 Minjing Zou1
Minjing Zou1 Suhad Al-Yahya2
Suhad Al-Yahya2 Monther Al-Alwan3
Monther Al-Alwan3 Huda A. BinEssa1
Huda A. BinEssa1 Khalid S. A. Khabar2
Khalid S. A. Khabar2 Falah Almohanna4
Falah Almohanna4 Abdullah M. Assiri4
Abdullah M. Assiri4 Abdulmohsen Altaweel5
Abdulmohsen Altaweel5 Amal Qattan1
Amal Qattan1 Brian F. Meyer6
Brian F. Meyer6 Ali S. Alzahrani7
Ali S. Alzahrani7 Yufei Shi1*
Yufei Shi1*- 1Department of Molecular Oncology, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
- 2Department of Molecular Biomedicine, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
- 3Department of Stem Cell & Tissue Re-engineering, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia and College of Medicine, Al-Faisal University, Riyadh, Saudi Arabia
- 4Department of Comparative Medicine, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
- 5Mawhiba, King Abdulaziz and His Companions Foundation for Giftedness and Creativity, Riyadh, Saudi Arabia
- 6Centre for Genomic Medicine, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
- 7Department of Medicine, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
Introduction: BRAFV600E mutations frequently occur in papillary thyroid cancer (PTC). β-catenin, encoded by CTNNB1, is a key downstream component of the canonical Wnt signaling pathway and is often overexpressed in PTC. BRAFV600E-driven PTC tumors rely on Wnt/β-catenin signaling to sustain growth and progression.
Methods: In the present study, we investigated the tumorigenicity of thyroid cancer cells derived from BRAFV600E PTC mice following Ctnnb1 ablation (BVE-Ctnnb1null).
Results: Remarkably, the tumorigenic potential of BVE-Ctnnb1null tumor cells was lost in nude mice. Global gene expression analysis of BVE-Ctnnb1null tumor cells showed up-regulation of NKG2D receptor activating ligands (H60a, H60b, H60c, Raet1a, Raet1b, Raet1c, Raet1d, Raet1e, and Ulbp1) and down-regulation of inhibitory MHC class I molecules H-2L and H-2K2 in BVE-Ctnnb1null tumor cells. In vitro cytotoxicity assay demonstrated that BVE-Ctnnb1wt tumor cells were resistant to NK cell-mediated cytotoxicity, whereas BVE-Ctnnb1null tumor cells were sensitive to NK cell-mediated killing. Furthermore, the overexpression of any one of these NKG2D ligands in the BVE-Ctnnb1wt cell line resulted in a significant reduction of tumor growth in nude mice.
Conclusions: Our results indicate that active β-catenin signaling inhibits NK cell-mediated immune responses against thyroid cancer cells. Targeting the β-catenin signaling pathway may have significant therapeutic benefits for BRAF-mutant thyroid cancer by not only inhibiting tumor growth but also enhancing host immune surveillance.
Introduction
Papillary thyroid cancer (PTC) is the most common type of thyroid cancer, accounting for more than 80% of all thyroid cancer cases (1). The BRAFV600E mutation is the most frequent genetic alteration in PTC ranging from 29% to 83% depending on the cohort studied with overall rate of approximately 45% (2, 3). Nearly 60% (59.7% of 496 cases) of PTC patients harbor BRAFV600E mutations from the TCGA thyroid cancer consortium (4). In some studies, BRAFV600E mutation has been shown to be one of the factors contributing to thyroid cancer recurrence and mortality (5, 6). The mutation constitutively activates the RAS–RAF–MEK–ERK mitogen-activated protein kinase (MAPK) signaling cascade and promotes the initiation and progression of PTC. It has been demonstrated that BrafV600E mutation drives oncogenic transformation of thyroid epithelial cells and development of PTC in transgenic mouse models (7, 8).
β-catenin is a downstream component of the Wnt/β-catenin pathway and plays an important role in embryonic development and tissue homeostasis (9). Dysregulation of its signaling is involved in many types of tumors, including thyroid cancer (10–13). Functional cross-talk between β-catenin and MAPK, PI3K/AKT, or CREB (cAMP-response element binding protein) signaling pathways has been demonstrated to maintain proliferation in thyroid cancer cell lines (12, 14). Recently, we have shown that BRAF-driven PTC requires active β-catenin signaling to sustain its growth (15). Deletion of β-catenin results in tumor regression and differentiation as well as increased sensitivity to the BRAFV600E inhibitor PLX4720.
In this study, we investigated the tumorigenicity of thyroid cancer cells derived from BrafV600E -induced PTC mice with wild-type Ctnnb1 (BVE-Ctnnb1wt) and deleted Ctnnb1 (BVE-Ctnnb1null). We found that β-catenin ablation led to complete loss of tumorigenicity of BVE-Ctnnb1null cells in nude mice, with increased expression of NKG2D ligands in BVE-Ctnnb1null cells. Our data strongly suggest that β-catenin exerts oncogenic effects by promoting tumor growth and evading NK cell-mediated immune surveillance.
Materials and methods
Animals
Athymic BALB/c-nu/nu (nude mice) and C57BL/6J mice (6–10 weeks of age) were acquired from Jackson Laboratory. Mice were provided with autoclaved food and water ad libitum. The study was approved by the Animal Care and Use Committee of the institution (RAC# 2190004) and was conducted in compliance with the Public Health Service Guidelines for the Care and Use of Animals in Research.
Thyroid tumor cell lines
BVE-Ctnnb1wt and BVE-Ctnnb1null were established from thyroid tumors collected aseptically from donor mice, as described previously (15) and maintained in DMEM/F12 growth medium containing 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin.
RNA sequencing for quantification of differentially expressed genes
Total RNA from BVE-Ctnnb1wt and BVE-Ctnnb1null cell lines was isolated using TRI Reagent® solution (#T9424, Sigma-Aldrich, St. Louis, MO, USA). Libraries were constructed using an Illumina TruSeq RNA Library Prep kit (San Diego, CA, USA) according to the manufacturer’s instructions. All sequencing was performed on an Illumina HiSeq 4000 platform with at least 20 million clean reads. Significant DEGs were selected based on the following criteria: Log2 fold change>2, false discovery rate (FDR) <0.001, and p-value from the difference test<0.01. Gene list annotation and enrichment of biological pathways were performed using Metascape (16).
Cytotoxicity assay and phenotyping of effector cells
A label-free cell-based assay (ACEA Biosystems, San Diego, CA, USA) was used to measure the cytotoxicity of spleen cells from C57BL/6J (effector) mice against tumor cells (target) at an effector/target ratio of 25:1, as previously described (17). The effector cells were immune phenotyped for the presence of surface markers: CD4, CD8, and asialo GM1 (aGM1) using complement-mediated (Cederlane Low-Tox®-M Rabbit Complement, #CL3051, 1:20 dilution) cytotoxicity assay in the presence or absence of appropriate blocking antibodies (18). Briefly, target cells (2000 cells/well) were seeded in electrode strip-coated 96-well electrode-integrated microplates. Spleen cells were harvested from two normal C57BL/6J mice. Splenocytes were added to the target cells at 50,000 cells/well in the presence of 5 µg/ml phytohemagglutinin (PHA) and 100 U/ml IL-2 for the activation of T and NK cells with or without anti-CD8 (#CL169AP, 1:20 dilution, plus 5ul complement), anti-CD4 (#CL012AP, 1:20 dilution, plus 5ul complement), or asialo GM1 (anti-NK cell) antibodies (#CL8955, 1:100 dilution, plus 5ul complement, Cedarlane, Ontario, Canada). The percentage of specific cytotoxicity was calculated based on the cell index (CI), which was derived from the changes in electrode impedance due to cell growth and attachment relative to that of the background (without cells). The following formula was used: Cytotoxicity = [(experimental CI – control CI)/(untreated CI – control CI)] × 100.
Cloning and expression of activating NKG2D ligands in BVE-Ctnnb1wt cells
The cDNA for each NKG2D ligand H60a (NM_010400.2), H60b (NM_001177775.1), H60c (ENSMUST00000170893.2), Rae-1α (Raet1a, NM_009016.1), Rae-1β (Raet1b, NM_009017.1), Rae-1γ (Raet1c, NM_009018.1), Rae-1δ (Raet1d, NM_020030.2), Rae-1ϵ (Raet1e, NM_001359808.1) or Ulbp1 (Mult1, NM_029975.2) was cloned into pcDNA3.1 expression vector and transfected into the BVE-Ctnnb1wt cell line using Lipofectamine (Invitrogen, CA, USA). The transfected cells were selected for 6 weeks with 100 µg/ml zeocin. Stable clones expressing each ligand were subcutaneously injected into nude mice to observe tumor growth. The overexpression of NKG2D ligands was confirmed by qRT-PCR.
Cloning and expression of inhibitory Ly49 ligands in BVE-Ctnnb1null cells
The cDNA for each Ly49 ligand H2-L, H2-K2, and H2-K1 was cloned into the pcDNA3.1 expression vector and transfected into the BVE-Ctnnb1null cell line using Lipofectamine (Invitrogen, CA, USA) and selected for 6 weeks with 100 µg/ml zeocin. Stable clones expressing each ligand were subcutaneously injected into nude mice to observe tumor growth. The overexpression of H2-L, H2-K2, and H2-K1 was confirmed by qRT-PCR.
Statistical analysis
Student’s t-test (two-tailed) was used to compare two groups and the area under curve (AUC) calculation was performed using GraphPad Prisim 8. A P value < 0.05 was considered significant.
Results
Tumorigenicity of BVE-Ctnnb1null cells
Tumorigenicity of BVE-Ctnnb1null and BVE-Ctnnb1wt cells was investigated in athymic nude mice. No tumor growth was observed after 4 weeks of subcutaneous injection of 1 × 106 BVE-Ctnnb1null cells into the right flank of the mice (n=8), except that a small nodule (0.5 × 0.5 cm) was formed at the injection site in one mouse (n=8). However, tumors (approximately 2 × 2 cm) developed in all mice 4 weeks following injection of 1 × 106 BVE-Ctnnb1wt cells. The nodule, which formed at the injection site of BVE-Ctnnb1null cells, was confirmed to be a tertiary lymph node by histology, with characteristic features of lymphoid follicles and germinal centers (Figures 1A-C; Supplementary Figure 1). Lymphoid tissues were also found within the tumors formed by BVE-Ctnnb1wt tumor cells, with heavy infiltration of lymphocytes and macrophages at the interface of the tumor and lymphoid tissue (Figures 1D-F; Supplementary Figure 1).
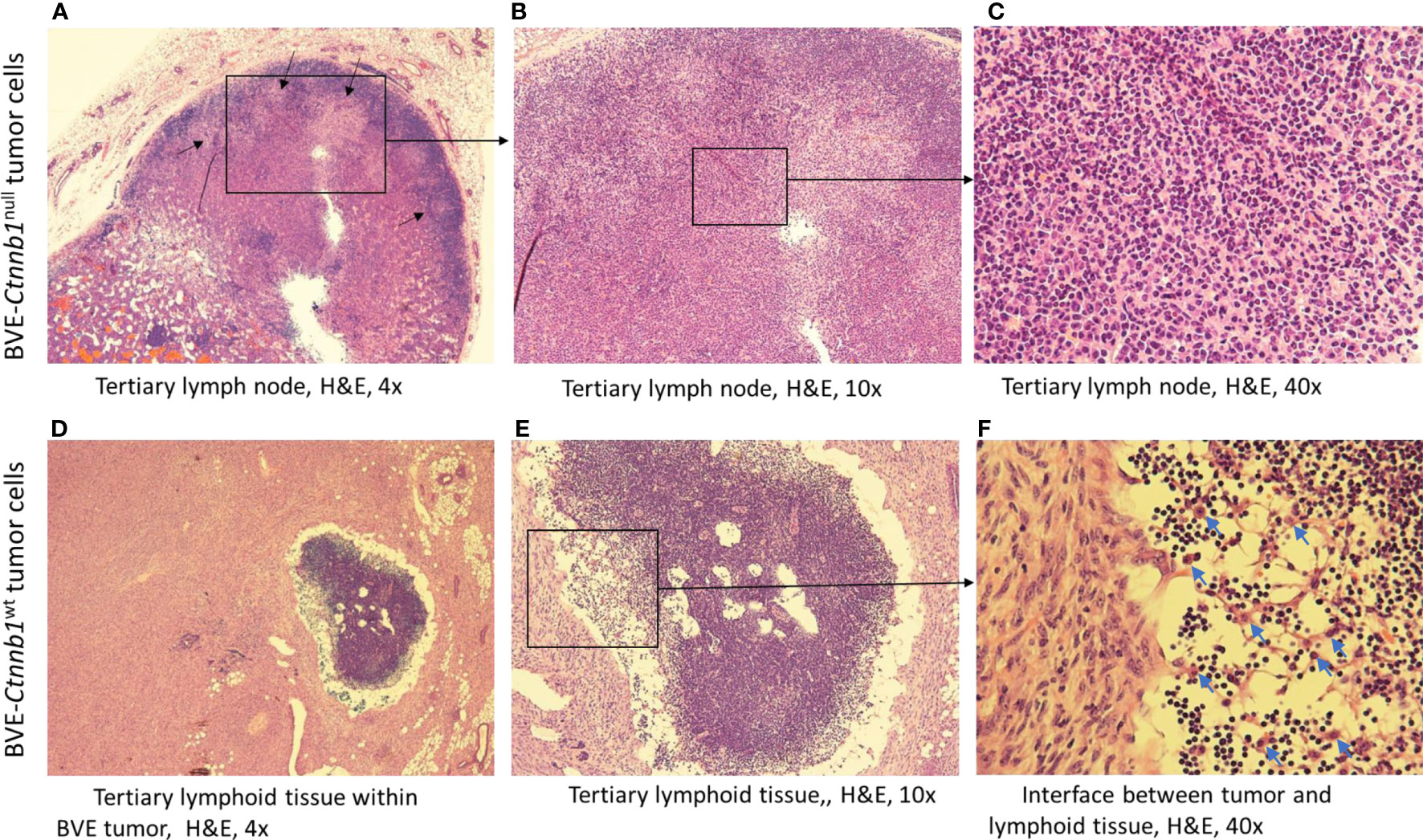
Figure 1 Tertiary lymph node formation at the injection site following subcutaneous injection of BVE-Ctnnb1null tumor cells into nude mice. Upper (A–C) (H&E staining): Characteristic features of lymphoid follicles and germinal centers are shown. Germinal centers are indicated by an arrow; Lower (D–F): Tertiary lymphoid tissue within a large tumor following subcutaneous injection of BVE-Ctnnb1wt tumor cells into nude mice. Heavy infiltration of lymphocytes and macrophages at the interface between tumor and lymphoid tissue are shown.
Overexpression of NKG2D receptor activating ligands in the BVE-Ctnnb1null cells
Since nude mice have no functional T cells, the rejection of BVE-Ctnnb1null cells is likely mediated by NK cells. To explore the mechanisms that resulted in BVE-Ctnnb1null tumor cell rejection, we compared the gene expression profiles of BVE-Ctnnb1wt and BVE-Ctnnb1null cells using RNA-Seq analysis of three biological replicates. Pathway analysis showed 833 up-regulated and 1407 down-regulated genes with a log2 fold-change >2 (Supplementary Table 1). Importantly, the genes involved in NK cell-mediated immunity were among the top 20 enriched ontology clusters that were significantly up-regulated in the BVE-Ctnnb1null cells (Figure 2A). The genes typically involved in cancer growth and progression, such as extracellular matrix organization, chemotaxis, vascular development, integrin cell surface interactions and signaling, and ERK-1/ERK2 signaling, were among the top 20 enriched ontology clusters that were significantly down-regulated in the BVE-Ctnnb1null cells (Figure 2B). These changes in biological pathways are consistent with our previous study showing that the β-catenin signaling pathway is required for the growth and progression of BRAFV600E-driven thyroid cancer (15). The gene list for each cluster is presented in Supplementary Tables 2 and 3.
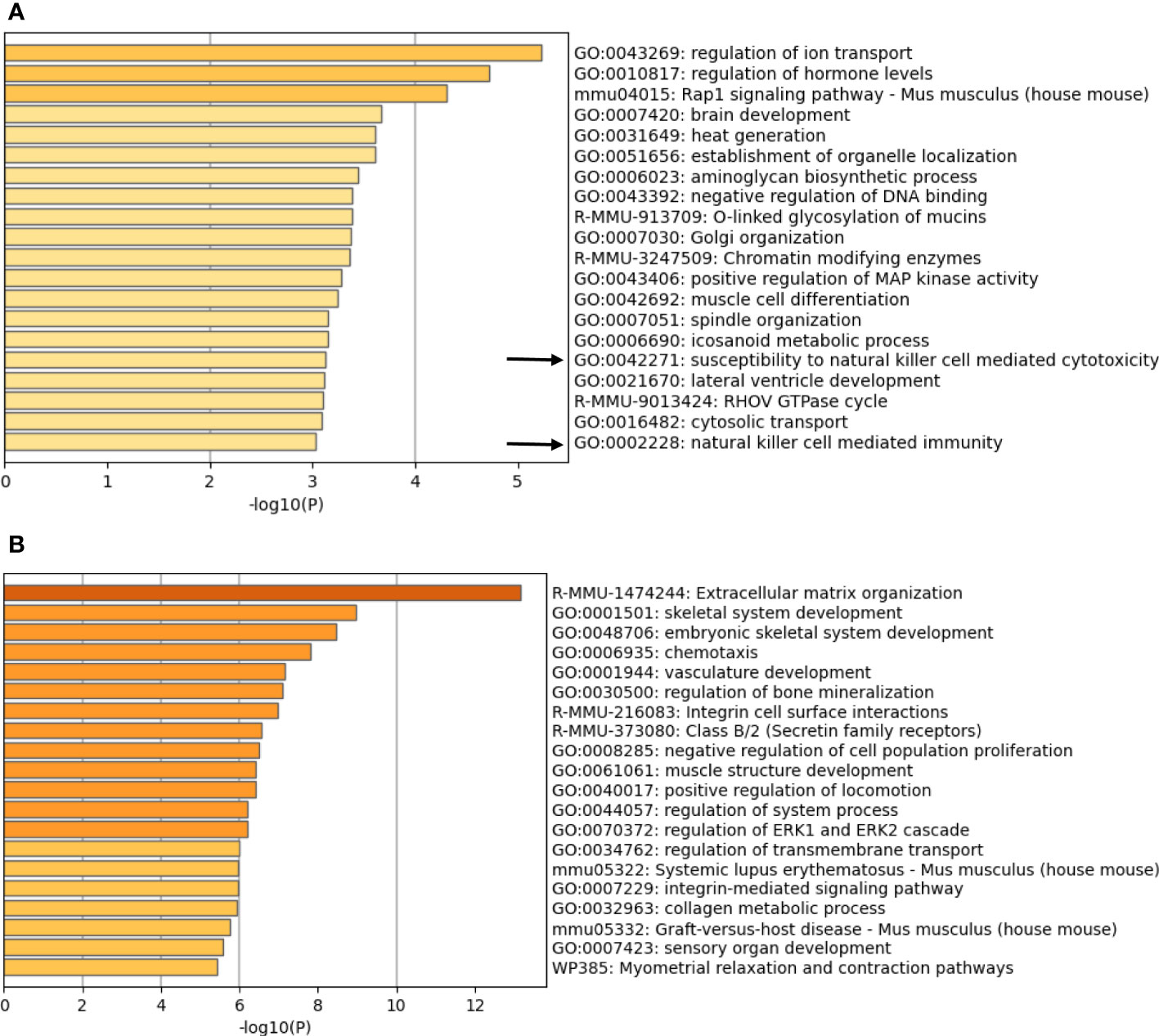
Figure 2 Enriched ontology clusters of differentially expressed genes. Pathway analysis of (A) up-regulated genes (833 genes) and (B) down-regulated genes (1407 genes) in the BVE-Ctnnb1null cells as compared to BVE-Ctnnb1wt cells. Top 20 enriched terms (GO/KEGG terms, canonical pathways, hall mark gene sets) are shown and colored by p-values. The gene clusters involving NK cell mediated immunity are indicated by an arrow.
We further analyzed the list of DEGs involved in NK cell activation. As shown in Figure 3, BVE-Ctnnb1null cells showed up-regulation of NKG2D activating ligands (H60a, H60b, H60c, Raet1a, Raet1b, Raet1c, Raet1d, Raet1e and Ulbp1) and down-regulation of inhibitory MHC class I molecules (H2-L, H2-K2, and H2-D1), which may lead to NK cell activation and tumor cell clearance. In contrast, the up-regulation of H2-L and H2-K2 and down-regulation of NKG2D ligands in BVE-Ctnnb1wt tumor cells would probably result in evasion of NK cell-mediated immune surveillance and promotion of tumor growth.
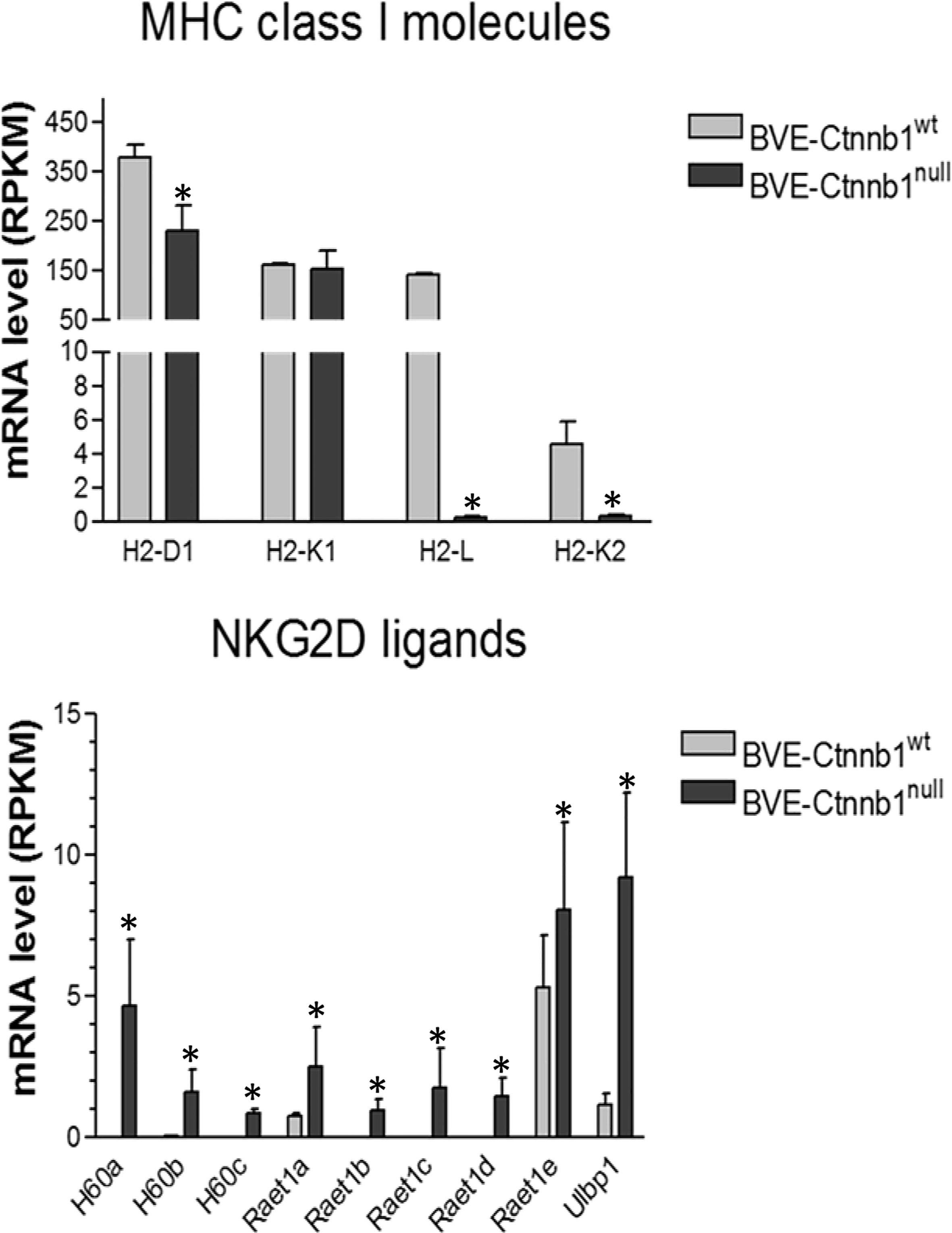
Figure 3 Expression of MHC class 1 molecules and NKG2D ligands in BVE-Ctnnb1wt and BVE-Ctnnb1null tumor cells. RNA-Seq analysis was performed on tumor cells and data are presented as mean ± SEM of three biological replicates. Down-regulation of MHC class 1 molecules (H2-L, H2-K2, and H2-D1), and up-regulation of NKG2D ligands. Asterisk indicates statistical significance as compared to BVE-Ctnnb1wt control. RPKM indicates Reads Per Kilobase Million.
NK-mediated cytotoxicity of BVE-Ctnnb1wt and BVE-Ctnnb1null cells
Next, we investigated the sensitivity of BVE-Ctnnb1null and BVE-Ctnnb1wt cells to NK cell-mediated cytotoxicity using in vitro cytotoxicity assay. As shown in Figure 4, activated splenocytes completely inhibited the replication of both BVE-Ctnnb1wt and BVE-Ctnnb1null tumor cells (top panel). However, BVE-Ctnnb1wt tumor cells were able to replicate at 67% efficiency at 120h in the presence of anti-CD8 T cell neutralizing antibodies, indicating their resistance to NK cell-mediated cytotoxicity. The area under curve (AUC) of BVE-Ctnnb1null, BVE-Ctnnb1wt, and BVE-Ctnnb1wt + splenocytes + anti-CD8 were 210.4 ± 3.2, 128.7 ± 2.4, and 42.5 ± 1.9, respectively. In contrast, BVE-Ctnnb1null tumor cells were unable to grow in the presence of anti-CD8 T cell neutralizing antibodies, indicating their sensitivity to NK killing (middle panel). Both BVE-Ctnnb1wt and BVE-Ctnnb1null tumor cells were unable to replicate in the presence of anti-NK cell neutralizing antibodies, indicating their sensitivity to CD8 T cell-mediated cytotoxicity (bottom panel).
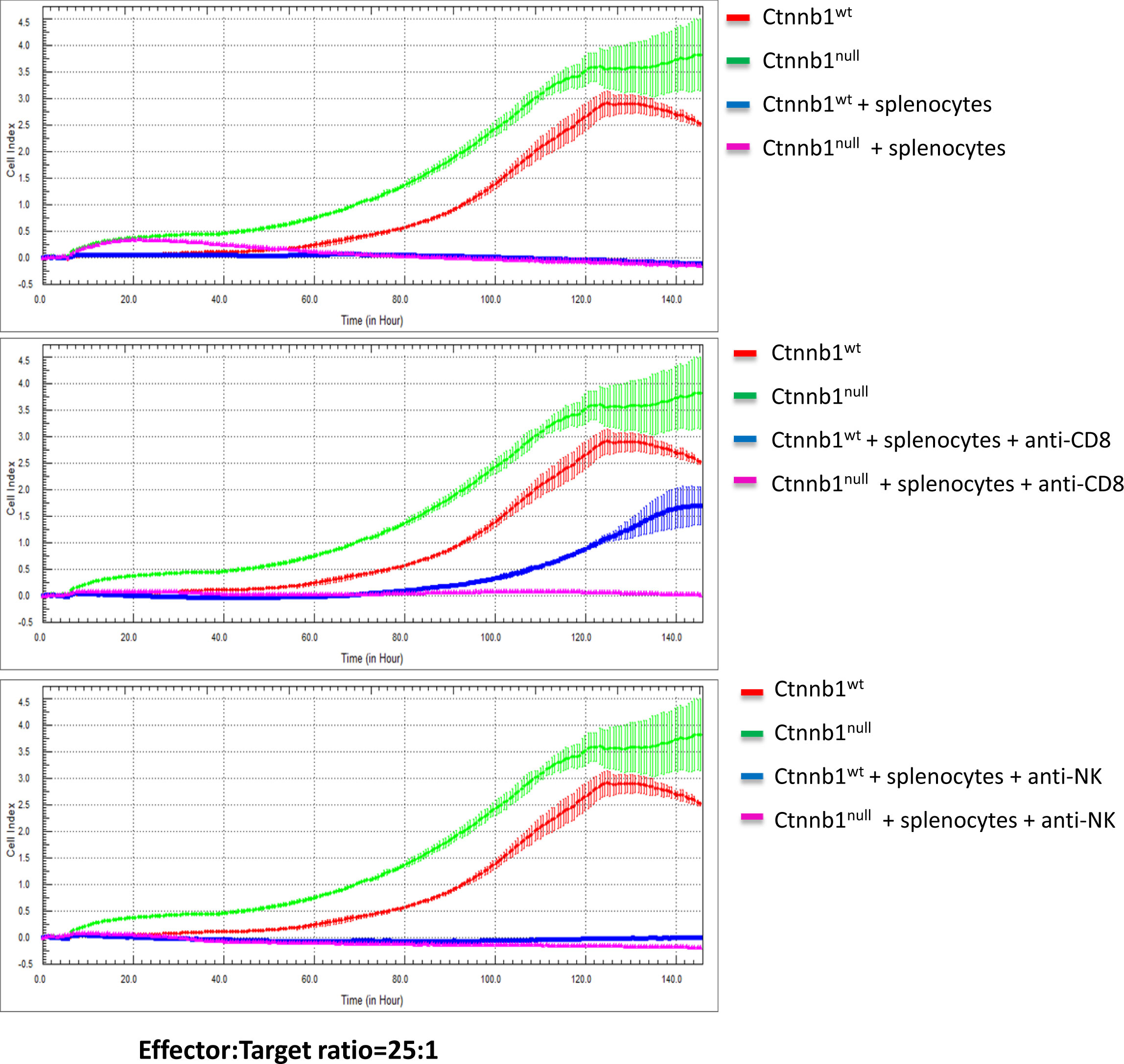
Figure 4 Cytotoxicity of splenocytes against BVE-Ctnnb1wt and BVE-Ctnnb1null tumor cells. The cytotoxicity of spleen cells (effector) against tumor cells (target) was measured at an effector/target ratio of 25:1 by real-time in vitro cytolytic assay in the presence or absence of anti-CD8 or anti-NK cell antibodies. The data are presented as the Cell Index over time. The target cell proliferation rate was registered as an increase in the impedance-related Cell Index over time. When effectors are added to target cells, their cytolytic activity causes adherent target cells to round up and detach, consequently reducing the Cell Index value.
Overexpression of NKG2D ligands in BVE-Ctnnb1wt cells reduces tumor growth
To further confirm the role of NKG2D activating ligands in tumor regression, we overexpressed each of these ligands by stable transfection of their cDNAs into BVE-Ctnnb1wt cells and then injected these ligand-expressing tumor cells (1 × 106) into nude mice (n=5) to observe tumor growth for 4 weeks. As shown in Figure 5, overexpression of these ligands caused a significant reduction in tumor growth (p<0.001). In contrast to the complete loss of tumorigenicity in BVE-Ctnnb1null cells, over-expression of a single NKG2D ligand could not eliminate BVE-Ctnnb1wt tumor cell growth in nude mice. This might be due to high expression of inhibitory H2-L and H2-K2 ligands in BVE-Ctnnb1wt tumor cells and/or tumor cell clearance may require the simultaneous expression of multiple NKG2D ligands for optimal NK cell activation.
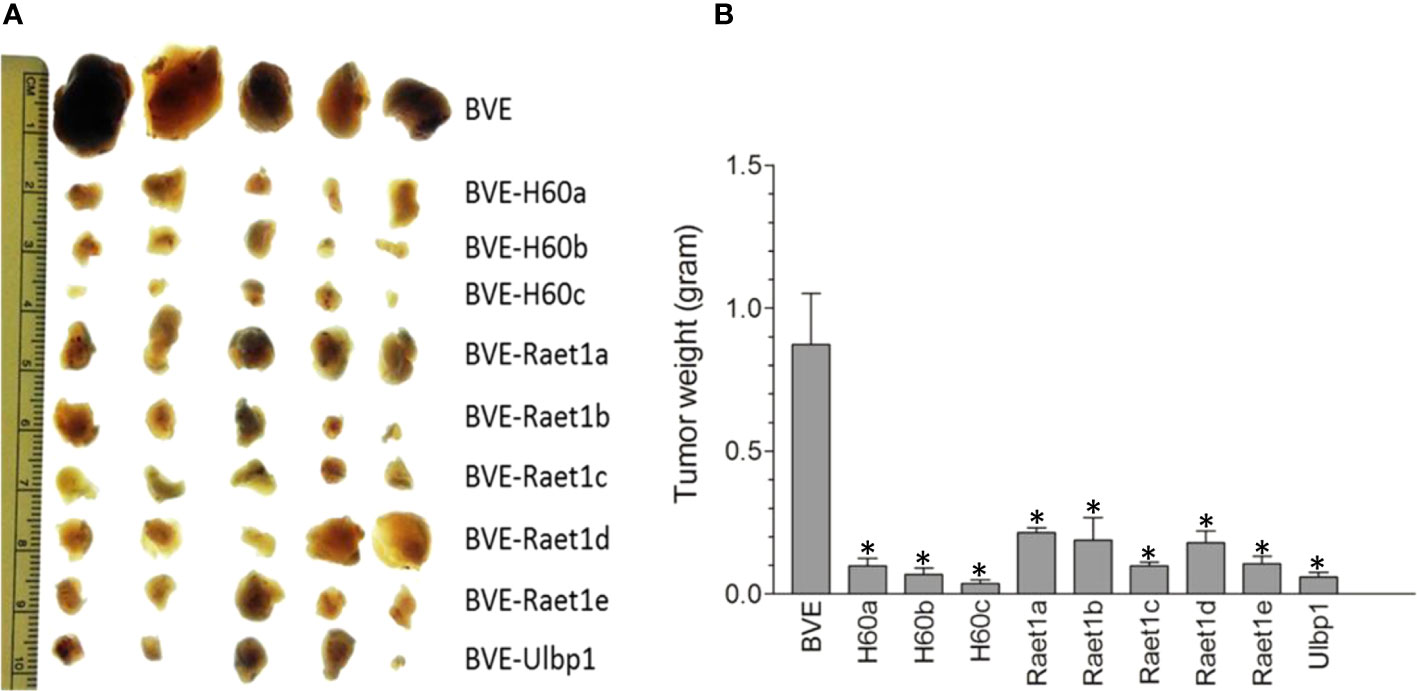
Figure 5 Regression of tumors after overexpression of NKG2D ligands in the BVE-Ctnnb1wt cells. (A) Each of NKG2D ligands was stably transfected under CMV promoter into BVE-Ctnnb1wt cells for stable expression. These NKG2D ligand-expressing cells (1 x 106) were injected subcutaneously into nude mice (n=5) to observe tumor growth for 4 weeks. (B) Quantification of tumor growth (weight) after subcutaneous injection of each NKG2D ligand-expressing BVE-Ctnnb1wt cells. Data are expressed as mean of tumor weight ± SEM of 5 tumors. Asterisk indicates statistical significance as compared to BVE-Ctnnb1wt control.
Overexpression of Ly49 ligands in BVE-Ctnnb1null cells partially rescue tumor growth
Since BVE-Ctnnb1null cells express high levels of NKG2D activating ligands and low levels of H2-L and H2-K2 inhibitory ligands, we investigated whether the loss of tumorigenicity in BVE-Ctnnb1null cells could be partially rescued by overexpression of H2-L or H2-K2 ligands in these cells. As shown in Figure 6, overexpression of these ligands resulted in tumor growth in nude mice, thus confirming the role of H2-L and H2-K2 ligands in NK cell inhibition. H2-K2 appeared to be more effective than H2-L in inhibiting NK cells. The potential mechanisms of NK cell-mediated rejection of BVE-Ctnnb1null cells are summarized in Figure 7.
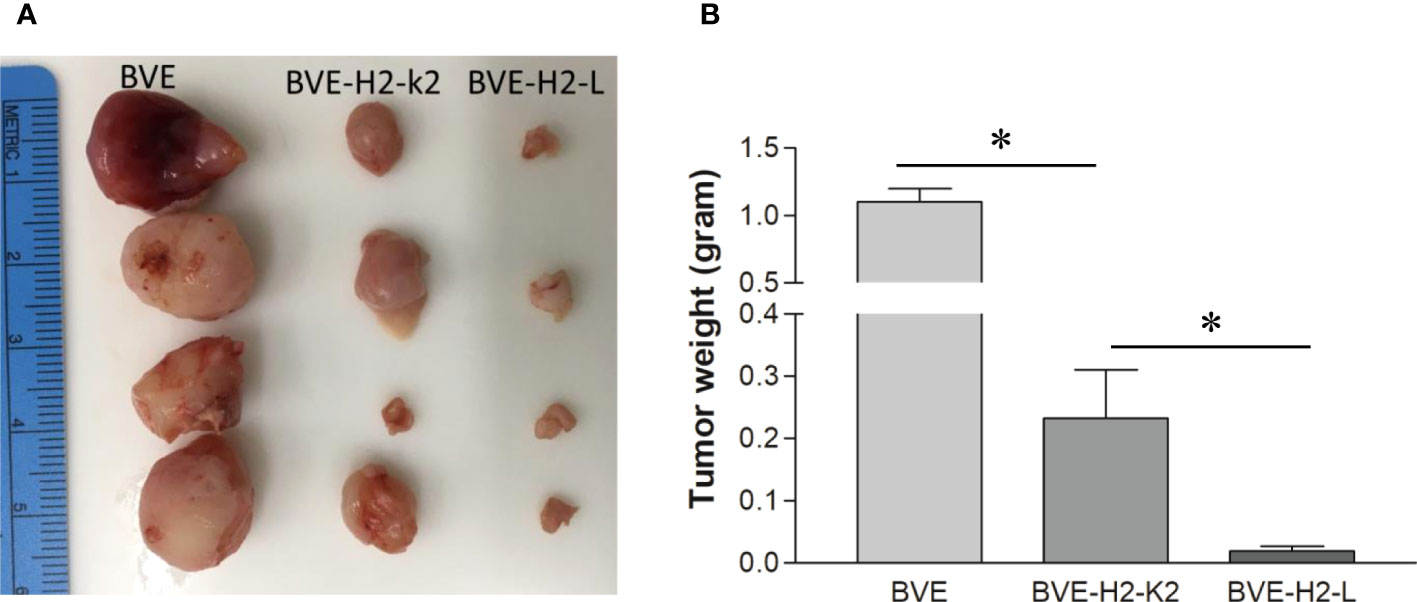
Figure 6 Tumor formation after overexpression of MHC class 1 molecules H2-L and H2-K2 in BVE-Ctnnb1null tumor cells. (A) Each of MHC class 1 molecules H2-L and H2-K2 was stably transfected under CMV promoter into BVE-Ctnnb1null cells. These H2-L and H2-K2-expressing cells (1 x 106) were injected subcutaneously into nude mice (n=4) to observe tumor growth for 6 weeks. BVE (BVE-Ctnnb1wt) cells were used as control. (B) Quantification of tumor weight after subcutaneous injection of H2-L and H2-K2-expressing BVE-Ctnnb1null cells to nude mice. Data are expressed as mean of tumor weight ± SEM of 4 tumors. Asterisk indicates statistical significance as compared to BVE-Ctnnb1wt control or between BVE-H2-K2 and BVE-H2-L.
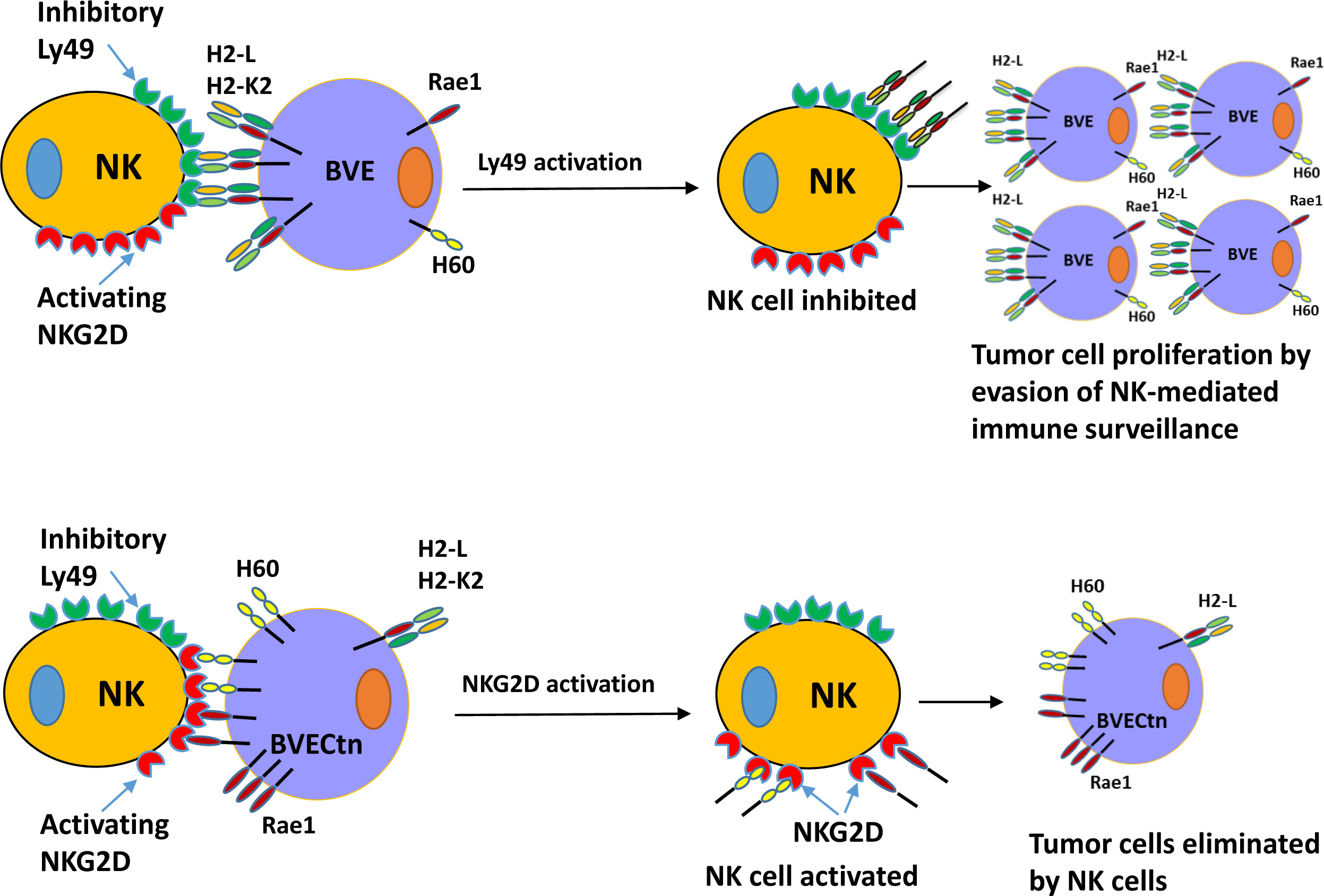
Figure 7 Schematic diagram of NK cell-mediated tumor rejection in nude mice. Top: BVE (BVE-Ctnnb1wt) tumor cells express high levels of MHC class I molecules H2-L and/or H2-K2, which bind to the inhibitory Ly49 receptor on the NK cells. This interaction results in NK cell inhibition and impaired immune surveillance. Bottom: BVECtn (BVE-Ctnnb1null) tumor cells express high levels of NKG2D receptor ligands H60 (a, b, and c) and Raet1 (a, b, c, d, and e), which bind to the activating NKG2D receptor. This binding leads to NK cell activation and tumor cell rejection.
Discussion
In the present study, we provided evidence of β-catenin in modulating NK cell activity against Braf-mutant PTC in mice. Down-regulation of β-catenin increased the expression of NKG2D ligands and decreased the expression of inhibitory MHC class I molecules, such as H-2L and H-2K2, in tumor cells, which triggered NK cell-mediated tumor rejection.
It has been reported that oncogenic activation of KRAS/BRAF/MEK signaling stimulates Wnt/β-catenin pathway by β-catenin/TCF4 complex activation via Frizzled co-receptor LRP6 phosphorylation, which in turn promotes tumor growth and invasion (19, 20). The LRP6 may be the link between MAPK and Wnt/β-catenin signaling cross-talk during oncogenesis (20). In the BrafV600E murine thyroid cancer, β-catenin expression is up-regulated and its knockout results in down-regulation of MARK signaling pathway, indicating a positive crosstalk between MAPK and Wnt/β-catenin signaling pathways. Active Wnt/β-catenin signaling in turn promotes BrafV600E-mediated tumor growth (15). Genetic alterations in β-catenin have not been identified in PTC. However, aberrant β-catenin expression or localization, such as the reduction of β-catenin plasma membrane levels and its aberrant nuclear localization, has been reported and is associated with c-Myc and cyclin D1 overexpression, loss of tumor differentiation, and poor prognosis in human PTC samples (13, 21).
The activation of tumor-intrinsic Wnt/β-catenin signaling is frequently associated with poor prognosis in most human cancers. Besides its intrinsic effects on tumor cell proliferation and apoptosis, the WntT/β-catenin signaling pathway has been found to act on the tumor microenvironment and lead to immune evasion by reducing tumor infiltration of dendritic cells (DCs), NK cells, and T cells via multiple mechanisms, such as inhibition of CCL4 expression on cytotoxic T lymphocytes, CXCL10 expression on DCs, or increased expression of dickkopf WNT signaling pathway inhibitor 1 (DKK1), which prevents NK cell-dependent cancer cell lysis by reducing the expression of NK cell-activating ligands (22–24).
Lee et al. reported that incubation of NK cells and lymphokine-activated killer cells with TGF-β1 resulted in a dramatic reduction in surface NKG2D expression associated with impaired NK cytotoxicity (25). They further demonstrated that the modulation of NKG2D by TGF-β1 was specific, as the expression of other NK receptors was not affected by the presence of TGF-β1. TGF-β1-mediated impairment of NK cell cytotoxicity was not due to alteration of other lytic moieties, such as perforin, Fas, or the apoptotic pathway. TGF-β1 signaling is activated in BRAFV600E-induced thyroid cancer (26). In our previous studies, we showed that TGF-β inhibited the anti-tumor activity of NK and CD8+ T cells in BrafV600E thyroid cancer mouse model. The immune suppression could be reversed by IL-12 treatment with increased lymphocyte and macrophage infiltration, resulting in a significant reduction in tumor load and an increase in survival (18). Increased infiltration of lymphocytes and macrophages has also been observed in the same thyroid cancer model following β-catenin ablation (15). Spranger et al. have reported that active β-catenin signaling in melanoma prevents anti-tumor immunity via depletion of T cells, linking β-catenin signaling to tumor evasion of immune surveillance (23, 27). Wang et al. have recently shown that β-catenin inhibition shifts the colorectal tumor microenvironment into a T cell-inflamed phenotype through the up-regulation of T/NK cell-recruiting CXCR3 chemokines CXCL9/10/11 (28).
In the present study, we have identified a novel mechanism by which BrafV600E-mutant thyroid cancer cells evade NK cell-mediated immune surveillance. This is achieved by down-regulation of stimulating NKG2D ligands and up-regulation of inhibitory MHC class I molecules. Given that the activation of Wnt/β-catenin signaling leads to increased TGF-β signaling, the down-regulation of stimulating NKG2D ligands and up-regulation of inhibitory MHC class I molecules may be due to increased TGF-β signaling. However, in contrast to the reduction in surface NKG2D expression reported by Lee et al, NKG2D expression was not significantly different between BVE-Ctnnb1wt and BVE-Ctnnb1null tumor cells.
NK cells play an important role in cancer immune surveillance because of their ability to recognize tumor cells through the interaction of several distinct cell surface receptors, with the net effect of NK cell activation and cytotoxic attack on tumor cells (29, 30). The activation of NK cells (CD56+ in humans and CD49b+ in mice) is balanced by anti- and pro-activation signals through inhibitory and activating receptors, such as Ly49 in mouse or killer cell Ig-like receptor (KIR in human) family receptors, NKG2 (CD159) family of C-type lectin-like receptors, including activating NKG2D and inhibitory NKG2A, and natural cytotoxicity receptors (NKp46, NKp44, and NKp30) (31–33). NKG2D is expressed in all NK cells and activated CD8+ T cells. Its ligands are not or rarely expressed in normal tissues, but are expressed in tumor cells and viral-infected tissues (34). Previous studies have shown that the expression of NKG2D ligands on tumor cells renders them susceptible to killing by NK cells in vitro and the rejection of tumor xenografts in vivo (35, 36). NKG2D-mediated killing is negatively regulated by MHC class I molecules on tumor cells, which bind to inhibitory Ly49 receptors on NK cells to maintain a balance between activating and inhibitory signals. Thus, high expression of MHC class I molecules in tumor cells prevents efficient NKG2D-mediated cytotoxicity against tumor cells (37, 38). Our data indicate that active β-catenin signaling in BrafV600E-mutant thyroid cancer cells inhibits NK cell-mediated immune response. Attenuation of Wnt/β-catenin signaling boosts the host immune response against thyroid cancer. To our knowledge, this is a first report describing the inhibitory effect of β-catenin expression on NK cell activity, linking oncogene activation to innate immune suppression. Given that overexpression of NKG2D ligands results in regression of thyroid cancer xenografts in vivo, these ligands could be used for cancer vaccine development or gene therapy to generate anti-cancer immune responses.
In summary, our results provide insights into β-catenin signaling in the NK cell-mediated immune response against BRAF-mutant thyroid cancer cells. β-catenin ablation results in increased expression of activating NKG2D ligands and decreased expression of inhibitory MHC class I molecules, H2-L and H2-K2. Targeting the β-catenin signaling pathway could not only inhibit tumor growth but also enhance host immune surveillance.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE207695.
Ethics statement
The animal study was reviewed and approved by The Animal Care and Use Committee of King Faisal Specialist Hospital and Research Centre.
Author contributions
MZ, ASA, MA-A and YS contributed to conception and design of the study. MZ, SA-Y, MA-A, HB, KK, FA, and AMA performed experiments. AQ organized the database. MZ and YS performed the statistical analysis. MZ wrote the first draft of the manuscript. MA-A, BM, ASA, and YS wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This study was supported by KACST grant 13-MED1765-20 and KFSHRC grant 2190004.
Acknowledgments
We would like to thank Drs. Shioko Kimura, Catrin Pritchard, and Rolf Kemler for generous gifts of TPO-Cre, LSL-BrafV600E, and floxed Ctnnb1 mice, respectively, and Mr. Cong Li and Kai Huang from BGI for their bioinformatics services.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1171816/full#supplementary-material
References
1. Mao Y, Xing M. Recent incidences and differential trends of thyroid cancer in the USA. Endocr Relat Cancer (2016) 23(4):313–22. doi: 10.1530/ERC-1515-0445
2. Xing M. BRAF mutation in thyroid cancer. Endocr Relat Cancer (2005) 12(2):245–62. doi: 10.1677/erc.1671.0978
3. Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer (2013) 13(3):184–99. doi: 10.1038/nrc3431
4. Agrawal N, Akbani R, Aksoy BA, Ally A, Arachchi H, Asa SL. Integrated genomic characterization of papillary thyroid carcinoma. Cell (2014) 159(3):676–90. doi: 10.1016/j.cell.2014.09.050
5. Xing M, Alzahrani AS, Carson KA, Shong YK, Kim TY, Viola D, et al. Association between BRAF V600E mutation and recurrence of papillary thyroid cancer. J Clin Oncol (2015) 33(1):42–50. doi: 10.1200/JCO.2014.1256.8253
6. Xing M, Alzahrani AS, Carson KA, Viola D, Elisei R, Bendlova B, et al. Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. Jama (2013) 309(14):1493–501. doi: 10.1001/jama.2013.3190
7. Knauf JA, Ma X, Smith EP, Zhang L, Mitsutake N, Liao XH, et al. Targeted expression of BRAFV600E in thyroid cells of transgenic mice results in papillary thyroid cancers that undergo dedifferentiation. Cancer Res (2005) 65(10):4238–45. doi: 10.1158/0008-5472.CAN-05-0047
8. Zou M, Baitei EY, BinEssa HA, Al-Mohanna FA, Parhar RS, St-Arnaud R, et al. Cyp24a1 attenuation limits progression of Braf(V600E) -induced papillary thyroid cancer cells and sensitizes them to BRAF(V600E) inhibitor PLX4720. Cancer Res (2017) 77(8):2161–72. doi: 10.1158/0008-5472.CAN-2116-2066
9. MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell (2009) 17(1):9–26. doi: 10.1016/j.devcel.2009.1006.1016
10. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell (2006) 127(3):469–80. doi: 10.1016/j.cell.2006.1010.1018
11. Chen G, Gao C, Gao X, Zhang DH, Kuan SF, Burns TF, et al. Wnt/beta-catenin pathway activation mediates adaptive resistance to BRAF inhibition in colorectal cancer. Mol Cancer Ther (2018) 17(4):806–13. doi: 10.1158/1535-7163.MCT-1117-0561
12. Castellone MD, De Falco V, Rao DM, Bellelli R, Muthu M, Basolo F, et al. The beta-catenin axis integrates multiple signals downstream from RET/papillary thyroid carcinoma leading to cell proliferation. Cancer Res (2009) 69(5):1867–76. doi: 10.1158/0008-5472.CAN-1808-1982
13. Ishigaki K, Namba H, Nakashima M, Nakayama T, Mitsutake N, Hayashi T, et al. Aberrant localization of beta-catenin correlates with overexpression of its target gene in human papillary thyroid cancer. J Clin Endocrinol Metab (2002) 87(7):3433–40. doi: 10.1210/jcem.3487.3437.8648
14. Sastre-Perona A, Riesco-Eizaguirre G, Zaballos MA, Santisteban P. Beta-catenin signaling is required for RAS-driven thyroid cancer through PI3K activation. Oncotarget (2016) 7(31):49435–49. doi: 10.18632/oncotarget.10356
15. Zou M, BinEssa HA, Al-Malki YH, Al-Yahya S, Al-Alwan M, Al-Jammaz I, et al. β-catenin attenuation inhibits tumor growth and promotes differentiation in a BRAF(V600E)-driven thyroid cancer animal model. Mol Cancer Ther (2021) 20(9):1603–13. doi: 10.1158/1535-7163.MCT-21-0037
16. Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun (2019) 10(1):1523. doi: 10.1038/s41467-019-09234-6
17. Al-Yahya S, Mahmoud L, Al-Zoghaibi F, Al-Tuhami A, Amer H, Almajhdi FN, et al. Human cytokinome analysis for interferon response. J Virol (2015) 89(14):7108–19. doi: 10.1128/JVI.03729-03714
18. Parhar RS, Zou M, Al-Mohanna FA, Baitei EY, Assiri AM, Meyer BF, et al. IL-12 immunotherapy of Braf(V600E)-induced papillary thyroid cancer in a mouse model. Lab Invest (2016) 96(1):89–97. doi: 10.1038/labinvest.2015.1126
19. Lemieux E, Cagnol S, Beaudry K, Carrier J, Rivard N. Oncogenic KRAS signalling promotes the wnt/β-catenin pathway through LRP6 in colorectal cancer. Oncogene (2015) 34(38):4914–27. doi: 10.1038/onc.2014.416
20. Krejci P, Aklian A, Kaucka M, Sevcikova E, Prochazkova J, Masek JK, et al. Receptor tyrosine kinases activate canonical WNT/β-catenin signaling via MAP kinase/LRP6 pathway and direct β-catenin phosphorylation. PloS One (2012) 7(4):e35826. doi: 10.1371/journal.pone.0035826
21. Garcia-Rostan G, Camp RL, Herrero A, Carcangiu ML, Rimm DL, Tallini G. Beta-catenin dysregulation in thyroid neoplasms: down-regulation, aberrant nuclear expression, and CTNNB1 exon 3 mutations are markers for aggressive tumor phenotypes and poor prognosis. Am J Pathol (2001) 158(3):987–96. doi: 10.1016/S0002-9440(10)64045-X
22. Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer (2018) 18(3):139–47. doi: 10.1038/nrc.2017.117
23. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature (2015) 523(7559):231–5. doi: 10.1038/nature14404
24. Galluzzi L, Spranger S, Fuchs E, López-Soto A. WNT signaling in cancer immunosurveillance. Trends Cell Biol (2019) 29(1):44–65. doi: 10.1016/j.tcb.2018.08.005
25. Lee JC, Lee KM, Kim DW, Heo DS. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol (Baltimore Md 1950) (2004) 172(12):7335–40. doi: 10.4049/jimmunol.172.12.7335
26. Knauf JA, Sartor MA, Medvedovic M, Lundsmith E, Ryder M, Salzano M, et al. Progression of BRAF-induced thyroid cancer is associated with epithelial-mesenchymal transition requiring concomitant MAP kinase and TGFβ signaling. Oncogene (2011) 30(28):3153–62. doi: 10.1038/onc.2011.44
27. Hu-Lieskovan S, Homet Moreno B, Ribas A. Excluding T cells: is beta-catenin the full story? Cancer Cell (2015) 27(6):749–50. doi: 10.1016/j.ccell.2015.1005.1014
28. Wang C, Yan J, Yin P, Gui L, Ji L, Ma B, et al. β-catenin inhibition shapes tumor immunity and synergizes with immunotherapy in colorectal cancer. Oncoimmunology (2020) 9(1):1809947. doi: 10.1080/2162402X.2020.1809947
29. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? the example of natural killer cells. Sci (New York NY) (2011) 331(6013):44–9. doi: 10.1126/science.1198687
30. Ben-Shmuel A, Biber G, Barda-Saad M. Unleashing natural killer cells in the tumor microenvironment-the next generation of immunotherapy? Front Immunol (2020) 11:275. doi: 10.3389/fimmu.2020.00275
31. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol (2019) 16(5):430–41. doi: 10.1038/s41423-019-0206-4
32. Krueger PD, Lassen MG, Qiao H, Hahn YS. Regulation of NK cell repertoire and function in the liver. Crit Rev In Immunol (2011) 31(1):43–52. doi: 10.1615/CritRevImmunol.v31.i1.40
33. Paul S, Lal G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front Immunol (2017) 8. doi: 10.3389/fimmu.2017.01124
34. Dhar P, Wu JD. NKG2D and its ligands in cancer. Curr Opin Immunol (2018) 51:55–61. doi: 10.1016/j.coi.2018.1002.1004
35. Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat Immunol (2000) 1(2):119–26. doi: 10.1038/77793
36. Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature (2001) 413(6852):165–71. doi: 10.1038/35093109
37. Malarkannan S. The balancing act: inhibitory Ly49 regulate NKG2D-mediated NK cell functions. Semin Immunol (2006) 18(3):186–92. doi: 10.1016/j.smim.2006.1004.1002
Keywords: β-catenin, CTNNB1, BRAFV600E, NKG2D ligands, NK cells, thyroid cancer
Citation: Zou M, Al-Yahya S, Al-Alwan M, BinEssa HA, Khabar KSA, Almohanna F, Assiri AM, Altaweel A, Qattan A, Meyer BF, Alzahrani AS and Shi Y (2023) β-catenin attenuation leads to up-regulation of activating NKG2D ligands and tumor regression in BrafV600E-driven thyroid cancer cells. Front. Immunol. 14:1171816. doi: 10.3389/fimmu.2023.1171816
Received: 22 February 2023; Accepted: 20 June 2023;
Published: 06 July 2023.
Edited by:
Giorgia Gri, University of Bologna, ItalyReviewed by:
Rengyun Liu, The First Affiliated Hospital of Sun Yat-sen University, ChinaVarun Sasidharan Nair, Helmholtz Association of German Research Centers (HZ), Germany
Khaled Murshed, Hamad Medical Corporation, Qatar
Copyright © 2023 Zou, Al-Yahya, Al-Alwan, BinEssa, Khabar, Almohanna, Assiri, Altaweel, Qattan, Meyer, Alzahrani and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yufei Shi, eXVmZWlAa2ZzaHJjLmVkdS5zYQ==