 Yuyan Yang
Yuyan Yang Wenling Zhao
Wenling Zhao Nan Yang
Nan Yang Shengnan Cui1
Shengnan Cui1 Hongzhong Jin
Hongzhong Jin Li Li
Li Li- 1Department of Dermatology, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Science and Peking Union Medical College, National Clinical Research Center for Dermatologic and Immunologic Diseases, Beijing, China
- 2Department of Dermatology, Shunyi Maternal and Children’s Hospital of Beijing Children’s Hospital, Beijing, China
- 3Department of Pharmacology, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences & School of Basic Medicine, Peking Union Medical College, Beijing, China
Bullous pemphigoid is an autoimmune blistering disorder that primarily occurs in elderly patients. Reports indicate that BP coexists with various hematological diseases, including acquired hemophilia A, hypereosinophilic syndrome, aplastic anemia, autoimmune thrombocytopenia, and hematological malignancies. Early identification of these comorbidities contributes to a better control and reduced mortality. This article details the atypical clinical manifestations of BP when associated with hematological diseases, specific diagnostic strategies, underlying mechanistic connections, and possible treatments. Cross-reactivity between autoantibodies and exposed abnormal epitopes, shared cytokines and immune cells, together with genetic susceptibility are the most common connections between BP and hematological diseases. Patients were most often successfully treated with oral steroids combined with medications specifically targeting the hematological disorders. However, the individual comorbidities require specific considerations.
1 Introduction
Bullous pemphigoid (BP) is an autoimmune blistering disease which is characterized by the presence of cutaneous bullae and autoantibody deposition at the epithelial basement membrane zone (1). BP affects predominantly elderly people while its mortality rate increases with each increasing decade of life (2). In European and Asian populations, the incidence of BP is 10.3 and 5.2 per million, respectively (3). Because of the development of improved diagnostic assays as well as aging populations, the annual incidence of BP is increasing (4). Classical manifestations of BP include tense blisters on the extremities and trunk with less mucosal involvement than the similar autoimmune blistering disease, pemphigus vulgaris (5, 6).
Autoantibodies against the auto-antigens BP180 (180kDa) and BP230 (230kDa) have been identified in BP patients. Epitope mapping indicated a relationship between autoantibody specificity and disease phenotype (7). The non-collagen extracellular domain of BP180, termed NC16A, is the primary binding epitope by anti-BP180 antibodies, correlated with typical severe erythematous inflammation and blister formation (8, 9). Noninflammatory bullous pemphigoid manifested as reduced erythema and sparse periblister eosinophilic infiltration is thought to be caused by autoantibodies targeted the midportion of BP180, probably associated with dipeptidyl peptidase- IV inhibitors (9). Furthermore, autoantibodies against multiple extracellular domains of BP180 may increase the morbidity of mucosal lesions (10). The typical binding site for BP230 is the globular C-terminal domain (11). BP severity and activity are reported to correlate directly with levels of anti-BP180 autoantibodies, but no consistent correlations with anti-BP230 autoantibody titers have yet been identified (1). However, because the phenomenon of epitope spreading occurs at an early stage of disease when BP230 is exposed, anti-BP230 autoantibodies also contribute to the development of skin lesions (11). IgG4 is the predominant subclass of BP autoantibodies, followed by IgG1 and IgG2. IgA and IgE autoantibodies directed at the basement membrane zone may also be present in BP (12).
BP has been reported in patients with concomitant hematological disorders, especially in older populations. Dysregulation of coagulation system results in abnormal bleeding issues, acquired hemophilia A and autoimmune thrombocytopenia (13, 14). Combination of these coagulation disorders with BP were reported to occur in elder populations, presenting as atypical hemorrhagic skin lesions. Changes in hematocytes and leukocytes usually destabilize the circulation and immune system, thus BP patients diagnosed with hypereosinophilic syndrome and aplastic anemia requires more concerns (15, 16). Hematological malignancies such as myelodysplastic syndrome and chronic lymphocytic leukemia are reported to be significantly associated with BP compared with other neoplasms (17–19). Acquired hemophilia A, autoimmune thrombocytopenia, hypereosinophilic syndrome and aplastic anemia are characterized by obscure onset and robust diagnostic assays. Early identification of these comorbidities contributes to a better control. On the other hand, detection of myelodysplastic syndrome and lymphoproliferative disorders in BP patients may conduce to better prognosis and mortality reduction.
The etiologies of BP and hematological disorders were proposed to be related rather than independent. Cross-immunoreactivity, specific cytokines and genetic predisposition were proposed to play important roles (5, 13). Most patients’ skin conditions were influenced by the additional hematological diseases. Hematological comorbidities and clinical features such as hematoma and eosinophilic dermatitis-like lesions make diagnosis of BP challenging without further examination. Previous studies of patients with BP and comorbidities such as neurologic diseases, neoplasms, cardiovascular diseases found that if these patients did not receive timely treatment, their mortality rates exceeded those of patients diagnosed with BP alone (18, 20). Thus, we assume that cases of BP associated with hematological diseases require more attention.
In this review, we discuss the atypical clinical manifestations associated with comorbidities, to aid diagnosis of BP in patients with hematological diseases. Furthermore, we describe the mechanisms present in BP cases complicated with hematological diseases. Finally, we present possible treatment strategies to improve the prognosis of these cases (concluded in Table 1). Details of all cases mentioned in this review were recorded in Table 2, especially the complex manifestations and therapeutic effects.
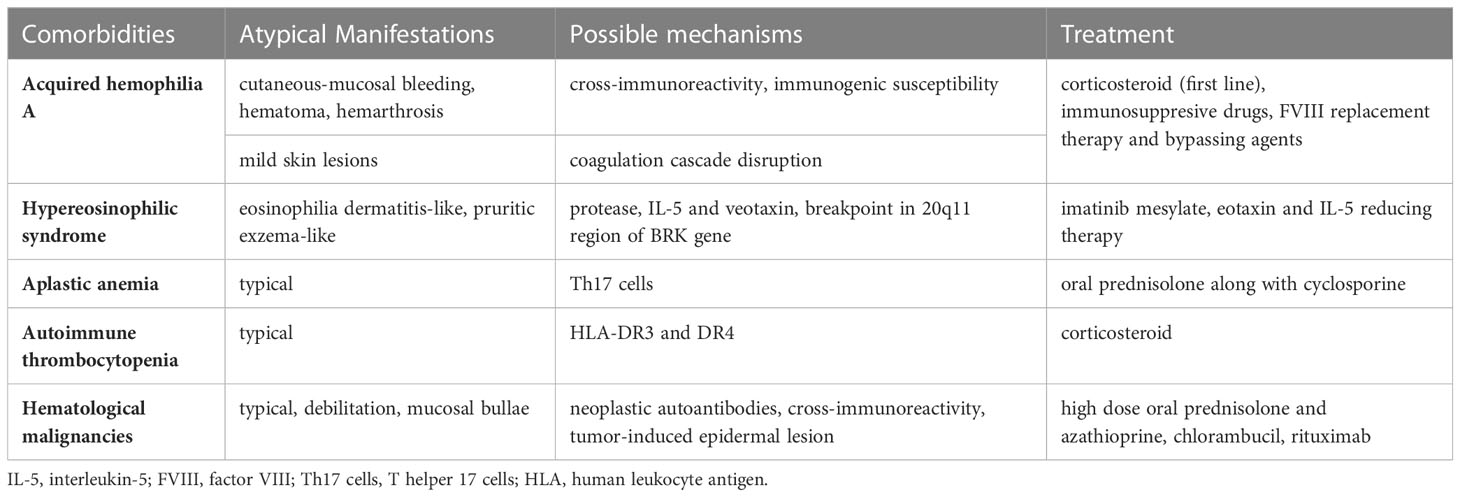
Table 1 Characteristics of patients diagnosed with bullous pemphigoids and hematological comorbidities.
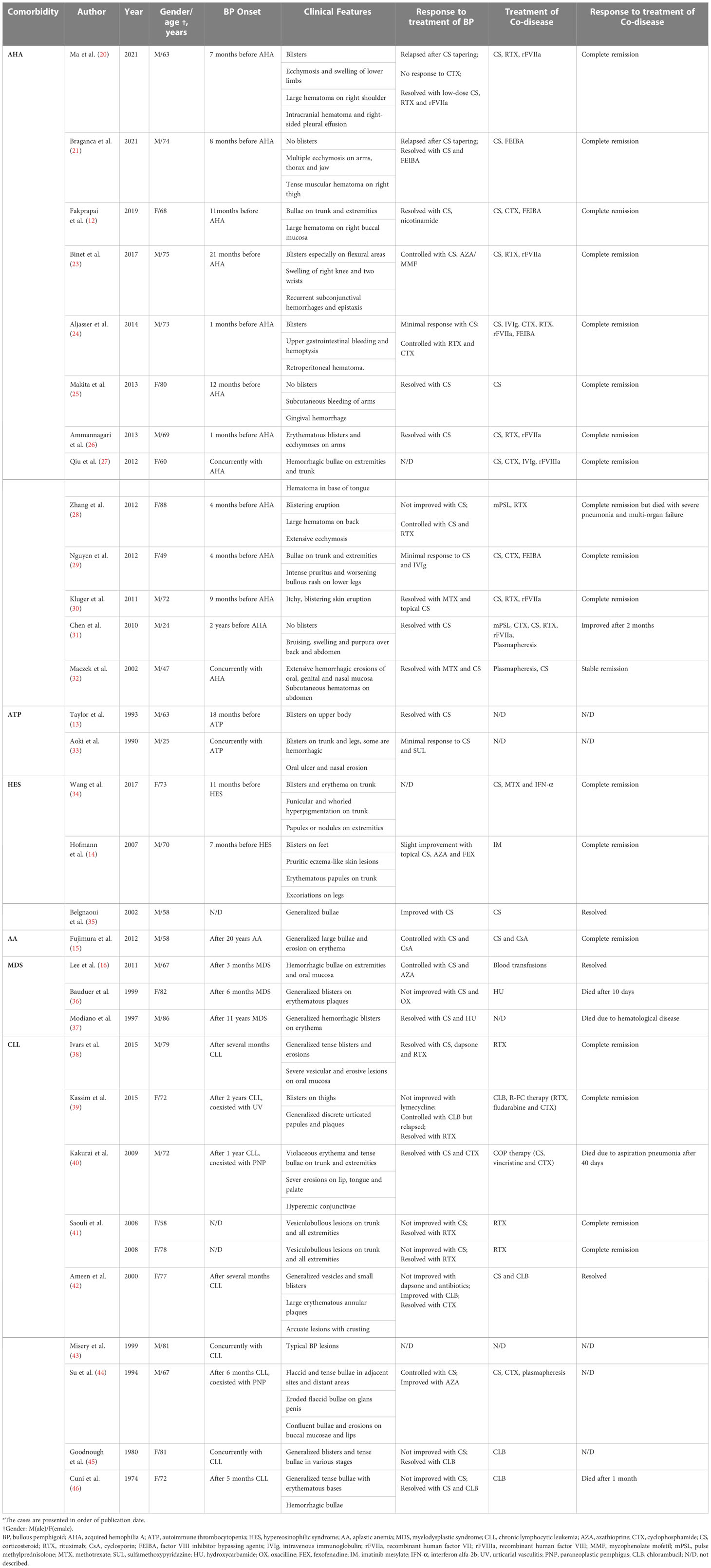
Table 2 Reported cases of bullous pemphigoid associated with various hematological diseases.
2 Acquired hemophilia A
Acquired hemophilia A (AHA) is a rare autoimmune disease with a high mortality rate ranges from 17-22% that results from the development of circulating autoantibodies against the endogenous coagulation factor VIII (47). AHA is characterized by spontaneous hemorrhage and excessive bleeding in the absence of a family history or childhood experience of disordered bleeding. Although activated partial thromboplastin time (APTT) is prolonged, thrombin times, prothrombin times, platelet count and function are usually normal (48).
Approximately 50% of AHA patients are concomitantly diagnosed with other diseases, especially autoimmune diseases including systemic lupus erythematosus, rheumatoid arthritis and BP (23). The combination of BP and AHA is relatively rare, with no more than 30 documented cases reported globally, and most of these patients had abnormally bleeding skin lesions (23). AHA often occurs in BP patients aged 24 to 88 years and is present in 68% patients aged over 65 years at diagnosis, with no sex predisposition (23). BP is typically diagnosed prior to AHA onset or they occur simultaneously, with the mean time between the diagnosis of the two diseases being 6 months (23). In none of the current reports does AHA occur prior to BP diagnosis.
Severe cutaneous-mucosal bleeding is seldom reported in BP patients (6). However, these symptoms usually indicate that there might be complicating AHA. Prior to the onset of AHA, hemorrhagic blisters rarely appear and APTT remains normal (49). Only after the development of AHA will extensive hemorrhagic bullae and large hematomas involving mucous membranes begin to appear (13). Hemarthrosis, which is not characteristically seen in AHA patients, has also been reported in a comorbidity case with BP (23). However, a minority of patients were reported to have unexpected mild skin lesions despite their high BP180 autoantibody titers (23). Because of this intersection of manifestation and immunological index, diagnosis of these patients could easily be missed and requires more clinical attention.
Apart from the clinical manifestations, diagnosis of the combination of AHA and BP further depends on laboratory tests. High anti-BP180 titers, prolonged APTT and increased FVIII inhibitor levels occurred simultaneously in an elderly patient as evidence of the comorbidity diagnosis (23). For most patients, laboratory tests indicate a parallel course of these two diseases. During remission of the two concurrent diseases, the eradication of FVIII inhibitors increased FVIII levels and shortened APTT, while anti-BP180 and BP230 IgG titers also decreased simultaneously (23).
Intrinsic risk factors for both AHA and BP might exist as there are abnormal clinical manifestations and parallel remission. One potential mechanism for the onset of comorbid disease is cross-reactivity between autoantibodies against the BP180 NC16A domain and factor VIII A2. This theory is strongly evidenced by sequence homology between the two binding domains (23). Autoantibodies found in patients with either disease predominantly belong to the IgG4 subtype, while a minority are of the IgG1 subtype (50). Though the case reported by Makita denied cross-activity between factor VIII and BP180, this negative result might be interfered by immunosuppressive therapies (25). However, no regions of high similarity have been identified between the NC16A domain of BP180 and factor VIII A2 (49). Furthermore, atypical clinical manifestations were also suggested to be associated with autoantibody cross-reactivity. Serum IgG extracted from a 47-year-old patient with AHA and rather mild BP targeted the mid-portion of extracellular domain of the BP180 antigen, which was reported to be related with reduced erythematous skin lesions and increased mucosal involvements (32).
A 78-year-old patient with BP and AHA was reported in 2006 and his medical history also included rheumatoid arthritis and vitiligo (49). His comorbidity was noted following persistent bleeding from buccal hemorrhagic swelling. As for those patients diagnosed with several simultaneous autoimmune diseases, studies inferred that there might be an underlying immunogenetic susceptibility to autoimmune diseases in them (49). BP patients diagnosed with other immunological abnormalities might have severe cutaneous-mucosal symptoms. However, no such genes have been discovered to date.
The reasons why mild cutaneous symptoms coexist with high autoantibody titers in a few patients with comorbidities remain unclear. According to previous researches, we inferred that this abnormality might be partially explained by a disruption in the coagulation cascade in patients with this comorbidity. Excessive coagulation activation has been demonstrated in BP and is associated with higher thrombotic risk (51). Infiltrating T cells, predominantly T helper type 2 (Th2) cells, in BP skin lesions can produce interleukin (IL)-5 and eotaxin to recruit and activate eosinophils (52). Eosinophils store and rapidly transfer tissue factors (TF) to the cell membrane (52). This signaling process leads to increased F1+2 and D-dimer levels, which initiate the coagulation cascade (51). This hypercoagulability usually contributes to tissue damage and blister formation in BP patients, thus forming a vicious cycle between cutaneous lesions and thrombosis.
Poor prognostic factors include elder age, other comorbidities, high anti-BP180 and anti-BP230 titers as well as high factor VIII inhibitor titers (53). It was reported that a patient with BP-induced AHA developed intracranial hematoma and hemothorax during the acute phase (21). Infections in continuously bleeding mucosal lesions should also be prevented. Thus, treatment of coexisting BP and AHA is primarily focused on autoantibody eradication and bleeding remission (13, 27). Immunosuppressive drugs have shown some benefit for both AHA and BP. The first-line therapy for this comorbidity is oral corticosteroids alone or with cyclophosphamide (13). Tapering of corticosteroids should be elaborated to avoid severe relapse (22). Rituximab was used in cases reported in 2012 and 2021 with successful disease management and is often used as a second-line therapy (13, 20, 21, 28). However, as another newly developed biologic, omalizumab was suspected a causality in AHA onset (54). Other treatments include cyclophosphamide, high-dose immunoglobulins and immunoabsorption (13, 23, 55). Tapering of immunosuppressive agents is more prudent because immune reconstitution inflammatory syndrome presenting as AHA has been reported in BP patients (56). Immune recovery, which occurs rapidly after inadequate withdrawal of the drug, may trigger subsequent AHA recurrence. Because of the persistent presence of circulating factor VIII autoantibodies, factor VIII remains insufficient. Thus, triggering the extrinsic coagulation pathway might be a targeted substitution for the disabled intrinsic pathway. Factor VIIa is a key component of the prothrombin activation complex in the bypass coagulation pathway. Consequently, recombinant activated factor VII and activated prothrombin complex concentrate were often prescribed. However, this dose should not exceed 200 units/kg daily as there is an increased risk of venous thromboembolism (13). When there was no elevated FVIII inhibitor titer, human FVIII replacement therapy may also be effective (13). Bypass therapies, including recombinant activated factor VIIa, activated prothrombin complex concentrate, and neutralization therapy mainly composed of factor VIII concentrates aim to control bleeding and maintaining effective hemostasis (25).
Non-specific adverse effects (e.g., infection, sepsis, neutropenia) are likely to occur in BP patients with comorbid AHA, especially those with severe skin lesions. Cutaneous relapses were also often seen after drug discontinuation (57, 58). Thus, an appropriate tapering dose should be administered for the treatment of this comorbidity.
Collectively, when BP patients developed extensive hemorrhagic bullae, large mucosal hematoma or even hemarthrosis, a combination with AHA should be investigated. BP180 titers only correlates partially with disease severity, with the linear correlation coefficient reported as 0.6 (59). However, if a patient with mild skin lesions and abnormally high autoantibody titers concomitantly complained about predisposed to easy bleeding, potential presence of comorbid AHA should be noticed. Clinicians are strongly advised to examine APTT and factor VIII inhibitors to diagnose the comorbidity and further adjust the current treatments.
3 Autoimmune thrombocytopenia
Autoimmune thrombocytopenia (ATP) is an autoimmune disease characterized by an autoantibody-mediated decrease in platelets (60).
Only a few patients have been reported to develop ATP after BP (14, 33). The interval course between BP and ATP ranged from 18 months to 3 years. Additionally, cutaneous manifestations were typical, presenting as pruritic bullous erythema on trunk and extremities.
Therapeutic agents, such as prednisolone and sulfamethoxypyridazine, have been associated with thrombocytopenia. Drug molecules are supposed to induce thrombocytopenia via direct cytotoxic effects on megakaryocytes and platelets, which leads to dysfunctional thrombopoiesis and increased platelet destruction (61). However, discontinuation of BP treatment did not resolve the thrombocytopenia (14). Thus, researchers have inferred that medications curing BP might be just triggers rather than the direct cause of the development of antiplatelet autoantibodies. Genetic predisposition may additionally play an important role as HLA-DR3 and DR4 haplotypes were found in a patient with both BP and autoimmune thrombocytopenia (14). Systematic corticosteroid therapy was suggested for skin lesions treatment because of its satisfactory therapeutic effect (14).
To summarize, it is important to monitor the development of thrombocytopenia after BP remission.
4 Hypereosinophilic syndrome
Hypereosinophilic syndrome (HES) is a myeloproliferative disorder characterized by idiopathic eosinophilia of at least 1500 cells/μl for more than 6 months with cutaneous or systemic involvement. Intense eosinophilic infiltration at the dermal–epidermal junction can be seen in skin biopsies (62). As a clonal hematopoietic disorder, two chromosomal abnormalities involving tyrosine kinases have been identified. Most HES patients carry an interstitial deletion on chromosome 4q12, resulting in the formation of a fusion gene termed FIPL1-PDGFRA. This gain-of-function mutation leads to the expression of constitutively active tyrosine kinase (63, 64). Deletion of the 20q11 region disrupts the nonreceptor tyrosine kinase gene BRK, which also causes an abnormal activation of tyrosine kinases (15). Both mutations lead to excessive peripheral and tissue eosinophils.
Comorbidities between BP and HES are extremely rare, with only six cases reported (15, 34, 35, 63). Because HES is usually diagnosed during routine follow-up rather than at the time of onset, determining the onset order of these two diseases is challenging. However, levels of eosinophils appeared to increase in more than 50% of BP patients, which could mask the coexisting HES if they exceeded 1500 cells/μl (65). Consequently, this comorbidity is particularly difficult to diagnose.
The clinical manifestations of patients with comorbid BP and HES were reportedly atypical. Skin lesions in patients with both BP and HES presented as eosinophilia dermatitis-like or pruritic eczema-like patterns instead of bullae (15, 34). These atypical manifestations often make the diagnosis of BP challenging. Direct immunofluorescence results have indicated that IgG or C3 deposition at the basement membrane zone and detection of BP180 antibodies could help avoid misdiagnosis. Furthermore, in patients with typical BP cutaneous symptoms, anomalously high levels of eosinophils may also be identified.
Eosinophils play an important role in BP pathogenesis (Figure 1). They predominantly infiltrate the dermal region and degranulate, releasing proteases, which induce the skin lesions. One of the most important proteases is matrix metalloproteinase 9 (MMP9), also known as gelatinase, which is capable of degrading extracellular matrix proteins and BP180 (1, 66). Hence, collagen fibers are degenerated and hemidesmosomes are disrupted. Eosinophil cationic protein (ECP) and eosinophil-derived neurotoxin (EDN) are ribonucleases produced by eosinophils, which trigger the apoptosis of keratinocytes around BP lesions. ECP also induces basal keratinocyte detachment (67). Through the activity of the ribonucleases, eosinophils contribute to dermal–epidermal separation and ultimately, blister formation (1). BP patients with comorbid HES have higher levels of blood eosinophilia, and thus may present with more severe skin lesions. However, although the six reported cases had more severe and extensive pruritus, the shape of bullae appeared to be intact without erosions or exudation, potentially because of limited eosinophil infiltration in the dermis and epidermis.
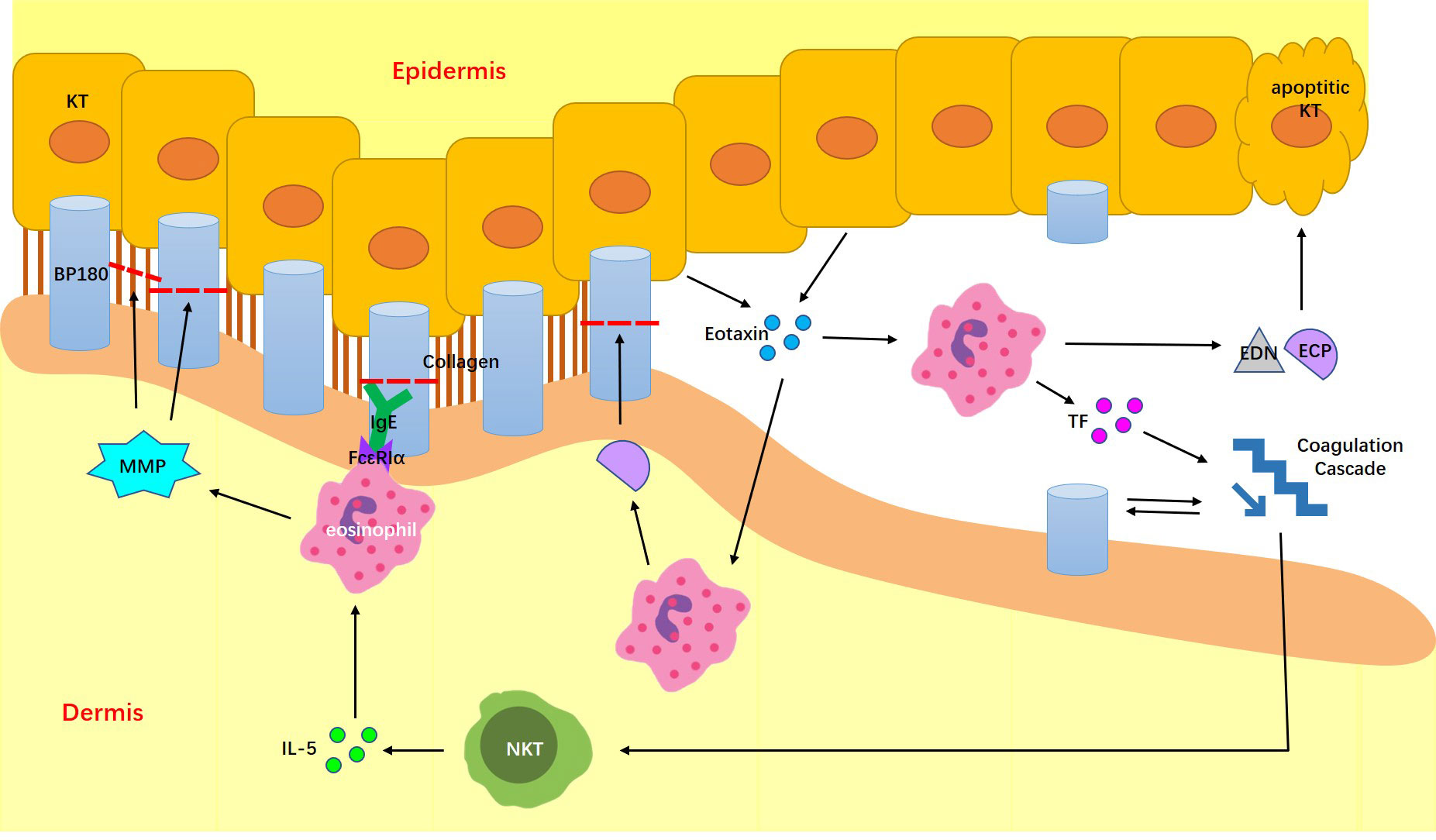
Figure 1 Possible mechanisms of BP combined with HES. IL-5, interleukin-5; MMP9, matrix metalloproteinase 9; EXM, extracellular matrix; ECP, Eosinophil cationic protein; EDN, eosinophil-derived neurotoxin; TF, transfer tissue factor.
BP triggers eosinophil accumulation mainly through the actions of cytokines, chemokines and autoantibodies. Natural killer T cells located in lesions and eosinophils gathering around BP blisters express excessive IL-5, which further contributes to eosinophil development, release, and degranulation. This mechanism is evidenced by a correlation between IL-5 expression, eosinophil activity and BP blister formation (68). Conversely, eotaxins, the major chemotaxin for eosinophils, were reportedly expressed in the epidermal keratinocytes around BP blisters. This abnormally increased release of eotaxins was possibly triggered by ECP and EDN (67). Eotaxins could recruit further eosinophil infiltration to the blisters. Furthermore, because eosinophils in BP patients were shown to express the high-affinity IgE receptor FcϵRIα, they may bind and be triggered by BP autoantibodies of the IgE subtype (67). The erythematous urticarial phenotype of BP is associated with increased IgE antibody levels (69).
Although elevated eosinophils play an important role in both BP and HES, these cases were shown to be comorbidities rather than complications (15). First, typical BP patients usually have lower levels of eosinophils than HES patients. Furthermore, no specific first-line medications used to treat BP are known to induce reactive eosinophilia. Moreover, the cause of HES is clonal proliferation of eosinophils.
This comorbidity was reportedly treated by imatinib mesylate, a novel tyrosine kinase inhibitor (15, 63). Imatinib mesylate is not a common drug for BP treatment; however, it still led to durable remission of both skin lesions and hypereosinophilia (15). Therapies aim to reduce eotaxin and IL-5 production to within normal ranges may also resolve blister formation and normalize eosinophil counts in BP patients diagnosed with HES. Simultaneous remission of these two diseases further supports the potential relationship between BP and HES.
In summary, in a BP patient presenting with eosinophilia dermatitis-like or pruritic eczema-like skin lesions who also has abnormally high eosinophilia, HES should be considered. In case that symptomatic therapy is unsatisfactory, we strongly recommend testing anti-BP180 titers to help rule out BP and HES comorbidity in HES patients with recalcitrant blood eosinophilia or refractory lesions.
5 Aplastic anemia
Aplastic anemia is an immune-mediated disease characterized by peripheral blood pancytopenia and bone marrow hypoplasia (70). Hematopoietic cells are actively destroyed by effector T cells, which results in a prominent decrease in the numbers of white blood cells, red blood cells and platelets. High levels of platelet-associated IgG have also been recognized (16).
Only one case has been reported with both BP and aplastic anemia (16). The aged patient had a 20-year history of uncured aplastic anemia and developed pruritic bullous erythema over his trunk and extremities. Although his clinical manifestations were atypical, the diagnosis was achieved based on high levels of serum BP180 autoantibodies and platelet-associated IgG (16).
IL-17-positive cells have been found infiltrating the BP blisters, which suggests an underlying association with T helper 17 (Th17) cells (16). Th17 cells are critical to the pathogenesis of autoimmune diseases (71). In both BP and aplastic anemia patients, Th17 cells are increased along with a reduction in regulatory T (Treg) cells (72, 73). Th17 cells act destructively in BP by mediating inflammation, while in aplastic anemia, they contribute to the recruitment of Th1 cells and regulation of proinflammatory cytokines in the bone marrow during the early disease stages (72, 73).
The patient with both BP and aplastic anemia was successfully treated with oral prednisolone and cyclosporine. The skin lesions disappeared and the numbers of blood cells returned to normal levels (16).
In summary, when patients with aplastic anemia develop pruritic erythema or bullous lesions, BP should be suspected. Even if anti-BP180 titers turn out to be negative, autoantibodies recognizing other BP related epitopes were recommended to be tested to avoid misdiagnosis.
6 Hematological malignancy
Myelodysplastic syndrome (MDS) is a hematological malignancy characterized by a heterogeneous group of malignant hematopoietic stem cell disorders (74).
Only three cases with both BP and MDS were reported (17, 36, 37). Clinical manifestations of patients combined with MDS were more severe than typical BP patients (17). Atypical skin lesions and examination results include multiple bullae on the oral mucosa and autoantibodies against desmoplakin (17, 37). The unusual autoantibodies may arise through recognition of common epitopes between desmoplakin and BP230 as their structures are similar. However, whether they are caused by MDS remains unclear (75).
While both diseases are common among older adults, the relationship between BP and MDS remains unknown. It has been suggested that a cross reaction between tumor-specific antigens and the basement membrane zone might lead to bullae development (76). Modiano reported a case in which tumor CD13+ and CD15+ cells infiltrated the dermal region and led to skin detachment (37). Subsequently, abnormal BP antigens were exposed, which induced further abnormal anti-BP180 expression. Linear IgG and C3 deposition along the basement membrane zone were found surrounding the area (37). Evidence also exists for the cross-reaction theory as BP erupted during the transformation of the previously refractory anemia to subacute myelomonocytic leukemia (36). Furthermore, genetic predisposition and external inducers such as radiation and chemicals are also implicated (77).
Lymphoproliferative disorders are a group of hematological tumors caused by proliferation of clonal malignant lymphoid stem cells (78). Among them, B-cell chronic lymphatic leukemia (CLL) is a the most common form with an annual incidence rate of 5.1/100,000 (79). According to previous studies, the incidence of autoimmune diseases in lymphoproliferative disorders is approximately 8% (42).
A nationwide record-linked study conducted in England (1999-2011) demonstrated elevated risk of BP in patients with lymphoid leukemia compared with a reference cohort (80). Lymphoproliferative disorder-related BP has been reported in 10 cases. All patients developed CLL before or concurrently with BP, with an interval course up to 2 years. Coexistence of paraneoplastic pemphigus was diagnosed in two men (40, 44). Two women also suffered from inflammatory arthritis (39, 45).
Cutaneous lesions in most patients diagnosed with BP and CLL were generalized pruritic blistering skin lesions, similar to those found in classic BP patients. Only a 79-year-old man developed atypical severe oral erosion (38). Discrete polymorphic skin lesions were reported in a 77-year-old man, mainly manifested as large erythematous annular plaques and arcuate lesions (42).
Both characterized by pruritus vesiculobullous lesions and dermal-epidermal detachment, BP-like pattern of Eosinophilic dermatosis of hematologic malignancies (EDHM) closely resembles malignancy-related BP (81, 82). Thus, differential diagnosis is important for precise treatment selection. In EDHM patients, both immunofluorescence and serum tests turned out to be autoantibody-negative against BP180 (83). Immunological examinations can help clinicians distinguish malignancy-related BP from EDHM before medication.
Paraneoplastic pemphigus (PNP) can also present as BP-like pattern while develop autoantibodies targeting BP180 or BP230 in patients with hematologic malignancies. Diagnosis of BP should not be made unless PNP has been excluded based on its diagnostic criteria (84). Recombinant protein containing envoplakin and periplakin are reported to be a sensitive and specific antigen for PNP diagnosis (85).
Lymphoproliferative disorders induce important alternations of the immune system by autoantibodies produced by neoplastic cells (79). It has been confirmed that neoplastic B cells are able to recognize self-antigens (86). Augmentation and hyperactivation of Th2 as well as IL-17-producing cells in B-CLL patients can further promote autoantibody production (79). P105 antigen, the targeted site of non-classic dermal-binding BP, exhibits 70% homology to tumor-associated antigen (39). However, whether the IgG autoantibodies against NC16A domain of BP180 or BP230 were produced by neoplastic cells has not been practically confirmed (40, 44). According to a research conducted by Misery, extracellular immortalized leukemic cells extracted from a patient diagnosed with BP and CLL failed to synthesize anti-230 autoantibody (43). On the other hand, pathogenic anti-BP180 antibodies might also be produced by donor-derived B lymphocytes in patients underwent hematopoietic stem cell transplantation, which need careful surveillance during treatment (87).
Other potential pathomechanisms include auto-reactive T-cell clones, which can be simulated by abnormal antigens presented by neoplastic cells from B-CLL patients (88). Besides, by producing IL-4 and IL-17, dysfunctional T-regulatory cells activated in CLL patients were reported to promote both tumor tolerance and peripheral inflammation (89). Chemotherapeutic agents, such as fludarabine, are also suggested to be associated with abnormal regulation of T-cells (88).
However, there is no direct validation of immunopathological relationship between BP and hematological malignancies, which still need further investigations.
Paraneoplastic dermatoses refer to cutaneous diseases secondary to malignancies (90). Patients with malignancy-associated BP usually have a former oncological medical history prior to BP onset. Furthermore, they usually develop BP at an early age (90). Atypical BP lesion, such as figurate erythema, might be a marker for hematological malignancy (76).
BP in MDS patients was reportedly difficult to control, probably related with immunological disorders synergized with other supposed pathomechanisms, such as hematological treatments, genetic and epigenetic factors. The skin lesions only responded to a combination of high dose oral prednisolone and azathioprine in one patient (17). The remaining two patients died before blister remission was achieved (36, 37).
Firstline treatment for malignancy-associated dermatoses usually referred to systemic steroids and immunosuppressants (79). However, steroids often failed to control BP skin lesions in LL patients (42, 45, 91). Early commencement of chemotherapy for CLL plays an important role in sustained resolution of cutaneous manifestations (39). Chlorambucil is also reported to improve skin lesions while help taper corticosteroids dosage rapidly (42, 45, 46). It is noteworthy that rituximab is specifically effective in both BP and CLL patients, thus result in a better prognosis for CLL-related BP patients (38, 41).
In summary, when BP presents with cachexia or inexplicable refractory course of disease, they should be screened for hematological malignancies. If the underlying tumor is left untreated, BP therapies alone cannot improve prognosis. In rare cases, dermatoses might reflect a potential unfavorable prognosis of the associated hematological malignancy (79).
7 Conclusion
The coexistence of bullous pemphigoid and hematological disorders is relatively rare compared with other comorbidities. According to the underlying pathological relationships occurring in both BP and hematological diseases, they may contribute to the symptoms seen in these patients. However, further research is required to illuminate the underlying mechanisms. Atypical clinical manifestations are often seen in patients diagnosed with two combined diseases. Skin lesions that are seldom present in typical BP patients, such as hematomas instead of characteristic bullae, make these comorbidities difficult to diagnose. Overlapping symptoms might result in mutual symptom concealment, where misdiagnosis is probable. Moreover, according to most reported cases, patients with comorbidities were curable following a timely diagnosis and treatment regime. Consequently, these rare comorbidities still require attention. More evidence will enable the prevention, diagnosis, specific treatment and health care of BP combined with hematological diseases.
Author contributions
LL, HJ, and YY contributed to conception of this review. SC and NY collected relevant resources. YY, WZ and NY performed the statistical analysis. YY and WZ wrote the first draft of the manuscript. YY and LL drew the figure. YY and SC performed the tables. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China [81972945 to LL]; the National Key Research and Development Program of China Grant [2016YEC0901500 to LL]; the Funds of Shunyi District for Health Improvement and Research [Wsjkfzkyzx-2019-y-07 to WZ].
Acknowledgments
We thank S. J. Win, PhD, from Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac) for editing a draft of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Miyamoto D, Santi CG, Aoki V, Maruta CW. Bullous pemphigoid. Bras Dermatol (2019) 94(2):133–46. doi: 10.1590/abd1806-4841.20199007
2. Ahmed AR, Anwar S, Reche PA. Molecular basis for global incidence of pemphigoid diseases and differences in phenutypes. Front Immunol (2022) 13:807173. doi: 10.3389/fimmu.2022.807173
3. Persson MSM, Begum N, Grainge MJ, Harman KE, Grindlay D, Gran S. The global incidence of bullous pemphigoid: a systematic review and meta-analysis. Br J Dermatol (2021) 21(10):4818–35. doi: 10.1111/bjd.20743
4. Egami S, Yamagami J, Amagai M. Autoimmune bullous skin diseases, pemphigus and pemphigoid. J Allergy Clin Immunol (2020) 145(4):1031–47. doi: 10.1016/j.jaci.2020.02.013
5. Moro F, Fania L, Sinagra JLM, Salemme A, Zenzo GD. Bullous pemphigoid: Trigger and predisposing factors. Biomolecules. (2020) 10(10):1432–59. doi: 10.3390/biom10101432
6. Chen X, Zhao W, Jin H, Li L. Risk factors for mucosal involvement in bullous pemphigoid and the possible mechanism: A review. Front Med (2021) 8:680871. doi: 10.3389/fmed.2021.680871
7. Hammers CM, Stanley JR. Recent advances in understanding pemphigus and bullous pemphigoid. J Invest Dermatol (2020) 140(4):733–41. doi: 10.1016/j.jid.2019.11.005
8. Nishie W. Collagen XVII processing and blistering skin diseases. Acta Derm Venereol (2020) 100(5):adv00054. doi: 10.2340/00015555-3399
9. Izumi K, Nishie W, Mai Y, Wada M, Natsuga K, Ujiie H, et al. Autoantibody profile differentiates between inflammatory and noninflammatory bullous pemphigoid. J Invest Dermatol (2016) 136(11):2201–10. doi: 10.1016/j.jid.2016.06.622
10. Di Zenzo G, Grosso F, Terracina M, Mariotti F, De Pita O, Owaribe K, et al. Characterization of the anti-BP180 autoantibody reactivity profile and epitope mapping in bullous pemphigoid patients. J Invest Dermatol (2004) 122(1):103–10. doi: 10.1046/j.0022-202X.2003.22126.x
11. Shih YC, Wang B, Yuan H, Zheng J, Pan M. Role of BP230 autoantibodies in bullous pemphigoid. J Dermatol (2020) 47(4):317–26. doi: 10.1111/1346-8138.15251
12. Zhou T, Peng B, Geng S. Emerging biomarkers and therapeutic strategies for refractory bullous pemphigoid. Front Immunol (2021) 12:718073. doi: 10.3389/fimmu.2021.718073
13. Fakprapai W, Wattanakrai P. Bullous pemphigoid associated with acquired hemophilia a: A case report and review of the literature. Case Rep Dermatol (2019) 11(2):130–9. doi: 10.1159/000499525
14. Taylor G, Venning V, Wojnarowska F. Bullous pemphigoid and associated autoimmune thrombocytopenia: two case reports. J Am Acad Dermatol (1993) 29(5 Pt 2):900–2. doi: 10.1016/0190-9622(93)70266-v
15. Hofmann SC, Technau K, Muller AM, Lubbert M, Bruckner-Tuderman L. Bullous pemphigoid associated with hypereosinophilic syndrome: simultaneous response to imatinib. J Am Acad Dermatol (2007) 56(5 Suppl):S68–72. doi: 10.1016/j.jaad.2006.02.059
16. Fujimura T, Kakizaki A, Kambayashi Y, Furudate S, Aiba S. Bullous pemphigoid accompanied by aplastic anemia: The induction of IL-17-Producing cells in the affected areas of the skin. Case Rep Dermatol (2012) 4(3):211–4. doi: 10.1159/000343882
17. Lee YY, Bee PC, Lee CK, Naiker M, Ismail R. Bullous pemphigoid in an elderly patient with myelodysplastic syndrome and refractory anemia coupled with excess of blast. Ann Dermatol (2011) 23(Suppl 3):S390–2. doi: 10.5021/ad.2011.23.S3.S390
18. Schulze F, Neumann K, Recke A, Zillikens D, Linder R, Schmidt E. Malignancies in pemphigus and pemphigoid diseases. J Invest Dermatol (2015) 135(5):1445–7. doi: 10.1038/jid.2014.547
19. Atzmony L, Mimouni I, Reiter O, Leshem YA, Taha O, Gdalevich M, et al. Association of bullous pemphigoid with malignancy: A systematic review and meta-analysis. J Am Acad Dermatol (2017) 77(4):691–9. doi: 10.1016/j.jaad.2017.05.006
20. Cai SC, Allen JC, Lim YL, Chua SH, Tan SH, Tang MB. Mortality of bullous pemphigoid in Singapore: risk factors and causes of death in 359 patients seen at the national skin centre. Br J Dermatol (2014) 170(6):1319–26. doi: 10.1111/bjd.12806
21. Ma H, Chang H. Life-threatening bleeding in a patient with pemphigoid-induced acquired hemophilia a and successfully treated with rituximab and rFVIIa: A case report. Medicine (2021) 100(3):e24025. doi: 10.1097/MD.0000000000024025
22. Braganca M, Valente C, Ferreira AI, Freitas-Silva M. Acquired hemophilia a associated with bullous pemphigoid: A rare combination. Transfus Apher Sci (2021) 61(2):103337. doi: 10.1016/j.transci.2021.103337
23. Binet Q, Lambert C, Sacre L, Eeckhoudt S, Hermans C. Successful management of acquired hemophilia a associated with bullous pemphigoid: A case report and review of the literature. Case Rep Hematol (2017) 2017:2057019. doi: 10.1155/2017/2057019
24. Aljasser MI, Sladden C, Crawford RI, Au S. Bullous pemphigoid associated with acquired hemophilia a: a rare association of autoimmune disease. J Cutan Med Surg (2014) 18(2):123–6. doi: 10.2310/7750.2013.13060
25. Makita S, Aoki T, Watarai A, Aida A, Katayama T, Danbara M, et al. Acquired hemophilia associated with autoimmune bullous diseases: a report of two cases and a review of the literature. Intern Med (2013) 52(7):807–10. doi: 10.2169/internalmedicine.52.9317
26. Ammannagari N, Laveaux K, Grethlein S. Acquired hemophilia in the setting of bullous pemphigoid: A case report. J Hematol (2013) 2(2):74–5. doi: 10.4021/jh74w
27. Qiu X, Zhang G, Xiao R, Zhang J, Zhou Y, Li G, et al. Acquired hemophilia associated with bullous pemphigoid: a case report. Int J Clin Exp Pathol (2012) 5(1):102–4.
28. Zhang X, Guo J, Guo X, Pan J. Successful treatment of acquired haemophilia in a patient with bullous pemphigoid with single-dosing regimen of rituximab. Haemophilia. (2012) 18(5):e393–5. doi: 10.1111/j.1365-2516.2012.02917.x
29. Nguyen C, Gordon JS, Chang AL. A little known but potentially life-threatening association of bullous pemphigoid and acquired hemophilia: Case report and review of the literature. J Clin Exp Dermatol Res (2012) S6:1–3. doi: 10.4172/2155-9554.S6-003
30. Kluger N, Navarro R, Pallure V, Guillot B. [Bullous pemphigoid and acquired haemophilia]. Ann Dermatol Venereol (2011) 138(5):422–3. doi: 10.1016/j.annder.2011.01.040
31. Chen C, Chen Y, Ho J, Wu C. Bullous pemphigoid associated with acquired hemophilia. Dermatologica Sinica (2010) 28(4):173–6. doi: 10.1016/S1027-8117(10)60038-9
32. Maczek C, Thoma-Uszynski S, Schuler G, Hertl M. [Simultaneous onset of pemphigoid and factor VIII antibody hemophilia]. Hautarzt. (2002) 53(6):412–5. doi: 10.1007/s001050100278
33. Aoki Y, Miyake N, Yamasowa M, Inoue F, Takamatsu T, Mizumoto T, et al. A case of autoimmune hemolytic anemia and bullous pemphigoid-like skin lesion combined with idiopathic thrombocytopenic purpura. Rinsho Ketsueki (1990) 31(3):346–51.
34. Wang FJ, Liu HH, Kong XX, Wu MJ. Report on a bullous pemphigoid case manifested as eosinophilia dermatitis. Chin J Lepr Skin Dis (2017) 33:37–8.
35. Belgnaoui F, Idrissi M, Benyoussef K, Loudiye T, Bella A, Senouci K, et al. Idiopathic hypereosinophilic syndrome and bullous pemphigoid. Ann Dermatol Venereol (2002) 129(11):1291–4.
36. Bauduer F, Barteau A, Truchet S, Massot-Bordenave J, Ducout L. Bullous pemphigoid associated with the transformation of a preexisting myelodysplastic syndrome. Leuk Lymphoma. (1999) 32(3-4):399–400. doi: 10.3109/10428199909167405
37. Modiano P, Reichert S, Barbaud A, Bernard P, Weber M, Schmutz JL. Bullous pemphigoid in association with cutaneous lesions specific to a myelodysplastic syndrome. Br J Dermatol (1997) 136(3):402–5.
38. Ivars M, Hashimoto T, Ishii N, Bernad I, Lecumberri R, Espana A. Atypical bullous pemphigoid with extensive cutaneous and mucosal erosions associated with chronic lymphocytic leukemia. J Dermatol (2015) 42(11):1128–9. doi: 10.1111/1346-8138.13067
39. Kassim JM, Igali L, Levell NJ. A 14-year paraneoplastic rash: urticarial vasculitis and dermal binding bullous pemphigoid secondary to chronic lymphocytic leukaemia. Clin Exp Dermatol (2015) 40(4):391–4. doi: 10.1111/ced.12553
40. Kakurai M, Demitsu T, Iida E, Umemoto N, Yamada T, Yoneda K, et al. Coexistence of paraneoplastic pemphigus and bullous pemphigoid. J Eur Acad Dermatol Venereol (2009) 23(8):962–4. doi: 10.1111/j.1468-3083.2008.03071.x
41. Saouli Z, Papadopoulos A, Kaiafa G, Girtovitis F, Kontoninas Z. A new approach on bullous pemphigoid therapy. Ann Oncol (2008) 19(4):825–6. doi: 10.1093/annonc/mdn046
42. Ameen M, Pembroke AC, Black MM, Russell-Jones R. Eosinophilic spongiosis in association with bullous pemphigoid and chronic lymphocytic leukaemia. Br J Dermatol (2000) 143(2):421–4. doi: 10.1046/j.1365-2133.2000.03674.x
43. Misery L, Cambazard F, Rimokh R, Ghohestani R, Magaud JP, Gaudillere A, et al. Bullous pemphigoid associated with chronic b-cell lymphatic leukaemia: the anti-230-kDa autoantibody is not synthesized by leukaemic cells. Br J Dermatol (1999) 141(1):155–7. doi: 10.1046/j.1365-2133.1999.02940.x
44. Su WP, Oursler JR, Muller SA. Paraneoplastic pemphigus: a case with high titer of circulating anti-basement membrane zone autoantibodies. J Am Acad Dermatol (1994) 30(5 Pt 2):841–4. doi: 10.1016/s0190-9622(94)70093-1
45. Goodnough LT, Muir WA. Bullous pemphigoid as a manifestation of chronic lymphocytic leukemia. Arch Intern Med (1980) 140(11):1526–7. doi: 10.1001/archinte.1980.00330220077028
46. Cuni LJ, Grunwald H, Rosner F. Bullous pemphigoid in chronic lymphocytic leukemia with the demonstration of antibasement membrane antibodies. Am J Med (1974) 57(6):987–92. doi: 10.1016/0002-9343(74)90179-x
47. Godaert L, Bartholet S, Colas S, Kanagaratnam L, Fanon JL, Drame M. Acquired hemophilia a in aged people: A systematic review of case reports and case series. Semin Hematol (2018) 55(4):197–201. doi: 10.1053/j.seminhematol.2018.02.004
48. Windyga J, Baran B, Odnoczko E, Buczma A, Drews K, Laudanski P, et al. Treatment guidelines for acquired hemophilia a. Ginekol Pol (2019) 90(6):353–64. doi: 10.5603/GP.2019.0063
49. Patel RS, Harman KE, Nichols C, Burd RM, Pavord S. Acquired haemophilia heralded by bleeding into the oral mucosa in a patient with bullous pemphigoid, rheumatoid arthritis, and vitiligo. Postgrad Med J (2006) 82(963):e3. doi: 10.1136/pgmj.2005.036483
50. Zuo WL, Zhang GS, Qing ZJ, Xu YX, Qin LX, Xu M. Identification of IgG subclass and FVIII binding epitope of an acquired FVIII inhibitor in a bullous pemphigoid patient. Zhonghua Xue Ye Xue Za Zhi (2006) 27(9):593–7. doi: 10.3760/cma.j.issn.0253-2727.2006.09.006
51. Marzano AV, Tedeschi A, Fanoni D, Bonanni E, Venegoni L, Berti E, et al. Activation of blood coagulation in bullous pemphigoid: role of eosinophils, and local and systemic implications. Br J Dermatol (2009) 160(2):266–72. doi: 10.1111/j.1365-2133.2008.08880.x
52. Lin L, Hwang BJ, Culton DA, Li N, Burette S, Koller BH, et al. Eosinophils mediate tissue injury in the autoimmune skin disease bullous pemphigoid. J Invest Dermatol (2018) 138(5):1032–43. doi: 10.1016/j.jid.2017.11.031
53. Eisenbarth GS, Gottlieb PA. Autoimmune polyendocrine syndromes. N Engl J Med (2004) 350(20):2068–79. doi: 10.1056/NEJMra030158
54. Mangin MA, Lienhart A, Gouraud A, Roux S, Hodique F, Jouen F, et al. Onset of acquired haemophilia a after omalizumab treatment in severe bullous pemphigoid - a report on two cases successfully treated with mycophenolate mofetil. Ann Dermatol Venereol (2021) 148(1):57–9. doi: 10.1016/j.annder.2020.09.577
55. Sourdeau E, Clauser S, Prud’Homme R, Bardet V, Calmette L. Acquired hemophilia a associated with bullous pemphigoid and multiple myeloma: a case report. Ann Biol Clin (Paris) (2019) 77(2):179–83. doi: 10.1684/abc.2018.1405
56. Sugiyama S, Tanaka R, Hayashi H, Izumi K, Nishie W, Aoyama Y. Acquired haemophilia a in DPP4 inhibitor-induced bullous pemphigoid as immune reconstitution syndrome. Acta Derm Venereol (2020) 100(13):adv00178. doi: 10.2340/00015555-3539
57. Kruse-Jarres R, Kempton CL, Baudo F, Collins PW, Knoebl P, Leissinger CA, et al. Acquired hemophilia a: Updated review of evidence and treatment guidance. Am J Hematol (2017) 92(7):695–705. doi: 10.1002/ajh.24777
58. Collins PW, Hirsch S, Baglin TP, Dolan G, Hanley J, Makris M, et al. Acquired hemophilia a in the united kingdom: a 2-year national surveillance study by the united kingdom haemophilia centre doctors’ organisation. Blood. (2007) 109(5):1870–7. doi: 10.1182/blood-2006-06-029850
59. Muhammed N, Korgaonkar S, Pradhan V, Khopkar US. A cross-sectional study to correlate disease severity in bullous pemphigoid patients with serum levels of autoantibodies against BP180 and BP230. Indian Dermatol Online J (2021) 12(5):696–700. doi: 10.4103/idoj.IDOJ_813_20
60. Lambert MP, Gernsheimer TB. Clinical updates in adult immune thrombocytopenia. Blood. (2017) 129(21):2829–35. doi: 10.1182/blood-2017-03-754119
61. Bakchoul T, Marini I. Drug-associated thrombocytopenia. Hematol Am Soc Hematol Educ Program (2018) 2018(1):576–83. doi: 10.1182/asheducation-2018.1.576
62. Noh HR, Magpantay GG. Hypereosinophilic syndrome. Allergy Asthma Proc (2017) 38(1):78–81. doi: 10.2500/aap.2017.38.3995
63. Kahn JE, Girszyn N, Bletry O. Diagnosis of non-parasitic hypereosinophilia. Presse Med (2006) 35(1 Pt 2):144–52. doi: 10.1016/s0755-4982(06)74537-7
64. Muller AM, Martens UM, Hofmann SC, Bruckner-Tuderman L, Mertelsmann R, Lubbert M. Imatinib mesylate as a novel treatment option for hypereosinophilic syndrome: two case reports and a comprehensive review of the literature. Ann Hematol (2006) 85(1):1–16. doi: 10.1007/s00277-005-1084-7
65. Kridin K. Peripheral eosinophilia in bullous pemphigoid: prevalence and influence on the clinical manifestation. Br J Dermatol (2018) 179(5):1141–7. doi: 10.1111/bjd.16679
66. Verraes S, Hornebeck W, Polette M, Borradori L, Bernard P. Respective contribution of neutrophil elastase and matrix metalloproteinase 9 in the degradation of BP180 (type XVII collagen) in human bullous pemphigoid. J Invest Dermatol (2001) 117(5):1091–6. doi: 10.1046/j.0022-202x.2001.01521.x
67. Amber KT, Valdebran M, Kridin K, Grando SA. The role of eosinophils in bullous pemphigoid: A developing model of eosinophil pathogenicity in mucocutaneous disease. Front Med (2018) 5:201. doi: 10.3389/fmed.2018.00201
68. Wakugawa M, Nakamura K, Hino H, Toyama K, Hattori N, Okochi H, et al. Elevated levels of eotaxin and interleukin-5 in blister fluid of bullous pemphigoid: correlation with tissue eosinophilia. Br J Dermatol (2000) 143(1):112–6. doi: 10.1046/j.1365-2133.2000.03599.x
69. Saniklidou AH, Tighe PJ, Fairclough LC, Todd I. IgE autoantibodies and their association with the disease activity and phenotype in bullous pemphigoid: a systematic review. Arch Dermatol Res (2018) 310(1):11–28. doi: 10.1007/s00403-017-1789-1
71. Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol (2007) 8(4):345–50. doi: 10.1038/ni0407-345
72. de Latour RP, Visconte V, Takaku T, Wu C, Erie AJ, Sarcon AK, et al. Th17 immune responses contribute to the pathophysiology of aplastic anemia. Blood. (2010) 116(20):4175–84. doi: 10.1182/blood-2010-01-266098
73. Arakawa M, Dainichi T, Ishii N, Hamada T, Karashima T, Nakama T, et al. Lesional Th17 cells and regulatory T cells in bullous pemphigoid. Exp Dermatol (2011) 20(12):1022–4. doi: 10.1111/j.1600-0625.2011.01378.x
74. Hasserjian RP. Myelodysplastic syndrome updated. Pathobiology. (2019) 86(1):7–13. doi: 10.1159/000489702
75. Green KJ, Virata ML, Elgart GW, Stanley JR, Parry DA. Comparative structural analysis of desmoplakin, bullous pemphigoid antigen and plectin: members of a new gene family involved in organization of intermediate filaments. Int J Biol Macromol (1992) 14(3):145–53. doi: 10.1016/s0141-8130(05)80004-2
76. Venning VA, Wojnarowska F. The association of bullous pemphigoid and malignant disease: a case control study. Br J Dermatol (1990) 123(4):439–45. doi: 10.1111/j.1365-2133.1990.tb01447.x
77. Iranzo P, Lopez I, Robles MT, Mascaro JM Jr., Campo E, Herrero C. Bullous pemphigoid associated with mantle cell lymphoma. Arch Dermatol (2004) 140(12):1496–9. doi: 10.1001/archderm.140.12.1496
78. Jung M, Rice L. Unusual autoimmune nonhematologic complications in chronic lymphocytic leukemia. Clin Lymphoma Myeloma Leuk (2011) 11 Suppl 1:S10–3. doi: 10.1016/j.clml.2011.02.005
79. Maglie R, Genovese G, Solimani F, Guglielmo A, Pileri A, Portelli F, et al. Immune-mediated dermatoses in patients with haematological malignancies: A comprehensive review. Am J Clin Dermatol (2020) 21(6):833–54. doi: 10.1007/s40257-020-00553-9
80. Ong E, Goldacre R, Hoang U, Sinclair R, Goldacre M. Associations between bullous pemphigoid and primary malignant cancers: an English national record linkage study, 1999-2011. Arch Dermatol Res (2014) 306(1):75–80. doi: 10.1007/s00403-013-1399-5
81. Maglie R, Ugolini F, De Logu F, Nassini R, Simi S, Nardiello P, et al. Overexpression of helper T cell type 2-related molecules in the skin of patients with eosinophilic dermatosis of hematologic malignancy. J Am Acad Dermatol (2022) 87(4):761–70. doi: 10.1016/j.jaad.2021.07.007
82. Qiao J, Sun CE, Zhu W, Zhu D, Fang H. Flame figures associated with eosinophilic dermatosis of hematologic malignancy: is it possible to distinguish the condition from eosinophilic cellulitis in patients with hematoproliferative disease? Int J Clin Exp Pathol (2013) 6(8):1683–7.
83. Maglie R, Antiga E, Vannucchi M, Del Bianco E, Bianchi B, Massi D, et al. Bullous eruption in a patient with b-cell chronic lymphocytic leukemia: a diagnostic challenge. Int J Dermatol (2017) 56(12):1445–7. doi: 10.1111/ijd.13807
84. Ouedraogo E, Gottlieb J, de Masson A, Lepelletier C, Jachiet M, Salle de Chou C, et al. Risk factors for death and survival in paraneoplastic pemphigus associated with hematologic malignancies in adults. J Am Acad Dermatol (2019) 80(6):1544–9. doi: 10.1016/j.jaad.2018.03.043
85. Huang Y, Li J, Zhu X. Detection of anti-envoplakin and anti-periplakin autoantibodies by ELISA in patients with paraneoplastic pemphigus. Arch Dermatol Res (2009) 301(10):703–9. doi: 10.1007/s00403-008-0901-y
86. Avalos AM, Meyer-Wentrup F, Ploegh HL. B-cell receptor signaling in lymphoid malignancies and autoimmunity. Adv Immunol (2014) 123:1–49. doi: 10.1016/B978-0-12-800266-7.00004-2
87. Kato K, Koike K, Kobayashi C, Iijima S, Hashimoto T, Tsuchida M. Bullous pemphigoid after allogeneic hematopoietic stem cell transplantation. Pediatr Int (2015) 57(3):480–3. doi: 10.1111/ped.12561
88. Maverakis E, Goodarzi H, Wehrli LN, Ono Y, Garcia MS. The etiology of paraneoplastic autoimmunity. Clin Rev Allergy Immunol (2012) 42(2):135–44. doi: 10.1007/s12016-010-8248-5
89. De Matteis S, Molinari C, Abbati G, Rossi T, Napolitano R, Ghetti M, et al. Immunosuppressive treg cells acquire the phenotype of effector-T cells in chronic lymphocytic leukemia patients. J Transl Med (2018) 16(1):172. doi: 10.1186/s12967-018-1545-0
90. Balestri R, Magnano M, La Placa M, Patrizi A, Angileri L, Tengattini V, et al. Malignancies in bullous pemphigoid: A controversial association. J Dermatol (2016) 43(2):125–33. doi: 10.1111/1346-8138.13079
Keywords: bullous pemphigoid, hematological diseases, mucosal hematoma, eosinophil, corticosteroid
Citation: Yang Y, Zhao W, Yang N, Cui S, Jin H and Li L (2023) Associations between bullous pemphigoid and hematological diseases: Literature review on mechanistic connections and possible treatments. Front. Immunol. 14:1155181. doi: 10.3389/fimmu.2023.1155181
Received: 31 January 2023; Accepted: 22 February 2023;
Published: 08 March 2023.
Edited by:
Takashi Hashimoto, Osaka City University, JapanReviewed by:
Kentaro Izumi, Hokkaido University, JapanRoberto Maglie, University of Florence, Italy
Monika Bowszyc-Dmochowska, Poznan University of Medical Sciences, Poland
Copyright © 2023 Yang, Zhao, Yang, Cui, Jin and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Li, bGlsaXB1bWNoMjAwN0BzaW5hLmNvbQ==