 Lincoln N. Strickland
Lincoln N. Strickland Erika Y. Faraoni
Erika Y. Faraoni Wei Ruan
Wei Ruan Xiaoyi Yuan
Xiaoyi Yuan Holger K. Eltzschig
Holger K. Eltzschig Jennifer M. Bailey-Lundberg*
Jennifer M. Bailey-Lundberg*- Department of Anesthesiology, Critical Care, and Pain Medicine, McGovern Medical School, The University of Texas Health Science Center at Houston, Houston, TX, United States
Pancreatic ductal adenocarcinoma (PDAC) is characterized by a dense desmoplastic stroma that impedes drug delivery, reduces parenchymal blood flow, and suppresses the anti-tumor immune response. The extracellular matrix and abundance of stromal cells result in severe hypoxia within the tumor microenvironment (TME), and emerging publications evaluating PDAC tumorigenesis have shown the adenosine signaling pathway promotes an immunosuppressive TME and contributes to the overall low survival rate. Hypoxia increases many elements of the adenosine signaling pathway, resulting in higher adenosine levels in the TME, further contributing to immune suppression. Extracellular adenosine signals through 4 adenosine receptors (Adora1, Adora2a, Adora2b, Adora3). Of the 4 receptors, Adora2b has the lowest affinity for adenosine and thus, has important consequences when stimulated by adenosine binding in the hypoxic TME. We and others have shown that Adora2b is present in normal pancreas tissue, and in injured or diseased pancreatic tissue, Adora2b levels are significantly elevated. The Adora2b receptor is present on many immune cells, including macrophages, dendritic cells, natural killer cells, natural killer T cells, γδ T cells, B cells, T cells, CD4+ T cells, and CD8+ T cells. In these immune cell types, adenosine signaling through Adora2b can reduce the adaptive anti-tumor response, augmenting immune suppression, or may contribute to transformation and changes in fibrosis, perineural invasion, or the vasculature by binding the Adora2b receptor on neoplastic epithelial cells, cancer-associated fibroblasts, blood vessels, lymphatic vessels, and nerves. In this review, we discuss the mechanistic consequences of Adora2b activation on cell types in the tumor microenvironment. As the cell-autonomous role of adenosine signaling through Adora2b has not been comprehensively studied in pancreatic cancer cells, we will also discuss published data from other malignancies to infer emerging therapeutic considerations for targeting the Adora2b adenosine receptor to reduce the proliferative, invasive, and metastatic potential of PDAC cells.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a lethal malignancy, with only a 3-13% 5-year survival rate, which is critically dependent on the stage at diagnosis. PDAC is characterized by a highly immunosuppressive and hypoxic tumor microenvironment. Risk factors include age, chronic pancreatitis, diabetes, genetic predisposition, obesity, and smoking (1, 2). Current therapeutic approaches including chemotherapy and radiation have not resulted in significant changes in overall survival, highlighting the continued need for testing new therapeutic strategies to treat PDAC patients. In this review, we will expand on an immune suppressive pathway in PDAC, the adenosine signaling pathway, with a focus on the role of the Adora2b receptor. Work from our lab and others has shown this pathway is elevated in a subset of patients with PDAC, and inhibition of extracellular adenosine generation augments anti-tumor immunity in several preclinical pancreatic cancer models (3–6). We will discuss the mechanistic consequences of elevated extracellular adenosine in the pancreatic cancer microenvironment and will emphasize emerging considerations for targeting the Adora2b receptor as a therapeutic target to improve outcomes for patients at high risk or who have been diagnosed with PDAC (7–9).
Heterocyclic aromatic molecules such as adenosine triphosphate (ATP), adenosine diphosphate (ADP), and adenosine are purines essential to life, indispensable for maintaining intracellular energy balance, cellular processes, and pathways (10). ATP is generated by glycolysis or oxidative phosphorylation and is commonly known as the principal molecule for storing and transferring energy in the cell (11). Within the cell, ATP molecules are transported by mitochondrial ADP/ATP carriers (AAC) proteins, major components of the inner mitochondrial membrane that regulate ATP synthesis by influencing ADP intake in the mitochondria. In the contexts of cellular injury, stress, hypoxia, or cell death, ATP can be secreted out of the cell in exosomes (exocytotic release), through connexin or pannexin channels, or by volume-regulated anion channels to the extracellular space, where it signals through purinergic receptors and participates in a broad range of cellular processes (12, 13). Some of the roles of extracellular ATP include the regulation of inflammation and fibrosis (14). Both ATP and extracellular ADP can be converted by an ectonucleotidase enzyme (CD39) into adenosine monophosphate (AMP), a molecule that can then be converted to adenosine by ecto-5’-nucleotidase (CD73) (Figure 1) (11, 15). Adenosine has been shown to participate in pro-inflammatory, anti-inflammatory, fibrotic, and immunosuppressive responses dependent on cell type activated, extracellular concentrations of ATP, ADP, and adenosine, degree of hypoxia, and availability and duration of binding to P1 receptors including Adora1, Adora3, Adora2a or Adora2b which can all be expressed on epithelial, stromal, or immune cells. Such responses vary depending on the P1 receptor involvement and intracellular signaling downstream of receptor activation (15–18). Extracellular adenosine signaling can be terminated through the uptake of adenosine into cells through two predominant equilibrative nucleoside transporters (ENTs), ENT1 and ENT2, which are bidirectional transport channels that allow transmembrane diffusion of nucleosides (19, 20). Termination of adenosine signaling can also occur when adenosine undergoes an irreversible termination process by the enzyme adenosine deaminase (ADA), which converts adenosine to inosine (21).
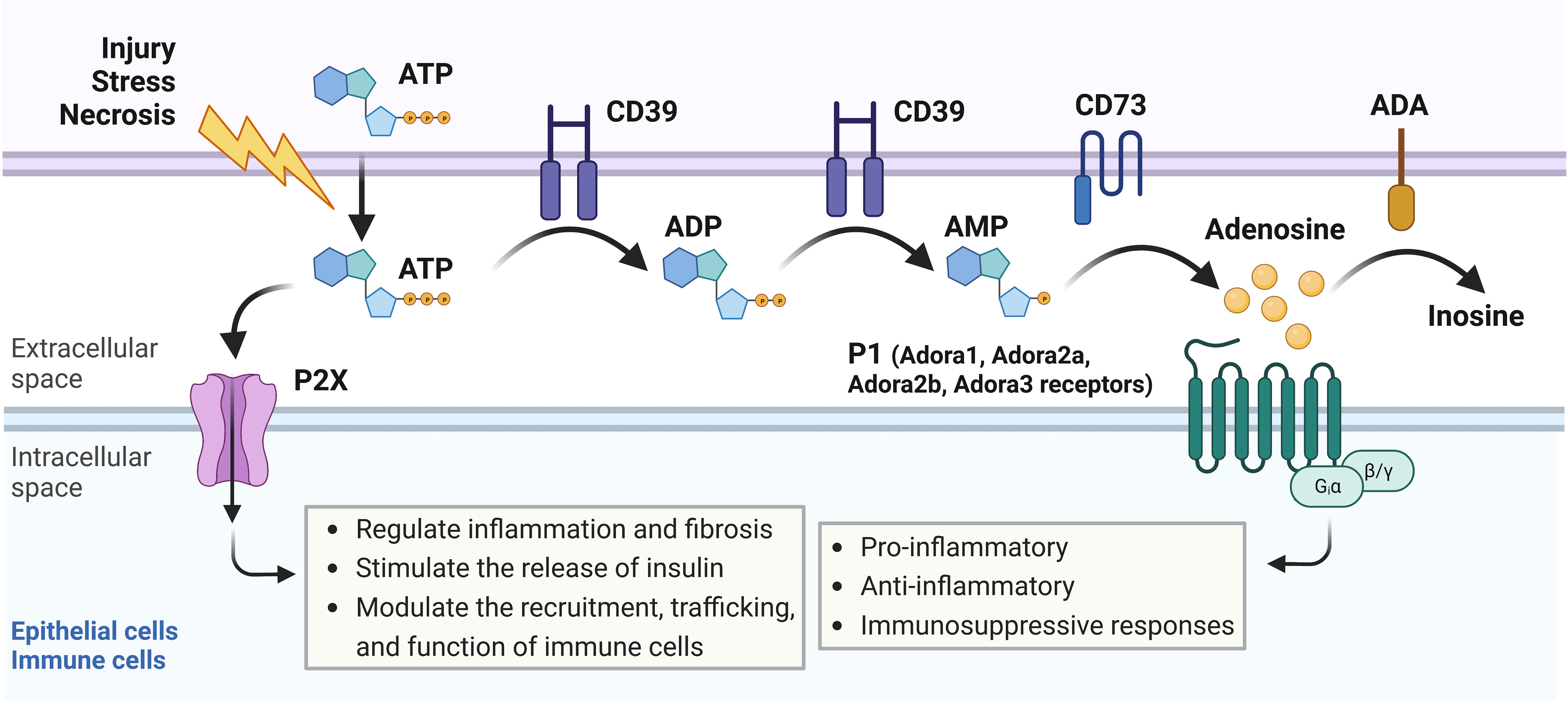
Figure 1 Adenosine signaling pathway overview. In response to cellular injury, stress, or necrosis, adenosine triphosphate (ATP) is released to the extracellular space, where it can signal through P2X receptors on epithelial or immune cells to regulate inflammation and fibrosis, stimulate the release of insulin, or modulate recruitment, trafficking, and function of immune cells. Extracellular ATP can also be converted by CD39, an ectonucleotidase enzyme, into adenosine diphosphate (ADP) or adenosine monophosphate (AMP). AMP can then be converted into adenosine by CD73, another ectonucleotidase enzyme. Adenosine binds to P1 receptors including Adora1, Adora3, Adora2a, or Adora2b, which can all be expressed on epithelial, stromal, or immune cells. Activation of the P1 receptors results in pro-inflammatory, anti-inflammatory, or immunosuppressive responses depending on which P1 receptor is involved and which intracellular signaling pathways are activated downstream. Adenosine can also be converted to inosine by adenosine deaminase (ADA) in an irreversible termination process.
Hypoxia-mediated adenosine signaling in inflammatory and tumor microenvironments
Hypoxia is a hallmark of chronic inflammatory conditions including several solid tumors; yet hypoxic conditions can occur in the early stages of inflammation due to the oxygen requirements of neutrophils and other immune cells, causing nearby epithelial and stromal cells to become oxygen-depleted (22). Chronic inflammation exacerbates this response resulting in hypoxia-inducible factor (HIF) activation in immune, stromal, and epithelial cells. Hypoxia-inducible factor 1-alpha (HIF-1α) is a well-known regulator of hypoxic cellular processes, and its activity is mainly controlled by post-translational rather than transcriptomic modifications. During normoxic conditions, HIF-1α levels are kept low by the Von Hippel-Lindau (VHL) tumor suppressor which targets HIF-1α for ubiquitin-mediated proteasomal degradation. However, when oxygen levels become depleted, HIF-1α starts to accumulate and HIF-1α stabilizes and binds to HIF-1β forming a complex that enters the nucleus and binds to hypoxia response elements (HRE) to either promote or repress genes (23, 24). In a mouse model of caerulean-induced acute pancreatitis, injured tissues presented high expression of HIF-1α, and inhibition of HIF-1α, through intraperitoneal injections of HIF-1α small molecule inhibitor PX478, reduced RIP3/p-MLKL expression and ROS production, mitigating acinar cell injury and necrosis (25). In the context of pancreatic cancer, HIF-1α levels are elevated in part due to the desmoplastic stroma and HIF-1α staining and expression strongly associates with PDAC lymph node metastasis, high tumor stage, poor prognosis, and immune evasion (26). A recent study in an autochthonous mouse model of PDAC with pancreas-specific expression of KrasG12D implicates HIF-1α may have a protective role, as genetic deletion of the gene promotes neoplasia. Immunohistochemical staining and ELISA analysis revealed that HIF-1α genetic deletion significantly increases secretion of the B-cell chemoattractant CXCL13, which increases the intrapancreatic accumulation of B cells, as shown through flow cytometry analysis. These data indicate HIF-1α prevents B cell infiltration into hypoxic regions and when B cells were depleted in mice, PanIN development was decreased, implicating B cells promote tumorigenesis in PDAC (27). The expression of Adora2b and its subsequent activation was shown to be elevated by HIF-1α in hepatic ischemia-reperfusion injury mouse models, acute lung injury, liver cancer, and breast cancer (28). During pancreatic diseases, hypoxic conditions tend to develop and both HIF-1α and Adora2b are elevated and involved in the inflammatory process (4, 29), yet, further analysis is needed to fully uncover the potential link between both molecules and their participation in the development of these diseases.
Studies of hypoxia-induced changes in gene expression identified a transcriptional program that promotes CD73 expression in the extracellular vicinity of inflamed tissues (Figure 2). In these studies, Adora2b gene expression is also elevated resulting in an endogenous feedback loop critical for injury resolution and ischemia tolerance under oxygen-deprived conditions (30–32). Transcription of CD73 is regulated by an HRE on the promoter in hypoxic epithelial cells and transcription of CD39 is either upregulated through Sp1 or downregulated through the formation of a HIF-1α and AHR complex with ARNT which decreases AHR recruitment to the CD39 promoter that has three AHR response elements (33–35). HIF-1α inhibits adenosine kinase and ENTs resulting in increased accumulation of adenosine in the tumor microenvironment (19, 20, 36). Another ligand for Adora2b is Netrin-1, a neuronal guidance molecule essential for the proper development of neurons. In PDAC, perineural infiltration is present in early and late stages of the disease and neuronal infiltration by tumor cells may contribute to pain and tumor progression indicating Netrin-1/Adora2b signaling could be evaluated as a therapeutic strategy to reduce perineural infiltration. In addition, signaling of Netrin-1 through the Adora2b receptor also inhibits immune cell infiltration into organs under hypoxic and inflammatory conditions (37–39) indicating several mechanistic consequences for Adora2b in pancreatic and other solid tumors. In addition to Netrin, in vitro data have shown that stimulation and activation of Adora2b by adenosine and NECA promotes cell proliferation and secretion of chromogranin A, a protein that is widely accepted as a biomarker for neuroendocrine tumors. Such findings suggest inhibition of the adenosine pathway, specifically targeting Adora2b receptors, may be of high interest in the therapeutic management of neuroendocrine tumors (40).
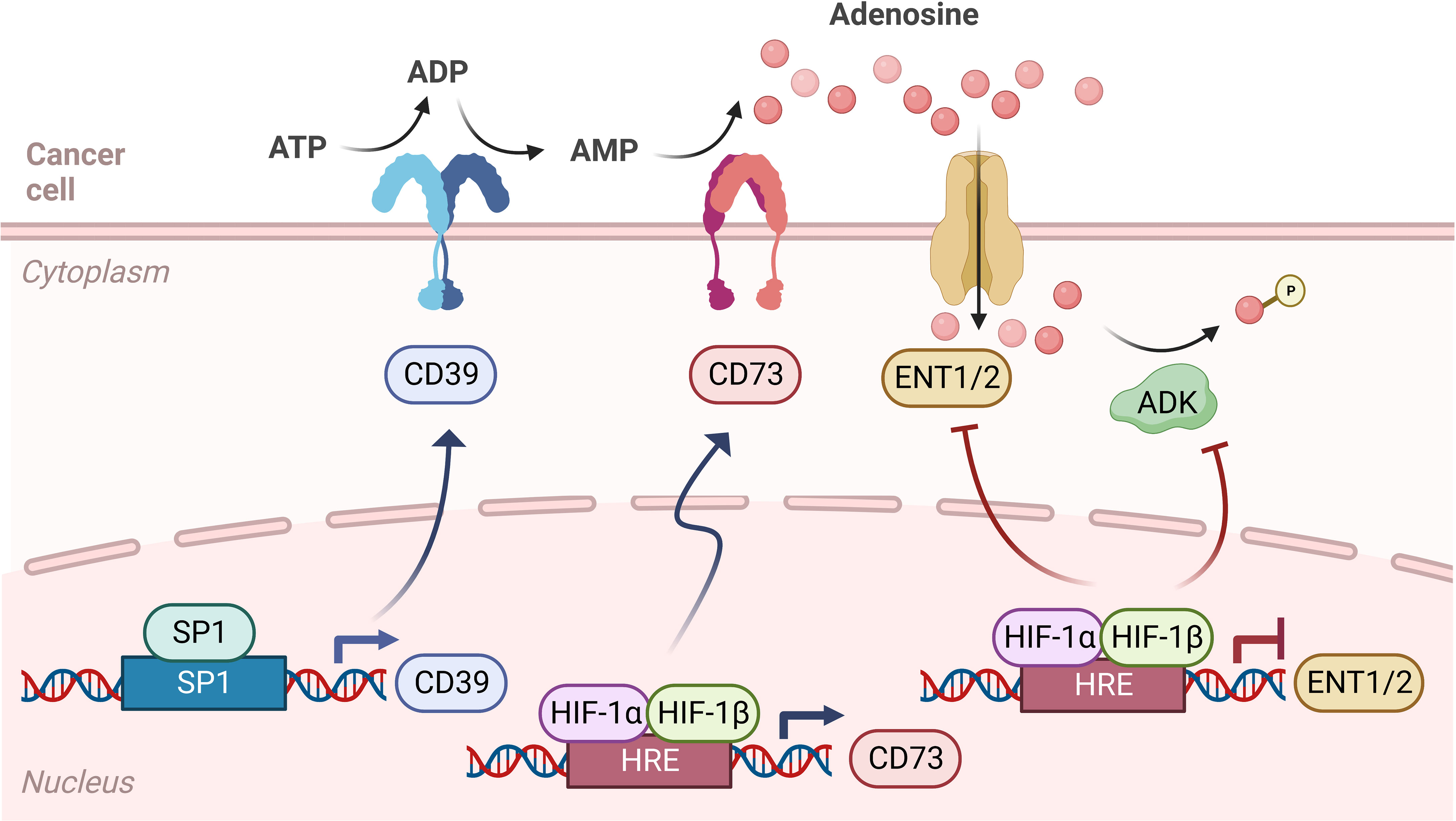
Figure 2 Adenosine signaling pathway during hypoxia. Adenosine signaling in hypoxia is similar to normoxia, as ATP is converted to ADP and AMP by CD39, then converted to adenosine by CD73. However, in hypoxic cancer cells, the transcription of CD39 is upregulated through Sp1, leading to more ADP and AMP in the tumor microenvironment (TME). Also, while levels of HIF-1α are kept low by the Von Hippel-Lindau (VHL) tumor suppressor in normoxic conditions, in hypoxia HIF-1α stabilizes and binds to HIF-1β, which forms a complex that enters the nucleus and binds to hypoxia response elements (HRE) on the gene promoter, therefore regulating the transcription of CD73 and equilibrative nucleoside transporters (ENT1/2). In hypoxia, CD73 transcription is upregulated, while ENT1 and ENT2 transcription is downregulated. HIF-1α also inhibits adenosine kinase and ENTs, leading to an accumulation of adenosine in the TME.
Another component of the PDAC TME is the vasculature, which is characterized by high microvascular density yet poor perfusing in the vessels and decreased vascular integrity. In PDAC patients, the superior mesenteric vessels are commonly involved, especially when tumors arise in the head of the pancreas. These clinical features of PDAC are notable in the context of adenosine signaling as hypoxia-mediated adenosine signaling influences vascular responses. In the context of inflammation, neutrophils exit the bloodstream through transendothelial migration (TEM) and secrete ATP and ADP resulting in high adenosine concentrations (41–44). Studies exploring the role of adenosine receptors in vascular leakage were completed in mice that were deficient in either Adora1, Adora3, Adora2a, or Adora2b, then subjected to hypoxia. While the Adora1, Adora3, or Adora2a mice did not have an increase in hypoxia-induced vascular leakage, the Adora2b deficient mice showed a significant increase in hypoxia-induced vascular leakage. Furthermore, administration of the Adora2b antagonist PSB1115 to wild-type mice also significantly increased neutrophil infiltration through TEM and worsened vascular leakage while administration of Adora2b agonist BAY-60-6583 reversed the hypoxia-induced vascular leakage. These findings suggest Adora2b has a key role in controlling hypoxia-associated vascular leak by increasing endothelial cell intracellular levels of cAMP which promotes vasculature resealing (31, 45). These studies suggest adenosine signaling events can be targeted to dampen hypoxia-induced inflammation and prevent excessive tissue damage (13, 30). In solid tumors with a hypoxic TME, Adora2b antagonists may promote increased infiltration of immune cells and anti-tumor immunity.
Functional consequences of adenosine receptor signaling in inflammation and cancer
Adora1 and Adora3 receptors
The Adora1, or adenosine A1 receptor, is a G protein-coupled receptor (GPCR) that, when bound to an agonist, causes Gi1,2,3 or G0 protein binding. Adora1 is ubiquitously expressed in the body and, when Gi1,2,3 is bound, adenylate cyclase is inhibited, and cAMP concentrations are decreased. This has important consequences in several fundamental biological contexts including slowing heart rate (46, 47), reducing glucose-induced insulin secretion (48), reducing blood flow, and promoting edema during acute pancreatitis (49). In the context of cancer, Adora1 overexpression has been published to facilitate the malignant progression of colorectal, kidney, and breast cancers, as well as glioblastoma and leukemia (50). Inhibition of Adora1 in combination with immune checkpoint blockade (ICB) therapy targeting PD-1 has shown promising therapeutic effects in non-small cell lung cancer and melanoma (51). In contrast, studies evaluating the role of hypoxia in the pancreas reveal Adora1 is downregulated during hypoxia (52) and analysis of RNA-seq data from The Cancer Genome Atlas (TCGA) database indicated this receptor was not associated with PDAC prognosis (48). Thus, the role of Adora1 in response to hypoxia or other environmental triggers of adenosine is dependent on tumor type and organ of origin.
The Adora3 or adenosine A3 receptor couples to Gi/Gq proteins. Like Adora1, Adora3 receptor activation promotes Gi protein binding and decreased adenylyl cyclase activity which reduces cAMP intracellular levels. Adenosine signaling through Adora3 has been shown to participate in the degranulation and activation of mast cells important in asthma pathogenesis (53–55). Adora3 also modulates cytokine release via T cell-mediated production of IL-10 which helps reverse neuropathic pain (56) and through down-regulation of nuclear factor-kappa B signaling results in the inhibition of inflammatory cytokine production in the colonic mucosa of patients with ulcerative colitis (57). Unlike the Adora1 receptor, hypoxic conditions do not affect Adora3 expression (52). In the context of the pancreas, low levels of Adora3 receptor expression have been reported and Adora3 is not associated with PDAC prognosis (48).
Adora2 receptors
Adora2 adenosine receptors consist of the adenosine A2A (Adora2a) and A2B (Adora2b) receptors, both of which are Gs-coupled GPCRs. In the pancreas, Adora2a and Adora2b have many similarities, as they both are present in the luminal membrane of ductal, insulin-positive beta, and PECAM-+ endothelial cells (11). Agonist binding to Adora2 receptors stimulates cAMP, a membrane-associated protein kinase A (type II PKA), and cAMP-activated Cl- channels which mediate critical pancreatic ductal secretions (48). Adora2a is the most abundant adenosine receptor in the pancreas and it participates in endocrine pancreatic functions as well as water and bicarbonate secretion responses (48). Adora2a is also a potent anti-inflammatory regulator as its activation limits immune cell activity during an inflammatory response preventing additional tissue damage (16, 58, 59). In studies carried out in mice lacking Adora2a receptors, behavioral alterations are present, suggesting the participation of Adora2a in regulating neuronal populations (60). In caerulein-mediated mouse models of pancreatitis, inhibition of adenosine uptake using a pharmacologic inhibitor enhanced stimulation of the Adora2a receptor, and was capable of reducing the severity of pancreatitis (61). Specifically, in pancreatic cancer patients, studies show CD73 and Adora2a expression on neoplastic or tumor cells correlates with divergent immune cell populations in the tumor microenvironment. In a publication by Sweed et al, when Adora2a is overexpressed in human PDAC patients, there are correlative high levels of tumor-infiltrating mononuclear cells (TIMC), associated with larger tumor sizes (62). Moreover, in an immunohistochemical study performed on 48 human PDAC tissues, Adora2a was overexpressed, and high Adora2a PDAC expression was associated with more aggressive cases and later tumor stages at the time of diagnosis (62). While no functional experiments were reported in this manuscript, these data indicate both autocrine and paracrine adenosine signaling through Adora2a are important in the pathogenesis of pancreatic cancer.
The Adora2b receptor is the only low-affinity adenosine receptor [Adora2b EC50 = 24 µM, Adora2a EC50 = 0.7 µM, Adora1 EC50 = 0.31 µM, Adora3 EC50 = 0.29 µM (63)], requiring high levels of extracellular adenosine to become activated rather than existing in a resting state (64). Adora2b is present in myocardial cells, epithelial cells, fibroblasts, and several immune cell types (65) and in many disease models is a potent anti-inflammatory regulator. However, controversial findings exist around its role in disease, fibrosis, and tumor development. Across several mouse models of acute injury, Adora2b activation has shown protective effects, either by modulating IL-10 production on the intestinal epithelium (66), stabilization of circadian rhythm protein (67) or enhancing alveolar fluid clearance in mice (68). Additionally, studies in Adora2b deficient mice showed enhanced pulmonary recruitment of effector T cells and failed induction of regulatory T cells during endotoxin-induced inflammation resulting in increased severity of the disease. Similarly, in a pulmonary disease mouse model, induction of Adora2b signaling attenuated inflammation and edema only in wild-type mice but not in mice lacking expression of the receptor (69, 70). Contrarily, the absence of Adora2b in an ulcerative colitis mouse model ameliorated acute intestinal inflammation, suggesting this receptor plays a pro-inflammatory role in the development of this disease (71, 72).
In cancer, there are also conflicting studies related to the function of Adora2b in the progression of different malignant diseases. High Adora2b levels are associated with a better prognosis in patients with ovarian cancer. In vitro pharmacological activation of Adora2b in ovarian carcinoma cells reduced cell migration and actin stress fiber expression (7). However, detrimental effects were observed for mammary carcinoma, hepatocellular carcinoma, lung adenocarcinoma (LUAD), and PDAC. Adenosine signaling through Adora2b in breast cancer cells regulates the tumor microenvironment and enhances pro-tumorigenic actions in cancer-associated fibroblasts, effects correlated with increased metastatic potential and poor prognosis (73). In hepatocellular carcinoma, Adora2b receptor blockage enhanced the benefits of sorafenib treatment by suppressing the inhibitory effects of adenosine on CD8+ T cells (74). Bioinformatic studies in LUAD and PDAC revealed Adora2b expression and associated signaling pathways predicted poor prognosis and significantly reduced overall survival (48, 75).
The function of Adora2b receptor on immune cells: implications for targeting to promote anti-cancer immunity
Adora2b in innate immunity
Comprised of many cell types including macrophages, dendritic cells, natural killer cells, natural killer T cells, γδ T cells, and more, the innate immune system provides a rapid response to foreign antigens, and the innate immunity antitumor response triggers effector mechanisms to contain the tumor. Adenosine binding to the Adora2b receptor has important functional consequences on innate immune cells (Figure 3). Macrophages impart critical functions in the resolution of inflammation and a return to normal tissue conditions. Their principal function is to clear dead cells from inflamed tissues through a process called efferocytosis, which also resolves inflammation by suppressing pro-inflammatory cytokines and stimulating anti-inflammatory cytokines. Adora2b on bone marrow-derived macrophages (BMDM) was discovered through flow cytometry experiments done in mice and functionality of the receptor was assessed by Adora2b agonist, 5’-N-ethylcarboxamidoadenosine (NECA), which resulted in increased cAMP levels in cultured BMDM (76, 77). Adora2b is upregulated on macrophages by IFN-γ and when Adora2b is activated, TNF production in infiltrating macrophages is suppressed, inhibiting their capacity to secrete cytokines important for anti-tumor immunity and promoting tumor growth (78).
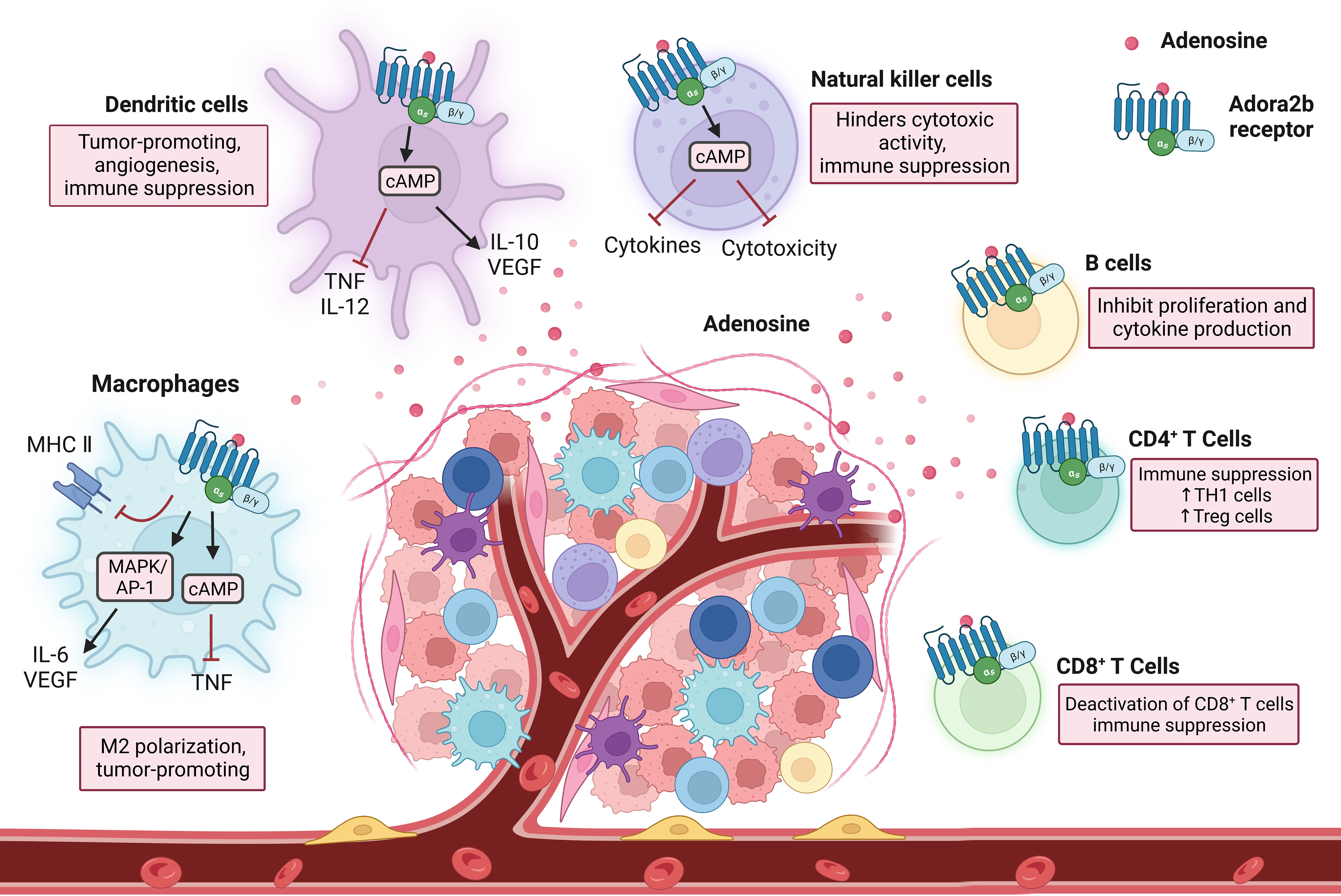
Figure 3 Immune cell interactions in response to Adora2b activation. As immune cells enter into the TME and encounter adenosine through the Adora2b receptor, they undergo changes resulting in immunosuppression. Adora2b activation on macrophages contributes to M2 polarization and tumorigenesis, as it inhibits MHC II expression, activates MAPK/AP-1 which increases IL-6 and vascular endothelial growth factor (VEGF) production, and increases cAMP levels which inhibits tumor necrosis factor (TNF) production. On dendritic cells, stimulation of Adora2b increases cAMP production which inhibits TNF and IL-12 production and increases IL-10 and VEGF release, resulting in tumorigenesis, angiogenesis, and immune suppression. Natural killer cells encounter adenosine through the Adora2b receptor and the cAMP pathway is activated resulting in blocked cytokine production and cytotoxicity, contributing to immunosuppression. On B cells, Adora2b activation results in the inhibition of proliferation and cytokine production. CD4+ T cell activation of Adora2b increases TH1 cell and Treg cell levels, as well as contributes to immune suppression. On CD8+ T cells, Adora2b activation results in the deactivation of CD8+ T cells and contributes to the suppression of the immune system.
Dendritic cells (DCs) are antigen-presenting cells and critical determinants of both innate and adaptive immunity. They dwell in peripheral tissues in an immature state and, when exposed to triggers, transform into differentiated and mature DCs. Stimulation of Adora2b on DCs stimulates maturation into a differentiated population with DC markers and monocyte or macrophage markers, allowing mature DCs to interact with T lymphocytes and promote CD4+ differentiation into Th1 cells through IL-12 production. DCs differentiated due to exposure to adenosine have decreased allostimulatory activity and express high levels of angiogenic, immune suppression, pro-inflammatory, and tolerogenic factors, such as COX-2, IDO, IL-6, IL-8, IL-10, TGF-β, and VEGF (79, 80).
Natural killer (NK) cells are critical in responses to stress and infections. Many types of NK cells have NK receptors (NKRs) that determine if a cell encountered by an NK cell becomes a target for destruction or is protected (81). When activated NK cells encounter adenosine through the Adora2b receptor, the cAMP pathway is activated and cytotoxic activity and cytokine production is blocked, contributing to reduced anti-tumor activity (82, 83). While NKs and natural killer T cells (NKTs) have many similarities, they are very different in the context of cancer. Both cell types display effector properties in early cancer stages and have impaired functionality in later stages. NKT cells become exhausted in advanced cancers and have an irregular metabolism. NKTs have exhaustion markers such as high CTLA4, PD1, and Tim3, as well as low granzyme B levels, and reduced cell numbers as cancer progresses further (84). Limited studies have been done assessing the role of the Adora2b receptor in NKT cells.
γδ T cells are a rare subtype of T cells, bridging the gap between the innate and adaptive immune system components, they possess both γ and δ T cell receptor chains. They have gained traction in the area of immunotherapy as they have an anti-tumor immune function and are critical in immune surveillance. Analysis of TCGA data has shown PDAC patients with high CD73 levels have lower amounts of γδ T cells (85). These cells are regulated by extracellular adenosine levels, and in mice treated with an Adora2b agonist, the DCs activate γδ T cells, elevating Th17 responses (86). When γδ T cells induce an elevated Th17 response, this contributes to the pathogenesis of autoimmune diseases and can be a target in inflammation-related diseases such as cancer. However, the specific role of the Adora2b receptor in this cell type is unknown and should be explored further.
Adora2b in adaptive immunity
Comprised of B cell and T cell subtypes, the adaptive immune system is responsible for recognizing and attacking specific antigens. B cells are lymphocytes that produce antibodies tagging specific antigens for destruction and play an important role in hypoxia and inflammation in the TME in PDAC. B cells express both CD73 and CD39, and the production of extracellular adenosine by B cells can inhibit T cell proliferation and the production of IL-10 cytokines. However, B cells have very low levels of the Adora2b receptor and few studies have been conducted to determine its role in B cell interactions (87).
T cells are a crucial group of cells in the immune system that generally express CD73, CD39, and the Adora2b receptor. The presence of Adora2b on T cells was confirmed through flow cytometry and the functionality of the receptor was determined by increased cAMP levels in the cells induced by an Adora2b agonist. Extracellular adenosine limits T cell mobility and increases cAMP levels in T cells, contributing to Adora2b-mediated immune suppression (87, 88). Helper T cells are CD4+ T lymphocytes that stimulate other immune cells to respond to infection and when activated, Adora2b receptor levels increase on the CD4+ T cell surface (88). In a model of endotoxin-induced pulmonary inflammation, mice with a genetic knockout of Adora2b had an enhanced CD4+ T cell response, resulting in increased inflammation (69). Adora2b on CD4+ T cells contributes to immunosuppression and could be a target in cancer, but additional studies are needed to learn more about the role of the receptor on CD4+ T cells. Cytotoxic T cells are CD8+ T cells that are important in protection against tumor growth, as they trigger apoptosis of pathogenic cells. In an in vitro experiment, activation of CD8+ T cells through an unspecific activation signal (phytohemagglutinin) and by a specific activation signal (the anti-T cell receptor/CD3 complex mAb, OKT3) triggers increased Adora2b levels and a decrease in IL-2 production (88). Through TCGA and The Cancer Immune Atlas analyses, PDAC patients with high CD73 levels had lower amounts of CD8+ T cells (4, 85). In studies performed in mice with genetic deletion of Adora2b, when murine PDAC cell lines derived from Pdx1:Cre; LsL-KrasG12D;LsL-Trp53R172H/+ (KPC) mice, were implanted subcutaneously, tumor growth was significantly reduced compared to implanted cells in WT mice and there was a significant increase in Granzyme B (GZM+) and CD8+ T cells in KPC-derived tumors implanted in Adora2b-/- mice (4). These data indicate paracrine adenosine Adora2b signaling restrains cytotoxic CD8+ T cell function. Also, in complimentary studies, wild-type mice treated with PSB1115, an Adora2b antagonist, had reduced KPC subcutaneous tumor growth compared to vehicle-treated KPC tumor-bearing mice. However, in wild-type mice without CD8+ T cells, treatment with the PSB1115 did not inhibit the growth of the KPC subcutaneous tumors indicating paracrine adenosine signaling through Adora2b on CD8+ T cells reduces their anti-tumor properties in PDAC (4). Future studies using genetic models or orthotopic implantation of KPC cells into the pancreas will aid in further delineating the role of Adora2b in pancreatic cancer.
Adora2b function in exocrine pancreatic diseases
The pancreas is comprised of both endocrine and exocrine cells. Specifically related to exocrine function, acinar cells organize into acini and constitute 70-90% of pancreatic cells while 5-25% of exocrine pancreatic cells are ducts. Acinar cells are responsible for releasing digestive enzymes and Cl- rich fluid, while ducts release bicarbonate pancreatic juice to neutralize stomach acidity and deliver acinar cell-derived enzymes to the duodenum (89, 90). The characteristic zymogen granules in acini store intracellular ATP at 10uM concentrations (91, 92). In a healthy pancreas, ATP is secreted by acinar cells into the ducts where P2 receptors regulate Cl- and K+ ion channels, cAMP signaling, and transporters resulting in ductal secretion of NaHCO3-rich fluid (93). Acini and ducts have both been shown to express CD39 and CD73 which generate luminal adenosine that signals through ductal P1 receptors Adora2a and Adora2b which stimulate the cystic fibrosis membrane conductance regulator Cl- channels important for ductal function (94). While less numerous, accounting for approximately 3-5% of pancreatic parenchyma, endocrine-functioning islet cells are critical for glucose homeostasis, and pancreatogenic (Type3c) diabetes can occur in a subset of patients with acute or recurrent acute pancreatitis (48, 95, 96). Both human and rodent ducts express adenosine receptors, with Adora2a and Adora2b being the most prevalent in these cells. When these receptors are stimulated, Cl- channels are opened and allow ductal secretions to occur indicating purinergic signaling is important for pancreas function and homeostasis (29, 48, 97, 98) (Figure 4, left panel).
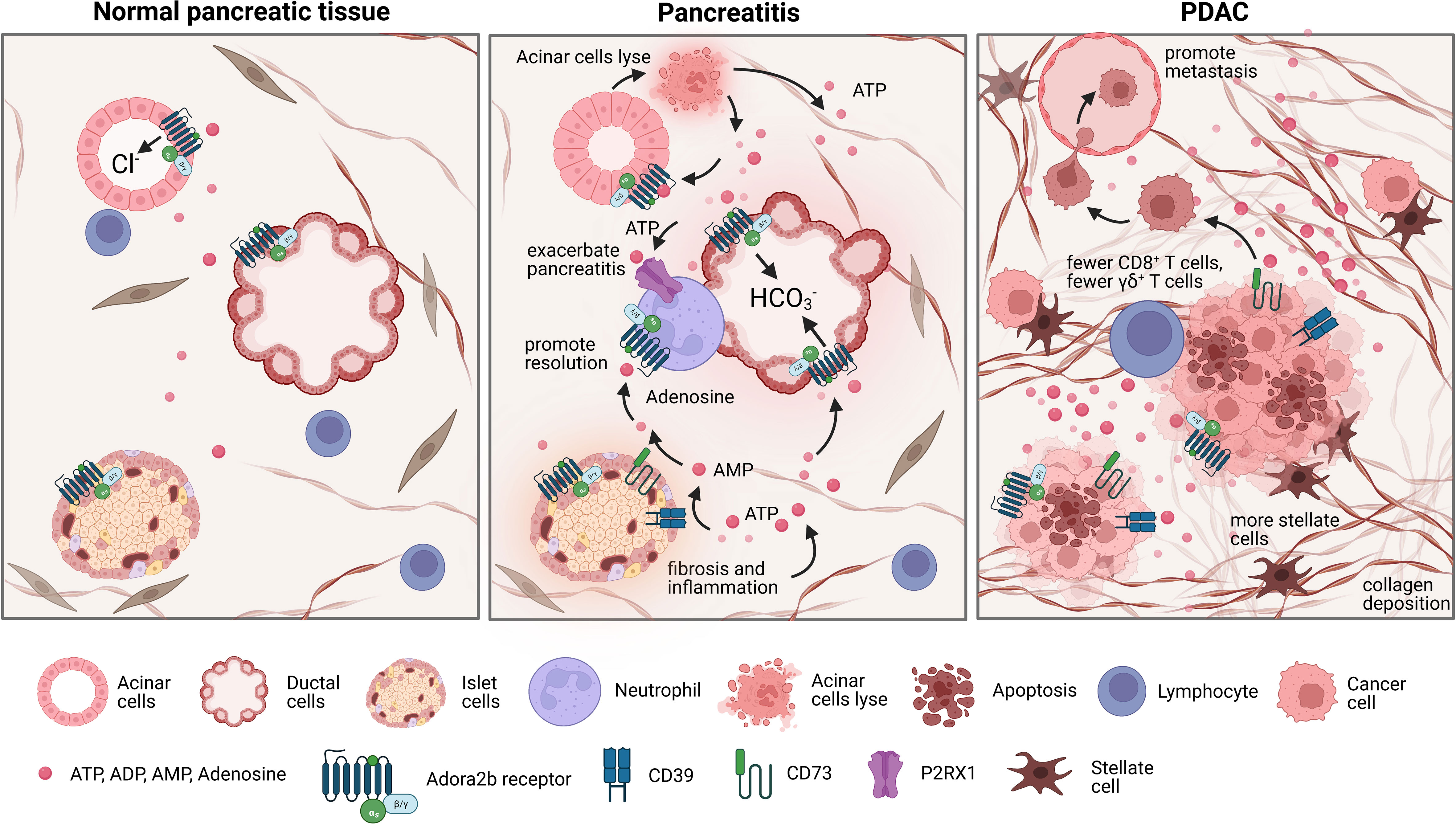
Figure 4 Changes in the pancreatic landscape in response to pancreatitis and pancreatic ductal adenocarcinoma (PDAC). Normal pancreas tissue is comprised of acinar cells that release digestive enzymes and Cl- rich fluid, ductal cells that release bicarbonate pancreatic juice, and islet cells that maintain glucose homeostasis. However, during pancreatitis acinar cells lyse, releasing ATP into the extracellular environment, promoting elevated purinergic signaling which leads to altered bicarbonate secretion levels and exacerbates inflammation. In chronic pancreatitis, damage to islet cells contributes to increased fibrosis and inflammation, promoting high extracellular ATP levels and increased adenosine signaling. Neutrophils also contribute to exacerbating pancreatitis by expressing P2RX1 which promotes glycolytic metabolism. Contrarily, adenosine can inhibit the inflammatory function of neutrophils through Adora2b mediated deactivation, which partially promotes the resolution of pancreatitis. PDAC is characteristically immunosuppressive and possesses a dense desmoplastic stroma with a hypoxic necrotic core. In the necrotic core, there are high levels of extracellular ATP and higher levels of CD39 and CD73, which leads to an accumulation of adenosine in the TME which can then bind to Adora2b and contribute to immunosuppression. This also leads to fewer γδ+ T cells, more collagen deposition, and more stellate cells.
In the pathophysiology of acute pancreatitis, the enzymes zymogen and trypsinogen are released due to premature activation of acinar cells resulting in local parenchymal destruction and activation of inflammatory pathways. When acinar cells prematurely secrete enzymes, they also secrete ATP and other inflammatory signals into the extracellular environment (99–101). High extracellular ATP levels promote elevated purinergic signaling which leads to altered bicarbonate secretion from pancreatic ducts, ductal dilation, infiltration of innate immune cells, and increased severity of pancreatitis (11). Purinergic receptors are expressed on neutrophils and are key chemoattracts for these cells, which elevate pancreatic inflammation and the severity of pancreatitis. Thus, the conversion of ATP by CD39 and CD73 to adenosine is an important anti-inflammatory mechanism to return the pancreas to normal homeostasis after acute injury (102). Recent studies using single-cell RNA sequencing reveal CD73 is expressed in T cells and ductal cells in murine and human models of chronic pancreatitis (103). During chronic pancreatitis, not only are acinar cells severely injured, but also islet cells, with increased fibrosis and inflammation. This causes an extreme accumulation of extracellular ATP and exacerbated purinergic signaling (11) as well as increased infiltration of P2RX1 expressing neutrophils (102). Anti-inflammatory adenosine Adora2b signaling on ducts, neutrophils, and insulin-producing beta cells is therefore critical to promote healing after acute and chronic pancreatic injury (Figure 4, middle panel). Adora2b signaling reduces netosis formation and reduces oxidative burst from neutrophils, critical functions that reduce neutrophil-mediated inflammation during pancreatitis (104, 105). Future studies to determine the exact role of Adora2b receptor signaling in acute and chronic pancreatitis are important for future therapeutic considerations.
PDAC has a characteristically immunosuppressive TME where tumor cells coexist with exhausted and deactivated immune cells within a dense hypoxic desmoplastic stroma and necrotic tumor core (27). Understanding and targeting mechanistic triggers of immune suppression is one therapeutic approach being testing in preclinical and clinical trials. In a recent immunohistochemical study on human PDAC tissues, Jacoberger-Foissac et al. found that worse prognosis occurred only when patients present with elevated expression of both CD39 and CD73. When CD39 levels are high but CD73 levels are low, there is an increase of CD8+ T cells; however, this effect is not present when CD73 levels are also high, reaffirming that production of adenosine limits CD8+ T cell infiltration into PDAC tumors (6). Elevated expression of CD39 and CD73 has also been associated with fewer γδ+ T cells, more collagen deposition, and more proliferation of stellate cells indicating adenosine signaling may also be a critical determinant of fibrosis and desmoplasia in pancreatitis and pancreatic cancer (85, 106, 107). Three recent publications have utilized preclinical mouse models to evaluate the role of adenosine signaling in pancreatic cancer and have collectively shown genetic deletion of CD73 or treatment with CD73 small molecule inhibitors in syngeneic or genetic mouse models significantly reduces the development and progression of pancreatic cancer and promotes increased anti-tumor immunity; however, there are some differences in the models and findings which we want to highlight (4–6). In a publication by King et al, the authors performed a metabolic screen and found elevated CD73 correlated with aggressiveness of disease. The authors genetically deleted Nt5e/CD73 in murine PDAC cells and used an orthotopic model to show deletion of CD73 significantly ablated tumor growth and reduced the abundance of infiltrating MDSCs. They further show the anti-tumor immune response in Nt5e depleted tumors was associated with CD4+ and CD8+ T cells expressing IFN γ and showed the response was dependent on CD4+ T cells, but not CD8+ T cells (5). In a second publication by Jacoberger-Foissac et al, CD39 expression on CD8+ T cells was shown to suppress IFN γ production by T cells and transplantation of murine KPC tumors, myeloid expression of CD39 and CD73 and tumor expression of CD73 promoted polarization of myeloid cells to an M2 phenotype, which promoted PDAC growth and targeting both CD73 and CD39 significantly enhanced the anti-tumor T cell response. These findings were both done in the transplanted or orthotopic setting. Similarly, in the publication by Faraoni et al, inhibition of CD73 in murine genetic (spontaneous) models of pancreatic cancer, significantly reduced cancer development in spontaneous models with higher expression of CD73 in the neoplastic and cancer cells. Notably, pharmacologic inhibition of CD73 correlated with a significant increase in activated CD8+GZM+ T cells and F4/80+ cells in both genetic models. The authors then expanded these studies to a subcutaneous model to show inhibition of CD73 or the Adora2b receptor reduced the growth rate of murine KPC tumors. A limitation of the subcutaneous model is it does not recapitulate the microenvironment of the pancreas or the desmoplastic response in the pancreas. However, in this model, Faraoni et al. show the reduction in tumor growth using a small molecule inhibitor of Adora2b is dependent on CD8+ T cells. These studies were conducted to expand beyond the findings using CD73 inhibitors in spontaneous, orthotopic and subcutaneous models as we show in the publication by Faraoni et al, that PDAC patients with high ADORA2b have reduced survival and poor prognosis. In addition, we have shown using Quantiseq and The Cancer Immune Atlas analysis that patients with high ADORA2b or high CD73 have decreased NK cells, CD8+ T cells, B cells, and M2 macrophages (4). In studies using implantation of murine KPC tumors into WT or Adora2b-/- mice, we show a significant reduction in tumor growth in tumors arising in Adora2b-/- mice compared to WT mice. Pharmacologic inhibition of Adora2b also restrained tumor growth in vivo; however, the effect of the small molecule inhibitor was not present in tumor growth in CD8KO mice indicating adenosine signaling through Adora2b significantly restrains CD8+ T cell anti-tumor activity in PDAC (4) (Figure 4, right panel). These data indicate that co-inhibition of CD73 and Adora2b may provide additional therapeutic targeting to activate anti-tumor immunity and improve outcomes for PDAC patients.
Adora2b function in metastasis
Greater than 90% of cancer-related deaths are due to metastasis, illustrating an urgent need for an improved understanding of mechanisms driving metastasis and ways to prevent metastases from forming. Traveling through the bloodstream, rogue cancer cells create metastatic cancer nodules that are highly resistant to therapies (108). In experimental mouse models of melanoma and triple-negative breast cancer metastasis, the incidence of metastasis is significantly decreased when mice are treated with an Adora2b antagonist (109). Similarly, genetic deletion of the Adora2b receptor in mouse and human triple-negative breast cancer cells reduces their metastatic capability in vivo (109), suggesting an important role for Adora2b in cancer metastasis. Recently, it was also shown that antagonizing Adora2b expression in gastric cancer cells increased the efficacy of cisplatin treatment (110). However, despite these promising results in melanoma, breast cancer, and gastric cancer cells, the specific role of Adora2b in metastatic development remains unknown. Metastasis is especially common in PDAC patients, due to the unfortunate ability of PDAC tumor cells to evade the exhausted and suppressed immune system. Future studies will be needed to further demonstrate the potential role of Adora2b in pancreatic cancer metastasis as well as their potential impact on this and other diseases.
Experimental considerations for targeting autocrine and paracrine Adora2b signaling
PDAC organoids and cell lines
Organoid models are a highly translational model system and provide an ex vivo approach to studying healthy pancreas and PDAC. Derived most from human or murine tissues, they are 3D and capable of self-renewal as well as spontaneous self-organization, providing a unique opportunity to study therapeutic approaches to augment personalized medicine, therapeutics, and mechanisms of resistance (111–114). Pancreatic organoids can also be orthotopically implanted after cryopreservation or genetic manipulation allowing more rapid studies of mechanistic drivers of PDAC development and metastasis in vivo. Noteworthy, it is important to mention that although organoids offer an interesting platform to test therapeutic drugs and can be applied to many different cell types and diseases, they still lack a high-fidelity cell type composition, have limited maturation, and have an atypical physiology which does not always can recapitulate or mimic interactions between molecules when compared to the physiologically normal and/or tumor microenvironments, which limit their applicability and reliability for certain tumor studies (115). If organoid models are not available, human PDAC cell lines can also be used as an in vitro mechanistic approach to study cell autonomous and non-cell autonomous purinergic signaling. Established cell lines from human PDAC primary tumors are BxPC-3, Capan-2, HPAC, MIA PaCa-2, and Panc-1. BxPC-3 is the only cell line mentioned which is wild type for KRAS and does not represent the majority of PDAC tumors, which have somatic mutations in KRAS (116). For each of these human cell lines, experiments can be done with Adora2b agonists, Adora2b antagonists, siRNA, or CRISPR/Cas9 mediated genetic deletions, to study the cell-autonomous upstream and downstream effects of adenosine signaling through the Adora2b receptor. The KPC cell line is also a very common murine PDAC cell line with mutations in Trp53 and Kras.
Mouse models
Mouse models are essential to studying pancreatic cancer and there are numerous models which would be useful to study the Adora2b receptor and its role in PDAC. First, there are syngeneic models utilizing subcutaneous or orthotopic implantation of KPC cells into the flank, pancreas, spleen, or any combination of these injection sites. These models are useful for studying treatment options using Adora2b antagonist compounds in primary tumors and metastatic sites (4). There are also genetically engineered mouse (GEM) models that can be used, such as the KPC and Pdx : Cre;LsL-KrasG12D (KC) models. The KPC mice have mutations in Kras, mutations or genetic deletion of Trp53, and use Cre-Lox technology through Cre recombinase gene insertion into Pdx-1 or Ptf1a (p48-Cre) coding exons. KPC mice begin to develop PDAC precursor lesions around 8-10 weeks of age and have PDAC by 4 months of age (117). KC mice are advantageous for prevention studies as they have slow development from PanIN to PDAC over a time frame of 12-15 months (118). Future studies in GEM models could also be used to test different Adora2b antagonist compounds in vivo and to study immune cell interactions in the preventive or therapeutic setting. Using cell-specific inducible CreER alleles crossed to an Adora2b floxed allele, genetically engineered mouse GEM models can be generated with genetic deletion of Adora2b in specific cells or tissues. Mice without Adora2b receptors in the defined immune cells, stromal cells, or vasculature could also be useful to study the role of the receptor in PDAC in the future.
Adora2b agonist and antagonist compounds
Selective adenosine agonists and antagonists have been described for the Adora2b receptor and support the protective and anti-inflammatory mechanistic consequences of Adora2b signaling. Particularly in pancreatic diseases, 5’-N-ethylcarboxamidoadenosine, commonly abbreviated as NECA, was recently administered in a model of pancreatitis and described as a suitable Adora2b agonist which may be involved in tissue regeneration and restraint of MPO accumulation and metaplasia during acute pancreatitis; however, no specific therapeutic applications of NECA have been described to date in the clinic (29). Though studies have shown short-term adenosine exposure is highly effective at reducing pain and inflammation, high levels of adenosine have been reported to increase tissue damage and may increase inflammation and potentiate protumor adenosine signaling (119). For these reasons, Adora2b antagonist compounds could be potential therapies in cancer (120). Notably, some of the Adora2b antagonists have been described to decrease the secretory rate of the pancreas by 25% and increase insulin production levels (48). Mice bearing KPC subcutaneous tumors treated with Adora2b antagonist PSB1115, presented with significantly decreased KPC tumor growth and significantly decreased fibrosis measured by IHC for α-SMA. These studies highlight the complex dynamics of this pathway and the urgent need for preclinical and clinical evaluation of targeting Adora2b receptor signaling to better deduce its role in immunity, fibrosis, and cancer (4) (Figure 5).
Figure 5 Commercially available compounds targeting Adora2b. (A, B) Chemical structures of Adora2b agonist compounds BAY-60-6583 and NECA. (C) Chemical structure of Adora2b antagonist compound PSB 1115.
Current therapeutic opportunities and clinical trials
Studies have shown there is an estimated time of 10 years between the moment at which a pancreatic epithelial cell undergoes an oncogenic hit and the time of diagnosis (121), which provides a wide window of opportunity for the detection and prevention of precancerous lesions including pancreatic intraepithelial neoplasia (PanIN). A recent study of healthy human pancreata has shown PanIN are present in individuals irrespective of age and these PanIN have transcriptional signatures that share similarities to cancer cells (122). Despite this recent finding, the lack of technology or systemic biomarkers available for performing early detection allows precursor lesions to progress to a point where, when detected, PDAC is diagnosed at advanced stages and is unresectable in 70-80% of patients diagnosed. Thus, there is a need to test and evaluate new approaches in patients with locally advanced or borderline resectable pancreatic cancer including the use of immunomodulators in the neoadjuvant setting. Considering recent publications showing Adora2b reduces the cytotoxic functionality of NKT and CD8+ T cells, there is an increased premise to evaluate inhibiting Adora2b signaling in the prevention setting. Targeting the adenosine signaling pathway at the preclinical stage has been an intense area of study in recent years and future studies in GEM models of PanIN initiation and progression to PDAC would aid in determining if targeting this pathway has clinical promise. Preclinical studies utilizing checkpoint blockade combined with ectoenzyme blockade approach through inhibition of CD39, CD73, PD-1/PDL-1, and the various adenosine receptors may show enhanced antitumor immunity, decreased tumor initiation, and metastasis, but have not yet been evaluated. As a tightly balanced extracellular amount of both adenosine and ATP is needed to maintain an adequate immune response, therapeutic combinations of CD39 with PD1/PDL1 with and without chemotherapy are being studied (123, 124). There are also ongoing clinical trials targeting the Adora2a receptor in combination with CD73 or PDL-1 inhibitors (Table 1) (125–128). Adora2a blockade studies are also ongoing in combination with PD-1, PDL-1, or chemotherapy (11). However, there are no current clinical trials specifically targeting the Adora2b receptor. It is important to consider the complex interactions between purinergic receptors and ATP/ADP/adenosine signaling, because receptor blockade may impact unwanted cell types and promote unintended effects on other receptors (11). For clinical and therapeutic considerations, there is also a need to evaluate the role of the Adora2b receptor in regulating perineural infiltration, fibrosis, and vasculature as the PDAC microenvironment is dynamic and recent studies have shown multiple subtypes of PDAC can co-exist in patients with pancreatic cancer.
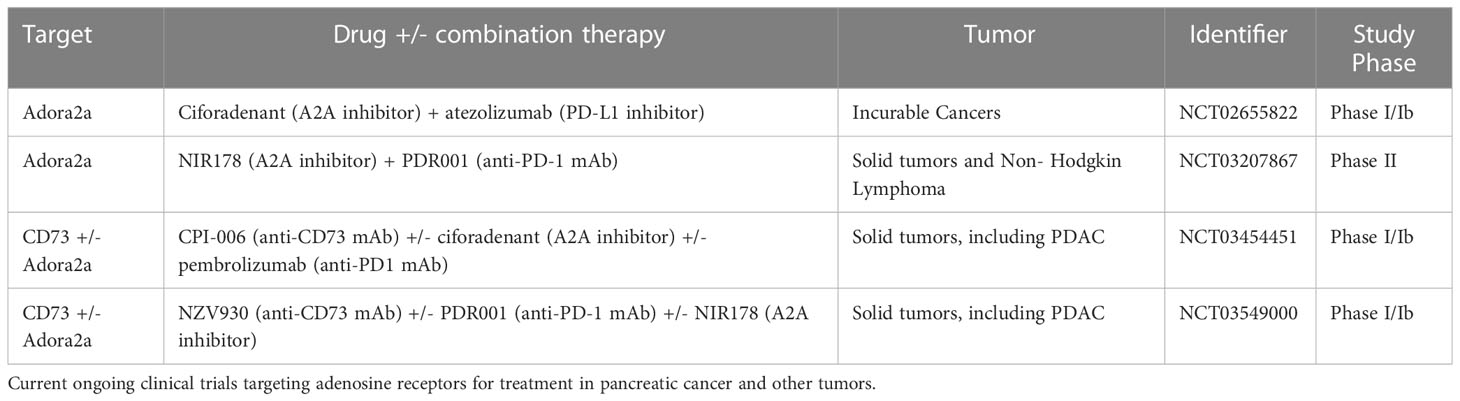
Table 1 Current clinical trials.
Challenges to the field
Pancreatic cancer is a particularly challenging field to study, as it is extremely complex, and tumor genetic and histologic heterogeneity is prominent when comparing patient tumor samples. The advent of sequencing human PDAC tumors has revealed PDAC subtypes (129–132) and Squamous and Basal subtypes have been reported to have the highest expression of CD73 (4) indicating they may have more pronounced intratumoral levels of adenosine. One of the challenges to this field is that most studies of adenosine receptor signaling, and interactions are performed in mouse models, which may not translate directly into humans. This limitation, while applicable to most, if not all preclinical studies, makes it difficult to accurately translate therapies targeting adenosine receptors into human patients, as there may be unintended side effects or limitations of small molecule inhibitor activity or delivery not observed in murine models. Another complication to using mouse models is the immense time requirement to breed genetically engineered mice that more accurately represent human PDAC progression. Despite these limitations, more preclinical and clinical studies need to be done to more accurately evaluate the role of adenosine signaling and possible resistance mechanisms to small molecular inhibitors targeting this pathway in cancer as most studies conducted on extracellular purinergic and adenosine signaling have been in diseases other than pancreatic cancer including acute lung disease, acute liver disease, asthma, diabetes, myocardial ischemia, sickle cell disease, and IBD. Another challenge related to the field of use of Adora2b small molecule inhibitors for immunotherapeutic consideration is that few studies have been performed exploring specifically the Adora2b receptor on individual tumor cells, fibroblasts, or immune cell types in the context of the tumor microenvironment. Studies using human or murine organoid cultures and genetic deletion of Adora2b or pharmacologic inhibition will aid in scientific understanding of the mechanistic consequences of Adora2b expression in pancreatic cancer and also help determine if different PDAC subtypes respond differently to Adora2b inhibition. In addition, the role of the gut microbiome or intrapancreatic bacteria or fungi may also elevate adenosine or inosine levels elevating the importance of targeting this pathway for cancer treatment (133, 134). Future studies evaluating the functional consequences of Adora2b receptor signaling in different innate and adaptive immune cell types and interactions are also desperately needed to advance immunotherapies in this field.
Discussion
Pancreatic ductal adenocarcinoma is aggressive, resistant to therapy, and successful treatments are desperately needed, as current options have not yet resulted in significant changes in overall survival. In this review, we discuss literature related to the function of Adora2b, a low-affinity adenosine receptor prominently known for its role in reducing inflammation. The hypoxic TME of PDAC creates a unique niche where CD73, CD39, and Adora2b are elevated resulting in dynamic changes in concentrations of ATP and extracellular adenosine. The ENT1 transporter promotes sensitivity to chemotherapy in PDAC patients and high expression has strong prognostic implications for improved outcomes in PDAC (135). ENT1 is critical for regulating nucleoside concentrations and under hypoxic conditions regulates adenosine receptor signaling (136) indicating another possible combination therapeutic approach, as ENT1 is important for the transport of nucleotides into and out of the cell. Future studies deducing the entire pathway in cancer development and metastasis will aid in determining the utility of targeting this pathway to improve patient outcomes.
Another important consideration is the four P1 adenosine receptors have divergent roles dependent on cell type expression and concentrations of ligands. Of the four receptors, Adora2a and Adora2b have been reported as high in PDAC and are overexpressed in the pancreas during pancreatic cancer; yet only high expression of Adora2b receptor was shown to correlate with significantly reduced survival in PDAC patients. We recently published that patients with high ADORA2B have reduced CD8+ T cells and NK cells indicating inhibiting this receptor may have utility in recruiting activated CD8+ T cells and NKT cells to target PDAC (4). However, these efforts are complicated by the fact that Adora2b is present on virtually all myeloid and lymphoid lineage cells, and activation of the receptor on these cells can alter their functionality and contribute to dynamic changes in immune cell function in the TME. A critical consideration for future trials is understanding patient-specific levels of CD73, Adora2b and ATP, ADP, and adenosine available to signal through P2 or P1 receptors. Adenosine is rapidly taken back into cells and converted to inosine by ADA, which has also been shown to have immunosuppressive consequences in cancer models (133). Thus, understanding the full context of this incredibly complex signaling pathway including Adora2b functionality warrants further consideration and research efforts. Clinical trials where patient samples are available pre and post-treatment are urgently needed to determine if targeting this pathway will improve overall survival. Trials in both the neoadjuvant and adjuvant setting should be conducted due to recent publications showing the Adora2 receptors can promote tumor growth, metastasis and reduce CD8+ T cell anti-tumor immunity predominantly in preclinical models (3, 4, 11, 120, 123–126, 137–144).
Author contributions
LS wrote the original draft. EF, XY, WR, HE, and JB-L edited the manuscript. WR made the Figures and LS made the Table. All authors contributed to the article and approved the submitted version.
Funding
Natural Science Foundation of Hunan Province Grant 2018JJ3736, Young Talent Foundation of Hunan Province Grant 2021RC3034, and 2022 International Anesthesia Research Society Mentored Research Award to WR. National Institute of Health Grants R01HL155950, and Parker B. Francis Fellowship to XY. National Institute of Health Grants R01HL154720, R01DK122796, R01HL133900, T32GM135118 and Department of Defense Grant W81XWH2110032 to HE; sponsored contract through Akebia Therapeutics, Inc. to HE. JB-L is funded by NIH grants Texas Medical Center Digestive Disease Center Pilot Award 2P30DK056338-16 and 1R21CA249924-01. The funder, Akebia Therapeutics Inc., was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Acknowledgments
Figures were created with BioRender and Chemdraw software.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Xiao A, Tan ML, Wu LM, Asrani VM, Windsor JA, Yadav D, et al. Global incidence and mortality of pancreatic diseases: a systematic review, meta-analysis, and meta-regression of population-based cohort studies. Lancet Gastroenterol Hepatol (2016) 1:45–55. doi: 10.1016/S2468-1253(16)30004-8
2. Hart PA, Conwell DL. Chronic pancreatitis: managing a difficult disease. Am J Gastroenterol (2020) 115:49–55. doi: 10.14309/ajg.0000000000000421
3. Faraoni EY, Strickland LN, O’Brien BJ, Barraza JF, Thosani NC, Wray CJ, et al. Radiofrequency ablation in combination with CD73 inhibitor AB680 reduces tumor growth and enhances anti-tumor immunity in a syngeneic model of pancreatic ductal adenocarcinoma. Front Oncol (2022) 12:995027. doi: 10.3389/fonc.2022.995027
4. Faraoni EY, Singh K, Chandra V, Le Roux O, Dai Y, Sahin I, et al. CD73-dependent adenosine signaling through Adora2b drives immunosuppression in ductal pancreatic cancer. Cancer Res (2023) 83(7):1111–27. doi: 10.1158/0008-5472.22361356.v1
5. King RJ, Shukla SK, He C, Vernucci E, Thakur R, Attri KS, et al. CD73 induces GM-CSF/MDSC-mediated suppression of T cells to accelerate pancreatic cancer pathogenesis. Oncogene (2022) 41(7):971–82. doi: 10.1038/s41388-021-02132-6
6. Jacoberger-Foissac C, Cousineau I, Bareche Y, Allard D, Chrobak P, Allard B, et al. CD73 inhibits cGAS-STING and cooperates with CD39 to promote pancreatic cancer. Cancer Immunol Res (2023) 11(1):56–71. doi: 10.1158/2326-6066.CIR-22-0260
7. Campos-Contreras ADR, González-Gallardo A, Díaz-Muñoz M, Vázquez-Cuevas FG. Adenosine receptor A2B negatively regulates cell migration in ovarian carcinoma cells. Int J Mol Sci (2022) 23(9):4585. doi: 10.3390/ijms23094585
8. Kitabatake K, Kaji T, Tsukimoto M. Involvement of CD73 and A2B receptor in radiation-induced DNA damage response and cell migration in human glioblastoma A172 cells. Biol Pharm Bull (2021) 44(2):197–210. doi: 10.1248/bpb.b20-00654
9. Wilkat M, Bast H, Drees R, Dünser J, Mahr A, Azoitei N, et al. Adenosine receptor 2B activity promotes autonomous growth, migration as well as vascularization of head and neck squamous cell carcinoma cells. Int J Cancer (2020) 147(1):202–17. doi: 10.1002/ijc.32835
10. Eltzschig HK. Extracellular adenosine signaling in molecular medicine. J Mol Med (Berl) (2013) 91(2):141–6. doi: 10.1007/s00109-013-0999-z
11. Faraoni EY, Ju C, Robson SC, EH K, Bailey-Lundberg JM. Purinergic and adenosinergic signaling in pancreatobiliary diseases. Front Physiol (2022) 13:849258. doi: 10.3389/fphys.2022.849258
12. Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer (2018) 18:601–18. doi: 10.1038/s41568-018-0037-0
13. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med (2012) 367(24):2322–33. doi: 10.1056/NEJMra1205750
14. Le TT, Berg NK, Harting MT, Li X, Eltzschig HK, Yuan X. Purinergic signaling in pulmonary inflammation. Front Immunol (2019) 10:1633. doi: 10.3389/fimmu.2019.01633
15. Wang W, Chen NY, Ren D, Davies J, Philip K, Eltzschig HK, et al. Enhancing extracellular adenosine levels restores barrier function in acute lung injury through expression of focal adhesion proteins. Front Mol Biosci (2021) 8:2021 doi: 10.3389/fmolb.2021.636678
16. Hasko G, Cronstein BN. Adenosine: an endogenous regulator of innate immunity. Trends Immunol (2004) 25:33–9. doi: 10.1016/j.it.2003.11.003
17. Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature (2014) 509(7500):310–7. doi: 10.1038/nature13085
18. Colgan SP. Neutrophils and inflammatory resolution in the mucosa. Semin Immunol (2015) 27(3):177–83. doi: 10.1016/j.smim.2015.03.007
19. Eltzschig HK, Abdulla P, Hoffman E, Hamilton KE, Daniels D, Schonfeld C, et al. HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J Exp Med (2005) 202:1493–505. doi: 10.1084/jem.20050177
20. Aherne CM, Collins CB, Rapp CR, Olli KE, Perrenoud L, Jedlicka P, et al. Coordination of ENT2-dependent adenosine transport and signaling dampens mucosal inflammation. JCI Insight (2018) 3(20):e121521. doi: 10.1172/jci.insight.121521
21. Wiginton DA, Coleman MS, Hutton JJ. Purification, characterization and radioimmunoassay of adenosine deaminase from human leukaemic granulocytes. Biochem J (1981) 195(2):389–97. doi: 10.1042/bj1950389
22. Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity (2014) 40(1):66–77. doi: 10.1016/j.immuni.2013.11.020
23. Eltzschig HK, Bratton DL, Colgan SP. Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat Rev Drug Discov (2014) 13:852–69. doi: 10.1038/nrd4422
24. Semenza GL. The genomics and genetics of oxygen homeostasis. Annu Rev Genomics Hum Genet (2020) 21:183–204. doi: 10.1146/annurev-genom-111119-073356
25. Shen Q, Shi X, Tao L, Zhu Q, Xiao W, Ding Y, et al. Inhibition of hypoxia-inducible factor-1α alleviates acinar cell necrosis in a mouse model of acute pancreatitis. Biochem Biophys Res Commun (2021) 572:72–9. doi: 10.1016/j.bbrc.2021.07.043
26. Lu Y, Hu J, Sun W, Duan X, Chen X. Hypoxia-mediated immune evasion of pancreatic carcinoma cells. Mol Med Rep (2015) 11(5):3666–72. doi: 10.3892/mmr.2015.3144
27. Lee KE, Spata M, Bayne LJ, Buza EL, Durham AC, Allman D, et al. Hif1a deletion reveals pro-neoplastic function of b cells in pancreatic neoplasia. Cancer Discovery (2016) 6(3):256–69. doi: 10.1158/2159-8290.CD-15-0822
28. Zhang X, Du P, Luo K, Li Y, Liu Z, Wang W, et al. Hypoxia-inducible factor-1alpha protects the liver against ischemia-reperfusion injury by regulating the A2B adenosine receptor. Bioengineered (2021) 12(1):3737–52. doi: 10.1080/21655979.2021.1953217
29. O’Brien BJ, Faraoni EY, Strickland LN, Ma Z, Mota V, Mota S, et al. CD73-generated extracellular adenosine promotes resolution of neutrophil-mediated tissue injury and restrains metaplasia in pancreatitis. FASEB J (2023) 37(1):e22684. doi: 10.1096/fj.202201537R
30. Poth JM, Brodsky K, Ehrentraut H, Grenz A, Eltzschig HK. Transcriptional control of adenosine signaling by hypoxia-inducible transcription factors during ischemic or inflammatory disease. J Mol Med (2013) 91:183–93. doi: 10.1007/s00109-012-0988-7
31. Kong T, Westerman KA, Faigle M, Eltzschig HK, Colgan SP. HIF-dependent induction of adenosine A2B receptor in hypoxia. FASEB J (2006) 20:2242–50. doi: 10.1096/fj.06-6419com
32. Koeppen M, Eckle T, Eltzschig HK. Interplay of hypoxia and A2B adenosine receptors in tissue protection. Adv Pharmacol (2011) 61:145–86. doi: 10.1016/B978-0-12-385526-8.00006-0
33. Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, et al. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest (2002) 110:993–1002. doi: 10.1172/JCI0215337
34. Hart ML, Gorzolla IC, Schittenhelm J, Robson SC, Eltzschig HK. SP1-dependent induction of CD39 facilitates hepatic ischemic preconditioning. J Immunol (2010) 184:4017–24. doi: 10.4049/jimmunol.0901851
35. Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-α. Nat Med (2015) 21:638–46. doi: 10.1038/nm.3868
36. Morote-Garcia JC, Rosenberger P, Nivillac NM, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology (2009) 136:607–18. doi: 10.1053/j.gastro.2008.10.037
37. Yuan X, Mills T, Doursout M, Evans SE, Vidal Melo MF, Eltzschig HK. Alternative adenosine receptor activation: the netrin-Adora2b link. Front Pharmacol (2022) 13. doi: 10.3389/fphar.2022.944994
38. Stein E, Zou Y, Poo M, Tessier-Lavigne M. Binding of DCC by netrin-1 to mediate axon guidance independent of adenosine A2B receptor activation. Science (2001) 291:1976–82. doi: 10.1126/science.1059391
39. Rosenberger P, Schwab JM, Mirakaj V, Masekowsky E, Mager A, Morote-Garcia JC, et al. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat Immunol (2009) 10:195–202. doi: 10.1038/ni.1683
40. Kalhan A, Gharibi B, Vazquez M, Jasani B, Neal J, Kidd M, et al. Adenosine A2A and A2B receptor expression in neuroendocrine tumours: potential targets for therapy. Purinergic Signal (2012) 8(2):265–74. doi: 10.1007/s11302-011-9280-5
41. Kong T, Eltzschig HK, Karhausen J, Colgan SP, Shelley CS. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of beta2 integrin gene expression. Proc Natl Acad Sci USA (2004) 101:10440–5. doi: 10.1073/pnas.0401339101
42. Lennon PF, Taylor CT, Stahl GL, Colgan SP. Neutrophil-derived 5 0 -adenosine monophosphate promotes endothelial barrier function via CD73-mediated conversion to adenosine and endothelial A2B receptor activation. J Exp Med (1998) 188:1433–43. doi: 10.1084/jem.188.8.1433
43. Eltzschig HK, Eckle T, Mager A, Kuper N, Karcher C, Weissmuller T, et al. ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ Res (2006) 99:1100–8. doi: 10.1161/01.RES.0000250174.31269.70
44. Cronstein BN. Adenosine, an endogenous anti-inflammatory agent. J Appl Physiol (1994) 76:5–13. doi: 10.1152/jappl.1994.76.1.5
45. Eckle T, Faigle M, Grenz A, Laucher S, Thompson LF, Eltzschig HK. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood (2008) 111:2024–35. doi: 10.1182/blood-2007-10-117044
46. Koeppen M ET, Eltzschig HK. Selective deletion of the A1 adenosine receptor abolishes heart-rate slowing effects of intravascular adenosine in vivo. PloS One (2009) 4:e6784. doi: 10.1371/journal.pone.0006784
47. Eltzschig H. Adenosine: an old drug newly rediscovered. Anesthesiology (2009) 111:904–15. doi: 10.1097/ALN.0b013e3181b060f2
48. Hayashi M. Expression of adenosine receptors in rodent pancreas. Int J Mol Sci (2019) 20(21):5329. doi: 10.3390/ijms20215329
49. Satoh A, Shimosegawa T, Satoh K, Ito H, Kohno Y, Masamune A, et al. Activation of adenosine A1-receptor pathway induces edema formation in the pancreas of rats. Gastroenterology (2000) 119:829–36. doi: 10.1053/gast.2000.16502
50. Lin X, Wang ZY, Xue G, Qin XJ, Wu JF, Zhang G. ADORA1 is a diagnostic-related biomarker and correlated with immune infiltrates in papillary thyroid carcinoma. J Cancer (2021) 12(13):3997–4010. doi: 10.7150/jca.50743
51. Liu H, Kuang X, Zhang Y, Ye Y, Li J, Liang L, et al. ADORA1 inhibition promotes tumor immune evasion by regulating the ATF3-PD-L1 axis. Cancer Cell (2020) 37(3):324–39.e8. doi: 10.1016/j.ccell.2020.02.006
52. Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthy- poxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med (2003) 198:783–96. doi: 10.1084/jem.20030891
53. Jin X, Shepherd RK, Duling BR, Linden J. Inosine binds to A3 adenosine receptors and stimulates mast cell degranulation. J Clin Invest (1997) 100:2849–57. doi: 10.1172/JCI119833
54. Zhong H, Shlykov SG, Molina JG, Sanborn BM, Jacobson MA, Tilley SL, et al. Activation of murine lung mast cells by the adenosine A3 receptor. J Immunol (2003) 171:338–45. doi: 10.4049/jimmunol.171.1.338
55. Gorzalczany Y, Akiva E, Klein O, Merimsky O, Sagi-Eisenberg R. Mast cells are directly activated by contact with cancer cells by a mechanism involving autocrine formation of adenosine and autocrine/paracrine signaling of the adenosine A3 receptor. Cancer Lett (2017) 397:23–32. doi: 10.1016/j.canlet.2017.03.026
56. Durante M, Squillace S, Lauro F, Giancotti LA, Coppi E, Cherchi F, et al. Adenosine A3 agonists reverse neuropathic pain via T cell-mediated production of IL-10. J Clin Invest (2021) 131(7):e139299. doi: 10.1172/JCI139299
57. Ren TH, Lv MM, An XM, Leung WK, Seto WK. Activation of adenosine A3 receptor inhibits inflammatory cytokine production in colonic mucosa of patients with ulcerative colitis by down-regulating the nuclear factor-kappa b signaling. J Dig Dis (2020) 21(1):38–45. doi: 10.1111/1751-2980.12831
58. Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature (2001) 414:916–20. doi: 10.1038/414916a
59. Sitkovsky MV, Lukashev D, Apasov S, Kojima H, Koshiba M, Caldwell C, et al. Physiological control of immune response and inflammatory tissue damage by hypoxia- inducible factors and adenosine A2A receptors. Annu Rev Immunol (2004) 22:657–82. doi: 10.1146/annurev.immunol.22.012703.104731
60. Ledent C, Vaugeois JM, Schiffmann SN, Pedrazzini T, El Yacoubi M, Vanderhaeghen JJ, et al. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature (1997) 388:674–8. doi: 10.1038/41771
61. Noji T, Nan-ya K, Mizutani M, Katagiri C, Sano J, Takada C, et al. KF24345, an adenosine uptake inhibitor, ameliorates the severity and mortality of lethal acute pancreatitis via endogenous adenosine in mice. Eur J Pharmacol (2002) 454:85–93. doi: 10.1016/S0014-2999(02)02476-7
62. Sweed D, Taha M, Abd Elhamed S, El Dein Mohamed AS. The prognostic role of CD73/A2AR expression and tumor immune response in periampullary carcinoma subtypes. Asian Pac J Cancer Prev (2022) 23(4):1239–46. doi: 10.31557/APJCP.2022.23.4.1239
63. Fredholm BB, Irenius E, Kull B, Schulte G. Comparison of the potency of adenosine as an agonist at human adenosine receptors expressed in Chinese hamster ovary cells. Biochem Pharmacol (2001) 61(4):443–8. doi: 10.1016/S0006-2952(00)00570-0
65. Aherne CM, Kewley EM, Eltzschig HK. The resurgence of A2B adenosine receptor signaling. Biochim Biophys Acta (2011) 1808(5):1329–39. doi: 10.1016/j.bbamem.2010.05.016
66. Frick JS, MacManus CF, Scully M, Glover LE, Eltzschig HK, Colgan SP. Contribution of adenosine A2B receptors to inflammatory parameters of experimental colitis. J Immunol (2009) 182:4957–64. doi: 10.4049/jimmunol.0801324
67. Eckle T, Hartmann K, Bonney S, Reithel S, Mittelbronn M, Walker LA, et al. Adora2b-elicited Per2 stabilization promotes a HIF-dependent metabolic switch crucial for myocardial adaptation to ischemia. Nat Med (2012) 18:774–82. doi: 10.1038/nm.2728
68. Eckle T, Grenz A, Laucher S, Eltzschig HK. A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J Clin Invest (2008) 118(10):3301–15. doi: 10.1172/JCI34203
69. Ehrentraut H, Westrich JA, Eltzschig HK, Clambey ET. Adora2b adenosine receptor engagement enhances regulatory T cell abundance during endotoxin-induced pulmonary inflammation. PloS One (2012) 7(2):e32416. doi: 10.1371/journal.pone.0032416
70. Schingnitz U, Hartmann K, Macmanus CF, Eckle T, Zug S, Colgan SP, et al. Signaling through the A2B adenosine receptor dampens endotoxin-induced acute lung injury. J Immunol (2010) 184:5271–9. doi: 10.4049/jimmunol.0903035
71. Kolachala VL, Vijay-Kumar M, Dalmasso G, Yang D, Linden J, Wang L, et al. A2B adenosine receptor gene deletion attenuates murine colitis. Gastroenterology (2008) 135:861–70. doi: 10.1053/j.gastro.2008.05.049
72. Kolachala V, Ruble B, Vijay-Kumar M, Wang L, Mwangi S, Figler H, et al. Blockade of adenosine A2B receptors ameliorates murine colitis. Br J Pharmacol (2008) 155:127–37. doi: 10.1038/bjp.2008.227
73. Vasiukov G, Novitskaya T, Zijlstra A, Owens P, Ye F, Zhao Z, et al. Myeloid cell-derived TGFβ signaling regulates ECM deposition in mammary carcinoma via adenosine-dependent mechanisms. Cancer Res (2020) 80(12):2628–38. doi: 10.1158/0008-5472.CAN-19-3954
74. Liao J, Zeng DN, Li JZ, Hua QM, Xiao Z, He C, et al. Targeting adenosinergic pathway enhances the anti-tumor efficacy of sorafenib in hepatocellular carcinoma. Hepatol Int (2020) 14(1):80–95. doi: 10.1007/s12072-019-10003-2
75. Sui Y, Liu J, Zhang J, Zheng Z, Wang Z, Jia Z, et al. Expression and gene regulation network of adenosine receptor A2B in lung adenocarcinoma: a potential diagnostic and prognostic biomarker. Front Mol Biosci (2021) 8:663011. doi: 10.3389/fmolb.2021.663011
76. Xaus J, Valledor AF, Cardó M, Marquès L, Beleta J, Palacios JM, et al. Adenosine inhibits macrophage colony-stimulating factor-dependent proliferation of macrophages through the induction of p27kip-1 expression. J Immunol (1999) 163(8):4140–9. doi: 10.4049/jimmunol.163.8.4140
77. Xaus MM J, Lloberas J, Soler C, Lluis C, Franco R. A. celada. IFN-gamma up-regulates the A2B adenosine receptor expression in macrophages: a mechanism of macrophage deactivation. J Immunol (1999) 162:3607–14. doi: 10.4049/jimmunol.162.6.3607
78. Murphy PS, Wang J, Bhagwat SP, Munger JC, Janssen WJ, Wright TW, et al. CD73 regulates anti-inflammatory signaling between apoptotic cells and endotoxin-conditioned tissue macrophages. Cell Death Differ (2017) 24:559–70. doi: 10.1038/cdd.2016.159
79. Ben Addi A, Lefort A, Hua X, Libert F, Communi D, Ledent C, et al. Modulation of murine dendritic cell function by adenine nucleotides and adenosine: involvement of the A(2B) receptor. Eur J Immunol (2008) 38:1610–20. doi: 10.1002/eji.200737781
80. Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood (2008) 112:1822–31. doi: 10.1182/blood-2008-02-136325
81. Victorino F, Sojka DK, Brodsky KS, McNamee EN, Masterson JC, Homann D, et al. Tissue-resident NK cells mediate ischemic kidney injury and are not depleted by anti-asialo-GM1 antibody. J Immunol (2015) 195:4973–85. doi: 10.4049/jimmunol.1500651
82. Bastid J, Regairaz A, Bonnefoy N, Déjou C, Giustiniani J, Laheurte C, et al. Inhibition of CD39 enzymatic function at the surface of tumor cells alleviates their immunosuppressive activity. Cancer Immunol Res (2015) 3(3):254–65. doi: 10.1158/2326-6066.CIR-14-0018
83. Raskovalova T, Huang X, Sitkovsky M, Zacharia LC, Jackson EK, Gorelik E. Gs protein-coupled adenosine receptor signaling and lytic function of activated NK cells. J Immunol (2005) 174:4383–91. doi: 10.4049/jimmunol.175.7.4383
84. Liu X, Li L, Si F, Huang L, Zhao Y, Zhang C, et al. NK and NKT cells have distinct properties and functions in cancer. Oncogene (2021) 40:4521–37. doi: 10.1038/s41388-021-01880-9
85. Chen Q, Pu N, Yin H, Zhang J, Zhao G, Lou W, et al. CD73 acts as a prognostic biomarker and promotes progression and immune escape in pancreatic cancer. J Cell Mol Med (2020) 24:8674–86. doi: 10.1111/jcmm.15500
86. Chen M, Liang D, Zuo A, Shao H, Kaplan HJ, Sun D. An A2B adenosine receptor agonist promotes Th17 autoimmune responses in experimental autoimmune uveitis (EAU) via dendritic cell activation. PloS One (2015) 10(7):e0132348. doi: 10.1371/journal.pone.0132348
87. Burnstock G, Boeynaems J. Purinergic signalling and immune cells. Purinergic Signalling (2014) 10:529–64. doi: 10.1007/s11302-014-9427-2
88. Mirabet M, Herrera C, Cordero OJ, Mallol J, Lluis C, Franco R. Expression of A2B adenosine receptors in human lymphocytes: their role in T cell activation. J Cell Sci (1999) 112:491–502. doi: 10.1242/jcs.112.4.491
89. Leung PS. Physiology of the pancreas. Adv Exp Med Biol (2010) 690:13–27. doi: 10.1007/978-90-481-9060-7_2
90. Grapin-Botton A. Ductal cells of the pancreas. Int J Biochem Cell Biol (2005) 37(3):504–10. doi: 10.1016/j.biocel.2004.07.010
91. Sorensen CE, Novak I. Visualization of ATP release in pancreatic acini in response to cholinergic stimulus. use of fluorescent probes and confocal microscopy. J Biol Chem (2001) 276(35):32925–32. doi: 10.1074/jbc.M103313200
92. Novak I, Nitschke R, Amstrup J. Purinergic receptors have different effects in rat exocrine pancreas. calcium signals monitored by fura-2 using confocal microscopy. Cell Physiol Biochem (2002) 12(2-3):83–92. doi: 10.1159/000063784
93. Novak I. Purinergic signalling in epithelial ion transport: regulation of secretion and absorption. Acta Physiol (Oxf) (2011) 202(3):501–22. doi: 10.1111/j.1748-1716.2010.02225.x
94. Yegutkin GG, Samburski SS, Jalkanen S, Novak I. ATP-consuming and ATP-generating enzymes secreted by pancreas. J Biol Chem (2006) 281(40):29441–7. doi: 10.1074/jbc.M602480200
95. Burnstock G, Novak I. Purinergic signalling in the pancreas in health and disease. J Endocrinology (2012) 213(2):123–41. doi: 10.1530/JOE-11-0434
96. Ewald N, Bretzel RG. Diabetes mellitus secondary to pancreatic diseases (Type 3c)–are we neglecting an important disease? Eur J Intern Med (2013) 24(3):203–6. doi: 10.1016/j.ejim.2012.12.017
97. Novak I. Purinergic receptors in the endocrine and exocrine pancreas. Purinergic Signalling (2008) 4:237–53. doi: 10.1007/s11302-007-9087-6
98. Hansen MR, Krabbe S, Novak I. Purinergic receptors and calcium signalling in human pancreatic duct cell lines. Cell Physiol Biochem (2008) 22(1-4):157–68. doi: 10.1159/000149793
99. Sendler M, Weiss FU, Golchert J, Homuth G, van den Brandt C, Mahajan UM, et al. Cathepsin b-mediated activation of trypsinogen in endocytosing macrophages increases severity of pancreatitis in mice. Gastroenterology (2018) 154:704.e10–18.e10. doi: 10.1053/j.gastro.2017.10.018
100. Mayerle J, Sendler M, Hegyi E, Beyer G, Lerch MM, Sahin-Tóth M. Genetics, cell biology, and pathophysiology of pancreatitis. Gastroenterology (2019) 156:1951.e1–68.e1. doi: 10.1053/j.gastro.2018.11.081
101. Saluja A, Dudeja V, Dawra R, Sah RP. Early intra-acinar events in pathogenesis of pancreatitis. Gastroenterology (2019) 156:1937–40. doi: 10.1053/j.gastro.2019.03.050
102. Wang X, Liu D, Qin W, Liu Y, Yuan X, Zhang X, et al. P2RX1-involved glycolytic metabolism supports neutrophil activation in acute pancreatitis. Front Immunol (2020) 11:549179. doi: 10.3389/fimmu.2020.549179
103. Ma Z, Lytle NK, Chen B, Jyotsana N, Novak SW, Cho CJ, et al. Single-cell transcriptomics reveals a conserved metaplasia program in pancreatic injury. Gastroenterology (2021) 162(2):604–20.e20. doi: 10.1101/2021.04.09.439243
104. Barletta KE, Cagnina RE, Burdick MD, Linden J, Mehrad B. Adenosine A(2B) receptor deficiency promotes host defenses against gram-negative bacterial pneumonia. Am J Respir Crit Care Med (2012) 186(10):1044–50. doi: 10.1164/rccm.201204-0622OC
105. Wang X, Chen D. Purinergic regulation of neutrophil function. Front Immunol (2018) 9:399. doi: 10.3389/fimmu.2018.00399
106. Künzli BM, Nuhn P, Enjyoji K, Banz Y, Smith RN, Csizmadia E, et al. Disordered pancreatic inflammatory responses and inhibition of fibrosis in CD39-null mice. Gastroenterology (2008) 134:292–305. doi: 10.1053/j.gastro.2007.10.030
107. Shevchenko I, Mathes A, Groth C, Karakhanova S, Müller V, Utikal J, et al. Enhanced expression of CD39 and CD73 on T cells in the regulation of anti-tumor immune responses. Oncoimmunology (2020) 9(1):1744946. doi: 10.1080/2162402X.2020.1744946
108. Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity (2019) 51(1):27–41. doi: 10.1016/j.immuni.2019.06.025
109. Mittal D, Sinha D, Barkauskas D, Young A, Kalimutho M, Stannard K, et al. Adenosine 2B receptor expression on cancer cells promotes metastasis. Cancer Res (2016) 76(15):4372–82. doi: 10.1158/0008-5472.CAN-16-0544
110. Wang H, Tan F, Xu Y, Ma Y, Li Y, Xiao H. Adenosine receptor A2B antagonist inhibits the metastasis of gastric cancer cells and enhances the efficacy of cisplatin. Technol Cancer Res Treat (2023) 22:15330338221150318. doi: 10.1177/15330338221150318
111. Casamitjana J, Espinet E, Rovira M. Pancreatic organoids for regenerative medicine and cancer research. Front Cell Dev Biol (2022) 10:886153. doi: 10.3389/fcell.2022.886153
112. Driehuis E, van Hoeck A, Moore K, Kolders S, Francies HE, Gulersonmez MC, et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc Natl Acad Sci USA (2019) 116(52):26580–90. doi: 10.1073/pnas.1911273116
113. Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell (2015) 160(1-2):324–38. doi: 10.1016/j.cell.2014.12.021
114. Ponz-Sarvise M, Corbo V, Tiriac H, Engle DD, Frese KK, Oni TE, et al. Identification of resistance pathways specific to malignancy using organoid models of pancreatic cancer. Clin Cancer Res (2019) 25(22):6742–55. doi: 10.1158/1078-0432.CCR-19-1398
115. Andrews MG, Kriegstein AR. Challenges of organoid research. Annu Rev Neurosci (2022) 45:23–39. doi: 10.1146/annurev-neuro-111020-090812
116. di Magliano MP, Logsdon CD. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology (2013) 144(6):1220–9. doi: 10.1053/j.gastro.2013.01.071
117. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell (2005) 7(5):469–83. doi: 10.1016/j.ccr.2005.04.023
118. Westphalen CB, Olive KP. Genetically engineered mouse models of pancreatic cancer. Cancer J (2012) 18(6):502–10. doi: 10.1097/PPO.0b013e31827ab4c4
119. Ingwersen J, Wingerath B, Graf J, Lepka K, Hofrichter M, Schröter F, et al. Dual roles of the adenosine A2a receptor in autoimmune neuroinflammation. J Neuroinflammation (2016) 13:48. doi: 10.1186/s12974-016-0512-z
120. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci USA (2006) 103(35):13132–7. doi: 10.1073/pnas.0605251103
121. Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature (2010) 467:1114–7. doi: 10.1038/nature09515
122. Carpenter ES, Elhossiny AM, Kadiyala P, Li J, McGue J, Griffith BD, et al. Analysis of donor pancreata defines the transcriptomic signature and microenvironment of early neoplastic lesions. Cancer Discov (2023). doi: 10.1158/2159-8290.CD-23-0013
123. Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, et al. Co-Inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell (2016) 30(3):391–403. doi: 10.1016/j.ccell.2016.06.025
124. Moesta AK, Li XY, Smyth MJ. Targeting CD39 in cancer. Nat Rev Immunol (2020) 20(12):739–55. doi: 10.1038/s41577-020-0376-4
125. Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology (2016) 5(8):e1208875. doi: 10.1080/2162402X.2016.1208875
126. Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S, et al. Adenosine receptor 2A blockade increases the efficacy of anti-PD-1 through enhanced antitumor T-cell responses. Cancer Immunol Res (2015) 3(5):506–17. doi: 10.1158/2326-6066.CIR-14-0211
127. Mittal D, Young A, Stannard K, Yong M, Teng MW, Allard B, et al. Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor. Cancer Res (2014) 74:3652–8. doi: 10.1158/0008-5472.CAN-14-0957
128. Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. J Clin Cancer Res (2013) 19:5626–35. doi: 10.1158/1078-0432.CCR-13-0545
129. Collisson EA, Maitra A. Pancreatic cancer genomics 2. 0: Profiling Metastases Cancer Cell (2017) 31(3):309–10. doi: 10.1016/j.ccell.2017.02.014
130. Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med (2011) 17(4):500–3. doi: 10.1038/nm.2344
131. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature (2016) 531(7592):47–52. doi: 10.1038/nature16965
132. Collisson EA, Bailey P, Chang DK, Biankin AV. Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol (2019) 16(4):207–20. doi: 10.1038/s41575-019-0109-y
133. Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science (2020) 369(6510):1481–9. doi: 10.1126/science.abc3421
134. He B, Hoang TK, Wang T, Ferris M, Taylor CM, Tian X, et al. Resetting microbiota by lactobacillus reuteri inhibits T reg deficiency-induced autoimmunity via adenosine A2A receptors. J Exp Med (2017) 214(1):107–23. doi: 10.1084/jem.20160961
135. Farrell JJ, Elsaleh H, Garcia M, Lai R, Ammar A, Regine WF, et al. Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology (2009) 136(1):187–95. doi: 10.1053/j.gastro.2008.09.067
136. Rose JB, Naydenova Z, Bang A, Ramadan A, Klawitter J, Schram K, et al. Absence of equilibrative nucleoside transporter 1 in ENT1 knockout mice leads to altered nucleoside levels following hypoxic challenge. Life Sci (2011) 89(17-18):621–30. doi: 10.1016/j.lfs.2011.08.007
137. Boison D, Yegutkin GG. Adenosine metabolism: emerging concepts for cancer therapy. Cancer Cell (2019) 36(6):582–96. doi: 10.1016/j.ccell.2019.10.007
138. Wang J, Lupo KB, Chambers AM, Matosevic S. Purinergic targeting enhances immunotherapy of CD73. J Immunother Cancer (2018) 6(1):136. doi: 10.1186/s40425-018-0441-8
139. Sek K, Mølck C, Stewart GD, Kats L, Darcy PK, Beavis PA. Targeting adenosine receptor signaling in cancer immunotherapy. Int J Mol Sci (2018) 19(12):3837. doi: 10.3390/ijms19123837
140. Ma SR, Deng WW, Liu JF, Mao L, Yu GT, Bu LL, et al. Blockade of adenosine A2A receptor enhances CD8+ T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol Cancer (2017) 16(1):99. doi: 10.1186/s12943-017-0665-0
141. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev (2017) 276(1):121–44. doi: 10.1111/imr.12528
142. Allard D, Allard B, Gaudreau PO, Chrobak P, Stagg J. CD73-adenosine: a next-generation target in immuno-oncology. Immunotherapy (2016) 8(2):145–63. doi: 10.2217/imt.15.106
143. Leone RD, Lo YC, Powell JD. A2aR antagonists: next generation checkpoint blockade for cancer immunotherapy. Comput Struct Biotechnol J (2015) 13:265–72. doi: 10.1016/j.csbj.2015.03.008
Keywords: immunotherapy, pancreatic adenocarcinoma, hypoxia, Adenosine receptor 2B, CD8+ T cell response
Citation: Strickland LN, Faraoni EY, Ruan W, Yuan X, Eltzschig HK and Bailey-Lundberg JM (2023) The resurgence of the Adora2b receptor as an immunotherapeutic target in pancreatic cancer. Front. Immunol. 14:1163585. doi: 10.3389/fimmu.2023.1163585
Received: 11 February 2023; Accepted: 19 April 2023;
Published: 28 April 2023.
Edited by:
Luca Antonioli, University of Pisa, ItalyReviewed by:
Chun Wai Mai, UCSI University, MalaysiaChang-il Hwang, University of California, Davis, United States
Copyright © 2023 Strickland, Faraoni, Ruan, Yuan, Eltzschig and Bailey-Lundberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jennifer M. Bailey-Lundberg, SmVubmlmZXIuTS5CYWlsZXlAdXRoLnRtYy5lZHU=