 Zhaoyun Liu
Zhaoyun Liu Chun Yang†
Chun Yang† Rong Fu
Rong Fu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 18 May 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1169541
This article is part of the Research Topic Identifying and developing novel therapeutic targets for multiple myeloma treatment View all 6 articles
Multiple myeloma (MM)as a haematological malignancy is still incurable. In addition to the presence of somatic genetic mutations in myeloma patients, the presence of immunosuppressive microenvironment greatly affects the outcome of treatment. Although the discovery of immunotherapy makes it possible to break the risk of high toxicity and side effects of traditional chemotherapeutic drugs, there are still obstacles of ineffective treatment or disease recurrence. In this review, we discuss therapeutic strategies to further enhance the specific anti-tumor immune response by activating the immunogenicity of MM cells themselves. New ideas for future myeloma therapeutic approaches are provided.
Multiple myeloma (MM) is a hematological malignancy due to a malignant clone of plasma cells and is highly heterogeneous, varying widely in clinical presentation, treatment options and prognosis. Myeloma is incurable and eventually relapses as the disease progresses (1). Over the past three decades there have been huge advances in the treatment of patients with MM. The advent of immunologic agents has revolutionized the treatment paradigm for patients while improving survival rates, with incredible results in both NDMM and relapsed refractory MM(RRMM) patients (2). Immunomodulators(IMIDs)-based therapy has become an essential ingredient in the treatment regimen for patients who are eligible or ineligible for transplantation (3). Monoclonal antibody(mAb) and bispecific antibodies and bispecific therapeutic engagers (BiTEs) therapy harness specific targets on the surface of myeloma cells to trigger an immune response, inducing disease remission. Immune checkpoint inhibitors (ICIs) therapy has shown outstanding performance in combination therapy. The emergence of CAR-T therapy in recent years has resulted numbers of clinical trials being conducted due to rapid onset of action and high response rates, but it cannot be denied the problems of high price and poor treatment persistence (4).
The key to the effectiveness of some immune agents is the expression of specific antigens on the surface of tumor cells (2). The inability to successfully activate antitumor immune responses due to antigen loss is a direct cause of the failure of these conventional immune agents. And how to sufficiently activate the specific anti-tumor immune response is exactly what we have listed in this review.
Generally, immunotherapies have an impact on tumor-host interactions, and effective treatment tilts the balance towards activating an immune response against malignant cells. The “resetting” effect of chemotherapeutic drugs on the immune system and the restoration of immune surveillance are discussed in the review by Zitvogel et al. (5) That successful antineoplastic drugs elicit therapeutic immune responses can be partly explained by enhancing malignant cells sensitivity to immune effect, increasing their antigenicity and immunogenicity (5). That is to say, the core of the therapeutic strategy proposed is to target the tumor cells themselves to increase their ability to elicit an effective immune response, rather than focusing on enhancing immune effector cells as traditional immunological agent model. But this therapy has not been discussed in the context of myeloma.
Here, we discussed immunotherapy strategies to enhance immune response based on MM cells. Immunogenic cell death (ICD) inducers target intact myeloma cells to improve their immunogenicity and achieving immunogenicity death of MM cells. Vaccines based on myeloma cell antigenic peptides or designed to directly load whole myeloma cells realize the purpose of actively releasing self-antigen. The advent of technology to detect neoantigens of myeloma has broken the immune tolerance dilemma and increased the autoantigenicity of MM cells, making them easier to be recognized by the immune system. Boosting signals from co-stimulatory molecules on the surface of myeloma cells appears to make them more sensitive to immune attack.
In this review, we outline the immunodeficiency in myeloma patients and main strategies currently available for immunotherapy. And these therapeutic approaches that target myeloma cells and up-regulate the body’s immune response are summarized.
It is now believed that the development of myeloma is the result of two simultaneous factors (6). One factor is the clonal evolution of tumor cells due to genetic mutations in myeloma patients. Another factor is the alteration in the composition and function of the immune system, leading to vandalized immune homeostasis as well as loss of normal immune surveillance function and formation of immunosuppressive microenvironment. Abnormalities in the immune machinery of myeloma involve many aspects, including immune effector cells deactivation, production of cytokines that promote tumor growth and form an immunosuppressive microenvironment, and the accumulation of myeloid-derived suppressor cells (MDSC) and tumor-associated macrophage (TAM) (7–9).
The specific mechanisms of immune dysregulation in myeloma patients are complex. T cells have numerical and functional defects and aberrant CD4/CD8 T cells ratios. Myeloma cells express programmed cell death ligand 1(PD-L1) and CD155 ligand, which directly lead to T cell depletion by binding to the T cell surface receptors programmed cell death protein 1(PD-1) and T cell immunoglobulin and ITIM domain(TIGIT), respectively (10). Transforming growth factor-β(TGF-β), secreted by MM cells, regulatory T cells(Tregs) and bone marrow mesenchymal stem cells (BMSCs), facilitates the differentiation and expansion of Tregs, which inhibit T-cell function. TGF-β can cause defects in the natural killer cells(NKs) number and function (11). MM cells expressing PD-L1 are also able to suppress NK cytotoxicity (12). Peripheral dendritic cells(DCs) exhibit an immature phenotype and impaired antigen presentation in MM, possibly associated with low expression of co-stimulatory molecules (13). TGF-β and IL-10 secreted from myeloma cells mediate the defective DC function, which could be restored by IL-12 and interferon-gamma (IFN-γ) (14).Indoleamine 2,3-dioxygenase(IDO) is produced by immature DCs and can deactivate T cells (15).
MDSCs and TAMs co-exist in the immunosuppressive microenvironment of myeloma. MDSCs are immature myeloid cells showing heterogeneity. The differentiation of MDSCs in myeloma is blocked by vascular endothelial growth factor(VEGF), granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-6 (16). Immature MDSCs accumulate in the microenvironment, inhibiting the cytotoxic effects of T cells by arginase, reactive oxygen (ROS) as well as nitric oxide and contributing to cancer progression and metastasis (16, 17). Macrophages can be divided into two subgroups according to their functions, i.e., M1 and M2 (18). M1 macrophages exert anti-tumor effects and act as APCs to activate cellular immune responses against MM antigens. Chemokine (C-X-C motif) ligand 12(CXCL12), chemokines ligand 2(CCL2), chemokines ligand 3(CCL3) and chemokines ligand 14(CCL14) produced by myeloma cells and BMSCs promote macrophage M2 polarization and imbalance the M1/M2 ratio (19, 20). M2 macrophages secrete or release cytokines that promote tumor neovascularization through direct or indirect action (21).
A variety of different immunotherapy strategies for MM are emerging, aiming to increase the depth and breadth of treatment while reducing the incidence of side effects. In recent years, the standard of care for myeloma has been further rewritten with advances in immunotherapy. We currently believe that myeloma immunotherapy focuses on two main areas. On the one hand, membrane surface molecules are used to build a bridge between tumor cells and effector cells, i.e., to provide a peptide chain or antibody to target and link myeloma cells and immune cells, narrowing the gap between them and achieving the specific targeting and killing effect. Examples include mAbs therapy, BiTEs therapy and CAR-T therapy. On the other hand, considering the existence of immunosuppressive microenvironment and immunosuppressed state in the patient’s body, immune cells frequently fail to perform their normal capacity of recognizing and killing tumor cells. ICIs and IMIDs can restore the depleted state of immune cells and strengthen the function of autoimmune cells. The following summarizes the general immunotherapy strategy for myeloma. (Figure 1)
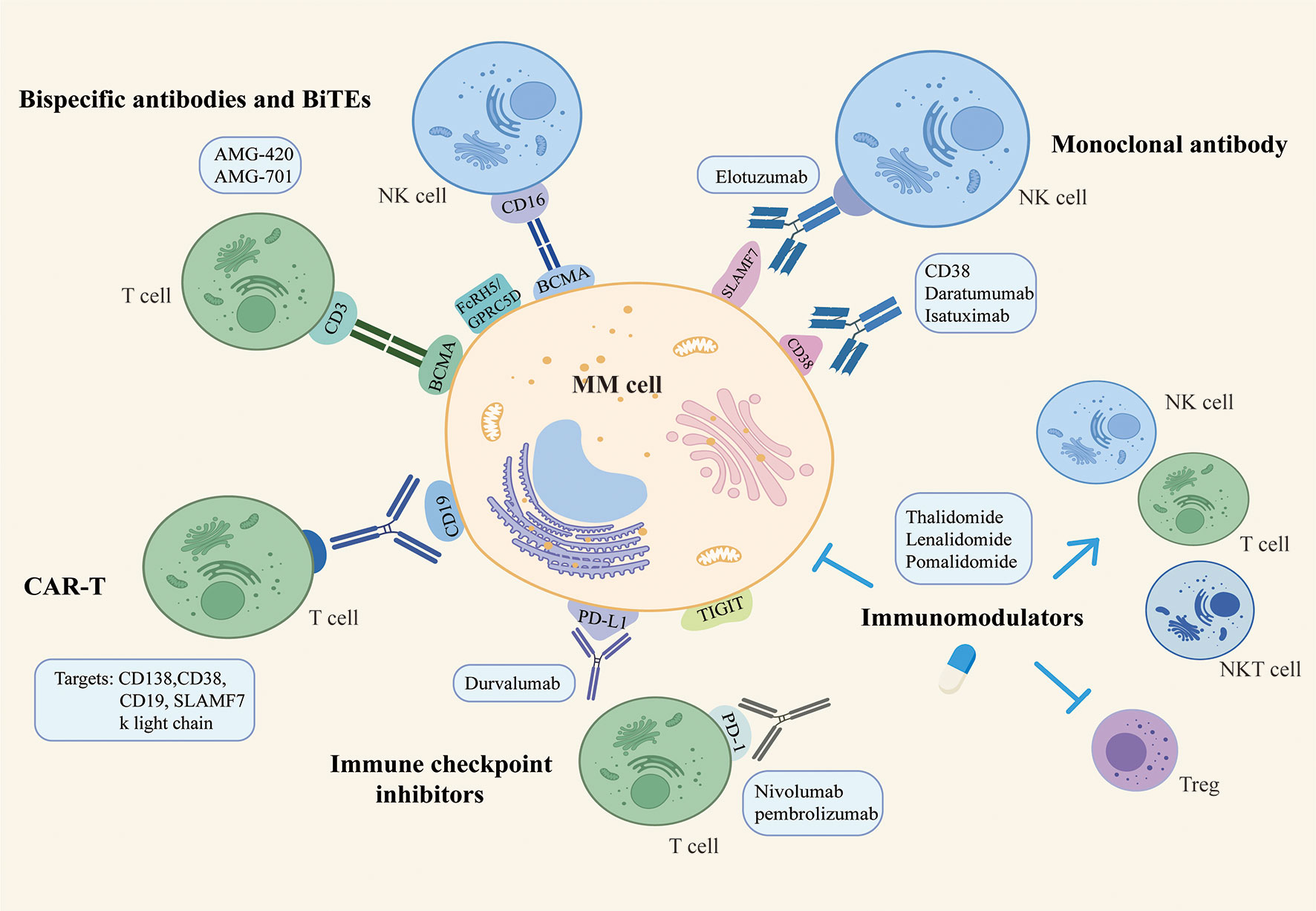
Figure 1 The general immunotherapy strategy for multiple myeloma.
Immunotherapy with mAb is clinically effective and can benefit even patients with advanced disease stages (22). Food and drug administration(FDA) has approved daratumumab and isatuximab targeting CD38 and elotuzumab targeting SLAM7 for the treatment of myeloma (23). Elotuzumab is an anti- signaling lymphocytes activating molecule factor 7(SLAMF7) mAb that exerts NK cell-mediated ADCC effects or directly activates NK cells to kill tumor cells (24, 25). The mechanism of Dara is more extensive. In addition to directly inducing apoptosis, macrophage-mediated phagocytosis and FC-dependent immune regulation, Daratumumab can also promote the activation and expansion of NK and T cells, restoring their anti-tumor ability (26, 27). Daratumumab has demonstrated outstanding clinical efficacy in myeloma patients, showing fast, in-depth and lasting responses even when administered as a single agent in relapse and refractory MM(RRMM) patients (28). Lenalidomide upregulated the expression of CD38 on the surface of myeloma cells (29), and the combination of Daratumumab with lenalidomide/dexamethasone (30) or other drugs such as bortezomib/dexamethasone are continuously carried out and they can significantly improve the clinical outcomes of patients (29).
Bispecific antibodies and BiTEs are emerging immunotherapeutic strategies that are thought to be somehow potentially superior to monoclonal antibodies. They have two binding sites, one end binds to tumor cell surface antigens, currently involved in B cell maturation antigen(BCMA), CD38, CD19, G protein-coupled receptor class C group 5 member D(GPRC5D) and Fc receptor-like 5(FCRL5), and the other end binds to molecules on immune effector cells, such as CD3 on T cell and CD16 on NK cell (31). In other words, the interaction between tumor cells and immune effector cells is no longer dependent on T cell receptor(TCR)-specific recognition and antigen presentation, but directly kills tumor cells while activating immune effector cells (32, 33). BiTEs refers to a structure consisting of single-chain variable fragment(scFV)-binding regions and a short linker, with lower molecular weight and relatively short half-life, thus requiring continuous infusion to maintain the desired therapeutic concentration (34). AMG420 is the first BiTE with confirmed clinical efficacy, targeting both CD3 and BCMA to induce TCR-independent immune response activation and tumor cell death (35). AMG-701 was developed to address the short half-life involved with AMG-420 (36). Anti-FcrH5/CD3 bispecific antibodies are capable of stimulating the formation of immune synapses (37). And the GPRC5D/CD3 bispecific antibodies constructed based on GPRC5D overexpression on myeloma cells can effectively recruit T cells to attack tumor cells (38). Moreover, Elranatamab was granted Orphan Drug Designation by the FDA as a humanized anti-BCMA/CD3 bispecific IgG2a antibody that has demonstrated a relatively high safety and durability of anti-myeloma effects (39). Talquetamab targets both MM cell surface GPRC5D and T cell surface CD3 to lyse target cells while activating cytotoxic T cells. Talquetamab has been approved by the FDA for the treatment of adult RRMM patients who have received at least 4 prior lines of therapy, including proteasome inhibitors, immunomodulators, and anti-CD38 antibodies, due to its significant clinical efficacy (40). Clinical and preclinical studies on the application of bispecific antibodies and BiTEs are being conducted and have shown potential clinical application value.
CAR is a chimeric protein that binding mAb-derived scFv to the T-cell receptor domain and co-stimulatory molecular signalling domain to enhance the T-cell immune response by mimicking T-cell activation in vivo (41). These patient-derived T cells are expanded and modified to selectively target tumor antigens and BCMA is most frequently designed for use with CART therapy owing to its high selectivity of expression on MM cells. Other targets include CD138, CD38, CD19, SLAMF7 and k light chain (42). Not only does CAR therapy recognize tumor antigens in a MHC-independent manner and reduced off-target effects, it also has a stronger affinity for antibody-antigen binding and a more rapid onset of action (4). The FDA has approved idecabtagene vicleucel (Abecma) and ciltacabtagene autoleucel (Carvykti) for the treatment of patients with heavily pretreated RRMM due to their high clinical response rate and MRD negativity (43). However, the sustainability of CAR-T treatment is problematic and the risk of relapse after treatment is significant. In addition, loss of myeloma cell surface antigens or relapsed disease that do not express the original CAR target are correlated with treatment ineffectiveness (44). The application of CAR-T also requires consideration of clinical toxicity, most commonly cytokine release syndrome and neurotoxicity due to the rapid expansion and activation of CAR-T cells (45).
IMIDs, which include thalidomide, lenalidomide and pomalidomide, have a significant role in improving the prognosis of patients with myeloma and are an essential component of existing treatment regimens. Research now generally agrees that IMIDs have both tumor and immune dual targeting effects (46).IMiDs have directed cytotoxic effects on myeloma cells, inducing growth arrest and apoptosis of MM cells, which is associated with downregulation of interferon regulatory factor 4(IRF4) and cereblon-dependent degradation of the transcription factors Ikaros/Aiolos (IKZF1/3) (47, 48). In terms of immunomodulation, IMiDs promote the immune system and improve the immunosuppressive microenvironment of myeloma by eliminating the adhesion between myeloma cells and BM, upregulating T, NK and NKT cells with the downregulation of Treg, as well as inhibiting angiogenesis (49).Additionally, IMiDs regulate the production of cytokines. Lenalidomide promotes the cytotoxicity and antibody-dependent cell-medicated cytotoxicity(ADCC) of NK and NKT cells by inducing IKZF1/3 degradation and IL-2 secretion in T cells (50, 51).Given that some patients remain sensitive to IMiDs even after relapse, combined IMiDs are also used as a preferred option for patients with RRMM (33). Pomalidomide was approved by the FDA in 2013 for refractory patients who had been received at least two therapeutic regimens, including bortezomib and lenalidomide (52).
PD1/PD-L1 are major immune checkpoints, and as negative regulatory axes of immune modulation, their overexpression would lead to immune escape and the formation of immune tolerance in myeloma cells (53). A remarkable increase in PD-L1 expression can be detected in RRMM patients (54). Another negatively regulated immune checkpoint is TIGIT, which inhibits the killing function of NK cells and the cytotoxic effect of CD8+ T cells, affecting antigen presentation and the release of anti-inflammatory factors (55). ICIs are specific mAbs that blocking PD-1/PD-L2 binding and acting by targeting immunosuppressive signals in the tumor microenvironment. However, monotherapy with ICIs has not seen remarkable benefits in the early stages. The PD-1 inhibitors pembrolizumab and nivolumab were not observed objective responses or significant clinical efficacy when used separately in patients with MM (56). These studies highlight that ICIs need to be used in combination with other therapeutic strategies to achieve translation of clinical outcomes. Based on the theoretical basis that Len in combination with ICI is capable of restoring the cytotoxic effects of depleted NK cells and preventing immune evasion by MM cells (57), the combination of ICIs and IMiDs has attracted many investigators. For example, pembrolizumab in combination with lenalidomide and dexamethasone for NDMM, or pembrolizumab in combination with pomalidomide and dexamethasone for RRMM (discontinued due to safety concerns) (58, 59). Furthermore, the strategy of combining multiple ICIs against diverse targets has a potentially synergistic therapeutic effect. In conclusion, ICIs are a viable treatment strategy for patients with MM, but are still being explored and a balance still needs to be found between immune efficacy and pharmacological toxicity.
The effects of immunological agents such as mABs, BITEs and CAR-T, which are now widely used in clinical practice, are dependent on the expression of specific antigens on myeloma cells. However, it is difficult to accurately and consistently monitor the expression of surface antigens in practical treatment. With the progress of treatment, the number of cell clones that do not express specific antigen will gradually increase, and the emergence of immune escape eventually lead to drug resistance. Patients have to switch to other drugs or take the next-line therapy. Therefore, we propose immunotherapeutic strategies that target myeloma cells with a view to circumventing the effects of antigen loss on immunotherapy efficacy.
Immunogenicity is an intrinsic property of antigen that reflects its ability to elicit an immune response. Myeloma cells can provoke some degree of immune response, however, most of the time immune cells are failing to kill tumor cells directly with the occurrence of malignant clones of tumor cells and cancer progression. Both ICD and vaccine treatment strategies aim to stimulate endogenous immune responses against malignant myeloma cells. The efficacy of immunotherapeutic agents would be enhanced by targeting tumor cells to increase their sensitivity to death and improve poorly immunogenic cells recognized by APCs, i.e. targeting ICD of tumors cells. Vaccine formulations can activate immune effector cells to continuously attack tumor cells, benefitting some patients with refractory disease and minimal residual disease (MRD). The identification of neoantigens by new technologies breaks the dilemma of immune tolerance of traditional chemotherapy and fills the gap of ICD and vaccine therapy. Therapeutic approaches that increase the expression of co-stimulatory molecules on the surface of myeloma cells complement the therapeutic strategies that target whole myeloma cells, allowing tumor cells to be successfully presented to DCs as APCs and activate effector T cells. Therapeutic strategies to enhance immune response induced by MM cells were summarized in Figure 2.
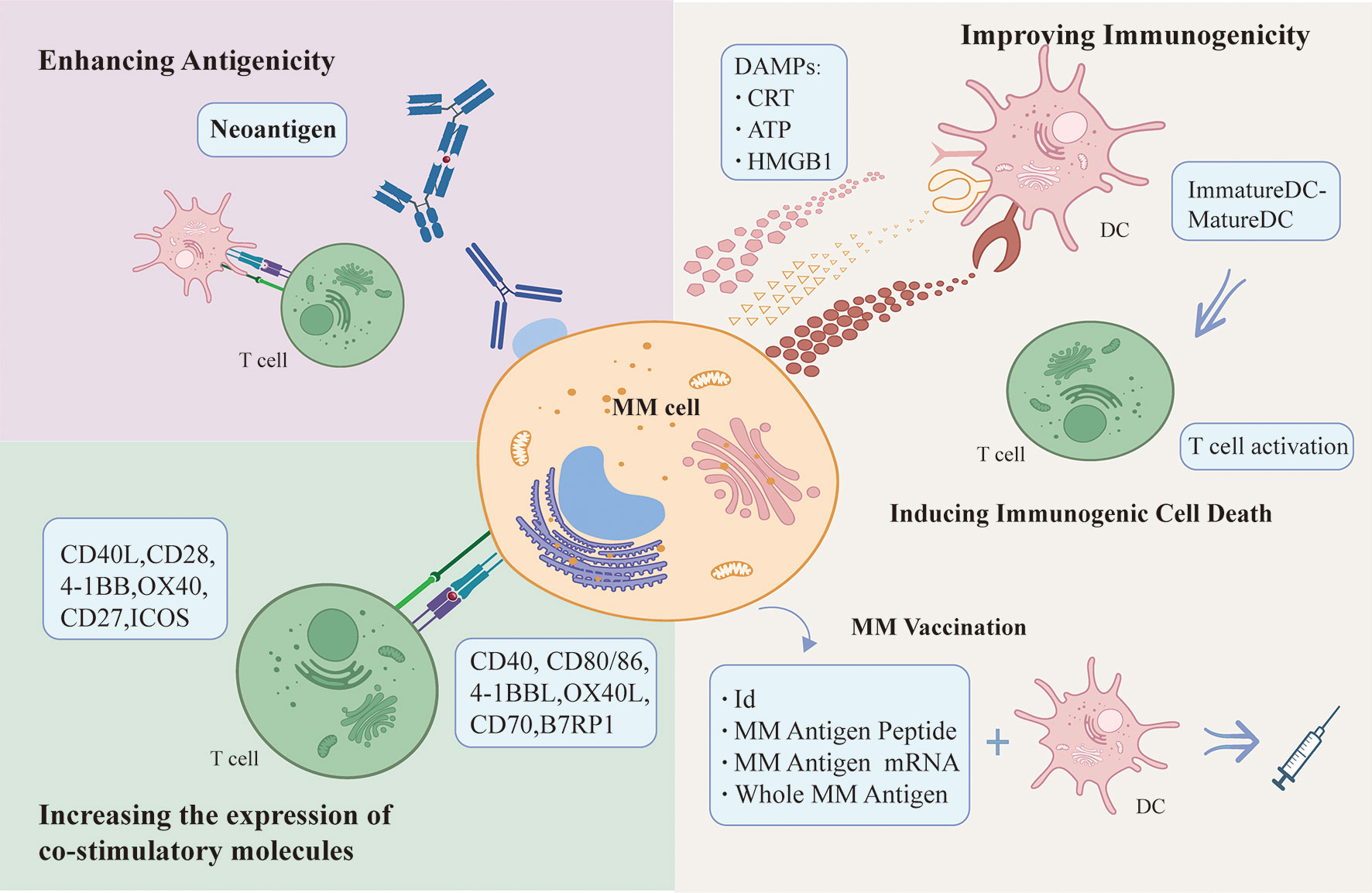
Figure 2 Therapeutic strategies to enhance immune response induced by MM cells.
The occurrence of ICD is dependent on sequential and concerted release of DAMPs, including the exposure of the calreticulin (CRT) and heat shock protein (HSP), the secretion of adenosine triphosphate (ATP), the release of the non-histone chromatin binding protein high mobility group box 1 (HMGB1) and cytokines such as type I interferon (INF I). DAMPs binding to specific pattern recognition receptors (PRRs) expressed by DCs recruits more APCs and activates their ability to process antigens. In the presence of activating co-stimulatory signals, T cells are successfully activated and immune responses are initiated. However, in the course of the ICD, innate immune responses are simultaneously initiated (60, 61).
Improved immunogenicity: Inducers acts on MM cells to increase MM cells immunogenicity and induce the release of DAMPs from dying MM cells, which bind to PRRs on DCs and initiate T cell activation and anti-tumor immune response. MM-derived DC vaccine can effectively activate immune effector cells to continuously attack MM cells and effectively initiate endogenous immune responses. Enhancing antigenicity: Neoantigens are promising in breaking the drug tolerance dilemma, as they are effectively recognized and killed by T cells, making them a good potential therapeutic target for immunotherapy. Up-regulating the expression of costimulatory molecules such as CD40, CD80/86, 4-1BBL, OX40L, CD70 and B7RP on the surface of myeloma cells can increase the susceptibility of myeloma cells to attack by immune effector cells.
ICD is a type of pro-inflammatory cell death, and the key factors are the generation of sustained reactive oxygen species (ROS) and endoplasmic reticulum stress (ERS). ER is associated with protein synthesis and undergoes an unfolded protein response (UPR), a homeostatic mechanism designed to rapidly detect and correct protein processing errors. When ICD occurs, over-activation of the UPR leads to ERS accompanied by the release of DAMPs to elicit immune responses and the development of memory immunity (62).
After the induction of ICD, dying cells expose and/or release DAMPs. What happens in the first is the transfer of CRT, which acts as an “eat me” signal, from the ER lumen to the cytoplasmic surface. Ecto-CRT exposure accompanied by co-translocation of ERp57 and ERS plays a significant role in it (63). Ecto-CRT interacts with the CD91 receptor on APC to enhance immunogenic recognition and phagocytosis of dead cells (64, 65). Dying tumor cells secrete ATP extracellularly via the autophagic pathway, bind to ionotropic (P2X7) and metabotropic (P2Y2) purinoceptors on the surface of APCs and acts as a “find me” signal to facilitate APCs recruitment (66). Extracellular ATP activates caspase 1-dependent NOD-like receptor thermal protein domain associated protein 3(NLRP3) inflammasome and secretes IL-1b for immunostimulatory effects (67). HMGB1 secreted by tumor cells binds to various PRRs (e.g., Toll-like receptor 4 (TLR4)) and promotes the release of cytokines such as IL-1b. Both the knockdown of HMGB1 and the application of TLR4-neutralising antibodies will deplete the efficacy of the immune response induced by the anthracyclines or cyclophosphamide in vivo models (68). Tumor cells undergoing ICD produce IFNI via RNA activation of the TLR3 or cyclic guanosine monophosphate-adenosine monophosphate synthase/stimulator of interferon genes (cGAS/STING) pathways. The IFNI response triggers the release of CXC-chemokine ligand 10 (CXCL10) when it binds to the interferon receptor (IFNAR I) on tumor cells, which exerts an immunostimulatory effect (69, 70).
Currently, there are type I and type II inducers. Type I inducer induces non-ER-targeted apoptosis, causing mild ERS, including anthracyclines, oxaliplatin, bortezomib, cyclophosphamide and radiation. Type II inducers selectively target ER and cause ROS-based ERS. Chrysin photodynamic therapy and lysovirus therapy, etc. are type II ICD inducers (60). Induction of ICDs is a very effective immunotherapeutic approach for targeting myeloma cells in MM as it enhances their immunogenicity and elicits a strong anti-tumor immune effect.
Bortezomib, a proteasome inhibitor (PI), is one of the representative drugs and it has been approved for first-line treatment of MM (71). There are a large amount of monoclonal proteins secreted by MM cells in the peripheral blood. Whereas protein overload in MM cells makes them dependent on proteasome activity. As a PI, bortezomib leads to accumulation of misfolded proteins and produces ROS, inducing immunogenic death of myeloma cells (72). It has been demonstrated that bortezomib induces immunogenicity in myeloma cells by activating the cGAS/STING pathway and producing type I interferon (73). Furthermore, the combination of bortezomib and STING agonists appears to induce a more intense ICD, providing a preclinical foundation for the combination of the two agents to improve the prognosis of MM patients (73). Bortezomib in combination with dexamethasone is currently the standard regimen for RRMM. Clinical trials combining mAbs (74), or a selection of different IMDs (75), are ongoing and some of them have shown excellent efficacy.
The second-generation PI carfilzomib is considered to be more effective and safer than bortezomib. Carfilzomib inhibits proteasome activity in an irreversible manner and misfolded proteins accumulate in the body, leading to significant ERS. A phase 2 study evaluated the efficacy of carfilzomib injection, involving 266 RRMM patients who had received median five prior treatments and mostly were intolerant to bortezomib and lenalidomide (76). ASPIRE (NCT01080391) study showed that combining carfilzomib with Lenalidomide and dexamethasone demonstrated the higher rates of overall response and improved PFS and 2-year overall survival (77). In a phase 3 multicenter randomized controlled study comparing the efficacy of carfilzomib and dexamethasone with bortezomib and dexamethasone for RRMM, the carfilzomib group showed longer progression-free survival(PFS), improved objective response rate(ORR), complete response (CR), very good partial response(VGPR), and median survival time with better clinical advantages than the bortezomib group (78).
Studies have demonstrated that irradiation, hyperthermia (HT), high hydrostatic pressure (HHP), bortezomib and lenalidomide as ICD inducers enhance the immunogenicity of dead tumor cells and augment anti-tumor T cell responses in vivo by improving DC function (79). Bortezomib induces HSP90 exposure on the surface of dying cells, which mediates intercellular contact between DCs and dying cells, delivers DC activation signals and successfully activates T cells to kill tumor cells. This targeting of tumor cells and enhancement of their immunogenicity by bortezomib, providing a unique immune activating stimulus has been demonstrated by Spisek et al. (80) Pomalidomide and lenalidomide have been proven to be common ICD inducers in MM. These immunomodulators enhance the uptake and presentation of tumor antigens by DCs and may play an adjuvant role in vaccine therapy (79, 81). In addition, experimental validation of the combination of the DNA methyltransferase inhibitor decitabine and the histone deacetylase inhibitor quisinostat acting on the murine immunocompetent 5T33MM model, vaccination of mice with epigenetic compounds effectively delayed the progression of tumors in vivo, activated DC cells and strengthened anti-tumor immunity in the form of inducing immunogenic death of myeloma cells (82). Mechanistically, epigenetic modulators can increase MM cell surface ectocalreticulin, reduce CD47 and PD-L1 expression, promote DC cell maturation and the persistence of immune effector CD4/8 cells (82).
ICD offers an approach to treat MM through combining immunogenic death of tumor cells with the initiation of specific anti-tumor immune responses. ICD makes it possible to achieve long-term anti-tumor effects by breaking down the immune tolerance and immunosuppressive microenvironment in patients. It is now widely accepted that ICD can transform dying tumor cells into a ‘vaccine’ that is able to induce anti-cancer immunity without the addition of any adjuvant (83). However, some questions remain to be addressed. For example, how to translate the results of in vitro studies into clinical efficacy (84). In addition, understanding the molecular mechanisms underlying inducers would be useful in transforming non-immunogenic cell death inducers into inducers that effectively induce immunogenicity in tumor cells and will guide the combination of drugs (83).
MM is featured by malignant clonal proliferation of the plasma cells and the secretion of idiotype antigens (Id), which is monoclonal immunoglobulins (Ig) or fragments (i.e., M proteins). Id has unique amino acid sequences in their variable regions, making them different from normal Ig and can be used as antigens for vaccination. Many researchers have conducted clinical trials of Id vaccines for MM therapy. Id is normally coupled with keyhole limpet hemocyanin (KLH) and supplemented with an immunostimulatory adjuvant such as GM-CSF for simultaneous injection. In some patients, researchers observed Id-specific immune responses, which do not always occur and are short-lived. Clinical responses are rarely observed in a small number of patients as well (85–88). These observations, however, disclose the low immunogenicity of Id and the efficacy needs to be improved.
The activation of initial T cells is more dependent on the presence of DC stimulatory signals, making DC the only dedicated antigen presenting cell (APC) that can directly activate initial T cells. As the most powerful APCs, DCs link innate and adaptive immune responses (89). However, MM patients have a quantitative and functional defect in DCs, affected by cytokines such as TGF-β and IL-10 (90). After antigen stimulation, DCs are unable to upregulate surface activating costimulatory molecules, thereby disrupting antigen presentation (91).
MM vaccines based on DCs are undergoing continuous advancements. The source of DCs, the method for maturation induction of DCs, the type of tumor antigens co-loaded with DCs and the technology, and the route of administration will have an impact on the clinical efficacy. Currently, the commonly used DCs are derived from in vitro preparations of circulating blood mononuclear DC (moDCs) or bone marrow progenitor cells (92). Maturation and activation of DCs are either accomplished through direct contact with antigens in vivo or mimicked in vitro by co-culture with cytokines such as prostaglandin E2, pathogen recognition receptor (PRR) agonists or TNF-a. DCs of allogeneic origin compensate for the DC number and defect in function due to autologous origin but may be limited by MHC molecules.
Many clinical studies have included post-HSCT patients in their study populations (93). Post-transplant vaccination may be effective in controlling residual lesions while the body rebuilds systemic immune system during the post-transplant period, providing an excellent immune microenvironment for the vaccination. Lacy et al. reported a phase II trial through which a comparison was conducted between 27 patients who treated with Id-DC vaccine after auto-HSCT and 124 patients who only received auto-HSCT during the same period. Although a discrepancy in PFS was not observed, a statistically and clinically significant improvement in overall survival(OS) was observed (94).
However, DC-based vaccines in populations with advanced disease progression and post-HSCT could not be successfully translated into clinical efficacy probably because of a higher tumor burden and/or immune compromise due to strong chemotherapy treatment (95). Röllig et al. used mature monocyte-derived Id-pulsed DCs and KLH to explore the potential of DC vaccines in stage I myeloma (96). Their results suggest that DC vaccine is a viable approach to elicit T-cell immune responses in patients with early stage myeloma.
The selection of appropriate tumor-associated antigens (TAAs) is critical for developing a vaccine therapy to maintain tumor specificity and immunologic efficacy. MUC1 is TAA expressed in all MM cells and in the serum of MM patients. ImMucin is a 21mer synthetic long peptide vaccine that encodes the signal peptide domain of the MUC1, possessing high density of T and B cell epitopes, and is able to elicit strong MUC1-specific T and B cell responses (97). New York esophageal squamous cell carcinoma 1(NY-ESO-1) is considered to be the most immunogenic cancer testicular antigen (CTA). In patients with advanced MM, NY-ESO-1 expression mediates spontaneous humoral and CD8+ T cell immunity (98). In contrast, PTD-NY-ESO-1 moDCs by proteins transduced appears to induce stronger CD8+ immune responses (99).
Researchers reported a CTA-mRNA-loaded vaccination for the treatment of MM. They extracted and prepared autologous moDCs pulsing KLH, and electroporated with mRNA for the melanoma antigen family A, 3 (MAGE-A3), SURVIVIN, and BCMA. This study showed that TAA-mRNA of mature electroporated DCs could induce TAA-T cell immune responses in myeloma patients after HSCT (100). CT7 and MAGE-A3 are the most common CTA in MM and WT1 is expressed in the bone marrow. They may be associated with high tumor load and disease recurrence. The Phase 1 clinical trial carried out by Chung et al. demonstrated the feasibility of preparing a DC vaccine from autologous Langerhans-type dendritic cells (LC) electroporated by CT7, MAGE-A3 and WT1 mRNA. It was observed that CD34+ haematopoietic progenitor cells (HPC)-derived LC could be more effective in activating cytotoxic T lymphocytes (CTL) to mount specific anti-myeloma immune responses (101).
Single-antigen DC vaccines have a potential limitation in that their antitumor effects can be affected by immune evasion if downregulation of antigen expression occurs. In myeloma, an alternative to target Id or TAA is the use of total MM-antigen spectrum loaded ex-vivo-generated moDCs. This approach makes it possible for multivalent immune responses to target tumor-specific neoantigens. Whole MM antigen-loaded DC technology for vaccine preparation includes the establishment of DC-tumor fusion cells, pulsing DCs with myeloma lysates, pulsing DCs with myeloma apoptotic bodies and loading of DCs with tumor exosomes or whole cell DNA or RNA (90).
Rosenblatt et al. reported outcomes in Phase II clinical trials in which DC-tumor fusion cells vaccines were used to target MRD in myeloma patients after HSCT. Although the treatment with DC-vaccines after HSCT promoted the tumor-specific T cells proliferate, no significant differences in T-cell responses were seen in patients who received vaccine therapy before transplantation from those who only received vaccine therapy after transplantation (102). The efficacy of vaccination using DCs pulsed with Id and tumor lysate were assessed in myeloma mouse model by Hong et al. They found that tumor lysate-pulsed DCs vaccines were more efficient in protecting mice against developing myeloma, delaying the progression of tumor, and inducing tumor regression against established tumor, suggesting a more pronounced advantage of myeloma cells themselves as a source of tumor antigens over Id proteins (103). Vasileiou et al. designed a preclinical study to explore alternative whole-tumor antigen approaches, i.e. phagocytosis of apoptotic bodies of autologous myeloma cells or total RNA transfection by electroporation. Both approaches are effective in inducing specific CD8 CTL and deserve further investigation in clinical trials (104). Table 1 summarizes the key clinical trials.
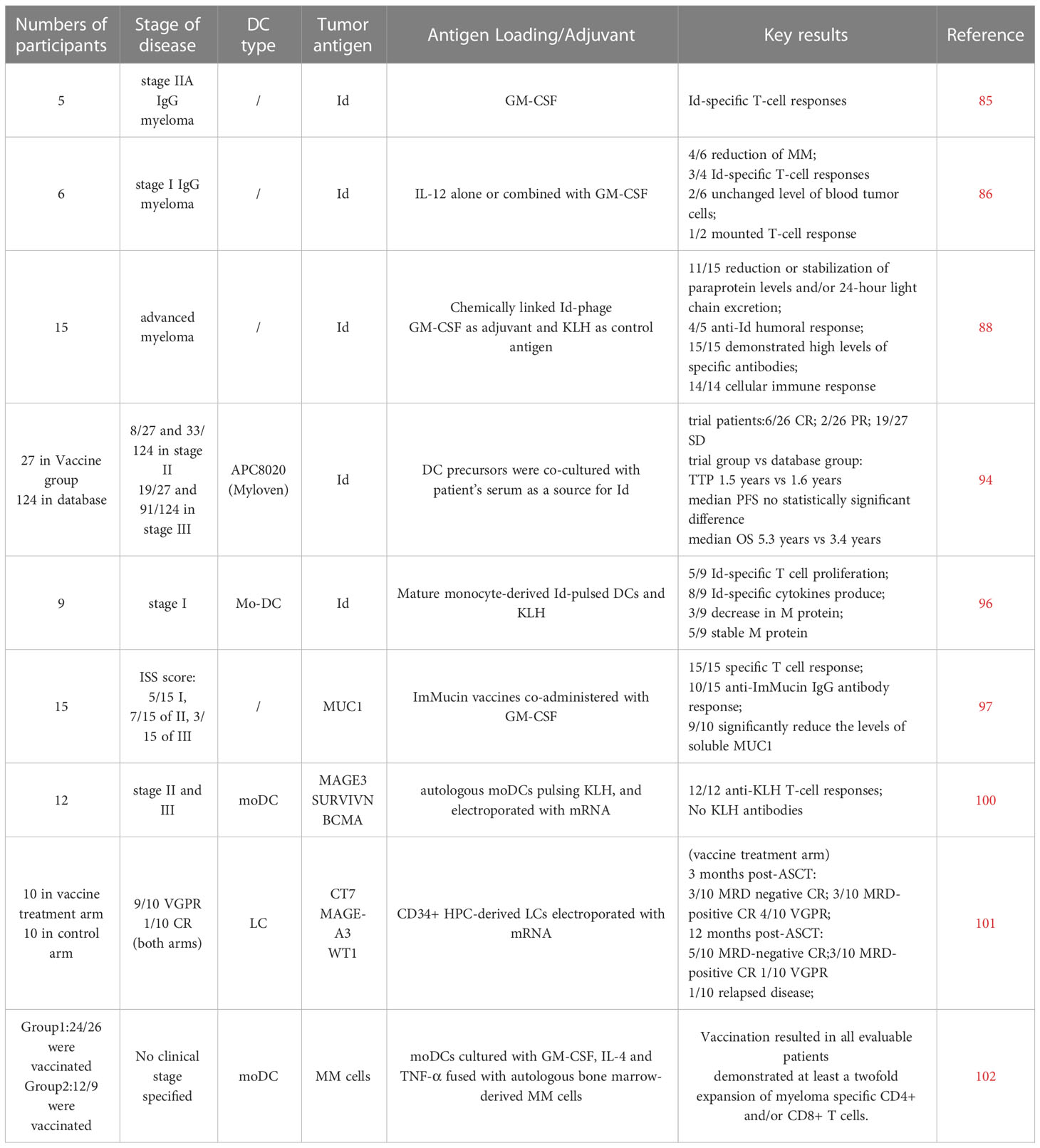
Table 1 MM vaccine clinical trials.
In MM, the combination of vaccines with existing treatment modalities is thought to play a synergistic role in improving the strength and duration of the immune response (105). We have explored some examples of vaccines combined with HSCT therapy above. The post-transplant status of MM patients provides a window for vaccine-stimulated anti-myeloma responses and targeting of MRD (102). Previous studies have shown that lenalidomide enhances vaccine anti-tumor responses and vaccine-specific cellular and humoral immunity, potentially as an adjuvant for cancer vaccines (106). In the mouse model, lenalidomide synergistically augments the efficacy of the DC vaccine by inhibiting the production of immunosuppressive cells, effectively inducing Th1-specific immune responses and reducing Th2-specific immune responses. In addition, lenalidomide enhances the activation and proliferation of NK cells in mice, obtaining higher levels of NK cell-mediated cytotoxicity in conjunction with the vaccine (107). Immune checkpoint PD-1/PD-L1 is an essential pathway for tumor-mediated immunosuppression. DC/myeloma cell fusion vaccines were investigated for high expression of PD-L1, which may provide inhibitory signaling and weaken vaccine immunity. In contrast, blocking PD-1 promotes vaccine-induced Th1 cell polarization, reverses the upregulation of PD-1 expression, reduces Treg cells and enhances anti-tumor immunity (108). Furthermore, the triple therapy of DC vaccine, PD-1 blockade and lenalidomide in the MM mouse model potently inhibited the growth of tumor cells, with enhancing functional activity of cytotoxic T lymphocytes and NK cells, synergistically enhancing anti-tumor immunity (109). The same results were observed in MM mouse models cotreatment with a triple combination of DC vaccine, PD-L1 blockade and pomalidomide (110).
In conclusion, from first- to second-generation vaccines, more and more vaccine preparation methods have emerged and strategies for recognizing tumor antigens have also been continually refined (90). However, it is difficult to standardize the vaccine preparation as well as the timing and route of vaccine use, due to the small number of patients included in the trials (111). With the complex immune microenvironment in MM patients, certain immune dysregulation and deficiencies in the quantity and function of immune cells affect the efficacy of vaccination. Based on the current use of vaccines in MM, we need to further refine our vaccine strategy, explore the combination with vaccines and other agents and achieve specialization and standardization of vaccine preparation to make it an effective clinical treatment.
MM is oncogene-dependent. Cytogenetics suggests that MM has a diverse genome, highlighted by structural rearrangements and copy number exceptions, and these exceptions determine the progression and final outcome of subsequent disease. Oncogenic mutations are more clonal while disordered driver genes represent a worse prognosis (112). Mutations in the coding region caused by genetic instability in carcinogenesis can cause amino acid sequence changes. New proteins, known as tumor antigens, can activate the body’s immune system and elicit effect immune responses (113). Increased expression or presentation of tumor antigens enhance tumor cells antigenicity. Based on their characteristics, tumor antigens are generally classified into three broad categories, i.e., tumor-associated antigens (TAAs), cancer testicular antigen (CTA) and tumor specific antigens (TSA). Most TAAs are embryonic antigens and might have induced immunological tolerance, making it difficult to develop a specific and durable immune response. Another risk of using TAAs as the immune target is the induction of autoimmunity against the corresponding normal tissues (114).
TSA, also known as neoantigen, is another effective target for tumor cell immunotherapy that has emerged in recent years. How neoantigen come to defined is still not well understood so far. Some researchers believe that these tumor neoantigens are generated by non-synonymous or other genetic changes (115). Another popular theory is that neoantigen come from peptides of tumor proteins with altered characteristics (116).
Somatic mutations caused by any mechanisms could generate tumor neoantigens. The RNA sequences from the MM Research Foundation CoMMpass Study were used for the identification of Intron retention (IR) events and the prediction of IR-neoantigens. It was found that high IR-neoantigens load was related to poor overall survival and unfavorable clinical outcome (117).
Wells et al. believe that reliable neoantigen predictions is dependent on the understanding of key parameters governing the immunogenic epitope (118). The prediction of neoantigen can be accomplished by using bioinformatics algorithms. Massively parallel sequencing (MPS) can identify specific somatic mutations by comparing DNA sequences of tumor cell and normal host cell origin. This process usually selects exon sequencing, in order to significantly reduce costs and complexity of analysis (114). However, due to human leukocyte antigen(HLA) restriction, not all mutations result in new epitopes that can be recognized by the immune system. Computer analysis is used to predict the affinity of new epitopes for HLA binding and to screen for neoantigens that most likely to induce potent T-cell immune response. Current predictions of antigenic epitopes focus on MHC I-binding epitopes, and the identification of major histocompatibility complex(MHC) II epitopes is complicated by the characteristics of MHC II peptide binding (119). From the outcomes of somatic mutations and gene-expression profiling, Jian et al. developed a neoantigen-prediction pipeline and constructed a neoantigen immune response score. This approach can be used to rapidly identify the new antigens created from somatic mutations and predict OS (120).
It is generally accepted that high somatic mutation rates are associated with increased genomic instability and reduced overall survival (121). The mutational load associated with MM is lower compared with other types of cancer. In a study involving 663 MM patients, a method combining exome sequencing and HLA binding prediction was used to determine mutational load and predict neoantigen load in MM patients. The mutational load was found to be directly proportional to the predicted neoantigen load. Survival analysis showed that PFS for the patients with higher somatic missense mutational load and predicted neoantigen load than mean values was significantly shortened and it was independent of disease stage and cytogenetic genetic abnormalities (122).
Permual et al. reported neoantigen-specific T cell responses in MM patients. This is the first study that experimentally validates the ability of triggering an immune response by neoantigens predicted from the next generation sequencing (NGS) in relapsed MM patients. Their data support that neoantigens are able to elicit cytotoxic CD8+ T cell activity in the setting of combination immunotherapy (123). Hence, neoantigens can form complex with human leucocyte antigen (HLA) molecules and presented to T cells, where the complex is recognized by TCR as “non-self”. Anti-tumor specific response is induced and not affected by central and peripheral immune tolerance and not causing damages to normal tissues. For this reason, tumor neoantigens are excellent targets for immunotherapy.
Studies have revealed that tumor-infiltrating lymphocytes(TILs) show strong neoantigen-killing ability once ICIs block the inhibitory signals from tumor cells, boosting the efficacy of ICIs (124). Strategies of combining targeted neoantigens can reduce relapse and side effects in patients undergoing bone marrow transplantation. Foglietta et al. immunized donors with recipient-derived Id, conjugated with KLH and observed anti-KLH and anti-Id cellular immune responses. Similarly, anti-neoantigen immune responses were detected in recipients after HSCT. These observations prove that neoantigen and tumor antigen-specific humoral and cellular immunity can be safely induced in HSCT donors and passively transferred to recipients (125).
It is well understood that tumor neoantigens are abundantly expressed in tumor cells, rarely expressed in normal somatic cells and are not subjected to thymic negative selection. Tumor antigens are natural targets for vaccine preparation. Bekri S. et al. experimentally proved that tumor antigen-specific CD4 (helper) T cells can provide protective antitumor immunity and against antigens that are not expressed by the vaccine through activating CD8 T cells (126). This may be associated with enhanced antigen cross-presentation by CD4+T, production of cytokines and upregulation of MHC II molecule protein (119).
Immunotherapy targeting neoantigens is an emerging strategy for personalized immunotherapy and has resulted in prolonged PFS and OS in clinical trials. However, these approaches still face a huge set of challenges such as the collection of more data on neoantigen mutations, the improvement of neoantigen prediction algorithms, and the exploration of optimal neoantigen targets. Studies on the combination of neoantigen-targeted therapies and other immunotherapies are being carried out and will definitely benefit more patients.
Sensitizes tumor cells to immune attack by enhancing the propensity of immune effector cells to recognize and kill tumor antigens. The main approach mentioned here is to enhance the expression of signals on the surface of tumor cells. The signaling molecules on the surface of tumor cells and their interaction with T cells determine whether T cells can be successfully activated to produce anti-tumor immunity.
The desire to generate effective anti-tumor immunity requires APCs present and process antigens to activate T cells, and differentiated antigen-specific CD8+ T cells migrate and persist in the tumor environment. APCs present antigens via the classical MHC class I and class II pathways. Besides, there is a cross-presentation pathway, which refers to the presentation of exogenous antigens on MHC I and is essential in anti-tumor responses and antigen tolerance (127).
In general, T cell activation requires three signals. TCR-CD3 + MHC I/II +CD8/CD4 is the first signal for T-cell activation. T cells interact with co-stimulatory molecules on the surface of the APC to generate a second signal (co-stimulatory signal) for activating various types of T cell responses. CD28 is a protein expressed as a homodimer on T cell and is the main family of co-stimulatory molecules, with receptor B7-1/2 (CD80/86) on APC (9). Besides, the cytokines produced during T cell activation act as a third signal to further promote T cell proliferation and differentiation and to generate immune memory, while IL-2 plays a key role in this process.
MM cells are generally considered to be weak APCs and express activating co-stimulatory signals on their surface, such as CD40,CD80/86,4-1BBL,OX40L,CD70,B7 relative protein1(B7RP1) molecules that binding to CD40L,CD28,4-1BB,OX40,CD27, inducible co-stimulator(ICOS) receptors on T cells, respectively (9). The inability of general tumor antigens to activate T cells are associated with low expression of activating co-stimulatory molecules (21, 128). Enhancing the expression of these signals and subsequently strengthening susceptibility to immune attacks appears to be a potential therapeutic approach.
DCs are thought to play a dual role and except uptaking and processing myeloma antigens and activating specific CD8+ T cells, DCs can also bind to CD28 molecules on the surface of non-apoptotic myeloma plasma cells through CD80/86 molecules, allowing myeloma plasma cells to evade killing by CD8+ T cells (129). Nair et al. demonstrated that CD28 expression may be associated with myeloma cell survival, induces IL-6 and IDO production upon binding to CD80/86 ligands, and is a target for myeloma therapy (130).
Another class of co-stimulatory molecules is the TNF/TNF receptor family. OX-40 ligand binds to the T cell surface receptor OX-40, leading to the expansion of CD4+ T cells (131). 4-1BBL binds to the T cell receptor not only induces the T cell activation, but also prevents apoptosis of activated T cell, enhancing the anti-tumor effect (132). MM cells express only the weaker 4-1BBL and B7 molecules on their surface. Transduction of B7-1 and 4-1BBL in MM cell lines makes it possible to activate and amplify T cell and stimulate anti-myeloma immune response (133). Given the ability of lysing viruses to enhance tumor cells immunogenicity and induce anti-tumor immune responses, Wenthe et al. infected myeloma cells with immunostimulatory Lokon lysing adenovirus (LOAd) carrying trimeric membrane-bound CD40L (LOAd700, LOAd703) and 4-1BBL (LOAd703).Their results showed that T cells are successfully activated (134).
CD40 is expressed on the surface of myeloma cells instead of on normal mature plasma cells. The interaction of CD40 with CD40L mediates the mechanism of angiogenesis in MM patients (135). CD40-CD40L pathway restores sensitivity to immune attack in malignant tumor cells associated with activation of the Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)-mediated signalling pathway (136). Thus, the CD40-CD40L pathway can effectively promote systemic anti-tumor immune responses, while targeting CD40 can directly inhibit tumor proliferation, and metastasis, and enhance tumor cells susceptibility. Advances in clinical trials and treatment of the CD40-CD40L pathway as an immune checkpoint have been discussed in detail in the review by Tang et al, and treatment targeting CD40/CD40L in MM still needs to be further explored (137).
Researching into the molecular mechanisms of immunodeficiency in MM provides more options for myeloma patients who are resistant to classical treatment. Nevertheless, due to the disease characteristics of myeloma itself, genomic and clonal evolution in the bone marrow immune microenvironment and the immune deficiency leading to eventual disease relapse, new treatment strategies are still imminent (138). As the continuous exploration of immunotherapy approaches, patients have led to prolonged PFS and overall remission rates. However, the problem of disease recurrence and treatment resistance still exists. Here we discuss the activation of specific anti-tumor immune responses mediated by targeting MM cells as a potential solution strategy to overcome the diminished effect of immunotherapy due to antigen loss. The therapeutic modality of targeting tumor cells is achieved by enhancing auto-immunogenicity of MM cells, increasing the expression of neoantigens and increasing the expression of co-stimulatory molecules. Some of these treatment strategies are still in the pre-clinical stage and lack large-scale clinical trials to prove that they can benefit the majority of patients.
In conclusion, we are summarized the therapeutic strategies to enhance the immune response based on multiple myeloma cells and these treatments have shown excellent clinical efficacy and deserve further exploration.
ZL: Conceptualization, Writing - Review and Editing, Project administration. CY: Writing - Original Draft, Visualization, XL: Resources. XX: Resources. XZ: Resources. RF: Supervision, Funding acquisition, Project administration. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China Youth Project (grant no.81900131,82000219), the National Natural Science Foundation of China Youth Project (grant no. 8210010129), the Tianjin Municipal Natural Science Foundation (grant no.18JCQNJC80400, 19JCZDJC32900), the Tianjin EducationCommission Research Project (grant no.2018KJ043,20140118),the Tianjin Education Commission Research Project (grant no. 2018KJ045), and the Tianjin Science and Technology Planning Project (no. 20YFZCSY00060).Tianjin Municipal Health Commission Youth Project (grant no. TJWJ2021QN001); Medjaden Academy & Research Foundation for Young Scientists (Grant No. MJR20221011);Tianjin Key Medical Discipline(Specialty) Construction project(GrantTJYXZDXK-028A).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Pinto V, Bergantim R, Caires HR, Seca H, Guimarães JE, Vasconcelos MH. Multiple myeloma: available therapies and causes of drug resistance. Cancers (2020) 12(2):407. doi: 10.3390/cancers12020407
2. Ackley J, Ochoa MA, Ghoshal D, Roy K, Lonial S, Boise LH. Keeping myeloma in check: the past, present and future of immunotherapy in multiple myeloma. Cancers (2021) 13(19):4787. doi: 10.3390/cancers13194787
3. Al Hamed R, Bazarbachi AH, Malard F, Harousseau J-L, Mohty M. Current status of autologous stem cell transplantation for multiple myeloma. Blood Cancer J (2019) 9(4):44. doi: 10.1038/s41408-019-0205-9
4. Vallet S, Pecherstorfer M, Podar K. Adoptive cell therapy in multiple myeloma. Expert Opin Biol Ther (2017) 17(12):1511–22. doi: 10.1080/14712598.2017.1375095
5. Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity (2013) 39(1):74–88. doi: 10.1016/j.immuni.2013.06.014
6. Krejcik J, Barnkob MB, Nyvold CG, Larsen TS, Barington T, Abildgaard N. Harnessing the immune system to fight multiple myeloma. Cancers (2021) 13(18):4546. doi: 10.3390/cancers13184546
7. Minnie SA, Hill GR. Immunotherapy of multiple myeloma. J Clin Invest (2020) 130(4):1565–75. doi: 10.1172/JCI129205
8. Rafei H, Haroun F, Tabbara IA. Novel immunotherapeutic agents for the treatment of multiple myeloma. Am J Clin Oncol (2019) 42(3):317–29. doi: 10.1097/COC.0000000000000506
9. Hoyos V, Borrello I. The immunotherapy era of myeloma: monoclonal antibodies, vaccines, and adoptive T-cell therapies. Blood (2016) 128(13):1679–87. doi: 10.1182/blood-2016-05-636357
10. Yousef S, Marvin J, Steinbach M, Langemo A, Kovacsovics T, Binder M, et al. Immunomodulatory molecule PD-L1 is expressed on malignant plasma cells and myeloma-propagating pre-plasma cells in the bone marrow of multiple myeloma patients. Blood Cancer J (2015) 5:e285. doi: 10.1038/bcj.2015.7
11. Trotta R, Dal Col J, Yu J, Ciarlariello D, Thomas B, Zhang X, et al. TGF-beta utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J Immunol Baltim Md 1950 (2008) 181(6):3784–92. doi: 10.4049/jimmunol.181.6.3784
12. Benson DM, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood (2010) 116(13):2286–94. doi: 10.1182/blood-2010-02-271874
13. Rutella S, Danese S, Leone G. Tolerogenic dendritic cells: cytokine modulation comes of age. Blood (2006) 108(5):1435–40. doi: 10.1182/blood-2006-03-006403
14. Brown R, Murray A, Pope B, Sze DM, Gibson J, Ho PJ, et al. Either interleukin-12 or interferon-gamma can correct the dendritic cell defect induced by transforming growth factor beta in patients with myeloma. Br J Haematol (2004) 125(6):743–8. doi: 10.1111/j.1365-2141.2004.04984.x
15. Bonanno G, Mariotti A, Procoli A, Folgiero V, Natale D, De Rosa L, et al. Indoleamine 2,3-dioxygenase 1 (IDO1) activity correlates with immune system abnormalities in multiple myeloma. J Transl Med (2012) 10:247. doi: 10.1186/1479-5876-10-247
16. Shay G, Hazlehurst L, Lynch CC. Dissecting the multiple myeloma-bone microenvironment reveals new therapeutic opportunities. J Mol Med Berl Ger (2016) 94(1):21–35. doi: 10.1007/s00109-015-1345-4
17. Botta C, Gullà A, Correale P, Tagliaferri P, Tassone P. Myeloid-derived suppressor cells in multiple myeloma: pre-clinical research and translational opportunities. Front Oncol (2014) 4:348. doi: 10.3389/fonc.2014.00348
18. García-Ortiz A, Rodríguez-García Y, Encinas J, Maroto-Martín E, Castellano E, Teixidó J, et al. The role of tumor microenvironment in multiple myeloma development and progression. Cancers (2021) 13(2):E217. doi: 10.3390/cancers13020217
19. Beider K, Bitner H, Leiba M, Gutwein O, Koren-Michowitz M, Ostrovsky O, et al. Multiple myeloma cells recruit tumor-supportive macrophages through the CXCR4/CXCL12 axis and promote their polarization toward the M2 phenotype. Oncotarget (2014) 5(22):11283–96. doi: 10.18632/oncotarget.2207
20. Chen H, Li M, Sanchez E, Soof CM, Bujarski S, Ng N, et al. JAK1/2 pathway inhibition suppresses M2 polarization and overcomes resistance of myeloma to lenalidomide by reducing TRIB1, MUC1, CD44, CXCL12, and CXCR4 expression. Br J Haematol (2020) 188(2):283–94. doi: 10.1111/bjh.16158
21. Leone P, Solimando AG, Malerba E, Fasano R, Buonavoglia A, Pappagallo F, et al. Actors on the scene: immune cells in the myeloma niche. Front Oncol (2020) 10:599098. doi: 10.3389/fonc.2020.599098
22. Richardson PG, Lonial S, Jakubowiak AJ, Harousseau J-L, Anderson KC. Monoclonal antibodies in the treatment of multiple myeloma. Br J Haematol (2011) 154(6):745–54. doi: 10.1111/j.1365-2141.2011.08790.x
23. Laubach JP, Paba Prada CE, Richardson PG, Longo DL. Daratumumab, elotuzumab, and the development of therapeutic monoclonal antibodies in multiple myeloma. Clin Pharmacol Ther (2017) 101(1):81–8. doi: 10.1002/cpt.550
24. Malaer JD, Mathew PA. CS1 (SLAMF7, CD319) is an effective immunotherapeutic target for multiple myeloma. Am J Cancer Res (2017) 7(8):1637–41.
25. Pazina T, James AM, MacFarlane AW, Bezman NA, Henning KA, Bee C, et al. The anti-SLAMF7 antibody elotuzumab mediates NK cell activation through both CD16-dependent and -independent mechanisms. Oncoimmunology (2017) 6(9):e1339853. doi: 10.1080/2162402X.2017.1339853
26. Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood (2016) 128(3):384–94. doi: 10.1182/blood-2015-12-687749
27. Overdijk MB, Verploegen S, Bögels M, van Egmond M, Lammerts van Bueren JJ, Mutis T, et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. mAbs (2015) 7(2):311–21. doi: 10.1080/19420862.2015.1007813
28. Lopes R, Ferreira BV, Caetano J, Barahona F, Carneiro EA, João C. Boosting immunity against multiple myeloma. Cancers (2021) 13(6):1221. doi: 10.3390/cancers13061221
29. van der Veer MS, de Weers M, van Kessel B, Bakker JM, Wittebol S, Parren PWHI, et al. Towards effective immunotherapy of myeloma: enhanced elimination of myeloma cells by combination of lenalidomide with the human CD38 monoclonal antibody daratumumab. Haematologica (2011) 96(2):284–90. doi: 10.3324/haematol.2010.030759
30. Plesner T, Arkenau H-T, Gimsing P, Krejcik J, Lemech C, Minnema MC, et al. Phase 1/2 study of daratumumab, lenalidomide, and dexamethasone for relapsed multiple myeloma. Blood (2016) 128(14):1821–8. doi: 10.1182/blood-2016-07-726729
31. Caraccio C, Krishna S, Phillips DJ, Schürch CM. Bispecific antibodies for multiple myeloma: a review of targets, drugs, clinical trials, and future directions. Front Immunol (2020) 11:501. doi: 10.3389/fimmu.2020.00501
32. Smits NC, Sentman CL. Bispecific T-cell engagers (BiTEs) as treatment of b-cell lymphoma. J Clin Oncol Off J Am Soc Clin Oncol (2016) 34(10):1131–3. doi: 10.1200/JCO.2015.64.9970
33. Shah Z, Malik MN, Batool SS, Kotapati S, Akhtar A, ur Rehman O, et al. Bispecific T-cell engager (BiTE) antibody based immunotherapy for treatment of relapsed refractory multiple myeloma RRMM): a systematic review of preclinical and clinical trials. Blood (2019) 134:5567. doi: 10.1182/blood-2019-129652
34. Wang Q, Chen Y, Park J, Liu X, Hu Y, Wang T, et al. Design and production of bispecific antibodies. Antibodies Basel Switz (2019) 8(3):43. doi: 10.3390/antib8030043
35. Hipp S, Tai Y-T, Blanset D, Deegen P, Wahl J, Thomas O, et al. Novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia (2017) 31(8):1743–51. doi: 10.1038/leu.2016.388
36. Cho S-F, Lin L, Xing L, Wen K, Yu T, Hsieh PA, et al. AMG 701 potently induces anti-multiple myeloma (MM) functions of T cells and IMiDs further enhance its efficacy to prevent MM relapse. In Vivo Blood (2019) 134:135. doi: 10.1182/blood-2019-128528
37. Li J, Stagg NJ, Johnston J, Harris MJ, Menzies SA, DiCara D, et al. Membrane-proximal epitope facilitates efficient T cell synapse formation by anti-FcRH5/CD3 and is a requirement for myeloma cell killing. Cancer Cell (2017) 31(3):383–95. doi: 10.1016/j.ccell.2017.02.001
38. Pillarisetti K, Edavettal S, Mendonça M, Li Y, Tornetta M, Babich A, et al. Gaudet, f. a T-Cell-Redirecting bispecific G-Protein-Coupled receptor class 5 member d x CD3 antibody to treat multiple myeloma. Blood (2020) 135(15):1232–43. doi: 10.1182/blood.2019003342
39. Cho S-F, Yeh T-J, Anderson KC, Tai Y-T. Bispecific antibodies in multiple myeloma treatment: a journey in progress. Front Oncol (2022) 12:1032775. doi: 10.3389/fonc.2022.1032775
40. Goldsmith SR, Streeter S, Covut F. Bispecific antibodies for the treatment of multiple myeloma. Curr Hematol Malig Rep (2022) 17(6):286–97. doi: 10.1007/s11899-022-00675-3
41. D’Agostino M, Raje N. Anti-BCMA CAR T-cell therapy in multiple myeloma: can we do better? Leukemia (2020) 34(1):21–34. doi: 10.1038/s41375-019-0669-4
42. Yang Y, Li Y, Gu H, Dong M, Cai Z. Emerging agents and regimens for multiple myeloma. J Hematol Oncol.J Hematol Oncol (2020) 13(1):150. doi: 10.1186/s13045-020-00980-5
43. Choi T, Kang Y. Chimeric antigen receptor (CAR) T-cell therapy for multiple myeloma. Pharmacol Ther (2022) 232:108007. doi: 10.1016/j.pharmthera.2021.108007
44. Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discovery (2018) 8(10):1219–26. doi: 10.1158/2159-8290.CD-18-0442
45. Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer (2021) 21(3):145–61. doi: 10.1038/s41568-020-00323-z
46. Quach H, Ritchie D, Stewart AK, Neeson P, Harrison S, Smyth MJ, et al. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia (2010) 24(1):22–32. doi: 10.1038/leu.2009.236
47. Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science (2014) 343(6168):301–5. doi: 10.1126/science.1244851
48. Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of ikaros proteins. Science (2014) 343(6168):305–9. doi: 10.1126/science.1244917
49. Davies F, Baz R. Lenalidomide mode of action: linking bench and clinical findings. Blood Rev (2010) 24 Suppl 1:S13–19. doi: 10.1016/S0268-960X(10)70004-7
50. Gandhi AK, Kang J, Havens CG, Conklin T, Ning Y, Wu L, et al. Immunomodulatory agents lenalidomide and pomalidomide Co-stimulate T cells by inducing degradation of T cell repressors ikaros and aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN.). Br J Haematol (2014) 164(6):811–21. doi: 10.1111/bjh.12708
51. Hideshima T, Ogiya D, Liu J, Harada T, Kurata K, Bae J, et al. Immunomodulatory drugs activate NK cells via both zap-70 and cereblon-dependent pathways. Leukemia (2021) 35(1):177–88. doi: 10.1038/s41375-020-0809-x
52. Dimopoulos MA, Leleu X, Palumbo A, Moreau P, Delforge M, Cavo M, et al. Expert panel consensus statement on the optimal use of pomalidomide in relapsed and refractory multiple myeloma. Leukemia (2014) 28(8):1573–85. doi: 10.1038/leu.2014.60
53. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
54. Tamura H, Ishibashi M, Yamashita T, Tanosaki S, Okuyama N, Kondo A, et al. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia (2013) 27(2):464–72. doi: 10.1038/leu.2012.213
55. Harjunpää H, Guillerey C. TIGIT as an emerging immune checkpoint. Clin Exp Immunol (2020) 200(2):108–19. doi: 10.1111/cei.13407
56. Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase ib study. J Clin Oncol Off J Am Soc Clin Oncol (2016) 34(23):2698–704. doi: 10.1200/JCO.2015.65.9789
57. Görgün G, Samur MK, Cowens KB, Paula S, Bianchi G, Anderson JE, et al. Lenalidomide enhances immune checkpoint blockade-induced immune response in multiple myeloma. Clin Cancer Res (2015) 21(20):4607–18. doi: 10.1158/1078-0432.CCR-15-0200
58. Usmani SZ, Schjesvold F, Oriol A, Karlin L, Cavo M, Rifkin RM, et al. KEYNOTE-185 investigators. pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): a randomised, open-label, phase 3 trial. Lancet Haematol (2019) 6(9):e448–58. doi: 10.1016/S2352-3026(19)30109-7
59. Mateos M-V, Blacklock H, Schjesvold F, Oriol A, Simpson D, George A, et al. KEYNOTE-183 investigators. pembrolizumab plus pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma (KEYNOTE-183): a randomised, open-label, phase 3 trial. Lancet Haematol (2019) 6(9):e459–69. doi: 10.1016/S2352-3026(19)30110-3
60. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer (2012) 12(12):860–75. doi: 10.1038/nrc3380
61. Serrano-del Valle A, Anel A, Naval J, Marzo I. Immunogenic cell death and immunotherapy of multiple myeloma. Front Cell Dev Biol (2019) 7:50. doi: 10.3389/fcell.2019.00050
62. Rufo N, Garg AD, Agostinis P. The unfolded protein response in immunogenic cell death and cancer immunotherapy. Trends Cancer (2017) 3(9):643–58. doi: 10.1016/j.trecan.2017.07.002
63. Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund A-C, Chapman DC, et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J (2009) 28(5):578–90. doi: 10.1038/emboj.2009.1
64. Sethuraman SN, Singh MP, Patil G, Li S, Fiering S, Hoopes PJ, et al. Novel calreticulin-nanoparticle in combination with focused ultrasound induces immunogenic cell death in melanoma to enhance antitumor immunity. Theranostics (2020) 10(8):3397–412. doi: 10.7150/thno.42243
65. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol (2013) 31:51–72. doi: 10.1146/annurev-immunol-032712-100008
66. Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ (2014) 21(1):79–91. doi: 10.1038/cdd.2013.75
67. Swanson KV, Deng M, Ting JP-Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
68. Fucikova J, Kepp O, Kasikova L, Petroni G, Yamazaki T, Liu P, et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis (2020) 11(11):1013. doi: 10.1038/s41419-020-03221-2
69. Hopfner K-P, Hornung V. Molecular mechanisms and cellular functions of CGAS-STING signalling. Nat Rev Mol Cell Biol (2020) 21(9):501–21. doi: 10.1038/s41580-020-0244-x
70. Gangaplara A, Martens C, Dahlstrom E, Metidji A, Gokhale AS, Glass DD, et al. Type I interferon signaling attenuates regulatory T cell function in viral infection and in the tumor microenvironment. PloS Pathog (2018) 14(4):e1006985. doi: 10.1371/journal.ppat.1006985
71. Arcuri LJ, Americo AD. Treatment of Relapsed/Refractory multiple myeloma in the bortezomib and lenalidomide era: a systematic review and network meta-analysis. Ann Hematol (2021) 100(3):725–34. doi: 10.1007/s00277-021-04404-3
72. Zitvogel L, Kroemer G. Bortezomib induces immunogenic cell death in multiple myeloma. Blood Cancer Discovery (2021) 2(5):405–7. doi: 10.1158/2643-3230.BCD-21-0059
73. Gulla A, Morelli E, Samur MK, Botta C, Hideshima T, Bianchi G, et al. Bortezomib induces anti–multiple myeloma immune response mediated by CGAS/STING pathway activation. Blood Cancer Discovery (2021) 2(5):468–83. doi: 10.1158/2643-3230.BCD-21-0047
74. Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med (2016) 375(8):754–66. doi: 10.1056/NEJMoa1606038
75. Moreau P, Attal M, Hulin C, Arnulf B, Belhadj K, Benboubker L, et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, phase 3 study. Lancet Lond Engl (2019) 394(10192):29–38. doi: 10.1016/S0140-6736(19)31240-1
76. Siegel DS, Martin T, Wang M, Vij R, Jakubowiak AJ, Lonial S, et al. Phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood (2012) 120(14):2817–25. doi: 10.1182/blood-2012-05-425934
77. Stewart AK, Rajkumar SV, Dimopoulos MA, Masszi T, Špička I, Oriol A, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med (2015) 372(2):142–52. doi: 10.1056/NEJMoa1411321
78. Dimopoulos MA, Moreau P, Palumbo A, Joshua D, Pour L, Hájek R, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol (2016) 17(1):27–38. doi: 10.1016/S1470-2045(15)00464-7
79. Liu Z, Xu X, Liu K, Zhang J, Ding D, Fu R. Immunogenic Cell Death in Hematological Malignancy Therapy. Adv Sci (Weinh) (2023) 10(13):e2207475. doi: 10.1002/advs.202207475
80. Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV. Bortezomib enhances dendritic cell (DC)–mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood (2007) 109(11):4839–45. doi: 10.1182/blood-2006-10-054221
81. Henry JY, Labarthe M-C, Meyer B, Dasgupta P, Dalgleish AG, Galustian C. Enhanced cross-priming of naive CD8+ T cells by dendritic cells treated by the IMiDs® immunomodulatory compounds lenalidomide and pomalidomide. Immunology (2013) 139(3):377–85. doi: 10.1111/imm.12087
82. De Beck L, Melhaoui S, De Veirman K, Menu E, De Bruyne E, Vanderkerken K, et al. Epigenetic treatment of multiple myeloma mediates tumor intrinsic and extrinsic immunomodulatory effects. OncoImmunology (2018) 7(10):e1484981. doi: 10.1080/2162402X.2018.1484981
83. Ahmed A, Tait SWG. Targeting immunogenic cell death in cancer. Mol Oncol (2020) 14(12):2994–3006. doi: 10.1002/1878-0261.12851
84. Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta (2010) 1805(1):53–71. doi: 10.1016/j.bbcan.2009.08.003
85. Osterborg A, Yi Q, Henriksson L, Fagerberg J, Bergenbrant S, Jeddi-Tehrani M, et al. Idiotype immunization combined with granulocyte-macrophage colony-stimulating factor in myeloma patients induced type I, major histocompatibility complex-restricted, CD8- and CD4-specific T-cell responses. Blood (1998) 91(7):2459–66. doi: 10.1182/blood.V91.7.2459
86. Rasmussen T, Hansson L, Osterborg A, Johnsen HE, Mellstedt H. Idiotype vaccination in multiple myeloma induced a reduction of circulating clonal tumor b cells. Blood (2003) 101(11):4607–10. doi: 10.1182/blood-2002-06-1925
87. Bocchia M, Defina M, Aprile L, Sicuranza A. Peptide vaccines for hematological malignancies: a missed promise? Int J Hematol (2014) 99(2):107–16. doi: 10.1007/s12185-013-1497-3
88. Roehnisch T, Then C, Nagel W, Blumenthal C, Braciak T, Donzeau M, et al. Phage idiotype vaccination: first phase I/II clinical trial in patients with multiple myeloma. J Transl Med (2014) 12:119. doi: 10.1186/1479-5876-12-119
89. Wang Y, Xiang Y, Xin VW, Wang X-W, Peng X-C, Liu X-Q, et al. Dendritic cell biology and its role in tumor immunotherapy. J Hematol Oncol.J Hematol Oncol (2020) 13 1):107. doi: 10.1186/s13045-020-00939-6
90. Verheye E, Bravo Melgar J, Deschoemaeker S, Raes G, Maes A, De Bruyne E, et al. Dendritic cell-based immunotherapy in multiple myeloma: challenges, opportunities, and future directions. Int J Mol Sci (2022) 23(2):904. doi: 10.3390/ijms23020904
91. Brown RD, Pope B, Murray A, Esdale W, Sze DM, Gibson J, et al. Dendritic cells from patients with myeloma are numerically normal but functionally defective as they fail to up-regulate CD80 (B7-1) expression after HuCD40LT stimulation because of inhibition by transforming growth factor-Beta1 and interleukin-10. Blood (2001) 98(10):2992–8. doi: 10.1182/blood.v98.10.2992
92. Nguyen-Pham T-N, Lee Y-K, Kim H-J, Lee J-J. Immunotherapy using dendritic cells against multiple myeloma: how to improve? Clin Dev Immunol (2012) 2012:397648. doi: 10.1155/2012/397648
93. Chung DJ, Pronschinske KB, Shyer JA, Sharma S, Leung S, Curran SA, et al. T-Cell exhaustion in multiple myeloma relapse after autotransplant: optimal timing of immunotherapy. Cancer Immunol Res (2016) 4(1):61–71. doi: 10.1158/2326-6066.CIR-15-0055
94. Lacy MQ, Mandrekar S, Dispenzieri A, Hayman S, Kumar S, Buadi F, et al. Idiotype-pulsed antigen-presenting cells following autologous transplantation for multiple myeloma may be associated with prolonged survival. Am J Hematol (2009) 84(12):799–802. doi: 10.1002/ajh.21560
95. Kitawaki T. DC-Based immunotherapy for hematological malignancies. Int J Hematol (2014) 99(2):117–22. doi: 10.1007/s12185-013-1496-4
96. Röllig C, Schmidt C, Bornhäuser M, Ehninger G, Schmitz M, Auffermann-Gretzinger S. Induction of cellular immune responses in patients with stage-I multiple myeloma after vaccination with autologous idiotype-pulsed dendritic cells. J Immunother Hagerstown Md 1997 (2011) 34(1):100–6. doi: 10.1097/CJI.0b013e3181facf48
97. Carmon L, Avivi I, Kovjazin R, Zuckerman T, Dray L, Gatt ME, et al. Phase I/II study exploring ImMucin, a pan-major histocompatibility complex, anti-MUC1 signal peptide vaccine, in multiple myeloma patients. Br J Haematol (2015) 169(1):44–56. doi: 10.1111/bjh.13245
98. van Rhee F, Szmania SM, Zhan F, Gupta SK, Pomtree M, Lin P, et al. NY-ESO-1 is highly expressed in poor-prognosis multiple myeloma and induces spontaneous humoral and cellular immune responses. Blood (2005) 105(10):3939–44. doi: 10.1182/blood-2004-09-3707
99. Batchu RB, Moreno AM, Szmania SM, Bennett G, Spagnoli GC, Ponnazhagan S, et al. Protein transduction of dendritic cells for NY-ESO-1-Based immunotherapy of myeloma. Cancer Res (2005) 65(21):10041–9. doi: 10.1158/0008-5472.CAN-05-1383
100. Hobo W, Strobbe L, Maas F, Fredrix H, Greupink-Draaisma A, Esendam B, et al. Immunogenicity of dendritic cells pulsed with MAGE3, survivin and b-cell maturation antigen MRNA for vaccination of multiple myeloma patients. Cancer Immunol Immunother CII (2013) 62(8):1381–92. doi: 10.1007/s00262-013-1438-2
101. Chung DJ, Sharma S, Rangesa M, DeWolf S, Elhanati Y, Perica K, et al. Langerhans dendritic cell vaccine bearing MRNA-encoded tumor antigens induces antimyeloma immunity after autotransplant. Blood Adv (2022) 6(5):1547–58. doi: 10.1182/bloodadvances.2021005941
102. Rosenblatt J, Avivi I, Vasir B, Uhl L, Munshi NC, Katz T, et al. Vaccination with dendritic Cell/Tumor fusions following autologous stem cell transplant induces immunologic and clinical responses in multiple myeloma patients. Clin Cancer Res (2013) 19(13):3640–8. doi: 10.1158/1078-0432.CCR-13-0282
103. Hong S, Li H, Qian J, Yang J, Lu Y, Yi Q. Optimizing dendritic cell vaccine for immunotherapy in multiple myeloma: tumour lysates are more potent tumour antigens than idiotype protein to promote anti-tumour immunity. Clin Exp Immunol (2012) 170(2):167–77. doi: 10.1111/j.1365-2249.2012.04642.x
104. Vasileiou S, Baltadakis I, Delimpasi S, Karatza M-H, Liapis K, Garofalaki M, et al. Ex vivo induction of multiple myeloma-specific immune responses by monocyte-derived dendritic cells following stimulation by whole-tumor antigen of autologous myeloma cells. J Immunother Hagerstown Md 1997 (2017) 40(7):253–64. doi: 10.1097/CJI.0000000000000182
105. Garfall AL, Stadtmauer EA. Cellular and vaccine immunotherapy for multiple myeloma. Hematol Am Soc Hematol Educ Program (2016) 2016(1):521–7. doi: 10.1182/asheducation-2016.1.521
106. Noonan K, Rudraraju L, Ferguson A, Emerling A, Pasetti MF, Huff CA, et al. Lenalidomide-induced immunomodulation in multiple myeloma: impact on vaccines and antitumor responses. Clin Cancer Res (2012) 18(5):1426–34. doi: 10.1158/1078-0432.CCR-11-1221
107. Nguyen-Pham T-N, Jung S-H, Vo M-C, Thanh-Tran H-T, Lee Y-K, Lee H-J, et al. Lenalidomide synergistically enhances the effect of dendritic cell vaccination in a model of murine multiple myeloma. J Immunother Hagerstown Md 1997 (2015) 38(8):330–9. doi: 10.1097/CJI.0000000000000097
108. Rosenblatt J, Glotzbecker B, Mills H, Vasir B, Tzachanis D, Levine JD, et al. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic Cell/Myeloma fusion vaccine. J Immunother Hagerstown Md 1997 (2011) 34(5):409–18. doi: 10.1097/CJI.0b013e31821ca6ce
109. Vo M-C, Jung S-H, Chu T-H, Lee H-J, Lakshmi TJ, Park H-S, et al. Lenalidomide and programmed death-1 blockade synergistically enhances the effects of dendritic cell vaccination in a model of murine myeloma. Front Immunol (2018) 9:1370. doi: 10.3389/fimmu.2018.01370
110. Chu T-H, Vo M-C, Park H-S, Lakshmi TJ, Jung S-H, Kim H-J, et al. Potent anti-myeloma efficacy of dendritic cell therapy in combination with pomalidomide and programmed death-ligand 1 blockade in a preclinical model of multiple myeloma. Cancer Immunol Immunother CII (2021) 70(1):31–45. doi: 10.1007/s00262-020-02654-0
111. Weinstock M, Rosenblatt J, Avigan D. Dendritic cell therapies for hematologic malignancies. Mol Ther Methods Clin Dev (2017) 5:66–75. doi: 10.1016/j.omtm.2017.03.004
112. Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies FE, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood (2018) 132(6):587–97. doi: 10.1182/blood-2018-03-840132
113. Zhang Z, Lu M, Qin Y, Gao W, Tao L, Su W, et al. Neoantigen: a new breakthrough in tumor immunotherapy. Front Immunol (2021) 12:672356. doi: 10.3389/fimmu.2021.672356
114. Yarchoan M, Johnson BA, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer (2017) 17(4):209–22. doi: 10.1038/nrc.2016.154
115. Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest (2015) 125(9):3413–21. doi: 10.1172/JCI80008
116. Sabdia MB, Patch A-M, Tsang H, Gandhi MK. Neoantigens - the next frontier in precision immunotherapy for b-cell lymphoproliferative disorders. Blood Rev (2022) 56:100969. doi: 10.1016/j.blre.2022.100969
117. Dong C, Cesarano A, Bombaci G, Reiter JL, Yu CY, Wang Y, et al. Intron retention-induced neoantigen load correlates with unfavorable prognosis in multiple myeloma. Oncogene (2021) 40(42):6130–8. doi: 10.1038/s41388-021-02005-y
118. Wells DK, van Buuren MM, Dang KK, Hubbard-Lucey VM, Sheehan KCF, Campbell KM, et al. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell (2020) 183(3):818–34. doi: 10.1016/j.cell.2020.09.015
119. Blass E, Ott PA. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol (2021) 18(4):215–29. doi: 10.1038/s41571-020-00460-2
120. Jian X, Xu L, Zhao J, Wang Y, Zhou W, Xie L. NAIRscore as a biomarker for the quality of immune response to neoantigens is related with an increased overall survival in multiple myeloma. Mol Ther Nucleic Acids (2022) 29:285–95. doi: 10.1016/j.omtn.2022.07.006
121. Walker BA, Boyle EM, Wardell CP, Murison A, Begum DB, Dahir NM, et al. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol (2015) 33(33):3911–20. doi: 10.1200/JCO.2014.59.1503
122. Miller A, Asmann Y, Cattaneo L, Braggio E, Keats J, Auclair D, et al. High somatic mutation and neoantigen burden are correlated with decreased progression-free survival in multiple myeloma. Blood Cancer J (2017) 7(9):e612. doi: 10.1038/bcj.2017.94
123. Perumal D, Imai N, Laganà A, Finnigan J, Melnekoff D, Leshchenko VV, et al. Mutation-derived neoantigen-specific T-cell responses in multiple myeloma. Clin Cancer Res (2020) 26(2):450–64. doi: 10.1158/1078-0432.CCR-19-2309
124. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature (2014) 515(7528):577–81. doi: 10.1038/nature13988
125. Foglietta M, Neelapu SS, Kwak LW, Jiang Y, Nattamai D, Lee S-T, et al. Neoantigen and tumor antigen-specific immunity transferred from immunized donors is detectable early after allogeneic transplantation in myeloma patients. Bone Marrow Transplant (2013) 48(2):269–77. doi: 10.1038/bmt.2012.132
126. Bekri S, Rodney-Sandy R, Gruenstein D, Mei A, Bogen B, Castle J, et al. Neoantigen vaccine-induced CD4 T cells confer protective immunity in a mouse model of multiple myeloma through activation of CD8 T cells against non-vaccine, tumor-associated antigens. J Immunother Cancer (2022) 10(2):e003572. doi: 10.1136/jitc-2021-003572
127. Kotsias F, Cebrian I, Alloatti A. Antigen processing and presentation. Int Rev Cell Mol Biol (2019) 348:69–121. doi: 10.1016/bs.ircmb.2019.07.005
128. Rutella S, Locatelli F. Targeting multiple-Myeloma-Induced immune dysfunction to improve immunotherapy outcomes. Clin Dev Immunol (2012) 2012:196063. doi: 10.1155/2012/196063
129. Leone P, Berardi S, Frassanito MA, Ria R, De Re V, Cicco S, et al. Dendritic cells accumulate in the bone marrow of myeloma patients where they protect tumor plasma cells from CD8+ T-cell killing. Blood (2015) 126(12):1443–51. doi: 10.1182/blood-2015-01-623975
130. Nair JR, Carlson LM, Koorella C, Rozanski CH, Byrne GE, Bergsagel PL, et al. CD28 expressed on malignant plasma cells induces a prosurvival and immunosuppressive microenvironment. J Immunol Baltim Md 1950 (2011) 187(3):1243–53. doi: 10.4049/jimmunol.1100016
131. Croft M. Co-Stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat Rev Immunol (2003) 3(8):609–20. doi: 10.1038/nri1148
132. Cheuk ATC, Mufti GJ, Guinn B. Role of 4-1BB:4-1BB ligand in cancer immunotherapy. Cancer Gene Ther (2004) 11(3):215–26. doi: 10.1038/sj.cgt.7700670
133. Lu Z-Y, Condomines M, Tarte K, Nadal L, Delteil MC, Rossi JF, et al. B7-1 and 4-1BB ligand expression on a myeloma cell line makes it possible to expand autologous tumor-specific cytotoxic T cells in vitro. Exp Hematol (2007) 35(3):443–53. doi: 10.1016/j.exphem.2006.11.002
134. Wenthe J, Naseri S, Hellström A-C, Wiklund HJ, Eriksson E, Loskog A. Immunostimulatory oncolytic virotherapy for multiple myeloma targeting 4-1BB and/or CD40. Cancer Gene Ther (2020) 27(12):948–59. doi: 10.1038/s41417-020-0176-9
135. Tsirakis G, Pappa CA, Psarakis FE, Fragioudaki M, Tsioutis C, Stavroulaki E, et al. Serum concentrations and clinical significance of soluble CD40 ligand in patients with multiple myeloma. Med Oncol Northwood Lond Engl (2012) 29(4):2396–401. doi: 10.1007/s12032-012-0203-2
136. Moschonas A, Kouraki M, Knox PG, Thymiakou E, Kardassis D, Eliopoulos AG. CD40 induces antigen transporter and immunoproteasome gene expression in carcinomas via the coordinated action of NF-KappaB and of NF-KappaB-Mediated de Novo synthesis of IRF-1. Mol Cell Biol (2008) 28(20):6208–22. doi: 10.1128/MCB.00611-08
137. Tang T, Cheng X, Truong B, Sun L, Yang X, Wang H. Molecular basis and therapeutic implications of CD40/CD40L immune checkpoint. Pharmacol Ther (2021) 219:107709. doi: 10.1016/j.pharmthera.2020.107709
Keywords: multiple myeloma, immunotherapy, immunogenic cell death, vaccine, neoantigen
Citation: Liu Z, Yang C, Liu X, Xu X, Zhao X and Fu R (2023) Therapeutic strategies to enhance immune response induced by multiple myeloma cells. Front. Immunol. 14:1169541. doi: 10.3389/fimmu.2023.1169541
Received: 19 February 2023; Accepted: 08 May 2023;
Published: 18 May 2023.
Edited by:
Ngoc Tung Tran, Indiana University, United StatesReviewed by:
Simrit Parmar, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2023 Liu, Yang, Liu, Xu, Zhao and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rong Fu, ZnVyb25nODM2OUB0bXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.