 Gábor Koncz
Gábor Koncz Viktória Jenei1
Viktória Jenei1 Eszter Váradi
Eszter Váradi Attila Bácsi
Attila Bácsi Anett Mázló
Anett Mázló
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 03 July 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1169560
This article is part of the Research TopicTranscriptional Regulation of Macrophage FunctionView all 16 articles
Most of the leading causes of death, such as cardiovascular diseases, cancer, dementia, neurodegenerative diseases, and many more, are associated with sterile inflammation, either as a cause or a consequence of these conditions. The ability to control the progression of inflammation toward tissue resolution before it becomes chronic holds significant clinical potential. During sterile inflammation, the initiation of inflammation occurs through damage-associated molecular patterns (DAMPs) in the absence of pathogen-associated molecules. Macrophages, which are primarily localized in the tissue, play a pivotal role in sensing DAMPs. Furthermore, macrophages can also detect and respond to resolution-associated molecular patterns (RAMPs) and specific pro-resolving mediators (SPMs) during sterile inflammation. Macrophages, being highly adaptable cells, are particularly influenced by changes in the microenvironment. In response to the tissue environment, monocytes, pro-inflammatory macrophages, and pro-resolution macrophages can modulate their differentiation state. Ultimately, DAMP and RAMP-primed macrophages, depending on the predominant subpopulation, regulate the balance between inflammatory and resolving processes. While sterile injury and pathogen-induced reactions may have distinct effects on macrophages, most studies have focused on macrophage responses induced by pathogens. In this review, which emphasizes available human data, we illustrate how macrophages sense these mediators by examining the expression of receptors for DAMPs, RAMPs, and SPMs. We also delve into the signaling pathways induced by DAMPs, RAMPs, and SPMs, which primarily contribute to the regulation of macrophage differentiation from a pro-inflammatory to a pro-resolution phenotype. Understanding the regulatory mechanisms behind the transition between macrophage subtypes can offer insights into manipulating the transition from inflammation to resolution in sterile inflammatory diseases.
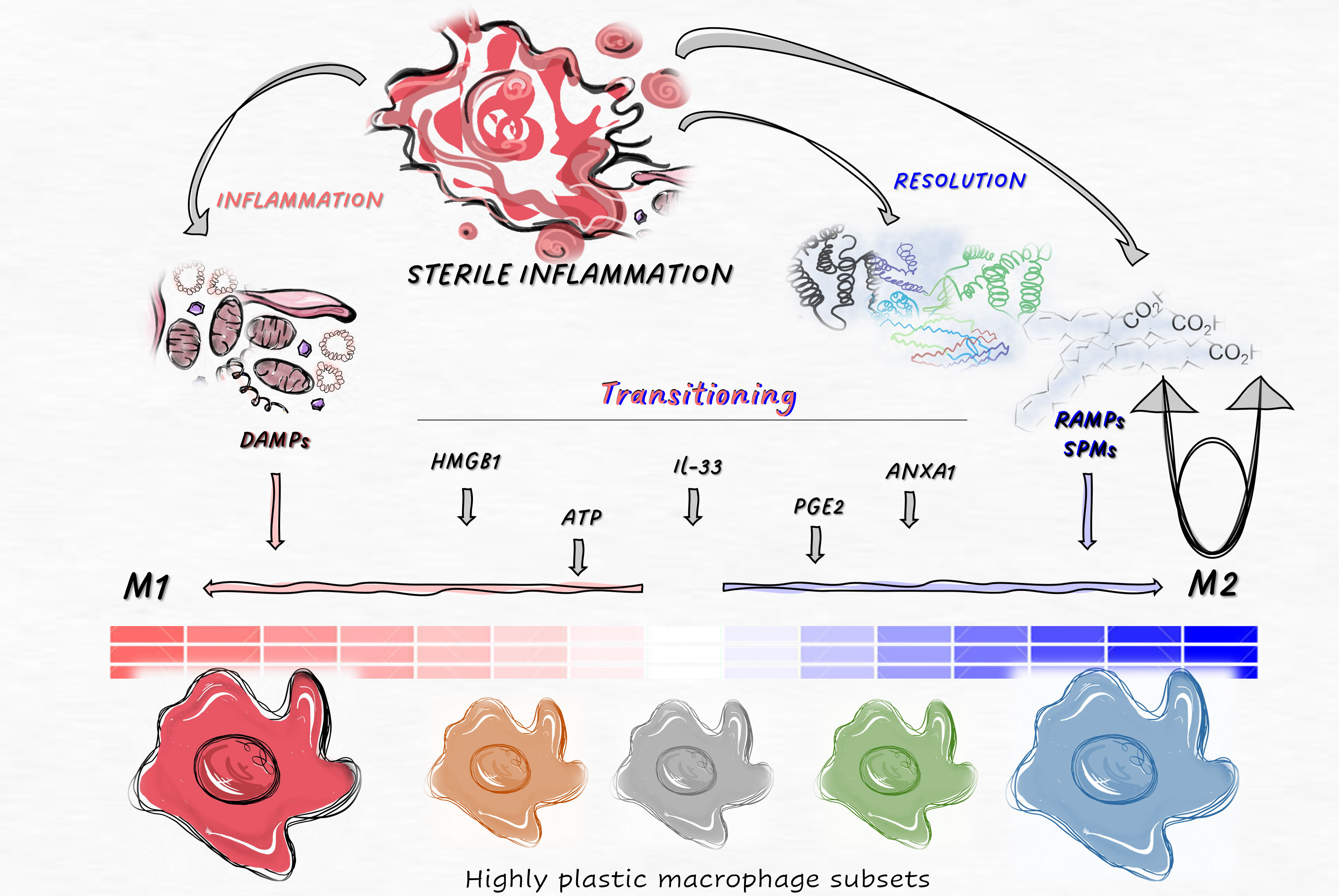
Graphical Abstract
Diseases associated with inflammation are major contributors to global mortality. Conditions such as cardiovascular and neurodegenerative diseases, cancer, various lung, kidney, and liver diseases, as well as autoimmune diseases, are all connected to sterile inflammation (1, 2). The social impact of these diseases and sterile inflammation surpasses that of infectious diseases. The inflammatory response is initiated by tissue-resident cells, leading to the infiltration of various cells and substances from the bloodstream. This complex immune response triggers the production of inflammatory mediators, resulting in tissue damage. However, over time, the inflammatory process transitions towards tissue regeneration, facilitated by the release of immunosuppressive cytokines, specialized pro-resolving mediators (SPMs), and growth factors (3, 4).
For decades, it has been known that sentinel cells of the immune system are capable of detecting and responding to various danger signals, not limited to those derived from pathogens. Consequently, both pathogen-associated molecular patterns (PAMPs) and damage-associated molecules (DAMPs) can activate tissue-resident cells. While the immune response to pathogenic invaders is primarily focused on eliminating microbes, sterile inflammation aims to resolve inflammation instead of removing the pathogen (5). Due to these distinct objectives, inflammatory responses induced solely by DAMPs in sterile conditions and those associated with pathogen-induced PAMPs may differ more than anticipated. Specifically, the kinetics and ratio of inflammation-to-resolution processes can significantly vary between sterile and pathogen-induced inflammation (6).
While sterile inflammation is typically dependent on damage-associated molecular patterns (DAMPs), the primary source of DAMPs is the release of intracellular components during cell death processes. Consequently, the initial stage of sterile inflammation often involves the activation of uncontrolled necrotic cell death or the newly described regulated necrotic cell death processes (7). During cell death, resolution-associated molecules (RAMPs) are simultaneously produced alongside DAMPs, thereby enhancing anti-inflammatory processes (4, 8). These regulated necrotic cell death pathways, which vary in terms of membrane permeabilization kinetics and mechanisms, secrete distinct combinations of RAMPs and DAMPs, as we have previously outlined (1). Since the ratio of these molecules influences tissue regeneration and can also trigger various innate and adaptive immune responses, the unique profile of these released mediators elicits diverse resolution and immune-related reactions depending on each specific cell death pathway (9).
Tissue-specific factors, such as the general regenerative capacity, immune and stem cell composition of the given tissue, the effects of the neuroendocrine system, etc. all influence the steps of the inflammatory and regenerative processes. Certain stimuli that trigger sterile inflammation, such as burn (10), cancer (11), trauma (12), autoimmunity (13), toxins, ischemia reperfusion injury and crystals (2), can induce different cell compositions and responses at the site of insults. However, according to currently accepted theory, the turning point from inflammatory responses to resolution depends on the macrophage population transitioning from a pro-inflammatory to a pro-resolution phenotype in each unique tissue environment (14).
Macrophages play crucial roles as primary responders, phagocytes, and professional antigen-presenting cells in the afferent, central, and efferent phases of immune responses. There are two distinct functional extremes within macrophage subtypes: the classical proinflammatory and the alternative pro-resolution subpopulations. In addition to these extremes, macrophage subsets with high plasticity exhibit functional and phenotypic differences (15). These different macrophages can function sequentially depending on the phase of inflammation, but they can also act concurrently. The relative ratio of these subtypes determines the balance between inflammation and resolution, ultimately affecting the homeostatic or pathological outcome.
Pathological sterile inflammation can arise from abnormal modes or intensities of cell death (9, 16), the dysregulated release of damage-associated molecular patterns (DAMPs) (as discussed in our review articles (1, 9)), or failure in transitioning from inflammation to resolution (17). In this review, we aim to provide a summary of how RAMPs and DAMPs - released during sterile tissue damage - influence the differentiation of monocytes into macrophages. Additionally, we examine how these factors regulate the function of inflammatory and tolerogenic macrophage subsets.
In the absence of pathogens, sterile inflammation is triggered by the release of damage-associated molecular patterns (DAMPs) associated with tissue damage (Table 1). DAMPs can originate from intracellular molecules released during necrotic processes, as well as from the extracellular matrix damage or the secretion of intracellular molecules through vesicles in response to cellular stress signals (22). Accordingly, modified matrix proteins, the extracellular appearance of cytosolic, nuclear, mitochondria-, ER-derived molecules, or vesicle-released proteins can activate the sentinel cells of innate immunity, resulting in the production of inflammatory mediators (23).
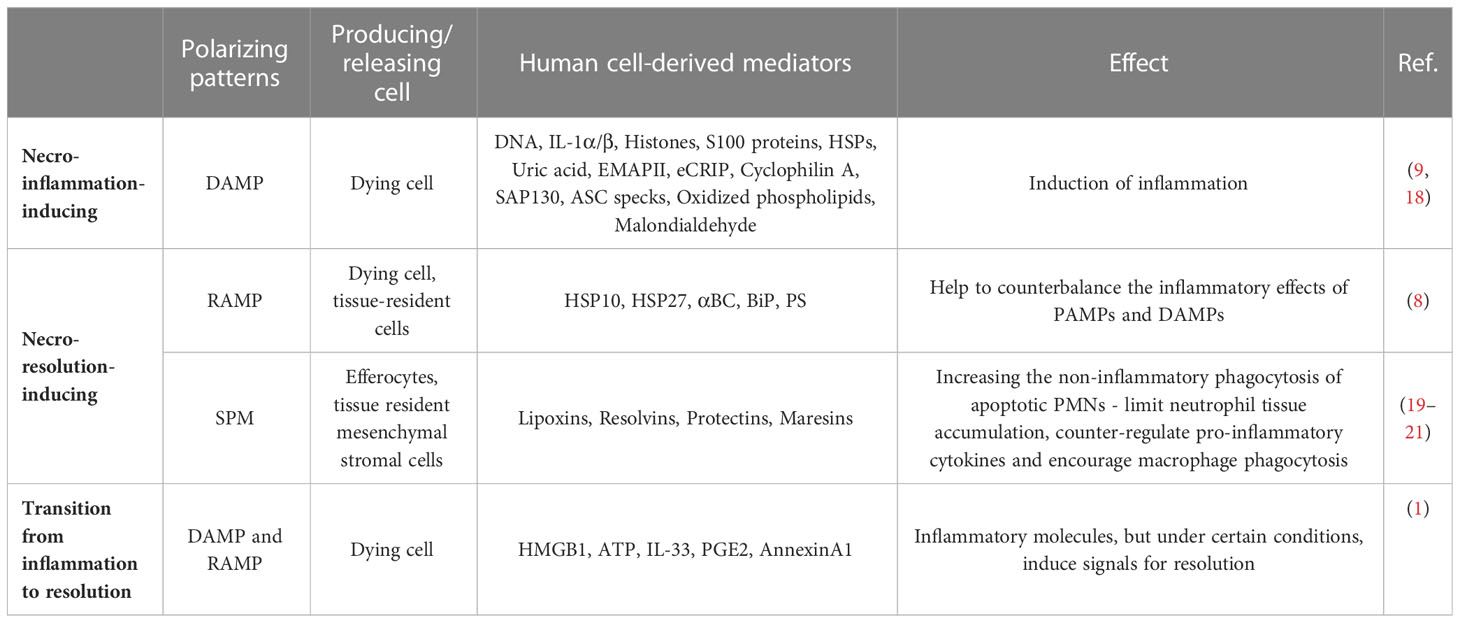
Table 1 DAMPs, RAMPs and SPMs as coordinators of necro-resolution and necro-inflammation.
Other dying cell-derived factors, RAMPs, and SPMs, such as protectins, resolvins, lipoxins and maresins, may also influence the inflammatory processes and induce necro-resolution. Immune cells – such as macrophages – sense the presence of dead cells, and engulf them, which activates immunosuppressive and tissue regenerative functions in a time-dependent manner by inducing the production of anti-inflammatory cytokines and further SPMs. The relative ratio of DAMPs/RAMPs and pro/anti-inflammatory mediators contribute to the formation of a polarizing microenvironment (Figure 1).
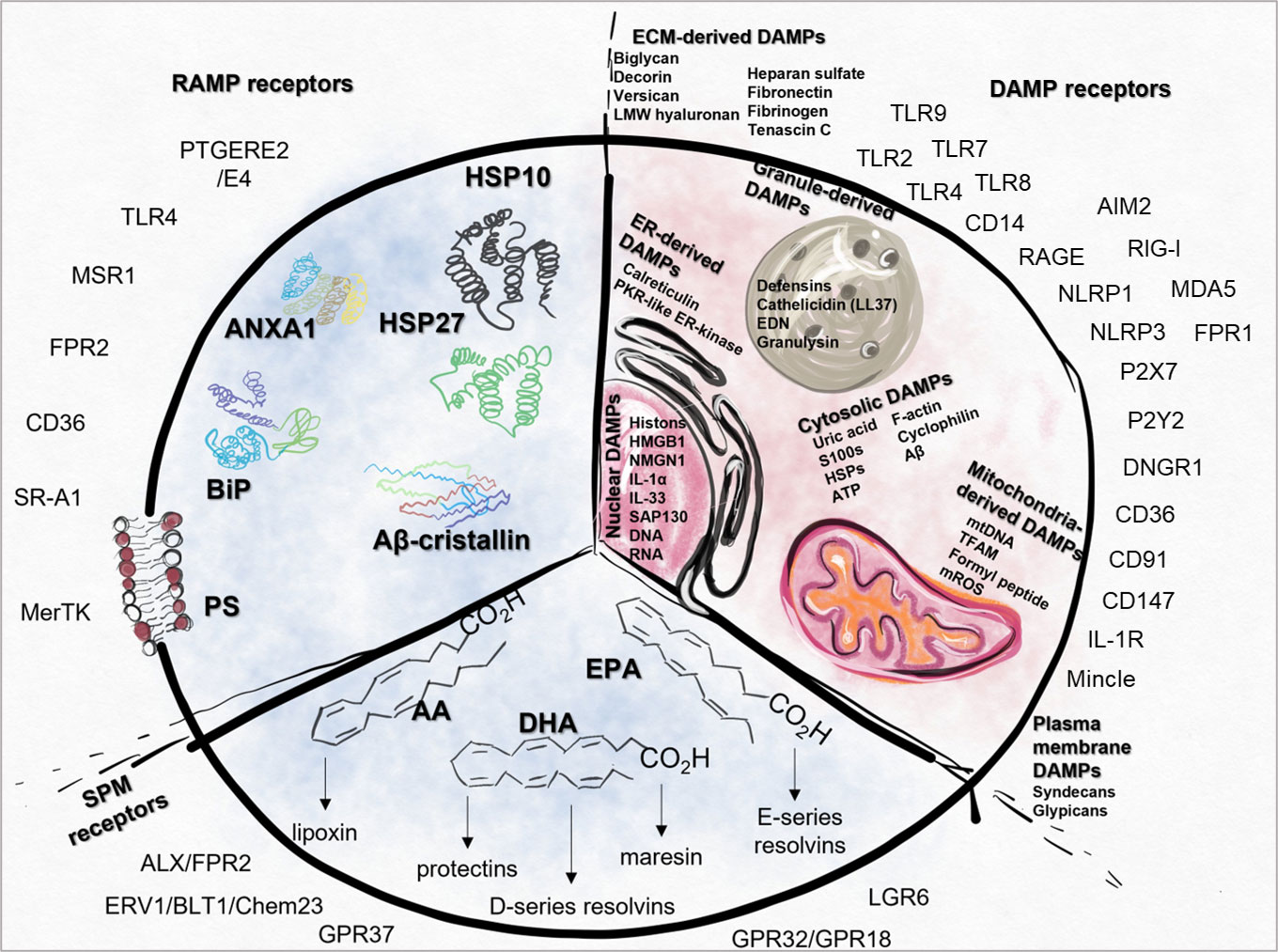
Figure 1 Summary of DAMPs, RAMPs, SPMs and their receptors. Intracellular (IC) DAMPs can be released from the cells during cell death or cellular stress and the damage of ECM also results in the generation of DAMPs (EC). DAMP-sensing receptors are responsible for the recognition of ECM, cytosolic, nuclear, mitochondria-, ER-, plasma membrane- or granule-derived DAMPs. In case of cell damage, RAMPs can also be released, therefore their receptors act simultaneously with the signals generated by DAMPs. SPMs can be produced from AA, DHA or EPA, which directs the resolution process by activating different SPM receptors. AA, arachidonic acid; AIM2, absent in melanoma 2; ALX, Lipoxin receptor; ANXA1, Annexin A1; Aβ, amyloid beta; BiPS, Binding immunoglobulin protein; CD, cluster of differentiation; CD14, cluster of differentiation 14; Chem23, Chemokine like receptor 23; CLEC4E, C-Type; Lectin Domain Family 4 Member E, CREB, Cyclic AMP-Responsive Element-Binding Protein; DAMP, danger associated molecular pattern; DHA, docosapentaenoic acid; ECM,, extracellular matrix; EPA, eicosapentaenoic acid; ERV1, human resolvin E1 receptor; FPR, Formyl peptide receptor; GPR37, G Protein-Coupled Receptor 37; HMGB1, High mobility group box 1 protein; HSP10, Heat shock protein 10; IC, intracellular; LGR6, Leucine-rich repeat-containing G protein-coupled receptor 6; LMW, Low-molecular-weight; LPS, Lipopolysaccharides; MDA5, melanoma differentiation-associated protein 5; MerTK, MER proto-oncogene, tyrosine kinase; Mincle, Macrophage Inducible C-Type Lectin; mtDNA, Mitochondrial DNA; MRC-1, Mannose-Receptor C type 1; MSR1, Macrophage scavenger receptor 1; NLRP1, NLR family pyrin domain containing 1; NLRP3, NLR family pyrin domain containing 3; PS, Phosphatidylserine; PTGERE2,, Prostaglandin E2 receptor; RAGE, Receptor for Advanced Glycation Endproducts; RAMP, resolution-associated molecular patterns; RIG-I, retinoic acid-inducible gene I; SAP130, Histone deacetylase complex subunit; SPM, Specialized pro-resolving mediators; SR-A1, Class, A1 scavenger receptors; ST2, Interleukin-1 receptor-like 1; TLR, Toll-like receptor; TREM1/2, Triggering receptor expressed on myeloid cells 1/2.
Importantly, the tipping point, when and how the inflammatory reactions turn into a resolution process, is poorly understood. Certain DAMPs, which originally trigger proinflammatory processes, can be converted into forms that trigger anti-inflammatory processes by further modifications, such as the oxidation of HMGB1 or the processing of ATP into adenosine. Certain receptors of dying cells (such as the “don’t eat me” signals CD24, CD31, and CD47) or soluble molecules that can bind to DAMPs (C1q, CD52) and stimulate the inhibitory receptors of efferocytes, thus transforming DAMP-mediated signaling into anti-inflammatory ones. Tissue damage increases the amount and proportion of dead cells. Phosphatidylserine (PS)-expressing cell corps activate MerTK-induced signaling in efferocytes, leading to SPM and anti-inflammatory cytokine production and polarization of macrophages to an M2 phenotype. The effects of additional DAMPs such as IL-33, Annexin A1 (ANXA1), and certain inflammatory mediators (PGE2) depend on the cells and/or receptors that sense them. Thus, alterations in the tissue environment can change their effect into an anti-inflammatory effect [reviewed in (1)].
The immune response is influenced not only by the types of RAMPs and DAMPs released but also by the cells responsible for sensing them. It is worth noting that the same RAMP or DAMP molecules can elicit different responses depending on the activation of either tolerogenic or inflammatory subpopulations of tissue-resident cells. Simultaneously, the recognition of these factors can impact the differentiation of monocytes, the polarization and the functional activity of macrophages, allowing them to finely adjust their activity and adapt to the current environment (24).
Macrophages that are in a resting state are referred to as inactive, non-polarized “M0” type macrophages. The M0 macrophages represent a diverse population characterized by their origin and tissue-specific functions. In this context, our focus is on the overall immune-related functions of macrophages, while a comprehensive summary of their specific characteristics can be found in reviews published elsewhere (3, 25, 26).
Functionally, classical macrophage-like cells (M1) exhibit pro-inflammatory, anti-microbial, and tumor-resistant properties. They are characterized by increased expression of CD80, CD86, and the production of immunostimulatory cytokines (TNFα, IL-1β, IL-6, IL-12, IL-23). M1 cells also secrete a wide range of chemokines (Figure 2 panel A), which facilitate the activation and proliferation of NK cells, CD4+ Th1 cells, and CD8+ T-lymphocytes.

Figure 2 Main characteristics of in vitro differentiated and polarized M1 and M2 macrophages. Each phenotype could be formed in response to certain inductors: M1 upon LPS and/or IFN-γ (A); М2а in the presence of IL4/IL13; M2b in response to immune complexes; M2c could be generated after binding of IL-10 or TGF-β snd M2d with IL6 and adenosine (B); The THP1 cell line is also often used for macrophage differentiation (C). Arg1- Arginase-1, BMP, Bone morphogenetic protein; CCL2, Monocyte chemoattractant protein-1; COX-2, cyclooxygenase-2; COX-2, cyclooxygenase-2; FABP4, Fatty acid binding protein 4; Fizz1, Resistin-like molecule alpha1; GM-CSF, Granulocyte-macrophage colony-stimulating factor; HIF-1-α, Hypoxia-Inducible Factor Promotes; IFN-γ, Interferon gamma; IL-1R, Interleukin-1 receptor; iNOS, Inducible nitric oxide synthase; iNOS, Inducible nitric oxide synthase; Klf4, Krüppel-like factor 4; M-CSF, macrophage colony-stimulating factor; MRC-1, Mannose-Receptor C type 1; PMA, Phorbol myristate acetate; PPARs, peroxisome proliferator-activated receptors; PPARs, peroxisome proliferator-activated receptors; PTEN, lipid and protein phosphatase and tensin homolog; SOCS3, suppressor of cytokine signaling-3; STAT1, Signal transducer and activator of transcription 1; TGF-beta, Transforming growth factor-beta; TNF, Tumor necrosis factor; VEGF, Vascular endothelial growth factor.
On the other hand, alternative macrophage-like cells (M2 cells) play a crucial role in anti-inflammatory and resolution processes. They are predominantly identified by the presence of CD163 and CD206 markers. M2 cells possess the ability to produce both inflammatory (IL-6, TNFα, IL-12) and anti-inflammatory (IL-10, TGFβ) cytokines, contributing to the restoration of homeostasis (Figure 2 panel B). Their functions as immunoregulators and promoters of tumor growth. M2 cells exhibit high phagocytic capacity and are involved in producing extracellular matrix (ECM) components, angiogenic factors, chemotactic factors, and various cytokines. In addition to their role in pathogen defense, they also participate in the clearance of apoptotic cells, suppression of inflammatory responses, and promotion of wound healing and tissue remodeling.
Within the two endpoints, between cells M1 and M2, several macrophage subpopulations can be created in vitro or identified in different tissues under steady state or different pathological conditions. M2 macrophages were further divided into M2a, M2b, M2c, and M2d subtypes based on applied stimuli and induced transcriptional changes (27) (Figure 2).
The transition from innate M1 to M2 states indicates that macrophage differentiation and activation occur along a spectrum, where distinct epigenetic, metabolic, phenotypic, morphological, and functional characteristics are acquired by different cell populations. The collective expression of macrophage-derived factors and cell surface markers determines the functional properties of macrophages. It is important to note that individual cells can co-express “markers” associated with both M1 (e.g., CD163, TNFα) and M2 (CD209, TGFβ) states, emphasizing the complexity of polarization (28). Furthermore, there can be the simultaneous presence of functionally distinct subpopulations of macrophages, enabling precise modulation of tissue responses.
The fate of monocyte/macrophage populations may also vary in the post-inflammatory phase; some of the cells are likely to die due to the inflammatory reaction. Surviving cells can undergo phenotype conversion in situ, leading to the appearance of M2-like resident macrophages, but a few of them, trained monocytes, preserve their phenotype for a longer period, even months, “remembering” previous inflammatory stimuli that affected them (29, 30).
In this chapter, we aim to provide a general overview of the commonly used and widely accepted methods for in vitro macrophage differentiation and polarization. In recent decades, various terms have been used to classify different types of macrophages exposed to cytokines or Toll-like receptor (TLR) agonists. In human studies, peripheral-blood monocytes or THP-1, a human monocyte-derived cell line from a patient with acute monocytic leukemia, have been commonly utilized as an in vitro system for generating macrophage subpopulations. Although not entirely representative of in vivo cell populations, this model system has proven to be one of the most reproducible experimental standards (29). Initially, the impact of interferon-γ (IFNγ) and/or lipopolysaccharide (LPS) on M1 macrophages and interleukin-4 (IL-4) on M2 macrophages was described (31). Another widely employed approach to differentiate polarized macrophages involves using GM-CSF for M1 cells and M-CSF for M2 cells alone (32). The essential protocols for in vitro macrophage differentiation and polarization, along with the key characteristics of the resulting M1 and M2 cells, are summarized in Figure 2.
Distinct differences have been observed in the transcriptomes of these generated cells. Stimulation of macrophages with IFNγ or IL-4 exclusively activates either STAT1 or STAT6 (33). Other genetic modifications also lead to shifts in macrophage polarization. For instance, IRF5 and SOCS3 drive M1 differentiation, whereas the transcription factors IRF4 or KLF4 and certain PPARs induce M2 differentiation (34). Notably, metabolic changes occur during differentiation as well. Molecules associated with anabolic growth, such as AKT2 or PTEN, promote an M1-like expression pattern, whereas the mTOR inhibitor TSC1 facilitates M2 formation (35). M2 macrophages exhibit more pronounced arginine metabolism compared to M1 macrophages, and this disparity disappears when the Slc7a2 promoter responsible for arginine transport is deleted (36).
It is important to note that in vitro models are not capable of modeling the complex interactions occurring within tissues, significant emphasis should be placed on considering results derived from in vivo systems. Although the polarization of macrophages into M1-like or M2-like states can also be identified in vivo, it requires a more precise characterization. Their characterization primarily relies on detailed phenotypic and functional studies, as well as single-cell analyses.
The in vivo differentiation of macrophages is a much more complex process influenced by numerous factors. Their characteristics are influenced by the individual’s general health condition; age (37, 38), potential pathological conditions (39–41), and the stage, flare-up, or resolution of diseases (14). There are numerous available in vivo model where sterile inflammation is artificially induced, resulting in tissue damage. In the case of immune reactions triggered by PAMPs, the inflammatory agent is known. However, in models that induce sterile inflammation, the question of which DAMP released due to injury serves as the initiator of immune reactions or its potential role in regulating the immune response is often not addressed (except, e.g (42–44). among others), even though DAMPs, RAMPs, and SPMs are part of the complex tissue microenvironment.
Here we collected models represent different experimental approaches used to study sterile inflammation in various contexts, but where the potential DAMPs are not identified. Sterile inflammation could be induced by overexposure to UVB irradiation (i.e., sunburn) in the mouse plantar skin (45), polyvinylpyrrolidone (PVP) in rat model (46), as well as with phorbol 12-myristate 13-acetate (PMA) to induce acute mouse ear inflammation (47). Another possibility is the stimulation with 2,6,10,14-tetramethylpentadecane (TMPD, also known as pristane) (48). Additionally, it can be evoked with cytodex bead slurry injection subcutaneously into the back space in mouse (49), furthermore, the hepatic I/R Injury also can be induced (50). The injection of cardiotoxin into the muscle (51) can be used to trigger sterile inflammation as well as the following method can be applied: injection of mice with heparin, and the use of an atraumatic clip to interrupt the arterial and portal venous blood supply to the cephalad lobes of the liver (52). In the case of autoimmune models, the DAMPs are also neglected, however their potential presence is also determining (53).
The presence of macrophages in different conditions is still a subject of conflicting studies, but the altered functions can be attributed at least in part to the changed or divergent DAMP/RAMP/SPM sensing-receptor expression patterns.
In addition to cytokines or PAMPs, macrophage polarization can also be induced by different types of endogenous damage-related molecules (DAMPs and RAMPs). Non-polarized macrophages express relevant receptors for DAMP-, RAMP-, and SPM-sensing which could influence their differentiation into proinflammatory or proresolution macrophage subtypes (54). The presence of these receptors on M0 cells shows that these cells are sensitive to DAMP, RAMP and SPM signals, indicating that these stimuli may direct macrophage differentiation.
The relevance of our question is supported by the fact that sterile inflammation is induced in many in vivo mouse studies (48, 55), yet the role of factors released from damaged or dying cells, such as DAMPs and RAMPs, in triggering inflammation during sterile inflammation is not widely investigated. The different phases involved in tissue damage and regeneration, which heavily rely on the innate immune system, particularly monocyte-derived macrophages. These macrophages play a dual role, first in reacting to the injury, and second by removing debris, promoting resolution of inflammation, and initiating tissue repair. At the end of the inflammation phase, the pro-inflammatory phase subsides, transitioning into a regenerative inflammation phase that leads to tissue repair. There is a dynamic nature ofmacrophage response and identifies specific molecular signatures characterizing inflammatory and repair macrophages during tissue injury and repair (14).
It is well known that besides IFNγ, PAMP signals, such as LPS, are the most important stimulus for inducing M1 macrophage polarization. Since DAMPs mostly stimulate a set of receptors overlapping with PAMPs (56), we can assume that DAMPs predominantly induce M1 differentiation. Accordingly, HMGB1 facilitates the reprogramming of macrophages towards an M1-like phenotype dependent on TLR4-mediated pathways (57, 58). Recombinant HSP90 expressed in extracellular vesicles shifts the differentiation of M0 macrophages towards the M1 phenotype (59). The presence of uric acid leads to the differentiation of monocytes into inflammatory M1-like macrophages through NLRP3 activation (60). Extracellular RNA (eRNA) released from different tissues induced the differentiation of M0 macrophages towards the M1 phenotype (61). Extracellular accumulation of the stress response protein eCIRP inhibited the polarization of M2 macrophages, thereby shifting the balance of macrophage subtypes towards an inflammatory phenotype (62). However, DAMP signals do not exclusively favor the formation of proinflammatory macrophages. Factors acting together with DAMPs can modify DAMP-related functions, so their effect also depends on the tissue environment. For example, DNA released from activated neutrophils during netosis induces a rapid inflammatory response, but DNAses are also secreted, eventually ceasing the proinflammatory effect of extracellular DNA (63). High doses of S100B may also contribute to subsequent tissue regeneration (64). In the presence of immune complexes, even the highly inflammatory LPS or IL-1β signals are modulated, leading to the differentiation of the M2 macrophage subtype (65).
Intracellular molecules released from dying cells that play a direct role in inducing resolution favor the differentiation of M2 macrophages over M1 cells. HSP27 secretion induces the differentiation of tolerogenic macrophages in the tumor microenvironment (8). The extracellular appearance of HSP10, HSP27, BiP and ANXA1 promotes autocrine IL-10 production driving the differentiation toward the M2 phenotype (66). SPM secretion, resolving D1/E1, lipoxin A4 and maresin1 from dying cells has been demonstrated, and efferocytes engulfing dead cells can also produce SPMs (9). The common effect of the different SPMs is the enforcement of the M2 differentiation (1). PS appearing on the surface of dead cells also increases the polarization in the M2 direction through the activation of MerTK receptors on the efferocytes (67). MerTK signaling also results in SMP production, which also enhances the differentiation of M2 cells. As MerTK and SPM expression increases in M2 cells, a complex positive feedback loop appears between MerTK, SPMs, and M2 cells in the presence of dead cell corps. An increase in the amount of dying cells or M2 cells therefore turns the inflammatory signals in the direction of resolution (1).
Depending on their environment or post-translational modifications, some molecules can function as both DAMPs and RAMPs, such as HMGB1, ATP, IL-33, prostaglandin E2 (PGE2) and ANXA1. These molecules, originally identified as DAMPs, appear to play a critical role in regulating the transition between pro-inflammatory and pro-resolution phases (1). Accordingly, they could have an ambivalent effect on macrophage polarization. The HMGB1 molecule has three functionally distinct isoforms based on the redox state of the three cysteines; disulfide-HMGB1, fully reduced, and sulfonyl-HMGB1 variations. Disulfide-HMGB1 induces inflammatory cytokine secretion and in accordance with this functionality, polarizes macrophages in the M1 direction, with a slightly different transcriptomic profile than LPS/IFNγ signals (68). Fully reduced HMGB1 is known to induce chemotaxis and has a limited effect on macrophage polarization (68). Sulfonyl-HMGB1 has been described as inactive in inducing inflammation and new observations also suggest that sulfonyl-HMGB1 increases the M2/M1 macrophage ratio (69). Extracellular ATP, another classic DAMP molecule, is converted to immunosuppressive adenosine under the action of CD39 and CD73 ectonucleotidases. Adenosine promotes differentiation towards M2 while inhibiting M1 polarization (70). Sensing ATP by any macrophage subtype leads to the release of anti-inflammatory proteins such as ANXA1, suggesting that intact ATP may also have a potential role in resolving inflammation (71). Extracellular ATP activates P2X7R on M1 polarized macrophages, leading to the release of pro-inflammatory IL-1β via activation of the caspase-1/NLRP3 inflammasome (72). IL-1β is an important polarizing cytokine for M2b cells under certain circumstances (65). IL-33, which belongs to the IL-1 cytokine family, it is not secreted by the ER-Golgi pathway, therefore it is mainly released from cells that lose membrane integrity. Although it is considered an alarmin molecule (with a DAMP-like function), it strongly supports the differentiation of M2 cells, thus also contributing to resolution (73). PGE2 plays a role in early inflammatory events, but the second wave of its production typically induces an anti-inflammatory process (74). Its effect on macrophage differentiation characteristically stimulates M2 polarization, while it blocks the function of M1 macrophages (75, 76). ANXA1 and its N-terminal cleavage product Ac2-26 induce differentiation of monocytes into M2a, M2c-like cells and transformation of macrophages into M2 subtypes (77). Accordingly, extracellular ANXA1 favors tissue regeneration (78, 79), while ANXA1 KO mice were characterized by impaired tissue regeneration with a predominance of the M1 phenotype (78, 80).
Cancer-driven immune editing limits or alters macrophage responsiveness to DAMPs, an example of how DAMP-mediated macrophage polarization depends on the tissue environment. The intense growth of tumors coupled with their resistance to apoptosis induces significant necrotic cell death, thus leading to intensive DAMP release. In this environment, the M1 phenotype-promoting effect of DAMP signals is abrogated and it even supports the differentiation of M2-like cells, the mechanism of which is not always clear (81). Hypoxia-induced macrophage polarization is HMGB1-dependent, significantly increasing the number of tumor-associated macrophages with an M2-like phenotype in HMGB1-positive murine and human melanomas (82). Pancreatic ductal adenocarcinoma cells with endothelial-mesenchymal transition secrete HSP90 to induce M2 polarization of macrophages (83). eRNA promotes the polarization of M2 macrophages in patients with colorectal cancer (84). S100A4 enhances protumor macrophage polarization by the upregulation of PPAR-γ (85). IL-33 enhances M2 polarization by inducing a metabolic shift toward increased cellular oxidative phosphorylation and ultimately promotes tumor growth in a B16 melanoma model (86).
Overall, it can be concluded that despite the presence of proinflammatory DAMPs, tissue resolution can also develop in special tissue microenvironments, highlighting the environmental effect on the functioning of DAMPs.
Depending on the differentiation state of the cells, different signaling pathways can be activated, which control whether pro- or anti-inflammatory mediators are produced in excess. If an excess of proinflammatory or proresolution macrophage subpopulation develops in the tissue environment as a result of previous stimuli, this significantly affects the immune response to a given stimulus. The different response ability of individual subpopulations may result from the non-identical expression of receptors or the inherent difference in signaling pathways. In the next chapters, we will review how DAMP and RAMP receptors are expressed in M1 and M2 cells and compare the signaling pathways activated in these cell populations.
Damage-related molecules, as a group of danger signals, are mostly recognized by the same PRRs as pathogen-related patterns. However, some receptors are specialized only for the detection of DAMPs (23). A surprisingly small number of comparative studies have examined PRR expression on M1 and M2 cells (15). Based on these studies, virtually all DAMP-sensing receptors are expressed by both M1 and M2 cells (Table 2).
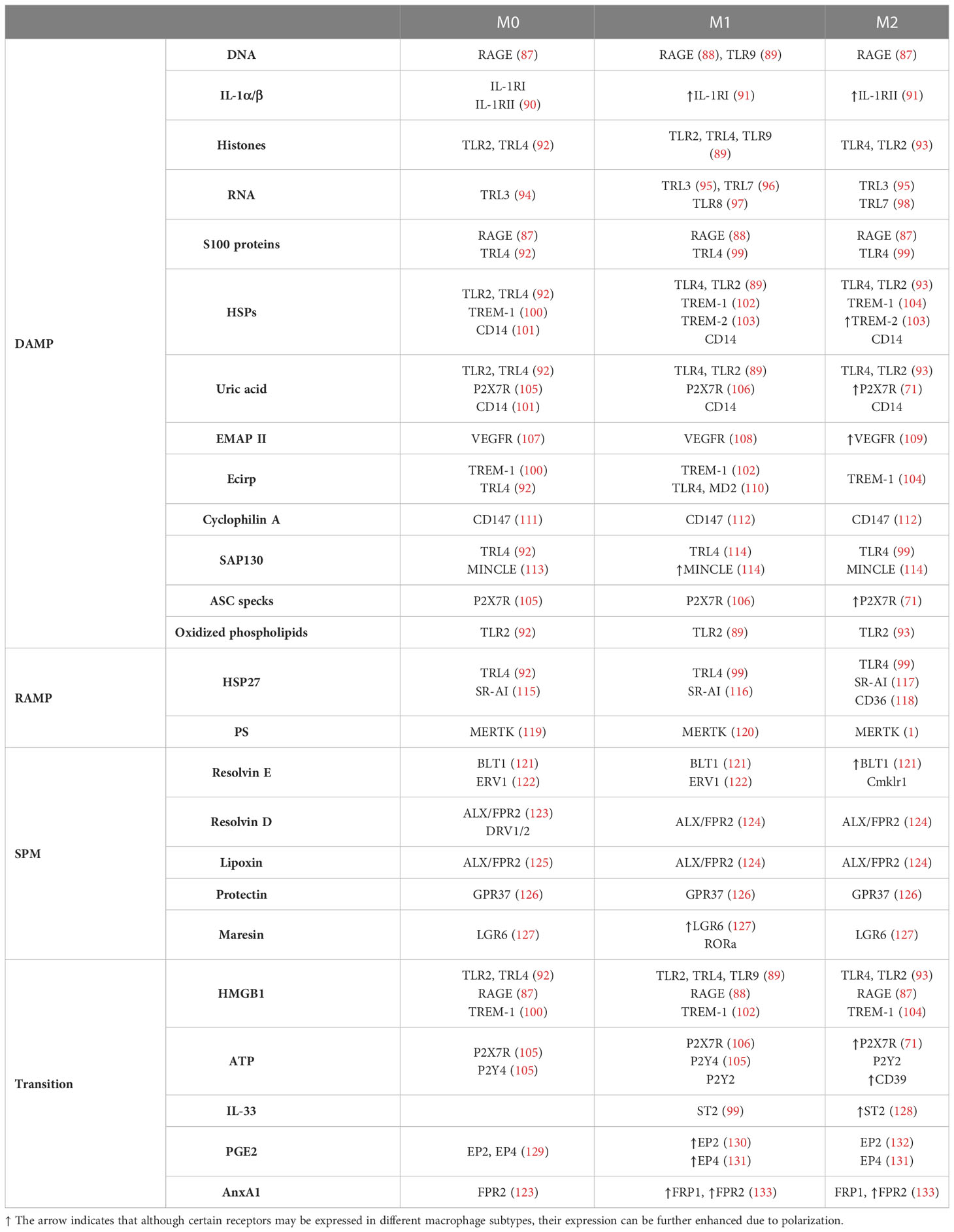
Table 2 DAMP, RAMP, SPM and “transitioning” molecule-sensing receptors on non-polarized, M1 and M2 cells.
Based on the literature, only the expression of Mincle (CLEC4E) (114), which recognizes bacterial glycolipids and SAP130 as a DAMP, and TLR2, which primarily recognizes bacterial lipids and various DAMPs, were markedly higher in M1 cells than in M2 macrophages. Thus, the different functional activity of pro- and anti-inflammatory macrophages presumably does not arise only from PRRs.
More dominant differences can be observed between the two macrophage subtypes in the expression of receptors, which can detect only danger signals, but not PAMPs, such as receptors for IL-1, heat shock proteins, VEGF, IL-33 and ATP (the last two are reviewed in the next chapter). Lower expression of IL-1RI, which has an inflammatory role, and higher expression of IL-1RII, a decoy receptor to block IL-1 signals has been published on M2 cells (27). The expression of the VEGF receptor, which detects extracellular cyclophilin A, was higher on M2 macrophages (109). Trigger receptors expressed on myeloid cells (TREM) are specialized to detect various DAMPs, such as extracellular HSP proteins, actin and cCIRP. TREM-1 is a well-known enhancer, while TREM-2 is a negative regulator of the inflammatory response (134). While TREM-1 expression is more dominant on M1 macrophages, TREM2 expression is the more typical feature of M2 macrophages based on literature data (103).
Examining the expression of the receptor, greater differences can be seen in the expression of receptors that sense DAMPs than those that sense PAMPs. However, further functional studies are needed to determine whether this divergence contributes to the functional differences between M1 and M2 macrophages.
Recognition of RAMPs and SPMs by diverse receptors (TLR4, SR-Al, CD36, PGE2R EP2/EP4, MERTK for RAMPs and ALX/FPR2, ChemR23, GPR32, GPR37, RORα, LGR6 for SPMs) are responsible for the initiation and completion of resolution process in a fine-tuned manner.
The receptors of secreted HSP proteins belonging to RAMPs, (HSP10, HSP27, BiP/HSPA5, αB-crystallin/HSPB5) are typically barely known (135). An interesting aspect of their anti-inflammatory effect is that HSPs involved in resolution can bind to the same receptors as HSPs with DAMP function, inhibiting those by competition. Hsp10 can interact with extracellular Hsp60, inhibiting inflammatory responses of this DAMP (136). Similarly, HSP27 binding may compete with LPS binding to TLR4 and oxLDL binding to SR-AI or CD36, thereby attenuating these activating signals (137). In addition, CD36 stimulation by HSP proteins resulted in PPARγ activation, indicating that high CD36 expression can convert the function of HSP proteins described as DAMPs into resolution-promoting activity (138). The expression of PS-recognizing MerTK receptors, which direct the efferocytosis of dead cells, is increased on M2 cells (67). As MerTK receptor signaling promotes the differentiation of macrophages to the M2 phenotype, this creates a positive feedback loop between efferocytosis and M2 differentiation (1, 139–141).
The receptors of individual SPMs are differentially expressed in M1 and M2 macrophages. Protectin and maresin receptors GPR37 and RORα are expressed at a higher level on M2 cells (126, 142), and the expression of the resolvin D and lipoxin-recognizing receptors FPR2 is also higher in THP-derived M2 than in M1 cells (133). In contrast, the expression of ChemR23 (resolvin E) and LGR6 (maresin) is higher on M1 cells (122, 127). The receptor for Resolvin D1, GPR32 is present on both LPS-activated M1 and IL-4-activated M2 macrophages, but its expression is reduced by TGFβ and IL-6 treatment (143).
We must underscore that SPMs even acting on M1 cells induce the differentiation of macrophages into the M2 phenotype so due to the widespread expression of SMP receptors facilitate resolution.
Over time, the signaling pathways that support inflammatory processes may weaken and those that promote dissolution and regeneration become more potent. The microenvironmental conditions promote the differentiation of M2 macrophages, while the functions of the M2 cells favor the resolution processes. This positive feedback accelerates the transition once the inflammation-resolution balance is tipped over (93). An important part of this regulation is that the receptors that convert DAMP signals into resolution signals are primarily expressed in M2 cells.
CD24 is considered a “don’t eat me” signal molecule. Siglec-10, the receptor for CD24, is expressed at a higher level in the M2 subtypes (the highest level in the M2c) (144). CD24 can be in complex with HMGB1 and induces the activation of Siglec-10, leading to anti-inflammatory signal transduction (145). By cooperating with CD24, Siglec-10 can also transform the inflammatory potential of other DAMPs, such as HSP70 or HSP90, into resolution (145). Similarly, HMGB1 loses its inflammatory potential in the presence of C1q. The C1q-HMGB1 complex activates the LAIR receptor of efferocytes, which inhibits inflammatory signals (146, 147). Like Siglec-10, LAIR1 expression is higher in M2 than in M1 macrophages (148). Thus, the engulfment of cellular debris by M2, but not by M1 cells, turns the effects of DAMPs into anti-inflammatory.
The expression of CD39 and CD73, the receptors responsible for the ATP-adenosine transition, is higher in the M2 than in the M1 phenotype based on published data (149). In contrast, some transcriptomic studies showed that the expression of CD73 was higher in M1 cells. Functionally, the hydrolysis of ATP to adenosine is more intense in M2 than in M1 cells (149). The generated adenosine inhibits the functions of M1 cells through A2A receptors and induces the polarization and activation of M2 cells through A2B receptors (70, 150). The ATP-P2X7Rs axis plays a role in inflammation in M1 cells, P2X7Rs might play an anti-inflammatory role in M2 macrophages (151). In intermediate M1/M2-polarized macrophages, extracellular ATP acts through its pyrophosphate chains, to inhibit IL-1β release (151), while P2X7R signaling does not induce NLRP3 activation in M2 macrophages (71).
The expression of the IL-33 receptor, ST2, is increased on M2 cells (128). In addition to the fact that IL-33 commonly induces M2 differentiation, IL-33 released from dying cells, or produced even by M2 cells (152), increases the expression of CD73, CD39 and MerTK, promoting other actors in tissue resolution (1).
PGE2 is known to have both a pro- and anti-inflammatory role. PGE2 binds to four different PGE2 receptors (EPs), of which EP4 mediates resolution signals. EP2 and EP4 receptors were detected on macrophages, but EP1 and EP3 receptors are barely or not expressed on these cells at all. LPS stimuli increase EP2 expression, thus modifying the EP2/EP4 ratio (130); however, increased expression of EP4 has also been demonstrated in M1 cells (131). In addition to SPMs,
Formyl peptide receptors (FPR) also recognize ANXA-1. FPR1 is mostly proinflammatory, while FPR2 has a pro-resolution effect. As mentioned, FPR2 expression is higher on M2 cells (133), moreover, IL-4 and IL-13 downregulate FPR1 expression on alternatively activated macrophages, providing a rather anti-inflammatory response to ANXA-1 on M2 cells (153). While the role of ANXA1 in promoting M2 differentiation is clear, further studies are needed to determine the ratio of FRP1/FRP2 receptors expressed on M1 and M2 cells and to demonstrate the potential differences in ANXA-mediated signaling in the two cell subtypes.
Overall, M2 macrophages, by expressing certain targeted receptors, have a high potential to convert the inflammation-inducing potential of DAMP molecules into an anti-inflammatory “RAMP-like” effect. After functional conversion, these DAMPs promote M2 differentiation, thereby accelerating the resolution process.
The pattern of DAMPs/RAMPs/SPMs released during sterile inflammation, depending on the different forms of cell death (1), results in the activation of many different receptors that can generate various stimuli. In the next chapter, we focus on the signaling initiated by DAMP/RAMP/SPM receptor ligation and the transcription factors activated.
As reviewed by Igor Malyshev et al. (154), there are two types of macrophage reprogramming signaling pathways: pathways that mainly program the M1 phenotype through the activity of Notch, TLR/NF-κB (р65/р50), PI3K/Akt2, JAK/STAT1, and HIF1α, as well as those pathways which mainly program the M2 phenotype through PI3K/Akt1, JAK/STAT3/6, TGFβ/SMAD, TLR/NF-κB (р50/р50), and HIF2α (154). Briefly, the profile of pro-inflammatory stimuli induces the polarization of M1 cells via the activation of transcription factors such as NF-κB, STAT1, STAT3, AP-1, SREBP-1, and HIF1α, while anti-inflammatory M2 macrophages are mainly controlled by transcription factors STAT6, GATA3, PPARγ, C/EBPββ, SP1, cMyc and LRX (155).
DAMP-recognizing PRRs could act in an additive or synergistic manner. TLRs can activate MyD88-IKK-NF-κB, and MyD88-MAPK-AP1 pathways, in addition, TLR3 and TLR4 signaling results in the TRIF-TBK1-IRF3/7 activation. Accordingly, DAMP-sensing receptors; TLR2, TLR3, TLR4 and TLR7 enhance the activity of AP-1 (156–158). TLRs, like TLR3, TLR4 and TLR9 induce the activation of M1-associated IRF3 and IRF5 transcription factors in macrophages (156, 159). Additionally, C-type lectin receptors through activating SYK kinase stimulate the MAPKs (AP1) and NF-κB pathways (160). NOD-like receptors with the help of adapter protein apoptosis-associated speck-like protein containing CARD, also PYCARD (ASC) activate inflammasome for caspase-1 activation resulting IL-1β and IL-18 release (161) (Figure 3).
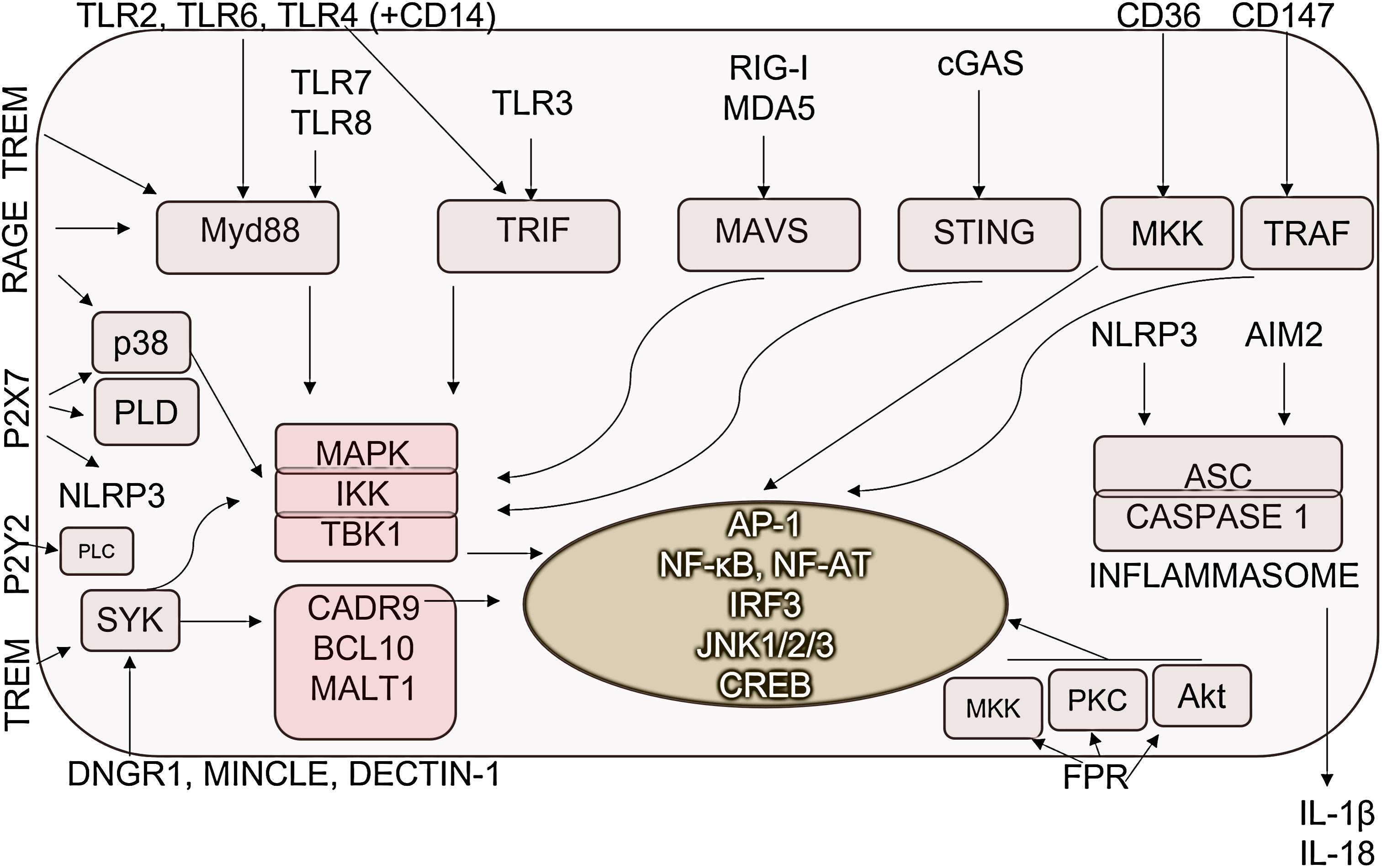
Figure 3 DAMP-sensing receptor-induced signaling pathways. DAMPs are recognized by pathogen-recognition receptors as well as various receptors specialized for the detection of human molecules. Their detection dominantly resulted in the activation of inflammation-promoting signaling pathways and transcription factors. AP-1, Activator protein 1; ASC, Apoptosis-associated speck-like protein containing a CARD; CD, cluster of differentiation; CREB, Cyclic AMP,Responsive Element-Binding Protein; DNGR-1, dendritic cell NK lectin group receptor-1; IKK, IκB kinase; JNKs- c,Jun N-terminal kinases; MAPKs, Mitogen-activated protein kinases; MALT1, mucosa-associated-lymphoid-tissue lymphoma-translocation gene 1; MAVS, Mitochondrial antiviral-signaling protein; MDA5, melanoma differentiation-associated protein 5; Mincle, Macrophage Inducible C-Type Lectin; MKK, mitogen-activated protein kinase kinase; MYD88, Myeloid differentiation primary response 88; NF-AT, Nuclear factor of activated T-cells; NF-κB, Nuclear factor-κB; NLRP3, NLR family pyrin domain containing 3; PKC, protein kinase C; PLC, Phospholipase C; PLD, Phospholipase D; RAGE, Receptor for Advanced Glycation Endproducts; RIG-I, retinoic acid-inducible gene I; STING, Stimulator of interferon genes; SYK, Spleen tyrosine kinase; TBK1, TANK-binding kinase 1; TLR, Toll-like receptor; TRAF, TNF receptor-associated factor; TREM1/2, Triggering receptor expressed on myeloid cells ½; TRIF, TIR-domain-containing adapter-inducing interferon-β.
Receptors specialized to recognize exclusively DAMPs also activate signaling pathways in macrophages. IL-1 induces similar signaling through the IL-1R as TLRs activating MyD88/IKK complex leading to NF-κB and MAPK activation. RAGE receptor activates multiple signaling pathways, such as NF-κB p65, MAP kinases, RAC1, PI3K, SMAD and the Wnt-β pathway (162, 163). M1-expressed STAT1/5 also can be triggered by RAGE and FPR2 (164, 165). TREM receptors activate MAPK-AP1 pathways. In addition, TREM1 receptors, which are mainly expressed on M1 cells, stimulate NF-κB (166), while TREM2, which is expressed on M2 cells, activates PI3K via DAP10 mediating anti-inflammatory signaling (167). Syk-mediated signaling increased Ca2+ concentration, thus contributing to M2-M1 macrophage polarization by activating NFAT (168, 169). MINCLE activates the HIF-1α transcription factor (170).
Focusing on M2-related signaling, TLR4 and MINCLE also activate the M2-specific transcription factor C/EBPββ (170), while VEGFR- and CD147-derived signals resulted in the activation of M2-associated SP1 (171, 172). TLR activation can result in the generation of p50/p50, NF-κB homodimers without transcriptional activity, determining macrophage polarization (93).
A more precise understanding of differentiation would require a description of the fine regulation of signaling pathways in macrophages upon DAMP stimuli. PI3K/Akt pathway could also be induced by DAMPs and has been generally considered as a negative regulator of TLR and NF-κB signaling in macrophages, resulting in M2 polarization (173).. However, each isoform of PI3K and the activated AKT can explain the dual role of this pathway in macrophage polarization (35). The switching between the Akt1-Akt2 is responsible for the macrophage plasticity, Akt1 promotes M2 and Akt2 promotes M1 phenotype formation (35). This process is regulated by miR-155 (174). In acute inflammation, the “positive signals”, namely, NF-κB, ERK, miR-155 and PTEN repress the inhibitory signals of PI3K/Akt/PDK-1 pathway, while during resolution the PI3K/Akt1 pathway excess initiates the activation/increased production of C/EBPββ, SOCS1, IRAK-M, miR-146a and IL-10 associated with M2 polarization (173).
Priming of cells can deterministically modify the outcome of signaling. The increased activity of STAT1 in the IFN-γ-reprogrammed macrophages produces a more intensive response to TLR4 ligands (175). IFN-γ and the TLR4 ligands activate the synthesis of SOCS3, which blocks STAT3, thereby preventing the formation of the M2 phenotype, but IL-4 activates the synthesis of SOCS1, which blocks STAT1, thereby preventing the generation of M1 cells (176).
Overall, DAMP sensing appears to predominantly, however not exclusively, reprogram macrophages to the M1 phenotype. Both increased expression of PRRs on M1 cells and activated signaling pathways contribute to M1 differentiation.
More RAMPs are likely to be released during necrotic cell death than during apoptosis, similar to what has been observed for DAMPs (reviewed in (1)). Released RAMPs act simultaneously with DAMPs to initiate the silencing of DAMP-induced signaling (177, 178) (Figure 4).
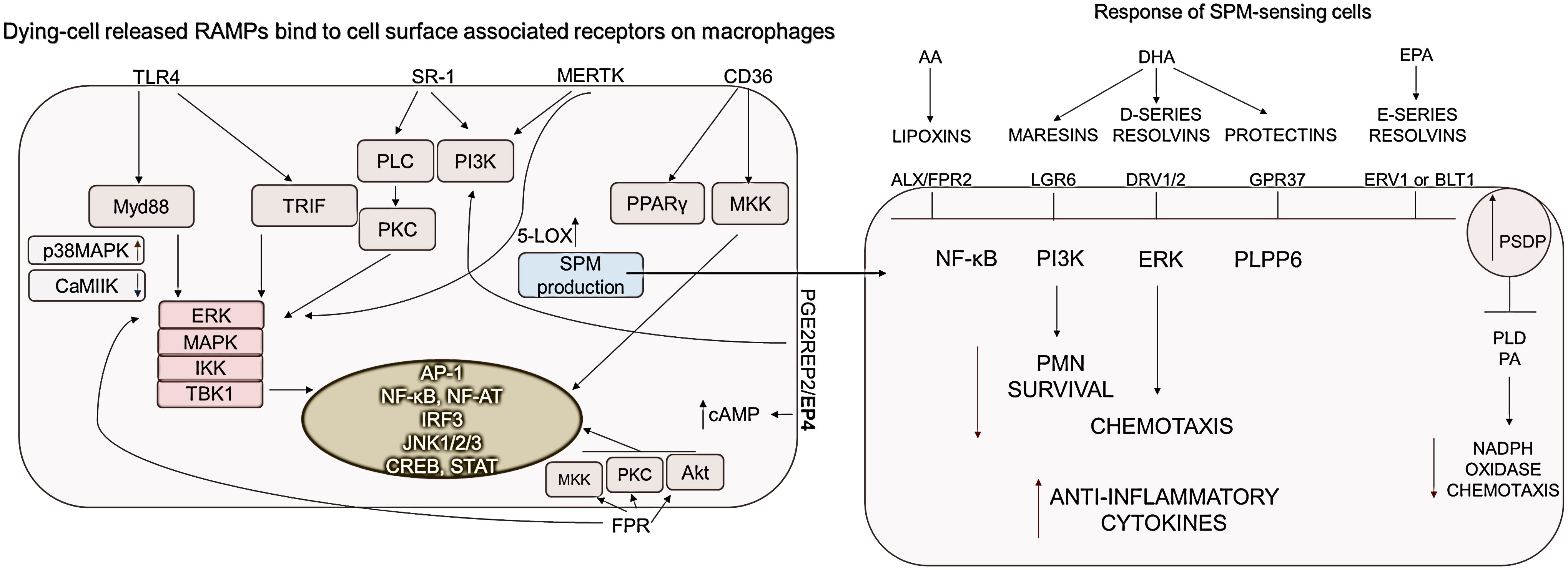
Figure 4 RAMP and SPM-sensing receptor-induced signaling pathways. Macrophages express receptors to sense RAMPs released from dying cells. CD36, PGE2R-derived signals, as well as MerTK activation, which detects phosphatidylserine on dying cells, are essential for increasing cAMP and SPM production. Accordingly to the DAMP/RAMP ratio and the generated signals, the SPMs decrease the survival of neutrophil granulocytes as well as their chemotaxis, while the production of anti-inflammatory cytokines is increased. ALX, Lipoxin receptor; BLT1, Leukotriene B4 receptor 1; CD, cluster of differentiation; DRV1, D-resolvin receptor 1; ERK, Extracellular signal-related kinase; ERV1, human resolvin E1 receptor; FPR, Formyl peptide receptor; GPR37, G Protein-Coupled Receptor 37; IKK, IκB kinase; JNKs- c,Jun N-terminal kinases; LGR6, Leucine-rich repeat-containing G protein-coupled receptor 6; 5-LOX, Expression of 5-lipoxygenase; MAPKs, Mitogen-activated protein kinases; MerTK, MER proto-oncogene, tyrosine kinase; MKK, mitogen-activated protein kinase kinase; MYD88, Myeloid differentiation primary response 88; NF-AT, Nuclear factor of activated T-cells; NF-κB, Nuclear factor-κB; PI3K, phosphoinositide-3 kinase; PKC, protein kinase C; PLC, Phospholipase C; PLD, Phospholipase D; PLPP6, phospholipid phosphatase 6; PPARs, peroxisome proliferator-activated receptors; SPM, Specialized pro-resolving mediators; SR-A1, Class A1 scavenger receptors; TBK1, TANK-binding kinase 1; TLR, Toll-like receptor; TRIF, TIR-domain-containing adapter-inducing interferon-β.
TLR4, SR-A, CD36, PGE2R EP2/EP4, MERTK are responsible for sensing the presence of HSP10, HSP27, αβ-crystallin, BiP, AnnexinA1 RAMPs (92, 115, 118, 135, 179–181). Signal transduction induced by extracellular HSPs is poorly understood. Hsp27 stimulation activates the ERK, c-Jun, and p38 MAPK pathways, of which only p38 was required for monocyte IL-10 production (182). Similarly, BiP-induced IL-10 production was reduced by the addition of the MAPK p38 pathway inhibitor SB203580, but unaffected by the ERK-1/2 inhibitor PD98059 (183, 184). This is supported by the fact that, among the MAPKs, p38 MAPK appears to play a dominant role in IL-4-induced alternative activation of macrophages (185). CD36 stimulation by HSP proteins results in PPARγ activation, indicating that high CD36 expression can convert the function of HSP proteins described as DAMPs into resolution-promoting activity. Although CD36 is rather expressed by M2 cells, it has been mentioned to induce the activation of STAT1/5 leading to the M1 cell polarization (138).
Clearance of dying cells by efferocytosis initiates a significant phenotypic shift within macrophages towards an anti-inflammatory, IL-10 and TGFβ producing phenotype. The activation of the most important receptor of efferocytosis, the MerTK receptor, either by Gas6, protein S or PS activates the MAPK, AKT, STAT6, pathways in macrophages, which results in M2 polarization (186, 187). In addition, MerTK reduces cytoplasmic calcium levels, thereby initiating two important pro-resolving processes (1): suppressed calcium levels reduce CaMKII activity, which results in MerTK synthesis, thus amplifying the pro-resolving program (188), and (2) suppressed CaMKII silences the MK2 activity which upregulates non-phosphorylated form of 5-LOX. This promotes its translocation from the nucleus into the cytoplasm, mediating the synthesis of SPMs (189).
SPMs could be recognized by receptors: ALX/FPR2, ChemR23, GPR32, GPR37, RORα, DRV1/2, ERV1 or BLT1, and LGR6. The ligation of LXA4 to ALX/FPR2 transmembrane receptor, as the binding of DHA- or EPA-derived mediators to their receptors, acts as a “stop signal” during inflammation by inhibiting NF-κB and ERK pathways among others (190)(Figure 4).
Changes in the cAMP levels are determined during the DAMP/RAMP/SPM-regulated resolution. Most SPMs, such as Resolvin D1, Resolvin D2, and N-3 docosapentaenoic acid–derived Resolvin D5, LXA4, maresin-1, increase intracellular levels of cAMP. In contrast, Resolvin E1 reduces cAMP levels via the activation of ERV-1/ChemR23 (191). The production of cAMP induces the activation of Epac1/2 and PKA signaling stimulating the p38-MAPK pathway, which results in the CREB activation. Consequently, PKA inhibits the GSK3, PI3K/Akt and NF-κB pathways (191). Stimulated Epac1/2 promotes the activation of SOCS3 via the stimulation of RAP1, c-Jun, and C/EBPβ (192). At the same time, cAMP-stimulated Epac1/2 also inhibits the NF-κB- and GSK3β-mediated pro-inflammatory cytokine production (193). In addition, an increase in cAMP level can promote IL-4-dependent M2 marker expression through a PKA/C/EBPββ/CREB-dependent pathway in murine macrophages (194). SPMs stimulate resolution in a positive feedback loop by activating the phosphorylation of 5-lipoxygenase (5-LOX), leading to further SPM production (191). SPMs (via DRV1/2, BLT1) dominantly induce the polarization of M2 cells after the activation of C/EBPββ and SP1 transcription factors (195, 196).
Macrophage phenotype switching has been proposed as an important step in the transition of a pro-inflammatory reaction into the resolution phase. The alterations of macrophage phenotypes are regulated by lipid mediators switching from pro-inflammatory lipid mediators such as leukotrienes and prostaglandins to SPMs (197). In addition to increased 5-LOX expression upon SPM stimuli, 12-LOX (198) and 15-LOX (178) expression levels are also elevated in M2 cells, and these enzymes are upregulated by SP1 and STAT6- dependent manner (199, 200).
Importantly, the levels of SPMs are much lower than typical pro-inflammatory mediators including the monohydroxylated fatty acid derivatives, leukotrienes, or certain prostaglandins. In the majority of the studies, the concentration is low (<50 pg/ml) which is close to the detection limit of several methods or below (178), drawing attention to the importance of further investigations.
In summary, SPMs typically promote alternative macrophage polarization, but the fact, that M1 cells could be also primed by these receptors, suggests the existence of M1 (associated with inflammatory or resolution-promoting) subpopulations similar to M2, again emphasizing that the functions of the macrophage populations are not sharply separated, in contrast to the M1 M2 classification. Overall RAMP signaling completed with SPM-induced changes is generally characterized by activation of cAMP and phosphorylation of p38 resulting in the M2-related transcription and a dominant inhibition of the NF-κB pathway to block macrophage polarization to M1 cells.
Extracellular HMGB1 can also activate TLR4 and RAGE receptors. While disulfide HMGB1 mostly stimulates TLR4, similar to LPS, resulting in inflammatory responses, sulfonyl HMGB1, on the other hand, leads to an anti-inflammatory response through RAGE (69).. The presence of soluble C1q or the “don’t eat me” signals remaining on the surface of necrotizing cells causes co-stimulation of Siglec-10 or LAIR receptors with HMGB1-mediated signaling (69). Both receptors contain an ITIM motif inducing the activation of SHP-1 and SHP2 phosphatases blocking both NF-κβ and IRF signaling and terminating the inflammatory effect of HMGB1 (201, 202).
ATP binding to P2X7R results in NLRP3 inflammasome activation and the elevated intracellular Ca2+ concentration leads to MAPK and calcineurin (NFAT) activation favoring M1 differentiation (203). In contrast, adenosine in macrophages through A2A and A2B receptors increases intracellular cAMP levels resulting in CREB and STAT3 stimulation and simultaneous inhibition of NF-κB (150). In addition, via phosphorylation of p38, activated SOCS3 up-regulates IL-10 production and induces VEGF production while phosphorylating STAT3 promotes the polarization into M2 phenotype (191, 204).
IL-33, as a member of the IL-1 family, binds to the ST2 (ILRL1) receptor and activates MyD88- AP1, NF-κB signaling through the intracellular Toll/IL-1 receptor (TIR) domain characteristic of the receptor family (205). Its role in resolution is primarily due to the fact that the ST2 receptor is dominantly found on the surface of mast cells, type 2 innate lymphoid cells (ILC2), Th2 and regulatory T cells, the activation of which results in the IL-4, IL-13 signal (73). However, the ST2 also stimulates the GATA3 transcription factor (158) and IL-33 inactivates GSK-3β through an ST2-independent pathway (206).
Among the PGE2 receptors, EP2 and EP4 receptor stimulation increases the intracellular cAMP level (207). EP4 is also able to induce tolerogenic signals via activating the phosphatidylinositol 3-kinase pathway and activating C/EBPββ which is an M2-associated transcription factor (130). PGE2 stimulation also enhances M2 polarization through Krupple-like factor 4 (KLF4), CREB and GATA3 factors (75, 208). As the production of PGE2 is increased as a result of the induction of cell death, it is an important negative regulator of the immune response during sterile inflammation (209).
As ANXA1 can also induce a proinflammatory signal, recombinant ANXA-1 promotes myeloid differentiation by activating the ERK1/2-NFAT2 pathway and increasing intracellular Ca2+ concentration (210). Although ANXA-1 binds to the same FRP2 receptor as LXA4, the effect of ANXA1 on directly increasing cAMP levels is still uncertain (191). Conversely, the elevated cAMP level increases ANXA-1 expression and activity, which promotes the efferocytosis of apoptotic leukocytes by macrophages (191). ANXA1 increases the expression of PPARγ by regulating the phosphorylation of STAT6 and as positive feedback, PPARγ upregulates the expression of ANXA1 (211).
Although the significance of sterile inflammation is considerable, most inflammation studies primarily focus on pathogen responses. Due to limited research, the phases of sterile inflammation, including cell death, DAMP release, inflammation, and resolution, have not been effectively interconnected. Some articles explore cell death and DAMP release, while others examine the role of macrophages in inflammation and resolution. However, the impact of cell death and DAMPs on macrophages during sterile inflammation remains poorly investigated. Numerous in vivo models induce sterile inflammation and muscle regeneration, but they often overlook the processes of cell death. Although various cell death pathways are well characterized, their immunological outcomes and especially their effects on resolution are inadequately studied. Further research is needed to establish connections between these isolated models.
The usage of M1 and M2 nomenclature is widespread. Cells obtained through different differentiation protocols are uniformly designated as M1 or M2, often without considering subpopulations. In vivo, macrophage subtypes exhibit greater complexity than the oversimplified M1/M2 classification based on in vitro studies. Within tissues, macrophage subsets exist on a continuum and are regulated differently based on the tissue type. Our review highlights the significant differences between DAMP-induced inflammation/resolution and PAMP-induced inflammation, which has been understudied thus far. This suggests the presence of distinct macrophage subpopulations and functionality, including various transitional forms.
The regulation of the transition between inflammation and resolution remains incompletely understood. The conversion of pro-inflammatory macrophages into resolution-inducing cells is a pivotal step in this process. Signaling mediated by resolution-promoting cells triggers positive feedback loops that determine the final outcome. Therefore, identifying the regulators of this tipping point holds great therapeutic potential. While M1 and M2 cells share certain functional elements, such as the expression of pattern recognition receptors (PRRs) and associated signaling pathways, more notable differences arise in their response to damage-associated molecular patterns (DAMPs) and resolution-associated molecular patterns (RAMPs) compared to pathogen-associated molecular patterns (PAMPs). Targeting the RAMP-DAMP pathway for optimal therapeutic intervention can pave new paths in inflammation and resolution regulation. Understanding the role of DAMPs and RAMPs released from dying cells during sterile inflammation is imperative. In-depth investigation using in vitro and, more importantly, complex in vivo models specifically designed to study sterile inflammation and its interdependent phases of cell death, DAMP release, and macrophage polarization is required. Exploring critical regulatory points and molecular mechanisms in the transition, such as HMGB1 and ATP conversion to an anti-inflammatory phenotype, as well as differences in AKT1/AKT2 and TREM1/TREM2 signaling, may offer insights to control sterile inflammatory pathological conditions effectively.
Conceptualization, AM, and GK. Writing—Original Draft Preparation, GK, AM, VJ, EV, MT, and BK. Writing—Review and Editing, GK, AB, VJ, MT and AM. Supervision, AM. Funding Acquisition, AM, VJ and GK. All authors contributed to the article and approved the submitted version.
Scholarship for New National Excellence Program of the Ministry for Innovation and Technology. (ÚNKP-22-3-I-DE-97) and for Young Talents of the Nation (NTP-NFTÖ-22-B-0129) are acknowledged for the financial support of this work. The publication is supported by the GINOP-2.3.2-15-2016-00050 project, the project is co-financed by the European Union and the European Regional Development Fund, National Research, Development and Innovation Office–NKFIH, PD 142930.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
2. Shen H, Kreisel D, Goldstein DR. Processes of sterile inflammation. J Immunol (2013) 191:2857–63. doi: 10.4049/jimmunol.1301539
3. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol (2014) 5:514. doi: 10.3389/fimmu.2014.00514
4. Chiang N, Serhan CN. Specialized pro-resolving mediator network: an update on production and actions. Essays Biochem (2020) 64:443–62. doi: 10.1042/EBC20200018
5. Austermann J, Roth J, Barczyk-Kahlert K. The good and the bad: monocytes' and macrophages' diverse functions in inflammation. Cells (2022) 11. doi: 10.3390/cells11121979
6. Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol (2010) 28:321–42. doi: 10.1146/annurev-immunol-030409-101311
7. Galluzzi L, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
8. Shields AM, Panayi GS, Corrigall VM. Resolution-associated molecular patterns (RAMP): RAMParts defending immunological homeostasis? Clin Exp Immunol (2011) 165:292–300. doi: 10.1111/j.1365-2249.2011.04433.x
9. Mazlo A, et al. Types of necroinflammation, the effect of cell death modalities on sterile inflammation. Cell Death Dis (2022) 13:423. doi: 10.1038/s41419-022-04883-w
10. Burgess M, Valdera F, Varon D, Kankuri E, Nuutila K. The immune and regenerative response to burn injury. Cells (2022) 11. doi: 10.3390/cells11193073
11. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
12. Peiseler M, Kubes P. Macrophages play an essential role in trauma-induced sterile inflammation and tissue repair. Eur J Trauma Emerg Surg (2018) 44:335–49. doi: 10.1007/s00068-018-0956-1
13. Hong Z, Mei J, Guo H, Zhu J, Wang C. Intervention of cGAS −STING signaling in sterile inflammatory diseases. J Mol Cell Biol (2022) 14.
14. Patsalos A, Tzerpos P, Wei X, Nagy L. Myeloid cell diversification during regenerative inflammation: lessons from skeletal muscle. Semin Cell Dev Biol (2021) 119:89–100. doi: 10.1016/j.semcdb.2021.05.005
15. Leuti A, et al. Macrophage plasticity and polarization are altered in the experimental model of multiple sclerosis. Biomolecules (2021) 11. doi: 10.3390/biom11060837
16. Julian L, et al. Defective apoptotic cell contractility provokes sterile inflammation, leading to liver damage and tumour suppression. Elife (2021) 10. doi: 10.7554/eLife.61983
17. Rothlin CV, Hille TD, Ghosh S. Determining the effector response to cell death. Nat Rev Immunol (2021) 21:292–304. doi: 10.1038/s41577-020-00456-0
18. Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol (2020) 15:493–518. doi: 10.1146/annurev-pathmechdis-012419-032847
19. Lavy M, Gauttier V, Poirier N, Barille-Nion S, Blanquart C. Specialized pro-resolving mediators mitigate cancer-related inflammation: role of tumor-associated macrophages and therapeutic opportunities. Front Immunol (2021) 12:702785. doi: 10.3389/fimmu.2021.702785
20. Basil MC, Levy BD. Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat Rev Immunol (2016) 16:51–67. doi: 10.1038/nri.2015.4
21. Fang X, et al. Human mesenchymal stem (Stromal) cells promote the resolution of acute lung injury in part through lipoxin A4. J Immunol (2015) 195:875–81. doi: 10.4049/jimmunol.1500244
22. Frevert CW, Felgenhauer J, Wygrecka M, Nastase MV, Schaefer L. Danger-associated molecular patterns derived from the extracellular matrix provide temporal control of innate immunity. J Histochem Cytochem (2018) 66:213–27. doi: 10.1369/0022155417740880
23. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol (2020) 20:95–112. doi: 10.1038/s41577-019-0215-7
24. Murray PJ. Macrophage polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
25. Yang S, Zhao M, Jia S. Macrophage: key player in the pathogenesis of autoimmune diseases. Front Immunol (2023) 14:1080310. doi: 10.3389/fimmu.2023.1080310
26. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discovery (2022) 21:799–820. doi: 10.1038/s41573-022-00520-5
27. Mantovani A, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25:677–86. doi: 10.1016/j.it.2004.09.015
28. Chong BF, et al. A subset of CD163+ macrophages displays mixed polarizations in discoid lupus skin. Arthritis Res Ther (2015) 17:324. doi: 10.1186/s13075-015-0839-3
29. Murray PJ, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
30. Chen L, Ozato K. Innate immune memory in hematopoietic Stem/Progenitor cells: myeloid-biased differentiation and the role of interferon. Front Immunol (2021) 12:621333. doi: 10.3389/fimmu.2021.621333
31. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med (1992) 176:287–92. doi: 10.1084/jem.176.1.287
32. Fleetwood AJ, Dinh H, Cook AD, Hertzog PJ, Hamilton JA. GM-CSF- and m-CSF-dependent macrophage phenotypes display differential dependence on type I interferon signaling. J Leukoc Biol (2009) 86:411–21. doi: 10.1189/jlb.1108702
33. Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol (2011) 11:750–61. doi: 10.1038/nri3088
34. Chawla A. Control of macrophage activation and function by PPARs. Circ Res (2010) 106:1559–69. doi: 10.1161/CIRCRESAHA.110.216523
35. Arranz A, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci U.S.A. (2012) 109:9517–22.
36. Mills CD. M1 and M2 macrophages: oracles of health and disease. Crit Rev Immunol (2012) 32:463–88. doi: 10.1615/CritRevImmunol.v32.i6.10
37. Mogilenko DA, Shchukina I, Artyomov MN. Immune ageing at single-cell resolution. Nat Rev Immunol (2022) 22:484–98. doi: 10.1038/s41577-021-00646-4
38. Duong L, et al. Macrophage function in the elderly and impact on injury repair and cancer. Immun Ageing (2021) 18:4. doi: 10.1186/s12979-021-00215-2
39. Chen AX, et al. Single-cell characterization of macrophages in glioblastoma reveals MARCO as a mesenchymal pro-tumor marker. Genome Med (2021) 13:88. doi: 10.1186/s13073-021-00906-x
40. Tiemeijer BM, Heester S, Sturtewagen AYW, Smits A, Tel J. Single-cell analysis reveals TLR-induced macrophage heterogeneity and quorum sensing dictate population wide anti-inflammatory feedback in response to LPS. Front Immunol (2023) 14:1135223. doi: 10.3389/fimmu.2023.1135223
41. Xiong J, et al. Bulk and single-cell characterisation of the immune heterogeneity of atherosclerosis identifies novel targets for immunotherapy. BMC Biol (2023) 21:46. doi: 10.1186/s12915-023-01540-2
42. Iyer SS, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U.S.A. (2009) 106:20388–93.
43. McDonald B, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science (2010) 330:362–6. doi: 10.1126/science.1195491
44. Rider P, et al. IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol (2011) 187:4835–43. doi: 10.4049/jimmunol.1102048
45. La Russa F, et al. Disruption of the sensory system affects sterile cutaneous inflammation. In Vivo. J Invest Dermatol (2019) 139:1936–1945 e1933. doi: 10.1016/j.jid.2019.01.037
46. Jovcic G, Stojanovic N, Kataranovski M, Petakov M, Obradovic P. Acute sterile inflammation–correlation between cellular changes and extramedullary-produced regulators. vivo. Ann Hematol (1993) 66:195–201. doi: 10.1007/BF01703235
47. Wu BC, Skovbakke SL, Masoudi H, Hancock REW, Franzyk H. In vivo anti-inflammatory activity of lipidated peptidomimetics Pam-(Lys-betaNspe)(6)-NH(2) and lau-(Lys-betaNspe)(6)-NH(2) against PMA-induced acute inflammation. Front Immunol (2020) 11:2102. doi: 10.3389/fimmu.2020.02102
48. Agliano F, Karlinsey KS, Ragazzi M, Menoret A, Vella AT. A benzimidazole inhibitor attenuates sterile inflammation induced in a model of systemic autoinflammation in female mice. Sci Rep (2020) 10:12100. doi: 10.1038/s41598-020-68985-1
49. Taylor KR, et al. Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on toll-like receptor 4, CD44, and MD-2. J Biol Chem (2007) 282:18265–75. doi: 10.1074/jbc.M606352200
50. Tseng A, Kim K, Li J, Cho J. Myeloperoxidase negatively regulates neutrophil-endothelial cell interactions by impairing alphaMbeta2 integrin function in sterile inflammation. Front Med (Lausanne) (2018) 5:134. doi: 10.3389/fmed.2018.00134
51. Patsalos A, et al. In situ macrophage phenotypic transition is affected by altered cellular composition prior to acute sterile muscle injury. J Physiol (2017) 595:5815–42. doi: 10.1113/JP274361
52. Zhai Y, et al. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response. via IFN Regul factor 3-dependent MyD88-independent pathway. J Immunol (2004) 173:7115–9. doi: 10.4049/jimmunol.173.12.7115
53. Seong SY, Matzinger P, Land WG. Editorial: DAMPs across the tree of life. Front Immunol (2021) 12:844315. doi: 10.3389/fimmu.2021.844315
54. He L, et al. Global characterization of macrophage polarization mechanisms and identification of M2-type polarization inhibitors. Cell Rep (2021) 37:109955. doi: 10.1016/j.celrep.2021.109955
55. Alakesh A, Jothiprakasam T, Raghavan JV, Jhunjhunwala S. Sterile inflammation alters neutrophil kinetics in mice. J Leukoc Biol (2022) 112:395–409. doi: 10.1002/JLB.1A0321-132RR
56. Raymond SL, et al. Microbial recognition and danger signals in sepsis and trauma. Biochim Biophys Acta Mol Basis Dis (2017) 1863:2564–73. doi: 10.1016/j.bbadis.2017.01.013
57. Schaper F, et al. High mobility group box 1 skews macrophage polarization and negatively influences phagocytosis of apoptotic cells. Rheumatol (Oxford) (2016) 55:2260–70. doi: 10.1093/rheumatology/kew324
58. Karuppagounder V, et al. Modulation of macrophage polarization and HMGB1-TLR2/TLR4 cascade plays a crucial role for cardiac remodeling in senescence-accelerated prone mice. PloS One (2016) 11:e0152922. doi: 10.1371/journal.pone.0152922
59. Saha B, et al. Extracellular vesicles from mice with alcoholic liver disease carry a distinct protein cargo and induce macrophage activation through heat shock protein 90. Hepatology (2018) 67:1986–2000. doi: 10.1002/hep.29732
60. Martin WJ, Shaw O, Liu X, Steiger S, Harper JL. Monosodium urate monohydrate crystal-recruited noninflammatory monocytes differentiate into M1-like proinflammatory macrophages in a peritoneal murine model of gout. Arthritis Rheum (2011) 63:1322–32. doi: 10.1002/art.30249
61. Cabrera-Fuentes HA, et al. Regulation of monocyte/macrophage polarisation by extracellular RNA. Thromb Haemost (2015) 113:473–81. doi: 10.1160/TH14-06-0507
62. Zhang W, et al. Extracellular CIRP-impaired Rab26 restrains EPOR-mediated macrophage polarization in acute lung injury. Front Immunol (2021) 12:768435. doi: 10.3389/fimmu.2021.768435
63. Nakazawa D, et al. The responses of macrophages in interaction with neutrophils that undergo NETosis. J Autoimmun (2016) 67:19–28. doi: 10.1016/j.jaut.2015.08.018
64. Riuzzi F, et al. Levels of S100B protein drive the reparative process in acute muscle injury and muscular dystrophy. Sci Rep (2017) 7:12537. doi: 10.1038/s41598-017-12880-9
65. Wang LX, Zhang SX, Wu HJ, Rong XL, Guo J. M2b macrophage polarization and its roles in diseases. J Leukoc Biol (2019) 106:345–58. doi: 10.1002/JLB.3RU1018-378RR
66. Chuang Y, Hung ME, Cangelose BK, Leonard JN. Regulation of the IL-10-driven macrophage phenotype under incoherent stimuli. Innate Immun (2016) 22:647–57. doi: 10.1177/1753425916668243
67. Zizzo G, Hilliard BA, Monestier M, Cohen PL. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J Immunol (2012) 189:3508–20. doi: 10.4049/jimmunol.1200662
68. Qu H, et al. Transcriptomic profiling reveals that HMGB1 induces macrophage polarization different from classical M1. Biomolecules (2022) 12. doi: 10.3390/biom12060779
69. Andersson U, Tracey KJ, Yang H. Post-translational modification of HMGB1 disulfide bonds in stimulating and inhibiting inflammation. Cells (2021) 10. doi: 10.3390/cells10123323
70. Hasko G, Pacher P. Regulation of macrophage function by adenosine. Arterioscler Thromb Vasc Biol (2012) 32:865–9. doi: 10.1161/ATVBAHA.111.226852
71. de Torre-Minguela C, Barbera-Cremades M, Gomez AI, Martin-Sanchez F, Pelegrin P. Macrophage activation and polarization modify P2X7 receptor secretome influencing the inflammatory process. Sci Rep (2016) 6:22586. doi: 10.1038/srep22586
72. Pelegrin P, Surprenant A. Dynamics of macrophage polarization reveal new mechanism to inhibit IL-1beta release through pyrophosphates. EMBO J (2009) 28:2114–27. doi: 10.1038/emboj.2009.163
73. Faas M, et al. IL-33-induced metabolic reprogramming controls the differentiation of alternatively activated macrophages and the resolution of inflammation. Immunity (2021) 54:2531–2546 e2535. doi: 10.1016/j.immuni.2021.09.010
74. Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol (2001) 2:612–9. doi: 10.1038/89759
75. Luan B, et al. CREB pathway links PGE2 signaling with macrophage polarization. Proc Natl Acad Sci U.S.A. (2015) 112:15642–7.
76. Tang T, et al. Macrophage responses to lipopolysaccharide are modulated by a feedback loop involving prostaglandin E(2), dual specificity phosphatase 1 and tristetraprolin. Sci Rep (2017) 7:4350. doi: 10.1038/s41598-017-04100-1
77. Li Y, et al. Pleiotropic regulation of macrophage polarization and tumorigenesis by formyl peptide receptor-2. Oncogene (2011) 30:3887–99. doi: 10.1038/onc.2011.112
78. McArthur S, et al. Annexin A1 drives macrophage skewing to accelerate muscle regeneration through AMPK activation. J Clin Invest (2020) 130:1156–67. doi: 10.1172/JCI124635
79. Moraes LA, et al. Annexin-A1 enhances breast cancer growth and migration by promoting alternative macrophage polarization in the tumour microenvironment. Sci Rep (2017) 7:17925. doi: 10.1038/s41598-017-17622-5
80. Locatelli I, et al. Endogenous annexin A1 is a novel protective determinant in nonalcoholic steatohepatitis in mice. Hepatology (2014) 60:531–44. doi: 10.1002/hep.27141
81. Ricketts TD, Prieto-Dominguez N, Gowda PS, Ubil E. Mechanisms of macrophage plasticity in the tumor environment: manipulating activation state to improve outcomes. Front Immunol (2021) 12:642285. doi: 10.3389/fimmu.2021.642285
82. Huber R, et al. Tumour hypoxia promotes melanoma growth and metastasis via high mobility group box-1 and M2-like macrophages. Sci Rep (2016) 6:29914. doi: 10.1038/srep29914
83. Fan CS, et al. Endothelial-mesenchymal transition harnesses HSP90alpha-secreting M2-macrophages to exacerbate pancreatic ductal adenocarcinoma. J Hematol Oncol (2019) 12:138. doi: 10.1186/s13045-019-0826-2
84. Zhou X, et al. Crosstalk between tumor-associated macrophages and MicroRNAs: a key role in tumor microenvironment. Int J Mol Sci (2022) 23. doi: 10.3390/ijms232113258
85. Liu S, et al. S100A4 enhances protumor macrophage polarization by control of PPAR-gamma-dependent induction of fatty acid oxidation. J Immunother Cancer (2021) 9. doi: 10.1136/jitc-2021-002548
86. Xu H, et al. The IL-33/ST2 axis affects tumor growth by regulating mitophagy in macrophages and reprogramming their polarization. Cancer Biol Med (2021) 18:172–83. doi: 10.20892/j.issn.2095-3941.2020.0211
87. Su C, et al. HMGB1 promotes lymphangiogenesis through the activation of RAGE on M2 macrophages in laryngeal squamous cell carcinoma. Dis Markers (2022) 2022:4487435. doi: 10.1155/2022/4487435
88. He C, Sun S, Zhang Y, Xie F, Li S. The role of irreversible electroporation in promoting M1 macrophage polarization via regulating the HMGB1-RAGE-MAPK axis in pancreatic cancer. Oncoimmunology (2021) 10:1897295. doi: 10.1080/2162402X.2021.1897295
89. Wang Y, et al. Extracellular HMGB1 as inflammatory mediator in the progression of mycoplasma gallisepticum infection. Cells (2022) 11. doi: 10.3390/cells11182817
90. Kalliolias GD, Gordon RA, Ivashkiv LB. Suppression of TNF-alpha and IL-1 signaling identifies a mechanism of homeostatic regulation of macrophages by IL-27. J Immunol (2010) 185:7047–56. doi: 10.4049/jimmunol.1001290
91. Spitzer P, et al. Analysis of surface levels of IL-1 receptors and macrophage scavenger receptor I in peripheral immune cells of patients with Alzheimer disease. J Geriatr Psychiatry Neurol (2019) 32:211–20. doi: 10.1177/0891988719841728
92. Quero L, Hanser E, Manigold T, Tiaden AN, Kyburz D. TLR2 stimulation impairs anti-inflammatory activity of M2-like macrophages, generating a chimeric M1/M2 phenotype. Arthritis Res Ther (2017) 19:245. doi: 10.1186/s13075-017-1447-1
93. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol (2014) 5:614. doi: 10.3389/fimmu.2014.00614
94. Vidyarthi A, et al. TLR-3 stimulation skews M2 macrophages to M1 through IFN-alphabeta signaling and restricts tumor progression. Front Immunol (2018) 9:1650. doi: 10.3389/fimmu.2018.01650
95. Su R, et al. TLR3 expression is a potential prognosis biomarker and shapes the immune-active tumor microenvironment in esophageal squamous cell carcinoma. J Inflammation Res (2022) 15:1437–56. doi: 10.2147/JIR.S348786
96. Lu CH, et al. Involvement of M1 macrophage polarization in endosomal toll-like receptors activated psoriatic inflammation. Mediators Inflammation (2018) 2018:3523642. doi: 10.1155/2018/3523642
97. Yang R, Yu S, Xu T, Zhang J, Wu S. Emerging role of RNA sensors in tumor microenvironment and immunotherapy. J Hematol Oncol (2022) 15:43. doi: 10.1186/s13045-022-01261-z
98. Karadimou G, et al. TLR7 expression is associated with M2 macrophage subset in calcific aortic valve stenosis. Cells (2020) 9. doi: 10.3390/cells9071710
99. Rai V, Dilisio MF, Samadi F, Agrawal DK. Counteractive effects of IL-33 and IL-37 on inflammation in osteoarthritis. Int J Environ Res Public Health (2022) 19. doi: 10.3390/ijerph19095690
100. Raggi F, et al. Regulation of human macrophage M1-M2 polarization balance by hypoxia and the triggering receptor expressed on myeloid cells-1. Front Immunol (2017) 8:1097. doi: 10.3389/fimmu.2017.01097
101. Vishnyakova P, et al. The response of two polar monocyte subsets to inflammation. BioMed Pharmacother (2021) 139:111614. doi: 10.1016/j.biopha.2021.111614
102. Lu Q, et al. TREM (Triggering receptor expressed on myeloid cells)-1 inhibition attenuates neuroinflammation via PKC (Protein kinase c) delta/CARD9 (Caspase recruitment domain family member 9) signaling pathway after intracerebral hemorrhage in mice. Stroke (2021) 52:2162–73. doi: 10.1161/STROKEAHA.120.032736
103. Turnbull IR, et al. Cutting edge: TREM-2 attenuates macrophage activation. J Immunol (2006) 177:3520–4. doi: 10.4049/jimmunol.177.6.3520
104. Syed MA, et al. TREM-1 attenuates RIPK3-mediated necroptosis in hyperoxia-induced lung injury in neonatal mice. Am J Respir Cell Mol Biol (2019) 60:308–22. doi: 10.1165/rcmb.2018-0219OC
105. Merz J, et al. Pro- and anti-inflammatory macrophages express a sub-type specific purinergic receptor profile. Purinergic Signal (2021) 17:481–92. doi: 10.1007/s11302-021-09798-3
106. Wu P, et al. P2X7 receptor induces microglia polarization to the M1 phenotype in cancer-induced bone pain rat models. Mol Pain (2022) 18:17448069211060962. doi: 10.1177/17448069211060962
107. Engel JE, et al. Recovery of renal function following kidney-specific VEGF therapy in experimental renovascular disease. Am J Nephrol (2020) 51:891–902. doi: 10.1159/000511260
108. Lee AS, et al. Vascular endothelial growth factor-c and -d are involved in lymphangiogenesis in mouse unilateral ureteral obstruction. Kidney Int (2013) 83:50–62. doi: 10.1038/ki.2012.312
109. Lai YS, Wahyuningtyas R, Aui SP, Chang KT. Autocrine VEGF signalling on M2 macrophages regulates PD-L1 expression for immunomodulation of T cells. J Cell Mol Med (2019) 23:1257–67.
110. Hernandez Baltazar D, et al. Does lipopolysaccharide-based neuroinflammation induce microglia polarization? Folia Neuropathol (2020) 58:113–22. doi: 10.5114/fn.2020.96755
111. Xiong L, Edwards CK, Zhou L. The biological function and clinical utilization of CD147 in human diseases: a review of the current scientific literature. Int J Mol Sci (2014) 15:17411–41. doi: 10.3390/ijms151017411
112. Zhang J, et al. Large-Scale single-cell and bulk sequencing analyses reveal the prognostic value and immune aspects of CD147 in pan-cancer. Front Immunol (2022) 13:810471. doi: 10.3389/fimmu.2022.810471
113. Trivedi NH, et al. Microbial co-infection alters macrophage polarization, phagosomal escape, and microbial killing. Innate Immun (2018) 24:152–62. doi: 10.1177/1753425918760180
114. Lv LL, et al. SAP130 released by damaged tubule drives necroinflammation via miRNA-219c/Mincle signaling in acute kidney injury. Cell Death Dis (2021) 12:866. doi: 10.1038/s41419-021-04131-7
115. Ahmed M, et al. Human serum albumin-based probes for molecular targeting of macrophage scavenger receptors. Int J Nanomedicine (2019) 14:3723–41. doi: 10.2147/IJN.S197990
116. Xie C, et al. Class A1 scavenger receptors mediated macrophages in impaired intestinal barrier of inflammatory bowel disease. Ann Transl Med (2020) 8:106. doi: 10.21037/atm.2019.12.107
117. Labonte AC, Sung SJ, Jennelle LT, Dandekar AP, Hahn YS. Expression of scavenger receptor-AI promotes alternative activation of murine macrophages to limit hepatic inflammation and fibrosis. Hepatology (2017) 65:32–43. doi: 10.1002/hep.28873
118. Pennathur S, et al. The macrophage phagocytic receptor CD36 promotes fibrogenic pathways on removal of apoptotic cells during chronic kidney injury. Am J Pathol (2015) 185:2232–45. doi: 10.1016/j.ajpath.2015.04.016
119. Wallet MA, et al. MerTK regulates thymic selection of autoreactive T cells. Proc Natl Acad Sci U.S.A. (2009) 106:4810–5.
120. Zhang C, Yang M, Ericsson AC. Function of macrophages in disease: current understanding on molecular mechanisms. Front Immunol (2021) 12:620510. doi: 10.3389/fimmu.2021.620510
121. Sasaki F, et al. Leukotriene B4 promotes neovascularization and macrophage recruitment in murine wet-type AMD models. JCI Insight (2018) 3. doi: 10.1172/jci.insight.96902
122. Herova M, Schmid M, Gemperle C, Hersberger M. ChemR23, the receptor for chemerin and resolvin E1, is expressed and functional on M1 but not on M2 macrophages. J Immunol (2015) 194:2330–7. doi: 10.4049/jimmunol.1402166
123. Amici SA, et al. CD38 is robustly induced in human macrophages and monocytes in inflammatory conditions. Front Immunol (2018) 9:1593. doi: 10.3389/fimmu.2018.01593
124. Chen YC, et al. Blood M2a monocyte polarization and increased formyl peptide receptor 1 expression are associated with progression from latent tuberculosis infection to active pulmonary tuberculosis disease. Int J Infect Dis (2020) 101:210–9. doi: 10.1016/j.ijid.2020.09.1056
125. Filep JG. Biasing the lipoxin A4/formyl peptide receptor 2 pushes inflammatory resolution. Proc Natl Acad Sci U.S.A. (2013) 110:18033–4.
126. Bang S, et al. GPR37 regulates macrophage phagocytosis and resolution of inflammatory pain. J Clin Invest (2018) 128:3568–82. doi: 10.1172/JCI99888
127. Chiang N, Libreros S, Norris PC, de la Rosa X, Serhan CN. Maresin 1 activates LGR6 receptor promoting phagocyte immunoresolvent functions. J Clin Invest (2019) 129:5294–311. doi: 10.1172/JCI129448
128. Hazlett LD, et al. IL-33 shifts macrophage polarization, promoting resistance against pseudomonas aeruginosa keratitis. Invest Ophthalmol Vis Sci (2010) 51:1524–32. doi: 10.1167/iovs.09-3983
129. Ganesh K, et al. Prostaglandin E(2) induces oncostatin m expression in human chronic wound macrophages through axl receptor tyrosine kinase pathway. J Immunol (2012) 189:2563–73. doi: 10.4049/jimmunol.1102762
130. Sugimoto Y, Narumiya S. Prostaglandin e receptors. J Biol Chem (2007) 282:11613–7. doi: 10.1074/jbc.R600038200
131. Lan Y, et al. Hepatocyte-derived prostaglandin E2-modulated macrophage M1-type polarization via mTOR-NPC1 axis-regulated cholesterol transport from lysosomes to the endoplasmic reticulum in hepatitis b virus x protein-related nonalcoholic steatohepatitis. Int J Mol Sci (2022) 23. doi: 10.3390/ijms231911660
132. Lu Y, et al. Bone marrow mesenchymal stem cell-derived exosomes improve renal fibrosis by reducing the polarisation of M1 and M2 macrophages through the activation of EP2 receptors. IET Nanobiotechnol (2022) 16:14–24. doi: 10.1049/nbt2.12071
133. Xie X, et al. FPR2 participates in epithelial ovarian cancer (EOC) progression through RhoA-mediated M2 macrophage polarization. J Ovarian Res (2021) 14:177. doi: 10.1186/s13048-021-00932-8
134. Singh H, Rai V, Nooti SK, Agrawal DK. Novel ligands and modulators of triggering receptor expressed on myeloid cells receptor family: 2015-2020 updates. Expert Opin Ther Pat (2021) 31:549–61. doi: 10.1080/13543776.2021.1883587
135. van Eden W, Spiering R, Broere F, van der Zee R. A case of mistaken identity: HSPs are no DAMPs but DAMPERs. Cell Stress Chaperones (2012) 17:281–92. doi: 10.1007/s12192-011-0311-5
136. Johnson BJ, et al. Heat shock protein 10 inhibits lipopolysaccharide-induced inflammatory mediator production. J Biol Chem (2005) 280:4037–47. doi: 10.1074/jbc.M411569200
137. Shi C, Deng J, Chiu M, Chen YX, O'Brien ER. Heat shock protein 27 immune complex altered signaling and transport (ICAST): novel mechanisms of attenuating inflammation. FASEB J (2020) 34:14287–301. doi: 10.1096/fj.202001389RR
138. Lee KJ, et al. CD36 signaling inhibits the translation of heat shock protein 70 induced by oxidized low density lipoprotein through activation of peroxisome proliferators-activated receptor gamma. Exp Mol Med (2008) 40:658–68. doi: 10.3858/emm.2008.40.6.658
139. Kim SY, et al. Liver X receptor and STAT1 cooperate downstream of Gas6/Mer to induce anti-inflammatory arginase 2 expression in macrophages. Sci Rep (2016) 6:29673. doi: 10.1038/srep29673
140. Shen Y, et al. Exogenous Gas6 attenuates silica-induced inflammation on differentiated THP-1 macrophages. Environ Toxicol Pharmacol (2016) 45:222–6. doi: 10.1016/j.etap.2016.05.029
141. Cai B, et al. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc Natl Acad Sci U.S.A. (2016) 113:6526–31.
142. Han YH, et al. A maresin 1/RORalpha/12-lipoxygenase autoregulatory circuit prevents inflammation and progression of nonalcoholic steatohepatitis. J Clin Invest (2019) 129:1684–98. doi: 10.1172/JCI124219
143. Schmid M, Gemperle C, Rimann N, Hersberger M. Resolvin D1 polarizes primary human macrophages toward a proresolution phenotype through GPR32. J Immunol (2016) 196:3429–37. doi: 10.4049/jimmunol.1501701
144. Freile JA, et al. CD24 is a potential immunotherapeutic target for mantle cell lymphoma. Biomedicines (2022) 10. doi: 10.3390/biomedicines10051175
145. Chen GY, Tang J, Zheng P, Liu Y. CD24 and siglec-10 selectively repress tissue damage-induced immune responses. Science (2009) 323:1722–5. doi: 10.1126/science.1168988
146. Son M, et al. C1q and HMGB1 reciprocally regulate human macrophage polarization. Blood (2016) 128:2218–28. doi: 10.1182/blood-2016-05-719757
147. Liu T, et al. HMGB1-C1q complexes regulate macrophage function by switching between leukotriene and specialized proresolving mediator biosynthesis. Proc Natl Acad Sci U.S.A. (2019) 116:23254–63.
148. Horn LA, et al. Remodeling the tumor microenvironment via blockade of LAIR-1 and TGF-beta signaling enables PD-L1-mediated tumor eradication. J Clin Invest (2022) 132.
149. Zanin RF, et al. Differential macrophage activation alters the expression profile of NTPDase and ecto-5'-nucleotidase. PloS One (2012) 7:e31205. doi: 10.1371/journal.pone.0031205
150. Csoka B, et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J (2012) 26:376–86. doi: 10.1096/fj.11-190934
151. Ren W, Rubini P, Tang Y, Engel T, Illes P. Inherent P2X7 receptors regulate macrophage functions during inflammatory diseases. Int J Mol Sci (2021) 23. doi: 10.3390/ijms23010232
152. Furukawa S, et al. Interleukin-33 produced by M2 macrophages and other immune cells contributes to Th2 immune reaction of IgG4-related disease. Sci Rep (2017) 7:42413. doi: 10.1038/srep42413
153. Gemperle C, et al. Regulation of the formyl peptide receptor 1 (FPR1) gene in primary human macrophages. PloS One (2012) 7:e50195. doi: 10.1371/journal.pone.0050195
154. Malyshev I, Malyshev Y. Current concept and update of the macrophage plasticity concept: intracellular mechanisms of reprogramming and M3 macrophage "Switch" phenotype. BioMed Res Int (2015) 2015:341308. doi: 10.1155/2015/341308
155. Viola A, Munari F, Sanchez-Rodriguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
156. Di Lorenzo A, Bolli E, Tarone L, Cavallo F, Conti L. Toll-like receptor 2 at the crossroad between cancer cells, the immune system, and the microbiota. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21249418
157. Yan ZQ. Regulation of TLR4 expression is a tale about tail. Arterioscler Thromb Vasc Biol (2006) 26:2582–4. doi: 10.1161/01.ATV.0000250933.92917.dd
158. Griesenauer B, Paczesny S. The ST2/IL-33 axis in immune cells during inflammatory diseases. Front Immunol (2017) 8:475. doi: 10.3389/fimmu.2017.00475
159. Tsuchiya M, et al. Emergent genome-wide control in wildtype and genetically mutated lipopolysaccarides-stimulated macrophages. PloS One (2009) 4:e4905. doi: 10.1371/journal.pone.0004905
160. Kingeter LM, Lin X. C-type lectin receptor-induced NF-kappaB activation in innate immune and inflammatory responses. Cell Mol Immunol (2012) 9:105–12. doi: 10.1038/cmi.2011.58
161. Saxena M, Yeretssian G. NOD-like receptors: master regulators of inflammation and cancer. Front Immunol (2014) 5:327. doi: 10.3389/fimmu.2014.00327
162. Azizian-Farsani F, et al. Receptor for advanced glycation end products acts as a fuel to colorectal cancer development. Front Oncol (2020) 10:552283. doi: 10.3389/fonc.2020.552283
163. Yan SF, Ramasamy R, Schmidt AM. The receptor for advanced glycation endproducts (RAGE) and cardiovascular disease. Expert Rev Mol Med (2009) 11:e9. doi: 10.1017/S146239940900101X
164. Kim HJ, Jeong MS, Jang SB. Molecular characteristics of RAGE and advances in small-molecule inhibitors. Int J Mol Sci (2021) 22. doi: 10.3390/ijms22136904
165. Ampomah PB, Moraes LA, Lukman HM, Lim LHK. Formyl peptide receptor 2 is regulated by RNA mimics and viruses through an IFN-beta-STAT3-dependent pathway. FASEB J (2018) 32:1468–78. doi: 10.1096/fj.201700584RR
166. Tammaro A, et al. TREM-1 and its potential ligands in non-infectious diseases: from biology to clinical perspectives. Pharmacol Ther (2017) 177:81–95. doi: 10.1016/j.pharmthera.2017.02.043
167. Peng Q, et al. TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal 3 ra38 (2010). doi: 10.1126/scisignal.2000500
168. Dominguez-Soto A, et al. Intravenous immunoglobulin promotes antitumor responses by modulating macrophage polarization. J Immunol (2014) 193:5181–9. doi: 10.4049/jimmunol.1303375
169. Chauhan A, et al. M1 macrophage polarization is dependent on TRPC1-mediated calcium entry. iScience (2018) 8:85–102. doi: 10.1016/j.isci.2018.09.014
170. Schoenen H, et al. Differential control of mincle-dependent cord factor recognition and macrophage responses by the transcription factors C/EBPbeta and HIF1alpha. J Immunol (2014) 193:3664–75. doi: 10.4049/jimmunol.1301593
171. Domingues I, Rino J, Demmers JA, de Lanerolle P, Santos SC. VEGFR2 translocates to the nucleus to regulate its own transcription. PloS One (2011) 6:e25668. doi: 10.1371/journal.pone.0025668
172. Wang P, et al. Psychological stress up-regulates CD147 expression through beta-Arrestin1/ERK to promote proliferation and invasiveness of glioma cells. Front Oncol (2020) 10:571181. doi: 10.3389/fonc.2020.571181
173. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol (2017) 198:1006–14. doi: 10.4049/jimmunol.1601515
174. Androulidaki A, et al. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity (2009) 31:220–31. doi: 10.1016/j.immuni.2009.06.024
175. Hu X, et al. Sensitization of IFN-gamma jak-STAT signaling during macrophage activation. Nat Immunol (2002) 3:859–66. doi: 10.1038/ni828
176. Qin H, et al. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J Immunol (2012) 189:3439–48. doi: 10.4049/jimmunol.1201168
177. Nijmeh J, Levy BD. Lipid-derived mediators are pivotal to leukocyte and lung cell responses in sepsis and ARDS. Cell Biochem Biophys (2021) 79:449–59. doi: 10.1007/s12013-021-01012-w
178. Schebb NH, et al. Formation, signaling and occurrence of specialized pro-resolving lipid mediators-what is the evidence so far? Front Pharmacol (2022) 13:838782. doi: 10.3389/fphar.2022.838782
179. Zhou Z, et al. Heat shock protein 10 of chlamydophila pneumoniae induces proinflammatory cytokines through toll-like receptor (TLR) 2 and TLR4 in human monocytes THP-1. In Vitro Cell Dev Biol Anim (2011) 47:541–9. doi: 10.1007/s11626-011-9441-4
180. Afrazi A, et al. Toll-like receptor 4-mediated endoplasmic reticulum stress in intestinal crypts induces necrotizing enterocolitis. J Biol Chem (2014) 289:9584–99. doi: 10.1074/jbc.M113.526517
181. Herbert SP, Odell AF, Ponnambalam S, Walker JH. The confluence-dependent interaction of cytosolic phospholipase A2-alpha with annexin A1 regulates endothelial cell prostaglandin E2 generation. J Biol Chem (2007) 282:34468–78. doi: 10.1074/jbc.M701541200
182. De AK, Kodys KM, Yeh BS, Miller-Graziano C. Exaggerated human monocyte IL-10 concomitant to minimal TNF-alpha induction by heat-shock protein 27 (Hsp27) suggests Hsp27 is primarily an antiinflammatory stimulus. J Immunol (2000) 165:3951–8. doi: 10.4049/jimmunol.165.7.3951
183. Corrigall VM, Bodman-Smith MD, Brunst M, Cornell H, Panayi GS. Inhibition of antigen-presenting cell function and stimulation of human peripheral blood mononuclear cells to express an antiinflammatory cytokine profile by the stress protein BiP: relevance to the treatment of inflammatory arthritis. Arthritis Rheum (2004) 50:1164–71. doi: 10.1002/art.20134
184. Shields AM, Panayi GS, Corrigall VM. A new-age for biologic therapies: long-term drug-free therapy with BiP? Front Immunol (2012) 3:17. doi: 10.3389/fimmu.2012.00017
185. Jimenez-Garcia L, Herranz S, Luque A, Hortelano S. Critical role of p38 MAPK in IL-4-induced alternative activation of peritoneal macrophages. Eur J Immunol (2015) 45:273–86. doi: 10.1002/eji.201444806
186. Nishi C, Yanagihashi Y, Segawa K, Nagata S. MERTK tyrosine kinase receptor together with TIM4 phosphatidylserine receptor mediates distinct signal transduction pathways for efferocytosis and cell proliferation. J Biol Chem (2019) 294:7221–30. doi: 10.1074/jbc.RA118.006628
187. Al-Zaeed N, Budai Z, Szondy Z, Sarang Z. TAM kinase signaling is indispensable for proper skeletal muscle regeneration in mice. Cell Death Dis (2021) 12:611. doi: 10.1038/s41419-021-03892-5
188. Doran AC, et al. CAMKIIgamma suppresses an efferocytosis pathway in macrophages and promotes atherosclerotic plaque necrosis. J Clin Invest (2017) 127:4075–89. doi: 10.1172/JCI94735
189. Cai B, et al. MerTK signaling in macrophages promotes the synthesis of inflammation resolution mediators by suppressing CaMKII activity. Sci Signal (2018) 11. doi: 10.1126/scisignal.aar3721
190. Roh J, Go EJ, Park JW, Kim YH, Park CK. Resolvins: potent pain inhibiting lipid mediators via transient receptor potential regulation. Front Cell Dev Biol (2020) 8:584206. doi: 10.3389/fcell.2020.584206
191. Tavares LP, et al. Blame the signaling: role of cAMP for the resolution of inflammation. Pharmacol Res (2020) 159:105030. doi: 10.1016/j.phrs.2020.105030
192. Pan Y, Liu J, Ren J, Luo Y, Sun X. Epac: a promising therapeutic target for vascular diseases: a review. Front Pharmacol (2022) 13:929152. doi: 10.3389/fphar.2022.929152
193. Kim S, Jee K, Kim D, Koh H, Chung J. Cyclic AMP inhibits akt activity by blocking the membrane localization of PDK1. J Biol Chem (2001) 276:12864–70. doi: 10.1074/jbc.M001492200
194. Polumuri S, Perkins DJ, Vogel SN. cAMP levels regulate macrophage alternative activation marker expression. Innate Immun (2021) 27:133–42. doi: 10.1177/1753425920975082
195. Cattaneo F, Parisi M, Ammendola R. Distinct signaling cascades elicited by different formyl peptide receptor 2 (FPR2) agonists. Int J Mol Sci (2013) 14:7193–230. doi: 10.3390/ijms14047193
196. Ge YJ, et al. Anti-inflammatory signaling through G protein-coupled receptors. Acta Pharmacol Sin (2020) 41:1531–8. doi: 10.1038/s41401-020-00523-1
197. Giannakis N, et al. Dynamic changes to lipid mediators support transitions among macrophage subtypes during muscle regeneration. Nat Immunol (2019) 20:626–36. doi: 10.1038/s41590-019-0356-7
198. Deng B, et al. Maresin biosynthesis and identification of maresin 2, a new anti-inflammatory and pro-resolving mediator from human macrophages. PloS One (2014) 9:e102362. doi: 10.1371/journal.pone.0102362
199. Hung JJ, Wang YT, Chang WC. Sp1 deacetylation induced by phorbol ester recruits p300 to activate 12(S)-lipoxygenase gene transcription. Mol Cell Biol (2006) 26:1770–85. doi: 10.1128/MCB.26.5.1770-1785.2006
200. Snodgrass RG, Brune B. Regulation and functions of 15-lipoxygenases in human macrophages. Front Pharmacol (2019) 10:719. doi: 10.3389/fphar.2019.00719
201. Liu YC, Yu MM, Chai YF, Shou ST. Sialic acids in the immune response during sepsis. Front Immunol (2017) 8:1601. doi: 10.3389/fimmu.2017.01601
202. Achieng AO, et al. Molecular basis of reduced LAIR1 expression in childhood severe malarial anaemia: implications for leukocyte inhibitory signalling. EBioMedicine (2019) 45:278–89. doi: 10.1016/j.ebiom.2019.06.040
203. Wang W, et al. Paxillin mediates ATP-induced activation of P2X7 receptor and NLRP3 inflammasome. BMC Biol (2020) 18:182. doi: 10.1186/s12915-020-00918-w
204. Allard B, Allard D, Buisseret L, Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol (2020) 17:611–29. doi: 10.1038/s41571-020-0382-2
205. Schmitz J, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
206. Nishizaki T. IL-33 suppresses GSK-3beta activation through an ST2-independent MyD88/TRAF6/RIP/PI3K/Akt pathway. Heliyon (2018) 4:e00971.
207. Sreeramkumar V, Fresno M, Cuesta N. Prostaglandin E2 and T cells: friends or foes? Immunol Cell Biol (2012) 90:579–86. doi: 10.1038/icb.2011.75
208. Liao X, et al. Kruppel-like factor 4 regulates macrophage polarization. J Clin Invest (2011) 121:2736–49. doi: 10.1172/JCI45444
209. Hangai S, et al. PGE2 induced in and released by dying cells functions as an inhibitory DAMP. Proc Natl Acad Sci U.S.A. (2016) 113:3844–9.
210. Barbosa CMV, et al. Extracellular annexin-A1 promotes myeloid/granulocytic differentiation of hematopoietic stem/progenitor cells via the Ca(2+)/MAPK signalling transduction pathway. Cell Death Discovery (2019) 5:135. doi: 10.1038/s41420-019-0215-1
Keywords: macrophage, polarization, differentiation, sterile inflammation, DAMP, RAMP, SPM
Citation: Koncz G, Jenei V, Tóth M, Váradi E, Kardos B, Bácsi A and Mázló A (2023) Damage-mediated macrophage polarization in sterile inflammation. Front. Immunol. 14:1169560. doi: 10.3389/fimmu.2023.1169560
Received: 19 February 2023; Accepted: 07 June 2023;
Published: 03 July 2023.
Edited by:
Marten A. Hoeksema, Amsterdam UMC, NetherlandsReviewed by:
Ajitha Thanabalasuriar, McGill University, CanadaCopyright © 2023 Koncz, Jenei, Tóth, Váradi, Kardos, Bácsi and Mázló. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anett Mázló, YW5ldHQubWF6bG9AZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.