 Chia-Ming Su
Chia-Ming Su Yijun Du
Yijun Du Raymond R. R. Rowland1
Raymond R. R. Rowland1 Qiuhong Wang
Qiuhong Wang Dongwan Yoo
Dongwan Yoo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 17 April 2023
Sec. Vaccines and Molecular Therapeutics
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1172000
This article is part of the Research TopicNew-generation vaccines and novel vaccinal strategies against infectious diseases of livestock, wild and companion animalsView all 11 articles
Type I interferons (IFNs-α/β) are antiviral cytokines that constitute the innate immunity of hosts to fight against viral infections. Recent studies, however, have revealed the pleiotropic functions of IFNs, in addition to their antiviral activities, for the priming of activation and maturation of adaptive immunity. In turn, many viruses have developed various strategies to counteract the IFN response and to evade the host immune system for their benefits. The inefficient innate immunity and delayed adaptive response fail to clear of invading viruses and negatively affect the efficacy of vaccines. A better understanding of evasion strategies will provide opportunities to revert the viral IFN antagonism. Furthermore, IFN antagonism-deficient viruses can be generated by reverse genetics technology. Such viruses can potentially serve as next-generation vaccines that can induce effective and broad-spectrum responses for both innate and adaptive immunities for various pathogens. This review describes the recent advances in developing IFN antagonism-deficient viruses, their immune evasion and attenuated phenotypes in natural host animal species, and future potential as veterinary vaccines.
Virus-host interactions play a vital role during infection and determine pathogenic consequences, including host and tissue tropisms, viral elimination and persistence, tumorigenesis, and clinical outcomes. Physical and chemical barriers, innate immune systems, and various types of immune cells are considered the first line of defense of a host against viral infections. Innate and adaptive immunities are two main surveillance systems to resolve viral infections. Immediately upon infection, the type I interferon (IFNs-α/β) production system is activated by recognizing viral components by host cell factors. It is the most effective cellular defense against viral infection in the early stage of infection (1). In turn, viruses have developed distinct immune-disarming abilities to facilitate replication. The modulation of IFN functions is an effective strategy for invading viruses to survive in the host (2). IFN suppression by viruses can also negatively impact the immunogenicity of live-attenuated vaccines (3). With the advances in the study of viral immune evasion mechanisms and reverse genetics technology, it is now possible to remove the IFN suppression function from target viruses and design new generation vaccines for diverse RNA viruses. Indeed, some engineered viruses have been constructed and examined in their natural host animal species for immunogenicity and clinical outcomes (4, 5). This article discusses the molecular mechanisms for viral IFN antagonism, generation of IFN antagonism-deficient viruses, their immunogenic consequences in animals following vaccination, and prospects as next-generation vaccines for veterinary diseases.
Immune cells involved in the innate immune system represent monocytes, macrophages, eosinophils, neutrophils, and natural killer (NK) cells. IFNs-α/β are antiviral cytokines and constitute one of the most critical components in the innate immune system against invading viruses. The IFNs-α/β signaling triggers expression of a series of IFN-stimulated genes (ISGs) and contributes to the antiviral state of the cell (6). All nucleated cells can produce IFNs-α/β, but plasmacytoid dendritic cells (pDC) are the main type that produce type I IFNs most abundantly (7).
Once an RNA virus enters the cell, viral components trigger a series of intricate recognition mechanisms within the cell (8). The innate immune signaling cascade starts with the recognition of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs) that are composed of fours groups of cellular proteins; nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), C-type lectin receptors (CLRs), and Toll-like receptors (TLRs). NLRs, RLRs, and CLRs reside in the cytoplasm, whereas TLRs locate in the endosomal membrane (9). The viral RNA binds to RIG-I or melanoma differentiation-associated gene 5 (MDA5) and triggers conformational changes to expose its caspase activation and recruitment domain (CARD) at the N-terminus. CARD-CARD dimers are then formed and recruit the IFN-β promoter stimulator-1 (IPS-1) [also known as CARD adaptor inducing IFN-β (Cardif), mitochondrial antiviral-signaling protein (MAVS), or virus-induced signaling adaptor (VISA)] (10, 11). IPS-1 recruits the NF-kappa-B essential modulator (NEMO), tumor necrosis factor receptor-associated factor (TRAF) 3, and TRAF family-member-associated NF-κB activator (TANK) proteins, and its complex activates TANK binding kinase 1 (TBK1) and inhibitor of kappa B (IκB) kinase ϵ (IKKϵ). Subsequently, IFN regulatory factor 3 (IRF3) and IRF7 are phosphorylated in the TBK1- and IKKϵ-dependent manners (12). Then, phosphorylated IRF3 and IRF7 form a homodimer or heterodimer for translocation to the nucleus (13), where the IRF3- or IRF7-dimer forms an enhanceosome which binds to the positive regulatory domains (PRDs) I-III regions in the IFN promoters for IFN gene transcriptions (14, 15). IPS-1 may also activate IKKα and IKKβ to trigger the degradation of IkB to free up NF-κB. The released NF-κB binds to PRDs of respective IFN genes and proinflammatory cytokine promoters (15–17). Besides the RIG-I-mediated signaling cascades, TLR3 and TLR7 also recognize double-stranded RNA and single-stranded RNA, respectively, in the endosome (18). TLR3 can recruit the TRIF, nucleosome assembly protein 1 (NAP1), and TRAF3 adaptors and activates IRF3 for IFN expression and downstream signaling for many antiviral proteins (9, 15).
Once IFNs are produced, they are secreted from the cell and bind to their receptors, interferon alpha and beta receptor subunit 1 (IFNAR1) and IFNAR2, on the surface of the same cell (autocrine) or adjacent cells (paracrine). The binding of IFNs to their receptors triggers the Janus kinase (JAK)-signal transducers and activator of transcription (STAT) signaling pathway. The activation of JAK1 and tyrosine kinase 2 (Tyk2) is the first response to the IFN signaling and results in phosphorylation and dimerization of STAT1 and STAT2, followed by the recruitment of IRF9 to form the IFN-stimulated gene factor 3 (ISGF3) complex. The ISGF3 complex then translocates to the nucleus. It induces the expression of ISGs by binding to the IFN-stimulated regulatory response elements (ISRE) in the promoters of ISGs (19). More than 300 ISGs have been identified so far, and they are the major executors of IFNs acting as effector molecules for the antiviral response (19, 20).
In addition to establishing an antiviral state, type I IFNs can enhance adaptive immunity by targeting dendritic cells (DCs), NK cells, T cells, and B cells (19). For DCs, type I IFNs play a key role in the generation and function of DCs (21), suggesting that type I IFNs represent a potent natural adjuvant for crossing the innate and adaptive immune systems. Type I IFNs enhance the ability of DCs to cross-present antigens to T cells and modulate antigen survival and processing (22, 23). IFNs regulate the DCs antigen presentation in an autocrine manner (24). Besides DCs, type I IFNs can promote the activation and survival of NK cells directly or indirectly via IL-15 cis and trans presentation (25) and regulate T cells directly through IFNARs on the surface of T cells. Type I IFNs are critical mediators for the spontaneous priming of antitumor CD8+ T cell response (26). In the herpesvirus-infected mouse model, type I IFNs have been shown to stimulate CD4+ T cells for undergoing clonal expansion (27). The T cells primed by type I IFNs also show an increased ability to help B cells to enhance antibody secretion (28). The studies using lymphocytic choriomeningitis virus or West Nile virus as models have also demonstrated that type I IFNs upregulate the survival, maturation, cytotoxicity, and clonal expansion of CD8+ T cells during infection (29–34). In addition to effector CD8+ T cells, memory CD8+ T cells are also regulated by type I IFNs. Studies using vaccinia virus, vesicular stomatitis virus, and lymphocytic choriomeningitis virus have shown that type I IFNs promote the differentiation of memory CD8+ T cells by affecting the initial clonal expansion (31, 35). Furthermore, type I IFNs regulate B cell activation, antibody secretion, and isotype switching during the vesicular stomatitis virus and West Nile virus infections (36–38). By increasing the level of B-cell survival factors, type I IFNs can promote survival and activation of B cells and enhance autoantibody production (39). Thus, it is evident that the immunological functions of type I IFNs are much broader than establishing an antiviral state and play an important role in regulating adaptive immunity.
Nuclear factor-κB (NF-κB) is a family of transcription factors and a key mediator for proinflammatory cytokine production. NF-κB consists of RelA (p65) and RelB, NF-κB1 (p50 and its precursor p105), NF-κB2 (p52 and its precursor p100), and c-Rel to form a homodimer or heterodimer with RelA or RelB. Numerous factors activate the NF-κB signaling including TLR ligands and cytokines such as tumor necrosis factor-α (TNF-α). Upon stimulation, upstream kinases IKKα and IKKβ are activated by phosphorylation. IKKβ then phosphorylates the IκB and degrades IκB through a proteasome-dependent manner. IκB is the negative regulator for NF-κB, and so the IκB degradation releases NF-κB, which is then phosphorylated. The activated NF-κB enters the nucleus and binds to the specific DNA locus, the κB site (40). NF-κB signaling triggers the expression of type I IFNs and various proinflammatory cytokines, including IL-1β, IL-6, IL-8, IL-15, and TNF-α (41). The expressed proinflammatory cytokines prompt the positive feedback loop to the NF-κB signaling in an autocrine manner (42). The protein inhibitor of activated STAT 1 (PIAS1) is the NF-κB negative regulator residing in the nucleus (43). PIAS1 prevents NF-κB dimers from binding to κB sites by binding to p65 in the nucleus (43). For respiratory viral infections, NF-κB is strongly activated in the lungs and results in the induction of proinflammatory cytokines and chemokines (44, 45).
Many viruses code for viral proteins to antagonize the IFN pathway and often produce more than one antagonist (6, 46, 47). Viruses have evolved to employ diverse strategies to evade the IFN system, and some of the viral strategies and mechanisms of action are discussed below (Figure 1). For convenience, the signaling cascade for production of type I IFNs in virus-infected cells will be referred to as ‘IFN production pathway’, and the signaling mediated by IFNs for production of ISGs will be referred to as ‘JAK-STAT signaling pathway’.
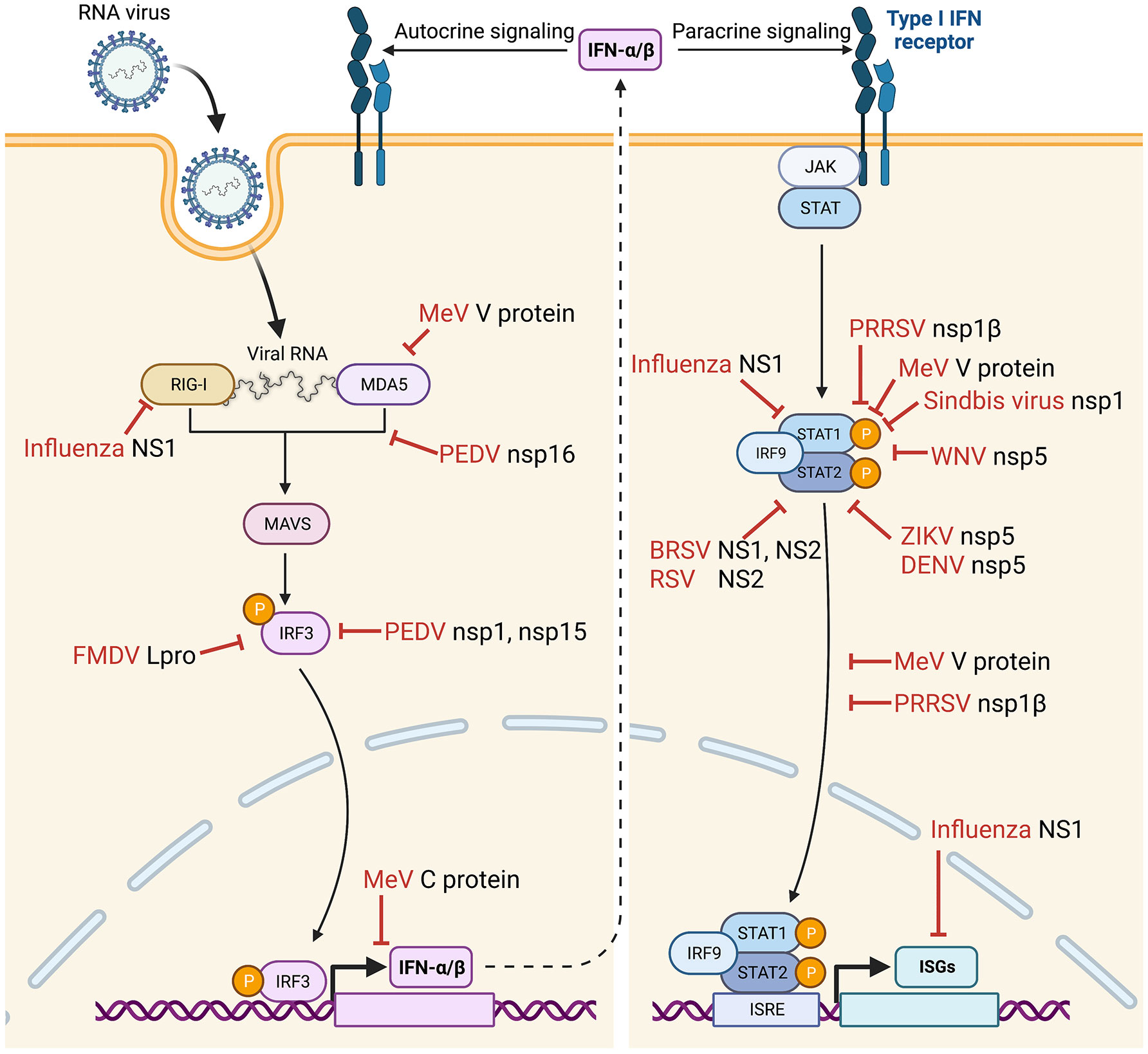
Figure 1 Suppression of IFNs-α/β production and IFN-stimulated gene (ISG) expression during infection by various animal viruses. IFN suppression-deficient viruses have been constructed, and their immunological and clinical outcomes have been examined in their natural host animal species. Vertical arrow (dotted line) divides the IFN signaling to “IFN production pathway” (left) and “IFN signaling JAK-STAT pathway” (right). Bars (red) indicate sites of action by specific viral proteins for IFN suppression. FMDV, foot-and-mouth disease virus (48–51); PEDV, porcine epidemic diarrhea virus (52); MeV, measles virus (53–59); PRRSV, porcine reproductive and respiratory syndrome virus (60–63); WNV, West Nile virus (64); ZIKV, Zika virus (65); DENV, Dengue virus (66); BRSV, bovine respiratory syncytial virus (67–70); RSV, respiratory syncytial virus (71, 72); IRF, interferon regulatory factor; ISRE, interferon stimulation response element; STAT, signal transducer and activator of transcription. Images were created with BioRender.com.
Some viruses suppress the IFN production pathway by disrupting IFN receptor functions, such as reducing receptor expression and degrading receptor molecules. For example, the influenza A virus disrupts IFN signaling by downregulating the expression of IFN receptor. The NS1 protein is a non-structural component encoded by influenza viruses A, B, and C. It counteracts the host antiviral responses by two primary mechanisms: inhibition of the activation of IRF3 and IFN transcription and inhibition of the processing of IFN pre-mRNAs (73). The NS1 protein binds the host cell RNA and forms complexes with RIG-I to antagonize IFN production (74). The NS1 protein also suppresses the expression of both chains of IFNAR receptor at the transcription level (75, 76). Epstein-Barr virus (EBV) establishes a latent infection in B lymphocytes. EBV expresses two isoforms of latent membrane protein 2 (LMP2). Of the two forms, LMP2A plays an essential role in viral latency and progression of EBV-related diseases such as Burkitt’s lymphoma, nasopharyngeal carcinoma, and Hodgkin’s lymphoma. The LMP2A and LMP2B proteins degrade IFN receptors and modulate both type I and type II IFN responses (77).
Activation of IRF3 is an essential step for downstream IFN production signaling (9, 15), and some viruses have developed mechanisms to manipulate the IRF3 activation. Hepatitis C virus NS3-4A is the viral serine protease and has a crucial role in the viral replication cycle. NS3-4A cleaves the polyprotein at four sequential sites to yield NS4A, NS4B, NS5A, and NS5B proteins. NS3-4A can cleave and inactivate the host proteins TRIF (TIR-domain-containing adapter-inducing interferon-β) and Cardif, which are essential adaptors in response to IFNs mediated by TLR3 and RIG-I, respectively. NS3/4A also cleaves IKK and inactivates IRF3 (78). Classical swine fever virus and bovine viral diarrhea virus are member viruses of the Pestivirus genus of the Flaviviridae family. For pestiviruses, Npro is the N-terminal viral protease of the polyprotein and can degrade IRF3 for inactivation (79–81). Besides the direct degradation, IRF3 function may be inhibited by some other viruses. The rabies virus P protein and the hantavirus G1 protein have been determined to suppress IFN signaling by inhibiting IRF3 phosphorylation (82, 83). Rabies virus infects all species warm-blooded animals. It replicates in the cytoplasm of the cell, and the viral RNA is tightly encapsidated by the viral nucleoprotein and the RNA polymerase complex which consists of the phosphoprotein (P). The P protein is a cofactor of RNA polymerase by participating in viral genome transcription and replication. The P protein prevents IFN response in virus-infected cells by targeting TANK-binding kinase-1 (TBK1) and inhibiting phosphorylation and activation of IRF3. Sin Nombre virus, carried by rodents, is an etiologic agent for hantavirus pulmonary syndrome. With a pathogenic strain of hantavirus, the cytoplasmic tail of the G1 glycoprotein was shown to regulate the IFN response by inhibiting IRF3 phosphorylation at the level of the TBK1 complex (84). In contrast, the hantavirus G1-mediated IFN downregulation was absent in the Prospect Hill nonpathogenic strain, indicating that the IFN suppression is an important virulence factor for hantaviruses.
Coronaviruses (CoV) carry diverse strategies to inhibit IRF3 activation. CoV genomes are the largest among all RNA viruses and contain the positive-sense RNA of 25-32 kb in length. The viral genome codes for two large polyproteins, and the nsp1 protein is the N-terminal cleavage product of the polyprotein. The nsp1 protein of SARS-CoV-1 suppresses the IFN response via IRF3-dependent downregulation (85, 86), whereas the ORF8b and ORF8ab proteins induce IRF3 degradation via the ubiquitin-dependent proteasomal pathway (87). SARS-CoV-2 is an emerging virus with an unknown animal origin. As the causative agent for COVID-19 in humans, it has crossed the species barrier to infect other animal species, including cats, dogs, tigers, lions, mink, white-tail deer, monkeys, ferrets, and hamsters, to name a few (88). Humans can facilitate reverse-zoonotic transmission to animals, and the virus in infected animals may evolve to spill back to humans and other animal species. SARS-CoV-2 codes for nsp3 papain-like protease and nsp5 3C-like protease, and these two viral proteases are responsible for cleaving viral polyproteins for replication. The nsp3 protein cleaves IRF3 directly, and this finding explains the blunted type I IFN response seen in COVID-19 patients during infection (89). Although the nsp12 viral RNA polymerase of SARS-CoV-2 does not impair IRF3 activation, it attenuates type I IFN production by inhibiting IRF3 nuclear translocation (90). Meanwhile, the ORF7a protein decreases IRF3 phosphorylation by downregulating TBK1 expression (91, 92). Similarly, the viral spike (S) protein binds directly to IRF3 and further diminishes its expression, whereas the levels of NF-κB and STAT1 transcription factors remain intact (93). The murine coronavirus mouse hepatitis virus can also delay ISG production by limiting the IRF3 transcription (94).
Besides the direct regulation of IRF3, some viruses inhibit signaling adaptors and regulators in the IFN production pathway. For example, the swine arterivirus, porcine reproductive and respiratory syndrome virus (PRRSV), has been found to inhibit the IFN promoter activity and impairs type I IFN production (60). The nsp1α protein of PRRSV downregulates IFN production by degrading the CREB (cyclic AMP response element-binding)-binding protein (CBP) via the ubiquitin-dependent proteasome pathway (95–97). PRRSV also suppresses the NF-κB activation and inhibits type I IFN production in the RIG-I dependent manner (98, 99). A recent study showed that nsp1β of PRRSV binds to nucleoporin 62 (Nup62) (100). Nup62 is a major structural component of the nuclear pore complex (NPC) on the nuclear membrane, and NPC functions as a gateway for nucleocytoplasmic trafficking of nuclear proteins and mRNAs. The binding of nsp1β to Nup62 disrupts the NPC integrity, causing the nuclear retention of host mRNAs and inhibiting the host cell protein production, including IFNs, ISGs, and IRF3 (100).
IFNs-α/β are secretory cytokines. Once expressed in virus-infected cells and secreted, they bind to their receptors on the same cell or neighbor cells and activate the IFN signaling pathway, namely the JAK-STAT signaling. Some viruses have developed mechanisms to impair the JAK-STAT pathway by targeting STAT1 and STAT2. Paramyxoviruses disturb the STAT signaling by using diverse mechanisms, such as the direct binding of viral protein to STAT to prevent phosphorylation (101). The paramyxovirus V protein is of particular interest since it regulates the JAK-STAT pathway by degrading STAT1 and STAT2 and mediating their nuclear accumulations (53, 102–104). Sendai virus, a paramyxovirus, produces a set of four C proteins by mRNA editing of the P gene and inhibits IFN-induced tyrosine phosphorylation of STATs (101, 105, 106). All four C proteins bind to STAT1, but only the largest form of C induces the mono-ubiquitination of STAT1 and its degradation (106). Dengue virus (DENV) and Zika virus (ZIKV) in the family Flaviviridae have shown the ability to reduce the concentration of STATs in the cells (101, 107, 108). NS5 protein of ZIKV induces the degradation of STAT2 via the proteasomal pathway (65, 109).
Respiratory syncytial virus (RSV) has diverse mechanisms to inhibit the type 1 IFN response in different cell types (110). RSV degrades STATs in epithelial cells, whereas, in DCs, it inhibits STAT1 and STAT2 phosphorylation and nuclear accumulation (110, 111). Furthermore, RSV induces STAT2 downregulation through ubiquitin-proteasome degradation by the Elongin-Cullin E3 ligase (112, 113). The porcine arterivirus PRRSV nsp1β protein inhibits STAT1 phosphorylation, leading to inhibition of the JAK-STAT signaling pathway, resulting in the suppression of ISG expressions (60, 61). Other viruses can also block the IFN signaling by targeting IRF9. Such mechanisms are usually found in viruses that cause persistent infections and oncogeneses, such as human papillomavirus, porcine bocavirus, and human T-cell leukemia virus (114–116).
Viruses can block the nuclear accumulation of activated STATs and inhibit IFN signaling. For example, the paramyxovirus virus V protein binds to MDA-5 and decreases the IFN promoter activation (117), as well as binding to STAT1 and STAT2 and preventing their nuclear accumulation (118). The P protein of the rabies virus binds to the DNA-binding and coiled-coil domains of STAT1 to influence the IFN-induced transcription and impairs its nuclear translocation (119). The porcine arterivirus nsp1β protein inhibits STAT1 phosphorylation and further affects the nuclear translocation of ISGF3, thus inhibiting the JAK-STAT signaling pathway (60, 61). PRRSV nsp1β also degrades karyopherin-α1, one of the nuclear transporter proteins, and blocks the ISGF3 nuclear translocation to suppress ISG expressions (120).
Of various host immune surveillance systems, the innate immune system builds on a series of antiviral responses. Although the IFN inhibitory ability of a virus is not the only determinant for virulence, it is often one of the most critical virulence factors. Type I IFNs can also prime the activation and maturation of adaptive immune responses. Disturbed and delayed host responses against viral infection are attributed to the suppression of type I IFNs, suggesting that viral inhibition of IFN response may cause the unsatisfactory efficacy of certain vaccines. Thus, future vaccines should stimulate both innate and adaptive immune response. A large body of evidence demonstrates the importance of type I IFNs not only for innate immunity and antiviral function but also for the development of adaptive immunity. Murine norovirus infection in mice, of which type I IFN receptors are deficient, transforms an acute infection into a systemically persistent infection (121). Nevertheless, CD8+ T cell and antibody responses are still enhanced during persistent norovirus infection, suggesting that the deficiency of IFNs leads to viral persistence despite enhanced adaptive immunity (121). Many viruses suppressing the IFN response are known to cause insufficient protection upon vaccination, which makes many vaccines less effective (122).
The potential of IFNs as vaccine adjuvants have been studied by many investigators. The results demonstrate that IFNs play a critical role in generating immune responses to vaccines. IFN treatment is the most effective therapy for controlling persistent hepatitis C virus infection (123–125). IFN-β is helpful for cancer vaccines that induce a greater expansion of tumor-specific CD8+ T cells (126). IFNs have been used as adjuvants coupled with veterinary vaccines. Replicating PRRSVs expressing various types of IFN increased the IFN levels in pigs and conferred protection against PRRSV challenges (127, 128). Recombinant porcine IFN (PoIFNα), used as an adjuvant in PRRSV vaccination, induces adaptive immune response in pigs (129). In other studies, PoIFNα, in combination with inactivated swine influenza virus, significantly upregulated the expression of various immunoregulatory cytokines and higher levels of HA-specific antibodies in 6-week-old pigs (130). Indeed, type I IFNs is a potent adjuvant for influenza vaccines to induce an effective humoral response in mice (131, 132). PoIFNα coupled with a foot-and-mouth disease virus (FMDV) protein vaccine generated more robust immunogenicity and complete protection of pigs from virulent challenge (133). In other FMDV studies, IFNα was endogenously expressed in mice using an adenovirus vector, which improved the generation of T helper cells and the production of all classes of IgG immunoglobulins, especially IgG1 and IgG2a subtypes (134, 135). Endogenous IFNα expressed using a plasmid linked to the equine encephalitis virus (EEV) vaccine showed protection after being administered 24 hours before the challenge (136, 137). This result highlights that up to 24 hours is required to develop the IFN-mediated antiviral response. An IFN inducer coupled with a non-replicating vaccine for the rabies virus provided better protection in rhesus monkeys than the vaccine alone (138).
Taken together, such studies provide insights into the potential application of IFNs as an adjuvant for improving vaccine efficacy. Moreover, reprogramming the viral IFN evasion strategy has allowed the conceptual development of new vaccine designs. The approaches have proven useful in enhancing IFN response during vaccination and improving innate and adaptive immunities. Live attenuated vaccines are generally preferred in veterinary medicine and are considered more protective than inactivated vaccines. Live attenuated vaccines elicit both humoral and cellular immunities, whereas inactivated vaccines induce mainly antibody response. Removing IFN antagonism from viruses has been a strategy for developing both human and veterinary vaccines to achieve better immunogenicity and improved protection. In this article, however, we limit our discussions to only viruses that have been examined in the natural host animals, excluding human trials, as potential vaccine candidates against viral infections in animals.
FMDV is the prototype member of the Aphthovirus genus in the Picornaviridae family. FMDV is a highly contagious disease of cloven-hoofed animals in many livestock-producing countries worldwide. Despite the importance of the disease, difficulties in inducing and maintaining an adequate immune response in vaccinated animals have been an issue in controlling FMDV infection. FMDV can suppress the type I IFN response and persists in the tonsils of infected animals for up to 2 years (139). Among the viral proteins, the leader protein Lpro stands out as the most effective IFN suppressor of FMDV (Figure 1) (48–51). Lpro of FMDV is also a deubiquitinase (DUB) and deISGylase and interacts with ISG15 to induce its degradation (140). The FMDV mutant with modified deISGylase activity showed viral attenuation in mice (141). Deletion of Lpro from FMDV resulted in a slightly slower growth than wild-type virus in cells and was attenuated in mice (142). Therefore, Lpro is the target of the attenuated vaccine design for FMDV in natural host animal species. After removing the IFN suppression function from Lpro, the IFN suppression-negative FMDV induces higher levels of IFNs and ISGs with a robust neutralizing antibody response in vaccinated pigs (143, 144). Furthermore, pigs were completely protected from the high dose challenges of wild-type virus (143). Besides swine, the virulence of FMDV with Lpro mutation was investigated in bovine (145). In this study, a 57 nucleotide in-frame insertion was made in the region between two functional AUG initiation codons of Lpro. Both wild-type and insertional Lpro mutant viruses established primary infection in the nasopharyngeal mucosa with subsequent dissemination to the lungs of the cattle. Insertional Lpro mutant FMDV, however, replicated slower and showed quantitatively lower viral loads in secretions and infected tissues and reduced clinical disease, indicating the Lpro mutant FMDV was attenuated in cattle. In another study, a recombinant FMDV was generated to completely delete the Lpro gene. The Lpro-deficient FMDV provided 100% protection in cattle from challenges with wild-type virus (146), demonstrating the potential use of Lpro mutants in developing live attenuated vaccines for FMD.
Alphavirus is the sole genus of the family Togaviridae, consisting of a group of positive-sense, single-stranded RNA viruses. Sindbis virus and Ross River virus are prototype viruses in the Alphavirus genus. Sindbis virus is one of the most widely distributed mosquito-borne viruses and is constantly found in insects and vertebrates. Sindbis virus infection causes polyarthritis, rash, and fever, although most infections are asymptomatic. It has been used as a model virus to study the molecular biology of togaviruses. The major virulence factor of the Sindbis virus is nsp1, which takes part in the inhibition of JAK-STAT signaling (Figure 1) (147). The neurovirulent strain AR86 inhibits tyrosine phosphorylation of STAT1 and STAT2, whereas two other closely-related strains, Girdwood and TR339, do not cause significant disease in adult mice and inhibit the JAK-STAT signaling pathway relatively inefficiently (147). Further, threonine at position 538 in the nsp1 protein of strain AR86 was identified as required for efficient disruption of STAT1 activation, while introducing threonine at position 538 to the Girdwood strain fully restored the ability to inhibit the JAK-STAT signaling and virulence in mice (147–149). A similar pathogenic effect has been demonstrated for the Ross River virus, a distantly related alphavirus, indicating that nsp1-mediated IFN suppression function can be removed from alphaviruses, and live-attenuated vaccine candidates can be developed (150–152).
Flaviviruses are vector-borne viruses transmitted by arthropods. Flaviviruses cause a tremendous disease burden for humans and animals, causing millions of human infections annually. West Nile virus (WNV) can cause acute encephalitis and high morbidity especially in horses and birds. The flaviviruses suppress host innate immune responses during infection, and the viral protein NS5 functions as an IFN antagonist by inhibiting the JAK-STAT signaling pathway (Figure 1) (64). NS5 inhibits IFN-dependent STAT1 phosphorylation or STAT2 degradation (153, 154). Kunjin virus is a naturally attenuated subtype of WNV. The molecular analyses of the Kunjin virus have identified serine 653 in NS5 as a potent amino acid residue that participates in viral attenuation and IFN modulation (154). After changing the amino acid residue S653 of NS5 to F653, the Kunjin virus restored the ability to downregulate JAK-STAT signaling. Similarly, the amino acid change from phenylalanine to serine in NS5 of the virulent strain of WNV resulted in the loss of the ability to inhibit the JAK-STAT signaling (154). In a mouse model, highly virulent WNV and WNV-like African Koutango virus infections produced severe neurological disease and higher morbidity. In addition, the enhanced virulence of WNV was associated with poor viral clearance and poor induction of neutralizing antibodies (155). Analogous to WNV, the virulence of the ZIKV results from the degradation of STAT2 mediated by NS5 (65). However, the IFN degradation by ZIKV NS5 is host species-restricted and functional for human and nonhuman primate but not for mouse (65). Like ZIKV, DENV NS5 does not antagonize IFN signaling in mice since the DENV NS5 protein does not bind to murine STAT2 (Figure 1) (66). Hence, the inability of the NS5 protein to bind to murine STAT2 induces IFN to greatly limit DENV replication in mice (66). Such studies highlight the importance of NS5 as a virulence factor and a target for constructing live attenuated flavivirus vaccines.
Paramyxoviruses constitute a group of negative-sense, single-stranded, non-segmented RNA viruses. Bovine respiratory syncytial virus (BRSV) is classified in the Orthopneumovirus genus of the Paramyxoviridae family. BRSV is a significant cause of respiratory disease in cattle and a major contributor to the bovine respiratory disease (BRD) complex (156). Cattle vaccinated with the formalin-inactivated virus show enhanced severity when infected post-vaccination, suggesting the need for alternative BRSV vaccines (157). The genome of RSV codes for ten genes. NS1 and NS2 proteins have been identified as viral antagonists for the IFN system of hosts (67–70) (Figure 1). Various strategies using reverse genetics to remove viral IFN antagonists are being considered to generate attenuated BRSV vaccines (71). BRSV was engineered to delete the NS1 or NS2 gene, and the NS1 or NS2 gene-lacking BRSV induced higher levels IFNs than wild-type BRSV in bovine nasal fibroblasts and bronchoalveolar macrophages of immunized cattle (157). Furthermore, the recombinant BRSV was attenuated in IFN-competent cells in vitro and in calves, demonstrating that the NS1 and NS2 proteins are the critical determinants for the virulence of BRSV virulence. Recombinant BRSV lacking either NS1 or NS2 also induced higher BRSV-specific antibody titers in calves and greater priming of BRSV-specific, IFN-gamma-producing CD4(+) T cells for the protection against challenges with virulent BRSV (157). This finding delivers a prospect for developing a live attenuated BRSV vaccine.
The IFN antagonism has also been studied for human RSV as well. Recombinant human RSV with the NS2 deletion was highly attenuated in the lower respiratory tract in chimpanzees and induced significant resistance to challenges with wild-type RSV (71, 72) (Figure 1). These findings demonstrate that the deletion of an IFN antagonist from RSV is clinically attenuated and may provide increased protective immunity. However, controversial findings were reported in African green monkeys after evaluation of a series of recombinant human RSV. RSV with the NS2 deletion was not attenuated, whereas RSV with a double deletion of both NS1 and NS2 was over-attenuated and did not provide sufficient protection against wild-type RSV challenge (158). However, recombinant RSV with a double-deletion of M2-2 and NS2 exhibited attenuation and protection in monkeys. Despite the conflicting data, the results implicate the potential of IFN-deficient RSV as a live attenuated vaccine candidate (158, 159). The vaccine potential of IFN-suppression-deficient RSVs was further supported by the assessment in children (160–162). RSV/ΔNS2/Δ1313/I1314L contains three attenuating elements: deletion of the NS2 gene, deletion of codon 1313 in the RSV polymerase gene, and stabilizing missense mutation of I1314L in the polymerase. This triple mutant RSV was evaluated in RSV-seronegative children and shown to be restricted in replication but immunogenic and primed for potent antibody responses after natural exposure to wild-type RSV (161, 162). Taken together, the deletion of the NS2 gene leads to attenuation and immunogenicity of RSV. It validates the strategy to develop live attenuated vaccines by deleting the IFN-modulating function from RSV using reverse genetics.
The measles virus (MeV) in the Morbillivirus genus is a highly immunotropic pathogen that can cause significant childhood morbidity and mortality worldwide. MeV infection induces immunosuppression in the host contributing to secondary infections and mortality (163). The MeV P gene codes for three proteins by mRNA editing; P as an essential polymerase cofactor, and V and C that function as viral antagonists of the IFN pathways (164, 165). The V protein is a multi-functional protein inhibiting IFN responses (Figure 1). The V protein interferes with IFN signaling by blocking STAT1/STAT2 nuclear translocation (53–56). In addition, it inactivates STAT1 and Tyk2 phosphorylation (57) and blocks the JAK1 function for IFN suppression (58, 59). The V protein also interacts with MDA5 and the RIG-I/TRIM25 regulatory complex in the IFN production pathway and inhibits downstream signaling (166–168). Besides the V protein, the C protein has also been determined to interfere with IFN transcription.
Furthermore, the nuclear localization signal and efficient nuclear accumulation are critical for the C protein to downregulate IFN-β production (Figure 1). Compared to the wild-type virus, a mutation in the nuclear localization signal of the C protein is a marker common to all vaccine strains of MeV (169). Amino acids essential for preventing STAT1 nuclear translocation were examined in the V and P proteins by screening the sequence of recombinant virus that could not antagonize STAT1 function, and three residues were identified in the shared domain of the P and V proteins; Y110, V112, and H115, with the Y110 being the most critical residue (164). A mutant virus was generated to harbor the three mutations and was used to assess the virulence in rhesus monkeys (170, 171). The inoculated monkeys showed a short duration of viremia and the absence of skin rash and other clinical signs observed with wild-type virus. This triple-amino acids mutant virus controlled the inflammatory response less efficiently, as measured by enhanced transcription of interleukin-6 and TNF-α in peripheral blood mononuclear cells from infected hosts. However, neutralizing antibody titers and virus-specific T-cell responses were equivalent in animals infected with either virus (172). These findings indicate that efficient MeV interactions with STAT1 are required to sustain virulence in a natural host by controlling the inflammatory response against the virus. Overall, these findings suggest that IFN-suppression defective MeV may have the potential as the vaccine for immunocompromised individuals.
Nipah virus is an emerging zoonotic pathogen, causing encephalitis and respiratory illness in humans and pigs. It belongs to the Henipavirus genus of the family Paramyxoviridae. Similar to MeV, the P gene of the Nipah virus codes for four proteins by mRNA editing; phosphoprotein P and three accessory proteins W, V, and C (173). These proteins possess a distinct IFN-antagonist activity, including the W protein acting as the inhibitor of the TLR-3 pathway, whereas proteins V and C function on the IFN signaling by interacting with STAT1/STAT2 (102, 174–177). However, the recombinant Nipah virus lacking the V, C or W protein still suppressed the IFN response as with the wild-type virus, indicating that the lack of each accessory protein does not significantly affect the inhibition of IFN signaling (174). Ferret challenge studies using a recombinant Nipha virus with deletion of the STAT1-binding motif also demonstrated the minor role of P, V, and W proteins in inhibiting IFN signaling (178, 179).
Influenza viruses belong to the family Orthomyxoviridae and contain single-stranded, negative-sense, eight segmented RNAs as the genome. Of seven genera of the family, influenza A and B viruses are of concern since they are frequently associated with respiratory disease in humans and animals. Two main subtypes are circulating in human population; H1N1 and H3N2. Of eight segments of the genomic RNA, segment 8 expresses two distinct proteins, nonstructural protein 1 (NS1) and nuclear export protein (NEP; also called NS2), using different reading frames from the same RNA segment. While NEP mediates the nuclear export of virion RNAs by acting as an adaptor between viral RNP complexes and the nuclear export machinery of the cell, NS1 is the viral antagonist for the IFN response of hosts (76, 180) (Figure 1). Influenza virus NS1 protein directly inhibits the production of IFNs by targeting RIG-I (180). In addition, the NS1 protein upregulates the inhibitors of JAK-STAT signaling and the suppressors of cytokine signaling (SOCS) family 1 and SOCS family 3 (76, 181), resulting in the downregulation of antiviral protein expression. NS1 protein also indirectly inhibits the IFN signal pathway and interacts with ISGs to antagonize the host’s antiviral response (180). Based on such information, influenza viruses were engineered to modify the NS1 functions. The NS1-modified viruses appeared clinically attenuated and retained the immunogenicity in various species of animals, including mice (182–185), pigs (186–188), horses (189, 190), poultry (191–194), macaques (195) as well as in humans (196–198). Influenza A virus containing a partial deletion in the NS1 gene provides solid evidence for clinical attenuation in animals while providing protective immunity against virulent challenges with the wild-type virus (199). Influenza virus A/WSN/33 (H1N1) expressing the mutant NS1 R38A/K41A showed a robust reduction of viral titers in the lungs of mice but triggered high levels of IFN-α/β production in the lung tissues (200). In addition, the NS1 R38A/K41A NS1 mutant virus induced high titers of neutralizing antibodies against heterologous influenza A virus and provided 100% protection in a mouse model against wild-type virus (200). Immunization of mice with the H1N1 influenza virus containing a shortened NS1 gene also showed the enhancement of influenza-specific CD8+ T-cellular response in the lungs and the reduction of proinflammatory cytokines with a lower extent of leukocyte infiltration after heterologous challenges, indicating that NS1-truncated influenza virus modifies not only effector T-cells but also specific immunoregulatory mechanisms (185). Influenza B viruses with NS1 mutations were also attenuated in animals while inducing adequate protection against both homologous and heterologous subtype challenges with influenza B viruses in BALB/c and C57BL/6 PKR(-/-) mice (201). Furthermore, the influenza B viruses with a truncated NS1 gene induced comparable cellular and humoral immune responses in both aged and young mice (202). It should be noted that most inbred mouse strains have deletions or point mutations in ISG Mx1 (203) which is an important antiviral factor in influenza virus infection. Therefore, using mice as a model to study IFN-influenza virus interaction should be of concern. Taken all together, these data demonstrated that the IFN-deficient system is applicable for manufacturing the IFN-sensitive influenza vaccine viruses.
Similar studies have been conducted for the swine influenza virus (SIV). Swine influenza is caused by the type A virus and regularly causes outbreaks in pigs. Swine influenza can cause high levels of illness in pig herds, but the mortality is low. Influenza viruses that circulate in swine are very different from influenza viruses circulating in people. Swine influenza viruses change genetically constantly. Pigs can be infected by avian influenza, human influenza, and swine influenza viruses. During the coinfection of pigs with influenza viruses from different species, the viruses can reassort, and new viruses can emerge as a mix of swine, human, and avian influenza viruses. Three main subtypes of influenza A virus have been isolated in pigs in the U.S.; H1N1, H3N2, and H1N2. Inactivated vaccines are less effective in protecting pigs, and thus, live-attenuated vaccines have been licensed and available in the U.S (204).. The SIV strain A/Swine/Texas/4199-2/98 (TX/98) H3N2 was used to investigate the role of NS1 protein for virulence, and the swine influenza virus NS1 mutants were shown to be attenuated in pigs (186, 187). Immunization of pigs with SIV NS1 mutant via the intranasal route provided complete protection against homologous and antigenically variant heterologous challenges (205, 206). Moreover, H3N2 NS1 mutant SIV-inoculated pigs displayed attenuated clinical symptoms and reduced viral titers despite a minor reduction in lung lesions when challenged with H1N1 heterosubtype SIV (205). A chimeric virus between the bat influenza virus and SIV was constructed to express a truncated NS1 protein. This virus induced remarkably higher levels of mucosal IgA response and antigen-specific IFN-γ secreting cells against the challenge virus in the lungs of immunized pigs (207).
The protective role of type I IFNs has also been studied for avian influenza virus in chickens. Avian influenza viruses continually circulate among wild birds and poultry worldwide, and the control of emerging influenza viruses for pandemic threats requires broadly protective vaccines. Influenza viruses with truncations in NS1 protein have shown broad-spectrum protection in birds and mammals, which has been correlated with the elevated IFN responses in vaccinated animals (208, 209). Immunologically improved strains of the avian influenza virus were identified by screening the subpopulation of viral vaccines (194). These strains appeared to have a small deletion in the NS1 protein or a single amino acid substitution in the polymerase 2 (PB2). These naturally occurring mutant viruses exhibited enhanced IFN-inducing phenotypes and protective immunity in chickens (194). Compared to an inactivated avian influenza vaccine, live-replicating NS1 mutant-avian influenza virus induced a more solid innate and highly cross-reactive serum antibody responses in immunologically immature chicken (210). Another study using H5N3 NS1 mutant virus also displayed similar levels of protection higher induction of IFN-β (211). However, the mutant virus reverted to wild-type phenotype within five back-passages in chickens, raising a concern about the stability of the NS1 mutant avian influenza vaccine (211). Taken all together, influenza viruses containing a truncated NS1 gene can boost a higher level of IFNs and adaptive immunity and confers protection from heterologous challenges in different species of animals. Such findings demonstrate that IFN-suppression negative mutant virus can be reprogramed to divert to an alternative vaccine candidate for veterinary diseases.
Coronaviruses (CoVs) infect humans and animals, causing a variety of diseases with varying severity. Emerging and reemerging CoVs include porcine epidemic diarrhea virus (PEDV), porcine delta-coronavirus (PDCoV), Middle East respiratory syndrome coronavirus (MERS-CoV), swine acute diarrhea syndrome coronavirus, canine-like alphacoronavirus, SARS-CoV-1, and SARS-CoV-2 (212, 213). CoVs are divided into four genera; Alpha-CoV, Beta-CoV, Gamma-CoV, and Delta-CoV. Among these, only alpha-CoVs and beta-CoVs harbor nonstructural protein 1 (nsp1), which inhibits antiviral host responses. The IFN antagonistic function has been removed from the Alpha-CoV transmissible gastroenteritis virus (TGEV) and PEDV and examined for the effect of IFN suppression on viral pathogenesis in pigs. A recombinant TGEV was constructed to alter the specific C-terminal motif of nsp1, based on the crystal structure. The nsp1 mutation did not affect viral replication in cells but significantly reduced clinical outcome and pathogenicity in pigs (214). PEDV is also a significant veterinary pathogen in swine that requires a better vaccine for control. PEDV infection results in enormous economic losses to the pork industry worldwide and has emerged in the U.S. in 2014. Multiple proteins of PEDV have been shown to inhibit IFN responses during infection (214–219). The nsp1 protein of PEDV is the most potent viral IFN antagonist (214), and three residues of F44, N93, and N95 of nsp1 are critical for both type I and type III IFN suppression (52, 220). PEDV nsp1 suppressed both types of IFN responses by interrupting the IRF3 and CREB-binding protein association and suppressing transcription factors (52) (Figure 1). A replication-competent PEDV mutant with an IFN inactive version of nsp1 induced IFN response in IFN-responsive cells (52). It was further demonstrated that PEDV carrying the nsp1 N93/95A mutation triggered a significantly higher level of IFN response and induced 100% protection from severe diarrhea and death in neonatal piglets post-challenge (220). These findings suggest that nsp1 is an essential virulence determinant for CoVs, providing a potential paradigm for the development of a new vaccine based on IFN modification.
Nonstructural protein 15 (nsp15) of PEDV is the viral endoribonuclease (EndoU) and has an additional function as the IFN antagonist. For PEDV nsp15, three residues of H226, H241, and K282 were identified as critical amino acids for endoribonuclease activity. PEDV nsp15 can directly degrade the RNA levels of TBK1 and IRF3 and suppress the production of IFNs and ISGs, demonstrating that PEDV antagonizes host’s innate response to facilitate its replication (Figure 1). The endoribonuclease activity was removed from PEDV. The nsp15-modified PEDV reduced IFN antagonism with enhanced production of both type I and type III IFNs in cells and was clinically attenuated in infected piglets (216).
Nonstructural protein 16 (nsp16) of CoVs is 2’-O-methyltransferase (2’-O-MTase), and the nsp16 protein of PEDV also contains the methyltransferase. The methyltransferase activity was removed from nsp16 of PEDV. The nsp16 mutant PEDV increased type I IFN production in pigs and conferred complete protection following virulent PEDV challenge (221). Furthermore, infection with the inactive version of nsp1, nsp15, and nsp16 induced total and neutralizing antibody responses, and upon challenge with wild-type PEDV, no detectable clinical symptoms were observed in pigs (52). The pathogenic significance of 2’-O-MTase was also studied for SARS-CoV-1 and MERS-CoV. Both viruses with the nsp16 mutation resulted in IFN-based virulence attenuation and conferred the protection of mice from parental virus challenges (222, 223).
Among animal CoVs, mouse hepatitis virus (MHV) has extensively been investigated (212). MHV is a beta-CoV and has been a research model to understand CoV genome replication, transcription, and pathogenesis. Studies showed that removal of IFN antagonism yielded stronger immunity to MHV. The nsp14 protein of MHV is a multifunctional protein with the N7-methyltransferase (N7-MTase) activity and is highly conserved among different CoVs. A N7-MTase-deficient recombinant MHV was constructed by replacing aspartic acid at position 330 and tyrosine at position 414 of nsp14, each with alanine (224). The N7-MTase-deficient MHV was highly attenuated in mice and showed delayed IFN production in vivo. Furthermore, this nsp14 mutant MHV induced an improved and long-term humoral immune responses and conferred complete protection against a lethal-dose of MHV (224). This study demonstrates the potential application of IFN antagonism to the design of live attenuated vaccines against CoVs circulating in humans and animals.
Arteriviruses in the order Nidovirales infect a diverse species of mammals, including horses, pigs, possums, primates, and rodents (225). Arteriviruses have positive-sense, single-stranded RNA genomes and produce enveloped spherical particles. Porcine reproductive and respiratory syndrome virus (PRRSV) has evolved to carry various strategies for type I IFN antagonism and to evade host immune response. In PRRSV-infected pigs, the IFN response is meager and remains low in the lungs where the virus actively replicates (226, 227). In controlling PRRSV infection in pigs, viral suppression of innate immunity, delayed response of the adaptive immunity, and antigenic heterogeneity of PRRSV are the keys (228). Studies attempted to express various types of IFNs using PRRSV as a vector. IFN-expressing PRRSV increased the IFN levels in pigs after immunization and enhanced the protection against the secondary PRRSV challenge (127, 128). The porcine IFN-α-expressing plasmid as an adjuvant for PRRSV vaccines also induced a prolonged adaptive immune response in pigs (129). Instead of exogenous administration of IFNs, endogenous expression of IFNs seems to be a better way to enhance adaptive immunity. Indeed, a PRRSV strain of IFN-inducing phenotype PRRSV has been shown to improve the neutralizing antibody production (229).
The nsp1β protein of PRRSV is a potent IFN antagonist inhibiting both types I IFN production and downstream signaling (60–63) (Figure 1). Mutant PRRSVs were generated to remove IFN-suppression function from nsp1β, and pigs infected with nsp1β-mutant PRRSVs induced higher levels of IFN-α and ISGs (230, 231). The NK cell function and the IFN-γ level were increased in the lungs of pigs infected with the nsp1β mutant virus (230). Moreover, pigs inoculated with a nsp1β-mutant PRRSV exhibited shorter duration and lower titers of viremia, and a more robust PRRSV-specific antibody response. The neutralizing antibody titers were also higher than those of control pigs, indicating that the IFN-suppression-negative PRRSV mutants are clinically attenuated (231). These studies demonstrate the role of type I IFNs in priming the adaptive immune response in pigs and provide evidence that IFN antagonism-deficient PRRSV may be developed as a new vaccine candidate. Removing IFN antagonism from the virus is a reasonable strategy for developing next-generation vaccines.
Evidence is available for another arterivirus, the equine arteritis virus (EAV), for the removal of type I IFNs in developing adaptive immunity and viral clearance. The nsp2 protein of EAV contains DUB activity and is responsible for the viral suppression of innate immunity (232). Thus, a DUB-negative mutant EAV was constructed. Vaccination of horses with the DUB-negative mutant virus induced higher levels of IFNs and adaptive immune responses (233). Taken together, the removal of IFN antagonism is an important strategy for developing novel vaccines for arteriviruses.
NF-κB is a transcription factor and functions as a hub of complex signaling networks in the cell. NF-κB signaling contributes to immunity, inflammation, cell growth, development, cancer, and other cellular processes. The NF-κB pathway links pathogen and cellular signals and organizes cell resistance to invading pathogens. Many viruses have developed distinct mechanisms to suppress or activate the NF-κB signaling pathway to promote virus replication or cell proliferation (234). Virus-induced NF-κB activation is linked to overproduction and uncontrolled release of proinflammatory markers resulting in a “cytokine storm”. SARS-CoV-2 activates the NF-κB signaling pathway and causes cytokine storm-like acute respiratory distress syndrome (235, 236). Targeting the NF-κB pathway has been considered a potential treatment for COVID-19 patients (236). The influenza virus can also activate NF-κB signaling and induces cytokine storm-like symptoms in infected hosts (237). Activation of NF-κB and production of proinflammatory cytokines are often synergistic during coinfection. Studies using PRRSV in pigs show that the virus can activate NF-κB signaling and elevate proinflammatory cytokine production when coinfected with Streptococcus suis. The cytokine storm-like production of inflammatory cytokines results in more severe clinical outcomes in coinfected animals (238). Thus, viral activation of NF-κB signaling and a cytokine storm-like event are additional concerns for attenuated viral vaccines. A study showed that the reduction of systemic inflammation and a boost of protective responses when combining an NF-κB inhibitor as an adjuvant with a vaccine in the influenza mice challenge model (239). Since the NF-κB signaling contributes to numerous cellular processes, potential off-target effects by NF-κB inhibition may be actuated and cause a systematically undesirable consequence. Thus, further research is required for developing NF-κB-activation-negative viruses as feasible live-attenuated vaccines. Nevertheless, by the fine-tuning removal of the NF-κB activation, a newly generated virus is anticipated to relieve the clinical severity attributed to cytokine upregulations. By reprogramming the NF-κB activation, the newly generated virus is expected to relieve the clinical severity attributed to cytokine upregulation.
This concept is useful for pigs with PRRSV as a new approach to vaccine developments. For activation of NF-κB, the nucleocapsid (N) protein is the sole viral protein for PRRSV (240, 241). Studies show that the nuclear localization signal (NLS) in N is the essential domain for binding to PIAS1. PIAS1 is the negative regulator for NF-κB, so the binding of viral N protein to PIAS1 results in the release of p65 from PIAS1 and renders NF-κB activation (241). The NLS-modified N protein loses the ability for NF-κB activation and induces significantly lower levels of NF-κB-mediated inflammatory cytokines in cells (241). Studies using the NLS-null PRRSV infection show milder clinical signs and a shorter duration of viremia with higher titers of antibodies in infected pigs, demonstrating the association between the NLS motif of N protein and clinical attenuation of PRRSV infection (242–244). These studies provide evidence that NF-κB-activation-negative virus can induce milder clinical symptoms and higher humoral immune response than wild-type virus during coinfection of pigs. NF-κB activation function can be eliminated from RNA viruses using reverse genetics tools, and clinically attenuated vaccine candidates can be developed.
Numerous mechanisms for how viruses fight against the host immune system have been identified. Since type I IFNs are critical components of the host innate immunity and for the development and maturation of adaptive immunity, many attempts have been made to remove IFN suppression functions from different viruses. Such approaches may enhance the IFN response upon immunization. With the help of recent advances in molecular virology and reverse genetics technology, it is possible to construct mutant viruses of which phenotypes are IFN-suppression-deficient. Subsequently, IFN-suppression-negative viruses have shown to induce better immune responses and confer partial to complete protections from both homologous and heterologous challenges in the respective animal species, such as mice, pigs, chickens, and cattle (Table 1). Still remaining challenges include:
1) Identification of IFN-suppressive genes and active sites from target viruses: It requires an in-depth understanding of the structure-function relationships of viral proteins and their role in the IFN signaling cascade.
2) Multigenic nature of viral IFN antagonists and introduction of multiple mutations: Viruses have evolved to equip with multiple proteins for IFN suppression such that their antagonism can be compensated even when one protein fails by evolutionary mutations. The strategy to overcome the multigenic nature of viral IFN antagonism is to introduce mutations to the most potent antagonist or simultaneous mutations to several antagonists.
3) Lethality of mutations for viral infectivity and viability: Many RNA viruses carry only a limited number of genes essential for replication and survival in a host. Deletion of a functional gene is often lethal for infectivity, and accurate measurements of the gene-wide mutations may be needed for the possible substitutions of amino acids and the removal of IFN antagonism.
4) Reversion of mutant viruses to virulence: RNA-dependent RNA polymerase lacks a proof-reading activity with an exception of coronaviruses that are found to contain an exonuclease as a proofreading enzyme. As a result, RNA viruses are prone to high rates of genetic mutations which may allow the reversion of IFN-negative attenuated viral mutants back to virulence. Deletion of the functional domain or amino acids instead of point mutations has been successful for some viruses to block their reversion to wild-type.
5) Over-attenuation of mutant viruses: Deletions or substitutions in a functional domain may confer over-attenuation of virus mutants. Continuous passages in cell culture have been attempted to increase viral titers. Cell culture passages in established lines may not provide immunological pressure but can allow viral adaptation for efficient replication and production of high viral titers.
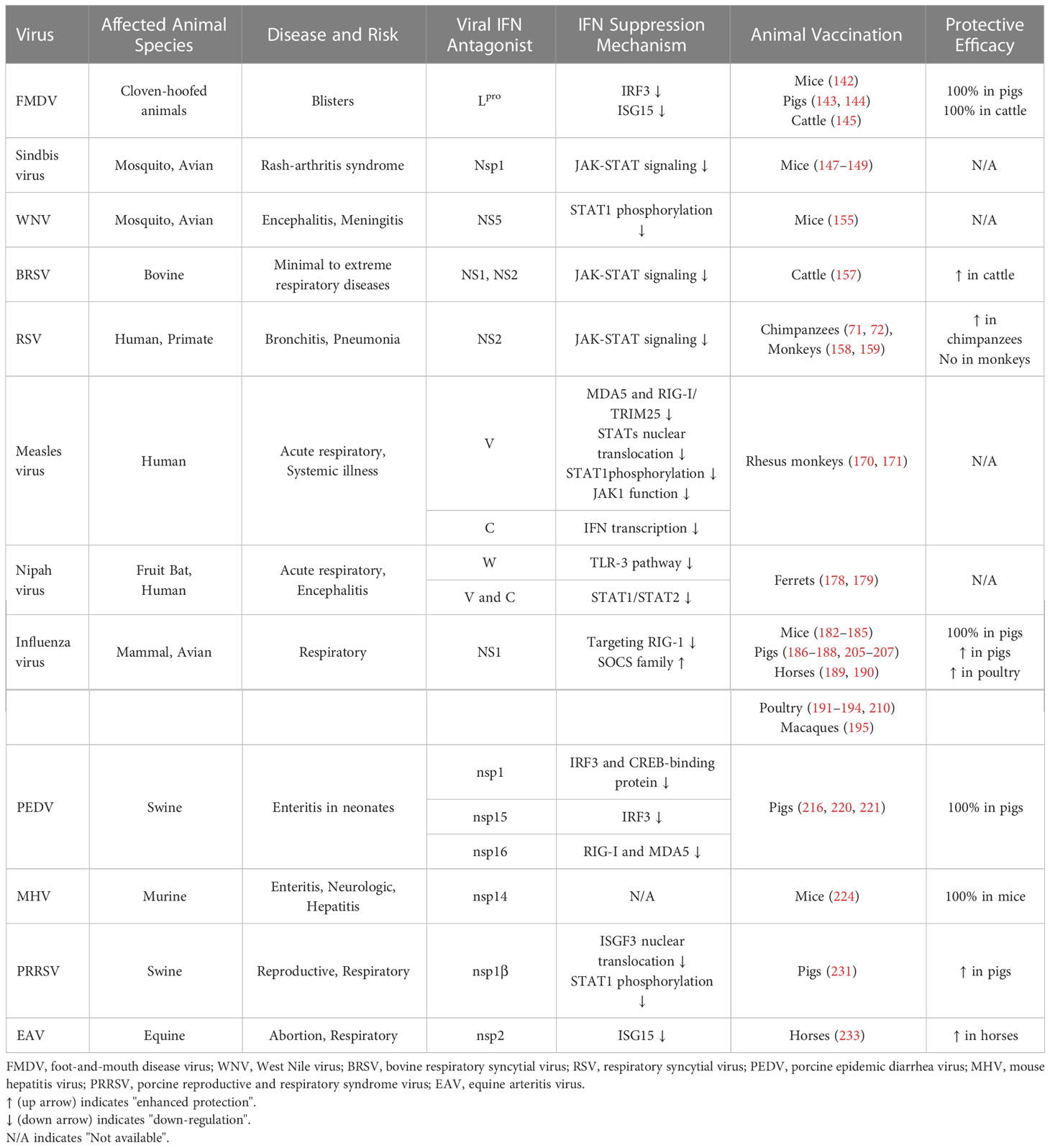
Table 1 The reprogramming of mutant viruses that their IFN evasion functions were eliminated by reverse genetics and the protective efficacies in natural host animal species following vaccination and virulent challenges.
The combination of research and new ideas will be vital in successfully developing future vaccines for veterinary diseases.
C-MS and DY contributed to developing concepts and design of the review article. C-MS performed the literature search, retrieved relevant articles, and prepared a draft of the original manuscript. C-MS and DY wrote the manuscript. YD, RR, and QW provided critical comments and revised the manuscript. All authors contributed to the article and approved the submitted version.
This project was supported by Agriculture and Food Research Initiative (AFRI) Competitive Grants nos. 2018-67015-28287 and 2023-67015-39710 from the U.S. Department of Agriculture (USDA) National Institute of Food and Agriculture (NIFA) to DY.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev (2001) 14:778–809. doi: 10.1128/CMR.14.4.778-809.2001
2. Katze MG, Fornek JL, Palermo RE, Walters K-A, Korth MJ. Innate immune modulation by RNA viruses: emerging insights from functional genomics. Nat Rev Immunol (2008) 8:644–54. doi: 10.1038/nri2377
3. Royer DJ, Carr MM, Chucair-Elliott AJ, Halford WP, Carr DJJ. Impact of type I interferon on the safety and immunogenicity of an experimental live-attenuated herpes simplex virus 1 vaccine in mice. J Virol (2017) 91:e02342–16. doi: 10.1128/JVI.02342-16
4. Ma Z, Li Z, Dong L, Yang T, Xiao S. Reverse genetic systems: rational design of coronavirus live attenuated vaccines with immune sequelae. Adv Virus Res (2020) 107:383–416. doi: 10.1016/bs.aivir.2020.06.003
5. Marsh G, Tannock G. The role of reverse genetics in the development of vaccines against respiratory viruses. Expert Opin Biol Ther (2005) 5:369–80. doi: 10.1517/14712598.5.3.369
6. Sadler AJ, Williams BRG. Interferon-inducible antiviral effectors. Nat Rev Immunol (2008) 8:559–68. doi: 10.1038/nri2314
7. Liu Y-J. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol (2005) 23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633
8. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev (2009) 22:240–73. doi: 10.1128/CMR.00046-08
9. Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol (2014) 32:461–88. doi: 10.1146/annurev-immunol-032713-120156
10. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, et al. IPS-1, an adaptor triggering RIG-i- and Mda5-mediated type I interferon induction. Nat Immunol (2005) 6:981–8. doi: 10.1038/ni1243
11. Xu L-G, Wang Y-Y, Han K-J, Li L-Y, Zhai Z, Shu H-B. VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol Cell (2005) 19:727–40. doi: 10.1016/j.molcel.2005.08.014
12. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124:783–801. doi: 10.1016/j.cell.2006.02.015
13. Lin R, Heylbroeck C, Pitha PM, Hiscott J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol (1998) 18:2986–96. doi: 10.1128/MCB.18.5.2986
14. Dragan AI, Hargreaves VV, Makeyeva EN, Privalov PL. Mechanisms of activation of interferon regulator factor 3: the role of c-terminal domain phosphorylation in IRF-3 dimerization and DNA binding. Nucleic Acids Res (2007) 35:3525–34. doi: 10.1093/nar/gkm142
15. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature (2005) 434:772–7. doi: 10.1038/nature03464
16. Caamanão J, Hunter CA. NF-κB family of transcription factors: central regulators of innate and adaptive immune functions. Clin Microbiol Rev (2002) 15:414–29. doi: 10.1128/CMR.15.3.414-429.2002
17. Napetschnig J, Wu H. Molecular basis of NF-κB signaling. Annu Rev Biophys (2013) 42:443–68. doi: 10.1146/annurev-biophys-083012-130338
18. Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat Med (2007) 13:543–51. doi: 10.1038/nm1590
19. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
20. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14:36–49. doi: 10.1038/nri3581
21. Gessani S, Conti L, Del Cornò M, Belardelli F. Type I interferons as regulators of human antigen presenting cell functions. Toxins (Basel) (2014) 6:1696–723. doi: 10.3390/toxins6061696
22. Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, et al. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol (2003) 4:1009–15. doi: 10.1038/ni978
23. Spadaro F, Lapenta C, Donati S, Abalsamo L, Barnaba V, Belardelli F, et al. IFN-α enhances cross-presentation in human dendritic cells by modulating antigen survival, endocytic routing, and processing. Blood (2012) 119:1407–17. doi: 10.1182/blood-2011-06-363564
24. Baranek T, Vu Manh T-P, Alexandre Y, Maqbool MA, Cabeza JZ, Tomasello E, et al. Differential responses of immune cells to type I interferon contribute to host resistance to viral infection. Cell Host Microbe (2012) 12:571–84. doi: 10.1016/j.chom.2012.09.002
25. Paolini R, Bernardini G, Molfetta R, Santoni A. NK cells and interferons. Cytokine Growth Fact Rev (2015) 26:113–20. doi: 10.1016/j.cytogfr.2014.11.003
26. Fuertes MB, Woo S-R, Burnett B, Fu Y-X, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends Immunol (2013) 34:67–73. doi: 10.1016/j.it.2012.10.004
27. Havenar-Daughton C, Kolumam GA, Murali-Krishna K. Cutting edge: the direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J Immunol (2006) 176:3315–9. doi: 10.4049/jimmunol.176.6.3315
28. Le Bon A, Thompson C, Kamphuis E, Durand V, Rossmann C, Kalinke U, et al. Cutting edge: enhancement of antibody responses through direct stimulation of b and T cells by type I IFN. J Immunol (2006) 176:2074–8. doi: 10.4049/jimmunol.176.4.2074
29. Aichele P, Unsoeld H, Koschella M, Schweier O, Kalinke U, Vucikuja S. Cutting edge: CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J Immunol (2006) 176:4525–9. doi: 10.4049/jimmunol.176.8.4525
30. Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Cutting edge: type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol (2005) 174:4465–9. doi: 10.4049/jimmunol.174.8.4465
31. Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med (2005) 202:637–50. doi: 10.1084/jem.20050821
32. Urban SL, Berg LJ, Welsh RM. Type 1 interferon licenses naïve CD8 T cells to mediate anti-viral cytotoxicity. Virology (2016) 493:52–9. doi: 10.1016/j.virol.2016.03.005
33. Pinto AK, Daffis S, Brien JD, Gainey MD, Yokoyama WM, Sheehan KCF, et al. A temporal role of type I interferon signaling in CD8+ T cell maturation during acute West Nile virus infection. PloS Pathog (2011) 7:e1002407. doi: 10.1371/journal.ppat.1002407
34. Agarwal P, Raghavan A, Nandiwada SL, Curtsinger JM, Bohjanen PR, Mueller DL, et al. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J Immunol (2009) 183:1695–704. doi: 10.4049/jimmunol.0900592
35. Thompson LJ, Kolumam GA, Thomas S, Murali-Krishna K. Innate inflammatory signals induced by various pathogens differentially dictate the IFN-I dependence of CD8 T cells for clonal expansion and memory formation. J Immunol (2006) 177:1746–54. doi: 10.4049/jimmunol.177.3.1746
36. Bach P, Kamphuis E, Odermatt B, Sutter G, Buchholz CJ, Kalinke U. Vesicular stomatitis virus glycoprotein displaying retrovirus-like particles induce a type I IFN receptor-dependent switch to neutralizing IgG antibodies. J Immunol (2007) 178:5839–47. doi: 10.4049/jimmunol.178.9.5839
37. Fink K, Lang KS, Manjarrez-Orduno N, Junt T, Senn BM, Holdener M, et al. Early type I interferon-mediated signals on b cells specifically enhance antiviral humoral responses. Eur J Immunol (2006) 36:2094–105. doi: 10.1002/eji.200635993
38. Purtha WE, Chachu KA, Virgin HW, Diamond MS. Early b-cell activation after West Nile virus infection requires Alpha/Beta interferon but not antigen receptor signaling. J Virol (2008) 82:10964–74. doi: 10.1128/JVI.01646-08
39. Kiefer K, Oropallo MA, Cancro MP, Marshak-Rothstein A. Role of type I interferons in the activation of autoreactive b cells. Immunol Cell Biol (2012) 90:498–504. doi: 10.1038/icb.2012.10
40. Chen L-F, Greene WC. Shaping the nuclear action of NF-κB. Nat Rev Mol Cell Biol (2004) 5:392–401. doi: 10.1038/nrm1368
41. Vallabhapurapu S, Karin M. Regulation and function of NF-κB transcription factors in the immune system. Annu Rev Immunol (2009) 27:693–733. doi: 10.1146/annurev.immunol.021908.132641
42. Kagoya Y, Yoshimi A, Kataoka K, Nakagawa M, Kumano K, Arai S, et al. Positive feedback between NF-κB and TNF-α promotes leukemia-initiating cell capacity. J Clin Invest (2014) 124:528–42. doi: 10.1172/JCI68101
43. Liu B, Yang R, Wong KA, Getman C, Stein N, Teitell MA, et al. Negative regulation of NF-κB signaling by PIAS1. Mol Cell Biol (2005) 25:1113–23. doi: 10.1128/MCB.25.3.1113-1123.2005
44. Haeberle HA, Casola A, Gatalica Z, Petronella S, Dieterich H-J, Ernst PB, et al. IκB kinase is a critical regulator of chemokine expression and lung inflammation in respiratory syncytial virus infection. J Virol (2004) 78:2232–41. doi: 10.1128/JVI.78.5.2232-2241.2004
45. Tian B, Zhang Y, Luxon BA, Garofalo RP, Casola A, Sinha M, et al. Identification of NF-κB-Dependent gene networks in respiratory syncytial virus-infected cells. J Virol (2002) 76:6800–14. doi: 10.1128/JVI.76.13.6800-6814.2002
46. Xu Q, Tang Y, Huang G. Innate immune responses in RNA viral infection. Front Med (2021) 15:333–46. doi: 10.1007/s11684-020-0776-7
47. Bowie AG, Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol (2008) 8:911–22. doi: 10.1038/nri2436
48. Medina GN, Segundo FD-S, Stenfeldt C, Arzt J, de los Santos T. The different tactics of foot-and-Mouth disease virus to evade innate immunity. Front Microbiol (2018) 9:2644. doi: 10.3389/fmicb.2018.02644
49. Rodríguez Pulido M, Sáiz M. Molecular mechanisms of foot-and-Mouth disease virus targeting the host antiviral response. Front Cell Infect Microbiol (2017) 7:252. doi: 10.3389/fcimb.2017.00252
50. Peng J, Yi J, Yang W, Ren J, Wen Y, Zheng H, et al. Advances in foot-and-Mouth disease virus proteins regulating host innate immunity. Front Microbiol (2020) 11:2046. doi: 10.3389/fmicb.2020.02046
51. Sui C, Jiang D, Wu X, Liu S, Li F, Pan L, et al. Inhibition of antiviral innate immunity by foot-and-Mouth disease virus l pro through interaction with the n-terminal domain of swine RNase l. J Virol (2021) 95:e0036121. doi: 10.1128/JVI.00361-21
52. Deng X, Buckley AC, Pillatzki A, Lager KM, Faaberg KS, Baker SC. Inactivating three interferon antagonists attenuates pathogenesis of an enteric coronavirus. J Virol (2020) 94:e00565–20. doi: 10.1128/JVI.00565-20
53. Palosaari H, Parisien J-P, Rodriguez JJ, Ulane CM, Horvath CM. STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J Virol (2003) 77:7635–44. doi: 10.1128/JVI.77.13.7635-7644.2003
54. Ramachandran A, Parisien J-P, Horvath CM. STAT2 is a primary target for measles virus V protein-mediated Alpha/Beta interferon signaling inhibition. J Virol (2008) 82:8330–8. doi: 10.1128/JVI.00831-08
55. Caignard G, Bouraï M, Jacob Y, Tangy F, Vidalain P-O. Inhibition of IFN-α/β signaling by two discrete peptides within measles virus V protein that specifically bind STAT1 and STAT2. Virology (2009) 383:112–20. doi: 10.1016/j.virol.2008.10.014
56. Nagano Y, Sugiyama A, Kimoto M, Wakahara T, Noguchi Y, Jiang X, et al. The measles virus V protein binding site to STAT2 overlaps that of IRF9. J Virol (2020) 94:e01169-20. doi: 10.1128/JVI.01169-20
57. Chinnakannan SK, Nanda SK, Baron MD. Morbillivirus V proteins exhibit multiple mechanisms to block type 1 and type 2 interferon signalling pathways. PloS One (2013) 8:e57063. doi: 10.1371/journal.pone.0057063
58. Caignard G, Guerbois M, Labernardière J-L, Jacob Y, Jones LM, Wild F, et al. Measles virus V protein blocks Jak1-mediated phosphorylation of STAT1 to escape IFN-α/β signaling. Virology (2007) 368:351–62. doi: 10.1016/j.virol.2007.06.037
59. Yokota S, Saito H, Kubota T, Yokosawa N, Amano K, Fujii N. Measles virus suppresses interferon-α signaling pathway: suppression of Jak1 phosphorylation and association of viral accessory proteins, c and V, with interferon-α receptor complex. Virology (2003) 306:135–46. doi: 10.1016/S0042-6822(02)00026-0
60. Chen Z, Lawson S, Sun Z, Zhou X, Guan X, Christopher-Hennings J, et al. Identification of two auto-cleavage products of nonstructural protein 1 (nsp1) in porcine reproductive and respiratory syndrome virus infected cells: nsp1 function as interferon antagonist. Virology (2010) 398:87–97. doi: 10.1016/j.virol.2009.11.033
61. Patel D, Nan Y, Shen M, Ritthipichai K, Zhu X, Zhang Y-J. Porcine reproductive and respiratory syndrome virus inhibits type I interferon signaling by blocking STAT1/STAT2 nuclear translocation. J Virol (2010) 84:11045–55. doi: 10.1128/JVI.00655-10
62. Beura LK, Sarkar SN, Kwon B, Subramaniam S, Jones C, Pattnaik AK, et al. Porcine reproductive and respiratory syndrome virus nonstructural protein 1β modulates host innate immune response by antagonizing IRF3 activation. J Virol (2010) 84:1574–84. doi: 10.1128/JVI.01326-09
63. Wang R, Nan Y, Yu Y, Zhang Y-J. Porcine reproductive and respiratory syndrome virus Nsp1 inhibits interferon-activated JAK/STAT signal transduction by inducing karyopherin-1 degradation. J Virol (2013) 87:5219–28. doi: 10.1128/JVI.02643-12
64. Best SM. The many faces of the flavivirus NS5 protein in antagonism of type I interferon signaling. J Virol (2017) 91:e01970-16. doi: 10.1128/JVI.01970-16
65. Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, et al. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe (2016) 19:882–90. doi: 10.1016/j.chom.2016.05.009
66. Ashour J, Morrison J, Laurent-Rolle M, Belicha-Villanueva A, Plumlee CR, Bernal-Rubio D, et al. Mouse STAT2 restricts early dengue virus replication. Cell Host Microbe (2010) 8:410–21. doi: 10.1016/j.chom.2010.10.007
67. Ramaswamy M, Shi L, Monick MM, Hunninghake GW, Look DC. Specific inhibition of type I interferon signal transduction by respiratory syncytial virus. Am J Respir Cell Mol Biol (2004) 30:893–900. doi: 10.1165/rcmb.2003-0410OC
68. Spann KM, Tran K-C, Chi B, Rabin RL, Collins PL. Suppression of the induction of alpha, beta, and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages. J Virol (2004) 78:6705–5. doi: 10.1128/JVI.78.12.6705.2004
69. Whelan JN, Tran KC, van Rossum DB, Teng MN. Identification of respiratory syncytial virus nonstructural protein 2 residues essential for exploitation of the host ubiquitin system and inhibition of innate immune responses. J Virol (2016) 90:6453–63. doi: 10.1128/JVI.00423-16
70. Yang P, Zheng J, Wang S, Liu P, Xie M, Zhao D. Respiratory syncytial virus nonstructural proteins 1 and 2 are crucial pathogenic factors that modulate interferon signaling and treg cell distribution in mice. Virology (2015) 485:223–32. doi: 10.1016/j.virol.2015.07.016
71. Collins PL. New generation live vaccines against human respiratory syncytial virus designed by reverse genetics. Proc Am Thorac Soc (2005) 2:166–73. doi: 10.1513/pats.200501-011AW
72. Whitehead SS, Bukreyev A, Teng MN, Firestone C-Y, St. Claire M, Elkins WR, et al. Recombinant respiratory syncytial virus bearing a deletion of either the NS2 or SH gene is attenuated in chimpanzees. J Virol (1999) 73:3438–42. doi: 10.1128/JVI.73.4.3438-3442.1999
73. Krug RM. Functions of the influenza a virus NS1 protein in antiviral defense. Curr Opin Virol (2015) 12:1–6. doi: 10.1016/j.coviro.2015.01.007
74. Min J-Y, Krug RM. The primary function of RNA binding by the influenza a virus NS1 protein in infected cells: inhibiting the 2′-5′ oligo (A) synthetase/RNase l pathway. Proc Natl Acad Sci (2006) 103:7100–5. doi: 10.1073/pnas.0602184103
75. Hale BG, Randall RE, Ortin J, Jackson D. The multifunctional NS1 protein of influenza a viruses. J Gen Virol (2008) 89:2359–76. doi: 10.1099/vir.0.2008/004606-0
76. Jia D, Rahbar R, Chan RWY, Lee SMY, Chan MCW, Wang BX, et al. Influenza virus non-structural protein 1 (NS1) disrupts interferon signaling. PloS One (2010) 5:e13927. doi: 10.1371/journal.pone.0013927
77. Shah KM, Stewart SE, Wei W, Woodman CBJ, O’Neil JD, Dawson CW, et al. The EBV-encoded latent membrane proteins, LMP2A and LMP2B, limit the actions of interferon by targeting interferon receptors for degradation. Oncogene (2009) 28:3903–14. doi: 10.1038/onc.2009.249
78. Luquin E, Larrea E, Civeira MP, Prieto J, Aldabe R. HCV structural proteins interfere with interferon-alpha Jak/STAT signalling pathway. Antiviral Res (2007) 76:194–7. doi: 10.1016/j.antiviral.2007.06.004
79. La Rocca SA, Herbert RJ, Crooke H, Drew TW, Wileman TE, Powell PP. Loss of interferon regulatory factor 3 in cells infected with classical swine fever virus involves the n-terminal protease, n pro. J Virol (2005) 79:7239–47. doi: 10.1128/JVI.79.11.7239-7247.2005
80. Baigent SJ, Zhang G, Fray MD, Flick-Smith H, Goodbourn S, McCauley JW. Inhibition of beta interferon transcription by noncytopathogenic bovine viral diarrhea virus is through an interferon regulatory factor 3-dependent mechanism. J Virol (2002) 76:8979–88. doi: 10.1128/JVI.76.18.8979-8988.2002
81. Gil LHVG, Ansari IH, Vassilev V, Liang D, Lai VCH, Zhong W, et al. The amino-terminal domain of bovine viral diarrhea virus n pro protein is necessary for Alpha/Beta interferon antagonism. J Virol (2006) 80:900–11. doi: 10.1128/JVI.80.2.900-911.2006
82. Brzoízka K, Finke S, Conzelmann K-K. Identification of the rabies virus Alpha/Beta interferon antagonist: phosphoprotein p interferes with phosphorylation of interferon regulatory factor 3. J Virol (2005) 79:7673–81. doi: 10.1128/JVI.79.12.7673-7681.2005
83. Prescott JB, Hall PR, Bondu-Hawkins VS, Ye C, Hjelle B. Early innate immune responses to sin nombre hantavirus occur independently of IFN regulatory factor 3, characterized pattern recognition receptors, and viral entry. J Immunol (2007) 179:1796–802. doi: 10.4049/jimmunol.179.3.1796
84. Alff PJ, Gavrilovskaya IN, Gorbunova E, Endriss K, Chong Y, Geimonen E, et al. The pathogenic NY-1 hantavirus G1 cytoplasmic tail inhibits RIG-i- and TBK-1-Directed interferon responses. J Virol (2006) 80:9676–86. doi: 10.1128/JVI.00508-06
85. Wathelet MG, Orr M, Frieman MB, Baric RS. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J Virol (2007) 81:11620–33. doi: 10.1128/JVI.00702-07
86. Devaraj SG, Wang N, Chen Z, Chen Z, Tseng M, Barretto N, et al. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J Biol Chem (2007) 282:32208–21. doi: 10.1074/jbc.M704870200
87. Frieman M, Ratia K, Johnston RE, Mesecar AD, Baric RS. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-κB signaling. J Virol (2009) 83:6689–705. doi: 10.1128/JVI.02220-08
88. Yoo HS, Yoo D. COVID-19 and veterinarians for one health, zoonotic- and reverse-zoonotic transmissions. J Vet Sci (2020) 21:e51. doi: 10.4142/jvs.2020.21.e51
89. Moustaqil M, Ollivier E, Chiu H-P, Van Tol S, Rudolffi-Soto P, Stevens C, et al. SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): implications for disease presentation across species. Emerg Microbes Infect (2021) 10:178–95. doi: 10.1080/22221751.2020.1870414
90. Wang W, Zhou Z, Xiao X, Tian Z, Dong X, Wang C, et al. SARS-CoV-2 nsp12 attenuates type I interferon production by inhibiting IRF3 nuclear translocation. Cell Mol Immunol (2021) 18:945–53. doi: 10.1038/s41423-020-00619-y
91. Kouwaki T, Nishimura T, Wang G, Oshiumi H. RIG-I-Like receptor-mediated recognition of viral genomic RNA of severe acute respiratory syndrome coronavirus-2 and viral escape from the host innate immune responses. Front Immunol (2021) 12:700926. doi: 10.3389/fimmu.2021.700926
92. Sui L, Zhao Y, Wang W, Wu P, Wang Z, Yu Y, et al. SARS-CoV-2 membrane protein inhibits type I interferon production through ubiquitin-mediated degradation of TBK1. Front Immunol (2021) 12:662989. doi: 10.3389/fimmu.2021.662989
93. Freitas RS, Crum TF, Parvatiyar K. SARS-CoV-2 spike antagonizes innate antiviral immunity by targeting interferon regulatory factor 3. Front Cell Infect Microbiol (2022) 11:789462. doi: 10.3389/fcimb.2021.789462
94. Rose KM, Elliott R, Martiínez-Sobrido L, Garciía-Sastre A, Weiss SR. Murine coronavirus delays expression of a subset of interferon-stimulated genes. J Virol (2010) 84:5656–69. doi: 10.1128/JVI.00211-10
95. Han M, Du Y, Song C, Yoo D. Degradation of CREB-binding protein and modulation of type I interferon induction by the zinc finger motif of the porcine reproductive and respiratory syndrome virus nsp1α subunit. Virus Res (2013) 172:54–65. doi: 10.1016/j.virusres.2012.12.012
96. Kim O, Sun Y, Lai FW, Song C, Yoo D. Modulation of type I interferon induction by porcine reproductive and respiratory syndrome virus and degradation of CREB-binding protein by non-structural protein 1 in MARC-145 and HeLa cells. Virology (2010) 402:315–26. doi: 10.1016/j.virol.2010.03.039
97. Chen Z, Liu S, Sun W, Chen L, Yoo D, Li F, et al. Nuclear export signal of PRRSV NSP1α is necessary for type I IFN inhibition. Virology (2016) 499:278–87. doi: 10.1016/j.virol.2016.07.008
98. Song C, Krell P, Yoo D. Nonstructural protein 1α subunit-based inhibition of NF-κB activation and suppression of interferon-β production by porcine reproductive and respiratory syndrome virus. Virology (2010) 407:268–80. doi: 10.1016/j.virol.2010.08.025
99. Subramaniam S, Kwon B, Beura LK, Kuszynski CA, Pattnaik AK, Osorio FA. Porcine reproductive and respiratory syndrome virus non-structural protein 1 suppresses tumor necrosis factor-alpha promoter activation by inhibiting NF-κB and Sp1. Virology (2010) 406:270–9. doi: 10.1016/j.virol.2010.07.016
100. Ke H, Han M, Kim J, Gustin KE, Yoo D. Porcine reproductive and respiratory syndrome virus nonstructural protein 1 beta interacts with nucleoporin 62 to promote viral replication and immune evasion. J Virol (2019) 93:e00469–19. doi: 10.1128/JVI.00469-19
101. Gotoh B, Komatsu T, Takeuchi K, Yokoo J. Paramyxovirus strategies for evading the interferon response. Rev Med Virol (2002) 12:337–57. doi: 10.1002/rmv.357
102. Rodriguez JJ, Parisien J-P, Horvath CM. Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J Virol (2002) 76:11476–83. doi: 10.1128/JVI.76.22.11476-11483.2002
103. Rodriguez JJ, Wang L-F, Horvath CM. Hendra virus V protein inhibits interferon signaling by preventing STAT1 and STAT2 nuclear accumulation. J Virol (2003) 77:11842–5. doi: 10.1128/JVI.77.21.11842-11845.2003
104. Didcock L, Young DF, Goodbourn S, Randall RE. Sendai Virus and simian virus 5 block activation of interferon-responsive genes: importance for virus pathogenesis. J Virol (1999) 73:3125–33. doi: 10.1128/JVI.73.4.3125-3133.1999
105. Garcin D, Latorre P, Kolakofsky D. Sendai Virus c proteins counteract the interferon-mediated induction of an antiviral state. J Virol (1999) 73:6559–65. doi: 10.1128/JVI.73.8.6559-6565.1999
106. Garcin D, Marq J-B, Strahle L, le Mercier P, Kolakofsky D. All four Sendai virus c proteins bind Stat1, but only the larger forms also induce its mono-ubiquitination and degradation. Virology (2002) 295:256–65. doi: 10.1006/viro.2001.1342
107. Jones M, Davidson A, Hibbert L, Gruenwald P, Schlaak J, Ball S, et al. Dengue virus inhibits alpha interferon signaling by reducing STAT2 expression. J Virol (2005) 79:5414–20. doi: 10.1128/JVI.79.9.5414-5420.2005
108. Parisien J-P, Lau JF, Horvath CM. STAT2 acts as a host range determinant for species-specific paramyxovirus interferon antagonism and simian virus 5 replication. J Virol (2002) 76:6435–41. doi: 10.1128/JVI.76.13.6435-6441.2002
109. Le VTK, Trilling M, Wilborn M, Hengel H, Zimmermann A. Human cytomegalovirus interferes with signal transducer and activator of transcription (STAT) 2 protein stability and tyrosine phosphorylation. J Gen Virol (2008) 89:2416–26. doi: 10.1099/vir.0.2008/001669-0
110. Senft AP, Taylor RH, Lei W, Campbell SA, Tipper JL, Martinez MJ, et al. Respiratory syncytial virus impairs macrophage IFN-α/β– and IFN-γ–stimulated transcription by distinct mechanisms. Am J Respir Cell Mol Biol (2010) 42:404–14. doi: 10.1165/rcmb.2008-0229OC
111. Jie Z, Dinwiddie DL, Senft AP, Harrod KS. Regulation of STAT signaling in mouse bone marrow derived dendritic cells by respiratory syncytial virus. Virus Res (2011) 156:127–33. doi: 10.1016/j.virusres.2011.01.007
112. Ramaswamy M, Shi L, Varga SM, Barik S, Behlke MA, Look DC. Respiratory syncytial virus nonstructural protein 2 specifically inhibits type I interferon signal transduction. Virology (2006) 344:328–39. doi: 10.1016/j.virol.2005.09.009
113. Elliott J, Lynch OT, Suessmuth Y, Qian P, Boyd CR, Burrows JF, et al. Respiratory syncytial virus NS1 protein degrades STAT2 by using the elongin-cullin E3 ligase. J Virol (2007) 81:3428–36. doi: 10.1128/JVI.02303-06
114. Barnard P, McMillan NAJ. The human papillomavirus E7 oncoprotein abrogates signaling mediated by interferon-α. Virology (1999) 259:305–13. doi: 10.1006/viro.1999.9771
115. Zhang R, Fang L, Wang D, Cai K, Zhang H, Xie L, et al. Porcine bocavirus NP1 negatively regulates interferon signaling pathway by targeting the DNA-binding domain of IRF9. Virology (2015) 485:414–21. doi: 10.1016/j.virol.2015.08.005
116. Verweij MC, Wellish M, Whitmer T, Malouli D, Lapel M, Jonjić S, et al. Varicella viruses inhibit interferon-stimulated JAK-STAT signaling through multiple mechanisms. PloS Pathog (2015) 11:e1004901. doi: 10.1371/journal.ppat.1004901
117. Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, et al. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN -β promoter. Proc Natl Acad Sci (2004) 101:17264–9. doi: 10.1073/pnas.0407639101
118. Hagmaier K, Stock N, Goodbourn S, Wang L-F, Randall R. A single amino acid substitution in the V protein of nipah virus alters its ability to block interferon signalling in cells from different species. J Gen Virol (2006) 87:3649–53. doi: 10.1099/vir.0.82261-0
119. Vidy A, Chelbi-Alix M, Blondel D. Rabies virus p protein interacts with STAT1 and inhibits interferon signal transduction pathways. J Virol (2005) 79:14411–20. doi: 10.1128/JVI.79.22.14411-14420.2005
120. Wang R, Nan Y, Yu Y, Zhang Y-J. Porcine reproductive and respiratory syndrome virus Nsp1β inhibits interferon-activated JAK/STAT signal transduction by inducing karyopherin-α1 degradation. J Virol (2013) 87:5219–28. doi: 10.1128/JVI.02643-12
121. Nice TJ, Osborne LC, Tomov VT, Artis D, Wherry EJ, Virgin HW. Type I interferon receptor deficiency in dendritic cells facilitates systemic murine norovirus persistence despite enhanced adaptive immunity. PloS Pathog (2016) 12:e1005684. doi: 10.1371/journal.ppat.1005684
122. Kimman TG, Cornelissen LA, Moormann RJ, Rebel JMJ, Stockhofe-Zurwieden N. Challenges for porcine reproductive and respiratory syndrome virus (PRRSV) vaccinology. Vaccine (2009) 27:3704–18. doi: 10.1016/j.vaccine.2009.04.022
123. Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis c: a randomised trial. Lancet (2001) 358:958–65. doi: 10.1016/S0140-6736(01)06102-5
124. Radkowski M, Gallegos-Orozco JF, Jablonska J, Colby TV, Walewska-Zielecka B, Kubicka J, et al. Persistence of hepatitis c virus in patients successfully treated for chronic hepatitis c. Hepatology (2005) 41:106–14. doi: 10.1002/hep.20518
125. Shiffman ML, Hofmann CM, Contos MJ, Luketic VA, Sanyal AJ, Sterling RK, et al. A randomized, controlled trial of maintenance interferon therapy for patients with chronic hepatitis c virus and persistent viremia. Gastroenterology (1999) 117:1164–72. doi: 10.1016/S0016-5085(99)70402-6
126. Audsley KM, Wagner T, Ta C, Newnes HV, Buzzai AC, Barnes SA, et al. IFNβ is a potent adjuvant for cancer vaccination strategies. Front Immunol (2021) 12:735133. doi: 10.3389/fimmu.2021.735133
127. Sang Y, Rowland RRR, Blecha F. Antiviral regulation in porcine monocytic cells at different activation states. J Virol (2014) 88:11395–410. doi: 10.1128/JVI.01714-14
128. Sang Y, Shi J, Sang W, Rowland RRR, Blecha F. Replication-competent recombinant porcine reproductive and respiratory syndrome (PRRS) viruses expressing indicator proteins and antiviral cytokines. Viruses (2012) 4:102–16. doi: 10.3390/v4010102
129. Meier WA, Husmann RJ, Schnitzlein WM, Osorio FA, Lunney JK, Zuckermann FA. Cytokines and synthetic double-stranded RNA augment the T helper 1 immune response of swine to porcine reproductive and respiratory syndrome virus. Vet Immunol Immunopathol (2004) 102:299–314. doi: 10.1016/j.vetimm.2004.09.012
130. Liu L, Fan W, Zhang H, Zhang S, Cui L, Wang M, et al. Interferon as a mucosal adjuvant for an influenza vaccine in pigs. Virol Sin (2019) 34:324–33. doi: 10.1007/s12250-019-00102-7
131. Bracci L, Canini I, Puzelli S, Sestili P, Venditti M, Spada M, et al. Type I IFN is a powerful mucosal adjuvant for a selective intranasal vaccination against influenza virus in mice and affects antigen capture at mucosal level. Vaccine (2005) 23:2994–3004. doi: 10.1016/j.vaccine.2004.12.006
132. Bracci L, Canini I, Venditti M, Spada M, Puzelli S, Donatelli I, et al. Type I IFN as a vaccine adjuvant for both systemic and mucosal vaccination against influenza virus. Vaccine (2006) 24:S56–7. doi: 10.1016/j.vaccine.2005.01.121
133. Cheng G, Zhao X, Yan W, Wang W, Zuo X, Huang K, et al. Alpha interferon is a powerful adjuvant for a recombinant protein vaccine against foot-and-mouth disease virus in swine, and an effective stimulus of in vivo immune response. Vaccine (2007) 25:5199–208. doi: 10.1016/j.vaccine.2007.04.089
134. Su C, Duan X, Zheng J, Liang L, Wang F, Guo L. IFN-α as an adjuvant for adenovirus-vectored FMDV subunit vaccine through improving the generation of T follicular helper cells. PloS One (2013) 8:e66134. doi: 10.1371/journal.pone.0066134
135. Chinsangaram J, Moraes MP, Koster M, Grubman MJ. Novel viral disease control strategy: adenovirus expressing alpha interferon rapidly protects swine from foot-and-Mouth disease. J Virol (2003) 77:1621–5. doi: 10.1128/JVI.77.2.1621-1625.2003
136. O’Brien L, Perkins S, Williams A, Eastaugh L, Phelps A, Wu J, et al. Alpha interferon as an adenovirus-vectored vaccine adjuvant and antiviral in Venezuelan equine encephalitis virus infection. J Gen Virol (2009) 90:874–82. doi: 10.1099/vir.0.006833-0
137. Wu JQH, Barabé ND, Huang Y-M, Rayner GA, Christopher ME, Schmaltz FL. Pre- and post-exposure protection against Western equine encephalitis virus after single inoculation with adenovirus vector expressing interferon alpha. Virology (2007) 369:206–13. doi: 10.1016/j.virol.2007.07.024
138. Baer GM, Moore SA, Shaddock JH, Levy HB. An effective rabies treatment in exposed monkeys: a single dose of interferon inducer and vaccine. Bull World Health Organ (1979) 57:807–13.
139. Hayer SS, Ranjan R, Biswal JK, Subramaniam S, Mohapatra JK, Sharma GK, et al. Quantitative characteristics of the foot-and-mouth disease carrier state under natural conditions in India. Transbound Emerg Dis (2018) 65:253–60. doi: 10.1111/tbed.12627
140. Swatek KN, Aumayr M, Pruneda JN, Visser LJ, Berryman S, Kueck AF, et al. Irreversible inactivation of ISG15 by a viral leader protease enables alternative infection detection strategies. Proc Natl Acad Sci (2018) 115:2371–6. doi: 10.1073/pnas.1710617115
141. Medina GN, Azzinaro P, Ramirez-Medina E, Gutkoska J, Fang Y, Diaz-San Segundo F, et al. Impairment of the DeISGylation activity of foot-and-Mouth disease virus lpro causes attenuation In vitro and In vivo. J Virol (2020) 94:e00341-20. doi: 10.1128/JVI.00341-20
142. Piccone ME, Rieder E, Mason PW, Grubman MJ. The foot-and-mouth disease virus leader proteinase gene is not required for viral replication. J Virol (1995) 69:5376–82. doi: 10.1128/jvi.69.9.5376-5382.1995
143. Díaz-San Segundo F, Weiss M, Pérez-Martín E, Dias CC, Grubman MJ, de los Santos T. Inoculation of swine with foot-and-Mouth disease SAP-mutant virus induces early protection against disease. J Virol (2012) 86:1316–27. doi: 10.1128/JVI.05941-11
144. Azzinaro PA, Medina GN, Rai D, Ramirez-Medina E, Spinard E, Rodriguez-Calzada M, et al. Mutation of FMDV lpro H138 residue drives viral attenuation in cell culture and in vivo in swine. Front Vet Sci (2022) 9:1028077. doi: 10.3389/fvets.2022.1028077
145. Arzt J, Pacheco JM, Stenfeldt C, Rodriguez LL. Pathogenesis of virulent and attenuated foot-and-mouth disease virus in cattle. Virol J (2017) 14:89. doi: 10.1186/s12985-017-0758-9
146. Uddowla S, Hollister J, Pacheco JM, Rodriguez LL, Rieder E. A safe foot-and-Mouth disease vaccine platform with two negative markers for differentiating infected from vaccinated animals. J Virol (2012) 86:11675–85. doi: 10.1128/JVI.01254-12
147. Simmons JD, Wollish AC, Heise MT. A determinant of sindbis virus neurovirulence enables efficient disruption of Jak/STAT signaling. J Virol (2010) 84:11429–39. doi: 10.1128/JVI.00577-10
148. Heise MT, Simpson DA, Johnston RE. A single amino acid change in nsP1 attenuates neurovirulence of the sindbis-group alphavirus S.A.AR86. J Virol (2000) 74:4207–13. doi: 10.1128/JVI.74.9.4207-4213.2000
149. Wollish AC, Ferris MT, Blevins LK, Loo Y-M, Gale M, Heise MT. An attenuating mutation in a neurovirulent sindbis virus strain interacts with the IPS-1 signaling pathway in vivo. Virology (2013) 435:269–80. doi: 10.1016/j.virol.2012.09.008
150. Cruz CC, Suthar MS, Montgomery SA, Shabman R, Simmons J, Johnston RE, et al. Modulation of type I IFN induction by a virulence determinant within the alphavirus nsP1 protein. Virology (2010) 399:1–10. doi: 10.1016/j.virol.2009.12.031
151. Jupille HJ, Oko L, Stoermer KA, Heise MT, Mahalingam S, Gunn BM, et al. Mutations in nsP1 and PE2 are critical determinants of Ross river virus-induced musculoskeletal inflammatory disease in a mouse model. Virology (2011) 410:216–27. doi: 10.1016/j.virol.2010.11.012
152. Stoermer Burrack KA, Hawman DW, Jupille HJ, Oko L, Minor M, Shives KD, et al. Attenuating mutations in nsP1 reveal tissue-specific mechanisms for control of Ross river virus infection. J Virol (2014) 88:3719–32. doi: 10.1128/JVI.02609-13
153. Best SM, Morris KL, Shannon JG, Robertson SJ, Mitzel DN, Park GS, et al. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J Virol (2005) 79:12828–39. doi: 10.1128/JVI.79.20.12828-12839.2005
154. Laurent-Rolle M, Boer EF, Lubick KJ, Wolfinbarger JB, Carmody AB, Rockx B, et al. The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAK-STAT signaling. J Virol (2010) 84:3503–15. doi: 10.1128/JVI.01161-09
155. Prow NA, Setoh YX, Biron RM, Sester DP, Kim KS, Hobson-Peters J, et al. The West Nile virus-like flavivirus koutango is highly virulent in mice due to delayed viral clearance and the induction of a poor neutralizing antibody response. J Virol (2014) 88:9947–62. doi: 10.1128/JVI.01304-14
156. Jim K. Impact of bovine respiratory disease (BRD) from the perspective of the Canadian beef producer. Anim Heal Res Rev (2009) 10:109–10. doi: 10.1017/S1466252309990119
157. Valarcher J-F, Furze J, Wyld S, Cook R, Conzelmann K-K, Taylor G. Role of Alpha/Beta interferons in the attenuation and immunogenicity of recombinant bovine respiratory syncytial viruses lacking NS proteins. J Virol (2003) 77:8426–39. doi: 10.1128/JVI.77.15.8426-8439.2003
158. Jin H, Cheng X, Traina-Dorge VL, Park HJ, Zhou H, Soike K, et al. Evaluation of recombinant respiratory syncytial virus gene deletion mutants in African green monkeys for their potential as live attenuated vaccine candidates. Vaccine (2003) 21:3647–52. doi: 10.1016/S0264-410X(03)00426-2
159. Jin H, Zhou H, Cheng X, Tang R, Munoz M, Nguyen N. Recombinant respiratory syncytial viruses with deletions in the NS1, NS2, SH, and M2-2 genes are attenuated in vitro and in vivo. Virology (2000) 273:210–8. doi: 10.1006/viro.2000.0393
160. Wright PF, Karron RA, Madhi SA, Treanor JJ, King JC, O’Shea A, et al. The interferon antagonist NS2 protein of respiratory syncytial virus is an important virulence determinant for humans. J Infect Dis (2006) 193:573–81. doi: 10.1086/499600
161. Karron RA, Luongo C, Mateo JS, Wanionek K, Collins PL, Buchholz UJ. Safety and immunogenicity of the respiratory syncytial virus vaccine RSV/ΔNS2/Δ1313/I1314L in RSV-seronegative children. J Infect Dis (2020) 222:82–91. doi: 10.1093/infdis/jiz408
162. Cunningham CK, Karron RA, Muresan P, Kelly MS, McFarland EJ, Perlowski C, et al. Evaluation of recombinant live-attenuated respiratory syncytial virus (RSV) vaccines RSV/ΔNS2/Δ1313/I1314L and RSV/276 in RSV-seronegative children. J Infect Dis (2022) 226:2069–78. doi: 10.1093/infdis/jiac253
163. Griffin DE. Measles virus-induced suppression of immune responses. Immunol Rev (2010) 236:176–89. doi: 10.1111/j.1600-065X.2010.00925.x
164. Devaux P, von Messling V, Songsungthong W, Springfeld C, Cattaneo R. Tyrosine 110 in the measles virus phosphoprotein is required to block STAT1 phosphorylation. Virology (2007) 360:72–83. doi: 10.1016/j.virol.2006.09.049
165. Gerlier D, Valentin H. Measles virus interaction with host cells and impact on innate immunity. Curr Top Microbiol Immunol (2009) 329:163–91. doi: 10.1007/978-3-540-70523-9_8
166. Parisien J-P, Bamming D, Komuro A, Ramachandran A, Rodriguez JJ, Barber G, et al. A shared interface mediates paramyxovirus interference with antiviral RNA helicases MDA5 and LGP2. J Virol (2009) 83:7252–60. doi: 10.1128/JVI.00153-09
167. Motz C, Schuhmann KM, Kirchhofer A, Moldt M, Witte G, Conzelmann K-K, et al. Paramyxovirus V proteins disrupt the fold of the RNA sensor MDA5 to inhibit antiviral signaling. Sci (80- ) (2013) 339:690–3. doi: 10.1126/science.1230949
168. Sánchez-Aparicio MT, Feinman LJ, García-Sastre A, Shaw ML. Paramyxovirus V proteins interact with the RIG-I/TRIM25 regulatory complex and inhibit RIG-I signaling. J Virol (2018) 92:e01960-17. doi: 10.1128/JVI.01960-17
169. Sparrer KMJ, Pfaller CK, Conzelmann K-K. Measles virus c protein interferes with beta interferon transcription in the nucleus. J Virol (2012) 86:796–805. doi: 10.1128/JVI.05899-11
170. de Vries RD, Lemon K, Ludlow M, McQuaid S, Yüksel S, van Amerongen G, et al. In vivo tropism of attenuated and pathogenic measles virus expressing green fluorescent protein in macaques. J Virol (2010) 84:4714–24. doi: 10.1128/JVI.02633-09
171. Devaux P, Hodge G, McChesney MB, Cattaneo R. Attenuation of V- or c-defective measles viruses: infection control by the inflammatory and interferon responses of rhesus monkeys. J Virol (2008) 82:5359–67. doi: 10.1128/JVI.00169-08
172. Devaux P, Hudacek AW, Hodge G, Reyes-del Valle J, McChesney MB, Cattaneo R. A recombinant measles virus unable to antagonize STAT1 function cannot control inflammation and is attenuated in rhesus monkeys. J Virol (2011) 85:348–56. doi: 10.1128/JVI.00802-10
173. Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, Lam SK, et al. Nipah virus: a recently emergent deadly paramyxovirus. Sci (80- ) (2000) 288:1432–5. doi: 10.1126/science.288.5470.1432
174. Yoneda M, Guillaume V, Sato H, Fujita K, Georges-Courbot M-C, Ikeda F, et al. The nonstructural proteins of nipah virus play a key role in pathogenicity in experimentally infected animals. PloS One (2010) 5:e12709. doi: 10.1371/journal.pone.0012709
175. Keiffer TR, Ciancanelli MJ, Edwards MR, Basler CF. Interactions of the nipah virus p, V, and W proteins across the STAT family of transcription factors. mSphere (2020) 5:e00449-20. doi: 10.1128/mSphere.00449-20
176. Uchida S, Horie R, Sato H, Kai C, Yoneda M. Possible role of the nipah virus V protein in the regulation of the interferon beta induction by interacting with UBX domain-containing protein1. Sci Rep (2018) 8:7682. doi: 10.1038/s41598-018-25815-9
177. Rodriguez JJ, Cruz CD, Horvath CM. Identification of the nuclear export signal and STAT-binding domains of the nipah virus V protein reveals mechanisms underlying interferon evasion. J Virol (2004) 78:5358–67. doi: 10.1128/JVI.78.10.5358-5367.2004
178. Satterfield BA, Cross RW, Fenton KA, Agans KN, Basler CF, Geisbert TW, et al. The immunomodulating V and W proteins of nipah virus determine disease course. Nat Commun (2015) 6:7483. doi: 10.1038/ncomms8483
179. Satterfield BA, Borisevich V, Foster SL, Rodriguez SE, Cross RW, Fenton KA, et al. Antagonism of STAT1 by nipah virus p gene products modulates disease course but not lethal outcome in the ferret model. Sci Rep (2019) 9:16710. doi: 10.1038/s41598-019-53037-0
180. Ji Z, Wang X, Liu X. NS1: a key protein in the “Game” between influenza a virus and host in innate immunity. Front Cell Infect Microbiol (2021) 11:670177. doi: 10.3389/fcimb.2021.670177
181. Pauli E-K, Schmolke M, Wolff T, Viemann D, Roth J, Bode JG, et al. Influenza a virus inhibits type I IFN signaling via NF-κB-Dependent induction of SOCS-3 expression. PloS Pathog (2008) 4:e1000196. doi: 10.1371/journal.ppat.1000196
182. Nogales A, Baker SF, Ortiz-Riaño E, Dewhurst S, Topham DJ, Martínez-Sobrido L. Influenza a virus attenuation by codon deoptimization of the NS gene for vaccine development. J Virol (2014) 88:10525–40. doi: 10.1128/JVI.01565-14
183. Mueller SN, Langley WA, Carnero E, Garciía-Sastre A, Ahmed R. Immunization with live attenuated influenza viruses that express altered NS1 proteins results in potent and protective memory CD8 + T-cell responses. J Virol (2010) 84:1847–55. doi: 10.1128/JVI.01317-09
184. Wang P, Zheng M, Lau S-Y, Chen P, Mok BW-Y, Liu S, et al. Generation of DelNS1 influenza viruses: a strategy for optimizing live attenuated influenza vaccines. MBio (2019) 10:e02180-19. doi: 10.1128/mBio.02180-19
185. Vasilyev K, Shurygina A-P, Sergeeva M, Stukova M, Egorov A. Intranasal immunization with the influenza a virus encoding truncated NS1 protein protects mice from heterologous challenge by restraining the inflammatory response in the lungs. Microorganisms (2021) 9:690. doi: 10.3390/microorganisms9040690
186. Soloírzano A, ebby WRJ, Lager KM, Janke BH, Garciía-Sastre A, Richt JA. Mutations in the NS1 protein of swine influenza virus impair anti-interferon activity and confer attenuation in pigs. J Virol (2005) 79:7535–43. doi: 10.1128/JVI.79.12.7535-7543.2005
187. Vandoorn E, Stadejek W, Parys A, Chepkwony S, Chiers K, Van Reeth K. Pathobiology of an NS1-truncated H3N2 swine influenza virus strain in pigs. J Virol (2022) 96:e00519–22. doi: 10.1128/jvi.00519-22
188. Nogales A, DeDiego ML, Martínez-Sobrido L. Live attenuated influenza a virus vaccines with modified NS1 proteins for veterinary use. Front Cell Infect Microbiol (2022) 12:954811. doi: 10.3389/fcimb.2022.954811
189. Chambers TM, Quinlivan M, Sturgill T, Cullinane A, Horohov DW, Zamarin D, et al. Influenza a viruses with truncated NS1 as modified live virus vaccines: pilot studies of safety and efficacy in horses. Equine Vet J (2009) 41:87–92. doi: 10.2746/042516408X371937
190. Quinlivan M, Zamarin D, Garciía-Sastre A, Cullinane A, Chambers T, Palese P. Attenuation of equine influenza viruses through truncations of the NS1 protein. J Virol (2005) 79:8431–9. doi: 10.1128/JVI.79.13.8431-8439.2005
191. Chen S, Zhu Y, Yang D, Yang Y, Shi S, Qin T, et al. Efficacy of live-attenuated H9N2 influenza vaccine candidates containing NS1 truncations against H9N2 avian influenza viruses. Front Microbiol (2017) 8:1086. doi: 10.3389/fmicb.2017.01086
192. Steel J, Lowen AC, Pena L, Angel M, Soloírzano A, Albrecht R, et al. Live attenuated influenza viruses containing NS1 truncations as vaccine candidates against H5N1 highly pathogenic avian influenza. J Virol (2009) 83:1742–53. doi: 10.1128/JVI.01920-08
193. Wang L, Suarez DL, Pantin-Jackwood M, Mibayashi M, García-Sastre A, Saif YM, et al. Characterization of influenza virus variants with different sizes of the non-structural (NS) genes and their potential as a live influenza vaccine in poultry. Vaccine (2008) 26:3580–6. doi: 10.1016/j.vaccine.2008.05.001
194. Ghorbani A, Abundo MC, Ji H, Taylor KJM, Ngunjiri JM, Lee C-W. Viral subpopulation screening guides in designing a high interferon-inducing live attenuated influenza vaccine by targeting rare mutations in NS1 and PB2 proteins. J Virol (2020) 95:e01722-20. doi: 10.1128/JVI.01722-20
195. Baskin CR, Bielefeldt-Ohmann H, Garciía-Sastre A, Tumpey TM, Van Hoeven N, Carter VS, et al. Functional genomic and serological analysis of the protective immune response resulting from vaccination of macaques with an NS1-truncated influenza virus. J Virol (2007) 81:11817–27. doi: 10.1128/JVI.00590-07
196. Wacheck V, Egorov A, Groiss F, Pfeiffer A, Fuereder T, Hoeflmayer D, et al. A novel type of influenza vaccine: safety and immunogenicity of replication-deficient influenza virus created by deletion of the interferon antagonist NS1. J Infect Dis (2010) 201:354–62. doi: 10.1086/649428
197. Mössler C, Groiss F, Wolzt M, Wolschek M, Seipelt J, Muster T. Phase I/II trial of a replication-deficient trivalent influenza virus vaccine lacking NS1. Vaccine (2013) 31:6194–200. doi: 10.1016/j.vaccine.2013.10.061
198. Nicolodi C, Groiss F, Kiselev O, Wolschek M, Seipelt J, Muster T. Safety and immunogenicity of a replication-deficient H5N1 influenza virus vaccine lacking NS1. Vaccine (2019) 37:3722–9. doi: 10.1016/j.vaccine.2019.05.013
199. Talon J, Salvatore M, O’Neill RE, Nakaya Y, Zheng H, Muster T, et al. Influenza a and b viruses expressing altered NS1 proteins: a vaccine approach. Proc Natl Acad Sci (2000) 97:4309–14. doi: 10.1073/pnas.070525997
200. Chen C, Fan W, Li J, Zheng W, Zhang S, Yang L, et al. A promising IFN-deficient system to manufacture IFN-sensitive influenza vaccine virus. Front Cell Infect Microbiol (2018) 8:127. doi: 10.3389/fcimb.2018.00127
201. Hai R, Martiínez-Sobrido L, Fraser KA, Ayllon J, Garciía-Sastre A, Palese P. Influenza b virus NS1-truncated mutants: live-attenuated vaccine approach. J Virol (2008) 82:10580–90. doi: 10.1128/JVI.01213-08
202. Pica N, Langlois RA, Krammer F, Margine I, Palese P. NS1-truncated live attenuated virus vaccine provides robust protection to aged mice from viral challenge. J Virol (2012) 86:10293–301. doi: 10.1128/JVI.01131-12
203. Staeheli P, Grob R, Meier E, Sutcliffe JG, Haller O. Influenza virus-susceptible mice carry mx genes with a large deletion or a nonsense mutation. Mol Cell Biol (1988) 8:4518–23. doi: 10.1128/mcb.8.10.4518-4523.1988
204. Loving CL, Lager KM, Vincent AL, Brockmeier SL, Gauger PC, Anderson TK, et al. Efficacy in pigs of inactivated and live attenuated influenza virus vaccines against infection and transmission of an emerging H3N2 similar to the 2011-2012 H3N2v. J Virol (2013) 87:9895–903. doi: 10.1128/JVI.01038-13
205. Vincent AL, Ma W, Lager KM, Janke BH, Webby RJ, García-Sastre A, et al. Efficacy of intranasal administration of a truncated NS1 modified live influenza virus vaccine in swine. Vaccine (2007) 25:7999–8009. doi: 10.1016/j.vaccine.2007.09.019
206. Artiaga BL, Morozov I, Ransburgh R, Kwon T, Balaraman V, Indran SV, et al. Evaluating α-galactosylceramide as an adjuvant for live attenuated influenza vaccines in pigs. Anim Dis (2022) 2:19. doi: 10.1186/s44149-022-00051-x
207. Lee J, Li Y, Li Y, Cino-Ozuna AG, Duff M, Lang Y, et al. Bat influenza vectored NS1-truncated live vaccine protects pigs against heterologous virus challenge. Vaccine (2021) 39:1943–50. doi: 10.1016/j.vaccine.2021.02.077
208. Jang H, Ngunjiri JM, Lee C-W. Association between interferon response and protective efficacy of NS1-truncated mutants as influenza vaccine candidates in chickens. PloS One (2016) 11:e0156603. doi: 10.1371/journal.pone.0156603
209. Ngunjiri JM, Ali A, Boyaka P, Marcus PI, Lee CW. In vivo assessment of NS1-truncated influenza virus with a novel SLSYSINWRH motif as a self-adjuvanting live attenuated vaccine. PloS One (2015) 10:e0118934. doi: 10.1371/journal.pone.0118934
210. Jang H, Elaish M KCM, Abundo MC, Ghorbani A, Ngunjiri JM, Lee C-W. Efficacy and synergy of live-attenuated and inactivated influenza vaccines in young chickens. PloS One (2018) 13:e0195285. doi: 10.1371/journal.pone.0195285
211. Brahmakshatriya VR, Lupiani B, Reddy SM. Characterization and evaluation of avian influenza NS1 mutant virus as a potential live and killed DIVA (differentiating between infected and vaccinated animals) vaccine for chickens. Vaccine (2010) 28:2388–96. doi: 10.1016/j.vaccine.2009.12.074
212. Zhang G, Li B, Yoo D, Qin T, Zhang X, Jia Y, et al. Animal coronaviruses and SARS-CoV-2. Transbound Emerg Dis (2021) 68:1097–110. doi: 10.1111/tbed.13791
213. Vlasova AN, Toh T-H, Lee JS-Y, Poovorawan Y, Davis P, Azevedo MSP, et al. Animal alphacoronaviruses found in human patients with acute respiratory illness in different countries. Emerg Microbes Infect (2022) 11:699–702. doi: 10.1080/22221751.2022.2040341
214. Zhang Q, Shi K, Yoo D. Suppression of type I interferon production by porcine epidemic diarrhea virus and degradation of CREB-binding protein by nsp1. Virology (2016) 489:252–68. doi: 10.1016/j.virol.2015.12.010
215. Zhang Q, Ke H, Blikslager A, Fujita T, Yoo D. Type III interferon restriction by porcine epidemic diarrhea virus and the role of viral protein nsp1 in IRF1 signaling. J Virol (2018) 92:e01677-17. doi: 10.1128/JVI.01677-17
216. Deng X, van Geelen A, Buckley AC, O’Brien A, Pillatzki A, Lager KM, et al. Coronavirus endoribonuclease activity in porcine epidemic diarrhea virus suppresses type I and type III interferon responses. J Virol (2019) 93:e02000-18. doi: 10.1128/JVI.02000-18
217. Xu X, Zhang H, Zhang Q, Dong J, Liang Y, Huang Y, et al. Porcine epidemic diarrhea virus e protein causes endoplasmic reticulum stress and up-regulates interleukin-8 expression. Virol J (2013) 10:26. doi: 10.1186/1743-422X-10-26
218. Ding Z, Fang L, Jing H, Zeng S, Wang D, Liu L, et al. Porcine epidemic diarrhea virus nucleocapsid protein antagonizes beta interferon production by sequestering the interaction between IRF3 and TBK1. J Virol (2014) 88:8936–45. doi: 10.1128/JVI.00700-14
219. Wang D, Fang L, Shi Y, Zhang H, Gao L, Peng G, et al. Porcine epidemic diarrhea virus 3C-like protease regulates its interferon antagonism by cleaving NEMO. J Virol (2016) 90:2090–101. doi: 10.1128/JVI.02514-15
220. Niu X, Kong F, Xu J, Liu M, Wang Q. Mutations in porcine epidemic diarrhea virus nsp1 cause increased viral sensitivity to host interferon responses and attenuation In vivo. J Virol (2022) 96:e0046922. doi: 10.1128/jvi.00469-22
221. Hou Y, Ke H, Kim J, Yoo D, Su Y, Boley P, et al. Engineering a live attenuated porcine epidemic diarrhea virus vaccine candidate via inactivation of the viral 2’- O -methyltransferase and the endocytosis signal of the spike protein. J Virol (2019) 93:e00406-19. doi: 10.1128/JVI.00406-19
222. Menachery VD, Gralinski LE, Mitchell HD, Dinnon KH, Leist SR, Yount BL, et al. Middle East respiratory syndrome coronavirus nonstructural protein 16 is necessary for interferon resistance and viral pathogenesis. mSphere (2017) 2:e00346-17. doi: 10.1128/mSphere.00346-17
223. Menachery VD, Yount BL, Josset L, Gralinski LE, Scobey T, Agnihothram S, et al. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2′-O-Methyltransferase activity. J Virol (2014) 88:4251–64. doi: 10.1128/JVI.03571-13
224. Zhang Z, Liu Q, Sun Y, Li J, Liu J, Pan R, et al. Live attenuated coronavirus vaccines deficient in N7-methyltransferase activity induce both humoral and cellular immune responses in mice. Emerg Microbes Infect (2021) 10:1626–37. doi: 10.1080/22221751.2021.1964385
225. Snijder EJ, Kikkert M, Fang Y. Arterivirus molecular biology and pathogenesis. J Gen Virol (2013) 94:2141–63. doi: 10.1099/vir.0.056341-0
226. Albina E, Piriou L, Hutet E, Cariolet R, L’Hospitalier R. Immune responses in pigs infected with porcine reproductive and respiratory syndrome virus (PRRSV). Vet Immunol Immunopathol (1998) 61:49–66. doi: 10.1016/S0165-2427(97)00134-7
227. Van Reeth K, Labarque G, Nauwynck H, Pensaert M. Differential production of proinflammatory cytokines in the pig lung during different respiratory virus infections: correlations with pathogenicity. Res Vet Sci (1999) 67:47–52. doi: 10.1053/rvsc.1998.0277
228. Ke H, Yoo D. The viral innate immune antagonism and an alternative vaccine design for PRRS virus. Vet Microbiol (2017) 209:75–89. doi: 10.1016/j.vetmic.2017.03.014
229. Wang R, Xiao Y, Opriessnig T, Ding Y, Yu Y, Nan Y, et al. Enhancing neutralizing antibody production by an interferon-inducing porcine reproductive and respiratory syndrome virus strain. Vaccine (2013) 31:5537–43. doi: 10.1016/j.vaccine.2013.09.023
230. Li Y, Shyu D-L, Shang P, Bai J, Ouyang K, Dhakal S, et al. Mutations in a highly conserved motif of nsp1β protein attenuate the innate immune suppression function of porcine reproductive and respiratory syndrome virus. J Virol (2016) 90:3584–99. doi: 10.1128/JVI.03069-15
231. Ke H, Han M, Zhang Q, Rowland R, Kerrigan M, Yoo D. Type I interferon suppression-negative and host mRNA nuclear retention-negative mutation in nsp1β confers attenuation of porcine reproductive and respiratory syndrome virus in pigs. Virology (2018) 517:177–87. doi: 10.1016/j.virol.2018.01.016
232. van Kasteren PB, Bailey-Elkin BA, James TW, Ninaber DK, Beugeling C, Khajehpour M, et al. Deubiquitinase function of arterivirus papain-like protease 2 suppresses the innate immune response in infected host cells. Proc Natl Acad Sci (2013) 110:E838–47. doi: 10.1073/pnas.1218464110
233. van Kasteren PB, Knaap RCM, van den Elzen P, Snijder EJ, Balasuriya UBR, van den Born E, et al. In vivo assessment of equine arteritis virus vaccine improvement by disabling the deubiquitinase activity of papain-like protease 2. Vet Microbiol (2015) 178:132–7. doi: 10.1016/j.vetmic.2015.04.018
234. Rahman MM, McFadden G. Modulation of NF-κB signalling by microbial pathogens. Nat Rev Microbiol (2011) 9:291–306. doi: 10.1038/nrmicro2539
235. Su C-M, Wang L, Yoo D. Activation of NF-κB and induction of proinflammatory cytokine expressions mediated by ORF7a protein of SARS-CoV-2. Sci Rep (2021) 11:13464. doi: 10.1038/s41598-021-92941-2
236. Kircheis R, Haasbach E, Lueftenegger D, Heyken WT, Ocker M, Planz O. NF-κB pathway as a potential target for treatment of critical stage COVID-19 patients. Front Immunol (2020) 11:598444. doi: 10.3389/fimmu.2020.598444
237. Gu Y, Zuo X, Zhang S, Ouyang Z, Jiang S, Wang F, et al. The mechanism behind influenza virus cytokine storm. Viruses (2021) 13:1362. doi: 10.3390/v13071362
238. Auray G, Lachance C, Wang Y, Gagnon CA, Segura M, Gottschalk M. Transcriptional analysis of PRRSV-infected porcine dendritic cell response to streptococcus suis infection reveals up-regulation of inflammatory-related genes expression. PloS One (2016) 11:e0156019. doi: 10.1371/journal.pone.0156019
239. Moser BA, Steinhardt RC, Escalante-Buendia Y, Boltz DA, Barker KM, Cassaidy BJ, et al. Increased vaccine tolerability and protection via NF-κB modulation. Sci Adv (2020) 6:eaaz8700. doi: 10.1126/sciadv.aaz8700
240. Wang G, Li L, Yu Y, Tu Y, Tong J, Zhang C, et al. Highly pathogenic porcine reproductive and respiratory syndrome virus infection and induction of apoptosis in bone marrow cells of infected piglets. J Gen Virol (2016) 97:1356–61. doi: 10.1099/jgv.0.000454
241. Ke H, Lee S, Kim J, Liu H-C, Yoo D. Interaction of PIAS1 with PRRS virus nucleocapsid protein mediates NF-κB activation and triggers proinflammatory mediators during viral infection. Sci Rep (2019) 9:11042. doi: 10.1038/s41598-019-47495-9
242. Lee C, Hodgins D, Calvert JG, Welch S-KW, Jolie R, Yoo D. Mutations within the nuclear localization signal of the porcine reproductive and respiratory syndrome virus nucleocapsid protein attenuate virus replication. Virology (2006) 346:238–50. doi: 10.1016/j.virol.2005.11.005
243. Lee C, Hodgins DC, Calvert JG, Welch S-KW, Jolie R, Yoo D. The nuclear localization signal of the PRRS virus nucleocapsid protein viral replication in vitro and antibody response in vivo. Adv Exp Med Biol (2006) 581:145–8. doi: 10.1007/978-0-387-33012-9_25
Keywords: type I interferons (IFNs), NF-kappa B (NF-κB), IFN antagonism, live-attenuated vaccine, veterinary vaccine, viral immune evasion, next-generation vaccines, veterinary virology
Citation: Su C-M, Du Y, Rowland RRR, Wang Q and Yoo D (2023) Reprogramming viral immune evasion for a rational design of next-generation vaccines for RNA viruses. Front. Immunol. 14:1172000. doi: 10.3389/fimmu.2023.1172000
Received: 22 February 2023; Accepted: 03 April 2023;
Published: 17 April 2023.
Edited by:
Denis Archambault, Université du Québec à Montréal, CanadaReviewed by:
Shafiqul Chowdhury, Louisiana State University, United StatesCopyright © 2023 Su, Du, Rowland, Wang and Yoo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongwan Yoo, ZHlvb0BpbGxpbm9pcy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.