 Alexander Glover
Alexander Glover Zhaoqin Zhang
Zhaoqin Zhang Claire Shannon-Lowe
Claire Shannon-Lowe
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 23 March 2023
Sec. Immunological Tolerance and Regulation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1161848
This article is part of the Research TopicMyeloid-Derived Suppressor Cells in Health and Diseases: Harnessing Their Roles in the Development of New Immunotherapy ApproachesView all 6 articles
Myeloid derived suppressor cells (MDSCs) are a heterogenous population of myeloid cells derived from monocyte and granulocyte precursors. They are pathologically expanded in conditions of ongoing inflammation where they function to suppress both innate and adaptive immunity. They are subdivided into three distinct subsets: monocytic (M-) MDSC, polymorphonuclear (or neutrophilic) (PMN-) MDSC and early-stage (e-) MDSC that may exhibit differential function in different pathological scenarios. However, in cancer they are associated with inhibition of the anti-tumour immune response and are universally associated with a poor prognosis. Seven human viruses classified as Group I carcinogenic agents are jointly responsible for nearly one fifth of all human cancers. These viruses represent a large diversity of species, including DNA, RNA and retroviridae. They include the human gammaherpesviruses (Epstein Barr virus (EBV) and Kaposi’s Sarcoma-Associated Herpesvirus (KSHV), members of the high-risk human papillomaviruses (HPVs), hepatitis B and C (HBV, HCV), Human T cell leukaemia virus (HTLV-1) and Merkel cell polyomavirus (MCPyV). Each of these viruses encode an array of different oncogenes that perturb numerous cellular pathways that ultimately, over time, lead to cancer. A prerequisite for oncogenesis is therefore establishment of chronic infection whereby the virus persists in the host cells without being eradicated by the antiviral immune response. Although some of the viruses can directly modulate the immune response to enable persistence, a growing body of evidence suggests the immune microenvironment is modulated by expansions of MDSCs, driven by viral persistence and oncogenesis. It is likely these MDSCs play a role in loss of immune recognition and function and it is therefore essential to understand their phenotype and function, particularly given the increasing importance of immunotherapy in the modern arsenal of anti-cancer therapies. This review will discuss the role of MDSCs in viral oncogenesis. In particular we will focus upon the mechanisms thought to drive the MDSC expansions, the subsets expanded and their impact upon the immune microenvironment. Importantly we will explore how MDSCs may modulate current immunotherapies and their impact upon the success of future immune-based therapies.
The concept that an infectious agent may be causal in oncogeneses dates back to 1842, when Domenico Rigoni-Stern described cervical cancer incidence as being highest in sex workers, lowest in nuns and at an intermediate level in married women (1). However, it took until 1911 for the first tumour-causing virus to be identified; discovered by the American pathologist Peyton Rous, the Rous sarcoma virus was shown to be responsible for the carcinogenesis of domestic chicken tumours (2). The first human cancer causing virus, the Epstein-Barr virus (EBV, was identified in 1964 as the infectious agent responsible for endemic Burkitt lymphoma (3). The later surprise discovery that EBV infects more than 95% of the world’s population and is the causal agent of infectious mononucleosis (IM) illustrates the concept that a common virus can trigger rare cancers (4). There are now seven known human oncogenic viruses (5, 6). They include the human gamma herpesviruses; EBV and Kaposi’s Sarcoma-Associated Herpesvirus (KSHV), members of the high-risk human papillomaviruses (HPVs), hepatitis B and C (HBV, HCV), Human T cell lymphotropic virus (HTLV-1) and Merkel cell polyomavirus (MCPyV). In no case does triggering cancer form part of the virus’ natural life cycle, yet all the viruses share the ability to cause chronic infection, which over time and with additional risk factors can lead to carcinogenesis (5).
Although it is well documented that virus-associated cancers are common in those with immunocompromise (7), particularly in conditions such as Kaposi’s sarcoma or post-transplant lymphoproliferative disease (PTLD), viral oncogenesis also occurs in those with an intact immune system. The mechanisms enabling viral oncogenesis are complex and varied but often involve modulation of the antiviral immune response, including limited expression of immunogenic viral antigens, or infection of tissues with reduced immune surveillance. There is, however, growing evidence that myeloid-derived suppressor cells (MDSCs) may play a role in this process.
MDSCs are a heterogeneous group of myeloid cells capable of suppressing antigen-specific immune function. There are three major classes of MDSCs the phenotype of which is illustrated in Table 1. Polymorphonuclear (PMN-MDSCs) resemble neutrophils but have a suppressive function. While key differences in function have been described between PMN-MDSC and neutrophils in both mice and humans (8), other groups have called into question if they can be truly be described as a differentiated population rather than an alternative activation state (9). Monocytic (M-MDSCs) are morphologically identical to monocytes but are immunosuppressive and HLA-DRlow. Early MDSCs (e-MDSCs) are the third group of suppressive myeloid cells; lacking mature monocyte or neutrophil markers, it is unclear if these cells could be the precursors for other MDSC classes (10). The study of MDSCs has been hampered by a lack of a specific phenotype to separate them from conventional neutrophils and monocytes (11). Accurate identification requires either demonstration of suppressive function, use of biochemical surrogate markers or density centrifugation in the case of PMN-MDSCs, making identifying populations in fixed tissues difficult (11). In addition, technical differences between laboratories can introduce variation in MDSC enumeration (12).
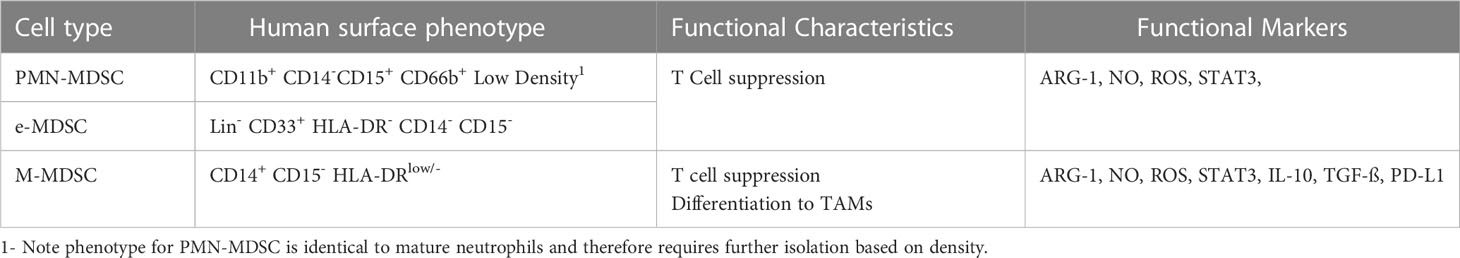
Table 1 Phenotype and functional markers by MDSC class.
MDSCs were first described in the context of cancer under a variety of names before the current terminology was settled upon (13). In cancer, MDSCs are associated with a reduction in overall (OS) and progression-free survival (PFS) and an increased rate of relapse (14). Over time they have been identified in a greater variety of disease states including infection (15), pregnancy (16), obesity (17) and sepsis (18). We will first discuss the mechanisms behind MDSC generation and how this can prevent viral clearance by the adaptive immune system. We will then discuss the evidence for MDSCs for each of the cancer-causing viruses. Finally, we will discuss what therapeutic options are available to eliminate MDSC populations.
While functionally distinct, PMN-MDSC are phenotypically almost identical to conventional neutrophils and morphologically resemble either immature band cells or have the segmented nucleus of a mature neutrophil. Neutrophils are the most abundant leukocyte in the blood and under steady-state conditions are produced in the marrow from lineage-committed precursors (19). The precise mechanisms by which PMN-MDSC are generated rather than inflammatory activated neutrophils are not fully understood.
Neutrophils are typically seen as short-lived cells, but their lifespan can be extended under inflammatory conditions. In particular, PGE2 can suppress neutrophil apoptosis and prolong survival through activation of protein kinase A (PKA) (20). Neutrophils have a large variety of cell surface receptors for chemokines, cytokines and immunoglobulin as well as innate immune receptors which act through diverse signalling pathways to trigger homing and activation (21). Classically, neutrophils extravasate at sites of inflammation following a chemokine gradient and are then activated after exposure to damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs). These activated, or effector, neutrophils then kill pathogens by phagocytosis, NETosis and degranulation (19, 22) and therefore constitute the immunological first line of defence against bacterial and fungal infections.
The process of MDSC generation appears to involve two distinct steps: the production of myeloid progenitors and then activation to a full PMN-MDSC phenotype. The relative contribution of each step may then impact on the morphological maturity of the resulting cells. Granulocyte differentiation is initially triggered by G-CSF which can result in emergency myelopoiesis and the release of immature myeloid cells (23). Thereafter, a number of factors have been suggested to trigger the generation of MDSCs from progenitors, including Stem cell factor (SCF) acting via C-KIT (24), combinations of G-CSF/IL-6 acting through C/EBPβ (25) and S100A8/A9 through NFκB. Furthermore, factors produced by MDSCs such as S100 family proteins can trigger an autocrine feedback loop leading to further MDSC generation (26). However, other studies propose mature blood neutrophils can develop an immunosuppressive phenotype induced by factors such as PGE2 (27) or endoplasmic reticulum (ER) stress (28) triggered by hypoxia, nutrient deprivation or increased ROS and NOS often associated with chronic inflammation and in a tumour microenvironment (29, 30).
PMN-MDSCs are usually characterised by their low density as opposed to the high density of classical mature neutrophils, although some activated pro-inflammatory neutrophils are also low density. Gene expression profiling of low-density neutrophils identified the expression of LOX-1 in the blood of cancer patients as well as in tumour tissue but not in healthy donors (28). Initially, due to their band-like immature morphology, similar to that observed in LOX1+ neutrophils, it was proposed that PMN-MDSCs have an immature phenotype (28). Yet, patients treated with high-dose GCSF for stem cell mobilisation exhibit an expansion of activated CD10+ neutrophils that are morphologically mature; inhibit T cell proliferation and release IFN-γ in-vitro, and have an activated phenotype (31). While this appears to be a contradiction, a further study correlated CD11b and CD16 expression with nuclear morphology and maturity demonstrating both mature and immature cells can be suppressive and meet the criteria to be defined as PMN-MDSCs. However, the greatest degree of immune suppression was seen in CD11b+CD16+ mature cells (32).
Monocytes differentiate from myeloid precursors in the marrow under the control of growth factors and are then released into the circulation. Conventional monocytes are divided into classical (CD14+CD16-), non-classical (CD14dimCD16+) and intermediate (CD14+CD16+) populations. Classical monocytes are produced from the marrow and are capable of phagocytosis and tissue migration. As they mature, they develop an intermediate phenotype which is strongly HLA-DR+ and can act as antigen-presenting cells. Non-classical monocytes principally function through complement and antibody-dependent phagocytosis (33). Monocytes can then enter tissues and further differentiate into macrophages and dendritic cells (34). However, M-MDSC arise from the CD14+ population and are HLA-DR negative (35). They express increased IL-10 and IL-8 but reduced IL-1β, IL-6 and TNF compared to classical monocytes (35).
Monocyte activation towards an inflammatory phenotype occurs after exposure to PAMPs and DAMPS. Conversely, M-MDSC generation appears to occur after weaker activation signals by cytokines, particularly TGF-β, or repeated low-intensity toll-like receptor (TLR) stimulation. TGF-β initially appears to act as a chemoattractant for CD14+CD33+ myeloid cells. Tumour tissue from multiple different cancers express TGF-β and this expression is closely associated with infiltration of CD14+CD33+ myeloid cells. In addition, TGF-β also appears to play a role in acquisition of the M-MDSC phenotype as demonstrated by in-vitro treatment of CD14+ monocytes with TGF-β, GM-CSF and IL-6, resulting in the development of the suppressive phenotype. Furthermore, repeated TLR 2/4 stimulation of the monocytes triggers downregulation of HLA-DR and secretion of IL-10 and TGF-β, thereby amplifying the M-MDSC phenotype and generating a TGF-β-mediated feedback loop (36, 37). In monocytes, TLR stimulation has been demonstrated by tumour-derived extracellular vesicles containing factors such as HSP72 that drive M-MDSC generation via MyD88/STAT3 requiring autocrine IL-6 production (38). In addition, WNT5a, S100A8/A9, Il-4 and PGE2 have variously been linked to the acquisition of an MDSC phenotype (39–42). However, maintenance and accumulation of the M-MDSC population appears to be mediated, at least in part, by PD-1 and TNF signalling respectively, whereby PD1-deletion in a murine model appears to prevent M-MDSC accumulation (43) and TNF signalling through TNFR-2 appears to prevent M-MDSC apoptosis in a c-FLIP-dependent manner.
M-MDSCs can further differentiate into tumour-associated macrophages (TAMs) which have an immunosuppressive M2 phenotype (44) that secrete IL-10, express PD-1 and contribute to tumour angiogenesis, and appears to depend on S100A9 expression and signalling acting via C/EBPbeta (39).
E-MDSC have been identified as myeloid cells lacking monocyte or neutrophil markers both in the peripheral blood and the tumour microenvironment. Due to the lack of mature lineage markers, they are thought to be immature and are potentially precursors for the other MDSC subsets. In-vitro e-MDSC show the least capacity to suppress T cell proliferation or IFN-γ release (32). No correlation between e-MDSC frequency and OS has been shown for either a head and neck or ovarian cancer (32, 45). One concern with e-MDSC enumeration is that mature basophils have a similar phenotype but are not immunosuppressive. Using a basophil marker, such as CD123high expression, allows more accurate identification of e-MDSC (10). More accurate identification of e-MDSC may help future studies to explore their functional effects.
The method by which MDSCs trigger immunosuppression depends on the class of cell involved, but common to both PMN-MDSCs and M-MDSCs is the secretion of soluble factors. This effect appears to be the primary method by which PMN-MDSCs mediate immunosuppression and is stronger on a per-cell basis compared to M-MDSCs. Arginase is located in the azurophilic granules of neutrophils and is secreted by both activated neutrophils and MDSCs (46). Arginine is essential for T cell proliferation and function (47). When PMN-MDSCs are cultured with T cells inhibition of arginase restores proliferation, enhances IFN-γ production and improves cytotoxic function (48) (32). Inhibiting the function of iNOS can similarly reverse the effect of PMN-MDSC and M-MDSCs on T cell proliferation (32, 49). ROS is produced by both M-MDSCs and PMN-MDSCs and can inhibit antigen-dependent T cell proliferation (50, 51). ROS is produced under the control of STAT3 and appears to function in the immunological synapse as the expression of the integrin MAC-1 is required for its effect on T cells (50).
M-MDSCs have several additional functional roles. M-MDSCs can induce TRegs through a CD40-CD40L dependent process (52) and trigger TReg and PMN-MDSC recruitment by secreting CCL2, CCL4 and CCL5 (53, 54). Secretion of cytokines including IL-10 and TGF-β promotes tolerance, suppresses cytotoxic function and contributes to further MDSC generation (42, 55). M-MDSCs also express PD-L1 and can adversely affect immune checkpoint therapy (56, 57)
MDSCs may have a physiological role to control inflammation, particularly in the lung and liver. Lung resident neutrophils have a reduced cytokine response to LPS challenge and can protect the lung from harmful inflammation (27). Furthermore, in a model of asthma, PMN-MDSCs can inhibit the function of Th2 cells and suppress inflammation (58) in a PGE2 and COX-1 dependent manner. However, in COVID-19 early expansion of MDSCs is predictive of a poor outcome by preventing viral clearance, yet in late disease, they can be beneficial by reducing secondary inflammation (59). As will be discussed later MDSCs can be beneficial in preventing inflammation in chronic viral hepatitis (60). Although, in the longer term this may be deleterious as it would inhibit viral clearance and promote carcinogenesis. Both examples illustrate the yin and yang of MDSC function depending upon the state of an infection.
MDSCs are generated in response to chronic inflammation, a state which is seen in all virally induced cancers. The action of cytokines, particularly IL-6 and TGF-β, as well as growth factors such as GM-CSF are important in both triggering emergency myelopoiesis and promoting the acquisition of an MDSC phenotype. Factors such as ER stress appear important in PMN-MDSC generation, whereas TLR stimulation and TGF-β are more important for M-MDSCs. MDSCs can also act via autocrine feedback loops to amplify signals leading to their generation and recruitment. MDSCs can then act to suppress T cell function through a variety of interconnected mechanisms. The nature of the underlying immune microenvironment can influence if M-MDSCs or PMN-MDSCs are predominant. Figure 1 links factors produced by oncogenic viruses or infected/transformed cells with the pathways which result in MDSC generation.
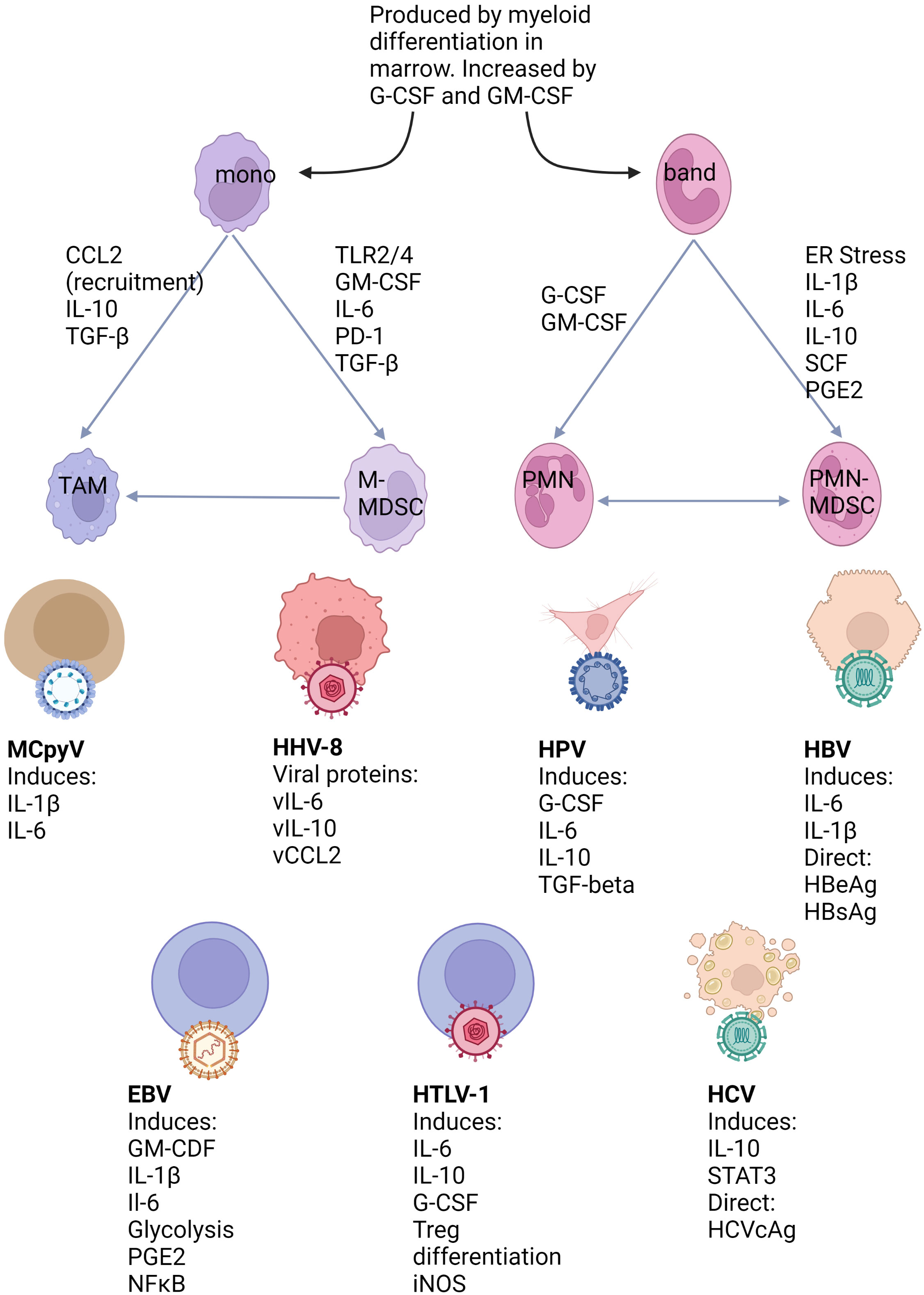
Figure 1 Mechanisms of MDSC generation and their relation to virus associated cancers. Myeloid cells are produced during myelopoiesis in the marrow in both steady state and inflammatory conditions. However, myeloid differentiation can be greatly expanded under growth factor control and lead to the release of immature cells into the circulation. Under conditions favouring MDSC development both bands and mature neutrophils can be induced to this phenotype. M-MDSCs arise from classical monocytes and can also differentiate to TAMs. The human oncogenic viruses illustrated below can either directly or indirectly produce factors known to be linked to MDSC differentiation pathways.
As discussed above identifying MDSCs can be difficult. PMN-MDSCs are short-lived labile cells that cannot be frozen so cannot be studied retrospectively (12). However, progress is being made to establish common flow cytometry protocols (12) and multiplex tissue staining panels (61) to ensure the unambiguous identification of the different MDSC populations, neutrophils and monocytes. This is particularly important when understanding the role of the different MDSC subsets in the context of different cancers. Although MDSCs have not yet been considered in many of the virus-associated cancers, the tumour microenvironment of these cancers actually exhibit the distinct characteristics that are both conducive to the generation of MDSCs and consistent with their presence, and therefore warrant further investigation.
Human T-lymphotropic virus-1 (HTLV-1) is a complex retrovirus which is prevalent worldwide in clusters of high-incidence areas, such as Japan and the Caribbean (62). Initial infection occurs either in infancy, due to spread in breastmilk, or as a sexually transmitted infection. Following a prolonged asymptomatic latency period, HTLV-1 can cause adult T cell leukaemia/lymphoma (ATL), occurring in 2-5% of carriers, as well as inflammatory diseases such as HTLV-1-associated myopathy (HAM).
HTLV-1 transmission is primarily dependent upon direct cell-to-cell contact via a virological synapse rather than infection of target cells with cell free virus. Following infection HTLV-1 integrates into the target cell DNA and is thought to persist by infectious (de novo) spread during early infection and thereafter by mitotic expansion (infected cell proliferation) of cells containing integrated HTLV-1 genome. Over time, the proviral load rises commensurate with the number of HTLV-1 infected clones and the subsequent risk of oncogenic transformation (63). HTLV-1 modifies the infected cell through the action of two genes, TAX and HBZ, that drive proliferation, inhibit apoptosis and promote tissue migration and are both required for oncogenesis (64). TAX is a multifunctional protein capable of driving profound changes to the immunological microenvironment.
ATL is a highly aggressive malignancy associated with profound immunosuppression, largely mediated through the action of TAX. Indeed TAX drives the acquisition of a CD4+CD25+ Treg phenotype of the infected transformed cells (65) and triggers the upregulation of MHC class II allowing infected cells to acquire a tolerogenic antigen-presenting cell function (66). TAX also drives changes to the cytokines secreted by the infected cells, leading to the production of inflammatory cytokines such as TNF-α and IL-6 as well as anti-inflammatory IL-10 through HBZ induction of TIGIT (67, 68). Much of the work looking at the tumour microenvironment has focused on lymphocytes where HTLV-1+ non-tumour cells assist in the growth of tumour cells particularly through IL-10 production (69). Checkpoint blockade, particularly the PD-1 axis leads to disease flair, presumably related to the Treg nature of the tumour (70). Allograft is the only curative treatment, implying that a graft versus tumour effect is important (71). Immunotherapy directly targeting viral antigens is at a much earlier stage, but animal studies show in principle this could be effective (72). Antiviral therapy with zidovudine and interferon is effective in ATL, particularly in non-lymphomatous disease (73) and although the mechanism behind this is not fully understand, it is likely mediated by reducing de novo infection of bystander cells and therefore modulating the TME.
There is very little data on MDSCs in HTLV-1 infection. Neutrophilia is common in ATL, particularly in the leukaemic form (74), and indeed neutrophils appear to show spontaneous activation in HTLV-1 infection with the production of IFN-γ, TNF-α and IL-12 and increased oxidative stress (75–77). Mechanistically, TAX is likely to drive both neutrophilia and neutrophil activation; TAX expressing cells produce G-CSF, leading to neutrophilia and splenomegaly in xenograft models (78) and TAX can also induce the expression of iNOS through an NFκB dependent pathway (79). M2 macrophages in the TME are associated with a worse prognosis in ATL and promote the growth of malignant cells in-vitro (80). However, there is almost no work focusing on MDSC in HTLV-1 infection or attempting to characterise the expanded myeloid compartment. Given the combination of chronic inflammation, production of regulatory cytokines and neutrophil expansion, the presence of MDSCs would certainly be very plausible. How these would function alongside a tumour composed of regulatory T cells is unclear but understanding this may help design more effective immunotherapy.
Merkel cell polyomavirus (MCPyV) is one of 14 human polyomaviruses, but surprisingly is the only one to drive carcinogenesis (81). MCPyV is responsible for up to 80% of cases of Merkel cell carcinoma (MCC), a rare and highly aggressive skin malignancy seen in older age. Initial infection with MCPyV appears to be completely asymptomatic and occurs during childhood, with a seroprevalence of around 70% (82). However, oncogenesis is always associated with viral integration into the host cell DNA, UV exposure and immune senescence. The MCPyV genome is always mutated in MCC resulting in truncation of the Large T (LT) gene that preserves the Retinoblastoma (RB) binding domain but removes its virus replication functions, resulting in a replication-deficient virus. Preservation of the LT RB binding domain and expression of Small T (ST) ensures continual inactivation of RB and degradation of P53 via ubiquitination by MDM2 respectively, leading to uncontrolled cell cycle, accumulation of mutations and development of MCC. Interestingly, both RB and P53 are highly mutated in MCPyV-negative MCC that more closely resemble the UV mutational signature observed in malignant melanoma (81) highlighting how the viral oncogenes mimic the mutational signature in the virus-negative carcinoma counterparts.
Immune control of MCPyV appears to be important in preventing carcinogenesis. Individuals with immunosuppression driven by factors such as HIV, ageing or medication exhibit significant increases in the frequency of MCC (81). However, MCPyV is largely controlled in healthy infected individuals who have readily detectable CD4+ T cells with specificity against LT and ST antigens (83). Conversely, although a clonal CD3+, CD8+ immune infiltrate into the tumour microenvironment is associated with a better prognosis (84), the T cells show signs of exhaustion with the expression of PD-1 on infiltrating lymphocytes and PD-L1 in the tumour microenvironment (85). However, reversing T cell anergy by blocking the PD-1 axis with pembrolizumab has revolutionised the treatment of MCC (86). In MCPyV+ MCC the response rate is around 70%, with 30% achieving CR and responders showing 89.6% survival at 3 years (87). Interestingly, while there was no correlation between CD8+ infiltrate, PD-L1 expression and response to PD-1 blockade a higher neutrophil-to-lymphocyte ratio in the blood was associated with a lower response rate and poorer overall survival (86). Unfortunately, neutrophil phenotyping was not performed in this study to assess if these cells were in fact PMN-MDSCs.
There are no studies, which have studied MCC with histological panels designed to enumerate MDSCs. Similarly in the blood, a high neutrophil to CD8+ ratio in the tumour is a poor prognostic marker (88). PD-L1 is expressed on tumour infiltrating cells expressing CD11c+, CD162+ and CD33+, attributed to DCs and macrophages but some CD33+ cells may be MDSCs (85, 89). PD-L1+CD33 positive cells appear to be enriched in the periphery of MCC tumours and may function to shield the tumour against infiltrating PD-1+ lymphocytes (89). In addition, MCC strongly express CD200R, which is associated with tumour-infiltrating myeloid cells including M2 macrophages and MDSCs (90, 91).
Overall, the understanding of non-lymphoid cells in the microenvironment of MCC is at a limited level, with evidence that myeloid cells in the form of circulating neutrophils and infiltrating PD-L1 cells may inhibit T cell response to tumour antigens. No current biomarkers accurately predict response to checkpoint inhibition therapy (92). Understanding the TME is likely to be of significance as there is currently work to target MCPyV antigens with therapeutic vaccination or adoptive immunotherapy, which has shown some success in small early-phase studies (93) (94).
Human herpesvirus-8, also known as the Kaposi sarcoma associated herpesvirus, is linked to two distinct human malignancies- Kaposi sarcoma (KS) and primary effusion lymphoma (PEL) (95). In endemic areas, HHV-8 infection usually occurs in childhood through infected saliva, but sexual transmission is an important route of spread in other populations. HHV-8 is endemic only in limited areas, particularly sub-Saharan Africa, the Mediterranean and south America (96). HHV-8 primarily infects B cells and epithelial cells before establishing latency in B lymphocytes and monocytes (97). Subsequent oncogenesis appears dependent on viral reactivation.
HHV-8 is a large double-stranded DNA virus that dedicates at least 20% of its genome to modulating the cell cycle and antiviral immunity and encoding numerous viral homologues of human genes. Latent cycle genes such as LANA-1, v-cyclin and v-FLIP as well as lytic cycles genes such as v-IL-6 and v-bcl-2 appear important in carcinogenesis, although the relative dependency on individual genes differs depending on the malignancy (97).
KS was historically a rare tumour but incidence rose rapidly in the 1980s with the emergence of HIV/AIDS, indicating the importance of immune control of the virus in preventing oncogenesis (95). HHV-8 is capable of marked modulation of the host immune system, in part due to the production of at least 10 genes with homology to human genes affecting immune function including IL-6, IL-10, TNF-alpha, CCL1, CCL2 and CCL3 (98, 99). Replication and transcription activator (RTA) protein down-regulates TLR signalling and viral interferon regulatory factors (vIRFs) block interferon signalling, reducing the innate response to the virus. vIL-6 acts via JAK to stimulate STAT3 and triggers a positive feedback loop generating endogenous IL-6 (99). As discussed, IL-6 is associated with MDSC generation and CCL2 with the recruitment of MDSC (100) (101).
There are relatively few studies that have looked at the immune environment in HHV-8-related tumours. In a mouse model vFLIP induced cytokine production leads to the expansion of myeloid cells with a PMN-MDSC phenotype (102). The TME in KS includes M2 macrophages, MDSCs and lymphocytes with high-level PD-L1 expression (103). In nodular KS, like in MCC, CD33+ PD-L1 positive cells have a peritumoral distribution and may function to protect the tumour from T lymphocyte infiltration (103). This study used CD33 as an MDSC marker making further phenotyping of the cells impossible and would not identify PMN-MDSC which do not act through PD-1L. Other groups have identified the PD1+ cells as being CD14+, therefore would this include monocytes and macrophages (104). In addition, HHV-8 can infect monocytes and modulate their function by upregulating PD-1 and increasing the production of a wide range of cytokines including IL-6, GM-CSF, IL-10 and IL-1beta (105). This may provide a mechanism by which HHV-8 infection of myeloid cells could trigger emergency myelopoiesis and MDSC generation.
The benefits of restoring the immune response to HHV-8 are clear; in AIDS-related KS, treating HIV with antiretroviral therapy leads to a response in 50%, although in around 10% there is a flare of disease (106). In those not responding to antiretrovirals, the standard treatment has been chemotherapy. However, there is growing evidence for immune modulation with imid drugs or PD-1 checkpoint inhibition (107). However, while HHV-8 can clearly cause conditions likely to lead to MDSC production, there is insufficient evidence of how this may impact the antiviral response or the effect of checkpoint blockade.
Human papilomaviridae (HPV) are a family of small non-enveloped double-stranded DNA viruses composed of over 450 distinct types that replicate in keratinocytes of squamous epithelia of either the cutaneous or mucosal surfaces. Most HPV infections are asymptomatic or cause self-limiting benign diseases, such as warts. However, a subset of HPV types, termed ‘high risk’ are the causative agents of 1/3 of all virally induced cancers, the most common of which are anogenital cancers and oropharyngeal cancer (108). All HPV types have a similar genome divided into the early open reading frames (ORFs); E1, E2, E4, E5, E6, E7, E8, (E5 and E8 are not present in all HPV types) and the late genes L2 and L2. The core genes, E1 and E2 are required for viral replication and the L1 and L2 are required for capsid formation. The accessory genes (E4-7) facilitate the different stages of the vegetative virus life cycle, mainly by altering the host cell environment to enable viral replication but also perturbing the host anti-viral defense mechanisms (109) (108). In cervical cancer, there is a prolonged progression through increasing grades of intraepithelial neoplasia (CIN) through to invasive cancer (ICC) during which immune control is gradually lost.
The principal mechanism by which HPV evades the immune system is by taking advantage of the complex renewal process in squamous epithelial tissues. Infected basal epithelial cells initially amplify and maintain the viral episomal genome at approximately 50 – 100 copies per cell where viral DNA replication occurs synchronously with the host DNA replication. As infected cells move from the basal lamina, the viral accessory proteins act in concert to delay differentiation by promoting cell cycle re-entry and proliferation. Viral DNA replication is induced by virus-mediated de-regulation of host cell control of the cell cycle. Ultimately, keratinocyte differentiation occurs with the production of the late structural proteins, production of virus particles and the sloughing off of the virus-laden squames (110). Limiting high expression levels of the viral proteins to the uppermost differentiated keratinocytes ensures the least exposure to the host immune pathways, reduces inflammation and delays the generation of an effective immune response, although this mechanism breaks down in CIN and ICC (110). In addition, the HPV proteins exhibit some immunomodulatory functions; E5 prevents effective antigen processing and E7 downregulates class I MHC and STAT1 preventing immune signalling (111). Indeed, the majority of those infected with HPV will clear the virus, which is associated with a CD4 and CD8 response (112). However, the risk of malignancy is increased with underlying immunosuppression (113). Amongst patients with HPV16+ ICC half have no CD4+ response to E2 or E6 with the other half having an impaired response despite having a normal response to other recall antigens (114). In contrast, strong IFN-γ and IL-5 responses are seen in patients who have cleared HPV16 without developing malignancy. Prophylactic HPV vaccination leads to a strong humeral immune response against L1, which is highly effective in preventing infection and can also reduce the relapse rate following surgical removal of CIN, but is ineffective in treating advanced disease (112, 115)
There has been significant research interest in the TME in CIN and ICC, in which multiple regulatory cells including TAMs, Tregs as well as MDSCs may play a deleterious role and help explain why some patients do not mount an immune response to the virus (116). The TME in ICC is enriched with cells expressing TGF-β and IL10 with lower expression of IL2 compared to cervicitis and CIN (117). The ratio of Treg to CD4+ and CD8+ cells is higher in advanced disease with very few regulatory cells in cervicitis (117). Infiltration with CD163+ TAMs is associated with advanced-stage ICC and increases with the progression of CIN (118). Total MDSCs are expanded in the blood of patients with ICC compared to healthy controls and increase with advanced stage (119). MDSCs are also found in tumour tissue in ICC and function ex vivo to inhibit T cell proliferation and cytokine production (119). In a cohort of 22 patients with ICC MDSC numbers in both blood and tissue were associated with poorer progression free survival, although given the association with advanced stage a more sophisticated multivariate analysis with greater numbers would give additional evidence of a causal relationship (119). PMN-MDSCs appear to be the predominant population found in the blood of patients with ICC compared to those with benign cervical lesions and secrete iNOS, IDO and arginase-1 (120). Based on RNA-seq data from tumour tissue in 306 patients there was an association between high MDSC infiltration and overall survival, although again this was not a multivariate analysis (120). A further study found PMN-MDSC numbers increased with disease stage in ICC and where negatively corelated with CD8+ cells in tumour tissues (121). The role of M-MDSCs is less clear, comparing all ICC patients to those with benign lesions appears to show no increase in numbers but when ICC are sorted by disease stage M-MDSC numbers appear to increase in metastatic ICC but not early or locally advanced tumours (57, 120). In CIN there is a strong negative correlation between increased neutrophil and reduced T cell infiltration which is not seen in cervicitis (122). This indicates that the microenvironment is different in CIN compared to the inflamed cervical mucosa.
MDSC production and activation is likely to be driven by cytokine production. ICC cells secrete cytokines associated with myeloid activation and differentiation including G-CSF, IL-6, IL-10 and TGF-beta (122) (122). Plasma G-CSF concentration is associated with increased numbers of MDSCs in the blood in CIN and ICC (122). A large cohort which only looked at leucocytosis, rather than more detailed immunophenotyping, showed this was associated with a worse prognosis and correlated with immunoreactivity for G-CSF in the tumour tissue (123). G-CSF production can be detected in around 85% of tumour biopsies and is associated with a poorer response to both radiotherapy and cisplatin chemotherapy (124, 125). TGF-β signalling is increased in HPV+ tumours compared to HPV- across a range of cancer types and is associated with an inflamed TME (126). IL-10 is upregulated in high-risk cervical lesions, although upregulation in early lesions does not predict which patients progress (127). Reduced CCL2 production in ICC is associated with increased overall survival and a lower infiltrate with immunosuppressive myeloid cells, although this study identified TAMs and did not stain for MDSCs (128). HPV-E6/E7 can induce STAT-3 production in transformed keratinocytes which leads to IL-6 production. This generates a positive feedback loop by inducing STAT3 activation in monocytes which leads to further myeloid accumulation (113).
MDSCs from the blood and tumour tissues of patients with ICC can inhibit T cell cytokine secretion and proliferation (119). When neutrophils are cultured with HPV+ ICC cell lines they become activated and express IL-6, IL-8 and CD62L. When T cells are cultured with SiHa spheroids, a HPV16+ cell line, they proliferate, secrete cytokines and eliminate tumour cells. However, when neutrophils are co-cultured at a 1:1 ratio with T cells they inhibit T cell function (122). This provides strong evidence that neutrophils, activated by tumour tissue to an MDSC phenotype, can inhibit anti-tumour T cell responses. MDSCs can also act via other mechanisms. PGE2 can enhance the stemness of cervical cancer cells (129). MDSCs in ICC also express B cell activating factor and can induce differentiation of IL-10 producing B regulatory cells. These can act then via IL-10 and STAT3 to create a positive feedback loop generating further MDSCs (120)
There is substantial interest in immunotherapy for HPV malignancies including checkpoint blockade and the development of therapeutic vaccines (130). PD-1 blockade shows benefit in treating ICC and HPV positive, as well as negative, head and neck cancers, however, overall response rates are low (131, 132). Response to therapeutic vaccination is less strong in advanced disease, potentially illustrating how the TME can inhibit the immune response (130). In a murine xenograft model, an E7-based DNA vaccine can produce a CD8+ effector memory response, however, this is insufficient to eliminate the tumour. When combined with MDSC reduction using an anti-GR-1 antibody complete responses were seen (133). Combining therapeutic vaccination with all trans-retinoic acid (ATRA) is also successful in a murine model in reducing MDSC number and increasing immunogenicity (134). ATRA alone can decrease MDSC numbers and increase CD8+ T cells in BALB/C mice with U14 cervical tumours and can enhance the efficacy of PD-1 checkpoint blockade (57). In an immunocompetent murine model of cervical cancer, MDSCs accumulate. Again, therapeutic E7 vaccination can induce antigen-specific T cells but does not lead to tumour regression, an effect not enhanced with checkpoint blockade. Ex vivo MDSCs but not Tregs could inhibit T cell and antigen-presenting cell function (135). This provides strong evidence of both the effect of MDSCs in HPV-positive tumours and illustrates the difficulties they can cause with immunotherapy. Immunotherapy to eliminate suppressors may also enhance the effect of standard cancer therapy. In a murine model, using cyclophosphamide to deplete Tregs and an iNOS inhibitor to reduce MDSC function alongside radiotherapy had a beneficial effect. This included a greater response to treatment as well as a significant increase in antigen specific CD8+ cells in the tumour (136).
Overall, the evidence for the role of MDSCs in HPV tumours is strong. In early-stage HPV infection virus can hide from the immune system due to its location combined with suppression of TLR and MHC expression. As CIN develops a cytokine milieu leads to the generation and invasion of regulatory cells including MDSCs. Most patients can clear the virus, presumably because the cytotoxic response can overpower immunosuppressive. However, in those with cancers, the immunosuppressive response predominates. There is still the need however for further data. There is no human data on the role of the MDSCs in predicting response to immunotherapy in HPV+ cancers, unlike what is seen in murine models. There is also insufficient data showing a clear correlation between MDSC expansion and disease progression, rather than an association with advanced disease.
Hepatitis B virus (HBV) is a member of Hepadnaviridae with a partially double-stranded DNA genome which is transmitted by infected bodily fluids and replicates via reverse transcription (137). In low-prevalence countries infection is usually sexually transmitted in adulthood, triggering a strong immune response. This leads to acute hepatitis followed by viral clearance in 99% of patients. In high-prevalence areas infection is perinatal, this leads to a tolerogenic immune response with viral persistence in 90% (138). Immune response to HBV in chronic infection is complex (139). Initially, the immune response is tolerogenic (IT) with high viral loads and low levels of CD8 T cell activation. Over decades the CD8 response becomes stronger leading to chronic hepatitis (CHB) with intermittent flairs of acute hepatitis (ACLF) and liver damage. Some patients clear most infected hepatocytes and seroconvert to produce anti-HBV E antigen (HBeAg) with low levels of HBV DNA and slowing of the progression of liver injury. In around 1% of anti-HBeAg positive patients per year, the viral DNA becomes undetectable, indicating a functional cure. The rest develop cirrhosis or hepatocellular carcinoma (HCC) many years after the initial infection (140).
HBV can directly lead to oncogenesis through random integrations into the human genome after reverse transcription, this increases genetic instability and prolongs viral gene expression (140). The HBV X antigen (HBx) can interact with transcription factors such as CREB promoting transformation (141, 142) and epigenetically silence tumour suppressors such as RUNX3, p16 and p21 through promoter hypermethylation (143, 144). Indirectly, HBV-associated chronic inflammation leads to oxidative stress, ROS production and altered cellular metabolism promoting DNA damage and leading to oncogenesis (145, 146).
The immune response to HBV includes innate immune IFN production, NK and T cells acting in both cytolytic and non-cytolytic manners against infected hepatocytes as well as the production of neutralizing antibodies by plasma cells (147). The tolerogenic state seen in chronic infection is characterized by regulatory and exhausted cells being enriched in the liver microenvironment including FOXP3+ T cells, PD-1/PD-L1 axis activation and the production of IL-10 and TGF-β with a weak CTL response to HBV antigens (148). However, the mechanisms behind this dysfunctional response are not fully understood. MDSCs are significantly expanded in CHB patients (60, 149–154), indicating a potential role in preventing viral clearance.
The relative importance of PMN- and M-MDSCs in CHB is debated, with divergent findings in different studies. Both the analytical strategies and dynamics during the progression of CHB might contribute to these discrepancies. M-MDSCs have been reported as being expanded in Indian and Chinese cohorts (155) (154, 156), whereas in a European cohort PMN-MDSCs were expanded more significantly (60). In paediatric, but not adult, CHB patients M-MDSC numbers correlated inversely with CD8 responses to HBsAg (157). These cells could home to the thymus and when transferred to mice could selectively inhibit HBsAg targeting CTLs. In ACLF M-MDSC expansion appears to correlate with markers of liver damage but not viral replication (153). Whereas in chronic infection DNA load and HBeAg positivity appears to be more significant (151, 154, 156). The relationship between MDSCs and liver damage is not clear with some groups showing a positive correlation with increasing hepatitis and others a negative implying a protective role (151, 154). MDSC phenotypes and function appear to dynamically change over time in different stages of HBV infection (60). Profound suppressive activities, including expression of Arginase, iNOS and PD-L1 are seen in active CHB, but much weaker suppressive functions, expressing only arginase, in the IT state (152). PMN-MDSCs expand transiently in acute HBV and are increased most in disease states with high viral replication but low levels of immune-mediated inflammation (60). This emphasizes that PMN-MDSCs may prevent immunopathology in CHB, but at the expense of poor viral control.
There is much less data on the role of MDSCs following the development of HCC. Elevated LOX-1+CD15+ PMN-MDSCs in both circulation and tumour tissues were correlated with poor clinical outcomes in HCC, but not with HBsAg expression (158). Expansion of M-MDSCs in HCC has also been associated with a poor prognosis after surgical or chemotherapeutic treatments (159–161). Only a single study has described increasing numbers of both PMN-MDSCs and M-MDSC on the progression of CHB to HCC with a positive correlation with increased liver damage (149). Interestingly, this study did not find any association of MDSC numbers with other clinical features or OS, which may be a function of relatively low patient numbers. More data are needed, with prospective follow-up, to examine if MDSC numbers in CHB are a risk factor for future HCC development.
MDSCs have been reported to act via several mechanisms in CHB infection. M-MDSCs can act via PD-1/PD-L1 and IL-10 to suppress HBsAg-specific cytotoxic function and proliferation in CD8+ T cells (154). PMN-MDSC from CHB patients inhibits T cell function in an arginase-dependent manner in-vitro. Arginase expressing MDSC can be identified in the blood and liver of CHB patients (60). In-vitro treatment of PBMC with HBeAg induces IDO expressing M-MDSCs which inhibit CD4+ and CD8+ T cell proliferation and IFN-γ production (156) Tregs can be induced by MDSCs in HBV infection in an IL-10 and TGF-β dependent process (155). In a mouse model, chemokine receptor CCR9 in M-MDSCs is induced by HBsAg via ERK/IL-6 pathway, and CCR9-CCL25 interaction mediates Gr1+Ly6ChighLy6G- M-MDSCs homing to the thymus, where HBsAg-specific CD8+CD4- T cells are killed by NOX1-expressing M-MDSCs (162). Another HBV-specific mechanism involves impaired CCR5-CCL5 interaction by TGF-β released from MDSCs, which affects the migration of HBV-specific T cells to the liver (152, 163, 164). This effect is seen during active hepatitis, but not during IT or after viral clearance (152).
Hepatitis antigens appear to be capable of directly inducing MDSC production. When PBMC are treated with HBeAg in-vitro there is induction of IL-1β, IL6 and IL-10 along with the accumulation of IDO producing MDSCs. Blocking IL-6 or IL-1β with neutralizing antibodies prevents MDSC generation (156). HBsAg can induce arginase and NOX-1 producing MDSC differentiation from mature monocytes via an ERK/IL-6/STAT-3 dependent process (165). It is interesting that different viral proteins can produce MDSC with altered function and illustrates the potential complexity of the immune microenvironment shaped by HBV infection. HBx and HBcAG can induce hepatocytes to secrete IL-6 which is likely to be capable of inducing MDSCs in a paracrine manner (166, 167). In a murine model of CHB, γδT cells can induce CD11b+Gr1+ M-MDSCs in mice through an IL-17-dependent manner, which then inhibit IFN-γ production by hepatic CD8+ T cells via ARG-I expression (168).
Therapy for HBV consists of either nucleoside analogue antiviral drugs such as entecavir, achieving viral clearance in 67% (169), or peg-IFN-α-a2 with a clearance rate of 25% (170). Even after viral clearance, Tenofovir does not normalize MDSC or Tregs numbers and HBV-specific CTL responses remain impaired (152, 155). This indicates that viral clearance is not necessarily correlated with recovered immune responses and may allow viral rebound at the end of treatment. This creates a rationale for combing immunotherapy with direct antivirals (171). Knocking down HBx expression with a siRNA can reduce MDSC frequency, IL-10 and ROS level in HBV-infected mice (172). Activating TLR-8 with GS-9688, a specific agonist decreases MDSC and Treg numbers and promotes a cellular antiviral response using ex vivo cells from CHB patients. This can reduce viral loads in a woodchuck HBV model (173). However, the residual MDSCs released more immunosuppressive gelactin-9 and PD-L1 after treatment (173). While IFN-α has been widely used in CHB treatment, its immunological effects are complex and may promote MDSC production (174). IFN-α can increase Tregs and MDSCs numbers with increased IL-10 and PD-1L expression, while the roles of Tregs and MDSCs promote tolerance in acute hepatitis B in a murine model (175). ATRA can reduce HBsAg-induced M-MDSC function and restore T cell function in a murine CHB model (165). Icaritin, a small molecule derived from horny goat weed, can prohibit the generation of MDSCs by disrupting STAT3 and AKT signalling. This can then modulate cytokine secretion in HCC patients (176). Again, in a murine model, depleting MDSCs with an anti-GR1 antibody or inhibiting their function with an arginase inhibitor can enhance CTL function and promote viral clearance (168).
Overall, there is convincing evidence for a functional role of MDSCs in HBV infection but the significance of this cell population in preventing viral clearance or allowing the development of HCC is unclear. Investigating MDSCs in HBV is difficult given the different stages of infection, where these cells may have different functional roles. Prospective trials where the same patients have measurements of MDSC phenotype and number in different phases of their disease using a standardized methodology may help answer these questions. This may also help identify if targeting MDSCs would be an effective method to help with viral clearance and prevent the development of HCC.
The Hepatitis C virus (HCV) belongs to Flaviviridae and has a single-stranded RNA genome (177). HCV is transmitted by infected blood exposure, which can be through transfusion, IV drug use or vertical transmission, and leads to chronic hepatitis (CHC) in 70-80% of those infected (178). 10-20% of those with CHC develop cirrhosis over 20-30 years and HCV is the second most common cause of HCC after HBV (179). There is no effective vaccine to prevent HCV infection, but treatment has been revolutionised over the last 10 years with direct-acting antivirals (DAA), which can cure 90% of patients (180, 181). Unlike HBV, HCV-associated HCC always develops occurs in the context of cirrhosis and HCV does not appear capable of direct transformation of hepatocytes (140). Instead, cytotoxicity mediated by type I IFN, NK cells and CD8+ effector T cells generates an inflammation-necrosis-regeneration cycle with ROS production, triggering lipid peroxidation, DNA damage and mitochondrial dysfunction (182, 183). Additionally, the HCV core antigen (HCVcAg) activates the Ras/MAPK and PI3K/Akt pathways which can promote transformation (184, 185). HCVcAg also plays a role in metabolic reprogramming with enhanced lipogenesis and impaired lipid degradation, this promotes steatosis and the development of cirrhosis (186).
During acute HCV infection, there is a robust IFN response triggered by HCV RNA and antigens through RIG-I and TLRs (187, 188). Viral clearance depends on a strong cellular and humoral adaptive immune response followed by the development of memory (189). However, in most patients a combination of immune exhaustion, regulatory cell expansion and viral escape mutations causes this process to fail (189).
MDSC may act as part of the expanded regulatory population in CHC (190–201). M-MDSC are the population most frequently reported as being elevated (190–195), with a few groups also observing PMN-MDSC expansion (196–200). Most studies have not attempted to study PMN-MDSCs, and others have carried out flow cytometry on frozen cells, which adversely affects PMN-MDSC enumeration as discussed earlier (190). It is unclear how much this variation is explained by biological variation in heterogenous patient populations or is a result of methodological differences
The clinical significance of MDSCs in CHC has also been explored. Many research groups identified positive correlations between MDSC frequencies and HCV genotypes (196), HCV RNA load (191, 192, 195, 196, 200, 201) or liver damage parameters including ALT and AST level (191, 195, 201), and negative correlation with CD4+ or CD8+ T cell counts and functions (191, 201). Although not all studies have found these associations (194, 197). It may be that MDSC populations change dynamically in response to viral factors, inflammation, development of cirrhosis and treatment as described in HBV (152). Longitudinal analysis of MDSC populations in different phases of disease and treatment may help better describe the significance of this population The role of HCV in producing MDSCs in HCC is another key question. One study compared PMN-MDSC numbers in HCC patients with and without HCV infection in an American patient cohort (202). MDSCs and Tregs were increased in HCC patients compared to healthy volunteers but there was no difference between virus positive and negative cases. This may relate to the presence of multiple factors capable of generating MDSCs being present in established HCC. It would be interesting to see similar work in other populations as well as to assess if this is also true for M-MDSCs.
PMBCs which have been exposed to HCV can develop an M-MDSC phenotype and reduce NK IFN production through arginase depletion which then suppresses mTOR signalling (203). Exposing CD33+ cells to HCVcAg or infected hepatocytes generates M-MDSCs, which upregulate ROS but not ARG-1 or iNOS through a p47-dependent process and then inhibit CD4 and CD8 T cells (204). HCV-induced M-MDSCs express high levels of pSTAT3 and Il-10 and can induce Treg function (194) MDSCs isolated from CHC patients can increase the ratio of follicular regulatory to follicular helper T cells in a cell contact-dependent process (205). This may then inhibit T cell-dependent B cell function.
PMN-MDSCs can be generated from PBMCs by culturing with HCVcAg in a process reliant on STAT3 which is activated by ERK 1/2 (201), However, M-MDSC generation is more reliant on the TLR2/PI3K/AKT/STAT3 axis, resulting in upregulated IDO, PD-L1 and IL-10 (206, 207). This suggests that different mechanisms are involved in M-MDSCs and PMN-MDSCs generation, although both are induced by HCVcAg. Interestingly, exosomes containing viral RNA generated by HCV-infected hepatocytes can also induce the generation of M-MDSCs (198, 199, 205). This process relies on modulating HOTAIRM1, HOXA and miR124 and leads to an M-MDSC phenotype with STAT3 activation and production of iNOS and ARG-1. An attempt to develop an HCV vaccine using mice mesenchymal stem cells transfected to express non-structural HCV proteins (NS3~NS5B) triggers strong anti-HCV immunity without expanding MDSC (208). Suggesting that this may be a successful approach for vaccine development.
During treatment with IFN and ribavirin, MDSC numbers decrease in proportion to HCV RNA load. This occurs as early as 4 weeks into therapy and is accompanied by increases in T cell function (191, 196, 197). After IFN therapy M-MDSC numbers rise with higher levels seen in non-responders versus responders (209). Although, the significance of this is less relevant as this therapy has been superseded by direct activating antivirals. Results with DAA have been mixed. A study looking at a European population did not detect significantly reduced M-MDSCs until 48 weeks post-therapy, along with scant recovery of T cell functions (193). However, in a Chinese cohort, there was a rapid reduction in M-MDSCs along with restoration of CD8+ T cells and NK cells with DAA therapy at as early as 12 weeks. The authors proposed the recovery of immune cells was associated with efficient viral clearance achieved by DAA therapy (195). In another Chinese cohort, PMN MDSCs but not M-MDSCs were elevated after DAA with levels higher in those not achieving a sustained antiviral response (201). These studies used different flow strategies, were carried out in different populations, may have involved infections with different strains of HCV and there was no detailed discussion in either paper of the degree of underlying liver damage in their respective cohorts. It may be that immune recovery in HCV requires both viral clearance and resolution of inflammation.
Overall, our current understanding of MDSCs in HCV infection, including their presence, clinical significance, immunosuppressive properties and generation are limited. Although there is data demonstrating the association between MDSC expansion and disease parameters, some results are conflicting. To build on this it will be necessary to use standardized MDSC detection techniques and correlate with clinical and virological features. There is also a need for further data on the link between MDSCs and HCC in CHC. This may better help understand the propensity of HCV to cause chronic infection. Furthermore, understanding the ability of HCV to cause sustained immune changes may help guide how antiviral and immunotherapy can be combined.
EBV is a γ-1 herpesvirus which infects 90-95% of individuals worldwide, establishing a lifelong latent infection. Primary infection is classically responsible for infectious mononucleosis, although infection in childhood is usually asymptomatic. EBV has a natural tropism for B cells but can also infect T cells and epithelial cells and is causally linked to a range of cancers including lymphoma, nasopharyngeal cancer (NPC) and gastric cancer (210).
After primary B cell infection, EBV initiates the expression of a unique set of growth transforming genes including the virus-encoded nuclear antigens EBNA 1, 2, 3A, 3B, 3C, and –LP, the latent membrane proteins LMPs 1 and 2A/2B, the non-coding EBER RNAs and two blocks of microRNAs (BHRF1- and BART-miRs). This growth transforming Latency III is highly immunogenic and vulnerable to immunosurveillance, therefore to establish lifelong persistence, the virus adopts a restricted Latency 0 expressing only the EBERs and BART-miRs (210). However, this reflects only two of several alternative forms of latency with increasingly restricted transcriptional programmes compared to Latency III. These alternate forms of latency, termed Latency II and I are observed during the establishment of Latency 0, however all forms of latency have been observed in the EBV-associated malignancies and are dependent upon the cell type infected and the class of tumour. Most EBV-related Hodgkin (HL) and non-Hodgkin lymphomas (NHL) as well as epithelial malignancies are latency II, (EBNA-1, LMP-1-2, EBER, BART-miRs), which rely on both EBV genes and somatic mutations to drive tumour growth (211). Importantly latency III tumours generally only occur in immunosuppressed states such as post-transplant lymphoproliferative disease (PTLD).
In healthy carriers, CD8+ responses to EBNA-3s are the dominant, with CD8+ responses to EBNA-1, EBNA-2 and LMP-2 subdominant and rare responses to LMP-1 (212). While latency I and II tumours are more common in immunosuppression, they also occur in those with intact immune systems where they virus employs a plethora of immune modulatory mechanisms. Such mechanisms include EBER-mediated induction of IL-10 in B cells (213); exosomes produced by NPC inducing Treg migration to the TME via CCL20 (214); inhibition of interferon signalling and reduction of class II MHC expression by BZLF1 and BRLF1 (215, 216), amongst many others. In addition, there is increasing evidence of a role for MDSC in driving immune evasion.
Both PMN-MDSCs and M-MDSCs have been identified in patients with EBV-related tumours. MDSCs, predominantly of a monocytic phenotype, are expanded in the blood of ENKTL patients (213). Ex vivo these cells can suppress T cell proliferation and IFN-γ production. In a multivariate analysis, high levels of MDSCs are associated with a worse prognosis in ENKTL (213). In chronic active EBV (CAEBV) PMN-MDSCs are expanded and again functionally inhibit T cell responses (217). Interestingly, while LMP-1-specific CTLs were isolated from these patients, no anti-tumour immune response was seen. MDSCs are expanded in NPC cancer patients and correlate with disease burden. In one study enrolling 49 patients’ total blood and tumour MDSC levels were associated with a worse prognosis (218). Unfortunately, this study did not use standardised flow to identify MDSC subtypes and did not carry out a multivariate analysis
There are multiple potential drivers of MDSC production and differentiation. High levels of the MDSC-associated cytokines GM-CSF, IL-1beta and IL-6 are seen in CAEBV patients with the same cytokines produced by CAEBV cell lines (217). In NPC, MDSC levels correlated with COX-2 production by the tumour and knocking down COX-2 could inhibit MDSC production in-vitro (218). High levels of the MDSC-associated cytokines GM-CSF, IL1β and IL-6 are seen in CAEBV patients with the same cytokines produced by CAEBV cell lines (217). In NPC, MDSC levels correlated with COX-2 production by the tumour and knocking down COX-2 could inhibit MDSC production in-vitro (218). In a murine model, MDSCs could stimulate further COX-2 expression by the NPC cells, an effect which could be blocked by inhibiting TGF-β or iNOS and enhanced through arginine supplementation. LMP-1 appears capable of inducing extra-mitochondrial glycolysis through a GLUT1-dependent process. In NPC tissues there is a strong correlation between LMP1 and GLUT1 with numbers of CD33+ MDSC. GLUT1 glycolysis activates the phosphorylation of NF-kB, induces COX-2 signalling pathways and activates the NLRp3 inflammasome leading to the production of IL-1β, IL-6 and GM-CSF (219). EBV may be able to directly modulate myeloid cells. EBV can infect monocyte-derived macrophages in-vitro and increases the production of IL-6 and TNF-α which then induces IDO (220). However, it should be noted that PMN-MDSCs and M-MDSCs are expanded in HL and NHL regardless of aetiology and are associated with a worse prognosis (221, 222). Therefore, the mechanism for MDSC generation cannot purely be a direct effect of the virus. Unfortunately, there have been no comparisons between EBV-positive and negative lymphoma or NPC which have looked at differences in MDSC populations or drivers for their differentiation.
There is the potential for crosstalk between cancer-causing viral infections which can drive MDSC generation. More NPC patients with CHB have EBV-positive tumours than those without this condition. CHB is associated with increased LOX1+ MDSCs and higher levels of EBV DNA (219). This implies that hepatitis B-induced MDSCs may inhibit the immune response to EBV, although further evidence would be needed to demonstrate this conclusively.
There is substantial interest in using EBV-targeted CTLs to treat EBV-positive tumours (223). However, response rates in advanced tumours other than PTLD may be limited (224). A clinical trial in NPC gave chemotherapy followed by multiple infusions of CTLs. EBV viral load and a high monocyte-to-lymphocyte ratio were associated with poor survival (225). The patients with the best responses showed a CD8+ cytotoxic response which was not seen in poor responders who had an expansion of M-MDSCs. Unfortunately, flow cytometry was performed retrospectively on frozen samples precluding analysis of PMN-MDSCs. Unfortunately, no other studies have examined MDSCs in the context of CTL therapy so the evidence for this is limited.
Overall, there is good evidence that MDSCs play a role in inhibiting the immune response to EBV in NPC and EBV-related T-cell lymphomas. The data for the role of EBV in modulating MDSCs in the TME of other lymphoma subtypes is much more limited. Answering these questions will require comparisons between EBV-positive and negative tumours. This may help guide the use of EBV-targeted immunotherapies.
Given their association with poor prognosis in multiple cancer types, there have been efforts to target MDSCs to improve the outcomes of cancer therapy. Potential strategies include depletion of MDSC populations, inhibiting recruitment to the TME, blocking suppressive functions, and differentiating away from a suppressive phenotype (226). The potential approaches, along with the evidence for them, are summarised in Table 2. However, very little of this research has been focused in the context of virally induced cancers or how therapy may impact the transformation of premalignant lesions.
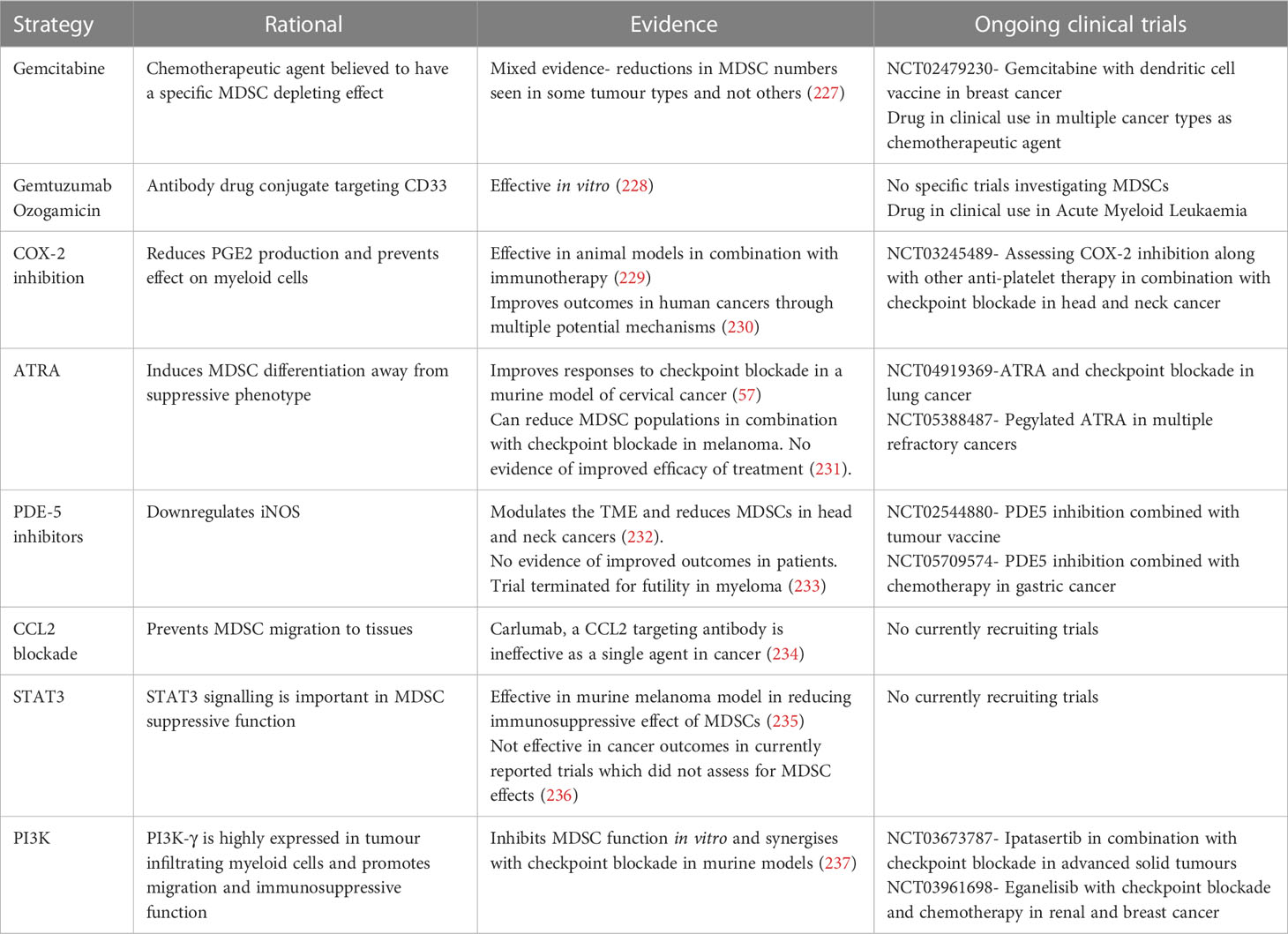
Table 2 Approaches to modulate MDSC populations in cancer.
Some chemotherapeutic agents may be able to reduce MDSC populations. Gemcitabine (GEM) appears capable of reducing PMN-MDSCs in patients with pancreatic cancer, although the effect is transient and M-MDSCs are not affected (238). Although, the evidence is mixed as GEM appears capable of enhancing the function of M-MDSCs in a mouse breast cancer model (239). The CD33 targeting antibody-drug conjugate gemtuzumab ozogamicin can efficiently eliminate MDSCs, however, it may be too toxic for easy combination with other treatments (228, 240)
In multiple cancer models including EBV-related NPC, COX appears to be a marker for immune evasion partially through PGE2-driven effects on myeloid cells (218, 241). COX-2 inhibition can decrease MDSC numbers and increase the efficacy of dendritic cell-based immunotherapy in the context of a murine model of mesothelioma (229). PGE2 induces arginase expression in a mouse model of lung cancer and inhibiting COX-2 can lead to a lymphocyte-mediated anti-tumour response (242). A large meta-analysis, across multiple tumour types, showed modest benefits (238) in adding the selective COX-2 inhibitor celecoxib to standard anticancer therapy with no concerning toxicity (230). Celecoxib has been used in a phase I trial combined with radiotherapy in NPC with encouraging outcomes and no clear toxicity, however, this trial did not report on the effect of EBV positive versus negative tumours or look at MDSC numbers or function (243). Overall, COX-2 inhibition is an interesting strategy given its low toxicity and data on combination with immunotherapy in virally induced cancers would be interesting.
As discussed earlier, ATRA can decrease the frequency of MDSCs, improving response to checkpoint blockade or therapeutic vaccination in murine models of cervical cancer (57, 134). One small clinical trial showed that ATRA can decrease the frequency of MDSCs and appears to increase T cell responses to checkpoint inhibition, although this was in the context of melanoma. Unfortunately, this study was too small to see if this led to improved tumour responses (231). ATRA acts by inducing MDSC differentiation away from a suppressive phenotype through the upregulation of glutathione synthesis (244). Other potential therapeutic approaches include phosphodiesterase-5 (PDE-5) inhibitors, which can reduce MDSC numbers in head and neck cancers through the downregulation of iNOS (232). Blocking chemokine signalling has the potential to prevent MDSC accumulation in the tissues. However, the CCL2 targeting MAB carlumab was unable to affect the CCL2/CCR2 axis sufficiently or have a single agent anti-tumour effect (234). Inhibiting STAT3 is another potential method to reduce the effect of MDSCs. The STAT3 inhibitor Napabucasin could eliminate the immunosuppressive effects of MDSCs in a murine model and in-vitro with human M-MDSCs (235). Although, this will require the use of highly selective inhibitors to avoid inhibiting STAT-dependent T cell function (245). PI3K, especially the PI3K-γ isoform is highly expressed on tumour infiltrating myeloid cells. Pharmacological inhibition of PI3K-γ can restore sensitivity to checkpoint blockade in murine cancer models (237). Although when targeting PI3K it is important to use specific inhibitors at appropriate doses to prevent off target inhibition of T cell function (246).
Targeting MDSCs appears to be an attractive therapeutic strategy. However, while there are options available, all are at a relatively early stage in development. The true potential in targeting MDSCs may come from a combination with other immunotherapy options.
There is growing evidence that MDSC expansion could help to explain how cancer-causing viruses can evade immune detection, trigger oncogenesis and prevent immune recognition of the resultant tumour, although the strength of evidence for this is highly dependent on the virus type. As illustrated in Figure 1, many factors released from virally transformed or infected cells along with substances directly produced by cancer-causing viruses are linked to the pathways known to generate MDSCs. There are, however, several unanswered questions. How much of the expansion in MDSCs is a direct result of the viral infection, which may occur at an early stage in infection, or a result of cellular transformation? For this question, comparisons of the TME between viral and non-viral mediated types of the same tumour may be beneficial. Is MDSC expansion a marker of advanced malignancy or a true independent marker of poor prognosis? How significant is the impact of MDSCs on targeted immunotherapy? This last question is becoming more significant given the development of therapeutic vaccines or cellular immunotherapies.
MDSCs remain challenging to study. In the blood human PMN-MDSCs can only be reliably identified with density gradient centrifugation followed by multicolour flow which has to be performed on fresh specimens. In tissues, MDSC identification requires the use of multiple different markers. There is also a need to correlate MDSC numbers and function with responses to treatments, particularly with novel immunotherapies. This will allow the potential benefits of MDSC manipulating treatments to be ascertained. However, doing this effectively will require the examination of larger cohorts of patients alongside other clinical data. However, there is scope for progress as newer highly multiplexed techniques may make the identification of MDSCs alongside the study of the tumour and other cells in the TME easier. Hopefully, this approach will help conclusively determine the role of MDSCs in virally induced cancers.
CSL and AG conceived the paper. All authors listed made a substantial, direct and intellectual contribution to the work and approved it for publication.
This work was funded by Cancer Research UK 1379789.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Scotto J, Bailar JC 3rd. Rigoni-stern and medical statistics. A nineteenth-century approach to cancer research. J Hist Med Allied Sci (1969) 24(1):65–75. doi: 10.1093/jhmas/xxiv.1.65
2. Weiss RA, Vogt PK. 100 years of rous sarcoma virus. J Exp Med (2011) 208(12):2351–5. doi: 10.1084/jem.20112160
3. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from burkitt's lymphoma. Lancet (1964) 1(7335):702–3. doi: 10.1016/s0140-6736(64)91524-7
4. Young LS, Yap LF, Murray PG. Epstein-barr virus: more than 50 years old and still providing surprises. Nat Rev Cancer (2016) 16(12):789–802. doi: 10.1038/nrc.2016.92
5. Chang Y, Moore PS, Weiss RA. Human oncogenic viruses: nature and discovery. Philos Trans R Soc Lond B Biol Sci (2017) 372(1732):20160264. doi: 10.1098/rstb.2016.0264
6. NTP (National Toxicology Program). Report on carcinogens, fourteenth edition. U.S: Department of Health and Human Services: Research Triangle Park (2016).
7. Grulich AE, Vajdic CM. The epidemiology of cancers in human immunodeficiency virus infection and after organ transplantation. Semin Oncol (2015) 42(2):247–57. doi: 10.1053/j.seminoncol.2014.12.029
8. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol (2018) 19(2):108–19. doi: 10.1038/s41590-017-0022-x
9. Quail DF, Amulic B, Aziz M, Barnes BJ, Eruslanov E, Fridlender ZG, et al. Neutrophil phenotypes and functions in cancer: A consensus statement. J Exp Med (2022) 219(6):e20220011. doi: 10.1084/jem.20220011
10. Khan ANH, Emmons TR, Wong JT, Alqassim E, Singel KL, Mark J, et al. Quantification of early-stage myeloid-derived suppressor cells in cancer requires excluding basophils. Cancer Immunol Res (2020) 8(6):819–28. doi: 10.1158/2326-6066.CIR-19-0556
11. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:12150. doi: 10.1038/ncomms12150
12. Cassetta L, Bruderek K, Skrzeczynska-Moncznik J, Osiecka O, Hu X, Rundgren IM, et al. Differential expansion of circulating human MDSC subsets in patients with cancer, infection and inflammation. J Immunother Cancer (2020) 8(2):e001223. doi: 10.1136/jitc-2020-001223
13. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res (2007) 67(1):425. doi: 10.1158/0008-5472.CAN-06-3037
14. Zhang S, Ma X, Zhu C, Liu L, Wang G, Yuan X. The role of myeloid-derived suppressor cells in patients with solid tumors: A meta-analysis. PloS One (2016) 11(10):e0164514. doi: 10.1371/journal.pone.0164514
15. Dorhoi A, Glaria E, Garcia-Tellez T, Nieuwenhuizen NE, Zelinskyy G, Favier B, et al. MDSCs in infectious diseases: regulation, roles, and readjustment. Cancer Immunol Immunother (2019) 68(4):673–85. doi: 10.1007/s00262-018-2277-y
16. Kostlin-Gille N, Gille C. Myeloid-derived suppressor cells in pregnancy and the neonatal period. Front Immunol (2020) 11:584712. doi: 10.3389/fimmu.2020.584712
17. Sanchez-Pino MD, Gilmore LA, Ochoa AC, Brown JC, Obesity-associated myeloid immunosuppressive cells. Key players in cancer risk and response to immunotherapy. Obes (Silver Spring) (2021) 29(6):944–53. doi: 10.1002/oby.23108
18. Schrijver IT, Theroude C, Roger T. Myeloid-derived suppressor cells in sepsis. Front Immunol (2019) 10:327. doi: 10.3389/fimmu.2019.00327
19. Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, et al. Neutrophils: New insights and open questions. Sci Immunol (2018) 3(30):eaat4579. doi: 10.1126/sciimmunol.aat4579
20. Prince LR, Prosseda SD, Higgins K, Carlring J, Prestwich EC, Ogryzko NV, et al. NR4A orphan nuclear receptor family members, NR4A2 and NR4A3, regulate neutrophil number and survival. Blood (2017) 130(8):1014–25. doi: 10.1182/blood-2017-03-770164
21. Futosi K, Fodor S, Mocsai A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol (2013) 17(3):638–50. doi: 10.1016/j.intimp.2013.06.034
22. Bergenfelz C, Leandersson K. The generation and identity of human myeloid-derived suppressor cells. Front Oncol (2020) 10:109. doi: 10.3389/fonc.2020.00109
23. Trikha P, Carson WE 3rd. Signaling pathways involved in MDSC regulation. Biochim Biophys Acta (2014) 1846(1):55–65. doi: 10.1016/j.bbcan.2014.04.003
24. Lee WC, Hsu PY, Hsu HY. Stem cell factor produced by tumor cells expands myeloid-derived suppressor cells in mice. Sci Rep (2020) 10(1):11257. doi: 10.1038/s41598-020-68061-8
25. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity (2010) 32(6):790–802. doi: 10.1016/j.immuni.2010.05.010
26. Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol (2008) 181(7):4666–75. doi: 10.4049/jimmunol.181.7.4666
27. Bae GH, Kim YS, Park JY, Lee M, Lee SK, Kim JC, et al. Unique characteristics of lung-resident neutrophils are maintained by PGE2/PKA/Tgm2-mediated signaling. Blood (2022) 140(8):889–99. doi: 10.1182/blood.2021014283
28. Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol (2016) 1(2):aaf8943. doi: 10.1126/sciimmunol.aaf8943
29. Kelly-Scumpia KM, Choi A, Shirazi R, Bersabe H, Park E, Scumpia PO, et al. ER stress regulates immunosuppressive function of myeloid derived suppressor cells in leprosy that can be overcome in the presence of IFN-gamma. iScience (2020) 23(5):101050. doi: 10.1016/j.isci.2020.101050
30. Mohamed E, Cao Y, Rodriguez PC. Endoplasmic reticulum stress regulates tumor growth and anti-tumor immunity: A promising opportunity for cancer immunotherapy. Cancer Immunol Immunother (2017) 66(8):1069–78. doi: 10.1007/s00262-017-2019-6
31. Marini O, Costa S, Bevilacqua D, Calzetti F, Tamassia N, Spina C, et al. Mature CD10(+) and immature CD10(-) neutrophils present in g-CSF-treated donors display opposite effects on T cells. Blood (2017) 129(10):1343–56. doi: 10.1182/blood-2016-04-713206
32. Lang S, Bruderek K, Kaspar C, Hoing B, Kanaan O, Dominas N, et al. Clinical relevance and suppressive capacity of human myeloid-derived suppressor cell subsets. Clin Cancer Res (2018) 24(19):4834–44. doi: 10.1158/1078-0432.CCR-17-3726
33. Kapellos TS, Bonaguro L, Gemund I, Reusch N, Saglam A, Hinkley ER, et al. Human monocyte subsets and phenotypes in major chronic inflammatory diseases. Front Immunol (2019) 10:2035. doi: 10.3389/fimmu.2019.02035
34. Boyette LB, Macedo C, Hadi K, Elinoff BD, Walters JT, Ramaswami B, et al. Phenotype, function, and differentiation potential of human monocyte subsets. PloS One (2017) 12(4):e0176460. doi: 10.1371/journal.pone.0176460
35. Bergenfelz C, Larsson AM, von Stedingk K, Gruvberger-Saal S, Aaltonen K, Jansson S, et al. Systemic monocytic-MDSCs are generated from monocytes and correlate with disease progression in breast cancer patients. PloS One (2015) 10(5):e0127028. doi: 10.1371/journal.pone.0127028
36. Gonzalez-Junca A, Driscoll KE, Pellicciotta I, Du S, Lo CH, Roy R, et al. Autocrine TGFbeta is a survival factor for monocytes and drives immunosuppressive lineage commitment. Cancer Immunol Res (2019) 7(2):306–20. doi: 10.1158/2326-6066.CIR-18-0310
37. Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol (2009) 30(10):475–87. doi: 10.1016/j.it.2009.07.009
38. Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest (2010) 120(2):457–71. doi: 10.1172/JCI40483
39. Kwak T, Wang F, Deng H, Condamine T, Kumar V, Perego M, et al. Distinct populations of immune-suppressive macrophages differentiate from monocytic myeloid-derived suppressor cells in cancer. Cell Rep (2020) 33(13):108571. doi: 10.1016/j.celrep.2020.108571
40. Mehmeti M, Bergenfelz C, Kallberg E, Millrud CR, Bjork P, Ivars F, et al. Wnt5a is a TLR2/4-ligand that induces tolerance in human myeloid cells. Commun Biol (2019) 2:176. doi: 10.1038/s42003-019-0432-4
41. Rodriguez-Ubreva J, Catala-Moll F, Obermajer N, Alvarez-Errico D, Ramirez RN, Company C, et al. Prostaglandin E2 leads to the acquisition of DNMT3A-dependent tolerogenic functions in human myeloid-derived suppressor cells. Cell Rep (2017) 21(1):154–67. doi: 10.1016/j.celrep.2017.09.018
42. Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res (2014) 20(15):4096–106. doi: 10.1158/1078-0432.CCR-14-0635
43. Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol (2020) 5(43):eaay1863. doi: 10.1126/sciimmunol.aay1863
44. Mantovani A, Sica A, Allavena P, Garlanda C, Locati M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum Immunol (2009) 70(5):325–30. doi: 10.1016/j.humimm.2009.02.008
45. Okla K, Czerwonka A, Wawruszak A, Bobinski M, Bilska M, Tarkowski R, et al. Clinical relevance and immunosuppressive pattern of circulating and infiltrating subsets of myeloid-derived suppressor cells (MDSCs) in epithelial ovarian cancer. Front Immunol (2019) 10:691. doi: 10.3389/fimmu.2019.00691
46. Paoletti S, Petkovic V, Sebastiani S, Danelon MG, Uguccioni M, Gerber BO. A rich chemokine environment strongly enhances leukocyte migration and activities. Blood (2005) 105(9):3405–12. doi: 10.1182/blood-2004-04-1648
47. Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates t-lymphocyte cell-cycle progression. Blood (2007) 109(4):1568–73. doi: 10.1182/blood-2006-06-031856
48. Sacchi A, Tumino N, Sabatini A, Cimini E, Casetti R, Bordoni V, et al. Myeloid-derived suppressor cells specifically suppress IFN-gamma production and antitumor cytotoxic activity of Vdelta2 T cells. Front Immunol (2018) 9:1271. doi: 10.3389/fimmu.2018.01271
49. Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int J Cancer (2014) 134(12):2853–64. doi: 10.1002/ijc.28622
50. Pillay J, Kamp VM, van Hoffen E, Visser T, Tak T, Lammers JW, et al. A subset of neutrophils in human systemic inflammation inhibits t cell responses through mac-1. J Clin Invest (2012) 122(1):327–36. doi: 10.1172/JCI57990
51. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol (2009) 182(9):5693–701. doi: 10.4049/jimmunol.0900092
52. Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, et al. Immune stimulatory receptor CD40 is required for t-cell suppression and t regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res (2010) 70(1):99–108. doi: 10.1158/0008-5472.CAN-09-1882
53. Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V, et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol (2012) 189(12):5602–11. doi: 10.4049/jimmunol.1201018
54. Jung YS, Lee HY, Kim SD, Park JS, Kim JK, Suh PG, et al. Wnt5a stimulates chemotactic migration and chemokine production in human neutrophils. Exp Mol Med (2013) 45:e27. doi: 10.1038/emm.2013.48
55. Wang L, Zhao J, Ren JP, Wu XY, Morrison ZD, Elgazzar MA, et al. Expansion of myeloid-derived suppressor cells promotes differentiation of regulatory t cells in HIV-1+ individuals. AIDS (2016) 30(10):1521–31. doi: 10.1097/QAD.0000000000001083
56. Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, et al. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front Immunol (2018) 9:1310. doi: 10.3389/fimmu.2018.01310
57. Liang Y, Wang W, Zhu X, Yu M, Zhou C. Inhibition of myeloid-derived suppressive cell function with all-trans retinoic acid enhanced anti-PD-L1 efficacy in cervical cancer. Sci Rep (2022) 12(1):9619. doi: 10.1038/s41598-022-13855-1
58. Cao Y, He Y, Wang X, Liu Y, Shi K, Zheng Z, et al. Polymorphonuclear myeloid-derived suppressor cells attenuate allergic airway inflammation by negatively regulating group 2 innate lymphoid cells. Immunology (2019) 156(4):402–12. doi: 10.1111/imm.13040
59. Schrijver IT, Karakike E, Theroude C, Baumgartner P, Harari A, Giamarellos-Bourboulis EJ, et al. High levels of monocytic myeloid-derived suppressor cells are associated with favorable outcome in patients with pneumonia and sepsis with multi-organ failure. Intensive Care Med Exp (2022) 10(1):5. doi: 10.1186/s40635-022-00431-0
60. Pallett LJ, Gill US, Quaglia A, Sinclair LV, Jover-Cobos M, Schurich A, et al. Metabolic regulation of hepatitis B immunopathology by myeloid-derived suppressor cells. Nat Med (2015) 21(6):591–600. doi: 10.1038/nm.3856
61. Si Y, Merz SF, Jansen P, Wang B, Bruderek K, Altenhoff P, et al. Multidimensional imaging provides evidence for down-regulation of T cell effector function by MDSC in human cancer tissue. Sci Immunol (2019) 4(40):eaaw9159. doi: 10.1126/sciimmunol.aaw9159
62. Gessain A, Cassar O. Epidemiological aspects and world distribution of HTLV-1 infection. Front Microbiol (2012) 3:388. doi: 10.3389/fmicb.2012.00388
63. Rowan AG, Dillon R, Witkover A, Melamed A, Demontis MA, Gillet NA, et al. Evolution of retrovirus-infected premalignant T-cell clones prior to adult t-cell leukemia/lymphoma diagnosis. Blood (2020) 135(23):2023–32. doi: 10.1182/blood.2019002665
64. Bangham CRM, Matsuoka M. Human t-cell leukaemia virus type 1: Parasitism and pathogenesis. Philos Trans R Soc Lond B Biol Sci (2017) 372(1732):20160272. doi: 10.1098/rstb.2016.0272
65. Toulza F, Nosaka K, Tanaka Y, Schioppa T, Balkwill F, Taylor GP, et al. Human t-lymphotropic virus type 1-induced CC chemokine ligand 22 maintains a high frequency of functional FoxP3+ regulatory T cells. J Immunol (2010) 185(1):183–9. doi: 10.4049/jimmunol.0903846
66. Tan BJ, Sugata K, Reda O, Matsuo M, Uchiyama K, Miyazato P, et al. HTLV-1 infection promotes excessive t cell activation and transformation into adult t cell leukemia/lymphoma. J Clin Invest (2021) 131(24):e150472. doi: 10.1172/JCI150472
67. Kagdi H, Demontis MA, Ramos JC, Taylor GP. Switching and loss of cellular cytokine producing capacity characterize in vivo viral infection and malignant transformation in human t- lymphotropic virus type 1 infection. PloS Pathog (2018) 14(2):e1006861. doi: 10.1371/journal.ppat.1006861
68. Yasuma K, Yasunaga J, Takemoto K, Sugata K, Mitobe Y, Takenouchi N, et al. HTLV-1 bZIP factor impairs anti-viral immunity by inducing co-inhibitory molecule, T cell immunoglobulin and ITIM domain (TIGIT). PloS Pathog (2016) 12(1):e1005372. doi: 10.1371/journal.ppat.1005372
69. El Hajj H, Hleihel R, El Sabban M, Bruneau J, Zaatari G, Cheminant M, et al. Loss of interleukin-10 activates innate immunity to eradicate adult T cell leukemia-initiating cells. Haematologica (2021) 106(5):1443–56. doi: 10.3324/haematol.2020.264523
70. Ratner L, Waldmann TA, Janakiram M, Brammer JE. Rapid progression of adult t-cell leukemia-lymphoma after PD-1 inhibitor therapy. N Engl J Med (2018) 378(20):1947–8. doi: 10.1056/NEJMc1803181
71. Bazarbachi A, Cwynarski K, Boumendil A, Finel H, Fields P, Raj K, et al. Outcome of patients with HTLV-1-associated adult T-cell leukemia/lymphoma after SCT: a retrospective study by the EBMT LWP. Bone Marrow Transplant (2014) 49(10):1266–8. doi: 10.1038/bmt.2014.143
72. Sugata K, Yasunaga J, Mitobe Y, Miura M, Miyazato P, Kohara M, et al. Protective effect of cytotoxic t lymphocytes targeting HTLV-1 bZIP factor. Blood (2015) 126(9):1095–105. doi: 10.1182/blood-2015-04-641118
73. Bazarbachi A, Plumelle Y, Carlos Ramos J, Tortevoye P, Otrock Z, Taylor G, et al. Meta-analysis on the use of zidovudine and interferon-alfa in adult t-cell leukemia/lymphoma showing improved survival in the leukemic subtypes. J Clin Oncol (2010) 28(27):4177–83. doi: 10.1200/JCO.2010.28.0669
74. Yamamoto S, Hattori T, Asou N, Nishimura H, Kawano F, Yodoi J, et al. Absolute neutrophilia in adult t cell leukemia. Jpn J Cancer Res (1986) 77(9):858–61.
75. Guerreiro JB, Porto MA, Santos SB, Lacerda L, Ho JL, Carvalho EM. Spontaneous neutrophil activation in HTLV-1 infected patients. Braz J Infect Dis (2005) 9(6):510–4. doi: 10.1590/s1413-86702005000600010
76. Brito-Melo GE, Peruhype-Magalhaes V, Teixeira-Carvalho A, Barbosa-Stancioli EF, Carneiro-Proietti AB, Catalan-Soares B, et al. IL-10 produced by CD4+ and CD8+ t cells emerge as a putative immunoregulatory mechanism to counterbalance the monocyte-derived TNF-alpha and guarantee asymptomatic clinical status during chronic HTLV-i infection. Clin Exp Immunol (2007) 147(1):35–44. doi: 10.1111/j.1365-2249.2006.03252.x
77. Hedayati-Moghadam M, Rezaee SAR, Boskabady MH, Mohamadian Roshan N, Saadat S, Bavarsad K, et al. Human T cell leukemia virus type 1 changes leukocyte number and oxidative stress in the lung and blood of female BALB/c mice. Adv BioMed Res (2021) 10:6. doi: 10.4103/abr.abr_117_20
78. Soda Y, Jinno A, Tanaka Y, Akagi T, Shimotohno K, Hoshino H. Rapid tumor formation and development of neutrophilia and splenomegaly in nude mice transplanted with human cells expressing human t cell leukemia virus type i or Tax1. Leukemia (2000) 14(8):1467–76. doi: 10.1038/sj.leu.2401844
79. Baydoun HH, Cherian MA, Green P, Ratner L. Inducible nitric oxide synthase mediates DNA double strand breaks in human T cell leukemia virus type 1-induced leukemia/lymphoma. Retrovirology (2015) 12:71. doi: 10.1186/s12977-015-0196-y
80. Komohara Y, Niino D, Saito Y, Ohnishi K, Horlad H, Ohshima K, et al. Clinical significance of CD163(+) tumor-associated macrophages in patients with adult T cell leukemia/lymphoma. Cancer Sci (2013) 104(7):945–51. doi: 10.1111/cas.12167
81. DeCaprio JA. Merkel cell polyomavirus and merkel cell carcinoma. Philos Trans R Soc Lond B Biol Sci (2017) 372(1732):20160276. doi: 10.1098/rstb.2016.0276
82. Gossai A, Waterboer T, Nelson HH, Michel A, Willhauck-Fleckenstein M, Farzan SF, et al. Seroepidemiology of human polyomaviruses in a US population. Am J Epidemiol (2016) 183(1):61–9. doi: 10.1093/aje/kwv155
83. Davies SI, Barrett J, Wong S, Chang MJ, Muranski PJ, Brownell I. Robust production of merkel cell polyomavirus oncogene specific t cells from healthy donors for adoptive transfer. Front Immunol (2020) 11:592721. doi: 10.3389/fimmu.2020.592721
84. Feldmeyer L, Hudgens CW, Ray-Lyons G, Nagarajan P, Aung PP, Curry JL, et al. Density, distribution, and composition of immune infiltrates correlate with survival in merkel cell carcinoma. Clin Cancer Res (2016) 22(22):5553–63. doi: 10.1158/1078-0432.CCR-16-0392
85. Dowlatshahi M, Huang V, Gehad AE, Jiang Y, Calarese A, Teague JE, et al. Tumor-specific t cells in human merkel cell carcinomas: a possible role for tregs and t-cell exhaustion in reducing t-cell responses. J Invest Dermatol (2013) 133(7):1879–89. doi: 10.1038/jid.2013.75
86. Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, et al. PD-1 blockade with pembrolizumab in advanced merkel-cell carcinoma. N Engl J Med (2016) 374(26):2542–52. doi: 10.1056/NEJMoa1603702
87. Nghiem P, Bhatia S, Lipson EJ, Sharfman WH, Kudchadkar RR, Brohl AS, et al. Three-year survival, correlates and salvage therapies in patients receiving first-line pembrolizumab for advanced merkel cell carcinoma. J Immunother Cancer (2021) 9(4):e002478. doi: 10.1136/jitc-2021-002478
88. Naseri S, Steiniche T, Georgsen JB, Thomsen R, Ladekarl M, Heje M, et al. Reduced infiltration of CD8-lymphocytes, high neutrophil-to-CD8-Lymphocyte ratio and absence of MC virus are negative prognostic markers for patients with merkel cell carcinoma. Cancers (Basel) (2020) 12(4):888. doi: 10.3390/cancers12040888
89. Mitteldorf C, Berisha A, Tronnier M, Pfaltz MC, Kempf W. PD-1 and PD-L1 in neoplastic cells and the tumor microenvironment of merkel cell carcinoma. J Cutan Pathol (2017) 44(9):740–6. doi: 10.1111/cup.12973
90. Gaiser MR, Weis CA, Gaiser T, Jiang H, Buder-Bakhaya K, Herpel E, et al. Merkel cell carcinoma expresses the immunoregulatory ligand CD200 and induces immunosuppressive macrophages and regulatory t cells. Oncoimmunology (2018) 7(5):e1426517. doi: 10.1080/2162402X.2018.1426517
91. Liu JQ, Hu A, Zhu J, Yu J, Talebian F, Bai XF. CD200-CD200R pathway in the regulation of tumor immune microenvironment and immunotherapy. Adv Exp Med Biol (2020) 1223:155–65. doi: 10.1007/978-3-030-35582-1_8
92. Gallo M, Guarnotta V, De Cicco F, Rubino M, Faggiano A, Colao A, et al. Immune checkpoint blockade for merkel cell carcinoma: actual findings and unanswered questions. J Cancer Res Clin Oncol (2019) 145(2):429–43. doi: 10.1007/s00432-019-02839-w
93. Chapuis AG, Afanasiev OK, Iyer JG, Paulson KG, Parvathaneni U, Hwang JH, et al. Regression of metastatic merkel cell carcinoma following transfer of polyomavirus-specific T cells and therapies capable of re-inducing HLA class-i. Cancer Immunol Res (2014) 2(1):27–36. doi: 10.1158/2326-6066.CIR-13-0087
94. Tabachnick-Cherny S, Pulliam T, Church C, Koelle DM, Nghiem P. Polyomavirus-driven merkel cell carcinoma: Prospects for therapeutic vaccine development. Mol Carcinog (2020) 59(7):807–21. doi: 10.1002/mc.23190
95. Mariggio G, Koch S, Schulz TF. Kaposi sarcoma herpesvirus pathogenesis. Philos Trans R Soc Lond B Biol Sci (2017) 372(1732):20160275. doi: 10.1098/rstb.2016.0275
96. Iarc Working Group on the Evaluation of Carcinogenic Risks to Humans. Biological agents. volume 100 b. A review of human carcinogens. IARC Monogr Eval Carcinog Risks Hum (2012) 100:1–441.
97. Jary A, Veyri M, Gothland A, Leducq V, Calvez V, Marcelin AG. Kaposi's sarcoma-associated herpesvirus, the etiological agent of all epidemiological forms of kaposi's sarcoma. Cancers (Basel) (2021) 13(24):6208. doi: 10.3390/cancers13246208
98. Broussard G, Damania B. KSHV: Immune modulation and immunotherapy. Front Immunol (2019) 10:3084. doi: 10.3389/fimmu.2019.03084
99. Suthaus J, Stuhlmann-Laeisz C, Tompkins VS, Rosean TR, Klapper W, Tosato G, et al. HHV-8-encoded viral IL-6 collaborates with mouse IL-6 in the development of multicentric castleman disease in mice. Blood (2012) 119(22):5173–81. doi: 10.1182/blood-2011-09-377705
100. Weber R, Groth C, Lasser S, Arkhypov I, Petrova V, Altevogt P, et al. IL-6 as a major regulator of MDSC activity and possible target for cancer immunotherapy. Cell Immunol (2021) 359:104254. doi: 10.1016/j.cellimm.2020.104254
101. Korbecki J, Kojder K, Siminska D, Bohatyrewicz R, Gutowska I, Chlubek D, et al. CC chemokines in a tumor: A review of pro-cancer and anti-cancer properties of the ligands of receptors CCR1, CCR2, CCR3, and CCR4. Int J Mol Sci (2020) 21(21):8412. doi: 10.3390/ijms21218412
102. Ballon G, Akar G, Cesarman E. Systemic expression of kaposi sarcoma herpesvirus (KSHV) vflip in endothelial cells leads to a profound proinflammatory phenotype and myeloid lineage remodeling in vivo. PloS Pathog (2015) 11(1):e1004581. doi: 10.1371/journal.ppat.1004581
103. Joest B, Kempf W, Berisha A, Peyk P, Tronnier M, Mitteldorf C. Stage-related PD-L1 expression in kaposi sarcoma tumor microenvironment. J Cutan Pathol (2020) 47(10):888–95. doi: 10.1111/cup.13716
104. Genovese G, Venegoni L, Fanoni D, Tourlaki A, Brambilla L, Berti E. PD-L1 expression in tumour microenvironment supports the rationale for immune checkpoint blockade in classic kaposi's sarcoma. J Eur Acad Dermatol Venereol (2019) 33(7):e269–71. doi: 10.1111/jdv.15543
105. Host KM, Jacobs SR, West JA, Zhang Z, Costantini LM, Stopford CM, et al. Kaposi's sarcoma-associated herpesvirus increases PD-L1 and proinflammatory cytokine expression in human monocytes. mBio (2017) 8(5):e00917–17. doi: 10.1128/mBio.00917-17
106. Schneider JW, Dittmer DP. Diagnosis and treatment of kaposi sarcoma. Am J Clin Dermatol (2017) 18(4):529–39. doi: 10.1007/s40257-017-0270-4
107. van Rhee F, Voorhees P, Dispenzieri A, Fossa A, Srkalovic G, Ide M, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric castleman disease. Blood (2018) 132(20):2115–24. doi: 10.1182/blood-2018-07-862334
108. Viens LJ, Henley SJ, Watson M, Markowitz LE, Thomas CC, Thompson TD, et al. Human papillomavirus-associated cancers - united states, 2008-2012. MMWR Morb Mortal Wkly Rep (2016) 65(26):661–6. doi: 10.15585/mmwr.mm6526a1
109. McBride AA. Oncogenic human papillomaviruses. Philos Trans R Soc Lond B Biol Sci (2017) 372(1732):20160273. doi: 10.1098/rstb.2016.0273
110. Doorbar J, Egawa N, Griffin H, Kranjec C, Murakami I. Human papillomavirus molecular biology and disease association. Rev Med Virol (2015) 25(Suppl 1):2–23. doi: 10.1002/rmv.1822
111. Tummers B, Burg SH. High-risk human papillomavirus targets crossroads in immune signaling. Viruses (2015) 7(5):2485–506. doi: 10.3390/v7052485
112. Di Donato V, Caruso G, Petrillo M, Kontopantelis E, Palaia I, Perniola G, et al. Adjuvant HPV vaccination to prevent recurrent cervical dysplasia after surgical treatment: A meta-analysis. Vaccines (Basel) (2021) 9(5):410. doi: 10.3390/vaccines9050410
113. Stern PL. Harnessing immunity for therapy in human papillomavirus driven cancers. Tumour Virus Res (2021) 11:200212. doi: 10.1016/j.tvr.2021.200212
114. de Jong A, van Poelgeest MI, van der Hulst JM, Drijfhout JW, Fleuren GJ, Melief CJ, et al. Human papillomavirus type 16-positive cervical cancer is associated with impaired CD4+ t-cell immunity against early antigens E2 and E6. Cancer Res (2004) 64(15):5449–55. doi: 10.1158/0008-5472.CAN-04-0831
115. Mo Y, Ma J, Zhang H, Shen J, Chen J, Hong J, et al. Prophylactic and therapeutic HPV vaccines: Current scenario and perspectives. Front Cell Infect Microbiol (2022) 12:909223. doi: 10.3389/fcimb.2022.909223
116. Jayshre R,S. The immune microenvironment in human papilloma virus-induced cervical lesions-evidence for estrogen as an immunomodulator. Front Cell Infect Microbiol (2021) 11:649815. doi: 10.3389/fcimb.2021.649815
117. Adurthi S, Krishna S, Mukherjee G, Bafna UD, Devi U, Jayshree RS. Regulatory T cells in a spectrum of HPV-induced cervical lesions: cervicitis, cervical intraepithelial neoplasia and squamous cell carcinoma. Am J Reprod Immunol (2008) 60(1):55–65. doi: 10.1111/j.1600-0897.2008.00590.x
118. Chen XJ, Han LF, Wu XG, Wei WF, Wu LF, Yi HY, et al. Clinical significance of CD163+ and CD68+ tumor-associated macrophages in high-risk HPV-related cervical cancer. J Cancer (2017) 8(18):3868–75. doi: 10.7150/jca.21444
119. Wu L, Liu H, Guo H, Wu Q, Yu S, Qin Y, et al. Circulating and tumor-infiltrating myeloid-derived suppressor cells in cervical carcinoma patients. Oncol Lett (2018) 15(6):9507–15. doi: 10.3892/ol.2018.8532
120. Jianyi D, Haili G, Bo Y, Meiqin Y, Baoyou H, Haoran H, et al. Myeloid-derived suppressor cells cross-talk with B10 cells by BAFF/BAFF-r pathway to promote immunosuppression in cervical cancer. Cancer Immunol Immunother (2022) 72(1):73–85. doi: 10.1007/s00262-022-03226-0
121. Liang Y, Lu B, Zhao P, Lu W. Increased circulating GrMyeloid-derived suppressor cells correlated with tumor burden and survival in locally advanced cervical cancer patient. J Cancer (2019) 10(6):1341–8. doi: 10.7150/jca.29647
122. Alvarez KLF, Beldi M, Sarmanho F, Rossetti RAM, Silveira CRF, Mota GR, et al. Local and systemic immunomodulatory mechanisms triggered by human papillomavirus transformed cells: a potential role for g-CSF and neutrophils. Sci Rep (2017) 7(1):9002. doi: 10.1038/s41598-017-09079-3
123. Mabuchi S, Matsumoto Y, Isohashi F, Yoshioka Y, Ohashi H, Morii E, et al. Pretreatment leukocytosis is an indicator of poor prognosis in patients with cervical cancer. Gynecol Oncol (2011) 122(1):25–32. doi: 10.1016/j.ygyno.2011.03.037
124. Kawano M, Mabuchi S, Matsumoto Y, Sasano T, Takahashi R, Kuroda H, et al. The significance of g-CSF expression and myeloid-derived suppressor cells in the chemoresistance of uterine cervical cancer. Sci Rep (2015) 5:18217. doi: 10.1038/srep18217
125. Mabuchi S, Matsumoto Y, Kawano M, Minami K, Seo Y, Sasano T, et al. Uterine cervical cancer displaying tumor-related leukocytosis: a distinct clinical entity with radioresistant feature. J Natl Cancer Inst (2014) 106(7):dju147. doi: 10.1093/jnci/dju147
126. Xia J, Zhang Q, Luan J, Min P, Zhang H, Chen G, et al. TGFbeta signaling activation correlates with immune-inflamed tumor microenvironment across human cancers and predicts response to immunotherapy. Cell Cycle (2022) 22(1):57–72. doi: 10.1080/15384101.2022.2109105
127. Syrjanen S, Naud P, Sarian L, Derchain S, Roteli-Martins C, Longatto-Filho A, et al. Immunosuppressive cytokine interleukin-10 (IL-10) is up-regulated in high-grade CIN but not associated with high-risk human papillomavirus (HPV) at baseline, outcomes of HR-HPV infections or incident CIN in the LAMS cohort. Virchows Arch (2009) 455(6):505–15. doi: 10.1007/s00428-009-0850-7
128. Zijlmans HJ, Fleuren GJ, Baelde HJ, Eilers PH, Kenter GG, Gorter A. The absence of CCL2 expression in cervical carcinoma is associated with increased survival and loss of heterozygosity at 17q11.2. J Pathol (2006) 208(4):507–17. doi: 10.1002/path.1918
129. Kuroda H, Mabuchi S, Yokoi E, Komura N, Kozasa K, Matsumoto Y, et al. Prostaglandin E2 produced by myeloid-derived suppressive cells induces cancer stem cells in uterine cervical cancer. Oncotarget (2018) 9(91):36317–30. doi: 10.18632/oncotarget.26347
130. Lee MY, Allen CT. Immunotherapy for HPV malignancies. Semin Radiat Oncol (2021) 31(4):361–70. doi: 10.1016/j.semradonc.2021.02.008
131. Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med (2016) 375(19):1856–67. doi: 10.1056/NEJMoa1602252
132. Chung HC, Ros W, Delord JP, Perets R, Italiano A, Shapira-Frommer R, et al. Efficacy and safety of pembrolizumab in previously treated advanced cervical cancer: Results from the phase II KEYNOTE-158 study. J Clin Oncol (2019) 37(17):1470–8. doi: 10.1200/JCO.18.01265
133. Diniz MO, Sales NS, Silva JR, Ferreira LC. Protection against HPV-16-Associated tumors requires the activation of CD8+ effector memory t cells and the control of myeloid-derived suppressor cells. Mol Cancer Ther (2016) 15(8):1920–30. doi: 10.1158/1535-7163.MCT-15-0742
134. Song X, Ye D, Liu B, Cui J, Zhao X, Yi L, et al. Combination of all-trans retinoic acid and a human papillomavirus therapeutic vaccine suppresses the number and function of immature myeloid cells and enhances antitumor immunity. Cancer Sci (2009) 100(2):334–40. doi: 10.1111/j.1349-7006.2008.01037.x
135. Galliverti G, Wullschleger S, Tichet M, Murugan D, Zangger N, Horton W, et al. Myeloid cells orchestrate systemic immunosuppression, impairing the efficacy of immunotherapy against HPV(+) cancers. Cancer Immunol Res (2020) 8(1):131–45. doi: 10.1158/2326-6066.CIR-19-0315
136. Hanoteau A, Newton JM, Krupar R, Huang C, Liu HC, Gaspero A, et al. Tumor microenvironment modulation enhances immunologic benefit of chemoradiotherapy. J Immunother Cancer (2019) 7(1):10. doi: 10.1186/s40425-018-0485-9
137. Summers J, O'Connell A, Millman I. Genome of hepatitis B virus: restriction enzyme cleavage and structure of DNA extracted from dane particles. Proc Natl Acad Sci U.S.A. (1975) 72(11):4597–601. doi: 10.1073/pnas.72.11.4597
138. Dienstag JL. Hepatitis B virus infection. N Engl J Med (2008) 359(14):1486–500. doi: 10.1056/NEJMra0801644
139. Guidotti LG, Chisari FV. Immunobiology and pathogenesis of viral hepatitis. Annu Rev Pathol (2006) 1:23–61. doi: 10.1146/annurev.pathol.1.110304.100230
140. Tu T, Budzinska MA, Shackel NA, Urban S. HBV DNA integration: Molecular mechanisms and clinical implications. Viruses (2017) 9:v9040075. doi: 10.3390/v9040075
141. Cougot D, Wu Y, Cairo S, Caramel J, Renard C-A, Lévy L, et al. The hepatitis B virus x protein functionally interacts with CREB-binding Protein/p300 in the regulation of CREB-mediated transcription*. J Biol Chem (2007) 282(7):4277–87. doi: 10.1074/jbc.M606774200
142. Truant R, Antunovic J, Greenblatt J, Prives C, Cromlish JA. Direct interaction of the hepatitis B virus HBx protein with p53 leads to inhibition by HBx of p53 response element-directed transactivation. J Virol (1995) 69(3):1851–9. doi: 10.1128/jvi.69.3.1851-1859.1995
143. Tian Y, Yang W, Song J, Wu Y, Ni B. Hepatitis B virus x protein-induced aberrant epigenetic modifications contributing to human hepatocellular carcinoma pathogenesis. Mol Cell Biol (2013) 33(15):2810–6. doi: 10.1128/mcb.00205-13
144. Zheng D-L, Zhang L, Cheng N, Xu X, Deng Q, Teng X-M, et al. Epigenetic modification induced by hepatitis B virus x protein via interaction with de novo DNA methyltransferase DNMT3A. J Hepatol (2009) 50(2):377–87. doi: 10.1016/j.jhep.2008.10.019
145. Kay J, Thadhani E, Samson L, Engelward B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair (Amst) (2019) 83:102673. doi: 10.1016/j.dnarep.2019.102673
146. Yu DY. Relevance of reactive oxygen species in liver disease observed in transgenic mice expressing the hepatitis B virus x protein. Lab Anim Res (2020) 36:6. doi: 10.1186/s42826-020-00037-1
147. Tan A, Koh S, Bertoletti A. Immune response in hepatitis B virus infection. Cold Spring Harb Perspect Med (2015) 5(8):a021428. doi: 10.1101/cshperspect.a021428
148. Li TY, Yang Y, Zhou G, Tu ZK. Immune suppression in chronic hepatitis B infection associated liver disease: A review. World J Gastroenterol (2019) 25(27):3527–37. doi: 10.3748/wjg.v25.i27.3527
149. Li T, Zhang X, Lv Z, Gao L, Yan H. Increased expression of myeloid-derived suppressor cells in patients with HBV-related hepatocellular carcinoma. BioMed Res Int (2020) 2020:6527192. doi: 10.1155/2020/6527192
150. Lu LR, Liu J, Xu Z, Zhang GL, Li DC, Lin CS. Expression and clinical significance of myeloid derived suppressor cells in chronic hepatitis B patients. Asian Pac J Cancer Prev (2014) 15(10):4367–72. doi: 10.7314/apjcp.2014.15.10.4367
151. Lv Y, Cui M, Lv Z, Lu J, Zhang X, Zhao Z, et al. Expression and significance of peripheral myeloid-derived suppressor cells in chronic hepatitis B patients. Clin Res Hepatol Gastroenterol (2018) 42(5):462–9. doi: 10.1016/j.clinre.2018.04.002
152. Pal S, Dey D, Chakraborty BC, Nandi M, Khatun M, Banerjee S, et al. Diverse facets of MDSC in different phases of chronic HBV infection: Impact on HBV-specific T cell response and homing. Hepatology (2022) 76(3):759–74. doi: 10.1002/hep.32331
153. Zeng Y, Li Y, Xu Z, Gan W, Lu L, Huang X, et al. Myeloid-derived suppressor cells expansion is closely associated with disease severity and progression in HBV-related acute-on-chronic liver failure. J Med Virol (2019) 91(8):1510–8. doi: 10.1002/jmv.25466
154. Huang A, Zhang B, Yan W, Wang B, Wei H, Zhang F, et al. Myeloid-derived suppressor cells regulate immune response in patients with chronic hepatitis B virus infection through PD-1-induced IL-10. J Immunol (2014) 193(11):5461–9. doi: 10.4049/jimmunol.1400849
155. Pal S, Nandi M, Dey D, Chakraborty BC, Shil A, Ghosh S, et al. Myeloid-derived suppressor cells induce regulatory t cells in chronically HBV infected patients with high levels of hepatitis B surface antigen and persist after antiviral therapy. Aliment Pharmacol Ther (2019) 49(10):1346–59. doi: 10.1111/apt.15226
156. Yang F, Yu X, Zhou C, Mao R, Zhu M, Zhu H, et al. Hepatitis B e antigen induces the expansion of monocytic myeloid-derived suppressor cells to dampen t-cell function in chronic hepatitis b virus infection. PloS Pathog (2019) 15(4):e1007690. doi: 10.1371/journal.ppat.1007690
157. Cassetta L, Baekkevold ES, Brandau S, Bujko A, Cassatella MA, Dorhoi A, et al. Deciphering myeloid-derived suppressor cells: isolation and markers in humans, mice and non-human primates. Cancer Immunol Immunother (2019) 68(4):687–97. doi: 10.1007/s00262-019-02302-2
158. Nan J, Xing YF, Hu B, Tang JX, Dong HM, He YM, et al. Endoplasmic reticulum stress induced LOX-1(+ ) CD15(+) polymorphonuclear myeloid-derived suppressor cells in hepatocellular carcinoma. Immunology (2018) 154(1):144–55. doi: 10.1111/imm.12876
159. Arihara F, Mizukoshi E, Kitahara M, Takata Y, Arai K, Yamashita T, et al. Increase in CD14+HLA-DR -/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol Immunother (2013) 62(8):1421–30. doi: 10.1007/s00262-013-1447-1
160. Gao XH, Tian L, Wu J, Ma XL, Zhang CY, Zhou Y, et al. Circulating CD14(+) HLA-DR(-/low) myeloid-derived suppressor cells predicted early recurrence of hepatocellular carcinoma after surgery. Hepatol Res (2017) 47(10):1061–71. doi: 10.1111/hepr.12831
161. Mizukoshi E, Yamashita T, Arai K, Terashima T, Kitahara M, Nakagawa H, et al. Myeloid-derived suppressor cells correlate with patient outcomes in hepatic arterial infusion chemotherapy for hepatocellular carcinoma. Cancer Immunol Immunother (2016) 65(6):715–25. doi: 10.1007/s00262-016-1837-2
162. Fang Z, Zhang Y, Zhu Z, Wang C, Hu Y, Peng X, et al. Monocytic MDSCs homing to thymus contribute to age-related CD8+ t cell tolerance of HBV. J Exp Med (2022) 219(4):e20211838. doi: 10.1084/jem.20211838
163. Ambade A, Lowe P, Kodys K, Catalano D, Gyongyosi B, Cho Y, et al. Pharmacological inhibition of CCR2/5 signaling prevents and reverses alcohol-induced liver damage, steatosis, and inflammation in mice. Hepatology (2019) 69(3):1105–21. doi: 10.1002/hep.30249
164. Moreno C, Gustot T, Nicaise C, Quertinmont E, Nagy N, Parmentier M, et al. CCR5 deficiency exacerbates t-cell-mediated hepatitis in mice. Hepatology (2005) 42(4):854–62. doi: 10.1002/hep.20865
165. Fang Z, Li J, Yu X, Zhang D, Ren G, Shi B, et al. Polarization of monocytic myeloid-derived suppressor cells by hepatitis B surface antigen is mediated via ERK/IL-6/STAT3 signaling feedback and restrains the activation of t cells in chronic hepatitis b virus infection. J Immunol (2015) 195(10):4873–83. doi: 10.4049/jimmunol.1501362
166. Xiang WQ, Feng WF, Ke W, Sun Z, Chen Z, Liu W. Hepatitis b virus x protein stimulates IL-6 expression in hepatocytes via a MyD88-dependent pathway. J Hepatol (2011) 54(1):26–33. doi: 10.1016/j.jhep.2010.08.006
167. Chen Z, Li YX, Fu HJ, Ren YL, Zou L, Shen SZ, et al. Hepatitis B virus core antigen stimulates IL-6 expression via p38, ERK and NF-κB pathways in hepatocytes. Cell Physiol Biochem (2017) 41(1):91–100. doi: 10.1159/000455954
168. Kong X, Sun R, Chen Y, Wei H, Tian Z. γδT cells drive myeloid-derived suppressor cell-mediated CD8+ T cell exhaustion in hepatitis B virus-induced immunotolerance. J Immunol (2014) 193(4):1645–53. doi: 10.4049/jimmunol.1303432
169. Chang TT, Gish RG, de Man R, Gadano A, Sollano J, Chao YC, et al. A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. N Engl J Med (2006) 354(10):1001–10. doi: 10.1056/NEJMoa051285
170. Lau GK, Piratvisuth T, Luo KX, Marcellin P, Thongsawat S, Cooksley G, et al. Peginterferon alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis b. N Engl J Med (2005) 352(26):2682–95. doi: 10.1056/NEJMoa043470
171. Bertoletti A, Le Bert N. Immunotherapy for chronic hepatitis B virus infection. Gut Liver (2018) 12(5):497–507. doi: 10.5009/gnl17233
172. Han Q, Hou Z, Yin C, Zhang C, Zhang J. 5'-triphosphate siRNA targeting HBx elicits a potent anti-HBV immune response in pAAV-HBV transfected mice. Antiviral Res (2019) 161:36–45. doi: 10.1016/j.antiviral.2018.11.006
173. Amin OE, Colbeck EJ, Daffis S, Khan S, Ramakrishnan D, Pattabiraman D, et al. Therapeutic potential of TLR8 agonist GS-9688 (Selgantolimod) in chronic hepatitis B: Remodeling of antiviral and regulatory mediators. Hepatology (2021) 74(1):55–71. doi: 10.1002/hep.31695
174. Taleb K, Auffray C, Villefroy P, Pereira A, Hosmalin A, Gaudry M, et al. Chronic type i IFN is sufficient to promote immunosuppression through accumulation of myeloid-derived suppressor cells. J Immunol (2017) 198(3):1156–63. doi: 10.4049/jimmunol.1502638
175. Zou S, Du Y, Huang S, Chen M, Yang X, Li S, et al. Simultaneous or prior activation of intrahepatic type i interferon signaling leads to hepatitis B virus persistence in a mouse model. J Virol (2021) 95(24):e0003421. doi: 10.1128/jvi.00034-21
176. Qin SK, Li Q, Ming Xu J, Liang J, Cheng Y, Fan Y, et al. Icaritin-induced immunomodulatory efficacy in advanced hepatitis B virus-related hepatocellular carcinoma: Immunodynamic biomarkers and overall survival. Cancer Sci (2020) 111(11):4218–31. doi: 10.1111/cas.14641
177. Lindenbach BD, Rice CM. Unravelling hepatitis C virus replication from genome to function. Nature (2005) 436(7053):933–8. doi: 10.1038/nature04077
178. Roudot-Thoraval F. Epidemiology of hepatitis C virus infection. Clin Res Hepatol Gastroenterol (2021) 45(3):101596. doi: 10.1016/j.clinre.2020.101596
179. McGlynn KA, London WT. The global epidemiology of hepatocellular carcinoma: Present and future. Clin Liver Dis (2011) 15(2):223–43. doi: 10.1016/j.cld.2011.03.006
180. Page K, Melia MT, Veenhuis RT, Winter M, Rousseau KE, Massaccesi G, et al. Randomized trial of a vaccine regimen to prevent chronic HCV infection. N Engl J Med (2021) 384(6):541–9. doi: 10.1056/NEJMoa2023345
181. Hill AM, Nath S, Simmons B. The road to elimination of hepatitis C: Analysis of cures versus new infections in 91 countries. J Virus Erad (2017) 3(3):117–23.
182. Lechner F, Wong DK, Dunbar PR, Chapman R, Chung RT, Dohrenwend P, et al. Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med (2000) 191(9):1499–512. doi: 10.1084/jem.191.9.1499
183. Bartsch H, Nair J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: role of lipid peroxidation, DNA damage, and repair. Langenbecks Arch Surg (2006) 391(5):499–510. doi: 10.1007/s00423-006-0073-1
184. Hayashi J, Aoki H, Kajino K, Moriyama M, Arakawa Y, Hino O. Hepatitis C virus core protein activates the MAPK/ERK cascade synergistically with tumor promoter TPA, but not with epidermal growth factor or transforming growth factor alpha. Hepatology (2000) 32(5):958–61. doi: 10.1053/jhep.2000.19343
185. Nakamura H, Aoki H, Hino O, Moriyama M. HCV core protein promotes heparin binding EGF-like growth factor expression and activates akt. Hepatol Res (2011) 41(5):455–62. doi: 10.1111/j.1872-034X.2011.00792.x
186. Syed GH, Amako Y, Siddiqui A. Hepatitis C virus hijacks host lipid metabolism. Trends Endocrinol Metab (2010) 21(1):33–40. doi: 10.1016/j.tem.2009.07.005
187. Foy E, Li K, Sumpter R Jr., Loo YM, Johnson CL, Wang C, et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-i signaling. Proc Natl Acad Sci U.S.A. (2005) 102(8):2986–91. doi: 10.1073/pnas.0408707102
188. Gao Y, Nepal N, Jin SZ. Toll-like receptors and hepatitis C virus infection. Hepatobiliary Pancreat Dis Int (2021) 20(6):521–9. doi: 10.1016/j.hbpd.2021.07.011
189. Kemming J, Thimme R, Neumann-Haefelin C. Adaptive immune response against hepatitis C virus. Int J Mol Sci (2020) 21(16):5644. doi: 10.3390/ijms21165644
190. Ning G, She L, Lu L, Liu Y, Zeng Y, Yan Y, et al. Analysis of monocytic and granulocytic myeloid-derived suppressor cells subsets in patients with hepatitis c virus infection and their clinical significance. BioMed Res Int (2015) 2015:385378. doi: 10.1155/2015/385378
191. Cai W, Qin A, Guo P, Yan D, Hu F, Yang Q, et al. Clinical significance and functional studies of myeloid-derived suppressor cells in chronic hepatitis C patients. J Clin Immunol (2013) 33(4):798–808. doi: 10.1007/s10875-012-9861-2
192. Lei AH, Yang Q, Cai WP, Liu YF, Lan Y, Qin AP, et al. Clinical significance of myeloid-derived suppressor cells in human immunodeficiency virus-1/ hepatitis C virus-coinfected patients. Scand J Immunol (2016) 83(6):438–44. doi: 10.1111/sji.12429
193. Telatin V, Nicoli F, Frasson C, Menegotto N, Barbaro F, Castelli E, et al. In chronic hepatitis c infection, myeloid-derived suppressor cell accumulation and t cell dysfunctions revert partially and late after successful direct-acting antiviral treatment. Front Cell Infect Microbiol (2019) 9:190. doi: 10.3389/fcimb.2019.00190
194. Ren JP, Zhao J, Dai J, Griffin JW, Wang L, Wu XY, et al. Hepatitis C virus-induced myeloid-derived suppressor cells regulate t-cell differentiation and function via the signal transducer and activator of transcription 3 pathway. Immunology (2016) 148(4):377–86. doi: 10.1111/imm.12616
195. Li Y, Zeng Y, Zeng G, Li J, Zhang X, Cai Q, et al. The effects of direct-acting antiviral agents on the frequency of myeloid-derived suppressor cells and natural killer cells in patients with chronic hepatitis C. J Med Virol (2019) 91(2):278–86. doi: 10.1002/jmv.25302
196. Zeng QL, Yang B, Sun HQ, Feng GH, Jin L, Zou ZS, et al. Myeloid-derived suppressor cells are associated with viral persistence and downregulation of TCR ζ chain expression on CD8(+) t cells in chronic hepatitis c patients. Mol Cells (2014) 37(1):66–73. doi: 10.14348/molcells.2014.2282
197. Liu Y, She LH, Wang XY, Zhang GL, Yan Y, Lin CS, et al. Expansion of myeloid-derived suppressor cells from peripheral blood decreases after 4-week antiviral treatment in patients with chronic hepatitis C. Int J Clin Exp Med (2014) 7(4):998–1004.
198. Thakuri BKC, Zhang J, Zhao J, Nguyen LN, Nguyen LNT, Schank M, et al. HCV-associated exosomes upregulate RUNXOR and RUNX1 expressions to promote MDSC expansion and suppressive functions through STAT3-miR124 axis. Cells (2020) 9(12):2715. doi: 10.3390/cells9122715
199. Thakuri BKC, Zhang J, Zhao J, Nguyen LN, Nguyen LNT, Khanal S, et al. LncRNA HOTAIRM1 promotes MDSC expansion and suppressive functions through the HOXA1-miR124 axis during HCV infection. Sci Rep (2020) 10(1):22033. doi: 10.1038/s41598-020-78786-1
200. Goh CC, Roggerson KM, Lee HC, Golden-Mason L, Rosen HR, Hahn YS. Hepatitis C virus-induced myeloid-derived suppressor cells suppress NK cell IFN-γ production by altering cellular metabolism via arginase-1. J Immunol (2016) 196(5):2283–92. doi: 10.4049/jimmunol.1501881
201. Wang M, Ping Y, Li Z, Li J, Zhang Z, Yue D, et al. Polarization of granulocytic myeloid-derived suppressor cells by hepatitis C core protein is mediated via IL-10/STAT3 signalling. J Viral Hepat (2019) 26(2):246–57. doi: 10.1111/jvh.13024
202. Kalathil S, Lugade AA, Miller A, Iyer R, Thanavala Y. Higher frequencies of GARP(+)CTLA-4(+)Foxp3(+) t regulatory cells and myeloid-derived suppressor cells in hepatocellular carcinoma patients are associated with impaired t-cell functionality. Cancer Res (2013) 73(8):2435–44. doi: 10.1158/0008-5472.CAN-12-3381
203. Goh CC, Roggerson KM, Lee HC, Golden-Mason L, Rosen HR, Hahn YS. Hepatitis C virus-induced myeloid-derived suppressor cells suppress NK cell IFN-gamma production by altering cellular metabolism via arginase-1. J Immunol (2016) 196(5):2283–92. doi: 10.4049/jimmunol.1501881
204. Tacke RS, Lee HC, Goh C, Courtney J, Polyak SJ, Rosen HR, et al. Myeloid suppressor cells induced by hepatitis C virus suppress t-cell responses through the production of reactive oxygen species. Hepatology (2012) 55(2):343–53. doi: 10.1002/hep.24700
205. Wang L, Cao D, Wang L, Zhao J, Nguyen LN, Dang X, et al. HCV-associated exosomes promote myeloid-derived suppressor cell expansion via inhibiting miR-124 to regulate t follicular cell differentiation and function. Cell Discovery (2018) 4:51. doi: 10.1038/s41421-018-0052-z
206. Zhai N, Li H, Song H, Yang Y, Cui A, Li T, et al. Hepatitis C virus induces MDSCs-like monocytes through TLR2/PI3K/AKT/STAT3 signaling. PloS One (2017) 12(1):e0170516. doi: 10.1371/journal.pone.0170516
207. Pang X, Song H, Zhang Q, Tu Z, Niu J. Hepatitis c virus regulates the production of monocytic myeloid-derived suppressor cells from peripheral blood mononuclear cells through PI3K pathway and autocrine signaling. Clin Immunol (2016) 164:57–64. doi: 10.1016/j.clim.2016.01.014
208. Masalova OV, Lesnova EI, Klimova RR, Momotyuk ED, Kozlov VV, Ivanova AM, et al. Genetically modified mouse mesenchymal stem cells expressing non-structural proteins of hepatitis c virus induce effective immune response. Vaccines (Basel) (2020) 8(1):62. doi: 10.3390/vaccines8010062
209. Salem ML, Zidan AA, Attia M, El-Naggar RE, Nassef M, Abou El-Azm AR, et al. IFN-α-based treatment of patients with chronic HCV show increased levels of cells with myeloid-derived suppressor cell phenotype and of IDO and NOS. Immunopharmacol Immunotoxicol (2017) 39(4):188–98. doi: 10.1080/08923973.2017.1320670
210. Shannon-Lowe C, Rickinson A. The global landscape of EBV-associated tumors. Front Oncol (2019) 9:713. doi: 10.3389/fonc.2019.00713
211. Shannon-Lowe C, Rickinson AB, Bell AI. Epstein-barr virus-associated lymphomas. Philos Trans R Soc Lond B Biol Sci (2017) 372(1732):20160271. doi: 10.1098/rstb.2016.0271
212. Long HM, Meckiff BJ, Taylor GS. The t-cell response to epstein-barr virus-new tricks from an old dog. Front Immunol (2019) 10:2193. doi: 10.3389/fimmu.2019.02193
213. Zhang H, Li ZL, Ye SB, Ouyang LY, Chen YS, He J, et al. Myeloid-derived suppressor cells inhibit t cell proliferation in human extranodal NK/T cell lymphoma: a novel prognostic indicator. Cancer Immunol Immunother (2015) 64(12):1587–99. doi: 10.1007/s00262-015-1765-6
214. Mrizak D, Martin N, Barjon C, Jimenez-Pailhes AS, Mustapha R, Niki T, et al. Effect of nasopharyngeal carcinoma-derived exosomes on human regulatory t cells. J Natl Cancer Inst (2015) 107(1):363. doi: 10.1093/jnci/dju363
215. Morrison TE, Mauser A, Wong A, Ting JP, Kenney SC. Inhibition of IFN-gamma signaling by an epstein-barr virus immediate-early protein. Immunity (2001) 15(5):787–99. doi: 10.1016/s1074-7613(01)00226-6
216. Bentz GL, Liu R, Hahn AM, Shackelford J, Pagano JS. Epstein-barr virus BRLF1 inhibits transcription of IRF3 and IRF7 and suppresses induction of interferon-beta. Virology (2010) 402(1):121–8. doi: 10.1016/j.virol.2010.03.014
217. Collins P, Fox CP, George LC, Pearce H, Ryan GB, De Santo C, et al. Characterising EBV-associated lymphoproliferative diseases and the role of myeloid-derived suppressor cells. Blood (2020) 137(2):203–15. doi: 10.1182/blood.2020005611
218. Li ZL, Ye SB, OuYang LY, Zhang H, Chen YS, He J, et al. COX-2 promotes metastasis in nasopharyngeal carcinoma by mediating interactions between cancer cells and myeloid-derived suppressor cells. Oncoimmunology (2015) 4(11):e1044712. doi: 10.1080/2162402X.2015.1044712
219. Cai TT, Ye SB, Liu YN, He J, Chen QY, Mai HQ, et al. LMP1-mediated glycolysis induces myeloid-derived suppressor cell expansion in nasopharyngeal carcinoma. PloS Pathog (2017) 13(7):e1006503. doi: 10.1371/journal.ppat.1006503
220. Liu WL, Lin YH, Xiao H, Xing S, Chen H, Chi PD, et al. Epstein-barr virus infection induces indoleamine 2,3-dioxygenase expression in human monocyte-derived macrophages through p38/mitogen-activated protein kinase and NF-kappaB pathways: impairment in t cell functions. J Virol (2014) 88(12):6660–71. doi: 10.1128/JVI.03678-13
221. Wang Y, Wang J, Zhu F, Wang H, Yi L, Huang K, et al. Elevated circulating myeloid-derived suppressor cells associated with poor prognosis in b-cell non-hodgkin's lymphoma patients. Immun Inflammation Dis (2022) 10(5):e616. doi: 10.1002/iid3.616
222. Marini O, Spina C, Mimiola E, Cassaro A, Malerba G, Todeschini G, et al. Identification of granulocytic myeloid-derived suppressor cells (G-MDSCs) in the peripheral blood of hodgkin and non-hodgkin lymphoma patients. Oncotarget (2016) 7(19):27676–88. doi: 10.18632/oncotarget.8507
223. Prockop S, Doubrovina E, Suser S, Heller G, Barker J, Dahi P, et al. Off-the-shelf EBV-specific t cell immunotherapy for rituximab-refractory EBV-associated lymphoma following transplantation. J Clin Invest (2020) 130(2):733–47. doi: 10.1172/JCI121127
224. McLaughlin LP, Rouce R, Gottschalk S, Torrano V, Carrum G, Wu MF, et al. EBV/LMP-specific t cells maintain remissions of t- and b-cell EBV lymphomas after allogeneic bone marrow transplantation. Blood (2018) 132(22):2351–61. doi: 10.1182/blood-2018-07-863654
225. Hopkins R, Xiang W, Marlier D, Au VB, Ching Q, Wu LX, et al. Monocytic myeloid-derived suppressor cells underpin resistance to adoptive T cell therapy in nasopharyngeal carcinoma. Mol Ther (2021) 29(2):734–43. doi: 10.1016/j.ymthe.2020.09.040
226. De Cicco P, Ercolano G, Ianaro A. The new era of cancer immunotherapy: Targeting myeloid-derived suppressor cells to overcome immune evasion. Front Immunol (2020) 11:1680. doi: 10.3389/fimmu.2020.01680
227. Goff PH, Riolobos L, LaFleur BJ, Spraker MB, Seo YD, Smythe KS, et al. Neoadjuvant therapy induces a potent immune response to sarcoma, dominated by myeloid and b cells. Clin Cancer Res (2022) 28(8):1701–11. doi: 10.1158/1078-0432.CCR-21-4239
228. Fultang L, Panetti S, Ng M, Collins P, Graef S, Rizkalla N, et al. MDSC targeting with gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. EBioMedicine (2019) 47:235–46. doi: 10.1016/j.ebiom.2019.08.025
229. Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC Funct BMC Cancer (2010) 10:464. doi: 10.1186/1471-2407-10-464
230. Ye SY, Li JY, Li TH, Song YX, Sun JX, Chen XW, et al. The efficacy and safety of celecoxib in addition to standard cancer therapy: A systematic review and meta-analysis of randomized controlled trials. Curr Oncol (2022) 29(9):6137–53. doi: 10.3390/curroncol29090482
231. Tobin RP, Jordan KR, Robinson WA, Davis D, Borges VF, Gonzalez R, et al. Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with ipilimumab. Int Immunopharmacol (2018) 63:282–91. doi: 10.1016/j.intimp.2018.08.007
232. Weed DT, Vella JL, Reis IM, de la Fuente AC, Gomez C, Sargi Z, et al. Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res (2015) 21(1):39–48. doi: 10.1158/1078-0432.CCR-14-1711
233. Buque A, Bloy N, Aranda F, Cremer I, Eggermont A, Fridman WH, et al. Trial watch-small molecules targeting the immunological tumor microenvironment for cancer therapy. Oncoimmunology (2016) 5(6):e1149674. doi: 10.1080/2162402X.2016.1149674
234. Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs (2013) 31(3):760–8. doi: 10.1007/s10637-012-9869-8
235. Bitsch R, Kurzay A, Ozbay Kurt F, de la Torre C, Lasser S, Lepper A, et al. STAT3 inhibitor napabucasin abrogates MDSC immunosuppressive capacity and prolongs survival of melanoma-bearing mice. J Immunother Cancer (2022) 10(3):e004384. doi: 10.1136/jitc-2021-004384
236. Jonker DJ, Nott L, Yoshino T, Gill S, Shapiro J, Ohtsu A, et al. Napabucasin versus placebo in refractory advanced colorectal cancer: a randomised phase 3 trial. Lancet Gastroenterol Hepatol (2018) 3(4):263–70. doi: 10.1016/S2468-1253(18)30009-8
237. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature (2016) 539(7629):443–7. doi: 10.1038/nature20554
238. Eriksson E, Wenthe J, Irenaeus S, Loskog A, Ullenhag G. Gemcitabine reduces MDSCs, tregs and TGFbeta-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J Transl Med (2016) 14(1):282. doi: 10.1186/s12967-016-1037-z
239. Wu C, Tan X, Hu X, Zhou M, Yan J, Ding C. Tumor microenvironment following gemcitabine treatment favors differentiation of immunosuppressive Ly6C(high) myeloid cells. J Immunol (2020) 204(1):212–23. doi: 10.4049/jimmunol.1900930
240. Leopold LH, Berger MS, Feingold J. Acute and long-term toxicities associated with gemtuzumab ozogamicin (Mylotarg) therapy of acute myeloid leukemia. Clin Lymphoma (2002) 2(Suppl 1):S29–34. doi: 10.3816/clm.2002.s.006
241. Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell (2015) 162(6):1257–70. doi: 10.1016/j.cell.2015.08.015
242. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase i in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med (2005) 202(7):931–9. doi: 10.1084/jem.20050715
243. Xue WP, Bai SM, Luo M, Bi ZF, Liu YM, Wu SK. Phase i clinical trial of nasopharyngeal radiotherapy and concurrent celecoxib for patients with locoregionally advanced nasopharyngeal carcinoma. Oral Oncol (2011) 47(8):753–7. doi: 10.1016/j.oraloncology.2011.06.002
244. Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res (2007) 67(22):11021–8. doi: 10.1158/0008-5472.CAN-07-2593
245. Seif F, Khoshmirsafa M, Aazami H, Mohsenzadegan M, Sedighi G, Bahar M. The role of JAK-STAT signaling pathway and its regulators in the fate of t helper cells. Cell Commun Signal (2017) 15(1):23. doi: 10.1186/s12964-017-0177-y
Keywords: myeloid derived suppressor cells (MDSC), epstein barr virus (EBV), human papillomavirus - HPV, viral hepatitis, viral oncogenesis, immunotherapy, neutrophils, monocytes
Citation: Glover A, Zhang Z and Shannon-Lowe C (2023) Deciphering the roles of myeloid derived suppressor cells in viral oncogenesis. Front. Immunol. 14:1161848. doi: 10.3389/fimmu.2023.1161848
Received: 08 February 2023; Accepted: 10 March 2023;
Published: 23 March 2023.
Edited by:
Shengjun Wang, Jiangsu University Affiliated People’s Hospital, ChinaReviewed by:
Stefania Canè, University of Verona, ItalyCopyright © 2023 Glover, Zhang and Shannon-Lowe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claire Shannon-Lowe, Yy5zaGFubm9ubG93ZUBiaGFtLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.