate genes that may contribute to disease onset and/or progression. Mutations were predominantly detected in the KLF2 and the NOTCH2 genes. KLF2 was inactivated by mutations in 42% of SMZL patients/cases (11). This is in line with findings that KLF2-deficient mice display a strong expansion of MZ B cells (16, 20, 22). Based on the mutations found in SMZL patients, expression constructs with genes encoding for different KLF2 mutant forms were generated. The effect of these KLF2 mutants on NF-κB activation was assessed in in vitro reporter assays in HEK293T cells and OCI-LY19 B-lymphoma cells. KLF2 mutants failed to suppress NF-κB activation in contrast to non-mutated KLF2 (11). Constitutive activation of the NF-κB signaling pathway contributes to SMZL pathogenesis by promoting MZ B cell survival and expansion (63, 64).
Multiple myeloma
The hallmark of Multiple Myeloma (MM), a malignant disease, is the expansion of plasma cells. Clinical signs include hypercalcemia, renal failure, anemia, and bone lesions. Moreover, MM is characterized by plasma cell expansion in the BM and the presence of free IgL chains, the so-called Bence Jones proteins that can be found in the serum and the urine of MM patients (65). Genetic predispositions such as mutations in the N-RAS, K-RAS or EGR1 genes as well as translocations are primary events in the onset of MM (65, 66). Deregulation of histone methylation can also contribute to MM. In this context, the chromosomal translocation t (4,14) (p16;q32) can be found in up to 20% of MM patients. This translocation results in the overexpression of WHSC1, a histone H3 lysine 36 (H3K36) methyltransferase (67). Furthermore, the KDM3a histone demethylase that catalyzes the removal of H3K9 mono- and di-methylations, is expressed in MM lines and was shown to be essential for MM cell proliferation and survival. KLF2 was identified as a target gene of KDM3a. KLF2 is highly expressed in MM cell lines (68). Downregulation of KLF2 resulted in an impairment of MM cell proliferation and in the induction of apoptosis. IRF-4 was identified a KLF2-regulated gene in MM cell lines. Together, KDM3a, KLF2, and IRF-4 regulate the expression of ITGβ7, an essential integrin for MM homing to and adhesion in the BM (69). As aforementioned, ITGβ7 is a crucially important KLF2-regulated target gene in healthy B cells and plasma cells. Therefore, KLF2 is involved in MM cell adhesion and BM homing. Moreover, KLF2 is involved in the regulation of the angiogenic factors EGFL7 and ITGβ3 in MM cells. KLF2 expression was increased by ITGβ3 signaling which in turn led to upregulation of EGFL7, thereby enhancing MM cell expansion (70). In contrast to naïve B cells, MM cells proliferate in the presence of KLF2. As aforementioned, KLF2 in MM cells promotes their proliferation and survival. Therefore, the complex interplay of the various signaling pathways implicated in the pathogenesis of MM (i.e., the RAS/RAF/MEK/ERK, the PI3K/AKT, the JAK/STAT, and the NF-κB pathways (71) with the KLF2 signaling network in MM cells needs to be further investigated.
IgA deficiencies
As aforementioned, B cell-specific deletion of Klf2 in the mouse resulted in a profound disturbance of the localization of IgA plasma cells concurrent with the absence of SIgA in the gut lumen and feces (37). These phenotypes are strikingly similar to those found in human IgA deficiencies (72). Loss of Itgβ7, a central player of IgA plasmablast/plasma cell homeostasis, is implicated in the human Kabuki syndrome. In a corresponding mouse model, deletion of the gene encoding for the Kmt2d histone methyltransferase led to a decrease of Itgβ7 expression, which consequently resulted in a defective homing of IgA plasmablasts to the gut (73). As Itgβ7 is also a direct target gene of KLF2, it will be of great interest to study the effect of KLF2 loss-of-function mutations on the onset and progression of gut-related diseases, such as Ulcerative colitis and Crohn’s disease.
B cell abnormalities
Recently, a novel mutation in the human KLF2 gene was discovered that leads to the disruption of the highly conserved zinc finger domain required for the nuclear transport and DNA-binding. The patients showed lymphopenia with decreased B cell numbers, lower numbers of switched memory B cells, and reduced serum IgG1. Moreover, L-Selectin on blood B cells was downregulated. In addition, this mutation also resulted in an imbalance of various T cell subsets (74).
Future perspectives
KLF2 is a central regulator of not only B cell and plasma cell differentiation, activation, and migration, but is equivalently important in other immune cells. KLF2 alterations have been associated with a multitude of diseases, such as adipogenesis, atherosclerosis, thrombosis, asthma, arthritis (3, 4, 7–9, 12). Thus, the challenge for further studies will be the identification and characterization of the KLF2-regulated signalosome, transcriptome, and proteome in various cell types in immune responses and diseases.
Author contributions
JW and WS conceptualized and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded in part by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) project grant TRR130 P09 (GEPRIS DFG project number: 215346292) to WS. We acknowledge financial support by Deutsche Forschungsgemeinschaft and Friedrich-Alexander-Universität Erlangen-Nürnberg within the funding programme “Open Access Publication Funding”.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in drosophila. Nature (1980) 287(5785):795–801. doi: 10.1038/287795a0
2. Preiss A, Rosenberg UB, Kienlin A, Seifert E, Jackle H. Molecular genetics of kruppel, a gene required for segmentation of the drosophila embryo. Nature (1985) 313(5997):27–32. doi: 10.1038/313027a0
3. McConnell BB, Yang VW. Mammalian kruppel-like factors in health and diseases. Physiol Rev (2010) 90(4):1337–81. doi: 10.1152/physrev.00058.2009
4. Wittner J, Schuh W. Kruppel-like factor 2 (KLF2) in immune cell migration. Vaccines (Basel). (2021) 9(10). doi: 10.3390/vaccines9101171
5. Anderson KP, Kern CB, Crable SC, Lingrel JB. Isolation of a gene encoding a functional zinc finger protein homologous to erythroid kruppel-like factor: identification of a new multigene family. Mol Cell Biol (1995) 15(11):5957–65. doi: 10.1128/MCB.15.11.5957
6. Kuo CT, Veselits ML, Barton KP, Lu MM, Clendenin C, Leiden JM. The LKLF transcription factor is required for normal tunica media formation and blood vessel stabilization during murine embryogenesis. Genes Dev (1997) 11(22):2996–3006. doi: 10.1101/gad.11.22.2996
7. Hart GT, Hogquist KA, Jameson SC. Kruppel-like factors in lymphocyte biology. J Immunol (2012) 188(2):521–6. doi: 10.4049/jimmunol.1101530
8. Sweet DR, Fan L, Hsieh PN, Jain MK. Kruppel-like factors in vascular inflammation: mechanistic insights and therapeutic potential. Front Cardiovasc Med (2018) 5:6. doi: 10.3389/fcvm.2018.00006
9. Tang X, Wang P, Zhang R, Watanabe I, Chang E, Vinayachandran V, et al. KLF2 regulates neutrophil activation and thrombosis in cardiac hypertrophy and heart failure progression. J Clin Invest (2022) 132(3). doi: 10.1172/JCI147191
10. Campos-Martin Y, Martinez N, Martinez-Lopez A, Cereceda L, Casado F, Algara P, et al. Clinical and diagnostic relevance of NOTCH2-and KLF2-mutations in splenic marginal zone lymphoma. Haematologica (2017) 102(8):e310–e2. doi: 10.3324/haematol.2016.161711
11. Clipson A, Wang M, de Leval L, Ashton-Key M, Wotherspoon A, Vassiliou G, et al. KLF2 mutation is the most frequent somatic change in splenic marginal zone lymphoma and identifies a subset with distinct genotype. Leukemia (2015) 29(5):1177–85. doi: 10.1038/leu.2014.330
12. Wu Z, Wang S. Role of kruppel-like transcription factors in adipogenesis. Dev Biol (2013) 373(2):235–43. doi: 10.1016/j.ydbio.2012.10.031
13. Schuh W, Meister S, Herrmann K, Bradl H, Jack H-M. Transcriptome analysis in primary b lymphoid precursors following induction of the pre-b cell receptor. Mol Immunol (2008) 45(2):362–75. doi: 10.1016/j.molimm.2007.06.154
14. Melchers F. The pre-b-cell receptor: selector of fitting immunoglobulin heavy chains for the b-cell repertoire. Nat Rev Immunol (2005) 5(7):578–84. doi: 10.1038/nri1649
15. Vettermann C, Jack HM. The pre-b cell receptor: turning autoreactivity into self-defense. Trends Immunol (2010) 31(5):176–83. doi: 10.1016/j.it.2010.02.004
16. Hart GT, Wang X, Hogquist KA, Jameson SC. Kruppel-like factor 2 (KLF2) regulates b-cell reactivity, subset differentiation, and trafficking molecule expression. Proc Natl Acad Sci USA. (2011) 108(2):716–21. doi: 10.1073/pnas.1013168108
17. Herglotz J, Unrau L, Hauschildt F, Fischer M, Kriebitzsch N, Alawi M, et al. Essential control of early b-cell development by Mef2 transcription factors. Blood (2016) 127(5):572–81. doi: 10.1182/blood-2015-04-643270
18. Ottens K, Satterthwaite AB. IRF4 has a unique role in early b cell development and acts prior to CD21 expression to control marginal zone b cell numbers. Front Immunol (2021) 12:779085. doi: 10.3389/fimmu.2021.779085
19. Winkelmann R, Sandrock L, Kirberg J, Jack H-M, Schuh W. KLF2–a negative regulator of pre-b cell clonal expansion and b cell activation. PloS One (2014) 9(5):e97953. doi: 10.1371/journal.pone.0097953
20. Winkelmann R, Sandrock L, Porstner M, Roth E, Mathews M, Hobeika E, et al. B cell homeostasis and plasma cell homing controlled by kruppel-like factor 2. Proc Natl Acad Sci United States America. (2011) 108(2):710–5. doi: 10.1073/pnas.1012858108
21. Ma S, Pathak S, Trinh L, Lu R. Interferon regulatory factors 4 and 8 induce the expression of ikaros and aiolos to down-regulate pre-b-cell receptor and promote cell-cycle withdrawal in pre-b-cell development. Blood (2008) 111(3):1396–403. doi: 10.1182/blood-2007-08-110106
22. Hoek KL, Gordy LE, Collins PL, Parekh VV, Aune TM, Joyce S, et al. Follicular b cell trafficking within the spleen actively restricts humoral immune responses. Immunity (2010) 33(2):254–65. doi: 10.1016/j.immuni.2010.07.016
23. Fruman DA, Ferl GZ, An SS, Donahue AC, Satterthwaite AB, Witte ON. Phosphoinositide 3-kinase and bruton’s tyrosine kinase regulate overlapping sets of genes in b lymphocytes. Proc Natl Acad Sci USA. (2002) 99(1):359–64. doi: 10.1073/pnas.012605099
24. Glynne R, Ghandour G, Rayner J, Mack DH, Goodnow CC. B-lymphocyte quiescence, tolerance and activation as viewed by global gene expression profiling on microarrays. Immunol Rev (2000) 176:216–46. doi: 10.1034/j.1600-065X.2000.00614.x
25. Yusuf I, Kharas MG, Chen J, Peralta RQ, Maruniak A, Sareen P, et al. KLF4 is a FOXO target gene that suppresses b cell proliferation. Int Immunol (2008) 20(5):671–81. doi: 10.1093/intimm/dxn024
26. Chen J, Limon JJ, Blanc C, Peng SL, Fruman DA. Foxo1 regulates marginal zone b-cell development. Eur J Immunol (2010) 40(7):1890–6. doi: 10.1002/eji.200939817
27. Yusuf I, Zhu X, Kharas MG, Chen J, Fruman DA. Optimal b-cell proliferation requires phosphoinositide 3-kinase-dependent inactivation of FOXO transcription factors. Blood (2004) 104(3):784–7. doi: 10.1182/blood-2003-09-3071
28. McHeyzer-Williams M, Okitsu S, Wang N, McHeyzer-Williams L. Molecular programming of b cell memory. Nat Rev Immunol (2011) 12(1):24–34. doi: 10.1038/nri3128
29. Stebegg M, Kumar SD, Silva-Cayetano A, Fonseca VR, Linterman MA, Graca L. Regulation of the germinal center response. Front Immunol (2018) 9:2469. doi: 10.3389/fimmu.2018.02469
30. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol (2022) 40:413–42. doi: 10.1146/annurev-immunol-120419-022408
31. Schuh W, Mielenz D, Jack H-M. Unraveling the mysteries of plasma cells. Adv Immunol (2020) 146:57–107. doi: 10.1016/bs.ai.2020.01.002
32. Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, et al. CD80 and PD-L2 define functionally distinct memory b cell subsets that are independent of antibody isotype. Nat Immunol (2014) 15(7):631–7. doi: 10.1038/ni.2914
33. Riedel R, Addo R, Ferreira-Gomes M, Heinz GA, Heinrich F, Kummer J, et al. Discrete populations of isotype-switched memory b lymphocytes are maintained in murine spleen and bone marrow. Nat Commun (2020) 11(1):2570. doi: 10.1038/s41467-020-16464-6
34. Bhattacharya D, Cheah MT, Franco CB, Hosen N, Pin CL, Sha WC, et al. Transcriptional profiling of antigen-dependent murine b cell differentiation and memory formation. J Immunol (2007) 179(10):6808–19. doi: 10.4049/jimmunol.179.10.6808
35. Pracht K, Meinzinger J, Daum P, Schulz SR, Reimer D, Hauke M, et al. A new staining protocol for detection of murine antibody-secreting plasma cell subsets by flow cytometry. Eur J Immunol (2017) 47(8):1389–92. doi: 10.1002/eji.201747019
36. Kabashima K, Haynes NM, Xu Y, Nutt SL, Allende ML, Proia RL, et al. Plasma cell S1P1 expression determines secondary lymphoid organ retention versus bone marrow tropism. J Exp Med (2006) 203(12):2683–90. doi: 10.1084/jem.20061289
37. Wittner J, Schulz SR, Steinmetz TD, Berges J, Hauke M, Channell WM, et al. Kruppel-like factor 2 controls IgA plasma cell compartmentalization and IgA responses. Mucosal Immunol (2022) 15:668–682. doi: 10.1038/s41385-022-00503-0
38. Jha P, Das H. KLF2 in regulation of NF-kappaB-Mediated immune cell function and inflammation. Int J Mol Sci (2017) 18(11). doi: 10.3390/ijms18112383
39. Cinamon G, Zachariah MA, Lam OM, Foss FW, Cyster JG. Follicular shuttling of marginal zone b cells facilitates antigen transport. Nat Immunol (2008) 9(1):54–62. doi: 10.1038/ni1542
40. Pillai S, Cariappa A. The follicular versus marginal zone b lymphocyte cell fate decision. Nat Rev Immunol (2009) 9(11):767–77. doi: 10.1038/nri2656
41. Saito T, Chiba S, Ichikawa M, Kunisato A, Asai T, Shimizu K, et al. Notch2 is preferentially expressed in mature b cells and indispensable for marginal zone b lineage development. Immunity (2003) 18(5):675–85. doi: 10.1016/S1074-7613(03)00111-0
42. Hozumi K, Negishi N, Suzuki D, Abe N, Sotomaru Y, Tamaoki N, et al. Delta-like 1 is necessary for the generation of marginal zone b cells but not T cells in vivo. Nat Immunol (2004) 5(6):638–44. doi: 10.1038/ni1075
43. Lechner M, Engleitner T, Babushku T, Schmidt-Supprian M, Rad R, Strobl LJ, et al. Notch2-mediated plasticity between marginal zone and follicular b cells. Nat Commun (2021) 12(1):1111. doi: 10.1038/s41467-021-21359-1
44. Hart GT, Peery SL, Hamilton SE, Jameson SC. Cutting edge: kruppel-like factor 2 is required for phenotypic maintenance but not development of B1 b cells. J Immunol (2012) 189(7):3293–7. doi: 10.4049/jimmunol.1201439
45. Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature (2004) 427(6972):355–60. doi: 10.1038/nature02284
46. Allende ML, Dreier JL, Mandala S, Proia RL. Expression of the sphingosine 1-phosphate receptor, S1P1, on T-cells controls thymic emigration. J Biol Chem (2004) 279(15):15396–401. doi: 10.1074/jbc.M314291200
47. Carlson CM, Endrizzi BT, Wu J, Ding X, Weinreich MA, Walsh ER, et al. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature (2006) 442(7100):299–302. doi: 10.1038/nature04882
48. Forster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. A putative chemokine receptor, BLR1, directs b cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell (1996) 87(6):1037–47. doi: 10.1016/S0092-8674(00)81798-5
49. Weber JP, Fuhrmann F, Feist RK, Lahmann A, Al Baz MS, Gentz L-J, et al. ICOS maintains the T follicular helper cell phenotype by down-regulating kruppel-like factor 2. J Exp Med (2015) 212(2):217–33. doi: 10.1084/jem.20141432
50. Alles M, Turchinovich G, Zhang P, Schuh W, Agenes F, Kirberg J. Leukocyte beta7 integrin targeted by kruppel-like factors. J Immunol (Baltimore Md: 1950). (2014) 193(4):1737–46. doi: 10.4049/jimmunol.1302613
51. Girkontaite I, Sakk V, Wagner M, Borggrefe T, Tedford K, Chun J, et al. The sphingosine-1-phosphate (S1P) lysophospholipid receptor S1P3 regulates MAdCAM-1+ endothelial cells in splenic marginal sinus organization. J Exp Med (2004) 200(11):1491–501. doi: 10.1084/jem.20041483
52. Lee JY, Skon CN, Lee YJ, Oh S, Taylor JJ, Malhotra D, et al. The transcription factor KLF2 restrains CD4(+) T follicular helper cell differentiation. Immunity (2015) 42(2):252–64. doi: 10.1016/j.immuni.2015.01.013
53. Pracht K, Wittner J, Kagerer F, Jäck H-M, Schuh W. The intestine: a highly dynamic microenvironment for IgA plasma cells. Front Immunol (2023) 14. doi: 10.3389/fimmu.2023.1114348
54. Miles A, Liaskou E, Eksteen B, Lalor PF, Adams DH. CCL25 and CCL28 promote alpha4 beta7-integrin-dependent adhesion of lymphocytes to MAdCAM-1 under shear flow. Am J Physiol Gastrointest Liver Physiol (2008) 294(5):G1257–67. doi: 10.1152/ajpgi.00266.2007
55. Wendt E, White GE, Ferry H, Huhn M, Greaves DR, Keshav S. Glucocorticoids suppress CCR9-mediated chemotaxis, calcium flux, and adhesion to MAdCAM-1 in human T cells. J Immunol (2016) 196(9):3910–9. doi: 10.4049/jimmunol.1500619
56. Guzman M, Lundborg LR, Yeasmin S, Tyler CJ, Zgajnar NR, Taupin V, et al. An integrin alphaEbeta7-dependent mechanism of IgA transcytosis requires direct plasma cell contact with intestinal epithelium. Mucosal Immunol (2021) 14(6):1347–57. doi: 10.1038/s41385-021-00439-x
57. Pabst O, Peters T, Czeloth N, Bernhardt G, Scharffetter-Kochanek K, Forster R. Cutting edge: egress of newly generated plasma cells from peripheral lymph nodes depends on beta 2 integrin. J Immunol (2005) 174(12):7492–5. doi: 10.4049/jimmunol.174.12.7492
58. Zibellini S, Capello D, Forconi F, Marcatili P, Rossi D, Rattotti S, et al. Stereotyped patterns of b-cell receptor in splenic marginal zone lymphoma. Haematologica (2010) 95(10):1792–6. doi: 10.3324/haematol.2010.025437
59. Bikos V, Darzentas N, Hadzidimitriou A, Davis Z, Hockley S, Traverse-Glehen A, et al. Over 30% of patients with splenic marginal zone lymphoma express the same immunoglobulin heavy variable gene: ontogenetic implications. Leukemia (2012) 26(7):1638–46. doi: 10.1038/leu.2012.3
60. Watkins AJ, Huang Y, Ye H, Chanudet E, Johnson N, Hamoudi R, et al. Splenic marginal zone lymphoma: characterization of 7q deletion and its value in diagnosis. J Pathol (2010) 220(4):461–74. doi: 10.1002/path.2665
61. Mateo M, Mollejo M, Villuendas R, Algara P, Sanchez-Beato M, Martinez P, et al. 7q31-32 allelic loss is a frequent finding in splenic marginal zone lymphoma. Am J Pathol (1999) 154(5):1583–9. doi: 10.1016/S0002-9440(10)65411-9
62. Gruszka-Westwood AM, Hamoudi R, Osborne L, Matutes E, Catovsky D. Deletion mapping on the long arm of chromosome 7 in splenic lymphoma with villous lymphocytes. Genes Chromosomes Cancer. (2003) 36(1):57–69. doi: 10.1002/gcc.10142
63. Spina V, Rossi D. NF-kappaB deregulation in splenic marginal zone lymphoma. Semin Cancer Biol (2016) 39:61–7. doi: 10.1016/j.semcancer.2016.08.002
64. Arcaini L, Rossi D. Nuclear factor-kappaB dysregulation in splenic marginal zone lymphoma: new therapeutic opportunities. Haematologica (2012) 97(5):638–40. doi: 10.3324/haematol.2011.058362
65. Kumar SK, Rajkumar V, Kyle RA, van Duin M, Sonneveld P, Mateos MV, et al. Multiple myeloma. Nat Rev Dis Primers. (2017) 3:17046. doi: 10.1038/nrdp.2017.46
66. Walker BA, Boyle EM, Wardell CP, Murison A, Begum DB, Dahir NM, et al. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol (2015) 33(33):3911–20. doi: 10.1200/JCO.2014.59.1503
67. Martinez-Garcia E, Popovic R, Min DJ, Sweet SM, Thomas PM, Zamdborg L, et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t (4,14) multiple myeloma cells. Blood (2011) 117(1):211–20. doi: 10.1182/blood-2010-07-298349
68. Ohguchi H, Hideshima T, Bhasin MK, Gorgun GT, Santo L, Cea M, et al. The KDM3A-KLF2-IRF4 axis maintains myeloma cell survival. Nat Commun (2016) 7:10258. doi: 10.1038/ncomms10258
69. Neri P, Ren L, Azab AK, Brentnall M, Gratton K, Klimowicz AC, et al. Integrin beta7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood (2011) 117(23):6202–13. doi: 10.1182/blood-2010-06-292243
70. Salama Y, Heida AH, Yokoyama K, Takahashi S, Hattori K, Heissig B. The EGFL7-ITGB3-KLF2 axis enhances survival of multiple myeloma in preclinical models. Blood Adv (2020) 4(6):1021–37. doi: 10.1182/bloodadvances.2019001002
71. Hideshima T, Anderson KC. Signaling pathway mediating myeloma cell growth and survival. Cancers (Basel). (2021) 13(2). doi: 10.3390/cancers13020216
72. Yel L. Selective IgA deficiency. J Clin Immunol (2010) 30(1):10–6. doi: 10.1007/s10875-009-9357-x
73. Pilarowski GO, Cazares T, Zhang L, Benjamin JS, Liu K, Jagannathan S, et al. Abnormal peyer patch development and b-cell gut homing drive IgA deficiency in kabuki syndrome. J Allergy Clin Immunol (2020) 145(3):982–92. doi: 10.1016/j.jaci.2019.11.034
Keywords: KLF2, plasma cells, quiescence, B cells, integrins, IgA, mucosal immunity, multiple myeloma
Citation: Wittner J and Schuh W (2023) Krüppel-like factor 2: a central regulator of B cell differentiation and plasma cell homing. Front. Immunol. 14:1172641. doi: 10.3389/fimmu.2023.1172641
Received: 23 February 2023; Accepted: 06 April 2023;
Published: 12 May 2023.
Edited by:
Stephen Nutt, The University of Melbourne, AustraliaReviewed by:
Zhoujie Ding, Monash University, AustraliaJasper Cornish, The University of Melbourne, Australia
Copyright © 2023 Wittner and Schuh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wolfgang Schuh, d29sZmdhbmcuc2NodWhAdWstZXJsYW5nZW4uZGU=