 Xun Liu1†
Xun Liu1† Yueyao Yang
Yueyao Yang Xiufeng Zheng
Xiufeng Zheng Ming Liu
Ming Liu Gang Wang
Gang Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 17 April 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1169850
This article is part of the Research Topic Advances of Novel Approaches to Enhance Therapeutic Efficacy and Safety in Human Solid Cold Tumor View all 12 articles
Immune check inhibitors (ICIs) have moderate response rates (~20%–30%) in some malignancies clinically, and, when used in combination with other immunotherapeutic strategies such as DNA tumor vaccines, there is evidence to suggest that they could optimize the efficacy of cancer treatment. In this study, we validated that intramuscular injection of plasmid DNA (pDNA) encoding OVA combined with pDNA encoding α-PD-1 (abbreviated as α-PD-1 in the following treatment groups) may enhance therapeutic efficacy by means of in situ gene delivery and enhanced muscle-specific potent promoter. Mice treated with pDNA-OVA or pDNA-α-PD-1 alone showed weak tumor inhibition in the MC38-OVA-bearing model. In comparison, the combined treatment of pDNA-OVA and pDNA-α-PD-1 resulted in superior tumor growth inhibition and a significantly improved survival rate of over 60% on day 45. In the B16-F10-OVA metastasis model, the addition of the DNA vaccine enhanced resistance to tumor metastasis and increased the populations of CD8+ T cells in blood and spleen. In conclusion, the current research shows that a combination of pDNA-encoded PD-1 antibody and DNA vaccine expressed in vivo is an efficient, safe, and economical strategy for tumor therapy.
An effective anti-tumor treatment requires not only the activation of immunity but also the inhibition of some inhibitory targets. Immunotherapy has attracted interest as a new method for cancer treatment. Blocking immune checkpoint molecules, including the CTLA4, PD-1, and PD-L1 molecules, was proposed as a highly effective anti-tumor treatment for most types of cancer (1, 2), with a 20%–50% success rate in different clinical trials (3). However, immune checkpoint inhibitor (ICI) therapy is still ineffective in most cancers, with the majority of cancer patients receiving little benefit from it. Nevertheless, its combination with other treatment methods is considered to be good treatment protocol and could have a tangible curative effect (4, 5).
In the growing field of immunotherapy, because of ability to promote extensive immune activation, DNA vaccines have good prospects as a treatment method for the prevention and cure of many cancers (6). Nevertheless, DNA vaccines have not received enough attention in clinical applications because of insufficient immune response as far. However, the application of new technologies has increased the immunogenicity of DNA vaccines; for instance, the optimization of plasmids and the utilization of gene delivery and genetic adjuvants has been improved through in vivo electrotransfer (7). The combined use of immune checkpoint inhibitors and DNA vaccines has a solid theoretical foundation. The transfection of plasmid DNA (pDNA) in vivo enables specific tissues to successfully produce desired proteins; pDNA can then either encode antigens or therapeutic proteins and is therefore a promising prospect in the treatment of various diseases. Recently, a number of studies have used the in vivo transfection of plasmids to produce therapeutic antibodies for tumor therapy (8–10).
To enhance this gene delivery efficiency, using an effective transport technique is of the utmost importance, as the phospholipid bilayer renders it impossible for any polar molecule, including DNA, to shuttle freely through the cell membrane. Numerous strategies have been explored to enhance the efficiency of penetrating cell membranes. In recent decades, researchers have made many efforts to boost the immunogenicity of DNA vaccines and gene delivery efficiency in bids to increase their clinical application potential. These strategies include the use of electroporation (EP) (7), which aims to increase the permeability of cell membranes and thus allow polar molecules to pass through.
Skeletal muscle is extensively distributed throughout the human body, and is both large in surface are and thick with blood vessels. To ensure that genes can be expressed at high levels in skeletal muscle without affecting the normal function of different tissues, we screened and developed muscle-specific promoters named efficient muscle-specific (EMS) promoters. By way of EMS promoters, the pDNA carrying the target gene is transferred to patien's skeletal muscle cells. The skeletal muscle cells function as “factories” that produce therapeutic proteins/polypeptides that are then secreted from cells, identifying their targets, and transported to them through blood vessels (11). A new finding related to the use of electroporation-mediated DNA shuttling is that several neo-antigens have been shown to effectively induce the anti-tumor function of CD8+ T cells in animals (12).
Here, we outline a method that utilizes DNA vectors as vaccine and immune checkpoint blockade (ICB) protein-encoding gene carriers. The pluronic L64 (L)/electroporation (E)/epigallocatechin gallate (G) (EGCG) (referred to as L/E/G) system was utilized to promote gene delivery efficiency. The aggregate of plasmid DNA encoding ovalbumin (OVA) (referred to as pOVA) vaccine with pVAX-α-PD-1 caused enormous tumor regression, which is often attributed to the elevated cytokine stage and proportion of CD8+ T and CD4+ T cells in tumors. We clearly demonstrate that this aggregate routine, via the L/E/G system, can prevent lung metastasis in B16F10-ovalbumin (OVA)-bearing mice models. In summary, we show that OVA vaccines with a modified delivery route can increase the efficacy of ICI, and that the plasmid encoding α-PD-1 provides a safer and more economical treatment choice for major cancer patients.
The published complementary DNA (cDNA) sequences were used to construct the mouse PD-1 antibody (EP 1445 264 A1), and the PD-1 antibody sequence was cloned onto the pVAX1 vector. The OVA gene was cloned on the pcDNA3.1(+) and efficient muscle-specific plasmid (pEMS) to construct the DNA vaccine. The pEMS is a reconstructed plasmid backbone, replacing the cytomegalovirus (CMV) promoter of pcDNA3.1(+) with EMS (Patent CN 113106094 B). In accordance with the manufacturer’s instructions, all plasmids were obtained using an Endofree Plasmid Extraction Kit (Cwbio, Jiangsu, China) and dissolved in phosphate-buffered saline (PBS). The purities and concentrations of plasmids were determined using agarose gel electrophoresis and spectrophotometry.
HEK293T, MC38, and B16F10 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA), and cultured in Dulbecco’s modified Eagle medium (DMEM) (Gibco, ThermoFisher, USA) containing 10% fetal bovine serum (FBS) (Sigma Aldrich, Taufkirchen, Germany) and 1% penicillin/streptomycin (Sigma Aldrich, Taufkirchen, Germany). MC38 cell line (MC38-OVA) and B16F10 cell line (B16F10-OVA) stably expressing ovalbumin were established by Lentivirus transfection. MC38-OVA and B16F10-OVA cells were cultured in the above-mentioned DMEM media and supplemented with 2 μg/mL puromycin at 37°C at a concentration of 5% CO2 to sustain the expression of OVA. All cell lines were routinely tested and confirmed as being free of Mycoplasma contamination.
C57BL/6J 6- to 8-week-old male mice were purchased from Sipeifu Biotechnology Company (Beijing, China). The mice were fed in a pathogen free environment, and kept in a room with a light/dark cycle of 12 hours. All animal experiments were approved by the Biomedical Research Ethics Committee of West China Hospital in Sichuan Universit in accordance with the guidelines set out by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC). MC38-OVA cells (5 × 105) resuspended in PBS were subcutaneously injected into C57BL/6 mice. In this experiment, the date of tumor inoculation was set as day 0. The tumor volume was measured once every 3 days with an electronic digital caliper, which calculated the volume as follows: (width2 × length)/2. Results were displayed with a Kaplan–Meier survival curve and tumor growth curve. The mice to which MC38-OVA tumors were attached were euthanized on the 14th day after tumor injection; after their death, tumor, blood, and spleen samples were collected for further analysis. The mice were euthanized when close to death or when their attached tumors were larger than 3,000 mm3 in size. To establish the lung metastasis model, B16F10-OVA (1 × 106) cells resuspended in PBS were inoculated on C57BL/6 mice via the caudal vein. The remaining mice were euthanized on day 29, and the organs needed for the experiment were collected.
Intramuscular gene delivery was administrated using the previously established L/E/G system (13). Briefly, L/E/G refers to a combined pluronic L64/electroporation/EGCG system. Following this system, pDNA was dissolved in EGCG (E4143, Sigma-Aldrich, Germany) working solution and incubated for 30 minutes at room temperature. Subsequently, the EGCG/pDNA complex was mixed with 0.4% (W/V) L64 (435449, Sigma-Aldrich, German) until L64 reached a concentration of 0.1%. Finally, the EGCG/pDNA/L64 complex was injected into the right anterior tibial muscle of the mice; 1 hour later, the mice were stimulated with an electrical pulse to promote gene transfer.
Proteins in the supernatant that was collected from the transfected HEK293T cells were separated by 10% SDS-PAGE gel using 100 V for 2 hours and transferred to a polyvinylidene difluoride (PVDF) membrane. The membranes were blocked with 5% skimmed milk (RM00014, ABclonal, China) for 1 hour at room temperature and then incubated with HRP goat anti-mouse lgG (H+L) secondary antibodies (31430, ThermoFisher, USA), which can bind the Fc region of α-PD-1 expressed protein in mice, at a 1:5,000 dilution overnight at 4°C. The next day, the membranes were washed three times with tris-buffered saline with 0.1% Tween® 20 detergent (TBST) and, subsequently, the protein bands were detected using enhanced chemiluminescence (ECL) with a ChemiDoc XRS machine and an image analyzer (Bio-Rad, USA).
Immunostaining of lymphocytes in excised tumor tissues was carried out in accordance with the method described in a previous study (14). The paraffin sections were probed with primary anti-CD4 antibody (14–0042–82, ThermoFisher, USA) and anti-CD8 antibodies (14–0081–82, ThermoFisher, USA) mAb.
Blood was collected and left to coagulate at room temperature for 30 minutes. The clotted blood was then centrifuged at 3,500 rpm for 15 minutes at room temperature. After the serum was collected and stored at –80°C, the frozen serum samples were shipped to Li lai Co. Ltd. (Chengdu, China) for aminotransferase (AST) and alanine aminotransferase (ALT) analysis. The weights of spleens, lungs, and tumors were measured on day 29.
The IFN-γ Single-Color enzyme-linked immunospot (ELISpot) assay was carried out on the mice samples in accordance with the manufacturer’s instructions (2210002, Dakewe Biotech, China). The procedure carried out was as follows: 1 × 105 fresh splenocytes were diluted in 100 µL of CTL-Test medium (Dakewe Biotech, China) and incubated overnight at 37°C on an anti-IFNγ-coated 96-well plate. For stimulation, 10 ng/µL of TRP2180–188 peptide (SVYDFFVWL) (GenScript, Nanjing, China) was added to the plate and cultured for 2 days. A cell stimulation cocktail (CT317-PR2, Dakewe Biotech, China) was used as a positive control and PBS and a P815 irrelevant peptide (LPYLGWLVF) were used as negative controls. Finally, the spots were counted using an ELISpot reader system (Xingjianya, China).
For the quantitative determination of cytokine levels in serum, the blood of mice was collected from the orbital veins, then serum was separated at room temperature and stored at -80°C. The concentrations of interleukin-2 (IL-2) (pg/mL) (RK00007, ABclonal, China), IL-4 (pg/mL) (RK00036, ABclonal, China), and IL-10 (pg/mL) (RK00016, ABclonal, China) were determined using commercial enzyme-linked immunosorbent assay (ELISA) kits.
Binding ELISA was conducted to verify the affinity of antibody in the supernatant from pVAX-α-PD-1-transfected cells. The procedure carried out was as follows: 96-well plates were pre-coated with 10 μg/mL of PD-1 protein (Sigma) overnight at 4°C. They were subsequently washed with 0.1% Tween 20/PBS and then blocked with 5% bovine serum albumin (BSA) in a humid chamber for 1 hour at room temperature. After blocking, plates were washed three times and incubated at room temperature with supernatant from cells for 1 hour. Next, plates were washed and incubated with HRP goat anti-mouse lgG (H+L) secondary antibody at a dilution of 1:10,000 for 1 hour. The optical density was measured at 450 nm with an automatic ELISA reader (Synergy H1 Microplate Reader, BioTek, USA).
Peripheral blood and spleens were collected 14 days after tumor implantation. Spleens were pressed into single-cell through a 70 μm cell strainer (251200, Sorfa, China), followed by removal of the red blood cells with RBC lysis buffer (00430054, ThermoFisher, USA). Cells were incubated for 10 minutes on ice with anti-mouse CD16/32 antibody (14–0161–82, Thermo Fisher, USA) for Fc blocking. For CD4+ and CD8+ T-cell detection, cells were stained with eBioscience™ Fixable Viability Dye eFluor™ 450 (65–0863–14, Thermo Fisher, USA), anti-CD45 (63–0451–82, Super Bright™ 600 rat anti-mouse CD45, Thermo Fisher, USA), anti-CD3 (11–0031–82, FITC rat anti-mouse CD3, Thermo Fisher, USA), anti-CD4 (17–0042–82, APC rat anti-Mouse CD4, Thermo Fisher, USA), and anti-CD8 (45–0081–80, PerCP-Cyanine5.5 rat anti-mouse, Thermo Fisher, USA) antibodies for 30 minutes at 4°C. Blood samples were stained directly, then mixed with 1 mL of RBC lysis buffer and incubated at 37°C for 5 minutes so that red blood cells could be lysed. Sample data were acquired with LSRFortessa (BD bioscience, NJ, USA) and analyzed with FlowJo software (FlowJo, Ashland, OR, USA).
Statistical analyses were carried out using GraphPad Prism software for Windows. For all experiments, statistical significance was determined using one-way analysis of variance (ANOVA) or a Student’s t-test. The survival rate of mice was calculated using the log-rank test. In all cases, differences between groups were considered significant when the p-value was < 0.05 (*p < 0.05, **p < 0.01, and ***p < 0.001); in some cases there was no significant difference (NS).
A DNA molecule capable of directing in vivo antibody production was designed by creating a cassette consisting of the single-chain variable fragment of anti-mouse PD-1(J43 clone) and the mouse Fc region with a D265A point mutation which leads to the total loss of cytolytic function and cloned onto pVAX plasmid (Figures 1A, B). To ensure that the plasmid production of PD-1 antibody, human 293T cells were transfected with pVAX and pVAX-α-PD-1-Fc. In the culture media of cells transfected with pVAX-α-PD-1-Fc, western blot assay under non-reducing conditions detected secretion of 120-KDa protein (Figure 1C). The binding affinity of α-PD-1 to the corresponding mouse antigen PD-1 was detected using an ELISA assay. The ELISA assay showed that α-PD-1 from cell culture media bound to PD-1 protein in a dose-dependent manner and that it had a significantly higher binding capacity than the control group (Figure 1D). The results of the assay also demonstrated that pVAX-α-PD-1 was capable of encoding functional mouse PD-1 antibodies.

Figure 1 Expression and binding of PD-1 antibody. (A) Schematic of DNA construct encoding PD-1 antibody. The α- PD-1-Fc gene was cloned onto the pVAX vector and driven by the CMV promoter. (B) Structure of PD-1 antibody. α-PD-1-Fc contained scFv of mouse PD-1 antibody and the Fc region of mouse IgG1 with a D265A mutation. (C) Western blot analysis of mouse IgG from supernatants of pVAX-α-PD-1-Fc transfected 293T under non-reduced conditions. The pVAX-empty vector served as a control. Numbers indicate molecular weight. (D) The binding ELISA of α-PD-1-Fc from cell culture supernatants with mouse PD-1 antibodies. Results are representative means ± SDs of three independent experiments.
We examined the tumor inhibition effect induced by the pCMV/EMS-OVA DNA vaccine combined with pVAX-α-PD-1 on the tumor progression in MC38-OVA-bearing mice. The mice were divided into three groups according to treatment type, i.e., the pCMV-empty vector, pCMV-OVA, and pEMS-OVA groups. These groups were evenly divided into two groups after tumor formation, and they were treated with pVAX-Ctrl or pVAX-α-PD-1, respectively. DNA vaccine injections, tumor inoculation, and pVAX-α-PD-1 injections were administered on different days (Figure 2A), and we examined the effects of these different vaccination pathways on tumor sizes. The pCMV-OVA vaccination showed little tumor inhibition effect, and the pEMS-OVA vaccination caused the greatest delay in tumor growth (Figure 2B). All mice that received the pVAX-α-PD-1 treatment displayed slower tumor growth than those administered with DNA-only vaccine treatment (Figures 2C–E). In particular, mice that were in the pEMS-OVA combined with pVAX-α-PD-1 treatment group showed significant tumor regression (Figure 2F). For safety reasons, we monitored the mice for changes in body weight after tumor inoculation. All mice, irrespective of group, displayed stable weight gain (Figure 2G), and this preliminarily finding indicated that these treatments were safe enough. However, mouse survival rate varied significantly across the groups, and all mice in the pCMV-empty vector combined with pVAX-Ctrl treatment group (EV) died within 23 days of tumor inoculation. The pEMS-OVA vaccine combined with pVAX-α-PD-1 treatment group (pEMS-OVA + p-α-PD-1) showed superior results, with about 60% of the mice surviving for more than 45 days. By contrast, approximately 40% of the mice in the pCMV OVA vaccine combined with pVAX-α-PD-1 treatment group (pCMV-OVA + p-α-PD-1) survived for more than 45 days and approximately 20% of the mice in the pVAX-α-PD-1 single treatment group (EV + p-α-PD-1) survived for more than 45 days (Figure 2H). The results above demonstrate that the pEMS-OVA vaccine combined with pVAX-α-PD-1 treatment provided the greatest therapeutic benefit.
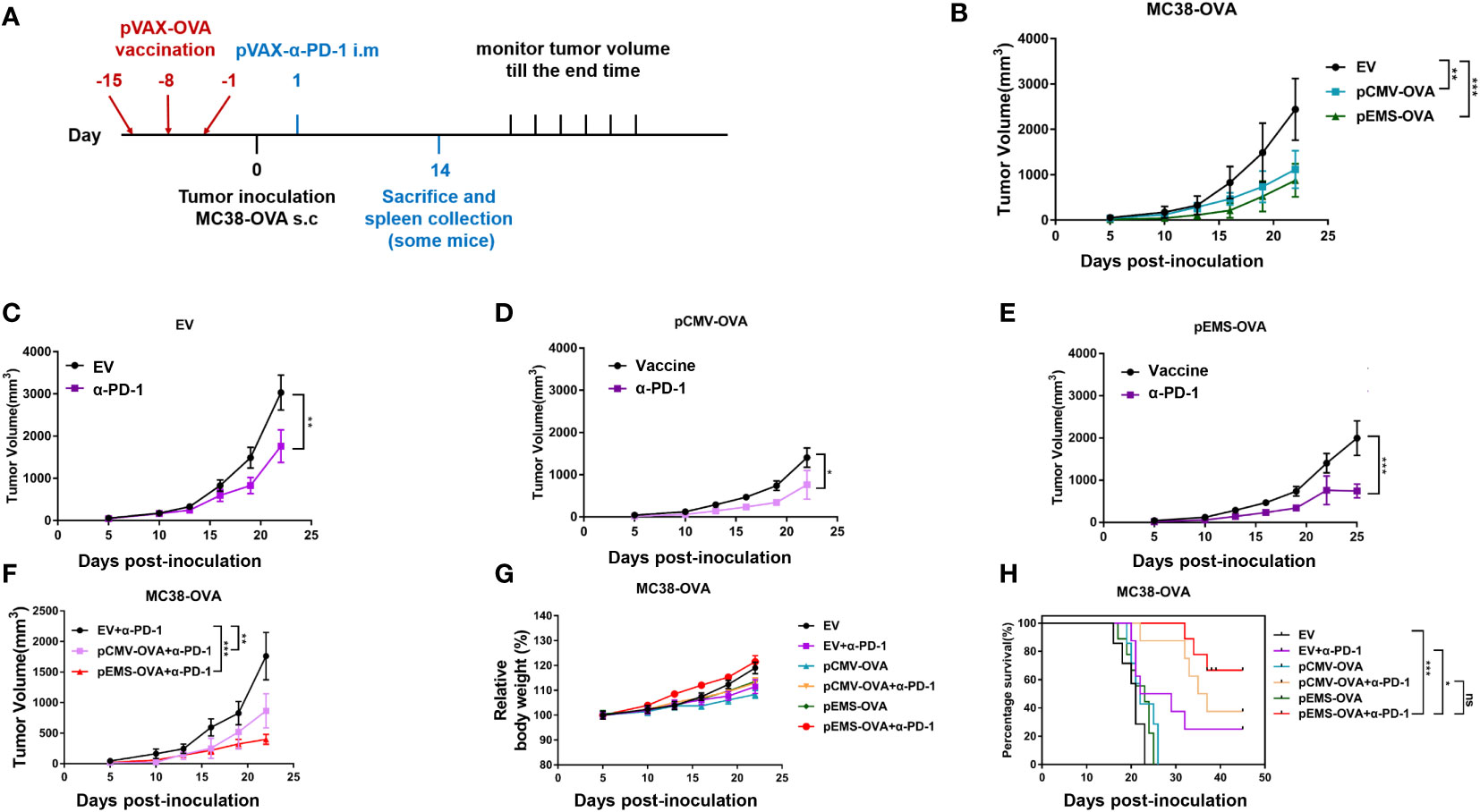
Figure 2 Anti-tumor activity of DNA vaccination and intramuscular gene transfer of PD-1 antibody combination in MC38-OVA model. (A) Schematic protocol of the tumor inoculation, DNA vaccination and pVAX-α-PD-1-Fc injection, and tumor measurement in MC38-OVA model. The DNA vaccines (pCMV-OVA or pEMS-OVA) were pre-intramuscularly electroporated 15 days, 8 days, and 1 day before 2 × 105 MC38-OVA tumor cells were subcutaneously injected into the right flanks of C57BL/6 mice. pVAX-α-PD-1-Fc were intramuscularly electroporated on day 1. Thereafter, the tumor volume was monitored every 3 days until mice were sacrificed. (B) Tumor growth curves of three different vaccine groups. The tumor growth curves of the empty vector (C), pCMV-OVA (D), and pEMS-OVA (E) DNA vaccination combined with pVAX-α-PD-1-Fc groups. (F) Tumor growth curves of three kinds in the pVAX-α-PD-1-Fc group. Relative body weight change (G) and percentage of survival curve (H) of mice receiving six different treatments. The error bars represent the mean ± SEM; n = 6 per group. Statistical analysis was carried out using one-way ANOVA with Dunnett’s multiple comparisons test or a Student’s t-test. The log-rank (Mantel-Cox) test was used to calculate the significance of survival (*p < 0.05, **p < 0.01, and ***p < 0.001). ANOVA, analysis of variance.
To study the activation levels of positive response in tumor microenvironment (TME) brought about by a combination of vaccines and pVAX-α-PD-1 therapy, the concentrations of IL-2, IL-4, and IL-10 cytokines in the serum were determined. The concentrations of IL-2, IL-4, and IL-10 were at very low levels in the vaccine-only treatment groups (i.e., the pCMV-empty vector, pCMV-OVA and pEMS-OVA groups). However, the concentration of IL-2 cytokines was slightly increased in the pCMV-empty combined with pVAX-α-PD-1 treatment group. Meanwhile, the concentrations of IL-2, IL-4, and IL-10 cytokines were significantly increased in the serum after the DNA vaccine combined with pVAX-α-PD-1 treatment, indicating that responses that were not triggered by DNA vaccine had been elicited (Figure 3A). We further evaluated the capacity of the DNA vaccine combined pVAX-α-PD-1 treatment to elicit a particular IFN-γ response by ELISpot. Without the vaccine, very few spots were detected, but the splenocytes from the DNA vaccine-treated mice showed a significant increase in the number of spots. In addition, the stimulation of immune cells with the DNA vaccine combined with pVAX–α-PD-1 treatment resulted in a significantly increased number of spots being observed (Figures 3B, C). To investigate whether the EMS promoter is more beneficial for immune activation after intramuscular injection of plasmids. In the next experiments, the mice were limited to three main combination groups that revealed significant efficacy after treatment. Fifteen days after tumor induction, MC38-OVA-bearing mice were euthanized and tumors were excised. The levels of CD8+ T and CD4+ T cells were higher in the tumor tissues of the therapeutic groups than those of the empty vector group (Figures 3D, E).
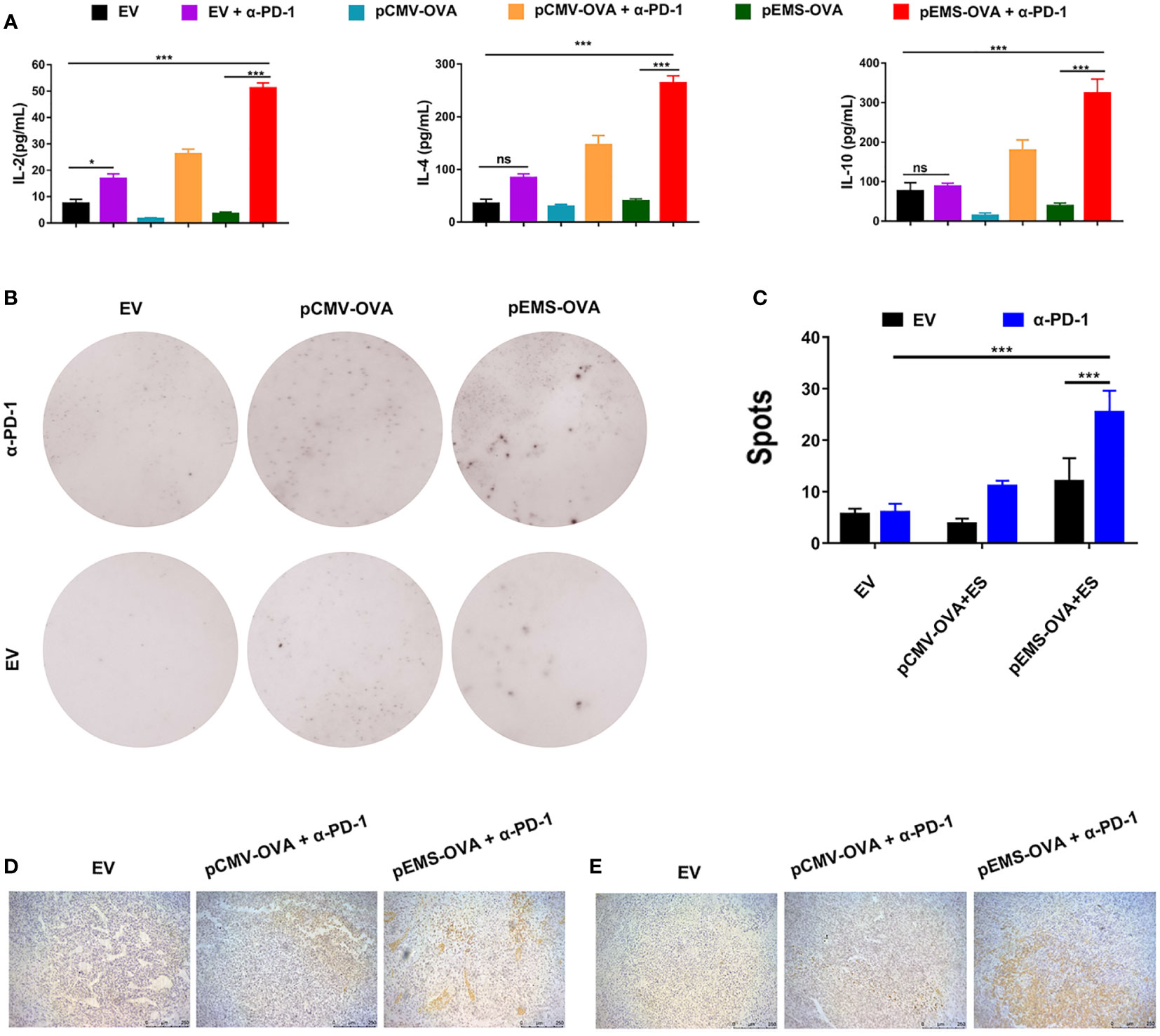
Figure 3 Immune response effect of DNA vaccination and intramuscular gene transfer of PD-1 antibody combination for MC38-OVA-bearing mice. (A) Levels of IL-2 (left), IL-4 (middle), and IL-10 (right) inflammatory mediators in the serum of mice receiving six different treatments were detected by ELISA on day 29. (B) Representative IFN-γ ELISPOT wells for splenocytes isolated from various six treatment groups. (C) Quantification of IFN-γ positive spots in the six treatment groups. Immunohistochemical analysis of CD4+ (D) and CD8+ (E) T-cell infiltration in MC38 tumors from mice treated with three kinds of vaccine combined pVAX-α-PD-1-Fc groups. Scale bars: 250 μm. Results are representative of three independent experiments and are expressed as the mean ± SD; n = 5 per group. One-way ANOVA was used to calculate the p-value, *p < 0.05, ***p < 0.001. ANOVA, analysis of variance; ELISPOT, enzyme-linked immunospot.
These results showed that the pEMS-OVA vaccine combined with pVAX-α-PD-1 treatment activated more antigen-specific lymphocyte cells than the pVAX-α-PD-1 treatment alone. In addition, increased PD-1 expression in cytotoxic T lymphocyte (CTL) indicated T-cell exhaustion and the loss of effector function (15, 16), and led to tumor growth. These results indicate that the combined pEMS-OVA vaccine and pVAX-α-PD-1 treatment leads to the increased recruitment of CTL into tumors, and thus has a significant anti-tumor effect.
Because cancer cells can spread from primitive tumors to different parts of the body even in the early stages, the inhibition of metastasis is an essential attribute of cancer vaccines. We tried to determine if pOVA vaccines (i.e., pCMV-OVA and pEMS-OVA) could enhance the inhibitory effect of intramuscular pDNA-based PD-1 antibody using a poorly immunogenic B16-F10 lung metastases model. We used the same treatment protocol as for the MC38-OVA model (Figure 4A). The weakest inhibitory effect on lung metastasis was observed in the empty vector treatment group. We also discovered the importance of the L/E/G system to vaccination, as we observed that, as in the empty vector group, there was weak suppression of lung metastasis in the DNA vaccination without L/E/G (pCMV-OVA-None + p-α-PD-1) treatment group. The pCMV-OVA combined with pVAX-α-PD-1 (pCMV-OVA + p-α-PD-1) therapy strongly suppressed the lung metastasis of tumors, which was consistent with findings in previous studies (17–19). The effect of α-PD-1 therapy is mainly dependent on innate immunity and the infiltration of lymphocytes in tumors (20, 21). We observed that pEMS-OVA combined with pVAX-α-PD-1 (pEMS-OVA + p-α-PD-1) therapy further prevents the formation of pulmonary nodules (Figures 4B, C).
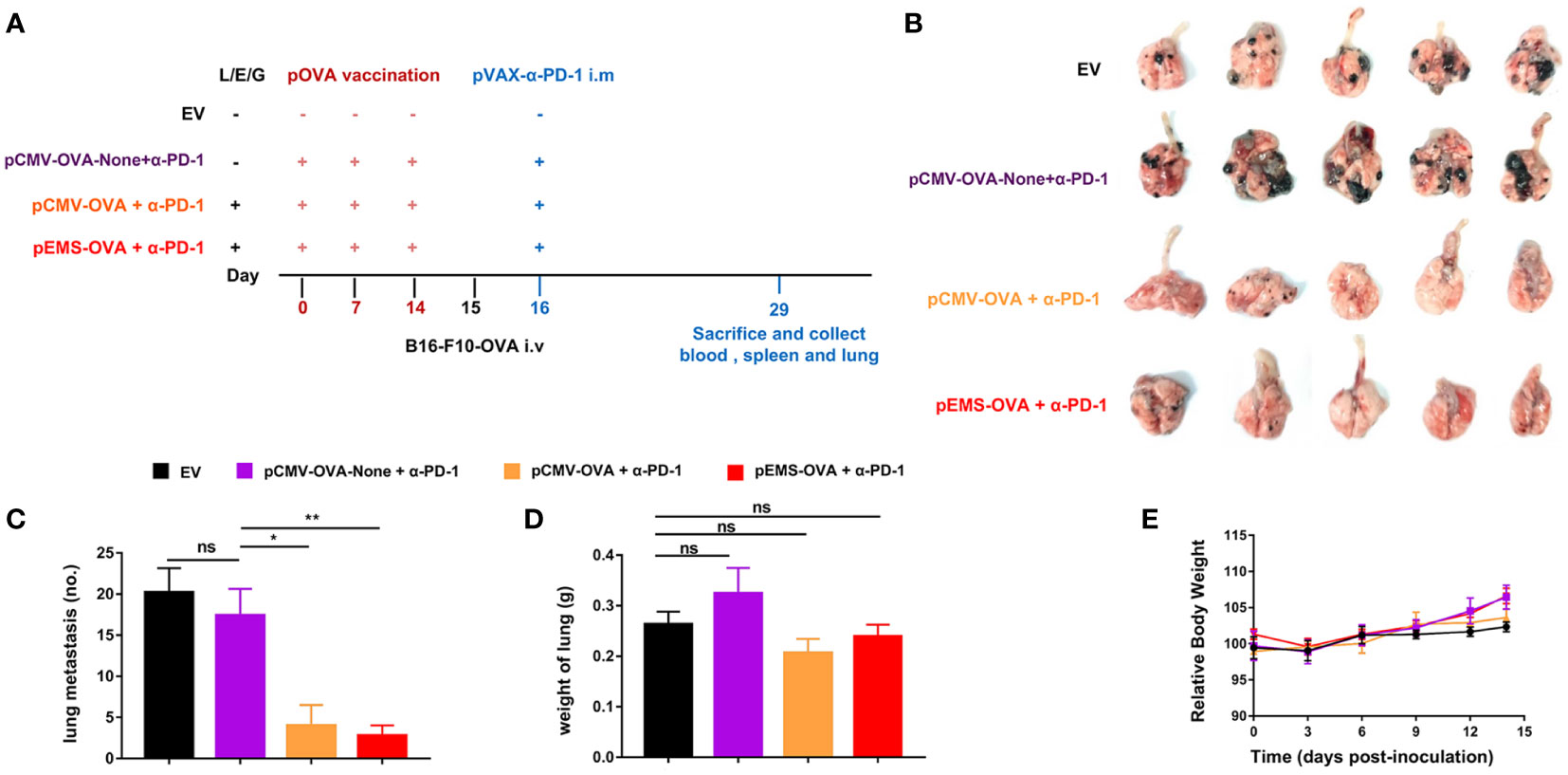
Figure 4 Lung metastasis inhibition of combination DNA vaccination and intramuscular gene transfer of PD-1 antibody in B16F10-OVA model. (A) Schematic protocol of the B16F10-OVA inoculation and combination therapy of DNA vaccination with pVAX-α-PD-1-Fc. C57BL/6 mice were divided into four groups: the combination of pOVA vaccine with α-PD-1 antibody (pCMV-OVA + α-PD-1 and pEMS-OVA + α-PD-1), the combination of vaccine without L/E/G and α-PD-1 (pCMV-OVA-none + α-PD-1) and no vaccine (empty vector). N = 5 per group. (B) Images of lung metastasis taken 2 weeks after B16F10-OVA inoculation. Number of metastases nodules on the surface of lung (C) and the wet weight of lungs (D) of the four groups. (E) Relative body weight change of mice receiving four different treatments. The data (C–E) represent the means ± SDs and were subjected to a one-way ANOVA; “NS” indicates no significant difference. *p < 0.05; **p < 0.01. ANOVA, analysis of variance; pluronic L64 (L)/electroporation (E)/epigallocatechin gallate (G) (EGCG) (referred to as L/E/G).
In excised lungs, a trend for increased lung weight was observed in the DNA vaccination without L/E/G (pCMV-OVA-None + p-α-PD-1) treatment group, although there was no statistical difference in comparison to the other groups (Figure 4D). Similarly, the hemoxylin and eosin (H&E) staining of lung tissues identified intensive metastases in control mice and mice in the DNA vaccination without L/E/G treatment group, whereas there were notably small numbers of nodules detected on the lung sections of mice in the pCMV-OVA combined with pVAX-α-PD-1 and the pEMS-OVA combined with pVAX-α-PD-1 treatment groups. Remarkably, the combination of pVAX-α-PD-1 and pEMS-OVA vaccine therapy brought about almost normal lung histopathology (Figure 5), and this efficacy may be attributed to increased CD8+ T-cell ratios in the blood and spleen (Figures 6B, D). There was no demonstrable difference, however, in the proportion of CD4+ T cells, both in the blood and spleen, across the treatment groups (Figures 6A, C). However, in the pEMS-OVA combined with pVAX-α-PD-1 treatment group, the proportion of CD8+ T cells was increased by up to 45%, and the proportion in the spleen was increased by up to 27%. The proportion of CD8+ T cells in the spleen and peripheral blood were also significantly elevated (Figures 6B, D).
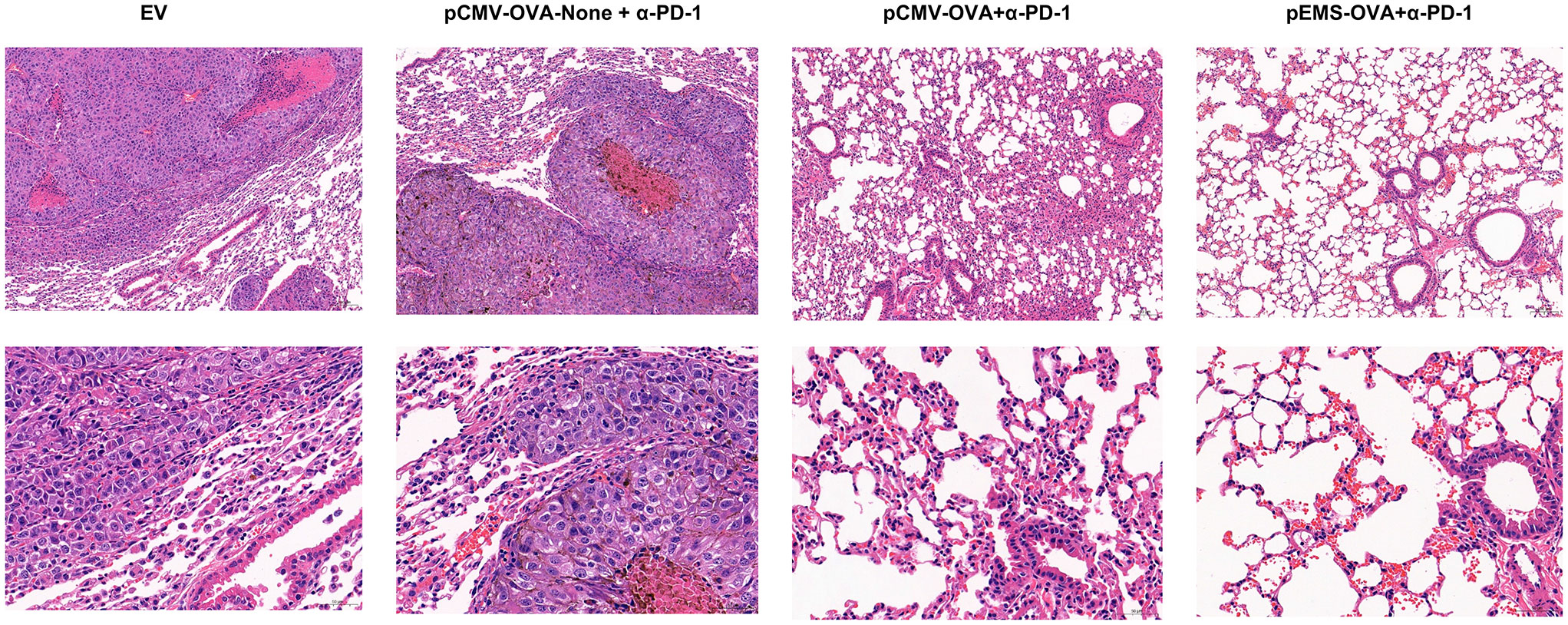
Figure 5 H&E staining of lungs collected from the treated B16F10-OVA-bearing mice. Representative H&E staining images of lungs isolated from B16F10-OVA-bearing mice following treatment with a combination of different DNA vaccination and intramuscular gene transfer of PD-1 antibody. H& E staining, hematoxylin and eosin staining.
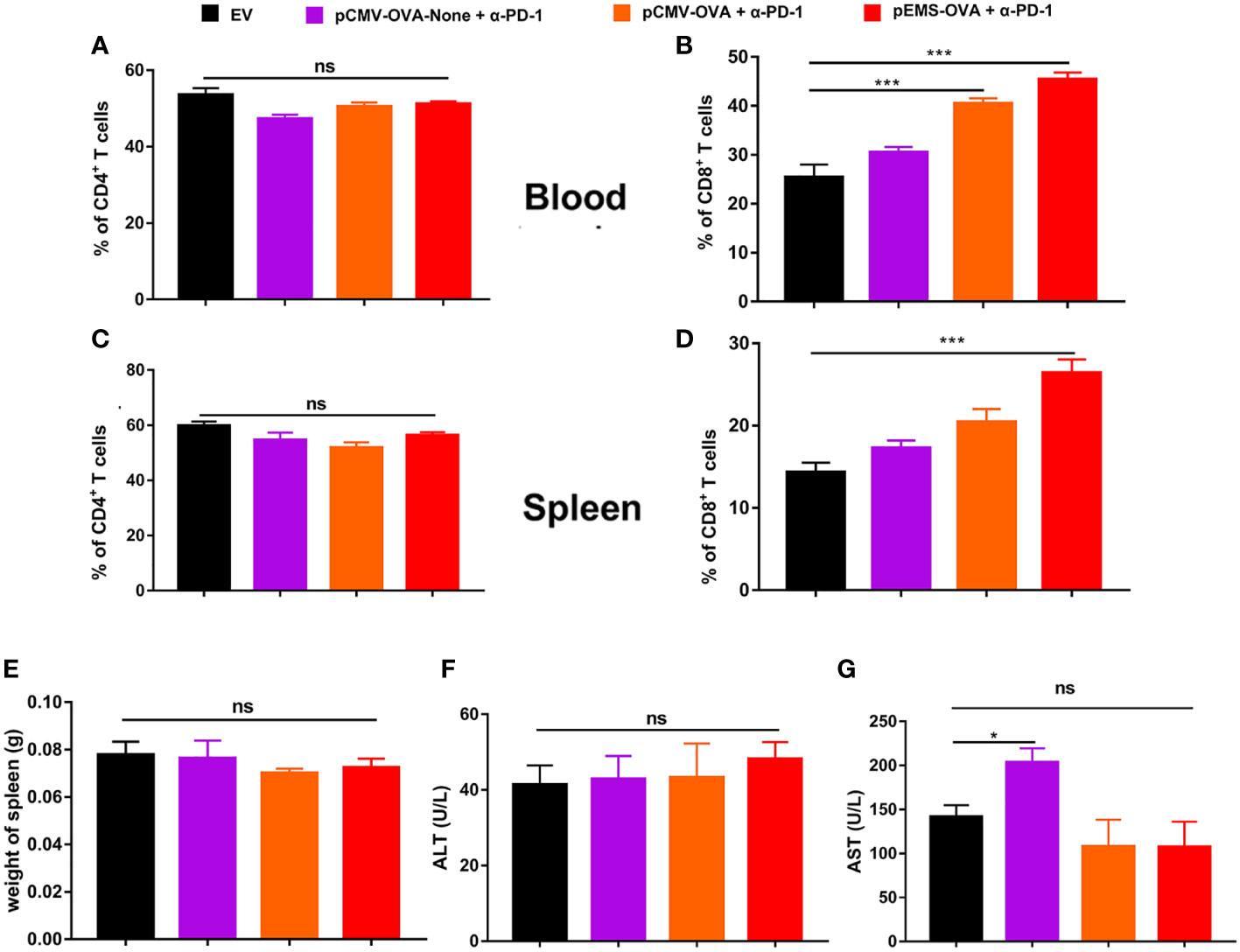
Figure 6 Immune cell populations and biochemical analysis of combination DNA vaccination and intramuscular gene transfer of PD-1 antibody in B16F10-OVA model. All mice were sacrificed at day 29. Immune cell populations analysis of spleen and blood from mice receiving four different treatments. (A) Percentage of CD4+ T cells in the blood. (B) Percentage of CD8+ T cells in the blood. (C) Percentage of CD4+ T cells in the spleen. (D) Percentage of CD8+ T cells in the spleen. (E) Spleen weight. (F) ALT level in the serum of mice. (G) AST level. Data are shown as means ± SDs. Statistical analyses were performed using one-way ANOVA with Dunnett’s multiple comparison test. *p < 0.05; **p < 0.01; ***p < 0.001. ANOVA, analysis of variance; AST, aminotransferase; ALT, alanine aminotransferase. .
Finally, we evaluated the safety of the treatments. The results showed that there was no increase in lung water content or splenomegaly after any treatment (Figures 4D, 6E). Moreover, mouse body weight increased slightly during the treatment course (Figure 4E). No significant difference was detected in ALT levels between the four groups (Figure 6F). However, AST levels were increased in the serum of the pCMV-OVA-none combined with pVAX-α-PD-1 treatment group when compared with the empty vector group, and this may be related to the malignant metastasis activity of the tumors. However, in the sera of the two vaccines with combined PD-1 antibody treatment groups, we observed decreased AST levels when compared with the empty vector group, but these differences were not significant (Figure 6G).
Overall, our data indicate that the pEMS-OVA combined with pVAX-α-PD-1 treatment had no apparent serious side effects and had enhanced anti-tumor effects. Potential mechanisms for synergistic effects of DNA vaccine may be attributed to the vaccine’s ability to boost CD8+ T cells, the exhaustion of which may have been prevented by the inhibition activity of PD-1 antibodies, expressed in vivo.
Immune checkpoint inhibitors, notably antibodies targeting PD-1 and CTLA4, have revolutionized the treatment strategies for many cancers, especially advanced melanoma, for which, currently, nearly 50% of tumors in patients can either regress or be controlled for long periods, compared to the historical figure of less than 10% (22). But achieving this ideal response with this treatment strategy is still difficult for many tumors. Recently, antibodies targeting PD-1, including pembrolizumab and nivolumab, have been approved as a second-line treatment plan for HCC patients. However, their objective response rates (ORR), being 20% and 16.9% for nivolumab and pembrolizumab, respectively, are not satisfactory (23, 24). This indicates that there are numerous patients with limited response to immunomonotherapy, and this has promoted the exploration of new tumor treatment strategies that may improve the effectiveness of immunotherapy for these unresponsive tumors.
Although satisfactory results have been obtained in preclinical models, the low immunogenicity of DNA vaccines and the adverse reactions of many patients to the blocking of immune checkpoints still limit their application in clinical environments. Previous studies have demonstrated that the intramuscular delivery of DNA is a promising therapeutic method. Because of its large therapeutic area, its accessibility, and the presence of many antigen-presenting cells within it, skeletal muscle was selected as an appropriate target, especially for the delivery of DNA plasmids encoding a variety of tumor antigens or immune checkpoint inhibitory antibodies. To make gene transfer by this means efficient, a variety of measures have been exploited to increase transfection efficiency (25).
Gene electrotransfer is a mature non-viral delivery strategy that has been used to deliver naked DNA or RNA to various types of tissue. As a commonly used delivery system, lipid nanoparticles (LNPs) provide support for the delivery and site-specific release of mRNA and DNA (26). An mRNA vaccine prepared with LNPs as carriers can therefore better resist the degradation of nuclease, making intravenous injection and other delivery routes possible. However, the clinical application potential of cationic liposomes is limited by their toxicity, non-specific immunogenicity, circulatory instability, and rapid clearance in vivo (26, 27). If LNPs are to be used more widely in clinical settings, more in-depth research is needed to solve the problems of biological distribution, insufficient immune cell recruitment, and clearance in vivo in target tissues (28, 29). mRNA is rapidly translated in the cytoplasm. The expression of therapeutic antibodies based on delivered RNA is more transient compared with that based on DNA or viral vector-mediated gene transfer (30). Although viral vectors can promote high levels of expression in the long term, it is difficult to repeatedly administer them and they may produce genetic markers in patients, and more attention ought be directed toward potential concerns with the safety of their use (31).
The L/E/G system has not been previously applied in the field of vaccine preparation. Under normal conditions, its components cause an immune reaction at the injection site and recruit cells such as macrophages, so it may be particularly suited to the production of vaccines. In this study, we used the L/E/G system to improve the transfer efficiency of plasmids. It can help plasmids to enter the nucleus efficiently. Traditional DNA vaccines must first be successfully transcribed in the nucleus before entering the cytoplasm to start the expression of antigen proteins. The nucleus's multi-layer barrier makes it difficult for DNA to enter the reaction site and therefore makes immune activation difficult. In this study, the L/E/G system could disturb the cell membrane and help plasmids to enter the muscle cells. In addition, it could deform pDNA so that it had a suitable architecture with an appropriate surface charge, prevent the degradation of nucleic acids, and improve delivery efficiency, thereby increasing the expression levels of exogenous genes in skeletal muscle cells (13). Using EMS promoter, OVA can be efficiently expressed in muscle, which is conducive to enhancing immune activation, thereby overcoming the obstacle of immune activation. Compared with the pathological alteration caused by the accumulation of lipids in liver of LNPs, L/E/G/does not cause pathological changes to main organs. Although the L/E/G system used here cannot overcome the relevant shortcomings of LNP, we still showed that it represents an available, economical, and efficient alternative to existing methods of gene delivery.
In this study, we explored the use DNA vaccine or plasmid encoded antibodies and the combined treatment of α-PD-1 with DNA vaccines through the L/E/G system, and found that the pEMS-OVA DNA vaccine can not only can prevent tumor attacks from occuring in the body, but also cooperate with pVAX-α-PD-1 plasmid to suppress tumors.
Based on studies related to neoantigen vaccination and immunomodulatory therapy, this study selected MC38 to construct a colon tumor model. It has been established that MC38 cells trigger spontaneous CD8+ T cell-induced immune responses in tumor-bearing mice. However, because of their immunosuppressive microenvironment, these immune cells are inactive and cannot eradicate tumor cells completely (32). In the early stages of tumor progression, the treatment of tumor-bearing animals with immunomodulatory antibodies targeting PD-L1 or PD-1 triggers effector T-cell responses, which can induce tumor cell death (33). In this study, we revealed that pOVA vaccines, in combination with α-PD-1 expressed by pVAX-α-PD-1, can trigger tumor death in a CD8+ T cell-dependent manner, and that treatment with α-PD-1 antibody alone moderately inhibited tumor growth.
In this study we also used a murine colon cancer model to determine what factors affected immune checkpoint inhibitor blockade, and, even though the vaccine vectors used were different, we observed that tumors were smaller in mice treated with pVAX-α-PD-1 than in those in the pVAX-empty vector group 15 days after tumor inoculation, which means that pVAX-α-PD-1 monotherapy can suppress tumor growth. In addition, tumor growth and the survival rate were substantially improved by pVAX-α-PD-1 treatment in the following experiment. Furthermore, when we examined different methods of immunization, we discovered that the pEMS-OVA expressed vaccine was associated with the highest tumor-suppressing efficiency. Our most important finding, however, was that the pEMS-OVA combined with pVAX-α-PD-1 treatment ameliorated the survival rate and reduced tumor growth. This synergistic effect originated from the ability of DNA vaccines and ICBs to stimulate T cells to fight specific cancer antigens, thereby increasing T-cell activity and tumor infiltration. Therefore, ICB creates a good microenvironment for the activity of cancer DNA vaccines. The pVAX-α-PD-1 treatment was found to significantly increase the quantity of IFN-γ-secreting CD8+ T cells. When used in combination, pOVA and pVAX-α-PD-1 not only increased levels of IL-2, IL-4, IL-10, and IFN-γ production but also demonstrably increased those of CD8+ and CD4+ T cells. IL-2 can not only adjust tolerance of the dysregulated immune system through regulatory T cells (Tregs) but also promote immune response through activating effector T cells and memory T cells (34). In early studies, IL-2 was co-administered with autologous lymphocytes for cancer treatment or for the amplification of patients’ natural killer (NK) cell and effector T-cell populations (35, 36). The combination of immunostimulatory plasmid DNA encoding IL-2 and siRNA delivered by TT-LDCP NPs targeting PD-L1 in the treatment of hepatocellular carcinoma (HCC) has also been shown to significantly increase tumor infiltration and CD8+ T-cell activation (37). The significant increase in the levels of IL-2 in the pEMS-OVA+α-PD-1 group may also indicate the up-regulated infiltration of CD8+ T cells. IL-4 is a Th2 cytokine that has a wide range of biological effects, especially with regard to immune responses (38). In the treatment of B-ALL with CD19/CD22/CD3 trispecific antibody, IL-4 was up-regulated in the treatment group (39). Another study showed that Man-CTS-TCL NPs increased the concentration of IL-4 concentration compared with the control group (40). After OVA stimulation, IL-4 levels in the spleen cells of mice increased significantly (41). Earlier studies have mainly focused on the immunosuppressive role of IL-10, but recent studies have shown that IL-10 can also reprogram the metabolic profile of T cells and restore the function of depleted T cells to produce a response (42). The tumor-triggered amplification of Tregs is an obvious obstacle to immunotherapy. Strikingly, however, AV-IL-27 treatment has been shown to induce the rapid elimination of Tregs in lymphoid organs and peripheral blood. AAV-IL-27 therapy also leads to tumor regression without significant adverse events, partly because of its induction of IL-10 (43). Further research has shown that IL-10 promotes the preservation of the effector function of antitumor CD8+ T cells (44). Interestingly, PD-1 blockade results in the compensatory release of IL-10 through tumor-infiltrating myeloid dendritic cells (45). Increased levels of IL-2/IL-4/IL-10 cytokine secretion after PD-1 antibody expressed by pVAX-α-PD-1 treatment were also observed in our study. The increased levels of IL-10 secretion in combination treatment groups may function through the the mechanism of compensatory IL-10 secretion by dendritic cells after PD-1 blockade. The production of cytokines is only one of many factors that affects tumor volume, so it is difficult to ensure that changes in tumor volumes are completely attributable to these observed trends. Immune activation starts to occur in the early phases of tumor development, especially when therapy combining DNA vaccines and ICBs is used.
In B16F10-OVA lung metastasis mice models, the combined use of ICBs and pOVA vaccines allowed us to reduce the doses of antibodies and induced a potentially more powerful immune response when compared with single therapy. The early stages of tumor development, that is up to 14 days after tumor injection, have been proven to be key to metastasis, and we therefore analyzed their role in the TME. The efficacy that suppresion of pulmonary metastases was benefit by an increase in the proportion of CD8. Consistent with these blood results, the combined use of ICBs and pOVA DNA vaccines can increase the percentage of CD8+ T cells in the spleen. In addition, pVAX-α-PD-1 without L/E/G treatment failed to inhibit lung metastases, indicating that the L/E/G system established in our previous study is sufficient to promote target gene expression and subsequent effects (13).
It is now clear that if a balance can be established between immune activation and immunosuppression, it is possible to achieve an effective anti-tumor response (46, 47) Therefore, the disadvantages of DNA vaccine or ICBs injection alone can be overcome by combining multiple treatment methods (48). In the case of cell vaccines, numerous in-depth studies have been conducted on the combined use of ICB and cancer vaccines (49, 50). Cell vaccine studies using triple therapy found that it is significantly superior to any dual or single therapies (49, 51). Strong anti-tumor effects were observed after treatment with a combination of cell-based vaccines and inhibitors. These can be attributed to antigen-specific effects, the increased proliferation of CD4+ T and CD8+ T cells, the release of antigen-specific cytokines, and the up-regulation of pivotal signaling molecules crucial to T-cell function (49). In contrast, few preclinical studies have tested the efficacy of DNA vaccines combined with immune checkpoint inhibitors (52). Published studies have explored the combination of single or dual CTLA4/PD-1 inhibitors and MYB oncoprotein targeting DNA vaccines (53), SSX2 cancer antigen (54), PSMA prostate-specific antigen (55), and TRP-2 and gp100 melanoma-specific antigens (56). The common characteristic of these studies is that combination therapy had better anti-tumor effects than single therapy. Some previous studies have demonstrated the feasibility of combined treatment of vaccines with ICI (57–60). As we know, the ICBs used in the above studies are antibodies (52, 61, 62), the costs of which are high, and the families of most cancer patients therefore cannot consider them as a treatment option. pVAX-α-PD-1 in this study utilized muscle cells as the production base of therapeutic antibodies. Currently, our research does not allow for the regulated expression of genes in vivo, and exploring gene switch platforms that control functional gene expression in vivo to enable self-regulatory therapy related to disease progression may provide a more valuable complement to this strategy (63). In addition, we can adjust the dosage of antibodies by changing the number of plasmids injected. Whether or not the effectiveness of this combined strategy is equally applicable to other tumor vaccines requires further study. The success of this strategy in preclinical research could make it possible for cancer patients to use cheap and affordable antibody drugs.
Our study reports on a specific combined treatment method. Driven by the muscle-specific promoter EMS, therapeutic proteins were specifically expressed in skeletal muscle to improve the effect of the vaccine. These observations show that the PD-1 antibody produced by intramuscular plasmid injection can synergize with DNA vaccines, most probably because of the highly increased effector function by expanding the frequencies of tumor-specific T cells. This combined strategy could represent a simple, convenient, economical, and effective means of oncology treatment.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by the West China Hospital of Sichuan University Biomedical Research Ethics Committee.
XL and YY performed the experiments, analyzed data, and wrote the manuscript. GW and ML were involved in study conception and design. XZ was involved in data analysis and contributed intellectually to the research. All authors contributed to manuscript editing. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Grant No. 31971390), Sichuan Science and Technology Program (Grant No. 2021YFH0142), and 1.3.5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (Grant No. ZYJC21043).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov (2019) 18(3):197–218. doi: 10.1038/s41573-018-0007-y
2. Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold tumors: A therapeutic challenge for immunotherapy. Front Immunol (2019) 10:168. doi: 10.3389/fimmu.2019.00168
3. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med (2015) 373(1):23–34. doi: 10.1056/NEJMoa1504030
4. Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JM, et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med (2016) 22(12):1402–10. doi: 10.1038/nm.4200
5. Yue W, Chen L, Yu L, Zhou B, Yin H, Ren W, et al. Checkpoint blockade and nanosonosensitizer-augmented noninvasive sonodynamic therapy combination reduces tumour growth and metastases in mice. Nat Commun (2019) 10(1):2025. doi: 10.1038/s41467-019-09760-3
6. Lopes A, Vanvarenberg K, Préat V, Vandermeulen G. Codon-optimized P1A-encoding DNA vaccine: Toward a therapeutic vaccination against P815 mastocytoma. Mol Ther Nucleic Acids (2017) 8:404–15. doi: 10.1016/j.omtn.2017.07.011
7. Lambricht L, Lopes A, Kos S, Sersa G, Préat V, Vandermeulen G. Clinical potential of electroporation for gene therapy and DNA vaccine delivery. Expert Opin Drug Deliv (2016) 13(2):295–310. doi: 10.1517/17425247.2016.1121990
8. Lallow EO, Jhumur NC, Ahmed I, Kudchodkar SB, Roberts CC, Jeong M, et al. Novel suction-based in vivo cutaneous DNA transfection platform. Sci Adv (2021) 7(45):eabj0611. doi: 10.1126/sciadv.abj0611
9. Spugnini EP, Scimeca M, Amadio B, Cortese G, Fanciulli M, Vincenzi B, et al. Definition of a novel plasmid-based gene transfection protocol of mammalian skeletal muscles by means of in vivo electroporation. Int J Mol Sci (2020) 21(18):6494. doi: 10.3390/ijms21186494
10. Yang R, Wang L, Xie J, Li X, Liu S, Qiu S, et al. Treatment of type 2 diabetes mellitus via reversing insulin resistance and regulating lipid homeostasis in vitro and in vivo using cajanonic acid a. Int J Mol Med (2018) 42(5):2329–42. doi: 10.3892/ijmm.2018.3836
11. Lu QL, Bou-Gharios G, Partridge TA. Non-viral gene delivery in skeletal muscle: a protein factory. Gene Ther (2003) 10(2):131–42. doi: 10.1038/sj.gt.3301874
12. Duperret EK, Perales-Puchalt A, Stoltz R, G HH, Mandloi N, Barlow J, et al. A Multi-neoantigen vaccine drives predominately MHC class I CD8(+) T-cell responses, impacting tumor challenge. Cancer Immunol Res (2019) 7(2):174–82. doi: 10.1158/2326-6066.CIR-18-0283
13. He Y, Liu Y, Sun Z, Han F, Tang JZ, Gao R, et al. The proper strategy to compress and protect plasmid DNA in the pluronic L64-electropulse system for enhanced intramuscular gene delivery. Regener Biomater (2019) 6(5):289–98. doi: 10.1093/rb/rby028
14. Peters T, Ausmeier K, Rüther U. Cloning of fatso (Fto), a novel gene deleted by the fused toes (Ft) mouse mutation. Mammalian genome : official journal of the International Mammalian Genome Society (1999) 10(10):983–6.
15. Shi L, Chen S, Yang L, Li Y. The role of PD-1 and PD-L1 in T-cell immune suppression in patients with hematological malignancies. J Hematol Oncol (2013) 6(1):74. doi: 10.1186/1756-8722-6-7
16. Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol Cancer (2019) 18(1):155. doi: 10.1186/s12943-019-1091-2
17. Gao L, Zhang C, Gao D, Liu H, Yu X, Lai J, et al. Enhanced anti-tumor efficacy through a combination of integrin αvβ6-targeted photodynamic therapy and immune checkpoint inhibition. Theranostics (2016) 6(5):627–37. doi: 10.7150/thno.14792
18. Gallotta M, Assi H, Degagné E, Kannan SK, Coffman RL, Guiducci C, et al. Inhaled TLR9 agonist renders lung tumors permissive to PD-1 blockade by promoting optimal CD4(+) and CD8(+) T-cell interplay. Cancer Res (2018) 78(17):4943–56. doi: 10.1158/0008-5472.CAN-18-0729
19. Ajona D, Ortiz-Espinosa S, Moreno H, Lozano T, Pajares MJ, Agorreta J, et al. A combined PD-1/C5a blockade synergistically protects against lung cancer growth and metastasis. Cancer Discov (2017) 7(7):694–703. doi: 10.1158/2159-8290.CD-16-1184
20. Taggart D, Andreou T, Scott KJ, Williams J, Rippaus N, Brownlie RJ, et al. Anti-PD-1/anti-CTLA-4 efficacy in melanoma brain metastases depends on extracranial disease and augmentation of CD8(+) T cell trafficking. Proc Natl Acad Sci USA (2018) 115(7):e1540-9. doi: 10.1073/pnas.1714089115
21. Kümpers C, Jokic M, Haase O, Offermann A, Vogel W, Grätz V, et al. Immune cell infiltration of the primary tumor, not PD-L1 status, is associated with improved response to checkpoint inhibition in metastatic melanoma. Front Med (Lausanne) (2019) 6:27. doi: 10.3389/fmed.2019.00027
22. Carlino MS, Larkin J, Long GV. Immune checkpoint inhibitors in melanoma. Lancet (London, England) (2021) 398(10304):1002–14. doi: 10.1016/S0140-6736(21)01206-X
23. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet (2017) 389(10088):2492–502. doi: 10.1016/S0140-6736(17)31046-2
24. Finn RS, Ryoo BY, Merle P, Kudo M, Bouattour M, Lim HY, et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: A randomized, double-blind, phase III trial. J Clin Oncol (2020) 38(3):193–202. doi: 10.1200/JCO.19.01307
25. Kaestner L, Scholz A, Lipp P. Conceptual and technical aspects of transfection and gene delivery. Bioorg Med Chem Lett (2015) 25(6):1171–6. doi: 10.1016/j.bmcl.2015.01.018
26. Miao L, Zhang Y, Huang L. mRNA vaccine for cancer immunotherapy. Mol Cancer (2021) 20(1):41. doi: 10.1186/s12943-021-01335-5
27. Qiu M, Li Y, Bloomer H, Xu Q. Developing biodegradable lipid nanoparticles for intracellular mRNA delivery and genome editing. Acc Chem Res (2021) 54(21):4001–11. doi: 10.1021/acs.accounts.1c00500
28. Cullis PR, Hope MJ. Lipid nanoparticle systems for enabling gene therapies. Mol Ther (2017) 25(7):1467–75. doi: 10.1016/j.ymthe.2017.03.013
29. Xiao Y, Shi K, Qu Y, Chu B, Qian Z. Engineering nanoparticles for targeted delivery of nucleic acid therapeutics in tumor. Mol Ther Methods Clin Dev (2019) 12:1–18. doi: 10.1016/j.omtm.2018.09.002
30. Rybakova Y, Kowalski PS, Huang Y, Gonzalez JT, Heartlein MW, DeRosa F, et al. mRNA delivery for therapeutic anti-HER2 antibody expression in vivo. Mol Ther (2019) 27(8):1415–23. doi: 10.1016/j.ymthe.2019.05.012
31. Hollevoet K, Declerck PJ. State of play and clinical prospects of antibody gene transfer. J Transl Med (2017) 15(1):131. doi: 10.1186/s12967-017-1234-4
32. Kleinovink JW, Marijt KA, Schoonderwoerd MJA, van Hall T, Ossendorp F, Fransen MF. PD-L1 expression on malignant cells is no prerequisite for checkpoint therapy. Oncoimmunology (2017) 6(4):e1294299. doi: 10.1080/2162402X.2017.1294299
33. Fransen MF, Schoonderwoerd M, Knopf P, Camps MG, Hawinkels LJ, Kneilling M, et al. Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI Insight (2018) 3(23):e124507. doi: 10.1172/jci.insight.124507
34. Hernandez R, Põder J, LaPorte KM, Malek TR. Engineering IL-2 for immunotherapy of autoimmunity and cancer. Nat Rev Immunol (2022) 22(10):614–28. doi: 10.1038/s41577-022-00680-w
35. Lotze MT, Frana LW, Sharrow SO, Robb RJ, Rosenberg SA. In vivo administration of purified human interleukin 2 And immunologic effects of the jurkat cell line-derived interleukin 2. J Immunol (1985) 134(1):157–66. doi: 10.4049/jimmunol.134.1.157
36. Rosenberg SA, Lotze MT, Muul LM, Chang AE, Avis FP, Leitman S, et al. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N Engl J Med (1987) 316(15):889–97. doi: 10.1056/NEJM198704093161501
37. Huang KW, Hsu FF, Qiu JT, Chern GJ, Lee YA, Chang CC, et al. Highly efficient and tumor-selective nanoparticles for dual-targeted immunogene therapy against cancer. Sci Adv (2020) 6(3):eaax5032. doi: 10.1126/sciadv.aax5032
38. Shi J, Song X, Traub B, Luxenhofer M, Kornmann M. Involvement of IL-4, IL-13 and their receptors in pancreatic cancer. Int J Mol Sci (2021) 22(6):2998. doi: 10.3390/ijms22062998
39. Zhao L, Li S, Wei X, Qi X, Liu D, Liu L, et al. A novel CD19/CD22/CD3 trispecific antibody enhances therapeutic efficacy and overcomes immune escape against b-ALL. Blood (2022) 140(16):1790–802. doi: 10.1182/blood.2022016243
40. Shi GN, Zhang CN, Xu R, Niu JF, Song HJ, Zhang XY, et al. Enhanced antitumor immunity by targeting dendritic cells with tumor cell lysate-loaded chitosan nanoparticles vaccine. Biomaterials (2017) 113:191–202. doi: 10.1016/j.biomaterials.2016.10.047
41. Jiang PL, Lin HJ, Wang HW, Tsai WY, Lin SF, Chien MY, et al. Galactosylated liposome as a dendritic cell-targeted mucosal vaccine for inducing protective anti-tumor immunity. Acta Biomater (2015) 11:356–67. doi: 10.1016/j.actbio.2014.09.019
42. Guo Y, Xie YQ, Gao M, Zhao Y, Franco F, Wenes M, et al. Metabolic reprogramming of terminally exhausted CD8(+) T cells by IL-10 enhances anti-tumor immunity. Nat Immunol (2021) 22(6):746–56. doi: 10.1038/s41590-021-00940-2
43. Zhu J, Liu JQ, Shi M, Cheng X, Ding M, Zhang JC, et al. IL-27 gene therapy induces depletion of tregs and enhances the efficacy of cancer immunotherapy. JCI Insight (2018) 3(7):e98745. doi: 10.1172/jci.insight.98745
44. Fujii S, Shimizu K, Shimizu T, Lotze MT. Interleukin-10 promotes the maintenance of antitumor CD8(+) T-cell effector function in situ. Blood (2001) 98(7):2143–51. doi: 10.1182/blood.V98.7.2143
45. Lamichhane P, Karyampudi L, Shreeder B, Krempski J, Bahr D, Daum J, et al. IL10 release upon PD-1 blockade sustains immunosuppression in ovarian cancer. Cancer Res (2017) 77(23):6667–78. doi: 10.1158/0008-5472.CAN-17-0740
46. Karaki S, Anson M, Tran T, Giusti D, Blanc C, Oudard S, et al. Is there still room for cancer vaccines at the era of checkpoint inhibitors. Vaccines (Basel) (2016) 4(4):37. doi: 10.3390/vaccines4040037
47. Swart M, Verbrugge I, Beltman JB. Combination approaches with immune-checkpoint blockade in cancer therapy. Front Oncol (2016) 6:233. doi: 10.3389/fonc.2016.00233
48. Kleponis J, Skelton R, Zheng L. Fueling the engine and releasing the break: combinational therapy of cancer vaccines and immune checkpoint inhibitors. Cancer Biol Med (2015) 12(3):201–8.
49. Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res (2013) 73(12):3591–603. doi: 10.1158/0008-5472.CAN-12-4100
50. Madan RA, Heery CR, Gulley JL. Combination of vaccine and immune checkpoint inhibitor is safe with encouraging clinical activity. Oncoimmunology (2012) 1(7):1167–8. doi: 10.4161/onci.20591
51. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA (2010) 107(9):4275–80. doi: 10.1073/pnas.0915174107
52. Duperret EK, Wise MC, Trautz A, Villarreal DO, Ferraro B, Walters J, et al. Synergy of immune checkpoint blockade with a novel synthetic consensus DNA vaccine targeting TERT. Mol Ther (2018) 26(2):435–45. doi: 10.1016/j.ymthe.2017.11.010
53. Cross RS, Malaterre J, Davenport AJ, Carpinteri S, Anderson RL, Darcy PK, et al. Therapeutic DNA vaccination against colorectal cancer by targeting the MYB oncoprotein. Clin Transl Immunol (2015) 4(1):e30. doi: 10.1038/cti.2014.29
54. Rekoske BT, Smith HA, Olson BM, Maricque BB, McNeel DG. PD-1 or PD-L1 blockade restores antitumor efficacy following SSX2 epitope-modified DNA vaccine immunization. Cancer Immunol Res (2015) 3(8):946–55. doi: 10.1158/2326-6066.CIR-14-0206
55. Muthumani K, Marnin L, Kudchodkar SB, Perales-Puchalt A, Choi H, Agarwal S, et al. Novel prostate cancer immunotherapy with a DNA-encoded anti-prostate-specific membrane antigen monoclonal antibody. Cancer Immunol Immunother (2017) 66(12):1577–88. doi: 10.1007/s00262-017-2042-7
56. Gregor PD, Wolchok JD, Ferrone CR, Buchinshky H, Guevara-Patiño JA, Perales MA, et al. CTLA-4 blockade in combination with xenogeneic DNA vaccines enhances T-cell responses, tumor immunity and autoimmunity to self antigens in animal and cellular model systems. Vaccine (2004) 22(13-14):1700–8. doi: 10.1016/j.vaccine.2003.10.048
57. Tondini E, Arakelian T, Oosterhuis K, Camps M, van Duikeren S, Han W, et al. A poly-neoantigen DNA vaccine synergizes with PD-1 blockade to induce T cell-mediated tumor control. Oncoimmunology (2019) 8(11):1652539. doi: 10.1080/2162402X.2019.1652539
58. Bausart M, Vanvarenberg K, Ucakar B, Lopes A, Vandermeulen G, Malfanti A, et al. Combination of DNA vaccine and immune checkpoint blockades improves the immune response in an orthotopic unresectable glioblastoma model. Pharmaceutics (2022) 14(5):1025. doi: 10.3390/pharmaceutics14051025
59. Lopes A, Vanvarenberg K, Kos Š, Lucas S, Colau D, Van den Eynde B, et al. Combination of immune checkpoint blockade with DNA cancer vaccine induces potent antitumor immunity against P815 mastocytoma. Sci Rep (2018) 8(1):15732. doi: 10.1038/s41598-018-33933-7
60. Kos S, Lopes A, Preat V, Cemazar M, Lampreht Tratar U, Ucakar B, et al. Intradermal DNA vaccination combined with dual CTLA-4 and PD-1 blockade provides robust tumor immunity in murine melanoma. PloS One (2019) 14(5):e0217762. doi: 10.1371/journal.pone.0217762
61. Miura R, Sawada SI, Mukai SA, Sasaki Y, Akiyoshi K. Synergistic anti-tumor efficacy by combination therapy of a self-assembled nanogel vaccine with an immune checkpoint anti-PD-1 antibody. RSC Adv (2020) 10(14):8074–9. doi: 10.1039/C9RA10066K
62. Lopes A, Bastiancich C, Bausart M, Ligot S, Lambricht L, Vanvarenberg K, et al. New generation of DNA-based immunotherapy induces a potent immune response and increases the survival in different tumor models. J Immunother Cancer (2021) 9(4):e001243. doi: 10.1136/jitc-2020-001243
Keywords: DNA vaccine, immune checkpoint inhibitors, intramuscular gene delivery, pDNA, L/E/G
Citation: Liu X, Yang Y, Zheng X, Liu M and Wang G (2023) Enhancedanti-tumor efficacy through a combination of intramuscularly expressed DNA vaccine and plasmid-encoded PD-1 antibody. Front. Immunol. 14:1169850. doi: 10.3389/fimmu.2023.1169850
Received: 20 February 2023; Accepted: 24 March 2023;
Published: 17 April 2023.
Edited by:
Lian Xiang Luo, Guangdong Medical University, ChinaReviewed by:
Chuqiao Dong, Nvidia, United StatesCopyright © 2023 Liu, Yang, Zheng, Liu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gang Wang, d2dhbmdAc2N1LmVkdS5jbg==; Ming Liu, bWluZ2xpdTcyMUBhbGl5dW4uY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.