 Thomas Schmitt1†
Thomas Schmitt1† Christoph Hudemann2†
Christoph Hudemann2† Sina Moztarzadeh1†
Sina Moztarzadeh1† Michael Hertl2†
Michael Hertl2† Ritva Tikkanen3†
Ritva Tikkanen3† Jens Waschke1*†
Jens Waschke1*†- 1Instiute of Anatomy, Faculty of Medicine, Chair of Vegetative Anatomy, Ludwig-Maximilian -Universität (LMU) Munich, München, Germany
- 2Department of Dermatology and Allergology, Philipps-University Marburg, Marburg, Germany
- 3Institute of Biochemistry, Medical Faculty, Justus-Liebig-University Giessen, Giessen, Germany
Introduction: Pemphigus is an autoantibody driven disease that impairs the barrier function of the skin and mucosa by disrupting desmosomes and thereby impeding cellular cohesion. It is known that the different clinical phenotypes of pemphigus vulgaris (PV) and pemphigus foliaceus (PF) are dependent on the autoantibody profile and target antigens that, amongst others, are primarily desmoglein (Dsg)1 and/or Dsg3 for PV and Dsg1 for PF. However, it was reported that autoantibodiesagainst different epitopes of Dsg1 and Dsg3 can be pathogenic or not. The underlying mechanisms are very complex and involve both direct inhibition of Dsg interactions and downstream signalling. The aim of this study was to find out whether there is target-epitope-specific Dsg3 signalling by comparing the effects of the two pathogenic murine IgGs, 2G4 and AK23.
Methods: Dispase-based dissociation assay, Western Blot analysis, Stimulated emission depletion microscopy, Fura-based Ca2+ flux measurements, Rho/Rac G-Protein-linked immunosorbent assay, Enzyme-linked immunosorbent assay.
Results: The IgGs are directed against the EC5 and EC1 domain of Dsg3, respectively. The data show that 2G4 was less effective in causing loss of cell adhesion, compared to AK23. STED imaging revealed that both autoantibodies had similar effects on keratin retraction and reduction of desmosome number whereas only AK23 induced Dsg3 depletion. Moreover, both antibodies induced phosphorylation of p38MAPK and Akt whereas Src was phosphorylated upon treatment with AK23 only. Interestingly, Src and Akt activation were p38MAPK-dependent. All pathogenic effects were rescued by p38MAPK inhibition and AK23-mediated effects were also ameliorated by Src inhibition.
Discussion: The results give first insights into pemphigus autoantibody-induced Dsg3 epitope-specific signalling which is involved in pathogenic events such as Dsg3 depletion.
Introduction
The skin and mucosa provide an effective barrier against external hazards such as pathogens or harmful chemicals. Pemphigus is an autoantibody driven disease that impairs barrier integrity by disturbing the turnover of desmosomes. Desmosomes provide mechanical cohesion between neighbouring epithelial cells (1). As a result, liquid-filled blisters are formed within the epithelia, which can affect a large portion of the skin or mucous membranes. The blisters can thus cause severe complications and high mortality, mostly due to barrier defects (2). Desmosomes consist of the cadherin adhesion molecules desmoglein (Dsg) and desmocollin (Dsc) subfamily. These are anchored to keratin filaments by adaptor proteins such as plakoglobin (Pg) and plakophilin (Pkp) and the linker protein desmoplakin (Dp). It is well known that the clinical pemphigus phenotypes correlate with the autoantibody profile. The most common pemphigus variants are pemphigus vulgaris (PV) characterized by autoantibodies against Dsg3, affecting the mucosa only or against Dsg1 and Dsg3, affecting the mucosa and epidermis and pemphigus foliaceus (PF) characterized by autoantibodies against Dsg1, affecting the epidermis exclusively (3–5). However, the pathomechanisms causing the loss of adhesion are quite complex (6). For anti-Dsg3 antibodies, it was shown that both direct inhibition of Dsg interaction (7–9) and signalling events induced by the autoantibody binding play a major role (10, 11). In this context, it was reported that several kinases and other signalling proteins relevant for pemphigus pathogenesis form complexes with desmosomal proteins (12, 13). Recently, interaction of Erk1 and p38MAPK with Dsg1 was confirmed by BIO-ID (14).
For anti-Dsg1 autoantibodies, direct inhibition was not found in all studies (7, 15–17). Frequently, autoantibodies against other antigens are also found (18). Autoantibodies against Dsc1, Dsc2 or Dsc3 were also reported to be pathogenic (19–26). However, the role of autoantibodies against other targets is less clear. It is hypothesised that they cause additive effects, thereby potentiating the pathogenic effects of the autoantibodies against the desmosomal cadherins (27–29).
It was reported that different anti-Dsg antibodies do not always show a similar degree of pathogenicity (7, 15–17). Dsg isoforms consist of five extracellular domains (EC1-EC5), similar to other cadherins. These share a high structural homology, except for the membrane proximal EC5 domain (30). Several studies reported that most pemphigus autoantibodies bind to the N-terminal EC1-domain, the major mediator of homo- and heterophilic interactions (31, 32). Approximately 50 % of PV patients exhibit detectable serum IgGs recognizing the EC1 domain, followed by EC2 (26 %), EC4 (21 %), EC5 (17%) and EC3 (15%) (33). Antibodies directed against EC1 or EC2 are known to exhibit pathogenic effects, whereas those targeting EC5 are reported to be mostly non-pathogenic (34, 35). It was reported that healthy individuals from endemic groups with an increased risk of PF onset often showed increased levels of non-pathogenic anti-Dsg1-IgGs (35–38). These were exclusively IgG2 whereas pathogenic autoantibodies were usually of IgG4 subtype (39). However, non-pathogenic and pathogenic antibodies can often share the same light or heavy chains (34). It is even hypothesised that pathogenic pemphigus autoantibodies can evolve from non-pathogenic antibodies via epitope spreading (34, 35, 40). Epitope spreading from Dsg3 to Dsg1 is described in the relatively frequent transformation from mucosal-dominant (anti-Dsg3-IgG only) to mucocutaneous PV (anti-Dsg1- and anti-Dsg3-IgG). The same is true for a less frequent transformation from PF (anti-Dsg1-IgG only) to PV (anti-Dsg1- and anti-Dsg3-IgG) (41, 42). A further piece of evidence is the observation that the introduction of a single Dsg3-reactive T-cell was sufficient to induce the expression of polyclonal anti-Dsg3 IgG in mice in vivo (43). Pemphigus patient sera usually display a complex polyclonal mixture of autoantibodies that in most, but not all cases show a good correlation with the disease severity (44, 45).
Due to their high prevalence in pemphigus patients and their high pathogenicity in several in vitro and in vivo studies, EC1-specific antibodies such as the mouse monoclonal IgG1 AK23 are typically used to investigate PV pathology (Figure 1A) (46). Recently, a pathogenic mouse monoclonal anti-Dsg3 IgG called 2G4, directed against the EC5 domain, was developed (47). The aim of this study was thus to directly compare the two antibodies to clarify if autoantibodies in PV can induce a Dsg3 epitope-specific signalling response.
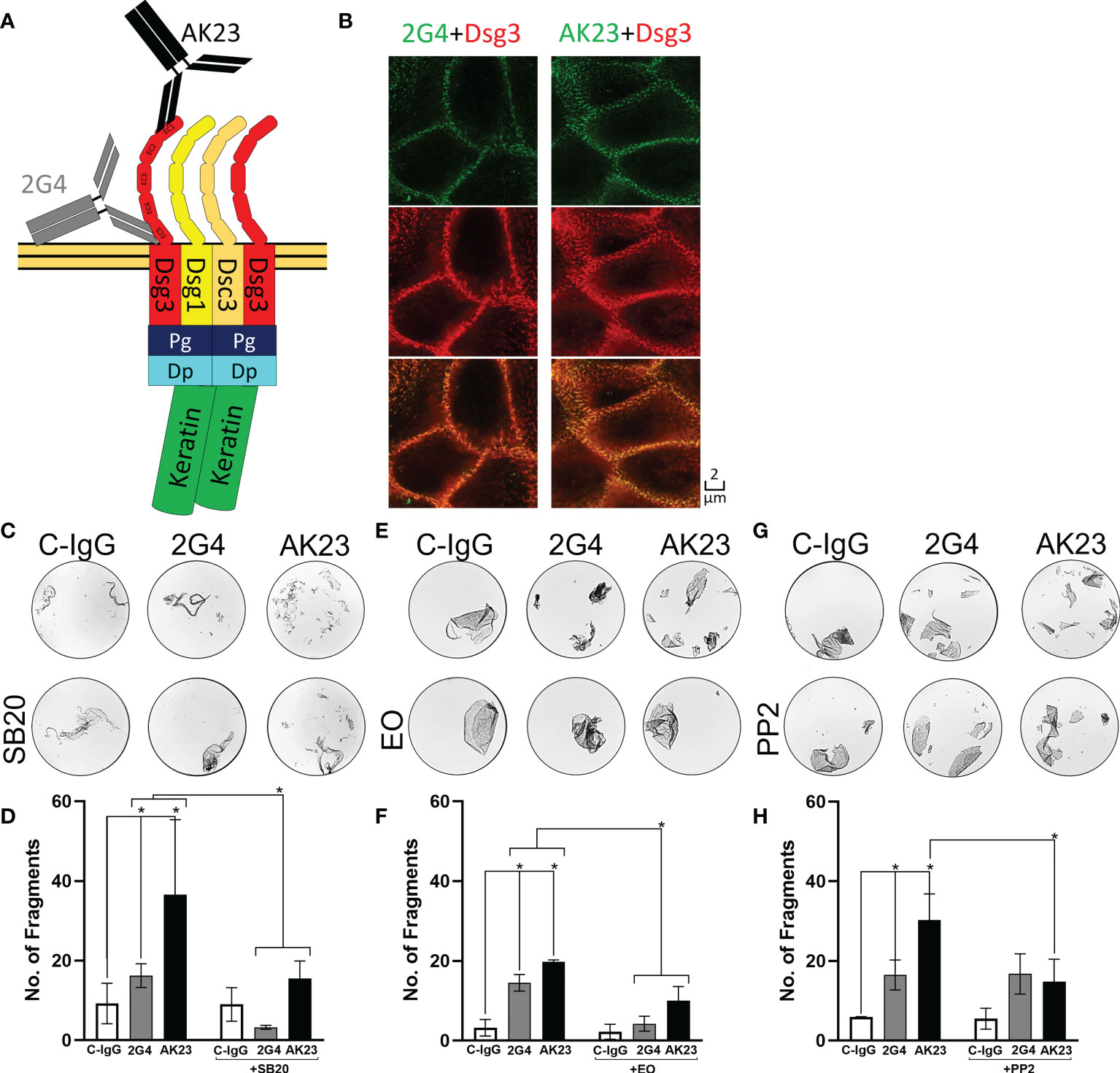
Figure 1 2G4 is a pathogenic Dsg3 specific IgG. (A) Schematic depiction of binding positions of AK23 (Black), directed against the EC1 domain and 2G4 (Grey) directed against the EC5 domain of Dsg3, on a desmosome-bound Dsg3. (B) Co-staining of either 2G4 or AK23 (green) with Dsg3 (intracellular domain, red) and an overlay of both channels in HaCaT cells after 24 hours incubation with the IgGs (1:50), imaged with the STED microscope (N=4). (C) Representative images of dispase-based dissociation assay results in HaCaT cells. Vehicle control (left) pre-treated with SB20 (60 µM, inhibiting p38MAPK, right). (D) Quantification of dispase-based dissociation assay results using SB20 (N=4). (E) Representative images of dispase-based dissociation assay results in HaCaT cells. Vehicle control (left) pre-treated with EO (40 nM, inhibiting p38MAPK, right). (F) Quantification of dispase-based dissociation assay results using EO (N=4). (G) Representative images of dispase-based dissociation assay results in HaCaT cells. Vehicle control (left) pre-treated with PP2 (10 µM, inhibiting Src, right). (H) Quantification of dispase-based dissociation assay results using PP2 (N=4). * indicates statistically significant differences in two-way-ANOVA with Sidaks correction for multiple comparisons p<0.05.
Results
AK23 is more effective than 2G4 in reducing cell-cell adhesion
Using immunostaining and super resolution stimulated emission depletion (STED) microscopy showed that both 2G4 and AK23 produced a Dsg3 staining pattern comparable to a commercial polyclonal IgG directed against the intracellular domain of Dsg3 (Figure 1B). In the dispase-based dissociation assay, 2G4 induced a significant loss of adhesion, compared to the control using IgG purified from the serum of healthy volunteers (C-IgG). However, 2G4 showed a 2.54-fold weaker effect on the adhesion of HaCaT cells than AK23 (1.98-fold without subtracting background fragments under control conditions). The effects of both IgGs were ameliorated by inhibition of p38MAPK, a kinase central for pemphigus pathogenesis (6), using SB202190 (SB20) (Figures 1C, D) or EO1428 (EO) (Figures 1E, F). Inhibition of Src, using PP2, on the other hand was effective in reducing AK23-induced effects, but had no effect on fragment numbers under 2G4 treatment (Figures 1G, H).
To check if there are additive and polyclonal effects potentially increasing the loss of adhesion, we used mixtures of AK23 and 2G4 in dispase dissociation assays. A mixture of AK23 and 2G4 at a concentration of 75 µg/ml each did not lead to a significant increase in the number of fragments, compared to AK23 alone at a concentration of 75 µg/ml (as applied in all other experiments). A mixture of AK23 and 2G4 at a concentration of 37.5 µg/ml for both led to a number of fragments similar to the number of fragments induced by 2G4 alone at a concentration of 75 µg/ml (Figures 2A, B).
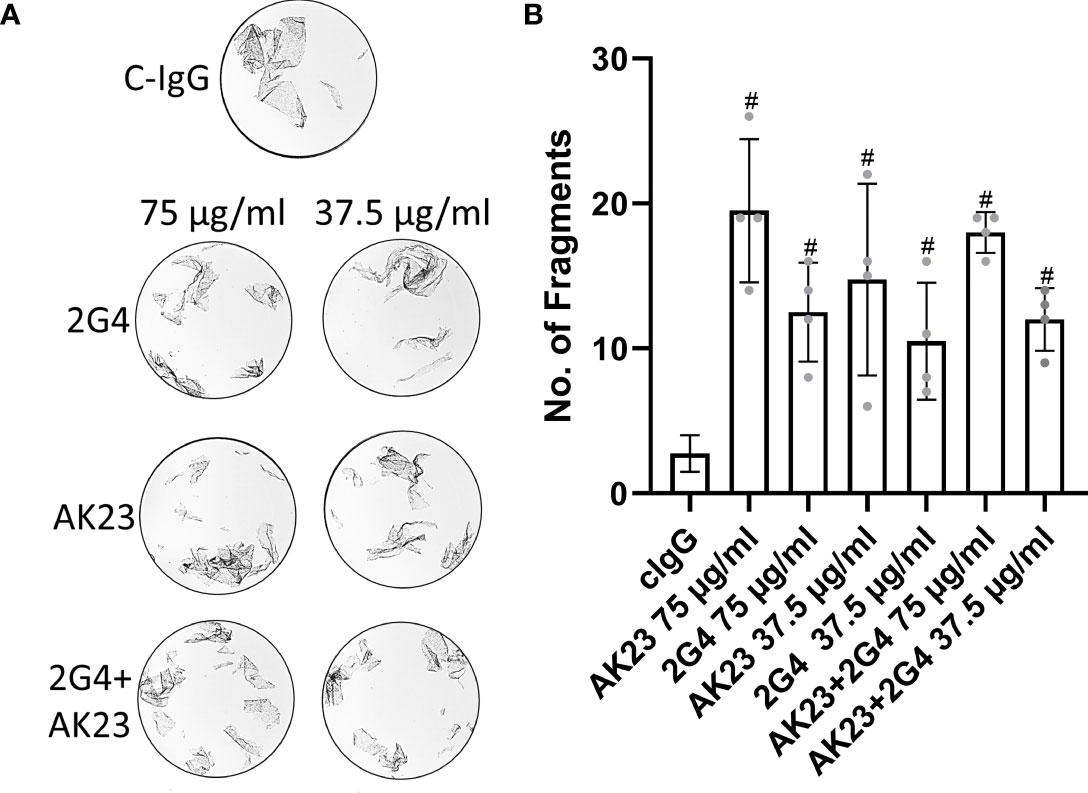
Figure 2 (A) Representative images of dispase-based dissociation assay results in HaCaT cells. Control, treated with C-IgG only (top), treated with either single 2G4, AK23 or a mixture of both at a concentration of 75 µg/ml eacht (left) and at half concentration of 37.5 µg/ml right. (B) Quantification of dispase-based dissociation assay (N=4). # indicates statistically significant differences towards control conditions, one-way-ANOVA with Dunett correction for multiple comparisons, p<0.05.
Effects of 2G4 and AK23 on the organization of keratin filaments and distribution of Dsg3 and Dp
Using STED microscopy, we observed that both 2G4 and AK23 caused fragmentation of Dsg3 staining along cell borders (Figure 3A, S1A). In addition, following incubation with AK23 the staining intensity of Dsg3 was significantly reduced, both along the cell borders and in the cytoplasm. This effect was not observed for 2G4, showing that AK23 led to a stronger redistribution and loss of Dsg3 than 2G4. The effects were ameliorated by inhibition of p38MAPK using either SB202190 (Figures 3A, D) or EO1428 (Figures S1A, D). p38MAPK inhibition with SB202190 under control conditions significantly increased Dsg3 staining intensity both at the cell border and in the cytoplasm whereas effects of EO1428 were not significant. This is indicating that p38MAPK is involved in Dsg3 turn-over under basal conditions as well (Figures 3A, D; S1A, D). Furthermore, a significant retraction of keratin filaments from the cell borders towards the nucleus, another typical pemphigus hallmark, was observed after incubation with either 2G4 or AK23. The effect was similar in extent for both IgGs and was also blocked by inhibition of p38MAPK with either SB202190 (Figures 3A, C) or EO1428 (Figures S1A, C).
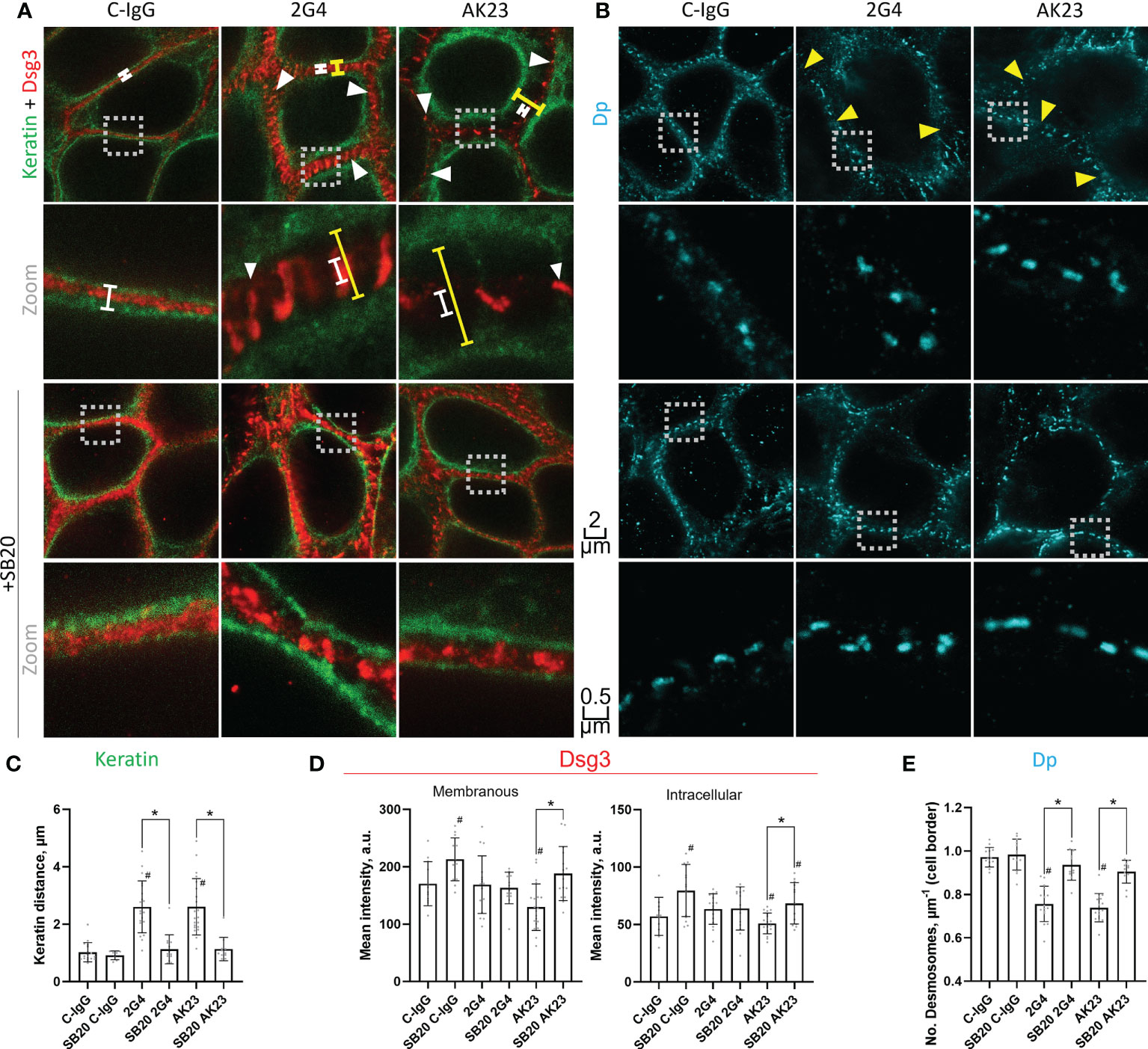
Figure 3 STED microscopy images reveal specific pathogenic effects of 2G4 and AK23. (A) Co-staining of Dsg3 (red) and pan-cytokeratin (green) in HaCaT cells treated with either C-IgG, AK23 or 2G4 for 24 h after 1 h preincubation with vehicle or SB20 (60 µM, inhibiting p38MAPK). Spans illustrate the degree of keratin filament retraction (yellow) compared to control (white). White arrowheads showcase the fragmented staining of Dsg3, which is mostly associated with keratin filaments. (B) Staining of Dp (with anti-Dp-pAb, A7169) after 24 h treatment with IgGs and 1 h preincubation with vehicle or SB20. Yellow arrowheads indicate reduced numbers of desmosomes per µm of cell border. Two different magnifications are shown for each condition (Scalebars as indicated, boxes with dotted outline indicate the area which was zoomed in on); Quantification of the STED images: (C) Keratin filament retraction form the cell borders towards the nucleus. (D) Dsg3 staining intensity at the cell borders/membranes (left) or in the cytoplasm (right). (E) Number of desmosomes per µm membrane (Experiments (N)=4-6, Evaluated images (n)=12-22). * marks statistically significant differences between the two conditions indicated, # indicates statistically significant differences towards control conditions, both in two-way-ANOVA with Sidaks correction for multiple comparisons, p<0.05.
In addition to Dsg3 and keratin filaments, the desmosomal plaque protein Dp was stained and investigated using STED microscopy. Neither of the two IgGs caused significant changes in desmosome morphology (Figures 3B; S1B). Due to the regular size and shape of desmosomal plaques, this staining was also utilized to determine the number of desmosomes per µm of cell border. Both 2G4 and AK23 caused a similar reduction of desmosome number, comparable to results for PV-patient skin (12). This effect was also abrogated by inhibition of p38MAPK with either SB202190 (Figure 3B, E) or EO1428 (Figure S1B, E).
2G4 and AK23 induce overlapping, yet distinct signalling responses
Next, we analysed the signalling pathways induced by 2G4 and AK23 respectively, using Western blot analyses after Triton X protein fractionation. Similar to previous studies, p38MAPK was significantly phosphorylated in the Triton X soluble (not cytoskeleton-bound fraction) and both IgGs induced a similar degree of p38MAPK phosphorylation. This effect was ameliorated upon treatment with the p38MAPK inhibitor SB202190 (Figures 4A, B) or EO (Figures S2A, N). Interestingly, Src, which is another well-known Dsg3-dependent signalling protein involved in loss of cell adhesion in pemphigus (48, 49), was significantly phosphorylated upon treatment with AK23 but not following 2G4 incubation. The effect was dependent on p38MAPK since treatment with SB202190 (Figures 4A, B) or EO 1428(Figures S2A, B) abolished this effect. As a third target of interest, Akt as downstream kinase of PI3-kinase, which is regulated by the Dsg3-dependent EGFR signalling pathway (6, 11, 50), was investigated. Akt phosphorylation was significantly increased to a similar degree after treatment with either 2G4 or AK23. Inhibition of p38MAPK with SB202190 (Figures 4A, B) or EO1428 (Figures S2A, B) abolished this effect. Neither of the two antibodies showed a significant impact on the activity of RhoA or Rac1 GTPases (Figure S3).
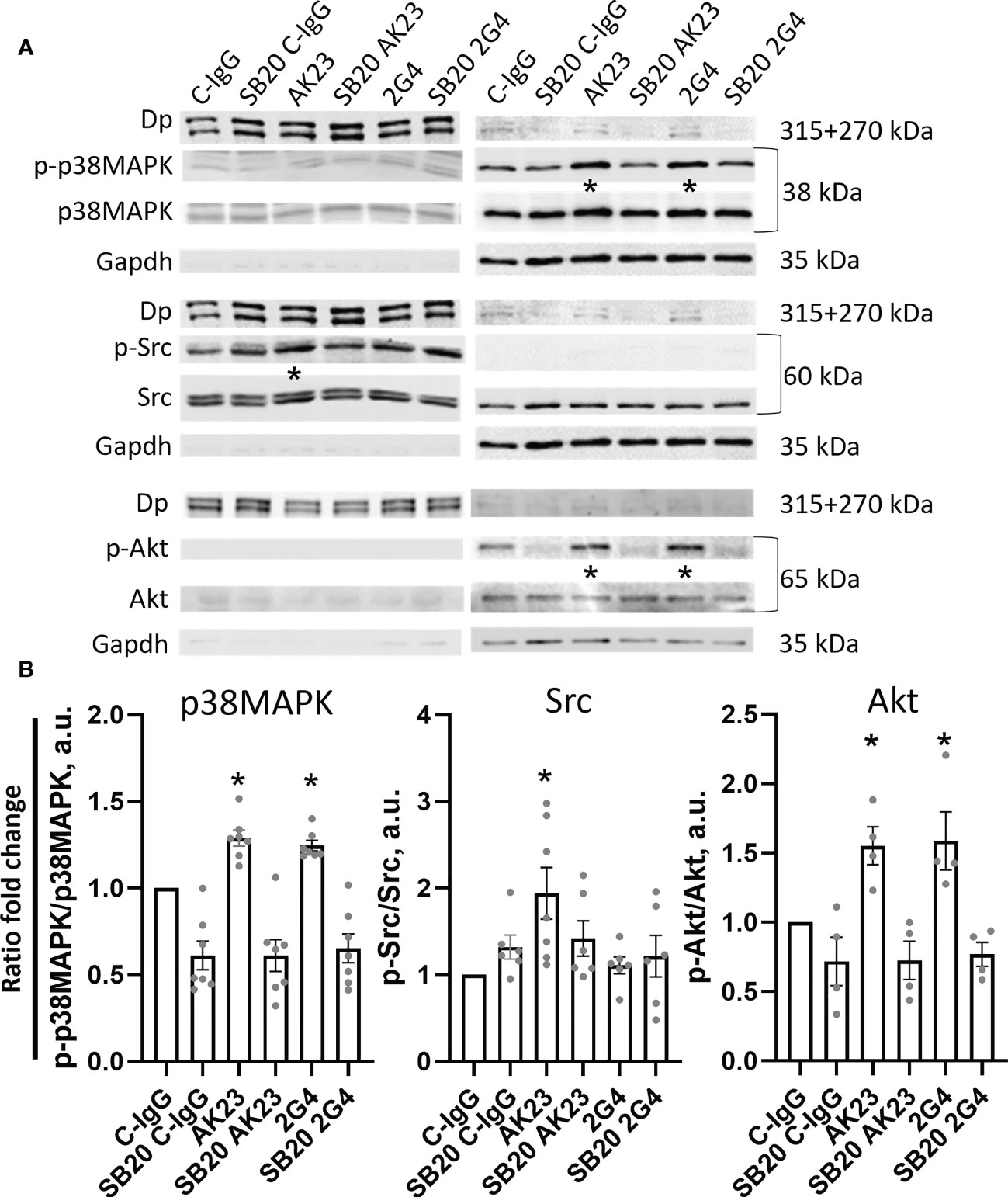
Figure 4 Western blot analysis reveals anti-Dsg3 epitope-specific kinase phosphorylation. (A) Representative Western blot images, with respective controls for Triton-soluble (membranous+cytosolic fraction, right) Gapdh and Triton-insoluble (cytoskeleton/desmosome-bound fraction, left) Dp. The cells were treated with either C-IgG, AK23 or 2G4 with or without pre-treatment with SB20 for 1 h, to inhibit p38 MAPK. (B) Quantification of differences in phosphorylation of the investigated proteins from the WB results. Significant changes were observed and quantified in the soluble fraction for p38MAPK and Akt and the insoluble fraction for Src (N=4-7). * marks statistically significant differences between the two conditions indicated in two-way-ANOVA with Sidaks correction for multiple comparisons, p<0.05.
In addition to protein phosphorylation, the Ca2+-influx after treatment with either 2G4 or AK23 was monitored. 2G4 induced a rapid influx of Ca2+ into the cells whereas AK23, similar to a previous report, did not (51) (Figure 5A). 2G4 did not show any signs of cross reactivity with Dsg1 (Figure S4), indicating a signalling mechanism dependent on Dsg3 in this case. This phenomenon was investigated further, employing mediators used in a previous study (51) (Table S2). However, in contrast to Dsg1-IgG-dependent signalling (51), inhibition of PI4K (with GSK-F1), PLC (with U-73122) or IP3R (with Xestospongin C) was not effective in preventing the 2G4-mediated Ca2+-influx. Rather, inhibition of the downstream calcium release-activated channel (CRAC) (with BTP-2) blocked Ca2+-influx, indicating that 2G4 induced a different pathway of Ca2+-influx into the endoplasmic reticulum (Figure 5B). Next, we tested whether this Ca2+-influx is involved in loss of cell adhesion caused by 2G4. However, none of the mediators including BTP-2 prevented any of the pathological changes, including loss of adhesion in the dispase-based dissociation assay (Figures 5C, D), keratin filament retraction and fragmentation of Dsg3 staining as revealed by STED microscopy (Figure 5E).
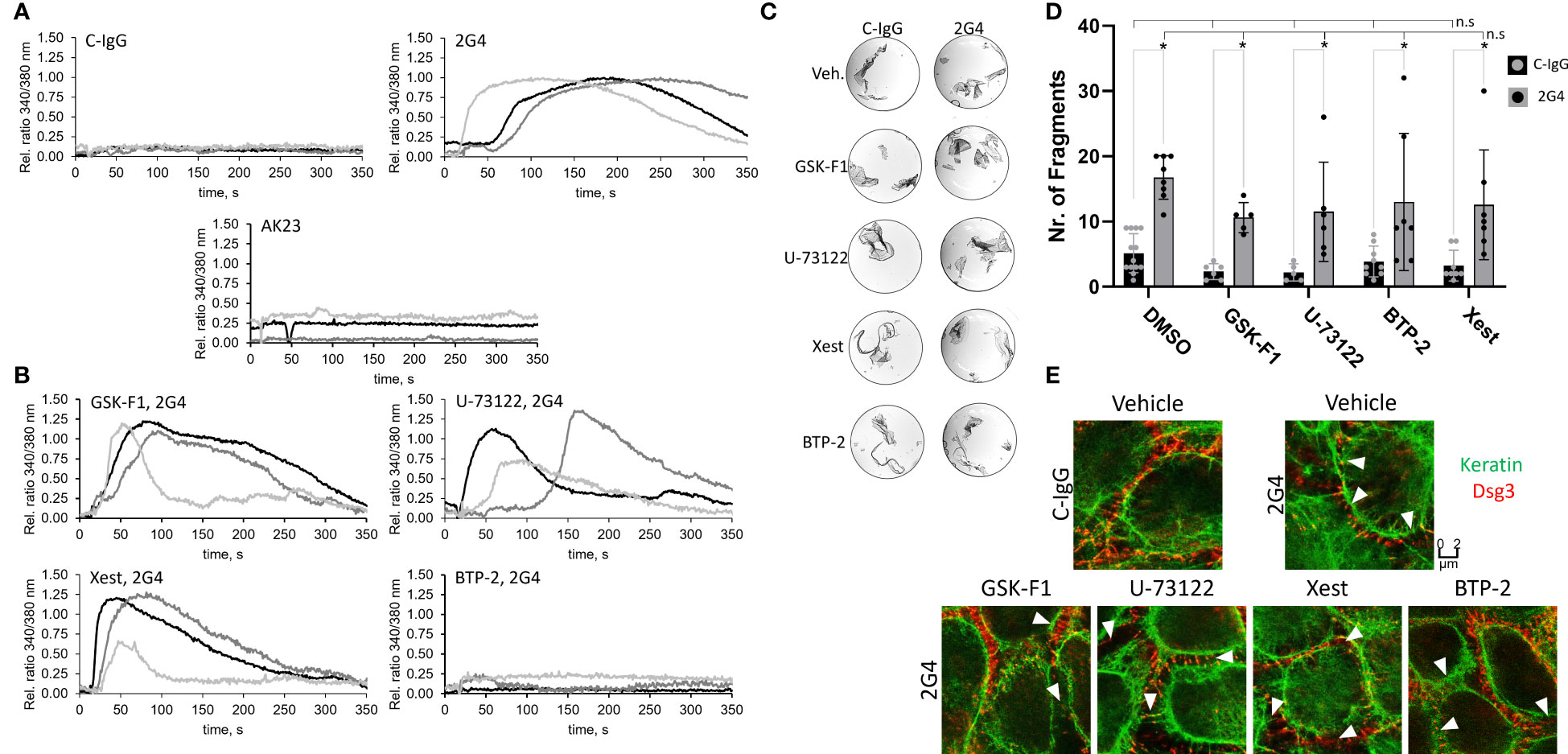
Figure 5 Alterations of intracellular Ca2+ signalling induced by 2G4. (A) Changes in intracellular Ca2+-concentration determined in HaCaT cells, after addition of 2G4, AK23 or C-IgG, by a measured change in ratio of 340/380 nm fluorescence of intracellular FURA-2AM upon Ca2+ binding (One curve represents one independent experiment, N=3, obtained from the mean of 8 cells, n=8). (B) Changes in intracellular Ca2+-concentration after addition of 2G4 to HaCaT cells previously treated with inhibitors of the anti-Dsg1-IgG-dependent Ca-influx pathway (see Table S2). (C) Representative images of dispase-based dissociation assay results after pre-treatment of HaCaT cells with Ca2+-influx pathway mediators for 1 h. (D) quantification of the dispase-based dissociation assay results (N=5-13, * makrs statistically significant differences between the two conditions indicated, n.s. (not significant) indicates, that there are no significant differences to the corresponding C-IgG (negative) or 2G4 (positive) control condition, both in two-way-ANOVA with Sidaks correction for multiple comparisons, p<0.05). (E) STED images of cells pre-treated with vehicle or Ca2+-influx pathway mediators for 1 h and C-IgG or 2G4 for 24 h. White arrowheads indicate fragmented Dsg3 staining.
Discussion
Differences in the pathogenic effects between 2G4 and AK23
We found that 2G4 induced effects typical for pemphigus pathogenesis including loss of adhesion, keratin filament retraction and fragmentation of Dsg3 staining, which is in line with previous data (47). In addition, we observed that 2G4, similar to AK23, induced a decrease in the number of desmosomes per µm of cell border. This is important because loss of desmosomes is a hallmark in pemphigus patient lesions (12, 52). The same was observed by using transmission electron microscopy of human skin treated with PV-IgG ex vivo. However, AK23 did not cause significant loss of desmosomes in intact full skin (53). The results of the current study show that 2G4 was more than two times less effective than AK23 in inducing loss of cell adhesion. Despite this, STED imaging revealed a similar degree of keratin filament retraction and loss of desmosomes for the two IgGs. The difference may still be explained in part by the loss of Dsg3 staining caused by AK23, but not 2G4, both along the cell membrane and within the cytosol, indicating Dsg3 degradation. In fact, a complete lack of extradesmosomal Dsg3 following incubation with AK23 was observed, whereas in 2G4-treated cells, Dsg3 was present in linear streaks between adjacent cells. This is also in agreement with previous findings, indicating that the extradesmosomal pool of Dsg3 is affected first, without major changes in desmosome composition (12, 54–57). It also fits to the finding that the loss of adhesion and the widening of the intercellular distance is actually initiated in the spaces between the desmosomes (58, 59) and the desmosomes are affected only later (12, 60–63). Consistent with these reports, the morphology of the desmosomes did not appear to be altered.
The fact that 2G4 induces the typical hallmarks of pemphigus pathology despite being directed against the EC5 domain is interesting because antibodies against most other domains than EC1 or EC2 and particularly those targeting the EC5 domain of Dsg3 or Dsg1, were reported to be mostly non-pathogenic (32, 34, 35, 64). For anti-Dsg1-IgGs found in individuals from an endemic population with high risk to develop PF, EC5-specific IgGs were reported to cause no symptoms. Individuals carrying anti-EC1 or anti-EC2 IgGs on the other hand suffered from clinically evident PF (39). Moreover, anti-Dsg3-IgGs isolated from two patients also demonstrated no pathogenic effects for any of the IgG fractions recognizing only EC5 (32). Furthermore, it was reported that IgGs against EC4 and EC5 are rarely found in clinically evident cases and only appear in rare atypical pemphigus cases, if at all. No patients were described to exclusively have IgGs against the EC5 domain, while patients with IgGs exclusively or mostly targeting EC1 and/or EC2 are common (33, 65). The lack of EC4 and EC5-specific IgGs in patients may be explained by the lower accessibility of these domains since the regions closer to the membrane are less exposed and are thus less likely to be subject to immunological processes, at least within mature desmosomes.
Differences in the pathogenic effects between 2G4 and AK23 are potentially caused by epitope-specific Dsg3 signalling
Signalling is known to be a major mechanism involved in pemphigus pathogenesis and was found to significantly contribute to blistering in human epidermis (66, 67). Stabilizing keratinocyte desmosomal adhesion may thus represent a novel additive treatment paradigm for pemphigus (68, 69). We proposed previously that autoantibody-specific signalling may contribute to the different clinical phenotypes of pemphigus (6). In line with this, we observed that 2G4 and AK23 induced p38MAPK activation. Akt as downstream target of EGFR and PI3-kinase was also phosphorylated and can thus also be considered to be activated in a Dsg3-dependent manner.
In addition to that, for the first time, we observed that Dsg3-mediated signalling is in part epitope-specific. Src was phosphorylated only after incubation with AK23 but was unaffected by 2G4. This difference in signalling response might also provide an explanation for the observed differences between the two IgGs in degree of loss of adhesion and their effect on the Dsg3 distribution and dynamics. It is likely that extradesmosomal Dsg complexes are degraded in response to AK23, since Dsg3, E-Cadherin and Pg containing complexes were reported to be regulated in a Src-dependent manner (48, 70, 71). This would impact adhesion via alterations in the actin cytoskeleton which was reported to stabilize extradesmosomal Dsg3 binding events (72).
Interestingly, the phosphorylation of both Src and Akt was dependent on p38MAPK activity. However, previous reports indicated that p38MAPK can also be regulated by Src, indicating a feedback loop (49, 73). Src seems to regulate the actin dynamics (48, 70, 71), p38MAPK on the other hand was reported to regulate keratin dynamics (74–77). However, p38MAPK was mostly found to be located in the soluble (non-desmosome-bound) pool and to be activated by depletion of Pg. Src on the other hand was confined mostly to the cytoskeleton bound (desmosomal) fraction, as also shown previously (78). Since it was proposed that autoantibodies in pemphigus first induce signalling via extradesmosomal Dsg molecules (79), it may be that Src activity is regulated by p38MAPK activity via keratin restructuring and p38MAPK activity at least in part by Src activity via actin restructuring.
These data support the hypothesis that p38MAPK is a central pathway in pemphigus pathogenesis, which in epidermal keratinocytes orchestrates other signalling events involved in skin blistering. The current results together with previous finding may explain, why p38MAPK inhibition alone was effective to abrogate PV-IgG-induced skin blistering in vivo and in human skin ex vivo (53, 80). Inhibition of Src alone on the other hand was protective against the effects of AK23 but not PV-IgG in human epidermis (49). This might be a result of the activation of Dsg1-dependent pathways via anti Dsg1-IgG, but it is now clear, that this effect could also be epitope specific and dependent on the presence of the right mixture of polyclonal IgGs. In contrast to that, for the formation of mucosal erosions, p38MAPK appears to not be critical. Inhibition of p38MAPK alone was not sufficient to prevent blister formation in human mucosa ex vivo (81). This indicates that the interplay of several different pathways is important for a significant loss of adhesion and blister formation in vivo. Both p38MAPK and Src are major players in this context and seem to be interdependent at least to some degree but can possibly also act independent of each other. This dynamic seems to be regulated at least in part in an epitope specific manner.
The second example for epitope-specific signalling we observed was that 2G4 induced a significant degree of rapid Ca2+-influx, whereas AK23, similar to previous findings, did not (17, 51). Since 2G4 showed no cross-reactivity with Dsg1, this most likely is a Dsg3-dependent mechanism. Inhibitors of the known Dsg1-dependent Ca2+-influx pathway revealed that CRAC was involved in this effect whereas PI4K, PLC and IP3R were not. Because inhibition of CRAC did not show any protective effect on 2G4-mediated loss of cell adhesion, we conclude that Ca2+-influx by this mechanism does not play a major role in pemphigus pathogenesis, which is different to the anti-Dsg1-induced Ca2+signalling, which reduced cell adhesion via PI4K, PLC, IP3R and CRAC (51).
Previous studies reported that the role of p38MAPK for loss of keratinocyte adhesion and epidermal blister formation might be dependent on polyclonal effects of anti-Dsg1- and/or anti-Dsg3-IgGs (82, 83). At least in combination with the bacterial exfoliative toxin A, which specifically cleaves Dsg1, much lower concentrations of AK23 of 10-15 µg/ml (compared to the 75 µg/ml used in the current study) were sufficient to cause loss of adhesion in a p38MAPK- and tyrosine kinase-independent manner. Moreover, a monoclonal PF-IgG was sufficient to cause epidermal blistering in a p38MAPK-independent manner. Therefore, we tested whether additive or polyclonal effects of AK23 and 2G4 on loss of cell adhesion were present when both antibodies were applied in combination. However, effects were similar when either the full or half of the concentration was used for both autoantibodies. These results indicate that polyclonal mixtures of these two antibodies did not induce additive effects on loss of adhesion and that the concentrations used are effective to induce maximal loss keratinocyte adhesion which is blunted by p38MAPK inhibition. Moreover, a single pathogenic autoantibody directed against Dsg3 is sufficient to induce an epitope-specific signalling response, at least at the concentrations used in this study. However, this does not rule out that combinations with other autoantibodies, which alone are considered to be non-pathogenic, may alter the signalling response or the extent of Dsg1/Dsg3 clustering and depletion (55, 82, 83).
Conclusion
The study supports the notion that monoclonal Dsg3-specific autoantibodies such as 2G4 and AK23 are pathogenic (47, 84) and are effective to cause loss of keratinocyte adhesion and induce reorganization of keratin filaments and loss of desmosomes. However, the pathogenicity is dependent on the target epitope and maybe explained in part by epitope-specific signalling including Src which may be involved in Dsg3 degradation. Further studies are required to determine how epitope-specific signalling is elicited upon autoantibody binding.
Materials and methods
Cell culture
The human epidermal keratinocyte cell line HaCaT was used for all experiments. The cells were cultivated in a humidified, 5% CO2 atmosphere at 37°C in Dulbecco’s Modified Eagle Medium (DMEM) (Life Technologies, CA) with 10 % FCS (Biochrom, DE) and 50 U/ml penicillin and 50 µg/ml streptomycin (both AppliChem, DE). They were passaged using a trypsin-EDTA solution (Merck, DE). For experiments, the cells were pre-incubated with the mediators (Table S2) for 1 h and with the IgGs for 1 h for signalling or 24 h for other experiments. Both AK23 and 2G4 were used in a concentration of 75 µg/ml, IgG purified from healthy volunteers was used 1:50.
Ratiometric intracellular Ca2+ measurements
Fura-2AM (Thermo Fisher, USA) was used to measure intracellular Ca2+ in real time. The cells were grown in an 8-well µ-slide (Ibidi, DE). Mediators (Table S2) were contained in all incubation steps and during the measurement. A mix of 1 µmol/l Fura-2AM and 0.02 % pluronic (Thermo Fisher, USA) was applied for 20 min in measurement buffer (140 mmol/l NaCl, 3.6 mmol/l KCl, 2.6 mmol/l CaCl2(H2O)2, 0.5 mmol/l MgSO4, 0.5 mmol/l NaH2PO4(H2O)2, 2 mmol/l NaHCO3, 5 mmol/l HEPES and 5 mmol/l D+Glucose, pH 7.35) at 37°C. The cells were washed twice with measurement buffer. Measurements were performed using MetaFluor (Moleculardevices, USA) on an Axio Observer A1 (Zeiss, DE) with a Polychrome V (Till Photonics, DE), a CoolSNAP-Hq2 digital camera (Photometrics, USA) and a Fura-2 filter set. For each independent experiment, the signals from 8 out of 15 randomly selected cells were evaluated (occasional non-responding cells were not included, very rare oscillating cells and weak responders were included, N = 4).
Cell lysis, gel electrophoresis and Western blotting
Cells were cultured in 24-well-plates. Lysates were fractioned into a soluble and insoluble fraction using Triton-extraction-buffer (0.5% Triton X-100, 50 mmol/l MES, 25 mmol/l EGTA, 5 mmol/l MgCl2, pH 6.8, 0.1% pepstatin+aprotinin+leupeptin, 1% Phenylmethylsulfonylfluorid) for 10 min on ice under gentle shaking. The pellet was separated at 14.000 rpm for 10 min at 4°C and the supernatant was retrieved. The pellet was washed once and lysed with ultrasound in SDS lysis buffer (25 mmol/l HEPES, 2 mmol/l EDTA, 25 mmol/l NaF, 1% SDS, pH 7.6, complete™ (Merk, USA)). The protein amount was determined with a commercial Pierce BCA protein assay kit. Western-blotting was performed, using a standard wet blotting protocol on nitrocellulose membranes (Life Technologies, USA). Membranes were blocked with ROTI®Block (Carl Roth, DE) 1:10 in Tris-buffered saline with 0.05% tween (TBST) for 1 h. The Antibodies (Table S1) were used overnight at 4°C in 5% BSA in TBST anti-rabbit/mouse horseradish-peroxidase-coupled secondary antibodies (Dianova, DE) were used 1:10.000 in TBST for 1 h and visualized with self-made ECL solution on a FluorchemE developer (Protein Simple, USA).
Dispase-based dissociation assay
After incubation as described above, confluent cell monolayers were washed with Hank´s buffered saline solution (HBSS) and subjected to 2.4 U/ml dispase II (Sigma Aldrich, USA) in HBSS for 20 min at 37 °C, 95% humidity and 5% CO2. After detachment of the monolayer the reaction was stopped by adding 200 µl HBSS. Defined shear stress was applied with an electrical pipette. Images for fragment counting were taken using a binocular microscope (Leica, DE) and an EOS 600D camera (Canon, Japan).
Immunostaining
Samples were washed and fixed with ethanol (-20°C) shaking on ice for 30 min followed by acetone (-20°C) for 3 min. The samples were blocked with 3% bovine serum albumin (BSA) and 1% normal goat serum in PBS for 30 min. Primary antibodies were applied over 3 h at RT STAR-RED- or Alexa 594-coupled goat-anti-rabbit/mouse secondary antibodies (Abberior GmbH, DE) were incubated for 1 h and DAPI 1:10.000 for 15 min. The coverslips were mounted on glass slides using Prolong™ Diamond Antifade Mountant (Thermo Fisher GmbH, DE).
STED microscopy
The samples were imaged using an Abberior 3D Stimulated emission depletion (STED) confocal microscope with IMMOIL-F30CC (Olympus GmbH, DE) Star Red and Alexa 594 were excited at 638 nm and 594 nm respectively, using pulsed diode lasers (PDL 594, Abberior Instruments; PiL063X, Advanced Laser Diode Systems). Fluorescent molecules were depleted at 775 nm with a pulsed fibre laser (PFL-P-30-775B1R, MPB Communications) and the emission was detected with an avalanche photodiode detector at 605-625 and 650-720 nm range.
Statistical analysis
Data were analysed using two-way-ANOVA followed by Sidak-post-hoc-test for multiple comparison or one-way-ANOVA followed by Dunnett-post-hoc-test for multiple comparison, using Graphpad Prism (Graphpad Software, USA). Error bars represent SEM. Significance was assumed with p ≤ 0.05. Data are shown as mean ± SEM. Each N represents one independent experiment, each n one technical replicate.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
TS: 65%, conceptualizing the project, performing experiments, performing data evaluation writing the manuscript, conceptualizing and designing the Figures, revising the manuscript. CH: 18%, providing materials, performing experiments, performing data evaluation, checking the manuscript, revising the manuscript. SM: 3%, performing experiments, performing data evaluation. MH: 2%, providing materials, checking the manuscript. RT: 2%, providing materials, checking the manuscript. JW: 10%, providing funding, conceptualizing the project, checking the manuscript, revising the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The research was supported by the Deutsche Forschungsgemeinschaft (DFG) FOR 2497/TP04 to RT (TI 291/10-2)/TP05 to JW and TP08 to MH (HE 1602/16-2) is acknowledged.
Acknowledgments
We thank Silke Gotschy, Martina Hitzenbichler and Manuel Schulze-Dasbeck for their excellent technical assistance. Special thanks also to Mariya Radeva for help with checking Figures.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1163066/full#supplementary-material
References
1. Spindler V, Gerull B, Green KJ, Kowalczyk AP, Leube R, Marian AJ, et al. Meeting report - desmosome dysfunction and disease: Alpine desmosome disease meeting. J Cell Sci (2023) 136(1):jcs260832. doi: 10.1242/jcs.260832
2. Carson PJ, Hameed A, Ahmed AR. Influence of treatment on the clinical course of pemphigus vulgaris. J Am Acad Dermatol (1996) 34:645–52. doi: 10.1016/s0190-9622(96)80066-1
3. Kasperkiewicz M, Ellebrecht CT, Takahashi H, Yamagami J, Zillikens D, Payne AS, et al. Pemphigus. Nat Rev Dis Primers (2017) 3:17026. doi: 10.1038/nrdp.2017.26
4. Schmidt E, Kasperkiewicz M, Joly P. Pemphigus. Lancet (2019) 394:882–94. doi: 10.1016/S0140-6736(19)31778-7
5. Amagai M, Stanley JR. Desmoglein as a target in skin disease and beyond. J Invest Dermatol (2012) 132:776–84. doi: 10.1038/jid.2011.390
6. Schmitt T, Waschke J. Autoantibody-specific signalling in pemphigus. Front Med (Lausanne) (2021) 8:701809. doi: 10.3389/fmed.2021.701809
7. Heupel W-M, Zillikens D, Drenckhahn D, Waschke J. Pemphigus vulgaris IgG directly inhibit desmoglein 3-mediated transinteraction. J Immunol (2008) 181:1825–34. doi: 10.4049/jimmunol.181.3.1825
8. Vielmuth F, Waschke J, Spindler V. Loss of desmoglein binding is not sufficient for keratinocyte dissociation in pemphigus. J Invest Dermatol (2015) 135:3068–77. doi: 10.1038/jid.2015.324
9. Ishii K, Yoshida K, Stanley JR, Yamagami J, Amagai M, Ishiko A. Pemphigus vulgaris and foliaceus IgG autoantibodies directly block heterophilic trans-interaction between desmoglein and desmocollin. J Invest Dermatol (2020) 140(10):1919–1926.e7. doi: 10.1016/j.jid.2020.02.010
10. Kitajima Y, Aoyama Y, Seishima M. Transmembrane signaling for adhesive regulation of desmosomes and hemidesmosomes, and for cell-cell datachment induced by pemphigus IgG in cultured keratinocytes: involvement of protein kinase c. J Investig Dermatol Symp Proc (1999) 4:137–44. doi: 10.1038/sj.jidsp.5640197
11. Burmester IA, Flaswinkel S, Thies C-S, Kasprick A, Kamaguchi M, Bumiller-Bini V, et al. Identification of novel therapeutic targets for blocking acantholysis in pemphigus. Br J Pharmacol (2020) 177(22):5114–30. doi: 10.1111/bph.15233
12. Schmitt T, Pircher J, Steinert L, Meier K, Ghoreschi K, Vielmuth F, et al. Dsg1 and Dsg3 composition of desmosomes across human epidermis and alterations in pemphigus vulgaris patient skin. Front Immunol (2022) 13:884241. doi: 10.3389/fimmu.2022.884241
13. Hartlieb E, Rötzer V, Radeva M, Spindler V, Waschke J. Desmoglein 2 compensates for desmoglein 3 but does not control cell adhesion via regulation of p38 mitogen-activated protein kinase in keratinocytes. J Biol Chem (2014) 289:17043–53. doi: 10.1074/jbc.M113.489336
14. Hegazy M, Koetsier JL, Huffine AL, Broussard JA, Godsel BM, Cohen-Barak E, et al. Epidermal stratification requires retromer-mediated desmoglein-1 recycling. Dev Cell (2022) 57:2683–2698.e8. doi: 10.1016/j.devcel.2022.11.010
15. Vielmuth F, Walter E, Fuchs M, Radeva MY, Buechau F, Magin TM, et al. Keratins regulate p38MAPK-dependent desmoglein binding properties in pemphigus. Front Immunol (2018) 9:528. doi: 10.3389/fimmu.2018.00528
16. Waschke J, Bruggeman P, Baumgartner W, Zillikens D, Drenckhahn D. Pemphigus foliaceus IgG causes dissociation of desmoglein 1-containing junctions without blocking desmoglein 1 transinteraction. J Clin Invest (2005) 115:3157–65. doi: 10.1172/JCI23475
17. Walter E, Vielmuth F, Rotkopf L, Sárdy M, Horváth ON, Goebeler M, et al. Different signaling patterns contribute to loss of keratinocyte cohesion dependent on autoantibody profile in pemphigus. Sci Rep (2017) 7:3579. doi: 10.1038/s41598-017-03697-7
18. Amber KT, Valdebran M, Grando SA. Non-desmoglein antibodies in patients with pemphigus vulgaris. Front Immunol (2018) 9:1190. doi: 10.3389/fimmu.2018.01190
19. Schmidt E, Goebeler M, Hertl M, Sárdy M, Sitaru C, Eming R, et al. S2k-leitlinie zur diagnostik des pemphigus vulgaris/foliaceus und des bullösen pemphigoids. JDDG: J der Deutschen Dermatologischen Gesellschaft (2015) 13:713–27. doi: 10.1111/ddg.40_12612
20. Kneisel A, Hertl M. Autoimmune bullous skin diseases. part 2: diagnosis and therapy. J Dtsch Dermatol Ges (2011) 9:927–47. doi: 10.1111/j.1610-0387.2011.07809.x
21. Mao X, Nagler AR, Farber SA, Choi EJ, Jackson LH, Leiferman KM, et al. Autoimmunity to desmocollin 3 in pemphigus vulgaris. Am J Pathol (2010) 177:2724–30. doi: 10.2353/ajpath.2010.100483
22. Rafei D, Müller R, Ishii N, Llamazares M, Hashimoto T, Hertl M, et al. IgG autoantibodies against desmocollin 3 in pemphigus sera induce loss of keratinocyte adhesion. Am J Pathol (2011) 178:718–23. doi: 10.1016/j.ajpath.2010.10.016
23. Spindler V, Heupel W-M, Efthymiadis A, Schmidt E, Eming R, Rankl C, et al. Desmocollin 3-mediated binding is crucial for keratinocyte cohesion and is impaired in pemphigus. J Biol Chem (2009) 284:30556–64. doi: 10.1074/jbc.M109.024810
24. Müller R, Heber B, Hashimoto T, Messer G, Müllegger R, Niedermeier A, et al. Autoantibodies against desmocollins in european patients with pemphigus. Clin Exp Dermatol (2009) 34:898–903. doi: 10.1111/j.1365-2230.2009.03241.x
25. Ivars M, España A, Alzuguren P, Pelacho B, Lasarte JJ, López-Zabalza MJ. The involvement of ADAM10 in acantholysis in mucocutaneous pemphigus vulgaris depends on the autoantibody profile of each patient. Br J Dermatol (2020) 182:1194–204. doi: 10.1111/bjd.18382
26. Geller S, Gat A, Zeeli T, Hafner A, Eming R, Hertl M, et al. The expanding spectrum of IgA pemphigus: a case report and review of the literature. Br J Dermatol (2014) 171:650–6. doi: 10.1111/bjd.12940
27. Grando SA, Pittelkow MR, Shultz LD, Dmochowski M, Nguyen VT. Pemphigus: an unfolding story. J Invest Dermatol (2001) 117 4:990–5. doi: 10.1046/j.0022-202x.2001.01489.x
28. Pan M, Liu X, Zheng J. The pathogenic role of autoantibodies in pemphigus vulgaris. Clin Exp Dermatol (2011) 36:703–7. doi: 10.1111/j.1365-2230.2011.04092.x
29. Kalantari-Dehaghi M, Anhalt GJ, Camilleri MJ, Chernyavsky AI, Chun S, Felgner PL, et al. Pemphigus vulgaris autoantibody profiling by proteomic technique. PloS One (2013) 8:e57587. doi: 10.1371/journal.pone.0057587
30. Koch PJ, Walsh MJ, Schmelz M, Goldschmidt MD, Zimbelmann R, Franke WW, et al. Identification of desmoglein, a constitutive desmosomal glycoprotein, as a member of the cadherin family of cell adhesion molecules // identification of desmoglein as a cadherin and analysis of desmoglein domain structure. Eur J Cell Biol (1990) 53:1–12. doi: 10.1038/sj.skinbio.6250001
31. Nose A, Tsuji K, Takeichi M. Localization of specificity determining sites in cadherin cell adhesion molecules. Cell (1990) 61:147–55. doi: 10.1016/0092-8674(90)90222-z
32. Di Zenzo G, Di Lullo G, Corti D, Calabresi V, Sinistro A, Vanzetta F, et al. Pemphigus autoantibodies generated through somatic mutations target the desmoglein-3 cis-interface. J Clin Invest (2012) 122:3781–90. doi: 10.1172/JCI64413
33. Müller R, Svoboda V, Wenzel E, Gebert S, Hunzelmann N, Müller H-H, et al. IgG reactivity against non-conformational NH-terminal epitopes of the desmoglein 3 ectodomain relates to clinical activity and phenotype of pemphigus vulgaris. Exp Dermatol (2006) 15:606–14. doi: 10.1111/j.1600-0625.2006.00451.x
34. Di Zenzo G, Amber KT, Sayar BS, Müller EJ, Borradori L. Immune response in pemphigus and beyond: progresses and emerging concepts. Semin Immunopathol (2016) 38:57–74. doi: 10.1007/s00281-015-0541-1
35. Kridin K. Pemphigus group: overview, epidemiology, mortality, and comorbidities. Immunol Res (2018) 66 2:255–70. doi: 10.1007/s12026-018-8986-7
36. Abida O, Masmoudi A, Rebaï A, Ben Ayed M, Mahfoudh N, Kallel-Sellami M, et al. The familial feature of tunisian endemic pemphigus foliaceus. Br J Dermatol (2009) 161:951–3. doi: 10.1111/j.1365-2133.2009.09386.x
37. Abida O, Zitouni M, Kallel-Sellami M, Mahfoudh N, Kammoun A, Ben Ayed M, et al. Tunisian endemic pemphigus foliaceus is associated with the HLA-DR3 gene: anti-desmoglein 1 antibody-positive healthy subjects bear protective alleles. Br J Dermatol (2009) 161:522–7. doi: 10.1111/j.1365-2133.2009.09207.x
38. Joly P, Mokhtar I, Gilbert D, Thomine E, Fazza B, Bardi R, et al. Immunoblot and immunoelectronmicroscopic analysis of endemic tunisian pemphigus. Br J Dermatol (1999) 140:44–9. doi: 10.1046/j.1365-2133.1999.02605.x
39. Kallel Sellami M, Ben Ayed M, Mouquet H, Drouot L, Zitouni M, Mokni M, et al. Anti-desmoglein 1 antibodies in tunisian healthy subjects: arguments for the role of environmental factors in the occurrence of tunisian pemphigus foliaceus. Clin Exp Immunol (2004) 137:195–200. doi: 10.1111/j.1365-2249.2004.02493.x
40. Amber KT, Staropoli P, Shiman MI, Elgart GW, Hertl M. Autoreactive t cells in the immune pathogenesis of pemphigus vulgaris. Exp Dermatol (2013) 22:699–704. doi: 10.1111/exd.12229
41. Hammers CM, Stanley JR. Mechanisms of disease: Pemphigus and bullous pemphigoid. Annu Rev Pathol (2016) 11:175–97. doi: 10.1146/annurev-pathol-012615-044313
42. Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis). Clin Dermatol (2011) 29:432–6. doi: 10.1016/j.clindermatol.2011.01.013
43. Takahashi H, Kuwana M, Amagai M. A single helper t cell clone is sufficient to commit polyclonal naive b cells to produce pathogenic IgG in experimental pemphigus vulgaris. J Immunol (2009) 182:1740–5. doi: 10.4049/jimmunol.182.3.1740
44. Aksu D, Peksari Y, Arica IE, Gurgey E. Assessing the autoantibody levels in relation to disease severity and therapy response in pemphigus patients. Indian J Dermatol (2010) 55:342–7. doi: 10.4103/0019-5154.74536
45. Weiss D, Ristl R, Griss J, Bangert C, Foedinger D, Stingl G, et al. Autoantibody levels and clinical disease severity in patients with pemphigus: Comparison of aggregated anti-desmoglein ELISA values and indirect immunofluorescence titres. Acta Derm Venereol (2015) 95:559–64. doi: 10.2340/00015555-2023
46. Mahoney MG, Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest (1999) 103:461–8. doi: 10.1172/JCI5252
47. Hudemann C, Exner Y, Pollmann R, Schneider K, Zakrzewicz A, Feldhoff S, et al. IgG against the membrane-proximal portion of the desmoglein 3 ectodomain induces loss of keratinocyte adhesion, a hallmark in pemphigus vulgaris. J Invest Dermatol (2022) 143:254–263.e3. doi: 10.1016/j.jid.2022.07.030
48. Kugelmann D, Rötzer V, Walter E, Egu DT, Fuchs MT, Vielmuth F, et al. Role of src and cortactin in pemphigus skin blistering. Front Immunol (2019) 10:626. doi: 10.3389/fimmu.2019.00626
49. Tsang SM, Brown L, Lin K, Liu L, Piper K, O'Toole EA, et al. Non-junctional human desmoglein 3 acts as an upstream regulator of src in e-cadherin adhesion, a pathway possibly involved in the pathogenesis of pemphigus vulgaris. J Pathol (2012) 227:81–93. doi: 10.1002/path.3982
50. Pretel M, España A, Marquina M, Pelacho B, López-Picazo JM, López-Zabalza MJ. An imbalance in Akt/mTOR is involved in the apoptotic and acantholytic processes in a mouse model of pemphigus vulgaris. Exp Dermatol (2009) 18:771–80. doi: 10.1111/j.1600-0625.2009.00893.x
51. Schmitt T, Egu DT, Walter E, Sigmund AM, Eichkorn R, Yazdi A, et al. Ca2+ signalling is critical for autoantibody-induced blistering of human epidermis in pemphigus. Br J Dermatol (2021) 185:595–604. doi: 10.1111/bjd.20091
52. Sokol E, Kramer D, Diercks GF, Kuipers J, Jonkman MF, Pas HH, et al. Large-scale electron microscopy maps of patient skin and mucosa provide insight into pathogenesis of blistering diseases. J Invest Dermatol (2015) 135:1763–70. doi: 10.1038/jid.2015.109
53. Egu DT, Walter E, Spindler V, Waschke J. Inhibition of p38MAPK signalling prevents epidermal blistering and alterations of desmosome structure induced by pemphigus autoantibodies in human epidermis. Br J Dermatol (2017) 177:1612–8. doi: 10.1111/bjd.15721
54. Mao X, Choi EJ, Payne AS. Disruption of desmosome assembly by monovalent human pemphigus vulgaris monoclonal antibodies. J Invest Dermatol (2009) 129:908–18. doi: 10.1038/jid.2008.339
55. Yamamoto Y, Aoyama Y, Shu E, Tsunoda K, Amagai M, Kitajima Y. Anti-desmoglein 3 (Dsg3) monoclonal antibodies deplete desmosomes of Dsg3 and differ in their Dsg3-depleting activities related to pathogenicity. J Biol Chem (2007) 282:17866–76. doi: 10.1074/jbc.M607963200
56. Aoyama Y, Kitajima Y. Pemphigus vulgaris-IgG causes a rapid depletion of desmoglein 3 (Dsg3) from the triton x-100 soluble pools, leading to the formation of Dsg3-depleted desmosomes in a human squamous carcinoma cell line, DJM-1 cells. J Invest Dermatol (1999) 112:67–71. doi: 10.1046/j.1523-1747.1999.00463.x
57. Jennings JM, Tucker DK, Kottke MD, Saito M, Delva E, Hanakawa Y, et al. Desmosome disassembly in response to pemphigus vulgaris IgG occurs in distinct phases and can be reversed by expression of exogenous Dsg3. J Invest Dermatol (2011) 131:706–18. doi: 10.1038/jid.2010.389
58. Takahashi Y, Patel HP, Labib RS, Diaz LA, Anhalt GJ. Experimentally induced pemphigus vulgaris in neonatal BALB/c mice: a time-course study of clinical, immunologic, ultrastructural, and cytochemical changes. J Invest Dermatol (1985) 84:41–6. doi: 10.1111/1523-1747.ep12274679
59. Hu CH, Michel B, Schiltz JR. Epidermal acantholysis induced in vitro by pemphigus autoantibody ultrastructural study. Am J Pathol (1978) 90:345–62.
60. Bystryn J-C, Grando SA. A novel explanation for acantholysis in pemphigus vulgaris: the basal cell shrinkage hypothesis. J Am Acad Dermatol (2006) 54:513–6. doi: 10.1016/j.jaad.2005.12.003
61. Spindler V, Waschke J. Desmosomal cadherins and signaling: lessons from autoimmune disease. Cell Commun Adhes (2014) 21:77–84. doi: 10.3109/15419061.2013.877000
62. Wilgram GF, Caulfield JB, Lever WF. An electron microscopic study of acantholysis in pemphigus vulgaris. J Invest Dermatol (1961) 36:373–82. doi: 10.1038/jid.1961.58
63. Sato M, Aoyama Y, Kitajima Y. Assembly pathway of desmoglein 3 to desmosomes and its perturbation by pemphigus vulgaris-IgG in cultured keratinocytes, as revealed by time-lapsed labeling immunoelectron microscopy. Lab Invest (2000) 80:1583–92. doi: 10.1038/labinvest.3780168
64. Kamiya K, Aoyama Y, Shirafuji Y, Hamada T, Morizane S, Fujii K, et al. Detection of antibodies against the non-calcium-dependent epitopes of desmoglein 3 in pemphigus vulgaris and their pathogenic significance. Br J Dermatol (2012) 167:252–61. doi: 10.1111/j.1365-2133.2012.10929.x
65. Ohyama B, Nishifuji K, Chan PT, Kawaguchi A, Yamashita T, Ishii N, et al. Epitope spreading is rarely found in pemphigus vulgaris by large-scale longitudinal study using desmoglein 2-based swapped molecules. J Invest Dermatol (2012) 132:1158–68. doi: 10.1038/jid.2011.448
66. Spindler V, Eming R, Schmidt E, Amagai M, Grando S, Jonkman MF, et al. Mechanisms causing loss of keratinocyte cohesion in pemphigus. J Invest Dermatol (2018) 138:32–7. doi: 10.1016/j.jid.2017.06.022
67. Egu DT, Schmitt T, Waschke J. Mechanisms causing acantholysis in pemphigus-lessons from human skin. Front Immunol (2022) 13:884067. doi: 10.3389/fimmu.2022.884067
68. Spindler V, Rötzer V, Dehner C, Kempf B, Gliem M, Radeva M, et al. Peptide-mediated desmoglein 3 crosslinking prevents pemphigus vulgaris autoantibody-induced skin blistering. J Clin Invest (2013) 123:800–11. doi: 10.1172/JCI60139
69. Sigmund AM, Winkler M, Engelmayer S, Kugelmann D, Egu DT, Steinert LS, et al. Apremilast prevents blistering in human epidermis and stabilizes keratinocyte adhesion in pemphigus. Nat Commun (2023) 14:116. doi: 10.1038/s41467-022-35741-0
70. Rötzer V, Hartlieb E, Vielmuth F, Gliem M, Spindler V, Waschke J. E-cadherin and src associate with extradesmosomal Dsg3 and modulate desmosome assembly and adhesion. Cell Mol Life Sci (2015) 72:4885–97. doi: 10.1007/s00018-015-1977-0
71. Shafraz O, Rübsam M, Stahley SN, Caldara AL, Kowalczyk AP, Niessen CM, et al. E-cadherin binds to desmoglein to facilitate desmosome assembly. Elife (2018) 7:e37629. doi: 10.7554/eLife.37629
72. Fuchs M, Radeva MY, Spindler V, Vielmuth F, Kugelmann D, Waschke J. Cytoskeletal anchorage of different Dsg3 pools revealed by combination of hybrid STED/SMFS-AFM. Cell Mol Life Sci (2023) 80:25. doi: 10.1007/s00018-022-04681-9
73. Chernyavsky AI, Arredondo J, Kitajima Y, Sato-Nagai M, Grando SA. Desmoglein versus non-desmoglein signaling in pemphigus acantholysis: characterization of novel signaling pathways downstream of pemphigus vulgaris antigens. J Biol Chem (2007) 282:13804–12. doi: 10.1074/jbc.M611365200
74. Sawant M, Schwarz N, Windoffer R, Magin TM, Krieger J, Mücke N, et al. Threonine 150 phosphorylation of keratin 5 is linked to epidermolysis bullosa simplex and regulates filament assembly and cell viability. J Invest Dermatol (2018) 138:627–36. doi: 10.1016/j.jid.2017.10.011
75. Meir M, Burkard N, Ungewiß H, Diefenbacher M, Flemming S, Kannapin F, et al. Neurotrophic factor GDNF regulates intestinal barrier function in inflammatory bowel disease. J Clin Invest (2019) 129:2824–40. doi: 10.1172/JCI120261
76. Wöll S, Windoffer R, Leube RE. p38 MAPK-dependent shaping of the keratin cytoskeleton in cultured cells. J Cell Biol (2007) 177:795–807. doi: 10.1083/jcb.200703174
77. Spindler V, Dehner C, Hübner S, Waschke J. Plakoglobin but not desmoplakin regulates keratinocyte cohesion via modulation of p38MAPK signaling. J Invest Dermatol (2014) 134:1655–64. doi: 10.1038/jid.2014.21
78. Tsang SM, Liu L, Teh M-T, Wheeler A, Grose R, Hart IR, et al. Desmoglein 3, via an interaction with e-cadherin, is associated with activation of src. PloS One (2010) 5:e14211. doi: 10.1371/journal.pone.0014211
79. Müller EJ, Williamson L, Kolly C, Suter MM. Outside-in signaling through integrins and cadherins: a central mechanism to control epidermal growth and differentiation? J Invest Dermatol (2008) 128:501–16. doi: 10.1038/sj.jid.5701248
80. Berkowitz P, Hu P, Liu Z, Diaz LA, Enghild JJ, Chua MP, et al. Desmosome signaling. inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J Biol Chem (2005) 280(25):23778–84. doi: 10.1074/jbc.M501365200
81. Egu DT, Sigmund AM, Schmidt E, Spindler V, Walter E, Waschke J. A new ex vivo human oral mucosa model reveals that p38MAPK inhibition is not effective in preventing autoantibody-induced mucosal blistering in pemphigus. Br J Dermatol (2020) 182:987–94. doi: 10.1111/bjd.18237
82. Saito M, Stahley SN, Caughman CY, Mao X, Tucker DK, Payne AS, et al. Signaling dependent and independent mechanisms in pemphigus vulgaris blister formation. PloS One (2012) 7:e50696. doi: 10.1371/journal.pone.0050696
83. Yoshida K, Ishii K, Shimizu A, Yokouchi M, Amagai M, Shiraishi K, et al. Non-pathogenic pemphigus foliaceus (PF) IgG acts synergistically with a directly pathogenic PF IgG to increase blistering by p38MAPK-dependent desmoglein 1 clustering. J Dermatol Sci (2017) 85:197–207. doi: 10.1016/j.jdermsci.2016.12.010
Keywords: pemphigus, autoimmune disease, skin, epidermis, desmosomes, adhesion, keratin, desmoglein (dsg)
Citation: Schmitt T, Hudemann C, Moztarzadeh S, Hertl M, Tikkanen R and Waschke J (2023) Dsg3 epitope-specific signalling in pemphigus. Front. Immunol. 14:1163066. doi: 10.3389/fimmu.2023.1163066
Received: 10 February 2023; Accepted: 31 March 2023;
Published: 18 April 2023.
Edited by:
Takashi Hashimoto, Osaka City University, JapanReviewed by:
Jong Hoon Kim, Yonsei University, Republic of KoreaGiovanni Di Zenzo, Istituto Dermopatico dell’Immacolata (IDI)-IRCCS, Italy
Hiroshi Koga, Kurume University School of Medicine, Japan
Copyright © 2023 Schmitt, Hudemann, Moztarzadeh, Hertl, Tikkanen and Waschke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jens Waschke, amVucy53YXNjaGtlQG1lZC51bmktbXVlbmNoZW4uZGU=
†ORCID: Thomas Schmitt, orcid.org/0000-0003-3781-7236
Christoph Hudemann, orcid.org/0000-0001-5807-6882
Sina Moztarzadeh, orcid.org/0000-0001-7519-598X
Michael Hertl, orcid.org/0000-0002-5494-2461
Ritva Tikkanen, orcid.org/0000-0002-8393-1825
Jens Waschke, orcid.org/0000-0003-1182-5422