
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 14 April 2023
Sec. Immunological Memory
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1168125
 Tom Hofland1
Tom Hofland1 Luca Danelli1†Georgina Cornish1†
Luca Danelli1†Georgina Cornish1† Tiziano Donnarumma1†Deborah M. Hunt2Luiz P. S. de Carvalho2†
Tiziano Donnarumma1†Deborah M. Hunt2Luiz P. S. de Carvalho2† George Kassiotis1,3*
George Kassiotis1,3*CD4+ T cells are typically considered as ‘helper’ or ‘regulatory’ populations that support and orchestrate the responses of other lymphocytes. However, they can also develop potent granzyme (Gzm)-mediated cytotoxic activity and CD4+ cytotoxic T cells (CTLs) have been amply documented both in humans and in mice, particularly in the context of human chronic infection and cancer. Despite the established description of CD4+ CTLs, as well as of the critical cytotoxic activity they exert against MHC class II-expressing targets, their developmental and memory maintenance requirements remain elusive. This is at least in part owing to the lack of a murine experimental system where CD4+ CTLs are stably induced. Here, we show that viral and bacterial vectors encoding the same epitope induce distinct CD4+ CTL responses in challenged mice, all of which are nevertheless transient in nature and lack recall properties. Consistent with prior reports, CD4+ CTL differentiation is accompanied by loss of TCF-1 expression, a transcription factor considered essential for memory T cell survival. Using genetic ablation of Tcf7, which encodes TCF-1, at the time of CD4+ T cell activation, we further show that, contrary to observations in CD8+ T cells, continued expression of TCF-1 is not required for CD4+ T cell memory survival. Whilst Tcf7-deficient CD4+ T cells persisted normally following retroviral infection, the CD4+ CTL subset still declined, precluding conclusive determination of the requirement for TCF-1 for murine CD4+ CTL survival. Using xenotransplantation of human CD4+ T cells into murine recipients, we demonstrate that human CD4+ CTLs develop and persist in the same experimental conditions where murine CD4+ CTLs fail to persist. These observations uncover a species-specific defect in murine CD4+ CTL persistence with implications for their use as a model system.
CD4+ T cells are classically defined as ‘helper’ populations that can differentiate into several different T helper subsets depending on the immune stimulus and environment (1, 2). Although CD4+ T cells were initially thought of as orchestrators of immune responses via the production of specific cytokines, more recently a subset of CD4+ T cells with cytotoxic potential has been described in both mice and human (3, 4). These CD4+ cytotoxic T cells (CTLs) display both lytic granule (via granzyme B (GzmB) and perforin) as well as Fas/FasL mediated cytotoxicity in an MHC class II-dependent manner, and develop especially in the context of prolonged antigen stimulation, such as chronic viral infection and cancer (3–7). Indeed, peptide-specific cytotoxic responses by CD4+ CTLs have been described towards multiple viruses and cancers (5, 7–12).
Because CD4+ CTLs can provide cytotoxic capacity, additionally to conventional NK cells and CD8+ T cells and independently of MHC class I-mediated presentation, there has been a growing interest in including or targeting CD4+ CTLs for immunotherapeutic and vaccine strategies. Consequently, since their discovery, there have been several studies that looked into the factors required for CD4+ CTL development. It has been shown that loss of the transcription factor ThPOK and upregulation of Runx3 can drive CD4+ CTL differentiation (13–15). This transcriptional reprogramming resembles that of CD8+ versus CD4+ lineage commitment during T cell development and induces expression of more genes classically involved in cytotoxic CD8+ T cell differentiation, including CD8α, T-bet, Eomes, and GzmB (13–16). Other factors that have been shown to stimulate CD4+ CTL differentiation include signaling via 4-1BB, OX40, and IL-2 receptors (17–21). In contrast, the T follicular helper (Tfh) differentiation program antagonizes CD4+ CTL differentiation (via upregulation of the transcription factor Bcl6), as does signalling mediated by the inhibitory receptors PD-1 and LAG3 (16).
Despite these studies that looked into the priming of CD4+ CTLs, there is less known about the longevity and memory formation of this population. In CD8+ T cells, cytotoxic differentiation is antagonistic with longevity and memory formation (22, 23). The transcription factor TCF-1 has been shown to be essential for CD8+ T cell memory formation, and the downregulation of TCF-1 in cytotoxic CD8+ T cell effector cells reduces their longevity and survival (24). Since CD4+ CTLs share some transcriptional programming with CD8+ T cells, including loss of TCF-1 expression, it is theoretically possible that induction of a cytotoxic program antagonizes the long-term survival of CD4+ CTLs in a similar fashion. A better understanding of the longevity and maintenance of CD4+ CTLs is required to determine their immunotherapeutic potential.
Here, we studied the long-term survival of cytotoxic CD4+ T cells during chronic infection. We report that GzmB+ CD4+ T cell populations are not stable in mice, and that cytotoxic differentiation hampers long-term persistence of CD4+ T cells. Surprisingly, we found that continued expression of the transcription factor TCF-1 plays no role in either memory formation or the persistence of CD4+ T cells. Finally, we found human CD4+ CTLs show a higher degree of long-term persistence, indicating a species-specific difference in CD4+ CTL survival.
Inbred C57BL/6J (B6), CD45.1+ congenic B6 (B6.SJL-Ptprca Pep3b/BoyJ) and Rag2-/-Il2rg-/-Cd47-/- compound deficient mice were originally obtained from The Jackson Laboratory (Bar Harbor, ME, USA). TCRβ-transgenic EF4.1 mice (25), Rag1-deficient (Rag1-/-) mice (26), Rag2-deficient (Rag2-/-) mice (27), GzmB-tdTomato reporter mice (28), Tnfrsf4Cre mice (29), Gt(ROSA)26SorYFP (R26YFP) reporter mice (30), and mice with a Cre-conditional Tcf7 allele (Tcf7tm1c(EUCOMM)Wtsi), referred to here as Tcf7fl/fl mice (31), were previously described and were maintained on the B6 genetic background at the Francis Crick Institute’s animal facilities. Mice with activation-induced deletion of Tcf7 in antigen-specific CD4+ T cells were obtained by combining the EF4.1, Tnfrsf4Cre, R26YFP and Tcf7fl/fl alleles, in a compound mutant strain. All animal experiments were approved by the ethical committee of the Francis Crick Institute, and conducted according to local guidelines and UK Home Office regulations under the Animals Scientific Procedures Act 1986 (ASPA).
Single-cell suspensions were prepared from the spleens of donor CD45.1+ or CD45.2+ TCRβ-transgenic EF4.1 mice and CD4+ T cells were enriched using an immunomagnetic positive selection kit (StemCell Technologies, Vancouver, Canada), at >90% purity. Donor transgenic CD4+ T cells (1×106 per recipient) were injected into recipient mice intravenously.
The Friend virus (FV) used in this study was a retroviral complex of a replication-competent B-tropic F-MLV (F-MLV-B) and a replication-defective spleen-focus forming virus (SFFV). Stocks were prepared as previously described (32). Mice were injected intravenously with 0.1 mL PBS containing 1,000 spleen focus-forming units (SFFU) of FV. Ad5.pIX-gp70 stocks were prepared at a titer of 9×109 viral genomes ml-1 by infection of 293A cells as previously described (33). Approximately 5×108 Ad5.pIX- gp70 viral genomes per mouse were administered intravenously. Recombinant Mycobacterium bovis bacille Calmette-Guérin (BCG) was generated by inserting the sequence encoding the F-MLV gp70 glycoprotein env122-141 epitope into the sodA gene, using previously described methodology (34). BCG.SOD-env122-141 was administered intravenously. Immunization with FBL-3 tumor cells was carried out by intravenous injection of 1.5×106 FBL-3 cells (35). For peptide immunization, mice received an intraperitoneal injection of a total of 12.5 nmol of synthetic env122-141 peptide mixed in Sigma Adjuvant System (Sigma-Aldrich, St. Louis, Missouri, USA).
The pMX plasmid encoding constitutively active STAT5a protein was kindly provided by Ben Seddon. Transfection of the Platinum-E retroviral packaging cell line and retroviral transduction of CD4+ T cells were performed as described previously (36).
Spleen and lung single-cell suspensions were stained for 20 min at room temperature or at 4°C with directly conjugated antibodies to surface markers. For detection of intracellular antigens, subsequent to surface staining, cells were fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific, Waltham, Massachusetts, USA) according to the manufacturer’s instructions. They were then incubated for 45 min at room temperature with directly conjugated antibodies to intracellular antigens. Zombie UV Fixable Viability Kit (BioLegend, San Diego, California, USA) or Fixable Live/Dead Near IR Dead Cell Stain kit (Invitrogen, Waltham, Massachusetts, USA) was used to label and exclude dead cells from analysis. The following anti-mouse antibodies were used: BV785-, BUV395- or BV711-anti-CD4 (clone GK1.5), PE/Cy7-, PerCP-Cy5.5-, or eFluor450-anti-CD45.1 (clone A20), PE/Cy7-anti-CD279 (PD-1, clone 29F.1A12), V500-anti-CD44 (clone IM7), BV421- or PerCP-Cy5.5-anti-CD162 (PSGL1, clone 2PH1), PE-anti-Bcl6 (clone K112-91), APC-eFluor-780 or BV711-anti-CD45.2 (clone 104), PE-anti-CD223 (LAG3, clone eBioC9B7W), FITC-, PerCP-Cy5.5-, AF700- or APC-anti-TCRβ (clone H57-597) (from Thermo Fisher Scientific, Waltham, Massachusetts, USA); Alexa(R)488- or Alexa(R)647-anti-TCF1 (clone C63D9) (from Cell Signaling Technology, Danvers, Massachusetts, USA), Alexa Fluor 647-anti-ThPOK (clone T43-94, BD Biosciences). For CXCR5 staining, splenocytes were incubated with biotin rat anti-mouse CXCR5 antibody (clone 2G8, BD Biosciences) at 37°C for 25 min, followed by incubation with APC- or PE-streptavidin (BioLegend) for 20 min at room temperature. Multi-colour cytometry was performed on LSRFortessa flow cytometers (from BD Biosciences), and analyzed with FlowJo v10.8.1 (Tree Star Inc., Ashland, OR, USA).
Statistical comparisons were made using SigmaPlot 13.0 (Systat Software Inc., Germany) or GraphPad Prism 9 (GraphPad Software, La Jolla, CA 92037 USA). Parametric comparisons of normally distributed values that satisfied the variance criteria were made by unpaired Student’s t-tests or One Way Analysis of variance (ANOVA) tests. Data that did not pass the variance test were compared with non-parametric two-tailed Mann-Whitney Rank Sum tests or ANOVA on Ranks tests. P values are indicated by asterisks as follows: * p<0.05; ** p<0.01; *** p<0.001, **** p<0.0001. Data were plotted as the mean ± standard error mean.
We have previously described an adoptive transfer system that allows the study of CD4+ T cell responses to a dominant H2-Ab-restricted env122-141 epitope within the F-MLV gp70 glycoprotein (16, 33, 37). In order to study CD4+ CTL differentiation and memory formation, we followed the response of CD4+ T cells to FV and various vectors of the env122-141 epitope, including Ad5.pIX-gp70 and BCG (which causes persistent infection). CD4+ CTLs were identified by staining for GzmB expression and separated from CD4+ Tfh counterparts by co-staining for TCF-1 expression (Figure 1A). We had previously demonstrated that GzmB+ CD4+ T cells generated following FV infection or Ad5.pIX-gp70 immunization in this model lose TCF-1 expression and exhibit GzmB-mediated cytotoxic activity against MHC class II-expressing targets, whereas CD4+ T cells that retain TCF-1 expression bear all the hallmarks of Tfh cells, including co-expression of CXCR5, BCL6 and PD-1, lack of PSGL1 and SLAM expression, and localization in B cell follicles and germinal centers (16, 38, 39). All immunizations gave rise to a GzmB+ CD4+ T cell population of varying sizes in the spleens of recipient mice, but in all cases this population was quickly lost in the weeks following the peak of the response (Figures 1A, B). BCG induced the biggest GzmB+ CD4+ T cell population and the most pronounced and persistent loss of TCF-1 in all CD4+ T cells, which was evident in ~50% of the cells in the chronic phase of infection (Figure 1B). Nevertheless, only 5% of EF4.1 CD4+ T cells expressed GzmB in the chronic phase of BCG infection (Figure 1B). The loss of GzmB+ CD4+ T cells from the spleens could not be explained by migration of GzmB+ CD4+ T cells to effector sites as similar loss of GzmB+ CD4+ T cells was observed within env-reactive CD4+ T cells also in the lung of FV and BCG infected mice (Figure S1).
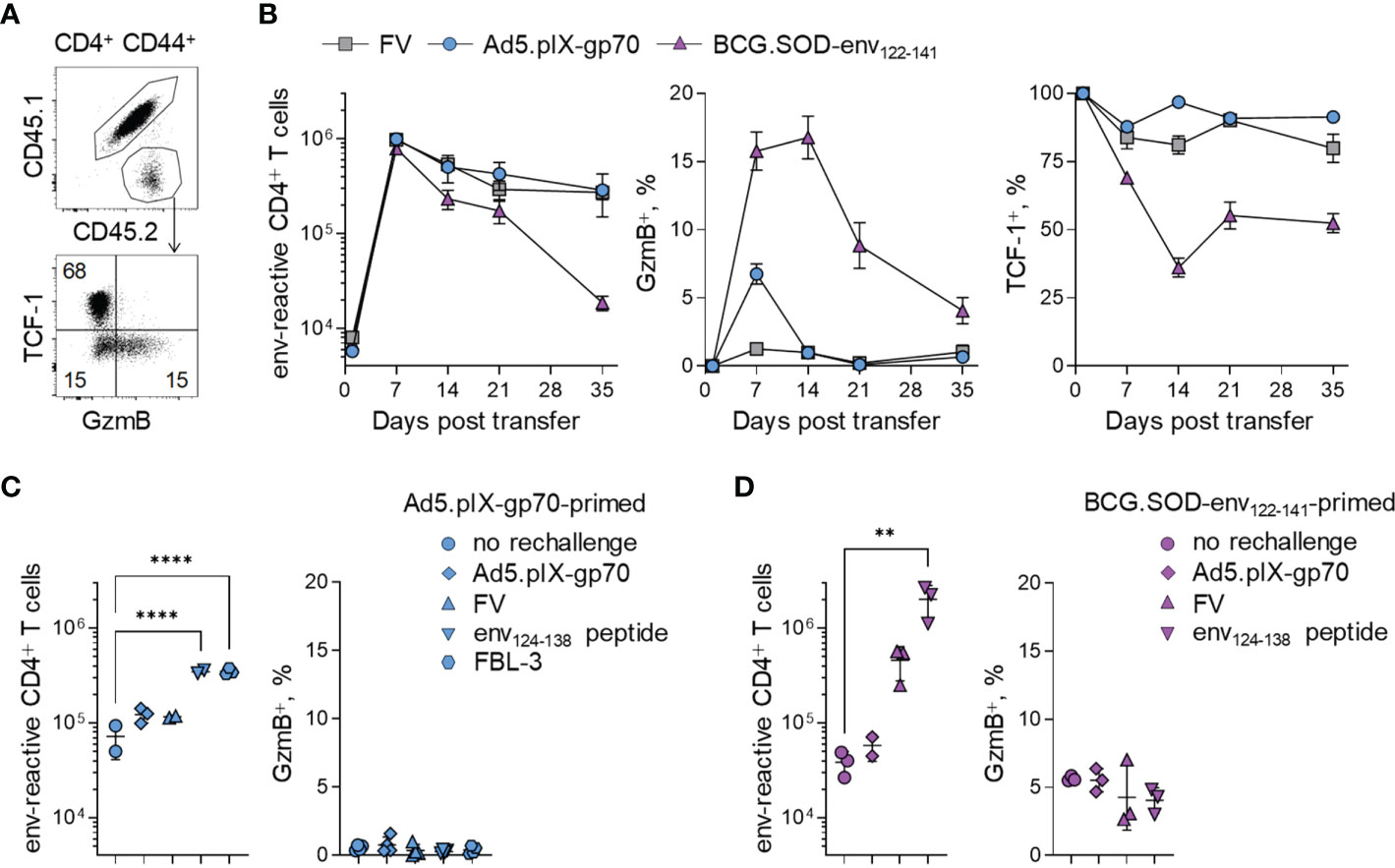
Figure 1 Persistence of env-reactive CD4+ CTLs following diverse challenges. (A) Flow-cytometric detection of CD44+ env-reactive donor CD4+ T cells (top) and TCF-1+ and GzmB+ CD4+ T cells (bottom) in spleens of WT recipient mice. (B) Absolute numbers (left), GzmB expression (middle) and TCF-1 expression (right) of env-reactive donor CD4+ T cells in the spleens of WT recipient mice at indicated time points after FV infection, or immunization with Ad5-pIX-gp70 or BCG.SOD-env122-141. (C) Absolute numbers (left) and GzmB expression (right) of env-reactive donor CD4+ T cells in the spleens of Ad5.pIX-gp70-primed mice at day 7 after re-challenge with indicated stimuli. (D) Absolute numbers (left) and GzmB expression (right) of env-reactive donor CD4+ T cells in the spleens of BCG.SOD-env122-141-primed mice at day 7 after re-challenge with indicated stimuli. **p < 0.01; ****p < 0.0001.
The lack of GzmB in memory CD4+ T cell populations could result either from loss of GzmB expression over time within memory CD4+ T cells, or the specific loss of GzmB+ CD4+ T cells themselves. To differentiate between the two possibilities, we rechallenged Ad5-pIX-gp70 or BCG.SOD-env122-141 immunized mice using different stimuli. We employed both homologous and heterologous rechallenges, including env122-138 peptide-based immunization, as heterologous rechallenges would not be affected by prior CD8+ T cell or antibody mediated immunity to the vectors and would recreate the priming environment. Although some stimuli induced bigger CD4+ T cell recall expansions, none of the rechallenges gave a GzmB recall response (Figures 1C, D, S2), indicating that a GzmB+ CD4+ T cell memory population had not formed following primary immunization.
To test more directly whether GzmB+ CD4+ T cells can seed a memory population, we transferred EF4.1 T cells into FV infected or uninfected Rag1-/- mice, deficient in T and B cells. Rag1-/- recipient mice that were not deliberately infected with FV carried related infectious murine leukemia viruses (MLVs), arising from recombination of defective endogenous retroviruses (40). These naturally occurring MLVs can also activate the EF4.1 T cells, albeit with lower avidity (41). In this setting, the reactive CD4+ T cells generated a very strong CTL response, with ~40% and ~60% of them producing GzmB at the peak of the response in FV infected or uninfected Rag1-/- recipients, respectively (Figures 2A, B). This heightened CTL differentiation seen in Rag1-/- recipients was due to the absence of competing CD8+ T cells and suppressive Treg cells, as previously described (39, 42). Nevertheless, similarly to those in WT recipients, GzmB+ CD4+ T cells disappeared over time in Rag1-/- recipients, despite persistent infection and TCF-1 loss (Figures 2A, B). To follow the fate of GzmB+ effector CD4+ T cells, we used a GzmB-tdTomato reporter to mark EF4.1 T cells (16, 28). Following transfer into primary FV-infected Rag1-/- recipients, GzmB-tdTomato+ and GzmB-tdTomato- effector CD4+ T cells were isolated at the peak of the response and re-transferred into secondary FV-infected Rag1-/- recipients (Figure 2C). Transferred GzmB-tdTomato+ CD4+ T cells showed no secondary expansion in secondary recipients and gradually lost GzmB expression over time. In contrast, GzmB-tdTomato- CD4+ T cells expanded considerably after transfer and gave a transient CTL response that was lost again over time, following kinetics similar to the primary response (Figures 2D, E). These results suggested that acquisition of the cytotoxic program by CD4+ T cells occurs only during the effector response and is incompatible with long-term persistence.
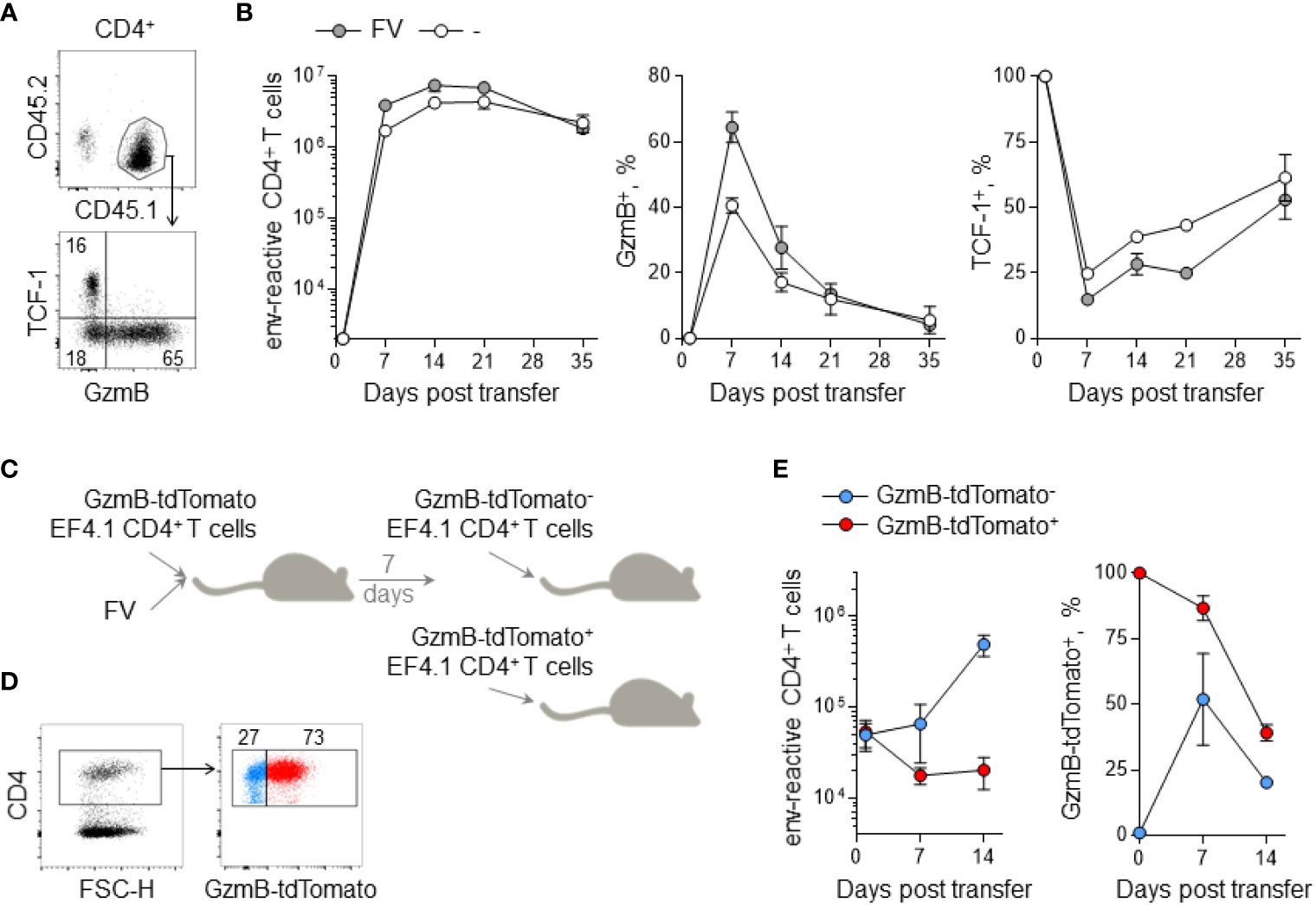
Figure 2 Persistence of env-reactive CD4+ CTLs in lymphocyte-deficient hosts. (A) Flow-cytometric detection of CD44+ env-reactive donor CD4+ T cells (top) and TCF-1+ and GzmB+ CD4+ T cells (bottom) in spleens of Rag1-/- recipient mice. (B) Absolute numbers (left), GzmB expression (middle) and TCF-1 expression (right) of env-reactive donor CD4+ T cells in the spleens of Rag1-/- recipient mice at indicated time points after adoptive transfer with or without FV infection. (C) EF4.1 GzmB-tdTomato CD4+ T cells were adoptively transferred into FV-infected Rag1-/- recipient mice. After 7 days, donor CD4+ T cells were sorted into either GzmB-tdTomato- or GzmB-tdTomato+ fractions and injected into new Rag1-/- recipient mice. (D) Flow-cytometric detection of GzmB-tdTomato+ donor CD4+ T cells for cell sorting. (E) Absolute numbers (left) and GzmB expression (right) of env-reactive donor CD4+ T cells in the spleens of Rag1-/- recipient mice at indicated time points after secondary adoptive transfer.
Expression of the cytotoxic program by CD4+ T cells during the effector but not the memory phase of the response to the diverse antigenic stimuli we have used here indicated that this program may rely on factors that were available only during the effector response. One such factor is IL-2, which is produced by effector CD4+ T cells. Indeed, IL-2 signaling has been reported to promote CD4+ CTL differentiation (20, 21). Furthermore, IL-2 signalling during CD4+ T cell priming has been shown to play an important role in memory formation (43, 44). Consistent with published reports, stimulation of GzmB-tdTomato EF4.1 T cells with IL-2 and IL-12 led to substantially increased GzmB expression (Figure S3A). It was therefore possible that reduced GzmB production at later time-points during the in vivo response or during recall responses was due to low IL-2 availability. To test if IL-2 signaling could increase the lifespan of GzmB+ CD4+ T cells, we transduced GzmB-tdTomato reporter EF4.1 T cells with a plasmid vector encoding constitutively active STAT5a (caSTAT5a) to mimic continuous IL-2 signaling. In FV infected Rag1-/- recipients, caSTAT5a-expressing CD4+ T cells showed similar expansion dynamics but increased GzmB expression compared to non-transduced CD4+ T cells at all time-points (Figures 3A, B). However, despite consistently inducing higher levels of GzmB expression compared to non-transduced cells, GzmB expression in caSTAT5a-expressing CD4+ T cells was lost over time with similar kinetics to non-transduced CD4+ T cells (Figure 3B). Similar results were obtained in WT recipient mice, although caSTAT5a-expressing CD4+ T cells were specifically lost over time compared to non-transduced CD4+ T cells (Figures 3C, S3B, E). These results indicated that IL-2 signalling boosted CD4+ CTL differentiation and GzmB expression, but it did not increase the stability or lifespan of GzmB+ CD4+ T cells.
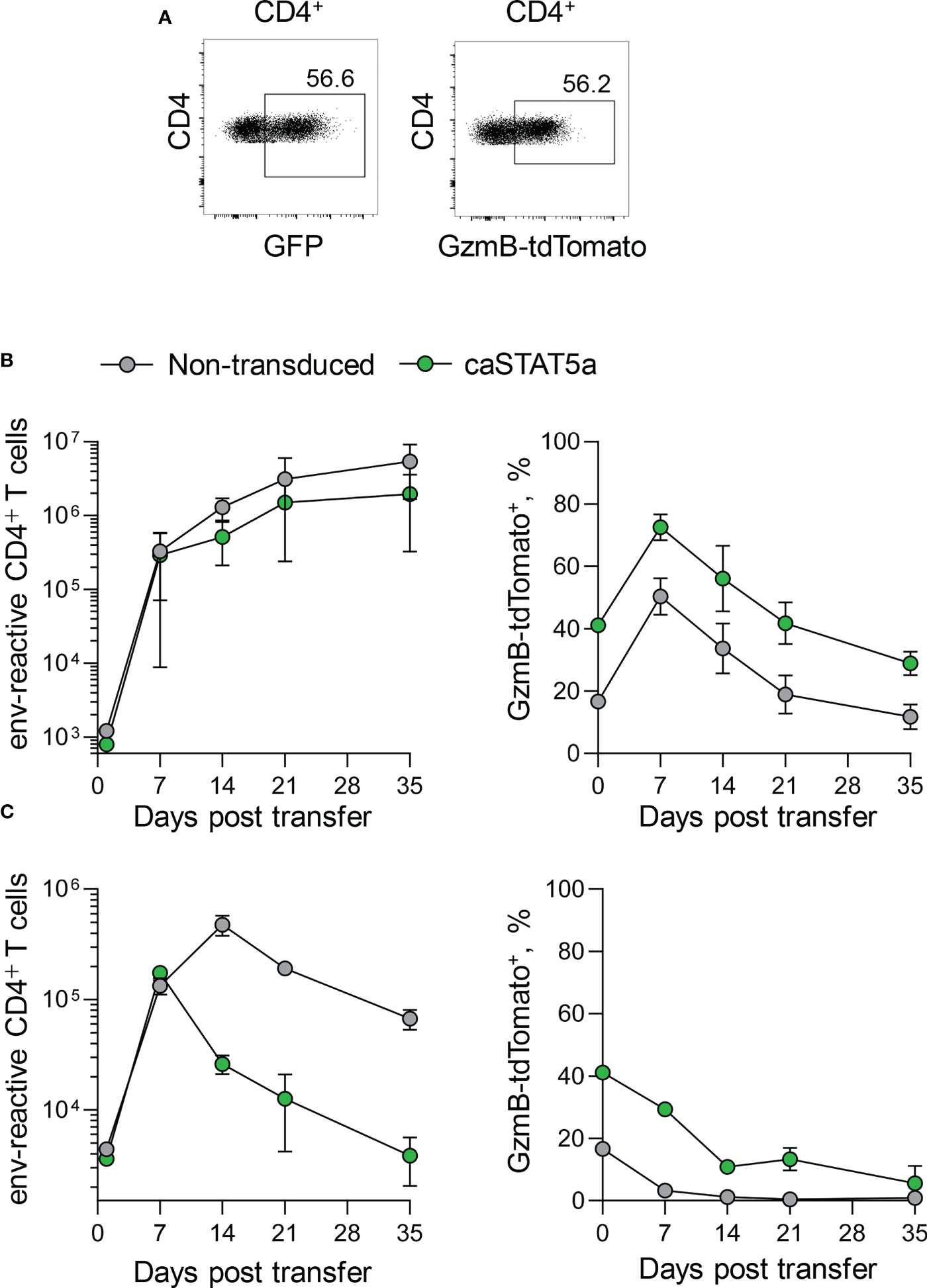
Figure 3 STAT5-dependence of GzmB expression in CD4+ T cells. (A) Flow-cytometric detection of GFP+ caSTAT5a-transduced (top) and GzmB-tdTomato+ (bottom) env-reactive donor CD4+ T cells in spleens of Rag1-/- recipient mice. (B) Absolute numbers (left) and GzmB-tdTomato expression (right) of non-transduced and caSTAT5a-transduced env-reactive donor CD4+ T cells in the spleens of Rag1-/- recipient mice at indicated time points after adoptive transfer. (C) Absolute numbers (left) and GzmB-tdTomato expression (right) of non-transduced and caSTAT5a-transduced env-reactive donor CD4+ T cells in the spleens of WT recipient mice at indicated time points after adoptive transfer.
An alternative explanation for the transient nature of the CD4+ CTL response we observed is that this transcriptional program requires loss of transcription factors thought to be required for CD4+ memory differentiation, such a ThPOK and TCF-1. Indeed, ThPOK downregulation has been shown to be associated with CD4+ CTL differentiation, and ThPOK deficient CD4+ T cells express more GzmB (13–15). However, loss of ThPOK is also associated with reduced memory formation in CD4+ T cells (45). Therefore, if loss or reduction in ThPOK expression were required for CD4+ CTL development, it could also compromise memory formation by GzmB+ CD4+ T cells in our model. To investigate if reduced ThPOK expression specifically in GzmB+ CD4+ T cells could underlie their reduced persistence, we measured ThPOK expression in EF4.1 CD4+ T cells after adoptive transfer in FV infected Rag1-/- mice by flow cytometry. At the peak of the CD4+ T cell response (day 7), we observed no difference in ThPOK expression between TCF-1+ and GzmB+ CD4+ T cells (Figure S4). Lack of ThPOK downregulation in this system indicated that reduction in ThPOK expression was neither required for the development of GzmB+ CD4+ T cells nor could it account for their transient nature.
GzmB+ CD4+ T cells are characterized by a number of transcriptional changes (13, 15, 16). These include the acquisition of CD8+ T cell markers, and the loss of TCF-1 (encoded by Tcf7). Although a role for continuous Tcf7 expression for T cell memory formation has not been investigated in CD4+ T cells at the level of detail that has been for CD8+ T cells, it is suggested in at least two experimental settings (46) and maintenance of TCF-1 expression in effector CD4+ T cells has been linked with enhance self-renewal capacity (47). It was therefore plausible that, firstly, TCF-1 was necessary for survival of all CD4+ memory T cells and, secondly, its loss specifically in GzmB+ CD4+ T cells was responsible for their reduced persistence into memory.
In order to investigate the role of TCF-1 in GzmB+ CD4+ T cells, we conditionally deleted Tcf7 upon CD4+ T cell activation using mice with a Cre-conditional Tcf7 allele (Tcf7fl/fl) crossed with mice expressing Cre under the control of the Tnfrsf4 locus (Tnfrsf4Cre), referred to here as Tcf7 activation-induced deletion (Tcf7AD) mice. Tcf7AD mice were additionally crossed with mice with a Cre-conditional YFP reporter allele (R26YFP mice) and with EF4.1 TCRβ transgenic mice, to allow tracking of Cre-mediated recombination and antigen-specific CD4+ T cells, respectively.
We first analyzed the degree of Cre-mediated recombination and potential effects on CD4+ T cell numbers and composition in naïve Tcf7AD mice, using littermates with one functional Tcf7 allele (Tcf7fl/wt) as controls. As previously described for the activity of the Tnfrsf4Cre driver in this system (37), a majority of memory-phenotype CD44+CD4+ T cells (60-70%) and of CD25+ Treg cells (80-90%) expressed YFP, indicating they had expressed Tnfrsf4, whereas only a minority of naïve CD44-CD25-CD4+ T cells (<10%) expressed YFP in control Tcf7fl/wt mice (Figure S5). Unexpectedly, these percentages were unaltered by deletion of Tcf7 in Tcf7fl/fl mice, arguing against an essential role for TCF-1 in maintaining the activated populations (Figure S5). Indeed, absolute numbers of naïve, memory-phenotype and Treg cells in the spleen or lymph nodes were indistinguishable between Tcf7fl/fl and control Tcf7fl/wt mice (Figure S5).
To examine a possible role for TCF-1 in a well-defined cohort of antigen-specific CD4+ T cells, we used Tcf7AD EF4.1 CD4+ T cells as donors for adoptive transfer into FV-infected recipients. Following transfer into Rag1-/- recipients, Tcf7AD CD4+ T cells showed comparable primary expansion, survival and memory formation when compared with WT EF4.1 CD4+ T cells, which were used as controls (Figures 4A, B).
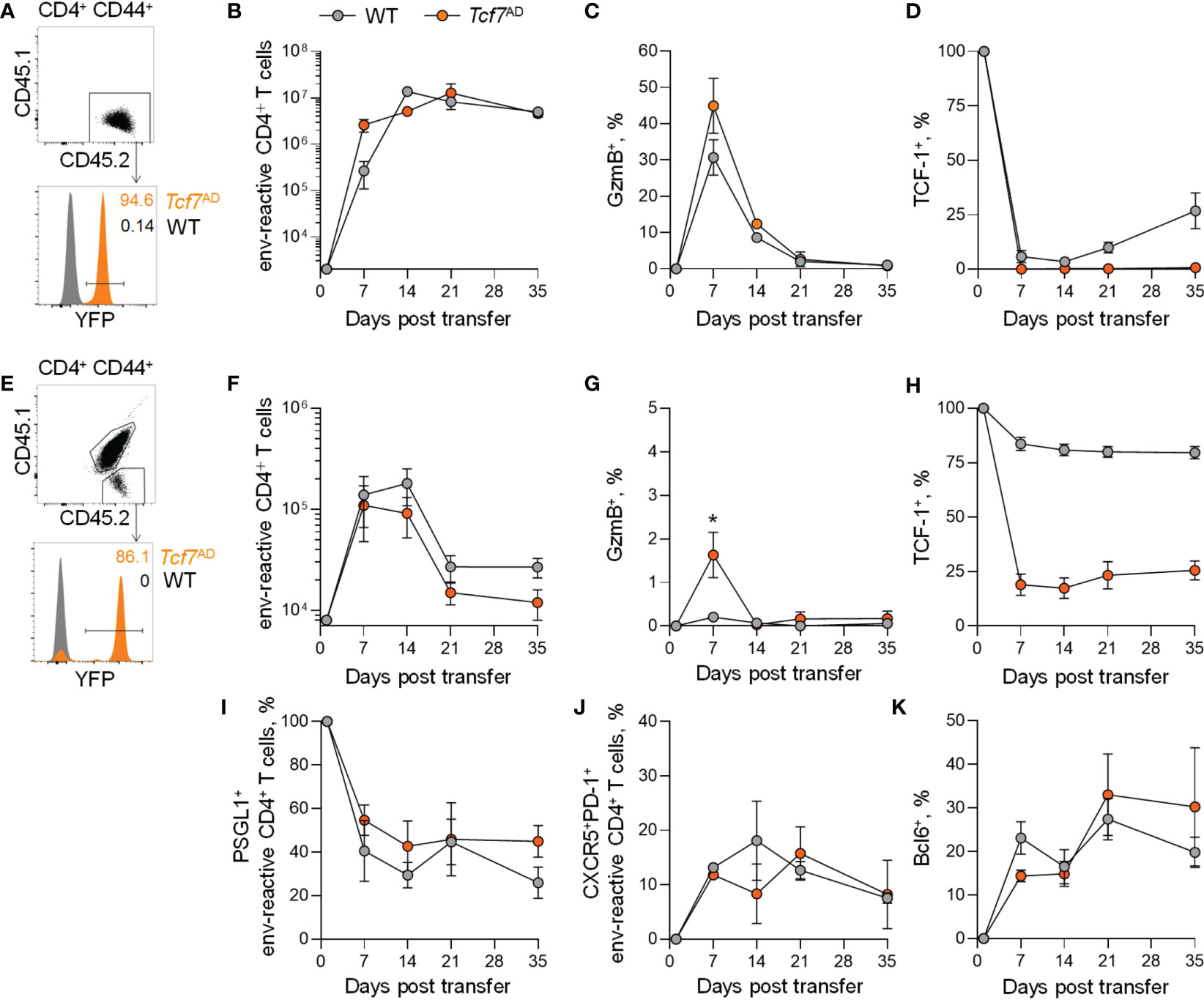
Figure 4 Memory CD4+ T cell, but not CTL maintenance independently of TCF-1. (A) Flow-cytometric detection of CD44+ env-reactive donor CD4+ T cells (top) and YFP+ CD4+ T cells (bottom) in spleens of Rag1-/- recipient mice. (B) Absolute numbers of WT EF4.1 and Tcf7AD EF4.1 env-reactive donor CD4+ T cells in the spleens of FV-infected Rag1-/- recipient mice at indicated time points after adoptive transfer. (C) GzmB expression and (D) TCF-1 expression of WT EF4.1 and Tcf7AD EF4.1 env-reactive donor CD4+ T cells in the spleens of FV-infected Rag1-/- recipient mice at indicated time points after adoptive transfer. (E) Flow-cytometric detection of CD44+ env-reactive donor CD4+ T cells (top) and YFP+ CD4+ T cells (bottom) in spleens of WT recipient mice. (F) Absolute numbers of WT EF4.1 and Tcf7AD EF4.1 env-reactive donor CD4+ T cells in the spleens of FV-infected WT recipient mice at indicated time points after adoptive transfer. (G) GzmB expression and (H) TCF-1 expression of WT EF4.1 and Tcf7AD EF4.1 env-reactive donor CD4+ T cells in the spleens of FV-infected WT recipient mice at indicated time points after adoptive transfer. (I) PSGL-1 expression of WT EF4.1 and Tcf7AD EF4.1 env-reactive donor CD4+ T cells in the spleens of FV-infected WT recipient mice at indicated time points after adoptive transfer. (J) Frequency of CXCR5+PD-1+ and (K) Bcl6+ cells within total WT EF4.1 and Tcf7AD EF4.1 env-reactive donor CD4+ T cells in the spleens of FV-infected WT recipient mice at indicated time points after adoptive transfer. *p < 0.05.
At the peak of the response, a moderately higher proportion of Tcf7AD than of WT donor EF4.1 CD4+ T cells differentiated into CTLs, as evidence by GzmB expression, but this proportion quickly declined thereafter with similar kinetics in both types for donor CD4+ T cell (Figure 4C). This unexpected finding indicated that TCF-1 does not play an important role for CD4+ T cell survival after T cell activation, which is in sharp contrast to the essential role of TCF-1 for the survival of CD8+ T cells. The unaffected survival of Tcf7AD EF4.1 CD4+ T cells, compared with WT counterparts, was not due to inefficient Tcf7 deletion or outgrowth of CD4+ T cells that has escaped deletion, as Tcf7AD donor EF4.1 CD4+ T cells remained negative for TCF-1 protein expression throughout the observation period, during which TCF-1 was re-expressed in a proportion of WT donor EF4.1 CD4+ T cells (Figure 4D).
Similar results were obtained when WT recipients were used, where donor EF4.1 CD4+ T cells would compete for space with host CD4+ T cells (Figures 4E, H), except smaller numbers of donor cells were recovered past the peak of the response. Although there was a trend of a smaller expansion of Tcf7AD EF4.1 CD4+ T cells in WT hosts, this did not reach statistical significance (Figure 4F). Despite smaller proportions of donor EF4.1 CD4+ T cells producing GzmB in WT hosts, this production was significantly higher in Tcf7AD than in WT EF4.1 CD4+ T cells at the peak and quickly declined thereafter (Figure 4G). Loss of TCF-1 was less complete in Tcf7AD donor EF4.1 CD4+ T cells transferred into WT than in Rag1-/- recipients, owing to reduced overall activation of donor EF4.1 CD4+ T cells in the T cell- and Treg-replete environment of WT recipient, but, importantly, we observed no preferential expansion or survival of TCF-1-expressing cells over time even within the Tcf7AD donor EF4.1 CD4+ T cell population (Figure 4H). This finding again argues against a requirement for TCF-1 for survival of primed CD4+ T cells. In addition to comparable persistence, despite higher peak GzmB production by Tcf7AD than by WT EF4.1 CD4+ T cells, loss of TCF-1 post activation did not influence overall differentiation of CD4+ T cells and the relative ratio of the mutually exclusive CTLs and Tfh subsets over time. Indeed, Tcf7AD CD4+ T cells differentiated into CXCR5+PD-1+PSGL-1- Tfh cells, and upregulated Bcl6 to a similar extent as WT EF4.1 CD4+ T cells (Figures 4I-K).
These data indicate that TCF-1 does not play a major role in CD4+ T cell differentiation and survival after T cell activation, and a lack of TCF-1 expression therefore cannot explain the loss of GzmB+ CD4+ T cells in the memory phase.
The lack of persistence of murine GzmB+ CD4+ T cells in our model is in stark contrast to what has been reported for human CD4+ CTLs, which have been shown to expand and persist, especially in the context of chronic viral infection (48, 49). This raised the possibility of a species difference in the ability of CD4+ CTLs to persist post activation or in settings of chronic antigenic stimulation. To test for a potential difference, we transferred human peripheral blood mononuclear cells (PBMCs) into Rag2-/-Il2rg-/-Cd47-/- compound mutant mice and followed human donor CD4+ T cells over time to analyse the persistence of human CD4+ CTLs, in the same settings where murine CD4+ CTLs failed to persist. Seven weeks after transfer, human CD4+ T cells established a stably engrafted population in recipient mice (Figures 5A, B). Similar to peak effector murine CD4+ T cells, human CD4+ T cells developed a population of GzmB+ CD4+ CTLs that lost expression of TCF-1 (Figure 5C). GzmB+ CD4+ T cells comprised between 5% and 15% of the total human donor CD4+ T cells, in line with estimates of the proportion of human CD4+ T cells xenoreactive with murine MHC-II molecules (50). These human CD4+ CTLs formed a stable population over a period of at least 6 weeks after initial differentiation (Figure 5C). This was in contrast to murine GzmB+ CD4+ T cells, which were lost almost completely after 3-5 weeks of adoptive transfer. The stability of human GzmB+ CD4+ T cells was intrinsically programmed, as similar results were obtained by adoptive transfer of isolated human CD4+ T cells (Figure S6). These data indicate there is a species-specific difference in the cytotoxic programming of human and murine CD4+ T cells, which prevents long-term persistence specifically in murine CD4+ CTLs.
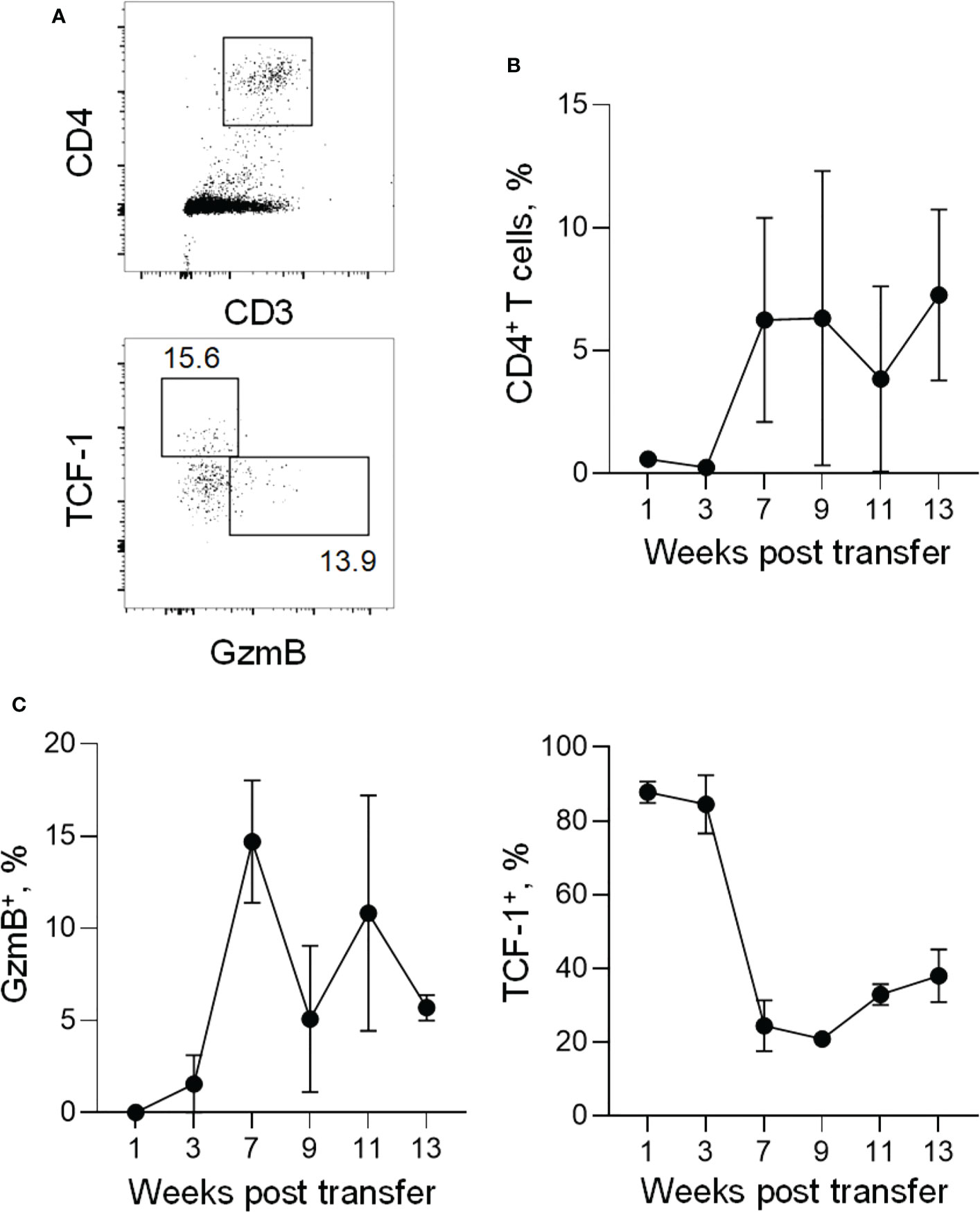
Figure 5 Expansion and persistence of xenografted human CD4+ CTLs. (A) Flow-cytometric detection of human CD4+ T cells (top) and GzmB+ and TCF-1+ CD4+ T cells (bottom) in peripheral blood of recipient triple KO mice injected with human PBMCs. (B) Frequency of human CD4+ T cells within single cells in peripheral blood (for weeks 1-11) or within splenocytes (week 13) of recipient triple KO mice at indicated time points after adoptive transfer. (C) Frequency of GzmB+ cells (left) or TCF-1+ cells (right) within total human CD4+ T cells in peripheral blood (weeks 1-11) or splenocytes (week 13) of recipient triple KO mice at indicated time points after adoptive transfer.
Cytotoxic CD4+ T cells have gained considerable interest in the last years as a new immune cell modality that could possibly be targeted for immunotherapeutic strategies. Here, we describe that the cytotoxic differentiation of CD4+ T cells antagonizes their longevity and memory formation in the context of chronic infection in mice.
The lack of memory formation by CD4+ CTLs could perhaps be a biological mechanism to protect the host from extensive immunopathology. CD4+ CTLs have been shown to exacerbate or be involved in multiple autoimmune diseases, including multiple sclerosis (MS) and rheumatoid arthritis (RA) (3, 51). Therefore, reducing the survival of CD4+ CTLs could be a strategy to limit the potential damaging effect of this population for the host. Interestingly, long-term effects of CD4+ CTL responses are not only limited by reduced survival of these cells, but also by a lack of cytotoxic differentiation of CD4+ T cells during recall responses. Although we did not investigate what limits cytotoxic differentiation during secondary stimulation, it could be explained by signalling via inhibitory receptors PD-1 and LAG-3, which we have shown are able to inhibit CD4+ CTL differentiation (16). Indeed, both PD-1 and LAG-3 are expressed by env-specific CD4+ T cells in the memory phase. Furthermore, transcriptional and epigenetic programming of memory CD4+ T cells could block the induction of cytotoxic differentiation in secondary immune responses. Similar mechanisms have been described in CD8+ T cells, where memory and cytotoxic effectors are characterised by divergent transcriptional and epigenetic states that determine which subset CD8+ T cell differentiate into and which prevent the acquisition of cytotoxic potential in memory CD8+ T cells (22, 52). Epigenetic programming has already been shown to regulate recall responses of memory CD4+ T cells, where memory cells formed by Tfh or Th1 cells induced a recall response of the same initial T cell subset (53). Therefore, lack of CD4+ CTL differentiation during recall responses could be explained by CD4+ CTL cells not seeding a memory population, which leads to a loss of this subset during a second stimulus. Interestingly, the mechanisms that prevent cytotoxic differentiation of CD4+ T cells in the memory phase seem to be very potent, since we show that they cannot be overcome by deliberate chronic activation of the cytotoxic differentiation pathway mediated by constitutive IL-2 signalling, and loss of CD4+ CTLs over time is not prevented.
CD4+ CTLs are characterized by loss of TCF-1, which is essential for CD8+ T cell memory survival, and is also expressed by memory CD4+ T cells. We therefore hypothesized that loss of TCF-1 in CD4+ CTLs could be responsible for their limited survival. However, here we show that following CD4+ T cell activation, loss of TCF-1 does not affect memory CD4+ T cell survival. In fact, we show that loss of TCF-1 has no effect at all on CD4+ T cells after T cell activation, since we find equal expansion and survival of CD4+ T cells, but also similar differentiation into CD4+ CTLs and Tfh cells in Tcf7AD mice. Previous studies on the role of TCF-1 in CD4+ T cells have shown that TCF-1 is essential for T cell development, Tfh differentiation and response to allogeneic MHC-II (24, 31, 54–56). However, most of these results were obtained using mouse models where TCF-1 was deleted before T cell stimulation. Our results now indicate TCF-1 only plays a role early after CD4+ T cell activation and quickly becomes redundant afterwards in the memory phase. This is in sharp contrast with CD8+ T cells, and clearly shows that the role of TCF-1 is different in CD4+ T cells. Although it is not possible from our experiments to rule out an effect of TCF-1 on the persistence of CD4+ CTLs, our observations that TCF-1 is redundant for survival of conventional murine memory CD4+ T cells and human CD4+ CTLs (which also lack TCF-1) may indicate that the lack of survival of murine CD4+ CTLs is unrelated to TCF-1 loss.
The lack of murine CD4+ CTL survival is in contrast to the enormous expansions of CD4+ CTLs in humans, where these cells persist over long periods of time (3–7, 48, 49). We show here that human CD4+ CTLs are able to survive much longer in the same environment as murine CD4+ CTLs. Although we did not investigate what causes this difference from a molecular standpoint, immunological differences between mice and men are well documented (including in CD4+ T cell responses) (57–59). It is interesting to ponder if survival of CD4+ CTLs has evolved in humans due to the beneficial properties of this immune cell population, or whether this quality has been lost in mice due to the possible detrimental effects to the host described above. Nevertheless, if CD4+ CTLs are to be used for immunotherapeutic strategies, the ability to survive for extended periods could be important. Indeed, in a recent study in leukemia patients that developed long time tumour remission after CAR therapy, the responsible CAR T cell population was formed almost completely of persisting cytotoxic CD4+ T cells (60). These cells showed similarities to the tumour-specific CD4+ CTLs that were found in bladder cancer patients (8). Although the anti-tumour response of CAR T cells is not within the natural MHC-II context of CD4+ T cells, it does clearly demonstrate the therapeutic potential of CD4+ CTLs as effector cells for anti-tumour immunotherapy. Future studies will need to show whether similar anti-tumour responses can be created using CD4+ CTLs within their natural MHC-II context, as seems to be the case for bladder cancer.
In conclusion, we show here that murine CD4+ CTLs are not able to form a memory population in the context of chronic viral infection. The lack of CD4+ CTL memory was not related to the loss ThPOK or TCF-1, and could not be prevented by chronic IL-2 signalling. Human CD4+ CTLs showed improved survival compared to their murine counterparts, which suggests that the mouse is not the ideal model to study the biology or the therapeutic potential of CD4+ CTLs. The improved survival of human CD4+ CTLs and the potential anti-tumor effects of this population indicates CD4+ CTLs could be promising targets for immunotherapeutic strategies.
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author/s.
All animal experiments were approved by the ethical committee of the Francis Crick Institute, and conducted according to local guidelines and UK Home Office regulations under the Animals Scientific Procedures Act 1986 (ASPA).
TH, LD, GC, TD and DH performed the experiments and analyzed the data. LD, and GK supervised the study. TH and GK wrote the manuscript. All authors contributed to the article and approved the submitted version.
We wish to thank Dr Kim Hasenkrug for FV and FBL-3 stocks, Dr Ulf Dittmer for the Ad5.pIX-gp70 stocks, and Dr Ben Seddon for providing the caSTAT5a plasmid. We are grateful for assistance from the Flow Cytometry and Biological Resource Facilities at the Francis Crick Institute. This work was supported by the Francis Crick Institute (CC2088, CC2000), which receives its core funding from Cancer Research UK, the UK Medical Research Council, and the Wellcome Trust. For the purpose of Open Access, the author has applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1168125/full#supplementary-material
1. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol (2010) 28:445–89. doi: 10.1146/annurev-immunol-030409-101212
2. O’shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science (2010) 327(5969):1098–102. doi: 10.1126/science.1178334
3. Preglej T, Ellmeier W. CD4(+) cytotoxic T cells - phenotype, function and transcriptional networks controlling their differentiation pathways. Immunol Lett (2022) 247:27–42. doi: 10.1016/j.imlet.2022.05.001
4. Hoeks C, Duran G, Hellings N, Broux B. When helpers go above and beyond: Development and characterization of cytotoxic CD4(+) T cells. Front Immunol (2022) 13:951900. doi: 10.3389/fimmu.2022.951900
5. Swain SL, Mckinstry KK, Strutt TM. Expanding roles for CD4+ T cells in immunity to viruses. Nat Rev Immunol (2012) 12(2):136–48. doi: 10.1038/nri3152
6. Van De Berg PJ, Van Leeuwen EM, Ten Berge IJ, Van Lier R. Cytotoxic human CD4(+) T cells. Curr Opin Immunol (2008) 20(3):339–43. doi: 10.1016/j.coi.2008.03.007
7. Cheroutre H, Husain MM. CD4 CTL: living up to the challenge. Semin Immunol (2013) 25(4):273–81. doi: 10.1016/j.smim.2013.10.022
8. Oh DY, Kwek SS, Raju SS, Li T, Mccarthy E, Chow E, et al. Intratumoral CD4(+) T cells mediate anti-tumor cytotoxicity in human bladder cancer. Cell (2020) 181(7):1612–1625.e1613. doi: 10.1016/j.cell.2020.05.017
9. Alspach E, Lussier DM, Miceli AP, Kizhvatov I, Dupage M, Luoma AM, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature (2019) 574(7780):696–701. doi: 10.1038/s41586-019-1671-8
10. Oliveira G, Stromhaug K, Cieri N, Iorgulescu JB, Klaeger S, Wolff JO, et al. Landscape of helper and regulatory antitumour CD4(+) T cells in melanoma. Nature (2022) 605(7910):532–8. doi: 10.1038/s41586-022-04682-5
11. Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med (2010) 207(3):637–50. doi: 10.1084/jem.20091918
12. Cachot A, Bilous M, Liu YC, Li X, Saillard M, Cenerenti M, et al. Tumor-specific cytolytic CD4 T cells mediate immunity against human cancer. Sci Adv (2021) 7(9). doi: 10.1126/sciadv.abe3348
13. Mucida D, Husain MM, Muroi S, Van Wijk F, Shinnakasu R, Naoe Y, et al. Transcriptional reprogramming of mature CD4+ helper T cells generates distinct MHC class II-restricted cytotoxic T lymphocytes. Nat Immunol (2013) 14(3):281–9. doi: 10.1038/ni.2523
14. Reis BS, Rogoz A, Costa-Pinto FA, Taniuchi I, Mucida D. Mutual expression of the transcription factors Runx3 and ThPOK regulates intestinal CD4+ T cell immunity. Nat Immunol (2013) 14(3):271–80. doi: 10.1038/ni.2518
15. Serroukh Y, Gu-Trantien C, Hooshiar Kashani B, Defrance M, Vu Manh TP, Azouz A, et al. The transcription factors Runx3 and ThPOK cross-regulate acquisition of cytotoxic function by human Th1 lymphocytes. Elife (2018) 7. doi: 10.7554/eLife.30496
16. Donnarumma T, Young GR, Merkenschlager J, Eksmond U, Bongard N, Nutt SL, et al. Opposing development of cytotoxic and follicular helper CD4 T cells controlled by the TCF-1-Bcl6 nexus. Cell Rep (2016) 17(6):1571–83. doi: 10.1016/j.celrep.2016.10.013
17. Qui HZ, Hagymasi AT, Bandyopadhyay S, St Rose MC, Ramanarasimhaiah R, Ménoret A, et al. CD134 plus CD137 dual costimulation induces eomesodermin in CD4 T cells to program cytotoxic Th1 differentiation. J Immunol (2011) 187(7):3555–64. doi: 10.4049/jimmunol.1101244
18. Curran MA, Geiger TL, Montalvo W, Kim M, Reiner SL, Al-Shamkhani A, et al. Systemic 4-1BB activation induces a novel T cell phenotype driven by high expression of eomesodermin. J Exp Med (2013) 210(4):743–55. doi: 10.1084/jem.20121190
19. Choi IK, Wang Z, Ke Q, Hong M, Qian Y, Zhao X, et al. Signaling by the Epstein-Barr virus LMP1 protein induces potent cytotoxic CD4(+) and CD8(+) T cell responses. Proc Natl Acad Sci U.S.A. (2018) 115(4):E686-e695. doi: 10.1073/pnas.1713607115
20. Ditoro D, Winstead CJ, Pham D, Witte S, Andargachew R, Singer JR, et al. Differential IL-2 expression defines developmental fates of follicular versus nonfollicular helper T cells. Science (2018) 361(6407). doi: 10.1126/science.aao2933
21. Śledzińska A, Vila De Mucha M, Bergerhoff K, Hotblack A, Demane DF, Ghorani E, et al. Regulatory T cells restrain interleukin-2- and blimp-1-Dependent acquisition of cytotoxic function by CD4(+) T cells. Immunity (2020) 52(1):151–166.e156. doi: 10.1016/j.immuni.2019.12.007
22. Cui W, Kaech SM. Generation of effector CD8+ T cells and their conversion to memory T cells. Immunol Rev (2010) 236:151–66. doi: 10.1111/j.1600-065X.2010.00926.x
23. Harty JT, Badovinac VP. Shaping and reshaping CD8+ T-cell memory. Nat Rev Immunol (2008) 8(2):107–19. doi: 10.1038/nri2251
24. Zhao X, Shan Q, Xue HH. TCF1 in T cell immunity: a broadened frontier. Nat Rev Immunol (2022) 22(3):147–57. doi: 10.1038/s41577-021-00563-6
25. Antunes I, Tolaini M, Kissenpfennig A, Iwashiro M, Kuribayashi K, Malissen B, et al. Retrovirus-specificity of regulatory T cells is neither present nor required in preventing retrovirus-induced bone marrow immune pathology. Immunity (2008) 29(5):782–94. doi: 10.1016/j.immuni.2008.09.016
26. Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature b and T lymphocytes. Cell (1992) 68(5):869–77. doi: 10.1016/0092-8674(92)90030-g
27. Monroe RJ, Seidl KJ, Gaertner F, Han S, Chen F, Sekiguchi J, et al. RAG2:GFP knockin mice reveal novel aspects of RAG2 expression in primary and peripheral lymphoid tissues. Immunity (1999) 11(2):201–12. doi: 10.1016/s1074-7613(00)80095-3
28. Mouchacca P, Schmitt-Verhulst AM, Boyer C. Visualization of cytolytic T cell differentiation and granule exocytosis with T cells from mice expressing active fluorescent granzyme b. PloS One (2013) 8(6):e67239. doi: 10.1371/journal.pone.0067239
29. Klinger M, Kim JK, Chmura SA, Barczak A, Erle DJ, Killeen N. Thymic OX40 expression discriminates cells undergoing strong responses to selection ligands. J Immunol (2009) 182(8):4581–9. doi: 10.4049/jimmunol.0900010
30. Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol (2001) 1):4. doi: 10.1186/1471-213x-1-4
31. Wu T, Shin HM, Moseman EA, Ji Y, Huang B, Harly C, et al. TCF1 is required for the T follicular helper cell response to viral infection. Cell Rep (2015) 12(12):2099–110. doi: 10.1016/j.celrep.2015.08.049
32. Marques R, Antunes I, Eksmond U, Stoye J, Hasenkrug K, Kassiotis G. B lymphocyte activation by coinfection prevents immune control of friend virus infection. J Immunol (2008) 181(5):3432–40. doi: 10.4049/jimmunol.181.5.3432
33. Thorborn G, Ploquin MJ, Eksmond U, Pike R, Bayer W, Dittmer U, et al. Clonotypic composition of the CD4+ T cell response to a vectored retroviral antigen is determined by its speed. J Immunol (2014) 193(4):1567–77. doi: 10.4049/jimmunol.1400667
34. Hetzel C, Janssen R, Ely SJ, Kristensen NM, Bunting K, Cooper JB, et al. An epitope delivery system for use with recombinant mycobacteria. Infect Immun (1998) 66(8):3643–8. doi: 10.1128/iai.66.8.3643-3648.1998
35. Klarnet JP, Kern DE, Okuno K, Holt C, Lilly F, Greenberg PD. FBL-reactive CD8+ cytotoxic and CD4+ helper T lymphocytes recognize distinct friend murine leukemia virus-encoded antigens. J Exp Med (1989) 169(2):457–67. doi: 10.1084/jem.169.2.457
36. Merkenschlager J, Eksmond U, Danelli L, Attig J, Young GR, Nowosad C, et al. MHC class II cell-autonomously regulates self-renewal and differentiation of normal and malignant b cells. Blood (2019) 133(10):1108–18. doi: 10.1182/blood-2018-11-885467
37. Merkenschlager J, Ploquin MJ, Eksmond U, Andargachew R, Thorborn G, Filby A, et al. Stepwise b-cell-dependent expansion of T helper clonotypes diversifies the T-cell response. Nat Commun (2016) 7):10281. doi: 10.1038/ncomms10281
38. Danelli L, Cornish G, Merkenschlager J, Kassiotis G. Default polyfunctional T helper 1 response to ample signal 1 alone. Cell Mol Immunol (2021) 18(7):1809–22. doi: 10.1038/s41423-020-0415-x
39. Danelli L, Donnarumma T, Kassiotis G. Correlates of follicular helper bias in the CD4 T cell response to a retroviral antigen. Front Immunol (2018) 9:1260. doi: 10.3389/fimmu.2018.01260
40. Young GR, Eksmond U, Salcedo R, Alexopoulou L, Stoye JP, Kassiotis G. Resurrection of endogenous retroviruses in antibody-deficient mice. Nature (2012) 491(7426):774–8. doi: 10.1038/nature11599
41. Young GR, Ploquin MJ, Eksmond U, Wadwa M, Stoye JP, Kassiotis G. Negative selection by an endogenous retrovirus promotes a higher-avidity CD4+ T cell response to retroviral infection. PloS Pathog (2012) 8(5):e1002709. doi: 10.1371/journal.ppat.1002709
42. Manzke N, Akhmetzyanova I, Hasenkrug KJ, Trilling M, Zelinskyy G, Dittmer U. CD4+ T cells develop antiretroviral cytotoxic activity in the absence of regulatory T cells and CD8+ T cells. J Virol (2013) 87(11):6306–13. doi: 10.1128/jvi.00432-13
43. Dooms H, Wolslegel K, Lin P, Abbas AK. Interleukin-2 enhances CD4+ T cell memory by promoting the generation of IL-7R alpha-expressing cells. J Exp Med (2007) 204(3):547–57. doi: 10.1084/jem.20062381
44. Fleury M, Vazquez-Mateo C, Hernandez-Escalante J. Dooms h. partial STAT5 signaling is sufficient for CD4(+) T cell priming but not memory formation. Cytokine (2022) 150:155770. doi: 10.1016/j.cyto.2021.155770
45. Ciucci T, Vacchio MS, Gao Y, Tomassoni Ardori F, Candia J, Mehta M, et al. The emergence and functional fitness of memory CD4(+) T cells require the transcription factor thpok. Immunity (2019) 50(1):91–105.e104. doi: 10.1016/j.immuni.2018.12.019
46. Gullicksrud JA, Li F, Xing S, Zeng Z, Peng W, Badovinac VP, et al. Differential requirements for Tcf1 long isoforms in CD8(+) and CD4(+) T cell responses to acute viral infection. J Immunol (2017) 199(3):911–9. doi: 10.4049/jimmunol.1700595
47. Nish SA, Zens KD, Kratchmarov R, Lin WW, Adams WC, Chen YH, et al. CD4+ T cell effector commitment coupled to self-renewal by asymmetric cell divisions. J Exp Med (2017) 214(1):39–47. doi: 10.1084/jem.20161046
48. Van Leeuwen EM, Remmerswaal EB, Vossen MT, Rowshani AT, Wertheim-Van Dillen PM, Van Lier RA, et al. Emergence of a CD4+CD28- granzyme b+, cytomegalovirus-specific T cell subset after recovery of primary cytomegalovirus infection. J Immunol (2004) 173(3):1834–41. doi: 10.4049/jimmunol.173.3.1834
49. Appay V, Zaunders JJ, Papagno L, Sutton J, Jaramillo A, Waters A, et al. Characterization of CD4(+) CTLs ex vivo. J Immunol (2002) 168(11):5954–8. doi: 10.4049/jimmunol.168.11.5954
50. Tahara H, Ide K, Basnet N, Tanaka Y, Ohdan H. Determination of the precursor frequency and the reaction intensity of xenoreactive human T lymphocytes. Xenotransplantation (2010) 17(3):188–96. doi: 10.1111/j.1399-3089.2010.00575.x
51. Dumitriu IE. The life (and death) of CD4+ CD28(null) T cells in inflammatory diseases. Immunology (2015) 146(2):185–93. doi: 10.1111/imm.12506
52. Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol (2018) 18(5):340–56. doi: 10.1038/nri.2017.146
53. Hale JS, Youngblood B, Latner DR, Mohammed AU, Ye L, Akondy RS, et al. Distinct memory CD4+ T cells with commitment to T follicular helper- and T helper 1-cell lineages are generated after acute viral infection. Immunity (2013) 38(4):805–17. doi: 10.1016/j.immuni.2013.02.020
54. Choi YS, Gullicksrud JA, Xing S, Zeng Z, Shan Q, Li F, et al. LEF-1 and TCF-1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat Immunol (2015) 16(9):980–90. doi: 10.1038/ni.3226
55. Xu L, Cao Y, Xie Z, Huang Q, Bai Q, Yang X, et al. The transcription factor TCF-1 initiates the differentiation of T(FH) cells during acute viral infection. Nat Immunol (2015) 16(9):991–9. doi: 10.1038/ni.3229
56. Mammadli M, Suo L, Sen JM, Karimi M. TCF-1 is required for CD4 T cell persistence functions during AlloImmunity. Int J Mol Sci (2023) 24(5). doi: 10.3390/ijms24054326
57. Medetgul-Ernar K, Davis MM. Standing on the shoulders of mice. Immunity (2022) 55(8):1343–53. doi: 10.1016/j.immuni.2022.07.008
58. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol (2004) 172(5):2731–8. doi: 10.4049/jimmunol.172.5.2731
59. Gros P, Casanova JL. Reconciling mouse and human immunology at the altar of genetics. Annu Rev Immunol (2022). doi: 10.1146/annurev-immunol-101721-065201
Keywords: CD4 T cell, granzyme B (GzmB), immunological memory, TCF-1, mouse, human
Citation: Hofland T, Danelli L, Cornish G, Donnarumma T, Hunt DM, de Carvalho LPS and Kassiotis G (2023) CD4+ T cell memory is impaired by species-specific cytotoxic differentiation, but not by TCF-1 loss. Front. Immunol. 14:1168125. doi: 10.3389/fimmu.2023.1168125
Received: 17 February 2023; Accepted: 30 March 2023;
Published: 14 April 2023.
Edited by:
Vandana Kalia, University of Washington, United StatesReviewed by:
Jorg Calis, University Medical Center Utrecht, NetherlandsCopyright © 2023 Hofland, Danelli, Cornish, Donnarumma, Hunt, de Carvalho and Kassiotis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George Kassiotis, Z2VvcmdlLmthc3Npb3Rpc0Bjcmljay5hYy51aw==
†Present addresses: Luca Danelli, Springer Nature, London, United Kingdom
Georgina Cornish, AstraZeneca, Great Abington, United Kingdom
Tiziano Donnarumma, CheckmAb, Milan, Italy
Luiz P. S. de Carvalho, Department of Chemistry, The Herbert Wertheim UF Scripps Institute for Biomedical Innovation & Technology, Jupiter, FL, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.