 Menglu Pan
Menglu Pan Huanhuan Zhao1†
Huanhuan Zhao1† Patrick S. C. Leung
Patrick S. C. Leung Zongwen Shuai
Zongwen Shuai- 1Department of Rheumatology and Immunology, First Affiliated Hospital of Anhui Medical University, Hefei, China
- 2Division of Rheumatology/Allergy and Clinical Immunology, University of California, Davis, Davis, CA, United States
- 3Inflammation and Immune Mediated Diseases Laboratory of Anhui Province, Hefei, China
Anti-neutrophil cytoplasmic autoantibodies (ANCA) associated vasculitis (AAV) is a necrotizing vasculitis mainly involving small blood vessels. It is demonstrated that T cells are important in the pathogenesis of AAV, including regulatory T cells (Treg) and helper T cells (Th), especially Th2, Th17, and follicular Th cells (Tfh). In addition, the exhaustion of T cells predicted the favorable prognosis of AAV. The immune checkpoints (ICs) consist of a group of co-stimulatory and co-inhibitory molecules expressed on the surface of T cells, which maintains a balance between the activation and exhaustion of T cells. CD28, inducible T-cell co-stimulator (ICOS), OX40, CD40L, glucocorticoid induced tumor necrosis factor receptor (GITR), and CD137 are the common co-stimulatory molecules, while the programmed cell death 1 (PD-1), cytotoxic T lymphocyte-associated molecule 4 (CTLA-4), T cell immunoglobulin (Ig) and mucin domain-containing protein 3 (TIM-3), B and T lymphocyte attenuator (BTLA), V-domain Ig suppressor of T cell activation (VISTA), T‐cell Ig and ITIM domain (TIGIT), CD200, and lymphocyte activation gene 3 (LAG-3) belong to co-inhibitory molecules. If this balance was disrupted and the activation of T cells was increased, autoimmune diseases (AIDs) might be induced. Even in the treatment of malignant tumors, activation of T cells by immune checkpoint inhibitors (ICIs) may result in AIDs known as rheumatic immune-related adverse events (Rh-irAEs), suggesting the importance of ICs in AIDs. In this review, we summarized the features of AAV induced by immunotherapy using ICIs in patients with malignant tumors, and then reviewed the biological characteristics of different ICs. Our aim was to explore potential targets in ICs for future treatment of AAV.
1 Introduction
Anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitis (AAV) is a group of life-threatening diseases, characterized by necrotizing inflammation of small blood vessels, with pauci-immune complex depositions. According to different clinical manifestations, it was mainly divided into three types, including granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA) (1–4). ANCA is composed of series of autoantibodies identifying their autoantigens in neutrophil plasma, including proteinase-3 (PR3) and myeloperoxidase (MPO) which may be expressed on activated neutrophils. GPA, mainly associated with PR3-ANCA, usually affects the sinuses, the lung, and the kidney with specific granulomatous inflammation. In contrast to GPA, MPA frequently damages the lung and the kidney with necrotizing vasculitis. In general, MPO-ANCA is predominantly detected in patients with MPA and EGPA. The clinical characteristics of EGPA include asthma, eosinophilia, and vasculitis (5, 6), but it is much less common than GPA and MPA. The distribution of AAV might be influenced by geographical and race. In east Asia, especially in China and Japan, MPA with MPO-ANCA is the predominant AAV, whereas in Europe, such as the UK and France, GPA with PR3-ANCA is the more common AAV (7, 8).
Although the exact etiopathogenesis of AAV remains unclear, studies have demonstrated that some factors, such as T and B cells, ANCA, the complement alternative pathway (cAP), and neutrophil extracellular traps (NETs), might play various important roles in the pathogenesis (Figure 1). On the genetic background, the interaction of infections, environmental and other factors might activate T cells, which could help B cells develop into plasma cells. Meanwhile, neutrophils could also be activated and express PR3 and MPO to combine with the ANCA secreted by plasma cells (9–11). The fragment a of fifth complement (C5a) produced by activating cAP could connect with the C5a receptor on neutrophils (12, 13). The combinations of these factors on neutrophils could activate the neutrophils further to promote their degranulation and NETs production, intensify respiratory burst, injury the vascular endothelium, accelerate the inflammatory response, and ultimately lead to clinical damages of AAV (11, 13–15).
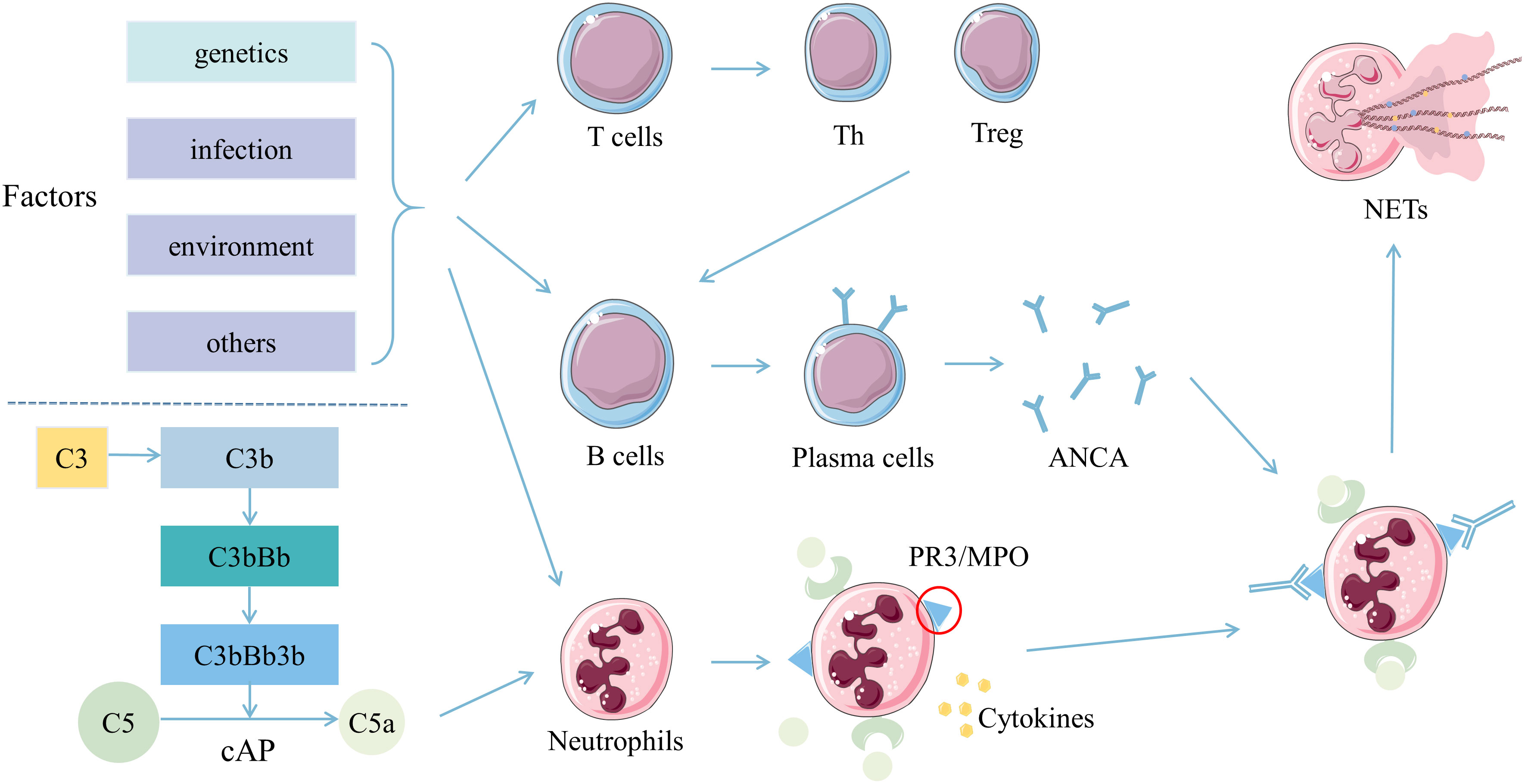
Figure 1 Pathogenesis of ANCA associated vasculitis. T cells, B cells, and neutrophils were activated by multiple stimulations in the background of genetic susceptibility. Th and Treg cells differentiated from T cells synergistically promote B cells to develop into plasma cells, and produce ANCA. ANCA then combined with PR3 or MPO expressed on neutrophils pre-activated by inflammatory cytokines. Also, C5a derived from activated cAP might combine with the C5a receptor on neutrophils. Neutrophils activated by ANCA, C5a and various cytokines might produce more NETs conducive for the inflammatory response and adaptive immunity, ultimately leading to clinical damages. Abbreviations: ANCA, anti-neutrophil cytoplasmic autoantibodies; C5a, fragment a of the fifth complement; cAP, complement alternative pathway; MPO, myeloperoxidase; NETs, neutrophil extracellular traps; PR3, proteinase-3; Th, helper T; Treg, regulatory T.
T cells play essential and pivotal roles in autoimmunity. Studies have revealed that subgroups of T cells, including regulatory T cells (Treg) and helper T cells (Th), especially follicular Th cells (Tfh), Th2 and Th17, were involved in the pathogenesis of AAV (16–19). The exhaustion of T cells could predict the favorable prognosis of AAV (20). Notably, the activation of T cells requires two signals. The first signal is traditional T cell receptor (TCR) signaling triggered by the recognition between TCR and specific peptides from the major histocompatibility complex (MHC) on the surface of antigen-presenting cells (APC). Immune checkpoint (IC) molecules transmit the second signal. ICs are a class of surface proteins to provide co-stimulatory or co-inhibitory signals by combining with the corresponding receptors or ligands on the surface of APCs (21, 22). Currently, immune checkpoint inhibitors (ICIs) have been used in treating various malignant tumors (23–26). However, 3.5% of patients treated with ICIs occurred rheumatic disease (27), suggesting that targeting ICs might have potential values in the treatment of rheumatic diseases. In this review, we summarized the association between ICIs and AAV, focused on the characteristics of ICs, and explored the potential therapeutic prospect of targeting ICs in AAV.
2 The association between ICIs and AAV
Currently, there are three types of ICIs in blocking co-inhibitory pathways, targeting programmed cell death 1 (PD-1) (nivolumab, pembrolizumab and cemiplimab), programmed death ligand 1 (PD-L1) (avelumab, atezolizumab and durvalumab), and cytotoxic T lymphocyte-associated molecule 4 (CTLA-4) (ipilimumab and tremelimumab). As the signal transductions of co-inhibitory were blocked, T cells could be activated, and then the tumor immunity could be enhanced (28, 29). Consequently, the inflammatory response might also be increased due to the activation of T cells, resulting in most patients developing immune-related adverse events (irAEs). The damages of irAEs could involve multiple organs, including but not limited to the skin, the gastrointestinal tract, the lung, and the kidney (29, 30). It is shown that CTLA-4 inhibitors may induce more irAEs than PD-1 inhibitors, and the combination of these two kinds of inhibitors can further increase the incidence of irAEs (31, 32).
Despite the rheumatic irAEs (Rh-irAEs) are not common in all irAEs, rheumatologists are still concerned about these Rh-irAEs. It was reported that the frequent Rh-irAEs were inflammatory arthritis and inflammatory myopathy. ICIs-induced vasculitis is less common than other rheumatic diseases and mainly affects the medium and large arteries (27, 29, 33). In Table 1, we summarized the cases of ICIs-induced AAV reported to date (23–25, 34–40). Interestingly, even with ICIs treatment, some AAV patients in remission did not relapse (41–43). Therefore, we speculated that the ICs molecules might be involved in the pathogenesis of AAV. What is more, different ICs might dominate in various stages.
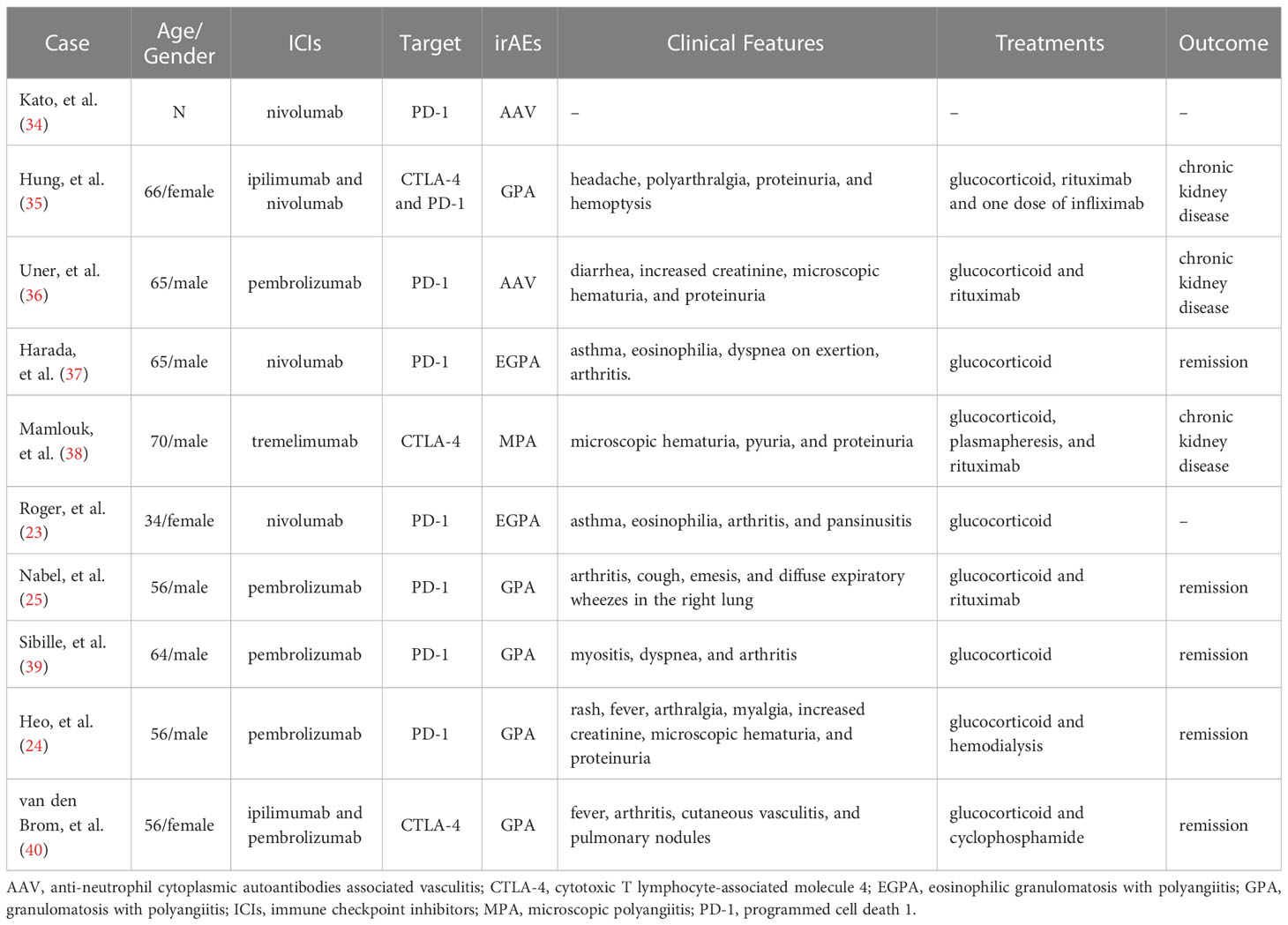
Table 1 Demographic characteristics and clinical data of patients with ICIs-induced AAV.
3 Co- stimulatory signal pathways
The co-stimulatory molecules expressed on the surface of T cells contained CD28, inducible T-cell co-stimulator (ICOS), OX40, and others (Figure 2). They will be discussed in detail below.
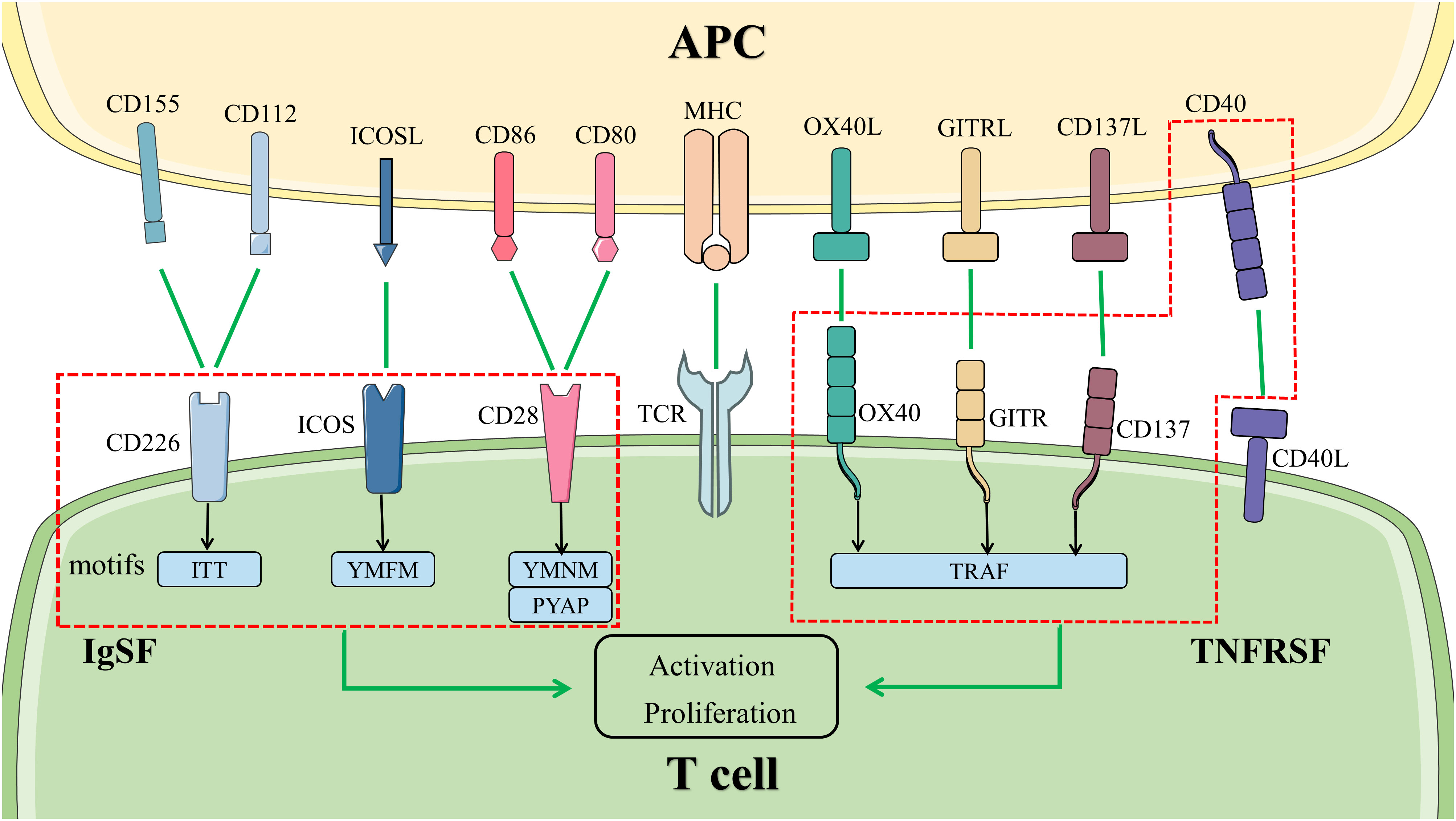
Figure 2 Co-stimulatory signal pathways in T cells. The activation and proliferation of T cells at least need two signals. The first signal is provided by the binding of TCR to MHC with antigenic polypeptide processed by APC. Co-stimulatory molecules bind to the ligand or receptor presented by APCs, transmitting the second signal. The molecules in the left box are members of IgSF, activating the signal pathways downstream by different motifs in the cytoplasmic tail. The members of TNFRSF are represented in the right box. They bind to TRAF to activate the signal pathways downstream. Abbreviations: APC, antigen-presenting cell; GITR, glucocorticoid induced tumor necrosis factor receptor; ICOS, inducible T-cell co-stimulator; IgSF, immunoglobulin superfamily; MHC, major histocompatibility complex; TCR, T cell receptor; TNFRSF, tumor necrosis factor receptor superfamily; TRAF, tumor necrosis factor receptor-associated factor.
3.1 CD28 signal pathway
CD28 is a member of the immunoglobulin (Ig) superfamily (IgSF) with typical Ig variable (IgV) domains. This transmembrane protein of 44 kDa is composed of a disulfide-linked homodimer. It was involved in the formation of immunologic synapses, the phosphorylation of proteins, and the remodeling of actin in T cells. Consequently, T cells were activated and produced cytokines (44–46). CD28 signal transduction is relied on two motifs in its cytoplasmic tail: YMNM and PYAP. The phosphorylated Src homology-2 (SH2) domain in the proximal YMNM motif binds the p85 subunit of phosphatidylinositol 3-kinase (PI3K), the growth factor receptor-bound protein 2 (GRB2), and the GRB2-related adapter protein 2. The distal PYAP motif combines with the lymphocyte cell-specific protein-tyrosine kinase (LCK) and GRB2. Their bindings could activate the downstream targets, including nuclear factor-κB (NF-κB), nuclear factor of activated T cells (NFAT), mammalian target of rapamycin (mTOR), and mitogen-activated protein kinase to affect cell cycle progression, apoptosis, and especially interleukin (IL)-2 transcription (47).
CD80 and CD86 are cognate ligands of CD28, which are mainly expressed on the surface of APCs, such as B cells, dendritic cells (DCs), and monocytes (17, 22, 48). However, there are some differences between their features and functions. CD80, which is existed as a dimer, is expressed rarely on resting B cells whereas the expression of CD86, which is in the form of a monomer, is higher than CD80. When B cells were activated, the density of CD86 on its surface increased early and even more compared with CD80 (49, 50). In animal experiments, it is demonstrated that in CD86-deficient mice, neither the antibody isotypes were switched nor germinal centers (GCs) were formed, whereas it is contrary in CD80-deficient mice (51). Furthermore, because CTLA-4, a co-stimulatory molecule with highly homologous to CD28, has a higher affinity for CD80 and CD86, it competitively inhibits the bindings of CD28 to CD80 and CD86, and actually restrains the cellular immune responses (52).
CD28 signal pathway plays a vital role in vasculitis. Zhang et al. constructed human artery-severe combined immunodeficiency mice chimeras with peripheral blood mononuclear cells from patients with giant cell arteritis (GCA) to induce vasculitis. Blocking the CD28 signal pathway significantly disrupted T-cell metabolic fitness and inhibited the remodeling of the vessel wall (53). In Takayasu’s arteritis (TAK), another large vasculitis, the active patients had higher mRNA levels of CD28 than the inactive patients (54). In patients with active GPA, T cells had a higher proliferative response to the stimulation of CD2/CD28 than healthy controls (HCs) (55). In addition, the expressions of CD80 and CD86 were also significantly increased in CD19+ B cells from patients with frequently relapsing EGPA (17). Besides ICs in cell membranes, soluble ICs have received attention gradually. Soluble ICs were produced by the proteolytic cleavage of extracellular regions, or by alternative splicing (56). Elevated soluble CD28(sCD28) levels was observed in sera samples in patients with active AAV. Such increase in was significantly positively correlated with disease activity markers, such as the Birmingham Vasculitis Activity Score, C-reactive protein, and erythrocyte sedimentation rate (57). Noteworthy, sCD28 level decreased when AAV patients from the active state became inactive after treatment (57), suggesting that sCD28 might play a potential immunopathological role in AAV and could be a novel biomarker to evaluate disease activity. Therefore, targeting the CD28 signal pathway may be effective for AAV. Abatacept, a CTLA-4-Ig fusion protein composed of the ligand-binding domain of CTLA-4 and the modified Fc portion of IgG, can block CD28 signal transduction by bindings to CD80 and CD86 (58). An open-label trial reported that Abatacept had improved the disease condition in patients with non-severe relapsing GPA (59). We prefer to inhibit the CD28 signal transduction directly. FR104 is a pegylated antigen-binding fragment (Fab) antibody. In the non-human primate (NHP) graft-versus-host disease (GVHD) model, the survival of GVHD-free was improved by FR104/sirolimus. Still, the overall survival was not improved because of the sepsis and a paralyzed interferon (IFN)-γ response in some patients without GVHD (60). In conclusion, it has clinical significance for targeting the CD28 signal pathway. Nevertheless, the challenge is to develop a more effective anti-CD28 antibody with fewer side effects.
3.2 ICOS signal pathway
Even though ICOS and CD28 belong to the same family, they still have several differences. Firstly, ICOS is expressed on the surface of activated T cells. The activation of T cells by TCR and CD28 signal is essential for the expression of ICOS. Then, ICOS can promote the activation of T cells further (61). Secondly, there is only one particular YMFM motif in the cytoplasmic tail of ICOS. YMFM binds to the p50α and p85 subunits of PI3K and tends to recruit the former. Subsequently, the AKT signal enhances markedly. The ICOS-PI3K-AKT pathway promotes the expression of cytokines as well as induces the formation of Tfh cells. Tfh cells migrate into the follicles, maintain in GCs, and promote the differentiation of B cells into plasma cells to secrete ANCA (21, 62–64). Thirdly, ICOS could combine with the ICOS ligand (ICOSL) instead of CD80 and CD86 (65).
ICOSL is expressed on the surface of B cells, macrophages, fibroblasts, muscle cells, podocytes, and other cells (65–68). Several factors regulated the expression of ICOSL on the surface of B cells. B cell receptor (BCR) signal reduced the expression of ICOSL on naïve B cells, which affected the formation of Tfh cells. The BCR signal was more substantial, and the reduction of ICOSL was more obvious. This inhibitory response could be reversed by the CD40 signal (69, 70). Similarly, in both NF-κB–inducing kinase (NIK) KO mice and B-cell activating factor belonging to tumor necrosis factor (TNF) receptor (TNFR) family receptor-deficient mice, Hu et al. revealed that the expression of ICOSL decreased significantly. They discovered the recombinant ICOSL-Fc fusion protein could increase the levels of Tfh cells in NIK KO mice, suggesting ICOSL is a target of the noncanonical NF-κB pathway (71).
Several studies focused on the differences of ICOS+ Tfh cells in vasculitis (19, 72, 73). Circulating CD4+ CXCR5+ ICOS+ Tfh cells were elevated and correlated with disease activity in patients with Henoch-Schönlein purpura (HSP) (72). The same results were observed in patients with active MPO-AAV (19) and Behcet’s disease (73). In patients with active AAV, the serum concentration of soluble ICOS was also higher than HCs (57). In addition, the production of pro-inflammatory factors was decreased by blocking the ICOS signal (73). AMG 557 is a fully human IgG2 monoclonal antibody (mAb) with a higher affinity to ICOSL that prevents the binding of ICOS and ICOSL. The safety and tolerability of AMG 557 are acceptable in patients with mild, stable systemic lupus erythematosus (SLE) (74). In another phase Ib, randomized, double-blind, placebo-controlled study, the potential efficacy of AMG 557 was evaluated (75). Fewer patients receiving AMG 577 (3 of 10 patients) or placebo (1 of 10 patients) achieved the primary efficacy endpoints. On day 169, compared with the placebo group, more patients in the AMG 557 group showed a ≥4-point improvement in the SLE disease activity index (SLEDAI) (70.0% vs. 20.0%, p=0.07), indicating the potential efficacy of AMG 557 (75). As mentioned above, ANCA is thought to be the pathogenic antibody of AAV, and the secretion of ANCA by plasma cells is regulated by Tfh cells. Therefore, the investigations on mAb targeting ICOS molecules on the surface of Tfh cells will be one of research directions of AAV treatment in the future.
3.3 OX40 signal pathway
The costimulatory molecule OX40 belongs to the TNFR superfamily (TNFRSF), also known as TNFRSF4 or CD134. Similar to other members of TNFRSF, OX40 is a type I transmembrane glycoprotein with four cysteine-rich domains in the extracellular region (76). Different from ICOS, OX40 expressed on CD4+ T cells was driven by the TCR signal. After the activation of T cells, the expression of OX40 is promoted by CD28 and CD40 signals (77). In the intracellular region, OX40 binds to TNF receptor-associated factor (TRAF) 2, TRAF3, TRAF 5, and TRAF6, which activates the NF-κB, PI3K-AKT, and NFAT signal pathways downstream. It promotes the survival of T cells and the secretion of cytokines (21, 78).
The only ligand of OX40 is OX40L (also known as TNFSF4, CD252), which is expressed on the surface of DCs, B cells, T cells, vascular endothelial cells (VECs), mast cells, Langerhans cells, and other types of cells (79–84). There is a conserved extracellular TNF homology domain on the OX40L for trimerization (76). OX40L trimer combines with three OX40 molecules to polarize T cells to Th cells, expand Treg cells, sustain the function of memory T cells, and promote the adhesion of activated T cells to VECs (77, 85).
The OX40 signal pathway is vital in rheumatic diseases. In patients with SLE or TAK, the expression of OX40L was enhanced on VECs (82, 86). Besides the expression of OX40, the soluble OX40L was increased in patients with HSP, and both of them were associated with disease activity (87). It was observed in patients with AAV that the expression of CD134 as well as the CD134+ T cells were increased. The majority of CD134+ T cells mainly secreted TNF-α (88, 89). The results were consistent with in vitro studies. So far, several mAbs against OX40 (KHK 4083 and GBR 830) and OX40L (KY 1005) have been developed. KHK 4083 demonstrated the safety and tolerability in patients with mild to moderate plaque psoriasis (90) and moderate to severe ulcerative colitis (UC) (91). In the phase II a study, GBR 830 showed the therapeutic potential for patients with moderate to severe atopic dermatitis (AD) (92). The pharmacological activity of KY 1005 in humans was evaluated. It is considered that targeting OX40L might be effective (93). These findings indicated that the OX40 signal pathway might be a potential therapeutic target in patients with AAV. The problem is that OX40 blockade might inhibit the function of Treg cells, which then leads to disease relapse. In the NHP GVAD model, Tkachev et al. reported that the combined administration with KY 1005 and sirolimus could control the activation of effector T cells while maintaining the reconstitution of Treg cells (94). It seems that combination treatments may be more promising.
3.4 Other co-stimulatory signal pathways
There are some other co-stimulatory molecules involving in the activation of T cells as well. We focused on the possibility of targeting CD40L, glucocorticoid induced TNF receptor (GITR), and CD137 for therapy in AAV.
CD40L (also known as TNFSF5 or CD154), mainly expressed in activated T cells and platelets, is the ligand of CD40 (95, 96). CD40, expressed in B cells, monocytes, DCs, and VECs, has similar structures to the other members of TNFRSF (96–99). The connection of CD40 and CD40L regulated Th cells differentiation, maintained GCs response, activated the CD8+ cytotoxic T lymphocytes (CTL), and sustained memory CTLs (100). Although the gene polymorphisms of CD40 were not related to the susceptibility of AAV (101), the levels of CD40L and soluble CD40L were raised in AAV patients, which was correlated with disease activity (102). It was a pity that the anti-CD40 mAb BI 655064 and the polyethylene glycol conjugated anti-CD40L Fab’ fragment dapirolizumab pegol (DZP) both did not achieve the expected clinical efficacy in the phase II studies (103, 104). Furthermore, blocking CD40L could lead to severe thromboembolic events. Because of the myocardial infarction and thromboembolic events occurring in patients, the study of BG9588 was terminated (105). In comparison, the CD40L binding protein VIB4920, which lacks an Fc domain, may have more therapeutic potential in AAV. By blocking the downstream CD40 signal, VIB4920 could inhibit the differentiation of plasma cells without platelet aggregation. The safety and efficacy of VIB4920 have been preliminarily demonstrated in patients with rheumatoid arthritis (RA) (106), and further exploration of the clinical efficacy is needed.
GITR (also known as TNFRSF18) and CD137 (also known as 4-1BB or TNFRSF9) are both members of TNFRSF (85). They are expressed in different types of activated T cells, that is, GITR is mainly expressed in Treg cells (107) while CD137 is primarily expressed in CD8+ T cells (108). The stimulations of the GITR signal as well as the CD137 signal enhanced T cells proliferation, raised the secretion of cytokines, and eliminated the suppressive effect of Treg cells (109–111). Compared to HCs, the expression of GITR was increased in patients with GPA and significantly correlated with disease activity (88). Giscombe et al. reported that, similar to animal tests, the expanded CD8+ T cells expressed more CD137 (89). The anti-GITR mAb exacerbated the disease severity in the murine model of collagen-induced arthritis (CIA) (112) and experimental autoimmune encephalomyelitis (EAE) (113). In contrast, the agonistic anti-CD137 mAb improved the CIA and EAE maybe by inducing expansion of CD11c+ CD8+ T cells (114, 115). Among the members of TNFRSF, CD137 was superior in increasing the secretion of cytokines by CD8+ T cells (116, 117). So far, the effects of inhibiting or stimulating GITR and anti-CD137 in the treatment of AAV are unknown. There is no doubt that this is a tempting field worthy of further exploration for AAV treatment.
4 Co-inhibitory signal pathways
The exhaustion of T cells in AAV and other autoimmune diseases (AIDs) predicts advantageous clinical outcomes. The expressions of co-inhibitory molecules restrain the differentiation of non-exhausted T cells, indicating the importance of co-inhibitory molecules in the exhaustion of T cells (20). At present, the research on PD-1 and CTLA-4 signal pathways is reported widely, whose blockers have been used in the treatment of many malignant tumors. In the previous section, we summarized the association between AAV and inhibitors of PD-1, PD-L1, and CTLA-4. In this section, we described more details on the mechanisms and roles of PD-1 and CTLA-4 signal pathways in AAV. We assessed the therapeutic potential of other co-inhibitory molecules associated with the exhaustion of T cells as well (Figure 3).
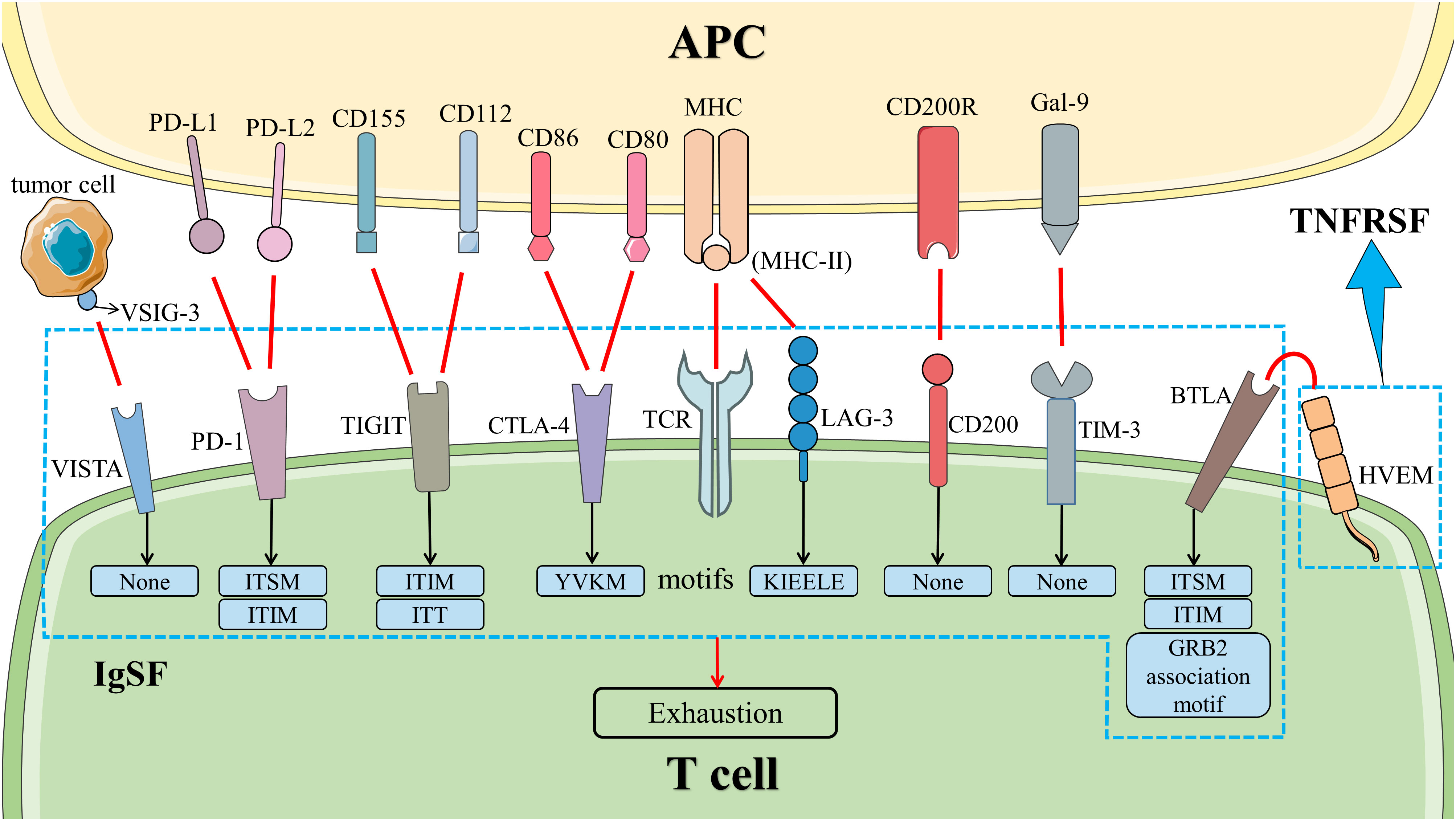
Figure 3 Co-inhibitory signal pathways in T cells. The co-inhibitory signal pathways induced the exhaustion of T cells. HVEM is a member of TNFRSF. Other molecules belong to IgSF. Except for LAG-3, VISTA, TIM-3, and CD200, all of them have inhibitory signal motifs in the cytoplasmic tail. LAG-3 has a unique KIEELE motif whose role is unclear, while VISTA, TIM-3, and CD200 do not have any motifs in the cytoplasmic tail. Abbreviation: APC, antigen-presenting cell; BTLA, B and T lymphocyte attenuator; CTLA-4, cytotoxic T lymphocyte-associated molecule 4; Gal-9, galactin-9; HVEM, herpesvirus entry mediator; IgSF, immunoglobulin superfamily; LAG-3, lymphocyte activation gene 3; MHC, major histocompatibility complex; PD-1, programmed cell death 1; PD-L1, programmed death ligand 1; TCR, T cell receptor; TIGIT, T‐cell immunoglobulin and ITIM domain; TIM-3, T cell immunoglobulin and mucin domain-containing protein 3; TNFRSF, tumor necrosis factor receptor superfamily; VISTA, V-domain immunoglobulin suppressor of T cell activation; VSIG-3, V-set and immunoglobulin domain containing 3. .
4.1 PD-1 signal pathway
PD-1 is a co-inhibitory receptor expressed in many types of activated or exhausted immune cells (118). PD-1, a transmembrane protein with 288 amino acids, consists of an extracellular domain, a transmembrane domain, and an intracellular cytoplasmic tail. Although PD-1 belongs to the CD28 family, it has unique molecular characteristics. PD-1 has an IgV-like domain in the extracellular domain whereas an immunoreceptor tyrosine-based inhibition motif (ITIM) and an immunoreceptor tyrosine-based switch motif (ITSM) in the cytoplasmic tail (119). ITIM and ITSM mediated the inhibition signal of PD-1, while ITSM might be more important. It has been reported that only the mutation of ITSM affected the transduction of the PD-1 signal (120–122). With the bindings of PD-1 and its ligand, ITSM was phosphorylated, the SH2-containing protein tyrosine phosphatase 2 (SHP-2) was recruited, and the PI3K/AKT signal pathway was inhibited, which suppressed the activation of T cells and the production of pro-inflammatory cytokines (119, 123). RAS/MEK/ERK signal pathway, which is responsible for activating T cells, is another target of the PD-1 signal to suppress the activity of T cells and concurrent inflammation (119).
PD-L1 and PD-L2 are the ligands of PD-1. Both of them are members of B7 family, which are known as B7-H1 and B7-DC, respectively. The expression of PD-L1 is detected on many types of cells (T and B cells, DCs, VECs, placenta, eyes, and others) whereas PD-L2 is mainly limited to express on the surface of macrophages, DCs, and mast cells (119, 124). As PD-L1 and PD-L2 are close in the distance on the same chromosome, they are regulated similarly by inflammatory factors (IFN-1, IFN-2, TNF-α and ILs). In addition, PD-L1 is regulated with post-translational regulation and microRNAs, including but not limited to miR-513, miR-155, miR-34a, miR-142-5p, and miR-93 (118, 125, 126). PD-1 is not the only receptor for PD-L1 and PD-L2. PD-L1 can also bind to CD80 to inhibit the immune response of T cells (127), while PD-L2 may combine with repulsive guidance molecule B (RGMB) to impair respiratory tolerance (128). Therefore, targeting PD-L1 or PD-L2 is also important immunotherapy, and actually, PD-L1 inhibitors have been developed to treat malignant tumors.
Zhang et al. demonstrated that, in contrast to patients with GCA, PD-1+ T cells were not enriched at renal lesions in patients with GPA, suggesting that PD-1 might play a different role in different diseases (129). Similarly, Zeisbrich et al. measured the expression of PD-L1 in monocytes and found that the frequency of PD-L1+ monocytes was not related to renal involvement although these monocytes tended to decrease in active patients with AAV (130). A previous study revealed that the expression of PD-1 on Th cells was lower in patients with localized GPA than that in patients with systemic GPA (131). We then detected the expression of PD-1 in Tfh cells. Compared to HCs, the expression intensity of PD-1 in Tfh cells was higher in patients with MPO-AAV. We also found that the expression of ICOS/PD-1 instead of PD-1 was associated with the levels of MPO-ANCA (19), indicating that co-stimulatory and co-inhibitory molecules were involved in the activation of T cells together. Although the fusion proteins containing an anti-PD-1 single-chain variable fragment could improve symptoms of type 1 diabetes (TID) and EAE models (132), its effects in AAV have yet to be explored. Soluble ICs in PD-1 signaling pathway also play an important role in AAV. It was observed that although the serum concentration of soluble PD-L2 in active AAV was lower than that in HCs, its level was significantly increased after treatment (57). In addition, the levels of serum soluble PD-1 (sPD-1) was higher in MPO-AAV (133). Noting that sPD-1 could restrain the exhaustion of T cells by binding to PD-L1 in cell membranes (134), sPD-1 is therefore likely an effective therapeutic target in MPO-AAV. Significantly, Targeting the PD-1 signal pathway alone may be less effective because of the poorer inhibitory impact on PD-1 of T cells (131). This may explain why PD-1 inhibitors induce fewer irAEs than CTLA-4 inhibitors. Compared to PD-1, PD-L1 may be a better target. The frequency of PD-L1+ monocytes is negatively correlated with the level of ANCA (130), so increasing the expression of PD-L1 may reduce the level of ANCA and improve disease activity. In lupus nephritis mice models, the recombinant adenovirus containing the full-length PD-L1 gene improved the renal lesions (135). In the same experiment, the anti-ICOSL mAb was also added to reinforce this process (135). As the results we found, targeting both ICOS and PD-1 in patients with AAV may be more effective.
4.2 CTLA-4 signal pathway
CTLA-4 is transiently expressed in the activated T cells. Beforehand, it localized in intracellular compartments of naïve T cells (136, 137). The gene expressing CTLA-4 is located in the same chromosomal region (2q33-34) as the gene expressing CD28, so CTLA-4 and CD28 exhibit a significant homology (138). CTLA-4 and CD28 bind to the same ligand but transduce an opposite signal. CTLA-4 leads to co-inhibitory signaling whereas CD28 provides the co-stimulatory signal. As a member of IgSF, CTLA-4 is also a transmembrane protein that contains an IgV-like domain in the extracellular domain and a YVKM motif in the intracellular cytoplasmic tail (139, 140). The IgV-like domain is the site that binding to CD80 and CD86 for CTLA-4, and the YVKM motif is important for CTLA-4 signal transduction in T cells. The interaction of the phosphorylated YVKM motif with SHP2 and serine/threonine protein phosphatase 2A (PP2A) dephosphorylated the TCR-CD3ζ complex so that the activated signal from TCR was suppressive (21). On the other hand, the phosphorylation of the YVKM motif inhibited the interaction with the clathrin-associated adaptor complex AP-2, resulting in the internalization of CTLA-4 (141). CTLA-4 expressed on the surface of activated T cells captured and degraded CD80 and CD86 by trans-endocytosis to inhibit the co-stimulatory signal from CD28 (142). Ultimately, CTLA-4 suppressed the activation of T cells.
In patients with GPA, the expression of CTLA-4 in T cells was found to be elevated, and to be related to disease severity. After stimulating with phytohaemagglutinin (PHA), the expression of CTLA-4 in T cells in patients with GPA did not increase, suggesting that the activation of T cells in patients with GPA was persistent (143). As mentioned above, Abatacept bind to CD80 and CD86 to inhibit the activation of T cells, and its efficacy in patients with GPA has been reported (59). Although agonistic CTLA-4 antibodies have not yet been successfully developed, directly targeting CTLA-4 may be less effective because of the CTLA-4 endocytosis. It is probably more effective to inhibit the bindings of CD28 to CD80 and CD86. In addition, polymorphisms of the CTLA-4 gene were associated with GPA (144–146), while they were not related to MPA in Japanese patients (147), indicating that there were race differences in CTLA-4 polymorphism. A meta-analysis showed that CTLA-4 (AT)86 and CTLA-4 (AT)106 were significantly associated with AAV in the Caucasian patients instead of the Asian patients (144). Therefore, when targeting CTLA-4, genetic variation should be considered to avoid invalid treatment.
4.3 Other co-inhibitory signal pathways
T cell Ig and mucin domain-containing protein 3 (TIM-3), as a co-inhibitory receptor for Th1, was reported in 2002 to be associated with the severity of EAE (148). After that, it was shown that TIM-3 was expressed in Treg cells, DCs, natural killer (NK) cells, and macrophages (149, 150). Similar to other members of IgSF, TIM-3 contains an IgV domain, a mucin domain, a transmembrane domain, and a cytoplasmic tail lacking inhibitory signaling motifs (151). TIM-3 binds to ligands through the IgV domain, and five conserved tyrosine residues at the cytoplasmic tails trigger the signaling downstream (152). Galactin-9 (Gal-9), phosphatidyl serine (PtdSer), high mobility group protein B1 (HMGB1), and carcinoembryonic antigen cell adhesion molecule 1 (CEACAM-1) are known ligands of TIM-3. Gal-9 is the earliest discovered as well as the most explored ligand. TIM-3-Gal-9 signal pathway induced intracellular calcium flux, apoptosis, and the suppression of Th1 (153). PtdSer and HMGB1 did not directly suppress the activation of T cells, but rather affected the immune responses of DCs (154, 155). CEACAM-1 promoted the exhaustion of T cells through cis- and trans-interactions with TIM-3 (156). In patients with AAV, the expression of TIM-3 was significantly reduced on DCs, and blocking TIM-3 enhanced the expression of DC cytokines. Also, there were no differences in the expression of TIM-3 on the surface of different T cell subtypes (CD4+ T cells and CD8+ T cells) between MPO-AAV and HCs (157). Further explorations might be needed to assess the expression of TIM-3 on Th and Treg cells. Studies have shown that the serum concentrations of soluble TIM-3 correlated with the diseases state of AAV (57, 133), i.e. it was increased in active AAV (57), and could predict the relapse in PR3-AAV with rituximab treatment (133). However, the specific mechanism of soluble TIM-3 in AAV remains to be further examined before considering as a therapeutic target. Yoon et al. demonstrated that the serum Gal-9 levels were independently related to disease activity in patients with AAV (158). In mice models of CIA and SLE, injection of Gal-9 improved symptoms (159, 160). Although targeting the TIM-3 signal pathway has a potential efficacy in AAV, the problem is the need to clarify in which immune cells the TIM-3 signal pathway is more dominant to confirm the effectiveness. Another problem is that TIM-3 is not the only receptor for its ligand, so targeting the ligands of TIM-3 may not completely enhance TIM-3 signaling.
B and T lymphocyte attenuator (BTLA, also known as CD272) is mainly expressed in B and T cells, especially in naïve B cells and Th1 (161). BTLA is a co-inhibitory molecule with similar structures to PD-1 and CTLA-4 (162). Similar to PD-1, ITSM and ITIM in the cytoplasmic tail of BTLA inhibit the activation of T cells. However, unlike PD-1, SHP-1 rather than SHP-2 is mainly recruited by BTLA (163, 164). The third signal motif in the cytoplasmic tail of BTLA is the GRB2 association motif, which binds to GRB2 and p85 subunits of PI3K and induces the activation of T cells (165). Accordingly, BTLA transmits bidirectional signaling. Herpesvirus entry mediator (HVEM, also known as TNFRSF14) is a ligand of BTLA. HVEM is a member of TNFRSF, which is expressed in T cells, B cells, NK cells, monocytes, and neutrophils (166). There are two types of interaction between HVEM and BTLA. When BTLA and HVEM interacted in the same T lymphocyte, a cis complex was formed, inhibiting HVEM-dependent NF-κB activation (167). When BTLA or HVEM was expressed in APCs, trans interaction provided a co-stimulatory signal (168). Therefore, the regulation of the BTLA-HVEM signal pathway in AIDs is complex. In patients with remission AAV, the expression of BTLA was decreased only on double negative T-cells (CD3+CD4-CD8-). In vitro experiments, it has been shown that agonistic anti-BTLA antibody inhibit the activation and proliferation of T cells, especially Th17 (169), suggesting that targeting BTLA to inhibit the activation of T cells may be one of the future therapeutic directions of AAV.
V-domain Ig suppressor of T cell activation (VISTA), also known as PD-1 homolog (PD-1H), is a member of IgSF first discovered in 2011 (170). Different from other members of IgSF, VISTA has four additional invariant cysteines (170, 171). Subsequently, the same laboratory confirmed that VISTA in humans is a co-inhibitory molecule, which inhibits the proliferation of T cells and the production of cytokines (172). VISTA is mainly expressed in hematopoietic cells, especially in CD11bhi myeloid cells. Within the T cell compartment, the expression of VISTA was highest in naïve T cells and FoxP3+ Treg cells (171, 172). It is certain that VISTA is a ligand for T cells (170, 172). Moreover, it may also be a receptor in T cells to transmit inhibitory signals (173). Wang et al. demonstrated that V-set and Ig domain containing 3 (VSIG-3) is a ligand of VISTA. The binding between VISTA and VSIG-3 induced the inhibitory effects (174). It is a pity that VSIG-3 is mainly expressed in tumor cells but not in normal immune cells (174). Currently, targeting VISTA is explored in AIDs. It was reported that VISTA KO mice developed SLE (175) and EAE (176). In mice models with SLE, agonistic VISTA mAb improved symptoms (175). In patients with AAV, VISTA was expressed in mononuclear phagocytes, CD4+ T cells, and CD8+ T cells. Compared to patients with a lower expression of VISTA, patients with a higher expression of VISTA might have a higher risk of renal progression (177). Therefore, targeting VISTA may inhibit disease severity in patients with AAV, which needs more experiments to confirm.
T‐cell Ig and ITIM domain (TIGIT), CD200, and lymphocyte activation gene 3 (LAG-3) are popular co-inhibitory molecules in recent studies. CD155 and CD112 are ligands of TIGIT, binding to CD226 as well (152). CD200 binds to CD200Rs, especially CD200R1 (178). Besides MHC-II, fibrinogen-like protein 1 (FGL-1) was also discovered to be a ligand for LAG-3 (179, 180). These signal pathways transmit inhibitory signals. As a result, their roles in AIDs are noted. TIGIT-Ig fusion protein has revealed the therapeutic effects in mice with SLE (181). In EAE mice models, agonistic TIGIT mAb as well as CD200-Fc fusion protein improved disease severity (182, 183). CD200-Fc fusion protein reduced the disease severity of CIA at the clinical and histologic levels (184). Remarkably, LAG-3-deficient mice do not induce AIDs. After exposure to mercury (Hg), it not only had increased susceptibility to AIDs but also did not respond to tolerance induction (185). In AAV, there are no relevant reports about the influences of TIGIT, CD200, and LAG-3, so it is unknown whether targeting these signal pathways will improve disease severity.
5 Conclusion
The pathogenesis of AAV involves multiple aspects of innate immunity and adaptive immunity in which the role of T cells is pivotal and complex. With the understanding of the IC molecules, its importance will be confirmed further in AAV. Firstly, some ICIs used for malignant tumors induced the attack or relapse of AAV. Secondly, inhibiting the co-stimulatory signal pathways or enhancing the co-inhibitory signal pathways inhibited the activation and proliferation of T cells so that AIDs could be improved. In AAV, although it is demonstrated that Abatacept is effective in clinical trials, the efficacy of targeting other ICs has not been demonstrated. Thirdly, since co-stimulatory and co-inhibitory molecules work together to regulate T cells, it may be more reasonable to target multiple ICs simultaneously in severe or refractory cases. The questions to be aware of are that targeting ICs may increase the risk of tumors and infections, and different ICs are dominant in different subsets of T cells, so it is required to precise dosing and localization. In conclusion, targeting ICs has therapeutic potential, and more preclinical research is needed to clarify their effectiveness and safety in AAV treatment.
Author contributions
PSCL originated the topic and revised the manuscript. ZS wrote the outline, organize and supervise the writing. MP and HZ wrote the original text and figures draft. RJ searched literatures and made the table. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Natural Science Foundation of China, Grant/Award Number: 81871296.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 Revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum (2013) 65(1):1–11. doi: 10.1002/art.37715
2. Grayson PC, Ponte C, Suppiah R, Robson JC, Craven A, Judge A, et al. 2022 American College of Rheumatology/European alliance of associations for rheumatology classification criteria for eosinophilic granulomatosis with polyangiitis. Ann Rheum Dis (2022) 81(3):309–14. doi: 10.1136/annrheumdis-2021-221794
3. Robson JC, Grayson PC, Ponte C, Suppiah R, Craven A, Judge A, et al. 2022 American College of Rheumatology/European alliance of associations for rheumatology classification criteria for granulomatosis with polyangiitis. Ann Rheum Dis (2022) 81(3):315–20. doi: 10.1136/annrheumdis-2021-221795
4. Suppiah R, Robson JC, Grayson PC, Ponte C, Craven A, Khalid S, et al. 2022 American College of Rheumatology/European alliance of associations for rheumatology classification criteria for microscopic polyangiitis. Ann Rheum Dis (2022) 81(3):321–6. doi: 10.1136/annrheumdis-2021-221796
5. Walker BS, Peterson LK, Koening C, White SK, Schmidt RL, Tebo AE. Performance of mpo-anca and Pr3-anca immunoassays for the stratification of specific anca-associated vasculitis: A systematic review and meta-analysis. Autoimmun Rev (2022) 21(6):103100. doi: 10.1016/j.autrev.2022.103100
6. Kitching AR, Anders HJ, Basu N, Brouwer E, Gordon J, Jayne DR, et al. Anca-associated vasculitis. Nat Rev Dis Primers (2020) 6(1):71. doi: 10.1038/s41572-020-0204-y
7. Pearce FA, Craven A, Merkel PA, Luqmani RA, Watts RA. Global ethnic and geographic differences in the clinical presentations of anti-neutrophil cytoplasm antibody-associated vasculitis. Rheumatol (Oxford) (2017) 56(11):1962–9. doi: 10.1093/rheumatology/kex293
8. Fujimoto S, Watts RA, Kobayashi S, Suzuki K, Jayne DR, Scott DG, et al. Comparison of the epidemiology of anti-neutrophil cytoplasmic antibody-associated vasculitis between Japan and the U.K. Rheumatol (Oxford) (2011) 50(10):1916–20. doi: 10.1093/rheumatology/ker205
9. Sharma RK, Lovstrom B, Gunnarsson I, Malmstrom V. Proteinase 3 autoreactivity in anti-neutrophil cytoplasmic antibody-associated vasculitis-immunological versus clinical features. Scand J Immunol (2020) 92(5):e12958. doi: 10.1111/sji.12958
10. London J, Dumoitier N, Lofek S, Dion J, Chaigne B, Mocek J, et al. Skewed peripheral b- and T-cell compartments in patients with anca-associated vasculitis. Rheumatol (Oxford) (2021) 60(5):2157–68. doi: 10.1093/rheumatology/keaa432
11. Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci U.S.A. (1990) 87(11):4115–9. doi: 10.1073/pnas.87.11.4115
12. Mathern DR, Heeger PS. Molecules great and small: The complement system. Clin J Am Soc Nephrol (2015) 10(9):1636–50. doi: 10.2215/CJN.06230614
13. Brilland B, Garnier AS, Chevailler A, Jeannin P, Subra JF, Augusto JF. Complement alternative pathway in anca-associated vasculitis: Two decades from bench to bedside. Autoimmun Rev (2020) 19(1):102424. doi: 10.1016/j.autrev.2019.102424
14. Massicotte-Azarniouch D, Herrera CA, Jennette JC, Falk RJ, Free ME. Mechanisms of vascular damage in anca vasculitis. Semin Immunopathol (2022) 44(3):325–45. doi: 10.1007/s00281-022-00920-0
15. Soderberg D, Segelmark M. Neutrophil extracellular traps in vasculitis, friend or foe? Curr Opin Rheumatol (2018) 30(1):16–23. doi: 10.1097/BOR.0000000000000450
16. Abdulahad WH, Stegeman CA, Limburg PC, Kallenberg CG. Skewed distribution of Th17 lymphocytes in patients with wegener's granulomatosis in remission. Arthritis Rheum (2008) 58(7):2196–205. doi: 10.1002/art.23557
17. Tsurikisawa N, Saito H, Oshikata C, Tsuburai T, Akiyama K. Decreases in the numbers of peripheral blood regulatory T cells, and increases in the levels of memory and activated b cells, in patients with active eosinophilic granulomatosis and polyangiitis. J Clin Immunol (2013) 33(5):965–76. doi: 10.1007/s10875-013-9898-x
18. Kerstein A, Schuler S, Cabral-Marques O, Fazio J, Hasler R, Muller A, et al. Environmental factor and inflammation-driven alteration of the total peripheral T-cell compartment in granulomatosis with polyangiitis. J Autoimmun (2017) 78:79–91. doi: 10.1016/j.jaut.2016.12.004
19. Xu J, Zhao H, Wang S, Zheng M, Shuai Z. Elevated level of serum interleukin-21 and its influence on disease activity in anti-neutrophil cytoplasmic antibodies against myeloperoxidase-associated vasculitis. J Interferon Cytokine Res (2022) 42(6):290–300. doi: 10.1089/jir.2022.0014
20. McKinney EF, Lee JC, Jayne DR, Lyons PA, Smith KG. T-Cell exhaustion, Co-stimulation and clinical outcome in autoimmunity and infection. Nature (2015) 523(7562):612–6. doi: 10.1038/nature14468
21. Chen L, Flies DB. Molecular mechanisms of T cell Co-stimulation and Co-inhibition. Nat Rev Immunol (2013) 13(4):227–42. doi: 10.1038/nri3405
22. Hid Cadena R, Reitsema RD, Huitema MG, van Sleen Y, van der Geest KSM, Heeringa P, et al. Decreased expression of negative immune checkpoint vista by Cd4+ T cells facilitates T helper 1, T helper 17, and T follicular helper lineage differentiation in gca. Front Immunol (2019) 10:1638. doi: 10.3389/fimmu.2019.01638
23. Roger A, Groh M, Lorillon G, Le Pendu C, Maillet J, Arangalage D, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) induced by immune checkpoint inhibitors. Ann Rheum Dis (2019) 78(8):e82. doi: 10.1136/annrheumdis-2018-213857
24. Heo MH, Kim HK, Lee H, Ahn MJ. Antineutrophil cytoplasmic antibody-associated rapid progressive glomerulonephritis after pembrolizumab treatment in thymic epithelial tumor: A case report. J Thorac Oncol (2017) 12(8):e103–e5. doi: 10.1016/j.jtho.2017.03.011
25. Nabel CS, Severgnini M, Hung YP, Cunningham-Bussel A, Gjini E, Kleinsteuber K, et al. Anti-Pd-1 immunotherapy-induced flare of a known underlying relapsing vasculitis mimicking recurrent cancer. Oncologist (2019) 24(8):1013–21. doi: 10.1634/theoncologist.2018-0633
26. Cano-Cruz LG, Barrera-Vargas A, Mateos-Soria A, Soto-Perez-de-Celis E, Merayo-Chalico J. Rheumatological adverse events of cancer therapy with immune checkpoint inhibitors. Arch Med Res (2022) 53(2):113–21. doi: 10.1016/j.arcmed.2021.09.004
27. Richter MD, Crowson C, Kottschade LA, Finnes HD, Markovic SN, Thanarajasingam U. Rheumatic syndromes associated with immune checkpoint inhibitors: A single-center cohort of sixty-one patients. Arthritis Rheumatol (2019) 71(3):468–75. doi: 10.1002/art.40745
28. Dulos J, Carven GJ, van Boxtel SJ, Evers S, Driessen-Engels LJ, Hobo W, et al. Pd-1 blockade augments Th1 and Th17 and suppresses Th2 responses in peripheral blood from patients with prostate and advanced melanoma cancer. J Immunother (2012) 35(2):169–78. doi: 10.1097/CJI.0b013e318247a4e7
29. Kostine M, Finckh A, Bingham CO, Visser K, Leipe J, Schulze-Koops H, et al. Eular points to consider for the diagnosis and management of rheumatic immune-related adverse events due to cancer immunotherapy with checkpoint inhibitors. Ann Rheum Dis (2021) 80(1):36–48. doi: 10.1136/annrheumdis-2020-217139
30. Crout TM, Lennep DS, Kishore S, Majithia V. Systemic vasculitis associated with immune check point inhibition: Analysis and review. Curr Rheumatol Rep (2019) 21(6):28. doi: 10.1007/s11926-019-0828-7
31. Tocut M, Brenner R, Zandman-Goddard G. Autoimmune phenomena and disease in cancer patients treated with immune checkpoint inhibitors. Autoimmun Rev (2018) 17(6):610–6. doi: 10.1016/j.autrev.2018.01.010
32. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med (2015) 372(26):2521–32. doi: 10.1056/NEJMoa1503093
33. Shen P, Deng X, Hu Z, Chen Z, Huang Y, Wang K, et al. Rheumatic manifestations and diseases from immune checkpoint inhibitors in cancer immunotherapy. Front Med (Lausanne) (2021) 8:762247. doi: 10.3389/fmed.2021.762247
34. Kato K, Mizuno T, Koseki T, Ito Y, Takahashi K, Tsuboi N, et al. Frequency of immune checkpoint inhibitor-induced vasculitides: An observational study using data from the Japanese adverse drug event report database. Front Pharmacol (2022) 13:803706. doi: 10.3389/fphar.2022.803706
35. Hung W, Cusnir I, Habib S, Smylie M, Solez K, Yacyshyn E. Immune checkpoint inhibitor-induced granulomatosis with polyangiitis. Rheumatol (Oxford) (2021) 60(6):e190–e1. doi: 10.1093/rheumatology/keaa818
36. Uner M, Alhasson B, Obhrai J, Bagnasco SM. Anca-associated pauci-immune necrotizing glomerulonephritis during the treatment with pembrolizumab. Virchows Arch (2021) 478(4):801–4. doi: 10.1007/s00428-020-02882-w
37. Harada M, Naoi H, Yasuda K, Ito Y, Kagoo N, Kubota T, et al. Programmed cell death-1 blockade in kidney carcinoma may induce eosinophilic granulomatosis with polyangiitis: A case report. BMC Pulm Med (2021) 21(1):6. doi: 10.1186/s12890-020-01375-5
38. Mamlouk O, Lin JS, Abdelrahim M, Tchakarov AS, Glass WF, Selamet U, et al. Checkpoint inhibitor-related renal vasculitis and use of rituximab. J Immunother Cancer (2020) 8(2):e000750. doi: 10.1136/jitc-2020-000750
39. Sibille A, Alfieri R, Malaise O, Detrembleur N, Pirotte M, Louis R, et al. Granulomatosis with polyangiitis in a patient on programmed death-1 inhibitor for advanced non-Small-Cell lung cancer. Front Oncol (2019) 9:478. doi: 10.3389/fonc.2019.00478
40. van den Brom RR, Abdulahad WH, Rutgers A, Kroesen BJ, Roozendaal C, de Groot DJ, et al. Rapid granulomatosis with polyangiitis induced by immune checkpoint inhibition. Rheumatol (Oxford) (2016) 55(6):1143–5. doi: 10.1093/rheumatology/kew063
41. Wulfken LM, Becker JC, Hayajneh R, Wagner AD, Schaper-Gerhardt K, Flatt N, et al. Case report: Sustained remission due to pd-1-Inhibition in a metastatic melanoma patient with depleted b cells. Front Immunol (2021) 12:733961. doi: 10.3389/fimmu.2021.733961
42. Yamada T, Masuda T, Yamaguchi K, Sakamoto S, Horimasu Y, Miyamoto S, et al. Non-small cell lung cancer treated by an anti-programmed cell death-1 antibody without a flare-up of preexisting granulomatosis with polyangiitis. Intern Med (2019) 58(21):3129–32. doi: 10.2169/internalmedicine.3018-19
43. Maul LV, Weichenthal M, Kahler KC, Hauschild A. Successful anti-Pd-1 antibody treatment in a metastatic melanoma patient with known severe autoimmune disease. J Immunother (2016) 39(4):188–90. doi: 10.1097/CJI.0000000000000118
44. Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. Cd28 costimulation: From mechanism to therapy. Immunity (2016) 44(5):973–88. doi: 10.1016/j.immuni.2016.04.020
45. June CH, Ledbetter JA, Gillespie MM, Lindsten T, Thompson CB. T-Cell proliferation involving the Cd28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Mol Cell Biol (1987) 7(12):4472–81. doi: 10.1128/mcb.7.12.4472-4481.1987
46. Martin PJ, Ledbetter JA, Morishita Y, June CH, Beatty PG, Hansen JA. A 44 kilodalton cell surface homodimer regulates interleukin 2 production by activated human T lymphocytes. J Immunol (1986) 136(9):3282–7.
47. Boomer JS, Green JM. An enigmatic tail of Cd28 signaling. Cold Spring Harb Perspect Biol (2010) 2(8):a002436. doi: 10.1101/cshperspect.a002436
48. Csernok E, Ai M, Gross WL, Wicklein D, Petersen A, Lindner B, et al. Wegener autoantigen induces maturation of dendritic cells and licenses them for Th1 priming Via the protease-activated receptor-2 pathway. Blood (2006) 107(11):4440–8. doi: 10.1182/blood-2005-05-1875
49. Bhatia S, Edidin M, Almo SC, Nathenson SG. Different cell surface oligomeric states of B7-1 and B7-2: Implications for signaling. Proc Natl Acad Sci U.S.A. (2005) 102(43):15569–74. doi: 10.1073/pnas.0507257102
50. MacPhee IA, Turner DR, Yagita H, Oliveira DB. Cd80(B7.1) and Cd86(B7.2) do not have distinct roles in setting the Th1/Th2 balance in autoimmunity in rats. Scand J Immunol (2001) 54(5):486–94. doi: 10.1046/j.1365-3083.2001.00998.x
51. Borriello F, Sethna MP, Boyd SD, Schweitzer AN, Tivol EA, Jacoby D, et al. B7-1 and B7-2 have overlapping, critical roles in immunoglobulin class switching and germinal center formation. Immunity (1997) 6(3):303–13. doi: 10.1016/s1074-7613(00)80333-7
52. Levine SM, Stone JH. New approaches to treatment in systemic vasculitis: Biological therapies. Best Pract Res Clin Rheumatol (2001) 15(2):315–33. doi: 10.1053/berh.2000.0146
53. Zhang H, Watanabe R, Berry GJ, Nadler SG, Goronzy JJ, Weyand CM. Cd28 signaling controls metabolic fitness of pathogenic T cells in medium and Large vessel vasculitis. J Am Coll Cardiol (2019) 73(14):1811–23. doi: 10.1016/j.jacc.2019.01.049
54. Tian Y, Li J, Tian X, Zeng X. Using the Co-expression network of T cell-Activation-Related genes to assess the disease activity in takayasu's arteritis patients. Arthritis Res Ther (2021) 23(1):303. doi: 10.1186/s13075-021-02636-2
55. Ludviksson BR, Sneller MC, Chua KS, Talar-Williams C, Langford CA, Ehrhardt RO, et al. Active wegener's granulomatosis is associated with hla-dr+ Cd4+ T cells exhibiting an unbalanced Th1-type T cell cytokine pattern: Reversal with il-10. J Immunol (1998) 160(7):3602–9.
56. Khan M, Arooj S, Wang H. Soluble B7-Cd28 family inhibitory immune checkpoint proteins and anti-cancer immunotherapy. Front Immunol (2021) 12:651634. doi: 10.3389/fimmu.2021.651634
57. Pyo JY, Yoon T, Ahn SS, Song JJ, Park YB, Lee SW. Soluble immune checkpoint molecules in patients with antineutrophil cytoplasmic antibody-associated vasculitis. Sci Rep (2022) 12(1):21319. doi: 10.1038/s41598-022-25466-x
58. Edner NM, Carlesso G, Rush JS, Walker LSK. Targeting Co-stimulatory molecules in autoimmune disease. Nat Rev Drug Discovery (2020) 19(12):860–83. doi: 10.1038/s41573-020-0081-9
59. Langford CA, Monach PA, Specks U, Seo P, Cuthbertson D, McAlear CA, et al. An open-label trial of abatacept (Ctla4-ig) in non-severe relapsing granulomatosis with polyangiitis (Wegener's). Ann Rheum Dis (2014) 73(7):1376–9. doi: 10.1136/annrheumdis-2013-204164
60. Watkins BK, Tkachev V, Furlan SN, Hunt DJ, Betz K, Yu A, et al. Cd28 blockade controls T cell activation to prevent graft-Versus-Host disease in primates. J Clin Invest (2018) 128(9):3991–4007. doi: 10.1172/JCI98793
61. Huang C, Zhu HX, Yao Y, Bian ZH, Zheng YJ, Li L, et al. Immune checkpoint molecules. possible future therapeutic implications in autoimmune diseases. J Autoimmun (2019) 104:102333. doi: 10.1016/j.jaut.2019.102333
62. Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. Icos receptor instructs T follicular helper cell versus effector cell differentiation Via induction of the transcriptional repressor Bcl6. Immunity (2011) 34(6):932–46. doi: 10.1016/j.immuni.2011.03.023
63. Xu H, Li X, Liu D, Li J, Zhang X, Chen X, et al. Follicular T-helper cell recruitment governed by bystander b cells and icos-driven motility. Nature (2013) 496(7446):523–7. doi: 10.1038/nature12058
64. Long Y, Feng J, Ma Y, Sun Y, Xu L, Song Y, et al. Altered follicular regulatory T (Tfr)- and helper T (Tfh)-cell subsets are associated with autoantibody levels in microscopic polyangiitis patients. Eur J Immunol (2021) 51(7):1809–23. doi: 10.1002/eji.202049093
65. Yoshinaga SK, Whoriskey JS, Khare SD, Sarmiento U, Guo J, Horan T, et al. T-Cell Co-stimulation through B7rp-1 and icos. Nature (1999) 402(6763):827–32. doi: 10.1038/45582
66. Koh KH, Cao Y, Mangos S, Tardi NJ, Dande RR, Lee HW, et al. Nonimmune cell-derived icos ligand functions as a renoprotective Alphavbeta3 integrin-selective antagonist. J Clin Invest (2019) 129(4):1713–26. doi: 10.1172/JCI123386
67. Wiendl H, Mitsdoerffer M, Schneider D, Melms A, Lochmuller H, Hohlfeld R, et al. Muscle fibres and cultured muscle cells express the B7.1/2-related inducible Co-stimulatory molecule, icosl: Implications for the pathogenesis of inflammatory myopathies. Brain (2003) 126(Pt 5):1026–35. doi: 10.1093/brain/awg114
68. Swallow MM, Wallin JJ, Sha WC. B7h, a novel costimulatory homolog of B7.1 and B7.2, is induced by tnfalpha. Immunity (1999) 11(4):423–32. doi: 10.1016/s1074-7613(00)80117-x
69. Liang L, Porter EM, Sha WC. Constitutive expression of the B7h ligand for inducible costimulator on naive b cells is extinguished after activation by distinct b cell receptor and interleukin 4 receptor-mediated pathways and can be rescued by Cd40 signaling. J Exp Med (2002) 196(1):97–108. doi: 10.1084/jem.20020298
70. Sacquin A, Gador M, Fazilleau N. The strength of bcr signaling shapes terminal development of follicular helper T cells in mice. Eur J Immunol (2017) 47(8):1295–304. doi: 10.1002/eji.201746952
71. Hu H, Wu X, Jin W, Chang M, Cheng X, Sun SC. Noncanonical nf-kappab regulates inducible costimulator (Icos) ligand expression and T follicular helper cell development. Proc Natl Acad Sci U.S.A. (2011) 108(31):12827–32. doi: 10.1073/pnas.1105774108
72. Zhang Z, Zhao S, Zhang L, Crew R, Zhang N, Sun X, et al. A higher frequency of Cd4(+)Cxcr5(+) T follicular helper cells in patients with newly diagnosed henoch-schonlein purpura nephritis. Int Immunopharmacol (2016) 32:8–15. doi: 10.1016/j.intimp.2015.12.037
73. Usui Y, Takeuchi M, Yamakawa N, Takeuchi A, Kezuka T, Ma J, et al. Expression and function of inducible costimulator on peripheral blood Cd4+ T cells in behcet's patients with uveitis: A new activity marker? Invest Ophthalmol Vis Sci (2010) 51(10):5099–104. doi: 10.1167/iovs.10-5286
74. Sullivan BA, Tsuji W, Kivitz A, Peng J, Arnold GE, Boedigheimer MJ, et al. Inducible T-cell Co-stimulator ligand (Icosl) blockade leads to selective inhibition of anti-klh igg responses in subjects with systemic lupus erythematosus. Lupus Sci Med (2016) 3(1):e000146. doi: 10.1136/lupus-2016-000146
75. Cheng LE, Amoura Z, Cheah B, Hiepe F, Sullivan BA, Zhou L, et al. Brief report: A randomized, double-blind, parallel-group, placebo-controlled, multiple-dose study to evaluate amg 557 in patients with systemic lupus erythematosus and active lupus arthritis. Arthritis Rheumatol (2018) 70(7):1071–6. doi: 10.1002/art.40479
76. Buchan SL, Rogel A, Al-Shamkhani A. The immunobiology of Cd27 and Ox40 and their potential as targets for cancer immunotherapy. Blood (2018) 131(1):39–48. doi: 10.1182/blood-2017-07-741025
77. Webb GJ, Hirschfield GM, Lane PJ. Ox40, Ox40l and autoimmunity: A comprehensive review. Clin Rev Allergy Immunol (2016) 50(3):312–32. doi: 10.1007/s12016-015-8498-3
78. Croft M. The role of tnf superfamily members in T-cell function and diseases. Nat Rev Immunol (2009) 9(4):271–85. doi: 10.1038/nri2526
79. Ohshima Y, Tanaka Y, Tozawa H, Takahashi Y, Maliszewski C, Delespesse G. Expression and function of Ox40 ligand on human dendritic cells. J Immunol (1997) 159(8):3838–48.
80. Stuber E, Neurath M, Calderhead D, Fell HP, Strober W. Cross-linking of Ox40 ligand, a member of the Tnf/Ngf cytokine family, induces proliferation and differentiation in murine splenic b cells. Immunity (1995) 2(5):507–21. doi: 10.1016/1074-7613(95)90031-4
81. Imura A, Hori T, Imada K, Ishikawa T, Tanaka Y, Maeda M, et al. The human Ox40/Gp34 system directly mediates adhesion of activated T cells to vascular endothelial cells. J Exp Med (1996) 183(5):2185–95. doi: 10.1084/jem.183.5.2185
82. Aten J, Roos A, Claessen N, Schilder-Tol EJM, Ten Berge IJM, Weening JJ. Strong and selective glomerular localization of Cd134 ligand and tnf receptor-1 in proliferative lupus nephritis. J Am Soc Nephrol (2000) 11(8):1426–38. doi: 10.1681/ASN.V1181426
83. Kashiwakura J, Yokoi H, Saito H, Okayama Y. T Cell proliferation by direct cross-talk between Ox40 ligand on human mast cells and Ox40 on human T cells: Comparison of gene expression profiles between human tonsillar and lung-cultured mast cells. J Immunol (2004) 173(8):5247–57. doi: 10.4049/jimmunol.173.8.5247
84. Sato T, Ishii N, Murata K, Kikuchi K, Nakagawa S, Ndhlovu LC, et al. Consequences of Ox40-Ox40 ligand interactions in langerhans cell function: Enhanced contact hypersensitivity responses in Ox40l-transgenic mice. Eur J Immunol (2002) 32(11):3326–35. doi: 10.1002/1521-4141(200211)32:11<3326::AID-IMMU3326>3.0.CO;2-9
85. Croft M, Siegel RM. Beyond tnf: Tnf superfamily cytokines as targets for the treatment of rheumatic diseases. Nat Rev Rheumatol (2017) 13(4):217–33. doi: 10.1038/nrrheum.2017.22
86. Seko Y, Takahashi N, Tada Y, Yagita H, Okumura K, Nagai R. Restricted usage of T-cell receptor vgamma-vdelta genes and expression of costimulatory molecules in takayasu's arteritis. Int J Cardiol (2000) 75 Suppl 1:S77–83. doi: 10.1016/s0167-5273(00)00194-7
87. Qin W, Hongya W, Yongjing C, Fang X, Yue M, Xuekun Z, et al. Increased Ox40 and soluble Ox40 ligands in children with henoch-schonlein purpura: Association with renal involvement. Pediatr Allergy Immunol (2011) 22(1 Pt 1):54–9. doi: 10.1111/j.1399-3038.2010.01111.x
88. Wilde B, Dolff S, Cai X, Specker C, Becker J, Totsch M, et al. Cd4+Cd25+ T-cell populations expressing Cd134 and gitr are associated with disease activity in patients with wegener's granulomatosis. Nephrol Dial Transplant (2009) 24(1):161–71. doi: 10.1093/ndt/gfn461
89. Giscombe R, Wang XB, Kakoulidou M, Lefvert AK. Characterization of the expanded T-cell populations in patients with wegener's granulomatosis. J Intern Med (2006) 260(3):224–30. doi: 10.1111/j.1365-2796.2006.01688.x
90. Papp KA, Gooderham MJ, Girard G, Raman M, Strout V. Phase I randomized study of Khk4083, an anti-Ox40 monoclonal antibody, in patients with mild to moderate plaque psoriasis. J Eur Acad Dermatol Venereol (2017) 31(8):1324–32. doi: 10.1111/jdv.14313
91. Furihata K, Ishiguro Y, Yoshimura N, Ito H, Katsushima S, Kaneko E, et al. A phase 1 study of Khk4083: A single-blind, randomized, placebo-controlled single-Ascending-Dose study in healthy adults and an open-label multiple-dose study in patients with ulcerative colitis. Clin Pharmacol Drug Dev (2021) 10(8):870–83. doi: 10.1002/cpdd.918
92. Guttman-Yassky E, Pavel AB, Zhou L, Estrada YD, Zhang N, Xu H, et al. Gbr 830, an anti-Ox40, improves skin gene signatures and clinical scores in patients with atopic dermatitis. J Allergy Clin Immunol (2019) 144(2):482–93 e7. doi: 10.1016/j.jaci.2018.11.053
93. Saghari M, Gal P, Gilbert S, Yateman M, Porter-Brown B, Brennan N, et al. Ox40l inhibition suppresses klh-driven immune responses in healthy volunteers: A randomized controlled trial demonstrating proof-of-Pharmacology for Ky1005. Clin Pharmacol Ther (2022) 111(5):1121–32. doi: 10.1002/cpt.2539
94. Tkachev V, Furlan SN, Watkins B, Hunt DJ, Zheng HB, Panoskaltsis-Mortari A, et al. Combined Ox40l and mtor blockade controls effector T cell activation while preserving treg reconstitution after transplant. Sci Transl Med (2017) 9(408):eaan3085. doi: 10.1126/scitranslmed.aan3085
95. Armitage RJ, Fanslow WC, Strockbine L, Sato TA, Clifford KN, Macduff BM, et al. Molecular and biological characterization of a murine ligand for Cd40. Nature (1992) 357(6373):80–2. doi: 10.1038/357080a0
96. Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, et al. Cd40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature (1998) 391(6667):591–4. doi: 10.1038/35393
97. Clark EA, Ledbetter JA. Activation of human b cells mediated through two distinct cell surface differentiation antigens, Bp35 and Bp50. Proc Natl Acad Sci U.S.A. (1986) 83(12):4494–8. doi: 10.1073/pnas.83.12.4494
98. Kiener PA, Moran-Davis P, Rankin BM, Wahl AF, Aruffo A, Hollenbaugh D. Stimulation of Cd40 with purified soluble Gp39 induces proinflammatory responses in human monocytes. J Immunol (1995) 155(10):4917–25.
99. Caux C, Massacrier C, Vanbervliet B, Dubois B, Van Kooten C, Durand I, et al. Activation of human dendritic cells through Cd40 cross-linking. J Exp Med (1994) 180(4):1263–72. doi: 10.1084/jem.180.4.1263
100. Tang T, Cheng X, Truong B, Sun L, Yang X, Wang H. Molecular basis and therapeutic implications of Cd40/Cd40l immune checkpoint. Pharmacol Ther (2021) 219:107709. doi: 10.1016/j.pharmthera.2020.107709
101. Wieczorek S, Holle JU, Muller S, Fricke H, Gross WL, Epplen JT. A functionally relevant Irf5 haplotype is associated with reduced risk to wegener's granulomatosis. J Mol Med (Berl) (2010) 88(4):413–21. doi: 10.1007/s00109-009-0580-y
102. Hoffmann JC, Patschan D, Dihazi H, Muller C, Schwarze K, Henze E, et al. Cytokine profiling in anti neutrophil cytoplasmic antibody-associated vasculitis: A cross-sectional cohort study. Rheumatol Int (2019) 39(11):1907–17. doi: 10.1007/s00296-019-04364-y
103. Visvanathan S, Daniluk S, Ptaszynski R, Muller-Ladner U, Ramanujam M, Rosenstock B, et al. Effects of bi 655064, an antagonistic anti-Cd40 antibody, on clinical and biomarker variables in patients with active rheumatoid arthritis: A randomised, double-blind, placebo-controlled, phase iia study. Ann Rheum Dis (2019) 78(6):754–60. doi: 10.1136/annrheumdis-2018-214729
104. Furie RA, Bruce IN, Dorner T, Leon MG, Leszczynski P, Urowitz M, et al. Phase 2, randomized, placebo-controlled trial of dapirolizumab pegol in patients with moderate-to-Severe active systemic lupus erythematosus. Rheumatol (Oxford) (2021) 60(11):5397–407. doi: 10.1093/rheumatology/keab381
105. Boumpas DT, Furie R, Manzi S, Illei GG, Wallace DJ, Balow JE, et al. A short course of Bg9588 (Anti-Cd40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum (2003) 48(3):719–27. doi: 10.1002/art.10856
106. Karnell JL, Albulescu M, Drabic S, Wang L, Moate R, Baca M, et al. A Cd40l-targeting protein reduces autoantibodies and improves disease activity in patients with autoimmunity. Sci Transl Med (2019) 11(489):eaar6584. doi: 10.1126/scitranslmed.aar6584
107. McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, et al. Cd4(+)Cd25(+) immunoregulatory T cells: Gene expression analysis reveals a functional role for the glucocorticoid-induced tnf receptor. Immunity (2002) 16(2):311–23. doi: 10.1016/s1074-7613(02)00280-7
108. Dawicki W, Watts TH. Expression and function of 4-1bb during Cd4 versus Cd8 T cell responses in vivo. Eur J Immunol (2004) 34(3):743–51. doi: 10.1002/eji.200324278
109. Tian J, Zhang B, Rui K, Wang S. The role of Gitr/Gitrl interaction in autoimmune diseases. Front Immunol (2020) 11:588682. doi: 10.3389/fimmu.2020.588682
110. Takahashi C, Mittler RS, Vella AT. Cutting edge: 4-1bb is a bona fide Cd8 T cell survival signal. J Immunol (1999) 162(9):5037–40.
111. Choi BK, Bae JS, Choi EM, Kang WJ, Sakaguchi S, Vinay DS, et al. 4-1bb-Dependent inhibition of immunosuppression by activated Cd4+Cd25+ T cells. J Leukoc Biol (2004) 75(5):785–91. doi: 10.1189/jlb.1003491
112. Patel M, Xu D, Kewin P, Choo-Kang B, McSharry C, Thomson NC, et al. Glucocorticoid-induced tnfr family-related protein (Gitr) activation exacerbates murine asthma and collagen-induced arthritis. Eur J Immunol (2005) 35(12):3581–90. doi: 10.1002/eji.200535421
113. Kohm AP, Williams JS, Miller SD. Cutting edge: Ligation of the glucocorticoid-induced tnf receptor enhances autoreactive Cd4+ T cell activation and experimental autoimmune encephalomyelitis. J Immunol (2004) 172(8):4686–90. doi: 10.4049/jimmunol.172.8.4686
114. Seo SK, Choi JH, Kim YH, Kang WJ, Park HY, Suh JH, et al. 4-1bb-Mediated immunotherapy of rheumatoid arthritis. Nat Med (2004) 10(10):1088–94. doi: 10.1038/nm1107
115. Kim YH, Choi BK, Shin SM, Kim CH, Oh HS, Park SH, et al. 4-1bb triggering ameliorates experimental autoimmune encephalomyelitis by modulating the balance between Th17 and regulatory T cells. J Immunol (2011) 187(3):1120–8. doi: 10.4049/jimmunol.1002681
116. Oh HS, Choi BK, Kim YH, Lee DG, Hwang S, Lee MJ, et al. 4-1bb signaling enhances primary and secondary population expansion of Cd8+ T cells by maximizing autocrine il-2/Il-2 receptor signaling. PloS One (2015) 10(5):e0126765. doi: 10.1371/journal.pone.0126765
117. Nguyen J, Pettmann J, Kruger P, Dushek O. Quantitative contributions of tnf receptor superfamily members to Cd8(+) T-cell responses. Mol Syst Biol (2021) 17(11):e10560. doi: 10.15252/msb.202110560
118. Kuchroo JR, Hafler DA, Sharpe AH, Lucca LE. The double-edged sword: Harnessing pd-1 blockade in tumor and autoimmunity. Sci Immunol (2021) 6(65):eabf4034. doi: 10.1126/sciimmunol.abf4034
119. Boussiotis VA. Molecular and biochemical aspects of the pd-1 checkpoint pathway. N Engl J Med (2016) 375(18):1767–78. doi: 10.1056/NEJMra1514296
120. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. Shp-1 and shp-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol (2004) 173(2):945–54. doi: 10.4049/jimmunol.173.2.945
121. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. Ctla-4 and pd-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25(21):9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
122. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase Shp2. J Exp Med (2012) 209(6):1201–17. doi: 10.1084/jem.20112741
123. Kreileder M, Barrett I, Bendtsen C, Brennan D, Kolch W. Signaling dynamics regulating crosstalks between T-cell activation and immune checkpoints. Trends Cell Biol (2021) 31(3):224–35. doi: 10.1016/j.tcb.2020.12.001
124. Sharpe AH, Pauken KE. The diverse functions of the Pd1 inhibitory pathway. Nat Rev Immunol (2018) 18(3):153–67. doi: 10.1038/nri.2017.108
125. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the pd-L1 checkpoint. Immunity (2018) 48(3):434–52. doi: 10.1016/j.immuni.2018.03.014
126. Ibanez-Vega J, Vilchez C, Jimenez K, Guevara C, Burgos PI, Naves R. Cellular and molecular regulation of the programmed death-1/Programmed death ligand system and its role in multiple sclerosis and other autoimmune diseases. J Autoimmun (2021) 123:102702. doi: 10.1016/j.jaut.2021.102702
127. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity (2007) 27(1):111–22. doi: 10.1016/j.immuni.2007.05.016
128. Xiao Y, Yu S, Zhu B, Bedoret D, Bu X, Francisco LM, et al. Rgmb is a novel binding partner for pd-L2 and its engagement with pd-L2 promotes respiratory tolerance. J Exp Med (2014) 211(5):943–59. doi: 10.1084/jem.20130790
129. Zhang H, Watanabe R, Berry GJ, Vaglio A, Liao YJ, Warrington KJ, et al. Immunoinhibitory checkpoint deficiency in medium and Large vessel vasculitis. Proc Natl Acad Sci U.S.A. (2017) 114(6):E970–E9. doi: 10.1073/pnas.1616848114
130. Zeisbrich M, Chevalier N, Sehnert B, Rizzi M, Venhoff N, Thiel J, et al. Cmtm6-deficient monocytes in anca-associated vasculitis fail to present the immune checkpoint pd-L1. Front Immunol (2021) 12:673912. doi: 10.3389/fimmu.2021.673912
131. Wilde B, Hua F, Dolff S, Jun C, Cai X, Specker C, et al. Aberrant expression of the negative costimulator pd-1 on T cells in granulomatosis with polyangiitis. Rheumatol (Oxford) (2012) 51(7):1188–97. doi: 10.1093/rheumatology/kes034
132. Zhao P, Wang P, Dong S, Zhou Z, Cao Y, Yagita H, et al. Depletion of pd-1-Positive cells ameliorates autoimmune disease. Nat BioMed Eng (2019) 3(4):292–305. doi: 10.1038/s41551-019-0360-0
133. Gamerith G, Mildner F, Merkel PA, Harris K, Cooney L, Lim N, et al. Association of baseline soluble immune checkpoints with the risk of relapse in Pr3-anca vasculitis following induction of remission. Ann Rheum Dis (2023) 82(2):253–61. doi: 10.1136/ard-2022-222479
134. Soltani-Zangbar MS, Parhizkar F, Abdollahi M, Shomali N, Aghebati-Maleki L, Shahmohammadi Farid S, et al. Immune system-related soluble mediators and covid-19: Basic mechanisms and clinical perspectives. Cell Commun Signal (2022) 20(1):131. doi: 10.1186/s12964-022-00948-7
135. Ding H, Wu X, Wu J, Yagita H, He Y, Zhang J, et al. Delivering pd-1 inhibitory signal concomitant with blocking icos Co-stimulation suppresses lupus-like syndrome in autoimmune bxsb mice. Clin Immunol (2006) 118(2-3):258–67. doi: 10.1016/j.clim.2005.10.017
136. He X, Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res (2020) 30(8):660–9. doi: 10.1038/s41422-020-0343-4
137. Nagai K. Co-Inhibitory receptor signaling in T-Cell-Mediated autoimmune glomerulonephritis. Front Med (Lausanne) (2020) 7:584382. doi: 10.3389/fmed.2020.584382
138. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. Ctla-4 is a second receptor for the b cell activation antigen B7. J Exp Med (1991) 174(3):561–9. doi: 10.1084/jem.174.3.561
139. Rowshanravan B, Halliday N, Sansom DM. Ctla-4: A moving target in immunotherapy. Blood (2018) 131(1):58–67. doi: 10.1182/blood-2017-06-741033
140. Kumar P, Bhattacharya P, Prabhakar BS. A comprehensive review on the role of Co-signaling receptors and treg homeostasis in autoimmunity and tumor immunity. J Autoimmun (2018) 95:77–99. doi: 10.1016/j.jaut.2018.08.007
141. Shiratori T, Miyatake S, Ohno H, Nakaseko C, Isono K, Bonifacino JS, et al. Tyrosine phosphorylation controls internalization of ctla-4 by regulating its interaction with clathrin-associated adaptor complex ap-2. Immunity (1997) 6(5):583–9. doi: 10.1016/s1074-7613(00)80346-5
142. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of Cd80 and Cd86: A molecular basis for the cell-extrinsic function of ctla-4. Science (2011) 332(6029):600–3. doi: 10.1126/science.1202947
143. Steiner K, Moosig F, Csernok E, Selleng K, Gross WL, Fleischer B, et al. Increased expression of ctla-4 (Cd152) by T and b lymphocytes in wegener's granulomatosis. Clin Exp Immunol (2001) 126(1):143–50. doi: 10.1046/j.1365-2249.2001.01575.x
144. Rahmattulla C, Mooyaart AL, van Hooven D, Schoones JW, Bruijn JA, Dekkers OM, et al. Genetic variants in anca-associated vasculitis: A meta-analysis. Ann Rheum Dis (2016) 75(9):1687–92. doi: 10.1136/annrheumdis-2015-207601
145. Chung SA, Xie G, Roshandel D, Sherva R, Edberg JC, Kravitz M, et al. Meta-analysis of genetic polymorphisms in granulomatosis with polyangiitis (Wegener's) reveals shared susceptibility loci with rheumatoid arthritis. Arthritis Rheum (2012) 64(10):3463–71. doi: 10.1002/art.34496
146. Spriewald BM, Witzke O, Wassmuth R, Wenzel RR, Arnold ML, Philipp T, et al. Distinct tumour necrosis factor alpha, interferon gamma, interleukin 10, and cytotoxic T cell antigen 4 gene polymorphisms in disease occurrence and end stage renal disease in wegener's granulomatosis. Ann Rheum Dis (2005) 64(3):457–61. doi: 10.1136/ard.2004.025809
147. Tsuchiya N, Kobayashi S, Kawasaki A, Kyogoku C, Arimura Y, Yoshida M, et al. Genetic background of Japanese patients with antineutrophil cytoplasmic antibody-associated vasculitis: Association of hla-Drb1*0901 with microscopic polyangiitis. J Rheumatol (2003) 30(7):1534–40.
148. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature (2002) 415(6871):536–41. doi: 10.1038/415536a
149. Gautron AS, Dominguez-Villar M, de Marcken M, Hafler DA. Enhanced suppressor function of Tim-3+ Foxp3+ regulatory T cells. Eur J Immunol (2014) 44(9):2703–11. doi: 10.1002/eji.201344392
150. Andrews LP, Yano H, Vignali DAA. Inhibitory receptors and ligands beyond pd-1, pd-L1 and ctla-4: Breakthroughs or backups. Nat Immunol (2019) 20(11):1425–34. doi: 10.1038/s41590-019-0512-0
151. Wolf Y, Anderson AC, Kuchroo VK. Tim3 comes of age as an inhibitory receptor. Nat Rev Immunol (2020) 20(3):173–85. doi: 10.1038/s41577-019-0224-6
152. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and tigit: Co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44(5):989–1004. doi: 10.1016/j.immuni.2016.05.001
153. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol (2005) 6(12):1245–52. doi: 10.1038/ni1271
154. Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood (2009) 113(16):3821–30. doi: 10.1182/blood-2008-10-185884
155. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating dcs suppress nucleic acid-mediated innate immune responses through interactions between the receptor Tim-3 and the alarmin Hmgb1. Nat Immunol (2012) 13(9):832–42. doi: 10.1038/ni.2376
156. Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. Ceacam1 regulates Tim-3-Mediated tolerance and exhaustion. Nature (2015) 517(7534):386–90. doi: 10.1038/nature13848
157. Su B, Mao X, Yin B, Chen C, Zhang M, Cui T, et al. Tim-3 regulates the nets-mediated dendritic cell activation in myeloperoxidase-Anca-Associated vasculitis. Clin Exp Rheumatol (2021) 39 Suppl 129(2):13–20. doi: 10.55563/clinexprheumatol/6y0bjb
158. Yoon T, Ahn SS, Pyo JY, Lee LE, Song JJ, Park YB, et al. Serum galectin-9 could be a potential biomarker in assessing the disease activity of antineutrophil cytoplasmic antibody-associated vasculitis. Clin Exp Rheumatol (2022) 40(4):779–86. doi: 10.55563/clinexprheumatol/xfqnx6
159. Seki M, Oomizu S, Sakata KM, Sakata A, Arikawa T, Watanabe K, et al. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin Immunol (2008) 127(1):78–88. doi: 10.1016/j.clim.2008.01.006
160. Moritoki M, Kadowaki T, Niki T, Nakano D, Soma G, Mori H, et al. Galectin-9 ameliorates clinical severity of Mrl/Lpr lupus-prone mice by inducing plasma cell apoptosis independently of Tim-3. PloS One (2013) 8(4):e60807. doi: 10.1371/journal.pone.0060807
161. Gaikwad S, Agrawal MY, Kaushik I, Ramachandran S, Srivastava SK. Immune checkpoint proteins: Signaling mechanisms and molecular interactions in cancer immunotherapy. Semin Cancer Biol (2022) 86(3):137–50. doi: 10.1016/j.semcancer.2022.03.014
162. Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, et al. Btla is a lymphocyte inhibitory receptor with similarities to ctla-4 and pd-1. Nat Immunol (2003) 4(7):670–9. doi: 10.1038/ni944
163. Gavrieli M, Watanabe N, Loftin SK, Murphy TL, Murphy KM. Characterization of phosphotyrosine binding motifs in the cytoplasmic domain of b and T lymphocyte attenuator required for association with protein tyrosine phosphatases shp-1 and shp-2. Biochem Biophys Res Commun (2003) 312(4):1236–43. doi: 10.1016/j.bbrc.2003.11.070
164. Celis-Gutierrez J, Blattmann P, Zhai Y, Jarmuzynski N, Ruminski K, Gregoire C, et al. Quantitative interactomics in primary T cells provides a rationale for concomitant pd-1 and btla coinhibitor blockade in cancer immunotherapy. Cell Rep (2019) 27(11):3315–30 e7. doi: 10.1016/j.celrep.2019.05.041
165. Gavrieli M, Murphy KM. Association of grb-2 and Pi3k P85 with phosphotyrosile peptides derived from btla. Biochem Biophys Res Commun (2006) 345(4):1440–5. doi: 10.1016/j.bbrc.2006.05.036
166. Wojciechowicz K, Spodzieja M, Lisowska KA, Wardowska A. The role of the btla-hvem complex in the pathogenesis of autoimmune diseases. Cell Immunol (2022) 376:104532. doi: 10.1016/j.cellimm.2022.104532
167. Cheung TC, Oborne LM, Steinberg MW, Macauley MG, Fukuyama S, Sanjo H, et al. T Cell intrinsic heterodimeric complexes between hvem and btla determine receptivity to the surrounding microenvironment. J Immunol (2009) 183(11):7286–96. doi: 10.4049/jimmunol.0902490
168. Cheung TC, Steinberg MW, Oborne LM, Macauley MG, Fukuyama S, Sanjo H, et al. Unconventional ligand activation of herpesvirus entry mediator signals cell survival. Proc Natl Acad Sci U.S.A. (2009) 106(15):6244–9. doi: 10.1073/pnas.0902115106
169. Werner K, Dolff S, Dai Y, Ma X, Brinkhoff A, Korth J, et al. The Co-inhibitor btla is functional in anca-associated vasculitis and suppresses Th17 cells. Front Immunol (2019) 10:2843. doi: 10.3389/fimmu.2019.02843
170. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. Vista, a novel mouse ig superfamily ligand that negatively regulates T cell responses. J Exp Med (2011) 208(3):577–92. doi: 10.1084/jem.20100619
171. Nowak EC, Lines JL, Varn FS, Deng J, Sarde A, Mabaera R, et al. Immunoregulatory functions of vista. Immunol Rev (2017) 276(1):66–79. doi: 10.1111/imr.12525
172. Lines JL, Pantazi E, Mak J, Sempere LF, Wang L, O'Connell S, et al. Vista is an immune checkpoint molecule for human T cells. Cancer Res (2014) 74(7):1924–32. doi: 10.1158/0008-5472.CAN-13-1504
173. Flies DB, Han X, Higuchi T, Zheng L, Sun J, Ye JJ, et al. Coinhibitory receptor pd-1h preferentially suppresses Cd4(+) T cell-mediated immunity. J Clin Invest (2014) 124(5):1966–75. doi: 10.1172/JCI74589
174. Wang J, Wu G, Manick B, Hernandez V, Renelt M, Erickson C, et al. Vsig-3 as a ligand of vista inhibits human T-cell function. Immunology (2019) 156(1):74–85. doi: 10.1111/imm.13001
175. Han X, Vesely MD, Yang W, Sanmamed MF, Badri T, Alawa J, et al. Pd-1h (Vista)-mediated suppression of autoimmunity in systemic and cutaneous lupus erythematosus. Sci Transl Med (2019) 11(522):eaax1159. doi: 10.1126/scitranslmed.aax1159
176. Wang L, Le Mercier I, Putra J, Chen W, Liu J, Schenk AD, et al. Disruption of the immune-checkpoint vista gene imparts a proinflammatory phenotype with predisposition to the development of autoimmunity. Proc Natl Acad Sci U.S.A. (2014) 111(41):14846–51. doi: 10.1073/pnas.1407447111
177. Kim MG, Yun D, Kang CL, Hong M, Hwang J, Moon KC, et al. Kidney vista prevents ifn-Gamma/Il-9 axis-mediated tubulointerstitial fibrosis after acute glomerular injury. J Clin Invest (2022) 132(1):e151189. doi: 10.1172/JCI151189
178. Gorczynski R, Chen Z, Kai Y, Lee L, Wong S, Marsden PA. Cd200 is a ligand for all members of the Cd200r family of immunoregulatory molecules. J Immunol (2004) 172(12):7744–9. doi: 10.4049/jimmunol.172.12.7744
179. Baixeras E, Huard B, Miossec C, Jitsukawa S, Martin M, Hercend T, et al. Characterization of the lymphocyte activation gene 3-encoded protein. a new ligand for human leukocyte antigen class ii antigens. J Exp Med (1992) 176(2):327–37. doi: 10.1084/jem.176.2.327
180. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of lag-3. Cell (2019) 176(1-2):334–47.e12. doi: 10.1016/j.cell.2018.11.010
181. Liu S, Sun L, Wang C, Cui Y, Ling Y, Li T, et al. Treatment of murine lupus with tigit-ig. Clin Immunol (2019) 203:72–80. doi: 10.1016/j.clim.2019.04.007
182. Dixon KO, Schorer M, Nevin J, Etminan Y, Amoozgar Z, Kondo T, et al. Functional anti-tigit antibodies regulate development of autoimmunity and antitumor immunity. J Immunol (2018) 200(8):3000–7. doi: 10.4049/jimmunol.1700407
183. Liu Y, Bando Y, Vargas-Lowy D, Elyaman W, Khoury SJ, Huang T, et al. Cd200r1 agonist attenuates mechanisms of chronic disease in a murine model of multiple sclerosis. J Neurosci (2010) 30(6):2025–38. doi: 10.1523/JNEUROSCI.4272-09.2010
184. Simelyte E, Criado G, Essex D, Uger RA, Feldmann M, Williams RO. Cd200-fc, a novel antiarthritic biologic agent that targets proinflammatory cytokine expression in the joints of mice with collagen-induced arthritis. Arthritis Rheum (2008) 58(4):1038–43. doi: 10.1002/art.23378
Keywords: antineutrophil cytoplasmic autoantibody associated vasculitis, immune checkpoint, co-stimulatory signal pathway, co-inhibitory signal pathway, T cells, immunotherapy
Citation: Pan M, Zhao H, Jin R, Leung PSC and Shuai Z (2023) Targeting immune checkpoints in anti-neutrophil cytoplasmic antibodies associated vasculitis: the potential therapeutic targets in the future. Front. Immunol. 14:1156212. doi: 10.3389/fimmu.2023.1156212
Received: 01 February 2023; Accepted: 27 March 2023;
Published: 06 April 2023.
Edited by:
Yuval Tal, Hadassah Medical Center, IsraelReviewed by:
Sung Soo Ahn, Yonsei University Health System, Republic of KoreaUma Sriram, Temple University, United States
Copyright © 2023 Pan, Zhao, Jin, Leung and Shuai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zongwen Shuai, c2h1YWl6b25nd2VuQGFobXUuZWR1LmNu; Patrick S. C. Leung, cHNsZXVuZ0B1Y2RhdmlzLmVkdQ==
†These authors have contributed equally to this work and share first authorship