 Miranda L. Curtiss1,2*
Miranda L. Curtiss1,2* Alexander F. Rosenberg3,4
Alexander F. Rosenberg3,4 Christopher D. Scharer5
Christopher D. Scharer5 Betty Mousseau3
Betty Mousseau3 Natalia A. Ballesteros Benavides1,3John E. Bradley6
Natalia A. Ballesteros Benavides1,3John E. Bradley6 Beatriz León3
Beatriz León3 Chad Steele7Troy D. Randall6Frances E. Lund3
Chad Steele7Troy D. Randall6Frances E. Lund3- 1Department of Medicine, Division of Pulmonary, Allergy and Critical Care, University of Alabama Birmingham (UAB), Birmingham, AL, United States
- 2Department of Medicine, Section of Allergy and Immunology, Birmingham VA Medical Center, Birmingham, AL, United States
- 3Department of Microbiology, University of Alabama Birmingham (UAB), Birmingham, AL, United States
- 4Informatics Institute, University of Alabama at Birmingham, Birmingham, AL, United States
- 5Department of Microbiology and Immunology, Emory University, Atlanta, GA, United States
- 6Department of Medicine, Division of Rheumatology, University of Alabama Birmingham (UAB), Birmingham, AL, United States
- 7Department of Microbiology and Immunology, Tulane University, New Orleans, LA, United States
Introduction: Data from patient cohorts and mouse models of atopic dermatitis, food allergy and asthma strongly support a role for chitinase-3-like-1 protein (CHI3L1) in allergic disease.
Methods: To address whether Chi3l1 also contributes to TH2 responses following nematode infection, we infected Chi3l1-/- mice with Heligmosomoides polygyrus (Hp) and analyzed T cell responses.
Results: As anticipated, we observed impaired TH2 responses in Hp-infected Chi3l1-/- mice. However, we also found that T cell intrinsic expression of Chi3l1 was required for ICOS upregulation following activation of naïve CD4 T cells and was necessary for the development of the IL-4+ TFH subset, which supports germinal center B cell reactions and IgE responses. We also observed roles for Chi3l1 in TFH, germinal center B cell, and IgE responses to alum-adjuvanted vaccination. While Chi3l1 was critical for IgE humoral responses it was not required for vaccine or infection-induced IgG1 responses.
Discussion: These results suggest that Chi3l1 modulates IgE responses, which are known to be highly dependent on IL-4-producing TFH cells.
Introduction
Allergens are broadly defined as non-pathogenic proteins that induce specific IgE in sensitized individuals (1). However, sensitized individuals contact allergenic proteins in the context of a complex molecular milieu derived from living organisms. For example, feces of house dust mite (HDM) and cockroach contain major allergens complexed to lipid-binding or chitin-binding proteins, and protease activity is a well-known feature of HDM allergens, mold-derived allergens and the industrial sensitizer papain (2). Thus, individuals are typically sensitized to allergens in the context of inflammatory stimuli that provide adjuvant-like effects. Chitins, which are polysaccharides found within the exoskeleton of arthropods, crustaceans and helminths, and the cell wall of bacteria and fungi (3, 4), are thought to promote TH2 allergic responses (5, 6). Chitinases, which degrade chitins, and chitinase-like binding proteins (CLP), which can bind and sequester chitin but cannot degrade chitin (7), are upregulated in the lungs of asthma patients and allergen-exposed mice (7–9). Chitinase deficient mice (Chit1-/- or Chia-/-) manifest enhanced airway inflammation in a HDM allergic airway disease model (10, 11), suggesting that chitinases function to attenuate allergic responses. In contrast, mice deficient in the CLP Chitinase-3-like 1 (Chi3l1-/-) make impaired allergic airway responses following HDM exposure (8, 9) and mice deficient in Chitinase-like protein 3 (Ym1) develop reduced lung eosinophilia and mucus production in an ovalbumin model of allergic airway disease (12). This suggests that CLPs may not prevent allergic responses by sequestering chitin but rather function to facilitate allergic responses.
CHI3L1 has been studied extensively in the setting of human allergic disease. Levels of YKL-40 (the protein product of the human CHI3L1 gene) and expression of CHI3L1 are increased in the lungs and serum of some asthma patient cohorts (13, 14). CHI3L1 SNPs are reported to confer risk of asthma development and airway remodeling (14–17) as well as increased serum IgE and atopy (18) in patient cohorts. Moreover, increased expression of YKL-40 and CHI3L1 have been linked to pathogenesis of allergic rhinitis (19, 20), atopic dermatitis (21–23) and food allergy (24). In agreement with human patient studies, experiments using Chi3l1-/- mice reveal that Chi3l1 regulates type 2 cytokines and IgE levels in mouse models of asthma, atopic dermatitis and food allergy (8, 23–26). Thus, both mouse and human data support a role for YKL-40/Chi3l1 in promoting atopy and allergic disease.
Parasitic infections are recognized as the likely impetus for the evolution of TH2 immunity in mammals, and studies of parasitic infections demonstrate that mediators of protective immunity to parasitic infections often contribute to pathologic responses to allergens (27–30). Chi3l1-/- mice, which make attenuated allergic responses (8, 23–26), did not manifest impaired clearance of the nematode Nippostrongylus brasiliensis (Nb) during early neutrophil-mediated stages of infection, in spite of reduced lung IL-17A levels (31). Since the Nb study did not measure TH2 or IgE responses following Nb infection, we used the helminth Heligmosomoides polygyrus bakerii (Hp) to address whether Chi3l1 is required for type 2 immunity in the setting of a strong TH2-driven enteric helminth infection. Hp primes a TH2 response (29, 32) and elicits IL-4 producing TFH cells (33–36) that initiate polyclonal IgE production by B cells (35, 37). Here, we show that, consistent with the prior allergy studies, Chi3l1 regulates the IL-4+IL-13+ TH2 response to Hp infection. In addition, we observed that Chi3l1 controls the size of the TFH compartment following both Hp infection and protein immunization. We demonstrate that Chi3l1-/- TFH cells express significantly lower levels of Inducible T cell costimulator (ICOS) – a key regulator of TFH development and maintenance in the B cell follicle (38, 39). RNA-seq analysis of the Chi3l1-/- TFH cells revealed that these cells express the canonical TFH transcriptional program but failed to acquire the normal transcriptional signature of IL-4 producing TFH cells (40). These deficits were associated with decreased IL-4 production by the remaining Chi3l1-/- TFH cells, decreased germinal center B cell responses and significantly decreased IgE+ ASCs and serum IgE following Hp infection or alum-adjuvanted vaccination. Thus, Chi3l1 plays a nonredundant, unexpected and critical role in promoting IL-4+ TFH cells that are known to amplify IgE antibody responses to both pathogens and allergens.
Methods
Mice
Animals were bred and maintained in the UAB animal facilities. All procedures were approved by the UAB IACUC and were conducted in accordance with the principles outlined by the National Research Council. BALB/cByJ mice (WT) and BALB/c CD45.1+ congenics (CByJ.SJL(B6)-Ptprca/J) were purchased from The Jackson Laboratory. BALB/c BRP-39 (Chi3l1-/-) mice (8) were provided by Dr. Allison Humbles (MedImmune). Bone marrow (BM) chimeras were generated by irradiating recipients with 850 Rads from a high-energy X-ray source (split dose 5 hours apart), and then reconstituting the recipients with 5x106 total BM cells delivered i.v. To generate 50:50 chimeras we reconstituted irradiated recipients (BALB/cByJ) with 2.5x106 BALB/c CD45.1+ (CByJ.SJL(B6)-Ptprca/J) + 2.5x106 Chi3l1-/- BM cells. Chimeras were used in experiments 8-12 weeks post-reconstitution. Both male and female mice were used and animals were matched for age and sex within an experiment. No gender-specific differences were observed.
Heligmosomoides polygyrus stock
H polygyrus (Hp) stocks were maintained as described previously (33, 34). Mice were gavaged with 200 Hp L3 larvae.
Flow cytometry
Mice were sacrificed at 8, 14 or 25 days post-infection (p.i.) for analysis of mesenteric lymph node (msLN) cells. Single cell suspensions from msLN, spleens or bone marrow (BM) were preincubated with FcR blocking antibody (2.4G2) after lysing red blood cells of spleens and BM with ACK lysis buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.1 mM EDTA), then stained with cocktails of labeled antibodies. Antibody clone names and vendor include the following. Antibodies from BD: CD3 (17A2), CD4 (GK1.5), CD11b (M1/70), CD25 (PC61 BD), CD21 (7G6), CD43 (S7), CD44 (IM7), CD45.1 (A20), CD45.2 (104), B220 (RA3-6B2), CD62L (MEL-14), CD138 (281-2). CXCR5 (2G8), Bcl-6 (K112-91), BP1 (63C), Ly-6G (1A8) and IgE (R35-72). Antibodies from eBioscience: CD19 (1D3), CD23 (MCD2305), CD24 (M1/69), CD38 (90), CD93 (AA4.1), CD279/PD-1 (J43), CD278/ICOS (7E.17G9 and C398.4A), Foxp3 (FJK-16s, IgD (11-26), IgM (II/41) and B220 (RA3-6B2). Antibodies from BioLegend: CD4 (GK1.5), CD49b (DX5), CD150/SLAMF1 (TC15-12F12.2), and Ly-76 (TER-119). Other reagents included: AF488-labeled PNA (Life Technologies) or PNA (Sigma) labeled with Pacific Blue (Invitrogen), 7AAD (Invitrogen™), and LIVE/DEAD® aqua or red (Life Technologies). Intracellular Bcl-6 and Foxp3 were detected using eBioscience™ Foxp3/Transcription Factor Staining Buffer set (Invitrogen™). To detect intracellular cytokines, cells were restimulated with 2.5 μg/mL plate-bound anti-CD3 (145-2C11, BioXcell) or 5 ng/mL PMA (Sigma) with 1.25 μM calcimycin (CalBiochem) plus Brefeldin A (BFA, 12.5 μg/mL, Sigma) for 4 hours. Cells were washed, incubated with FcBlock, surface stained and washed. Cells were fixed in formalin, permeabilized with 0.1% NP-40, washed, incubated with anti-cytokine antibodies (IL-4 (11B11, BD and Invitrogen), IL-13 (eBio13A, eBioscience), IFNγ (XMG1.2 eBioscience) and IL-17A (TC11-18H10, BD Pharmingen) in permeabilization buffer and washed. All incubations prior to fixation were performed with BFA. All flow analysis was performed using the BD Canto.
In vitro activation of CD4 T cells
CD4+ T cells were purified from spleens of uninfected BALB/c CD45.1+ (CByJ.SJL(B6)-Ptprca/J), BALB/c and Chi3l1-/- mice using CD4 MACS L3T4 beads (Miltenyi Biotech) and stimulated for 48 hours as previously described (41) with 2.5 μg/mL plate-bound anti-CD3 (145-2C11, BioXcell) and 2.5μg/ml plate-bound anti-CD28 (37.51, eBioscience). To mimic early TFH development cells were cultured in the presence of 10 μg/ml rIL-6 (R&D) and neutralizing antibodies to IL-2 (JES6-1A12, BioXcell, 5 μg/ml and S4B6-1 BioXcell, 5 μg/mL). For CD4 T cell co-cultures, BALB/c or Chi3l-/- CD45.2+ T cells were mixed at a 1:1 ratio with CD45.1+ T cells and then stimulated for 48 hours with CD3 and CD28 antibodies.
Cell sorting
14 days after oral gavage with 200 L3 Hp (D14), infected BALB/c ByJ and Chi3l1-/- mice were sacrificed and msLN were collected and pooled. Single cell suspensions of msLN cells were incubated with FcBlock (BD), enriched with CD4 microbeads (Miltenyi), and stained with fluorochrome-conjugated antibodies. CD4+CXCR5+PD-1hiLinneg cells were sorted (BD Aria, UAB Flow Cytometry Core), pelleted, lysed in TRIzol (ThermoFisher), and stored at -80°C until used.
Production of recombinant influenza NS1 antigen and NS1 tetramers
The influenza NS1 gene, modified to contain a 3’ in frame addition of the BirA enzymatic biotinylation site and the 6X-His purification tag (GeneArt), was mutated at R38A and K41A (to prevent aggregation of NS1 at high concentrations (42)), then cloned into the pTRC–His2c expression vector (Invitrogen) and expressed in the BirA-enzyme containing E. coli strain CVB101 (Avidity). Biotinylated recombinant NS1 was purified by FPLC and then tetramerized to fluorochrome-conjugated streptavidin (Prozyme). To detect NS1-specific B cells, PE-labeled NS1 tetramers (1:100) were incubated with cells for 30 min at 4°C.
NP(15)-OVA or NS1 immunization
50 μg biotinylated NS1 protein or 50 μg (4-hydroxy-3-nitrophenyl)-acetyl(15)-OVA (NP-OVA) was diluted to 1 mg/mL in PBS and adsorbed to 100 μg alhydrogel alum (InVivoGen) in a total of 200 μL per mouse for 30 min at room temperature, then injected i.p. on day 1 and analyzed on day 12 (D12) (splenic T and B cell responses) or day 14 (D14) (serum antibody).
ELISAs
IgG1 detection
Serum from uninfected mice was analyzed for total IgG1 using a mouse clonotyping kit and standards (Southern Biotech) according to manufacturer’s recommendations. Hp-specific IgG1 was detected as previously described (43) using plates coated with Hp extract and detected using rat anti-mouse IgG1-HRP antibody (Southern Biotech). NP-OVA specific IgG1 was detected as previously described (44) using plates coated with NP(5)-BSA and detected using goat anti-mouse IgG1-HRP (Southern Biotech).
IgE detection
For analysis of total IgE in serum from uninfected, D21 Hp-infected mice, or NS1 immunized mice, ELISAs were performed using paired rat anti-mouse IgE antibodies (capture antibody: 1 μg/mL purified R35-72, detect antibody biotinylated R35-118, BD) and streptavidin-HRP (BD). Purified mouse IgE K was used as a standard (C38-2, BD). To detect NP-OVA specific IgE, plates were coated with 1 μg/mL purified anti-IgE (R35-72, BD). Samples were applied by 2-fold dilutions to the coated plates. Specific IgE was detected by biotinylated NP(5)-BSA followed by streptavidin-HRP.
RNA-seq analysis
RNA was isolated (RNeasy micro column (Qiagen)) from TFH cells sorted into TRIzol as previously described (45) and then enriched with Oligo(dT) beads. Sequencing libraries (NEBNext Ultra II RNA Library Prep Kit for Illumina (NEB, Ipswitch, MA, USA)) were prepared by GeneWiz LLC (South Plainfield, NJ, USA) and sequenced with a 2x150bp Paired End configuration on an Illumina HiSeq 4000. Image analysis and base calling were conducted using Hiseq Control Software (HCS). Raw sequence data (.bcl files) generated from Illumina HiSeq was converted into fastq files and de-multiplexed using Illumina’s bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification. Adapter content was removed from fastq files using Skewer 0.2.2 (46) and data aligned with STAR 2.5.3a (47) to the ENSEMBLE BALB/c/j GCA_001632525 reference mouse genome and transcriptome. PCR duplicate reads were flagged using PICARD MarkDupilicates 1.127 (http://broadinstitute.github.io/picard), gene counts for each sample were computed using GenomicRanges 1.34.0 (48), and reads per kilobase per million (RPKM) normalized in R 3.5.2. Genes with at least 3 reads per million (RPM) in all samples of either the WT and/or Chi3l1-/- TFH groups were considered detected. After confirming that the first 8 exons of the Chi3l1 gene, which were deleted in the Chi3l1-/- mouse strain, were not expressed in the Chi3l1-/- TFH samples, we removed the Chi3l1 gene (also called Chil1) from the RNA-seq analysis as exons 8-10 of the Chi3l1 gene, which are directly downstream from the inserted promoter + neomycin cassette (8), were expressed, presumably due to the insertion of the neomycin deletion cassette.
9853 expressed genes (defined as genes with at least 3 reads per million (RPM) in all samples of either the WT and/or Chi3l1-/- TFH) were identified. Differential expression between groups was analyzed using DESeq2 1.26.0 (49) and 1465 genes were identified as significantly different between WT and Chi3l1-/- TFH cells using an FDR cutoff of q<0.05. See Table S1 for gene expression levels and detailed methods. 193 genes with average reads per kilobase of transcript per million (RPKM) > 1 in either group and meeting a criteria of FDR p < 0.05 and threshold of ±0.3785 log2 fold-change (FC) (1.3-fold) were submitted to Ingenuity Pathway Analysis (IPA, QIAGEN Digital Insights), of which 182 were used by IPA to identify significantly enriched pathways. For gene set enrichment analysis (GSEA), expressed genes from WT and Chi31l-/- TFH cells were ranked by multiplying the -log10 of the P-value from DESeq2 (49) by the sign of the fold change and then used as input in the GSEA (50) PreRanked analysis program (http://software.broadinstitute.org/gsea/index.jsp). RNA-seq data sets were deposited in the NCBI Gene Expression Omnibus (GSE203113). RNA-seq processing code is available at https://github.com/cdschar/Curtiss_Tfh_RNAseq/.
Statistical analysis
Statistical details of all experiments including tests used, n, and number of experimental repeats are provided in figure legends. FlowJo (version 9, Tree Star) was used for flow cytometric analyses. Prism Graphpad (version 9) was used for statistical analyses of flow cytometry experiments. Statistical analysis of RNA-seq experiments is summarized within the text of the RNA-seq experimental design in Table S1.
Results
Chi3l1 regulates IL-4 production by restimulated LN CD4+ T effectors from Hp-infected mice
Murine allergic airway disease models revealed that type 2 cytokine responses are blunted in Chi3l1-/- animals (8) and that Chi3l1-/- CD4 T cells primed in vitro with antibodies to CD3 and CD28 in the presence of TH2-polarizing conditions are impaired in IL-4 production (51). To test whether Chi3l1 regulates TH2 responses to helminth infection, we measured mesenteric LN (msLN) CD4 T cell responses in wild-type (WT) BALB/c and Chi3l1-/- BALB/c mice that were orally infected with Hp. Consistent with previous studies (51), uninfected Chi3l1-/- mice did not exhibit changes in the numbers of total msLN cells or CD19 B cells (Figures S1A–C). However, the msLN total CD4 and activated CD44hiCD62Llo CD4 T cell responses were attenuated in Hp-infected Chi3l1-/- mice (Figures S1D–F). To determine whether the remaining Chi3l1-/- CD4 T cells were competent to produce TH2 cytokines, we analyzed intracellular cytokine levels in anti-CD3 restimulated msLN CD44hi CD4 T cells from D8 Hp-infected mice. We observed that the percentage and number of CD44hi Chi3l1-/- CD4 TH2 cells making both IL-4 and IL-13 were significantly decreased in Chi3l1-/- T cells relative to the restimulated WT T cells (Figures S2A–C). This impairment in type 2 cytokine production was not rescued with PMA+calcimycin stimulation (Figures S2D–G) and, consistent with the dominant TH2 response to Hp infection (52), few CD4 T cells from BALB/c or Chi3l1-/- mice produced IFNγ (Figures S2H–J) or IL-17A (Figures S2K–M) following anti-CD3 stimulation. These data therefore suggest that the development or effector potential of parasite-elicited CD4 T cells was compromised in Chi3l1-/- mice.
Chi3l1 regulates TFH responses to Hp infection
Helminth infections induce robust T follicular helper (TFH) responses (33, 53). Since TFH responses have not been analyzed in Chi3l1-/- mice, we enumerated TFH (CXCR5+PD-1hi) cells in the msLN of uninfected and Hp-infected animals. TFH cells (Figure 1A) were detected at low levels in the msLN of uninfected BALB/c mice and increased in both frequency and number (Figures 1B, C) over the first two weeks following Hp infection. Although the kinetics of the TFH responses were similar between the Hp-infected BALB/c and Chi3l1-/- mice, the magnitude of the Chi3l1-/- TFH response was significantly decreased (Figures 1B, C). Moreover, while the TFH master regulator (54) Bcl-6 (Figures 1D, E) and TFH-supporting receptor SLAMF1 (CD150) (55) (Figure 1F) were expressed at similar levels in the msLN BALB/c and Chi3l1-/- TFH cells, expression of ICOS, a key co-stimulatory molecule for TFH cells (39, 54, 56–58), was significantly decreased on the Chi3l1-/- CXCR5+PD-1hi TFH cells throughout infection (Figures 1G, H). To assess whether the reduction in ICOS expression in Chi3l1-/- TFH cells was likely to be cell intrinsic, we activated splenic CD4 T cells from uninfected WT and Chi3l1-/- mice under early TFH differentiation conditions (41) with platebound anti-CD3 + anti-CD28 and IL-6 plus IL-2 blocking antibodies. We observed decreased ICOS upregulation in the in vitro activated Chi3l1-/- CD4 T cells (Figure 1I). Together, these data suggest that Chi3l1 does not control T cell commitment to the TFH lineage but might be important for the expansion, maintenance, or function of this T cell subset.
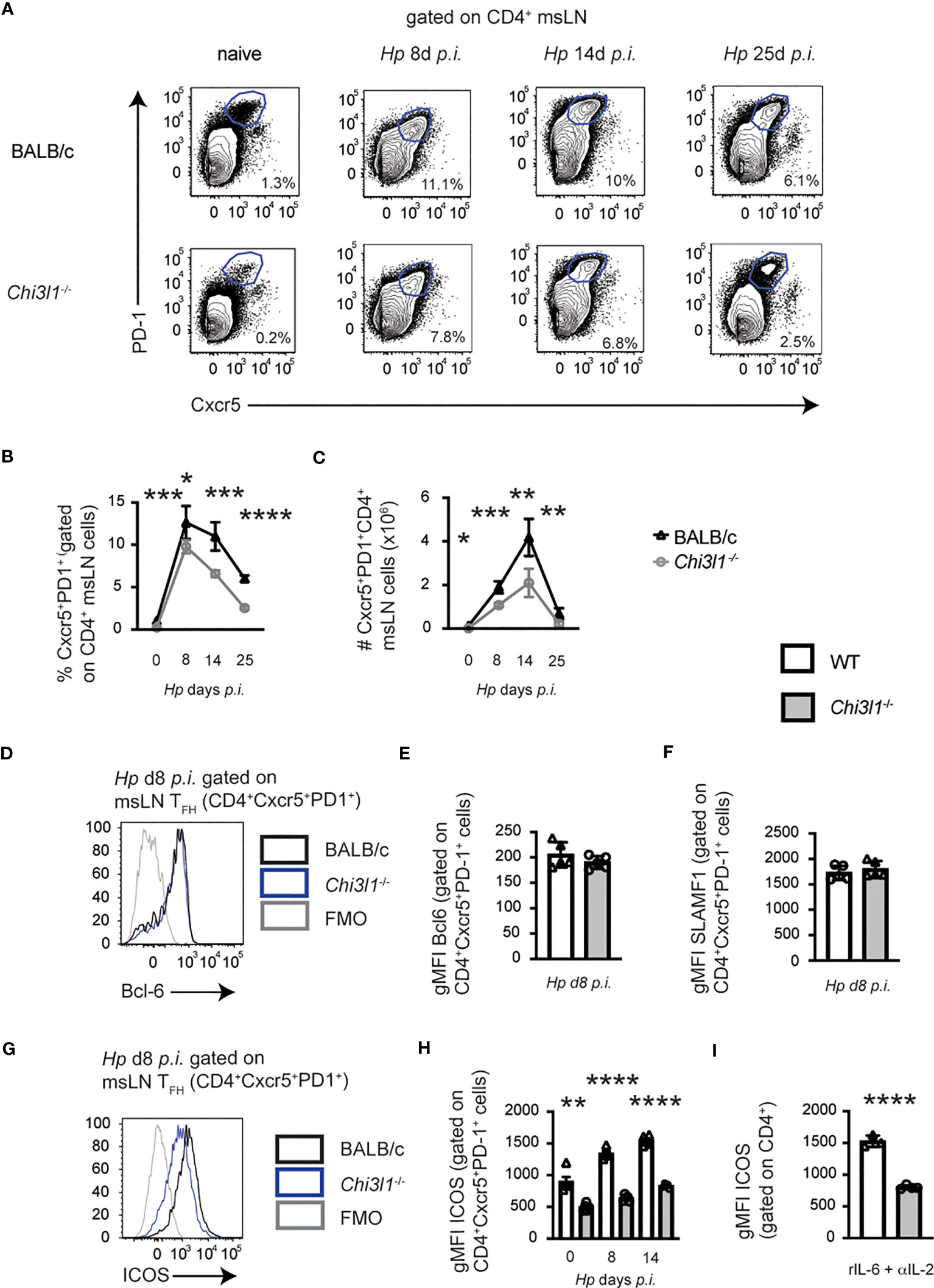
Figure 1 Chi3l1 regulates TFH responses to Hp infection. (A–H) Enumeration and characterization of msLN TFH cells from uninfected (n=5/group) or Hp-infected BALB/c (white bars) and Chi3l1-/- (grey bars) mice (n=5/group). (A–C) TFH response to Hp infection with flow plots (A) showing CXCR5+PD-1+ TFH cells between D0 (uninfected) and D25 post-Hp with frequency (B) and number (C) of TFH cells. Kinetics of the msLN response (enumeration of total msLN cells, CD19 B cells, CD4 T cell and activated CD44hiCD62Llo) shown in Figures S1A–F. Cytokine production by CD44hi CD4 T cells shown in Figure S2. (D–H) TFH phenotype in msLN of uninfected (D0) and Hp-infected mice. Bcl-6 (D, E), SLAM (F) and ICOS (G, H) expression by TFH on D8 (D–G) or between D0-D14 (H). Flow cytometry plots for Bcl-6 (D) and ICOS (G) are shown. Expression levels of Bcl-6 (E), SLAM (F) or ICOS (G) in TFH cells presented as the geometric Mean Fluorescence Intensity (gMFI). (I) gMFI of ICOS expression by purified splenic CD4+ T cells from uninfected BALB/c (white bars) and Chi3l1-/- (grey bars) mice (n=4/group) that were stimulated in vitro for 48 hours in triplicate with anti-CD3 plus anti-CD28 in the presence of rIL-6 plus anti-IL-2. Data representative of 2 (D–F) or ≥ 3 independent experiments (all others). (A–H) Data displayed as the mean ± SD of each group with individual animals depicted as circles or triangles. (I) Data displayed as the mean ± SD of each group with individual animals (assayed in triplicate) depicted as circles or triangles. Statistical significance determined using unpaired 2-tailed student’s t test. *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001.
ICOS expression by Foxp3+CD25+ TREG does not require Chi3l1
Since ICOS levels were decreased in the Chi3l1-/- TFH cells, we asked whether ICOS expression was altered in Foxp3+CD25+CD4+ cells, since ICOS function is critical in both TFH and TREG development during Hp infection (59). We examined mice on D25 post Hp-infection – a timepoint when the CD4 T cell response is contracting and TREG responses are induced to facilitate establishment of a chronic infection (29, 60, 61). The numbers of CD25+Foxp3+ TREG (Figures S1G–H) were similar between the groups. Moreover, we did not observe reduced ICOS expression by Chi3l1-/- CD25+Foxp3+ TREGs (Figures S1I–J). Thus, Chi3l1 modulates ICOS expression by Hp-infection elicited TFH cells but not TREG cells.
Hematopoietic cell expression of Chi3l1 regulates TH2 and TFH responses to Hp
Transgene-directed Chi3l1 expression by epithelial cells is reported to restore lung TH2 cytokine levels in allergen-exposed Chi3l1-/- mice (8). To assess whether the attenuated TH2 and TFH responses to Hp were due to Chi3l1 expression by radiation resistant cells, like epithelial cells, or to Chi3l1 expression by radiation sensitive cells, like bone marrow (BM)-derived immune cells, we analyzed Hp-elicited T cell responses in BALB/c and Chi3l1-/- mice that were lethally irradiated and reconstituted with either BALB/c BM or Chi3l1-/- BM. msLN cells of BM chimeras that were competent to express Chi3l1 in all cell types (WT donor/WT recipient), lacked Chi3l1 in all cell types (KO donor/KO recipient), lacked expression of Chi3l1 specifically in the hematopoietic compartment (KO donor/WT recipient) or lacked expression of Chi3l1 in the radiation resistant compartment (WT donor/KO recipient) were compared 8 days after Hp infection. We found no statistically significant differences in total numbers of msLN cells, B cells, or CD4 T cells in any of the groups (Figures S3A–C). However, and consistent with our analysis of non-chimeric Chi3l1-/- mice, we observed a significant reduction in the numbers of total activated CD62LloCD44hi CD4 cells (Figure S3D) in the KO donor/KO recipient mice compared to the WT donor/WT recipient mice. Similarly, the day 8 Hp-infected KO donor/KO recipient mice had decreased frequencies and numbers of CXCR5+PD-1+ TFH cells (Figures S3E–G), which expressed lower levels of ICOS (Figure S3H). Moreover, the T cells from the KO donor/KO recipients produced significantly less IL-4 and IL-13 following anti-CD3 restimulation when compared to the T cells from the WT donor/WT recipients (Figures S3I–K). When we compared the data from these chimeras to the other two groups of chimeras, we found no differences in any of the T cell responses between the KO donor/WT recipients and the KO donor/KO recipients (Figures S3D–K). These results indicated that Chi3l1 expression by BM-derived cells was necessary for optimal CD4 T cell responses to Hp infection. Consistent with this idea, the CD4 T cell responses were largely, although not completely, rescued (Figures S3D–K) in Chi3l1-/- mice reconstituted with WT BM (WT donor/KO recipient). Thus, while we could not completely rule out a role for Chi3l1 expressing non-hematopoietic cells in regulating CD4 T cell responses to Hp, our data indicated that CD4 T cell responses to Hp minimally require hematopoietic cell expression of Chi3l1.
To further assess the contribution of Chi3l1-expressing hematopoietic cells to Hp-induced CD4 T cell responses, we directly compared Hp-elicited T cell responses in BALB/c and Chi3l1-/- mice that were lethally irradiated and reconstituted with either BALB/c BM or Chi3l1-/- BM (Figure 2A). We observed a significant reduction in the numbers of total activated CD62LloCD44hi CD4 cells (Figure 2B) and CXCR5hiPD-1hi TFH cells (Figures 2C–E) in the Hp-infected BALB/c recipients reconstituted with Chi3l1-/- BM relative to animals reconstituted with BALB/c BM. Expression of ICOS on CXCR5+PD-1hi TFH cells was reduced in mice reconstituted with Chi3l1-/- BM (Figure 2F). We confirmed the effector potential of CD4 T cells from BALB/c mice reconstituted with Chi3l1-/- BM chimeras was impaired, as the frequencies and numbers of IL-4+ single producers (Figures 2G–I) and IL-4+IL-13+ double producers (Figures 2J, K) were decreased following anti-CD3 stimulation. Thus, Chi3l1 expression within BM derived radiation sensitive immune cells regulates ICOS expression by TFH cells and is required for development of TFH responses and TH2 cells with effector potential following Hp infection.
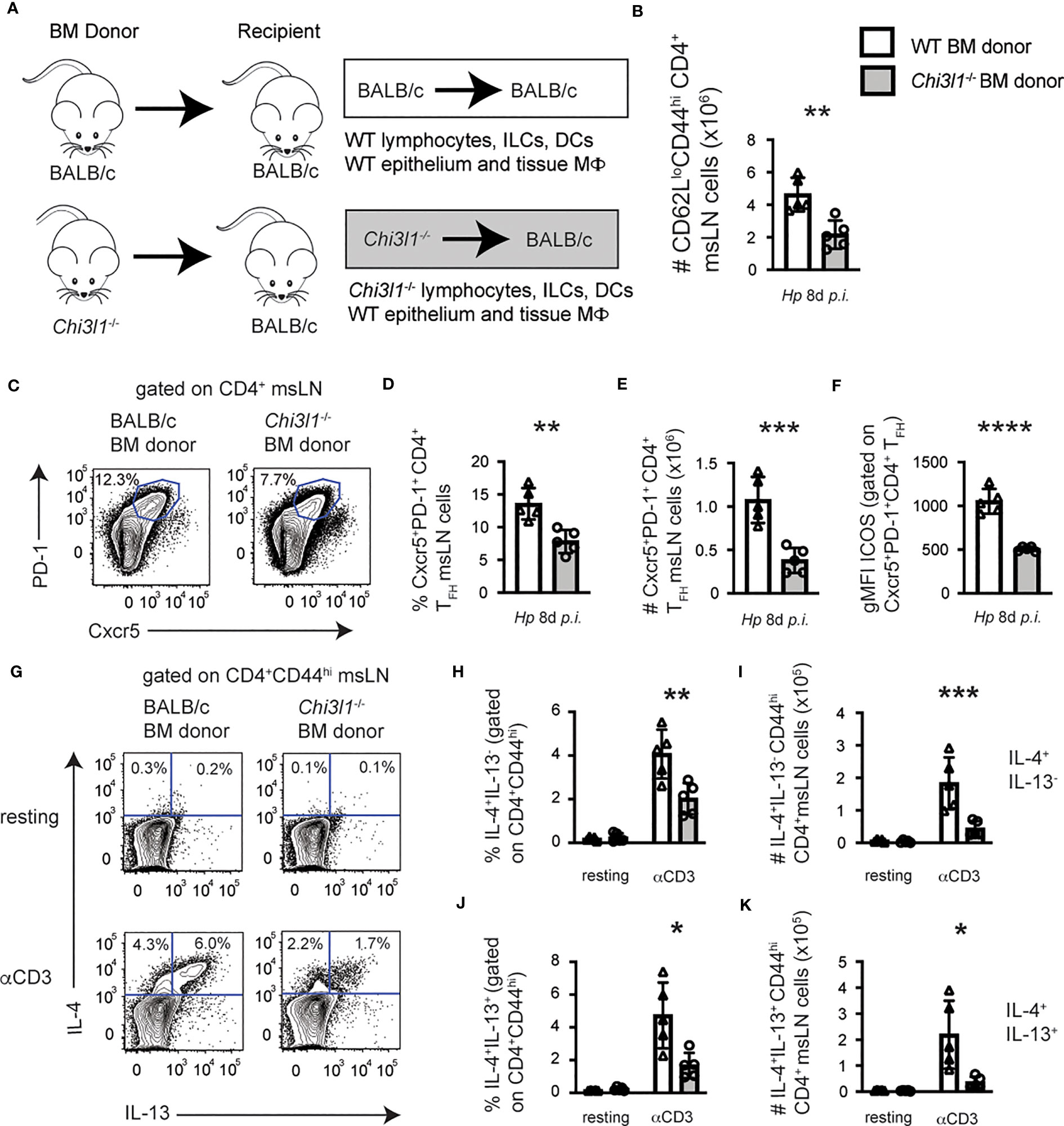
Figure 2 Chi3l1 expressing hematopoietic cells are necessary and sufficient for TFH and TH2 responses to Hp. Enumeration of T cell responses in msLNs of D8 Hp-infected BM chimeric mice (n=5 mice/group) that were generated (A) by reconstituting lethally-irradiated BALB/c recipient mice with BALB/c (white bars) or Chi3l1-/- (grey bars) BM. msLN CD4+ CD62LloCD44hi cells from D8 Hp-infected chimeric mice were analyzed directly ex vivo (C–F) or following restimulation for 4 hours with anti-CD3 in the presence of BFA (G–K). (B–E) Number (B) of activated CD4 T cells in msLN from D8 Hp-infected mice. Representative flow cytometry plot (C) showing CXCR5+PD-1+ TFH cells in D8 msLNs with the percentages (D) and numbers (E) of TFH cells and ICOS expression levels (F) by TFH cells represented as gMFI. (G–K) IL-4 and IL-13 production by resting and anti-CD3 stimulated msLN cells. Representative flow plot (G) showing IL-4 and IL-13 expression by CD44hi CD4 cells from D8 Hp-infected chimeras with the percentage and number of IL-4+IL-13neg (H–I) and IL-4+IL-13+ (J–K) producers. Analysis of TFH responses and CD4 T cell responses in reciprocal BM chimeras is shown in Figure S3. Data representative of ≥ 3 independent experiments displayed as mean ± SD of each group with cells from individual animals depicted as circles or triangles. Unpaired 2-tailed student’s t-test was used to assess statistical significance. *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001.
Cell intrinsic expression of Chi3l1 regulates B cell development but appears not critical for TFH development or expansion
CHI3L1 is a secreted protein (62, 63) and may regulate T cell responses to Hp via autocrine and/or paracrine mechanisms. To address whether T cell-intrinsic expression of Chi3l1 regulates TH2 and TFH responses to Hp, we measured CD4 T cell responses in Hp-infected BM chimeras (Figure 3A) that were reconstituted with a 1:1 mixture of BM derived from CD45.1+ WT BALB/c congenic animals and CD45.2+ Chi3l1-/- mice (Figures 3B, C). Although the percentage (Figures 3D, E) of Chi3l1-/- CD4 msLN cells in naïve chimeras was modestly increased relative to the BALB/c CD4 T cells present in the same animal, the numbers (Figure 3F) of WT and Chi3l1-/- CD4 T cells were similar in uninfected 50:50 chimeras. This was not limited to CD4 T cells in the msLN as the ratios of WT to Chi3l1-/- splenic CD4 T cells, CD8 T cells and myeloid cells (CD19negCD3negDX5neg) cells were similar (Figures S4A–E), even after correcting for BM input (Figure S4F). Moreover, the percentages (Figures 3D, E) and numbers (Figure 3F) of msLN Chi3l1-/- and BALB/c CD4 cells isolated from the same chimeric recipient on day 8 post-Hp infection were not significantly different. Similarly, we observed no significant differences in either the frequency (Figures 3G, H) or number (Figure 3I) of TFH cells of each genotype present in same host on D0 or 8 days post-infection.
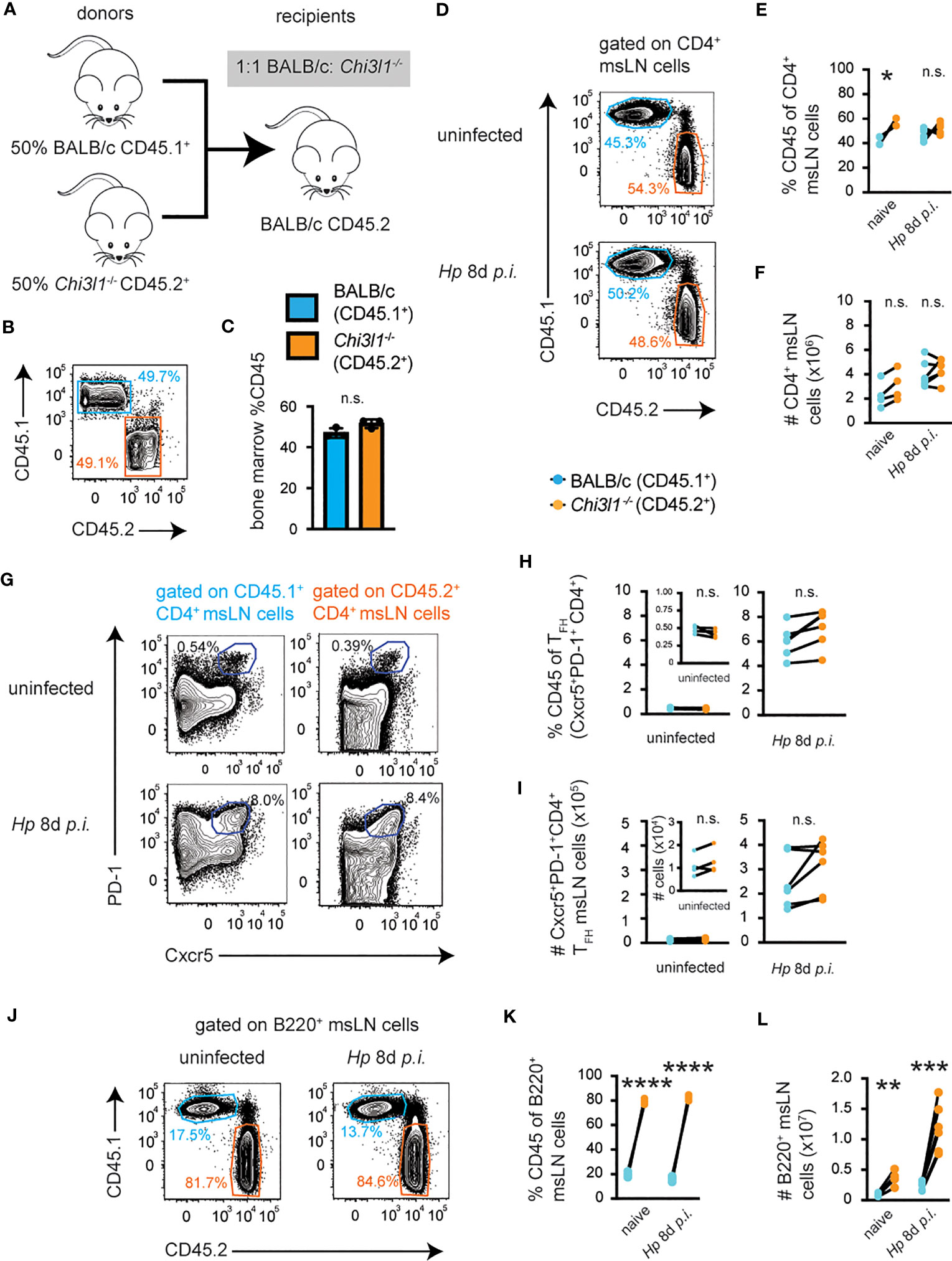
Figure 3 Chi3l1 regulates TFH development and expansion via a cell-extrinsic mechanism. Enumeration of T and B cell responses in mLNs from Hp-infected mixed 50:50 BM chimeras that were generated (A) by reconstituting lethally-irradiated BALB/c recipient mice with a 1:1 ratio (B, C) of BALB/c (CD45.1+, teal bars) and Chi3l1-/- (CD45.2+, orange bars) BM. Chimeras were analyzed before infection (n=4/group), on D8 (n=6/group) or on D14 (n=5/group) post-Hp infection. Cells were analyzed directly ex vivo. (D–I) Flow plots (D) showing BALB/c CD45.1+ (teal) and Chi3l1-/- CD45.2+ (orange) donor-derived msLN CD4 T cell populations before and on D8 post-infection. Frequency (E) and number (F) of CD4 cells of each genotype derived from each animal at each timepoint. Flow plots (G) showing both donor-derived TFH populations in uninfected and D8 post-infection with frequency (H) and number (I) of TFH cells of each genotype derived from each animal at each timepoint. (J–L) Flow plots (J) showing BALB/c CD45.1+ (teal) and Chi3l1-/- CD45.2+ (orange) donor-derived msLN B220+ B cell populations before and on D8 post-infection. Frequency (K) and number (L) of B220+ B cells of each genotype derived from each animal at each timepoint. Analysis of B cell development in BM and spleen in 50:50 chimeras is shown in Figure S4. Data representative of ≥3 independent experiments. Data displayed as mean ± SD in triplicate shown as bars (C) or as cell populations derived from each genotype from individual animals shown as paired lines (E–L). Statistical analysis was performed with unpaired 2-tailed student’s t-test (C) or paired 2-tailed student’s t-test (all others). *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001. *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001.
While T cell intrinsic expression of Chi3l1 did not appear to be required for the development of the TFH compartment, we did observe that the percentage (Figures 3J, K) and number (Figure 3L) of Chi3l1-/- B220+ msLN cells was significantly increased relative to BALB/c B220+ B cells in both uninfected and Hp-infected 50:50 chimeric mice. The same phenotype was also observed in the spleen (Figures S4C–E), even after normalizing for the input BM cells in the 50:50 chimeras (Figure S4F). To see if this effect was limited to mature B cells in the periphery, we examined B cell development in the BM of the 50:50 chimeras. We found that the BM of these chimeras was heavily skewed toward the Chi3l1-/- B cell progenitors at multiple stages of B cell development, including the pre-B cell stage (Fraction C-C’) in the BM (Figures S4G–K), and between Fraction E of the BM and development of mature follicular and marginal zone B cells in the spleen (Figures S4L–N). Since the number of mature B cells was not altered in non-chimeric Chi3l1-/- mice (Figure S1C), or in reciprocal BM chimeras (Figure S3B) we concluded that a B cell intrinsic role for Chi3l1 during B cell development was revealed when WT and Chi3l1-/- B lineage precursors were forced to compete in the 50:50 BM chimeras.
CD4 T cell-intrinsic Chi3l1 is required for functional T cell responses to Hp
Although T cell intrinsic expression of Chi3l1 did not appear to be required for development or expansion of the CD4 T cell compartments following Hp infection, we considered the possibility that T cell intrinsic expression of Chi3l1 might instead be required for the functional or effector attributes of the T cells. To test this hypothesis, we analyzed cytokine responses by the CD4 cells from the Hp-infected 50:50 chimeras. We observed that the proportion of Chi3l1-/- CD44hi CD4 cells that were competent to produce IL-4 following restimulation was significantly decreased relative to the frequency of IL-4 producing BALB/c CD4 T cells derived from the same animals (Figures 4A, B). Moreover, consistent with a prior publication reporting a TH1 differentiation bias in Chi3l1-/- mice (51), we observed that the proportion of CD44hi Chi3l1-/- CD4 cells that produced IFNγ following anti-CD3 restimulation was significantly increased relative to the frequency of IFNγ-producing BALB/c CD4 T cells derived from the same animals (Figures 4C, D). Next, we assessed ICOS expression by the TFH cells present in the Hp-infected 50:50 chimeras. We observed significantly decreased ICOS expression (Figures 4E, F) in the Chi3l1-/- TFH cells compared to the BALB/c TFH cells from the same infected animal. This was not a transient defect as it was seen even at the peak of the TFH response on D14 post-infection (Figure 4G). These results therefore suggest that T cell intrinsic expression of Chi3l1 was important for the functional potential of the CD4 cells from Hp-infected mice.
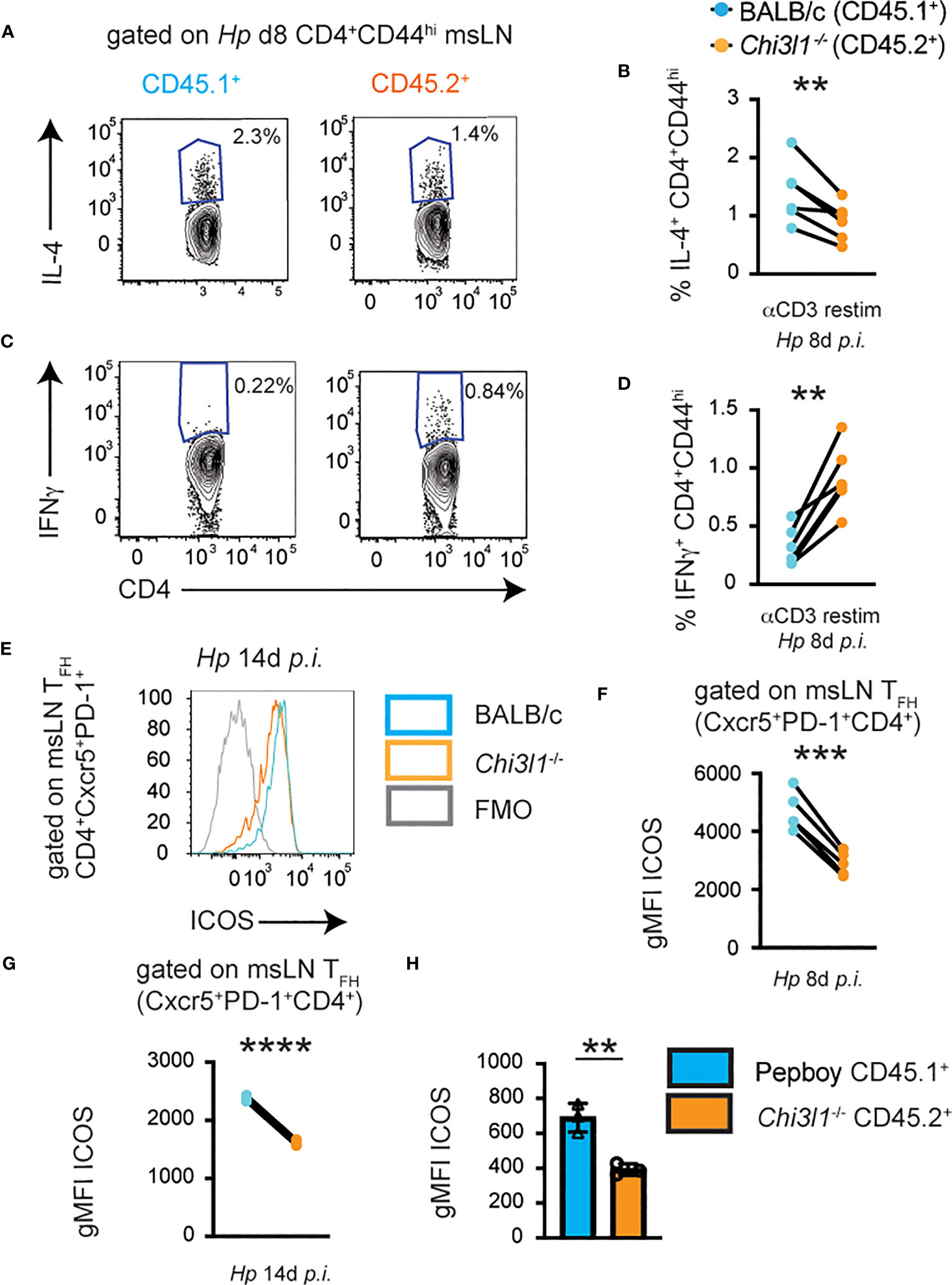
Figure 4 Chi3l1 regulates cytokine production and ICOS expression in T cells by a cell intrinsic mechanism. (A–G) CD4 T responses in mLNs from Hp-infected mixed 50:50 BM chimeras that were generated as described in Figure 3 with 50% BALB/c (CD45.1+) BM plus 50% Chi3l1-/- (CD45.2+) BM. (A–D) Cytokine production by anti-CD3 restimulated donor D8 BALB/c CD45.1+ (teal) and Chi3l1-/- CD45.2+ (orange) CD44hi CD4 cells. Flow plots showing IL-4 (A) and IFNγ (C) production with the percentage of IL-4 (B) and IFNγ (D) producers of each genotype derived from each D8 Hp-infected animal. (E–G) ICOS expression by donor BALB/c CD45.1+ (teal) and Chi3l1-/- CD45.2+ (orange) CXCR5+PD-1+ TFH cells on D8 and D14 post-Hp infection. Flow plots (E) showing ICOS expression by D14 TFH cells. gMFI of ICOS expression levels on D8 (F) and D14 (G) post-infection by BALB/c or Chi3l1-/- donor-derived TFH cells from the same animal. (H) gMFI of ICOS expression by purified splenic CD4+ T cells from uninfected BALB/c CD45.1+ mice (teal bars) that were co-cultured in triplicate at a 1:1 ratio with CD4 T cells from Chi3l1-/- CD45.2+ (orange bars) mice (n=3/group) and stimulated for 48 hours with anti-CD3 plus anti-CD28. Data representative of ≥3 (A-G) or 2 (H) independent experiments. Data in (A–G) represent cell populations derived from each genotype from individual animals shown as paired lines. Data in (H) displayed as the mean ± SD of each group with individual animals (assayed in triplicate) depicted as circles or triangles. Statistical analysis was performed with paired 2-tailed student’s t-test (A–G) or unpaired 2-tailed student’s t-test (H). **p≤0.01, ***p≤0.001, ****p≤0.0001.
One potential caveat to the 50:50 chimera experiments was that the CD45.2+ hematopoietic compartment would include Chi3l1-/- cells as well as any radiation resistant hematopoietic cells from the CD45.2+ WT host. The presence of these radiation resistant WT cells could potentially mask Chi3l1-/- cell intrinsic deficits. Although this is not a major concern for B cells, which are radiation sensitive (64), some T cell subsets are reported to be more resistant to radiation than others (reviewed in (65)). Given that we observed no differences in the frequencies or numbers of WT and Chi3l1-/- TFH cells in the Hp-infected 50:50 chimeras (Figures 3D, E), yet we observed a reduction in ICOS levels in CD45.2+ TFH cells from these same animals (Figures 4E–G), we suspected that the defect in ICOS upregulation was likely a Chi3l1-dependent T cell-intrinsic defect. Our in vitro data (Figure 1I) examining ICOS upregulation in activated Chi3l1-/- T cells also suggested a T cell intrinsic role for Chi3l1 in regulating ICOS expression. To further confirm this conclusion, we co-cultured purified splenic CD4+ T cells from uninfected BALB/c CD45.1+ mice at a 1:1 ratio with either purified BALB/c CD45.2+ splenic CD4+ T cells or with Chi3l1-/- CD45.2+ CD4+ T cells in the presence of platebound anti-CD3 + anti-CD28. Again, we observed that ICOS expression by Chi3l1-/- CD4 T cells was significantly decreased and was not rescued in trans by the presence of the WT CD4+ T cells (Figure 4H). Taken together, these data indicate that T cell intrinsic expression of Chi3l1 does play a role in ICOS upregulation early after TCR activation, in ICOS expression by Hp-induced TFH cells and in IL-4 production by restimulated Hp-elicited CD4 T cells.
Chi3l1 regulates B cell responses to Hp infection and immunization
Our data showed that Chi3L1 regulates expression of ICOS by Hp infection elicited TFH cells and we know that ICOS is a key co-stimulatory molecule that modulates TFH-B cell interactions, particularly during the germinal center B cell (GCB) response (38, 39). We therefore hypothesized that Chi3l1-/- mice would mount impaired B cell responses to Hp. To test this, we analyzed B cell subsets in BALB/c and Chi3l1-/- msLNs before and on D14 after Hp-infection mice (Figure 5A; Figures S5A–C). The numbers of total msLN cells (Figure S5D) and B220+CD138- B cells (Figure S5E) were decreased in the D14 Hp-infected Chi3l1-/- mice relative to WT mice. This reduction was not due to changes in the number of naïve msLN B cells (Figure S5F). Instead, antigen-experienced isotype-switched B cells (Figure 5B), isotype-switched GCB cells (Figure 5C), and antibody secreting cells (ASCs) (Figure 5D) were decreased in the D14 Hp-infected Chi3l1-/- msLNs. Interestingly, and unlike the phenotype of GCB cells from spleen (Figure S7C), msLN GCB cells in infected mice included both a CD38loPNA+ and CD38intPNA+ population (Figure S5G). Both msLN GCB populations expressed similarly high levels of PNA (Figure S5G) and both GCB populations were present in Hp-infected WT and Chi3l1-/- msLNs (Figure S5C).
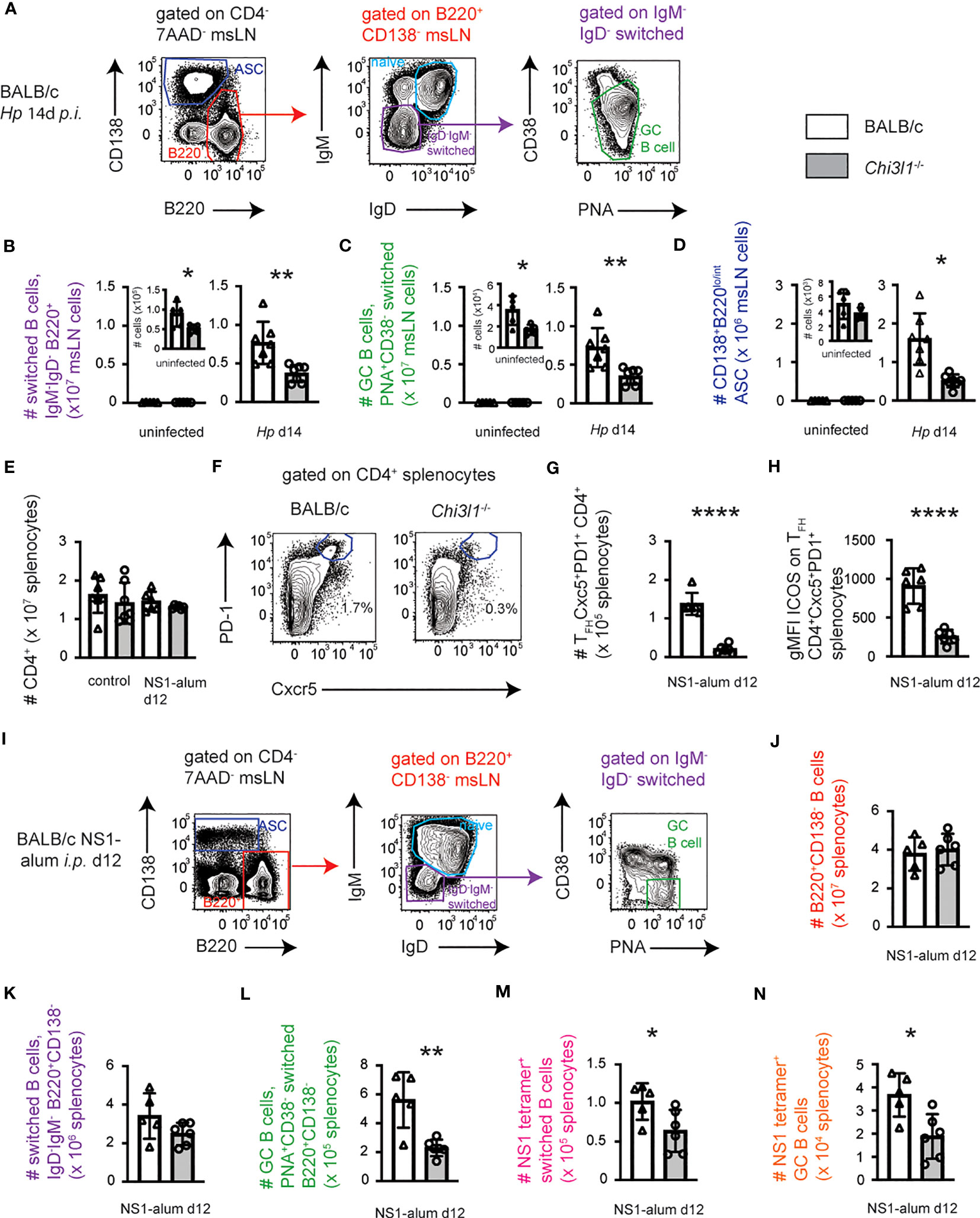
Figure 5 Chi3l1 regulates B cell responses to Hp infection and alum-adjuvanted protein immunization. Enumeration of B cell responses in BALB/c (white bars) and Chi3l1-/- (grey bars) mice following infection with Hp (A–D) or vaccination with NS1 protein in alum (E–N). (A–D) Characterization of B cell responses in msLNs of uninfected (n=5/group) and D14 Hp-infected (n=7/group) BALB/c and Chi3l1-/- mice. Flow plots (A) showing the gating strategy to identify B220loCD138hi ASCs (blue gate), total B cells (red gate), IgDnegIgMneg isotype-switched B cells (purple gate), naïve B cells (cerulean gate) and PNAhiCD38lo GCB cells (green gate). Numbers of isotype-switched B cells (B), GCB cells (C), and ASCS (D) in msLNs of uninfected and D14 Hp-infected mice are shown. Additional analyses of msLN B cell and ASC responses in Hp-infected WT and Chi3l1-/- mice (Figure S5) and 50:50 BM chimeras (Figure S6) are provided. (E–N) Analysis of splenic CD4 T cell (E–H) and B cell (F–N) responses in BALB/c and Chi3l1-/- mice (n=5-6/group) at baseline and on D12 post-immunization with influenza NS1 protein adsorbed to alum. Enumeration (E) of splenic CD4 T cells with representative flow plots showing the gating strategy (F) to identify and quantitate (G) splenic CXCR5+PD-1+ TFH cells. ICOS expression (H) by TFH cells reported as gMFI. Representative gating of splenic B cell subsets (I) and enumeration of total splenic B cells (J) from immunized mice. Enumeration of total splenic IgD-IgM- isotype-switched B cells (K) and PNA+CD38- GCB cells (L). Numbers of NS1+ isotype-switched cells (M) and NS1+ GCB cells (N). Representative flow plots with gating strategy to identify B cell subsets and NS1-specific B cells in spleens of control and NS1-immunized WT and Chi3l1-/- mice provided in Figure S7. Data is representative of 3 independent experiments. Data displayed as the mean ± SD of each group with individual animals depicted as circles or triangles. Statistical analysis was performed with unpaired 2-tailed student’s t-test. *p≤0.05, **p≤0.01, ****p≤0.0001.
Since our data using 50:50 BM chimeras revealed that B cell intrinsic expression of Chi3l1 can globally alter BM B cell development (Figure S4) in the setting of competition, we addressed whether B cell intrinsic expression of Chi3l1 might be necessary for optimal GCB and ASC responses to Hp by analyzing D14-infected 50:50 BM chimera msLNs. In contrast to our results showing attenuated GCB and ASC responses in Hp-infected Chi3l1-/- mice (Figures 5C, D), we observed that Chi3l1-/- B cells in the 50:50 chimeras were competent to generate both ASC (Figures S6A–C) and GCB (Figures S6D–K) responses to Hp. This was not simply due to the presence of increased numbers of Chi3l1-/- B cells in the Hp-infected 50:50 chimeras (Figures S6D–E) as the frequencies of GCB cells present within the WT and Chi3l1-/- B cell compartments of Hp-infected 50:50 chimeras were elevated within the Chi3l1-/- B cell compartment (Figures S6F–H; reciprocal gating approach shown in Figures S6I–K). This result suggested that the reduction in the B cell responses to Hp in the Chi3l1-/- global knockout mice was not due to an intrinsic inability of the Chi3l1-/- B cells to enter the GCB pathway or differentiate into ASCs. Rather the defect in the Chi3l1-/- Hp-elicited B cell response in animals with a global deficiency in Chi3l1 was likely due to the loss of Chi3l1 by other cell types.
Next, we assessed whether the defective B cell response observed in the Hp-infected Chi3l1-/- mice was limited to the setting of helminth infection. We therefore immunized mice with recombinant protein (influenza NS1) that was adsorbed to the TH2-biasing adjuvant alum and used flow cytometry to measure splenic polyclonal CD4 T cell and B cell responses as well as NS1-specific B cell responses on D12 post-immunization (B cell subset flow gating strategies shown in Figures S7A–C). We first confirmed that numbers of splenocytes (Figure S7D) and splenic CD4 T cells (Figure 5E) did not differ in unvaccinated wildtype and Chi3l1-/- control mice or at D12 post-vaccination with NS1. However, the number of splenic TFH cells in immunized Chi3l1-/- mice was significantly reduced (Figures 5F, G) and the remaining Ch3l1-/- TFH cells expressed decreased levels of ICOS (Figure 5H). The number of splenic ASC (Figure S7E), percentage and number of isotype-switched B cells (Figures S7F–G), and number of GCB cells (Figures S7H–I) did not differ between WT and Chi3l1-/- unvaccinated control mice. Likewise, the numbers of total B cells (Figures 5I, J), ASC (Figure S7E) and switched B cells (Figure 5K) did not differ in vaccinated mice. In contrast, the percentages and numbers of splenic GCB cells were reduced in vaccinated Chi3l1-/- mice (Figure 5L; Figures S7H–I). Finally, using fluorochrome-labeled NS1 protein tetramers, we identified NS1-specific switched B cells (Figure S7J) and GCB cells (Figure S7L) in control and vaccinated WT and Chi3l1-/- mice. Although the frequency of NS1-specific isotype-switched (Figure S7K) and GCB cells (Figure S7L) was unchanged between the vaccinated WT and Chi3l1-/- mice, the numbers of NS1-specific isotype-switched B cells (Figure 5M) and NS1-specific GCB cells (Figure 5N) were significantly decreased in vaccinated Chi3l1-/- mice. Thus, Chi3l1 regulates the magnitude of TFH and antigen-specific B cell responses to both infection and immunization.
Chi3l1 regulates IgE responses
Given our data and published reports showing that Hp-induced IgE responses depend on TFH cells (35), we next analyzed IgE responses in the Hp-infected Chi3l1-/- mice. We observed a significant reduction in the frequency and number of IgE+ ASCs in the msLNs from Hp-infected Chi3l1-/- mice (Figures 6A–C) compared to the WT animals. Moreover, the impaired IgE+ ASC response was accompanied by a significant reduction in total IgE levels in the serum of D21 Hp-infected Chi3l1-/- mice (Figure 6D). In contrast, Hp-specific IgG1 serum antibody titers were equivalent between Chi3l1-/- and WT mice (Figure 6E). To determine whether the loss of the IgE+ ASCs in the Chi3l1-/- Hp-infected mice was due to a B cell intrinsic defect, we enumerated IgE-expressing ASCs within the msLN of D14 Hp-infected 50:50 chimeras. Consistent with the increase in Chi3l1-/- mature B cells in the 50:50 chimeras, we found significantly more Chi3l1-/- ASCs in the msLN of Hp-infected 50:50 chimeras (Figure S6L). However, the frequency of IgE ASCs within the Chi3l1-/- ASC compartment was equivalent to the frequency of IgE+ ASCs within the WT ASC compartment (Figures S6M–N). These data suggested that Chi3l1 expression by non-B cells likely regulated IgE+ ASC responses to Hp.
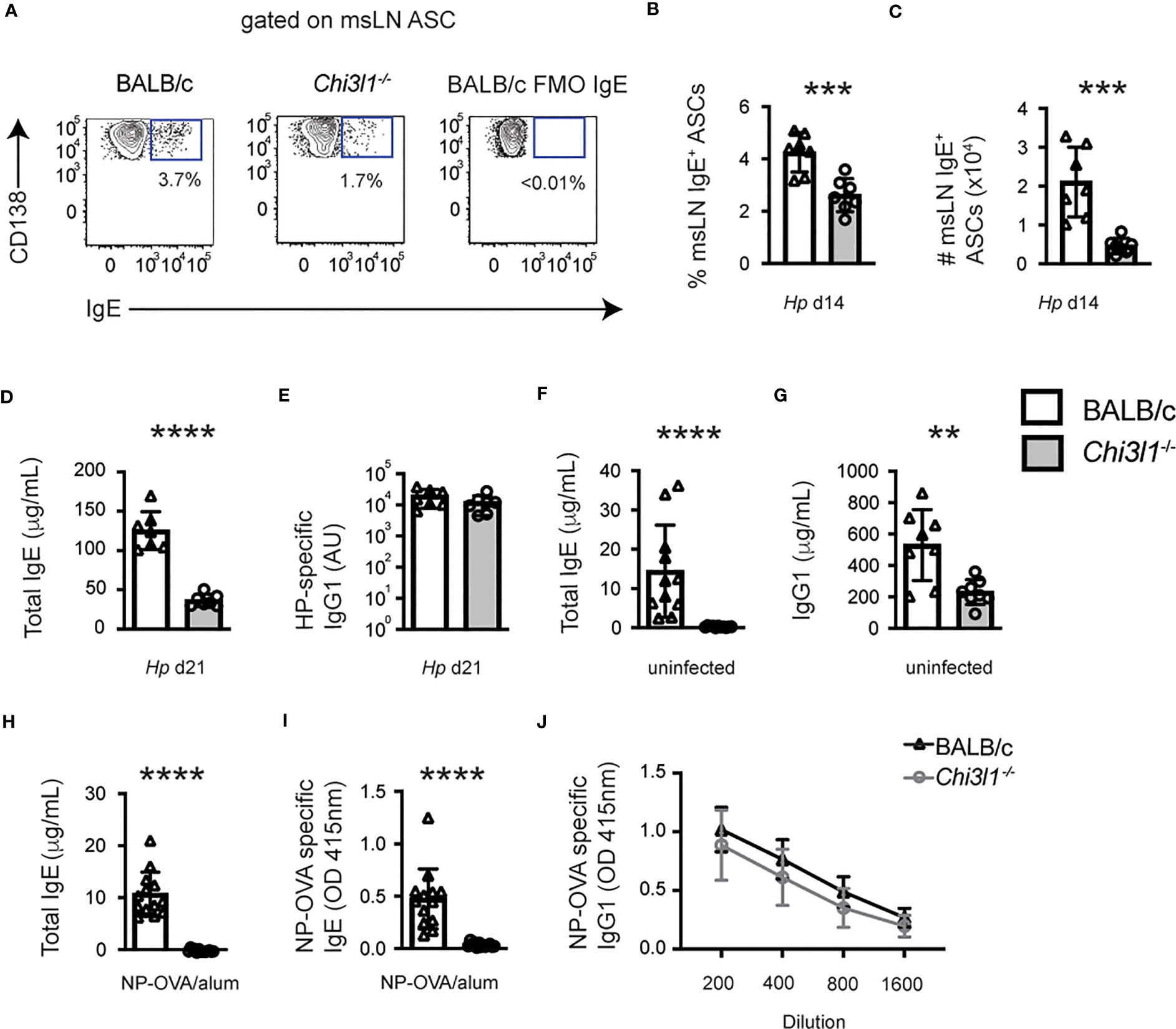
Figure 6 Chi3l1 regulates IgE responses to infection and immunization. Evaluation of IgE and IgG1 antibody responses in Hp-infected (A–E), naïve (F–G) and NP-OVA immunized (H–J) BALB/c (white bars) and Chi3l1-/- (grey bars) mice. (A–C) Quantitation of D14 IgE-expressing ASCs (A–C) from Hp-infected mice (n=7 mice/group). Representative flow plots showing msLN IgE-expressing ASCs (A) with the frequency (B) and number (C) of IgE-expressing ASCs. Hp-induced IgE+ ASCs in 50:50 BM chimeras are shown in Figures S6L–N. (D, E) Serum antibody levels in Hp-infected mice. ELISA quantitation of D21 total serum IgE (D) and Hp-specific IgG1 (E). (F, G) Serum IgE (F) and IgG1 (G) antibody levels in uninfected mice (n=8/group). (H–J) Serum antibody levels in mice immunized with NP-OVA adsorbed to alum (n=13/group). Data is reported in μg/ml for total IgE (H) and OD values for NP-specific IgE (I, serum at 1:10 dilution) and NP-OVA specific IgG1 (J, serum diluted 1:200 to 1:1600). Data representative of 1 (G), 2 (H–J), 3 (A–E), or 5 (F) independent experiments. Data displayed as the mean ± SD of each group with individual animals depicted as circles or triangles. Statistical analysis was performed with unpaired 2-tailed student’s t-test (A–H) or 2-way ANOVA (J). P values of (J) is not significant (P > 0.05). **p≤0.01, ***p≤0.001, ****p≤0.0001.
Next, to assess whether the defective IgE response in Chi3l1-/- mice was restricted to Hp infection, we first examined serum IgE levels in non-infected animals. Although serum IgE levels were low in WT naïve mice, IgE was detected (Figure 6F). In contrast, IgE levels were undetectable (at least 100-fold lower) in the Chi3l1-/- serum (Figure 6F). However, total IgG1 in serum from uninfected Chi3l1-/- mice was only decreased ~2.5 fold relative to the WT animals (Figure 6G), again suggesting a more profound deficit in IgE production by Chi3l1-/- mice. To address whether antigen-specific IgE responses are also dependent on Chi3l1, we vaccinated mice with nitrophenyl haptenated ovalbumin (NP-OVA) adsorbed to alum and measured total and NP-specific IgE on D14. Again, total IgE levels were significantly lower in the vaccinated Chi3l1-/- mice (Figure 6H). Moreover, NP-specific IgE, which was easily measured in vaccinated WT mice, was undetectable in Chi3l1-/- serum (Figure 6I). In contrast, NP-specific IgG1 levels were not significant different between Chi3l1-/- and WT mice. (Figure 6J). Thus, Chi3l1 plays a central role in IgE responses but is dispensable for IgG1 responses.
Chi3l1 regulates IL-4+ TFH programming
IL-4 producing TFH cells are required for the polyclonal IgE response to Hp (35). Since B cell intrinsic expression of Chi3l1 was not required for the IgE ASC response to Hp (Figures S6L–N) and TFH cells were decreased in the Hp-infected Chi3l1-/- mice, we suspected that the large reduction in IgE responses seen in these mice might reflect additional functional impairments in the Chi3l1-/- TFH cells. To assess this in an unbiased fashion, we compared the transcriptome of TFH cells from msLNs of WT and Chi3l1-/- mice on D14 post Hp infection. We identified 9853 expressed genes and 1465 differentially expressed genes (DEG) between the two TFH populations that met an FDR q<0.05 cutoff (Table S1). 252 DEG exhibited at least a ±0.3785 log2 fold-change (log2FC) (Figure 7A), with 193 of these genes expressed at >1 mean RPKM in either BALB/c or Chi3l1-/- TFH. Principal component analysis based on these 193 DEGs shows clear sample group separation (Figure 7B). Since the differences in expression levels between most of the genes expressed by WT and Chi3l1-/- TFH cells was relatively modest, with only 24 of the DEG with RPKM > 1 meeting a ± 1 log2FC threshold (Figures 7A, C), we reasoned that we might observe smaller changes in gene expression across many genes within specific pathways. To test this, we performed Gene Set Enrichment Analysis (GSEA) (50) comparing the rank-ordered WT and Chi3l1-/- TFH gene set to the 5219 C7 Immunologic Signature Gene Sets from MSigDB. None of the C7 gene sets were preferentially enriched (FDR q<0.05) in the WT or Chi3l1-/- TFH transcriptomes, including the 54 C7 gene sets derived from TFH cells (data not shown). This result, which was consistent with normal expression of the TFH lineage master regulator Bcl-6 by Chi3l1-/- TFH cells (Figures 1D, E), indicated that Chi3l1 is not required for establishment of the core TFH transcriptional program (66). We next performed Ingenuity Pathway Analysis (IPA) with the 193 genes meeting an FDR q<0.05 and ± 0.3785 log2FC threshold to assess whether changes in expression of suites of genes might reflect alterations in Chi3l1-/- TFH cell function or signaling. IPA-defined pathways that were predicted to be most significantly different (B-H p < 0.05) between the WT and Chi3l1-/- TFH cells included TH2 and TH1/TH2 activation (Figure 7D).
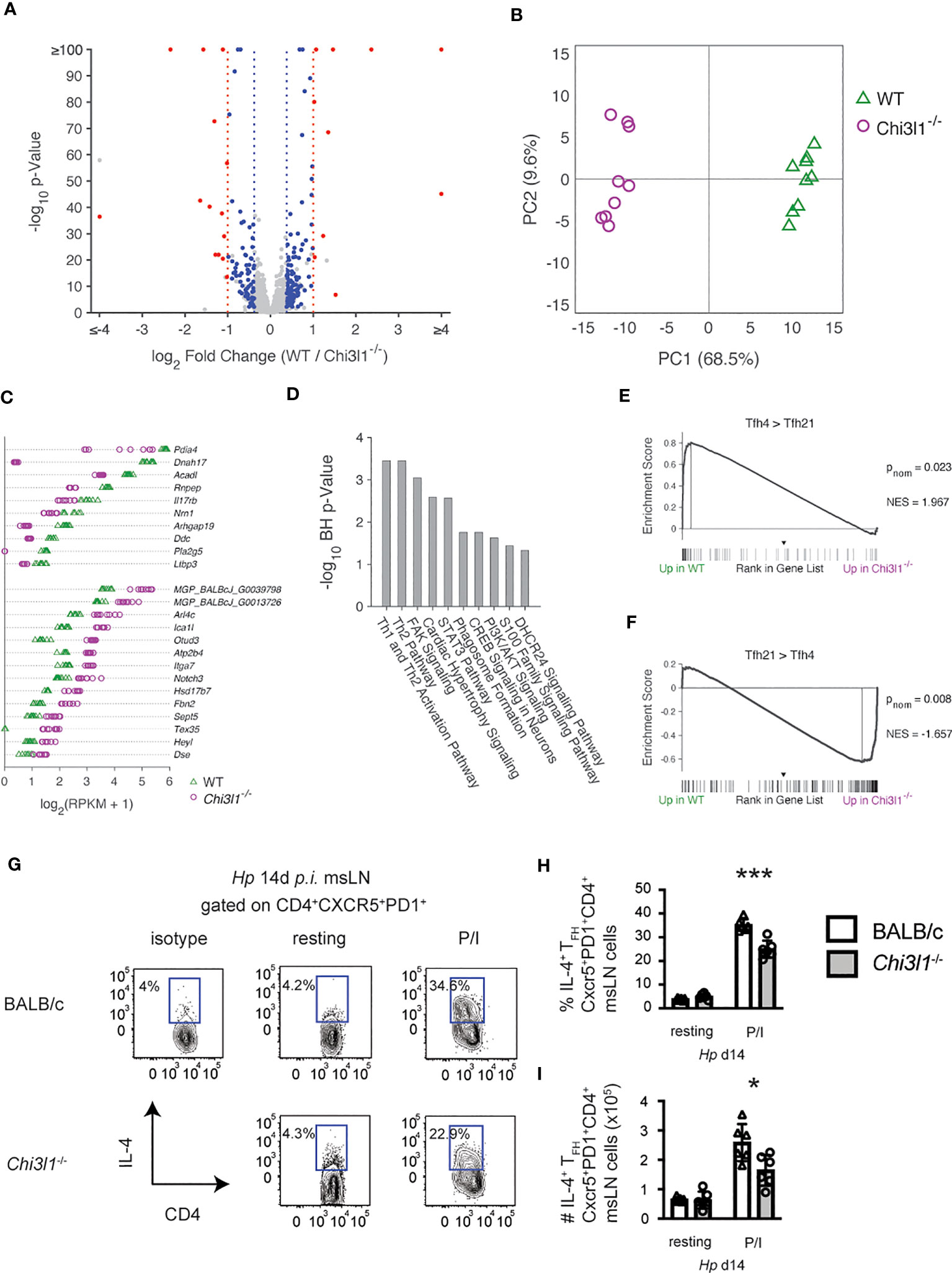
Figure 7 Chi3l1 regulates TFH4 programming. (A–G) RNA-seq analysis of sorted-purified CXCR5+PD-1+ msLN TFH isolated from D14 Hp-infected WT and Chi3l1-/- mice (n=9 independently pooled samples/group). See Table S1 for gene expression levels. (A) Volcano plot showing 9853 expressed genes (defined as genes with at least 3 RPM in all samples of either the WT and/or Chi3l1-/- TFH). 1465 genes were expressed at significantly different levels (FDR q<0.05) between WT and Chi3l1-/- TFH cells. Of those, genes meeting criteria of >1 RPKM in at least one group and log2FC of ±0.3785 (193 genes) are indicated with blue or red symbols. Red symbols indicate the 24 genes meeting a threshold of abs(log2FC) of ±1, blue indicate the 169 genes with abs(log2FC) between 0.3785 and 1.0. (B) Principal component analysis of 193 genes with > 1 mean RPKM in at least one group, ± 0.3785 log2FC (1.3-fold) and FDR q<0.05. (C) Absolute expression values of 24 genes with > 1 mean RPKM in at least one group, ± 1.0 log2FC (2-fold) and FDR q<0.05. (D) The gene list containing 193 genes > 1 RPKM in at least one group meeting an FDR q< 0.05 and ± 0.3785 log2FC cutoff were imported into Ingenuity Pathway Analysis (IPA, QIAGEN Digital Insights) to identify significant predicted signaling pathways. 182 genes were analyzed by IPA. Pathways with a Benjamini-Hochberg (B–H) corrected overlap p< 0.05 are shown. (E, F) Gene set enrichment analysis (GSEA) (50) using the ranked gene list of WT and Chi3l1-/- TFH cells and DEG identified as up in IL4+IL-21neg TFH cells (TFH4, panel E) or up in IL-4negIL-21+ TFH cells (TFH21, panel F) isolated from Nippostrongylus brasiliensis-infected mice (40). pnom and NES are provided. (G–I) Cytokine production by D14 Hp-infected WT and Chi3l1-/- TFH cells (N=5 mice/group) following in vitro PMA+ionomycin restimulation. Flow plots (G) measuring IL-4 production and the percentage (H) and number (I) of TFH4 cells in each culture. Data representative of 3 independent experiments (G–I) or one experiment (A–F) with 9 independent RNA-seq samples/group. Data in (G–I) displayed as the mean ± SD of each group with individual animals depicted as circles or triangles. Statistical tests for RNA-seq data described in the legends. Statistical analysis for (G–I) was performed with unpaired 2-tailed student’s t-test. *p≤0.05, ***p≤0.001.
IL-4 produced by TFH cells is required for B cell class switching to IgE following Hp infection (35, 67) and even modest reductions in the amount of IL-4 present can prevent B cell switching to IgE while having no impact on the IgG1 response (68). Prior studies examining the TFH response after infection with the nematode Nb showed that TFH cells change over time following infection and proceed from producing IL-21 alone (TFH21 cells) to producing primarily IL-4 (TFH4 cells) (40). This change in the TFH cytokine profile from IL-21 to IL-4 is associated with migration of TFH cells into the B cell follicle (69), formation of ICOS-regulated TFH/B cell conjugates that support TFH cell survival and expansion (70), and acquisition of a transcriptionally distinct TFH4 program (40). Given our data, we hypothesized that Chi3l1 might regulate acquisition of the TFH4 transcriptional program. To assess this, we performed GSEA using published lists of genes that are differentially expressed in TFH4 and TFH21 cells from Nb-infected mice (40). We found that DEG that are normally upregulated in Nb TFH4 cells compared to Nb TFH21 cells (40) were significantly enriched in Hp WT TFH cells compared to Hp Chi3l1-/- TFH cells (Figure 7E). In contrast, we observed significant enrichment in the transcriptome of the Hp-induced Chi3l1-/- TFH cells for genes that are increased in Nb TFH21 cells relative to Nb TFH4 cells (Figure 7F). Finally, to address whether loss of Chi3l1 also altered IL-4 production specifically by TFH cells, we measured IL-4 in PMA+ionomycin- restimulated CXCR5+PD-1hi TFH cells from D14 Hp-infected WT and Chi3l1-/- mice. Consistent with the GSEA data, we observed a significant decrease in frequency and number (Figures 7G–I) of Chi3l1-/- TFH4 cells relative to WT TFH4 cells. Together, the data support the conclusion that Chi3l1 contributes to the development or maintenance of the ICOS-expressing TFH4 subset that facilitates B cell IgE responses.
Discussion
Studies of human allergic diseases reveal that expression of YKL-40 protein and CHI3L1 transcripts are increased in the lungs and serum of some asthma cohorts (13, 14). Increased expression of YKL-40/CHI3L1 is positively associated with pathogenesis in allergic rhinitis (19, 20), atopic dermatitis (21–23) and food allergy (24) and CHI3L1 SNPs confer risk of asthma development and airway remodeling (14–17) as well as increased serum IgE and atopy (18). Similarly, experiments using Chi3l1-/- mice show that Chi3l1 regulates type 2 cytokines and IgE in models of asthma, atopic dermatitis and food allergy (8, 23–26). Thus, both mouse and human data support a role for Chi3l1 in promoting atopic disease. Here, we extend these prior studies to show that Chi3l1 also facilitates TH2 responses to the pathogen Hp. In addition, we identify Chi3l1 as a key regulator of TFH responses following both Hp infection and alum-adjuvanted vaccination and demonstrate that Chi3l1, while not required for TFH development, plays critical roles in TFH expansion, ICOS expression by TFH cells, IL-4 production by TFH cells and TFH-dependent IgE production.
Lower organisms utilize chitinases and chitinase-like proteins (CLPs) in innate host defense against chitin-containing pathogens. In Drosophila, the transport of secreted CLP homologues via the hemolymph as well as expression of CLP by hemocytes, the evolutionary precursors of mammalian leukocytes, mediate host defense (71). Following infection, the CLP homologue is transcriptionally upregulated (72), resulting in increased hemolymph levels (73, 74). In parallel, mammalian CLPs are theorized to function as soluble mediators that can be expressed and secreted by activated hematopoietic and damaged mesenchymal cells (75, 76). Consistent with this, our data suggested that Chi3l1 can function as a cytokine-like paracrine regulator in some settings. However, we also observed cell-intrinsic roles for Chi3l1 in CD4 T cell function and progenitor B cell development, suggesting that Chi3l1 might signal in an autocrine manner. Consistent with prior reports (51), we observed no difference in the size of the mature CD4 T cell in mice globally deficient in Chi3l1. Likewise, the number of WT and Chi3l1-/- mature msLN CD4 T cells and splenic CD4 T cells, CD8 T cells and myeloid cells were similar in uninfected 50WT:50Chi3l1-/- chimeras, indicating that Chi3l1 expression by T and myeloid cells is not required for their development. In contrast, when Chi3l1-/- B lineage BM precursors were placed in direct competition with WT B lineage precursors, we observed that the mature peripheral naïve B cell compartment was dominated by Chi3l1-/- B cells. This effect was evident as early as the pre-B cell stage in the BM, suggesting that Chi3l1 can play a B cell intrinsic role as a negative regulator of B cell development and that this intrinsic property becomes evident when WT and Chi3l1-/- B cell precursors are forced to compete in the competitive BM chimera setting.
Following Hp infection, we observed significant decreases in size of the CD4 T cell, TFH cell, and GCB cell responses in the Chi3l1 global knockouts. These defects appeared to be rescued in the LNs of Hp-infected 50WT:50Chi3l1-/- BM chimeras, suggesting that intrinsic expression of Chi3l1 by T cells is not important for the development or expansion of TFH cells. However, given the way that we generated the 50WT:50Chi3l1-/- BM chimeras, we can’t absolutely exclude a T cell-intrinsic role for Chi3l1 in TFH development. What we do know is that Chi3l1 expression by hematopoietic cells was both necessary and sufficient to elicit CD4 T cells responses to Hp. These results, which are complimentary to a study showing that hematopoietic lineage expression of Chi3l1 was sufficient to rescue airway inflammation in Chi3l1-/- mice sensitized to Aspergillus fumigatus conidia (25), suggest that Chi3l1-producing hematopoietic cells support expansion of Hp-induced TFH and CD4 T cells.
Although Chi3l1 expression by T cells does not control the size of the CD4 T cell and TFH response to Hp, our data show that multiple attributes of TFH cells are dependent on T cell intrinsic expression of Chi3l1. Indeed, despite equivalent numbers of WT and Chi3l1-/- CD4 T cells and TFH cells in Hp-infected 50WT:50Chi3l1-/- BM chimeras, the frequency of CD4 T cells competent to produce IL-4 within the Chi3l1-/- CD4 compartment was decreased by almost 50% relative to the WT CD4 T cell compartment. These data fit well with prior studies showing that Chi3l1-/- CD4 T cells primed in vitro with antibodies to CD3 and CD28 in the presence of TH2-polarizing conditions are impaired in IL-4 production (51). Moreover, both our in vitro and in vivo experiments reveal that ICOS upregulation by naïve T cells and TFH cells requires T cell-specific expression of Chi3l1. Given that both ICOS and Chi3l1 are upregulated by WT CD4 T cells following TCR and CD28 engagement (51, 77–79) and Chi3l1-/- CD4 T cells are impaired in ICOS expression after in vitro stimulation with plate-bound antibodies to CD3 and CD28, we think it likely that Chi3l1 enhances ICOS expression by modulating signaling downstream of TCR and/or CD28 engagement. Chi3l1-/- CD4 T cells are reported to be hyperresponsive to anti-CD3+CD28 ligation in vitro (51), suggesting that Chi3l1 functions as a negative regulator. This is intriguing given a recent report showing that low tonic TCR signaling is associated with increased ICOS expression by pre-TFH, enhanced TFH development and more robust GCB cell responses (80).
While TFH responses to Hp infection and vaccination are clearly impaired in Chi3l1-/- mice, we do not think that this is due to a requirement for Chi3l1 during commitment to the TFH lineage as the remaining msLN Chi3l1-/- TFH cells express normal levels of the master TFH regulator Bcl-6 (66) and the core TFH transcriptional program (66) appears intact. Instead, we find that fewer Chi3l1-/- TFH cells progress to becoming functional IL-4 producing TFH cells (TFH4 cells), which are normally induced following Hp infection (35). This is due, at least in part, to an inability of these cells to acquire the mature TFH4 transcriptional program (40). Instead, Chi3l1-/- TFH cells appear to be transcriptionally enriched in genes expressed by IL-21 and the IL-21 plus IL-4 producing TFH cells (40). Prior studies examining the TFH response after infection with the nematode Nippostrongylus brasiliensis showed that TFH cells change over time following infection and proceed from producing IL-21 alone to producing primarily IL-4 (40). This change in the TFH cytokine profile from IL-21 to IL-4 coincides with the migration of TFH cells into the B cell follicle (69) and the formation of TFH/B cell conjugates (70). Our data therefore suggest that Chi3l1 is required for the full maturation of the TFH response from TFH21 to B cell-engaged TFH4 cells.
Although our data do not specify the mechanism by which Chi3l1 controls the expansion and maturation of the TFH4 response to Hp infection, we suspect that it is related to decreased expression of ICOS by Chi3l1-/- CD4 TFH cells. It is well appreciated that ICOS/ICOSL interactions between B cells and TFH cells is required for the maintenance of the TFH population in the B cell follicle and GC (81) and that blockade of ICOS/ICOSL in established allergic responses is sufficient to ablate ongoing TFH and GC responses (82). Moreover, data from parasite infection models indicate that IL-4 production by TFH cells not only requires B cells (34) but is dependent on ICOS/ICOSL interactions within the B cell follicle (70). Thus, we think that Chi3l1, which is upregulated in CD4 T cells upon TCR stimulation (51), induces ICOS expression in a cell intrinsic manner. ICOS-expressing TFH cells can then productively engage with ICOSL-expressing B cells, which in turn provide TFH cells with appropriate co-stimulatory signals to support their expansion, maintenance and acquisition of the TFH4 transcriptional program within the B cell follicle (70, 81–83). Given the known role for ICOS-ICOSL interactions in supporting TFH-dependent B cell responses (81), we argue that the decrease in antigen-specific B cell responses in vaccinated Chi3l1-/- mice is also due, in large part, to the poor expansion of the TFH compartment and the reduction in ICOS expression by the remaining Chi3l1-/- TFH cells.
One of the most striking defects in naïve, vaccinated and Hp-infected Chi3l1-/- mice is the near total ablation of secreted IgE and IgE-expressing ASCs. The data from the Hp-infected 50WT:50Chi3l1-/- mice indicate that this is not due to the loss of Chi3l1 expression by B lineage cells. We propose the loss of the IgE response in vaccinated and infected Chi3l1-/- mice is most likely due to the impaired IL-4 production by the TFH as it was reported that IL-4 produced specifically by TFH cells is required for B cell class switching to IgE following Hp infection (35, 67). We think that poor conversion of TFH21 cells to the TFH4 subset may also explain why Hp-infected Chi3l1-/- mice make relatively normal IgG1 responses. It is reported that IL-21 promotes switching to IgG1 and actively inhibits switching to IgE (84) and that even modest reductions in the amount of IL-4 produced can prevent B cell switching to IgE while having no impact on the IgG1 response (68). Although we think that the loss of the IgE response in Chi3l1-/- mice is due to the impaired TFH4 response, ICOS/ICOSL interactions also contribute to the development of both IL-4 producing T cells and IgE responses following allergen sensitization and challenge (83) and blockade of ICOS/ICOSL in established allergic responses is sufficient to impair IgE production (82). Therefore, it is possible that Chi3l1 tunes ICOS expression levels, which favors IL-4 production by TFH cells and supports isotype switching of the ICOSL-expressing B cells to IgE.
In summary, our data reveal new roles for Chi3l1 in controlling upregulation of ICOS by TFH cells, expansion of the TFH compartment, acquisition of the TFH4 transcriptional program, establishment of TFH-dependent B cell responses and production of secreted IgE. We show that these effects are due to paracrine hematopoietic cell derived Chi3l1 as well as T cell intrinsic Chi3l1. These data provide avenues for future research into the mechanisms by which Chi3l1 exerts its various functions in allergic responses and suggest that directed approaches that block Chi3l1 activity or function specifically within the T cell compartment may attenuate both TH2 responses and TFH-driven IgE responses in pathologic settings such as atopy and allergic airway disease.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GSE203113 (GEO); https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE203113.
Ethics statement
The animal study was reviewed and approved by University of Alabama Birmingham Institutional Animal Care and Use Committee (IACUC).
Author contributions
Conceptualization, MC and FL, Methodology, MC and JB, Investigation and Analysis, MC, NB, AR, CDS, JB, and BM, Writing – Original Draft: MC and FL, Writing – Review and Editing: MC, FL, CDS, and AR, Funding Acquisition: FL, TR, and MC, Resources, CDS, BL, TR, and AR, Supervision, MC and FL. All authors contributed to the article and approved the submitted version.
Funding
Financial support provided by R01 AI104725 (FL), R01 AI50740 (FL), R01 AI153413 (TR), R01 AI52476 (TR), T32 2T32HL105346 (VT), UAB Frommeyer Investigative Fellowship (MC), UAB Dixon Award (MC), and AAAAI/ALA Allergic Respiratory Disease Research Award (MC).
Acknowledgments
We thank Scott Simpler, Rebecca Burnham, and Uma Mudunuru for mouse colony support, Sithy Allie and Amber Papillion for thoughtful comments, and Jessica Peel and Chris Risley for advice designing B cell panels. Allison Humbles (MEDIMMUNE) and Astra Zeneca provided mice. We thank Vidya Sigar Hanumanthu and the UAB Flow Cytometry Core Facility for sorting. Portions of this manuscript were previously uploaded to the biorxiv preprint server (biorxiv.org).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1158493/full#supplementary-material
References
1. Hazebrouck S, Canon N, Dreskin SC. The effector function of allergens. Front Allergy (2022) 3:818732. doi: 10.3389/falgy.2022.818732
2. Matsumura Y. Role of allergen source-derived proteases in sensitization via airway epithelial cells. J Allergy (Cairo) (2012) 2012:903659. doi: 10.1155/2012/903659
3. Tharanathan RN, Kittur FS. Chitin–the undisputed biomolecule of great potential. Crit Rev Food Sci Nutr (2003) 43:61–87. doi: 10.1080/10408690390826455
4. Foster JM, Zhang Y, Kumar S, Carlow CK. Parasitic nematodes have two distinct chitin synthases. Mol Biochem Parasitol (2005) 142:126–32. doi: 10.1016/j.molbiopara.2005.03.011
5. Da Silva CA, Pochard P, Lee CG, Elias JA. Chitin particles are multifaceted immune adjuvants. Am J Respir Crit Care Med (2010) 182:1482–91. doi: 10.1164/rccm.200912-1877OC
6. Arae K, Morita H, Unno H, Motomura K, Toyama S, Okada N, et al. Chitin promotes antigen-specific Th2 cell-mediated murine asthma through induction of IL-33-mediated IL-1beta production by DCs. Sci Rep (2018) 8:11721. doi: 10.1038/s41598-018-30259-2
7. Lee CG, Da Silva CA, Dela Cruz CS, Ahangari F, Ma B, Kang MJ, et al. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu Rev Physiol (2011) 73:479–501. doi: 10.1146/annurev-physiol-012110-142250
8. Lee CG, Hartl D, Lee GR, Koller B, Matsuura H, Da Silva CA, et al. Role of breast regression protein 39 (BRP-39)/chitinase 3-like-1 in Th2 and IL-13-induced tissue responses and apoptosis. J Exp Med (2009) 206:1149–66. doi: 10.1084/jem.20081271
9. Nikota JK, Botelho FM, Bauer CM, Jordana M, Coyle AJ, Humbles AA, et al. Differential expression and function of breast regression protein 39 (BRP-39) in murine models of subacute cigarette smoke exposure and allergic airway inflammation. Respir Res (2011) 12:39. doi: 10.1186/1465-9921-12-39
10. Kim LK, Morita R, Kobayashi Y, Eisenbarth SC, Lee CG, Elias J, et al. AMCase is a crucial regulator of type 2 immune responses to inhaled house dust mites. Proc Natl Acad Sci U.S.A. (2015) 112:E2891–2899. doi: 10.1073/pnas.1507393112
11. Hong JY, Kim M, Sol IS, Kim KW, Lee CM, Elias JA, et al. Chitotriosidase inhibits allergic asthmatic airways via regulation of TGF-beta expression and Foxp3(+) Treg cells. Allergy (2018) 73:1686–99. doi: 10.1111/all.13426
12. Zhu W, Lonnblom E, Forster M, Johannesson M, Tao P, Meng L, et al. Natural polymorphism of Ym1 regulates pneumonitis through alternative activation of macrophages. Sci Adv (2020) 6:1–12. doi: 10.1126/sciadv.aba9337
13. Chupp GL, Lee CG, Jarjour N, Shim YM, Holm CT, He S, et al. A chitinase-like protein in the lung and circulation of patients with severe asthma. N Engl J Med (2007) 357:2016–27. doi: 10.1056/NEJMoa073600
14. Ober C, Tan Z, Sun Y, Possick JD, Pan L, Nicolae R, et al. Effect of variation in CHI3L1 on serum YKL-40 level, risk of asthma, and lung function. N Engl J Med (2008) 358:1682–91. doi: 10.1056/NEJMoa0708801
15. Gomez JL, Crisafi GM, Holm CT, Meyers DA, Hawkins GA, Bleecker ER, et al. Genetic variation in chitinase 3-like 1 (CHI3L1) contributes to asthma severity and airway expression of YKL-40. J Allergy Clin Immunol (2015) 136:51–58 e10. doi: 10.1016/j.jaci.2014.11.027
16. Gomez JL, Yan X, Holm CT, Grant N, Liu Q, Cohn L, et al. Characterisation of asthma subgroups associated with circulating YKL-40 levels. Eur Respir J (2017) 4(1700800):1–12. doi: 10.1183/13993003.00800-2017
17. Liu L, Zhang X, Liu Y, Zhang L, Zheng J, Wang J, et al. Chitinase-like protein YKL-40 correlates with inflammatory phenotypes, anti-asthma responsiveness and future exacerbations. Respir Res (2019) 20:95. doi: 10.1186/s12931-019-1051-9
18. Sohn MH, Lee JH, Kim KW, Kim SW, Lee SH, Kim KE, et al. Genetic variation in the promoter region of chitinase 3-like 1 is associated with atopy. Am J Respir Crit Care Med (2009) 179:449–56. doi: 10.1164/rccm.200809-1422OC
19. Kwon JW, Kim TW, Cho SH, Min KU, Park HW. Serum YKL-40 levels are correlated with symptom severity in patients with allergic rhinitis. Allergy (2011) 66:1252–3. doi: 10.1111/j.1398-9995.2011.02596.x
20. Acevedo N, Bornacelly A, Mercado D, Unneberg P, Mittermann I, Valenta R, et al. Genetic variants in CHIA and CHI3L1 are associated with the IgE response to the ascaris resistance marker ABA-1 and the birch pollen allergen bet v 1. PloS One (2016) 11:e0167453. doi: 10.1371/journal.pone.0167453
21. Dezman K, Korosec P, Rupnik H, Rijavec M. SPINK5 is associated with early-onset and CHI3L1 with late-onset atopic dermatitis. Int J Immunogenet (2017) 44:212–8. doi: 10.1111/iji.12327
22. Salomon J, Matusiak L, Nowicka-Suszko D, Szepietowski JC. Chitinase-3-like protein 1 (YKL-40) reflects the severity of symptoms in atopic dermatitis. J Immunol Res (2017) 2017:5746031. doi: 10.1155/2017/5746031
23. Kwak EJ, Hong JY, Kim MN, Kim SY, Kim SH, Park CO, et al. Chitinase 3-like 1 drives allergic skin inflammation via Th2 immunity and M2 macrophage activation. Clin Exp Allergy (2019) 49(11:1464–1474. doi: 10.1111/cea.13478
24. Kim EG, Kim MN, Hong JY, Lee JW, Kim SY, Kim KW, et al. Chitinase 3-like 1 contributes to food allergy via M2 macrophage polarization. Allergy Asthma Immunol Res (2020) 12:1012–28. doi: 10.4168/aair.2020.12.6.1012
25. Mackel JJ, Garth JM, Jones M, Ellis DA, Blackburn JP, Yu Z, et al. Chitinase 3-like-1 protects airway function despite promoting type 2 inflammation during fungal-associated allergic airway inflammation. Am J Physiol Lung Cell Mol Physiol (2021) 320:L615–26. doi: 10.1152/ajplung.00528.2020
26. Lee YS, Yu JE, Kim MJ, Ham HJ, Jeon SH, Yun J, et al. New therapeutic strategy for atopic dermatitis by targeting CHI3L1/ITGA5 axis. Clin Transl Med (2022) 12:e739. doi: 10.1002/ctm2.739
27. Allen JE, Maizels RM. Diversity and dialogue in immunity to helminths. Nat Rev Immunol (2011) 11:375–88. doi: 10.1038/nri2992
28. Artis D, Maizels RM, Finkelman FD. Forum: immunology: allergy challenged. Nature (2012) 484:458–9. doi: 10.1038/484458a
29. Reynolds LA, Filbey KJ, Maizels RM. Immunity to the model intestinal helminth parasite Heligmosomoides polygyrus. Semin Immunopathol (2012) 34:829–46. doi: 10.1007/s00281-012-0347-3
30. Fitzsimmons CM, Falcone FH, Dunne DW. Helminth allergens, parasite-specific IgE, and its protective role in human immunity. Front Immunol (2014) 5:61. doi: 10.3389/fimmu.2014.00061
31. Sutherland TE, Logan N, Ruckerl D, Humbles AA, Allan SM, Papayannopoulos V, et al. Chitinase-like proteins promote IL-17-mediated neutrophilia in a tradeoff between nematode killing and host damage. Nat Immunol (2014) 15:1116–25. doi: 10.1038/ni.3023
32. Urban JF Jr., Katona IM, Finkelman FD. Heligmosomoides polygyrus: CD4+ but not CD8+ T cells regulate the IgE response and protective immunity in mice. Exp Parasitol (1991) 73:500–11. doi: 10.1016/0014-4894(91)90074-7
33. King IL, Mohrs M. IL-4-producing CD4+ T cells in reactive lymph nodes during helminth infection are T follicular helper cells. J Exp Med (2009) 206:1001–7. doi: 10.1084/jem.20090313
34. Leon B, Ballesteros-Tato A, Browning JL, Dunn R, Randall TD, Lund FE. Regulation of T(H)2 development by CXCR5+ dendritic cells and lymphotoxin-expressing B cells. Nat Immunol (2012) 13:681–90. doi: 10.1038/ni.2309
35. Meli AP, Fontes G, Leung Soo C, King IL. T follicular helper cell-derived IL-4 is required for IgE production during intestinal helminth infection. J Immunol (2017) 199:244–52. doi: 10.4049/jimmunol.1700141
36. Gowthaman U, Chen JS, Zhang B, Flynn WF, Lu Y, Song W, et al. Identification of a T follicular helper cell subset that drives anaphylactic IgE. Science. (2019) 365(6456):1–14. doi: 10.1126/science.aaw6433
37. McCoy KD, Stoel M, Stettler R, Merky P, Fink K, Senn BM, et al. Polyclonal and specific antibodies mediate protective immunity against enteric helminth infection. Cell Host Microbe (2008) 4:362–73. doi: 10.1016/j.chom.2008.08.014
38. Akiba H, Takeda K, Kojima Y, Usui Y, Harada N, Yamazaki T, et al. The role of ICOS in the CXCR5+ follicular B helper T cell maintenance in vivo. J Immunol (2005) 175:2340–8. doi: 10.4049/jimmunol.175.4.2340
39. Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity (2011) 34:932–46. doi: 10.1016/j.immuni.2011.03.023
40. Weinstein JS, Herman EI, Lainez B, Licona-Limon P, Esplugues E, Flavell R, et al. TFH cells progressively differentiate to regulate the germinal center response. Nat Immunol (2016) 17:1197–205. doi: 10.1038/ni.3554
41. Papillion A, Powell MD, Chisolm DA, Bachus H, Fuller MJ, Weinmann AS, et al. Inhibition of IL-2 responsiveness by IL-6 is required for the generation of GC-TFH cells. Sci Immunol (2019) 4:1–11. doi: 10.1126/sciimmunol.aaw7636
42. Bornholdt ZA, Prasad BV. X-ray structure of NS1 from a highly pathogenic H5N1 influenza virus. Nature (2008) 456:985–8. doi: 10.1038/nature07444
43. Wojciechowski W, Harris DP, Sprague F, Mousseau B, Makris M, Kusser K, et al. Cytokine-producing effector B cells regulate type 2 immunity to H. polygyrus. Immunity (2009) 30:421–33. doi: 10.1016/j.immuni.2009.01.006
44. Rangel-Moreno J, Moyron-Quiroz JE, Carragher DM, Kusser K, Hartson L, Moquin A, et al. Omental milky spots develop in the absence of lymphoid tissue-inducer cells and support B and T cell responses to peritoneal antigens. Immunity (2009) 30:731–43. doi: 10.1016/j.immuni.2009.03.014
45. Stone SL, Peel JN, Scharer CD, Risley CA, Chisolm DA, Schultz MD, et al. T-bet transcription factor promotes antibody-secreting cell differentiation by limiting the inflammatory effects of IFN-gamma on B cells. Immunity (2019) 50:1172–1187.e1177. doi: 10.1016/j.immuni.2019.04.004
46. Jiang H, Lei R, Ding SW, Zhu S. Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinf (2014) 15:182. doi: 10.1186/1471-2105-15-182
47. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics (2013) 29:15–21. doi: 10.1093/bioinformatics/bts635
48. Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, et al. Software for computing and annotating genomic ranges. PloS Comput Biol (2013) 9:e1003118. doi: 10.1371/journal.pcbi.1003118
49. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol (2014) 15:550. doi: 10.1186/s13059-014-0550-8
50. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U.S.A. (2005) 102:15545–50. doi: 10.1073/pnas.0506580102
51. Kim DH, Park HJ, Lim S, Koo JH, Lee HG, Choi JO, et al. Regulation of chitinase-3-like-1 in T cell elicits Th1 and cytotoxic responses to inhibit lung metastasis. Nat Commun (2018) 9:503. doi: 10.1038/s41467-017-02731-6
52. Svetic A, Madden KB, Zhou XD, Lu P, Katona IM, Finkelman FD, et al. A primary intestinal helminthic infection rapidly induces a gut-associated elevation of Th2-associated cytokines and IL-3. J Immunol (1993) 150:3434–41. doi: 10.4049/jimmunol.150.8.3434
53. Glatman Zaretsky A, Taylor JJ, King IL, Marshall FA, Mohrs M, Pearce EJ. T follicular helper cells differentiate from Th2 cells in response to helminth antigens. J Exp Med (2009) 206:991–9. doi: 10.1084/jem.20090303
54. Johnston RJ, Poholek AC, Ditoro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science (2009) 325:1006–10. doi: 10.1126/science.1175870
55. Yusuf I, Kageyama R, Monticelli L, Johnston RJ, Ditoro D, Hansen K, et al. Germinal center T follicular helper cell IL-4 production is dependent on signaling lymphocytic activation molecule receptor (CD150). J Immunol (2010) 185:190–202. doi: 10.4049/jimmunol.0903505
56. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 mediates the development of T follicular helper cells. Science (2009) 325:1001–5. doi: 10.1126/science.1176676
57. Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity (2009) 31:457–68. doi: 10.1016/j.immuni.2009.07.002
58. Crotty S, Johnston RJ, Schoenberger SP. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol (2010) 11:114–20. doi: 10.1038/ni.1837
59. Redpath SA, van der Werf N, Cervera AM, Macdonald AS, Gray D, Maizels RM, et al. ICOS controls Foxp3(+) regulatory T-cell expansion, maintenance and IL-10 production during helminth infection. Eur J Immunol (2013) 43:705–15. doi: 10.1002/eji.201242794
60. Wilson MS, Taylor MD, Balic A, Finney CA, Lamb JR, Maizels RM. Suppression of allergic airway inflammation by helminth-induced regulatory T cells. J Exp Med (2005) 202:1199–212. doi: 10.1084/jem.20042572
61. Rausch S, Huehn J, Loddenkemper C, Hepworth MR, Klotz C, Sparwasser T, et al. Establishment of nematode infection despite increased Th2 responses and immunopathology after selective depletion of Foxp3+ cells. Eur J Immunol (2009) 39:3066–77. doi: 10.1002/eji.200939644
62. Johansen JS, Williamson MK, Rice JS, Price PA. Identification of proteins secreted by human osteoblastic cells in culture. J Bone Miner Res (1992) 7:501–12. doi: 10.1002/jbmr.5650070506
63. Johansen JS. Studies on serum YKL-40 as a biomarker in diseases with inflammation, tissue remodelling, fibroses and cancer. Dan Med Bull (2006) 53:172–209. Available at: https://www.ncbi.nlm.nih.gov/pubmed/17087877
64. Eric HK, Melba LA, Jun L, Xiao Wen M, Michael FM, Gregory AN, et al. Acute effects of whole-body proton irradiation on the immune system of the mouse. Radiat Res (2000) 153:587–94. doi: 10.1667/0033-7587(2000)153[0587:AEOWBP]2.0.CO;2
65. Heylmann D, Rödel F, Kindler T, Kaina B. Radiation sensitivity of human and murine peripheral blood lymphocytes, stem and progenitor cells. Biochim Biophys Acta (2014) 1846:121–9. doi: 10.1016/j.bbcan.2014.04.009
66. Hart AP, Laufer TM. A review of signaling and transcriptional control in T follicular helper cell differentiation. J Leukoc Biol (2022) 111:173–95. doi: 10.1002/JLB.1RI0121-066R
67. King IL, Mohrs K, Meli AP, Downey J, Lanthier P, Tzelepis F, et al. Intestinal helminth infection impacts the systemic distribution and function of the naive lymphocyte pool. Mucosal Immunol (2017) 10:1160–8. doi: 10.1038/mi.2016.127
68. Robinson MJ, Prout M, Mearns H, Kyle R, Camberis M, Forbes-Blom EE, et al. IL-4 haploinsufficiency specifically impairs IgE responses against allergens in mice. J Immunol (2017) 198:1815–22. doi: 10.4049/jimmunol.1601434
69. Gonzalez DG, Cote CM, Patel JR, Smith CB, Zhang Y, Nickerson KM, et al. Nonredundant roles of IL-21 and IL-4 in the phased initiation of germinal center B cells and subsequent self-renewal transitions. J Immunol (2018) 201:3569–79. doi: 10.4049/jimmunol.1500497
70. Reinhardt RL, Liang HE, Locksley RM. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol (2009) 10:385–93. doi: 10.1038/ni.1715
71. Broz V, Kucerova L, Rouhova L, Fleischmannova J, Strnad H, Bryant PJ, et al. Drosophila imaginal disc growth factor 2 is a trophic factor involved in energy balance, detoxification, and innate immunity. Sci Rep (2017) 7:43273. doi: 10.1038/srep43273
72. De Gregorio E, Spellman PT, Rubin GM, Lemaitre B. Genome-wide analysis of the Drosophila immune response by using oligonucleotide microarrays. Proc Natl Acad Sci U.S.A. (2001) 98:12590–5. doi: 10.1073/pnas.221458698
73. Vierstraete E, Verleyen P, Sas F, Van Den Bergh G, De Loof A, Arckens L, et al. The instantly released Drosophila immune proteome is infection-specific. Biochem Biophys Res Commun (2004) 317:1052–60. doi: 10.1016/j.bbrc.2004.03.150
74. Arefin B, Kucerova L, Dobes P, Markus R, Strnad H, Wang Z, et al. Genome-wide transcriptional analysis of Drosophila larvae infected by entomopathogenic nematodes shows involvement of complement, recognition and extracellular matrix proteins. J Innate Immun (2014) 6:192–204. doi: 10.1159/000353734
75. Owhashi M, Arita H, Hayai N. Identification of a novel eosinophil chemotactic cytokine (ECF-L) as a chitinase family protein. J Biol Chem (2000) 275:1279–86. doi: 10.1074/jbc.275.2.1279
76. Houston DR, Recklies AD, Krupa JC, Van Aalten DM. Structure and ligand-induced conformational change of the 39-kDa glycoprotein from human articular chondrocytes. J Biol Chem (2003) 278:30206–12. doi: 10.1074/jbc.M303371200
77. McAdam AJ, Chang TT, Lumelsky AE, Greenfield EA, Boussiotis VA, Duke-Cohan JS, et al. Mouse inducible costimulatory molecule (ICOS) expression is enhanced by CD28 costimulation and regulates differentiation of CD4+ T cells. J Immunol (2000) 165:5035–40. doi: 10.4049/jimmunol.165.9.5035
78. Tan AH, Wong SC, Lam KP. Regulation of mouse inducible costimulator (ICOS) expression by Fyn-NFATc2 and ERK signaling in T cells. J Biol Chem (2006) 281:28666–78. doi: 10.1074/jbc.M604081200
79. Yang PS, Yu MH, Hou YC, Chang CP, Lin SC, Kuo IY, et al. Targeting protumor factor chitinase-3-like-1 secreted by Rab37 vesicles for cancer immunotherapy. Theranostics (2022) 12:340–61. doi: 10.7150/thno.65522
80. Bartleson JM, Viehmann Milam AA, Donermeyer DL, Horvath S, Xia Y, Egawa T, et al. Strength of tonic T cell receptor signaling instructs T follicular helper cell-fate decisions. Nat Immunol (2020) 21:1384–96. doi: 10.1038/s41590-020-0781-7
81. Liu D, Xu H, Shih C, Wan Z, Ma X, Ma W, et al. T-B-cell entanglement and ICOSL-driven feed-forward regulation of germinal centre reaction. Nature (2015) 517:214–8. doi: 10.1038/nature13803
82. Uwadiae FI, Pyle CJ, Walker SA, Lloyd CM, Harker JA. Targeting the ICOS/ICOS-L pathway in a mouse model of established allergic asthma disrupts T follicular helper cell responses and ameliorates disease. Allergy (2019) 74:650–62. doi: 10.1111/all.13602
83. Zhang X, Hu X, Tian T, Pang W. The role of ICOS in allergic disease: Positive or Negative? Int Immunopharmacol (2022) 103:108394. doi: 10.1016/j.intimp.2021.108394
Keywords: IgE (immunoglobulin E), helminth, germinal center, T follicular helper (T cell FH, T helper 2 (Th2) cells, enteric, IL-4, germinal center (GC) B cells
Citation: Curtiss ML, Rosenberg AF, Scharer CD, Mousseau B, Benavides NAB, Bradley JE, León B, Steele C, Randall TD and Lund FE (2023) Chitinase-3-like 1 regulates TH2 cells, TFH cells and IgE responses to helminth infection. Front. Immunol. 14:1158493. doi: 10.3389/fimmu.2023.1158493
Received: 04 February 2023; Accepted: 12 July 2023;
Published: 27 July 2023.
Edited by:
Alisa Gruden-Movsesijan, Institute for the Application of Nuclear Energy (INEP), SerbiaReviewed by:
Svetlana P. Chapoval, University of Maryland, United StatesDominik Rückerl, The University of Manchester, United Kingdom
Christopher Allen, University of California, San Francisco, United States
Copyright © 2023 Curtiss, Rosenberg, Scharer, Mousseau, Benavides, Bradley, León, Steele, Randall and Lund. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miranda L. Curtiss, bWxjdXJ0aXNzQHVhYm1jLmVkdQ==