 Dengqiang Wu
Dengqiang Wu Yujie Li
Yujie Li Ranxing Xu1*
Ranxing Xu1*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 22 June 2023
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1155606
Rheumatoid arthritis (RA) is a chronic systemic autoimmune disease of undefined etiology, with persistent synovial inflammation and destruction of articular cartilage and bone. Current clinical drugs for RA mainly include non-steroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, disease modifying anti-rheumatic drugs (DMARDs) and so on, which can relieve patients’ joint symptoms. If we want to have a complete cure for RA, there are still some limitations of these drugs. Therefore, we need to explore new mechanisms of RA to prevent and treat RA radically. Pyroptosis is a newly discovered programmed cell death (PCD) in recent years, which is characterized by the appearance of holes in cell membranes, cell swelling and rupture, and the release of intracellular pro-inflammatory factors into the extracellular space, resulting in a strong inflammatory response. The nature of pyroptosis is pro-inflammatory, and whether it is participating in the development of RA has attracted a wide interest among scholars. This review describes the discovery and mechanism of pyroptosis, the main therapeutic strategies for RA, and the role of pyroptosis in the mechanism of RA development. From the perspective of pyroptosis, the study of new mechanisms of RA may provide a potential target for the treatment of RA and the development of new drugs in the clinics.
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by symmetrical pain and swelling of multiple joints in the hands, wrists and feet. The pathology is characterized by persistent synovial inflammation, which can lead to articular cartilage erosion and joint deformity, is one of the main factors of disability (1–4). The global prevalence of RA is about 0.5-1.0% on average, and the quality of life of RA patients is lower than that of common diseases such as diabetes, myocardial infarction, hypertension and so on (5). Patients also suffer a huge financial burden of treatment.
Clinically available western medicines for RA include non-steroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, disease-modifying anti-rheumatic drugs (DMARDs), biologics, botanicals, analgesics, etc. Most of these drugs can alleviate the symptoms of RA patients to a certain extent, but there are still many limitations such as adverse drug reactions and individual differences in drug efficacy (6–8). Therefore, the key to treat RA is to study the mechanism of RA and stop its development at the root. Recently, there is a large number of researches reporting that some Chinese medicines such as punicalagin, celastrol and BaiHu GuiZhi can treat RA through the pyroptosis pathway (9–12). Pyroptosis is a rapidly studied PCD in recent years, which is a form of cell death mediated by gasdermin protein, usually activated by cysteinyl aspartate specific proteinase (caspase), and accompanied with a strong inflammatory response (13–15). Morphologically, it appear holes in the plasma membrane, subsequently swelling with “light bulb” under the microscope (15–17). It has been demonstrated that chondrocytes, fibroblast-like synoviocytes (FLS), and monocytes undergo pyroptosis in RA patients (18). And high level of pyroptosis-related molecules such as NOD-like receptor thermal protein domain associated protein 3 (NLRP3), caspases, IL-18, IL-1β were detected in serum and synovial fluid (19, 20). All evidence suggests that pyroptosis is involved in regulating the pathogenesis of RA. Its role in RA has gradually attracted the attention of scholars (18, 21, 22). Therefore, this review describes the role of pyroptosis in the pathogenesis of RA and also provides new possible targets for the treatment of RA.
Pyroptosis is a kind of PCD which has been studied deeply in recent years (13, 23, 24). It is an important non-specific defense mechanism in body, which plays a certain role in antagonizing the invasion of external pathogens and sensing endogenous danger signals (13, 23, 25, 26). In 1992, scholars such as Zychlinsky et al. observed that Shigellaflexneri could induce lytic death in infected host macrophages (27). This form of PCD was partly similar to apoptosis and was considered to be apoptosis at that time (27).
In 2001, Cookson BT et al. first described this mode of cell death as “pyroptosis”. “pyro” means “burning” in Greek and is used to indicate that the occurrence of such PCD accompanied by an inflammatory response, formally distinguishing it from apoptosis (28). The gasdermin (GSDM) family, core protein of pyroptosis was discovered in 2015 (16, 29). The study of pyroptosis has entered a whole new field since then. In 2018, the Nomenclature Committee on Cell Death (NCCD) recommended that the definition of pyroptosis be revised to a regulated cell death that depends on the GSDM protein family to form plasma membrane pores, which is often the result of inflammatory caspase activation (30).
Gasdermin protein is the key of pyroptosis, including gasdermin A (GSDMA), gasdermin B (GSDMB), gasdermin C (GSDMC), gasdermin D (GSDMD), gasdermin E (GSDME, also known as DFNA5) and Pejvakin (PJVK, also known as DFNB59) (13). With the exception of PJVK, all other gasdermins consist of C-terminal domain (GSDM-CT) and N-terminal domain (GSDM-NT). In the resting state, GSDM-CT binds to GSDM-NT to inhibit the drilling activity of GSDM-N (31, 32). GSDM-NT will be released and freed to the inner surface of the cell membrane to bind to the phosphatidylinositol in the plasma membrane, forming a pore with an inner diameter of approximately 12-14 nm when the pyroptosis activation signal is triggered (29). Cell contents such as IL-18, IL-1 β and LDH are released to the outside of the cell through pores (16, 29, 33, 34). Due to the osmotic pressure inside and outside, the cells will swell with the fluid infiltration and eventually rupture and die. The activation pathway of pyroptosis can be divided into the canonical inflammasome pathway of caspase-1 activated by inflammasome and the noncanonical pathway of caspase-4/5/11 activated by lipopolysaccharide (LPS) (13, 29). GSDMD is the executive protein of pyroptosis, the common substrate of inflammatory caspase-1/4/5/11 enzymes, and the core of the two pathways (16, 29, 35, 36).
Inflammasome is a class of multi-protein complexes, the main types of which including NLRP1, NLRP3, NLRC4, AIM2 and so on (37–40). The basic structure of inflammasome includes three parts: pattern recognition receptor (PRR), apoptosis-associated speck-like protein containing CARD (ASC) and pro-caspase-1 (41, 42). PRRs recognize dangerous signals. Different inflammasome will be activated by a variety of corresponding activation ways when they are stimulated by different external signals. ASC will recruit PRRs and pro-caspase-1 through the oligomerization of pyrin domain (PYD) and C-terminal caspase recruitment domain (CARD) (41, 43, 44), then assemble inflammasome complexes for downstream signal transduction. As an effector molecule, pro-caspase-1 is recruited by ASC to form activated caspase-1 (41, 45, 46).
In the canonical inflammasome pathway, NLRP1 is activated by anthrax toxin and Toxoplasma gondii infection (47); NLRP3 is activated by various membrane injuries; NLRC4 inflammasome recognizes bacterial flagellin (48, 49); AIM2 acts as a DNA-binding receptor to recognize double-stranded DNA (dsDNA) (40). Then ASC recruited pro-caspase-1 to develop inflammasome, and the activated caspase-1 further cleave the downstream GSDMD protein to promote the generation of pyroptosis (16, 36, 46). Different from the canonical pathway of pyroptosis, caspase-11 (mouse) or caspase-4/5 (human) can start the activation process by sensing the expression of LPS directly, which form pores in the cell membrane through GSDMD cleavage and promote GSDMD-mediated pyroptosis, a noncanonical pathway of pyroptosis (29). In addition, the researchers found that K+ efflux can activate NLRP3 inflammasome (50), which further induce caspase-1-mediated pyroptosis (50, 51) (Figure 1).
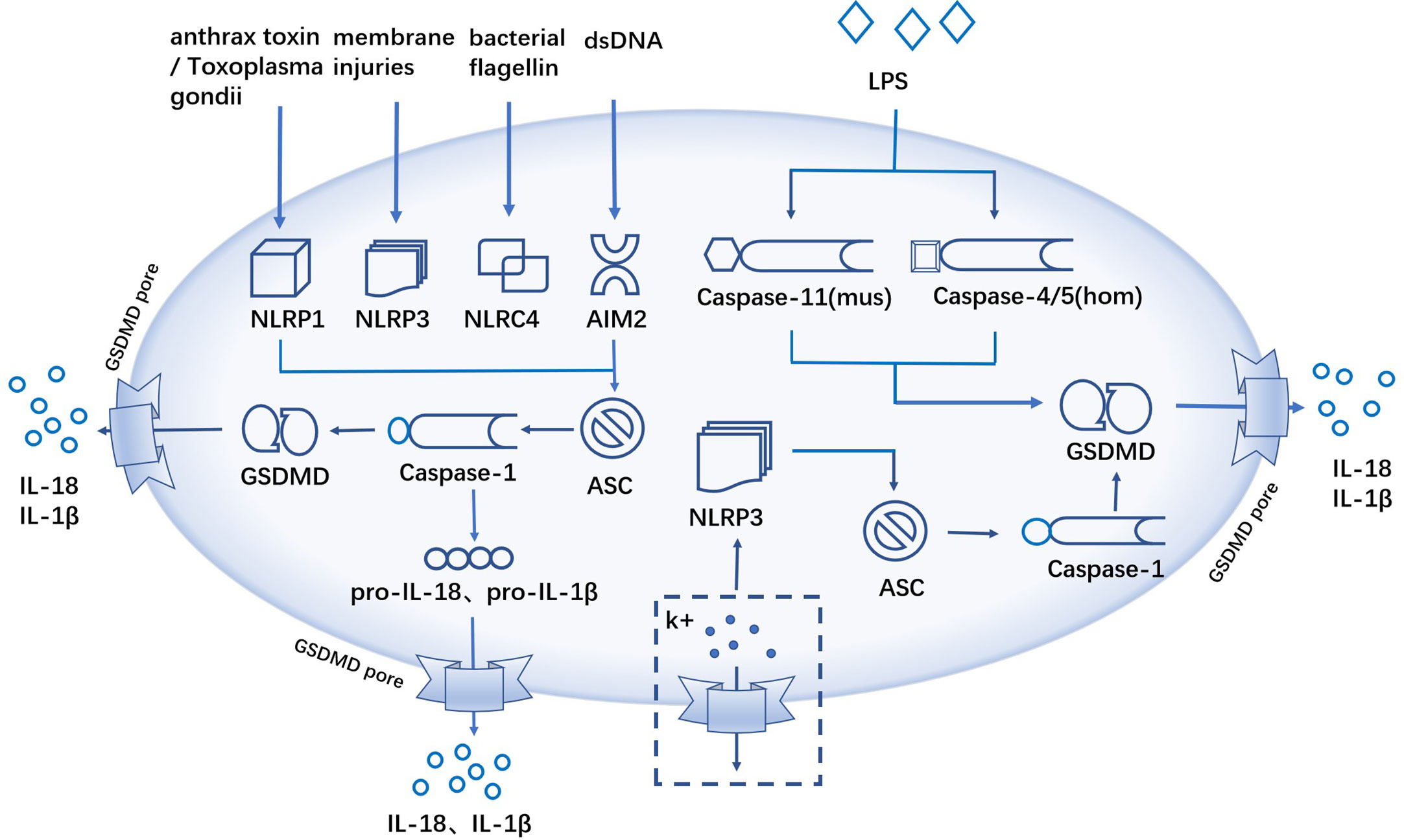
Figure 1 The canonical and noncanonical inflammasome pathway. The canonical inflammasome pathway, anthrax toxin/Toxoplasma gondii infections, membrane injuries, bacterial flagellin and dsDNA activate inflammatory and ASC recruits pro-caspase-1 to form inflammatory complexes, activated caspase-1 cleaves GSDMD, and the cellular contents like IL-18, IL-1β and LDH are released extracellularly through the GSDMD pores. The non-canonical inflammasome pathway, caspase-11 (mouse) or caspase-4/5 (human) sensing LPS, promotes pyroptosis via GSDMD cleavage. K+ efflux activates NLRP3 inflammatory and activated caspase-1 induces pyroptosis.
In addition to the canonical and noncanonical pyroptosis pathway, there are other activation types of gasdermins-mediated pyroptosis. Chemotherapeutic drugs such as paclitaxel and cisplatin can activate pro-caspase-3, which in turn mediates pyroptosis by cleaving GSDME (15). This study also showed that more “apoptosis” can be transformed into “pyroptosis” in the regulation of GSDME (15, 52). Granzyme B is a serine protease that is mainly expressed by NK cells or cytotoxic T lymphocytes (53, 54). Granzyme B can cut GSDME at the same site like caspase-3 and induce GSDME-mediated pyroptosis (55). A study on melanoma cells found that iron significantly increased chemotherapy-triggered reactive oxygen species (ROS), promoting the oxidation and oligomerization of the outer mitochondrial membrane protein Tom20. Activated Tom20 recruited Bax to the mitochondria, which then induced further activation of caspase-3 by cytochrome C and ultimately trigger GSDME-mediated pyroptosis in melanoma cells (56). Granzyme A can enter target cells via perforin and induce pyroptosis by cleaving GSDMB protein at the Lys244 site. And GSDMB protein is currently recognized as an executor protein that can be cleaved by Granzyme A (57). In hypoxic environment, p-STAT3 interacts with PD-L1 physically to enhance the transcription of GSDMC. At the same time, under the treatment of TNF-α, activated caspase-8 cleaves GSDMC in MDA-MB-231 breast cancer cells to induce GSDMC-mediated pyroptosis (58).
Rheumatoid arthritis is a chronic systemic autoimmune disease, and its pathogenesis is not clear yet (59, 60). The main clinical manifestations are symmetrical and invasive inflammation of small joints such as hands and feet, which can lead to joint deformities and function loss, and even involve heart and lung (5, 61, 62). From a pathological point of view, RA is a widespread inflammatory disease that mainly involves articular synovium, which can spread to articular cartilage, bone tissue and joint ligament (62–64). Western medicines and Chinese herbs are available for the treatment of RA.
At present, western medical treatment is mainly based on NSAIDs, disease-modifying antirheumatic drugs (DMARDs) and glucocorticoids (65–68). The main mechanism of NSAIDs is to inhibit cyclooxygenase (COX) to prevent the synthesis of inflammatory mediators such as prostaglandins and thromboxane, thus exerting anti-inflammatory and analgesic effects. Representative drugs include meloxicam tablets, diclofenac, aspirin and so on (69–71). Considerable progress has been made in the development of DMARDs, which are effective in preventing joint imaging damage in patients with RA. This kind of drugs include conventional synthetic disease modifying anti-rheumatic drugs(csDMARDs), biological disease modifying anti-rheumatic drugs(bDMARDs) and target synthetic disease modifying anti-rheumatic drugs(tsDMARDs) (66, 72, 73). The commonly used csDMARDs include methotrexate (MTX), sulfasalazine (SASP), leflunomide (LEF), penicillamine and so on. MTX is generally the first choice for the treatment of RA, mainly through the inhibition of dihydrofolate reductase, reducing the production of inflammatory factors to achieve immunosuppressive effects (74–76). SASP can reduce the level of immunoglobulin, reduce the secretion of IL-8 and monocyte chemoattractant protein, thereby interfering with T-cell function (77). LET is a new type of antimetabolic immunosuppressant, whose mechanism is to inhibit dihydroorotic acid dehydrogenase and tyrosine kinase activity and to attenuate the activation of lymphocyte growth (78, 79). Tofacitab and baricitinib are tsDMARDs, which interfere with the production of downstream inflammatory cytokines by inhibiting Janus kinase (JAK), thereby preventing the further development of RA (80–82). The main purpose of bDMARDs is to inhibit various core inflammatory factors of RA to achieve the purpose of treating and alleviating joint injury. Etanercept is a common TNF-α inhibitor that competitively binds to TNF-α and exerting anti-inflammatory effects (83). Anakinra, a human-derived IL-1 receptor antagonist, is greatly limited in the treatment of RA due to its short half-life (84). Sarilumab is a humanized monoclonal antibody that inhibits IL-6-mediated signal transduction and mitigates the progression of RA (85, 86). Rituximab is a human-mouse chimeric anti-CD20 monoclonal antibody, which can significantly reducing the secretion of autoantibodies and rheumatoid factors in the body (87, 88). The above-mentioned bDMARDs drugs are targeted and effective in relieving RA symptoms, but also have the disadvantage of increasing the risk of infection (89, 90). Glucocorticoids have powerful anti-inflammatory effects and are mainly used clinically in small doses and short courses for the treatment of RA, providing rapid relief of joint swelling and inflammation (91–93). The main clinically applied glucocorticoids are hydrocortisone, dexamethasone and prednisone. However, glucocorticoid cannot completely treat RA and has serious side effects such as atherosclerosis, hypertension, infection, osteoporosis and so on (89, 90).
Chinese medicine has a good efficacy in treating RA (94). Recent studies have found that some Chinese medicines can be mediated through pyroptosis pathway in the prevention and treatment of RA. Therefore, it may provide some new targets for clinical drug development of RA if scholars can study the mechanism of Chinese medicine against RA from the perspective of pyroptosis.
Ling et al. found that Hmong Jin Wu Jian Wu Bone Capsules were able to inhibit fibroblast-like synoviocyte (FLS) pyroptosis through the NLRP3/caspase/GSDMD pathway in the treatment of RA (95). Ge et al. found that punicalagin was able to reduce the expression of NLRP3 inflammatory in collagen-induced arthritis (CIA) mice and inhibit the occurrence of pyroptosis, resulting in an anti-inflammatory effect (9). Celastrol is a quinone methylated triterpenoid that is used in the treatment of RA. Studies have shown that celastrol can block the nuclear factor kappa-B (NF-κB) signaling pathway, inhibiting the activation of NLRP3 inflammatory. In addition, it also inhibits LPS and adenosine triphosphate (ATP)-induced ROS. Therefore, celastrol may play a role in the treatment of RA by inhibiting the ROS/NF-κB/NLRP3 signaling pathway (10). The Chinese medicine BaiHu GuiZhi can attenuate the inflammatory response of RA by inhibiting the activation of Toll-like receptor 4(TLR4)-mediated NLRP3 inflammasome. The increased levels of NLRP3, caspase-1, LDH, IL-1β and IL-18 were significantly reversed by Baihu Gui Zhi, confirming the immunomodulatory and anti-inflammatory activities of Baihu GuiZhi especially in pyroptosis (11, 12). Cinnamaldehyde, an important component of cinnamon, inhibits the expression of NLRP3 and HIF-1α to reduce the level of IL-1β, thereby alleviating the synovial inflammatory response in adjuvant arthritis (AA) rats (96). Paeoniflorin was extracted from Paeonia lactiflora and its monomeric derivatives were found to exert therapeutic effects in rats with AA by regulating TLR4/NLRP3/GSDMD pathway and inhibiting macrophage pyroptosis (97). Chinese herb Taraxasterol can inhibit FLS pyroptosis by regulating the NLRP3 inflammatory to play an anti-RA role (98).
Fibroblast-like synoviocyte (FLS) is the main cell forming the synovial structure and has an important role in the inflammatory response to RA and joint damage. Abnormally activated FLS exhibits “tumor-like” properties and is in an activated proliferative state. Overproliferation and insufficient apoptosis of FLS play a key role in the destruction and persistent inflammation of RA (99, 100). FLS in RA patients can cause synovial proliferation, inflammatory response and bone destruction by producing large amounts of pro-inflammatory cytokines, matrix metalloproteinases (MMPs) (101). Abnormally overproliferating FLS migrates to unaffected joints, attaches to bone tissue and resists apoptosis (102, 103). Therefore the proliferation of FLS contributes to the development of RA.
pyroptosis is a PCD accompanied by inflammatory response, and recent studies have found that pyroptosis in FLS promote the progression of RA. It has been shown that increased GSDME expression in RA synovial tissue and tumor necrosis factor-α (TNF-α) plus hypoxia induce GSDME-mediated pyroptosis in FLS, with a concomitant increase of migration and invasion (104). This study links aberrantly activated FLS to the inflammatory and hypoxic synovial microenvironment and provides a new idea for fls-targeted therapy in RA. Another research, under hypoxic conditions, ROS levels within FLS increased dramatically. Excess ROS promotes the expression of GRK2, which increases the synthesis of HIF-1α. HIF-1α is translocated to the nucleus and initiates transcription of NLRP3 inflammasomes. High levels of NLRP3 inflammasomes, activate caspase-1, which in turn cuts the GSDMD thereby triggering FLS pyroptosis (105) (Figure 2).
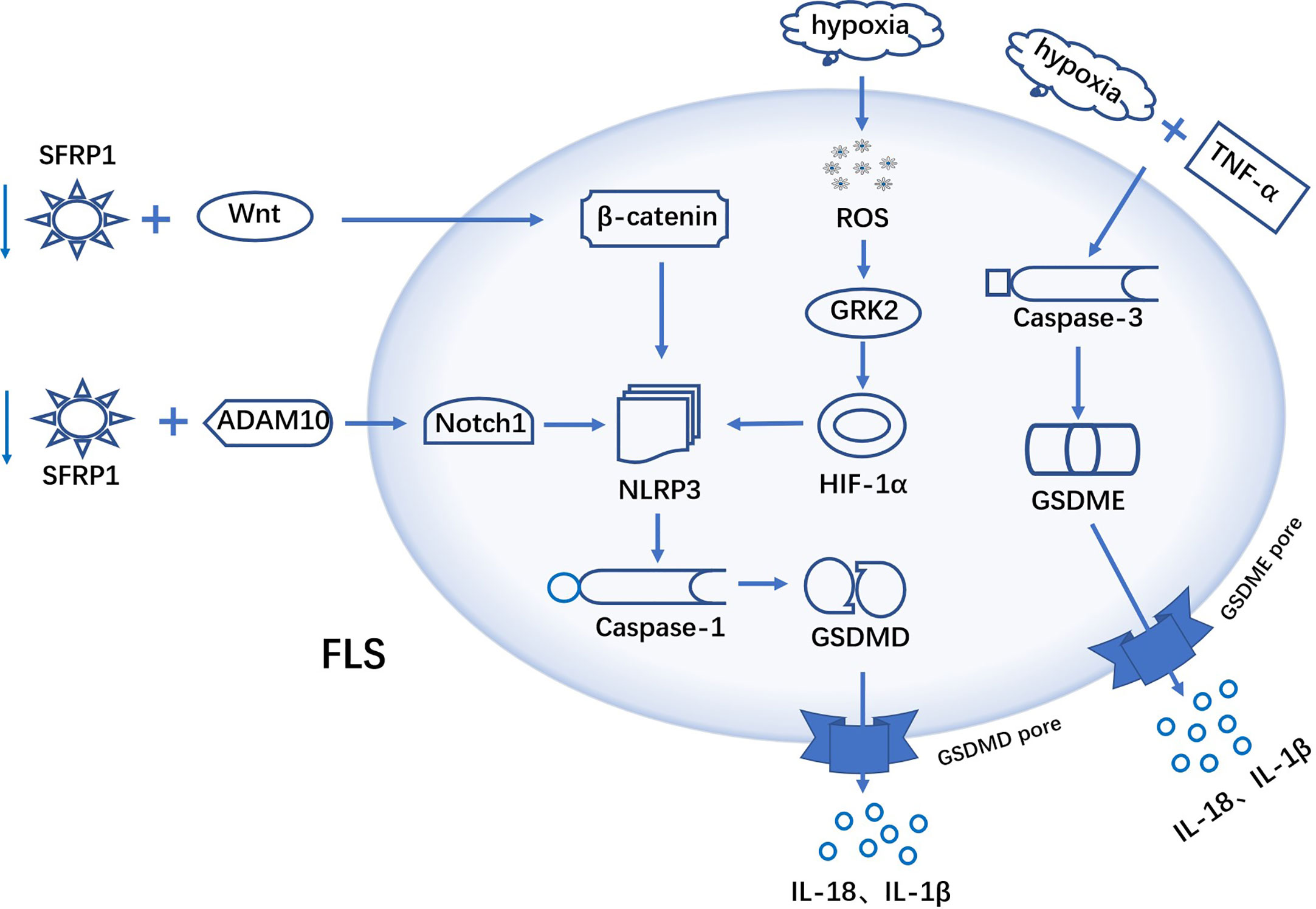
Figure 2 Mechanism of FLS pyroptosis in RA TNF-α plus hypoxia induces GSDME-mediated pyroptosis in FLS. ROS in FLS were elevated under hypoxic conditions. Excess ROS promoted GRK2 expression, which in turn increased the synthesis of HIF-1α. HIF-1α activates the transcription of NLRP3 inflammasome, excess NLRP3 activate caspase-1, and activated caspase-1 cuts GSDMD. b-catenin interacts with NLRP3 to activate NLRP3 inflammasome and initiate pyroptosis. SFRP1 binds to ADAM10 metalloprotein and down-regulates its activity, leading to blocking the activation of Notch signaling. A hypothesis: SFRP1 negatively regulates NLRP3-mediated pyroptosis via wnt/b-catenin and Notch signaling pathways in RA-FLS.
β-catenin interacts with NLRP3 and promotes its conjugation to ASC, which activates NLRP3 inflammatory and triggers pyroptosis (106). Notch1 is a novel factor that mediates the activation of NLRP3 inflammasomes. Secreted Frizzled-Related Protein 1(SFRP1) can bind to ADAM10 metalloprotein and down-regulate its activity, thereby inhibiting the activation of Notch signaling (107, 108). In the study by Jiang et al, they hypothesized that in RA-FLS, SFRP1 can negatively regulate NLRP3-mediated pyroptosis through Wnt/b-catenin and Notch signaling pathways (109). Therefore, this implies that blocking the activation of downstream signaling by upregulating the expression of SFRP1 in RA-FLS would contribute to the treatment of RA (Figure 2). In addition, Yang et al. have shown that LPS could induce FLS pyroptosis through two signaling pathways, NLRP-3/caspase-1/GSDMD and caspase-3/GSDME. NF-κB, as an upstream molecule of these two signaling pathways, is also involved in the activation of FLS pyroptosis (110).
Circular RNAs (CircRNAs) is a class of non-coding RNA molecules that do not have a 5’ end cap and a 3’ end poly(A) and are covalently bonded to form a circular structure. Hsa_circ_0044235 is expressed at lower levels in RA patients than in normal (111). Overexpression of Hsa_circ_0044235 inhibits NLRP3-mediated FLS pyroptosis, and this regulation may be achieved through the miR-135b-5p-SIRT1 signaling axis (112).
Pentraxin 3(PTX3), an important component of innate immunity (113), is elevated in the serum of RA patients and is considered as a new marker for the diagnosis of rheumatoid arthritis (114). In RA patients, complement 1q (C1q) and PTX3 act synergistically to induce GSDMD-dependent pyroptosis in CD14+ monocytes, while promoting the release of inflammatory cytokines (tumor necrosis factor-α, IL-1β and IL-6). And the released IL-6 further promotes PTX3 plus C1q-induced pyroptosis in normal monocytes (21) (Figure 3).
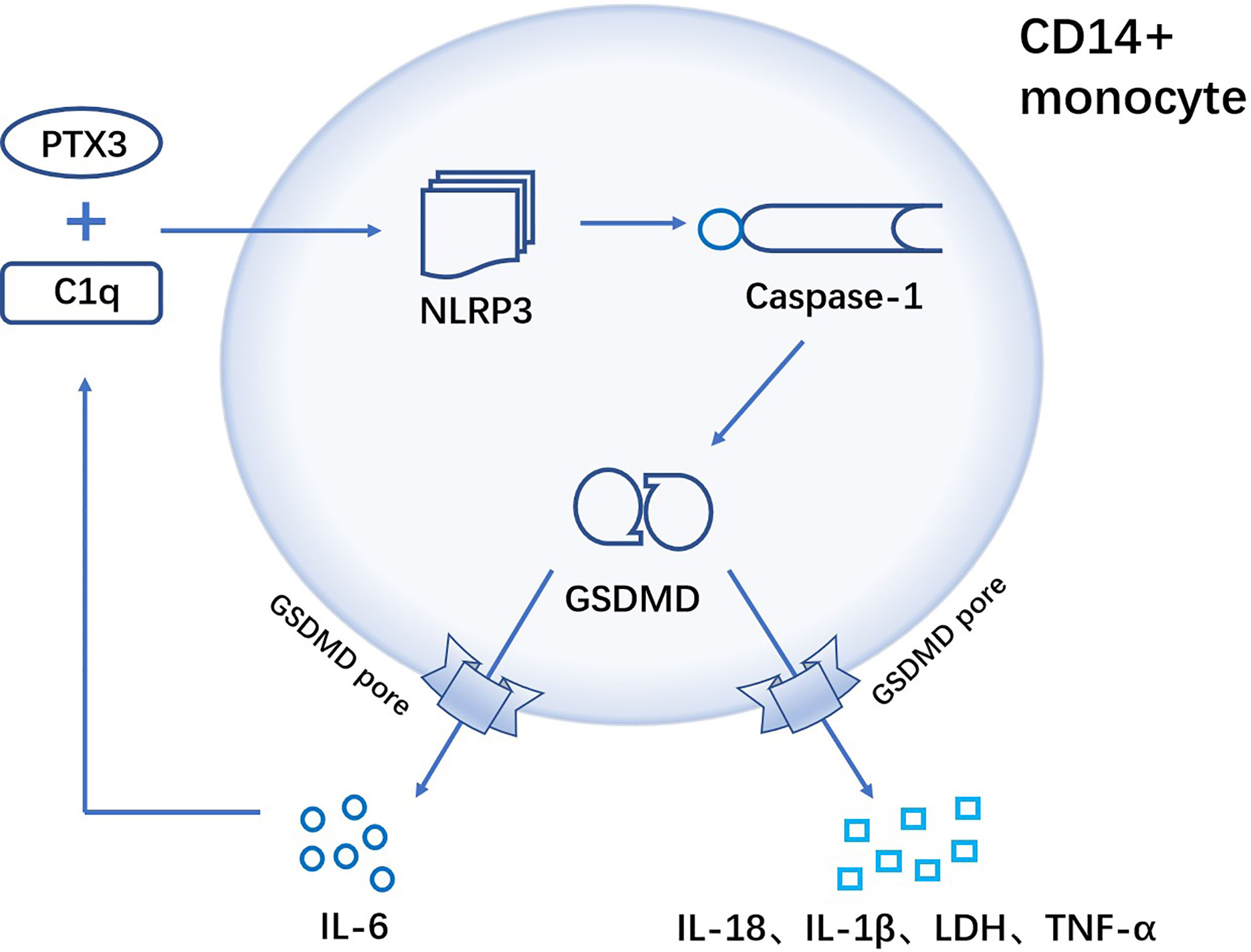
Figure 3 C1q and PTX3 act synergistically to induce GSDMD-dependent pyroptosis of CD14+ monocytes in RA. In CD14+ monocytes, the combination of C1q and PTX3 induces NLRP3 inflammasome, followed by the activation of caspase-1. Activated caspase-1 further cleaves the pyroptosis execution protein GSDMD, and GSDMD-N frees to the cell membrane to form pores that result in pyroptosis. Cellular contents are released from the GSDMD pore (TNF-α, IL-1β, IL-18, IL-6 and LDH), where IL-6 can positively enhance C1q+PTX3 combination to mediate GSDMD-dependent pyroptosis.
The expression of pyroptosis-related proteins caspase3 and GSDME-N were increased in monocytes and macrophages of RA patients. And by activated caspase3-GSDME signaling pathway, tumor necrosis factor-α (TNF-α) could induce pyroptosis of monocytes and macrophages (Figure 4). In CIA mice, GSDME deficiency effectively attenuated the degree of joint damage (22), so targeted inhibition of GSDME may be a potential treatment for RA.
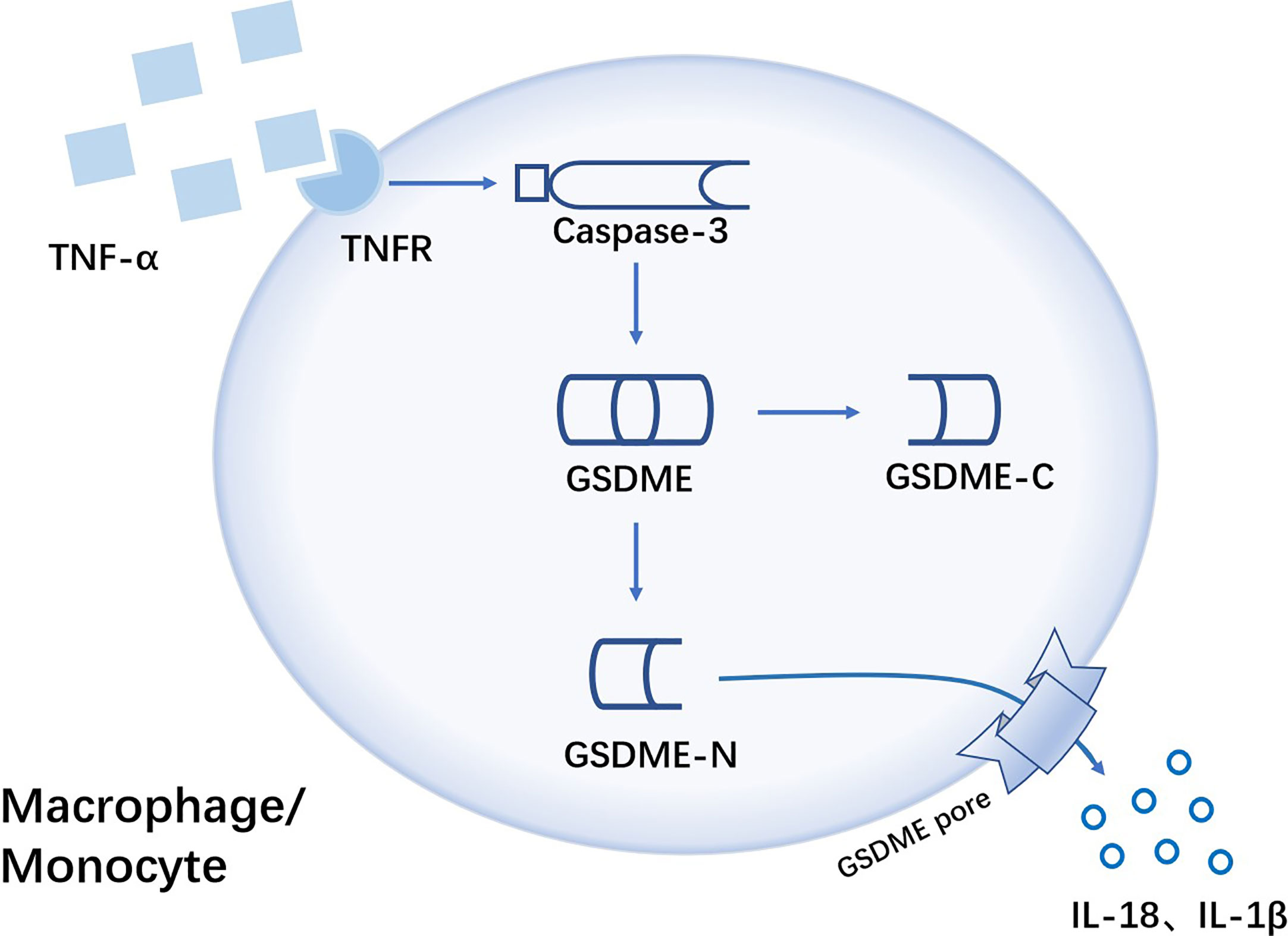
Figure 4 TNF-α induces GSDME-mediated pyroptosis of monocytes/macrophages in RA TNF-α is sensed by Tumor necrosis factor-α receptor(TNFR) on the surface of monocytes/macrophages, with the activation of caspase-3. Then activated caspase-3 cleaves GSDME, followed by free GSDME-N playing a pore-forming role leading to GSDME-dependent pyroptosis. Pro-inflammatory cytokines like IL-18 and IL-1β are released from GSDME pore.
DNA polymerase β (POL β), which is a key enzyme for base excision repair (115), has been shown to play a critical role in the pathogenesis of RA. POL β was lowly expressed in peripheral blood mononuclear cells (PBMC) of RA patients and CIA mice. Pol β deficiency promoted LPS plus ATP-induced pyroptosis in macrophage. And polβ knockdown promoted pyroptosis by activating the cGAS-STING-NF-κB signaling pathway, which upregulated NLRP3, IL-1β and IL-18 (116). As a new mechanism of macrophage pyroptosis in RA patients, it provides a new direction for the development of anti-RA drugs and treatment.
Acid-sensitive ion channels(ASICs) is a class of extracellular proton-activated cation channels whose main role is to permeabilize Na+ and Ca2+ (117). There are 7 subunits: ASIC1a, ASIC1b, ASIC1b2, ASIC2a, ASIC2b, ASIC3, ASIC4, of which ASIC1a is the only subunit that transports Ca2+, while the others are all Na+ permeable channels (118, 119). Acid-base balance is one of the most significant requirements for the maintenance of normal physiological activity (120). Almost all diseases, such as inflammation, hypoxia, cancer, etc., can cause pH changes to different degrees. Changes in pH can be sensed by the body through ASICs, which in turn regulate the corresponding tissue changes (121, 122).
The main pathological feature of RA is persistent synovitis, which leads to the destruction of articular cartilage and bone, and the accumulation of inflammatory metabolites in the joints of RA patients leads to localized tissue acidification. Acidified joint fluid can lead to apoptosis of chondrocytes, which can exacerbate the symptoms of RA (123). Acidification of joint fluid can significantly induce apoptosis in articular chondrocytes, and Wu et al. found that pyroptosis is also involved in the pathological changes. They found that extracellular acidosis significantly increased the expression of pyroptosis related protein such as ASIC1a, ASC, NLRP3 and caspase-1 in chondrocytes, while promoting the release of LDH, IL-1b and IL-18. The expression of pyroptosis associated molecules was decreased with the ASIC1a inhibitors amiloride and Psalmotoxin-1 (Pctx-1) (124). These results suggest that ASIC1a may trigger chondrocyte pyroptosis in adjuvant arthritis (AA) rat by elevating [Ca2+]i.
The inhibition of ASICs may provide a new idea for the treatment of RA, but the drugs amiloride has been found to be non-selective for ASICs, while the animal toxin represented by Pctx1 is selective but with high toxicity (125, 126), so finding a suitable ASICs inhibitor will determine whether the ASICs therapy can be applied clinically.
MicroRNAs play a very important role in the regulation of gene expression as non-coding single-stranded RNAs (127). A number of studies have shown an association between microRNAs and the development of RA (128–131). The expression of NLRP3, GSDMD, cleaved caspase-1 and cleaved caspase-3 in N1511, a mouse chondrocyte, was upregulated after IL-1β stimulation. Pyroptosis was inhibited by downregulation of miR-144-3p in N1511 cells, and miR-144-3p-activated chondrocyte pyroptosis was regulated by the PTEN/PINK1/Parkin signaling axis (132).
MRE11A is an enzyme that helps to repair single and double-stranded nucleic acid breaks (133–135). Decreased MRE11A expression of CD4+ T cells in RA patients results in diminished mitochondrial capacity (136) and release of mtDNA (mitochondrial DNA) into the cytoplasm and triggers inflammation. In a humanized NSG mouse model, MRE11A low T cells exhibit tissue invasiveness and strong pro-inflammatory properties, while recombinant MRE11A alleviates the symptoms of tissue synovitis (137). Mechanistic studies show that in MRE11A low T cells, oxidized mtDNA leaks into the cytoplasm, triggering NLRP3 and AIM2 inflammasomes assembly (138), with caspase-1 activation, leading to GSDM-dependent pyroptosis (Figure 5).
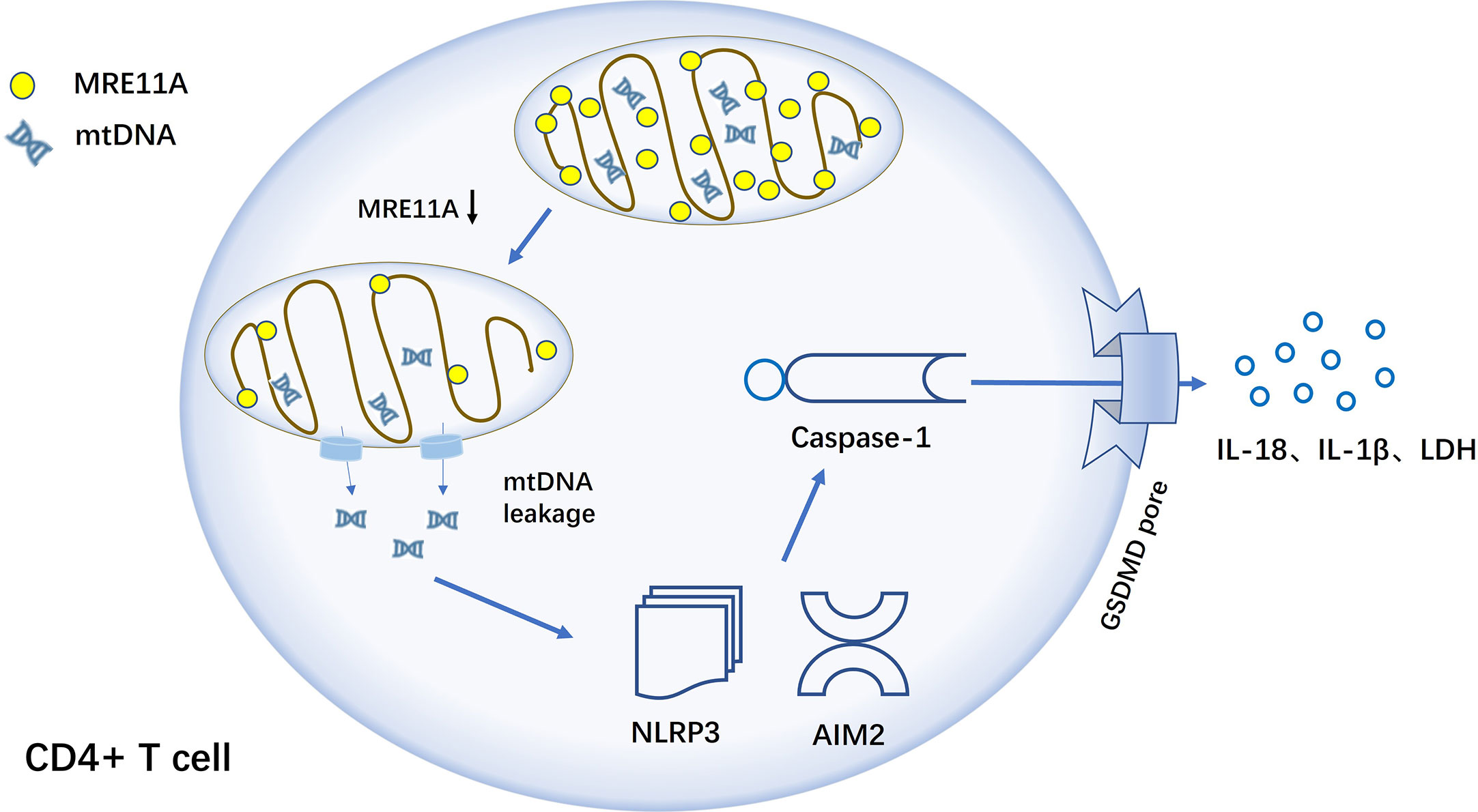
Figure 5 MRE11A loss of function results in caspase-1-induced pyroptosis of CD4+ T cell in RA T cells in RA lack MRE11A and mtDNA leak into the cytoplasm, activating inflammasome NLRP3 and AIM2, followed by further caspase-1 activation, triggering caspase-1-mediated pyroptosis and causing tissue inflammation.
In RA, T cells are converted into effector cells with an inflammatory response that promotes the progression of the disease. Therefore, up-regulating the expression of MRE11A in RA CD4+ T cells to reduce caspase-1 activation and the generation of pyroptosis, which can achieve the effect of slowing down the progression of synovitis in patients (133). MRE11A, as a repair enzyme for mtDNA, may play a key central role in the development of RA and is one more potential target for the clinical treatment of RA. A new, reliable and effective RA drug can be developed by blocking a link in the pyroptosis activation pathway from the perspective of CD4+ T cell death form, prolonging T cell lifespan and avoiding T cell-mediated inflammatory responses.
Inhibitors of NLRP3, a key protein in the pyroptosis pathway, such as MCC950, OLT1177, CY-09, and the FDA-approved drug disulfiram (GSDMD inhibitor), we summarize the current mechanism of these drugs and their exploration in the clinical trials (Table 1). The discovery of these drugs provides a reference for their future clinical application against inflammatory diseases caused by pyroptosis.
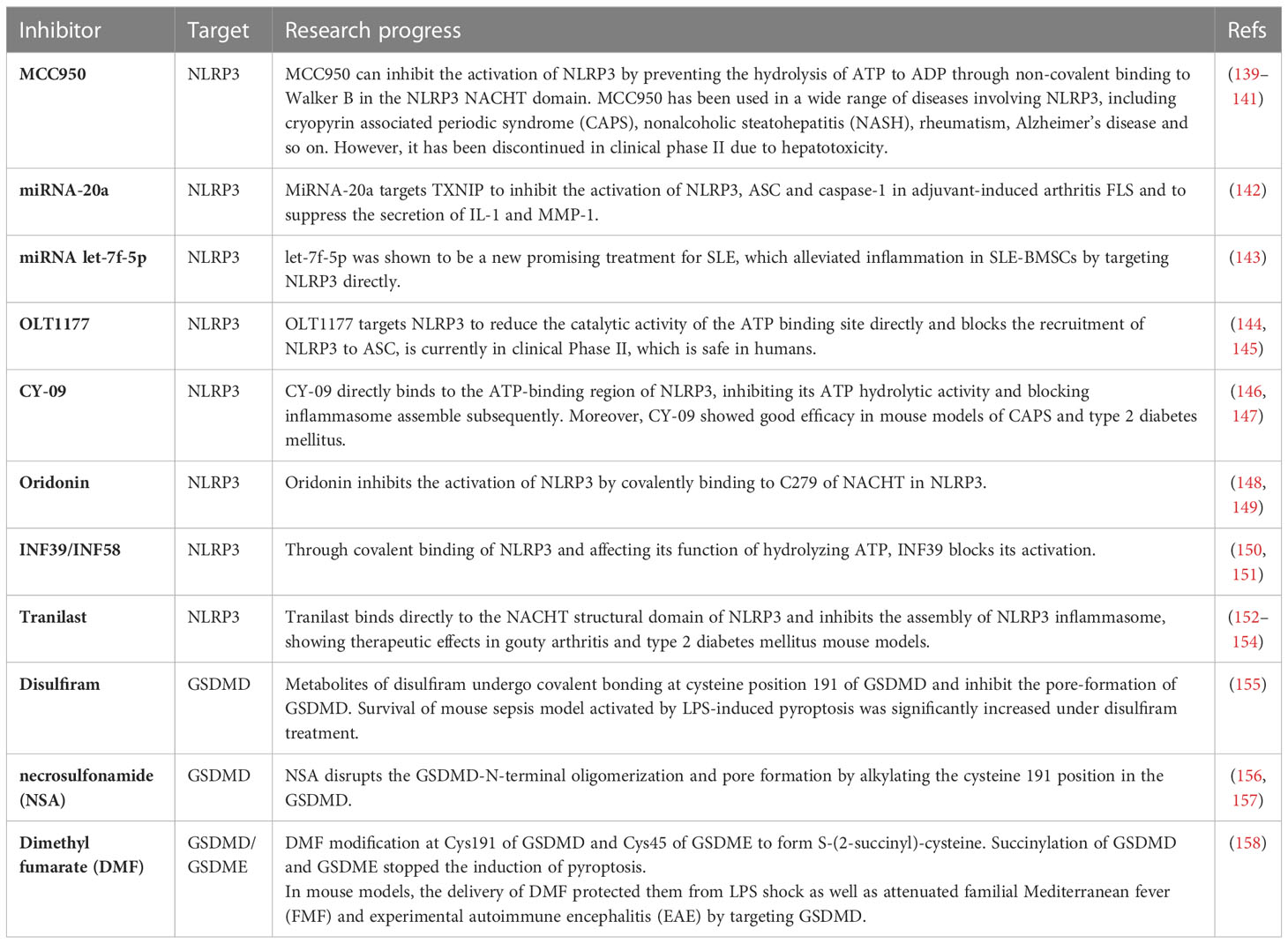
Table 1 Research progress of pyroptosis key protein inhibitors.
PCD has been extensively studied and identified as one of the essential pathological mechanisms of RA, and there is a close association between multiple types of PCD and RA. In addition to pyroptosis, other forms of PCD such as ferroptosis, apoptosis, necroptosis and autophagy have been shown to be involved in the pathogenesis of RA (18, 160, 161). Therefore, targeting PCD therapy could be a potential therapeutic strategy.
FLS exhibits resistance to apoptosis during the progression of RA, leading to synovial proliferation and promoting the release of associated cytokines through diverse mechanisms (162, 163). RA-FLS is characterized by tumor-like behaviors such as activation, invasion and migration, as well as promoting the destruction of bone and cartilage, so apoptosis-induction in RA-FLS is an effective strategy for the treatment of RA (164). Bcl-2 and Mcl-2 anti-apoptosis proteins are up-regulated in the FLS of RA patients, indicating that apoptosis is inhibited in the development of RA, and how to effectively promote FLS apoptosis and inhibit synovial proliferation has important clinical implications for RA treatment (165). Necroptosis is a mode of PCD distinct from apoptosis and necrosis. Similar to apoptosis, it strictly follows the regulation of intracellular signaling pathways and possesses the morphological characteristics of necrosis (166–168). As a non-caspase-dependent mode PCD, it can be triggered by the binding of death receptors such as TNFR or Toll-like receptors to ligands in the absence of intracellular caspase signaling factors. And the mechanism is related to the cascade activation of receptor-interacting protein kinase 1(RIPK1), receptor-interacting protein kinase 3 (RIPK3) and the downstream mixed lineage kinase domain-like protein (MLKL) (167, 169–171). Neutrophils in the joints of RA patients undergo necroptosis with upregulated expression of RIPK1, RIPK3 and MLKL (172). Studies in the articular cartilage of AA rats have shown increased expression of RIPK1, RIPK3 and P-MLKL mediated by ASIC-1a. The use of Necrostatin-1, an inhibitor of RIPK1, reduced joint damage in AA rats (173).
Ferroptosis is an iron-dependent PCD accompanied by iron accumulation and lipid peroxidation (160, 174). ROS, GPX4 and iron accumulation have been found to be closely associated with the development of RA in recent studies, indicating a likely link between RA and ferroptosis-related pathological processes (175). In the development of RA, FLS produces the pro-inflammatory cytokine TNF-α, which leads to synovitis and bone destruction (176, 177). As a product of oxidative stress, ROS are present in large quantities in the joint lumen of RA and pathological ROS imbalance affects the progression of RA through ferroptosis (178, 179). GPX4, a core molecule of ferroptosis, can effectively inhibit lipid peroxidation and block ferroptosis in cells (180, 181). In several drug studies for RA treatment, a decrease in inflammatory factors, mainly TNF-α, was observed in conjunction with an increase in GPX4 expression (161, 175). Drugs targeting ferroptosis could be a potential strategy for RA treatment.
Autophagy is a non-apoptosis form of PCD, which ensures the maintenance of cellular homeostasis by removing damaged organelles and waste proteins through the autophagosome-lysosome pathway (182, 183). Patient with RA have a significant increase in the expression of autophagy-related proteins such as Beclin1, ATG5 and LC3 in synovial tissue (184). By expressing different molecular patterns, FLS enhances autophagy and anti-apoptosis to promote its proliferation. In addition, the development of autophagy by T cells, osteoblasts and osteoclasts has been connected to RA (163, 185–187). Thus, the phenomenon of autophagy is strongly associated with the progression of RA.
The various PCD do not exist in isolation in the normal organism. Though the signaling pathways between pyroptosis, apoptosis, necroptosis and ferroptosis are different, they are not unrelated and these different PCD pathways are closely linked and regulated by each other (18, 160, 168). For example, ROS plays a key role in regulating iron overload-induced necroptosis in osteoblasts, while RIPK1/RIPK3/MLKL is a key pathway in iron-induced necroptosis (188). Iron-mediated ferroptosis can be involved in the induction of pyroptosis. The combination of iron and ROS-inducing drugs can cause a significant increase of ROS levels in melanoma cells, leading to pyroptosis via the ROS-Tom20-Caspase 3-GSDME signaling pathway (56). The study by wang et al. found that the presence of GSDME promotes a shift from TNFα-induced apoptosis to pyroptosis (15), and though this study was conducted on tumor cells, RA-associated cells may have similar mechanistic links and changes. Several PCDs involved in the pathogenesis of RA cooperate with each other and synergistically contribute to the multiple pathological processes of RA. Several PCDs involved in the pathogenesis of rheumatoid arthritis cooperate with each other and contribute to the multiple pathological processes of rheumatoid arthritis. Further study on the role of these PCDs in RA will have profound implications for the early diagnosis, treatment and prognosis of RA.
RA is a multisystemic inflammatory autoimmune disease involving peripheral joints, and the pathogenesis is still unclear. While greater progress has been made in the treatment of RA in recent years, such as the clinical application of some DMARDs, the pathogenesis of RA remains to be further explored. Both canonical and noncanonical activation pathways of pyroptosis as a pro-inflammatory death modality can induce strong inflammatory responses in body. Scientists have found in the results of numerous experimental studies that the progression of RA is closely associated with pyroptosis. IL-18 and IL-1β are pro-inflammatory cytokines released by cells undergoing pyroptosis, and they both belong to the interleukin-1 family, which has biologically similar inflammatory functions. IL-1β, as a product after inflammatory vesicle activation, promotes the binding of receptor activator of NF-kB (RANK) to ligands and promotes overactivation of RA-osteoclasts (189). IL-18 is secreted at significantly higher level in the synovial fluid and serum of RA patients than in normal, where it acts as an angiogenic mediator and leukocyte chemotactic agent, mediating the inflammatory process in RA (190–193). Knockdown of IL-18 gene resulted in reduced arthritic symptoms and decreased pro-inflammatory factors in RA mouse models, and inhibitors of caspase-1 reduced serum concentrations of IL-18 and IL-1β in CIA mice (194). All signs indicate the involvement of pyroptosis in the progression of RA. Absent in melanoma 2 (AIM2) is a member of pyroptosis and PANoptosis pathway. The latest studies have found that it is aberrantly expressed in a variety of immune cells such as T cells, B cells, FLS, etc. and that it mediates RA through a related molecular mechanism. There are several targeted inhibitors of AIM2, but their effectiveness in treating RA is unknown (195). Certain Chinese medicines, such as punicalagin, Celastrol, BaiHu GuiZhi, Cinnamaldehyde, etc., have obvious effects in anti-RA process, and mechanistic studies have revealed that their prevention and treatment of RA is through the pyroptosis pathway. There is also abundant evidence showing that chondrocytes, FLS, monocytes and CD4+ T cells in the RA organism undergo pyroptosis (Table 2). Therefore, inhibition of cell pyroptosis is very important for the treatment of RA, which can be used as a new drug target to control RA by blocking a certain part of pyroptosis. In addition to pyroptosis, a number of other PCDs including necroptosis, autophagy and ferroptosis are also involved in the pathogenesis and molecular regulation of RA. And these PCDs are connected and interact with each other, but the clear mechanisms and pathways involved are not fully clarified.

Table 2 Mechanism of the key cells in RA development.
Although pyroptosis is implicated in rheumatoid arthritis, the definite mechanism and pathway involved are not fully understood, and all these clinical drug targeting pyroptosis in RA require extensive preclinical studies. Just like the NLRP3 inhibitor MCC950 was stopped in clinical phase II due to its hepatotoxicity. Currently, there are few studies on targeted therapy drugs in pyroptosis, so we expect more studies to elaborate the molecular mechanism of PCD in RA, and we also expect that target therapy can open up a whole new way for the treatment of RA.
DW and YL drafted the manuscript. DW, YL, and RX discussed and revised the manuscript. RX designed the research. These authors contributed equally: DW and YL. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Otón T, Carmona L. The epidemiology of established rheumatoid arthritis. Best Pract Res Clin Rheumatol (2019) 33:101477. doi: 10.1016/j.berh.2019.101477
2. Lieben L, Marshall L. Rheumatoid arthritis. Nat Rev Dis Primers (2018) 4:18002. doi: 10.1038/nrdp.2018.2
3. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
4. Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol (2017) 17:151–64. doi: 10.1038/nri.2016.147
5. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet (2016) 388:2023–38. doi: 10.1016/S0140-6736(16)30173-8
6. Cohen SB. Rheumatoid arthritis: threshold for success in RA drug development. Nat Rev Rheumatol (2012) 8:567–8. doi: 10.1038/nrrheum.2012.150
7. Ferraccioli G, Gremese E. Pathogenetic, clinical and pharmaco-economic assessment in rheumatoid arthritis (RA). Intern Emerg Med (2011) 6(Suppl 1):11–5. doi: 10.1007/s11739-011-0668-6
8. Collison J. Therapy: retreatment with rituximab is beneficial in RA. Nat Rev Rheumatol (2017) 13:130. doi: 10.1038/nrrheum.2017.12
9. Ge G, Bai J, Wang Q, Liang X, Tao H, Chen H, et al. Punicalagin ameliorates collagen-induced arthritis by downregulating M1 macrophage and pyroptosis via NF-κB signaling pathway. Sci China Life Sci (2022) 65:588–603. doi: 10.1007/s11427-020-1939-1
10. Jing M, Yang J, Zhang L, Liu J, Xu S, Wang M, et al. Celastrol inhibits rheumatoid arthritis through the ROS-NF-κB-NLRP3 inflammasome axis. Int Immunopharmacol (2021) 98:107879. doi: 10.1016/j.intimp.2021.107879
11. Li W, Mao X, Wang X, Liu Y, Wang K, Li C, et al. Disease-modifying anti-rheumatic drug prescription baihu-guizhi decoction attenuates rheumatoid arthritis via suppressing toll-like receptor 4-mediated NLRP3 inflammasome activation. Front Pharmacol (2021) 12:743086. doi: 10.3389/fphar.2021.743086
12. Li W, Wang K, Liu Y, Wu H, He Y, Li C, et al. A novel drug combination of mangiferin and cinnamic acid alleviates rheumatoid arthritis by inhibiting TLR4/NFκB/NLRP3 activation-induced pyroptosis. Front Immunol (2022) 13:912933. doi: 10.3389/fimmu.2022.912933
13. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci (2017) 42:245–54. doi: 10.1016/j.tibs.2016.10.004
14. Ruan J, Wang S, Wang J. Mechanism and regulation of pyroptosis-mediated in cancer cell death. Chem Biol Interact (2020) 323:109052. doi: 10.1016/j.cbi.2020.109052
15. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature (2017) 547:99–103. doi: 10.1038/nature22393
16. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
17. Wang K, Sun Q, Zhong X, Zeng M, Zeng H, Shi X, et al. Structural mechanism for GSDMD targeting by autoprocessed caspases in pyroptosis. Cell (2020) 180:941–55.e20. doi: 10.1016/j.cell.2020.02.002
18. Zhao J, Jiang P, Guo S, Schrodi SJ, He D. Apoptosis, autophagy, NETosis, necroptosis, and pyroptosis mediated programmed cell death as targets for innovative therapy in rheumatoid arthritis. Front Immunol (2021) 12:809806. doi: 10.3389/fimmu.2021.809806
19. Burska A, Boissinot M, Ponchel F. Cytokines as biomarkers in rheumatoid arthritis. Mediators Inflammation (2014) 2014:545493. doi: 10.1155/2014/545493
20. Mathews RJ, Robinson JI, Battellino M, Wong C, Taylor JC, Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate (BRAGGSS), et al. Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment. Ann Rheum Dis (2014) 73:1202–10. doi: 10.1136/annrheumdis-2013-203276
21. Wu X-Y, Li K-T, Yang H-X, Yang B, Lu X, Zhao L-D, et al. Complement C1q synergizes with PTX3 in promoting NLRP3 inflammasome over-activation and pyroptosis in rheumatoid arthritis. J Autoimmun (2020) 106:102336. doi: 10.1016/j.jaut.2019.102336
22. Zhai Z, Yang F, Xu W, Han J, Luo G, Li Y, et al. Attenuation of rheumatoid arthritis through the inhibition of tumor necrosis factor-induced caspase 3/Gasdermin e-mediated pyroptosis. Arthritis Rheumatol (2022) 74:427–40. doi: 10.1002/art.41963
23. Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol (2017) 27:673–84. doi: 10.1016/j.tcb.2017.05.005
24. Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, et al. Pyroptosis: a new frontier in cancer. BioMed Pharmacother (2020) 121:109595. doi: 10.1016/j.biopha.2019.109595
25. Loveless R, Bloomquist R, Teng Y. Pyroptosis at the forefront of anticancer immunity. J Exp Clin Cancer Res (2021) 40:264. doi: 10.1186/s13046-021-02065-8
26. Case CL. Regulating caspase-1 during infection: roles of NLRs, AIM2, and ASC. Yale J Biol Med (2011) 84:333–43.
27. Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature (1992) 358:167–9. doi: 10.1038/358167a0
28. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol (2001) 9:113–4. doi: 10.1016/s0966-842x(00)01936-3
29. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin d for non-canonical inflammasome signalling. Nature (2015) 526:666–71. doi: 10.1038/nature15541
30. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
31. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535:111–6. doi: 10.1038/nature18590
32. Kuang S, Zheng J, Yang H, Li S, Duan S, Shen Y, et al. Structure insight of GSDMD reveals the basis of GSDMD autoinhibition in cell pyroptosis. Proc Natl Acad Sci U.S.A. (2017) 114:10642–7. doi: 10.1073/pnas.1708194114
33. Tsuchiya K, Hosojima S, Hara H, Kushiyama H, Mahib MR, Kinoshita T, et al. Gasdermin d mediates the maturation and release of IL-1α downstream of inflammasomes. Cell Rep (2021) 34:108887. doi: 10.1016/j.celrep.2021.108887
34. Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, et al. Gasdermin d pore structure reveals preferential release of mature interleukin-1. Nature (2021) 593:607–11. doi: 10.1038/s41586-021-03478-3
35. Karmakar M, Minns M, Greenberg EN, Diaz-Aponte J, Pestonjamasp K, Johnson JL, et al. N-GSDMD trafficking to neutrophil organelles facilitates IL-1β release independently of plasma membrane pores and pyroptosis. Nat Commun (2020) 11:2212. doi: 10.1038/s41467-020-16043-9
36. He W, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin d is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res (2015) 25:1285–98. doi: 10.1038/cr.2015.139
37. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol (2013) 13:397–411. doi: 10.1038/nri3452
38. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10:417–26. doi: 10.1016/s1097-2765(02)00599-3
39. Chavarría-Smith J, Vance RE. The NLRP1 inflammasomes. Immunol Rev (2015) 265:22–34. doi: 10.1111/imr.12283
40. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458:514–8. doi: 10.1038/nature07725
41. Srinivasula SM, Poyet J-L, Razmara M, Datta P, Zhang Z, Alnemri ES. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem (2002) 277:21119–22. doi: 10.1074/jbc.C200179200
42. Bryan NB, Dorfleutner A, Kramer SJ, Yun C, Rojanasakul Y, Stehlik C. Differential splicing of the apoptosis-associated speck like protein containing a caspase recruitment domain (ASC) regulates inflammasomes. J Inflammation (Lond) (2010) 7:23. doi: 10.1186/1476-9255-7-23
43. Rathinam VAK, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell (2016) 165:792–800. doi: 10.1016/j.cell.2016.03.046
44. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci (2016) 41:1012–21. doi: 10.1016/j.tibs.2016.09.002
45. Hwang I, Park S, Hong S, Kim E-H, Yu J-W. Salmonella promotes ASC oligomerization-dependent caspase-1 activation. Immune Netw (2012) 12:284–90. doi: 10.4110/in.2012.12.6.284
46. Motani K, Kushiyama H, Imamura R, Kinoshita T, Nishiuchi T, Suda T. Caspase-1 protein induces apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC)-mediated necrosis independently of its catalytic activity. J Biol Chem (2011) 286:33963–72. doi: 10.1074/jbc.M111.286823
47. Ewald SE, Chavarria-Smith J, Boothroyd JC. NLRP1 is an inflammasome sensor for toxoplasma gondii. Infect Immun (2014) 82:460–8. doi: 10.1128/IAI.01170-13
48. Zhao Y, Yang J, Shi J, Gong Y-N, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature (2011) 477:596–600. doi: 10.1038/nature10510
49. Yang J, Hwang I, Lee E, Shin SJ, Lee E-J, Rhee JH, et al. Bacterial outer membrane vesicle-mediated cytosolic delivery of flagellin triggers host NLRC4 canonical inflammasome signaling. Front Immunol (2020) 11:581165. doi: 10.3389/fimmu.2020.581165
50. Kanneganti T-D, Lamkanfi M. K+ drops tilt the NLRP3 inflammasome. Immunity (2013) 38:1085–8. doi: 10.1016/j.immuni.2013.06.001
51. Rühl S, Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting k(+) efflux. Eur J Immunol (2015) 45:2927–36. doi: 10.1002/eji.201545772
52. Wang Y, Yin B, Li D, Wang G, Han X, Sun X. GSDME mediates caspase-3-dependent pyroptosis in gastric cancer. Biochem Biophys Res Commun (2018) 495:1418–25. doi: 10.1016/j.bbrc.2017.11.156
53. Hagn M, Sontheimer K, Dahlke K, Brueggemann S, Kaltenmeier C, Beyer T, et al. Human b cells differentiate into granzyme b-secreting cytotoxic b lymphocytes upon incomplete T-cell help. Immunol Cell Biol (2012) 90:457–67. doi: 10.1038/icb.2011.64
54. Afonina IS, Cullen SP, Martin SJ. Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme b. Immunol Rev (2010) 235:105–16. doi: 10.1111/j.0105-2896.2010.00908.x
55. Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin e suppresses tumour growth by activating anti-tumour immunity. Nature (2020) 579:415–20. doi: 10.1038/s41586-020-2071-9
56. Zhou B, Zhang J-Y, Liu X-S, Chen H-Z, Ai Y-L, Cheng K, et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res (2018) 28:1171–85. doi: 10.1038/s41422-018-0090-y
57. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme a from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science (2020) 368:eaaz7548. doi: 10.1126/science.aaz7548
58. Hou J, Zhao R, Xia W, Chang C-W, You Y, Hsu J-M, et al. PD-L1-mediated gasdermin c expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol (2020) 22:1264–75. doi: 10.1038/s41556-020-0575-z
59. Chemin K, Klareskog L, Malmström V. Is rheumatoid arthritis an autoimmune disease? Curr Opin Rheumatol (2016) 28:181–8. doi: 10.1097/BOR.0000000000000253
60. Aringer M, Tony H-P. Systemic disease rheumatoid arthritis. Z Rheumatol (2012) 71:840. doi: 10.1007/s00393-011-0924-1
61. Rose J. Autoimmune connective tissue diseases: systemic lupus erythematosus and rheumatoid arthritis. Emerg Med Clin North Am (2022) 40:179–91. doi: 10.1016/j.emc.2021.09.003
62. Miossec P. Rheumatoid arthritis: still a chronic disease. Lancet (2013) 381:884–6. doi: 10.1016/S0140-6736(12)62192-8
63. Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ, Xu J. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res (2018) 6:15. doi: 10.1038/s41413-018-0016-9
64. Uzuki M, Murakami K, Kamataki A, Kubota A, Suguro T, Sawai T. Rheumatoid arthritis:progress in diagnosis and treatment topics: II. pathophysiology; 1. pathological findings of rheumatoid arthritis. Nihon Naika Gakkai Zasshi (2012) 101:2830–8. doi: 10.2169/naika.101.2830
65. Khoshroo A, Ramezani K, Moghimi N, Bonakdar M, Ramezani N. The effect of disease-modifying antirheumatic drugs (DMARDs) on bone homeostasis in rheumatoid arthritis (RA) patients. Inflammopharmacology (2023) 31:689–97. doi: 10.1007/s10787-022-01088-7
66. Tanaka E, Yamanaka H. DMARDs (disease-modifying antirheumatic drugs)]. Nihon Rinsho (2013) 71:1199–206.
67. Gorter SL. Rheumatoid arthritis and glucocorticoids; the contribution of a literature search to the development of a EULAR recommendation on treatment with glucocorticoids in RA. Clin Exp Rheumatol (2011) 29:S77–80.
68. Thakur S, Riyaz B, Patil A, Kaur A, Kapoor B, Mishra V. Novel drug delivery systems for NSAIDs in management of rheumatoid arthritis: an overview. BioMed Pharmacother (2018) 106:1011–23. doi: 10.1016/j.biopha.2018.07.027
69. Kurth T, Hennekens CH, Buring JE, Gaziano JM. Aspirin, NSAIDs, and COX-2 inhibitors in cardiovascular disease: possible interactions and implications for treatment of rheumatoid arthritis. Curr Rheumatol Rep (2004) 6:351–6. doi: 10.1007/s11926-004-0009-0
70. Smucny J, Chai G. Are selective COX-2 inhibitors as effective as NSAIDs in patients with rheumatoid arthritis? Am Fam Physician (2004) 69:595–6.
71. Lo V, Meadows SE, Saseen J. When should COX-2 selective NSAIDs be used for osteoarthritis and rheumatoid arthritis? J Fam Pract (2006) 55:260–2.
72. Araújo F, Gonçalves J, Fonseca JE. Biosimilar DMARDs: what does the future hold? Drugs (2016) 76:629–37. doi: 10.1007/s40265-016-0556-5
73. Fleischmann R. Don’t forget traditional DMARDs. Rheumatol (Oxford) (2011) 50:429–30. doi: 10.1093/rheumatology/keq382
74. Wang W, Zhou H, Liu L. Side effects of methotrexate therapy for rheumatoid arthritis: a systematic review. Eur J Med Chem (2018) 158:502–16. doi: 10.1016/j.ejmech.2018.09.027
75. Zhao Z, Hua Z, Luo X, Li Y, Yu L, Li M, et al. Application and pharmacological mechanism of methotrexate in rheumatoid arthritis. BioMed Pharmacother (2022) 150:113074. doi: 10.1016/j.biopha.2022.113074
76. Kremer JM. Let’s re-examine these MTX points once again. Ann Rheum Dis (2016) 75:e54. doi: 10.1136/annrheumdis-2016-209834
77. Volin MV, Campbell PL, Connors MA, Woodruff DC, Koch AE. The effect of sulfasalazine on rheumatoid arthritic synovial tissue chemokine production. Exp Mol Pathol (2002) 73:84–92. doi: 10.1006/exmp.2002.2460
78. Soukup T, Dosedel M, Nekvindova J, Toms J, Vlcek J, Pavek P. Genetic polymorphisms in metabolic pathways of leflunomide in the treatment of rheumatoid arthritis. Clin Exp Rheumatol (2015) 33:426–32.
80. Keeling S, Maksymowych WP. JAK inhibitors, psoriatic arthritis, and axial spondyloarthritis: a critical review of clinical trials. Expert Rev Clin Immunol (2021) 17:701–15. doi: 10.1080/1744666X.2021.1925541
81. Dhillon S. Tofacitinib: a review in rheumatoid arthritis. Drugs (2017) 77:1987–2001. doi: 10.1007/s40265-017-0835-9
82. Al-Salama ZT, Scott LJ. Baricitinib: a review in rheumatoid arthritis. Drugs (2018) 78:761–72. doi: 10.1007/s40265-018-0908-4
83. Zhao S, Mysler E, Moots RJ. Etanercept for the treatment of rheumatoid arthritis. Immunotherapy (2018) 10:433–45. doi: 10.2217/imt-2017-0155
84. Yamanaka G, Ishida Y, Kanou K, Suzuki S, Watanabe Y, Takamatsu T, et al. Towards a treatment for neuroinflammation in epilepsy: interleukin-1 receptor antagonist, anakinra, as a potential treatment in intractable epilepsy. Int J Mol Sci (2021) 22:6282. doi: 10.3390/ijms22126282
85. Onuora S. Rheumatoid arthritis: sarilumab more effective than adalimumab. Nat Rev Rheumatol (2017) 13:2. doi: 10.1038/nrrheum.2016.207
86. Lamb YN, Deeks ED. Sarilumab: a review in moderate to severe rheumatoid arthritis. Drugs (2018) 78:929–40. doi: 10.1007/s40265-018-0929-z
87. Tavakolpour S, Alesaeidi S, Darvishi M, GhasemiAdl M, Darabi-Monadi S, Akhlaghdoust M, et al. A comprehensive review of rituximab therapy in rheumatoid arthritis patients. Clin Rheumatol (2019) 38:2977–94. doi: 10.1007/s10067-019-04699-8
88. Salles G, Barrett M, Foà R, Maurer J, O’Brien S, Valente N, et al. Rituximab in b-cell hematologic malignancies: a review of 20 years of clinical experience. Adv Ther (2017) 34:2232–73. doi: 10.1007/s12325-017-0612-x
89. Spies CM, Bijlsma JWJ, Burmester G-R, Buttgereit F. Pharmacology of glucocorticoids in rheumatoid arthritis. Curr Opin Pharmacol (2010) 10:302–7. doi: 10.1016/j.coph.2010.02.001
90. Keyßer G. Safety aspects of the treatment with glucocorticoids for rheumatoid arthritis]. Z Rheumatol (2021) 80:295–304. doi: 10.1007/s00393-021-00972-x
91. Volkmann ER. Tapering glucocorticoids in rheumatoid arthritis. Lancet (2020) 396:218–9. doi: 10.1016/S0140-6736(20)30761-3
92. Akama H. Topics of glucocorticoids–centered on therapy for rheumatoid arthritis. Nihon Rinsho Meneki Gakkai Kaishi (2011) 34:464–75. doi: 10.2177/jsci.34.464
93. Giangreco D, Cutolo M. Morning glucocorticoids versus night glucocorticoids: the role of low-dose glucocorticoid chronotherapy in rheumatoid arthritis. J Clin Rheumatol (2014) 20:437–9. doi: 10.1097/RHU.0000000000000182
94. Wang W, Zhou H, Liu L. The role of Chinese herbal medicine in the management of adverse drug reactions of leflunomide in treating rheumatoid arthritis. Phytomedicine (2020) 68:153136. doi: 10.1016/j.phymed.2019.153136
95. Ling Y, Xiao M, Huang Z-W, Xu H, Huang F-Q, Ren N-N, et al. Jinwujiangu capsule treats fibroblast-like synoviocytes of rheumatoid arthritis by inhibiting pyroptosis via the NLRP3/CAPSES/GSDMD pathway. Evid Based Complement Alternat Med (2021) 2021:4836992. doi: 10.1155/2021/4836992
96. Liu P, Wang J, Wen W, Pan T, Chen H, Fu Y, et al. Cinnamaldehyde suppresses NLRP3 derived IL-1β via activating succinate/HIF-1 in rheumatoid arthritis rats. Int Immunopharmacol (2020) 84:106570. doi: 10.1016/j.intimp.2020.106570
97. Xu L, Wang H, Yu Q-Q, Ge J-R, Zhang X-Z, Mei D, et al. The monomer derivative of paeoniflorin inhibits macrophage pyroptosis via regulating TLR4/ NLRP3/ GSDMD signaling pathway in adjuvant arthritis rats. Int Immunopharmacol (2021) 101:108169. doi: 10.1016/j.intimp.2021.108169
98. Chen J, Wu W, Zhang M, Chen C. Taraxasterol suppresses inflammation in IL-1β-induced rheumatoid arthritis fibroblast-like synoviocytes and rheumatoid arthritis progression in mice. Int Immunopharmacol (2019) 70:274–83. doi: 10.1016/j.intimp.2019.02.029
99. Matsuo Y, Mizoguchi F, Saito T, Kawahata K, Ueha S, Matsushima K, et al. Local fibroblast proliferation but not influx is responsible for synovial hyperplasia in a murine model of rheumatoid arthritis. Biochem Biophys Res Commun (2016) 470:504–9. doi: 10.1016/j.bbrc.2016.01.121
100. Mousavi MJ, Karami J, Aslani S, Tahmasebi MN, Vaziri AS, Jamshidi A, et al. Transformation of fibroblast-like synoviocytes in rheumatoid arthritis; from a friend to foe. Auto Immun Highlights (2021) 12:3. doi: 10.1186/s13317-020-00145-x
101. Bustamante MF, Garcia-Carbonell R, Whisenant KD, Guma M. Fibroblast-like synoviocyte metabolism in the pathogenesis of rheumatoid arthritis. Arthritis Res Ther (2017) 19:110. doi: 10.1186/s13075-017-1303-3
102. Liu J, Zhang Q, Li R-L, Wei S-J, Gao Y-X, Ai L, et al. Anti-proliferation and anti-migration effects of an aqueous extract of cinnamomi ramulus on MH7A rheumatoid arthritis-derived fibroblast-like synoviocytes through induction of apoptosis, cell arrest and suppression of matrix metalloproteinase. Pharm Biol (2020) 58:863–77. doi: 10.1080/13880209.2020.1810287
103. Zhang K, Gao S, Guo J, Ni G, Chen Z, Li F, et al. Hypericin-photodynamic therapy inhibits proliferation and induces apoptosis in human rheumatoid arthritis fibroblast-like synoviocytes cell line MH7A. Iran J Basic Med Sci (2018) 21:130–7. doi: 10.22038/IJBMS.2018.23871.5991
104. Wu T, Zhang X-P, Zhang Q, Zou Y-Y, Ma J-D, Chen L-F, et al. Gasdermin-e mediated pyroptosis-a novel mechanism regulating migration, invasion and release of inflammatory cytokines in rheumatoid arthritis fibroblast-like synoviocytes. Front Cell Dev Biol (2021) 9:810635. doi: 10.3389/fcell.2021.810635
105. Hong Z, Zhang X, Zhang T, Hu L, Liu R, Wang P, et al. The ROS/GRK2/HIF-1α/NLRP3 pathway mediates pyroptosis of fibroblast-like synoviocytes and the regulation of monomer derivatives of paeoniflorin. Oxid Med Cell Longev (2022) 2022:4566851. doi: 10.1155/2022/4566851
106. Huang L, Luo R, Li J, Wang D, Zhang Y, Liu L, et al. β-catenin promotes NLRP3 inflammasome activation via increasing the association between NLRP3 and ASC. Mol Immunol (2020) 121:186–94. doi: 10.1016/j.molimm.2020.02.017
107. Lee S, Kim SK, Park H, Lee YJ, Park SH, Lee KJ, et al. Contribution of autophagy-Notch1-Mediated NLRP3 inflammasome activation to chronic inflammation and fibrosis in keloid fibroblasts. Int J Mol Sci (2020) 21:8050. doi: 10.3390/ijms21218050
108. Esteve P, Sandonìs A, Cardozo M, Malapeira J, Ibañez C, Crespo I, et al. SFRPs act as negative modulators of ADAM10 to regulate retinal neurogenesis. Nat Neurosci (2011) 14:562–9. doi: 10.1038/nn.2794
109. Jiang P, Wei K, Chang C, Zhao J, Zhang R, Xu L, et al. SFRP1 negatively modulates pyroptosis of fibroblast-like synoviocytes in rheumatoid arthritis: a review. Front Immunol (2022) 13:903475. doi: 10.3389/fimmu.2022.903475
110. Yang P, Feng W, Li C, Kou Y, Li D, Liu S, et al. LPS induces fibroblast-like synoviocytes RSC-364 cells to pyroptosis through NF-κB mediated dual signalling pathway. J Mol Histol (2021) 52:661–9. doi: 10.1007/s10735-021-09988-8
111. Luo Q, Zhang L, Li X, Fu B, Deng Z, Qing C, et al. Identification of circular RNAs hsa_circ_0044235 in peripheral blood as novel biomarkers for rheumatoid arthritis. Clin Exp Immunol (2018) 194:118–24. doi: 10.1111/cei.13181
112. Chen S, Luo Z, Chen X. Hsa_circ_0044235 regulates the pyroptosis of rheumatoid arthritis via MiR-135b-5p-SIRT1 axis. Cell Cycle (2021) 20:1107–21. doi: 10.1080/15384101.2021.1916272
113. Inforzato A, Doni A, Barajon I, Leone R, Garlanda C, Bottazzi B, et al. PTX3 as a paradigm for the interaction of pentraxins with the complement system. Semin Immunol (2013) 25:79–85. doi: 10.1016/j.smim.2013.05.002
114. Sharma A, Khan R, Gupta N, Sharma A, Zaheer MS, Abbas M, et al. Acute phase reactant, pentraxin 3, as a novel marker for the diagnosis of rheumatoid arthritis. Clin Chim Acta (2018) 480:65–70. doi: 10.1016/j.cca.2018.01.035
115. Nicolay NH, Helleday T, Sharma RA. Biological relevance of DNA polymerase β and translesion synthesis polymerases to cancer and its treatment. Curr Mol Pharmacol (2012) 5:54–67.
116. Gu L, Sun Y, Wu T, Chen G, Tang X, Zhao L, et al. A novel mechanism for macrophage pyroptosis in rheumatoid arthritis induced by pol β deficiency. Cell Death Dis (2022) 13:583. doi: 10.1038/s41419-022-05047-6
117. Gründer S, Pusch M. Biophysical properties of acid-sensing ion channels (ASICs). Neuropharmacology (2015) 94:9–18. doi: 10.1016/j.neuropharm.2014.12.016
118. Bhowmick S, Moore JT, Kirschner DL, Curry MC, Westbrook EG, Rasley BT, et al. Acidotoxicity via ASIC1a mediates cell death during oxygen glucose deprivation and abolishes excitotoxicity. ACS Chem Neurosci (2017) 8:1204–12. doi: 10.1021/acschemneuro.6b00355
119. Lin Y-C, Liu Y-C, Huang Y-Y, Lien C-C. High-density expression of Ca2+-permeable ASIC1a channels in NG2 glia of rat hippocampus. PloS One (2010) 5:e12665. doi: 10.1371/journal.pone.0012665
120. Allison SJ. Acid-base disorders: paracrine regulation of acid-base balance. Nat Rev Nephrol (2013) 9:493. doi: 10.1038/nrneph.2013.138
121. Wu J, Wang J-J, Liu T-T, Zhou Y-M, Qiu C-Y, Shen D-W, et al. PPAR-α acutely inhibits functional activity of ASICs in rat dorsal root ganglion neurons. Oncotarget (2017) 8:93051–62. doi: 10.18632/oncotarget.21805
122. Deval E, Gasull X, Noël J, Salinas M, Baron A, Diochot S, et al. Acid-sensing ion channels (ASICs): pharmacology and implication in pain. Pharmacol Ther (2010) 128:549–58. doi: 10.1016/j.pharmthera.2010.08.006
123. Ahn H, Kim JM, Lee K, Kim H, Jeong D. Extracellular acidosis accelerates bone resorption by enhancing osteoclast survival, adhesion, and migration. Biochem Biophys Res Commun (2012) 418:144–8. doi: 10.1016/j.bbrc.2011.12.149
124. Wu X, Ren G, Zhou R, Ge J, Chen F-H. The role of Ca2+ in acid-sensing ion channel 1a-mediated chondrocyte pyroptosis in rat adjuvant arthritis. Lab Invest (2019) 99:499–513. doi: 10.1038/s41374-018-0135-3
125. Heusser SA, Borg CB, Colding JM, Pless SA. Conformational decoupling in acid-sensing ion channels uncovers mechanism and stoichiometry of PcTx1-mediated inhibition. Elife (2022) 11:e73384. doi: 10.7554/eLife.73384
126. McCarthy CA, Rash LD, Chassagnon IR, King GF, Widdop RE. PcTx1 affords neuroprotection in a conscious model of stroke in hypertensive rats via selective inhibition of ASIC1a. Neuropharmacology (2015) 99:650–7. doi: 10.1016/j.neuropharm.2015.08.040
127. Cloney R. Non-coding RNA: deciphering the rules of microRNA targeting. Nat Rev Genet (2016) 17:718. doi: 10.1038/nrg.2016.148
128. Najm A, Masson F-M, Preuss P, Georges S, Ory B, Quillard T, et al. MicroRNA-17-5p reduces inflammation and bone erosions in mice with collagen-induced arthritis and directly targets the JAK/STAT pathway in rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheumatol (2020) 72:2030–9. doi: 10.1002/art.41441
129. Kmiołek T, Paradowska-Gorycka A. miRNAs as biomarkers and possible therapeutic strategies in rheumatoid arthritis. Cells (2022) 11:452. doi: 10.3390/cells11030452
130. Safari F, Damavandi E, Rostamian A-R, Movassaghi S, Imani-Saber Z, Saffari M, et al. Plasma levels of MicroRNA-146a-5p, MicroRNA-24-3p, and MicroRNA-125a-5p as potential diagnostic biomarkers for rheumatoid arthris. Iran J Allergy Asthma Immunol (2021) 20:326–37. doi: 10.18502/ijaai.v20i3.6334
131. Evangelatos G, Fragoulis GE, Koulouri V, Lambrou GI. MicroRNAs in rheumatoid arthritis: from pathogenesis to clinical impact. Autoimmun Rev (2019) 18:102391. doi: 10.1016/j.autrev.2019.102391
132. Jiang J-M, Mo M-L, Long X-P, Xie L-H. MiR-144-3p induced by SP1 promotes IL-1β-induced pyroptosis in chondrocytes via PTEN/PINK1/Parkin axis. Autoimmunity (2022) 55:21–31. doi: 10.1080/08916934.2021.1983802
133. Li Y, Shen Y, Jin K, Wen Z, Cao W, Wu B, et al. The DNA repair nuclease MRE11A functions as a mitochondrial protector and prevents T cell pyroptosis and tissue inflammation. Cell Metab (2019) 30:477–492.e6. doi: 10.1016/j.cmet.2019.06.016
134. Syed A, Tainer JA. The MRE11-RAD50-NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu Rev Biochem (2018) 87:263–94. doi: 10.1146/annurev-biochem-062917-012415
135. Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol (2009) 16:814–8. doi: 10.1038/nsmb.1640
136. Koetz K, Bryl E, Spickschen K, O’Fallon WM, Goronzy JJ, Weyand CM. T Cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci U.S.A. (2000) 97:9203–8. doi: 10.1073/pnas.97.16.9203
137. Li Y, Shen Y, Hohensinner P, Ju J, Wen Z, Goodman SB, et al. Deficient activity of the nuclease MRE11A induces T cell aging and promotes arthritogenic effector functions in patients with rheumatoid arthritis. Immunity (2016) 45:903–16. doi: 10.1016/j.immuni.2016.09.013
138. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin X-J, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature (2018) 560:198–203. doi: 10.1038/s41586-018-0372-z
139. Tapia-Abellán A, Angosto-Bazarra D, Martínez-Banaclocha H, de Torre-Minguela C, Cerón-Carrasco JP, Pérez-Sánchez H, et al. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat Chem Biol (2019) 15:560–4. doi: 10.1038/s41589-019-0278-6
140. Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol (2019) 15:556–9. doi: 10.1038/s41589-019-0277-7
141. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discovery (2018) 17:588–606. doi: 10.1038/nrd.2018.97
142. Li X-F, Shen W-W, Sun Y-Y, Li W-X, Sun Z-H, Liu Y-H, et al. MicroRNA-20a negatively regulates expression of NLRP3-inflammasome by targeting TXNIP in adjuvant-induced arthritis fibroblast-like synoviocytes. Joint Bone Spine (2016) 83:695–700. doi: 10.1016/j.jbspin.2015.10.007
143. Tan W, Gu Z, Leng J, Zou X, Chen H, Min F, et al. Let-7f-5p ameliorates inflammation by targeting NLRP3 in bone marrow-derived mesenchymal stem cells in patients with systemic lupus erythematosus. BioMed Pharmacother (2019) 118:109313. doi: 10.1016/j.biopha.2019.109313
144. Sánchez-Fernández A, Skouras DB, Dinarello CA, López-Vales R. OLT1177 (Dapansutrile), a selective NLRP3 inflammasome inhibitor, ameliorates experimental autoimmune encephalomyelitis pathogenesis. Front Immunol (2019) 10:2578. doi: 10.3389/fimmu.2019.02578
145. Marchetti C, Swartzwelter B, Gamboni F, Neff CP, Richter K, Azam T, et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci U.S.A. (2018) 115:E1530–9. doi: 10.1073/pnas.1716095115
146. Wang Y, Liu Y-J, Zhang M-M, Zhou H, Gao Y-H, Cheng W-J, et al. CY-09 alleviates the depression-like behaviors via inhibiting NLRP3 inflammasome-mediated neuroinflammation in lipopolysaccharide-induced mice. ACS Chem Neurosci (2022) 13:3291–302. doi: 10.1021/acschemneuro.2c00348
147. Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J, et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med (2017) 214:3219–38. doi: 10.1084/jem.20171419
148. He H, Jiang H, Chen Y, Ye J, Wang A, Wang C, et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun (2018) 9:2550. doi: 10.1038/s41467-018-04947-6
149. Lin J, Lai X, Fan X, Ye B, Zhong L, Zhang Y, et al. Oridonin protects against myocardial ischemia-reperfusion injury by inhibiting GSDMD-mediated pyroptosis. Genes (Basel) (2022) 13:2133. doi: 10.3390/genes13112133
150. Chen W, Tang P, Fan S, Jiang X. A novel inhibitor INF 39 promotes osteogenesis via blocking the NLRP3/IL-1β axis. BioMed Res Int (2022) 2022:7250578. doi: 10.1155/2022/7250578
151. Shi Y, Lv Q, Zheng M, Sun H, Shi F. NLRP3 inflammasome inhibitor INF39 attenuated NLRP3 assembly in macrophages. Int Immunopharmacol (2021) 92:107358. doi: 10.1016/j.intimp.2020.107358
152. Saeedi-Boroujeni A, Mahmoudian-Sani M-R, Nashibi R, Houshmandfar S, Tahmaseby Gandomkari S, Khodadadi A. Tranilast: a potential anti-inflammatory and NLRP3 inflammasome inhibitor drug for COVID-19. Immunopharmacol Immunotoxicol (2021) 43:247–58. doi: 10.1080/08923973.2021.1925293
153. Pfab T, Hocher B. Tranilast and hypertensive heart disease: further insights into mechanisms of an anti-inflammatory and anti-fibrotic drug. J Hypertens (2004) 22:883–6. doi: 10.1097/00004872-200405000-00006
154. Huang Y, Jiang H, Chen Y, Wang X, Yang Y, Tao J, et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med (2018) 10:e8689. doi: 10.15252/emmm.201708689
155. Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-Approved disulfiram inhibits pyroptosis by blocking gasdermin d pore formation. Nat Immunol (2020) 21:736–45. doi: 10.1038/s41590-020-0669-6
156. Yang W, Tao K, Wang Y, Huang Y, Duan C, Wang T, et al. Necrosulfonamide ameliorates intestinal inflammation via inhibiting GSDMD-medicated pyroptosis and MLKL-mediated necroptosis. Biochem Pharmacol (2022) 206:115338. doi: 10.1016/j.bcp.2022.115338
157. Zhang J, Wei K. Necrosulfonamide reverses pyroptosis-induced inhibition of proliferation and differentiation of osteoblasts through the NLRP3/caspase-1/GSDMD pathway. Exp Cell Res (2021) 405:112648. doi: 10.1016/j.yexcr.2021.112648
158. Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination inactivates gasdermin d and blocks pyroptosis. Science (2020) 369:1633–7. doi: 10.1126/science.abb9818
159. Xie Q, Wei M, Zhang B, Kang X, Liu D, Zheng W, et al. MicroRNA−33 regulates the NLRP3 inflammasome signaling pathway in macrophages. Mol Med Rep (2018) 17:3318–27. doi: 10.3892/mmr.2017.8224
160. Zhao T, Yang Q, Xi Y, Xie Z, Shen J, Li Z, et al. Ferroptosis in rheumatoid arthritis: a potential therapeutic strategy. Front Immunol (2022) 13:779585. doi: 10.3389/fimmu.2022.779585
161. Xie Z, Hou H, Luo D, An R, Zhao Y, Qiu C. ROS-dependent lipid peroxidation and reliant antioxidant ferroptosis-Suppressor-Protein 1 in rheumatoid arthritis: a covert clue for potential therapy. Inflammation (2021) 44:35–47. doi: 10.1007/s10753-020-01338-2
162. Li Y, Xing Q, Wei Y, Zhao L, Zhang P, Han X, et al. Activation of RXR by bexarotene inhibits inflammatory conditions in human rheumatoid arthritis fibroblast−like synoviocytes. Int J Mol Med (2019) 44:1963–70. doi: 10.3892/ijmm.2019.4336
163. Kim EK, Kwon J-E, Lee S-Y, Lee E-J, Kim DS, Moon S-J, et al. IL-17-mediated mitochondrial dysfunction impairs apoptosis in rheumatoid arthritis synovial fibroblasts through activation of autophagy. Cell Death Dis (2017) 8:e2565. doi: 10.1038/cddis.2016.490
164. Audo R, Combe B, Coulet B, Morel J, Hahne M. The pleiotropic effect of TRAIL on tumor-like synovial fibroblasts from rheumatoid arthritis patients is mediated by caspases. Cell Death Differ (2009) 16:1227–37. doi: 10.1038/cdd.2009.38
165. Zhang Q, Liu J, Zhang M, Wei S, Li R, Gao Y, et al. Apoptosis induction of fibroblast-like synoviocytes is an important molecular-mechanism for herbal medicine along with its active components in treating rheumatoid arthritis. Biomolecules (2019) 9:795. doi: 10.3390/biom9120795
166. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol (2021) 18:1106–21. doi: 10.1038/s41423-020-00630-3
167. Khoury MK, Gupta K, Franco SR, Liu B. Necroptosis in the pathophysiology of disease. Am J Pathol (2020) 190:272–85. doi: 10.1016/j.ajpath.2019.10.012
168. Ketelut-Carneiro N, Fitzgerald KA. Apoptosis, pyroptosis, and necroptosis-oh my! the many ways a cell can die. J Mol Biol (2022) 434:167378. doi: 10.1016/j.jmb.2021.167378
169. Zhan C, Huang M, Yang X, Hou J. MLKL: functions beyond serving as the executioner of necroptosis. Theranostics (2021) 11:4759–69. doi: 10.7150/thno.54072
170. Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci (2019) 20:19–33. doi: 10.1038/s41583-018-0093-1
171. Wang Y, Zhao M, He S, Luo Y, Zhao Y, Cheng J, et al. Necroptosis regulates tumor repopulation after radiotherapy via RIP1/RIP3/MLKL/JNK/IL8 pathway. J Exp Clin Cancer Res (2019) 38:461. doi: 10.1186/s13046-019-1423-5
172. Wang X, Gessier F, Perozzo R, Stojkov D, Hosseini A, Amirshahrokhi K, et al. RIPK3-MLKL-Mediated neutrophil death requires concurrent activation of fibroblast activation protein-α. J Immunol (2020) 205:1653–63. doi: 10.4049/jimmunol.2000113
173. Chen Y, Zhu C-J, Zhu F, Dai B-B, Song S-J, Wang Z-Q, et al. Necrostatin-1 ameliorates adjuvant arthritis rat articular chondrocyte injury via inhibiting ASIC1a-mediated necroptosis. Biochem Biophys Res Commun (2018) 504:843–50. doi: 10.1016/j.bbrc.2018.09.031
174. Gu X, Liu Y, Dai X, Yang Y-G, Zhang X. Deciphering the potential roles of ferroptosis in regulating tumor immunity and tumor immunotherapy. Front Immunol (2023) 14:1137107. doi: 10.3389/fimmu.2023.1137107
175. Hu J, Zhang R, Chang Q, Ji M, Zhang H, Geng R, et al. p53: a regulator of ferroptosis induced by galectin-1 derived peptide 3 in MH7A cells. Front Genet (2022) 13:920273. doi: 10.3389/fgene.2022.920273
176. Wong CK, Chen DP, Tam LS, Li EK, Yin YB, Lam CWK. Effects of inflammatory cytokine IL-27 on the activation of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res Ther (2010) 12:R129. doi: 10.1186/ar3067
177. Badot V, Durez P, Van den Eynde BJ, Nzeusseu-Toukap A, Houssiau FA, Lauwerys BR. Rheumatoid arthritis synovial fibroblasts produce a soluble form of the interleukin-7 receptor in response to pro-inflammatory cytokines. J Cell Mol Med (2011) 15:2335–42. doi: 10.1111/j.1582-4934.2010.01228.x
178. Smallwood MJ, Nissim A, Knight AR, Whiteman M, Haigh R, Winyard PG. Oxidative stress in autoimmune rheumatic diseases. Free Radic Biol Med (2018) 125:3–14. doi: 10.1016/j.freeradbiomed.2018.05.086
179. Datta S, Kundu S, Ghosh P, De S, Ghosh A, Chatterjee M. Correlation of oxidant status with oxidative tissue damage in patients with rheumatoid arthritis. Clin Rheumatol (2014) 33:1557–64. doi: 10.1007/s10067-014-2597-z
180. Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med (2019) 133:144–52. doi: 10.1016/j.freeradbiomed.2018.09.014
181. Forcina GC, Dixon SJ. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics (2019) 19:e1800311. doi: 10.1002/pmic.201800311
182. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol (2010) 221:3–12. doi: 10.1002/path.2697
183. Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal (2014) 20:460–73. doi: 10.1089/ars.2013.5371
184. Zhu L, Wang H, Wu Y, He Z, Qin Y, Shen Q. The autophagy level is increased in the synovial tissues of patients with active rheumatoid arthritis and is correlated with disease severity. Mediators Inflammation (2017) 2017:7623145. doi: 10.1155/2017/7623145
185. Karami J, Masoumi M, Khorramdelazad H, Bashiri H, Darvishi P, Sereshki HA, et al. Role of autophagy in the pathogenesis of rheumatoid arthritis: latest evidence and therapeutic approaches. Life Sci (2020) 254:117734. doi: 10.1016/j.lfs.2020.117734
186. Ding J-T, Hong F-F, Yang S-L. Roles of autophagy in rheumatoid arthritis. Clin Exp Rheumatol (2022) 40:2179–87. doi: 10.55563/clinexprheumatol/exg1ic
187. Misra S, Bagchi A, Sarkar A, Niyogi S, Bhattacharjee D, Chatterjee S, et al. Methotrexate and theaflavin-3, 3’-digallate synergistically restore the balance between apoptosis and autophagy in synovial fibroblast of RA: an ex vivo approach with cultured human RA FLS. Inflammopharmacology (2021) 29:1427–42. doi: 10.1007/s10787-021-00857-0
188. Tian Q, Qin B, Gu Y, Zhou L, Chen S, Zhang S, et al. ROS-mediated necroptosis is involved in iron overload-induced osteoblastic cell death. Oxid Med Cell Longev (2020) 2020:1295382. doi: 10.1155/2020/1295382
189. Jung SM, Kim KW, Yang C-W, Park S-H, Ju JH. Cytokine-mediated bone destruction in rheumatoid arthritis. J Immunol Res (2014) 2014:263625. doi: 10.1155/2014/263625
190. Gracie JA, Forsey RJ, Chan WL, Gilmour A, Leung BP, Greer MR, et al. A proinflammatory role for IL-18 in rheumatoid arthritis. J Clin Invest (1999) 104:1393–401. doi: 10.1172/JCI7317
191. Ruth JH, Park CC, Amin MA, Lesch C, Marotte H, Shahrara S, et al. Interleukin-18 as an in vivo mediator of monocyte recruitment in rodent models of rheumatoid arthritis. Arthritis Res Ther (2010) 12:R118. doi: 10.1186/ar3055
192. Min HK, Kim S, Lee J-Y, Kim K-W, Lee S-H, Kim H-R. IL-18 binding protein suppresses IL-17-induced osteoclastogenesis and rectifies type 17 helper T cell / regulatory T cell imbalance in rheumatoid arthritis. J Transl Med (2021) 19:392. doi: 10.1186/s12967-021-03071-2
193. Volin MV, Koch AE. Interleukin-18: a mediator of inflammation and angiogenesis in rheumatoid arthritis. J Interferon Cytokine Res (2011) 31:745–51. doi: 10.1089/jir.2011.0050
194. Kim HW, Kwon Y-J, Park BW, Song JJ, Park Y-B, Park MC. Differential expressions of NOD-like receptors and their associations with inflammatory responses in rheumatoid arthritis. Clin Exp Rheumatol (2017) 35:630–7.
Keywords: pyroptosis, rheumatoid arthritis treatment, inflammasome, gasdermin protein, caspase
Citation: Wu D, Li Y and Xu R (2023) Can pyroptosis be a new target in rheumatoid arthritis treatment?. Front. Immunol. 14:1155606. doi: 10.3389/fimmu.2023.1155606
Received: 31 January 2023; Accepted: 07 June 2023;
Published: 22 June 2023.
Edited by:
Gerald Nabozny, Boehringer Ingelheim, United StatesReviewed by:
Sarang Tartey, IGM Biosciences, United StatesCopyright © 2023 Wu, Li and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ranxing Xu, eHJ4MDEyMUAxNjMuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.