 Yoshihito Nihei
Yoshihito Nihei Hitoshi Suzuki
Hitoshi Suzuki Yusuke Suzuki
Yusuke Suzuki
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 11 April 2023
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1165394
This article is part of the Research TopicImmunoglobulin A in Infection and InflammationView all 8 articles
Immunoglobulin A (IgA) is the most abundant isotype of antibodies, provides a first line of defense at mucosal surfaces against pathogens, and thereby contributes to mucosal homeostasis. IgA is generally considered as a non-inflammatory antibody because of its main function, neutralizing pathogenic virus or bacteria. Meanwhile, IgA can induce IgA-mediated diseases, such as IgA nephropathy (IgAN) and IgA vasculitis. IgAN is characterized by the deposition of IgA and complement C3, often with IgG and/or IgM, in the glomerular mesangial region, followed by mesangial cell proliferation and excessive synthesis of extracellular matrix in glomeruli. Almost half a century has passed since the first report of patients with IgAN; it remains debatable about the mechanism how IgA antibodies selectively bind to mesangial region—a hallmark of IgAN—and cause glomerular injuries in IgAN. Previous lectin- and mass-spectrometry-based analysis have revealed that IgAN patients showed elevated serum level of undergalactosylated IgA1 in O-linked glycans of its hinge region, called galactose-deficient IgA1 (Gd-IgA1). Thereafter, numerous studies have confirmed that the glomerular IgA from IgAN patients are enriched with Gd-IgA1; thus, the first hit of the current pathogenesis of IgAN has been considered to increase circulating levels of Gd-IgA1. Recent studies, however, demonstrated that this aberrant glycosylation alone is not sufficient to disease onset and progression, suggesting that several additional factors are required for the selective deposition of IgA in the mesangial region and induce nephritis. Herein, we discuss the current understanding of the characteristics of pathogenic IgA and its mechanism of inducing inflammation in IgAN.
Immunoglobulin A (IgA) is the most abundant isotype of antibodies (Abs); approximately 66 mg/kg IgA Abs is generated daily from antibody-secreting cells (ASCs) that reside mainly in the mucosal lumen (1, 2). There are two types of subclasses in human, namely, IgA1 and IgA2; the most significant structural difference between IgA1 and IgA2 is that IgA1 has a longer hinge region, while IgA2 lacks 13 amino acids compared to IgA1 (3). While approximately 90% of serum IgA is IgA1 subclass in its monomeric form, secretary IgA (sIgA), which are produced from plasma cells as dimeric or further polymeric forms, can be found predominantly in the mucosal lumen. The proportion of subclass of IgA is different depending on which segment of the mucosal lumen (4). The shorter hinge region of IgA2 than IgA1 makes it less susceptible to degradation by bacterial proteases, which would explain the higher prevalence of IgA2 in the lower gastrointestinal tract (GI). Monomeric IgA has an anti-inflammatory effector function, and sIgA (polymeric IgA) acts as neutralizing Abs against pathogenic virus or bacteria at mucosal surfaces (1, 5, 6). Therefore, IgA is generally considered as non-inflammatory Abs. However, IgA is sometimes involved in IgA-mediated diseases, including IgA nephropathy (IgAN) (7) and IgA vasculitis (8).
IgAN is the most common type of primary glomerulonephritis worldwide, with a global prevalence of 2.5 cases per 100,000 adults per year, and one of the first causes of end-stage renal disease (ESRD) (9). IgAN is characterized by the deposition of IgA and complement C3 in the glomerular mesangial region, often with co-deposition of IgG and/or IgM (10). Histologically, mesangial cell proliferation and expansion of extracellular matrix are observed (10). Although over half a century has passed since the first report of patients with IgAN by Berger et al. (11), no specific and causal treatment strategies have been developed, leading to ESRD in 30%–40% of cases within 10–20 years after disease onset (12). This is largely due to the lack of understanding of the pathogenesis of IgAN, particularly the characteristics of nephritogenic IgA. Herein, we summarize and discuss the current understanding of the characteristics of pathogenic IgA and its mechanism of inducing inflammation in IgAN.
In the 1980s, characteristics of IgA Abs deposited in glomeruli in IgAN began to be investigated. Monteiro et al. examined the mesangial IgA eluted from glomeruli of percutaneous renal biopsies of 20 patients with IgAN and directly demonstrated that mesangial IgA are predominantly polymeric and anionic (13). In early 2000s, the lectin- and mass-spectrometry-based analysis have revealed that IgAN patients showed elevated serum level of aberrantly glycosylated, specifically galactose-deficient, IgA1 in O-linked glycans of its hinge region (galactose-deficient IgA1: Gd-IgA1) (14, 15); more than 70% of patients with IgAN showed increased serum Gd-IgA1 levels above the 90th percentile of that in healthy controls (16). The amino acid sequence of the human IgA1 hinge region is proline-rich and consists of nine serine/threonines. O-Glycosylation usually occurs in three to six of these sites (17). B cells from IgAN patients have been reported to show decreased levels of core 1 β1,3-galactosyltransferase, the enzyme that attaches galactose to N-acetylgalactosamine (GalNAc) and molecular chaperon (Cosmc), which is necessary for galactosyltransferase stabilization (18). As a result, serum IgA1 with decreased galactose in O-linked glycans is increased in patients with IgAN. Thereafter, numerous studies have demonstrated that the glomerular IgA from IgAN patients are enriched with these galactose-deficient IgA1 (Gd-IgA1). This was further confirmed by the results of immunofluorescent (IF) staining of the kidney using KM55, an anti-Gd-IgA1 monoclonal Abs that we recently established (19, 20). Thus, Gd-IgA1 have been thought to play a central role in the pathogenesis of IgAN, although other glycosylation patterns of IgA1 have been reported, including the reduced sialylation of O-glycans and modified N-glycosylation of IgA1 (21).
This aberrant glycosylation in IgA alone, however, cannot be sufficient for the selective deposition of IgA in the mesangial region. In this regard, Berthelot et al. have reported that transferrin receptor 1 (TfR1) and transglutaminase 2 (TG2) expressed on mesangial cells are important for the IgA binding to the mesangial region (22). Meanwhile, Zeng et al. have reported the possibility that J chain expressed in mesangial cells might be involved in the selective deposition of IgA in the mesangial region (23). They performed a single-cell RNA sequencing using cells from the renal biopsies of 13 IgAN patients and normal tissues from six patients who had undergone nephrectomy and found significant upregulation of JCHAIN in mesangial cells. However, they have not directly demonstrated that J chain is involved in selective IgA deposition in the mesangial region, and it is unclear whether J chain expression precedes IgA deposition. Although the region-specific Ab deposition implies auto-Ab recognition of self-antigen(s), IgA-type auto-Abs against self-antigen in mesangial cells have not yet been identified. Therefore, the detailed mechanism by which IgA is specifically deposited in the mesangial region in patients with IgAN remains unclear.
In the pathogenesis of IgAN, the involvement of Gd-IgA1 production and mucosal immunity has been discussed (24, 25). This is because some of patients with IgAN show gross hematuria and exacerbation of nephritis after upper respiratory tract infection or colitis (10), and polymeric IgA containing J chains, generally found in the mucosa lumen, were detected in glomeruli from patients with IgAN (26). Moreover, recent genome-wide association studies provided the evidence for an association between IgAN and genes related to mucosal IgA production (LIF, OSM, TNFSF13, and DEFA) (27), deducing the connection between Gd-IgA1 production and mucosal immunity. However, it is still inconclusive where Gd-IgA1-producing cells are generated. Plasma cells in nasal-associated lymphoid tissue (NALT) predominantly produce IgA1 subclass (IgA1:IgA2 = 9:1), whereas those derived from gut-associated lymphoid tissue (GALT) generate a high amount of IgA2, although the IgA1/IgA2 ratio differs depending on which segment of the GI tract. Given that the subclass of IgA deposited in the kidney in IgAN is IgA1, it is speculated that Gd-IgA1 is derived from NALT. This theory is supported mainly in Japan, where tonsillectomy has been used to treat IgAN and showed clinical efficacy (28). However, the report from a subanalysis of the VALIGA study showed no efficacy of tonsillectomy in European patients with IgAN (29). Thus, tonsils are currently considered to be involved in the pathogenesis of IgAN only in a limited group of patients, such as Japanese subjects. On the other hand, in IgAN in Caucasian, the involvement of GALT in the production of Gd-IgA1 is strongly supported (24, 30). In Europe, there are relatively many reports showing the association of IgAN and celiac disease (31, 32) or inflammatory bowel disease (IBD) (33). The recent NEFIGAN Trial using intestinal selective steroids (Nefecon) in patients with IgAN, which was reported to reduce proteinuria in the treatment group (34), also supports the involvement of GALT in the pathogenesis of IgAN in Caucasian. Thus, the responsible mucosa that produces Gd-IgA1 may differ in different ethnicities. Details on the association of Gd-IgA1 production and mucosal immunity are described elsewhere (24, 25).
Since mucosal IgA are generated by commensal bacteria, it is conceivable that certain bacteria are involved in Gd-IgA1 production. Although several intestinal or oral bacteria have been reported to affect clinical parameters in patients with IgAN (35, 36), specific bacteria directly involved in Gd-IgA1 production have not been identified. Instead, several types of toll-like receptor (TLR), a family of pattern recognition receptors that recognize pathogen-associated molecular patterns and play a central role in sensing microbial infection, have been reported to be involved in the pathogenesis of IgAN (24). Coppo’s group reported that TLR4 expression in circulating mononuclear cells from patients with IgAN was correlated with proteinuria (37), whereas Yu’s group recently showed that TLR7 expression was significantly increased in peripheral blood B cells from patients with IgAN (38). We have reported that TLR9, which recognizes unmethylated CpG motifs abundant in bacterial DNA, is involved in the production of abnormal glycosylated IgA. This is evident from our studies using IgAN model mice. We have shown that the ddY mouse strain, spontaneous IgAN model mice, could be classified into three groups based on the timing of disease onset: early-onset, late-onset, and quiescent groups. We performed a genome-wide scan of the early- and late-onset ddY mice and found that MyD88, an adaptor protein required for signaling of some TLRs, was a candidate gene for progression of renal injury. Analysis of mRNA expression of TLRs associated with MyD88 in splenocytes from ddY mice revealed a significant increase in TLR9 gene (39). In vivo, intranasal and systemic administration of CpG DNA into early-onset ddY mice aggravated nephritis (39, 40), suggesting that activation of TLR9 is involved at least in the pathogenesis of mouse IgAN. In human IgAN, we reported that polymorphisms of the TLR9 gene are associated with histological severity of patients with IgAN (39), indicating the involvement of TLR9 in the pathogenesis of human IgAN. Which types of TLR are involved in Gg-IgA1 production needs to be further investigated.
The IgA deposition in mesangial region is a hallmark of IgAN; however, the deposition of IgA alone is not sufficient for the subsequent glomerular inflammation. It was reported that renal transplant donors and necropsy individuals who died from traumatic injuries with glomerular IgA deposition were asymptomatic (41–43). Although these reports do not confirm that the glomerular IgA in these individuals were Gd-IgA1, it is likely that at least IgA deposition alone does not lead to induce nephritis. Hereafter, we will focus on the possible mechanism how IgA—generally considered as neutralizing Abs—induce glomerular inflammation in IgAN (Figure 1).
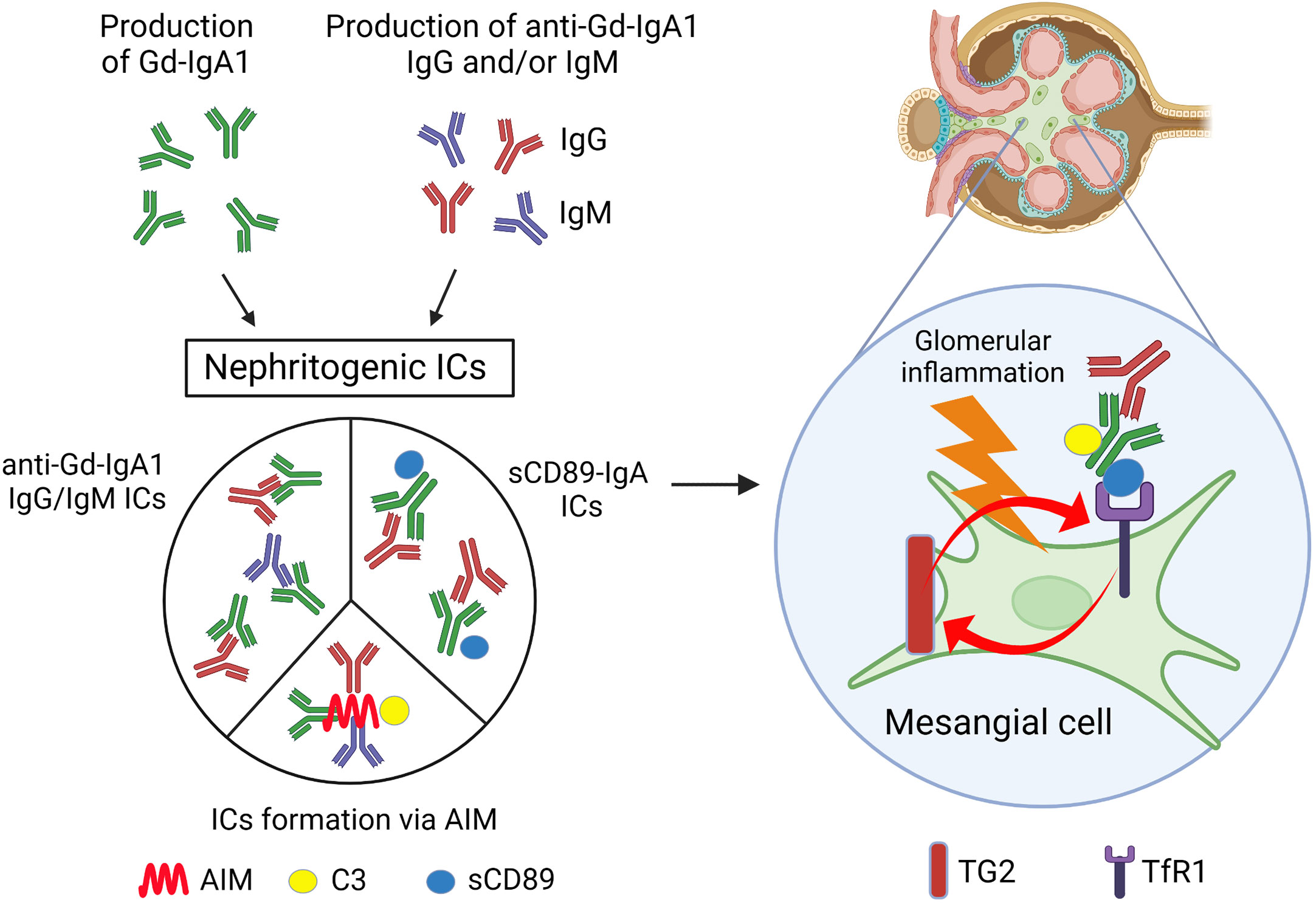
Figure 1 Scheme of the possible mechanism for glomerular inflammation in IgAN. Patients with IgAN show increased levels of serum Gd-IgA1 and anti-Gd-IgA1 IgG and/or IgM Abs. Nephritogenic ICs including these Abs, generated by various mechanisms as depicted, deposit mesangial region in glomeruli and induce mesangial inflammation. Gd-IgA1, galactose-deficient IgA1; ICs, immune complexes; AIM, apoptosis inhibitor of macrophages; sCD89, soluble fragment of CD89; C3, complement C3; TG2, transglutaminase 2; TfR1, transferrin receptor 1. Created with BioRender.com.
In IgAN, immune complex (IC) formation is thought to be important in initiating glomerular inflammation. This has been experimentally demonstrated; both in vitro and in vivo experiments have shown that activation of human mesangial cells requires ICs containing IgG, not IgA alone (44, 45). The glomerular ICs in IgAN contain Gd-IgA1 and complement C3, IgG, and/or IgM. Although C3 is often co-deposited, IgG and IgM are not always detectable in routine IF microscopy. Recently, however, Rizk et al. have shown that IgG could be detectable even in the IgAN kidney-biopsy specimens without IgG by routine IF microscopy by extracting deposited Abs in glomeruli from remnant IgAN kidney-biopsy specimens (46), suggesting that IF microscopy using routine Abs is only under a sensitivity of detection, but in fact most patients have co-deposition of IgG. In the current widely accepted pathogenesis of IgAN, these IgG are thought to be auto-Abs that recognize Gd-IgA1 as a self-antigen. Tomana et al. reported that the reactivity of serum IgG extracted from IgAN patients to IgA1 was severely reduced when the O-linked glycans of the IgA1 hinge region was enzymatically removed, suggesting that sera from patients with IgAN contain IgG Abs against the O-linked glycans in the hinge region of IgA1 (47). In addition, we have reported that the affinity of patients’ IgG to IgA1 was enhanced when GalNAc was exposed by specifically removing sialic acid and galactose from the O-linked glycans of the IgA1 hinge region. Sequence analysis of the variable region of the heavy and light chains of these IgG revealed that the amino acid in the third position in the complementarity-determining region (CDR) 3 was serine in patients with IgAN rather than alanine in the healthy individuals (48). Further experiments revealed that this serine residue in CDR3 is required for effective binding to Gd-IgA1. These results suggest that endogenous anti-Gd-IgA1 IgG Abs recognize GalNAc as their antigen. It remains to be determined whether these IgG Abs are produced in an antigen (Gd-IgA1)-dependent manner or whether they are part of the existing natural Abs. Regarding IgM and IgG, Matsumoto et al. have recently demonstrated that anti-Gd-IgA1 IgM are present in circulating ICs from patients with IgAN, and these ICs contain high levels of complement C3 (49). These data may have identified one possible reason for the glomerular IgM co-deposition with IgA in IgAN. On the other hand, we demonstrated that ICs can be formed by mechanisms other than those described above, using gddY mice, an isolated group of ddY (grouped ddY: gddY) mice with a 100% incidence by selective mating of an early-onset group of ddY mice (50, 51). We have shown that the molecule apoptosis inhibitor of macrophages (AIM) is required to form ICs (52). We found that AIM accumulation in glomeruli colocalized with IgA in gddY mice and patients with IgAN. AIM-deficient (AIM−/−) gddY mice showed glomerular IgA deposition but no IgG, IgM, or C3 co-deposition and no proteinuria. Administration of recombinant AIM to AIM−/− gddY resulted in IgG, IgM, and C3 co-deposition and subsequent proteinuria. These results suggest that ICs can be formed via AIM; AIM is responsible for binding glomerular IgA to other immunoglobulins and complement C3, although the mechanism is still unknown. In any case, through these mechanisms, Gd-IgA1 forms high-molecular nephritogenic ICs with IgG, IgM, and C3, thereby escaping clearance by the liver, the normal catabolic pathway for circulatory IgA1, and deposit in the glomeruli, causing subsequent inflammation in glomeruli.
IgA can interact with several receptors, including polymeric immunoglobulin receptor, TfR1 and FcαRI (CD89) (6). TfR was identified as IgA1 receptor expressed on human mesangial cells (53). CD89 is a member of the Fc receptor immunoglobulin superfamily and expressed on various blood myeloid cells including monocytes/macrophages, dendritic cells, neutrophils, and eosinophils (54). While CD89 binds monomeric IgA with a low affinity and generally exhibits anti-inflammatory functions following binding of serum monomeric IgA, CD89 binds polymeric IgA and IgA-antigen complexes with a higher avidity (55). Their binding induces phagocytosis (56), antibody-dependent cellular cytotoxicity (57), or release of inflammatory mediators (58). In IgAN, Monteiro et al. have reported the role of CD89 in the pathogenesis of IgAN. They found that circulating soluble fragment of CD89 (sCD89)-IgA complexes are present in adult patients with IgAN and further demonstrated that these complexes are involved in the pathogenesis of IgAN using the transgenic mice expressing human CD89 on macrophages and monocytes. In this mouse model, sCD89-IgA complexes were deposited in the mesangial region and induced glomerular inflammation (59). Moreover, recently, they reported that the levels of circulating sCD89-IgA complexes and free sCD89 were increased in childhood IgAN, and they were associated with proteinuria and MEST pathological score. In this report, they found that not only sCD89-IgA complexes but also sCD89 itself could directly induce mesangial cell proliferation both in vitro and in vivo (60). IgA-sCD89 ICs were deposited to glomeruli by sCD89 binding to TfR1 on mesangial cells (22). Gd-IgA1-sCD89 complexes may be easily deposited on mesangial cells, since IgA1 ICs and hypo-galactosylated IgA1 show higher affinity to TfR1 (61, 62). sCD89-TfR1 interaction has been shown to be impaired in the absence of TG2 (22), of which expression on glomeruli was related to the clinical and histological severity in IgAN patients (63). SCD89-IgA complexes–TfR1 interaction can be one of the explanations for the binding of IgA1 in the mesangial region in IgAN (Figure 1).
Complement activation has a pivotal role in triggering glomerular inflammation in IgAN (64). It occurs via three different pathways: the classical, the alternative, and the lectin pathways. In patients with IgAN, C3 and C4d mesangial depositions were observed in >90% and 18.9%–56%, respectively (64, 65), while C1q deposition is rarely observed (64). Glomerular C3 and C4d deposition were reported to be predictors of renal outcome in patients with IgAN (66, 67). Thus, complement activation in IgAN is considered to take place through the alternative and the lectin pathway. Although it was reported that IgA can activate both the alternative and the lectin pathway in vitro (68, 69), it is still unknown where Gd-IgA1 containing ICs activate the complement pathway, in circulation and/or after deposition in the mesangial region. In any case, the alternative and the lectin pathways lead to the formation of membrane attack complex (C5b-9) and initiate glomerular injury. In this process, anaphylatoxins C3a and C5a, which are produced by C3 activation, was also reported to be involved in glomerular inflammation (70, 71). Prolonged local inflammation resulting from these complement activations leads to chronic glomerular injury culminating in glomerulosclerosis.
Although IgA is generally considered as non-inflammatory Abs, however, IgA can induce IgA-mediated disease. Among them, IgAN is the most common. Although the key feature of nephritogenic IgA in IgAN is that they are under-galactosylated, the deposition of these IgA1 on glomeruli alone does not induce inflammation. Gd-IgA1 is likely to be generated via TLR (and/or other molecules) signaling and forms ICs with IgG, IgM, and complement C3 to induce nephritis; however, the detailed mechanism of these ICs formation remains unclear. Mesangial IgA deposition cannot solely be explained by the formation of these ICs containing Gd-IgA1, suggesting that other reasons for the selective deposition of IgA are present. Identification of the characteristics of such IgA might provide the key insights in the pathogenesis of IgAN.
HS and YS were responsible for the conception and design of the review. YN and HS drafted and revised the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor RMC declared a past collaboration with the author YS.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Keppler SJ, Goess MC, Heinze JM. The wanderings of gut-derived IgA plasma cells: Impact on systemic immune responses. Front Immunol (2021) 12:670290. doi: 10.3389/fimmu.2021.670290
2. Leusen JH. IgA as therapeutic antibody. Mol Immunol (2015) 68(1):35–9. doi: 10.1016/j.molimm.2015.09.005
3. Hansen IS, Baeten DLP, den Dunnen J. The inflammatory function of human IgA. Cell Mol Life Sci (2019) 76(6):1041–55. doi: 10.1007/s00018-018-2976-8
4. Perše M, Večerić-Haler Ž. The role of IgA in the pathogenesis of IgA nephropathy. Int J Mol Sci (2019) 20(24):6199. doi: 10.3390/ijms20246199
5. Corthésy B. Multi-faceted functions of secretory IgA at mucosal surfaces. Front Immunol (2013) 4:185. doi: 10.3389/fimmu.2013.00185
6. Breedveld A, van Egmond M. IgA and fcαri: Pathological roles and therapeutic opportunities. Front Immunol (2019) 10:553. doi: 10.3389/fimmu.2019.00553
7. Suzuki H, Kiryluk K, Novak J, Moldoveanu Z, Herr AB, Renfrow MB, et al. The pathophysiology of IgA nephropathy. J Am Soc Nephrol (2011) 22(10):1795–803. doi: 10.1681/asn.2011050464
8. Pillebout E, Sunderkötter C. IgA vasculitis. Semin Immunopathol (2021) 43(5):729–38. doi: 10.1007/s00281-021-00874-9
9. McGrogan A, Franssen CF, de Vries CS. The incidence of primary glomerulonephritis worldwide: A systematic review of the literature. Nephrol Dial Transplant (2011) 26(2):414–30. doi: 10.1093/ndt/gfq665
10. Wyatt RJ, Julian BA. IgA nephropathy. N Engl J Med (2013) 368(25):2402–14. doi: 10.1056/NEJMra1206793
12. Lai KN, Tang SC, Schena FP, Novak J, Tomino Y, Fogo AB, et al. IgA nephropathy. Nat Rev Dis Primers (2016) 2:16001. doi: 10.1038/nrdp.2016.1
13. Monteiro RC, Halbwachs-Mecarelli L, Roque-Barreira MC, Noel LH, Berger J, Lesavre P. Charge and size of mesangial IgA in IgA nephropathy. Kidney Int (1985) 28(4):666–71. doi: 10.1038/ki.1985.181
14. Allen AC, Bailey EM, Brenchley PE, Buck KS, Barratt J, Feehally J. Mesangial IgA1 in IgA nephropathy exhibits aberrant O-glycosylation: Observations in three patients. Kidney Int (2001) 60(3):969–73. doi: 10.1046/j.1523-1755.2001.060003969.x
15. Hiki Y, Odani H, Takahashi M, Yasuda Y, Nishimoto A, Iwase H, et al. Mass spectrometry proves under-O-Glycosylation of glomerular IgA1 in iga nephropathy. Kidney Int (2001) 59(3):1077–85. doi: 10.1046/j.1523-1755.2001.0590031077.x
16. Moldoveanu Z, Wyatt RJ, Lee JY, Tomana M, Julian BA, Mestecky J, et al. Patients with IgA nephropathy have increased serum galactose-deficient IgA1 levels. Kidney Int (2007) 71(11):1148–54. doi: 10.1038/sj.ki.5002185
17. Novak J, Barratt J, Julian BA, Renfrow MB. Aberrant glycosylation of the IgA1 molecule in IgA nephropathy. Semin Nephrol (2018) 38(5):461–76. doi: 10.1016/j.semnephrol.2018.05.016
18. Suzuki H, Moldoveanu Z, Hall S, Brown R, Vu HL, Novak L, et al. IgA1-secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J Clin Invest (2008) 118(2):629–39. doi: 10.1172/jci33189
19. Yasutake J, Suzuki Y, Suzuki H, Hiura N, Yanagawa H, Makita Y, et al. Novel lectin-independent approach to detect galactose-deficient IgA1 in iga nephropathy. Nephrol Dial Transplant (2015) 30(8):1315–21. doi: 10.1093/ndt/gfv221
20. Suzuki H, Yasutake J, Makita Y, Tanbo Y, Yamasaki K, Sofue T, et al. IgA nephropathy and IgA vasculitis with nephritis have a shared feature involving galactose-deficient IgA1-oriented pathogenesis. Kidney Int (2018) 93(3):700–5. doi: 10.1016/j.kint.2017.10.019
21. Dotz V, Visconti A, Lomax-Browne HJ, Clerc F, Hipgrave Ederveen AL, Medjeral-Thomas NR, et al. O- and n-glycosylation of serum immunoglobulin a is associated with IgA nephropathy and glomerular function. J Am Soc Nephrol (2021) 32(10):2455–65. doi: 10.1681/asn.2020081208
22. Berthelot L, Papista C, Maciel TT, Biarnes-Pelicot M, Tissandie E, Wang PH, et al. Transglutaminase is essential for IgA nephropathy development acting through IgA receptors. J Exp Med (2012) 209(4):793–806. doi: 10.1084/jem.20112005
23. Zheng Y, Lu P, Deng Y, Wen L, Wang Y, Ma X, et al. Single-cell transcriptomics reveal immune mechanisms of the onset and progression of IgA nephropathy. Cell Rep (2020) 33(12):108525. doi: 10.1016/j.celrep.2020.108525
24. Gesualdo L, Di Leo V, Coppo R. The mucosal immune system and IgA nephropathy. Semin Immunopathol (2021) 43(5):657–68. doi: 10.1007/s00281-021-00871-y
25. Kano T, Suzuki H, Makita Y, Nihei Y, Fukao Y, Nakayama M, et al. Mucosal immune system dysregulation in the pathogenesis of IgA nephropathy. Biomedicines (2022) 10(12):3027. doi: 10.3390/biomedicines10123027
26. Tomino Y, Sakai H, Miura M, Endoh M, Nomoto Y. Detection of polymeric IgA in glomeruli from patients with iga nephropathy. Clin Exp Immunol (1982) 49(2):419–25.
27. Kiryluk K, Novak J, Gharavi AG. Pathogenesis of immunoglobulin a nephropathy: Recent insight from genetic studies. Annu Rev Med (2013) 64:339–56. doi: 10.1146/annurev-med-041811-142014
28. Hirano K, Matsuzaki K, Yasuda T, Nishikawa M, Yasuda Y, Koike K, et al. Association between tonsillectomy and outcomes in patients with immunoglobulin a nephropathy. JAMA Netw Open (2019) 2(5):e194772. doi: 10.1001/jamanetworkopen.2019.4772
29. Feehally J, Coppo R, Troyanov S, Bellur SS, Cattran D, Cook T, et al. Tonsillectomy in a European cohort of 1,147 patients with IgA nephropathy. Nephron (2016) 132(1):15–24. doi: 10.1159/000441852
30. Barratt J, Rovin BH, Cattran D, Floege J, Lafayette R, Tesar V, et al. Why target the gut to treat IgA nephropathy? Kidney Int Rep (2020) 5(10):1620–4. doi: 10.1016/j.ekir.2020.08.009
31. Collin P, Syrjänen J, Partanen J, Pasternack A, Kaukinen K, Mustonen J. Celiac disease and hla dq in patients with IgA nephropathy. Am J Gastroenterol (2002) 97(10):2572–6. doi: 10.1111/j.1572-0241.2002.06025.x
32. Nurmi R, Metso M, Pörsti I, Niemelä O, Huhtala H, Mustonen J, et al. Celiac disease or positive tissue transglutaminase antibodies in patients undergoing renal biopsies. Dig Liver Dis (2018) 50(1):27–31. doi: 10.1016/j.dld.2017.09.131
33. Pohjonen J, Nurmi R, Metso M, Oksanen P, Huhtala H, Pörsti I, et al. Inflammatory bowel disease in patients undergoing renal biopsies. Clin Kidney J (2019) 12(5):645–51. doi: 10.1093/ckj/sfz004
34. Barratt J, Lafayette R, Kristensen J, Stone A, Cattran D, Floege J, et al. Results from part a of the multi-center, double-blind, randomized, placebo-controlled nefigard trial, which evaluated targeted-release formulation of budesonide for the treatment of primary immunoglobulin a nephropathy. Kidney Int (2023) 103(2):391–402. doi: 10.1016/j.kint.2022.09.017
35. De Angelis M, Montemurno E, Piccolo M, Vannini L, Lauriero G, Maranzano V, et al. Microbiota and metabolome associated with immunoglobulin a nephropathy (Igan). PloS One (2014) 9(6):e99006. doi: 10.1371/journal.pone.0099006
36. Khasnobish A, Takayasu L, Watanabe KI, Nguyen TTT, Arakawa K, Hotta O, et al. Dysbiosis in the salivary microbiome associated with IgA nephropathy-a Japanese cohort study. Microbes Environ (2021) 36(2):ME21006. doi: 10.1264/jsme2.ME21006
37. Coppo R, Camilla R, Amore A, Peruzzi L, Daprà V, Loiacono E, et al. Toll-like receptor 4 expression is increased in circulating mononuclear cells of patients with immunoglobulin a nephropathy. Clin Exp Immunol (2010) 159(1):73–81. doi: 10.1111/j.1365-2249.2009.04045.x
38. Zheng N, Xie K, Ye H, Dong Y, Wang B, Luo N, et al. TLR7 in b cells promotes renal inflammation and gd-IgA1 synthesis in IgA nephropathy. JCI Insight (2020) 5(14):e136965. doi: 10.1172/jci.insight.136965
39. Suzuki H, Suzuki Y, Narita I, Aizawa M, Kihara M, Yamanaka T, et al. Toll-like receptor 9 affects severity of IgA nephropathy. J Am Soc Nephrol (2008) 19(12):2384–95. doi: 10.1681/asn.2007121311
40. Makita Y, Suzuki H, Kano T, Takahata A, Julian BA, Novak J, et al. TLR9 activation induces aberrant IgA glycosylation via APRIL- and IL-6-Mediated pathways in IgA nephropathy. Kidney Int (2020) 97(2):340–9. doi: 10.1016/j.kint.2019.08.022
41. Suzuki K, Honda K, Tanabe K, Toma H, Nihei H, Yamaguchi Y. Incidence of latent mesangial IgA deposition in renal allograft donors in Japan. Kidney Int (2003) 63(6):2286–94. doi: 10.1046/j.1523-1755.63.6s.2.x
42. Varis J, Rantala I, Pasternack A, Oksa H, Jäntti M, Paunu ES, et al. Immunoglobulin and complement deposition in glomeruli of 756 subjects who had committed suicide or met with a violent death. J Clin Pathol (1993) 46(7):607–10. doi: 10.1136/jcp.46.7.607
43. Sinniah R. Occurrence of mesangial IgA and IgM deposits in a control necropsy population. J Clin Pathol (1983) 36(3):276–9. doi: 10.1136/jcp.36.3.276
44. Zhao YF, Zhu L, Liu LJ, Shi SF, Lv JC, Zhang H. Pathogenic role of glycan-specific IgG antibodies in IgA nephropathy. BMC Nephrol (2017) 18(1):301. doi: 10.1186/s12882-017-0722-3
45. Moldoveanu Z, Suzuki H, Reily C, Satake K, Novak L, Xu N, et al. Experimental evidence of pathogenic role of IgG autoantibodies in IgA nephropathy. J Autoimmun (2021) 118:102593. doi: 10.1016/j.jaut.2021.102593
46. Rizk DV, Saha MK, Hall S, Novak L, Brown R, Huang ZQ, et al. Glomerular immunodeposits of patients with IgA nephropathy are enriched for IgG autoantibodies specific for galactose-deficient IgA1. J Am Soc Nephrol (2019) 30(10):2017–26. doi: 10.1681/asn.2018111156
47. Tomana M, Novak J, Julian BA, Matousovic K, Konecny K, Mestecky J. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Invest (1999) 104(1):73–81. doi: 10.1172/jci5535
48. Suzuki H, Fan R, Zhang Z, Brown R, Hall S, Julian BA, et al. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J Clin Invest (2009) 119(6):1668–77. doi: 10.1172/jci38468
49. Matsumoto Y, Aryal RP, Heimburg-Molinaro J, Park SS, Wever WJ, Lehoux S, et al. Identification and characterization of circulating immune complexes in IgA nephropathy. Sci Adv (2022) 8(43):eabm8783. doi: 10.1126/sciadv.abm8783
50. Suzuki H, Suzuki Y, Yamanaka T, Hirose S, Nishimura H, Toei J, et al. Genome-wide scan in a novel IgA nephropathy model identifies a susceptibility locus on murine chromosome 10, in a region syntenic to human Igan1 on chromosome 6q22-23. J Am Soc Nephrol (2005) 16(5):1289–99. doi: 10.1681/asn.2004030219
51. Okazaki K, Suzuki Y, Otsuji M, Suzuki H, Kihara M, Kajiyama T, et al. Development of a model of early-onset IgA nephropathy. J Am Soc Nephrol (2012) 23(8):1364–74. doi: 10.1681/asn.2011121160
52. Takahata A, Arai S, Hiramoto E, Kitada K, Kato R, Makita Y, et al. Crucial role of AIM/CD5l in the development of glomerular inflammation in IgA nephropathy. J Am Soc Nephrol (2020) 31(9):2013–24. doi: 10.1681/asn.2019100987
53. Moura IC, Centelles MN, Arcos-Fajardo M, Malheiros DM, Collawn JF, Cooper MD, et al. Identification of the transferrin receptor as a novel immunoglobulin (Ig)A1 receptor and its enhanced expression on mesangial cells in IgA nephropathy. J Exp Med (2001) 194(4):417–25. doi: 10.1084/jem.194.4.417
54. Lechner SM, Papista C, Chemouny JM, Berthelot L, Monteiro RC. Role of IgA receptors in the pathogenesis of IgA nephropathy. J Nephrol (2016) 29(1):5–11. doi: 10.1007/s40620-015-0246-5
55. Monteiro RC, Van De Winkel JG. IgA fc receptors. Annu Rev Immunol (2003) 21:177–204. doi: 10.1146/annurev.immunol.21.120601.141011
56. Gorter A, Hiemstra PS, Leijh PC, van der Sluys ME, van den Barselaar MT, van Es LA, et al. IgA- and secretory IgA-opsonized s. aureus induce a respiratory burst and phagocytosis by polymorphonuclear leucocytes. Immunology (1987) 61(3):303–9.
57. Otten MA, Rudolph E, Dechant M, Tuk CW, Reijmers RM, Beelen RH, et al. Immature neutrophils mediate tumor cell killing via IgA but not IgG fc receptors. J Immunol (2005) 174(9):5472–80. doi: 10.4049/jimmunol.174.9.5472
58. Aleyd E, van Hout MW, Ganzevles SH, Hoeben KA, Everts V, Bakema JE, et al. IgA enhances netosis and release of neutrophil extracellular traps by polymorphonuclear cells via fcα receptor I. J Immunol (2014) 192(5):2374–83. doi: 10.4049/jimmunol.1300261
59. Launay P, Grossetête B, Arcos-Fajardo M, Gaudin E, Torres SP, Beaudoin L, et al. Fcalpha receptor (CD89) mediates the development of immunoglobulin a (IgA) nephropathy (Berger's disease). evidence for pathogenic soluble receptor-IgA complexes in patients and CD89 transgenic mice. J Exp Med (2000) 191(11):1999–2009. doi: 10.1084/jem.191.11.1999
60. Cambier A, Gleeson PJ, Abbad L, Canesi F, da Silva J, Bex-Coudrat J, et al. Soluble CD89 is a critical factor for mesangial proliferation in childhood IgA nephropathy. Kidney Int (2022) 101(2):274–87. doi: 10.1016/j.kint.2021.09.023
61. Moura IC, Arcos-Fajardo M, Sadaka C, Leroy V, Benhamou M, Novak J, et al. Glycosylation and size of IgA1 are essential for interaction with mesangial transferrin receptor in IgA nephropathy. J Am Soc Nephrol (2004) 15(3):622–34. doi: 10.1097/01.asn.0000115401.07980.0c
62. Moura IC, Arcos-Fajardo M, Gdoura A, Leroy V, Sadaka C, Mahlaoui N, et al. Engagement of transferrin receptor by polymeric IgA1: Evidence for a positive feedback loop involving increased receptor expression and mesangial cell proliferation in IgA nephropathy. J Am Soc Nephrol (2005) 16(9):2667–76. doi: 10.1681/asn.2004111006
63. Ikee R, Kobayashi S, Hemmi N, Saigusa T, Namikoshi T, Yamada M, et al. Involvement of transglutaminase-2 in pathological changes in renal disease. Nephron Clin Pract (2007) 105(3):c139–46. doi: 10.1159/000098646
64. Maillard N, Wyatt RJ, Julian BA, Kiryluk K, Gharavi A, Fremeaux-Bacchi V, et al. Current understanding of the role of complement in IgA nephropathy. J Am Soc Nephrol (2015) 26(7):1503–12. doi: 10.1681/asn.2014101000
65. Jiang Y, Zan J, Shi S, Hou W, Zhao W, Zhong X, et al. Glomerular C4d deposition and kidney disease progression in IgA nephropathy: A systematic review and meta-analysis. Kidney Med (2021) 3(6):1014–21. doi: 10.1016/j.xkme.2021.06.009
66. Wu D, Li X, Yao X, Zhang N, Lei L, Zhang H, et al. Mesangial C3 deposition and serum C3 levels predict renal outcome in IgA nephropathy. Clin Exp Nephrol (2021) 25(6):641–51. doi: 10.1007/s10157-021-02034-7
67. Espinosa M, Ortega R, Sánchez M, Segarra A, Salcedo MT, González F, et al. Association of C4d deposition with clinical outcomes in IgA nephropathy. Clin J Am Soc Nephrol (2014) 9(5):897–904. doi: 10.2215/cjn.09710913
68. Hiemstra PS, Gorter A, Stuurman ME, Van Es LA, Daha MR. Activation of the alternative pathway of complement by human serum IgA. Eur J Immunol (1987) 17(3):321–6. doi: 10.1002/eji.1830170304
69. Roos A, Bouwman LH, van Gijlswijk-Janssen DJ, Faber-Krol MC, Stahl GL, Daha MR. Human IgA activates the complement system via the mannan-binding lectin pathway. J Immunol (2001) 167(5):2861–8. doi: 10.4049/jimmunol.167.5.2861
70. Liu L, Zhang Y, Duan X, Peng Q, Liu Q, Zhou Y, et al. C3a, C5a renal expression and their receptors are correlated to severity of IgA nephropathy. J Clin Immunol (2014) 34(2):224–32. doi: 10.1007/s10875-013-9970-6
Keywords: IgA, IgA nephropathy, galactose deficient IgA1, immune complex, pathogenesis
Citation: Nihei Y, Suzuki H and Suzuki Y (2023) Current understanding of IgA antibodies in the pathogenesis of IgA nephropathy. Front. Immunol. 14:1165394. doi: 10.3389/fimmu.2023.1165394
Received: 14 February 2023; Accepted: 27 March 2023;
Published: 11 April 2023.
Edited by:
Renato C. Monteiro, Université de Paris, FranceReviewed by:
Patrick James Gleeson, University College Cork, IrelandCopyright © 2023 Nihei, Suzuki and Suzuki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yusuke Suzuki, eXVzdWtlQGp1bnRlbmRvLmFjLmpw; Hitoshi Suzuki, c2hpdG9zaGlAanVudGVuZG8uYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.